

Abstract
Water is essential to protein structure and stability, yet our understanding of how water shapes proteins is far from thorough. Our incomplete knowledge of protein–water interactions is due in part to a long-standing technological inability to assess experimentally how water removal impacts local protein structure. It is now possible to obtain residue-level information on dehydrated protein structures via liquid-observed vapor exchange (LOVE) NMR, a solution NMR technique that quantifies the extent of hydrogen–deuterium exchange between unprotected amide protons of a dehydrated protein and D2O vapor. Here, we apply LOVE NMR, Fourier transform infrared spectroscopy, and solution hydrogen–deuterium exchange to globular proteins GB1, CI2, and two variants thereof to link mutation-induced changes in the dehydrated protein structure to changes in solution structure and stability. We find that a mutation that destabilizes GB1 in solution does not affect its dehydrated structure, whereas a mutation that stabilizes CI2 in solution makes several regions of the protein more susceptible to dehydration-induced unfolding, suggesting that water is primarily responsible for the destabilization of the GB1 variant but plays a stabilizing role in the CI2 variant. Our results indicate that changes in dehydrated protein structure cannot be predicted from changes in solution stability alone and demonstrate the ability of LOVE NMR to uncover the variable role of water in protein stability. Further application of LOVE NMR to other proteins and their variants will improve the ability to predict and modulate protein structure and stability in both the hydrated and dehydrated states for applications in medicine and biotechnology.
Kauzmann’s 1959 review, “Some Factors in the Interpretation of Protein Denaturation”, brought to light the key role of water in determining protein structure, stability, and function.1,2 Yet, despite over six decades of research into protein–water interactions, our ability to predict, let alone experimentally assess, the contribution of water to local protein structure and stability remains limited.3
Our inadequate understanding of how protein–water interactions shape and stabilize native protein structure stems in part from the technological inability to observe the impact of dehydration on local protein structure experimentally. For years, Fourier transform infrared (FTIR) spectroscopy was one of a few techniques capable of assessing dehydrated protein structures, yet this and most other techniques can only provide information on the global secondary structure composition.4 However, with the recent development of liquid-observed vapor exchange nuclear magnetic resonance (LOVE NMR),5 it is now possible to localize, at the residue-level, protein regions that lose structure upon dehydration.
Based on the well-established principle that hydrogen bonds are essential to protein structures6,7 and that amide protons are less likely to exchange with deuterons from the environment if involved in intra- or intermolecular H-bonds,8 LOVE NMR5 uses solution NMR spectroscopy to quantify the extent of hydrogen–deuterium exchange (HDX) between D2O vapor and the amide protons of a dried protein. For a given residue, the percent of the amide-proton signal remaining after vapor exchange (%Protected) reflects the fraction of the dry protein population trapped in a conformation in which that residue is protected from exchange. Given that dried protein samples are conformationally heterogeneous,9 the fraction of the population in which a residue is protected likely comprises an ensemble of conformations. Thus, LOVE NMR pinpoints where, and with what probability, dehydration causes unfolding, i.e., which protein regions depend most on interactions with water to maintain native structure.
Here, we use an improved version of LOVE NMR (Figure S1), FTIR spectroscopy, and NMR-detected solution HDX to link mutation-induced changes in dehydrated protein structure to changes in solution stability. We apply this methodology to two recombinant proteins: the 6 kDa B1 domain of staphylococcal protein G (GB1) and 7 kDa chymotrypsin inhibitor 2 (CI2) from barley (Figure 1), and two variants: I6L GB1 and I20V CI2. The variants were selected because both perturb a “global unfolding residue”, i.e., a residue whose opening free energy is comparable to the global stability of the protein.10–12
Figure 1.
