

Abstract
The global propagation of SARS-CoV-2 leads to an unprecedented public health emergency. Despite that the lungs are the primary organ targeted by COVID-19, systemic endothelial inflammation and dysfunction is observed particularly in patients with severe COVID-19, manifested by elevated endothelial injury markers, endotheliitis, and coagulopathy. Here, we review the clinical characteristics of COVID-19 associated endothelial dysfunction; and the likely pathological mechanisms underlying the disease including direct cell entry or indirect immune overreactions after SARS-CoV-2 infection. In addition, we discuss potential biomarkers that might indicate the disease severity, particularly related to the abnormal development of thrombosis that is a fatal vascular complication of severe COVID-19. Furthermore, we summarize clinical trials targeting the direct and indirect pathological pathways after SARS-CoV-2 infection to prevent or inhibit the virus induced endothelial disorders.
Keywords: COVID-19, SARS-CoV-2, endothelial dysfunction, immunity, thrombosis
Graphical abstract

1. Introduction
The global pandemic of coronavirus disease 2019 (COVID-19) is caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Although it is generally acknowledged that COVID-19 associated endothelial disorders are rare complications, symptoms of endothelial activation are observed particularly in critically ill COVID-19 patients including endotheliitis, coagulopathy, and thrombosis [[1], [2], [3], [4]]. Pediatric patients who developed multisystem inflammatory syndrome in children (MIS-C) after SARS-CoV-2 infection also show signs of endothelial dysfunction and arterial stiffness [5]. Thus, COVID-19 related endothelial injury can be found in patients at all ages (see Table 1 ). In addition to clinical cases, animal studies also demonstrated endothelial disruption and vascular thrombosis in the lungs of rhesus macaques infected by SARS-CoV-2 [6]. Nevertheless, human features of COVID-19 related endothelial dysfunction were hardly recapitulated in mice [7]. Whether the virus can directly induce pathological events in human endothelial cells (ECs) or endothelial activation and inflammation is only secondary to an overreactive immune response to SARS-CoV-2 remains to be investigated.
Table 1.
Characteristics of endothelial inflammation and dysfunction in COVID-19 patients
| Patient age (years old) | Manifestations | Tissues collected | Presence of SARS-CoV-2 in ECs? | Ref |
|---|---|---|---|---|
| 15 | Elevated inflammation and endothelial injury | Blood | N/A | [31] |
| 24 | Changes in vascular reactivity and arterial stiffness | Examination by FMD, NMD, PWV, Aix, and cIMT | N/A | [35] |
| 24 | Elevated factor VIII, vWF, ischemic stroke | Blood, examination by MRI of the brain | N/A | [32] |
| 31 | Endotheliitis and vasculitis of small cardiac vessels | Heart (autopsy) | N/A | [28] |
| 32 | Impaired endothelium-dependent microvascular reactivity | Skin, examination of microvascular flow by LDPM | N/A | [34] |
| 40 | Multifocal vasculitis, arteriolitis | Bowel (biopsy) | Positive | [26] |
| 43 | Endotheliitis in venous vessels | Specimen from hemicolectomy | Positive | [27] |
| 72 | Elevated D-dimers, vWF, factor VIII | Blood | N/A | [30] |
| 79 | Leukocytoclastic vasculitis | Epidermis and dermis (biopsy) | N/A | [29] |
| Pediatrics, adults | Chilblain-like skin lesion, lymphocytic vasculitis | Skin (biopsy) | Inconclusive | [[19], [20], [21], [22], [23], [24]] |
| Aged adults | Endotheliitis | Kidney, heart, small intestine, lung, liver (autopsy), resected small intestine | Positive | [3] |
| Aged adults | Thrombosis, microangiopathy, increased angiogenesis | Lung (autopsy) | Positive | [4,25] |
| Aged adults | Limb ischemia, femoropopliteal occlusion | Resected arterial segments | Negative (arterial wall) | [33] |
Abbreviations: EC endothelial cell, vWF von Willebrand factor, LDPM laser doppler perfusion monitoring, FMD flow-mediated dilation, NMD nitroglycerin-mediated dilation, PWV pulse wave velocity, Aix augmentation index, cIMT carotic intima-mediated thickness, MRI magnetic resonance imaging
It is generally believed that ECs express ACE2, which is the major entry receptor of SARS-CoV-2 [[8], [9], [10]]. Accordingly, the prevalent view holds that COVID-19 related endothelial dysfunction is resulted from direct infection of the endothelium by SARS-CoV-2. Nevertheless, some studies argue against this hypothesis as the basal expression levels of endothelial ACE2 are so low that may escape from SARS-CoV-2 mediated cell entry [11]. The significantly increased expression of proinflammatory cytokines including interleukin-6 (IL-6), IL-1β, tumor necrosis factor α (TNF-α), and interferon γ (IFN-γ); and chemokines in critically ill patients with COVID-19 may also suggest that a cytokine release storm occurs in the severe cases [12,13]. Indeed, postmortem studies demonstrate widespread inflammation in the specimens collected from patients succumbed to COVID-19 [3,4,14], indicating the overreactive immune responses in severe COVID-19 patients. In this regard, the dysregulated immune responses are also believed to be the leading cause of COVID-19 related endothelial dysfunction and mortality.
Recently, researchers have been endeavoring to develop new technologies and study the potential mechanisms by which SARS-CoV-2 induces endothelial injury. For instances, in vitro platforms including human organoids and organ-on-chip have emerged [[15], [16], [17], [18]]. In one study, human embryonic stem cells (hESCs) were differentiated into lung and colonic organoids to screen for SARS-CoV-2 inhibitors [15]. In another study, human blood vessel and kidney organoids were generated from pluripotent stem cells (iPSCs) for investigating the potential function of human recombinant soluble ACE2 (hrsACE2) in the inhibition of SARS-CoV-2 infection by competitive binding [16]. Moreover, the coculture system of epithelial cells, ECs, and immune cells in a human lung-on-chip model allows researchers to analyze the response of distinct cell types after SARS-CoV-2 infection at the organ level, recapitulating in vivo viral infection [17,18]. These emerging technologies aiming at developing in vitro human cell-based platforms for disease modeling and drug screening will aid the development of new treatment options against COVID-19.
Here, we summarize the most recent updates regarding the clinical characteristics of COVID-19 related endothelial pathologies. We discuss possible direct and indirect mechanisms underlying these endothelial disorders. In addition, we review the biomarkers that might indicate the disease severity, particularly related to the abnormal development of coagulation in thrombosis that is a fatal vascular complication of severe COVID-19. Lastly, we summarize clinical trials and management strategies targeting COVID-19 related endothelial disorders.
2. Clinical characteristics of endothelial inflammation and dysfunction in COVID-19 patients
The clinical signs of endothelial inflammation and dysfunction induced by SARS-CoV-2 are displayed by a wide range of organs (see Table 1). For instance, chilblain-like skin lesion was the skin manifestation of lymphocytic vasculitis in COVID-19 patients at different ages [[19], [20], [21], [22], [23], [24]]. Besides, COVID-19 related endotheliitis and vasculitis in multiple organs including the lungs [3,4,25], intestine [3,26,27], heart [3,28], kidney [3], liver [3], and skin [29] have been reported. Moreover, elevated endothelial injury biomarkers in the blood [[30], [31], [32]], vascular occlusion [32,33], and altered vascular reactivity and stiffness [34,35] were observed in both mild and severe COVID-19 patients.
Clinically, COVID-19 related endothelial dysfunction is also characterized by well-defined endothelial injury biomarkers. Circulating endothelial cell (CEC), a cell-based biomarker shed from injured vessels, was identified to be elevated in critically ill COVID-19 patients, and patients with pre-existing conditions [36]. Of note, even the convalescent COVID-19 patients displayed significantly increased amount of CEC compared with the healthy group, suggesting persistent vascular dysfunction after recovery from COVID-19 [37]. In addition, molecular biomarkers of endothelial dysfunction such as von Willebrand factor (vWF) have been found increased continuously with the development of acute respiratory distress syndrome (ARDS) after SARS-CoV-2 infection [30]. P-selectin and E-selectin were also observed to be elevated in COVID-19 patients admitted to intensive care unit (ICU) or developed ARDS [38,39]. Higher levels of Angiopoietin-2 (Ang-2), which implied aggravated pulmonary vascular leakage; and soluble intercellular adhesion molecule-1 (ICAM-1), which was expressed by activated ECs in response to inflammation, were also detected in the plasma of non-survivors [40]. All these observations reflect the characteristics of endothelial injury and dysfunction in patients with COVID-19 particularly in the severe conditions. Nevertheless, whether there is a strain dependent effect of SARS-CoV-2 in endothelial inflammation, activation and dysfunction requires further investigations.
3. Evidence of direct infection of ECs by SARS-CoV-2
In fact, only a few studies have examined the presence of viral protein within the endothelium of different organs (Table 1), therefore, whether there is a direct infection of ECs by SARS-CoV-2 remains unclear. We next summarize the studies on potential receptors that could mediate cell entry of SARS-CoV-2 into the endothelium or that are activated after interacting with the virus (Fig. 1 ).
Fig. 1.

Potential mechanisms by which SARS-CoV-2 activates endothelial cells. The virus could infect, enter or activate endothelial cells (EC) by (A) binding to the extracellular domain of ACE2 via S protein; (B) binding to neuropilin-1 (NRP1) to promote the infection with the presence of ACE2 and TMPRSS2; (C) binding to high density lipoprotein (HDL) that facilitates viral entry through the HDL receptor SR-B1 in an ACE2-dependent manner; (D) endocytosis through CD147/peptidylprolyl isomerase B(A); and/or (E) activating the toll-like receptor (TLR) 2 signaling pathway. Solid lines indicate direct binding; and dash lines indicate interactions.
3.1. Possible infection of ECs by SARS-CoV-2 via ACE2
As the first discovered human homolog of angiotensin converting enzyme (ACE), angiotensin-converting enzyme 2 (ACE2) was initially identified to negatively regulate canonical renin-angiotensin system (RAS), the overstimulation of which exerts detrimental impacts on the cardiovascular system, including hypertension, fibrosis, and inflammation [8,41]. In canonical RAS, angiotensinogen is cleaved by protease renin to generate angiotensin I (Ang I), which is further processed by ACE and converted to Ang II [42]. Ang II is the first end product of canonical RAS, functioning via interacting with Ang II receptor type 1 receptor (AT1R) and Ang II receptor type 2 receptor (AT2R). ACE2 antagonizes RAS primarily through degrading Ang I and Ang II into Ang 1-9 and Ang 1-7, respectively. Ang 1-9 can be further catalyzed by ACE to generate Ang 1-7. Ang 1-7 is known for anti-fibrosis, anti-inflammation, and vasodilation induction by counterposing Ang II and AT1R related actions [43]. Thus, as a negative regulator of canonical RAS, ACE2 mitigates the cardiovascular damage resulting from chronic RAS activation.
Another notable function of ACE2 is the receptor mediated host cell entry of infective agents such as SARS-CoV and SARS-CoV-2 responsible for severe acute respiratory syndrome (SARS) and COVID-19, respectively [9,[44], [45], [46]]. Viral entry relies on the interaction of ACE2 with viral spike (S) protein (Fig. 1A), which contains the S1 and S2 units [44,[47], [48], [49], [50]]. Compared with SARS-CoV, SARS-CoV-2 possesses a S protein with significantly higher affinity to ACE2, and SARS-CoV-2 can utilize a wider range of host proteases to prime viral penetration [51]. Two critical steps are required for the entry of SARS-CoV-2. The first step is the binding of S1 to the extracellular domain of the transmembrane ACE2; and the second step is the cleavage between viral S1 and S2, primarily accomplished by host protease transmembrane serine protease 2 (TMPRSS2) [49,50]. The detachment of S1 from S2 allows the conformational transition of S2, facilitating viral internalization [44,49,50].
It is widely acknowledged from previous work that expression of surface ACE2 is abundant in the pulmonary epithelium [45] and small intestinal epithelium [52], which explains the vulnerability of the lungs to SARS-CoV-2; and diarrhea is also frequently presented in COVID-19 cases [9,53]. In addition to the epithelium, ACE2 is also believed to be abundantly expressed by the endothelium [4,[8], [9], [10],16,54] . To date, Donoghue et al cloned the human ACE2 gene in year 2000 and identified the ACE2 protein in human coronary and renal ECs [8]. After that, the propagation of SARS-CoV and SARS-CoV-2 in subsequent years accelerates the research on endothelial expression and function of ACE2, aiming at understanding the mechanisms underlying endothelial injuries accompanied with the pathological progression of severe SARS and COVID-19. In 2004, Hamming et al delineated a comprehensive tissue and cellular distribution of ACE2; and demonstrated the high expression levels of ACE2 in the vascular endothelium by immunohistochemistry, despite the lack of clinical evidence supporting the direct infection of SARS-CoV on the endothelium [9,10]. With the outbreak of COVID-19, more groups have investigated the role of endothelial ACE2 after SARS-CoV-2 infection. Ackermann et al provided some clinical evidences that viral infection can increase endothelial ACE2 expression as demonstrated by the elevated levels of ACE2-positive ECs in the lungs of COVID-19 and influenza A (H1N1) infected patients, respectively, compared with the uninfected lungs through examining the pulmonary autopsies derived from patients who died from COVID-19 [4]. Importantly, the electron micrographs demonstrated disrupted endothelial structure with intracellular SARS-CoV-2, indicating that the endothelial injury was possibly resulted from direct viral infection [4]. However, only 7 specimens were examined in this study, and the relatively small sample size could not draw a solid conclusion [4]. Furthermore, Monteil et al also found the presence of viral RNA in human capillary organoids after SARS-CoV-2 infection [16]. In another study, Ahmetaj-Shala et al reported the detectable levels of ACE2 transcripts in human ECs by reanalyzing the human tissue transcriptomic databases [54]. In line with these observations in human, some animal and in vitro cellular experiments also suggested that SARS-CoV-2 can dampen EC function through targeting ACE2. A representative work conducted by Lei et al showed that the S protein of SARS-CoV-2 alone was able to downregulate ACE2 and AMPK activity in hamsters as well as in human pulmonary arterial endothelial cells (PAECs) [55]. Moreover, this group has further recapitulated this by expressing a mutated and unstable ACE2 in ECs and demonstrated increased mitochondrial fragmentation mediated by S protein in these ECs [55]. However, interpretation of these results is limited by the lack of a rescue experiment in which overexpression of a wildtype ACE2 should compensate the S protein-induced endothelial mitochondrial impairment.
In fact, recent work has argued against the degree of contribution of ACE2 to the endothelial pathogenesis associated with COVID-19. The other prevalent perspective against the role of endothelial ACE2 in COVID-19 associated endothelial pathogenesis is based on the conflicting observation of endothelial ACE2 expression as recent reports have revealed that human ECs express very low or only a basal level of ACE2 which is insufficient to mediate the direct infection of SARS-CoV-2. For instance, McCracken et al reported that only scarce endothelial ACE2 transcripts could be detected in human hearts by reanalyzing the publically available single-cell RNA sequencing data and the authors believed that the detectable ACE2 transcripts could also be the contaminated ones from pericytes [11]. Besides, the authors revealed inactive ACE2 transcriptional status in ECs by showing the enrichment of repressive histone markers and the lack of active histone modifications at the ACE2 gene locus [11]. More convincingly, McCracken et al also demonstrated extremely low viral replication in primary human cardiac and pulmonary ECs after exposure to high concentrations of SARS-CoV-2 [11]. Similarly, Conde et al demonstrated that primary human ECs isolated from multiple tissues were capable of being infected by SARS-CoV-2 only when recombinant ACE2 was introduced as they lacked ACE2 expression [56]. In another study, it has been demonstrated that the endothelial injury caused by COVID-19 could be the secondary effect after alveolar epithelial infection [57]. Compared with human pulmonary alveolar epithelial cell (HPAEpiC), human lung microvascular endothelial cell (HULEC-5a) was almost insusceptible to SARS-CoV-2 infection; however, these cells displayed mitochondrial fragmentation and decreased Golgi apparatus after treatment with the culture supernatant of HPAEpiC infected by SARS-CoV-2, indicating an epithelium-dependent infection of SARS-CoV-2 in ECs [57]. Similarly, a German research team also demonstrated that SARS-CoV-2 was not able to infect mono-cultured ECs, it was epithelial cells rather than ECs being susceptible to SARS-CoV-2 inoculation in a human lung-on-chip model [17]. When ECs were cocultured with epithelial and mononuclear cells in a biochip, viral protein synthesis, robust interferon release, and cell death were only detected in epithelial cells in the beginning of infection, even though the epithelial-endothelial barrier displayed significantly higher permeability with the extended infection period [17]. Altogether, these evidences suggest that direct infection of ECs through ACE2 is less likely to occur. COVID-19 related endothelial injury could be mediated by other entry receptors or induced by the inflammatory factors released from infected cells such as epithelial cells adjacent to ECs.
