

Abstract
Aberrant expression of long non-coding RNAs (lncRNAs) has been reported in multiple cancers. However, the underlying mechanisms mediated by super-enhancers remain elusive. Here we sought to define the role of a novel lncRNA termed lncRNA-DAW in tumorigenesis. Our results revealed that lncRNA-DAW was driven by a liver-specific super-enhancer and transcriptionally activated by HNF4G, leading to frequent elevation in hepatocellular carcinoma (HCC) specimens. Ectopic expression of lncRNA-DAW promoted both in vivo and in vitro tumor growth. By using RNA sequencing, Wnt2 was screened out as a downstream effector of lncRNA-DAW. We next found that lncRNA-DAW physically interacted with EZH2, a negative regulator of Wnt2. This interplay subsequently potentiated CDK1-EZH2 interaction, leading to the phosphorylation and ubiquitination of EZH2. The lncRNA-DAW-mediated EZH2 degradation facilitated the de-repression of Wnt2 transcription, which eventually activated the Wnt/β-catenin pathway. Furthermore, we verified that Wnt2 potentiated in vitro and in vivo cancer cell growth by activating the Wnt/β-catenin pathway. Finally, Wnt2 amplification was confirmed as a common event in liver cancer, and the expression of lncRNA-DAW was positively correlated with Wnt2 in HCC specimens. Collectively, we are the first to identify lncRNA-DAW as a novel candidate oncogene in liver cancer, and this lncRNA may serve as a novel clinical diagnosis biomarker for liver cancer.
Keywords: cell growth, lncRNA, super-enhancer, Wnt/β-catenin pathway, liver cancer
Graphical abstract
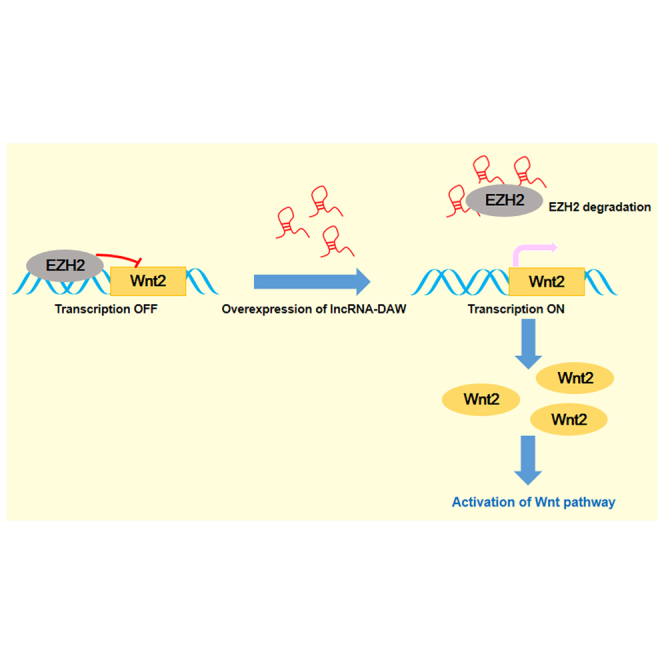
lncRNA-DAW was driven by a liver-specific super-enhancer and frequently upregulated in liver cancer. lncRNA-DAW diminished PRC2-mediated repression of Wnt2 by antagonizing the function of EZH2, eventually leading to the activation of the Wnt/β-catenin pathway and cancer cell proliferation.
Introduction
As the second leading cause of cancer-related death globally, hepatocellular carcinoma (HCC) is a devastating disease with limited therapeutic approaches and relatively low 5-year survival rate.1 Although some HCC-related risk factors have been identified so far, a large portion of cancer patients are still diagnosed in advanced stage.2 In this stage, the conventional therapies such as liver resection and chemotherapy are less effective compared with early tumor stage. Despite recent progress in the diagnosis of primary liver cancer, the diagnostic tools remain limited and novel tumor markers are urgently needed.2 Thus, detailed illustration of the genetic landscape during hepatocarcinogenesis is of great importance to develop innovative diagnostic and therapeutic strategies to improve the clinical outcome of these cancer patients.
In the past decades, emerging innovative molecular approaches have provided mechanistic insights into the pathogenesis of HCC. It is well characterized that aberrant gene expression is essential for cancer cells to acquire multiple cancer hallmarks. In liver cancer, the oncogenic cancer driver genes are extensively activated, leading to the hyper-activation of key oncogenic signaling pathways, such as PI3K/AKT and Wnt/β-catenin pathways.3, 4, 5, 6 Recent advance indicates that the robust gene transcription is frequently driven by a cluster of enhancer regions, namely, the super-enhancer. An enhancer is a distant non-coding DNA element that cooperates with a promoter to reinforce the expression of a target gene.7 A super-enhancer is an exceptionally large cluster of enhancers that synergistically drives gene transcriptions. As a common trait, super-enhancers typically exhibited an enrichment of histone-3-lysine-27 acetylation (H3K27Ac), which was densely occupied by various transcription factors.8 Emerging evidence indicates that cancer cells may utilize the super-enhancers surrounding the cancer driver genes, ranging from protein-coding genes to non-coding RNAs.8
It is known that the vast majority of the human genome is pervasively transcribed into a wide spectrum of non-coding RNAs, such as microRNA (miRNA), piwi-interacting RNA (piRNA), and long non-coding RNA (lncRNA).9,10 Recent research progress has highlighted the central roles of non-coding RNA in orchestrating gene expression. As an active participant in gene regulation, lncRNAs are a class of the non-coding RNA transcripts that surpass 200 nucleotides in length. Emerging evidence indicates that lncRNAs are actively involved in many important biological events, such as immune response, cell fate determination, genome imprinting, as well as chromatin structure remodeling.11
In the present study, we identified a novel non-coding RNA driven by a liver-specific super-enhancer, which was termed lncRNA-DAW (Distant Activator of Wnt2). lncRNA-DAW was frequently upregulated in liver cancer tissues, and its overexpression accelerated tumor growth as well as cancer metastasis. Further mechanistic studies discovered that lncRNA-DAW directly associated with EZH2, which potentiated the EZH2-CDK1 interaction and triggered the phosphorylation and ubiquitination of EZH2. Subsequent study showed that lncRNA-DAW antagonized the suppressive effect of EZH2 on Wnt2 expression, leading to the activation of the Wnt/β-catenin pathway. Further clinical investigation revealed that Wnt2 was significantly upregulated in liver patients and high expression of Wnt2 indicated poor prognosis. Taken together, our study reveals a novel mechanism by which lncRNA modulates the Wnt/β-catenin pathway through epigenetically activating Wnt2, which might provide mechanistic insights into the missing link between lncRNAs and cancer progression.
Results
lncRNA-DAW was driven by a liver-specific super-enhancer
In order to identify the potent super-enhancers in liver cancer, we searched the public databases dbSUPER (https://asntech.org/dbsuper/index.php) and Cistrome (http://cistrome.org/db/#/). According to these two databases, we found that chromosome 20q13.12 is one of the top chromosomal segments and it harbored strong H3K27ac signal in normal hepatocytes compared with other normal organs (Figure 1A). We next compared the hallmarks of super-enhancers. We found that, in the normal hepatocytes, this region harbored weak H3K27me3 signal plus strong H3K4me1 and H3K27ac signal (Figure 1B), suggesting that this locus might harbor a liver-specific super-enhancer.
