

ABSTRACT
Thermophilic Methanothermobacter spp. are used as model microbes to study the physiology and biochemistry of the conversion of molecular hydrogen and carbon dioxide into methane (i.e., hydrogenotrophic methanogenesis). Yet, a genetic system for these model microbes was missing despite intensive work for four decades. Here, we report the successful implementation of genetic tools for Methanothermobacter thermautotrophicus ΔH. We developed shuttle vectors that replicated in Escherichia coli and M. thermautotrophicus ΔH. For M. thermautotrophicus ΔH, a thermostable neomycin resistance cassette served as the selectable marker for positive selection with neomycin, and the cryptic plasmid pME2001 from Methanothermobacter marburgensis served as the replicon. The shuttle-vector DNA was transferred from E. coli into M. thermautotrophicus ΔH via interdomain conjugation. After the successful validation of DNA transfer and positive selection in M. thermautotrophicus ΔH, we demonstrated heterologous gene expression of a thermostable β-galactosidase-encoding gene (bgaB) from Geobacillus stearothermophilus under the expression control of four distinct synthetic and native promoters. In quantitative in-vitro enzyme activity assay, we found significantly different β-galactosidase activity with these distinct promoters. With a formate dehydrogenase operon-encoding shuttle vector, we allowed growth of M. thermautotrophicus ΔH on formate as the sole growth substrate, while this was not possible for the empty-vector control.
KEYWORDS: Archaea, genetics, Methanothermobacter, shuttle vector, β-galactosidase, formate, beta-galactosidase
INTRODUCTION
Methanogenesis is the biological production of methane, which is catalyzed by methane-producing archaea (methanogens). Hydrogenotrophic methanogens grow with molecular hydrogen (electron donor) and carbon dioxide (electron acceptor and carbon source) as the substrates (1). The Methanobacteriales species Methanothermobacter thermautotrophicus and Methanothermobacter marburgensis have been studied as model microbes for the biochemistry of hydrogenotrophic methanogenesis, and deep insights into their energy and carbon metabolism have been acquired (1–3). For example, comparative genome analyses of M. thermautotrophicus and M. marburgensis revealed the genes that are most likely required for hydrogenotrophic methanogenesis (4), and a plethora of studies unraveled the mechanism of key enzymes such as the methyl-coenzyme M reductase (reviewed in reference 5). Furthermore, the pseudomurein-containing cell wall, which is specific to Methanobacteriales, has attracted research on Methanothermobacter spp. (6, 7).
Additionally, Methanothermobacter spp. have been implemented as biocatalysts in the power-to-gas platform on a large scale already (8, 9), because high cell densities and high methane production rates can be achieved (10, 11). In the power-to-gas platform, molecular hydrogen from the electrolysis of water with surplus electric power from renewable resources is combined with carbon dioxide, and these gases are converted into methane (8). This methane (i.e., renewable natural gas) can be introduced into the natural gas grid with large storage capacities to substitute for fossil natural gas. With genetic engineering of Methanothermobacter spp., the metabolism of the biocatalysts can, for example, be improved to maximize methane production rates or be amended for an expanded substrate or product spectrum, which would allow conversion of the power-to-gas platform into a broader power-to-chemical (i.e., power-to-x) platform (9).
This long-lasting interest in the biochemistry, physiology, and application of Methanothermobacter spp. led to extensive attempts to establish genetic tools in the past (12–14). Successes were reported in M. marburgensis for reversing spontaneous amino acid-auxotrophic phenotypes via generalized transduction from the wild-type strain with the virus ΨM2 (15) or via the uptake of free DNA (i.e., natural competence) with high-molecular-weight genomic DNA (16–18). In addition, 5-fluorouracil-resistant phenotypes were reported to be conferred with high-molecular-weight genomic DNA from a spontaneously resistant strain via natural competence in M. marburgensis with a higher frequency than the rate for the occurrence of spontaneous resistance (16). Other spontaneously antibiotic-resistant Methanothermobacter strains were investigated as potential sources for genes that can be used as selectable markers, including pseudomonic acid (17)- and neomycin (19)-resistant strains. Furthermore, several potential Escherichia coli-Methanothermobacter shuttle vectors have been constructed and replicated in E. coli, which were based on the cryptic plasmid (i.e., no physiological function has been assigned to the plasmid) pME2001 from M. marburgensis (12). Nevertheless, until now none of these approaches was translated into a reliable genetic system for Methanothermobacter spp.
Spurred by the recurring interest in Methanothermobacter spp. as model microbes for methanogenic biochemistry and physiology with a long history of fundamental research, and as biotechnologically relevant microbes in power-to-gas processes, we set out to utilize modern molecular biology tools and the knowledge of genetic tools for other methanogens and thermophilic microbes to develop a genetic system for M. thermautotrophicus ΔH. We report the successful establishment of genetic tools for M. thermautotrophicus ΔH, which now provide the basis to investigate hypotheses from four decades of research on the evolution, physiology, and biochemistry of Methanothermobacter spp. on a genetic level.
RESULTS
Clonal populations of M. thermautotrophicus ΔH can be obtained on solidified medium plates as individual colonies with high plating efficiencies.
The first requirement to allow genetic work with any given microbe is the capability to isolate clonal populations. This is typically achieved by plating microbial cultures on solidified medium plates and by selecting individual colonies. Therefore, we first reproduced the high plating efficiencies that have been reported in the literature for Methanothermobacter spp. (20). We investigated three common plating techniques (spot, spread, and pour plating) and compared factors that influenced plating efficiency (i.e., individual colonies per cell count of plated microbial cells [Materials and Methods]). We achieved dense growth with spot plating, but individual colonies were barely distinguishable with this plating technique, while we achieved plating efficiencies between 1 and 5% with spread plating, and 50% and higher with pour plating (see Text S1A and Fig. S1 in the supplemental material).
Supplemental results (A to I) and discussion (J). (A) Differences in plating efficiencies for various plating techniques. (B) Description of growth-inhibiting effects of antibiotics. (C) Details on the design of the modular shuttle-vector system pMVS. (D) Analysis of successful DNA transfer via site-specific PCR. (E) Retransformation of E. coli with plasmid extracts from M. thermautotrophicus ΔH. (F) Approximation of the conjugation frequency. (G) Segregational stability of shuttle vector under nonselective conditions. (H) Control experiments for conjugational DNA transfer. (I) Quantitative β-galactosidase enzyme activity assay. (J) Discussion on plating efficiency of M. thermautotrophicus ΔH. Download Text S1, DOCX file, 0.06 MB (66.1KB, docx) .
Copyright © 2021 Fink et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
(A) Spot plating of M. thermautotrophicus ΔH cells on solidified medium plates. (B) Pour plating of 1 × 104 M. thermautotrophicus ΔH cells on solidified medium plates. (C) Spread plating of 1 × 104 M. thermautotrophicus ΔH cells on solidified medium plates. (D) Comparison of the influence from various factors on the plating efficiency with spread plating of 1 × 104 M. thermautotrophicus ΔH cells and pour plating of 1 × 103 M. thermautotrophicus ΔH cells, including the influence of paper clips, 0.1 vol% hydrogen sulfide (H2S) in the headspace gas mixture, exponential growth phase (Exp), and stationary growth phase (Stat). The bars give the average percentage (log scale) of individual colonies per cell count of initial microbial cells used for plating from technical replicates (n = 3; n = 4 for spread plating with paper clips and H2S in exponential growth phase) with error bars indicating standard deviation. (E) Influence of paper clips on the lid elevation. The arrow demonstrates the increase of lid/plate space compared for a plate without paper clips (upper picture) and one with paper clips (lower picture). Download FIG S1, TIF file, 0.3 MB (341.8KB, tif) .
Copyright © 2021 Fink et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
M. thermautotrophicus ΔH is sensitive to antibiotics commonly used in methanogen genetic systems.
To find a suitable selection pressure for the positive selection of genetically modified cells, we analyzed several antibiotics such as simvastatin and neomycin (Text S1B). For both antibiotics, thermostable selectable markers are available, which have been successfully used in thermophilic nonmethanogenic microbes, such as Pyrococcus furiosus and Thermococcus kodakarensis (simvastatin) (21, 22), as well as Thermoanaerobacter spp. (neomycin) (23), but recently also in the thermophilic methanogens Methanocaldococcus jannaschii (simvastatin) and Methanoculleus thermophilus (neomycin) (24, 25). Both simvastatin and neomycin efficiently inhibited growth of M. thermautotrophicus ΔH cells in liquid culture at concentrations of 21.5 μg/ml and 250 μg/ml, respectively (Text S1B and Fig. S2 and S3). While these antibiotics inhibited growth of M. thermautotrophicus ΔH, we observed the appearance of spontaneously resistant M. thermautotrophicus ΔH cells for both antibiotics (Text S1B and Fig. S2 and S3). The incubation period for the appearance of spontaneously resistant M. thermautotrophicus ΔH cells differed for each antibiotic compared to the incubation period for nonselective growth of wild-type cells (16 to 24 h). We observed inhibition of growth for at least 48 h with simvastatin and 60 h with neomycin at the concentrations indicated above (Text S1B). Because we found that neomycin inhibits growth of M. thermautotrophicus ΔH for a longer incubation period than that for simvastatin, we decided to focus on neomycin as the selection pressure to develop the first selectable marker for M. thermautotrophicus ΔH.
Measurement of OD600 after different incubation periods at 60°C (0 h, 24 h, 48 h, and 60 h) with initially 5 × 105 cells/ml of wild-type M. thermautotrophicus ΔH in liquid mineral medium containing neomycin concentrations of 0 μg/ml (long-dashed line, n = 1), 50 μg/ml (dotted line), 100 μg/ml (short-dashed line), and 250 μg/ml (black solid line). For comparison to wild-type M. thermautotrophicus ΔH, initially 5 × 105 cells/ml of pMVS-V1-carrying M. thermautotrophicus ΔH were incubated in selective liquid mineral medium with 250 μg/ml neomycin (gray solid line, n = 1) and analyzed after the same incubation periods. Average (n = 3) with error bars indicating standard deviation. Download FIG S2, EPS file, 1.3 MB (1.3MB, eps) .
Copyright © 2021 Fink et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
(A) Individual colonies from 1 × 108 wild-type M. thermautotrophicus ΔH cells (gray bars), which were pour plated on solidified medium plates containing simvastatin concentrations ranging from 13 μg/ml to 21.5 μg/ml. (B) Individual colonies from 1 × 103 wild-type M. thermautotrophicus ΔH cells (gray bars) and pMVS-V1-carrying M. thermautotrophicus ΔH (black bars, n = 1), which were pour plated on solidified medium plates containing neomycin concentrations ranging from 0 μg/ml to 250 μg/ml. Average (n = 3) with error bars indicating standard deviation for wild-type M. thermautotrophicus ΔH. Download FIG S3, EPS file, 1.3 MB (1.3MB, eps) .
Copyright © 2021 Fink et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
A modular plasmid design enables the fast generation of shuttle vectors to test genetic elements for functionality in M. thermautotrophicus ΔH.
Before we focused on the neomycin-selectable marker, we had constructed a subset of plasmids that would allow us to test various approaches for the transfer of DNA into M. thermautotrophicus ΔH and for positive selection (Materials and Methods). To ease exchangeability of genetic elements in shuttle vectors, and to allow fast adaptation to new findings, we decided on a modular plasmid design. Inspired by the pSEVA system for Gram-negative bacteria (26), and the pMTL80000 system for Clostridia (27), we established the Methanothermobacter vector system (pMVS) design.
The pMVS design consists of five modules, which are separated by rare 8-bp recognition sequences for the restriction enzymes PmeI, AsiSI, FseI, AscI, and PacI (Fig. 1). To follow the pMVS design, these rare restriction enzyme-recognition sequences need to stay unique to grant exchangeability of the modules by restriction/ligation cloning. The five modules are (restriction enzyme boundaries are given in parentheses) (i) the replicon for E. coli (AsiSI, PmeI), (ii) the selectable marker for E. coli (PmeI, FseI), (iii) the replicon for M. thermautotrophicus ΔH (AsiSI, PacI), (iv) the selectable marker for M. thermautotrophicus ΔH (AscI, FseI), and (v) an application module that can be used to include any genetic cargo such as a reporter gene or another gene of interest (PacI, AscI) (Fig. 1 and Text S1C).
FIG 1.

Plasmid maps of the Methanothermobacter vector system (pMVS). (A) pMVS-V1 consists of four modules, which are intersected by the 8-bp restriction enzyme recognition sites AscI, FseI, PmeI, and AsiSI. The four modules are the replicon for E. coli (ColE1, tra), the selectable marker for E. coli (Camr), the replicon for M. thermautotrophicus ΔH (pME2001), and the selectable marker for M. thermautotrophicus ΔH (Neor). (B) pMVS1111A:Psynth-bgaB consists of five modules, which are intersected by the 8-bp restriction enzyme recognition sites AscI, FseI, PmeI, AsiSI, and PacI. The five modules are as in pMVS-V1 with the following differences: the replicon for M. thermautotrophicus ΔH is flanked by AsiSI and PacI, and the shuttle vector contains the bgaB gene in the application module, which is flanked by PacI and AscI. Psynth, synthetic promoter sequence, which is based on PhmtB; Tmcr, terminator sequence from the mcr operon of M. voltae. The nomenclature for the pMVS design is realized by adding a four-digit code after pMVS for the definition of the first four modules, which defines the plasmid backbone with basic functions for replication and selection in E. coli (modules 1 and 2) and M. thermautotrophicus ΔH (modules 3 and 4). Additional large capital letters can be amended to each digit to define differences, such as varying promoter sequences, in a given module. For the fifth (application) module, a descriptive name is added after the four-digit code, which allows staying as flexible as possible with the genetic cargo used (without a limitation to nine digits), while staying within the pMVS design boundaries.
