

SUMMARY
The rich functional diversity of the nervous system is founded in the specific connectivity of the underlying neural circuitry. Neurons are often preprogrammed to respond to multiple axon guidance signals because they use sequential guideposts along their pathways, but this necessitates a strict spatiotemporal regulation of intracellular signaling to ensure the cues are detected in the correct order. We performed a mouse mutagenesis screen and identified the Rho GTPase antagonist p190RhoGAP as a critical regulator of motor axon guidance. Rather than acting as a compulsory signal relay, p190RhoGAP uses a nonconventional GAP-independent mode to transiently suppress attraction to Netrin-1 while motor axons exit the spinal cord. Once in the periphery, a subset of axons requires p190RhoGAP-mediated inhibition of Rho signaling to target specific muscles. Thus, the multifunctional activity of p190RhoGAP emerges from its modular design. Our findings reveal a cell-intrinsic gate that filters conflicting signals, establishing temporal windows of signal detection.
In Brief
Axon growth requires an enormous signaling matrix as the underlying code for building the nervous system. In a genetic screen, Bonanomi et al. identify p190RhoGAP as a critical regulatory switch that silences contextually inappropriate guidance signals for precise wiring of neuronal connections.
Graphical Abstract
INTRODUCTION
Cellular communication is controlled by signaling processes that are inherently context dependent and temporally regulated. An extreme example of signaling complexity is manifested in the way axon growth is directed. During embryonic development, neurons build intricate connectivity networks through combinatorial, synergistic, and sequential activation of a conserved but remarkably limited set of ligand-receptor systems (Bonanomi et al., 2012; Dudanova and Klein, 2013; Kolodkin and Tessier-Lavigne, 2011; Morales and Kania, 2017). Although elegant, this design strategy risks introducing signaling noise due to the temporal and spatial overlap of competing guidance signals. Nevertheless, the intrinsic filters that sort potentially conflicting instructions within neurons are poorly understood.
Axon trajectories are not simply defined by the complement of guidance systems within each neuronal population; rather, they also depend on tight control of receptor deployment through differential splicing, localized synthesis, trafficking, and proteolytic processing that enable neurons to adjust their sensitivity to directional cues at intermediate steps along their projection pathways (Bai and Pfaff, 2011; Jung et al., 2012; O’Donnell et al., 2009). Despite these multiple layers of control, growing axons are often equipped ahead of time with receptors that will be used at later steps of navigation, presumably to ensure timely activation of guidance responses (Dickson and Zou, 2010; Keleman et al., 2002). The risk associated with this preparedness is accidental or premature detection of contradictory instructions from opposing guidance signals, leading to axon targeting errors.
While many control mechanisms that operate at the receptor level have been documented, the degree to which intracellular components contribute to correct detection of guidance cues at specific times and places during pathfinding is less clear. A few signaling regulators preferentially associated with individual guidance receptors have been identified (Bashaw and Klein, 2010; O’Donnell et al., 2009); yet, for the most part the downstream nodes of axon guidance pathways appear to be relatively unspecialized compared to cell-surface receptors and ligands. Thus, a common view is that neuron-type-specific connectivity is primarily determined by the selective expression of receptor systems that impinge on a somewhat “generic” obligatory but non-instructive signal transduction machinery, shared among many cell types for the control of basic cell motility and chemotaxis. However, because there is remarkable conservation among the intrinsic components of signaling pathways it has been challenging to distinguish their roles as core effectors versus instructive regulators.
We conducted a mouse ENU (N-ethyl-N-nitrosourea) mutagenesis screen designed to identify instructive determinants of axon guidance in spinal motor neurons (Lewcock et al., 2007). The cell bodies of motor neurons reside in the ventral spinal cord, while their axons exit the CNS to connect with target muscles (Bonanomi, 2019; Jessell, 2000). From our forward genetic mouse screen, we isolated the Cassin mutant, in which separate classes of motor neurons make guidance errors at multiple choice-points for axon turning. The Cassin mutation was mapped to the Arhgap35 gene, which encodes the GTPase-activating protein p190RhoGAP, a negative regulator of RhoA family GTPases required for tissue morphogenesis with evolutionarily conserved roles in axon branching and fasciculation (Billuart et al., 2001; Brouns et al., 2000, 2001; Jeong et al., 2012). We show that p190RhoGAP prevents inappropriate activation of Netrin-DCC signaling when motor axons navigate out of the spinal cord. Despite the classification of p190 as a RhoGAP, gating of Netrin-DCC signaling was independent of the control of Rho GTPases. Our findings identify an intracellular signaling regulator that imposes a context-dependent switch upon the timing of guidance receptor activation to ensure axons ignore incongruous directional signals that would divert their normal path causing miswiring of neural circuits.
RESULTS
Cassin Mutants Exhibit Defects in Motor Axon Guidance
In order to identify recessive genes involved in the development of spinal motor neurons, we conducted a forward genetic screen in which mice expressing a motor neuron-specific farnesylated-GFP reporter (ISLMN::fGFP) were mutagenized with ENU, and the offspring were intercrossed to generate homozygous mutants (Lewcock et al., 2007). fGFP-labeled motor projections were visualized in whole embryo flat mounts, which were used to screen for defects in motor axon development (Figure 1A).
Figure 1. Cassin ENU Mutants Display Defects in Motor Axon Exit from the Spinal Cord.

(A–D) Whole mounts of ISLMN::fGFP+ mouse embryos (E12.5, dorsal views) show motor neuron cell bodies (MN) in the spinal cord (SC) and their axons exiting through the ventral roots (VR) in wild-type (A). Aberrant bundles of motor axons that fail to leave the spinal cord are visible in Cassin (B and C) and p190 mutants (D) (arrowheads). A, anterior; P, posterior; M, medial; L, lateral.
(E–H) ISLMN::fGFP+ motor neuron cell bodies (MN) and axons (arrowheads) in E11.5 transverse sections. Laminin-β1 (E and F) marks the spinal cord basal lamina (dashed line in G and H). Isl1/2 labels motor neurons and dorsal root ganglia (DRG). Motor axons select motor exit points in wild-type embryos (arrowheads in E and G) but form ectopic dorsally projecting bundles in Cassin mutants (arrowheads in F and H). ISLMN::fGFP signal is higher in medially positioned motor neurons.
(I–K) E12 spinal cord “open-book.” Bilateral columns of motor neuron somata (MN) separated by floor plate (FP), but no axons, are present in wild-types (I), whereas numerous intraspinal axon bundles spanning the intermediate (I) through dorsal (D) regions are detected in Cassin mutants (asterisks in J). (K) Ectopic intraspinal bundle (arrowheads) formed by convergence of misrouted axons in spinal open-book of Cassin embryo.
(L) Schematic of motor axon “exiting” phenotype of Cassin mutants.
(M) RNA-seq reads from spinal cord and adjacent tissues of E12.5 wild-type (blue trace) and Cassin (red trace) embryos aligned at the Arhgap35 locus (p190RhoGAP). Mammalian conservation track is reported. Exon coverage and levels of p190 mRNA are normal in mutants. RNA-seq did not detect global changes in gene expression or abnormal splicing in Cassin mutants.
(N) Schematic of p190RhoGAP and amino acid sequence alignment around isoleucine 602 (green) showing high evolutionary conservation and ENU-induced mutation I602N (red). p190 consists of a GTPase-like N-terminal domain competent for GTP binding, a “middle domain” with multiple protein-protein interaction motifs, and a C-terminal catalytic GAP domain.
(O) Genomic DNA sequencing identifies homozygous T > A substitution in p190 exon 1 of Cassin embryos, leading to I602N mutation.
(P) Decreased levels of p190RhoGAP in total lysates of E13.5 spinal cords of p190I602N/I602N (Cassin) mutant embryos compared to wild-type (+/+) and heterozygous (p190I602N/+) littermates. Levels of p120RasGAP, a known interactor of p190, are unchanged. ERK is a loading control.
(Q) Equal amounts of p190 were immunoprecipitated (IP) from spinal cord lysates used in (P). The levels of tyrosine-phosphorylated (P-Tyr) p190 and co-immunoprecipitated p120RasGAP detected by western blotting are reduced in p190I602N/I602N mutants.
(R) ncrease in GTP-bound RhoA (active form) pulled down from lysates of E13.5 p190I602N/I602N spinal cords compared to wild-type and heterozygous littermates (10.7- ± 2.2-fold increase p190I602N/I602N [n = 3] versus controls [n = 5]; mean ± SEM, Mann-Whitney U test p < 0.05).
Scale bars in (A), (B), and (D), 200 μm; in (C), 100 μm, in (F) and (G), 100 μm, in (H), 20 μm, and in (I)–(L), 50 μm. See also Figure S1.
To increase the probability of unmasking regulatory molecular switches of guidance pathways rather than structural or permissive components, we focused on mutants in which motor axons did not simply display abnormal morphogenesis (e.g., arrested growth, impaired branching) but were diverted from their normal trajectories. With these criteria, we identified a mutant line, named Cassin after the Italian mountaineering explorer, which displayed severe motor axon projection errors characterized by ectopic axon bundles at the pial surface (i.e., the boundary between the CNS and periphery) (Figures 1B, 1C, and S1A–S1F). Rather than leaving the spinal cord through the ventral root exit points, Cassin mutants formed aberrant motor fascicles that extended dorsally in close apposition to the limiting membrane of the spinal cord (Figures 1E–1H). Defects in motor axon exiting were highly penetrant, observed at every spinal cord level, and resulted in the corresponding depletion and disorganization of the ventral roots (Figures S1B, S1C, and S1F). In some instances, ectopic bundles grew in the interstitial space between the sensory dorsal root ganglia and the outer surface of the spinal cord, which is covered by meningeal membranes, but unlike EphA3/A4 mutants were never observed invading the sensory ganglia (Gallarda et al., 2008). Because motor axons were rerouted onto an ectopic path in Cassin embryos, we reasoned that the ENU mutation might have affected pathfinding rather than a more general aspect of axon growth.
Defects in motor projection were not accompanied by a change in total motor neuron number or cell body positioning (Figures 1E–1H and S1H–S1J). Cassin mutants were born at the expected Mendelian ratio but died soon after birth with cyanosis and respiratory failure. Although a recessive mutation, occasionally heterozygous Cassin mutants had small ectopic motor axon bundles, and a minority of homozygous mutant embryos (<2%) displayed neural tube closure defects and exencephaly (Figures S1E, S1G, and 2N).
Figure 2. p190 Is Required within Motor Neurons for Axon Pathfinding.

(A) Transgenic mouse crosses to test complementation of ENU allele p190I602N and p190 knockout allele.
(B–G) Whole-mount images of ISLMN::fGFP in E12.5 single (p190+/−) (B, D, and F) or compound heterozygous (p190I602N/−) (C, E, and G) embryos. Compound heterozygotes display ectopic motor axon bundles (arrowheads in C), aberrant “bridging” of intercostal nerves (arrowheads in E) and hindlimb nerve guidance errors (arrow in G) as found in both Cassin (p190I602N/I602N) and p190 homozygous mutants (see Figures 1B and 1D). Arrowheads in (G) point to ectopic bundles.
(H) Transgenic strategy for conditional expression of p190. Cre removes the poly(A)-STOP cassette allowing expression of RFP-tagged p190.
(I) Mouse crosses to test the requirement of p190 in motor neurons. Transgenic p190-RFP is driven by Olig2MN::Cre in ISLMN::fGFP+ motor neurons of p190 knockout embryos.
(J and K) Restricted expression of p190-RFP in motor neuron cell bodies (arrow) and axons (arrowhead) in transverse sections of Olig2MN::Cre-positive (K) but not in Cre-negative control embryos (E12.5) (J). SC, spinal cord; DRG, dorsal root ganglia.
(L and M) Whole-mount images of ISLMN::fGFP reporter (E12.5). Motor axon exiting defects of p190−/− embryos (arrowheads in L) are corrected by expression of p190-RFP in motor neurons (M).
(N) Quantification of ectopic ISLMN::fGFP+ motor axon bundles in embryo whole mounts. Mean ± SEM; Mann-Whitney U test, ***p < 0.001 p190I602N/I602N and p190−/− versus heterozygous; p190−/− versus p190−/−;Olig2MN::Cre;p190-RFPLSL; Tukey’s multiple comparisons test, ns, p > 0.05 p190I602N/− versus p190I602N/I602N and p190−/−. The number of analyzed embryos is reported in the Supplemental Information.
Scale bars in (B)–(M), 200 μm, and in (J) and (K), 50 μm. See also Figure S2.
To visualize the aberrant intraspinal motor axons at higher resolution, we imaged spinal cord “open-book” preparations (Figure S1K). While in wild-type embryos ISLMN::fGFP exclusively labeled the bilateral columns of motor neuron cell bodies, in Cassin mutants numerous ectopic GFP+ axons bundles were found throughout the intermediate region of the spinal cord (Figures 1I–1J). These intraspinal fascicles were formed by axons that converged within each spinal segment (Figures 1K, S1N, and S1O). Although the majority of misprojecting axons remained inside the spinal cord, some were found in the surrounding meninges (Figures S1L and S1M). Thus, a unique hallmark of motor neurons—their ability to project axons through the basal boundary of the neuroepithelium and into the periphery—is severely affected in Cassin mutants (Figure 1L). In addition, Cassin mutants displayed discrete defects in distal projection of subsets of motor neurons. Specifically, thoracic intercostal nerves formed by hypaxial motor neurons were defasciculated (Figures S1P–S1R), and the hindlimb-innervating peroneal nerve displayed an aberrant proximal branch resulting in thinning or ablation of the main nerve tract (Figures S1S–S1U). Taken together, the Cassin mutation affects both proximal (axon exiting) and distal (muscle targeting) pathfinding decisions but does not appear to disrupt axon growth per se.
Cassin Is a p190RhoGAP Mutant
To identify the Cassin mutation with single-base pair resolution, we performed deep sequencing of mRNAs extracted from the spinal cord and surrounding mesenchyme of mutant embryos and control littermates. SNP-discovery identified an A > T transversion in the first exon of the Arhgap35 gene that encodes the Rho GTPase-activating protein p190RhoGAP (hereafter p190). The novel SNP causes the amino acid substitution I602N, which alters a highly conserved residue and is predicted as “deleterious” by in silico tools (see STAR Methods) (Figures 1M–1O). All affected embryos, but none of the phenotypically normal littermates, were homozygous for the mutation.
Compared to wild-type embryos, p190 protein levels were strongly reduced in Cassin (p190I602N/I602N) mutants (Figure 1P). When equal amounts of p190 were immunoprecipitated from mutant and control embryonic spinal cords using limiting antibody, p190I602N had lower stoichiometry of tyrosine-phosphorylation and impaired association with the binding partner p120RasGAP—both are indications of altered signaling competence (Figure 1Q) (Bouton et al., 1991; Settleman et al., 1992b). p190 inhibits RhoA by promoting GTP hydrolysis and contributes to a substantial fraction of Rho-GAP activity in the cell (Settleman et al., 1992a; Vincent and Settleman, 1999). As expected, higher amounts of GTP-bound (active) RhoA were pulled down from the spinal cord of p190I602N/I602N embryos (Figure 1R). These results indicate that the Cassin mutation severely affects p190 protein levels and activity.
To confirm that the motor axon guidance defects observed in Cassin mutants were due to p190 loss of function, we crossed the ISLMN::fGFP reporter into p190 knockout mice (Brouns et al., 2000). Embryos with a targeted disruption of p190 exhibited motor axon guidance phenotypes similar to Cassin mutants, including failure to exit the spinal cord (Figure 1D), aberrant “bridging” of intercostal nerves (Figure S2K), and limb innervation defects (Figure S2M). As observed for Cassin mutants, p190 knockout mice died at birth, and a subset exhibited neural tube closure defects (Brouns et al., 2000).
Next, we crossed the Cassin and p190 knockout alleles to generate embryos heterozygous for both mutations. Compound Cassin and p190 hemizygotes displayed motor projection phenotypes virtually identical to homozygous mutants of the two alleles (Figures 2A–2G and 2N). Lack of complementation between the two recessive mutations demonstrates that the ENU mutant Cassin is a new loss-of-function allele of p190. These findings highlight the critical role of p190 for a discrete subset of motor axon guidance decisions. The specificity of these phenotypes is remarkable in light of the wholesale increase in RhoA activation observed in p190 mutants.
p190 Is Required in Motor Neurons to Control Axon Pathfinding
p190 is expressed ubiquitously, with enrichment in the developing CNS (Brouns et al., 2000) (Figures S2A and S2B). Despite the widespread distribution, the overall pattern of peripheral nerves comprising both sensory and motor axons appeared largely intact in mutant embryos (Figures S2C and S2D; data not shown). Thus, identification of the p190 mutant was likely only possible because of the motor neuron-specific GFP reporter employed in our screen.
The broad expression of p190 raised the question of whether the motor phenotypes observed in mutant mice reflected a requirement in motor neurons or in other cells that may indirectly influence motor axon targeting. To distinguish between these possibilities, we generated a transgenic mouse line (p190-RFPLSL) that enables Cre-dependent expression of RFP-tagged p190 upon removal of a “floxable” STOP cassette (Figure 2H). Olig2MN::Cre (Dessaud et al., 2007) was used to achieve restricted expression of p190-RFP in motor neurons at levels ~2.5-fold higher than endogenous p190 (Figures 2J, 2K, and S2E). Next, we combined p190-RFPLSL and Olig2MN::Cre with the p190 knockout allele and visualized motor projections in embryos that lack p190 in all tissues except motor neurons expressing exogenous p190-RFP (Figure 2I). Reestablishing p190 expression in motor neurons was sufficient to correct all motor axon guidance phenotypes of p190 mutants, including spinal cord exiting defects and abnormal projections of intercostal and limb nerves (Figures 2L–2N and S2F–S2N). Therefore, p190 functions intrinsically within motor neurons for proper axonal targeting.
The GAP Activity of p190 Is Dispensable for Motor Axon Exiting from the Spinal Cord
In Cassin mutants, p190 loss of function leads to an abnormal increase in active RhoA. To address whether the control of motor axon targeting by p190 was mediated by its inhibitory effect on Rho, we employed CRISPR/Cas-mediated gene targeting to generate mice harboring a point mutation in p190 that disrupts the catalytic arginine R1284 required for GAP activity (Li et al., 1997) (Figure 3A).
Figure 3. The GAP Activity of p190 Is Dispensable for Motor Axon Exiting but Required for Distal Pathfinding.

(A) Schematic of p190 and protein sequence alignment highlighting the catalytic arginine R1284. The targeted mutation R1284A generates GAP-inactive p190. Direct cDNA sequencing demonstrates CGG (Arg-1284) > GCA (Ala-1284) substitution. The synonymous C > T substitution in the third position of Tyr-1283 was introduced to facilitate annealing of allele-specific genotyping primers.
(B–G) Whole-mount images of ISLMN::fGFP+ p190+/− heterozygotes (controls; B, D, and F) or p190R1284/− compound heterozygous embryos (C, E, and G), carrying one copy of R1284A allele and one copy of the knockout allele. (B and C) Motor axon exiting defects are absent in p190R1284/− heterozygotes (C, arrowhead to a residual short ectopic bundle). p190R1284/− embryos display normal fasciculation of intercostal nerves (E) while the peroneal nerve in the hindlimb is severely stunted (arrow in G).
(H) Quantification of ectopic GFP+ motor axon bundles in embryo whole mounts. Mean ± SEM; Mann-Whitney U test, ***p < 0.001 p190−/− versus p190R1284/−; ns, p > 0.05 p190+/− versus p190R1284/−. p190R1284A/− embryos only display rare short ectopic bundles, similarly to p190+/− controls.