Figure 1.

lncRNA-DAW was driven by a liver-specific super-enhancer
(A) The H3K27ac ChIP-seq data of lncRNA-DAW (LINC01430) locus in a panel of normal human tissues. The yellow region indicates the gene locus of lncRNA-DAW. (B) The ChIP-seq data of super-enhancer histone markers of lncRNA-DAW locus in normal hepatocytes. (C) The RNA-seq data from NCBI showed that lncRNA-DAW (LINC01430) was highly expressed in human liver tissues. (D) The RNA levels of lncRNA-DAW were measured across a panel of adult human tissues by using qRT-PCR assays. (E) Cellular localization of lncRNA-DAW. The relative levels of cytoplasmic RNA transcript β-actin, nuclear RNA transcript U1, and lncRNA-DAW from different cell compartments were assessed by qRT-PCR assays. The total levels for each gene were considered as 100% and results were presented as a relative percentage of total levels (n = 4). (F) Compared with LO2 cells, lncRNA-DAW was upregulated in most of the HCC cell lines. (G) The expression levels of lncRNA-DAW in 50 pairs of HCC and adjacent normal tissues were determined by qRT-PCR assays.
To identify the potent genes driven by this liver-specific super-enhancer, we scanned this locus and identified eight previously unreported lncRNAs within this territory (Figure 1C). We first examined the expression profiles of these lncRNAs across a panel of human tissues. Among these candidate lncRNAs, linc01430, which was termed lncRNA-DAW in our study, was found to be highly expressed in liver tissues (Figure 1C). To further verify this finding, qRT-PCR was conducted and the results showed that this lncRNA was a liver-enriched RNA transcript among a variety of human tissues (Figure 1D). In accordance with this finding, another high-throughput RNA sequencing (RNA-seq) dataset also showed that lncRNA-DAW was highly expressed in liver compared with other tissues (Figure S1). Subsequent analysis found that lncRNA-DAW was composed of three exons with a full length of 442 nt. Because some lncRNAs possess short open reading frames and encode functional peptides,12 we thus conducted protein-coding potential analysis. According to the bioinformatics analysis, the exon sequence of lncRNA-DAW was found to be less conserved across different species (Figure S2A). Subsequent protein-coding potential analysis showed that the positive strand of lncRNA-DAW might encode three peptides, but these peptides are quite short and no similar experimentally verified protein sequence was found in the human protein database UniProtKB/Swiss-Prot, suggesting that lncRNA-DAW has a weak coding potential (Figure S2B). To gain sights of the biological importance of lncRNA-DAW in liver cancer, we investigated the expression profiles of lncRNA-DAW in liver cancer cell lines as well as liver cancer tissues. We then determined the cellular distribution of this lncRNA and found that this lncRNA was predominantly accumulated in the nucleus (Figure 1E). We found that lncRNA-DAW was upregulated in a panel of liver cancer cell lines compared with normal liver cell line LO2 (Figure 1F). To examine its clinical significance, we monitored its expression in 50 paired HCC specimens and found that this lncRNA was significantly upregulated in HCC tissues (Figure 1G).
HNF4G cooperated with super-enhancer to activate gene transcription
As super-enhancer was frequently occupied by various transcription factors, we next tried to screen out the candidate transcription factors that contribute to aberrant activation of lncRNA-DAW in liver cancer. According to the chromatin immunoprecipitation sequencing (ChIP-seq) data of HepG2 from UCSC Genome Browser (https://genome.ucsc.edu/) and Cistrome (http://cistrome.org/db/#/), we found that this gene locus was occupied by HNF4G, a member of the hepatocyte nuclear factor family (Figure 2A). We then used ChIP experiments and found that HNF4G physically interacted with super-enhancer SE-1 and SE2 (Figure 2B). To further verify it, we inserted the SE-1 and SE-2 DNA fragments into the pGL-3 control vector. The luciferase assay showed that transfection with pGL-3 vector harboring enhancer element significantly increased the luciferase activity (Figure 2C). To confirm the regulatory role of HNF4G in these two elements, we knocked down HNF4G and then transfected the pGL-3 vector harboring enhancer element in HepG2 cells. We found that silencing of HNF4G significantly impaired the luciferase activity compared with control groups (Figure 2D). Subsequently, RT-PCR experiments further confirmed the siHNF4G-mediated suppressive effects on lncRNA-DAW expression (Figure 2E).
Figure 2.

Knockdown of HNF4G suppressed gene transcription of lncRNA-DAW
(A) The ChIP-seq data showed that HNF4G interacted with the super-enhancer region. (B) The binding affinity of HNF4G to super-enhancer region was determined by ChIP experiments. (C) The luciferase activities of SE-1 and SE-2 were evaluated by luciferase assays (n = 3). (D) After silencing of HNF4G, the luciferase activities of SE-1 and SE-2 were evaluated by luciferase assays. (E) After silencing of HNF4G, the expression levels of lncRNA-DAW were determined by qRT-PCR assays.
lncRNA-DAW accelerated in vivo and in vitro tumor growth
In order to examine the functional characterization of lncRNA-DAW in tumor growth, we first generated lncRNA-DAW-overexpressing stable cell lines (Figure 3A). According to 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays, ectopic expression of lncRNA-DAW promoted cell proliferation (Figure 3B). The subsequent study also confirmed that lncRNA-DAW enhanced colony formation capacity (Figure 3C) and anchorage-independent growth (Figure 3D). Consistent with the results of gain-of-function study, we found that knockdown of lncRNA-DAW by small interfering RNA (siRNA) impaired cancer cell growth based on MTT assay and colony formation assay (Figure S3). In addition to the in vitro study, we also analyzed the in vivo cell growth capacity by xenograft tumor model. The lncRNA-DAW-overexpressing cells were subcutaneously injected into immunodeficient nude mice, and overexpression of lncRNA-DAW could remarkably potentiate in vivo tumor growth (Figure 3E; Figures S4A–S4D). In an attempt to evaluate the in vivo metastatic capacity, we delivered the cancer cells to lung tissues through hydrodynamic tail vein injection in nude mice. Mice were sacrificed and the metastatic nodules within indicated tissues were counted (Figure 3F). More metastatic nodules were observed within lung in the lncRNA-DAW-overexpressing groups (Figure 3G). Similar results were also displayed in the in vivo study of lncRNA-DAW-overexpressing B16-F10 cells (Figures S4E and S4F). Further study indicated that ectopic expression of lncRNA-DAW increased the tumor-induced mortality (Figure 3H), suggesting that lncRNA-DAW might serve as a novel oncogene in liver cancer.
Figure 3.