Based on the first shuttle vector (pMVS-V1) that led to successful DNA transfer and selection protocols, as described below, we defined pMVS-V1 as our archetype shuttle vector (Fig. 1A), with a combination of (i) the ColE1-derived replicon for E. coli in combination with the conjugational transfer function (tra region) from RK2 (27), (ii) the chloramphenicol-selectable marker (Camr) for E. coli (27), (iii) the entire cryptic plasmid pME2001 from M. marburgensis as the replicon for M. thermautotrophicus ΔH (14), and (iv) the thermostable neomycin-selectable marker (Neor) (28) for M. thermautotrophicus ΔH under the control of the Psynth promoter sequence (22), but without an application module and a PacI recognition sequence (Fig. 1A). After we had demonstrated the functionality of pMVS-V1, our first complete shuttle vector, pMVS1111A-Psynth-bgaB, was then constructed based on this archetype shuttle vector. pMVS1111A-Psynth-bgaB contains the β-galactosidase-encoding gene bgaB (see below) and an additional PacI site, which completes the application module (Fig. 1B, Materials and Methods, and Text S1C).
DNA transfer into M. thermautotrophicus ΔH is possible via interdomain conjugation with E. coli S17-1.
Following our broad approach, we investigated several published protocols (and modified versions) to transfer DNA into M. thermautotrophicus ΔH by using our various plasmids and shuttle vectors (Materials and Methods). These DNA transfer protocols included (i) natural competence protocols (16, 24), (ii) chemically/physically induced transformation protocols, such as with elevated calcium and magnesium concentrations in the mineral medium or with low-temperature incubation of precultures to induce stress conditions, (iii) an electroporation protocol (29), and (iv) an interdomain conjugation protocol with E. coli (30). Most attempts did not result in cells with the expected selectable phenotype and, if so, could not be linked to the respective anticipated genotype and appeared to be spontaneously resistant cells. The protocol that finally led to a successful DNA transfer into M. thermautotrophicus ΔH was an interdomain conjugation protocol with E. coli S17-1 (Fig. 2 and Materials and Methods), which was a modified version of the protocol for conjugational DNA transfer into Methanococcus maripaludis (30).
FIG 2.
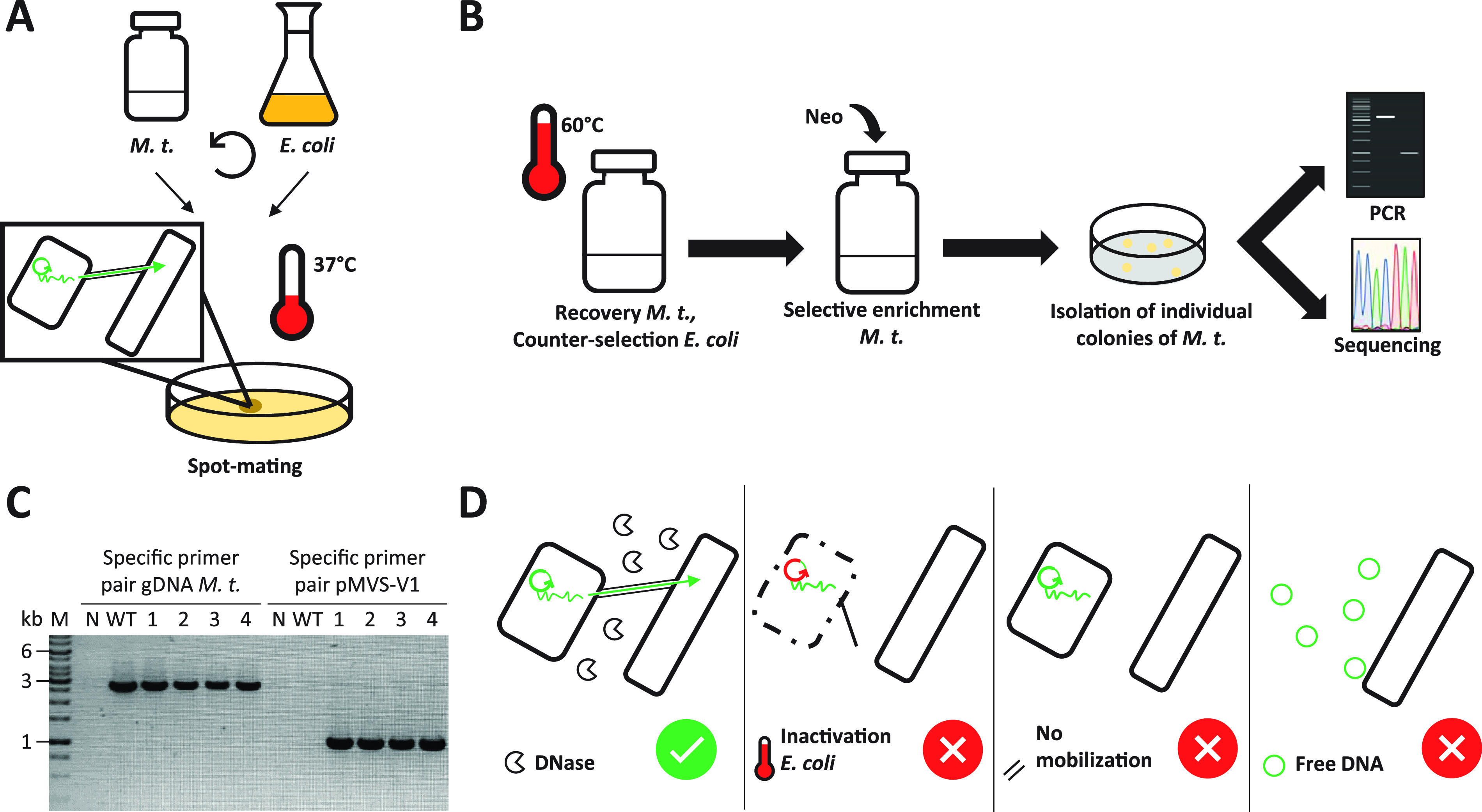
Schematic depiction and analysis of interdomain conjugation between E. coli S17-1 and M. thermautotrophicus ΔH. (A) Wild-type M. thermautotrophicus ΔH (M. t.) and the shuttle vector-carrying E. coli were harvested by centrifugation, mixed, and spotted on solidified medium plates that support growth of both microbes. During the spot-mating step at 37°C, the DNA transfer process via conjugation takes place (small scheme). (B) The process to isolate and identify individual colonies of genetically modified M. thermautotrophicus ΔH in the standard protocol. After the spot-mating step, M. thermautotrophicus ΔH cells were recovered in nonselective liquid mineral medium at 60°C, and afterward, transconjugants were enriched in neomycin (Neo)-containing selective liquid mineral medium at 60°C. Individual colonies were obtained from plating the enrichment culture. Those colonies were analyzed by PCR and Sanger sequencing. (C) PCR analysis of four respective transconjugants (1 to 4) with primer combinations specific for the shuttle vector pMVS-V1 replicon (1-kb fragment) and for genomic DNA (gDNA) of M. thermautotrophicus ΔH (2.8-kb fragment). N, water negative control; WT, control with wild-type M. thermautotrophicus ΔH; M, GeneRuler 1-kb DNA ladder (Thermo Scientific, Waltham, MA, USA). (D) Experimental conditions for the confirmation of conjugation as the mechanism for DNA transfer were (from left to right) DNase I treatment, heat inactivation of E. coli S17-1, conjugation with nonconjugative E. coli NEB stable, and addition of free plasmid DNA directly to M. thermautotrophicus ΔH cell culture.
To achieve a successful conjugational DNA transfer of the archetype shuttle vector pMVS-V1 from E. coli S17-1 (donor) to M. thermautotrophicus ΔH (recipient), we increased the recipient cell concentration to ∼1.6 × 109 cells from a culture in the early stationary growth phase, which is a 5-fold-higher cell concentration than that in the work of Dodsworth et al. (30). Furthermore, we used a spot-mating procedure to allow close physical contact between donor and recipient cells during an incubation period at 37°C on solidified medium plates, which supported metabolic activity of E. coli S17-1 (Fig. 2A), in contrast to direct spread plating as in the work of Dodsworth et al. (30). After resuspending the spot-mated cells (donor + recipient) from the solidified medium plate, we recovered M. thermautotrophicus ΔH in liquid mineral medium without any complex medium additions (no organic carbon source) and without antibiotic additions at 60°C under a molecular hydrogen/carbon dioxide atmosphere (Fig. 2B and Materials and Methods). These incubation conditions decreased the viability of E. coli S17-1 to a very minimum, and therefore, no counterselection with antibiotics against donor cells was required. After a short incubation period (3 to 4 h) to recover M. thermautotrophicus ΔH under nonselective conditions, the cells were transferred to liquid medium, which contained neomycin for a selective-enrichment step. Importantly, the required incubation period for the cells to grow in this step was key for a successful identification of transconjugants (i.e., recipient cells that received the shuttle vector). Neomycin at a concentration of 250 μg/ml inhibits growth of M. thermautotrophicus ΔH for at least 60 h in liquid medium (Text S1B). Therefore, when the cells did not grow within less than 60 h (typically growth appeared after 24 to 48 h in a successful experiment) in the selective-enrichment step, the conjugation experiment was regarded as unsuccessful, because the number of spontaneously resistant cells is considerably higher than potential transconjugants, which renders screening essentially impossible.
The shuttle-vector DNA confers the observed antibiotic-resistant phenotype and is maintained in M. thermautotrophicus ΔH with high segregational stability.
After we found selective growth of putative transconjugant cells, we confirmed the successful DNA transfer into M. thermautotrophicus ΔH via two site-specific PCRs, which amplified a fragment of the pMVS-V1 shuttle vector and a fragment of the M. thermautotrophicus ΔH genomic DNA, respectively, with liquid cultures derived from individual colonies (Fig. 2C). In addition, we extracted plasmid DNA from several independent M. thermautotrophicus ΔH transconjugant cultures, transformed E. coli with this plasmid DNA extract, reextracted the plasmid DNA again from E. coli, and finally, performed restriction-enzyme digestions and Sanger sequencing to confirm the plasmid DNA integrity and sequence, without finding any differences from the original pMVS-V1 shuttle vector (Text S1D and Fig. S4). With different shuttle vectors in independent experiments, we achieved reliable DNA transfer into M. thermautotrophicus ΔH with our standard protocol (Materials and Methods), which includes a selective-enrichment step (Fig. 2B). However, this protocol was not suitable to calculate the conjugation frequency, due to the selective-enrichment step. To determine the conjugation frequencies, we performed experiments without the selective-enrichment step but with a prolonged nonselective-recovery step, which resulted in 5 ± 4 colonies (n = 6) and from which we calculated conjugation frequencies of approximately 4 × 10−9 to 6 × 10−6 transconjugants per initial recipients (see equation S1 in Text S1F). With this assessment, we showed that experimental variations considerably influenced the conjugation frequency (Text S1F and Table S1). Nevertheless, when following our standard protocol, a reliable transfer of plasmid DNA was achieved. Once the plasmid DNA was transferred into M. thermautotrophicus ΔH, it was maintained with high segregational stability over many cell divisions in an experiment under nonselective growth conditions, and we did not observe loss of pMVS-V1 (Text S1G and Fig. S5).
(A) Restriction-enzyme digestion of plasmid DNA, which was extracted from three independent E. coli pMVS-V1 enrichment cultures (1 to 3). The E. coli strains were generated with plasmid DNA, which was extracted from genetically modified M. thermautotrophicus ΔH. The unique cutter KpnI (K), resulting in one 8.2-kb fragment, and the dual cutter NdeI (N), resulting in 2.7-kb and 5.5-kb fragments, were used. M, GeneRuler 1-kb DNA ladder (Thermo Scientific, Waltham, MA, USA). (B) Exemplified sequence alignment of Sanger sequences from retransformed E. coli plasmid DNA to the original pMVS-V1 sequence (main text and Fig. 1 for further explanations). Red arrows indicate the sequence and its direction. No deletions, insertions, or nucleotide exchanges were detected. Download FIG S4, TIF file, 0.1 MB (120.9KB, tif) .
Copyright © 2021 Fink et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
(A) PCR analysis of individual colonies from selective solidified medium plates after one selective transfer of pMVS-V1-carrying M. thermautotrophicus ΔH in liquid mineral medium. (B) PCR analysis of individual colonies from nonselective solidified medium plates after three nonselective transfers (∼21 to 28 cell divisions) in liquid mineral medium (PCR analysis of the first and second transfer is not shown but gave the same results). A 1-kb fragment results from the specific primer pair for the pME2001 replicon for M. thermautotrophicus ΔH (a) and a 1.5-kb fragment from the specific primer pair for M. thermautotrophicus ΔH genomic DNA (b). All 16 colonies each result in positive PCR for both primer combinations. Wild-type M. thermautotrophicus ΔH (WT) does not result in PCR signal for pMVS-V1, and water as negative control (N) does not result in any PCR signal. M, GeneRuler 1-kb DNA ladder (Thermo Scientific, Waltham, MA, USA). Download FIG S5, TIF file, 0.4 MB (385.4KB, tif) .
Copyright © 2021 Fink et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Example calculations for conjugation frequencies for DNA transfer into M. thermautotrophicus ΔH based on equation S1. Download Table S1, DOCX file, 0.01 MB (13.3KB, docx) .
Copyright © 2021 Fink et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Free plasmid DNA is not resulting in DNA transfer into M. thermautotrophicus ΔH.