(I) Incidence of peroneal nerve defect in p190R1284/− mutants and p190R1284/+ controls with color-coded phenotypic classes: normal, green; moderate thinning, blue; severely stunted, red.
Scale bars, 200 μm. See also Figure S3.
The p190R1284A variant lacking GAP activity was expressed at levels comparable to the wild-type protein (Figure S3A). Embryos expressing exclusively p190R1284A (p190R1284A/−) were compared to p190+/− controls (see STAR Methods). As expected, loss of GAP activity resulted in a substantial increase in GTP-bound (active) RhoA in p190R1284A embryo spinal cords (Figure S3B). Strikingly, motor axon exiting and intercostal nerves were normal in p190R1284A mutants, whereas the peroneal nerve in the hindlimb was severely affected (Figures 3B–3I). We conclude that distinct motor axon pathways display differential dependence on the GAP activity of p190. These results argue for a non-canonical function of p190 in mediating motor axon exiting from the spinal cord.
p190 contains additional signaling motifs besides the GAP domain, including an N-terminal GTP-binding domain (Foster et al., 1994). Knockin mice containing a point mutation (S36N) that impairs GTP binding display all three distinctive phenotypes of p190 mutants (defective axon exiting, intercostal bridging, and peroneal nerve mistargeting), albeit with reduced expressivity (Figures S3C, S3D, and S3G–S3K, cf. Figures 2N and 7P). Although p190S36N transcript levels were comparable to those of the wild-type allele, p190S36N protein levels were lower (Figures S3E and S3F). Because p190S36N is a hypomorphic mutant, it remains unclear whether GTP binding is required per se for motor pathfinding. Nevertheless, these results provide evidence that motor axons are sensitive to lowering of p190 levels beyond a critical threshold.
Figure 7. Rescue of Motor Exiting Defects by Inactivation of Netrin-1/DCC.

(A–E) Whole mount of ISLMN::fGFP+ embryos (E12.5, dorsal views). Numerous ectopic intraspinal motor axon bundles are detected in p190 homozygous mutants (B, arrowheads) but are nearly absent in p190−/−;Netrin-1−/− double-knockout embryos (C) and p190 mutants carrying a single (D, p190−/−;DCC+/−) or two (E, p190−/−;DCC−/−) targeted DCC knockout alleles. (A) Control embryo (DCC+/−).
(F) Quantification of ectopic ISLMN::fGFP+ motor axon bundles. Motor axon exiting defects observed in p190 mutants depend on Netrin-1/DCC signaling. The phenotype is not rescued by Unc5C deletion. Mean ± SEM; ns, p > 0.05, ***p < 0.001 one-way ANOVA, Dunnet’s multiple comparisons test versus p190−/−.
(G) Schematic of the phenotypic rescue by either Netrin-1 or DCC deletion in p190 mutants.
(H–K) Intercostal nerves in ISLMN::fGFP+ embryo whole mounts (E12.5, ventral views). Aberrant nerve “bridging” found in p190 mutants (arrowheads in I) is still present in p190/Netrin-1 (J) and p190/DCC (K) double knockouts (arrowheads). (H) Control embryo (p190+/−).
(L–O) Limb-innervating peroneal nerve in ISLMN::fGFP+ embryo whole mounts (E12.5, dorsal views). The peroneal nerve projection phenotype detected in p190 mutants (arrowheads in M) is not rescued in p190/Netrin-1 (N) and p190/DCC (O) double knockouts (arrowheads). (L) Control embryo (p190+/−).
(P) Incidence of intercostal and limb nerve targeting defects with color-coded phenotypic classes: normal, green; moderate, blue; severe, red. Both phenotypes are detected with comparable expressivity in p190−/−, p190−/−;Netrin-1−/− and p190−/−;DCC+/− mutants.
Scale bars in (A)–(E), 100 μm, and in (H)–(O), 200 μm. See also Figure S7.
p190 Controls Motor Axon Exiting Independently of Cxcr4
The proximal projection phenotype of p190 mutants was reminiscent of the defects observed in knockout embryos for the chemokine receptor Cxcr4 (Lieberam et al., 2005). This similarity led us to hypothesize that p190 might mediate Cxcr4 signaling required for motor axon exiting. To study epistatic interactions, we generated an allelic series of compound p190/Cxcr4 mutants and visualized motor projections with ISLMN::fGFP (Figures 4A–4H and S4E–S4I). Cxcr4−/− embryos displayed ectopic motor axon fascicles at the spinal cord interface, which were more irregular and abundant compared to p190 knockouts, possibly reflecting guidance errors immediately past the exit points (Figures 4F, 4I, and S4A–S4C) (Lieberam et al., 2005). The number of ectopic axon bundles was markedly increased in embryos harboring a combination of p190 and Cxcr4 mutant alleles compared to single mutants (Figure 4I). Strikingly, with the removal of one or two p190 alleles from Cxcr4 knockouts (p190+/−;Cxcr4−/− or p190−/−;Cxcr4−/−) the vast majority of motor axons failed to reach the periphery (Figures 4G–4K′, S4D, and S4G–S4I).
Figure 4. p190 and Cxcr4 Control Motor Axon Exiting via Parallel Synergistic Pathways.

(A–H) Whole-mount images of ISLMN::fGFP in allelic series of p190 and Cxcr4 mutant embryos (E12.5, dorsal views): (A) p190+/−; (B) Cxcr4+/−; (C) p190+/−; Cxcr4+/−; (D) p190−/−; (E) p190−/−; Cxcr4+/−; (F) Cxcr4−/−.; (G) p190+/−; Cxcr4−/−; (H) p190−/−; Cxcr4−/−. Arrowheads point to ectopic axon bundles. Asterisks mark axial nerves, which are progressively depleted with the worsening of motor axon exiting phenotype in compound p190;Cxcr4 mutants.
(I) Quantification of ectopic ISLMN::fGFP+ motor axon bundles in embryo whole mounts. Mean ± SEM; Tukey’s multiple comparisons test, ***p < 0.001 p190+/−; Cxcr4+/− versus p190+/− and Cxcr4+/−; Dunnet’s multiple comparisons test, *p < 0.05 p190+/− versus p190−/−;Cxcr4+/; ***p < 0.001 p190−/− versus p190−/−; Cxcr4−/−; Cxcr4−/− versus p190+/−; Cxcr4−/− and p190−/−; Cxcr4−/−.
(J–K′) Transverse sections of p190;Cxcr4 double mutant (K and K′) and control (J and J′) embryos (E12.5) stained for motor neuron marker Isl1/2 (blue) and pan-axonal marker bIII-tubulin (red). Arrowheads point to aberrant motor bundles (K and K′).
(L–O) Neurospheres of ISLMN::fGFP+ wild-type (L and M) or p190−/− (N and O) ESCMN exhibit increased axonal growth after treatment with Cxcl12 for 15 h. (P) Quantification of axon outgrowth. Cxcl12-induced axon outgrowth in p190−/− ESCMN is comparable to wild-type (p > 0.05). Mean ± SEM, n = 18–23 neurospheres per condition. Mann-Whitney U test, ***p < 0.001 treated versus untreated.
Scale bars in (A)–(H), 200 μm, in (J)–(K′), 100 μm, and in (L)–(O), 200 μm. See also Figure S4.
To further examine the relationship between p190 and Cxcr4 signaling, we used in vitro assays. Since the transient expression of Cxcr4 in motor neurons makes the use of primary cells impractical (Lieberam et al., 2005), we differentiated motor neurons from either wild-type or p190 mouse embryonic stem cells (ESCMN). ESCMN expressed Cxcr4 and activated signaling in response to Cxcl12 (Figure S4J; data not shown). Cxcl12 treatment enhanced motor axon outgrowth in both wild-type and p190-null ESCMN neurospheres to a comparable extent (Figures 4L–4P). In addition, chemokine signaling was normal in p190−/− ESCMN (Figure S4J), and the levels of tyrosine-phosphorylated p190 did not change after Cxcl12 stimulation (Figure S4K). Thus, Cxcl12/Cxcr4 signaling promotes motor axon growth independently of p190. Counter to our initial hypothesis, these results indicate that p190 and Cxcr4 function in parallel pathways that synergize to instruct motor axon projection from the spinal cord (Figure S4L).
p190 Interacts with Netrin Receptor DCC
p190 has been linked to semaphorin signaling in other systems, and mouse mutants with impaired Sema3A and/or Sema3F activity display intercostal nerve defasciculation similar to p190 mutants (Barberis et al., 2005; Huber et al., 2005; Jeong et al., 2012). However, we found that motor axon exiting was normal in either Sema3A or Sema3F mutant embryos. In addition, the severity of the p190−/− phenotype was not significantly altered in p190/Sema3A and p190/Sema3F double knockouts, indicating that these semaphorins are dispensable for motor axon exiting and are not implicated in the defects observed in p190 mutants (Figure S4M)
In order to gain insight into the signaling pathways that require p190 to control motor axon guidance, motor neurons isolated from p190−/− embryos were gene profiled using RNA sequencing (RNA-seq). This analysis did not reveal gene expression changes that could underlie defects in motor neuron development (Figure S5A). Next, p190 was immunoprecipitated from the ventral spinal cord of e12.5 embryos, and the associated proteins were identified by tandem mass spectrometry. Bioinformatic analysis of p190-interacting proteins revealed enrichment in regulators of cell morphogenesis, cytoskeletal organization, and axon guidance (Figure S5B). Of particular interest was the identification of components of Netrin signaling, a conserved guidance pathway that influences motor neuron projection (Poliak et al., 2015; Serafini et al., 1994). Moreover, the prominent p190 interactor p120RasGAP functions downstream of the Netrin receptor DCC in cortical neurons (Antoine-Bertrand et al., 2016). These findings prompted us to examine whether p190 participates in Netrin-1/DCC signaling and whether this guidance system is required for motor axon exiting.
DCC was detected on motor axons emerging from ventral exit points (Figures 5A and 5B) (Keino-Masu et al., 1996). At the same embryonic stages, Netrin-1 transcript was abundant in the ventricular zone, floor plate, and the paraxial mesenchyme (Figure 5C) (Serafini et al., 1996), whereas Netrin-1 protein accumulated on the laminin-rich pial surface of the spinal cord (Figures 5D and 5F) (Dominici et al., 2017; Varadarajan et al., 2017). In p190 mutant embryos, ectopic motor axon bundles extend dorsally along the pial surface encountering high levels of Netrin-1 (Figures 5E and 5G).
Figure 5. p190 Associates with Netrin-1 Receptor DCC.

(A and B) DCC (red) on Hb9MN::GFP+ motor axons extending from the spinal cord (arrowheads) in E10 (A) and E10.5 (B) embryos.
(C) Netrin-1 mRNA in the ventricular zone (VZ), floor plate (FP), and somites (S) in E10.5 transverse section.
(D and E) Netrin-1 (red) protein accumulation on the pial surface (arrowheads in D) in E11.5 ISLMN::fGFP+ embryos. Ectopic motor axon bundles in p190−/− extend dorsally in close association with Netrin-1+ basal membrane (arrowhead in E).
(F and G) Laminin-β1 staining (red) on the spinal cord basal membrane in E11.5 transverse sections of control (F) and p190−/− (G) embryos. Motor axons grow aberrantly along the laminin-β1+ (red) pial-surface in p190 mutants (arrowheads in G).
(H) DCC is detected in p190 immunocomplexes. Protein lysates of E12.5 ventral spinal cords were subjected to immunoprecipitation (IP) with anti-p190 antibody or species-matched IgG control and analyzed by western blotting with antibodies to p190 and DCC. </p/> (I) p190 is detected in DCC immunocomplexes. DCC was immunoprecipitated with an antibody to the extracellular domain from protein lysates of either control(wild-type or p190+/−) or p190−/− E12.5 ventral spinal cords. Immunocomplexes were probed with antibodies to p190 and DCC.
(J) Schematic of DCC receptor showing three conserved motifs (P1, P2, P3) in the cytoplasmic domain. Truncations of the cytoplasmic domain and deletions of the P2 motif and P2-P3 linker sequence (L2–3) were introduced in DCC fused to extracellular FLAG tag. The ability of each DCC mutant to associate (+) or not (−) with p190 is reported.
(K) Immunoprecipitation of full-length DCC (FL) and deletion mutants with anti-FLAG antibody in AD293 cells followed by immunoblot for endogenous p190 (asterisk) and transfected p190-RFP (arrowhead). Deletion of the C-terminal region encompassing the P2 and P3 motifs (DCC 1–1327) abolishes p190 interaction.
(L) Detection of endogenous p190 (asterisk) and transfected p190-RFP (arrowhead) in DCC immunocomplexes from AD293 cells treated with Netrin-1 for the indicated times. Total lysates were probed with anti-phospho-AKT antibody to monitor the intracellular signaling response to Netrin-1. p190 pull-down is increased moderately at the peak of Netrin-1 stimulation (1.78- ± 0.17-fold 5 min versus untreated, mean ± SEM, p < 0.05 paired t test, n = 3).
(M–O) p190-RFP (M) and DCC (N) colocalize (arrowheads) in the axonal growth cone of primary mouse motor neurons expressing the MN(218−2)::GFP reporter (blue in O) (Pearson’s co-localization coefficient: 0.82 ± 0.015 [mean ± SEM]; n = 15 growth cones). Transgenic p190-RFP is driven in motor neurons by Olig2MN::Cre.
(P–R′) Close association between DCC (endogenous) and p190-RFP (driven by Olig2MN::Cre) revealed by proximity ligation assay (PLA, red) in MN(218−2)::GFP+ motor axons before (P and P′) and after stimulation with Netrin-1 (Q and Q′). Background signal is negligible in controls in which the anti-DCC antibody is omitted (R and R′).
(S) Quantification of PLA signal. Mean ± SEM; n, fields of view encompassing comparable GFP+ axon area. Untreated (17), Netrin-treated (16); Mann-Whitney U test, ns, p > 0.05.
Scale bars in (A), 25 μm, in (B)–(D), 100 μm, in (E)–(G), 50 μm, and in (M)–(R′), 10 μm. See also Figure S5 and Tables S1 and S2.
DCC was pulled down with p190 from ventral spinal cord lysates (Figure 5H), and conversely p190 was present in DCC immunocomplexes (Figure 5I). The intracellular domain of DCC contains evolutionary conserved motifs (P1, P2, and P3) that mediate protein interactions critical for signaling (Lai Wing Sun et al., 2011). A series of deletion mutants spanning the intracellular domain of DCC were co-transfected with RFP-tagged p190 in AD293 cells to assess the competence of each truncated variant to pull down p190 (either endogenous and/or exogenous) (Figures 5J, 5K, and S5C). Deletion of the P3 motif (DCC 1–1421) did not affect interaction with p190, whereas a truncation that eliminates the P2+P3 motifs (DCC 1–1327) abolished binding. However, p190 association with P2 deletion mutants was partially restored in the presence of the P3 sequence, indicating that p190 binds to the C-terminal region of DCC encompassing the P2-P3 motifs. The interaction between p190 and DCC was detected in basal conditions, with a moderate increase after Netrin-1 stimulation (Figure 5L).
Next, we investigated the relative distribution of p190 and DCC in motor axons. Primary motor neurons were cultured from Olig2MN::Cre; p190-RFPLSL embryos carrying the MN(218–2)::GFP reporter (Amin et al., 2015) to achieve restricted expression of p190-RFP in GFP+ motor neurons. DCC and p190-RFP colocalized in the axonal growth cones before and after Netrin-1 treatment (Figures 5M–5O; data not shown). In transfected COS7 cells, p190 predominantly localized to the plasma membrane when co-expressed with wild-type DCC, but not with a truncated DCC variant lacking the P2-P3 domains (Figures S5D–S5E″). A proximity ligation assay (PLA), which probes close association (<30–40 nm) between protein pairs (Söderberg et al., 2006), revealed fluorescent puncta corresponding to sites of p190/DCC interaction in growth cones and axons in basal conditions. PLA puncta did not change significantly in number after Netrin-1 treatment, although they became mostly associated with the distal region of the growth cone (Figures 5P–5S).
Because targeting of DCC to the plasma membrane is influenced by Rho signaling (Moore et al., 2008), we examined whether the surface levels of the receptor changed in motor neurons lacking p190. Staining for surface-associated DCC did not reveal differences between p190−/− and control motor axons in the presence or absence of Netrin-1 (Figures S5F–S5J). In addition, no changes in total DCC protein levels were detected in p190 mutant embryos (Figure 5I; data not shown). These results reveal a constitutive physical interaction between p190 and conserved intracellular motifs of DCC.
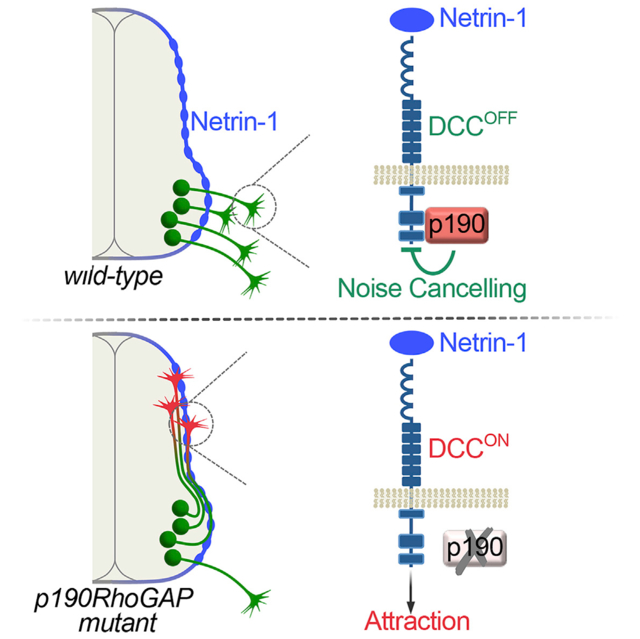
p190 Silences Netrin-1 Attraction
In p190 mutant embryos, aberrant motor axons project in close apposition to the Netrin-rich pial surface (Figure 5E). This observation prompted us to examine how p190 mutant axons respond to Netrin-1. Motor explants from p190 mutants or controls (a mix of heterozygous and wild-type embryos) harboring the Hb9MN::GFP transgene were cultured in 3D matrices next to cell aggregates releasing Netrin-1, and GFP+ motor axons extending on the side facing toward (proximal) and away (distal) from the aggregates were quantified (Figure 6A). Consistent with the limited sensitivity of motor neurons to Netrin-1 at early developmental stages (Bai et al., 2011; Varela-Echavarría et al., 1997), Netrin-1 failed to promote axon outgrowth from control explants (Figures 6B–6D). In contrast, p190−/− explants gained responsiveness to Netrin-1, leading to a marked increase in axonal outgrowth toward the point source (Figures 6B, 6E, and 6F). Next, we sought to mimic the distribution of Netrin-1 at the spinal cord interface by culturing motor explants on Netrin-coated surfaces (Figure 6G). Control explants exhibited negligible axon growth on either control (immunoglobulin G [IgG]-Fc) or Netrin-1 substrates (Figures 6H–6J), whereas outgrowth of p190−/− motor neurons on Netrin-1 was robust (Figures 6H, 6K, and 6L).
Figure 6. p190 Curbs Axon Attraction to Netrin-1.

(A) Schematic of 3D-motor explant attraction assay. Hb9MN::GFP+ explants are co-cultured with cell aggregates in collagen-Matrigel matrices for ~15 h, which is insufficient time for motor axon outgrowth unless cell aggregates express a chemoattractant. The proximal (P) and distal (D) sides of the explant correspond to the two halves of the ventral spinal cord, containing the left and right motor columns (MC) and the floor plate (FP) in between.