lncRNA-DAW potentiated in vitro and in vivo tumor growth as well as cancer metastasis
(A) The RNA levels of lncRNA-DAW were examined in the lncRNA-DAW-overexpressing stable cell lines. (B) The effect of lncRNA-DAW on cell growth was determined by MTT assay (n = 4). (C) The effect of lncRNA-DAW on colony formation was evaluated by colony formation assay (n = 4). (D) The anchorage-independent growth was monitored by soft agar assay (n = 4). (E) The lncRNA-DAW and corresponding control cells were subcutaneously injected into nude mice. The nude mice were sacrificed and the tumor tissues were collected. Tumor weight and tumor volumes were measured and calculated (n = 5). (F) The lncRNA-DAW and vector-transfected stable cells were introduced into nude mice through tail vein. The lung tissues were collected at the indicated time points. (G) The lung tissues were subjected to hematoxylin-eosin (H&E) staining. Representative images were captured and the metastatic lesions from the indicated tissues were calculated (n = 5). (H) Survival rate of animals receiving tail vein injection of B16-F10 cancer cells overexpressing lncRNA-DAW and corresponding control cells (n = 10). ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Ectopic expression of lncRNA-DAW activated Wnt/β-catenin pathway through promoting Wnt2 expression
To investigate the mechanistic role of lncRNA-DAW in HCC, we conducted transcriptome study in lncRNA-DAW-overexpressing cells. The bioinformatics analysis of the RNA-seq data showed that enforced expression of lncRNA-DAW affected several signaling pathways (Figures 4A and S5). Considering that the cell surface receptor-linked signal transduction is essential for the development of liver cancer, several candidate genes from this category were chosen for further investigation, including Wnt2, Wnt9b, CCL8, and DKK4 (Figure 4A). Based on the qRT-PCR results, among the selected candidate genes, Wnt2 gene displayed the most remarkable change after overexpression of lncRNA-DAW (Figure 4B). To further validate this finding, we designed the siRNA targeting lncRNA-DAW, and silencing of lncRNA-DAW inhibited the Wnt2 RNA level (Figure 4C). Consistently, the western blotting results also confirmed the overexpression of lncRNA-DAW increased Wnt2 protein levels while knockdown of lncRNA-DAW inhibited Wnt2 expression (Figure 4D). The expression of Wnt2 was also increased in lncRNA-DAW-overexpressing tumor tissues derived from xenograft tumor model (Figure 4E).
Figure 4.

Wnt2 was a potential downstream target of lncRNA-DAW
(A) Gene Ontology enrichment analysis in the lncRNA-DAW-overexpressing cells. (B) qRT-PCR was applied to validate the expression alteration from RNA-seq data. (C) Silencing of lncRNA-DAW resulted in downregulation of Wnt2 (n = 4). (D) The protein levels of Wnt2 after ectopic expression or gene silencing of lncRNA-DAW were monitored by western blotting (n = 4). (E) The immunohistochemical staining of Wnt2 in lncRNA-DAW-overexpressing tumor tissues (n = 4). (F) The protein levels of total and phosphorylated β-catenin as well as GSK3β were examined by western blotting (n = 4). (G) The luciferase assay was used to monitor the effects of lncRNA-DAW in TOPflash and FOPflash vector (n = 4). (H) The RNA levels of β-catenin downstream target genes were determined by qRT-PCR (n = 4). ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Increased expression of Wnt ligand can attenuate the inhibitory phosphorylation of β-catenin and thus activate this signaling. In order to elucidate the effect of lncRNA-DAW on the Wnt/β-catenin pathway, we evaluated the protein levels of phosphorylated and total β-catenin. Ectopic expression of lncRNA-DAW repressed the inhibitory phosphorylation of β-catenin and increased β-catenin expression (Figure 4F), while opposite results were achieved by silencing lncRNA-DAW (Figure S6). Consistently, the luciferase assay showed that lncRNA-DAW significantly activated the luciferase activity of TOPflash vector with β-catenin/LEF1 binding sites, while it did not affect the luciferase activity of FOPflash vector with mutated β-catenin binding sites (Figure 4G). Furthermore, ectopic expression of lncRNA-DAW also activated the expression of several known β-catenin target genes (Figure 4H). All these data suggest that lncRNA-DAW overexpression activates Wnt/β-catenin signaling.
Wnt2 expression was suppressed by EZH2 in HCC cells
Previous study showed that lncRNAs might exert their function through physically interacting with Polycomb Repressive Complex 1 (PRC1), Polycomb Repressive Complex 2 (PRC2), or other RNA binding proteins.10,11 Thus, we monitored the Wnt2 mRNA level after ectopic expression of these proteins and found that overexpression of the core components from PRC2, especially EZH2, significantly suppressed Wnt2 mRNA expression (Figure 5A). Consistently, western blot also validated that Wnt2 was decreased after upregulation of EZH2 (Figure 5B). Given that EZH2 epigenetically silences gene expression through specifically inducing methylation on Lys-27 of histone 3 (H3K27me3), we conducted a ChIP assay using EZH2 and H3K27me3 antibodies and designed specific primers targeting Wnt2 promoter region. According to qRT-PCR results, these two antibodies significantly enriched the DNA fragment from the Wnt2 promoter compared with the negative control glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody (Figures 5C and 5D). To further evaluate the regulatory role of EZH2 in Wnt2 expression, we employed the EZH2 inhibitor 3-deazaneplanocin A (DZNep) to perform a loss-of-function study. It was shown that DZNep could attenuate the transcriptional suppression on Wnt2 by EZH2 (Figure 5E) and increase Wnt2 expression (Figure 5F). On the other hand, treatment with DZNep also significantly reduced the EZH2's occupancy of Wnt2 promoter (Figure 5G), suggesting that EZH2 epigenetically suppresses Wnt2 expression.
Figure 5.

EZH2 epigenetically silenced Wnt2 expression
(A) The expression of Wnt2 was determined by qRT-PCR after ectopic expression of several epigenetic regulators (n = 4). (B) The protein level of Wnt2 was determined by western blotting (n = 4). (C) Left: illustration of ChIP primer design within Wnt2 proximal promoter. Right: ChIP assay showed enrichment of EZH2 on Wnt2 promoter (n = 4), and GAPDH antibody was used as a negative control. (D) ChIP assay data showed enrichment of H3K27me3 modification on Wnt2 promoter (n = 4), and GAPDH antibody was used as a negative control. (E) The RNA level of Wnt2 after treatment with EZH2 inhibitor DZNep (n = 4). (F) The expression of Wnt2 was determined by qRT-PCR assay after treatment with DZNep (n = 4). (G) ChIP assay showed enrichment of EZH2 on Wnt2 promoter after treatment with DZNep (n = 4), and GAPDH antibody was used as a negative control. ∗∗p < 0.01; ∗∗∗p < 0.001.
lncRNA-DAW recruited EZH2 and promoted EZH2 phosphorylation and degradation
Currently, it is recognized that EZH2 might physically interact with several lncRNAs and trigger gene silencing.13 We wondered whether lncRNA-DAW might directly associate with EZH2 and thus regulate Wnt2 expression. To testify this hypothesis, we performed an RNA immunoprecipitation (RIP) assay and found that EZH2 successfully pulled down the lncRNA-DAW RNA transcript (Figure 6A) as well as a previously reported RNA partner, HOX Transcript Antisense RNA (HOTAIR), which has been demonstrated to recruit EZH2 in breast cancer. Next, to better understand how Wnt2 expression was regulated by lncRNA-DAW:EZH2 interaction, we conducted a ChIP assay to examine the EZH2's occupancy on the Wnt2 promoter. Interestingly, overexpression of lncRNA-DAW significantly reduced EZH2's binding to the Wnt2 promoter (Figure 6B), indicating that lncRNA-DAW attenuated EZH2's suppressive effect on the Wnt2 promoter.