3.2. Possible infection of ECs by SARS-CoV-2 via cell surface receptors beyond ACE2
The single transmembrane glycoproteins neuropilins (NRPs) are involved in diverse biological processes, such as angiogenesis, vascular permeability, immune regulation, and neuron development, through serving as co-receptors for various molecules [58]. Two studies identified neuropilin-1 (NRP1) as a host factor that enhanced SARS-CoV-2 uptake via interacting with a conserved C-terminal motif ([R/K]-XX-[R/K], CendR motif) of SARS-CoV-2 S1 generated by furin cleavage, based on the presence of ACE2 and TMPRSS2 [59,60] (Fig. 1B). Given that the furin cleavage site was possessed by SARS-CoV-2 S protein rather than SARS-CoV S protein, the above findings explained the enhanced spread of COVID-19 compared with SARS. More importantly, the tissue wide distribution and abundant expression levels of NRP1 accounted for the extrapulmonary manifestations of COVID-19, as ACE2 exhibited a rather low expression level in most tissues [61]. Though ECs were not included in the cell line experiments, the scRNA-seq data from human lung and olfactory epithelium emphasized the highest levels of NRP1 in ECs, implying that NRP1 may play a potential role in the reciprocal action between SARS-CoV-2 and endothelium [59]. In fact, NRP1 is a well-characterized co-receptor of vascular endothelial growth factor (VEGF) in ECs [62,63], its cell surface expression on ECs has been well acknowledged.
Scavenger receptor B type 1 (SR-B1), a cell surface receptor of high-density lipoprotein (HDL) expressed by a variety of cell types including ECs [64], has also been reported to facilitate ACE2-mediated cell entry of SARS-CoV-2 [65] (Fig. 1C). Increased viral particles attaching to the cell surface and enhanced viral entry were observed with the presence of HDL. Mechanistically, SARS-CoV-2 S1 can bind to HDL, facilitating viral entry through SR-B1 in an ACE2-dependent manner [65]. In addition, CD147 (BSG) was demonstrated as a potential host cell receptor involved in the infection of SARS-CoV-2 (Fig. 1D). Wang et al first discovered that human T cells, which lack ACE2, could be infected by SARS-CoV-2 in a dose-dependent manner via CD147-mediated endocytosis [66]. CD147 was also identified to be expressed by ECs, and CD147/peptidylprolyl isomerase B(A) could be a potential pathway employed by SARS-CoV-2 to invade the endothelium [54]. Furthermore, Toll-like receptor 2 (TLR2) was also found to participate in endothelial activation in the context of SARS-CoV-2 infection without host cell entry (Fig. 1E). Qian et al reported that the nucleocapsid (N) protein of SARS-CoV-2 was able to trigger endothelial inflammation by activating NFκB and MAPK signal pathway via TLR2 [67]. Altogether, these evidences have demonstrated the possibility of host cell entry or activation through expression of EC surface receptors; however, more work is needed to validate these findings in vivo. Whether blocking these pathways would prevent COVID-19 associated endothelial dysfunction requires future investigations.
4. Evidence of endothelial dysfunction in severe COVID-19 patients as a result of the over-reactive immune responses
Endothelial dysfunction generally refers to abnormal endothelial activation, and vascular endothelial balance shifting toward vasoconstriction and inflammation, causing endotheliitis, vessel leakage, and thrombosis [68,69]. Sustained endothelial dysfunction in COVID-19 was observed with an elevated level of CECs in convalescent patients [37]. Importantly, CEC counts and markers in convalescent patients were positively correlated with increased release of cytokines such as IL-18, C-X-C motif chemokine ligand 10 (CXCL10), and CXCL12, which are either related to the migration or activation of T cells and B cells [37]. In addition, expression of the receptors marking cytotoxic effector lymphocytes including Integrin Subunit Alpha L (ITGAL), Selectin P Ligand (SELPLG), and C-X3-C Motif Chemokine Receptor 1 (CX3CR1) were also positively correlated with increased expression of their corresponding ligands such as ICAM, Selectin P (SELP) and C-X3-C Motif Chemokine Ligand 1 (CX3CL1), respectively, by CECs derived from peripheral blood mononuclear cells (PBMCs) of both mild and severe COVID-19 patients [37] (Fig. 2 ). These observations highlight a potentially causative relationship between persistent immune activation and endothelial dysfunction in the pathological development of COVID-19. Nevertheless, whether the reparative endothelial progenitor cells also expressed these markers and their activation following interaction with the activated immune cells might impair the endothelial repair process leading to endothelial dysfunction; and/or they are activated cells released from the damaged endothelium after interaction with the activated immune cells remains to be determined.
Fig. 2.

Potential mechanisms by which circulating endothelial cells are activated by lymphocytes or their cytokines during COVID-19 progression. The positive correlation between the expression of CEC activation markers such as (A) ICAM1, (B) SELP, and (C) CX3CL1 and their corresponding counter receptors ITGAL, SELPLG, and CX3CR1 on lymphocytes collected from COVID-19 PBMCs implicates the interaction of activated ECs and effector lymphocytes. The high frequency of cytotoxic effector cells in COVID-19 patients may also explain the susceptibility of COVID-19 patients to endothelial injury.
The progression of COVID-19 can be divided into three phases, the early infection phase exhibiting mild constitutional symptoms and lymphopenia, the pulmonary phase characterized by moderate pulmonary symptoms, and the severe phase showing elevated inflammatory markers resulted from respiratory failure and multiple organ dysfunction [70]. Despite that hyperinflammation was most common in severe COVID-19 patients [70], it has been demonstrated that distinct cytokine patterns displayed by individual COVID-19 patients at different disease stages were associated with the disease severity [71]. For example, the levels of IFNα in early COVID-19 plasma predicted the duration of disease course, and the continuously elevated expression levels of IL-6, IL-8, and IL-5 throughout the whole diseased period indicated high risk of mortality [71].
Infiltration of neutrophils and monocytes in air spaces, accumulation of cytokines and macrophages in the lungs, and the aggregations of T cells surrounding blood vessels implied widespread inflammation triggered by SARS-CoV-2 [3,14,[72], [73], [74]]. Correspondingly, the damage caused by SARS-CoV-2 to immune organs was second only to lung infection [75]. Enhanced expression levels of IL-1β, IL-7, IL-8, IL-9, IL-10, IFNγ, TNFα, along with granulocyte-colony stimulating factor (GCSF) were detected in the plasma collected from COVID-19 patients of varying disease severity [13]. Expression levels of IL-2, IL-7, IL-10, TNFα, and GCSF were also higher in ICU patients than that of non-ICU patients [13]. Additionally, hyperactivated T lymphocytes and high concentrations of cytotoxic granules in CD8+ T cells have been detected in COVID-19 cases [75]. Thus, an overreactive immune system was seen in patients with a severity correlated signature during COVID-19 progression.
The autopsies collected from patients succumbed to COVID-19 revealed that diffuse alveolar damage, airway inflammation, and vascular endotheliitis were leading processes in all cases [3,4,14,74]. In multiple organs of patients who died from COVID-19, dead inflammatory cells were frequently observed to be accumulated around vessel wall and dead ECs [26]. The synchronization of hyperinflammation and endothelial dysfunction indicated the crosstalk between overreactive immune system and impaired endothelium at the severe stage of COVID-19. Generally speaking, the active innate and adaptive immune system could contribute to the direct exposure of vascular endothelium to cytokines following SARS-CoV-2 infection [76,77]. The ECs exposed to proinflammatory cytokines transformed to an active state with increased expression levels of cellular adhesive molecules, recruiting leucocytes and enhancing inflammation [76]. This amplifying cascade could result in disrupted endothelial integrity and function, even EC death, finally reflected by vascular permeability and thrombosis [76]. Moreover, the damaged ECs can in turn secret proinflammatory cytokines, boosting intense inflammation [78].
Several cytokines and chemokines have been identified as the leading causes of the inflammation that induced endothelial dysfunction of COVID-19 [79]. Mounting clinical evidences suggested that the activated IL-6 signaling in COVID-19 patients was closely correlated with the poor clinical outcome [12,13,78,[80], [81], [82], [83]]. In a cohort of 1,484 COVID-19 patients in the New York city, their circulating IL-6, IL-8, and TNF-α levels were significantly higher than that of healthy donors and patients with cancer who did not develop cytokine release storm after chimeric antigen receptor-engineered T (CAR-T) cell therapies [12]. High serum IL-6 and TNF-α levels were identified to be predominant predictors of COVID-19 severity and death [12]. Consistently, Coomes et al demonstrated that the mean concentrations of IL-6 in patients with complicated COVID-19 was 2.9-fold higher than patients with noncomplicated COVID-19 [81]. Moreover, the administration of Tocilizumab, which blocks IL-6 receptor, alleviated the infections brought by SARS-CoV-2 [80,84]. IL-6 triggers EC activation during the early phase of inflammation [85]. It has been shown that human ECs expressed a higher level of adhesion molecules when stimulated by IL-6, tending to attract more lymphocytes and transform to an active state [86]. In turn, the impaired ECs can produce IL-6 by themselves, exaggerating the inflammation and inflammation-based endothelial injury in COVID-19 [78]. Consequently, IL-6 participates in the positive feedback loop promoting overreactive immune response and endothelial dysfunction in COVID-19.
Another elevated circulating marker in severe COVID-19 patients is IL-1β [87]. IL-1β was released by activated alveolar macrophages and pulmonary epithelial cells due to the lung infection by SARS-CoV-2; and could stimulate vascular endothelium to promote the release of cell adhesion molecules by ECs [87]. Moreover, IL-1β has been found to down-regulate VE-cadherin, which is an element of endothelial cell-to-cell adherens junctions [88]. Xiong et al recently demonstrated that the increased levels of IL-1β in ARDS inhibited cyclic adenosine monophosphate (cAMP) generation and cAMP response element binding (CREB) mediated transcription of VE-cadherin, thereby impairing endothelial barrier integrity [88]. In addition, TNF-α and IFN-γ also play critical roles in inflammation-induced endothelial dysfunction [12,13]. Karki et al indicated that in COVID-19, TNF-α and IFN-γ acted synergistically to induce PANoptosis, which is an inflammatory programmed cell death process involving pyroptosis, apoptosis, and necroptosis [89]. PANoptosis caused by the combination of TNF-α and IFN-γ perpetuated cytokine storm, leading to COVID-19 related ARDS and multi-organ failure. It is noteworthy that co-treatment of TNF-α and IFN-γ on primary umbilical vein endothelial cells (HUVECs) contributed to robust cell death, suggesting that TNF-α and IFN-γ not only mediated endothelial dysfunction through cytokine-storm, but also directly eliminated ECs [89]. Mechanistically, TNF-α could also regulate endothelial barrier integrity by mediating morphological remodeling of ECs through activating Rho GTPases and reactive oxygen species (ROS), and by promoting the release of endothelial adhesion molecules [90]. Taken together, the above work has revealed that the activation and dysfunction of ECs in severe COVID-19 can be associated with overreactive immune responses induced by SARS-CoV-2 infection.
5. Signaling pathways possibly involved in COVID-19 associated endothelial dysfunction
There are a number of review articles summarizing the potential mechanisms that could be involved in endothelial dysfunction as a result of SARS-CoV-2 infection [[90], [91], [92], [93], [94], [95]], here, we focus on the ROS, VEGFA and HMGB1-RAGE/TLR4 pathways that are more recently identified as key signaling events potentially involved in COVID-19 mediated endothelial dysfunction (Fig. 3 ).
Fig. 3.

Potential mechanisms underlying the pathogenesis of SARS-CoV-2 induced endothelial dysfunction. (A) Reactive oxygen species (ROS) and oxidative stress: in COVID-19 patients, the stimulated NADPH oxidase induced by TNF-α leads to ROS accumulation. Additionally, elevated IFN reduces expression of functional ACE2 leading to imbalanced RAS signaling and increased ROS. Excessive ROS generation then disturbs vascular tone and increases endothelial permeability; (B) The VEGFA/VEGFR2 pathway: inflammatory cytokines can activate the VEGFA/VEGFR2 signaling that promotes the phosphorylation of endothelial adhesion molecules and eNOS by activating the corresponding kinases, thereby increasing vascular permeability; or (C) The HMGB1-RAGE/TLR4 pathway: the binding of HMGB1 to RAGE or TLR4 up-regulates adhesion molecules and chemokines mainly through MAPK and NFκB pathway, respectively, thereby promoting vascular leakiness. In addition, the property of HMGB1 to rupture lysosomal membrane could activate inflammasome that eventually leads to pyroptosis of ECs. Black solid lines indicate direct binding or interaction, pink solid or dash lines indicate activation or stimulation, green solid or dash lines indicate inhibition, and black dash lines indicate a consequence.
5.1. ROS and oxidative stress
ROS are byproducts of oxygen metabolism. At moderate concentrations, ROS are important signaling factors in maintaining tissue homeostasis [92]. However, uncontrolled ROS accumulation caused by either excessive production or inefficient elimination after infection by respiratory viruses leads to oxidative stress and endothelial dysfunction [92,96] (Fig. 3A).
In patients contracting SARS-CoV-2, the elevated levels of pro-inflammatory factor TNF-α could greatly contribute to ROS generation [13,89]. TNF-α was demonstrated to induce oxidative stress by stimulating NADPH-oxidase in ECs, further up-regulating the expression of endothelial adhesion molecules such as ICAM-1 and increasing cell permeability in a NFκB-dependent manner [97,98]. Another potential trigger of excessive ROS production following COVID-19 is the deficiency of functional ACE2 after SARS-CoV-2 infection [[99], [100], [101]]. Although some evidences showed that ACE2 was up-regulated by IFN signaling in COVID-19 [45,102], it has recently been shown that the IFN-stimulated ACE2 was actually a truncated isoform with no function [103,104]. Thus, rather than being increased, the expression level of functional ACE2 could be reduced along with internalization of the SARS-CoV-2/ACE2 complex in COVID-19 [[99], [100], [101]]. As a consequence, the balance of RAS signaling could have been shifted to the accumulation of Ang II and AT1R that enhances the generation of ROS in ECs [105]. In addition to increased endothelial permeability, oxidative stress followed by SARS-CoV-2 infection could also disturb vascular tone [91,106]. Healthy vascular tone relies on the balance of vasodilators such as NO, and vasoconstrictors such as ROS [93]. The excessive generation of ROS after viral infection could not only lead to the accumulation of vasoconstrictors, but also reduce the bioavailability of NO, thereby exacerbating the disruption of vascular tone [91].
5.2. VEGFA/VEGFR2 pathway
In patients with COVID-19, it is possible that the elevated levels of cytokines during inflammation could up-regulate and activate the vascular endothelial growth factor-A (VEGFA) signaling pathway through binding to its main receptor vascular endothelial growth factor receptor2 (VEGFR2) [107]. VEGFA/VEGFR2 is a predominant ligand-receptor complex in the VEGF system, which promotes angiogenesis by regulating EC proliferation, migration, and survival [94]. Nevertheless, VEGFA/VEGFR2 signaling can also contribute to endothelial inflammation by increasing vascular permeability potentially related to endothelial dysfunction in COVID-19 [94,107] (Fig. 3B).