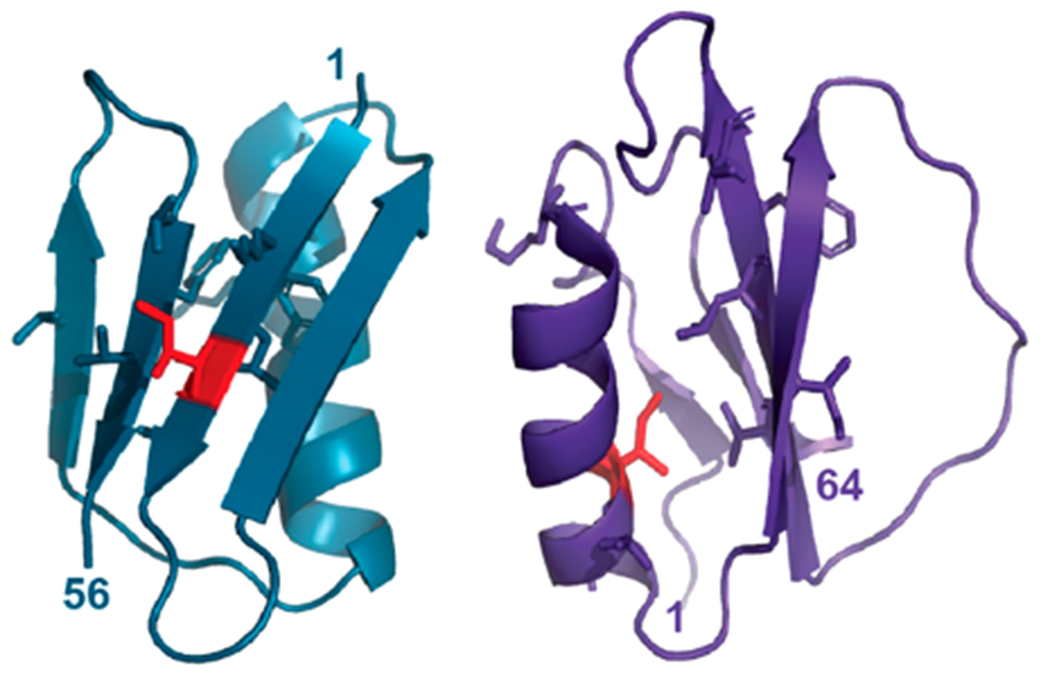
Structures of GB1 (left, PDB 2QMT) and CI2 (PDB 2CI2). Side chains are shown for global unfolding residues, with mutated residues in red.
To enable direct comparison of hydrated and dehydrated structures and provide a reference for the LOVE NMR data, we also employed attenuated total reflection FTIR (ATRFTIR) spectroscopy. Secondary structure information (Figure 2) was derived from the fitting of the amide 1 band (Figure S2, Table S1).
Figure 2.
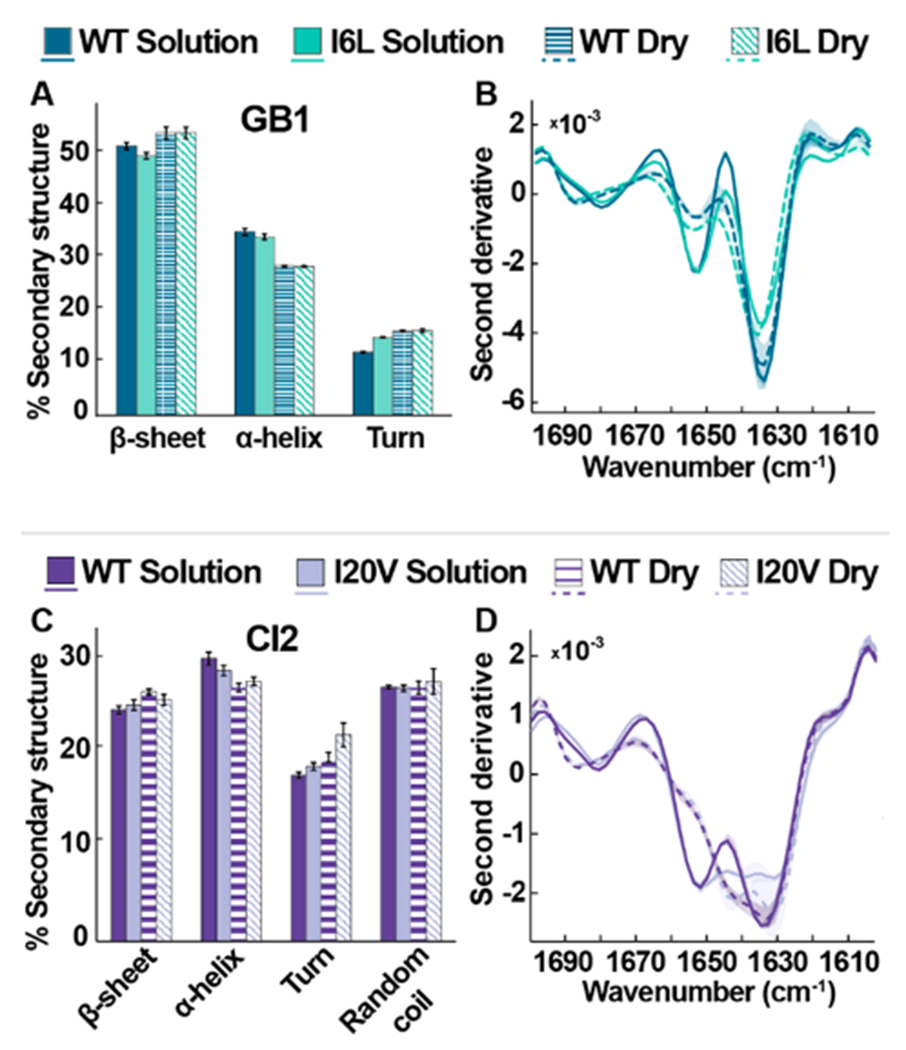
FTIR studies of wild-type and variant proteins in solution (7.5 mM HEPES, pH 6.5) and the lyophilized state. (A, C) Average secondary structures of GB1 and CI2 variants. Error bars represent standard deviations from the mean. (B, D) Averaged second derivative spectra. Shadows represent the range of data from three independent experiments.
Upon lyophilization, all samples exhibit an increase in β-sheet character and a decrease in α-helical character (Figure 2A,C). Inspection of the second derivative plots (Figure 2B,D) reveals that upon dehydration, the proteins exhibit a frequency upshift in the 1680 cm−1 β-sheet region, indicating increased amide-bond strength and/or a change in bond orientation.13
In solution, I6L GB1 exhibits slightly less β- and more turn character than the wild-type (WT) protein (Figure 2A), but the differences are lost upon lyophilization. Inspection of the second derivative data (Figure 2B) reveals that, in the dry state, both GB1 proteins exhibit an intensity decrease in the 1652 cm−1 helix region, suggesting a broader variety of helix conformations in the lyophilized form.14–16
In solution, WT and I20V CI2 possess similar secondary structure profiles, with I20V exhibiting only a slight decrease in α-helical character relative to WT (Figure 2C). Second derivative solution spectra of I20V exhibit reduced intensity at 1630 cm−1 (Figure 2D), suggesting reduced β-sheet interactions, while the uniformity of shape implies an overall consistent conformation.14 Differences between WT and I20V CI2 become more apparent in the dry state, with I20V exhibiting slightly less β-sheet character and more turn character than WT (Figure 2C). Like the GB1 proteins, second derivative spectra of the CI2 proteins reveal that dehydration decreases the intensity at 1655 cm−1 (Figure 2D), again indicative of a broader variety of helix conformations. Relative to GB1, however, the FTIR data suggest that the CI2 is less sensitive to dehydration, as the proteins exhibit a smaller secondary structure change between the solution and dry state (Figure 2C).