Figure 6.

lncRNA-DAW alleviated the EZH2-mediated suppression on Wnt2 promoter by inducing EZH2 phosphorylation and degradation
(A) The RNA IP assay was conducted to investigate the interaction between lncRNA-DAW and EZH2 (n = 4). (B) ChIP assay showed the relative enrichment of EZH2 on Wnt2 promoter after overexpression of lncRNA-DAW (n = 4). (C) Western blot analysis of total EZH2 and phosphorylated EZH2 proteins in control and lncRNA-DAW-overexpressing cells (n = 4). (D) The protein levels of endogenous EZH2-associated CDK1 and Akt in control and lncRNA-DAW-overexpressing cells were determined by coIP assay in HepG2 cells (n = 4). (E) Western blots of endogenous EZH2-associated ubiquitination in control and lncRNA-DAW cells after IP with anti-EZH2 antibody in HepG2 cells (n = 4). (F) The EZH2 protein level was detected by western blot in the indicated cells after treatment with MG132 (10 μM) for 6 h (n = 4). ∗p < 0.05; ∗∗p < 0.01.
As many lncRNAs are found to modulate the phosphorylation and stability of the RNA binding proteins,14 we also examined the protein levels of total and phosphorylated EZH2 mediated by lncRNA-DAW. As expected, we found that EZH2 phosphorylation was enhanced while total EZH2 protein was reduced in lncRNA-DAW-overexpressing cells (Figure 6C). It was shown that the phosphorylation of EZH2 is crucial for modulating its protein stability. CDK1 and Akt are two major kinases that mediate the phosphorylation of EZH2 and suppress its methyltransferase activity,15,16 so we next examined the CDK1-EZH2 and Akt-EZH2 interaction by co-immunoprecipitation (coIP). We then immunoprecipitated endogenous EZH2 protein from lncRNA-DAW-expressing HepG2 cells and control cells to examine the CDK1-EZH2 and Akt-EZH2 interaction. It was found that EZH2 pulled down more CDK1 protein in the lncRNA-DAW-expressing cells compared with control cells (Figure 6D). However, enhanced expression of lncRNA-DAW did not affect the mutual interaction between Akt and EZH2 (Figure 6D).
Given that CDK1-mediated phosphorylation of EZH2 might trigger EZH2 ubiquitination, we utilized EZH2 antibody to pull down endogenous EZH2 proteins and examined their modification by ubiquitin antibody. According to the coIP results, we detected an increased EZH2 ubiquitination in lncRNA-DAW-expressing cells (Figure 6E), suggesting that lncRNA-DAW might modulate EZH2 stability by enhancing its ubiquitination. Because the protein stability of EZH2 is tightly modulated by the ubiquitin-proteasome pathway, we treated lncRNA-DAW-expressing cells or control cells with the proteasome inhibitor MG132 to investigate the protein stability of EZH2 in the presence or absence of MG132. Immunoblotting showed that the transient overexpression of lncRNA-DAW decreased endogenous EZH2 in a dose-dependent manner, whereas this decrease was suppressed by MG132 treatment (Figure 6F). This result proposed that destabilization of EZH2 mediated by lncRNA-DAW may depend on the proteasomal function. According to these results, lncRNA-DAW recruits EZH2 and induces EZH2 phosphorylation and degradation, which leads to promoting Wnt2 expression.
Wnt2 promoted tumor growth through activation of Wnt/β-catenin pathway
To disclose the regulatory role of Wnt2 in liver cancer cells, we evaluated the overexpression effect of Wnt2 on HepG2 and PLC/PRF/5 cells. The overexpression of Wnt2 was evaluated by qRT-PCR assays (Figure 7A), and forced expression of Wnt2 significantly promoted in vitro cell proliferation (Figure 7B) as well as colony formation capacity (Figure 7C). On the other hand, we also monitored Wnt2-mediated in vivo tumor growth and found that ectopic expression of Wnt2 enhanced tumor growth (Figure 7D; Figure S7). Moreover, increased expression of cell proliferation marker Ki-67 was observed in Wnt2-overexpressing tumor cells (Figure 7E).
Figure 7.

Wnt2 accelerated tumor growth via activation of Wnt/β-catenin pathway
(A) The RNA levels of Wnt2 were examined in the Wnt2-overexpressing stable cell lines (n = 3). (B) The effect of Wnt2 on cell growth was determined by MTT assay (n = 4). (C) The effect of Wnt2 in colony formation was evaluated by colony formation assay (n = 4). (D) 1 × 107 Wnt2 and corresponding control cells were subcutaneously injected into nude mice. The tumor weight and tumor volumes were measured and calculated (n = 5). (E) The Ki-67 staining in Wnt2-overexpressing tissues. (F) The effect of Wnt2 in modulating core components of the Wnt/β-catenin pathway (n = 4). (G) The luciferase assay was used to monitor the effect of Wnt2 in TOPflash and FOPflash vector (n = 4). (H) The RNA levels of β-catenin downstream target genes were determined by qRT-PCR (n = 4). ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
As a ligand, Wnt2 mediates tumor formation via Wnt/β-catenin signaling. To explore the underlying molecular mechanism of the proliferation-enhancing effect of Wnt2, the Wnt/β-catenin pathway in liver cancer cells was characterized. Western blotting results revealed that Wnt2 overexpression suppressed inhibitory phosphorylation of β-catenin and increased total β-catenin level (Figure 7F). To further evaluate the effect of Wnt2 on Wnt/β-catenin signaling, we utilized the TOPflash and FOPflash vector as described in Figure 4G. In addition, the luciferase assay showed that Wnt2 enhanced the luciferase activity of TOPflash vector but did not affect the luciferase activity of FOPflash vector (Figure 7G). Consistently, further studies demonstrated that Wnt2 also potentiated β-catenin's transcriptional activity and activated the gene expression of β-catenin downstream targets (Figure 7H; Figure S8). To sum up, these findings indicated that Wnt2 exerted its oncogenic function via direct activation of the canonical Wnt/β-catenin pathway.
Clinical association between lncRNA-DAW and Wnt2 was identified in liver cancer tissues
To further evaluate the clinical implication of Wnt2 in human cancer, we obtained its chromosome alteration status from the cBioPortal website. It was found that the DNA copy number of Wnt2 was frequently amplified across a variety of human cancer types (Figure 8A). Then we focused on the 50 pairs of liver cancer tissues and their adjacent non-cancerous tissues and verified that Wnt2 was significantly upregulated in liver cancer tissues (Figure 8B). In accordance with our results, the dataset extracted from Oncomine website confirmed that upregulation of Wnt2 was a frequent event in liver cancer tissues (Figure 8C). Furthermore, Kaplan-Meier analysis obtained from the PROGgene website revealed that patients with higher Wnt2 expression usually had a poor survival rate (Figure 8D).