By having demonstrated DNA transfer into M. thermautotrophicus ΔH, we further analyzed whether this transfer was indeed depending on conjugational DNA transfer from E. coli or whether it was rather by uptake of free DNA under the utilized cultivation conditions during the conjugation protocol (Materials and Methods). E. coli S17-1 donor cells contain a large amount of pMVS-V1, because of the high-copy-number ColE1 replicon, which might be released into the liquid medium from lysing cells. Therefore, we conducted control experiments with free pMVS-V1 plasmid DNA, heat-inactivated E. coli S17-1 cells, and nonconjugative E. coli NEB stable cells that carry pMVS-V1 (Fig. 2D). None of these experiments resulted in DNA transfer into M. thermautotrophicus ΔH (Fig. 2D, Text S1H, and Fig. S6). In contrast, DNase I treatment of the E. coli S17-1 donor cells did not negatively influence the success of a conjugational DNA transfer into M. thermautotrophicus ΔH (Fig. 2D, Text S1H, and Fig. S6). Thus, we concluded that DNA transfer occurs due to conjugational mobilization activity from E. coli S17-1 to M. thermautotrophicus ΔH.
(A) Enrichment cultures of genetically modified M. thermautotrophicus ΔH in 250-μg/ml neomycin-containing selective liquid mineral medium from conjugation/DNA transfer experiments with the standard protocol (1), the DNase I-treated E. coli S17-1 (2), the heat-inactivated E. coli S17-1 (3), and NEB stable as nonconjugative E. coli (4). Slight turbidity in cultures 3 and 4 is caused due to high initial cell count. Enrichment is visible in cultures 1 and 2. (B) Spread-plated M. thermautotrophicus ΔH using culture 1 from panel A (standard protocol). Black circles represent colonies, which were used for PCR analysis. (C) Spread-plated M. thermautotrophicus ΔH using culture 2 from panel A (DNase I treatment). Black circles represent colonies, which were used for PCR analysis. (D) PCR analysis of the four colonies from panels B and C (1 to 4), wild-type M. thermautotrophicus ΔH (5), purified shuttle-vector DNA as positive control (6), and water as negative control (7). A primer combination which amplifies a 1-kb fragment from the pME2001 replicon was used for PCR amplification. M, GeneRuler 1-kb DNA ladder (Thermo Scientific, Waltham, MA, USA). Download FIG S6, TIF file, 0.4 MB (389.5KB, tif) .
Copyright © 2021 Fink et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
A thermostable β-galactosidase (BgaB) from Geobacillus stearothermophilus is a functional reporter to investigate promoter sequences in M. thermautotrophicus ΔH.
With a DNA transfer protocol and a functional shuttle vector at hand, we proceeded with adding a genetic cargo (i.e., gene of interest) to the application module of the archetype pMVS-V1 shuttle vector. To enable the analysis of the effects from different promoter sequences on gene expression in M. thermautotrophicus ΔH, we decided to implement a reporter gene as our first gene of interest. We chose the bgaB gene from G. stearothermophilus, which encodes a thermostable β-galactosidase (31). We placed a codon-optimized version of the bgaB gene under the control of the nonnative Psynth promoter (Materials and Methods). We transferred the resulting shuttle vector pMVS1111A:Psynth-bgaB (Fig. 1B) into M. thermautotrophicus ΔH via conjugation. In a qualitative preliminary experiment with cell lysate from pMVS1111A:Psynth-bgaB-carrying M. thermautotrophicus ΔH cells (and pMVS-V1-carrying cells as an empty-vector negative control), we found that, indeed, the β-galactosidase BgaB is produced in M. thermautotrophicus ΔH and results in a color reaction in an enzyme assay with 3,4-cyclohexenoesculetin-β-d-galactopyranoside (S-Gal) only in the presence of the bgaB gene (Fig. 3B and Materials and Methods).
FIG 3.

Enzyme activity assays with M. thermautotrophicus ΔH strains that carry a thermostable β-galactosidase (BgaB)-encoding gene under the control of four distinct promoter sequences. (A) Sequence alignment of distinct putative promoter sequences that we analyzed for activity to drive the expression of a thermostable β-galactosidase (bgaB) gene. Sequence repeats in Pmrt(M.t.) are underlined. The transcription start site is indicated by “+1,” highlighted in bold, and underlined. TATA box sequences of Psynth, Psynth(BRE), and PhmtB are surrounded by a box. BRE sequences are highlighted in italics and ribosome-binding sites in red. Dashes are used as spacers, while dots indicate additional base pairs, which are left out here for visualization. Differences between Psynth, Psynth(BRE), and PhmtB between the TATA box sequence and transcription start site are highlighted in bold. (B) Qualitative analysis of BgaB activity with S-Gal as chromogenic substance in an in vitro assay with cell lysate of empty-vector-carrying M. thermautotrophicus ΔH (pMVS-V1) or pMVS1111A:Psynth-bgaB-carrying M. thermautotrophicus ΔH (Psynth-bgaB) cells. (C) Quantitative analysis of BgaB activity with ONPG as chromogenic substance in an in vitro assay with cell lysate of M. thermautotrophicus ΔH strains that carry plasmids with the bgaB gene under the control of the four distinct promoters [pMVS-V1, empty-vector control; Pmrt(M.t.)-bgaB, pMVS1111A:Pmrt(M.t.)-bgaB; Psynth-bgaB, pMVS1111A:Psynth-bgaB; Psynth(BRE)-bgaB, pMVS1111A:Psynth(BRE)-bgaB; PhmtB-bgaB pMVS1111A:PhmtB-bgaB]. Average (n = 3) with error bars indicating standard deviation. Significance was tested with Student’s t test (two-tailed): *, significant difference (P < 0.05); **, highly significant difference (P < 0.01); n.s., no significant difference (P > 0.05).
This result sparked us to establish a quantitative β-galactosidase enzyme activity assay with o-nitrophenyl-β-d-galactopyranoside (ONPG) as the chromogenic substrate for the β-galactosidase (Text S1I), which allowed us to investigate promoter sequences for their relative in-vivo effect on gene expression in M. thermautotrophicus ΔH during a growth experiment (Fig. S1I). Overall, we selected four distinct promoter sequences [Psynth, Psynth(BRE), PhmtB, and Pmrt(M.t.)] based on our previous results and the peer-reviewed literature and compared the effects of these promoters on gene expression with the established enzyme assay (Fig. 3 and Text S1I) (22, 32). With our optimized quantitative β-galactosidase enzyme activity assay, we found that the Psynth promoter, without a transcription factor B recognition element (BRE) sequence, resulted in a significantly higher β-galactosidase enzyme activity (510 ± 50 Miller units) than did the empty-vector control (46 ± 5 Miller units; P < 0.01) (Fig. 3C). However, significantly lower enzyme activity was measured with this promoter than with the Psynth(BRE) (1,350 ± 140 Miller units; P < 0.05) and the PhmtB (5,000 ± 100 Miller units; P < 0.01) (Fig. 3C) promoters, which both contained a BRE sequence (Fig. 3A). The Pmrt(M.t.) promoter resulted only in a β-galactosidase activity (65 ± 5 Miller units) that was comparable to and not significantly different from the empty-vector negative control in the enzyme assay (45 ± 5 Miller units) (Fig. 3C). Therefore, this promoter has to be considered inactive under the tested conditions (Fig. 3C). We did not include the commonly used PmcrB(M.v.) promoter for methanogen genetic systems in this comparison, because we already had found that this promoter is not functional in driving the neomycin-selectable marker (Text S1I).
The metabolism of M. thermautotrophicus ΔH can be amended to enable growth on formate as an alternative substrate.
Formate as the sole carbon and energy substrate can be utilized by several methanogens, such as Methanococcus spp. (33), Methanobacterium spp., and also Methanothermobacter spp. (34). For example, the strain M. thermautotrophicus Z-245 can grow with only formate, instead of molecular hydrogen and carbon dioxide, while M. thermautotrophicus ΔH is not able to grow with only formate (Fig. 4 and Fig. S8) (34, 35). It was hypothesized by Nölling and Reeve (35) that the genetic reason for this is the missing formate dehydrogenase (fdh) operon in the genome of M. thermautotrophicus ΔH compared to the same genomic region in M. thermautotrophicus Z-245. We argued that we can test this hypothesis in vivo, by providing the fdh operon as a genetic cargo in the application module of our shuttle vector. Thus, we constructed the shuttle vector pMVS1111A:PhmtB-fdhZ-245 that contains the entire fdh operon from M. thermautotrophicus Z-245, including the genes (in this order) fdhC, fdhA, and fdhB and additionally an open reading frame with unknown function (orf3), as indicated in the work of Nölling and Reeve (35), under the control of the constitutive PhmtB promoter (Fig. 3). In control experiments with M. thermautotrophicus Z-245, we confirmed growth of this microbe with either formate or molecular hydrogen and carbon dioxide as substrates (Fig. S8). Growth on formate was possible with M. thermautotrophicus ΔH cells that carry pMVS1111A:PhmtB-fdhZ-245 but not with cells that carry the empty-vector control pMVS-V1, and thus, no growth was observed from the small amount of yeast extract that we had added to provide any potentially missing micronutrients (Fig. 4 and Materials and Methods). The formate dehydrogenase-producing strain had a prolonged lag phase with formate but reached a final optical density at 600 nm (OD600) comparable to growth on molecular hydrogen and carbon dioxide. Hence, the hypothesis of Nölling and Reeve (35) was proven to be correct.
FIG 4.

Analysis of genetically modified M. thermautotrophicus ΔH strains for growth on formate. (A) Growth behavior of M. thermautotrophicus pMVS-V1 on molecular hydrogen and carbon dioxide (gray) and on formate (black) as the carbon and energy source. Average (n = 3) with error bars indicating standard deviation. The dotted line indicates the remaining amount of formate in the medium of M. thermautotrophicus pMVS-V1 grown on formate determined by HPLC measurements. (B) Growth behavior of M. thermautotrophicus pMVS1111A:PhmtB-fdhZ-245 with either molecular hydrogen and carbon dioxide (gray) or with formate (black) as the carbon and energy source. Average (n = 3) with error bars indicating standard deviation. The dotted line indicates the remaining amount of formate in the medium of M. thermautotrophicus pMVS1111A:PhmtB-fdhZ-245 grown on formate determined by HPLC measurements.
Analysis of wild-type M. thermautotrophicus Z-245 growth either with molecular hydrogen and carbon dioxide (black solid line) or with formate (gray solid line) as the carbon and energy source. Average (n = 3) with error bars indicating standard deviation. Download FIG S8, EPS file, 1.2 MB (1.2MB, eps) .
Copyright © 2021 Fink et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DISCUSSION
Here, we reported a robust method for genetic manipulation of M. thermautotrophicus ΔH (Fig. 2), including replicating shuttle vectors, which we based on the cryptic plasmid pME2001 (12). To achieve this, in a first step, we investigated different plating protocols to obtain clonal populations. Importantly, while we achieved plating efficiencies of up to 100%, the plating conditions considerably influenced the outcome over a range of 3 orders of magnitude (see Text S1J in the supplemental material). Thus, for specific purposes the plating technique has to be carefully considered to avoid misleading results, such as false interpretations of DNA transfer events. At a too-high plating efficiency, spontaneously resistant cells might overgrow genetically modified M. thermautotrophicus ΔH.
In general, it was noticeable that spontaneously resistant M. thermautotrophicus ΔH cells appeared readily with the antibiotics simvastatin and neomycin. Once those spontaneously resistant cells appeared, they were not inhibited or delayed in growth in subsequent transfers in liquid medium as well as on solidified medium plates. This has been already reported with neomycin for M. thermautotrophicus ΔH (19), M. maripaludis, and Methanococcus vannielii (36). However, in comparison to M. maripaludis for which spontaneously neomycin-resistant colonies appeared to be smaller than genetically modified colonies (which were found to carry a neomycin-selectable marker) (37), we cannot see this difference in colony size for M. thermautotrophicus ΔH. Therefore, we implemented a selective-enrichment step in liquid medium, which provided enough time for growth of the genetically modified M. thermautotrophicus ΔH, while at the same time it excluded the onset of growth of the spontaneously resistant cells by substrate limitation in the gas phase, because those cells appeared only after a longer incubation period (Fig. S2). After the selective-enrichment step, the genetically modified M. thermautotrophicus ΔH cells outnumbered the spontaneously resistant cells sufficiently to obtain and select genetically modified individual colonies on selective solidified medium plates.
Yet, we were not successful with genetically manipulating M. thermautotrophicus ΔH when we fused various potential selectable markers with the PmcrB(M.v.) promoter, which is commonly utilized in other methanogen genetic systems. However, after we switched to the Psynth promoter to drive the neomycin-selectable marker, positive selection of shuttle vector-carrying cells with neomycin was possible in combination with the selective-enrichment step. Taken together, we believe that the most important parameters for the successful implementation of our genetic tools for M. thermautotrophicus ΔH, compared to previous attempts during the last four decades, were (i) the construction of the shuttle vector with restriction/ligation-independent cloning to fuse the pME2001 replicon with the other modules precisely at the IF5 sequence, to not interrupt any open reading frame or potential sequence of the origin of replication, (ii) the adaptations to the published conjugation protocol for Methanococcus spp. by Dodsworth et al. (30), specifically the applied temperature, medium, and headspace gas conditions during the spot-mating procedure, in combination with the selective-enrichment step with limited gas supply that facilitated the selection for genetically modified cells over spontaneously resistant cells, and (iii) the utilization of a constitutive native promoter sequence, which was demonstrated to be functional in vitro and in other thermophilic archaea, because the classical PmcrB(M.v.) promoter turned out to be inactive in M. thermautotrophicus ΔH.