To gain more information on how mutation affects the solution and solid states, we used HDX to acquire residue-level solution opening free energies 17 and LOVE NMR to acquire %Protected values in the dry state. For GB1 in solution, the I6L substitution is generally destabilizing, reducing the average measurable value by 0.3 ± 0.4 kcal/mol and the of global unfolding residues by 0.7 ± 0.2 kcal/mol (Figure 3A), where the uncertainty is the sample standard deviation of the mean. Residues preceding or following a global-unfolder experience small stability increases, with L7 witnessing the largest increase (0.52 ± 0.04 kcal/mol). Despite these changes in solution stability, the GB1 proteins exhibit nearly identical LOVE profiles (Figure 3B), in agreement with our FTIR analysis.
Figure 3.
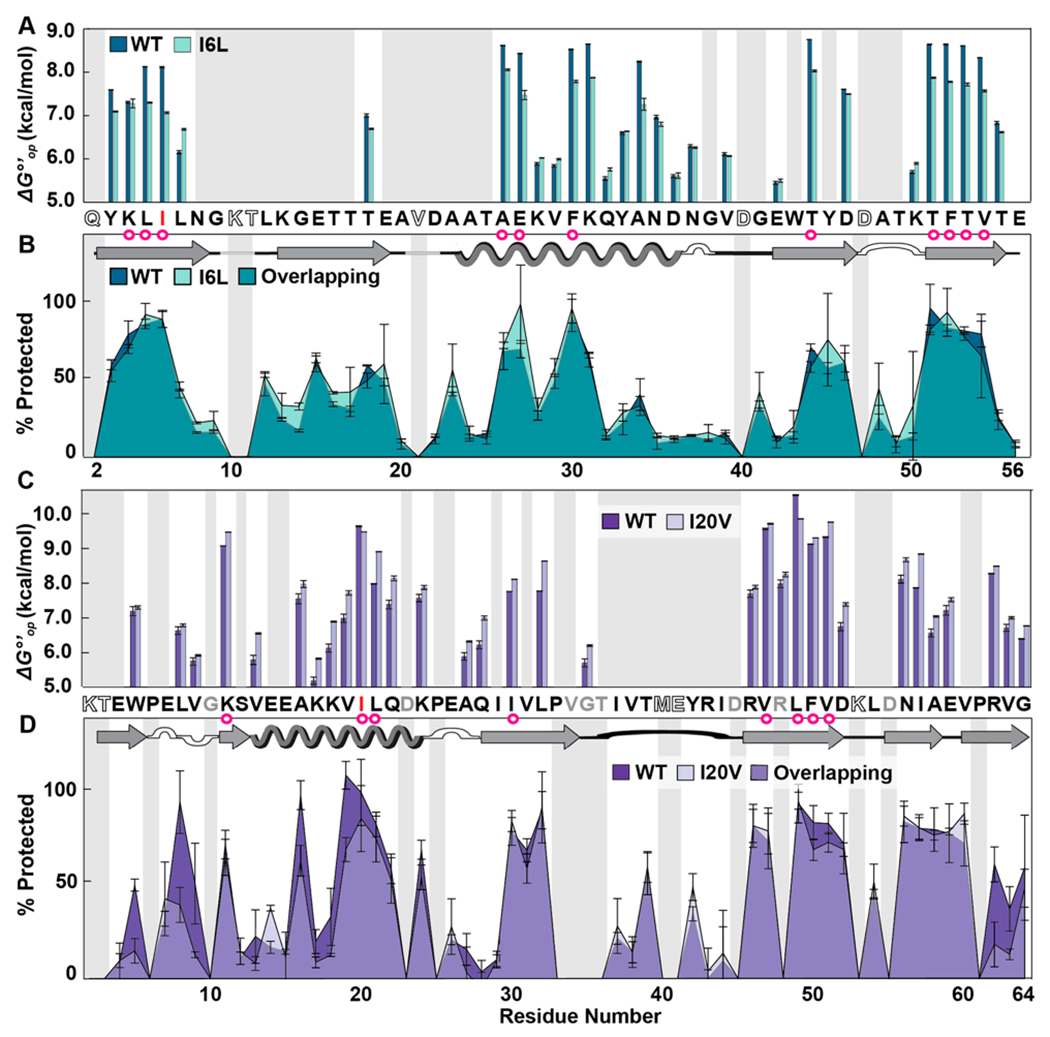
Residue-level solution stabilities and dry state structures of wild-type (WT) and variant proteins. (A) Opening free energies (7.5 mM HEPES, pH 7.5, 22 °C). (B) Overlaid LOVE profiles of WT and I6L GB1 freeze-dried in 1.5 mM HEPES pH 6.5. (C) Opening free energies CI2. (D) Overlaid LOVE profiles of WT and I20V CI2 freeze-dried in 1.5 mM HEPES pH 6.5. Primary and secondary structures of the WT proteins are shown between panels. In both primary structures, red letters indicate which residue was mutated. Magenta circles indicate solution global-unfolding residues of WT proteins. Gray boxes indicate data that are missing due to rapid back exchange (open letters in primary structure) and/or the inability to reliably integrate peak volumes due to overlapping resonances (gray letters in primary structure). Error bars represent standard deviations from the mean calculated from triplicate analysis.
In solution, the I20V CI2 substitution is, at best, marginally stabilizing, increasing the average measurable by 0.4 ± 0.3 kcal/mol and the average of global unfolders by 0.2 ± 0.5 kcal/mol (Figure 3C). Only the site of the mutation (V20) and L49, a residue within the hydrophobic core, are less stable in the variant.
We expected the isoleucine-to-valine change to be slightly destabilizing because less hydrophobic surface is buried in the variant. Specifically, pure surface area arguments suggest a reduction of 0.9 kcal/mol,18 but the decrease could be much smaller because side chains are never completely exposed in the unfolded state.19 This disparity is probably caused by a small stress-relieving conformational change.