(B) Quantification of directional axon growth (GFP+ pixels on the P versus D sides of the explant). p190−/− motor axons are attracted to Netrin-1. Mean ± SEM, n explants: control AP-Fc (11); control Netrin-1 (49); p190−/−/AP-Fc (18); p190−/−/AP-Fc (54). Mann-Whitney U test, ns, p > 0.05; ***p < 0.001 Netrin-1 versus AP-Fc control.
(C–F) Motor axon growth is minimal in the presence of aggregates releasing control AP-Fc (C and E), or in control explants exposed to Netrin-1 (D). In contrast, numerous GFP+ motor axons extend from the P side in p190−/− explants (F).
(G) Schematic of 2D motor axon growth assay. Hb9MN::GFP+ motor neuron explants are cultured on surfaces coated with laminin and either recombinant Netrin-1 or control IgG-Fc.
(H) Quantification of GFP+ motor axons on Netrin-1 relative to control IgG-Fc. Control explants are insensitive to Netrin-1, while p190−/− explants show increased outgrowth. Mean ± SEM, n explants: control IgG (23); control Netrin-1 (47); p190−/−/IgG (22); p190−/−/Netrin-1 (38). Mann-Whitney U test, ns, p > 0.05; ***p < 0.001 Netrin-treated versus IgG control.
(I–L) p190−/− explants (L) display enhanced motor axon outgrowth on Netrin-1 substrate compared to control explants (J). Axon outgrowth is minimal on IgG-Fc substrate (I and K).
(M) Quantification of GFP+ motor axons. Mean ± SEM, n explants: p190−/−/IgG (7); p190−/−/Netrin-1 (13); p190R1284/−/IgG (7); p190R1284/−/Netrin-1 (9). Mann-Whitney U test, ns, p > 0.05; ***p < 0.001 Netrin-treated versus IgG control.
(N–Q) ISLMN::fGFP+ motor explants grown on either Netrin-1 (O and Q) or control IgG-Fc substrates (N and P). p190−/− motor axons display enhanced outgrowth on Netrin-1+ (O) while axon growth is minimal for p190R1284/− explants (Q).
Scale bars, 200 μm. See also Figure S6.
We conclude that p190 normally attenuates Netrin-1 signaling in motor neurons; conversely, axons lacking p190 acquire a premature attraction to Netrin-1. The unmasking of a positive axon outgrowth response to Netrin-1 in p190 mutants was unexpected because the elevated levels of active-RhoA in neurons lacking p190 is viewed as inhibitory for cytoskeletal growth in conventional signaling schema (Hall and Lalli, 2010; Moore et al., 2008). However, motor explants derived from p190R1284A embryos lacking GAP activity did not exhibit enhanced outgrowth on a Netrin-coated substrate, unlike p190−/− explants, indicating that p190 silences Netrin-1 attraction in motor neurons through a non-canonical signaling mode independent of its GAP activity (Figures 6M–6Q).
Midline Axon Guidance Is Normal in p190 Mutants
Commissural neurons in the dorsal spinal cord depend on Netrin-1 to reach the midline (Serafini et al., 1996). Therefore, abnormal responsiveness of these cells to Netrin-1 in p190 mutants might affect their intraspinal course. However, the trajectory and area occupied by precrossing commissural axons was similar in p190−/− and control embryos, indicating that p190 is not required for proper fasciculation and pathfinding of this class of neurons (Figures S6A–S6C).
Defective DCC receptor processing leads to enhanced attraction to Netrin-1, causing aberrant motor axon growth into the midline floor plate (Bai et al., 2011). Surprisingly, despite the increase in Netrin-1 responses revealed by our in vitro assays, we did not detect inappropriate motor axon growth toward the midline in any of the available p190 mutants (ENU-induced or targeted alleles) (Figures S6D and S6E; data not shown). We reasoned that p190-deficient motor neurons might still be sensitive to midline-derived repulsive cues. Indeed, both p190−/− and control motor explants cultured next to wild-type floor plate tissue extended significantly more axons away from the floor plate explant, demonstrating that they are equally repelled (Figures S6F–S6I).
Genetic Inactivation of Netrin-1/DCC Signaling Rescues Motor Axon Exiting Defects
Our explant assays indicate that p190 suppresses Netrin-1 attraction in motor neurons. To gain insight into the silencing mechanism, we examined whether p190 influenced the composition of DCC receptor complexes. DCC was immunoprecipitated from lysates of either wild-type or p190−/− ESCMN and the associated proteins were identified by tandem mass spectrometry. DCC interactomes revealed distinct changes in p190 mutants (Figure S7A). Of note, many of the proteins that became associated with DCC in p190-null cells were involved in cytoskeletal remodeling, GTPase activity, G-protein-coupled receptor signaling, and vesicle trafficking, whereas those detected only in wild-type cells were linked to transcription and translation, two known outputs of DCC signaling (Neuhaus-Follini and Bashaw, 2015; Tcherkezian et al., 2010) (Figure S7B). These results indicate that p190 affects the configuration of DCC receptor complexes and might compete with signaling effectors for binding.
We reasoned that abnormal attraction to Netrin-1 might contribute to the motor pathfinding defects observed in p190 mutants. To test this possibility, we crossed p190 knockout mice to Netrin-1 mutants to generate double-mutant embryos. We found that the number of ectopic motor axon bundles at the spinal cord interface was markedly reduced after inactivation of Netrin-1 (Figures 7A–7C and 7F). To determine which Netrin-1 receptor system within motor neurons was responsible for the phenotypic rescue, p190 mutants were crossed to mice harboring targeted deletions of either DCC, which mediates chemoattraction, or Unc5C, which mediates repulsion. Ectopic intraspinal motor axon fascicles were nearly absent in p190/DCC double-knockout embryos but were still present in p190/Unc5C compound mutants (Figures 7E, 7F, S7C, and S7D). Dependency of the motor exit phenotype on DCC signaling is further demonstrated by the significant reduction in aberrant axon bundles in p190 mutants after deletion of a single allele of DCC (p190−/−;DCC+/−) (Figures 7D and 7F). Therefore, inhibition of Netrin-1/DCC signaling corrects the motor axon exiting phenotype of p190 mutants (Figure 7G). Nevertheless, the distinctive intercostal and limb nerve targeting phenotypes of p190−/− embryos were still present with comparable penetrance and expressivity in p190/Netrin and p190/DCC double mutants—as well as in p190/Unc5C knockouts (Figures 7H–7P, S7E, and S7F). Of note, none of the motor guidance phenotypes observed in p190 mutants were present in Netrin-1, DCC, or Unc5C mutants alone, indicating these signaling systems are normally not involved (Figure 7F; data not shown). However, Netrin-1/DCC signaling becomes inappropriately activated in the absence of p190, causing intraspinal projection errors. Interestingly, distal motor pathfinding defects that arise from lack of p190 were independent of Netrin-1.
DISCUSSION
The intricate connectivity of the nervous system is based on a very elaborate temporal and spatial distribution of guidance cues that are arrayed at high density within the pathways traversed by multiple classes of growing axons. Our studies reveal that neurons are confronted with a “noisy” environment that they must navigate through, and that their signaling pathways contain control features that permit rapid and decisive responsiveness to the relevant instructive cues while simultaneously filtering out potentially detrimental cues that would lead to misdirected axon targeting. Using genetic screens, we show that the Rho GTPase regulator p190RhoGAP acts as a context-dependent filter that interacts with DCC and suppresses detection of Netrin-1 as motor axons leave the spinal cord. In the absence of p190, motor axons become strongly attracted to Netrin-1 deposited on the pial surface of the spinal cord and are diverted to grow inappropriately along this structure instead of extending into the periphery. At more distal pathfinding steps, p190 functions independently of Netrin-1/DCC to control the projection of genetically defined motor neuron subclasses that target intercostal and limb muscles (Figure 8).
Figure 8. Model for Temporal and Context-Dependent Control of Motor Axon Guidance by p190.

(A) (Left) p190 suppresses Netrin-1/DCC attractive signaling in a GAP-independent manner and collaborates with Cxcl12/Cxcr4 pathway to control motor axon exiting from the spinal cord. Netrin-1 deposited on the pial surface of the spinal cord is a substrate for dorsal commissural neurons (dCN) that project to the midline but must be ignored by motor axons that select ventral exit points in response to Cxcl12/Cxcr4 chemoattraction. (Right) DCC attractive signaling is turned-on in p190 mutants, causing inappropriate motor axon attraction to pial-associated Netrin-1 that results in intra-spinal axon growth. Cxcr4 is unable to compensate for derepression of DCC signaling. (B) (Left) In the periphery, motor axon targeting to limb muscles depends on suppression of Rho signaling by p190-GAP activity. DCC signaling is switched on enabling axons to respond to target-derived Netrin-1 (Poliak et al., 2015). (Right) Rho signaling is abnormally enhanced in the absence of p190, causing mistargeting of motor neurons that innervate the dorsal limb muscles (LMCL). Motor axons that innervate ventral muscles (LMCM) are unaffected.
The selective phenotypes we observed were unexpected given the widespread tissue distribution of p190 and the potential pleiotropic effects of RhoA inhibition. Despite this, axon guidance defects were rescued by selective re-expression of p190 in motor neurons. Of the ~70 GAPs—and as many Rho-activating GEFs (Guanine nucleotide Exchange Factors)—several have been found to link individual cell surface receptors to Rho signaling for the control of axon targeting and synapse formation (Bashaw and Klein, 2010; Hall and Lalli, 2010). Our findings extend these observations in two important ways. First, we show that sequential steps of motor axon targeting are dependent upon p190. Second, using structure-function mapping, we found that the temporally distinct activities associated with p190 depend on different domains: GAP activity is required for limb navigation, whereas axon-exiting from the spinal cord relies upon a nonconventional signaling mode independent of Rho inhibition (Figure 8). Thus, the modular organization of p190 contributes to its functionally expanded role in axon targeting.
p190RhoGAP Controls Motor Axon Exiting from the Spinal Cord
Motor neurons are the only class of neurons in the CNS to send axons across the border of the neuroepithelium to establish the essential connection between the brain and peripheral organs (Bonanomi and Pfaff, 2010). How this fundamental developmental step is controlled has remained incompletely understood, but it has been shown to require axonal Cxcr4 and its cognate ligand Cxcl12 expressed by tissues surrounding the neural tube (Lieberam et al., 2005). Because of the apparent similarities in proximal motor projection phenotypes between p190 and Cxcr4 mutants, we hypothesized that p190 might act as a Cxcr4 effector in motor neurons. However, studies in compound mouse mutants demonstrate that the two genes have non-redundant roles in motor axon exiting (Figure 8A). In addition, while the exiting phenotype of p190 mutants is rescued by deletion of either Netrin-1 or DCC, the projection defects of Cxcr4 mutants are independent of Netrin-1 (Figure S7G), further indicating that Cxcr4 and p190 use different mechanisms to control motor axon outgrowth from the spinal cord. Therefore, motor axon growth into the periphery is most likely based on p190 and Cxcr4 signal-pathway integration.
Although Netrins are secreted molecules, they bind avidly to the extracellular matrix and can function as short-range adhesive cues (Akin and Zipursky, 2016; Brankatschk and Dickson, 2006; Kennedy et al., 1994; Serafini et al., 1994). Netrin-1 derived from neural progenitors accumulates on the pial surface creating an adhesive substrate that directs ventral projection of commissural neurons by haptotaxis (Dominici et al., 2017; Moreno-Bravo et al., 2019; Varadarajan et al., 2017; Wu et al., 2019). Therefore, Netrin-1 localized at the lateral margins of the spinal cord is a critical growth-promoting substrate for commissural neurons but represents a “mock” guidance signal for DCC+ motor neurons that need to pierce through the basement membrane to reach peripheral targets. Our findings are consistent with the model that motor axons use an intrinsic silencing mechanism centered on p190 interaction with DCC to avoid attraction to pial-associated Netrin-1, which would prevent them from leaving the spinal cord. When Netrin-1/DCC signaling is inappropriately activated in p190 mutants, motor axons select an aberrant path along the margin of the neuroepithelium (Figure 8A). Thus, the defining feature of motor neuron connectivity—the projection from the CNS—depends on precise temporal coordination of both acquisition of Cxcl12/Cxcr4 chemoattraction and suppression of Netrin-1 attraction mediated by p190 silencing of DCC (Figure 8A).
Silencing of Netrin-1 Signaling by p190RhoGAP
Motor neurons that select ventral exit points at spinal and hind-brain levels are insensitive to Netrin-1 despite expression of DCC and Unc5 receptors (Bai et al., 2011; Colamarino and Tessier-Lavigne, 1995; Varela-Echavarría et al., 1997). In contrast, motor axons lacking p190 gain aberrant attraction to Netrin-1. We found that p190 is not required to transmit Netrin-1 signals per se but rather suppresses attraction of early-extending motor axons to Netrin-1 through an unconventional GAP-independent process. Our results indicate that p190 binds to intracellular motifs of DCC that are known docking sites for signaling effectors and that the composition of DCC signaling complexes is influenced by p190. These findings are consistent with a model whereby p190 competes with Netrin-1 effectors for binding to DCC, thus maintaining the receptor in a poised non-signaling state.
p190 might regulate motor axon pathfinding by impinging on adhesive interactions mediated by Netrin-DCC, either directly or through crosstalk with cell-adhesion complexes (Akin and Zipursky, 2016; Brankatschk and Dickson, 2006; Moore et al., 2009). Interestingly, laminin was shown to convert Netrin attraction to repulsion in retinal axons (Höpker et al., 1999) and p190A functions downstream of laminin-receptor integrins (Billuart et al., 2001; Nakahara et al., 1998). It is possible that p190 mediates the inhibitory activity of laminin on Netrin attraction, thus enabling motor axons to project through the pial surface, where the two ligands are co-expressed. Alternatively, p190 might control Netrin responsiveness at the level of DCC recyclingtrafficking (Lai Wing Sun et al., 2011; Moore et al., 2008), although we show that surface levels of the receptor are unchanged in motor axons lacking p190.
Silencing of Netrin-DCC by p190 needs to be dynamically regulated to enable motor axons to respond to Netrin-1 once they reach the periphery. Indeed, Netrin-1 is expressed in several motor neuron targets and influences limb innervation (Poliak et al., 2015). Gating of Netrin-1 responsiveness through p190 may be controlled by intrinsic time-dependent changes in key regulatory molecule(s) (Shewan et al., 2002) or derepression of DCC by peripheral signals.
Motor neurons employ at least two separate silencing mechanisms to ensure that Netrin-1/DCC signaling remains latent until axons have left the spinal cord. In addition to p190-mediated silencing, the Presenilin-dependent interaction between the Slit-Robo and Netrin-DCC receptor systems suppresses Netrin-1 responses in early-extending motor axons (Bai et al., 2011; Stein and Tessier-Lavigne, 2001). Surprisingly, while disruption of Robo-mediated silencing leads to aberrant attraction to the Netrin-1+ floor plate (Bai et al., 2011), motor axons in p190 mutants were repelled away from the floor plate. The different phenotypic outcomes might arise from a temporal separation between the two DCC-silencing modalities. For example, Robo could act early to influence the initial orientation of motor axons, whereas p190 might function at a later step when axons have reached their exit points. Alternatively, the elevated levels of active-RhoA in p190 mutants may enhance the inhibitory growth effect of repulsive cues located within the floor plate, thereby preventing motor axons from entering the midline despite the presence of Netrin-1.
Context-Dependent Suppression of Incongruous Guidance Signals
Multiple neuronal classes establish axonal trajectories concomitantly during embryonic development, often crossing paths and sharing navigational guideposts. This overlap raises the question of how neurons filter out directional signals that are irrelevant for their own class-specific projection but act as potent cues that guide the connectivity of other neuronal subtypes. Far from being inconsequential, inappropriate detection of spurious cues generates “noise” that interferes with proper axon targeting. One level of “noise cancellation” is at the level of regulated receptor expression. However, many classes of projection neurons use sequential guideposts and are equipped to respond to multiple signals, thereby necessitating the use of post-translational mechanisms that include hierarchical receptor interactions and trafficking (Bashaw and Klein, 2010; Dickson and Zou, 2010). In this study, we propose that Netrin-1 deposited on the basal lamina—a substrate for commissural neurons—becomes a barrier for DCC-expressing motor axons if they are not made refractory to this mock cue by p190, which clamps Netrin-1 signaling in a dormant state.
While axon guidance is traditionally viewed as the result of productive responses to instructive signals, our findings suggest that equally important for brain wiring is the suppression of irrelevant signals that axons would be otherwise competent to detect. Although energetically expensive, the use of repressor modules to gain control over biological systems kept in a silent but poised state is widespread and contributes to the robustness, fast activation, and reversibility of cellular processes (Lim et al., 2015). This regulatory logic offers clear advantages for neuronal wiring, enabling axons to operate in a noisy molecular environment, filtering out unwanted signals and rapidly switching responsiveness to instructive cues at sequential choice points during pathfinding. We expect similar discriminatory mechanisms govern cell behavior in a variety of settings where detection of potent but potentially detrimental stimuli must be turned off to prevent erroneous activation of pathological states (Sever and Brugge, 2015).
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Samuel Pfaff (pfaff@salk.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse lines
ISLMN::fGFP (Jax stock# 017952); Olig2::Cre (Dessaud et al., 2007); SE1MN::GFP (Shirasaki et al., 2006); Hb9MN::GFP (Lee et al., 2004); MN(218−2)::GFP (Amin et al., 2015); p190RhoGAP−/− (Jax stock# 020997); Cxcr4−/− (Jax stock# 004341); Netrin-1−/− (Serafini et al., 1996); DCC−/− (MMRRC stock# 030626); Unc5C−/− (Burgess et al., 2006); Sema3A−/− (Jax stock# 014646); Sema3F−/− (Jax stock# 006710).
p190-RFPLSL transgenic mice: monomeric Cherry cDNA was added to the 5′ of N-terminally HA-tagged p190A (Rat, M94721) (Hu and Settleman, 1997). The Cherry-HA-p190A cDNA was inserted in a modified pCAGEN plasmid, under the CAGGS promoter, and preceded by a 3× poly-A STOP cassette flanked by LoxP sites. A 3′ WPRE-SV40 poly(A) cassette was added to improve transgene expression. The linearized transgenic construct, with backbone removed, was diluted to 2 ng/ml in RNase free microinjection buffer (10 mM Tris-HCl pH 7.4, 0.2 mM EDTA) and 1–2 pl were injected into the male pronucleus of B6D2F1 zygotes (Jackson Labs). Microinjected embryos were transferred into the oviducts of 0.5 dpc pseudopregnant CD1 females (Charles River Laboratories). Microinjections were performed at the Salk Transgenic Core Facility.
p190R1284A knockin mice were generated with CRISPR/Cas9-mediated homologous recombination of a 200bp single-stranded oligodeoxynucleotide (ssODN) containing the R1284A point mutation. A guide RNA targeting exon-4 of Arhgap35 (5′-AAGCACT GAAGGCATCTACC) was designed using https://zlab.bio/guide-design-resources to minimize the possibility of off-target effects and were transcribed in vitro with T7 Quick High Yield RNA Synthesis Kit (New England Biolabs Cat# E2050S). The ssODN had 98bp homology arms around the CGG > GCA (R1284A) mutation which disrupts the PAM sequence, and contained a synonymous C > T change to facilitate annealing of genotyping primer (see Figure 3). Freshly prepared microinjection mixture containing 5 ng/μl Cas9 mRNA (Thermo Scientific Cat# A29378):2.5 ng/μl guide-RNA:10 ng/μl ssODN (PAGE-purified Ultramer, IDT) in microinjection buffer was injected into the cytoplasm and pronucleus of B6D2F1 zygotes (~2–3 pl per embryo), which were reimplanted into CD1 recipient females. Founders were screened by PCR genotyping and confirmed by direct Sanger sequencing. Hemizygous founders were bred for > 4 generations with ISLMN::fGFP and wild-type CB6F1 mice to obtain p190R1284A/+;ISLMN::fGFP heterozygous males that were used in timed matings with p190+/−;ISLMN::fGFP females to generate compound heterozygous embryos for the analysis. Most p190+/R1284A heterozygous were born runt but recovered normal weight past weaning age. Males were fertile but the majority of females displayed genital malformations that prevented breeding. For this reason, the study was conducted on compound heterozygous p190R1284A/− embryos obtained by crossing p190+/R1284A males to p190+/− females.
p190S36N knockin mice were generated with CRISPR/Cas9-mediated ssODN homologous recombination with guide RNA 5′-AG AAAGGCCAGTGCGGCATT targeting exon-2 of Arhgap35. The ssODN had 101–96bp homology arms around the TCT > AAT (S36N) mutation. Three additional synonymous nucleotide changes were introduced to destroy PAM sequence (Gly-34 G > A), facilitate annealing of allele-specific genotyping primers (Lys-35 G > A) and abolish a BglII site (Gly-32 C > A) for genotyping purporses (Figure S3). Hemizygous founders were bred for > 4 generations with ISLMN::fGFP and wild-type CB6F1 mice to generate p190S36N/+;ISLMN::fGFP heterozygous males that were used in timed matings with either p190+/−;ISLMN::fGFP females, to obtain compound heterozygous embryos, or with p190S36N/+;ISLMN::fGFP females to obtain homozygous mutant embryos.