Mechanistically, VEGFA/VEGFR2 could lead to increased endothelial permeability of the lungs after SARS-CoV-2 infection through mediating the phosphorylation states of endothelial adhesion proteins and the activation of NO production [[107], [108], [109], [110], [111], [112], [113], [114]]. VE-cadherin is an important component of endothelial adherens junctions, the phosphorylation of which is closely associated with the endothelial barrier integrity [108]. The binding of VEGFA to VEGFR2 leads to the phosphorylation of human VEGFR2 at Y951, allowing the interaction of T cell specific adaptor (TSAd) and Src kinase to promote the phosphorylation of VE-cadherin at Y658 and Y685 [108]. In addition to VE-Cadherin, VEGFA also regulates the tyrosine phosphorylation of endothelial adhesion molecule β-catenin [109,110]. It has been demonstrated that VEGFA/VEGFR2 signaling can activate focal adhesion tyrosine kinase (FAK), resulting in the FAK mediated phosphorylation of β-catenin at Y142 and the dissociation of β-catenin from VE-Cadherin that disrupts EC junction [109]. Besides, phosphorylation and destabilization of other endothelial junctional proteins such as occludin and connexin 43 can also be resulted from the activated VEGFA/VEGFR2 signaling [111,112]. Furthermore, endothelial NO synthase (eNOS) was suggested to be activated by Akt phosphorylation is induced by VEGFA [113,114]. Increased NO production during inflammation could then contribute to impaired vessel barrier integrity [115].
5.3. HMGB1/RAGE/TLR4 pathway
The high mobility group box 1 protein (HMGB1) is a damage-associated molecular pattern (DAMP) [116]. HMGB1 released by dying or stressed cells functions as proinflammatory molecule through interacting with multiple receptors based on its redox state, including the receptor for advanced glycation endproducts (RAGE) and toll like receptor 4 (TLR4) [116]. The activation of the HMGB1-RAGE/TLR4 axis, which is common in pulmonary inflammation [95], causes endothelial dysfunction mainly by directly inducing EC death [117], increasing endothelial permeability [118], and exaggerating inflammation [119,120].
RAGE is predominantly expressed by type 1 alveolar epithelial cells [121], as well as inflammation-activated vascular ECs and leukocytes [122]. The interaction between HMGB1 and RAGE is involved in inflammation and cell migration by activating pathways such as NFκB and MAPK [119,123], and increasing the levels of adhesion molecules ICAM and VCAM [118], as well as chemokine CXCL12 [120]. Disulfide HMGB1, a redox isoform of HMGB1, can bind to TLR4 co-receptor MD-2 [124]. In macrophages, the HMGB1-dependent activation of the TNF release relies on the interaction of HMGB1 and TLR4, suggesting that the pro-inflammatory response could be triggered by the HMGB1/TLR4 axis [124]. Moreover, there is an interplay between RAGE and TLR4 in the regulation of HMGB1-induced inflammation. Specifically, activation of the HMGB1/TLR4 axis enhances the expression of RAGE, while the interaction between HMGB1 and RAGE increases the cell surface distribution of TLR4 [125].
Another potential proinflammatory mechanism involving HMGB1, RAGE, and TLR4 depends on the detergent property of HMGB1 to permeabilize the phospholipid bilayer under acidic conditions [126,127]. With the help of cell surface RAGE and TLR4 located at the endosomal membrane, DAMPs containing extracellular HMGB1 are able to translocate into lysosomes. However, instead of being degraded in the lysosomes, DAMPs may translocate to the cytosol through HMGB1 mediated degradation of the lysosomal membrane in the acidic lysosome. This process could further amplify the inflammatory cascades and induce pyroptosis [[126], [127], [128]]. Indeed, evidences from Kawasaki disease suggested that the lysosomal rupture triggered by HMGB1/RAGE activates NLRP3 inflammasome in ECs causing pyroptosis [117]. Thus, the activation of HMGB1/RAGE/TLR4 axis might provide an explanation underlying the overactivated immune response and endothelial dysfunction in COVID-19 [95]. Importantly, it has been proposed that targeting HMGB1 could alleviate its proinflammatory activities in exaggerated pulmonary inflammation [95]. Particularly, chloroquine that can neutralize the acidic environment of the endolysosomal system was shown to improve the outcome of COVID-19 infections [129]. Nevertheless, the therapeutic efficacy targeting this pathway in larger controlled clinical studies requires further investigations.
6. Endothelial dysfunction and thrombosis in COVID-19 patients
Abnormal coagulation and thrombotic complications are perceived as dominant manifestations of endothelial dysfunction in severe COVID-19. For instance, thrombotic vessel occlusions and high levels of circulating coagulant markers have been detected in specimen of patients suffered from COVID-19, highlighting the coagulant activation following SARS-CoV-2 infection [[130], [131], [132]]. D-dimer, produced by cleaved cross-linked fibrin, was an indicator for coagulation [133]. Plasma D-dimer levels in patients with severe COVID-19 were usually higher than that of mild patients [132,134,135]. And D-dimer continued to increase with the disease exacerbation in patients who did not survive COVID-19 [132,[135], [136], [137]]. These clinical observations suggested that coagulation activation was positively correlated with COVID-19 severity and mortality. Indeed, the propensity of SARS-CoV-2 to cause vascular complications such as stroke, ischemia, and thrombosis was always a leading threat of COVID-19 [138].
At the cellular level, dysregulated ECs, platelets, and immune system are currently perceived as the primary causes of COVID-19 associated coagulopathy. Healthy endothelium possesses antithrombotic ability through preventing platelet aggregation and fibrin formation via expressing tissue factor pathway inhibitors, recruiting anticoagulant molecules, and activating anticoagulant factors [[139], [140], [141]]. However, under pathological conditions, ECs can be activated by proinflammatory cytokines and pathogen-associated molecular patterns (PAMPs), thereby releasing procoagulant factors and contributing to both the initiation and propagation of thrombus generation [139]. The adhesion glycoprotein vWF stored in Weibel-Palade bodies of ECs plays a critical role in thrombosis [142]. Mechanistically, vWF serves as a mediator for platelet adhesion and aggregation primarily by shaping into large multimers that form high-strength bonds with platelets via platelet receptors [143]. Of note, clinical observations highlighted considerably elevated levels of vWF in COVID-19 patients, particularly in the critically ill patients admitted to ICU, reflecting the hypercoagulable milieu caused by infection-mediated endothelial injury due to SARS-CoV-2 invasion [142,[144], [145], [146], [147], [148], [149], [150], [151], [152], [153], [154], [155], [156], [157], [158], [159]]. Besides vWF, the impaired endothelium also released tissue factor and adhesion molecules such as ICAM, P-selectin, and E-selectin, all of which supported the development of thrombosis [[160], [161], [162]].
Platelet activation also participates in coagulation and clot formation. Increasing evidences showed activated platelets in COVID-19 patients [163,164]. Dysfunctional ECs possessing the coagulant potential could stimulate platelets and accelerate thrombosis during SARS-CoV-2 infection [139]. Moreover, in the case of COVID-19, platelet activation may also be directly stimulated by SARS-CoV-2. Zhang et al reported that platelets expressed endogenous ACE2 and TMPRSS2, which allowed SARS-CoV-2 to activate platelets [164]. However, a recent study argued that the ACE2 mRNA was not detected in platelets, even though the SARS-CoV-2 gene was found in platelets of 2 of 25 patients with COVID-19 [163]. Although it remains unclear whether platelets expressed ACE2, platelets were endowed with a spectrum of receptors that can recognize single-stranded RNA virus [165]. Like in encephalomyocarditis, platelets can be directly stimulated by encephalomyocarditis virus (EMCV) via platelet TLR-7, promoting the interaction between platelets and neutrophils [166]. Thus, it is reasonable to assume that the direct activation of platelets by SARS-CoV-2 also contributed to the initiation of thrombosis in COVID-19.
In patients with COVID-19, elevated neutrophil counts and the major neutrophil chemotactic factor IL-8 were linked to poor prognosis [72,74,136,162,[167], [168], [169], [170]]. A notable feature of neutrophils is to form neutrophil extracellular traps (NETs), which are extracellular de-condensed DNA in complex with histones and granule proteins released from activated neutrophils [171]. NETosis, the process of NETs formation, has been initially identified as a defense event to entrap pathogens [171]. Nevertheless, accumulating studies emphasize the pivotal contribution of NETs to atherosclerosis and thrombosis [171]. Currently, NETs have been known as the driver of thrombus initiation and progression in severe COVID-19 [72,167,168,[172], [173], [174], [175]]. The infiltration of NETs was ubiquitously detected in the postmortem lung biopsies from patients succumbed to COVID-19, involving airway, interstitial, and vascular compartments [72,173]. In the sera collected from COVID-19 patients, the levels of cell-free DNA and the well-defined markers of NETs including myeloperoxidase-DNA (MPO-DNA) and citrullinated histone H3 (Cit-H3) were increased with the disease severity [174]. Mounting evidence suggested that the milieu promoted NETosis following SARS-CoV-2 infection. Middleton et al showed that not only MPO-DNA and Cit-H3 were increased in sera of COVID-19 patients, but also platelet factor 4 (PF4), RANTES (regulated on activation, normal T cell expressed and secreted), and platelet-neutrophil aggregates, all of which could trigger NETosis [172]. Moreover, higher titers of antiphospholipid antibodies (aPL) presented in COVID-19 patients promoted NETosis [176]. The injection of anticardiolipin IgG purified from COVID-19 patient intensified venous thrombosis in mice, demonstrating the procoagulant feature of aPL antibodies induced by SARS-CoV-2 [176]. Mechanistically, Skendros et al proved that NETs dependent thrombosis in COVID-19 was based on platelet-neutrophil interaction, the release of tissue factor, and complement activation [162]. Of note, Skendros et al also proved the interaction of NETs with platelets and ECs in the context of COVID-19 by showing that the human arterial endothelial cells (HAECs) treated with the NETs generated from neutrophils which have been exposed to platelet-rich plasma (PRP) from COVID-19 patients exhibited thrombotic potential [162]. Conclusively, COVID-19 associated thrombosis could be a comprehensive reflection of injured endothelium, dysregulated platelets, and disordered immune system resulted from SARS-CoV-2 infection.
7. Current clinical trials targeting COVID-19 related endothelial disorders
As aforementioned, it has been generally acknowledged that COVID-19 related endothelial dysfunction could be attributed to direct infection of SARS-CoV-2 through the entry receptors such as ACE2; and/or indirect consequences following over-reactive immune responses against SARS-CoV-2 infection. Treatment options targeting the entry receptors or uncontrolled inflammation could be effective in relieving endothelial disorders following SARS-CoV-2 infection. Moreover, there was a positive correlation between the initiation/progression of thrombosis and the survival rate of severe COVID19 cases; therefore, anti-coagulation therapy has been tested. In this section, we discuss about the clinical management and outcomes of trials addressing the causes of endothelial disorders with pros and cons of each approach (also see Table 2 ).
Table 2.
Advantages and disadvantages of the proposed clinical approaches in the prevention and management of COVID-19 associated endothelial dysfunction
| Approach | Advantages | Disadvantages | Ref |
|---|---|---|---|
| rhACE2 |
|
|
[179] |
| Anti-inflammation therapies |
|
|
[[180], [181], [182], [183], [184], [185], [186]] |
| Cell-based therapies |
|
|
[[188], [189], [190], [191], [192]] |
| Anti-coagulation therapies |
|
|
[[197], [198], [199], [200], [201], [202], [203]] |
Given that ACE2 is the main cell entry receptor of SARS-CoV-2, it has suggested as a COVID-19 drug target. The therapeutic potential of hrsACE2 in COVID-19 was demonstrated by inhibition of SARS-CoV-2 infection to human blood vessel organoids [16] (Fig. 4 ). Clinically, in a case of 45-year-old woman with severe COVID-19, a marked reduction of Ang-2, IL-6, IL-8, and inflammation marker ferritin was observed following the administration of hrsACE2 [177]. Indeed, hrsACE2 was able to neutralize SARS-CoV-2 through binding to the viral S protein, protecting organs from injury due to the overactivated RAS signal in COVID-19 [[177], [178], [179]]. Nevertheless, ACE2 targeting is still an underexplored strategy in clinical setting. The drawbacks of this approach include the short half-life of soluble ACE2 and the potential of contributing to an unbalanced RAS after an excessive administration of rhACE2.
Fig. 4.
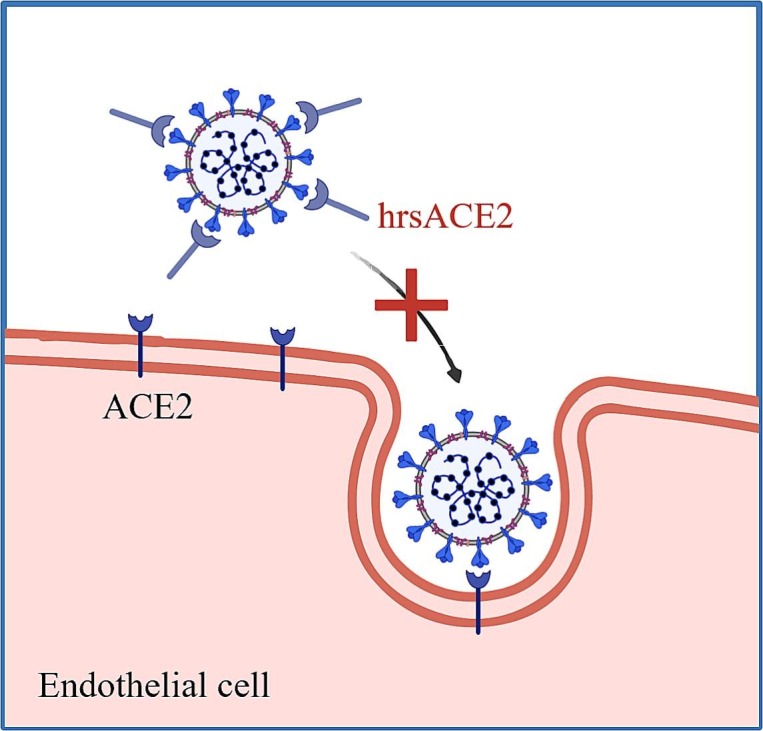
Therapeutic strategies targeting the entry of SARS-CoV-2 into endothelial cells by competitive binding through recombinant soluble ACE2. Soluble human recombinant ACE2 (hrsACE2) neutralizes SARS-CoV-2 through binding to the viral S protein.
Another key to successful treatment of COVID-19 related endothelial dysfunction is the control of systemic inflammation. Tocilizumab, a monoclonal antibody blocking the binding of IL-6 to IL-6 receptor (IL-6R), has been utilized for the treatment of COVID-19 (Fig. 5A). A cohort study of 4,485 adults admitted to ICUs due to severe COVID-19 suggested that the early treatment with tocilizumab could lower the in-hospital mortality of critical COVID-19 [180,181]. Known for anti-inflammation, glucocorticoids have been widely used in the management of SARS and Middle East respiratory syndrome (MERS), both of which are also RNA viruses [182]. In an open-label Randomized Evaluation of COVID-19 Therapy (RECOVERY) trial involving 6,425 patients, dexamethasone displayed its benefits in mitigating inflammation and reducing the incidence of death among patients receiving respiratory support [183] (Fig. 5B). Given the critical roles of NETs and complement activation in the progression of severe COVID-19 and COVID-19 related immunothrombosis, drugs targeting NETosis and complement activation have also become the attractive candidates with a goal for managing dysregulated immune systems following SARS-CoV-2 infection (Fig. 5C). Recombinant DNase I (dornase alfa), which clears extracellular DNA fragments and NETs, has been demonstrated to improve the ventilation of COVID-19 patients in two small scale clinical investigations [184,185]. Moreover, neonatal NET-inhibitory factor (nNIF) that impedes NETosis has been found to block in vitro NET formation induced by plasma from COVID-19 patients, even though the mechanisms of actions require further investigations [172]. Therefore, more studies are required to demonstrate the clinical outcomes of using NETs inhibitors on potential treatment against COVID-19. Furthermore, complement inhibitor eculizumab that blocks the generation of C5a from C5 has been approved by the FDA for administrating on patients with COVID-19, contributing to mitigated inflammation and improvement on the overall survival rate [186,187] (Fig. 5D). Nonetheless, anti-inflammatory regimen would need to be tightly regulated to avoid side effects including systemic immunosuppression that could lead to infections and allergic reactions.
Fig. 5.

Therapeutic strategies targeting over-activation of the immune responses during COVID-19 progression. Over-activated immune response caused by SARS-CoV-2 infection could be mitigated by (A) blocking the interaction between IL-6 and IL-6R through Tocilizumab; (B) controlling cytokine release storm from leukocytes through dexamethasone; (C) inhibiting formation of neutrophil extracellular traps (NETs) and promoting clearance of NETs through DNase I (Dornase alfa) or neonatal NET inhibitory factor (nNIF); (D) suppressing complement activation through Eculizumab; or (E) controlling inflammation after infection by implantation of mesenchymal stem cells (MSCs).