Figure 8.

Wnt2 was upregulated and lncRNA-DAW expression was positively correlated with Wnt2 in HCC specimens
(A) The chromosome alteration of Wnt2 in different cancer types. (B) Expression profiles of Wnt2 in paired HCC and adjacent normal liver tissues were monitored by qRT-PCR. (C) The expression profiles of Wnt2 in liver cancer patients were obtained from the Oncomine database. (D) The Kaplan-Meier analysis of Wnt2 in liver cancer patients was obtained from the PROGgene website. (E) Expression profiles of lncRNA-DAW and Wnt2 in paired HCC and adjacent normal tissues. (F) The statistical association between lncRNA-DAW and Wnt2 expression in HCC specimens.
On the other hand, we then evaluated the expression profiles of lncRNA-DAW and Wnt2 in 50 paired liver cancer tissues. Both lncRNA-DAW and Wnt2 were upregulated in most of the HCC specimens in our study (Figure 8E). More importantly, a significantly positive association between the expression of lncRNA-DAW and Wnt2 was identified in these HCC specimens (Figure 8F). To sum up, these results indicated that increased lncRNA-DAW and Wnt2 expression may be a frequent event in human HCC tissues (Figure 9), indicating that lncRNA-DAW and Wnt2 might serve as putative therapeutic targets for liver cancer patients.
Figure 9.

Hypothetical model for lncRNA-DAW/EZH2/Wnt2 axis
Transcription factor HNF4G binds to the liver-specific super-enhancer and activates the expression of lncRNA-DAW. lncRNA-DAW physically interacts with EZH2, a negative regulator of Wnt2. This interplay subsequently leads to the degradation of EZH2. lncRNA-DAW-mediated EZH2 degradation facilitates the de-repression of Wnt2 transcription, which eventually activates the Wnt/β-catenin pathway.
Discussion
Hepatocarcinogenesis is regarded as a complicated and multi-step process including both genetic and epigenetic alterations within the human genome. Numerous oncogenes, including c-MYC, RAF1, CCND1, AKT, and ERBB2, are frequently amplified in a wide spectrum of human cancer types, suggesting that gene amplification is an important event that occurs in the genetic landscape alteration during hepatocarcinogenesis.17,18 Similar to other solid tumors, amplification of genomic DNA copy number is also frequently detected in HCC, including the amplification of 20q13.12. Previous investigation showed that gain of 20q13.12 segment was significantly associated with hepatic metastasis, overall survival, as well as unfavorable outcomes in patients with HCC.19 By integration of array comparative genomic hybridization (aCGH) and expression data on the 20q13.12-13.33 locus in liver cancer patients, a panel of candidate genes correlated with 20q13.12-13.33 gain were identified.19 For instance, RNF114 is one of the candidate oncogenes within this locus and it may contribute to the unfavorable outcomes and cancer recurrence. RNF114 belongs to the ubiquitin ligase family and has a critical regulatory role in modulating cell cycle progression, differentiation, and apoptosis.20, 21, 22 RNF114 also displays frequent copy number amplification and is commonly upregulated in a variety of human cancer types,19,21 suggesting its oncogenic potential during hepatocarcinogenesis. Moreover, in addition to a vital role in carcinogenesis, the 20q13.12 region has been implicated in several human diseases like type II diabetes and obesity.23 It was found that this genomic locus harbored novel promoters and distal regulatory elements, which might play a role in the development of diabetes and obesity. Taken together, these findings highlight the physiological and pathological importance of the 20q13.12 region in promoting cancer progression and maintaining the balanced harmony of cellular homeostasis.
In the present study, we characterized a novel lncRNA from the 20q13.12 region, which was termed lncRNA-DAW. Consistent with the previous cytogenetic study, lncRNA-DAW was frequently upregulated in liver cancer tissues. Subsequent high-throughput screening and functional studies identified Wnt2 as an important target of lncRNA-DAW in mediating its oncogenic function. Wnt2, a member of the WNT family, plays a critical role in orchestrating multiple biological events. In mouse, Wnt2 is essential for the proper vascularization of placenta, and approximately 50% of Wnt2-deficient mice died perinatally due to vascular defects.24 In Drosophila models, Wnt2 is required for cell fate determination during the development of the male reproductive tract, and the male Wnt2 null mutant flies have development defects in the reproductive tract.25 Intriguingly, Wnt2 upregulation was a common event in various human cancers.26, 27, 28 It was demonstrated that Wnt2 played pro-oncogene roles in many solid tumors, including non-small-cell lung cancer, esophageal cancer, pancreatic cancer, liver cancer, and gastro-intestinal cancer. Previous functional study verified that enhanced expression of Wnt2 promoted anchorage-independent cell growth, cancer metastasis, and tumor invasion.26,27,29,30 Subsequent mechanistic study demonstrated that the expression of Wnt2 was implicated in stabilizing β-catenin protein by antagonizing the function of the β-catenin-destruction complex, leading to the activation of the canonical Wnt/β-catenin pathway.26 In spite of the significant role of Wnt2 in malignant cancer, it remains largely unknown how Wnt2 is regulated under the epigenetic landscape.
Recent advances of genome-wide high-throughput sequencing data have revealed the abundance of lncRNA elements in mammalian transcriptomes. Surprisingly, many lncRNAs appear less conserved than the protein-encoding genes. Therefore, due to the lack of functional studies and sequence conservation, it is a great challenge to precisely predict the functional roles of lncRNAs under physiological or pathological conditions. Nevertheless, a recent breakthrough on RNA secondary structure indicates the importance of secondary structural elements within lncRNAs. In spite of poor sequence conservation, some lncRNAs display similar secondary structures. Therefore, it is speculated that RNA secondary structures might be evolutionarily conserved and the functional similarity of interspecies lncRNAs might be structure dependent rather than sequence dependent. However, in spite of these recent advances, many further investigations are still required to better illustrate the biological function of lncRNAs based on evolutionary conservation and RNA secondary structures.
Based on our results, we demonstrated that lncRNA-DAW could alleviate the suppressive effect of EZH2 on Wnt2 promoter, ultimately leading to the transcriptional activation of Wnt2. As a core component of PRC2, EZH2 epigenetically silences its target gene transcription by catalyzing the addition of methyl groups to histone H3 at lysine 27. In accordance with our results, previous study showed that EZH2 specifically occupied the promoter of Wnt2 and thus initiated histone modification H3K27me3 in HEK293T cells, which resulted in transcriptional inactivation of Wnt2.31 Nevertheless, whether any other player may get involved in this process remains elusive. Emerging as a novel modulator in a wide range of cellular events, lncRNAs exert their function through epigenetically modulating the gene expression of oncogene or tumor suppressor. For instance, the lncRNA HOTAIR promotes breast cancer metastasis by physically interacting with PRC2 and subsequently epigenetically silencing the HOXD gene cluster.32 These findings indicated that lncRNA-DAW might follow a similar regulatory mechanism and thus contribute to hepatocarcinogenesis. In this study, we identified lncRNA-DAW as a novel pattern of EZH2 and revealed that their mutual interaction may facilitate release of EZH2 from the Wnt2 proximal promoter, which shed light on the epigenetic dysregulation of Wnt2 in liver cancer cells.