With the reporter gene bgaB, which encodes a thermostable β-galactosidase, we further confirmed the activity of this Psynth promoter, as well as the activity of the Psynth(BRE) and PhmtB promoters, which are all based on the promoter region upstream of the hmtB gene of M. thermautotrophicus ΔH. The BRE sequence upstream of the Psynth sequence, which we had implemented in Psynth(BRE), and differences in the transcription-initiation region in PhmtB compared to both Psynth and Psynth(BRE) influenced the promoter strength significantly (Fig. 3). This was not surprising, because the critical role of the BRE sequence in archaea is well known, and similar observations were made with archaeal promoters in Saccharolobus solfataricus and Haloferax volcanii, where promoter strength was influenced by the presence or absence of a BRE sequence (38, 39). Furthermore, modifications in the transcription-initiation region in the promoter sequence were shown to influence the strength of gene expression in Sulfolobus acidocaldarius and S. solfataricus (38, 40). Thus, we now have established constitutive promoters of different strength. The thermostable β-galactosidase as a reporter provides an adequate basis to further investigate promoter sequences of M. thermautotrophicus ΔH and to establish inducible-promoter systems.
In addition to demonstrating heterologous production of functional β-galactosidase enzymes in M. thermautotrophicus ΔH, we demonstrated the production of an active formate dehydrogenase enzyme complex from the fdh operon from M. thermautotrophicus Z-245 (35), which amended the metabolism of M. thermautotrophicus ΔH for the ability to utilize formate as an alternative growth substrate (Fig. 4). These results are a cornerstone for heterologous and homologous (over)expression of genes in M. thermautotrophicus ΔH and enable researchers to further expand the genetic toolbox with methodology for chromosomal gene deletions and integrations such as allelic exchange, markerless mutagenesis (41), and CRISPR/Cas technology (42). We will now be able to target modifications in the metabolism of M. thermautotrophicus ΔH not only on the substrate but also on the product side. The possibility to change the product spectrum of hydrogenotrophic methanogens has been demonstrated already for M. maripaludis with a genetic modification that resulted in geraniol production (43). Our genetic tools for heterologous gene expression enable us to broaden the product spectrum of M. thermautotrophicus ΔH and to utilize this industrially relevant and robust microbe for power-to-chemicals (i.e., power-to-x) applications.
MATERIALS AND METHODS
Microbial strains, media, and cultivation conditions.
M. thermautotrophicus ΔH (DSM 1053), M. thermautotrophicus Z-245 (DSM 3720), and M. marburgensis (DSM 2133) were obtained from the DSMZ (Braunschweig, Germany) and were cultivated in mineral medium according to basic principles for methanogen cultivation as stated in the work of Balch et al. (44), with modifications to the medium composition when required and with adjustments to state-of-the-art anaerobic handling equipment. The mineral medium contained (per liter) sodium chloride, 0.45 g; sodium hydrogen carbonate, 6.00 g; dipotassium hydrogen phosphate, 0.17 g; potassium dihydrogen phosphate, 0.23; ammonium chloride, 0.19 g; magnesium chloride hexahydrate, 0.08 g; calcium chloride dihydrate, 0.06 g; ammonium nickel sulfate, 1 ml (0.2 wt%); iron(II)chloride pentahydrate, 1 ml (0.2 wt%); resazurin indicator solution, 4 ml (0.025 wt%); and trace element solution, 1 ml (10-fold as stated in the work of Balch et al. [44]). All chemicals were per analysis grade. No vitamins were added. For the formate growth experiments, the mineral medium was supplemented with 200 mM sodium formate, 10 μM sodium molybdate, 1 μM sodium selenite, and 0.125 wt% yeast extract for all tested strains, including the empty-vector-carrying M. thermautotrophicus ΔH strain, to provide a source of potentially limiting micronutrients. The medium was gassed using N2/CO2 (80/20 vol%) to eliminate dissolved oxygen. The pH value was adjusted to 7.2 using hydrochloric acid. As reducing agent and sulfur source, 0.5 g/liter cysteine hydrochloride, and for solid mineral medium additionally 0.3 g/liter sodium sulfide monohydrate, was added. Afterward, the mineral medium was dispensed into serum bottles inside an anaerobic chamber with a 100% N2 atmosphere (UniLab Pro Eco; MBraun, Garching, Germany). The headspace of the serum bottles was exchanged to 200 kPa H2/CO2 (80/20 vol%) and autoclaved (100 kPa, 20 min, 121°C). For the formate growth experiments, the headspace of the serum bottles was exchanged to 152 kPa H2/CO2 (80/20 vol%) as positive control, which provides the same electron equivalents as 200 mM formate, or 152 kPa N2/CO2 (80/20 vol%), when formate was the substrate. All Methanothermobacter strains in liquid medium were incubated at 60°C with shaking at 150 rpm (Lab Companion ISS-7100R; Jeio Tech, Republic of Korea). For cultivation of genetically modified M. thermautotrophicus ΔH strains, 250 μg/ml neomycin sodium salt was added. For solidified mineral medium, 1.5 wt% Bacto agar (BD Life Science, Berkshire, United Kingdom) was added as a supplement prior to autoclaving. Afterward, solidified medium plates were poured and dried for 2 h inside the anaerobic chamber. M. thermautotrophicus ΔH was applied to solidified medium plates by spot plating, spread plating, or pour plating. For spot plating, 50 μl of M. thermautotrophicus ΔH culture was spotted on a solidified medium plate. Incubation was started after the drop was completely absorbed. For spread plating, 50 μl of diluted or undiluted liquid culture was applied to a solidified medium plate and spread out with a Drigalski spatula until the liquid was completely absorbed. For pour plating, 5 ml mineral medium containing 0.8 wt% Bacto agar (soft agar) was mixed with a liquid M. thermautotrophicus ΔH culture and poured on top of a solidified medium plate, which contained 1.5 wt% Bacto agar. For better gas-solid mass transfer and to avoid sealing of the plates by water, paper clips were added to the petri dish prior to incubation in a custom-made stainless-steel jar (Raff + Grund, Freiburg am Neckar, Germany) inspired by the work of Balch et al. (44). The gas phase of the stainless-steel jar was exchanged to 200 kPa of H2/CO2/H2S (79.9/20/0.1 vol%). The pressurized anaerobic jar was incubated without shaking at 60°C (Memmert, Schwabach, Germany).
For general cloning and gene manipulation, E. coli NEB stable (New England Biolabs, Frankfurt/Main, Germany) was used. E. coli S17-1 for conjugational DNA transfer was kindly provided by Wolfgang Wohlleben of the Department for Biotechnology at the University of Tübingen, Germany (45). E. coli BL21(DE3) with pME2508 (Archaea Center of the University of Regensburg, Germany) was used to produce recombinant pseudomurein endoisopeptidase (PeiP) enzyme. E. coli was cultivated in LB medium, which contained (per liter) sodium chloride, 10 g; tryptone, 10 g; and yeast extract, 5 g, and which was supplemented with appropriate amounts of chloramphenicol (30 μg/ml), ampicillin (100 μg/ml), or kanamycin (50 μg/ml). For cultivation of E. coli S17-1, trimethoprim (10 μg/ml) was added to stabilize the genome-integrated tra module, which is responsible for mobilization of plasmid DNA (45). Solidified LB medium plates contained 1.5 wt% of Kobe I agar (Carl Roth, Karlsruhe, Germany) and were incubated at 37°C. Liquid E. coli cultures were incubated at 37°C with shaking at 150 rpm.
Molecular cloning and plasmid construction.
All primers, gBlock DNA fragments (IDT, Coralville, IA, USA), and plasmids used in this study are summarized in Tables 1 to 3. PCR was performed with Q5 Hot Start high-fidelity polymerase (NEB, Ipswich, MA, USA) according to the manufacturer’s guidelines and with the required primer combinations (Table 1). Primer concentrations were reduced 10-fold, and elongation time was prolonged by 1 min. Resulting PCR products were DpnI digested when required and purified using a PCR purification kit (Qiagen, Hilden, Germany). For initial fusion of the first shuttle-vector construct pSV1_1, as described below, we used the Gibson Assembly Ultra kit (Synthetic Genomics, La Jolla, CA, USA). All follow-up constructs were assembled with Gibson Assembly master mix (New England Biolabs [NEB], Ipswich, MA, USA) or restriction/ligation cloning with the aid of the implemented modular restriction enzyme-recognition sites (Fig. 1). E. coli cells were transformed with DNA via chemical transformation by following a standard heat shock protocol (46). All plasmids were confirmed by Sanger sequencing (MPI Genomics Center, Tübingen, Germany).
TABLE 1.
List of primers used in this study
| Name | Purposea | Sequence (5′→3′) | Reference |
|---|---|---|---|
| Gib_CF1 | pCF203 | ATAAAATGCTTGGGAGATGACGCCCGCCCCAC | This study |
| Gib_CF2 | pCF203 | ATAATCTCCTCTATTTCCATGAGAATCACTCCTATTTTTTTGATATATACATCATAACATTAC | This study |
| Gib_CF3 | pCF203 | AAAAAATAGGAGTGATTCTCATGGAAATAGAGGAGATTATAGAGAAAGTTGCTAGG | This study |
| Gib_CF4 | pCF203 | TGCGGGTCGTGGGGCGGGCGTCATCTCCCAAGCATTTTATGAGCCCTAGC | This study |
| Gib_CF5 | pSB1 | GCCGGTGGTTACCGTGATATTATCTATTACTATATCCCTATATAAAGAATACTCAAAAAATGGGC | This study |
| Gib_CF6 | pSB1 | GTAATAGATAATATCACGGTAACCACCGGCTAGCAGGTGATGCATATGGCTAAAATGAGAATATCAC | This study |
| Gib_CF7 | pSV1_1 | TATTTTGAATCCATTGCGTTGCGCTCACTG | This study |
| Gib_CF8 | pSV1_1 | TGGGCGGCCGGCCGCGTTAATATTTTGTTAAAATTCGCGTTAAATTTTTGTTAAATCAG | This study |
| Gib_CF9 | pSV1_1 | AAATATTAACGCGGCCGGCCGCCC | This study |
| Gib_CF10 | pSV1_1 | TGCATTTTTTTGCGGCGCGCCCTGACA | This study |
| Gib_CF11 | pSV1_1 | AGCGCAACGCAATGGATTCAAAATAGATTCATAATGGAGTCATCCACG | This study |
| Gib_CF12 | pSV1_1 | TCAGGGCGCGCCGCAAAAAAATGCAAATAAAATTTGGGGTGG | This study |
| Res_CF1 | pSV1_2 | CGTACTGCAGCGATCGCGGTCATATGGATACAGCGGCC | This study |
| Res_CF2 | pSV1_2 | GTTATGGATTATAAGCGGCCGGC | This study |
| Gib_CF13 | pMVS1111A:Psynth-bgaB | CCACCCTGCCACCCCAAATTTTATTTGCATTTTTTTGCGGGTTAATTAAGCCTGGAGGAATGCCTTTATATAGG | This study |
| Gib_CF14 | pMVS1111A:Psynth-bgaB | TTTATATATTTTTAATTCACTGGGGGCAATTCTGTCAGGGCGCGCCTGGGGTCGTGCGCTC | This study |
| Gib_CF15 | pMVS1111A:PhmtB/Pmrt(M.t.)-bgaB | CGGCTCTAGCTATGTCCGATC | This study |
| Gib_CF16 | pMVS1111A:PhmtB/Pmrt(M.t.)-bgaB | CACTGGGGGCAATTCTGTCAG | This study |
| Gib_CF21 | pCF201 | AATACAAGAAAGGCGCGCCAAATCATTATATAGGACCTTGATAAAATTTTTTAGAGGGC | This study |
| Gib_CF22 | pCF201 | ACCTGACGTGTGGCCGGCCCGATTCAAATATAACAGCCGTTATAACACCGC | This study |
| Gib_CF23 | pCF201 | AATCGGGCCGGCCACACGTCAGGTGGCACTTTTCG | This study |
| Gib_CF24 | pCF201 | CATCCACGGATGCGATCGCCTAAGAAACCATTATTATCATGACATTAACCTATAAAAATAGGC | This study |
| Gib_CF17 | pCF202 | AAAAAATAGGAGTGATTCTCATGGCTAAAATGAGAATATCACCGGAAT | This study |
| Gib_CF18 | pCF202 | TGCGGGTCGTGGGGCGGGCGCTAAAACAATTCATCCAGTAAAATATAATATTTTATTTTCTCCCAAT | This study |
| Gib_CF19 | pCF202 | GATATTCTCATTTTAGCCATGAGAATCACTCCTATTTTTTTGATATATACATCATAACATTAC | This study |
| Gib_CF20 | pCF202 | TACTGGATGAATTGTTTTAGCGCCCGCCCCACG | This study |
| Res_LM1 | pMVS1111A:PhmtB-fdhZ-245 | ATCGGCTTAGGCGCGCCTGCTCATCGTCAATTCTAGTAGAGTCATGAATCATTATGCAGG | This study |
| Res_LM2 | pMVS1111A:PhmtB-fdhZ-245 | GCTTAGCGCATTAATTAACCGCCCATTTTTTGAGTATTC | This study |
| Gib_LM1 | pLM201 | TAACAGCGGCGCTATCAAGGTCCTGCATAATGATTCATGACGCCCGCCCCACG | This study |
| Gib_LM2 | pLM201 | TTTGCCGTATCTGCAGGCGATTTAAAAGATGATCCCATGAGAATCACTCCTATTTTTTTG | This study |
| Gib_LM3 | pLM201 | TTATGATGTATATATCAAAAAAATAGGAGTGATTCTCATGGGATCATCTTTTAAATCGCC | This study |
| Gib_LM4 | pLM201 | TCCTTTCGGTCGGGCGCTGCGGGTCGTGGGGCGGGCGTCATGAATCATTATGCAGGACC | This study |
| Gib_LM5 | pLM202 | ATCCTATATAAATATATCGCTAATTTTAAGGTTTTTCTGAGCCATCGGTTGGTTCATGGGTTAATTAAGAATACTCAAAAAATGGGCG | This study |
| Gib_LM6 | pLM202 | CGATATATTTATATAGGATTATATGAATAGATAATATCACATAAAATGAGGTGGTTAATTATGGGATCATCTTTTAAATCGCCTGCAG | This study |
| Seq_CF1 | Specific for gDNA M. t.1.5 kb | CCACCAGTTCGACTCCCTGG | This study |
| Seq_CF2 | Specific for gDNA M. t.1.5 kb | CTGTTAAAGGCGGGGGTGG | This study |
| Seq_CF3 | Specific for gDNA M. t. 2.8 kb | CTTGGGTGATGATGGGATGTATTG | This study |
| Seq_CF4 | Specific for gDNA M. t. 2.8 kb | CGAGGAGAAACACATCCAGCTG | This study |
| Seq_CF5 | Specific for pME2001 replicon | GTTAATCCAGCACATCCTCC | This study |
| Seq_CF6 | Specific for pME2001 replicon | CCTGTCCAACTTATACCTTTGG | This study |
| Seq_CF7 | Analysis of bgaB constructs | CCCCATAACATCGGCACAGTAC | This study |
| Seq_CF8 | Analysis of bgaB constructs | CCTGGCTGGGGTTAATAAATGTTG | This study |
| Seq_LM1 | Analysis of fdhZ-245 constructs | GATTTCTGGAATCCGCCATGGG | This study |
| Seq_LM2 | Analysis of fdhZ-245 constructs | CTAATAGTCGCCGATCCAAG | This study |
| Seq_LM3 | Analysis of fdhZ-245 constructs | GGTTCCTGGCTTGAATG | This study |
| Seq_LM4 | Analysis of fdhZ-245 constructs | GAGAAGCAAAGGATGACTG | This study |
| Seq_LM5 | Analysis of fdhZ-245 constructs | CAGCACCCATCTTATTCG | This study |
| Seq_LM6 | Analysis of fdhZ-245 constructs | GCAGTTAAGAAGGGTTCG | This study |
| Seq_LM7 | Analysis of fdhZ-245 constructs | GGCTCCGTTATAAGGGTTG | This study |
| Seq_LM8 | Analysis of fdhZ-245 constructs | CTGAATGGATCGAGAAAGG | This study |
| Seq_LM9 | Analysis of fdhZ-245 constructs | CATTCTTTCGAGATGGAAG | This study |
| Seq_LM10 | Analysis of fdhZ-245 constructs | CCTATATTCGCATTCGTGG | This study |
| Seq_LM11 | Analysis of fdhZ-245 constructs | ATGTTTGCCACACTGTG | This study |
| Seq_LM12 | Analysis of fdhZ-245 constructs | GGTGGGGTTTTGGTGTGCG | This study |
Abbreviations: gDNA, genomic DNA; M. t., M. thermautotrophicus.