Cell-based approaches have also gained attention as a potential treatment strategy targeting COVID-19 related endothelial dysfunction. By virtue of its anti-viral properties and the lack of expression of ACE2 and SARS-CoV-2 priming enzymes make mesenchymal stem cells (MSCs) a suitable candidate as a potential treatment option [[188], [189], [190]] (Fig. 5E). In particular, MSCs can enhance pathogen engulfment, assist in the clearance of infected host cells, and release anti-inflammatory factors that improve the inflammatory conditions after viral infection [191]. Besides, most MSCs are present in the pulmonary vascular bed following intravenous delivery to the patients, thus restoring endothelial integrity while mitigating inflammation [192]. Clinical trials using MSCs against SARS-CoV-2 have been conducted worldwide. The initial trial was launched in China including 7 COVID-19 patients diagnosed with varied disease severities. After being transplanted with MSCs originated from undisclosed tissues, all patients showed significantly improved pulmonary function and alleviated symptoms as evident by decreased inflammatory markers and increased numbers of regulatory T cells and regulatory dendritic cells [190]. Moreover, another report also documented that implantation of allogeneic MSCs derived from umbilical cord (hUCMSCs) successfully led to remission of severe inflammation in a 65-year-old woman who eventually recovered from COVID-19 [193]. MSC derivatives including its conditioned media and exosomes, which retain the bioactivity of factors secreted by MSCs and even contain organelles such as mitochondria, were also applied in the MSC-based trials targeting COVID-19 [194]. MSC derivatives provide similar protective effects as MSCs to the patients with an added benefit of reduced tumorigenicity that is associated with living MSCs [195]. In addition to MSCs, Wu et al also demonstrated the safety use of clinical-grade immunity- and matrix-regulatory cells (IMRCs) differentiated from hESCs in the improvement of fibrosis and inflammatory lung disorders in animal studies, indicating the potential of hESCs in the treatment of COVID-19 and its complications [196].
Due to the fatal threat of thrombotic complications after SARS-CoV-2 infection, anticoagulation therapy has been utilized for the treatment of COVID-19 (Fig. 6 ). The majority of clinical evidences showed that therapeutic and prophylactic anticoagulation led to lower in-hospital mortality and the need for intubation among patients with COVID-19, suggesting that anticoagulation could be associated with better prognosis [197]. Heparin was recommended to be a potential therapeutic strategy for COVID-19 related coagulation. A clinical study involving 449 patients with severe COVID-19 indicated that anticoagulant therapy mainly through low molecular weight heparin (LMWH) contributed to decreased mortality of patients with sepsis-induced coagulopathy (SIC) and significantly elevated D-dimer, suggesting that LMWH was beneficial to COVID-19 patients with thrombotic symptoms [198]. Moreover, the feature of LMWH to inhibit IL-6 and the replication of coronavirus may also contribute to its beneficial function as an anticoagulation drug [[199], [200], [201]]. Although unfractionated heparin and LMWH were considered as promising drugs targeting COVID-19 related coagulopathy, the adverse effects should be taken seriously. One primary concern is bleeding [202]. Besides, heparin may disturb the protective process in the lungs as the coagulation within alveoli and airways was believed to improve the survival of COVID-19 patients [203]. Thus, how to keep the balance between the benefits and potential risks of using anticoagulation therapies is a key question regarding the treatment strategy for COVID-19 related coagulopathy.
Fig. 6.

Therapeutic strategies targeting thrombosis during COVID-19 progression. Low molecular weight heparin (LMWH) has been used as an antithrombotic drug to inhibit thrombin that is essential in blood clot formation; to inhibit the release of IL-6 from activated leukocytes; and to inhibit the replication of SARS-CoV-2.
8. Conclusions and future perspectives
Taken together, there are some evidences to suggest that targeting the host cell entry pathway or the control of over-reactive immune responses following SARS-CoV-2 infection could provide some clinical benefits to COVID-19 patients. Further research is needed to study the therapeutic efficacy of these approaches in a larger population with multicenter trials. Moreover, given that it is controversial in the expression levels and/or contribution of endothelial ACE2 to SARS-CoV-2 induced endothelial dysfunction, future research direction should focus on alternative pathways by which the virus can directly induce endothelial pathologies independent of ACE2. Currently, the emergence of various SARS-CoV-2 variants raise some concerns over the preventive measures as well as therapeutic treatment strategies targeting COVID-19 and its associated complications.. Essentially, the therapeutic efficiency and efficacy targeting COVID-19 associated endothelial pathologies induced by different strains of SARS-CoV-2 also require future investigations. Furthermore, one of the major concerns limiting research progress on understanding the COVID-19 related endothelial pathogenesis is the lack of animal models that can recapitulate all aspects of COVID-19 in humans especially the unusual features including endothelial dysfunction after SARS-CoV-2 infection in vivo [7]. Moreover, hamsters might represent a better animal model than mouse to study human physiological responses after SARS-CoV-2 infection but deeper mechanistic investigations are limited by the lack of commercially available tools such as specific detection antibodies for immunohistochemistry studies. Therefore, future efforts are required to build more animal models that display a similar degree of susceptibility and immune responses following SARS-CoV-2 infection compared to human; and to develop and share reagents that allow researchers for conducting more basic research into investigation of the COVID-19 related endothelial pathogenesis.
Author contributions
Z.M. and K.O.L. conceptualized this work; Z.M. wrote the original draft; K.Y.Y. made the figures; H.Y. contributed valuable suggestions; K.O.L. reviewed and edited the manuscript.
Disclosures
None.
Acknowledgments
This work was supported by National Natural Science Foundation of China (81922077; 82070494); Research Grants Council of Hong Kong (14100021, 14108420, C4026-17WF, M-402-20); Croucher Foundation Innovation Award; University Grants Committee Research Matching Grant Scheme (2019, 2020, 2021); Faculty of Medicine Innovation Award, Young Researcher Award, Research Committee Funding, Direct Grants, and postgraduate studentship (Z.M.) from CUHK.
References
- 1.Middeldorp S., Coppens M., van Haaps T.F., Foppen M., Vlaar A.P., Muller M.C.A., Bouman C.C.S., Beenen L.F.M., Kootte R.S., Heijmans J., Smits L.P., Bonta P.I., van Es N. Incidence of venous thromboembolism in hospitalized patients with COVID-19. J. Thromb. Haemost. 2020;18(8):1995–2002. doi: 10.1111/jth.14888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nahum J., Morichau-Beauchant T., Daviaud F., Echegut P., Fichet J.M., Maillet J.M., Thierry S. Venous thrombosis among critically ill patients with coronavirus disease 2019 (COVID-19) JAMA Netw. Open. 2020;3(5) doi: 10.1001/jamanetworkopen.2020.10478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Varga Z., Flammer A.J., Steiger P., Haberecker M., Andermatt R., Zinkernagel A.S., Mehra M.R., Schuepbach R.A., Ruschitzka F., Moch H. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395(10234):1417–1418. doi: 10.1016/S0140-6736(20)30937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ackermann M., Verleden S.E., Kuehnel M., Haverich A., Welte T., Laenger F., Vanstapel A., Werlein C., Stark H., Tzankov A., Li W.W., Li V.W., Mentzer S.J., Jonigk D. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in covid-19. N. Engl. J. Med. 2020;383(2):120–128. doi: 10.1056/NEJMoa2015432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ciftel M., Ates N., Yilmaz O. Investigation of endothelial dysfunction and arterial stiffness in multisystem inflammatory syndrome in children. Eur. J. Pediatr. 2021 doi: 10.1007/s00431-021-04136-6. online epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aid M., Busman-Sahay K., Vidal S.J., Maliga Z., Bondoc S., Starke C., Terry M., Jacobson C.A., Wrijil L., Ducat S., Brook O.R., Miller A.D., Porto M., Pellegrini K.L., Pino M., Hoang T.N., Chandrashekar A., Patel S., Stephenson K., Bosinger S.E., Andersen H., Lewis M.G., Hecht J.L., Sorger P.K., Martinot A.J., Estes J.D., Barouch D.H. Vascular disease and thrombosis in SARS-CoV-2-infected rhesus macaques. Cell. 2020;183(5) doi: 10.1016/j.cell.2020.10.005. 1354–1366 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Munoz-Fontela C., Dowling W.E., Funnell S.G.P., Gsell P.S., Riveros-Balta A.X., Albrecht R.A., Andersen H., Baric R.S., Carroll M.W., Cavaleri M., Qin C., Crozier I., Dallmeier K., de Waal L., de Wit E., Delang L., Dohm E., Duprex W.P., Falzarano D., Finch C.L., Frieman M.B., Graham B.S., Gralinski L.E., Guilfoyle K., Haagmans B.L., Hamilton G.A., Hartman A.L., Herfst S., Kaptein S.J.F., Klimstra W.B., Knezevic I., Krause P.R., Kuhn J.H., Le Grand R., Lewis M.G., Liu W.C., Maisonnasse P., McElroy A.K., Munster V., Oreshkova N., Rasmussen A.L., Rocha-Pereira J., Rockx B., Rodriguez E., Rogers T.F., Salguero F.J., Schotsaert M., Stittelaar K.J., Thibaut H.J., Tseng C.T., Vergara-Alert J., Beer M., Brasel T., Chan J.F.W., Garcia-Sastre A., Neyts J., Perlman S., Reed D.S., Richt J.A., Roy C.J., Segales J., Vasan S.S., Henao-Restrepo A.M., Barouch D.H. Animal models for COVID-19. Nature. 2020;586(7830):509–515. doi: 10.1038/s41586-020-2787-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Donoghue M., Hsieh F., Baronas E., Godbout K., Gosselin M., Stagliano N., Donovan M., Woolf B., Robison K., Jeyaseelan R., Breitbart R.E., Acton S. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000;87(5):E1–E9. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 9.Hamming I., Timens W., Bulthuis M.L., Lely A.T., Navis G., van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004;203(2):631–637. doi: 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.To K.F., Lo A.W. Exploring the pathogenesis of severe acute respiratory syndrome (SARS): the tissue distribution of the coronavirus (SARS-CoV) and its putative receptor, angiotensin-converting enzyme 2 (ACE2) J. Pathol. 2004;203(3):740–743. doi: 10.1002/path.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCracken I.R., Saginc G., He L., Huseynov A., Daniels A., Fletcher S., Peghaire C., Kalna V., Andaloussi-Mae M., Muhl L., Craig N.M., Griffiths S.J., Haas J.G., Tait-Burkard C., Lendahl U., Birdsey G.M., Betsholtz C., Noseda M., Baker A.H., Randi A.M. Lack of evidence of angiotensin-converting enzyme 2 expression and replicative infection by SARS-CoV-2 in human endothelial cells. Circulation. 2021;143(8):865–868. doi: 10.1161/CIRCULATIONAHA.120.052824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Del Valle D.M., Kim-Schulze S., Huang H.H., Beckmann N.D., Nirenberg S., Wang B., Lavin Y., Swartz T.H., Madduri D., Stock A., Marron T.U., Xie H., Patel M., Tuballes K., Van Oekelen O., Rahman A., Kovatch P., Aberg J.A., Schadt E., Jagannath S., Mazumdar M., Charney A.W., Firpo-Betancourt A., Mendu D.R., Jhang J., Reich D., Sigel K., Cordon-Cardo C., Feldmann M., Parekh S., Merad M., Gnjatic S. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020;26(10):1636–1643. doi: 10.1038/s41591-020-1051-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., Cheng Z., Yu T., Xia J., Wei Y., Wu W., Xie X., Yin W., Li H., Liu M., Xiao Y., Gao H., Guo L., Xie J., Wang G., Jiang R., Gao Z., Jin Q., Wang J., Cao B. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barton L.M., Duval E.J., Stroberg E., Ghosh S., Mukhopadhyay S. COVID-19 autopsies, Oklahoma, USA. Am. J. Clin. Pathol. 2020;153(6):725–733. doi: 10.1093/ajcp/aqaa062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han Y., Duan X., Yang L., Nilsson-Payant B.E., Wang P., Duan F., Tang X., Yaron T.M., Zhang T., Uhl S., Bram Y., Richardson C., Zhu J., Zhao Z., Redmond D., Houghton S., Nguyen D.T., Xu D., Wang X., Jessurun J., Borczuk A., Huang Y., Johnson J.L., Liu Y., Xiang J., Wang H., Cantley L.C., tenOever B.R., Ho D.D., Pan F.C., Evans T., Chen H.J., Schwartz R.E., Chen S. Identification of SARS-CoV-2 inhibitors using lung and colonic organoids. Nature. 2021;589(7841):270–275. doi: 10.1038/s41586-020-2901-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Monteil V., Kwon H., Prado P., Hagelkruys A., Wimmer R.A., Stahl M., Leopoldi A., Garreta E., Hurtado Del Pozo C., Prosper F., Romero J.P., Wirnsberger G., Zhang H., Slutsky A.S., Conder R., Montserrat N., Mirazimi A., Penninger J.M. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell. 2020;181(4) doi: 10.1016/j.cell.2020.04.004. 905–913 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deinhardt-Emmer S., Bottcher S., Haring C., Giebeler L., Henke A., Zell R., Jungwirth J., Jordan P.M., Werz O., Hornung F., Brandt C., Marquet M., Mosig A.S., Pletz M.W., Schacke M., Rodel J., Heller R., Nietzsche S., Loffler B., Ehrhardt C. SARS-CoV-2 causes severe epithelial inflammation and barrier dysfunction. J. Virol. 2021;95 doi: 10.1128/JVI.00110-21. e00110-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang M., Wang P., Luo R., Wang Y., Li Z., Guo Y., Yao Y., Li M., Tao T., Chen W., Han J., Liu H., Cui K., Zhang X., Zheng Y., Qin J. Biomimetic human disease model of SARS-CoV-2 induced lung injury and immune responses on organ chip system. Adv. Sci. 2020:2002928. doi: 10.1002/advs.202002928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baeck M., Hoton D., Marot L., Herman A. Chilblains and COVID-19: why SARS-CoV-2 endothelial infection is questioned. Brit. J. Dermatol. 2020;183(6):1152–1153. doi: 10.1111/bjd.19489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Colmenero I., Santonja C., Alonso-Riano M., Andina D., Rodriguez-Peralto J.L., Requena L., Torrelo A. Chilblains and COVID-19: why SARS-CoV-2 endothelial infection is questioned. Reply from the authors. Brit. J. Dermatol. 2020;183(6):1153–1154. doi: 10.1111/bjd.19491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Colmenero I., Santonja C., Alonso-Riano M., Noguera-Morel L., Hernandez-Martin A., Andina D., Wiesner T., Rodriguez-Peralto J.L., Requena L., Torrelo A. SARS-CoV-2 endothelial infection causes COVID-19 chilblains: histopathological, immunohistochemical and ultrastructural study of seven paediatric cases. Br. J. Dermatol. 