Although the I20V mutation is marginally stabilizing in solution, LOVE NMR data show that the mutation reduces dry-state protection in certain regions (Figure 3D). Specifically, the terminal β-sheets of dehydrated I20V CI2 are less than half as protected as they are in the WT protein. In addition, V19, the residue preceding the mutation; A16, the native H-bond acceptor of I20; and L8, the H-bond donor of one of the residues in the N-terminal β-sheet, all experience reduced dry-state protection.
Our results indicate that changes in dehydrated protein structure are not predicted by changes in solution stability alone, pointing to a variable role for water in maintaining local protein structure and stability. Although both the I6L and I20V substitutions perturb global unfolding residues, there are two key structural differences. First, in GB1, the side chain of isoleucine 6 faces outward, while in CI2, that of isoleucine 20 faces inward toward the hydrophobic core (Figure 1). Second, the I6L substitution occurs in a β-strand, while the I20V substitution occurs in an α-helix. These distinguishing characteristics may help explain the contrasting LOVE NMR data.
The observation that the I6L substitution destabilizes GB1 in solution but has almost no effect on dehydrated structure suggests that water is primarily responsible for the reduced stability of the variant. Given the water-facing location of the position-6 side chain, one mechanism of water-mediated destabilization could reduce the “side chain blocking” of hydration by leucine relative to isoleucine. This mechanism of β-sheet destabilization, which was suggested by Bai and Englander,20 would affect the protein in solution but not in the dry state.
The observation that the I20V substitution slightly stabilizes CI2 in solution but makes the protein more susceptible to dehydration suggests that water plays an enhanced stabilizing role in the I20V variant, particularly at the terminal β-sheets. The origin of this stabilization is unclear, but the results are consistent with those of Ladurner et al., who found strain in the α-helix of WT CI2 from nonoptimal core packing;21 perhaps alleviating some of the steric strain in the core via a volume-reducing mutation allows tighter core packing and an enhanced hydrophobic effect. Alternatively, the mutation could lead to an increase in the number or strength of hydrogen bonds with water.