TABLE 2.
List of gBlocks used in this study
| Name | Sequence (5′→3′) | Reference |
|---|---|---|
| gBlock PmcrB-pac-Tmcr | GGTACCGAAAAGTGCCACCTGACCGATGGCCGGCCGCCCATTTTTTGAGTATTCAAATTCAAATTATTGTGTTATTAACATCTTATATATAAACTTTTCTATTTAATGTTAATGAAAAAGTGAATATATATACATAGAGTAATGTTATGATGTATATATCAAAAAAATAGGAGTGATTCTCATGACCGAGTACAAGCCCACCGTTAGGCTCGCAACCAGGGATGATGTTCCCAGGGCAGTTAGGACCCTCGCAGCAGCATTCGCAGATTACCCCGCAACCAGGCACACCGTTGATCCCGATAGGCACATAGAGAGGGTTACCGAGCTCCAGGAGCTCTTCCTCACCAGGGTTGGTCTCGATATAGGTAAGGTTTGGGTTGCAGATGATGGTGCAGCAGTTGCAGTTTGGACCACCCCCGAGTCAGTTGAGGCAGGTGCAGTTTTCGCAGAGATAGGTCCCAGGATGGCAGAGCTCTCAGGTTCAAGGCTCGCAGCACAGCAGCAGATGGAGGGTCTCCTCGCACCCCACAGGCCCAAGGAGCCCGCATGGTTCCTCGCAACCGTTGGTGTTTCACCCGATCACCAGGGTAAGGGTCTCGGTTCAGCAGTTGTTCTCCCCGGTGTTGAGGCAGCAGAGAGGGCAGGTGTTCCCGCATTCCTCGAGACCTCAGCACCCAGGAACCTCCCCTTCTACGAGAGGCTCGGTTTCACCGTTACCGCAGATGTTGAGTGCCCCAAGGATAGGGCAACCTGGTGCATGACCAGGAAGCCCGGTGCATGACGCCCGCCCCACGACCCGCAGCGCCCGACCGAAAGGAGCGCACGACCCCATGGCTCCGACCGAAGCCACCCGGGGCGGCCCCGCCGACCCCGCACCCGCCCCCGAGGCCCACCGCGGGGGACACACCGAACACGCCGACCCTGCTGAACACGCGGCGCAGTTCGGTGCCCAGGAGCGGATCGGGAATTAATTCGAAGCTGCTGGTGAAAGAGACCCTATCTTACCTGCTAAAATCTAAGTTAATTACTAATTTATTATTAATTTATTATTAGATTGGGCAAAATAGTAAAAGAAAACTAAAGGAAACCTAATATGGTTTCCTTTTTTTATATATTTTTAATTCACTGGGGGCAATTCTGTCAGGGCGCGCCTTCGGGCCATCGGGCCC | This study |
| gBlock PhmtB-PacI-bgaB | CGGCTCTAGCTATGTCCGATCAATCTTAATTAAGCCTGGAGGAATGCCCCCATGAACCAACCGATGGCTCAGAAAAACCTTAAAATTAGCGATATATTTATATAGGATTATATGAATAGATAATATCACATAAAATGAGGTGGTTAATTATGAACGTTCTCAGTTCCATCTGCTATGGGGGGGATTACAAC | This study |
| gBlock Pmrt(M.t.)-PacI-bgaB-cor | CGGCTCTAGCTATGTCCGATCAATCTTAATTAAGCCTGGAGGAATGCCCCATTTCCATGGATTATCGCTGGCAATCCCATAACCCCATCAGTTTTATTAATAAAATAGTAAATTTTATTAATAAATAAATAAAACAAGAGGTGTGAATACCATGAACGTTCTCAGTTCCATCTGCTATGGGGGGGATTACAAC | This study |
| gBlock codon-optimized bgaB | CTGACAGAATTGCCCCCAGTGAATTAAAAATATATAAAAAAAGGAAACCATATTAGGTTTCCTTTAGTTTTCTTTTACTATTTTGCCCAATCTAATAATAAATTAATAATAAATTAGTAATTAACTTAGATTTTAGCAGGTAAGTGGGGTCGTGCGCTCCTTTCGGTCGGGCGCTGCGGGTCGTGGGGCGGGCGCTAGACCTTGCCGGCTTCGTCGTGTTCCCTAAGGACAGCGACGTCGACGCCCTGAATCCTGAGTTCACCCCCCCTGAAGCATTTGCCATCTATCATATTCTGGTAGATCTTATCTTCCGGAAGGGAGAGTGTGACCTCATAGTCGTTGTGGTTAATTATAATAAGGTACTTCCATTCATCGGTCTCCCTCTGCTGAACTTCGACATTCTCAGCAACCTCCAGTATAGGATTTATGTGGTGTTTAGCAAACACCTGTTCGAGAAGCCTGCCAAGGTAGTTGCTGTCAGGGTATGTTCCTACGTATATGCCCTCCCCCTTTCCGTAGCAGTTCCTGGTAACAGCAGGAAGGCCGGCATACCAATCACCTTTGAATGTGGCGAGAGGCTCAGCACCTTCCAGCCTTATTATATCGGCCCATGTGGTACAGTCATACTCGCCGTCGTTTGAGTAGATCTTATTCACCTTTGTCTCGGGATAAGGAACGAATTCCTCCACGAAGATGCCGAGAATGTCCCTCAGCGGTCCTGGATATCCCCCGAGGTGCACTCTATCGTTCTCATCGACTATCACACTGAAAAAGCTTACAATCAGGGTTCCGCCGTTTGCGACAAACTGCCTAAGGTTCTCATCTTCTCCCTCTTTCACCATATACAGCATCGGTGCAATAACAACCTTATATTTTGTGAGATCGTCGGACGGTCTTACAAAGTCGACTGCTATGTTTCTCTTGTAAAGCTCTCTATAATATGCCTCTACTATGGGAATATATCTGAGCTTGTTGTGCGGTTTGGAACTGAGCTCAACTGCCCACCAGTTTTCCCAGTCAAAGATAATTGCCACCTCTGCCTTTATTCTACTCCCCACGAGGCAGTCAAGTTTTTTCAGCTCCTGGCCAAGCTGGGTAACTTCCCTGTATATTCTATTGTTTTCGTTAAGAAAGTGGGGCACCATTGCTCCGTGAAACTTCTCAGCTCCTGCTCTGGACTGCCTCCACTGAAAGAACATTATCCCATCGGCACCCCTGGCGATTGTTGCGTAACTCCAGAGTCTCATAACCCCCGGCGGCTTTGGCACATTGATATCTCTCCAATTAACGTGACTGGTGACCTGCTCCATAAGAATGAACGGCTGCCCCTTCCTAAGTGACCTCATGAGGTCATTCATCATTGCGTGCTGTATAGGGAGTCCCTCCCTGGGATCTGGGTAGCTATCCCAGGTAACGATATCTACGTGCTGAGCCCACTGAAAGTAGTTGAGTGGCTTGAATGATCCCATGAAATTTGTGGAGACCGGGATATCGGGGGTTACTTCCCTGAGGATCTCCTTTTCTGTAAGGAAGAGTTTGAGGATTGAATCATTCATGAATCTGTAGTAATCAAGCTCCTGGCTGGGGTTAATAAATGTTGGTGCCTTCCTAGGGGGATTAATCTCATCCCAGTGGTTATATCTCTGGCCCCAGAAGTTTGTACCCCATCTTTCATTAAGTTCATCAATGGTCTTATACCTTTCTTTAAGCCATTTTCTGAAAGCAACTGCGCAATTCTCACAGAAACACTTACTTACATGGCAAGCGTATTCGTTATTTACGTGCCACATTTTGAGGGCTGGATGATTTTTGTATCTCTCAGCTATAGCCCTTACCAGCCTCTTTATATGTGTTATAAGCTGAGGGTGATTTGGGCAATAATGCTGTCTACTCCCGAAACTCAGTATCACACCGGACTCGTCAATAGGGAGTGAATCAGGGTATTTCTTCACGAACCAGGCGGGTGTGGTTGCGGTGGCGGTCCCCAGATTTATGTATACCCCATGATCGTAGAGGATGTCTATCACTTTGTCGAGCCATTCAAAATCAAATACACCGTCTGATGGCTCGATTTTGGACCAGCTAAAGATTCCGAGTGAAACAAGATTAACACCGGCCTTCTGCATAAGTTTTGCGTCCTCGTACCATATCTCCTCGGGCCACTGTTCTGGGTTGTAATCCCCCCCATAGCAGATGGAACTGAGAACGTTCATATGCATCACCTGCTAGCCGGTGGTTACCGTGATATTATCTATTACTATATCCCTATATAAAGGCATTCCTCCAGGCTTAATTAAC | This study |
TABLE 3.