In conclusion, our study revealed the oncogenic role of a previously unknown lncRNA in liver cancer. lncRNA-DAW was a liver-enriched lncRNA and it was frequently upregulated in liver cancer tissues. Subsequent functional studies demonstrated that overexpression of lncRNA-DAW significantly potentiated tumor growth and in vivo cancer metastasis. Further investigation revealed the molecular mechanism by which lncRNA-DAW diminished PRC2-mediated repression of Wnt2 by antagonizing the function of EZH2, eventually leading to the activation of the Wnt/β-catenin pathway and tumorigenesis. This finding might provide mechanistic insights into the molecular mechanisms underlying hepatocarcinogenesis, and lncRNA-DAW may serve as a therapeutic target for patients with HCC.
Materials and methods
Cell culture and retrovirus transduction
The HEK293T, B16-F10, LO2, Hep3B, Huh7, HepG2, and PLC/PRF/5 cells were all maintained in Dulbecco's modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum plus streptomycin (100 mg/mL) and penicillin (100 U/mL). The lncRNA-DAW-overexpressing cells were produced by using the retroviral gene delivery method. To generate virus particles, equal amounts of pBABE-lncRNA-DAW plasmid and virus packaging vector pCL-Ampho were co-transfected into HEK293T cells. The supernatant of culture medium was harvested and the virus particles were enriched by 0.45-μm pore size nitrocellulose membranes (Millipore, United States) at day 2. The virus concentration was determined by virus titer assay under the microscope. The virus was diluted to 1 × 106 TU/mL and used for infection. The indicated cell lines were then incubated with the above supernatant plus hexadimethrine bromide (Sigma-Aldrich, United States). Then the retrovirus-infected cells were selected with puromycin (Sigma-Aldrich, United States) at day 4. The virus-infected cells were harvested after antibiotic selection and the relative RNA levels were determined by qRT-PCR. As for the proteasome inhibitor experiment, HepG2 cells were transiently transfected with or without lncRNA-DAW expression plasmid. HepG2 cells were exposed to the proteasome inhibitor MG132 (20 μM) for 6 h before harvest.
Plasmid construction and siRNA synthesis
The lncRNA-DAW sequence was obtained from NCBI (http://www.ncbi.nlm.nih.gov/) and the chemically synthesized DNA fragment was inserted into pBabe-puro vector. The siRNAs against lncRNA-DAW were synthesized by RiboBio Company (Guangzhou, China). The related sequences are listed in Table S1.
RNA extraction and RT-PCR
The RNA samples were prepared as previously described.33, 34, 35 The total RNA of various human tissues was purchased from Clontech Company (TaKaRa, Japan). The RNA samples were extracted by the FavorPrep Tissue Total RNA Mini Kit (Favorgen, Hong Kong) following the manufacturer's instructions. The isolated RNA samples were subsequently converted into cDNA using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, United States). The relative expression levels of indicated genes were calculated by the 2−ΔΔCT method. The primer sequences are listed in Table S1.
Western blot
The protein samples were harvested by Radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldrich, United States) supplemented with Complete Protease Inhibitor Cocktail (Roche, United States). SDS-PAGE was conducted to separate the cellular proteins, and the polyvinylidene fluoride (PVDF) membranes were then probed with the indicated antibodies. The immunoblots were visualized on Kodak film developer (Fujifilm, Japan).
Colony formation assay
The lncRNA-DAW-overexpressing stable cells and corresponding negative control cells were plated into 12-well plates. After culturing in the incubator for 2 weeks, cells were visualized by crystal violet solution. The colony numbers were counted by ImageJ software (National Institutes of Health, United States).
RNA immunoprecipitation and ChIP
RNA immunoprecipitation (IP) and ChIP were performed as previously described.13,36,37 For RNA IP, the cells were fixed by formaldehyde and collected by NP-40 lysis buffer (Sigma-Aldrich, United States) supplemented with 1 mM PMSF, 1 mM DTT, 1% Protease Inhibitor Cocktail (Sigma-Aldrich, United States), as well as RNase Inhibitor (Life Technologies, United States). The supernatant of cell lysates was incubated with Protein G Sepharose 4 Fast Flow bead slurry (GE Healthcare, United States) pre-coated with the indicated antibody. The co-precipitated RNAs were extracted using TRIzol reagent (Invitrogen, United States) and the target genes were detected by qRT-PCR.
For ChIP assay, after cross-linking with 1% formaldehyde, the cells were lysed by cold IP buffer (150 mM NaCl, 50 mM Tris-HCl [pH 7.5], 5 mM EDTA, 0.5% NP-40, 1% Triton X-100) and the chromatin samples were isolated by centrifuge. After sonication, chromatin samples were incubated with the EZH2 or H2K27me3 antibodies overnight at 4°C with gentle shaking. Then, the samples were mixed with protein G beads at 4°C for 2 h. After several washes with cold IP buffer, 10% Chelex 100 slurry suspension was added to the beads and boiled for 10 min. The samples were centrifuged and the supernatant was collected for subsequent PCR analysis. The ChIP primer sequences are listed in Table S1.
CoIP
The cells were lysed by cold lysis buffer (1% Triton X-100, 50 mM Tris-7.5, 1 mM EDTA, 150 mM NaCl, and protease inhibitors). The cell lysates were sonicated and centrifuged. The supernatant was collected and incubated with indicated antibodies overnight at 4°C with gentle shaking. Then, the protein G beads were added to the lysates at 4°C. After 2 h of incubation, the beads were washed by cold lysis buffer. The washing buffer was removed and the protein loading buffer was added to beads. The beads and protein loading buffer were boiled and subjected to western blotting analysis.
Luciferase reporter assay
HEK293T cells were cultured and co-transfected with indicated luciferase reporter plasmids. Then the HEK293T cells were lysed by lysis buffer and the luciferase activity was measured by the Dual-Luciferase Reporter Assay System (Promega, Hong Kong).
Animal study
For the xenograft tumor model, 1 × 107 lncRNA-DAW-overexpressing cells and their parallel control cells were subcutaneously implanted into nude mice. The tumor volume was calculated through using the following formula: Volume = (Length × Width2)/2. The nude mice were sacrificed at the indicated time points and the tumor tissues were harvested for further investigation. For the in vivo metastasis assay, the indicated cancer cells were introduced into C57BL/6 mice through hydrodynamic tail vein injection with slight modification. Two milliliters of PBS solution containing tumor cells was introduced to mice through the tail vein within 10 s. The C57BL/6 mice were sacrificed and the indicated tissues were excised and subsequently fixed in paraformaldehyde solution for further investigation. The animal handling procedure was conducted under the institutional Laboratory Animal Services Centre (LASEC) Guidelines in The Chinese University of Hong Kong.