All mice were immune competent, SFP free, handled and housed in accordance with IACUC and AAALAC guidelines of the Salk Institute for Biological Studies and Ospedale San Raffaele. All mouse lines were maintained by crossing to CB6F1 mice. Embryos were generated by timed matings, and the day on of vaginal plug was designated as embryonic day 0.5 (E0.5).
Cell lines
Immortalized cell lines were maintained at 37C with 5% CO2 in DMEM supplemented with 10% FBS, 1% L-Glutamine, 1% Penicillin/Streptomycin (GIBCO/Thermo Scientific). AD293 cells are a derivative of the HEK293 cell line (Stratagene Cat# 240085). Cos7 (Cat# CRL-1651) and HeLa (Cat# CCL-2) were from the American Type Culture Collection (ATCC).
Mouse ES cell lines were derived from p190−/−; ISLMN::fGFP and control ISLMN::fGFP mouse blastocysts 3.5 days post-fertilization as described (Sternfeld et al., 2017). ES cells were differentiated into neurospheres (embryoid bodies) containing ~40%–50% GFP+ motor neurons using established protocols (Sternfeld et al., 2017; Wichterle et al., 2002). To test responsiveness to Cxcl12 (Figure 4), embryoid bodies from 3 distinct p190−/− ESC lines and 2 wild-type lines were cultured for 15 h on coverslips coated with PDL/laminin (3 mg/ml) in MN media with or without 100ng/ml recombinant Cxcl12/SDF1 (R&D Systems Cat# 460-SD-010).
Primary motor neuron cultures
Motor neuron explants dissected from E12.5 mouse embryos were maintained in motor neuron (MN) media: Neurobasal media containing B27 supplement (GIBCO/Thermo Scientific), 2mM L-Glutamine (GIBCO/Thermo Scientific), 1% Penicillin/Streptomycin (GIBCO/Thermo Scientific), 50 μM Glutamic Acid (Sigma-Aldrich). See Method Details for specific procedures.
METHODS DETAILS
ENU screen and mapping of Cassin mutant
The design of the ENU-mutagenesis screen was previously described (Lewcock et al., 2007). Briefly, fifty DBA/2J mouse males (Jackson Labs) were given intraperitoneal injections of 100 mg/kg ENU (Sigma-Aldrich) once a week for 3 consecutive weeks starting at 8 weeks of age. Eight to ten weeks after injection, animals regained fertility and were mated to ISLMN::fGFP mice (maintained on CB6F1/J background, an F1 hybrid between BALB/cJ and C57BL/6J strains). Eighty-eight GFP+ F1 males were generated and used in the screen. Six to eight G3 litters derived from each F1 male were examined. Embryos at E12.5 were fixed for 2 h in 4% paraformaldehyde in PBS, transferred to PBS overnight, then eviscerated and cleared by consecutive 2–4 h-incubations (4°C) in 30%, 50%, 80% glycerol (in PBS) and flat-mounted between coverslips for visualization of GFP under a fluorescence stereomicroscope. A panel of 40 MIT markers was used for the initial mapping of ENU mutations (Lewcock et al., 2007). After the Cassin mutation was localized to chromosome 7, SNP markers were used to further refine the position. The selected SNP markers were different between DBA/2J and either C57/Bl6 or BALB/c strains. After > 6 generations of backcrosses, the genomic region containing the mutation was restricted to a ~5Mb segment between dbSNps rs3687759 (12.9Mbp) and rs13479144 (18.3Mbp) that contains 128 annotated genes.
Individual libraries for mRNA sequencing were prepared from the brachial spinal cord region and surrounding mesenchymal tissue from three G6 mutant embryos (E12.5), three unaffected (i.e., control) littermates, and one stage-matched DBA/2J embryo using Illumina mRNA-Seq Sample Prep Kit (RS-100–0801). Genome Analyzer II (Illumina) was used to perform 2×36 cycles of single-end sequencing.
Reads were aligned to the reference genome with MOSAIK software to detect SNPs. To identify the ENU-associated base-change, the SNPs detected in chromosome 7 in Cassin mutants were filtered to remove (1) reference SNPs (from Single Nucleotide Polymorphism database – dbSNP), (2) SNPs found in CB6F1/J control littermate samples, (3) SNPs found in the DBA/2J sample. The A > T transversion at 16,563,294 Mbp of chromosome 7 (GRCm38/mm10 Assembly) in Arhgap35 exon-1 was the only novel homozygous SNPs detected in all reads aligned at that location (n = 66) and it was found in all Arhgap35 transcripts from Cassin embryos but never in the parental mouse strain. The resulting I602N mutation in p190RhoGAP is predicted as “deleterious” by PROVEAN (score: −6; cutoff for neutral variants: −2.5) and SIFT (score: 0.00; cutoff for deleterious variants:0.1).
Purification and RNA-sequencing of primary motor neurons
Ventral spinal cord regions micro-dissected from E12.5 p190−/− embryos or control littermates (wild-type or p190+/−) were pulled and dissociated with papain (Worthington Biochemical Cat# LK003153) according to the manufacturer’s instructions. After 10 min rotation at 37°C, the tissue samples were mechanically triturated and centrifuged at 1000 rpm for 5 min. Dissociated cells were resuspended in Neurobasal media (without phenol red) with 2% Horse Serum, passed through a 35μm cell strainer (BD Falcon Cat# 08–771-23) and sorted on a Becton Dickinson FACS Vantage SE. DiVa using Coherent Sapphire 488 nm solid state laser. Cells were collected directly into RLT lysis buffer containing β-mercaptoethanol (QIAGEN). RNA was isolated using the RNeasy Mini Kit (QIAGEN) with on-column DNase digestion (QIAGEN RNase-Free DNase Set Cat# 79254). Each sample contained between 100,000 and 200,000 GFP+ motor neurons isolated from 2–5 spinal cords during a single experimental session. Five p190−/− and five control RNA samples were quantified with Agilent 2200 TapeStation system prior to preparation of mRNA-sequencing libraries (50 bp single-end) using the Illumina TruSeq RNA Library Preparation Kit (v2) according to the manufacturer’s instructions. Libraries were sequenced with Illumina HiSeq 2500 platform at the Salk NGS Core.
DNA constructs
A FLAG-tag was inserted between the signal sequence and the start codon of rat DCC cDNA (NM_012841.1) in a modified pN1-CMV-polyA vector (Clontech). A series of DCC mutant constructs with intracellular deletions were generated from this template vector using In-Fusion HD cloning (Clontech). Cherry-HA-p190 (referred as p190-RFP) was generated as described above and cloned in a modified pN1-CMV-polyA vector.
Explant assays
For 2D axon outgrowth assay, explants prepared from individually dissected motor columns at lumbar level were cultured for 15–18 h in MN media on Poly-D-Lysine (PDL)-coverslips (BD Biosciences Cat #354086) coated with 5 mg/ml laminin (GIBCO/Thermo Scientific Cat# 23017015) and 5 μg/ml of either recombinant Netrin-1 (R&D Systems Cat# 1109N1025) or control IgG-Fc (Jackson ImmunoResearch Cat# NC9712967).
For 3D axon attraction assay, explants containing both motor columns with floor plate in between were embedded ventral side down in a 1:1 mix of rat tail collagen (Corning Cat# 354236) and Matrigel (Corning Cat# 356234) for ~15 h in MN media next to aggregates of AD293 cells transfected with expression vectors encoding either Netrin-1 or control Alkaline Phosphatase-Fc using Lipofectamine 2000 (Invitrogen/Thermo Scientific Cat# 11668027). Alternatively, motor explants were co-cultured for ~24 h in MN media supplemented with 1ng/ml GDNF (R&D Systems Cat# 212-GD-010) next to floor plate dissected from wild-type embryo littermates.
Immunostaining and in situ Hybridization
For immunostaining and PLA assay, motor explants were cultured for ~15 h on PDL/lamininhigh (30–50 μg/ml)-coated coverslips in MN media with 1ng/ml GDNF. When indicated, explants were stimulated with 1 μg/ml Netrin-1 for 10 min prior to fixation in PBS containing 4% PFA and 4% sucrose for 20 min at RT. Since none of the p190RhoGAP antibodies tested was suitable for immunostaining (i.e., non-specific signal was detected in p190−/− neurons), the subcellular distribution of p190 was examined in primary motor neurons derived from Olig2MN::Cre; p190-RFPLSL embryos in which p190 double-tagged with RFP and HA is selectively expressed in these cells (Figures 5M–5R′). For PLA (Duolink, Sigma-Aldrich), explants prepared from Olig2MN::Cre;p190-RFPLSL; MN(218−2)::GFP embryos (E12.5) were fixed, washed twice in PBS, pre-blocked with 1% BSA/2% Donkey Serum/0.1% Tx-100 in PBS for 15 min, incubated with mouse anti-HA (1:1000; HA.11 Covance Cat# MMS-101R) to HA-tagged p190-RFP and goat anti-DCC (1:300; R&D Systems Cat# AF844) for 45 min (RT) in the same blocking buffer, followed by PLA probes according to the manufacturer’s instructions. For detection of surface-associated DCC, after fixation, explants were incubated with goat anti-DCC antibody to the extracellular domain (R&D Systems Cat# AF844) in blocking buffer without detergent for 30 min (RT) followed by Alexa Fluor conjugated secondary antibodies (1:750, Invitrogen/Thermo Scientific) for 30 min (RT) in the same buffer.
For cryosectioning, embryos were fixed in 4% paraformaldehyde diluted in PBS for 2–3 h (4°C), washed extensively in PBS, cryoprotected in 30% sucrose for 2–4 h at 4°C, and embedded in OTC compound. Cryosections (30–40 μm) were incubated with primary (ON, 4°C) and secondary antibodies (2–3 h, RT) in PBS containing 1% BSA and 0.2% Tx-100.
Whole mount immunohistochemistry of e12.5 embryos (Figures S2C and S2D) was performed with anti-neurofilament 2H3 supernatant (Developmental Hybridoma Bank, 1:50).
Stained cells and tissue sections were mounted with Vectashield (Vector Labs) or PermaFluor (Thermo Scientific Scientific).
Primary antibodies used for immunostaining were:
rabbit anti-Isl1/2 [1:2,500] (Ericson et al., 1992), rat Laminin β1 [1:1000; Chemicon/Millipore Cat# MAB1928], rabbit anti-RFP [1:3000; MBL International Cat# PM005], mouse anti-β3 tubulin (1:2000; Novus Biologicals Cat# NB120–11314), goat anti-Netrin-1 [1:2000; R&D Systems Cat# AF1109; after heat-induced antigen retrieval with 10mM citrate buffer pH 6, for 1–2 min], goat anti-DCC [1:1000 for IHC; R&D Systems Cat# AF844], goat anti-Robo3 [1:2000; R&D Systems Cat# AF3076], guinea pig anti-FoxP1 [1:10,000] (Dasen et al., 2008), rabbit anti-GFP [1:2000; Invitrogen/Thermo Scientific Cat# A6455]. Secondary antibodies were: goat or donkey anti-rabbit/mouse/goat/guinea pig Alexa Fluor 488-, 555-, 647-conjugated (1:1000; Invitrogen/Thermo Scientific).
In situ hybridization probes were: mouse p190A (NM_172739; 1156–1837bp) cloned in pCR2.1TOPO (Invitrogen/Thermo Scientific), mouse Netrin-1 (NM_008744.2; 1823–2680bp) cloned in pCRII-Topo (Invitrogen/Thermo Scientific).
Microscopes
Fluorescence-assisted microdissections were performed with a Zeiss Lumar V12 microscope. The same instrument was used for imaging neurofilament staining in embryo whole-mounts. Representative images of the ISLMN::fGFP+ embryo flat-mounts shown in Figures S1A–S1C were acquired with a Prairie Technologies Ultima two-photon microscope. Other images were acquired with an Olympus Fluoview 1000 confocal microscope. All images correspond to maximal Z-projections of confocal stacks.
Immunoprecipitation and western blotting
For p190A pull-down from Cassin E13.5 spinal cords (Figure 1Q), freshly dissected individual spinal cords were homogenized immediately using 10–15 strokes of a microcentrifuge tube pestle in cold modified RIPA buffer (150mM NaCl, 50mM TrisHCl pH7.4, 10mM EDTA, 1% NP40, 0.5% Sodium Deoxycholate) supplemented with protease (Roche Applied Science Cat# 11836170001) and phosphatase (Thermo Scientific Cat# 78420) inhibitor cocktails, followed by 1 h on a nutator mixer at 4°C. After clarification by centrifugation at 13,000 ×g for 15 min, 500 μg of total protein lysate diluted in ~250 μL of lysis buffer were immunoprecipitated with of rabbit anti-p190A antibody (1:200; Cell Signaling Tech. Cat# 2860) ON (4°C), followed by incubation with 40 mL of protein A/G bead slurry (Santa Cruz Biotech. Cat# sc-2003) for 2 h (4°C). The beads were washed 4 times with 0.5% NP-40 lysis buffer without Sodium Deoxycholate (Wash Buffer) and resuspended in NuPAGE LDS sample buffer (Invitrogen/Thermo Scientific). Precipitates were subjected to western blotting with NuPAGE 4%–12% Bis-Tris gels (Invitrogen/Thermo Scientific) and probed with the indicated antibodies. To detect tyrosine-phosphorylated p190A, the membranes were first incubated with mouse anti-phospo Tyr antibody (1:10,000; clone 4G10, Millipore Cat# 05–321). After removal of primary/secondary antibodies with stripping buffer (Thermo Scientific Cat# 21059), the filters were incubated with rabbit anti-p190A antibody (1:2000; Cell Signaling Tech. Cat# 2860). Western blot for “Total Lysates” was performed with 15–30 μg of total protein per sample.
The levels of active RhoA in Cassin (Figure 1R) or p190AR1284A (Figure S3B) embryo spinal cord were assessed by pull-down of GTP-bound RhoA with Rhotekin-Rho binding domain beads followed by probing with mouse anti-RhoA antibody (Rho Activation Assay Kit, Millipore Cat# 17–294). 500 mg of E12.5 spinal cord homogenates diluted in 500 μL of lysis buffer were incubated with 20 μL of Rhotekin-RBD beads and processed according to the manufacturer’s instructions.
To assess activation of Cxcl12 signaling (Figure S4K), HeLa cells were starved for 8 h prior to stimulation with 100ng/ml recombinant Cxcl12/SDF1 for the indicated times and lysed with rotation for 1 h at 4°C in modified RIPA buffer. After clarification by centrifugation, 500 μg of total protein lysates diluted in 250 μL of buffer were incubated ON (4°C) with rabbit anti-p190A antibody (1:200; Cell Signaling Tech.), followed by incubation with 40 μL of protein A/G beads for 2 h (4°C).
ESCMN embryoid bodies at 7 days of in vitro differentiation were starved ON in MN media before treatment with 100ng/ml Cxcl12/SDF1 (Figure S4J), lysed by sonication in RIPA buffer (140 mM NaCl, 10 mM Tris-Cl pH 7.4, 1 mM EDTA, 1% Triton X-100, 0.1% SDS), cleared by centrifugation 15,000 ×g for 10 min and resuspended in NuPAGE LDS sample buffer.
To quantify the levels of endogenous and transgenic p190A in Olig2MN::Cre; p190-RFPLSL E12.5 embryos (Figure S2E), the dorsal and ventral spinal cord regions were dissected from transgenic and control (Cre-negative) embryo littermates, lysed by sonication in RIPA buffer, cleared by centrifugation and resuspended in sample buffer. Membranes were probed with rabbit anti-RFP antibody (1:2000; MBL) after stripping of rabbit anti-190 antibody (Cell Signaling Tech.).
Affinity purification of protein complexes for Mass Spectrometry (MS) was carried out with M-270 Epoxy magnetic beads (Invitrogen/Thermo Scientific Cat# 14301) based on previous protocols with modifications (Cristea et al., 2005). Two independent experiments were conducted for IP-MS analysis of p190 interacting proteins in the ventral embryonic spinal cord (Figure S5B). For each experiment, 25 mg of M-270 Epoxy magnetic beads were conjugated with 200 μg of either mouse anti-p190A (clone D2D6, Millipore Cat# 05–378), previously concentrated on Microcon-10kDa units (Millipore), or mouse IgG (Jackson ImmunoResearch, Cat# 015–000-003) ON at 30°C. In each experiment, an average of ~65 E12.5 ventral spinal cords (with floor plate in between) were collected in batches, homogenized immediately with 20 strokes of a microcentrifuge tube pestle in cold modified RIPA buffer followed by 1 h on a nutator at 4°C, cleared by centrifugation at 13,000 ×g for 15 min (4°C) and kept on ice until ready for incubation with antibody-conjugated beads. Approximately 4mg of total protein lysate diluted in 1ml of buffer were incubated with 15mg of either anti-p190- or IgG-conjugated beads (~109 beads/ml) for 45 min at 4°C with rotation. After 6 washes in Wash Buffer, proteins were eluted from the beads with 500 μL of 0.5N NH4OH, 0.5mM EDTA solution for 20min on a shaker (RT). The eluate was snap-frozen in liquid nitrogen and dried in a vacuum evaporator for > 8 h. Dried samples were processed from MS as described below. 2% of the eluate was processed for silver staining (Invitrogen/Thermo Scientific Cat# LC6070).
M-270 Epoxy beads conjugated with anti-190A antibody were used for the p190A-DCC co-immunoprecipitation from E12.5 ventral spinal cord homogenates (Figure 5H). After clarification, 650 μg of total protein lysate were incubated with 2mg of either anti-p190A- or control IgG-conjugated beads in 120 μL of lysis buffer and processed as described above. Dried samples were resuspended in NuPAGE LDS sample buffer and subjected to western blotting. Membranes were probed with goat anti-DCC antibody (1:2000; R&D Systems Cat# AF844) and, after stripping, with rabbit anti-190A antibody (1:3000; Cell Signaling Tech.)