List of plasmids used in this study
| Name | Function | Reference | M. thermautotrophicus straina |
|---|---|---|---|
| pMTL83151 | Shuttle vector for Clostridia spp. | Heap et al. (27) | - |
| pMU131 | Shuttle vector for Thermoanaerobacter spp. | Shaw et al. (23) | - |
| pME2001 | Cryptic plasmid of M. marburgensis | Bokranz et al. (50) | - |
| pBBR1-MCS2 | Standard cloning vector in E. coli | Kovach et al. (51) | - |
| pUC19 | Standard cloning vector in E. coli | Yanisch-Perron et al. (52) | - |
| pYS3 | Shuttle vector for Pyrococcus furiosus including Simr | Waege et al. (21) | - |
| pME2508 | PeiP production in E. coli | Luo et al. (48) | - |
| pCF200 | pUC57 vector including synthesized PmcrB(M.v.)_Purr codon-optimized for M. thermautotrophicus, Tmcr | This study | - |
| pCF201 | pUC19 vector including native M. thermautotrophicus Z-245 fdhZ-245 operon with putative promoter region | This study | - |
| pLM201 | Exchange of Neor to coding region of fdhZ-245 from pCF201 in pCF204 | This study | - |
| pLM202 | Exchange of PmcrB(M.v.) to PhmtB in pLM201 | This study | - |
| pCF203 | Exchange of Purr to Simr in pCF200 | This study | - |
| pCF204 | Exchange of Purr to Neor in pCF200 | This study | - |
| pCF404 | pUC57 including 1 kb up- and downstream of annotated pyrF gene (MTH_RS00570) and PmcrB(M.v.)_Purr | This study | - |
| pCF407 | Exchange of PmcrB(M.v.)_Purr to PmcrB(M.v.)_Neor in pCF404 | This study | - |
| pSB1 | Exchange of PmcrB(M.v.) promoter to Psynth in pCF407 | This study | - |
| pSV1_1 | Shuttle vector construct containing PmcrB(M.v.)_Simr and pBBR1MCS2 backbone and pME2001 replicon | This study | - |
| pSV1_2 | Shuttle vector construct containing PmcrB(M.v.)_Simr and pMTL backbone and pME2001 replicon | This study | - |
| pSV1_3 | Shuttle vector construct containing PmcrB(M.v.)_Neor and pMTL80151 backbone and pME2001 replicon | This study | - |
| pMVS-V1 | Shuttle vector construct containing Psynth_Neor and pMTL backbone and pME2001 replicon | This study | x |
| pMVS1111A:Psynth-bgaB | Shuttle vector construct pMVS-V1 including β-galactosidase (bgaB) gene and promoter Psynth | This study | x |
| pMVS1111A:PhmtB-bgaB | Shuttle vector construct pMVS-V1 including β-galactosidase (bgaB) gene and promoter PhmtB | This study | x |
| pMVS1111A:Pmrt(M.t.)-bgaB | Shuttle vector construct pMVS-V1 including β-galactosidase (bgaB) gene and promoter Pmrt(M.t.) | This study | x |
| pMVS1111A:Psynth(BRE)-bgaB | Shuttle vector construct pMVS-V1 including β-galactosidase (bgaB) gene and promoter Psynth(BRE) | This study | x |
| pMVS1111A:PhmtB-fdhZ-245 | Shuttle vector construct pMVS-V1 including fdhZ-245 operon from M. thermautotrophicus Z-245 and promoter PhmtB | This study | x |
-, strain not available; x, strain available.
pCF200, which contains the puromycin acetyltransferase (pac) gene (Purr) from Streptomyces alboniger as a codon-optimized version for M. thermautotrophicus ΔH under the control of the PmcrB(M.v.) promoter and the Tmcr terminator from Methanococcus voltae (47), was completely synthesized (BioCat, Heidelberg, Germany). The pac gene in pCF200 was exchanged to the 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HmgA)-encoding gene (Simr) from Thermococcus kodakarensis by using pYS3 (21) and pCF200 as the templates for Gibson Assembly, resulting in pCF203. pCF203, pME2001 (extracted from wild-type M. marburgensis), and pBBR1-MCS2 (Addgene catalog no. 85168) were used as the templates for Gibson Assembly with the Gibson Assembly Ultra kit (Synthetic Genomics, La Jolla, CA, USA) and resulted in the putative shuttle vector pSV1_1. pSV1_1 was the basis for further shuttle vectors. For the introduction of a high-copy-number replicon for E. coli, a tra region for plasmid mobilization, and an additional AsiSI restriction enzyme-recognition sequence, the pBBR1-MCS2 backbone was exchanged to the E. coli vector backbone from pMTL83151 (27), including Camr, ColE1, and the tra minigene for mobilization, via Gibson Assembly resulting in pSV1_2. To implement the thermostable neomycin phosphotransferase gene (Neor) (28), the Purr from pCF200 was exchanged to Neor from pMU131 (23) by Gibson Assembly. Afterward, based on pCF404, the Neor under the control of the PmcrB(M.v.) promoter and the Tmcr terminator was used to construct a putative integration plasmid for the exchange of an annotated pyrF gene in M. thermautotrophicus ΔH, using AscI and FseI as restriction enzymes and T4 ligase for ligation, resulting in pCF407. In pCF407 the PmcrB(M.v.) promoter was exchanged to Psynth by inverse PCR resulting in pSB1. The fragments PmcrB_Neor_Tmcr from pCF407 and Psynth_Neor_Tmcr from pSB1 were used to substitute for the Simr in pSV1_2 by restriction-ligation cloning using AscI and FseI, resulting in pSV1_3 and pMVS-V1, respectively. To generate pMVS1111A:Psynth-bgaB, the PCR-amplified gBlock with the thermostable β-galactosidase (bgaB) gene, which was codon optimized for M. thermautotrophicus ΔH and which was placed under the control of the Psynth promoter, was fused to AscI-digested pMVS-V1 with Gibson Assembly. The AscI restriction enzyme-recognition sequence was recovered at the intersection with the selectable-marker module, and a PacI sequence was introduced at the intersection with the M. thermautotrophicus ΔH replicon module. Further promoters [Psynth(BRE), PhmtB, Pmrt(M.t.)] were amplified via overlap-extension PCR of the β-galactosidase gBlock and promoter gBlock and inserted by restriction/ligation cloning using restriction enzymes PacI and AscI. pCF201 was constructed by amplifying the fdhZ-245 operon from M. thermautotrophicus Z-245 genomic DNA and introducing the fragment into pUC19 by Gibson Assembly. Gibson Assembly was used to exchange the Neor-coding region in pCF204 with the fdhZ-245 operon from pCF201, resulting in plasmid pLM201. The promoter PmcrB(M.v.) in pLM201 was exchanged to PhmtB by inverse PCR of the complete plasmid, except of the PmcrB(M.v.) sequence, with primers containing overlapping parts of PhmtB in the overhangs, and direct transformation of E. coli with the linear PCR product. The resulting plasmid pLM202 was used to amplify the PhmtB-fdhZ-245 cassette by PCR to include PacI and AscI restriction enzyme-recognition sequences. Restriction/ligation cloning with the restriction enzymes PacI and AscI was used to exchange the β-galactosidase gene in pMVS1111A:Psynth-bgaB for the PhmtB-fdhZ-245 cassette to give pMVS1111A:PhmtB-fdhZ-245.
Plasmid DNA extraction from Methanothermobacter spp.
For plasmid DNA extraction from Methanothermobacter spp., 10 ml of liquid cell culture was centrifuged at 3,700 rpm for 15 min at room temperature (Centrifuge 5920 R, rotor S-4x1000; Eppendorf, Hamburg, Germany). The supernatant was discarded, and the cell pellet was resuspended in 150 μl of sucrose (30 wt%)-containing buffer P1 (from QIAprep Spin miniprep kit; Qiagen, Hilden, Germany). For lysis of Methanothermobacter cells, alkaline lysis was combined with enzymatic lysis by adding a final concentration of 100 ng/ml of pseudomurein endoisopeptidase (PeiP) to the sample prior to incubation for 1 h at 60°C. The pseudomurein-degrading enzyme PeiP, which lyses pseudomurein-containing Methanobacteriales cell walls, was produced as a heterologous 6×His-tagged protein from pME2508-carrying E. coli BL21(DE3) as described previously (48). The recombinant protein was purified via a Protino Ni-TED column according to the manufacturer’s guidelines (Macherey+Nagel, Düren, Germany). After the PeiP treatment, the QIAprep Spin miniprep kit (Qiagen, Hilden, Germany) manufacturer’s guidelines were followed with final elution in 40 μl nuclease-free water.
Interdomain conjugational DNA transfer.
DNA transfer via interdomain conjugation was achieved between E. coli S17-1 and M. thermautotrophicus ΔH. E. coli S17-1 was transformed with the respective shuttle vector. Overnight cultures of the respective E. coli S17-1 donor strains were inoculated. At the same time, 20 ml of liquid mineral medium was inoculated with wild-type M. thermautotrophicus ΔH (recipient). The overnight culture of E. coli S17-1, which contained the shuttle vector, was diluted into 10 ml of fresh LB medium in a sterile 50-ml baffled flask for better aeration to give an OD600 of 0.3 to 0.5. When this culture reached an OD600 of 2.0 to 2.5, the incubation was stopped and the culture was harvested aerobically at 3,700 rpm for 10 min at room temperature (Centrifuge 5920 R, rotor S-4x1000; Eppendorf, Hamburg, Germany). The supernatant was discarded, and the E. coli S17-1 pellet was transferred into the anaerobic chamber. Wild-type M. thermautotrophicus ΔH was grown to early stationary growth phase (OD600 of 0.25 to 0.35). 8 ml of M. thermautotrophicus ΔH culture were centrifuged stepwise at 12,500 rpm for 4 min at room temperature (MySPIN 12 minicentrifuge; Thermo Scientific, Waltham, MA, USA) inside the anaerobic chamber. The final pellet was resuspended in 250 μl of the original nonconcentrated M. thermautotrophicus ΔH culture and gently mixed with the E. coli S17-1 pellet. 100 μl of cell suspension were anaerobically spotted on solid LB-MS medium, which was a mixture that consisted of 50 vol% of mineral medium and 50 vol% of LB medium without the 10 g/liter sodium chloride. The spot was dried, while the lid of the petri dish was kept slightly open for 1 h at 37°C in the incubator (Coy Laboratory Products, Grass Lake, MI, USA) within the anaerobic chamber. When the spot was completely absorbed, the plates were provided with paper clips and transferred to a stainless-steel jar. The gas phase of the jar was exchanged to 200 kPa H2/CO2/H2S (79.9/20/0.1 vol%) and incubated at 37°C without shaking for 16 to 20 h. The spot-mated E. coli S17-1 and M. thermautotrophicus ΔH cells were washed from the LB-MS plates using 1 ml nonselective mineral medium and transferred to 4 ml nonselective mineral medium in a 50-ml serum bottle with an H2/CO2 (80/20 vol%) gas phase. After recovery for 3 to 4 h at 60°C with shaking at 150 rpm, 1 ml of the culture was transferred to 20 ml selective liquid mineral medium in a 100-ml serum bottle and incubated at 60°C with shaking at 150 rpm. Growth of M. thermautotrophicus ΔH after 24 to 48 h of incubation indicated successful DNA transfer into M. thermautotrophicus ΔH, while growth only later than 48 h indicated the appearance of spontaneously neomycin-resistant M. thermautotrophicus ΔH cells (these cultures can be discarded as unsuccessful). Fifty microliters from this selective-enrichment culture was spread plated on selective solidified medium plates, and individual colonies were analyzed after 2 days of incubation at 60°C. A larger amount of individual colonies can be obtained by pour plating, but the analysis of spread-plated cells is easier from an experimental handling point of view (discussed below).
To determine the conjugation frequency, the following modifications to the standard protocol were made: (i) the cell count of M. thermautotrophicus ΔH in liquid culture was determined by counting in a Petroff counting chamber (the initial recipient cell number in 100 μl of the stepwise-concentrated culture was calculated based on this cell count), (ii) the 5-ml nonselective recovery culture from the washed spot after spot mating was incubated for ∼16 to 20 h instead of 3 to 4 h, and (iii) 100 μl of the nonselective recovery culture was directly spread plated on selective solidified medium plates, without a liquid selective-enrichment step.
Molecular methods for analysis of genetically modified M. thermautotrophicus ΔH.
PCR analysis was performed from liquid cultures and directly from individual colonies. 100 μl of liquid culture or one individual colony, which was resuspended in 40 μl of deionized water, was boiled for 12 min at 100°C (ThermoMixer C; Eppendorf, Hamburg, Germany). After cooling the sample on ice, 1 μl was added to a 10-μl PCR mix. PCR was performed using Phire plant PCR master mix (Thermo Scientific, Waltham, MA, USA). The denaturation and annealing times were increased from 5 s to 20 s and to 10 s, respectively. Thirty cycles were performed for all analyses. We observed false-positive PCR signals for shuttle-vector DNA due to plasmid DNA carryover from E. coli for two transfers after the nonselective liquid recovery step. After the third transfer, plasmid DNA from E. coli was not detectable anymore in any of our experiments. For robust PCR amplifications of individual colonies from M. thermautotrophicus ΔH, it was crucial to keep the agar contamination of the PCR sample as low as possible. Therefore, even though the plating efficiency is higher with pour plating, genetically modified M. thermautotrophicus ΔH strains were spread plated instead of pour plated. This led to a lower total colony count but to more reliable results.
Additional to PCR analysis, plasmid DNA from genetically modified M. thermautotrophicus ΔH strains was extracted as described above. The purified plasmid DNA was used for retransformation of E. coli NEB stable. Analysis of E. coli NEB stable colonies was performed via test restriction digestions and Sanger sequencing for further confirmation of stable replication of shuttle vectors in M. thermautotrophicus ΔH.
β-Galactosidase enzyme activity assays.
For a qualitative β-galactosidase enzyme activity assay with the lactose analogue S-Gal, 2 ml of overnight cell cultures that carry pMVS-V1 or pMVS1111A:PSynth-bgaB was harvested by centrifugation for 4 min at 13,000 rpm at room temperature (Centrifuge 5424, rotor FA-45-24-11; Eppendorf, Hamburg, Germany). The supernatant was discarded, and the samples were stored at −20°C until further use. All samples were resuspended in 100 μl buffer P1 (from Qiagen QIAprep Spin miniprep kit) containing sucrose (30 wt%) and lysed by adding 100 ng/ml PeiP, followed by incubation for 30 min at 60°C. Fifty microliters of the cell lysate was incubated with 250 μg/ml S-Gal and 250 μg/ml ammonium ferric citrate in 1 ml LB medium, which provided any potentially required trace compounds. The samples were incubated for 1 h at 60°C. After ∼30 min, a color change was visible.