2020;183(4):729–737. doi: 10.1111/bjd.19327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Santonja C., Heras F., Nunez L., Requena L. COVID-19 chilblain-like lesion: immunohistochemical demonstration of SARS-CoV-2 spike protein in blood vessel endothelium and sweat gland epithelium in a polymerase chain reaction-negative patient. Brit. J. Dermatol. 2020;183(4):778–780. doi: 10.1111/bjd.19338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brealey J.K., Miller S.E. SARS-CoV-2 has not been detected directly by electron microscopy in the endothelium of chilblain lesions. Br. J. Dermatol. 2021;184(1):186. doi: 10.1111/bjd.19572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gambichler T., Reuther J., Stucker M., Stranzenbach R., Torres-Reyes C., Schlottmann R., Schmidt W.E., Hayajneh R., Sriram A., Becker J.C. SARS-CoV-2 spike protein is present in both endothelial and eccrine cells of a chilblain-like skin lesion. J. Eur. Acad. Dermatol. 2021;35(3):E187–E189. doi: 10.1111/jdv.16970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perez-Mies B., Gomez-Rojo M., Carretero-Barrio I., Bardi T., Benito A., Garcia-Cosio M., Caballero A., de Pablo R., Galan J.C., Pestana D., Palacios J. Pulmonary vascular proliferation in patients with severe COVID-19: an autopsy study. Thorax. 2021;76:1044–1046. doi: 10.1136/thoraxjnl-2020-216714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carnevale S., Beretta P., Morbini P. Direct endothelial damage and vasculitis due to SARS-CoV-2 in small bowel submucosa of COVID-19 patient with diarrhea. J. Med. Virol. 2021;93(1):61–63. doi: 10.1002/jmv.26119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stahl K., Brasen J.H., Hoeper M.M., David S. Direct evidence of SARS-CoV-2 in gut endothelium. Intensive Care Med. 2020;46(11):2081–2082. doi: 10.1007/s00134-020-06237-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fox S.E., Lameira F.S., Rinker E.B., Vander Heide R.S. Cardiac endotheliitis and multisystem inflammatory syndrome after COVID-19. Ann. Intern. Med. 2020;173(12):1025–1027. doi: 10.7326/L20-0882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Negrini S., Guadagno A., Greco M., Parodi A., Burlando M. An unusual case of bullous haemorrhagic vasculitis in a COVID-19 patient. J. Eur. Acad. Dermatol. Venereol. 2020;34(11):e675–e676. doi: 10.1111/jdv.16760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Escher R., Breakey N., Lammle B. Severe COVID-19 infection associated with endothelial activation. Thromb. Res. 2020;190:62. doi: 10.1016/j.thromres.2020.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fraser D.D., Patterson E.K., Daley M., Cepinskas G., L.C.-S. Team Case report: inflammation and endothelial injury profiling of COVID-19 pediatric multisystem inflammatory syndrome (MIS-C) Front. Pediatr. 2021;9 doi: 10.3389/fped.2021.597926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crippa S., Kagi G., Graf L., Meyer Sauteur P.M., Kohler P. Stroke in a young adult with mild COVID-19 suggesting endotheliitis. New Microbes New Infect. 2020;38 doi: 10.1016/j.nmni.2020.100781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gonzalez Canas E., Gimenez Gaibar A., Rodriguez Lorenzo L., Castro Rios J.G., Martinez Toiran A., Bella Cueto M.R., Bella Burgos B., Espasa Soley M. Acute peripheral arterial thrombosis in COVID-19. Role of endothelial inflammation. Br. J. Surg. 2020;107(10):e444–e445. doi: 10.1002/bjs.11904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sabioni L.R., Tibirica E., Lamas C.C., Amorim G.D., De Lorenzo A. Systemic microvascular dysfunction in COVID-19. Am. J. Cardiovasc. Dis. 2020;10(4):386–391. [PMC free article] [PubMed] [Google Scholar]
- 35.Jud P., Kessler H.H., Brodmann M. Case report: changes of vascular reactivity and arterial stiffness in a patient with covid-19 infection. Front Cardiovasc. Med. 2021;8 doi: 10.3389/fcvm.2021.671669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guervilly C., Burtey S., Sabatier F., Cauchois R., Lano G., Abdili E., Daviet F., Arnaud L., Brunet P., Hraiech S., Jourde-Chiche N., Koubi M., Lacroix R., Pietri L., Berda Y., Robert T., Degioanni C., Velier M., Papazian L., Kaplanski G., Dignat-George F. Circulating endothelial cells as a marker of endothelial injury in severe COVID-19. J. Infect. Dis. 2020;222(11):1789–1793. doi: 10.1093/infdis/jiaa528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chioh F.W., Fong S.W., Young B.E., Wu K.X., Siau A., Krishnan S., Chan Y.H., Carissimo G., Teo L.L., Gao F., Tan R.S., Zhong L., Koh A.S., Tan S.Y., Tambyah P.A., Renia L., Ng L.F., Lye D.C., Cheung C. Convalescent COVID-19 patients are susceptible to endothelial dysfunction due to persistent immune activation. Elife. 2021;10 doi: 10.7554/eLife.64909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neri T., Nieri D., Celi A. P-selectin blockade in COVID-19-related ARDS. Am. J. Phys. Lung Cell. Mol. Phys. 2020;318(6):L1237–L1238. doi: 10.1152/ajplung.00202.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smadja D.M., Guerin C.L., Chocron R., Yatim N., Boussier J., Gendron N., Khider L., Hadjadj J., Goudot G., Debuc B., Juvin P., Hauw-Berlemont C., Augy J.L., Peron N., Messas E., Planquette B., Sanchez O., Charbit B., Gaussem P., Duffy D., Terrier B., Mirault T., Diehl J.L. Angiopoietin-2 as a marker of endothelial activation is a good predictor factor for intensive care unit admission of COVID-19 patients. Angiogenesis. 2020;23(4):611–620. doi: 10.1007/s10456-020-09730-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spadaro S., Fogagnolo A., Campo G., Zucchetti O., Verri M., Ottaviani I., Tunstall T., Grasso S., Scaramuzzo V., Murgolo F., Marangoni E., Sega F. Vieceli Dalla, Fortini F., Pavasini R., Rizzo P., Ferrari R., Papi A., Volta C.A., Contoli M. Markers of endothelial and epithelial pulmonary injury in mechanically ventilated COVID-19 ICU patients. Crit. Care. 2021;25(1):74. doi: 10.1186/s13054-021-03499-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tipnis S.R., Hooper N.M., Hyde R., Karran E., Christie G., Turner A.J. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000;275(43):33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- 42.Paz Ocaranza M., Riquelme J.A., Garcia L., Jalil J.E., Chiong M., Santos R.A.S., Lavandero S. Counter-regulatory renin-angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 2020;17(2):116–129. doi: 10.1038/s41569-019-0244-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Santos R.A.S., Sampaio W.O., Alzamora A.C., Motta-Santos D., Alenina N., Bader M., Campagnole-Santos M.J. The ACE2/angiotensin-(1-7)/MAS axis of the renin-angiotensin system: focus on angiotensin-(1-7) Physiol. Rev. 2018;98(1):505–553. doi: 10.1152/physrev.00023.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scialo F., Daniele A., Amato F., Pastore L., Matera M.G., Cazzola M., Castaldo G., Bianco A. ACE2: the major cell entry receptor for SARS-CoV-2. Lung. 2020;198(6):867–877. doi: 10.1007/s00408-020-00408-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ziegler C.G.K., Allon S.J., Nyquist S.K., Mbano I.M., Miao V.N., Tzouanas C.N., Cao Y., Yousif A.S., Bals J., Hauser B.M., Feldman J., Muus C., Wadsworth M.H., 2nd, Kazer S.W., Hughes T.K., Doran B., Gatter G.J., Vukovic M., Taliaferro F., Mead B.E., Guo Z., Wang J.P., Gras D., Plaisant M., Ansari M., Angelidis I., Adler H., Sucre J.M.S., Taylor C.J., Lin B., Waghray A., Mitsialis V., Dwyer D.F., Buchheit K.M., Boyce J.A., Barrett N.A., Laidlaw T.M., Carroll S.L., Colonna L., Tkachev V., Peterson C.W., Yu A., Zheng H.B., Gideon H.P., Winchell C.G., Lin P.L., Bingle C.D., Snapper S.B., Kropski J.A., Theis F.J., Schiller H.B., Zaragosi L.E., Barbry P., Leslie A., Kiem H.P., Flynn J.L., Fortune S.M., Berger B., Finberg R.W., Kean L.S., Garber M., Schmidt A.G., Lingwood D., Shalek A.K., Ordovas-Montanes J., H.C.A.L.B.N.E.a. lung-network@humancellatlas.org, H.C.A.L.B. Network SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell Subsets across tissues. Cell. 2020;181(5) doi: 10.1016/j.cell.2020.04.035. 1016–1035 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C., Choe H., Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang L., Jackson C.B., Mou H., Ojha A., Peng H., Quinlan B.D., Rangarajan E.S., Pan A., Vanderheiden A., Suthar M.S., Li W., Izard T., Rader C., Farzan M., Choe H. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun. 2020;11(1):6013. doi: 10.1038/s41467-020-19808-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Zhang Q., Shi X., Wang Q., Zhang L., Wang X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581(7807):215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 49.Hoffmann M., Kleine-Weber H., Schroeder S., Kruger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.H., Nitsche A., Muller M.A., Drosten C., Pohlmann S. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2) doi: 10.1016/j.cell.2020.02.052. 271–280 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Matsuyama S., Nao N., Shirato K., Kawase M., Saito S., Takayama I., Nagata N., Sekizuka T., Katoh H., Kato F., Sakata M., Tahara M., Kutsuna S., Ohmagari N., Kuroda M., Suzuki T., Kageyama T., Takeda M. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc. Natl. Acad. Sci. U. S. A. 2020;117(13):7001–7003. doi: 10.1073/pnas.2002589117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.V'Kovski P., Kratzel A., Steiner S., Stalder H., Thiel V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021;19(3):155–170. doi: 10.1038/s41579-020-00468-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dhar D., Mohanty A. Gut microbiota and Covid-19 - possible link and implications. Virus Res. 2020;285 doi: 10.1016/j.virusres.2020.198018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.D'Amico F., Baumgart D.C., Danese S., Peyrin-Biroulet L. Diarrhea during COVID-19 infection: pathogenesis, epidemiology, prevention, and management. Clin. Gastroenterol. Hepatol. 2020;18(8):1663–1672. doi: 10.1016/j.cgh.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ahmetaj-Shala B., Vaja R., Atanur S.S., George P.M., Kirkby N.S., Mitchell J.A. Cardiorenal tissues express SARS-CoV-2 entry genes and basigin (BSG/CD147) increases with age in endothelial cells. JACC Basic Transl. Sci. 2020;5(11):1111–1123. doi: 10.1016/j.jacbts.2020.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lei Y., Zhang J., Schiavon C.R., He M., Chen L., Shen H., Zhang Y., Yin Q., Cho Y., Andrade L., Shadel G.S., Hepokoski M., Lei T., Wang H., Zhang J., Yuan J.X., Malhotra A., Manor U., Wang S., Yuan Z.Y., Shyy J.Y. SARS-CoV-2 spike protein impairs endothelial function via downregulation of ACE 2. Circ. Res. 2021;128(9):1323–1326. doi: 10.1161/CIRCRESAHA.121.318902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Conde J.N., Schutt W.R., Gorbunova E.E., Mackow E.R. Recombinant ACE2 expression is required for SARS-CoV-2 to infect primary human endothelial cells and induce inflammatory and procoagulative responses. Mbio. 2020;11(6) doi: 10.1128/mBio.03185-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang P., Luo R., Zhang M., Wang Y., Song T., Tao T., Li Z., Jin L., Zheng H., Chen W., Zhao M., Zheng Y., Qin J. A cross-talk between epithelium and endothelium mediates human alveolar-capillary injury during SARS-CoV-2 infection. Cell Death Dis. 2020;11(12):1042. doi: 10.1038/s41419-020-03252-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mayi B.S., Leibowitz J.A., Woods A.T., Ammon K.A., Liu A.E., Raja A. The role of Neuropilin-1 in COVID-19. PLoS Pathog. 2021;17(1) doi: 10.1371/journal.ppat.1009153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cantuti-Castelvetri L., Ojha R., Pedro L.D., Djannatian M., Franz J., Kuivanen S., van der Meer F., Kallio K., Kaya T., Anastasina M., Smura T., Levanov L., Szirovicza L., Tobi A., Kallio-Kokko H., Osterlund P., Joensuu M., Meunier F.A., Butcher S.J., Winkler M.S., Mollenhauer B., Helenius A., Gokce O., Teesalu T., Hepojoki J., Vapalahti O., Stadelmann C., Balistreri G., Simons M. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science. 2020;370(6518):856–860. doi: 10.1126/science.abd2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Daly J.L., Simonetti B., Klein K., Chen K.E., Williamson M.K., Anton-Plagaro C., Shoemark D.K., Simon-Gracia L., Bauer M., Hollandi R., Greber U.F., Horvath P., Sessions R.B., Helenius A., Hiscox J.A., Teesalu T., Matthews D.A., Davidson A.D., Collins B.M., Cullen P.J., Yamauchi Y. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science. 2020;370(6518):861–865. doi: 10.1126/science.abd3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kyrou I., Randeva H.S., Spandidos D.A., Karteris E. Not only ACE2-the quest for additional host cell mediators of SARS-CoV-2 infection: neuropilin-1 (NRP1) as a novel SARS-CoV-2 host cell entry mediator implicated in COVID-19. Signal Transduct. Target Ther. 2021;6(1):21. doi: 10.1038/s41392-020-00460-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Melincovici C.S., Bosca A.B., Susman S., Marginean M., Mihu C., Istrate M., Moldovan I.M., Roman A.L., Mihu C.M. Vascular endothelial growth factor (VEGF) - key factor in normal and pathological angiogenesis. Romanian J. Morphol. Embryol. 2018;59(2):455–467. [PubMed] [Google Scholar]
- 63.Mone P., Gambardella J., Wang X., Jankauskas S.S., Matarese A., Santulli G. miR-24 targets SARS-CoV-2 co-factor Neuropilin-1 in human brain microvascular endothelial cells: insights for COVID-19 neurological manifestations. Res. Sq. 2021 doi: 10.3390/ncrna7010009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huang L., Chambliss K.L., Gao X., Yuhanna I.S., Behling-Kelly E., Bergaya S., Ahmed M., Michaely P., Luby-Phelps K., Darehshouri A., Xu L., Fisher E.A., Ge W.P., Mineo C., Shaul P.W. SR-B1 drives endothelial cell LDL transcytosis via DOCK4 to promote atherosclerosis. Nature. 2019;569(7757):565–569. doi: 10.1038/s41586-019-1140-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wei C., Wan L., Yan Q., Wang X., Zhang J., Yang X., Zhang Y., Fan C., Li D., Deng Y., Sun J., Gong J., Yang X., Wang Y., Wang X., Li J., Yang H., Li H., Zhang Z., Wang R., Du P., Zong Y., Yin F., Zhang W., Wang N., Peng Y., Lin H., Feng J., Qin C., Chen W., Gao Q., Zhang R., Cao Y., Zhong H. HDL-scavenger receptor B type 1 facilitates SARS-CoV-2 entry. Nat. Metab. 2020;2(12):1391–1400. doi: 10.1038/s42255-020-00324-0. [DOI] [PubMed] [Google Scholar]