In summary, comparisons of mutation-induced changes in protein solution stability to changes in dehydrated protein structure suggest that water plays a dominant role in the destabilization of the I6L variant of GB1 and a more nuanced, stabilizing role in the I20V variant of CI2. Our results illustrate the complex nature of protein–water interactions and demonstrate the ability of LOVE NMR to localize structural changes in the dry state.
We envision that the residue-level information on the dehydrated protein structure provided by LOVE NMR can be combined with other techniques to gain a deeper understanding of water’s role in determining protein structure and stability. Understanding the fundamental interaction between proteins and water will, in turn, improve the ability to predict protein structure and stability, enabling the de novo design of protein-based therapeutics and industrial enzymes.
Supplementary Material
ACKNOWLEDGMENTS
We thank Stu Parnham (UNC Biomolecular NMR Core) and Brandie Ehrmann (UNC Chemistry Mass Spectrometry Core) for equipment maintenance and advice. We also thank the Pielak lab for the helpful discussion and Elizabeth Pielak for comments on the manuscript.
Funding
This research was supported by NIH grant R01GM127291 to G.J.P. and a NIH training grant (T32GM008570) to C.J.C.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.1c00552.
Assigned 1H–15N heteronuclear single quantum coherence (HSQC) spectra (pH 4.5, 4 °C and pH 7.5, 22 °C); ATR-FTIR spectra in the solution and dry states; peak fits derived from FTIR; %Protected and ΔGop°′ values (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.biochem.1c00552
The authors declare no competing financial interest.
Contributor Information
Candice J. Crilly, Department of Chemistry, University of North Carolina at Chapel Hill (UNC–CH), Chapel Hill, North Carolina 27599-3290, United States
Jonathan E. Eicher, Department of Chemistry, University of North Carolina at Chapel Hill (UNC–CH), Chapel Hill, North Carolina 27599-3290, United States
Owen Warmuth, Department of Chemistry, University of North Carolina at Chapel Hill (UNC–CH), Chapel Hill, North Carolina 27599-3290, United States.
Joanna M. Atkin, Department of Chemistry, University of North Carolina at Chapel Hill (UNC–CH), Chapel Hill, North Carolina 27599-3290, United States
Gary J. Pielak, Department of Chemistry, University of North Carolina at Chapel Hill (UNC–CH), Chapel Hill, North Carolina 27599-3290, United States; Department of Biochemistry & Biophysics, Lineberger Cancer Center, and Integrative Program for Biological and Genome Sciences, UNC–CH, Chapel Hill, North Carolina 27599, United States.
REFERENCES
- (1).Kauzmann W Some Factors in the Interpretation of Protein Denaturation. Adv. Protein Chem 1959, 14, 1–63. [DOI] [PubMed] [Google Scholar]
- (2).Bellissent-Funel M-C; Hassanali A; Havenith M; Henchman R; Pohl P; Sterpone F; van der Spoel D; Xu Y; Garcia AE Water Determines the Structure and Dynamics of Proteins. Chem. Rev 2016, 116 (13), 7673–7697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Anandakrishnan R; Izadi S; Onufriev AV Why Computed Protein Folding Landscapes Are Sensitive to the Water Model. J. Chem. Theory Comput 2019, 15 (1), 625–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Moorthy BS; Iyer LK; Topp EM Characterizing Protein Structure, Dynamics and Conformation in Lyophilized Solids. Curr. Pharm. Des 2015, 21 (40), 