For a quantitative β-galactosidase enzyme activity assay with the lactose analogue ONPG, 4 ml of cell culture was harvested anaerobically by stepwise centrifugation (Centrifuge 5424, rotor FA-45-24-11; Eppendorf, Hamburg, Germany). Afterward, the same lysis procedure for samples was applied as for the S-Gal assay. The resulting cell lysate was used for a quantitative in vitro β-galactosidase enzyme activity assay with ONPG as chromogenic substance according to the method of Jensen et al. (31). In brief, 12.5 μl of cell lysate (equal to 0.5 ml of original cell culture) was mixed with 600 μl of ONPG (1 mg/ml)-containing substrate solution. The mixture was incubated for 2 h at 60°C. Afterward, 200 μl was added to 200 μl of 1 M sodium bicarbonate stop solution in a 96-well plate. The absorbance at 420 nm was measured in a microplate reader (Multiskan Go; Thermo Scientific, Waltham, MA, USA). For the preliminary experiment (see Fig. S7 in the supplemental material), 25 μl of cell lysate was mixed with 675 μl of ONPG substrate solution instead. After the incubation for 2 h at 60°C, 350 μl of substrate solution was added to 350 μl of stop solution, and the absorbance at 420 nm was measured in a cuvette (1-cm path length) with a spectrophotometer (NP80; Implen, Munich, Germany). Enzyme activity was defined in Miller units as change in absorbance at 420 nm per assay time in hours, optical density at 600 nm, and volume of M. thermautotrophicus ΔH cell culture [ΔA420 × (h × OD600 × liter)−1].
(A to D) The measurements of absorbance at 420 nm (A420) of the enzyme activity assay (bars) and optical density at 600 nm (OD600) of the corresponding genetically engineered M. thermautotrophicus ΔH strain (dots) after different incubation periods [M. thermautotrophicus ΔH with pMVS-V1 (A), pMVS1111A:Pmrt(M.t.)-bgaB (B), pMVS1111A:Psynth-bgaB (C), or pMVS1111A:PhmtB-bgaB (D)]. (E) The resulting Miller units from panels A to D for the four different M. thermautotrophicus ΔH strains are given for the incubation periods 15 h, 19 h, 24 h, and 36 h for each strain from left to right. Average [pMVS-V1 (n = 2), pMVS1111A:Pmrt(M.t.)-bgaB (n = 3), pMVS1111A:Psynth-bgaB (n = 2), and pMVS1111A:PhmtB-bgaB (n = 3)] with error bars indicating standard deviation. The Miller units in this experiment are lower than in the experiment in Fig. 3C because of differences in the experimental parameters (Materials and Methods). Download FIG S7, EPS file, 1.9 MB (1.9MB, eps) .
Copyright © 2021 Fink et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
High-performance liquid chromatography for formate analysis.
Formate concentrations were analyzed via a high-pressure liquid chromatography (HPLC) system as described in the work of Klask et al. (49). For HPLC sample preparation, all culture samples were centrifuged for 5 min at 13,000 rpm (Centrifuge 5424; Eppendorf, Germany) in 2-ml reaction tubes. 450 μl of the supernatant were transferred into clean reaction tubes and stored at −20°C until use. Frozen samples were thawed at room temperature. The samples were vortexed and centrifuged again, and 400 μl of the supernatant was transferred into short-thread HPLC/gas chromatography (GC) vials (glass vial ND9; VWR, Germany) and sealed with short screw caps, which contained rubber septa (6 mm for ND9; VWR, Germany). Formate standards (1 to 500 μM) were prepared freshly for the analysis. All HPLC samples were randomized.
ACKNOWLEDGMENTS
We are grateful to John Reeve, William W. Metcalf, Rudolf K. Thauer, and Michael Rother for helpful discussions. We acknowledge Caroline Schlaiß, Sylvia Lemke, and Gabriela Contreras-Arriagada for performing supportive experiments, Luis Antoniotti from the Max Planck Institute for Developmental Biology workshop for his technical input during the design of the stainless-steel jars, and the Archaea centre of the University of Regensburg for kindly providing the plasmids pYS3 (Winfried Hausner) and pME2508.
The work was funded by the Alexander von Humboldt Foundation in the framework of the Alexander von Humboldt Professorship (L.T.A.) and the U.S. Office of Naval Research Global (ONRG, N62909-19-1-2076; L.T.A., B.M.). Additional funding sources were the German Federal Ministry of Education and Research (MethanoPEP, 031B0851C; B.M.) and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy – EXC 2124 – 390838134 (L.T.A., B.M.).
B.M. and L.T.A. initiated the work. C.F. and B.M. designed the experiments. C.F., S.B., A.M.E., and L.M. performed laboratory experiments and analyzed the data. L.T.A. and B.M. supervised the project. C.F. and B.M. wrote the manuscript, while all edited the paper and approved the final version.
We declare no conflict of interest.
Contributor Information
Bastian Molitor, Email: bastian.molitor@uni-tuebingen.de.
Christa M. Schleper, University of Vienna
REFERENCES
- 1.Thauer RK, Kaster AK, Seedorf H, Buckel W, Hedderich R. 2008. Methanogenic archaea: ecologically relevant differences in energy conservation. Nat Rev Microbiol 6:579–591. doi: 10.1038/nrmicro1931. [DOI] [PubMed] [Google Scholar]
- 2.Thauer RK. 1998. Biochemistry of methanogenesis: a tribute to Marjory Stephenson: 1998 Marjory Stephenson prize lecture. Microbiology 144:2377–2406. doi: 10.1099/00221287-144-9-2377. [DOI] [PubMed] [Google Scholar]
- 3.Thauer RK. 2015. My lifelong passion for biochemistry and anaerobic microorganisms. Annu Rev Microbiol 69:1–30. doi: 10.1146/annurev-micro-091014-104344. [DOI] [PubMed] [Google Scholar]
- 4.Kaster A-K, Goenrich M, Seedorf H, Liesegang H, Wollherr A, Gottschalk G, Thauer RK. 2011. More than 200 genes required for methane formation from H2 and CO2 and energy conservation are present in Methanothermobacter marburgensis and Methanothermobacter thermautotrophicus. Archaea 2011:973848. doi: 10.1155/2011/973848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thauer RK. 2019. Methyl (alkyl)-coenzyme M reductases: nickel F-430-containing enzymes involved in anaerobic methane formation and in anaerobic oxidation of methane or of short chain alkanes. Biochemistry 58:5198–5220. doi: 10.1021/acs.biochem.9b00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pende N, Sogues A, Megrian D, Sartori-Rupp A, England P, Palabikyan H, Rittmann SK-MR, Graña M, Wehenkel AM, Alzari PM, Gribaldo S. 2021. SepF is the FtsZ anchor in archaea, with features of an ancestral cell division system. Nat Commun 12:3214. doi: 10.1038/s41467-021-23099-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kandler O, König H. 1998. Cell wall polymers in archaea (archaebacteria). Cell Mol Life Sci 54:305–308. doi: 10.1007/s000180050156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thema M, Weidlich T, Hörl M, Bellack A, Mörs F, Hackl F, Kohlmayer M, Gleich J, Stabenau C, Trabold T, Neubert M, Ortloff F, Brotsack R, Schmack D, Huber H, Hafenbradl D, Karl J, Sterner M. 2019. Biological CO2-methanation: an approach to standardization. Energies 12:1670. doi: 10.3390/en12091670. [DOI] [Google Scholar]
- 9.Pfeifer K, Ergal İ, Koller M, Basen M, Schuster B, Rittmann SK-MR. 2021. Archaea biotechnology. Biotechnol Adv 47:107668. doi: 10.1016/j.biotechadv.2020.107668. [DOI] [PubMed] [Google Scholar]
- 10.Martin MR, Fornero JJ, Stark R, Mets L, Angenent LT. 2013. A single-culture bioprocess of Methanothermobacter thermautotrophicus to upgrade digester biogas by CO2-to-CH4 conversion with H2. Archaea 2013:157529. doi: 10.1155/2013/157529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seifert AH, Rittmann S, Herwig C. 2014. Analysis of process related factors to increase volumetric productivity and quality of biomethane with Methanothermobacter marburgensis. Appl Energy 132:155–162. doi: 10.1016/j.apenergy.2014.07.002. [DOI] [Google Scholar]
- 12.Meile L, Reeve JN. 1985. Potential shuttle vectors based on the methanogen plasmid pME2001. Nat Biotechnol 3:69–72. doi: 10.1038/nbt0185-69. [DOI] [Google Scholar]
- 13.Leisinger T, Meile L. 1993. Plasmids, phages, and gene transfer in methanogenic bacteria, p 1–12. In Sebald M (ed), Genetics and molecular biology of anaerobic bacteria. Springer, New York, NY. [Google Scholar]
- 14.Luo Y, Leisinger T, Wasserfallen A. 2001. Comparative sequence analysis of plasmids pME2001 and pME2200 of Methanothermobacter marburgensis strains Marburg and ZH3. Plasmid 45:18–30. doi: 10.1006/plas.2000.1493. [DOI] [PubMed] [Google Scholar]
- 15.Meile L, Abendschein P, Leisinger T. 1990. Transduction in the archaebacterium Methanobacterium thermoautotrophicum Marburg. J Bacteriol 172:3507–3508. doi: 10.1128/jb.172.6.3507-3508.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Worrell VE, Nagle DP, McCarthy D, Eisenbraun A. 1988. Genetic transformation system in the archaebacterium Methanobacterium thermoautotrophicum Marburg. J Bacteriol 170:653–656. doi: 10.1128/jb.170.2.653-656.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jenal U, Rechsteiner T, Tan PY, Bühlmann E, Meile L, Leisinger T. 1991. Isoleucyl-tRNA synthetase of Methanobacterium thermoautotrophicum Marburg. Cloning of the gene, nucleotide sequence, and localization of a base change conferring resistance to pseudomonic acid. J Biol Chem 266:10570–10577. doi: 10.1016/S0021-9258(18)99261-6. [DOI] [PubMed] [Google Scholar]
- 18.Meile L, Stettler R, Banholzer R, Kotik M, Leisinger T. 1991. Tryptophan gene cluster of Methanobacterium thermoautotrophicum Marburg: molecular cloning and nucleotide sequence of a putative trpEGCFBAD operon. J Bacteriol 173:5017–5023. doi: 10.1128/jb.173.16.5017-5023.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Majernik A, Cubonova L, Polak P, Smigan P, Greksak M. 2003. Biochemical analysis of neomycin-resistance in the methanoarchaeon Methanothermobacter thermautotrophicus and some implications for energetic processes in this strain. Anaerobe 9:31–38. doi: 10.1016/S1075-9964(03)00042-8. [DOI] [PubMed] [Google Scholar]
- 20.Rospert S, Linder D, Ellermann J, Thauer RK. 1990. Two genetically distinct methyl‐coenzyme M reductases in Methanobacterium thermoautotrophicum strain Marburg and ΔH. Eur J Biochem 194:871–877. doi: 10.1111/j.1432-1033.1990.tb19481.x. [DOI] [PubMed] [Google Scholar]
- 21.Waege I, Schmid G, Thumann S, Thomm M, Hausner W. 2010. Shuttle vector-based transformation system for Pyrococcus furiosus. Appl Environ Microbiol 76:3308–3313. doi: 10.1128/AEM.01951-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Santangelo TJ, Cubonová L, Matsumi R, Atomi H, Imanaka T, Reeve JN. 2008. Polarity in archaeal operon transcription in Thermococcus kodakaraensis. J Bacteriol 190:2244–2248. doi: 10.1128/JB.01811-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shaw AJ, Hogsett DA, Lynd LR. 2010. Natural competence in Thermoanaerobacter and Thermoanaerobacterium species. Appl Environ Microbiol 76:4713–4719. doi: 10.1128/AEM.00402-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Susanti D, Frazier MC, Mukhopadhyay B. 2019. A genetic system for Methanocaldococcus jannaschii: an evolutionary deeply rooted hyperthermophilic methanarchaeon. Front Microbiol 10:1256. doi: 10.3389/fmicb.2019.01256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fonseca DR, Halim MFA, Holten MP, Costa KC. 2020. Type IV-like pili facilitate transformation in naturally competent archaea. J Bacteriol 202:e00355-20. doi: 10.1128/JB.00355-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martinez-Garcia E, Aparicio T, Goni-Moreno A, Fraile S, de Lorenzo V. 2015. SEVA 2.0: an update of the Standard European Vector Architecture for de-/re-construction of bacterial functionalities. Nucleic Acids Res 43:D1183–D1189. doi: 10.1093/nar/gku1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heap JT, Pennington OJ, Cartman ST, Minton NP. 2009. A modular system for Clostridium shuttle plasmids. J Microbiol Methods 78:79–85. doi: 10.1016/j.mimet.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 28.Hoseki J, Yano T, Koyama Y, Kuramitsu S, Kagamiyama H. 1999. Directed evolution of thermostable kanamycin-resistance gene: a convenient selection marker for Thermus thermophilus. J Biochem 126:951–956. doi: 10.1093/oxfordjournals.jbchem.a022539. [DOI] [PubMed] [Google Scholar]
- 29.Molitor B, Kirchner K, Henrich AW, Schmitz S, Rosenbaum MA. 2016. Expanding the molecular toolkit for the homoacetogen Clostridium ljungdahlii. Sci Rep 6:31518. doi: 10.1038/srep31518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dodsworth JA, Li L, Wei S, Hedlund BP, Leigh JA, de Figueiredo P. 2010. Interdomain conjugal transfer of DNA from bacteria to archaea. Appl Environ Microbiol 76:5644–5647. doi: 10.1128/AEM.00967-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jensen TO, Pogrebnyakov I, Falkenberg KB, Redl S, Nielsen AT. 2017. Application of the thermostable β-galactosidase, BgaB, from Geobacillus stearothermophilus as a versatile reporter under anaerobic and aerobic conditions. AMB Express 7:169–179. doi: 10.1186/s13568-017-0469-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Darcy TJ, Hausner W, Awery DE, Edwards AM, Thomm M, Reeve JN. 1999. Methanobacterium thermoautotrophicum RNA polymerase and transcription in vitro. J Bacteriol 181:4424–4429. doi: 10.1128/JB.181.14.4424-4429.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wood GE, Haydock AK, Leigh JA. 2003. Function and regulation of the formate dehydrogenase genes of the methanogenic archaeon Methanococcus maripaludis. J Bacteriol 185:2548–2554. doi: 10.1128/JB.185.8.2548-2554.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wasserfallen A, Nölling J, Pfister P, Reeve J, De Macario EC. 2000. Phylogenetic analysis of 18 thermophilic Methanobacterium isolates supports the proposals to create a new genus, Methanothermobacter gen. nov., and to reclassify several isolates in three species, Methanothermobacter thermautotrophicus comb. nov., Methanothermobacter wolfeii comb. nov., and Methanothermobacter marburgensis sp. nov. Int J Syst Evol Microbiol 50:43–53. doi: 10.1099/00207713-50-1-43. [DOI] [PubMed] [Google Scholar]