Clinical tissue sample analysis
Fifty paired tumor tissues and normal adjacent tissues were collected from tumor resection in the Prince of Wales Hospital (The Chinese University of Hong Kong, Hong Kong) as previously described.13 All the human tissues were collected with informed consent from patients, and this investigation was approved by The Joint Chinese University of Hong Kong-New Territories Ease Cluster Clinical Research Ethics Committee.
Bioinformatics analysis
A total of eight RefSeq-validated long non-protein-coding RNA genes were found in the chromosome 20q13.12 region. We retrieved the reads per kilobase per million mapped reads (RPKM) of the eight genes in 20 human tissues from NCBI BioProject RNA sequencing of total RNA from 20 human tissues (PRJNA280600). Cross-tissue expression patterns of the eight genes were illustrated in heatmap, where the raw RPKMs were scaled by the tissue type. The plot was generated in R with function heatmap.2. The online bioinformatics programs, cBioPortal (http://www.cbioportal.org/index.do), Oncomine (https://www.oncomine.org) and PROGgene (http://watson.compbio.iupui.edu/chirayu/proggene/database/?url=proggene) were applied to analyze the clinical implication of Wnt2 in liver cancer patients.
Ethics approval and consent to participate
The animal handling procedures were approved by the institutional LASEC Guidelines in The Chinese University of Hong Kong. All the tumor tissues were collected with consent forms prior to enrollment, and this clinical study was approved by The Joint Chinese University of Hong Kong-New Territories Ease Cluster Clinical Research Ethics Committee. The experimental methods complied with the Helsinki Declaration.
Statistics
Experimental data are expressed as mean ± SD. Significance of difference was evaluated using two-tail Student’s t test. The correlation between lncRNA-DAW and Wnt2 in HCC specimens was performed by using Pearson's correlation in GraphPad Prism 5.0. Differences were considered to be statistically significant when p < 0.05.
Acknowledgments
We are grateful to the patients for their contributions to this study. This work was supported by grants from the National Natural Science Foundation of China (81902886 to W.L., 81773066 to W.F., and 81772404 to J.Z.), Medical Scientific Research Foundation of Guangdong Province of China (A2021134 to P.L.), and the Fundamental Research Funds for the Central Universities, Sun Yat-sen University (2021qntd34).
Author contributions
W.L., L.L., and J.Z. conceived the project and designed the experiments. W.L., C.S., W.H., and P.L. conducted the experiments. X.Z. and W.F. did data analysis. W.L. wrote the manuscript. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.10.028.
Contributor Information
Lizhu Lin, Email: lizhulin_llz@163.com.
Jinfang Zhang, Email: zhangjf06@gzucm.edu.cn.
Supplemental information
References
- 1.Hu J.J., Che L., Li L., Pilo M.G., Cigliano A., Ribback S., Li X.L., Latte G., Mela M., Evert M., et al. Co-activation of AKT and c-Met triggers rapid hepatocellular carcinoma development via the mTORC1/FASN pathway in mice. Sci. Rep. 2016;6:20484. doi: 10.1038/Srep20484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qi W., Liang W.C., Waye M.M., Jiang H.Q. The function of miRNA in hepatic cancer stem cell. Biomed. Res. Int. 2013;2013:358902. doi: 10.1155/2013/358902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang C.M., Cigliano A., Delogu S., Armbruster J., Dombrowski F., Evert M., Chen X., Calvisi D.F. Functional crosstalk between AKT/mTOR and Ras/MAPK pathways in hepatocarcinogenesis implications for the treatment of human liver cancer. Cell Cycle. 2013;12:1999–2010. doi: 10.4161/cc.25099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takigawa Y., Brown A.M.C. Wnt signaling in liver cancer. Curr. Drug Targets. 2008;9:1013–1024. doi: 10.2174/138945008786786127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Waisberg J., Saba G.T. Wnt-/-beta-catenin pathway signaling in human hepatocellular carcinoma. World J. Hepatol. 2015;7:2631–2635. doi: 10.4254/wjh.v7.i26.2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu W.Q., Kimelman D. Mechanistic insights from structural studies of beta-catenin and its binding partners. J. Cell Sci. 2007;120:3337–3344. doi: 10.1242/jcs.013771. [DOI] [PubMed] [Google Scholar]
- 7.Zhang T., Song X., Zhang Z., Mao Q., Xia W., Xu L., Jiang F., Dong G. Aberrant super-enhancer landscape reveals core transcriptional regulatory circuitry in lung adenocarcinoma. Oncogenesis. 2020;9:92. doi: 10.1038/s41389-020-00277-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie J.J., Jiang Y.Y., Jiang Y., Li C.Q., Lim M.C., An O., Mayakonda A., Ding L.W., Long L., Sun C., et al. Super-enhancer-driven long non-coding RNA LINC01503, regulated by TP63, is over-expressed and oncogenic in squamous cell carcinoma. Gastroenterology. 2018;154:2137–2151.e1. doi: 10.1053/j.gastro.2018.02.018. [DOI] [PubMed] [Google Scholar]
- 9.Huarte M. The emerging role of lncRNAs in cancer. Nat. Med. 2015;21:1253–1261. doi: 10.1038/nm.3981. [DOI] [PubMed] [Google Scholar]
- 10.Rinn J.L., Chang H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012;81:145–166. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Quinn J.J., Chang H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016;17:47–62. doi: 10.1038/nrg.2015.10. [DOI] [PubMed] [Google Scholar]
- 12.Andrews S.J., Rothnagel J.A. Emerging evidence for functional peptides encoded by short open reading frames. Nat. Rev. Genet. 2014;15:193–204. doi: 10.1038/nrg3520. [DOI] [PubMed] [Google Scholar]
- 13.Fu W.M., Zhu X., Wang W.M., Lu Y.F., Hu B.G., Wang H., Liang W.C., Wang S.S., Ko C.H., Waye M.M., et al. Hotair mediates hepatocarcinogenesis through suppressing miRNA-218 expression and activating P14 and P16 signaling. J. Hepatol. 2015;63:886–895. doi: 10.1016/j.jhep.2015.05.016. [DOI] [PubMed] [Google Scholar]
- 14.Li Z.W., Hou P.F., Fan D.M., Dong M.C., Ma M.S., Li H.Y., Yao R.S., Li Y.X., Wang G.N., Geng P.Y., et al. The degradation of EZH2 mediated by lncRNA ANCR attenuated the invasion and metastasis of breast cancer. Cell Death Differ. 2017;24:59–71. doi: 10.1038/cdd.2016.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cha T.L., Zhou B.H.P., Xia W.Y., Wu Y.D., Yang C.C., Chen C.T., Ping B., Otte A.P., Hung M.C. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science. 2005;310:306–310. doi: 10.1126/science.1118947. [DOI] [PubMed] [Google Scholar]