For MS analysis of DCC interacting proteins in ESCMN (Figures S7A and S7B), 25 mg of M-270 Epoxy magnetic beads were conjugated with 200 μg of goat anti-DCC antibody to the extracellular domain (a 1:1 mix of R&D System Cat# AF844 and Sigma-Aldrich Cat# D3441) as described above. Three distinct p190−/− ESC lines and 3 wild-type lines were combined in four 150mm dishes per genotype (a total of 12×10^6 cells per genotype) and differentiated for 6 days into floating motor neuron embryoid bodies as described (Sternfeld et al., 2017) before the media was replaced with MN media for an additional day. For each genotype, 4mg of embryoid body protein lysate were incubated with 12.5mg of anti-DCC-conjugated beads (~109 beads/ml) for 45 min at 4°C with rotation and processed as described above.
For DCC-p190A co-immunoprecipitation from E12.5 ventral spinal cords (Figure 5I), 600 μg of total protein lysate prepared as described above were incubated on rotation with 3 μg of goat anti-DCC antibody (R&D System) ON (4°C), followed by incubation with 40 μL of protein A/G bead slurry for 2 h (4°C). Washed beads were resuspended in NuPAGE LDS sample buffer.
For immunoprecipitation assays in cell lines (Figures 5K, 5L, and S5C), AD293 cells grown to > 80% confluency in 60 mm dishes were transfected with 8 μg of plasmid DNA and 16 μL of Lipofectamine 2000 (Invitrogen/Thermo Scientific) according to the manufacturer’s instructions for ~36 h prior to lysis with modified RIPA buffer for 1 h at 4°C with rotation. After clarification at 13,000 ×g for 10 min, 500–600 μg of protein homogenates diluted to 500–600 μL in lysis buffer were pre-cleared by incubation with 20 μL of protein A/G bead slurry for 30 min (4°C). Lysates were immunoprecipitated ON (4°C) with 1.5 μg of mouse anti-FLAG antibody (M2, Sigma-Aldrich Cat# F1804) to tagged DCC constructs, followed by incubation with 30 μL protein A/G bead slurry for 2 h (4°C). Beads were washed 3 times in Wash Buffer, with 5 min rotation each time (4°C) and resuspended in NuPAGE LDS sample buffer. For Netrin stimulation (Figure 5L), cells were starved overnight in serum-free media before treatment with recombinant Netrin-1 (500 μg/ml) for the indicated times. Protein lysates were immunoblotted with anti-phospho-AKT antibody (1:1000; Cell Signaling Tech. Cat# 4058) to detect intracellular signaling responses to Netrin-1 stimulation (Xie et al., 2006)
Other antibodies used for immunoblotting were: mouse anti-p120RasGAP (1:2000; clone B4F8, AbCAM Cat# ab2922), rabbit-anti actin (1:5000; Cell Signaling Tech. Cat# 8456), rabbit anti-Phospho-Erk1/2 (Thr202/Tyr204) (1:2000; Cell Signaling Tech. Cat# 4370), rabbit anti-Erk1/2 (1:3000; Cell Signaling Tech. Cat# 9102), rabbit anti-GAPDH (1:5000; Cell Signaling Tech. Cat# 2118).
Sample preparation for Mass Spectrometry, Data Acquisition and Analysis
Immunoprecipitated protein samples prepared as described above were first denatured in 8M urea and then reduced and alkylated with 10 mM Tris(2-carboxyethyl)phosphine hydrochloride (Roche Applied Science) and 55 mM iodoacetamide (Sigma-Aldrich), respectively. The samples were then digested over-night with trypsin (Promega) according to the manufacturer’s specifications.
The protein digests were pressure-loaded onto 250 micron i.d. fused silica capillary (Polymicro Technologies) columns with a Kasil frit packed with 3cm of 5 micron Partisphere strong cation exchange (SCX) resin (Whatman) and 3 cm of 5 micron C18 resin (Phenomenex). After desalting, each bi-phasic column was connected to a 100 micron i.d. fused silica capillary (Polymicro Technologies) analytical column with a 5 micron pulled-tip, packed with 10 cm of 5 micron C18 resin (Phenomenex).
Each MudPIT column was placed inline with a 1200 quaternary HPLC pump (Agilent Technologies) and the eluted peptides were electrosprayed directly into an LTQ Orbitrap Velos mass spectrometer (Thermo Scientific). The buffer solutions used were 5% acetonitrile/0.1% formic acid (buffer A), 80% acetonitrile/0.1% formic acid (buffer B) and 500 mM ammonium acetate/5% acetonitrile/0.1% formic acid (buffer C). A eleven-step MudPIT was run with salt pulses of 0%, 10%, 20%, 30%, 40%, 50%, 60%, 70% and 100% buffer C and 90% buffer C/10% buffer B twice. The 120 min elution gradient had the following profile: 10% buffer B beginning at 10 min to 45% buffer B at 90 min to 100% buffer B from 100 min to 110 min. A cycle consisted of one full scan mass spectrum (300–1600 m/z) followed by 20 data-dependent collision induced dissociation (CID) MS/MS spectra. Application of mass spectrometer scan functions and HPLC solvent gradients were controlled by the Xcalibur data system (Thermo Scientific).
MS/MS spectra were extracted using RawXtract (version 1.9.9). MS/MS spectra were searched with the Sequest algorithm against a mouse IPI database concatenated to a decoy database in which the sequence for each entry in the original database was reversed. The Sequest search was performed using no enzyme specificity and static modification of cysteine due to carboxyamidomethylation (57.02146). Sequest search results were assembled and filtered using the DTASelect (version 2.0) algorithm, requiring peptides to be at least half tryptic and a minimum of two peptides per protein identification. The protein identification false positive rate was kept below one percent and all peptide-spectra matches had less than 10ppm mass error.
Gene Ontology and pathway analysis
p190A interacting proteins (604 proteins from two independent IP-MS experiments) for which spectra were detected in p190-immunoprecipitates but not in IgG-controls were filtered against the CRAPome database (http://www.crapome.org/) to remove protein “contaminants” that were identified in over 65% control experiments. Pathway annotation was conducted with Reactome (https://www.reactome.org) and GeneCodis software (http://genecodis.cnb.csic.es) on 519 proteins identified by at least two spectra (See Table S3). The same parameters applied to DCC-interacting proteins detected after IP-MS yielded 589 proteins that were detected either exclusively in wild-type (175) or p190−/− ESCMN (204), or present in both conditions (210). p190 was identified in DCC-immunocomplexes from wild-type ESCMN with 59 spectra counts (See Tables S4 and S5). Gene Ontology annotation was conducted with GO, KEGG and REACTOME terms. Associations between GO and REACTOME terms and genes were obtained through R packages org.Mm.eg.db and reactome.db, respectively. KEGG associations were obtained through the live KEGG REST online resource. To test for term enrichment of a gene set we used Fisher’s Exact Test at each term to compare the proportion of the gene set that was annotated with a term to the proportion of a background gene set that was annotated with the same term. A one-sided test was used to specifically test for enrichment in the gene set relative to background. In the case of a two-condition comparison we used the two-tailed Fisher’s Exact Test to compare the proportion of genes annotated with a term in each condition to see if the proportion was different. Terms were considered significant at p < 0.1.
Ontology term networks were constructed from thresholded pairwise similarity matrices of the terms. Term-to-term similarity was calculated by comparing the word content of the term names as well as the genes associated with the terms making terms with similar words and genes more similar to one another than terms with few or no common words or associated genes. The similarity scores were thresholded by finding each term’s maximum similarity score and using the 0.1 quantile of those, which ensured that at least 90% of the terms would have at least one connection in the network. The similarity scores between terms were converted to dissimilarity by subtracting them from 1. The dissimilarity scores were used as edge weights for the clustering step. Clustering of the network was performed using the edge betweenness community detection algorithm implemented in the igraph package for R.
QUANTIFICATION AND STATISTICAL ANALYSIS
Image analysis and Quantification
Whenever possible, images used for quantification were acquired using the same microscope settings and adjusted to the same background levels. Ectopic axon bundles were manually counted in flat-mounted ISLMN::fGFP+ embryos inspected at a fluorescence stereomicroscope (Zeiss Lumar V12). Number of embryos analyzed:
Figure 2N: wild-type (> 50); p190+/I602N (40); p190I602N/I602N (44); p190+/− (20); p190−/− (51); p190I602N/− (6); p190−/−;Olig2MN::Cre;p190-RFPLSL (8).
Figure 3H: p190+/− (20); p190−/− (51); p190R1284/+ (7); p190R1284/− (12).
Figure 3I: p190R1284/+ (14 limbs); p190R1284/− (24 limbs).
Figure 4I: p190+/− (20); p190−/− (51); Cxcr4+/− (4); Cxcr4−/− (26); p190+/−;Cxcr4+/− (25); p190−/−;Cxcr4+/− (9); p190+/−;Cxcr4−/− (7); p190−/−;Cxcr4−/− (> 10).
Figure 7F: Netrin-1−/− (Ntn1−/−) (> 10); DCC+/− (> 10); Unc5C−/− (5); p190−/ − (51); p190−/−;Netrin-1+/− (7); p190−/−;Netrin-1−/− (5); p190−/−;DCC+/− (19); p190−/−;DCC−/− (8); p190−/−;Unc5C+/− (5); p190−/−;Unc5C−/ − (4).
Figure 7P: Intercostal nerve phenotype, n embryos: p190−/− (29), p190−/−;Netrin-1−/− (5), p190−/−;DCC+/− (8). Limb nerve phenotype, n limbs: p190−/− (64), p190−/−;Netrin-1−/− (20), p190−/−; DCC+/− (16).
Figure S3J: p190S36N/+ (16); p190S36N/− (12).
Figure S3K: Intercostal nerve phenotype, n embryos: p190S36N/+ (16); p190S36N/− (12). Limb nerve phenotype, n limbs: p190S36N/+ (32); p190S36N/− (24).
Figure S4M: Sema3A−/− (7); Sema3F−/− (6); p190−/− (51); p190−/−;Sema3A+/− (7); p190−/−;Sema3A−/− (3); p190−/−;Sema3F+/− (10); p190−/−;Sema3F−/− (3).
Figure S7G: Cxcr4+/− (4); Cxcr4−/− (26); Cxcr4−/−;Netrin+/− (8); Cxcr4−/−;Netrin−/− (5)
A semi-automated quantification of axon outgrowth from motor explants was performed using the FeatureJ plugin of NIH ImageJ. To measure the total axonal area (#pixels), the algorithm was applied to images of GFP+ axons after removal of the signal associated with the cell bodies.
Pearson’s correlation coefficients were calculated using the Coloc2 plugin of NIH ImageJ/Fiji.
PLA puncta with minimum size of 2 px^2 overlapping to GFP+ motor axons were counted on thresholded images (maximum projection of confocal stacks) using the “analyze particle” command of NIH ImageJ.
The intensity of surface-DCC signal (# pixels × mean fluorescence) in GFP+ axons was measured using Photoshop software on projected confocal stacks.
For quantification of Robo3+ signal on commissural axons, the pixel area occupied by Robo3+ axons was expressed as percentage of the total spinal cord area and normalized to the values obtained in controls (Ruiz de Almodovar et al., 2011). Measurements were performed on maximum projection images of confocal stacks of E11.5 transverse sections using Photoshop software.
Nonparametric Mann–Whitney U test (two-tailed), Dunnett’s multiple comparison test (after one-way ANOVA) and Tukey’s multiple comparison test (after one-way ANOVA) were performed with GraphPad Prism software.
Gene Expression Quantification and Differential Expression Testing
Single-end RNA-sequencing reads were quantified using Kallisto software with default settings. We used transcripts annotated by Gencode for the mm10 mouse genome build. Transcript level read counts and TPM expressions were summed by gene name to generate gene-level data. All downstream analysis was performed on the gene-level data. Differential gene expression testing was performed using edgeR’s negative binomial GLM method. Genes were called significant with post hoc corrected p values < 0.05.
DATA AND SOFTWARE AVAILABILITY
The accession number for the RNA-sequencing data of FACS-sorted WT and p190 knockout motor neurons reported in this paper is GEO: GSE126534.
Supplementary Material
Figure S3. A p190 mutant incompetent for GTP-binding exhibits motor axon pathfinding defects. Related to Figure 3 M
(A) Immunoblotting for p190 in brain lysates of 3 p190R1284/−embryos and 3 p190+/− control littermates (E12.5). GADPH is a loading control. The R1284A mutation does not affect p190 protein levels.
(B) Increase in GTP-bound RhoA (active form) pulled-down from spinal cords homogenates of E12.5 p190R1284/− embryos compared to p190R1284/+ (HET) and wild-type littermates [9.3 ± 1.8-fold increase p190R1284/− (n=3) vs. controls (wild-type/heterozygous; n=3); Mean ± SEM, Mann–Whitney U test p<0.05].
(C) Schematic of p190 highlighting the S36N mutation that prevents GTP-binding.
(D) Direct cDNA sequencing demonstrates TCT (Ser-36)>AAT (Asn-36) substitution. The other synonymous substitutions were introduced to destroy the PAM site (Gly-34 G>A) and facilitate genotyping of the mutant allele (Gly-32 C>A and Lys-35 G>A).
(E) p190 gene expression is not affected by the S36N mutation. Transcript levels were assessed by quantitative PCR on brain cDNA prepared from p190S36N/S36N homozygous embryos, p190S36N/+ heterozygotes or wild-type littermates (e12.5) using two different primer sets annealing at either ends of the coding sequence.
(F) p190 protein levels are reduced in p190S36N mutants. Immunoblot analysis of p190 from brain lysates of p190S36N/S36N homozygous mutants, p190S36N/+ heterozygotes, p190S36N/− compound heterozygotes carrying one copy of the knockout allele and one copy of S36N allele, p190+/− heterozygotes, or wild-type littermates. GADPH is a loading control.
(G-I) Whole-mount images of ISLMN::fGFP reporter in E12.5 p190S36N/− compound heterozygous embryos. Embryo expressing p190S36N exhibit defects in motor axon exiting (G), intercostal nerve fasciculation (H) and peroneal nerve projection (I) qualitatively similar to p190-null embryos.
(J) Quantification of ectopic ISLMN::fGFP+ motor axon bundles in embryo whole-mounts. Mean ± SEM; Mann–Whitney U test (***) p<0.001.
(K) Incidence of intercostal (Left) and peroneal (Right) nerve defects in p190S36N/− mutants and p190S36N/+ controls with color-coded phenotypic classes: normal, green; moderate, blue; severe, red.
Scale bar: 200μm
Figure S2. p190 is expressed broadly but controls selective steps of motor pathfinding. Related to Figure 2
(A) In situ hybridization for p190 mRNA in transverse section of wild-type E11 embryo reveals widespread expression throughout the mesenchyme and neuronal tissues, with moderate enrichment in the ventral half of the spinal cord, motor columns (MN, outlined) and dorsal root ganglia (DRG).
(B) Normalized RNA-seq read counts for p190 in motor neurons FACS-purified from E10.5 or E12.5 embryos (MN E10.5, E12.5), compared to cells in the motor neuron-depleted fraction of whole (SC MN− E10.5) or ventral-only (vSC MN− E12.5) spinal cord, and sensory neurons (DRG, E12.5). p190 is not enriched in motor neurons. Mean ± SEM, N sequenced RNA samples (each containing cells from >8 embryos): MN e10.5 (1); SC MN− E10.5 (1); MN E12.5 (14); vSC MN− E12.5 (10); DRG (4).
(C-D) Whole mount neurofilament staining of E12.5 embryos. The pattern of cranial and brachial peripheral nerves is intact in p190I602N/I602N mutants.
(E) Western blotting of total lysates of motor columns (MN) or dorsal spinal cord (dSC) of E12.5 Olig2MN::Cre; p190-RFPLSL or p190-RFPLSL (Cre-negative) embryos. Exogenous p190-RFP is detected exclusively in Olig2MN::Cre+ motor neurons with both anti-p190 and anti-RFP antibodies. Anti-p190 antibody also detects equal levels of the endogenous protein across samples. The levels of p190-RFP are 2.5 ± 0.23 higher than endogenous p190 (Mean ± SEM, n=3 embryos). Actin is a loading control.
(F-G) Dorsal views of ISLMN::fGFP+ whole-mounts show rescue of motor axon exiting defects in p190−/−;Olig2MN::Cre;p190-RFPLSL embryos (E12.5).
(H-J) High-magnification ventral views of the boxed area in (G). Transgenic p190-RFP is expressed selectively in the motor column (asterisks in I).
(K-L) Intercostal nerves in E12.5 p190−/− or p190−/−;Olig2MN::Cre;p190-RFPLSL embryos. The intercostal “bridging” phenotype of p190 mutants (arrowheads in K) is rescued after expression of p190-RFP in motor neurons (L).
(M-N) Motor axon projections into the hindlimb in E12.5 embryo whole-mounts. The peroneal nerve phenotype detected in p190 mutants (arrowheads in M) is rescued in p190−/−;Olig2MN::Cre;p190-RFPLSL embryos, reestablishing a normal size of the main nerve tract (arrow in N, compare to M).
Scale bars: A,B: 500μm; C: 100μm; F,G: 200μm; K-L: 200μm; M-N: 200μm
Figure S1. The Cassin ENU mutation causes motor axon pathfinding defects. Related to Figure 1
(A-C) 3D views of ISLMN::fGFP 2-photon imaging of E12.5 wild-type and Cassin embryo whole-mounts showing spinal motor neuron columns (MN) and motor axons extending through ventral roots (VR). Arrows point to axial nerves that target muscles of the back. Ectopic intraspinal axon bundles (asterisks) and corresponding depletion of axons in ventral roots (arrowheads) are visible in Cassin mutants (B, C). A, anterior; P, posterior; M, medial; L, lateral; D, dorsal; V, ventral.
(D-F) Low magnification dorsal views of ISLMN::fGFP+ motor projections in embryo whole mounts (E12.5). Ectopic motor axons bundles are present at every level of the spinal cord in Cassin homozygous embryos (arrowheads in F). Only a few thin ectopic bundles are found in heterozygous embryos (arrowheads in E), and none are visible in wild-type littermates. Arrows point to axial nerves.
(G) A subset of Cassin homozygous mutants show exencephaly (arrowhead)
(H-I’) Transverse sections of wild-type and Cassin mutant spinal cords of E10.5 ISLMN::fGFP+ (green) embryos stained for the motor neuron nuclear marker Hb9 (red). Hb9 staining extracted from the merged images is shown in H’-I’. Motor neuron numbers, estimated by counting total Hb9+ pixels, are unaffected in Cassin embryos (Mann–Whitney U test p=0.81 mutant vs. wild-type). (J) Plot profiles of average pixel intensity of Hb9 immunoreactivity across a 200 pixel-wide rectangular selection spanning spinal cord hemisections of Cassin mutant (n=6, red trace) and wild-type (n=4, blue trace) embryos as shown in H’ and I’. The relative distribution of motor neuron cell bodies along the mediolateral axis of the spinal cord is unchanged in Cassin mutants (2way ANOVA p=0.37 mutant vs. wild-type). Misguided motor axons are visible in mutant embryos (arrow in I). M, medial; L, lateral.
(K) Schematic of the “open-book” spinal cord preparation. Motor axons that have exited the spinal cord are removed, while columns of motor neuron cell bodies are visible in the ventral region (V) adjacent to the floor plate (FP). RP, roof plate; I, intermediate region; D, dorsal region.