- 35.Nölling J, Reeve JN. 1997. Growth-and substrate-dependent transcription of the formate dehydrogenase (fdhCAB) operon in Methanobacterium thermoformicicum Z-245. J Bacteriol 179:899–908. doi: 10.1128/jb.179.3.899-908.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Argyle JL, Tumbula DL, Leigh JA. 1996. Neomycin resistance as a selectable marker in Methanococcus maripaludis. Appl Environ Microbiol 62:4233–4237. doi: 10.1128/aem.62.11.4233-4237.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones WJ, Whitman WB, Fields RD, Wolfe RS. 1983. Growth and plating efficiency of Methanococci on agar media. Appl Environ Microbiol 46:220–226. doi: 10.1128/aem.46.1.220-226.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ao X, Li Y, Wang F, Feng M, Lin Y, Zhao S, Liang Y, Peng N. 2013. The Sulfolobus initiator element is an important contributor to promoter strength. J Bacteriol 195:5216–5222. doi: 10.1128/JB.00768-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gregor D, Pfeifer F. 2005. In vivo analyses of constitutive and regulated promoters in halophilic archaea. Microbiology (Reading) 151:25–33. doi: 10.1099/mic.0.27541-0. [DOI] [PubMed] [Google Scholar]
- 40.Peng N, Xia Q, Chen Z, Liang YX, She Q. 2009. An upstream activation element exerting differential transcriptional activation on an archaeal promoter. Mol Microbiol 74:928–939. doi: 10.1111/j.1365-2958.2009.06908.x. [DOI] [PubMed] [Google Scholar]
- 41.Enzmann F, Mayer F, Rother M, Holtmann D. 2018. Methanogens: biochemical background and biotechnological applications. AMB Express 8:1. doi: 10.1186/s13568-017-0531-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nayak DD, Metcalf WW. 2017. Cas9-mediated genome editing in the methanogenic archaeon Methanosarcina acetivorans. Proc Natl Acad Sci USA 114:2976–2981. doi: 10.1073/pnas.1618596114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lyu Z, Jain R, Smith P, Fetchko T, Yan Y, Whitman WB. 2016. Engineering the autotroph Methanococcus maripaludis for geraniol production. ACS Synth Biol 5:577–581. doi: 10.1021/acssynbio.5b00267. [DOI] [PubMed] [Google Scholar]
- 44.Balch W, Fox G, Magrum L, Woese C, Wolfe R. 1979. Methanogens: reevaluation of a unique biological group. Microbiol Rev 43:260–296. doi: 10.1128/mr.43.2.260-296.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pozzi R, Coles M, Linke D, Kulik A, Nega M, Wohlleben W, Stegmann E. 2016. Distinct mechanisms contribute to immunity in the lantibiotic NAI-107 producer strain Microbispora ATCC PTA-5024. Environ Microbiol 18:118–132. doi: 10.1111/1462-2920.12892. [DOI] [PubMed] [Google Scholar]
- 46.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 47.Sarmiento F, Leigh JA, Whitman WB. 2011. Genetic systems for hydrogenotrophic methanogens. Methods Enzymol 494:43–73. doi: 10.1016/B978-0-12-385112-3.00003-2. [DOI] [PubMed] [Google Scholar]
- 48.Luo Y, Pfister P, Leisinger T, Wasserfallen A. 2002. Pseudomurein endoisopeptidases PeiW and PeiP, two moderately related members of a novel family of proteases produced in Methanothermobacter strains. FEMS Microbiol Lett 208:47–51. doi: 10.1111/j.1574-6968.2002.tb11059.x. [DOI] [PubMed] [Google Scholar]
- 49.Klask C-M, Kliem-Kuster N, Molitor B, Angenent LT. 2020. Nitrate feed improves growth and ethanol production of Clostridium ljungdahlii with CO2 and H2, but results in stochastic inhibition events. Front Microbiol 11:724. doi: 10.3389/fmicb.2020.00724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bokranz M, Klein A, Meile L. 1990. Complete nucleotide sequence of plasmid pME2001 of Methanobacterium thermoautotrophicum (Marburg). Nucleic Acids Res 18:363. doi: 10.1093/nar/18.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kovach ME, Elzer PH, Steven Hill D, Robertson GT, Farris MA, Roop RM, Peterson KM. 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166:175–176. doi: 10.1016/0378-1119(95)00584-1. [DOI] [PubMed] [Google Scholar]
- 52.Yanisch-Perron C, Vieira J, Messing J. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mpl8 and pUC19 vectors. Gene 33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental results (A to I) and discussion (J). (A) Differences in plating efficiencies for various plating techniques. (B) Description of growth-inhibiting effects of antibiotics. (C) Details on the design of the modular shuttle-vector system pMVS. (D) Analysis of successful DNA transfer via site-specific PCR. (E) Retransformation of E. coli with plasmid extracts from M. thermautotrophicus ΔH. (F) Approximation of the conjugation frequency. (G) Segregational stability of shuttle vector under nonselective conditions. (H) Control experiments for conjugational DNA transfer. (I) Quantitative β-galactosidase enzyme activity assay. (J) Discussion on plating efficiency of M. thermautotrophicus ΔH. Download Text S1, DOCX file, 0.06 MB (66.1KB, docx) .
Copyright © 2021 Fink et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
(A) Spot plating of M. thermautotrophicus ΔH cells on solidified medium plates. (B) Pour plating of 1 × 104 M. thermautotrophicus ΔH cells on solidified medium plates. (C) Spread plating of 1 × 104 M. thermautotrophicus ΔH cells on solidified medium plates. (D) Comparison of the influence from various factors on the plating efficiency with spread plating of 1 × 104 M. thermautotrophicus ΔH cells and pour plating of 1 × 103 M. thermautotrophicus ΔH cells, including the influence of paper clips, 0.1 vol% hydrogen sulfide (H2S) in the headspace gas mixture, exponential growth phase (Exp), and stationary growth phase (Stat). The bars give the average percentage (log scale) of individual colonies per cell count of initial microbial cells used for plating from technical replicates (n = 3; n = 4 for spread plating with paper clips and H2S in exponential growth phase) with error bars indicating standard deviation. (E) Influence of paper clips on the lid elevation. The arrow demonstrates the increase of lid/plate space compared for a plate without paper clips (upper picture) and one with paper clips (lower picture). Download FIG S1, TIF file, 0.3 MB (341.8KB, tif) .
Copyright © 2021 Fink et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Measurement of OD600 after different incubation periods at 60°C (0 h, 24 h, 48 h, and 60 h) with initially 5 × 105 cells/ml of wild-type M. thermautotrophicus ΔH in liquid mineral medium containing neomycin concentrations of 0 μg/ml (long-dashed line, n = 1), 50 μg/ml (dotted line), 100 μg/ml (short-dashed line), and 250 μg/ml (black solid line). For comparison to wild-type M. thermautotrophicus ΔH, initially 5 × 105 cells/ml of pMVS-V1-carrying M. thermautotrophicus ΔH were incubated in selective liquid mineral medium with 250 μg/ml neomycin (gray solid line, n = 1) and analyzed after the same incubation periods. Average (n = 3) with error bars indicating standard deviation. Download FIG S2, EPS file, 1.3 MB (1.3MB, eps) .
Copyright © 2021 Fink et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
(A) Individual colonies from 1 × 108 wild-type M. thermautotrophicus ΔH cells (gray bars), which were pour plated on solidified medium plates containing simvastatin concentrations ranging from 13 μg/ml to 21.5 μg/ml. (B) Individual colonies from 1 × 103 wild-type M. thermautotrophicus ΔH cells (gray bars) and pMVS-V1-carrying M. thermautotrophicus ΔH (black bars, n = 1), which were pour plated on solidified medium plates containing neomycin concentrations ranging from 0 μg/ml to 250 μg/ml. Average (n = 3) with error bars indicating standard deviation for wild-type M. thermautotrophicus ΔH. Download FIG S3, EPS file, 1.3 MB (1.3MB, eps) .
Copyright © 2021 Fink et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
(A) Restriction-enzyme digestion of plasmid DNA, which was extracted from three independent E. coli pMVS-V1 enrichment cultures (1 to 3). The E. coli strains were generated with plasmid DNA, which was extracted from genetically modified M. thermautotrophicus ΔH. The unique cutter KpnI (K), resulting in one 8.2-kb fragment, and the dual cutter NdeI (N), resulting in 2.7-kb and 5.5-kb fragments, were used. M, GeneRuler 1-kb DNA ladder (Thermo Scientific, Waltham, MA, USA). (B) Exemplified sequence alignment of Sanger sequences from retransformed E. coli plasmid DNA to the original pMVS-V1 sequence (main text and Fig. 1 for further explanations). Red arrows indicate the sequence and its direction. No deletions, insertions, or nucleotide exchanges were detected. Download FIG S4, TIF file, 0.1 MB (120.9KB, tif) .
Copyright © 2021 Fink et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
(A) PCR analysis of individual colonies from selective solidified medium plates after one selective transfer of pMVS-V1-carrying M. thermautotrophicus ΔH in liquid mineral medium. (B) PCR analysis of individual colonies from nonselective solidified medium plates after three nonselective transfers (∼21 to 28 cell divisions) in liquid mineral medium (PCR analysis of the first and second transfer is not shown but gave the same results). A 1-kb fragment results from the specific primer pair for the pME2001 replicon for M. thermautotrophicus ΔH (a) and a 1.5-kb fragment from the specific primer pair for M. thermautotrophicus ΔH genomic DNA (b). All 16 colonies each result in positive PCR for both primer combinations. Wild-type M. thermautotrophicus ΔH (WT) does not result in PCR signal for pMVS-V1, and water as negative control (N) does not result in any PCR signal. M, GeneRuler 1-kb DNA ladder (Thermo Scientific, Waltham, MA, USA). Download FIG S5, TIF file, 0.4 MB (385.4KB, tif) .
Copyright © 2021 Fink et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Example calculations for conjugation frequencies for DNA transfer into M. thermautotrophicus ΔH based on equation S1. Download Table S1, DOCX file, 0.01 MB (13.3KB, docx) .
Copyright © 2021 Fink et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
(A) Enrichment cultures of genetically modified M. thermautotrophicus ΔH in 250-μg/ml neomycin-containing selective liquid mineral medium from conjugation/DNA transfer experiments with the standard protocol (1), the DNase I-treated E. coli S17-1 (2), the heat-inactivated E. coli S17-1 (3), and NEB stable as nonconjugative E. coli (4). Slight turbidity in cultures 3 and 4 is caused due to high initial cell count. Enrichment is visible in cultures 1 and 2. (B) Spread-plated M. thermautotrophicus ΔH using culture 1 from panel A (standard protocol). Black circles represent colonies, which were used for PCR analysis. (C) Spread-plated M. thermautotrophicus ΔH using culture 2 from panel A (DNase I treatment). Black circles represent colonies, which were used for PCR analysis. (D) PCR analysis of the four colonies from panels B and C (1 to 4), wild-type M. thermautotrophicus ΔH (5), purified shuttle-vector DNA as positive control (6), and water as negative control (7). A primer combination which amplifies a 1-kb fragment from the pME2001 replicon was used for PCR amplification. M, GeneRuler 1-kb DNA ladder (Thermo Scientific, Waltham, MA, USA). Download FIG S6, TIF file, 0.4 MB (389.5KB, tif) .
Copyright © 2021 Fink et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Analysis of wild-type M. thermautotrophicus Z-245 growth either with molecular hydrogen and carbon dioxide (black solid line) or with formate (gray solid line) as the carbon and energy source. Average (n = 3) with error bars indicating standard deviation. Download FIG S8, EPS file, 1.2 MB (1.2MB, eps) .
Copyright © 2021 Fink et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
(A to D) The measurements of absorbance at 420 nm (A420) of the enzyme activity assay (bars) and optical density at 600 nm (OD600) of the corresponding genetically engineered M. thermautotrophicus ΔH strain (dots) after different incubation periods [M. thermautotrophicus ΔH with pMVS-V1 (A), pMVS1111A:Pmrt(M.t.)-bgaB (B), pMVS1111A:Psynth-bgaB (C), or pMVS1111A:PhmtB-bgaB (D)]. (E) The resulting Miller units from panels A to D for the four different M. thermautotrophicus ΔH strains are given for the incubation periods 15 h, 19 h, 24 h, and 36 h for each strain from left to right. Average [pMVS-V1 (n = 2), pMVS1111A:Pmrt(M.t.)-bgaB (n = 3), pMVS1111A:Psynth-bgaB (n = 2), and pMVS1111A:PhmtB-bgaB (n = 3)] with error bars indicating standard deviation. The Miller units in this experiment are lower than in the experiment in Fig. 3C because of differences in the experimental parameters (Materials and Methods). Download FIG S7, EPS file, 1.9 MB (1.9MB, eps) .
Copyright © 2021 Fink et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.