- 66.Wang K., Chen W., Zhang Z., Deng Y.Q., Lian J.Q., Du P., Wei D., Zhang Y., Sun X.X., Gong L., Yang X., He L., Zhang L., Yang Z.W., Geng J.J., Chen R., Zhang H., Wang B., Zhu Y.M., Nan G., Jiang J.L., Li L., Wu J., Lin P., Huang W., Xie L.Z., Zheng Z.H., Zhang K., Miao J.L., Cui H.Y., Huang M., Zhang J., Fu L., Yang X.M., Zhao Z.P., Sun S.H., Gu H.J., Wang Z., Wang C.F., Lu Y.C., Liu Y.Y., Wang Q.Y., Bian H.J., Zhu P., Chen Z.N. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct. Target. 2020;5(1) doi: 10.1038/s41392-020-00426-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Qian Y., Lei T., Patel P.S., Lee C.H., Monaghan-Nichols P., Xin H.B., Qiu J., Fu M. Direct activation of endothelial cells by SARS-CoV-2 nucleocapsid protein is blocked by Simvastatin. bioRxiv. 2021;95:e0139621. doi: 10.1128/JVI.01396-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rajendran P., Rengarajan T., Thangavel J., Nishigaki Y., Sakthisekaran D., Sethi G., Nishigaki I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013;9(10):1057–1069. doi: 10.7150/ijbs.7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Deanfield J.E., Halcox J.P., Rabelink T.J. Endothelial function and dysfunction: testing and clinical relevance. Circulation. 2007;115(10):1285–1295. doi: 10.1161/CIRCULATIONAHA.106.652859. [DOI] [PubMed] [Google Scholar]
- 70.Siddiqi H.K., Mehra M.R. COVID-19 illness in native and immunosuppressed states: a clinical-therapeutic staging proposal. J. Heart Lung Transplant. 2020;39(5):405–407. doi: 10.1016/j.healun.2020.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu Y., Chen D., Hou J., Li H., Cao D., Guo M., Ling Y., Gao M., Zhou Y., Wan Y., Zhu Z. An inter-correlated cytokine network identified at the center of cytokine storm predicted COVID-19 prognosis. Cytokine. 2021;138 doi: 10.1016/j.cyto.2020.155365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Barnes B.J., Adrover J.M., Baxter-Stoltzfus A., Borczuk A., Cools-Lartigue J., Crawford J.M., Dassler-Plenker J., Guerci P., Huynh C., Knight J.S., Loda M., Looney M.R., McAllister F., Rayes R., Renaud S., Rousseau S., Salvatore S., Schwartz R.E., Spicer J.D., Yost C.C., Weber A., Zuo Y., Egeblad M. Targeting potential drivers of COVID-19: neutrophil extracellular traps. J. Exp. Med. 2020;217(6) doi: 10.1084/jem.20200652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang C., Xie J., Zhao L., Fei X., Zhang H., Tan Y., Nie X., Zhou L., Liu Z., Ren Y., Yuan L., Zhang Y., Zhang J., Liang L., Chen X., Liu X., Wang P., Han X., Weng X., Chen Y., Yu T., Zhang X., Cai J., Chen R., Shi Z.L., Bian X.W. Alveolar macrophage dysfunction and cytokine storm in the pathogenesis of two severe COVID-19 patients. EBioMedicine. 2020;57 doi: 10.1016/j.ebiom.2020.102833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fox S.E., Akmatbekov A., Harbert J.L., Li G., Quincy Brown J., Vander Heide R.S. Pulmonary and cardiac pathology in African American patients with COVID-19: an autopsy series from New Orleans, Lancet. Respir. Med. 2020;8(7):681–686. doi: 10.1016/S2213-2600(20)30243-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guzik T.J., Mohiddin S.A., Dimarco A., Patel V., Savvatis K., Marelli-Berg F.M., Madhur M.S., Tomaszewski M., Maffia P., D'Acquisto F., Nicklin S.A., Marian A.J., Nosalski R., Murray E.C., Guzik B., Berry C., Touyz R.M., Kreutz R., Wang D.W., Bhella D., Sagliocco O., Crea F., Thomson E.C., McInnes I.B. COVID-19 and the cardiovascular system: implications for risk assessment, diagnosis, and treatment options. Cardiovasc. Res. 2020;116(10):1666–1687. doi: 10.1093/cvr/cvaa106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Evans P.C., Rainger G.E., Mason J.C., Guzik T.J., Osto E., Stamataki Z., Neil D., Hoefer I.E., Fragiadaki M., Waltenberger J., Weber C., Bochaton-Piallat M.L., Back M. Endothelial dysfunction in COVID-19: a position paper of the ESC Working Group for Atherosclerosis and Vascular Biology, and the ESC Council of Basic Cardiovascular Science. Cardiovasc. Res. 2020;116(14):2177–2184. doi: 10.1093/cvr/cvaa230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kaur S., Bansal R., Kollimuttathuillam S., Gowda A.M., Singh B., Mehta D., Maroules M. The looming storm: blood and cytokines in COVID-19. Blood Rev. 2021;46 doi: 10.1016/j.blre.2020.100743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen X., Zhao B., Qu Y., Chen Y., Xiong J., Feng Y., Men D., Huang Q., Liu Y., Yang B., Ding J., Li F. Detectable serum severe acute respiratory syndrome coronavirus 2 viral load (RNAemia) is closely correlated with drastically elevated interleukin 6 level in critically ill patients with coronavirus disease 2019. Clin. Infect. Dis. 2020;71(8):1937–1942. doi: 10.1093/cid/ciaa449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mehta P., McAuley D.F., Brown M., Sanchez E., Tattersall R.S., Manson J.J., U.K. Hlh Across Speciality Collaboration COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395(10229):1033–1034. doi: 10.1016/S0140-6736(20)30628-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Galvan-Roman J.M., Rodriguez-Garcia S.C., Roy-Vallejo E., Marcos-Jimenez A., Sanchez-Alonso S., Fernandez-Diaz C., Alcaraz-Serna A., Mateu-Albero T., Rodriguez-Cortes P., Sanchez-Cerrillo I., Esparcia L., Martinez-Fleta P., Lopez-Sanz C., Gabrie L., Guerola L. Del Campo, Suarez-Fernandez C., Ancochea J., Canabal A., Albert P., Rodriguez-Serrano D.A., Aguilar J.M., Del Arco C., de Los Santos I., Garcia-Fraile L., de la Camara R., Serra J.M., Ramirez E., Alonso T., Landete P., Soriano J.B., Martin-Gayo E., Torres A. Fraile, Cruz N.D. Zurita, Garcia-Vicuna R., Cardenoso L., Sanchez-Madrid F., Alfranca A., Munoz-Calleja C., Gonzalez-Alvaro I., R.-C. Group IL-6 serum levels predict severity and response to tocilizumab in COVID-19: an observational study. J. Allergy Clin. Immunol. 2021;147(1) doi: 10.1016/j.jaci.2020.09.018. 72–80 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Coomes E.A., Haghbayan H. Interleukin-6 in Covid-19: a systematic review and meta-analysis. Rev. Med. Virol. 2020;30(6):1–9. doi: 10.1002/rmv.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen G., Wu D., Guo W., Cao Y., Huang D., Wang H., Wang T., Zhang X., Chen H., Yu H., Zhang X., Zhang M., Wu S., Song J., Chen T., Han M., Li S., Luo X., Zhao J., Ning Q. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Invest. 2020;130(5):2620–2629. doi: 10.1172/JCI137244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tjendra Y., Al Mana A.F., Espejo A.P., Akgun Y., Millan N.C., Gomez-Fernandez C., Cray C. Predicting disease severity and outcome in COVID-19 patients: a review of multiple biomarkers. Arch. Pathol. Lab. Med. 2020;144(12):1465–1474. doi: 10.5858/arpa.2020-0471-SA. [DOI] [PubMed] [Google Scholar]
- 84.Stone J.H., Frigault M.J., Serling-Boyd N.J., Fernandes A.D., Harvey L., Foulkes A.S., Horick N.K., Healy B.C., Shah R., Bensaci A.M., Woolley A.E., Nikiforow S., Lin N., Sagar M., Schrager H., Huckins D.S., Axelrod M., Pincus M.D., Fleisher J., Sacks C.A., Dougan M., North C.M., Halvorsen Y.D., Thurber T.K., Dagher Z., Scherer A., Wallwork R.S., Kim A.Y., Schoenfeld S., Sen P., Neilan T.G., Perugino C.A., Unizony S.H., Collier D.S., Matza M.A., Yinh J.M., Bowman K.A., Meyerowitz E., Zafar A., Drobni Z.D., Bolster M.B., Kohler M., D'Silva K.M., Dau J., Lockwood M.M., Cubbison C., Weber B.N., Mansour M.K., Investigators B.B.T.T. Efficacy of tocilizumab in patients hospitalized with covid-19. N. Engl. J. Med. 2020;383(24):2333–2344. doi: 10.1056/NEJMoa2028836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Desai T.R., Leeper N.J., Hynes K.L., Gewertz B.L. Interleukin-6 causes endothelial barrier dysfunction via the protein kinase C pathway. J. Surg. Res. 2002;104(2):118–123. doi: 10.1006/jsre.2002.6415. [DOI] [PubMed] [Google Scholar]
- 86.Watson C., Whittaker S., Smith N., Vora A.J., Dumonde D.C., Brown K.A. IL-6 acts on endothelial cells to preferentially increase their adherence for lymphocytes. Clin. Exp. Immunol. 1996;105(1):112–119. doi: 10.1046/j.1365-2249.1996.d01-717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liao M., Liu Y., Yuan J., Wen Y., Xu G., Zhao J., Cheng L., Li J., Wang X., Wang F., Liu L., Amit I., Zhang S., Zhang Z. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020;26(6):842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- 88.Xiong S., Hong Z., Huang L.S., Tsukasaki Y., Nepal S., Di A., Zhong M., Wu W., Ye Z., Gao X., Rao G.N., Mehta D., Rehman J., Malik A.B. IL-1beta suppression of VE-cadherin transcription underlies sepsis-induced inflammatory lung injury. J. Clin. Invest. 2020;130(7):3684–3698. doi: 10.1172/JCI136908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Karki R., Sharma B.R., Tuladhar S., Williams E.P., Zalduondo L., Samir P., Zheng M., Sundaram B., Banoth B., Malireddi R.K.S., Schreiner P., Neale G., Vogel P., Webby R., Jonsson C.B., Kanneganti T.D. Synergism of TNF-alpha and IFN-gamma triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell. 2021;184(1) doi: 10.1016/j.cell.2020.11.025. 149–168 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Marcos-Ramiro B., Garcia-Weber D., Millan J. TNF-induced endothelial barrier disruption: beyond actin and Rho. Thromb. Haemost. 2014;112(6):1088–1102. doi: 10.1160/TH14-04-0299. [DOI] [PubMed] [Google Scholar]
- 91.Incalza M.A., D'Oria R., Natalicchio A., Perrini S., Laviola L., Giorgino F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018;100:1–19. doi: 10.1016/j.vph.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 92.Forstermann U., Xia N., Li H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ. Res. 2017;120(4):713–735. doi: 10.1161/CIRCRESAHA.116.309326. [DOI] [PubMed] [Google Scholar]
- 93.Higashi Y., Noma K., Yoshizumi M., Kihara Y. Endothelial function and oxidative stress in cardiovascular diseases. Circ. J. 2009;73(3):411–418. doi: 10.1253/circj.cj-08-1102. [DOI] [PubMed] [Google Scholar]
- 94.Simons M., Gordon E., Claesson-Welsh L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016;17(10):611–625. doi: 10.1038/nrm.2016.87. [DOI] [PubMed] [Google Scholar]
- 95.Andersson U., Ottestad W., Tracey K.J. Extracellular HMGB1: a therapeutic target in severe pulmonary inflammation including COVID-19? Mol. Med. 2020;26(1):42. doi: 10.1186/s10020-020-00172-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hendricks K., To E., Broughton B., Peshavariya H., Vlahos R., Selemidis S. Influenza a virus (Iav) causes vascular endothelial cell oxidative stress via Nox2 nadph oxidase. Respirology. 2016;21:82. 48: PA3967. [Google Scholar]
- 97.Roebuck K.A. Oxidant stress regulation of IL-8 and ICAM-1 gene expression: differential activation and binding of the transcription factors AP-1 and NF-kappaB (Review) Int. J. Mol. Med. 1999;4(3):223–230. doi: 10.3892/ijmm.4.3.223. [DOI] [PubMed] [Google Scholar]
- 98.Min J.K., Kim Y.M., Kim S.W., Kwon M.C., Kong Y.Y., Hwang I.K., Won M.H., Rho J., Kwon Y.G. TNF-related activation-induced cytokine enhances leukocyte adhesiveness: induction of ICAM-1 and VCAM-1 via TNF receptor-associated factor and protein kinase C-dependent NF-kappaB activation in endothelial cells. J. Immunol. 2005;175(1):531–540. doi: 10.4049/jimmunol.175.1.531. [DOI] [PubMed] [Google Scholar]
- 99.Verdecchia P., Cavallini C., Spanevello A., Angeli F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 2020;76:14–20. doi: 10.1016/j.ejim.2020.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gan R., Rosoman N.P., Henshaw D.J.E., Noble E.P., Georgius P., Sommerfeld N. COVID-19 as a viral functional ACE2 deficiency disorder with ACE2 related multi-organ disease. Med. Hypotheses. 2020;144 doi: 10.1016/j.mehy.2020.110024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cook J.R., Ausiello J. Functional ACE2 deficiency leading to angiotensin imbalance in the pathophysiology of COVID-19. Rev. Endocr. Metab. Disord. 2021;1:1–20. doi: 10.1007/s11154-021-09663-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Salka K., Abutaleb K., Chorvinsky E., Thiruvengadam G., Arroyo M., Gomez J.L., Gutierrez M.J., Pillai D.K., Jaiswal J.K., Nino G. IFN stimulates ACE2 expression in pediatric airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2021;64(4):515–518. doi: 10.1165/rcmb.2020-0352LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ng K.W., Attig J., Bolland W., Young G.R., Major J., Wrobel A.G., Gamblin S., Wack A., Kassiotis G. Tissue-specific and interferon-inducible expression of nonfunctional ACE2 through endogenous retroelement co-option. Nat. Genet. 2020;52(12):1294–1302. doi: 10.1038/s41588-020-00732-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Onabajo O.O., Banday A.R., Stanifer M.L., Yan W.S., Obajemu A., Santer D.M., Florez-Vargas O., Piontkivska H., Vargas J.M., Ring T.M.J., Kee C., Doldan P., Tyrrell D.L., Mendoza J.L., Boulant S., Prokunina-Olsson L. Interferons and viruses induce a novel truncated ACE2 isoform and not the full-length SARS-CoV-2 receptor. Nat. Genet. 2020;52(12):1283–1293. doi: 10.1038/s41588-020-00731-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ding J., Yu M., Jiang J., Luo Y., Zhang Q., Wang S., Yang F., Wang A., Wang L., Zhuang M., Wu S., Zhang Q., Xia Y., Lu D. Angiotensin II decreases endothelial nitric oxide synthase phosphorylation via AT1R Nox/ROS/PP2A pathway. Front. Physiol. 2020;11 doi: 10.3389/fphys.2020.566410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mather K.J., Mirzamohammadi B., Lteif A., Steinberg H.O., Baron A.D. Endothelin contributes to basal vascular tone and endothelial dysfunction in human obesity and type 2 diabetes. Diabetes. 2002;51(12):3517–3523. doi: 10.2337/diabetes.51.12.3517. [DOI] [PubMed] [Google Scholar]
- 107.Polidoro R.B., Hagan R.S., de Santis Santiago R., Schmidt N.W. Overview: systemic inflammatory response derived from lung injury caused by SARS-CoV-2 infection explains severe outcomes in COVID-19. Front. Immunol. 2020;11:1626. doi: 10.3389/fimmu.2020.01626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lampugnani M.G., Dejana E., Giampietro C. Vascular endothelial (VE)-cadherin, endothelial adherens junctions, and vascular disease. Cold Spring Harb. Perspect. Biol. 2018;10(10) doi: 10.1101/cshperspect.a029322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chen X.L., Nam J.O., Jean C., Lawson C., Walsh C.T., Goka E., Lim S.T., Tomar A., Tancioni I., Uryu S., Guan J.L., Acevedo L.M., Weis S.M., Cheresh D.A., Schlaepfer D.D. VEGF-induced vascular permeability is mediated by FAK. Dev. Cell. 2012;22(1):146–157. doi: 10.1016/j.devcel.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cohen A.W., Carbajal J.M., Schaeffer R.C., Jr. VEGF stimulates tyrosine phosphorylation of beta-catenin and small-pore endothelial barrier dysfunction. Am. J. Phys. 1999;277(5):H2038–H2049. doi: 10.1152/ajpheart.1999.277.5.H2038. [DOI] [PubMed] [Google Scholar]
- 111.Kevil C.G., Payne D.K., Mire E., Alexander J.S. Vascular permeability factor/vascular endothelial cell growth factor-mediated permeability occurs through disorganization of endothelial junctional proteins. J. Biol. Chem. 1998;273(24):15099–15103. doi: 10.1074/jbc.273.24.15099. [DOI] [PubMed] [Google Scholar]
- 112.Suarez S., Ballmer-Hofer K. VEGF transiently disrupts gap junctional communication in endothelial cells. J. Cell Sci. 2001;114(6):1229–1235. doi: 10.1242/jcs.114.6.1229. [DOI] [PubMed] [Google Scholar]