(L) Ectopic ISLMN::fGFP+ intraspinal motor axon bundles in Cassin mutant “open-book” spinal cord (E12) (asterisks).
(M) Flat-mount image of spinal meninges removed from the mutant embryo shown in (L). A subset of aberrant motor axon bundles are seen attached to the meningeal membranes (asterisks).
(N-O) Open-book spinal cord preparations of E12 wild-type and Cassin homozygous embryos expressing the SE1MN::GFP reporter specific for axial muscle-innervating neurons of the medial division of the Medial Motor Column (MMCM) (Shirasaki et al., 2006). In mutants, individual MMCm neurons positioned at different rostrocaudal levels (arrowheads) form dorsal intraspinal projections merging in large axon bundles (arrows). V, ventral region; I, intermediate region.
(P-Q) Intercostal nerves labeled by ISLMN::fGFP in wild-type and Cassin embryo flat-mounts (E12.5, ventral views). In wild-type embryos, intercostal nerves formed by motor neurons of the lateral division of the MMC (MMCL) were tightly fasciculated, while many nerve branches crossed between main nerve tracts in Cassin mutants.
(R) Schematic of intercostal nerve fasciculation (“bridging”) defects in Cassin mutants.
(S-T) Motor axon projections into the hindlimb labeled by Hb9MN::GFP motor neuron reporter in embryo whole-mounts (E12.5, dorsal view). In wild-type embryos, the peroneal nerve formed by motor neurons located in the lateral division of the Lateral Motor Column (LMCL) selects a dorsal trajectory at the base of the limb and gives rise to secondary branches at stereotypical proximodistal positions along its course. The peroneal nerve is affected in Cassin mutants: an aberrant proximal nerve branch extends rostrally (arrowheads) depleting the main nerve tract (arrow). Gl, Gluteus nerve; Dp, Deep peroneal; Sp, Superficial peroneal; A, anterior; P, posterior; M, medial; L, lateral.
(U) Schematic of abnormal hindlimb peroneal nerve targeting in Cassin mutants.
Scale bars: A-C: 50μm; D-F: 200μm; H-I’: 100μm; L-M: 100μm; N-O: 50μm; P-Q: 100μm; S-T: 100μm
Figure S4. p190 and Cxcr4 cooperate to instruct motor projection into the periphery. Related to Figure 4
(A-D) Transverse sections of ISLMN::fGFP+ (green) embryos (E12.5) stained for the motor neuron marker Isl1/2 (blue) and the pan-axonal marker bIII-tubulin (red). (B, C) In Cxcr4−/− embryos, ectopic axon bundles extend dorsally along the limiting membrane of the spinal cord, leading to corresponding thinning of peripheral nerves (asterisk, compare with A). The boxed region in B is enlarged in C. (D) In p190;Cxcr4 double mutants, virtually all motor axons form bundles within the spinal cord (arrowhead) and peripheral nerves are undetectable (asterisk). The boxed area in D in enlarged in Figure 3K.
(E-I) Low-magnification views of p190;Cxcr4 compound mutant embryos whole-mounts (E12.5). Ectopic bundles of motor axons that fail to project into the periphery are detected in Cxcr4−/− embryos (F, arrowheads). The phenotype is exacerbated in the presence of one (G, arrowheads) or two copies of p190 knockout allele (H, I, arrowheads). The boxed area in H is enlarged in I.
(J) Cxcr4 receptor signaling monitored by activation of ERK1/2 phosphorylation in wild-type and p190−/− ESCMN neurospheres stimulated with Cxcl12 for the reported times. Total lysates were analyzed by western blotting with the indicated antibodies.
(K) (Upper panels) Tyrosine-phosphorylated p190 detected after immunoprecipitation (IP) and western blotting from lysates of HeLa cells stimulated with Cxcl12 for the indicated times. (Bottom panel) Western blotting for phospho-ERK1/2. Cxcl12 triggers robust activation of ERK1/2 phosphorylation but no changes p190 tyrosine-phosphorylation.
(L) Model of cooperative interaction between p190- and Cxcr4-dependent signaling for the control of motor axon extension from the spinal cord. p190 and Cxcr4 operate in parallel synergistic pathways.
(M) Quantification of ectopic ISLMN::fGFP+ motor axon bundles in embryo whole-mounts (E12.5). The motor axon exiting phenotype of p190 mutants is not rescued by knockout of either Sema3A or Sema3F. Mean ± SEM; Dunnetś multiple comparisons test (ns) p>0.05 vs. p190−/−.
Scale bars: A,B,D,I: 100μm; C: 50μm; E-H: 200μm
Figure S6. p190 is dispensable for midline axon guidance. Related to Figure 6
(A-B’) Transverse sections of ISLMN::fGFP+ E11.5 embryos stained for commissural axon marker Robo3 (red). The trajectory and fasciculation of commissural axons extending toward the floor plate (FP) is unchanged in p190−/− embryos (arrows). Intraspinal motor axon bundles are visible in mutants (arrowheads in B, B’). The boxed areas in A, B are enlarged in A’, B’.
(C) Quantification of the area occupied by commissural axons (% of total spinal cord area), normalized to the values obtained in control embryos (wild-type or p190+/−). Mean ± SEM, n embryos: control (9); p190+/− (5). Mann–Whitney U test (ns) p>0.05 p190+/− vs. control.
(D-E) Images of ISLMN::fGFP+ motor columns (MN) separated by floor plate (FP) in transverse sections of control and p190I602N/I602N (Cassin) embryos (E12.5). No aberrant crossing of the floor plate by motor axons is observed.
(F) Schematic of 3D-motor explant repulsion assay. Hb9MN::GFP+ motor explants are co-cultured with floor plate tissue in collagen/matrigel matrices for ~24 hrs. Chemorepellents released from the floor plate inhibit axon growth from the proximal side (P) while axons extend normally on the distal side (D). (G) Quantification of motor axon repulsion reported as fraction of GFP+ motor axons present on the P (red bars) or D (grey bars) sides. Mean ± SEM, n explants: control (23); p190−/− (22). Both control and p190−/− explants display significant reduction of axon growth on the P side. Mann–Whitney U test (***) p<0.001. The extent of growth inhibition on the P side is comparable between control and p190−/− explants; (ns) p>0.05.
(H-I) Inhibition of motor axon growth from the side closer to floor plate (P) is observed in both control and p190−/− explants. Axon growth is abundant on the distal side.
Scale bars: A-B: 100μm; A’-B’: 25μm; D-E: 20μm; G-H: 200μm
Figure S5. Identification of p190 interacting proteins and association with DCC receptor. Related to Figure 5
(A) MA plot of fold-change in gene expression in p190−/− vs. control (p190+/− or wild-type) motor neurons (y-axis) against average expression level (x-axis). RNA-seq was conducted on Hb9MN::GFP+ motor neurons purified by FACS from E12.5 spinal cords. Differentially expressed genes are highlighted in red (upregulated) and green (downregulated). The modest decrease in Arhgap35 expression (logFC −1.08) in p190 knockout cells is explained by normal levels of the mutant transcript lacking a 5’ sequence corresponding to the targeted deletion in exon-1. The other four genes with small but significant changes in expression were functionally unrelated and were all located within 3Mbp from the Arhgap35 locus suggesting that they might be affected by unintended positional effects of gene targeting. Of note, the expression of these genes was normal in Cassin mutant spinal cords analyzed by RNA-seq.
(B) (Top) Gene Ontology network for proteins detected by mass spectrometry in p190 immunocomplexes (but not IgG controls) affinity-purified from the ventral spinal cord of E12.5 embryos. Circle size is proportional to the relative enrichment of the corresponding GO term. Representative biological processes for the main modules are indicated. (Bottom) Unsupervised annotation of the p190 interactome reveals enrichment in axon guidance and cell motility pathways, including Netrin-1. Additional Netrin-1 signaling components were identified through manual curation of the protein dataset (stacked bar, striped) [See Tables S1–S3].
(C) Co-immunoprecipitation of DCC full-length (FL) and deletion mutants (See schematics in Figure 5J) with p190-RFP in transfected AD293 cells. The P3 domain restored p190 binding to P2-deletion mutants.
(D-E”) Confocal images of Cos-7 cells transfected with p190-RFP and either full length DCC (D-D”) or a deletion mutant lacking the P2+P3 motifs required for interaction with p190 (DCC 1–1327) (E-E”). Surface-associated DCC (DCCsurf, green) was detected with an antibody to the extracellular domain under non-permeabilizing conditions after Netrin-1 treatment. Targeting of p190-RFP (red) to the plasma membrane is prominent in the presence of wild-type DCC (arrowheads) but strongly reduced with the truncated receptor. The boxed areas in D’ and E’ are enlarged in D” and E”.
(F-J) Surface-associated DCC (DCCsurf, red) detected in cultured Hb9MN::GFP+ motor neurons derived from either control (p190+/− or wild-type) or p190−/− embryos. DCCsurf decreases after Netrin-1 treatment. Arrowheads point to growth cones. (J) Quantification of DCCsurf signal. The levels of DCC on the axon surface are unchanged in p190−/− motor neurons. Mean ± SEM, n=12 fields of view encompassing comparable GFP+ axon area. Mann–Whitney U test (ns) p>0.05 control/untreated vs. p190−/−/untreated and control/Netrin-1 vs. p190−/−/Netrin-1; (***) p<0.001 treated vs. untreated.
Scale bars: D,D’,E,E’: 10μm; D”,E”: 4μm; F-I’: 20μm
Figure S7. Effect of p190 on DCC receptor complexes and specificity of p190/Netrin genetic interaction. Related to Figure 7
(A) Heat map display of spectral counts for DCC interacting proteins revealed in either wild-type or p190−/− ESCMN by mass spectrometry analysis of affinity-purified DCC complexes. Many interactors are detected exclusively in one condition. ESCMN were used in lieu of primary motor neurons to obtain sufficient material from knockout cells. p190 was detected in DCC immunocomplexes [See Table S4].
(B) Gene Ontology network of DCC interactomes showing clusters of terms (circles) significantly enriched among DCC-interacting proteins detected in either wild-type (blue) or p190−/− cells (red), or found in both conditions (gray). The circle size of “wild-type” and “knockout” modules is proportional to the relative enrichment of the GO term. Representative biological processes for the main modules are shown [See Table S5].
(C-F) Whole-mount images of ISLMN::fGFP+ Unc5C and p190/Unc5C mutants (E12.5, dorsal views). Motor axon exit is normal in Unc5C knockouts (C) while aberrant intraspinal bundles are visible in p190/Unc5C double mutants (arrowheads in D). Likewise, intercostal bridging (arrowheads in E) and limb nerve defects (arrowheads in F) observed in p190 knockouts are still present in p190/Unc5C compound mutants (See Figure 7F for quantification).
(G) Quantification of ectopic ISLMN::fGFP+ motor axon bundles in embryo whole-mounts of Cxcr4/Netrin-1 compound mutants (E12.5). Motor axon exiting defects observed in Cxcr4 mutants are not rescued by Netrin-1 gene deletion. Mean ± SEM; Dunnetś multiple comparisons test (ns) p>0.05 vs. Cxcr4−/−.
Scale bars: A-D: 200μm
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti-Isl1/2 | Ericson et al., 1992 | N/A |
| rat anti-Laminin β1 | Millipore | Cat# MAB1928; RRID: AB_11213381 |
| rabbit anti-RFP | MBL International | Cat# PM005; RRID: AB_591279 |
| mouse anti-β3 tubulin | Novus | Cat# NB120-11314; RRID: AB_792496 |
| goat anti-Netrin-1 | R&D Systems | Cat# AF1109; RRID: AB_2298775 |
| goat anti-DCC | Sigma-Aldrich | Cat# D3441; RRID: AB_476903 |
| goat anti-DCC | R&D Systems | Cat# AF844; RRID: AB_2089765 |
| goat anti-Robo3 | R&D Systems | Cat# AF3076; RRID: AB_2181865 |
| guinea pig anti-FoxP1 | Dasen et al., 2008 | N/A |
| rabbit anti-GFP | Invitrogen | Cat# A-6455; RRID: AB_221570 |
| donkey anti-goat IgG, Alexa Fluor 488 | Invitrogen | Cat# A-11055; RRID: AB_2534102 |
| donkey anti-goat IgG, Alexa Fluor 555 | Invitrogen | Cat# A-21432; RRID: AB_2535853 |
| donkey anti-goat IgG, Alexa Fluor 647 | Invitrogen | Cat# A-21447; RRID: AB_2535864 |
| donkey anti-mouse IgG, Alexa Fluor 488 | Invitrogen | Cat# A-21202; RRID: AB_141607 |
| donkey anti-mouse IgG, Alexa Fluor 555 | Invitrogen | Cat# 31570; RRID: AB_2536180 |
| donkey anti-mouse IgG, Alexa Fluor 647 | Invitrogen | Cat# A-31571; RRID: AB_162542 |
| donkey anti-rabbit IgG, Alexa Fluor 488 | Invitrogen | Cat# A-21206; RRID: AB_2535792 |
| donkey anti-rabbit IgG, Alexa Fluor 555 | Invitrogen | Cat# A-31572; RRID: AB_162543 |
| donkey anti-rabbit IgG, Alexa Fluor 647 | Invitrogen | Cat# A-31573; RRID: AB_253618 |
| goat anti-guinea pig IgG, Alexa Fluor 647 | Invitrogen | Cat# A-21450; RRID: AB_141882 |
| mouse anti-neurofilament (NF-M), clone 2H3 | Developmental Hybridoma Bank | Cat# 2H3 s; RRID: AB_2314897 |
| rabbit anti-p190A antibody | Cell Signaling Technology | Cat# 2860S; RRID: AB_2263862 |
| mouse anti-RhoGAP p190, clone D2D6 | Millipore | Cat# 05-378; RRID: AB_309704 |
| mouse anti-phosphotyrosine, clone 4G10 | Millipore | Cat# 05-321; RRID: AB_309678 |
| mouse anti-FLAG M2 | Sigma-Aldrich | Cat# F1804; RRID: AB_262044 |
| mouse anti-GAP, clone B4F8 | Abcam | Cat# ab2922; RRID: AB_303418 |
| rabbit anti-Actin, clone D18C11 | Cell Signaling Technology | Cat# 8456S; RRID: AB_10998774 |
| rabbit anti-phospho-p44/42 MAPK(Erk1/2) (Thr202/Tyr204), clone D13.14.4E | Cell Signaling Technology | Cat# 4094S; RRID: AB_10694057 |
| rabbit anti-p44/42 MAPK (Erk1/2) | Cell Signaling Technology | Cat# 9102; RRID: AB_330744 |
| rabbit anti-phospho Akt (Ser473), clone 193H12 | Cell Signaling Technology | Cat# 4058; RRID: AB_331168 |
| rabbit anti-GAPDH, clone 14C10 | Cell Signaling Technology | Cat# 2118; RRID: AB_561053 |
| mouse anti-HA.11, clone 16B12 | Covance Research Products Inc | Cat# MMS-101R-1000; RRID: AB_291262 |
| ChromPure Mouse IgG, whole molecule antibody | Jackson ImmunoResearch | Cat# 015-000-003; RRID: AB_2337188 |
| AffiniPure Goat Anti-Rabbit IgG, Fc fragment specific | Jackson ImmunoResearch | Cat# NC9712967 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| ENU, N-Ethyl-N-nitrosourea | Sigma-Aldrich | Cat# N3385 |
| Laminin Mouse Protein | GIBCO | Cat# #23017015 |
| Collagen I, Rat Tail | Corning | Cat# #354236 |
| Matrigel Basement Membrane Matrix | Corning | Cat# 356234 |
| Recombinant Mouse Netrin-1 Protein | R&D Systems | Cat# 1109-N1-025 |
| Recombinant Human GDNF Protein | R&D Systems | Cat# 212-GD-010 |
| Recombinant Mouse CXCL12/SDF-1 alpha | R&D Systems | Cat# 460-SD-010 |
| Duolink In Situ Orange Starter Kit Mouse/Goat | Sigma-Aldrich | Cat# DUO92104 |
| Rho Activation Assay Kit | Millipore | Cat# 17-294 |
| Dynabeads M-270 Epoxy | Invitrogen | Cat# 14301 |
| SilverQuest Silver Staining Kit | Invitrogen | Cat# LC6070 |
| Lipofectamine 2000 Transfection Reagent | Invitrogen | Cat# 11668027 |
| Critical Commercial Assays | ||
| Papain | Worthington Biochemical | Cat# LK003153 |
| T7 Quick High Yield RNA Synthesis Kit | New England Biolabs | Cat# E2050S |
| Cas9 mRNA | Invitrogen | Cat# A29378 |
| RNeasy Mini Kit | QIAGEN | Cat# 74104 |
| RNase-Free DNase Set | QIAGEN | Cat# 79254 |
| Illumina mRNA-Seq Sample Prep Kit | Illumina | Cat# RS-100-0801 |
| TruSeq RNA Library Preparation Kit v2 | Illumina | Cat# RS-122-2001 |
| Deposited Data | ||
| RNA-seq WT and p190 knockout motor neurons | this paper | GEO: GSE126534 |
| Experimental Models: Cell Lines | ||
| AD-293 | Stratagene | Cat# 240085 |
| COS-7 | ATCC | Cat# CRL-1651 |
| HeLa | ATCC | Cat# CCL-2 |
| Mouse embryonic stem cell line ISLMN::fGFP | this paper | N/A |
| Mouse embryonic stem cell line p190−/−; ISLMN::fGFP | this paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: ISLMN::fGFP: Tg(Isl1-EGFP*)1Slp/J | The Jackson Laboratory | JAX:017952 |
| Mouse: Olig2::Cre | Dessaud et al., 2007 | N/A |
| Mouse: SE1MN::GFP | Shirasaki et al., 2006 | N/A |
| Mouse: Hb9MN::GFP | Lee et al., 2004 | N/A |
| Mouse: p190I602N: Arhgap35m201Slp/J | this paper/The Jackson Laboratory | JAX:017951 |
| Mouse: p190RhoGAP−/−: B6;129-Arhgap35tm1Jset/AjkJ | The Jackson Laboratory | JAX:020997 |
| Mouse: Cxcr4−/−: B6.129X-Cxcr4tm1Qma/J | The Jackson Laboratory | JAX:004341 |
| Mouse: Netrìn-1−/− | Serafini et al., 1996 | N/A |
| Mouse: DCC−/−: B6.129S2-Dcctm1Wbg/Mmmh | MMRRC | MMRRC:030626-MU |
| Mouse: Unc5C−/− | Burgess et al., 2006 | N/A |
| Mouse: Sema3A−/−: C3;B6-Sema3am808Ddg/J | The Jackson Laboratory | JAX:014646 |
| Mouse: Sema3F−/−: B6;129P2-Sema3fm1Mom/MomJ | The Jackson Laboratory | JAX:006710 |
| Mouse: MN(218-2)::GFP | Amin et al., 2015 | N/A |
| Mouse: p190-RFPLSL | this paper | N/A |
| Mouse: p190R1284A | this paper | N/A |
| Mouse: p190S36N | this paper | N/A |
| Oligonucleotides | ||
| gRNA Arhgap35 exon4: AAGCACTGAAGGCATCTACC | this paper | N/A |
| gRNA Arhgap35 exon2: AGAAAGGCCAGTGCGGCATT | this paper | N/A |
| single-stranded oligodeoxynucleotide_R1284A | this paper | N/A |
| single-stranded oligodeoxynucleotide_S36N | this paper | N/A |
| Recombinant DNA | ||
| Plasmid: pCAG-LoxSTOPLox-WPRE-polyA | Bonanomi et al., 2012 | N/A |
| Plasmid: pCAG-LoxSTOPLox-RFP-p190A | this paper | N/A |
| Plasmid: pCMV-RFP-p190A | this paper | N/A |
| Plasmid: pCMV-FLAG-DCC | this paper | N/A |
| Plasmid: pCMV-FLAG-DCC 1-1421 (ΔΠ3) | this paper | N/A |
| Plasmid: pCMV-FLAG-DCC 1-1363 | this paper | N/A |
| Plasmid: pCMV-FLAG-DCC 1-1327 | this paper | N/A |
| Plasmid: pCMV-FLAG-DCC 1-1247 | this paper | N/A |
| Plasmid: pCMV-FLAG-DCC 1-1171 | this paper | N/A |
| Plasmid: pCMV-FLAG-DCC 1-1120 | this paper | N/A |
| Plasmid: pCMV-FLAG-DCC ΔΠ3+ΔL2-3 | this paper | N/A |
| Plasmid: pCMV-FLAG-DCC ΔL2-3 | this paper | N/A |
| Plasmid: pCMV-FLAG-DCC ΔΠ2 | this paper | N/A |
| Plasmid for in situ probe: pCR2.1TOPO-p190A (NM_172739; 1156-1837bp) | this paper | N/A |
| Plasmid for in situ probe: pCRII-Topo-Netrin-1 (Mouse, NM_008744.2; 1823-2680bp) | this paper | N/A |
| Arhgap35 cDNA (Rat, M94721) | Hu and Settleman, 1997 | N/A |
| DCC cDNA (Rat, NM_012841.1) | Tcherkezian et al., 2010 | N/A |
| Software and Algorithms | ||
| GraphPad Prism5 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/; RRID: SCR_002798 |
| GeneCodis | http://genecodis.cnb.csic.es | |
| Reactome | https://reactome.org/ | |
| CRAPome | https//www.crapome.org | |
| Bioconductor | https://bioconductor.org | |
| ImageJ | https://imagej.nih.gov/ij/; RRID: SCR_003070 | |
| Fiji | https://fiji.sc/ | |
| FeatureJ | https://imagescience.org/meijering/ | https://imagescience.org/meijering/software/featurej/ |
| Coloc2 | GitHub | https://imagej.net/Coloc_2 |
| Photoshop CS4 | Adobe | https://www.adobe.com/products/photoshop.html |
| CRISPR Tool | Zhang Lab, MIT | http://zlab.bio/guide-design-resources |
| Kallisto | http://pachterlab.github.io | |
| PROVEAN | http://provean.jcvi.org/index.php | |
| SIFT | http://sift.bii.a-star.edu.sg/ | |
| MOSAIK | https://github.com/wanpinglee/MOSAIK | |
Highlights.