- 16.Zeng X.Z., Chen S.A., Huang H.J. Phosphorylation of EZH2 by CDK1 and CDK2 A possible regulatory mechanism of transmission of the H3K27me3 epigenetic mark through cell divisions. Cell Cycle. 2011;10:579–583. doi: 10.4161/cc.10.4.14722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li L., Pilo G.M., Li X.L., Cigliano A., Latte G., Che L., Joseph C., Mela M., Wang C.M., Jiang L.J., et al. Inactivation of fatty acid synthase impairs hepatocarcinogenesis driven by AKT in mice and humans. J. Hepatol. 2016;64:333–341. doi: 10.1016/j.jhep.2015.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beroukhim R., Mermel C.H., Porter D., Wei G., Raychaudhuri S., Donovan J., Barretina J., Boehm J.S., Dobson J., Urashima M., et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang D., Zhu Z.Z., Jiang H.M., Zhu J.Y., Cong W.M., Wen B.J., He S.Q., Liu S.F. Multiple genes identified as targets for 20q13.12-13.33 gain contributing to unfavorable clinical outcomes in patients with hepatocellular carcinoma. Hepatol. Int. 2015;9:438–446. doi: 10.1007/s12072-015-9642-0. [DOI] [PubMed] [Google Scholar]
- 20.Rodriguez M.S., Egana I., Lopitz-Otsoa F., Aillet F., Lopez-Mato M.P., Dorronsoro A., Lobato-Gil S., Sutherland J.D., Barrio R., Trigueros C., Lang V. The RING ubiquitin E3 RNF114 interacts with A20 and modulates NF-kappa B activity and T-cell activation. Cell Death Dis. 2014;5:e1399. doi: 10.1038/cddis.2014.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han J., Kim Y.L., Lee K.W., Her N.G., Ha T.K., Yoon S., Jeong S.I., Lee J.H., Kang M.J., Lee M.G., et al. ZNF313 is a novel cell cycle activator with an E3 ligase activity inhibiting cellular senescence by destabilizing p21(WAF1) Cell Death Differ. 2013;20:1055–1067. doi: 10.1038/cdd.2013.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee M.G., Han J., Jeong S.I., Her N.G., Lee J.H., Ha T.K., Kang M.J., Ryu B.K., Chi S.G. XAF1 directs apoptotic switch of p53 signaling through activation of HIPK2 and ZNF313. Proc. Natl. Acad. Sci. U S A. 2014;111:15532–15537. doi: 10.1073/pnas.1411746111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akan P., Sahlen M., Deloukas P. A histone map of human chromosome 20q13.12. PLoS One. 2009;4:e4479. doi: 10.1371/journal.pone.0004479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Monkley S.J., Delaney S.J., Pennisi D.J., Christiansen J.H., Wainwright B.J. Targeted disruption of the Wnt2 gene results in placentation defects. Development. 1996;122:3343–3353. doi: 10.1242/dev.122.11.3343. [DOI] [PubMed] [Google Scholar]
- 25.Kozopas K.M., Samos C.H., Nusse R. DWnt-2, a Drosophila Wnt gene required for the development of the male reproductive tract, specifies a sexually dimorphic cell fate. Genes Dev. 1998;12:1155–1165. doi: 10.1101/gad.12.8.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fu L., Zhang C.Y., Zhang L.Y., Dong S.S., Lu L.H., Chen J., Dai Y.D., Li Y., Kong K.L., Kwong D.L., Guan X.Y. Wnt2 secreted by tumour fibroblasts promotes tumour progression in oesophageal cancer by activation of the Wnt/beta-catenin signalling pathway. Gut. 2011;60:1635–1643. doi: 10.1136/gut.2011.241638. [DOI] [PubMed] [Google Scholar]
- 27.Katoh M. Frequent up-regulation of WNT2 in primary gastric cancer and colorectal cancer. Int. J. Oncol. 2001;19:1003–1007. doi: 10.3892/ijo.19.5.1003. [DOI] [PubMed] [Google Scholar]
- 28.Yu M., Ting D.T., Stott S.L., Wittner B.S., Ozsolak F., Paul S., Ciciliano J.C., Smas M.E., Winokur D., Gilman A.J., et al. RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature. 2012;487:510–513. doi: 10.1038/nature11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bravo D.T., Yang Y.L., Kuchenbecker K., Hung M.S., Xu Z.D., Jablons D.M., You L. Frizzled-8 receptor is activated by the Wnt-2 ligand in non-small cell lung cancer. BMC Cancer. 2013;13:316. doi: 10.1186/1471-2407-13-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vider B.Z., Zimber A., Chastre E., Prevot S., Gespach C., Estlein D., Wolloch Y., Tronick S.R., Gazit A., Yaniv A. Evidence for the involvement of the Wnt 2 gene in human colorectal cancer. Oncogene. 1996;12:153–158. [PubMed] [Google Scholar]
- 31.Jung Y.S., Jun S., Lee S.H., Sharma A., Park J.I. Wnt2 complements Wnt/beta-catenin signaling in colorectal cancer. Oncotarget. 2015;6:37257–37268. doi: 10.18632/oncotarget.6133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gupta R.A., Shah N., Wang K.C., Kim J., Horlings H.M., Wong D.J., Tsai M.C., Hung T., Argani P., Rinn J.L., et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–1076. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liang W.C., Liang P.P., Wong C.W., Ng T.B., Huang J.J., Zhang J.F., Waye M.M., Fu W.M. CRISPR/Cas9 technology targeting Fas gene protects mice from Concanavalin-A induced fulminant hepatic failure. J. Cell Biochem. 2017;118:530–536. doi: 10.1002/jcb.25722. [DOI] [PubMed] [Google Scholar]
- 34.Liang W.C., Wang Y., Liang P.P., Pan X.Q., Fu W.M., Yeung V.S., Lu Y.F., Wan D.C., Tsui S.K., Tsang S.Y., et al. MiR-25 suppresses 3T3-L1 adipogenesis by directly targeting KLF4 and C/EBPalpha. J. Cell Biochem. 2015;116:2658–2666. doi: 10.1002/jcb.25214. [DOI] [PubMed] [Google Scholar]
- 35.Liang W.C., Wang Y., Xiao L.J., Wang Y.B., Fu W.M., Wang W.M., Jiang H.Q., Qi W., Wan D.C., Zhang J.F., Waye M.M. Identification of miRNAs that specifically target tumor suppressive KLF6-FL rather than oncogenic KLF6-SV1 isoform. RNA Biol. 2014;11:845–854. doi: 10.4161/rna.29356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liang W.C., Fu W.M., Wang Y.B., Sun Y.X., Xu L.L., Wong C.W., Chan K.M., Li G., Waye M.M., Zhang J.F. H19 activates Wnt signaling and promotes osteoblast differentiation by functioning as a competing endogenous RNA. Sci. Rep. 2016;6:20121. doi: 10.1038/srep20121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liang W.C., Fu W.M., Wong C.W., Wang Y., Wang W.M., Hu G.X., Zhang L., Xiao L.J., Wan D.C., Zhang J.F., Waye M.M. The lncRNA H19 promotes epithelial to mesenchymal transition by functioning as miRNA sponges in colorectal cancer. Oncotarget. 2015;6:22513–22525. doi: 10.18632/oncotarget.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.