5845–5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Crilly CJ; Brom JA; Kowalewski ME; Piszkiewicz S; Pielak GJ Dried Protein Structure Revealed at the Residue Level by Liquid-Observed Vapor Exchange NMR. Biochemistry 2021, 60 (2), 152–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Pauling L; Corey RB; Branson HR The Structure of Proteins: Two Hydrogen-Bonded Helical Configurations of the Polypeptide Chain. Proc. Natl. Acad. Sci. U. S. A 1951, 37 (4), 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Rose GD Protein Folding - Seeing Is Deceiving. Protein Sci. 2021, 30 (8), 1606–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hvidt A; Nielsen SO Hydrogen Exchange in Proteins. Adv. Protein Chem 1966, 21, 287–386. [DOI] [PubMed] [Google Scholar]
- (9).Moorthy BS; Schultz SG; Kim SG; Topp EM Predicting Protein Aggregation During Storage in Lyophilized Solids Using Solid State Amide Hydrogen/Deuterium Exchange with Mass Spectrometric Analysis (ssHDX-MS). Mol. Pharmaceutics 2014, 11 (6), 1869–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Huyghues-Despointes BMP; Scholtz JM; Pace CN Protein Conformational Stabilities Can Be Determined from Hydrogen Exchange Rates. Nat. Struct. Biol 1999, 6 (10), 910–912. [DOI] [PubMed] [Google Scholar]
- (11).Smith CK; Withka JM; Regan L A Thermodynamic Scale for the Beta-Sheet Forming Tendencies of the Amino Acids. Biochemistry 1994, 33 (18), 5510–5517. [DOI] [PubMed] [Google Scholar]
- (12).Itzhaki LS; Neira JL; Fersht AR Hydrogen Exchange in Chymotrypsin Inhibitor 2 Probed by Denaturants and Temperature. J. Mol. Biol 1997, 270 (1), 89–98. [DOI] [PubMed] [Google Scholar]
- (13).Pézolet M; Bonenfant S; Dousseau F; Popineau Y Conformation of Wheat Gluten Proteins Comparison between Functional and Solution States as Determined by Infrared Spectroscopy. FEBS Lett. 1992, 299 (3), 247–250. [DOI] [PubMed] [Google Scholar]
- (14).Sadat A; Joye IJ Peak Fitting Applied to Fourier Transform Infrared and Raman Spectroscopic Analysis of Proteins. Appl. Sci 2020, 10 (17), 5918. [Google Scholar]
- (15).Athokpam B; Ramesh SG; McKenzie RH Effect of Hydrogen Bonding on the Infrared Absorption Intensity of OH Stretch Vibrations. Chem. Phys 2017, 488–489, 43–54. [Google Scholar]
- (16).Allison SD; Chang B; Randolph TW; Carpenter JF Hydrogen Bonding between Sugar and Protein Is Responsible for Inhibition of Dehydration-Induced Protein Unfolding. Arch. Biochem. Biophys 1999, 365 (2), 289–298. [DOI] [PubMed] [Google Scholar]
- (17).Englander SW; Kallenbach NR Hydrogen Exchange and Structural Dynamics of Proteins and Nucleic Acids. Q. Rev. Biophys 1983, 16 (4), 521–655. [DOI] [PubMed] [Google Scholar]
- (18).Radzicka A; Wolfenden R Comparing the Polarities of the Amino Acids: Side-Chain Distribution Coefficients between the Vapor Phase, Cyclohexane, 1-Octanol, and Neutral Aqueous Solution. Biochemistry 1988, 27 (5), 1664–1670. [Google Scholar]
- (19).Creamer TP; Srinivasan R; Rose GD Modeling Unfolded States of Peptides and Proteins. Biochemistry 1995, 34 (50), 16245–16250. [DOI] [PubMed] [Google Scholar]
- (20).Bai Y; Englander SW Hydrogen Bond Strength and β-Sheet Propensities: The Role of a Side Chain Blocking Effect. Proteins: Struct., Funct., Genet 1994, 18 (3), 262–266. [DOI] [PubMed] [Google Scholar]
- (21).Ladurner AG; Itzhaki LS; Fersht AR Strain in the Folding Nucleus of Chymotrypsin Inhibitor 2. Folding Des. 1997, 2 (6), 363–368. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.