p190 is a context-dependent gate for Netrin-DCC signalling
Distinct temporal activities of p190 emerge from a modular domain organization
p190 is a cell intrinsic signal-detection filter
Suppression of incongruous directional signals prevents miswiring
ACKNOWLEDGMENTS
We thank Christopher Hinckley for two-photon imaging; Andrea Dal Mas for targeting constructs; Matteo Puglisi for data analysis; Jeffrey Settleman and Anthony Koleske for 190 mice and plasmids; Bennett Novitch and Thomas Jessell for Olig2::Cre mice; Marc Tessier-Lavigne for Netrin-1 mice; Susan Ackerman for Unc5C mice; Qing Ma and Marcus Kaul for Cxcr4 mice; John Flanagan for DCC plasmids; Yelena Dayn (Salk Transgenic Core), James Moresco (Salk Mass Spectrometry Core), and the Advanced Light and Electron Microscopy Bioimaging Center of San Raffaele Scientific Institute for technical support. S.L.P. is supported as a Howard Hughes Medical Institute Investigator and as a Benjamin H. Lewis chair in neuroscience. D.B. is supported by the European Research Council Starting Grant 335590 and a Career Development Award from the Giovanni Armenise-Harvard Foundation.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found with this article online at https://doi.org/10.1016/j.neuron.2019.02.034.
REFERENCES
- Akin O, and Zipursky SL (2016). Frazzled promotes growth cone attachment at the source of a Netrin gradient in the Drosophila visual system. eLife. Published online October 15, 2016. 10.7554/eLife.20762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin ND, Bai G, Klug JR, Bonanomi D, Pankratz MT, Gifford WD, Hinckley CA, Sternfeld MJ, Driscoll SP, Dominguez B, et al. (2015). Loss of motoneuron-specific microRNA-218 causes systemic neuromuscular failure. Science 350, 1525–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoine-Bertrand J, Duquette PM, Alchini R, Kennedy TE, Fournier AE, and Lamarche-Vane N (2016). p120RasGAP protein mediates Netrin-1 protein-induced cortical axon outgrowth and guidance. J. Biol. Chem. 291, 4589–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai G, and Pfaff SL (2011). Protease regulation: the Yin and Yang of neural development and disease. Neuron 72, 9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai G, Chivatakarn O, Bonanomi D, Lettieri K, Franco L, Xia C, Stein E, Ma L, Lewcock JW, and Pfaff SL (2011). Presenilin-dependent receptor processing is required for axon guidance. Cell 144, 106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barberis D, Casazza A, Sordella R, Corso S, Artigiani S, Settleman J, Comoglio PM, and Tamagnone L (2005). p190 Rho-GTPase activating protein associates with plexins and it is required for semaphorin signalling. J. Cell Sci. 118, 4689–4700. [DOI] [PubMed] [Google Scholar]
- Bashaw GJ, and Klein R (2010). Signaling from axon guidance receptors. Cold Spring Harb. Perspect. Biol. 2, a001941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billuart P, Winter CG, Maresh A, Zhao X, and Luo L (2001). Regulating axon branch stability: the role of p190 RhoGAP in repressing a retraction signaling pathway. Cell 107, 195–207. [DOI] [PubMed] [Google Scholar]
- Bonanomi D (2019). Axon pathfinding for locomotion. Semin. Cell Dev. Biol. 85, 26–35. [DOI] [PubMed] [Google Scholar]
- Bonanomi D, and Pfaff SL (2010). Motor axon pathfinding. Cold Spring Harb. Perspect. Biol. 2, a001735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonanomi D, Chivatakarn O, Bai G, Abdesselem H, Lettieri K, Marquardt T, Pierchala BA, and Pfaff SL (2012). Ret is a multifunctional coreceptor that integrates diffusible- and contact-axon guidance signals. Cell 148, 568–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouton AH, Kanner SB, Vines RR, Wang HC, Gibbs JB, and Parsons JT (1991). Transformation by pp60src or stimulation of cells with epidermal growth factor induces the stable association of tyrosine-phosphorylated cellular proteins with GTPase-activating protein. Mol. Cell. Biol. 11, 945–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brankatschk M, and Dickson BJ (2006). Netrins guide Drosophila commissural axons at short range. Nat. Neurosci. 9, 188–194. [DOI] [PubMed] [Google Scholar]
- Brouns MR, Matheson SF, Hu KQ, Delalle I, Caviness VS, Silver J, Bronson RT, and Settleman J (2000). The adhesion signaling molecule p190 RhoGAP is required for morphogenetic processes in neural development. Development 127, 4891–4903. [DOI] [PubMed] [Google Scholar]
- Brouns MR, Matheson SF, and Settleman J (2001). p190 RhoGAP is the principal Src substrate in brain and regulates axon outgrowth, guidance and fasciculation. Nat. Cell Biol. 3, 361–367. [DOI] [PubMed] [Google Scholar]
- Burgess RW, Jucius TJ, and Ackerman SL (2006). Motor axon guidance of the mammalian trochlear and phrenic nerves: dependence on the netrin receptor Unc5c and modifier loci. J. Neurosci. 26, 5756–5766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colamarino SA, and Tessier-Lavigne M (1995). The axonal chemoattractant netrin-1 is also a chemorepellent for trochlear motor axons. Cell 81, 621–629. [DOI] [PubMed] [Google Scholar]
- Cristea IM, Williams R, Chait BT, and Rout MP (2005). Fluorescent proteins as proteomic probes. Mol. Cell. Proteomics 4, 1933–1941. [DOI] [PubMed] [Google Scholar]
- Dasen JS, De Camilli A, Wang B, Tucker PW, and Jessell TM (2008). Hox repertoires for motor neuron diversity and connectivity gated by a single accessory factor, FoxP1. Cell 134, 304–316. [DOI] [PubMed] [Google Scholar]
- Dessaud E, Yang LL, Hill K, Cox B, Ulloa F, Ribeiro A, Mynett A, Novitch BG, and Briscoe J (2007). Interpretation of the sonic hedgehog morphogen gradient by a temporal adaptation mechanism. Nature 450, 717–720. [DOI] [PubMed] [Google Scholar]
- Dickson BJ, and Zou Y (2010). Navigating intermediate targets: the nervous system midline. Cold Spring Harb. Perspect. Biol. 2, a002055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominici C, Moreno-Bravo JA, Puiggros SR, Rappeneau Q, Rama N, Vieugue P, Bernet A, Mehlen P, and Chédotal A (2017). Floor-plate-derived netrin-1 is dispensable for commissural axon guidance. Nature 545, 350–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudanova I, and Klein R (2013). Integration of guidance cues: parallel signaling and crosstalk. Trends Neurosci. 36, 295–304. [DOI] [PubMed] [Google Scholar]
- Ericson J, Thor S, Edlund T, Jessell TM, and Yamada T (1992). Early stages of motor neuron differentiation revealed by expression of homeobox gene Islet-1. Science 256, 1555–1560. [DOI] [PubMed] [Google Scholar]
- Foster R, Hu KQ, Shaywitz DA, and Settleman J (1994). p190 RhoGAP, the major RasGAP-associated protein, binds GTP directly. Mol. Cell. Biol. 14, 7173–7181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallarda BW, Bonanomi D, Müller D, Brown A, Alaynick WA, Andrews SE, Lemke G, Pfaff SL, and Marquardt T (2008). Segregation of axial motor and sensory pathways via heterotypic trans-axonal signaling. Science 320, 233–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A, and Lalli G (2010). Rho and Ras GTPases in axon growth, guidance, and branching. Cold Spring Harb. Perspect. Biol. 2, a001818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höpker VH, Shewan D, Tessier-Lavigne M, Poo M, and Holt C (1999). Growth-cone attraction to netrin-1 is converted to repulsion by laminin-1. Nature 401, 69–73. [DOI] [PubMed] [Google Scholar]
- Hu KQ, and Settleman J (1997). Tandem SH2 binding sites mediate the RasGAP-RhoGAP interaction: a conformational mechanism for SH3 domain regulation. EMBO J. 16, 473–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber AB, Kania A, Tran TS, Gu C, De Marco Garcia N, Lieberam I, Johnson D, Jessell TM, Ginty DD, and Kolodkin AL (2005). Distinct roles for secreted semaphorin signaling in spinal motor axon guidance. Neuron 48, 949–964. [DOI] [PubMed] [Google Scholar]
- Jeong S, Juhaszova K, and Kolodkin AL (2012). The control of semaphoring-1a-mediated reverse signaling by opposing pebble and RhoGAPp190 functions in Drosophila. Neuron 76, 721–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessell TM (2000). Neuronal specification in the spinal cord: inductive signals and transcriptional codes. Nat. Rev. Genet. 1, 20–29. [DOI] [PubMed] [Google Scholar]
- Jung H, Yoon BC, and Holt CE (2012). Axonal mRNA localization and local protein synthesis in nervous system assembly, maintenance and repair. Nat. Rev. Neurosci. 13, 308–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keino-Masu K, Masu M, Hinck L, Leonardo ED, Chan SS, Culotti JG, and Tessier-Lavigne M (1996). Deleted in Colorectal Cancer (DCC) encodes a netrin receptor. Cell 87, 175–185. [DOI] [PubMed] [Google Scholar]
- Keleman K, Rajagopalan S, Cleppien D, Teis D, Paiha K, Huber LA, Technau GM, and Dickson BJ (2002). Comm sorts robo to control axon guidance at the Drosophila midline. Cell 110, 415–427. [DOI] [PubMed] [Google Scholar]
- Kennedy TE, Serafini T, de la Torre JR, and Tessier-Lavigne M (1994). Netrins are diffusible chemotropic factors for commissural axons in the embryonic spinal cord. Cell 78, 425–435. [DOI] [PubMed] [Google Scholar]
- Kolodkin AL, and Tessier-Lavigne M (2011). Mechanisms and molecules of neuronal wiring: a primer. Cold Spring Harb. Perspect. Biol. Published online June 1, 2011. 10.1101/cshperspect.a001727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Wing Sun K, Correia JP, and Kennedy TE (2011). Netrins: versatile extracellular cues with diverse functions. Development 138, 2153–2169. [DOI] [PubMed] [Google Scholar]
- Lee SK, Jurata LW, Funahashi J, Ruiz EC, and Pfaff SL (2004). Analysis of embryonic motoneuron gene regulation: derepression of general activators function in concert with enhancer factors. Development 131, 3295–3306. [DOI] [PubMed] [Google Scholar]
- Lewcock JW, Genoud N, Lettieri K, and Pfaff SL (2007). The ubiquitin ligase Phr1 regulates axon outgrowth through modulation of microtubule dynamics. Neuron 56, 604–620. [DOI] [PubMed] [Google Scholar]
- Li R, Zhang B, and Zheng Y (1997). Structural determinants required for the interaction between Rho GTPase and the GTPase-activating domain of p190. J. Biol. Chem. 272, 32830–32835. [DOI] [PubMed] [Google Scholar]
- Lieberam I, Agalliu D, Nagasawa T, Ericson J, and Jessell TM (2005). A Cxcl12-CXCR4 chemokine signaling pathway defines the initial trajectory of mammalian motor axons. Neuron 47, 667–679. [DOI] [PubMed] [Google Scholar]
- Lim W, Mayer B, and Pawson T (2015). Cell Signaling: Principles and Mechanisms (Garland Science, Taylor & Francis Group; ). [Google Scholar]
- Moore SW, Correia JP, Lai Wing Sun K, Pool M, Fournier AE, and Kennedy TE (2008). Rho inhibition recruits DCC to the neuronal plasma membrane and enhances axon chemoattraction to netrin 1. Development 135, 2855–2864. [DOI] [PubMed] [Google Scholar]
- Moore SW, Biais N, and Sheetz MP (2009). Traction on immobilized netrin-1 is sufficient to reorient axons. Science 325, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales D, and Kania A (2017). Cooperation and crosstalk in axon guidance cue integration: Additivity, synergy, and fine-tuning in combinatorial signaling. Dev. Neurobiol. 77, 891–904. [DOI] [PubMed] [Google Scholar]
- Moreno-Bravo JA, Roig Puiggros S, Mehlen P, and Chédotal A (2019). Synergistic activity of floor-plate- and ventricular-zone-derived Netrin-1 in spinal cord commissural axon guidance. Neuron 101, 625–634.e3. [DOI] [PubMed] [Google Scholar]
- Nakahara H, Mueller SC, Nomizu M, Yamada Y, Yeh Y, and Chen WT (1998). Activation of beta1 integrin signaling stimulates tyrosine phosphorylation of p190RhoGAP and membrane-protrusive activities at invadopodia. J. Biol. Chem. 273, 9–12. [DOI] [PubMed] [Google Scholar]
- Neuhaus-Follini A, and Bashaw GJ (2015). The intracellular domain of the Frazzled/DCC receptor is a transcription factor required for commissural axon guidance. Neuron 87, 751–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell M, Chance RK, and Bashaw GJ (2009). Axon growth and guidance: receptor regulation and signal transduction. Annu. Rev. Neurosci. 32, 383–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliak S, Morales D, Croteau LP, Krawchuk D, Palmesino E, Morton S, Cloutier JF, Charron F, Dalva MB, Ackerman SL, et al. (2015). Synergistic integration of Netrin and ephrin axon guidance signals by spinal motor neurons. eLife. Published online December 3, 2015. 10.7554/eLife.10841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz de Almodovar C, Fabre PJ, Knevels E, Coulon C, Segura I, Haddick PC, Aerts L, Delattin N, Strasser G, Oh WJ, et al. (2011). VEGF mediates commissural axon chemoattraction through its receptor Flk1. Neuron 70, 966–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini T, Kennedy TE, Galko MJ, Mirzayan C, Jessell TM, and Tessier-Lavigne M (1994). The netrins define a family of axon outgrowthpromoting proteins homologous to C. elegans UNC-6. Cell 78, 409–424. [DOI] [PubMed] [Google Scholar]
- Serafini T, Colamarino SA, Leonardo ED, Wang H, Beddington R, Skarnes WC, and Tessier-Lavigne M (1996). Netrin-1 is required for commissural axon guidance in the developing vertebrate nervous system. Cell 87, 1001–1014. [DOI] [PubMed] [Google Scholar]
- Settleman J, Albright CF, Foster LC, and Weinberg RA (1992a). Association between GTPase activators for Rho and Ras families. Nature 359, 153–154. [DOI] [PubMed] [Google Scholar]
- Settleman J, Narasimhan V, Foster LC, and Weinberg RA (1992b). Molecular cloning of cDNAs encoding the GAP-associated protein p190: implications for a signaling pathway from ras to the nucleus. Cell 69, 539–549. [DOI] [PubMed] [Google Scholar]
- Sever R, and Brugge JS (2015). Signal transduction in cancer. Cold Spring Harb. Perspect. Med. Published online April 1, 2015. 10.1101/cshperspect.a006098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shewan D, Dwivedy A, Anderson R, and Holt CE (2002). Age-related changes underlie switch in netrin-1 responsiveness as growth cones advance along visual pathway. Nat. Neurosci. 5, 955–962. [DOI] [PubMed] [Google Scholar]
- Shirasaki R, Lewcock JW, Lettieri K, and Pfaff SL (2006). FGF as a target-derived chemoattractant for developing motor axons genetically programmed by the LIM code. Neuron 50, 841–853. [DOI] [PubMed] [Google Scholar]
- Söderberg O, Gullberg M, Jarvius M, Ridderstråle K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, and Landegren U (2006). Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000. [DOI] [PubMed] [Google Scholar]
- Stein E, and Tessier-Lavigne M (2001). Hierarchical organization of guidance receptors: silencing of netrin attraction by slit through a Robo/DCC receptor complex. Science 291, 1928–1938. [DOI] [PubMed] [Google Scholar]
- Sternfeld MJ, Hinckley CA, Moore NJ, Pankratz MT, Hilde KL, Driscoll SP, Hayashi M, Amin ND, Bonanomi D, Gifford WD, et al. (2017). Speed and segmentation control mechanisms characterized in rhythmically-active circuits created from spinal neurons produced from genetically-tagged embryonic stem cells. eLife. Published online February 14, 2017. 10.7554/eLife.21540.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tcherkezian J, Brittis PA, Thomas F, Roux PP, and Flanagan JG (2010). Transmembrane receptor DCC associates with protein synthesis machinery and regulates translation. Cell 141, 632–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadarajan SG, Kong JH, Phan KD, Kao TJ, Panaitof SC, Cardin J, Eltzschig H, Kania A, Novitch BG, and Butler SJ (2017). Netrin1 produced by neural progenitors, not floor plate cells, is required for axon guidance in the spinal cord. Neuron 94, 790–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela-Echavarría A, Tucker A, Püschel AW, and Guthrie S (1997). Motor axon subpopulations respond differentially to the chemorepellents netrin-1 and semaphorin D. Neuron 18, 193–207. [DOI] [PubMed] [Google Scholar]
- Vincent S, and Settleman J (1999). Inhibition of RhoGAP activity is sufficient for the induction of Rho-mediated actin reorganization. Eur. J. Cell Biol. 78, 539–548. [DOI] [PubMed] [Google Scholar]
- Wichterle H, Lieberam I, Porter JA, and Jessell TM (2002). Directed differentiation of embryonic stem cells into motor neurons. Cell 110, 385–397. [DOI] [PubMed] [Google Scholar]
- Wu Z, Makihara S, Yam PT, Teo S, Renier N, Balekoglu N, Moreno-Bravo JA, Olsen O, Chédotal A, Charron F, and Tessier-Lavigne M (2019). Long-range guidance of spinal commissural axons by Netrin1 and Sonic Hedgehog from midline floor plate cells. Neuron 101, 635–647. [DOI] [PubMed] [Google Scholar]
- Xie Y, Hong Y, Ma XY, Ren XR, Ackerman S, Mei L, and Xiong WC (2006). DCC-dependent phospholipase C signaling in netrin-1-induced neurite elongation. J. Biol. Chem. 281, 2605–2611. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S3. A p190 mutant incompetent for GTP-binding exhibits motor axon pathfinding defects. Related to Figure 3 M
(A) Immunoblotting for p190 in brain lysates of 3 p190R1284/−embryos and 3 p190+/− control littermates (E12.5). GADPH is a loading control. The R1284A mutation does not affect p190 protein levels.