- 113.Fulton D., Gratton J.P., McCabe T.J., Fontana J., Fujio Y., Walsh K., Franke T.F., Papapetropoulos A., Sessa W.C. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399(6736):597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Murohara T., Horowitz J.R., Silver M., Tsurumi Y., Chen D., Sullivan A., Isner J.M. Vascular endothelial growth factor/vascular permeability factor enhances vascular permeability via nitric oxide and prostacyclin. Circulation. 1998;97(1):99–107. doi: 10.1161/01.cir.97.1.99. [DOI] [PubMed] [Google Scholar]
- 115.Filep J.G., Foldes-Filep E., Sirois P. Nitric oxide modulates vascular permeability in the rat coronary circulation. Br. J. Pharmacol. 1993;108(2):323–326. doi: 10.1111/j.1476-5381.1993.tb12803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Venereau E., Ceriotti C., Bianchi M.E. DAMPs from cell death to new life. Front. Immunol. 2015;6:422. doi: 10.3389/fimmu.2015.00422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jia C., Zhang J., Chen H.W., Zhuge Y.Z., Chen H.Q., Qian F.Y., Zhou K.L., Niu C., Wang F.Y., Qiu H.X., Wang Z.Q., Xiao J., Rong X., Chu M.P. Endothelial cell pyroptosis plays an important role in Kawasaki disease via HMGB1/RAGE/cathespin B signaling pathway and NLRP3 inflammasome activation. Cell Death Dis. 2019;10 doi: 10.1038/s41419-019-2021-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fiuza C., Bustin M., Talwar S., Tropea M., Gerstenberger E., Shelhamer J.H., Suffredini A.F. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. 2003;101(7):2652–2660. doi: 10.1182/blood-2002-05-1300. [DOI] [PubMed] [Google Scholar]
- 119.Luan Z.G., Zhang H., Yang P.T., Ma X.C., Zhang C., Guo R.X. HMGB1 activates nuclear factor-kappaB signaling by RAGE and increases the production of TNF-alpha in human umbilical vein endothelial cells. Immunobiology. 2010;215(12):956–962. doi: 10.1016/j.imbio.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 120.Schiraldi M., Raucci A., Munoz L.M., Livoti E., Celona B., Venereau E., Apuzzo T., De Marchis F., Pedotti M., Bachi A., Thelen M., Varani L., Mellado M., Proudfoot A., Bianchi M.E., Uguccioni M. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J. Exp. Med. 2012;209(3):551–563. doi: 10.1084/jem.20111739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ota C., Ishizawa K., Yamada M., Tando Y., He M., Takahashi T., Yamaya M., Yamamoto Y., Yamamoto H., Kure S., Kubo H. Receptor for advanced glycation end products expressed on alveolar epithelial cells is the main target for hyperoxia-induced lung injury. Respir. Investig. 2016;54(2):98–108. doi: 10.1016/j.resinv.2015.08.009. [DOI] [PubMed] [Google Scholar]
- 122.Kierdorf K., Fritz G. RAGE regulation and signaling in inflammation and beyond. J. Leukoc. Biol. 2013;94(1):55–68. doi: 10.1189/jlb.1012519. [DOI] [PubMed] [Google Scholar]
- 123.Kim S.W., Lim C.M., Kim J.B., Shin J.H., Lee S., Lee M., Lee J.K. Extracellular HMGB1 released by NMDA treatment confers neuronal apoptosis via RAGE-p38 MAPK/ERK signaling pathway. Neurotox. Res. 2011;20(2):159–169. doi: 10.1007/s12640-010-9231-x. [DOI] [PubMed] [Google Scholar]
- 124.Yang H.A., Hreggvidsdottir H.S., Palmblad K., Wang H.C., Ochani M., Li J.H., Lu B., Chavan S., Rosas-Ballina M., Al-Abed Y., Akira S., Bierhaus A., Erlandsson-Harris H., Andersson U., Tracey K.J. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. P. Natl. Acad. Sci. USA. 2010;107(26):11942–11947. doi: 10.1073/pnas.1003893107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhong H., Li X., Zhou S., Jiang P., Liu X., Ouyang M., Nie Y., Chen X., Zhang L., Liu Y., Tao T., Tang J. Interplay between RAGE and TLR4 regulates HMGB1-induced inflammation by promoting cell surface expression of RAGE and TLR4. J. Immunol. 2020;205(3):767–775. doi: 10.4049/jimmunol.1900860. [DOI] [PubMed] [Google Scholar]
- 126.Deng M., Tang Y., Li W., Wang X., Zhang R., Zhang X., Zhao X., Liu J., Tang C., Liu Z., Huang Y., Peng H., Xiao L., Tang D., Scott M.J., Wang Q., Liu J., Xiao X., Watkins S., Li J., Yang H., Wang H., Chen F., Tracey K.J., Billiar T.R., Lu B. The endotoxin delivery protein HMGB1 mediates caspase-11-dependent lethality in sepsis. Immunity. 2018;49(4) doi: 10.1016/j.immuni.2018.08.016. 740–753 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yuan X., Bhat O.M., Lohner H., Zhang Y., Li P.L. Downregulation of lysosomal acid ceramidase mediates HMGB1-induced migration and proliferation of mouse coronary arterial myocytes. Front. Cell Dev. Biol. 2020;8:111. doi: 10.3389/fcell.2020.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Xu J., Jiang Y., Wang J., Shi X., Liu Q., Liu Z., Li Y., Scott M.J., Xiao G., Li S., Fan L., Billiar T.R., Wilson M.A., Fan J. Macrophage endocytosis of high-mobility group box 1 triggers pyroptosis. Cell Death Differ. 2014;21(8):1229–1239. doi: 10.1038/cdd.2014.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gao J.J., Tian Z.X., Yang X. Breakthrough: chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Biosci. Trends. 2020;14(1):72–73. doi: 10.5582/bst.2020.01047. [DOI] [PubMed] [Google Scholar]
- 130.Nicolai L., Leunig A., Brambs S., Kaiser R., Joppich M., Hoffknecht M.L., Gold C., Engel A., Polewka V., Muenchhoff M., Hellmuth J.C., Ruhle A., Ledderose S., Weinberger T., Schulz H., Scherer C., Rudelius M., Zoller M., Keppler O.T., Zwissler B., von Bergwelt-Baildon M., Kaab S., Zimmer R., Bulow R.D., von Stillfried S., Boor P., Massberg S., Pekayvaz K., Stark K. Vascular neutrophilic inflammation and immunothrombosis distinguish severe COVID-19 from influenza pneumonia. J. Thromb. Haemost. 2021;19(2):574–581. doi: 10.1111/jth.15179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Nicolai L., Leunig A., Brambs S., Kaiser R., Weinberger T., Weigand M., Muenchhoff M., Hellmuth J.C., Ledderose S., Schulz H., Scherer C., Rudelius M., Zoller M., Hochter D., Keppler O., Teupser D., Zwissler B., von Bergwelt-Baildon M., Kaab S., Massberg S., Pekayvaz K., Stark K. Immunothrombotic dysregulation in COVID-19 pneumonia is associated with respiratory failure and coagulopathy. Circulation. 2020;142(12):1176–1189. doi: 10.1161/CIRCULATIONAHA.120.048488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhou F., Yu T., Du R., Fan G., Liu Y., Liu Z., Xiang J., Wang Y., Song B., Gu X., Guan L., Wei Y., Li H., Wu X., Xu J., Tu S., Zhang Y., Chen H., Cao B. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054–1062. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rostami M., Mansouritorghabeh H. D-dimer level in COVID-19 infection: a systematic review. Expert. Rev. Hematol. 2020;13(11):1265–1275. doi: 10.1080/17474086.2020.1831383. [DOI] [PubMed] [Google Scholar]
- 134.Han H., Yang L., Liu R., Liu F., Wu K.L., Li J., Liu X.H., Zhu C.L. Prominent changes in blood coagulation of patients with SARS-CoV-2 infection. Clin. Chem. Lab. Med. 2020;58(7):1116–1120. doi: 10.1515/cclm-2020-0188. [DOI] [PubMed] [Google Scholar]
- 135.Tang N., Li D., Wang X., Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020;18(4):844–847. doi: 10.1111/jth.14768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wang D., Hu B., Hu C., Zhu F., Liu X., Zhang J., Wang B., Xiang H., Cheng Z., Xiong Y., Zhao Y., Li Y., Wang X., Peng Z. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA. 2020;323(11):1061–1069. doi: 10.1001/jama.2020.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhang L., Yan X., Fan Q., Liu H., Liu X., Liu Z., Zhang Z. D-dimer levels on admission to predict in-hospital mortality in patients with Covid-19. J. Thromb. Haemost. 2020;18(6):1324–1329. doi: 10.1111/jth.14859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.McFadyen J.D., Stevens H., Peter K. The emerging threat of (micro)thrombosis in COVID-19 and its therapeutic implications. Circ. Res. 2020;127(4):571–587. doi: 10.1161/CIRCRESAHA.120.317447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yau J.W., Teoh H., Verma S. Endothelial cell control of thrombosis. BMC Cardiovasc. Disord. 2015;15:130. doi: 10.1186/s12872-015-0124-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pober J.S., Sessa W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007;7(10):803–815. doi: 10.1038/nri2171. [DOI] [PubMed] [Google Scholar]
- 141.Steffel J., Luscher T.F., Tanner F.C. Tissue factor in cardiovascular diseases: molecular mechanisms and clinical implications. Circulation. 2006;113(5):722–731. doi: 10.1161/CIRCULATIONAHA.105.567297. [DOI] [PubMed] [Google Scholar]
- 142.Ward S.E., Curley G.F., Lavin M., Fogarty H., Karampini E., McEvoy N.L., Clarke J., Boylan M., Alalqam R., Worrall A.P., Kelly C., de Barra E., Glavey S., Ni Cheallaigh C., Bergin C., Martin-Loeches I., Townsend L., Mallon P.W., O'Sullivan J.M., O'Donnell J.S., Irish C.-V.S.I. Von Willebrand factor propeptide in severe coronavirus disease 2019 (COVID-19): evidence of acute and sustained endothelial cell activation. Br. J. Haematol. 2021;192(4):714–719. doi: 10.1111/bjh.17273. [DOI] [PubMed] [Google Scholar]
- 143.Moake J.L., Turner N.A., Stathopoulos N.A., Nolasco L.H., Hellums J.D. Involvement of large plasma von Willebrand factor (vWF) multimers and unusually large vWF forms derived from endothelial cells in shear stress-induced platelet aggregation. J. Clin. Invest. 1986;78(6):1456–1461. doi: 10.1172/JCI112736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ladikou E.E., Sivaloganathan H., Milne K.M., Arter W.E., Ramasamy R., Saad R., Stoneham S.M., Philips B., Eziefula A.C., Chevassut T. Von Willebrand factor (vWF): marker of endothelial damage and thrombotic risk in COVID-19? Clin. Med. 2020;20(5):e178–e182. doi: 10.7861/clinmed.2020-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Helms J., Tacquard C., Severac F., Leonard-Lorant I., Ohana M., Delabranche X., Merdji H., Clere-Jehl R., Schenck M., Gandet F. Fagot, Fafi-Kremer S., Castelain V., Schneider F., Grunebaum L., Angles-Cano E., Sattler L., Mertes P.M., Meziani F., C.T. Group High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med. 2020;46(6):1089–1098. doi: 10.1007/s00134-020-06062-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Goshua G., Pine A.B., Meizlish M.L., Chang C.H., Zhang H., Bahel P., Baluha A., Bar N., Bona R.D., Burns A.J., Dela Cruz C.S., Dumont A., Halene S., Hwa J., Koff J., Menninger H., Neparidze N., Price C., Siner J.M., Tormey C., Rinder H.M., Chun H.J., Lee A.I. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020;7(8):e575–e582. doi: 10.1016/S2352-3026(20)30216-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Mancini I., Baronciani L., Artoni A., Colpani P., Biganzoli M., Cozzi G., Novembrino C., Boscolo Anzoletti M., De Zan V., Pagliari M.T., Gualtierotti R., Aliberti S., Panigada M., Grasselli G., Blasi F., Peyvandi F. The ADAMTS13-von Willebrand factor axis in COVID-19 patients. J. Thromb. Haemost. 2021;19(2):513–521. doi: 10.1111/jth.15191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cugno M., Meroni P.L., Gualtierotti R., Griffini S., Grovetti E., Torri A., Lonati P., Grossi C., Borghi M.O., Novembrino C., Boscolo M., Uceda Renteria S.C., Valenti L., Lamorte G., Manunta M., Prati D., Pesenti A., Blasi F., Costantino G., Gori A., Bandera A., Tedesco F., Peyvandi F. Complement activation and endothelial perturbation parallel COVID-19 severity and activity. J. Autoimmun. 2021;116 doi: 10.1016/j.jaut.2020.102560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hoechter D.J., Becker-Pennrich A., Langrehr J., Bruegel M., Zwissler B., Schaefer S., Spannagl M., Hinske L.C., Zoller M. Higher procoagulatory potential but lower DIC score in COVID-19 ARDS patients compared to non-COVID-19 ARDS patients. Thromb. Res. 2020;196:186–192. doi: 10.1016/j.thromres.2020.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Philippe A., Chocron R., Gendron N., Bory O., Beauvais A., Peron N., Khider L., Guerin C.L., Goudot G., Levasseur F., Peronino C., Duchemin J., Brichet J., Sourdeau E., Desvard F., Bertil S., Pene F., Cheurfa C., Szwebel T.A., Planquette B., Rivet N., Jourdi G., Hauw-Berlemont C., Hermann B., Gaussem P., Mirault T., Terrier B., Sanchez O., Diehl J.L., Fontenay M., Smadja D.M. Circulating Von Willebrand factor and high molecular weight multimers as markers of endothelial injury predict COVID-19 in-hospital mortality. Angiogenesis. 2021;24(3):505–517. doi: 10.1007/s10456-020-09762-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Henry B.M., Benoit S.W., de Oliveira M.H.S., Lippi G., Favaloro E.J., Benoit J.L. ADAMTS13 activity to von Willebrand factor antigen ratio predicts acute kidney injury in patients with COVID-19: evidence of SARS-CoV-2 induced secondary thrombotic microangiopathy. Int. J. Lab. Hematol. 2021;43(Suppl. 1):129–136. doi: 10.1111/ijlh.13415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Vassiliou A.G., Keskinidou C., Jahaj E., Gallos P., Dimopoulou I., Kotanidou A., Orfanos S.E. ICU admission levels of endothelial biomarkers as predictors of mortality in critically ill COVID-19 patients. Cells. 2021;10(1) doi: 10.3390/cells10010186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ward S.E., Fogarty H., Karampini E., Lavin M., Schneppenheim S., Dittmer R., Morrin H., Glavey S., Ni Cheallaigh C., Bergin C., Martin-Loeches I., Mallon P.W., Curley G.F., Baker R.I., Budde U., O'Sullivan J.M., O'Donnell J.S., C.-V.S.i. Irish ADAMTS13 regulation of VWF multimer distribution in severe COVID-19. J. Thromb. Haemost. 2021;19:1914–1921. doi: 10.1111/jth.15409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Philippe A., Gendron N., Bory O., Beauvais A., Mirault T., Planquette B., Sanchez O., Diehl J.L., Chocron R., Smadja D.M. Von Willebrand factor collagen-binding capacity predicts in-hospital mortality in COVID-19 patients: insight from VWF/ADAMTS13 ratio imbalance. Angiogenesis. 2021;24(3):407–411. doi: 10.1007/s10456-021-09789-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Mei Z.W., van Wijk X.M.R., Pham H.P., Marin M.J. Role of von Willebrand factor in COVID-19 associated coagulopathy. J. Appl. Lab. Med. 2021;6:1305–1315. doi: 10.1093/jalm/jfab042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.De Cristofaro R., Liuzzo G., Sacco M., Lancellotti S., Pedicino D., Andreotti F. Marked von Willebrand factor and factor VIII elevations in severe acute respiratory syndrome coronavirus-2-positive, but not severe acute respiratory syndrome coronavirus-2-negative, pneumonia: a case-control study. Blood Coagul. Fibrinolysis. 2021;32(4):285–289. doi: 10.1097/MBC.0000000000000998. [DOI] [PubMed] [Google Scholar]