(B) Increase in GTP-bound RhoA (active form) pulled-down from spinal cords homogenates of E12.5 p190R1284/− embryos compared to p190R1284/+ (HET) and wild-type littermates [9.3 ± 1.8-fold increase p190R1284/− (n=3) vs. controls (wild-type/heterozygous; n=3); Mean ± SEM, Mann–Whitney U test p<0.05].
(C) Schematic of p190 highlighting the S36N mutation that prevents GTP-binding.
(D) Direct cDNA sequencing demonstrates TCT (Ser-36)>AAT (Asn-36) substitution. The other synonymous substitutions were introduced to destroy the PAM site (Gly-34 G>A) and facilitate genotyping of the mutant allele (Gly-32 C>A and Lys-35 G>A).
(E) p190 gene expression is not affected by the S36N mutation. Transcript levels were assessed by quantitative PCR on brain cDNA prepared from p190S36N/S36N homozygous embryos, p190S36N/+ heterozygotes or wild-type littermates (e12.5) using two different primer sets annealing at either ends of the coding sequence.
(F) p190 protein levels are reduced in p190S36N mutants. Immunoblot analysis of p190 from brain lysates of p190S36N/S36N homozygous mutants, p190S36N/+ heterozygotes, p190S36N/− compound heterozygotes carrying one copy of the knockout allele and one copy of S36N allele, p190+/− heterozygotes, or wild-type littermates. GADPH is a loading control.
(G-I) Whole-mount images of ISLMN::fGFP reporter in E12.5 p190S36N/− compound heterozygous embryos. Embryo expressing p190S36N exhibit defects in motor axon exiting (G), intercostal nerve fasciculation (H) and peroneal nerve projection (I) qualitatively similar to p190-null embryos.
(J) Quantification of ectopic ISLMN::fGFP+ motor axon bundles in embryo whole-mounts. Mean ± SEM; Mann–Whitney U test (***) p<0.001.
(K) Incidence of intercostal (Left) and peroneal (Right) nerve defects in p190S36N/− mutants and p190S36N/+ controls with color-coded phenotypic classes: normal, green; moderate, blue; severe, red.
Scale bar: 200μm
Figure S2. p190 is expressed broadly but controls selective steps of motor pathfinding. Related to Figure 2
(A) In situ hybridization for p190 mRNA in transverse section of wild-type E11 embryo reveals widespread expression throughout the mesenchyme and neuronal tissues, with moderate enrichment in the ventral half of the spinal cord, motor columns (MN, outlined) and dorsal root ganglia (DRG).
(B) Normalized RNA-seq read counts for p190 in motor neurons FACS-purified from E10.5 or E12.5 embryos (MN E10.5, E12.5), compared to cells in the motor neuron-depleted fraction of whole (SC MN− E10.5) or ventral-only (vSC MN− E12.5) spinal cord, and sensory neurons (DRG, E12.5). p190 is not enriched in motor neurons. Mean ± SEM, N sequenced RNA samples (each containing cells from >8 embryos): MN e10.5 (1); SC MN− E10.5 (1); MN E12.5 (14); vSC MN− E12.5 (10); DRG (4).
(C-D) Whole mount neurofilament staining of E12.5 embryos. The pattern of cranial and brachial peripheral nerves is intact in p190I602N/I602N mutants.
(E) Western blotting of total lysates of motor columns (MN) or dorsal spinal cord (dSC) of E12.5 Olig2MN::Cre; p190-RFPLSL or p190-RFPLSL (Cre-negative) embryos. Exogenous p190-RFP is detected exclusively in Olig2MN::Cre+ motor neurons with both anti-p190 and anti-RFP antibodies. Anti-p190 antibody also detects equal levels of the endogenous protein across samples. The levels of p190-RFP are 2.5 ± 0.23 higher than endogenous p190 (Mean ± SEM, n=3 embryos). Actin is a loading control.
(F-G) Dorsal views of ISLMN::fGFP+ whole-mounts show rescue of motor axon exiting defects in p190−/−;Olig2MN::Cre;p190-RFPLSL embryos (E12.5).
(H-J) High-magnification ventral views of the boxed area in (G). Transgenic p190-RFP is expressed selectively in the motor column (asterisks in I).
(K-L) Intercostal nerves in E12.5 p190−/− or p190−/−;Olig2MN::Cre;p190-RFPLSL embryos. The intercostal “bridging” phenotype of p190 mutants (arrowheads in K) is rescued after expression of p190-RFP in motor neurons (L).
(M-N) Motor axon projections into the hindlimb in E12.5 embryo whole-mounts. The peroneal nerve phenotype detected in p190 mutants (arrowheads in M) is rescued in p190−/−;Olig2MN::Cre;p190-RFPLSL embryos, reestablishing a normal size of the main nerve tract (arrow in N, compare to M).
Scale bars: A,B: 500μm; C: 100μm; F,G: 200μm; K-L: 200μm; M-N: 200μm
Figure S1. The Cassin ENU mutation causes motor axon pathfinding defects. Related to Figure 1
(A-C) 3D views of ISLMN::fGFP 2-photon imaging of E12.5 wild-type and Cassin embryo whole-mounts showing spinal motor neuron columns (MN) and motor axons extending through ventral roots (VR). Arrows point to axial nerves that target muscles of the back. Ectopic intraspinal axon bundles (asterisks) and corresponding depletion of axons in ventral roots (arrowheads) are visible in Cassin mutants (B, C). A, anterior; P, posterior; M, medial; L, lateral; D, dorsal; V, ventral.
(D-F) Low magnification dorsal views of ISLMN::fGFP+ motor projections in embryo whole mounts (E12.5). Ectopic motor axons bundles are present at every level of the spinal cord in Cassin homozygous embryos (arrowheads in F). Only a few thin ectopic bundles are found in heterozygous embryos (arrowheads in E), and none are visible in wild-type littermates. Arrows point to axial nerves.
(G) A subset of Cassin homozygous mutants show exencephaly (arrowhead)
(H-I’) Transverse sections of wild-type and Cassin mutant spinal cords of E10.5 ISLMN::fGFP+ (green) embryos stained for the motor neuron nuclear marker Hb9 (red). Hb9 staining extracted from the merged images is shown in H’-I’. Motor neuron numbers, estimated by counting total Hb9+ pixels, are unaffected in Cassin embryos (Mann–Whitney U test p=0.81 mutant vs. wild-type). (J) Plot profiles of average pixel intensity of Hb9 immunoreactivity across a 200 pixel-wide rectangular selection spanning spinal cord hemisections of Cassin mutant (n=6, red trace) and wild-type (n=4, blue trace) embryos as shown in H’ and I’. The relative distribution of motor neuron cell bodies along the mediolateral axis of the spinal cord is unchanged in Cassin mutants (2way ANOVA p=0.37 mutant vs. wild-type). Misguided motor axons are visible in mutant embryos (arrow in I). M, medial; L, lateral.
(K) Schematic of the “open-book” spinal cord preparation. Motor axons that have exited the spinal cord are removed, while columns of motor neuron cell bodies are visible in the ventral region (V) adjacent to the floor plate (FP). RP, roof plate; I, intermediate region; D, dorsal region.
(L) Ectopic ISLMN::fGFP+ intraspinal motor axon bundles in Cassin mutant “open-book” spinal cord (E12) (asterisks).
(M) Flat-mount image of spinal meninges removed from the mutant embryo shown in (L). A subset of aberrant motor axon bundles are seen attached to the meningeal membranes (asterisks).
(N-O) Open-book spinal cord preparations of E12 wild-type and Cassin homozygous embryos expressing the SE1MN::GFP reporter specific for axial muscle-innervating neurons of the medial division of the Medial Motor Column (MMCM) (Shirasaki et al., 2006). In mutants, individual MMCm neurons positioned at different rostrocaudal levels (arrowheads) form dorsal intraspinal projections merging in large axon bundles (arrows). V, ventral region; I, intermediate region.
(P-Q) Intercostal nerves labeled by ISLMN::fGFP in wild-type and Cassin embryo flat-mounts (E12.5, ventral views). In wild-type embryos, intercostal nerves formed by motor neurons of the lateral division of the MMC (MMCL) were tightly fasciculated, while many nerve branches crossed between main nerve tracts in Cassin mutants.
(R) Schematic of intercostal nerve fasciculation (“bridging”) defects in Cassin mutants.
(S-T) Motor axon projections into the hindlimb labeled by Hb9MN::GFP motor neuron reporter in embryo whole-mounts (E12.5, dorsal view). In wild-type embryos, the peroneal nerve formed by motor neurons located in the lateral division of the Lateral Motor Column (LMCL) selects a dorsal trajectory at the base of the limb and gives rise to secondary branches at stereotypical proximodistal positions along its course. The peroneal nerve is affected in Cassin mutants: an aberrant proximal nerve branch extends rostrally (arrowheads) depleting the main nerve tract (arrow). Gl, Gluteus nerve; Dp, Deep peroneal; Sp, Superficial peroneal; A, anterior; P, posterior; M, medial; L, lateral.
(U) Schematic of abnormal hindlimb peroneal nerve targeting in Cassin mutants.
Scale bars: A-C: 50μm; D-F: 200μm; H-I’: 100μm; L-M: 100μm; N-O: 50μm; P-Q: 100μm; S-T: 100μm
Figure S4. p190 and Cxcr4 cooperate to instruct motor projection into the periphery. Related to Figure 4
(A-D) Transverse sections of ISLMN::fGFP+ (green) embryos (E12.5) stained for the motor neuron marker Isl1/2 (blue) and the pan-axonal marker bIII-tubulin (red). (B, C) In Cxcr4−/− embryos, ectopic axon bundles extend dorsally along the limiting membrane of the spinal cord, leading to corresponding thinning of peripheral nerves (asterisk, compare with A). The boxed region in B is enlarged in C. (D) In p190;Cxcr4 double mutants, virtually all motor axons form bundles within the spinal cord (arrowhead) and peripheral nerves are undetectable (asterisk). The boxed area in D in enlarged in Figure 3K.
(E-I) Low-magnification views of p190;Cxcr4 compound mutant embryos whole-mounts (E12.5). Ectopic bundles of motor axons that fail to project into the periphery are detected in Cxcr4−/− embryos (F, arrowheads). The phenotype is exacerbated in the presence of one (G, arrowheads) or two copies of p190 knockout allele (H, I, arrowheads). The boxed area in H is enlarged in I.
(J) Cxcr4 receptor signaling monitored by activation of ERK1/2 phosphorylation in wild-type and p190−/− ESCMN neurospheres stimulated with Cxcl12 for the reported times. Total lysates were analyzed by western blotting with the indicated antibodies.
(K) (Upper panels) Tyrosine-phosphorylated p190 detected after immunoprecipitation (IP) and western blotting from lysates of HeLa cells stimulated with Cxcl12 for the indicated times. (Bottom panel) Western blotting for phospho-ERK1/2. Cxcl12 triggers robust activation of ERK1/2 phosphorylation but no changes p190 tyrosine-phosphorylation.
(L) Model of cooperative interaction between p190- and Cxcr4-dependent signaling for the control of motor axon extension from the spinal cord. p190 and Cxcr4 operate in parallel synergistic pathways.
(M) Quantification of ectopic ISLMN::fGFP+ motor axon bundles in embryo whole-mounts (E12.5). The motor axon exiting phenotype of p190 mutants is not rescued by knockout of either Sema3A or Sema3F. Mean ± SEM; Dunnetś multiple comparisons test (ns) p>0.05 vs. p190−/−.
Scale bars: A,B,D,I: 100μm; C: 50μm; E-H: 200μm
Figure S6. p190 is dispensable for midline axon guidance. Related to Figure 6
(A-B’) Transverse sections of ISLMN::fGFP+ E11.5 embryos stained for commissural axon marker Robo3 (red). The trajectory and fasciculation of commissural axons extending toward the floor plate (FP) is unchanged in p190−/− embryos (arrows). Intraspinal motor axon bundles are visible in mutants (arrowheads in B, B’). The boxed areas in A, B are enlarged in A’, B’.
(C) Quantification of the area occupied by commissural axons (% of total spinal cord area), normalized to the values obtained in control embryos (wild-type or p190+/−). Mean ± SEM, n embryos: control (9); p190+/− (5). Mann–Whitney U test (ns) p>0.05 p190+/− vs. control.
(D-E) Images of ISLMN::fGFP+ motor columns (MN) separated by floor plate (FP) in transverse sections of control and p190I602N/I602N (Cassin) embryos (E12.5). No aberrant crossing of the floor plate by motor axons is observed.
(F) Schematic of 3D-motor explant repulsion assay. Hb9MN::GFP+ motor explants are co-cultured with floor plate tissue in collagen/matrigel matrices for ~24 hrs. Chemorepellents released from the floor plate inhibit axon growth from the proximal side (P) while axons extend normally on the distal side (D). (G) Quantification of motor axon repulsion reported as fraction of GFP+ motor axons present on the P (red bars) or D (grey bars) sides. Mean ± SEM, n explants: control (23); p190−/− (22). Both control and p190−/− explants display significant reduction of axon growth on the P side. Mann–Whitney U test (***) p<0.001. The extent of growth inhibition on the P side is comparable between control and p190−/− explants; (ns) p>0.05.
(H-I) Inhibition of motor axon growth from the side closer to floor plate (P) is observed in both control and p190−/− explants. Axon growth is abundant on the distal side.
Scale bars: A-B: 100μm; A’-B’: 25μm; D-E: 20μm; G-H: 200μm
Figure S5. Identification of p190 interacting proteins and association with DCC receptor. Related to Figure 5
(A) MA plot of fold-change in gene expression in p190−/− vs. control (p190+/− or wild-type) motor neurons (y-axis) against average expression level (x-axis). RNA-seq was conducted on Hb9MN::GFP+ motor neurons purified by FACS from E12.5 spinal cords. Differentially expressed genes are highlighted in red (upregulated) and green (downregulated). The modest decrease in Arhgap35 expression (logFC −1.08) in p190 knockout cells is explained by normal levels of the mutant transcript lacking a 5’ sequence corresponding to the targeted deletion in exon-1. The other four genes with small but significant changes in expression were functionally unrelated and were all located within 3Mbp from the Arhgap35 locus suggesting that they might be affected by unintended positional effects of gene targeting. Of note, the expression of these genes was normal in Cassin mutant spinal cords analyzed by RNA-seq.
(B) (Top) Gene Ontology network for proteins detected by mass spectrometry in p190 immunocomplexes (but not IgG controls) affinity-purified from the ventral spinal cord of E12.5 embryos. Circle size is proportional to the relative enrichment of the corresponding GO term. Representative biological processes for the main modules are indicated. (Bottom) Unsupervised annotation of the p190 interactome reveals enrichment in axon guidance and cell motility pathways, including Netrin-1. Additional Netrin-1 signaling components were identified through manual curation of the protein dataset (stacked bar, striped) [See Tables S1–S3].
(C) Co-immunoprecipitation of DCC full-length (FL) and deletion mutants (See schematics in Figure 5J) with p190-RFP in transfected AD293 cells. The P3 domain restored p190 binding to P2-deletion mutants.
(D-E”) Confocal images of Cos-7 cells transfected with p190-RFP and either full length DCC (D-D”) or a deletion mutant lacking the P2+P3 motifs required for interaction with p190 (DCC 1–1327) (E-E”). Surface-associated DCC (DCCsurf, green) was detected with an antibody to the extracellular domain under non-permeabilizing conditions after Netrin-1 treatment. Targeting of p190-RFP (red) to the plasma membrane is prominent in the presence of wild-type DCC (arrowheads) but strongly reduced with the truncated receptor. The boxed areas in D’ and E’ are enlarged in D” and E”.
(F-J) Surface-associated DCC (DCCsurf, red) detected in cultured Hb9MN::GFP+ motor neurons derived from either control (p190+/− or wild-type) or p190−/− embryos. DCCsurf decreases after Netrin-1 treatment. Arrowheads point to growth cones. (J) Quantification of DCCsurf signal. The levels of DCC on the axon surface are unchanged in p190−/− motor neurons. Mean ± SEM, n=12 fields of view encompassing comparable GFP+ axon area. Mann–Whitney U test (ns) p>0.05 control/untreated vs. p190−/−/untreated and control/Netrin-1 vs. p190−/−/Netrin-1; (***) p<0.001 treated vs. untreated.
Scale bars: D,D’,E,E’: 10μm; D”,E”: 4μm; F-I’: 20μm
Figure S7. Effect of p190 on DCC receptor complexes and specificity of p190/Netrin genetic interaction. Related to Figure 7
(A) Heat map display of spectral counts for DCC interacting proteins revealed in either wild-type or p190−/− ESCMN by mass spectrometry analysis of affinity-purified DCC complexes. Many interactors are detected exclusively in one condition. ESCMN were used in lieu of primary motor neurons to obtain sufficient material from knockout cells. p190 was detected in DCC immunocomplexes [See Table S4].
(B) Gene Ontology network of DCC interactomes showing clusters of terms (circles) significantly enriched among DCC-interacting proteins detected in either wild-type (blue) or p190−/− cells (red), or found in both conditions (gray). The circle size of “wild-type” and “knockout” modules is proportional to the relative enrichment of the GO term. Representative biological processes for the main modules are shown [See Table S5].
(C-F) Whole-mount images of ISLMN::fGFP+ Unc5C and p190/Unc5C mutants (E12.5, dorsal views). Motor axon exit is normal in Unc5C knockouts (C) while aberrant intraspinal bundles are visible in p190/Unc5C double mutants (arrowheads in D). Likewise, intercostal bridging (arrowheads in E) and limb nerve defects (arrowheads in F) observed in p190 knockouts are still present in p190/Unc5C compound mutants (See Figure 7F for quantification).
(G) Quantification of ectopic ISLMN::fGFP+ motor axon bundles in embryo whole-mounts of Cxcr4/Netrin-1 compound mutants (E12.5). Motor axon exiting defects observed in Cxcr4 mutants are not rescued by Netrin-1 gene deletion. Mean ± SEM; Dunnetś multiple comparisons test (ns) p>0.05 vs. Cxcr4−/−.
Scale bars: A-D: 200μm
Data Availability Statement
The accession number for the RNA-sequencing data of FACS-sorted WT and p190 knockout motor neurons reported in this paper is GEO: GSE126534.