- 157.Wibowo A., Pranata R., Lim M.A., Akbar M.R., Martha J.W. Endotheliopathy marked by high von Willebrand factor (vWF) antigen in COVID-19 is associated with poor outcome: a systematic review and meta-analysis. Int. J. Infect. Dis. 2021 doi: 10.1016/j.ijid.2021.06.051. S1201-9712(21)00540-3, (Online ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Marchetti M., Gomez-Rosas P., Sanga E., Gamba S., Verzeroli C., Russo L., Restuccia F., Schieppati F., Bonanomi E., Rizzi M., Fagiuoli S., D'Alessio A., Lorini L., Falanga A. Endothelium activation markers in severe hospitalized COVID-19 patients: role in mortality risk prediction. TH Open. 2021;5(3):e253–e263. doi: 10.1055/s-0041-1731711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Thomas V.V., Kumar S.E., Alexander V., Nadaraj A., Vijayalekshmi B., Prabhu S., Kumar S., Murugabharathy K., Thomas S.M., Hansdak S., Carey R., Iyyadurai R., Pichamuthu K., Abhilash K.P.P., Varghese G.M., Nair S., Goel A., Jeyaseelan L., Zachariah U., Zachariah A., Eapen C.E. Plasma Von Willebrand factor levels predict survival in COVID-19 patients across the entire spectrum of disease severity. Indian J. Hematol. Blood Transfus. 2021:1–8. doi: 10.1007/s12288-021-01459-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kayal S., Jais J.P., Aguini N., Chaudiere J., Labrousse J. Elevated circulating E-selectin, intercellular adhesion molecule 1, and von Willebrand factor in patients with severe infection. Am. J. Respir. Crit. Care Med. 1998;157(3 Pt 1):776–784. doi: 10.1164/ajrccm.157.3.9705034. [DOI] [PubMed] [Google Scholar]
- 161.Polgar J., Matuskova J., Wagner D.D. The P-selectin, tissue factor, coagulation triad. J. Thromb. Haemost. 2005;3(8):1590–1596. doi: 10.1111/j.1538-7836.2005.01373.x. [DOI] [PubMed] [Google Scholar]
- 162.Skendros P., Mitsios A., Chrysanthopoulou A., Mastellos D.C., Metallidis S., Rafailidis P., Ntinopoulou M., Sertaridou E., Tsironidou V., Tsigalou C., Tektonidou M., Konstantinidis T., Papagoras C., Mitroulis I., Germanidis G., Lambris J.D., Ritis K. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Invest. 2020;130(11):6151–6157. doi: 10.1172/JCI141374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Manne B.K., Denorme F., Middleton E.A., Portier I., Rowley J.W., Stubben C., Petrey A.C., Tolley N.D., Guo L., Cody M., Weyrich A.S., Yost C.C., Rondina M.T., Campbell R.A. Platelet gene expression and function in patients with COVID-19. Blood. 2020;136(11):1317–1329. doi: 10.1182/blood.2020007214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Zhang S., Liu Y., Wang X., Yang L., Li H., Wang Y., Liu M., Zhao X., Xie Y., Yang Y., Zhang S., Fan Z., Dong J., Yuan Z., Ding Z., Zhang Y., Hu L. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J. Hematol. Oncol. 2020;13(1):120. doi: 10.1186/s13045-020-00954-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Koupenova M., Clancy L., Corkrey H.A., Freedman J.E. Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ. Res. 2018;122(2):337–351. doi: 10.1161/CIRCRESAHA.117.310795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Koupenova M., Vitseva O., MacKay C.R., Beaulieu L.M., Benjamin E.J., Mick E., Kurt-Jones E.A., Ravid K., Freedman J.E. Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood. 2014;124(5):791–802. doi: 10.1182/blood-2013-11-536003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Laforge M., Elbim C., Frere C., Hemadi M., Massaad C., Nuss P., Benoliel J.J., Becker C. Tissue damage from neutrophil-induced oxidative stress in COVID-19. Nat. Rev. Immunol. 2020;20(9):515–516. doi: 10.1038/s41577-020-0407-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Leppkes M., Knopf J., Naschberger E., Lindemann A., Singh J., Herrmann I., Sturzl M., Staats L., Mahajan A., Schauer C., Kremer A.N., Volkl S., Amann K., Evert K., Falkeis C., Wehrfritz A., Rieker R.J., Hartmann A., Kremer A.E., Neurath M.F., Munoz L.E., Schett G., Herrmann M. Vascular occlusion by neutrophil extracellular traps in COVID-19. EBioMedicine. 2020;58 doi: 10.1016/j.ebiom.2020.102925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Li S., Jiang L., Li X., Lin F., Wang Y., Li B., Jiang T., An W., Liu S., Liu H., Xu P., Zhao L., Zhang L., Mu J., Wang H., Kang J., Li Y., Huang L., Zhu C., Zhao S., Lu J., Ji J., Zhao J. Clinical and pathological investigation of patients with severe COVID-19. JCI Insight. 2020;5(12) doi: 10.1172/jci.insight.138070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zhang X., Tan Y., Ling Y., Lu G., Liu F., Yi Z., Jia X., Wu M., Shi B., Xu S., Chen J., Wang W., Chen B., Jiang L., Yu S., Lu J., Wang J., Xu M., Yuan Z., Zhang Q., Zhang X., Zhao G., Wang S., Chen S., Lu H. Viral and host factors related to the clinical outcome of COVID-19. Nature. 2020;583(7816):437–440. doi: 10.1038/s41586-020-2355-0. [DOI] [PubMed] [Google Scholar]
- 171.Martinod K., Wagner D.D. Thrombosis: tangled up in NETs. Blood. 2014;123(18):2768–2776. doi: 10.1182/blood-2013-10-463646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Middleton E.A., He X.Y., Denorme F., Campbell R.A., Ng D., Salvatore S.P., Mostyka M., Baxter-Stoltzfus A., Borczuk A.C., Loda M., Cody M.J., Manne B.K., Portier I., Harris E.S., Petrey A.C., Beswick E.J., Caulin A.F., Iovino A., Abegglen L.M., Weyrich A.S., Rondina M.T., Egeblad M., Schiffman J.D., Yost C.C. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood. 2020;136(10):1169–1179. doi: 10.1182/blood.2020007008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Radermecker C., Detrembleur N., Guiot J., Cavalier E., Henket M., d'Emal C., Vanwinge C., Cataldo D., Oury C., Delvenne P., Marichal T. Neutrophil extracellular traps infiltrate the lung airway, interstitial, and vascular compartments in severe COVID-19. J. Exp. Med. 2020;217(12) doi: 10.1084/jem.20201012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Zuo Y., Yalavarthi S., Shi H., Gockman K., Zuo M., Madison J.A., Blair C., Weber A., Barnes B.J., Egeblad M., Woods R.J., Kanthi Y., Knight J.S. Neutrophil extracellular traps in COVID-19. JCI Insight. 2020;5(11) doi: 10.1172/jci.insight.138999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ng H., Havervall S., Rosell A., Aguilera K., Parv K., von Meijenfeldt F.A., Lisman T., Mackman N., Thalin C., Phillipson M. Circulating markers of neutrophil extracellular traps are of prognostic value in patients with COVID-19. Arterioscler. Thromb. Vasc. Biol. 2021;41(2):988–994. doi: 10.1161/ATVBAHA.120.315267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zuo Y., Estes S.K., Ali R.A., Gandhi A.A., Yalavarthi S., Shi H., Sule G., Gockman K., Madison J.A., Zuo M., Yadav V., Wang J., Woodard W., Lezak S.P., Lugogo N.L., Smith S.A., Morrissey J.H., Kanthi Y., Knight J.S. Prothrombotic autoantibodies in serum from patients hospitalized with COVID-19. Sci. Transl. Med. 2020;12(570) doi: 10.1126/scitranslmed.abd3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Zoufaly A., Poglitsch M., Aberle J.H., Hoepler W., Seitz T., Traugott M., Grieb A., Pawelka E., Laferl H., Wenisch C., Neuhold S., Haider D., Stiasny K., Bergthaler A., Puchhammer-Stoeckl E., Mirazimi A., Montserrat N., Zhang H., Slutsky A.S., Penninger J.M. Human recombinant soluble ACE2 in severe COVID-19. Lancet Respir. Med. 2020;8(11):1154–1158. doi: 10.1016/S2213-2600(20)30418-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Abd El-Aziz T.M., Al-Sabi A., Stockand J.D. Human recombinant soluble ACE2 (hrsACE2) shows promise for treating severe COVID-19. Signal Transduct. Target Ther. 2020;5(1):258. doi: 10.1038/s41392-020-00374-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Pang X., Cui Y., Zhu Y. Recombinant human ACE2: potential therapeutics of SARS-CoV-2 infection and its complication. Acta Pharmacol. Sin. 2020;41(9):1255–1257. doi: 10.1038/s41401-020-0430-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Gupta S., Wang W., Hayek S.S., Chan L., Mathews K.S., Melamed M.L., Brenner S.K., Leonberg-Yoo A., Schenck E.J., Radbel J., Reiser J., Bansal A., Srivastava A., Zhou Y., Finkel D., Green A., Mallappallil M., Faugno A.J., Zhang J., Velez J.C.Q., Shaefi S., Parikh C.R., Charytan D.M., Athavale A.M., Friedman A.N., Redfern R.E., Short S.A.P., Correa S., Pokharel K.K., Admon A.J., Donnelly J.P., Gershengorn H.B., Douin D.J., Semler M.W., Hernan M.A., Leaf D.E., Investigators S.-C. Association between early treatment with tocilizumab and mortality among critically ill patients with COVID-19. JAMA Intern. Med. 2021;181(1):41–51. doi: 10.1001/jamainternmed.2020.6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Leaf D.E., Gupta S., Wang W. Tocilizumab in covid-19. N. Engl. J. Med. 2021;384(1):86–87. doi: 10.1056/NEJMc2032911. [DOI] [PubMed] [Google Scholar]
- 182.Solinas C., Perra L., Aiello M., Migliori E., Petrosillo N. A critical evaluation of glucocorticoids in the management of severe COVID-19. Cytokine Growth Factor Rev. 2020;54:8–23. doi: 10.1016/j.cytogfr.2020.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.R.C. Group, Horby P., Lim W.S., Emberson J.R., Mafham M., Bell J.L., Linsell L., Staplin N., Brightling C., Ustianowski A., Elmahi E., Prudon B., Green C., Felton T., Chadwick D., Rege K., Fegan C., Chappell L.C., Faust S.N., Jaki T., Jeffery K., Montgomery A., Rowan K., Juszczak E., Baillie J.K., Haynes R., Landray M.J. Dexamethasone in hospitalized patients with covid-19. N. Engl. J. Med. 2021;384(8):693–704. doi: 10.1056/NEJMoa2021436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Okur H.K., Yalcin K., Tastan C., Demir S., Yurtsever B., Karakus G.S., Kancagi D.D., Abanuz S., Seyis U., Zengin R., Hemsinlioglu C., Kara M., Yildiz M.E., Deliceo E., Birgen N., Pelit N.B., Cuhadaroglu C., Kocagoz A.S., Ovali E. Preliminary report of in vitro and in vivo effectiveness of dornase alfa on SARS-CoV-2 infection. New Microbes New Infect. 2020;37 doi: 10.1016/j.nmni.2020.100756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Weber A.G., Chau A.S., Egeblad M., Barnes B.J., Janowitz T. Nebulized in-line endotracheal dornase alfa and albuterol administered to mechanically ventilated COVID-19 patients: a case series. Mol. Med. 2020;26(1):91. doi: 10.1186/s10020-020-00215-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Diurno F., Numis F.G., Porta G., Cirillo F., Maddaluno S., Ragozzino A., De Negri P., Di Gennaro C., Pagano A., Allegorico E., Bressy L., Bosso G., Ferrara A., Serra C., Montisci A., D'Amico M., Morello S. Schiano Lo, Di Costanzo G., Tucci A.G., Marchetti P., Di Vincenzo U., Sorrentino I., Casciotta A., Fusco M., Buonerba C., Berretta M., Ceccarelli M., Nunnari G., Diessa Y., Cicala S., Facchini G. Eculizumab treatment in patients with COVID-19: preliminary results from real life ASL Napoli 2 Nord experience. Eur. Rev. Med. Pharmacol. Sci. 2020;24(7):4040–4047. doi: 10.26355/eurrev_202004_20875. [DOI] [PubMed] [Google Scholar]
- 187.Annane D., Heming N., Grimaldi-Bensouda L., Fremeaux-Bacchi V., Vigan M., Roux A.L., Marchal A., Michelon H., Rottman M., Moine P., Garches C.C.G. Eculizumab as an emergency treatment for adult patients with severe COVID-19 in the intensive care unit: a proof-of-concept study. EClinicalMedicine. 2020;28 doi: 10.1016/j.eclinm.2020.100590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Wu X., Dao Thi V.L., Huang Y., Billerbeck E., Saha D., Hoffmann H.H., Wang Y., Silva L.A.V., Sarbanes S., Sun T., Andrus L., Yu Y., Quirk C., Li M., MacDonald M.R., Schneider W.M., An X., Rosenberg B.R., Rice C.M. Intrinsic immunity shapes viral resistance of stem cells. Cell. 2018;172(3) doi: 10.1016/j.cell.2017.11.018. 423–438 e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Avanzini M.A., Mura M., Percivalle E., Bastaroli F., Croce S., Valsecchi C., Lenta E., Nykjaer G., Cassaniti I., Bagnarino J., Baldanti F., Zecca M., Comoli P., Gnecchi M. Human mesenchymal stromal cells do not express ACE2 and TMPRSS2 and are not permissive to SARS-CoV-2 infection. Stem Cells Transl. Med. 2021;10(4):636–642. doi: 10.1002/sctm.20-0385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Leng Z., Zhu R., Hou W., Feng Y., Yang Y., Han Q., Shan G., Meng F., Du D., Wang S., Fan J., Wang W., Deng L., Shi H., Li H., Hu Z., Zhang F., Gao J., Liu H., Li X., Zhao Y., Yin K., He X., Gao Z., Wang Y., Yang B., Jin R., Stambler I., Lim L.W., Su H., Moskalev A., Cano A., Chakrabarti S., Min K.J., Ellison-Hughes G., Caruso C., Jin K., Zhao R.C. Transplantation of ACE2(-) mesenchymal stem cells improves the outcome of patients with COVID-19 pneumonia. Aging Dis. 2020;11(2):216–228. doi: 10.14336/AD.2020.0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Bernardo M.E., Fibbe W.E. Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell. 2013;13(4):392–402. doi: 10.1016/j.stem.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 192.Lee J.W., Fang X., Gupta N., Serikov V., Matthay M.A. Allogeneic human mesenchymal stem cells for treatment of E. coli endotoxin-induced acute lung injury in the ex vivo perfused human lung. Proc. Natl. Acad. Sci. U. S. A. 2009;106(38):16357–16362. doi: 10.1073/pnas.0907996106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Liang B., Chen J., Li T., Wu H., Yang W., Li Y., Li J., Yu C., Nie F., Ma Z., Yang M., Xiao M., Nie P., Gao Y., Qian C., Hu M. Clinical remission of a critically ill COVID-19 patient treated by human umbilical cord mesenchymal stem cells: a case report. Medicine (Baltimore) 2020;99(31) doi: 10.1097/MD.0000000000021429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Khoury M., Cuenca J., Cruz F.F., Figueroa F.E., Rocco P.R.M., Weiss D.J. Current status of cell-based therapies for respiratory virus infections: applicability to COVID-19. Eur. Respir. J. 2020;55(6) doi: 10.1183/13993003.00858-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Abraham A., Krasnodembskaya A. Mesenchymal stem cell-derived extracellular vesicles for the treatment of acute respiratory distress syndrome. Stem Cells Transl. Med. 2020;9(1):28–38. doi: 10.1002/sctm.19-0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Wu J., Song D., Li Z., Guo B., Xiao Y., Liu W., Liang L., Feng C., Gao T., Chen Y., Li Y., Wang Z., Wen J., Yang S., Liu P., Wang L., Wang Y., Peng L., Stacey G.N., Hu Z., Feng G., Li W., Huo Y., Jin R., Shyh-Chang N., Zhou Q., Wang L., Hu B., Dai H., Hao J. Immunity-and-matrix-regulatory cells derived from human embryonic stem cells safely and effectively treat mouse lung injury and fibrosis. Cell Res. 2020;30(9):794–809. doi: 10.1038/s41422-020-0354-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Nadkarni G.N., Lala A., Bagiella E., Chang H.L., Moreno P.R., Pujadas E., Arvind V., Bose S., Charney A.W., Chen M.D., Cordon-Cardo C., Dunn A.S., Farkouh M.E., Glicksberg B.S., Kia A., Kohli-Seth R., Levin M.A., Timsina P., Zhao S., Fayad Z.A., Fuster V. Anticoagulation, bleeding, mortality, and pathology in hospitalized patients with COVID-19. J. Am. Coll. Cardiol. 2020;76(16):1815–1826. doi: 10.1016/j.jacc.2020.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Tang N., Bai H., Chen X., Gong J., Li D., Sun Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. 2020;18(5):1094–1099. doi: 10.1111/jth.14817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Mummery R.S., Rider C.C. Characterization of the heparin-binding properties of IL-6. J. Immunol. 2000;165(10):5671–5679. doi: 10.4049/jimmunol.165.10.5671. [DOI] [PubMed] [Google Scholar]
- 200.Lang J., Yang N., Deng J., Liu K., Yang P., Zhang G., Jiang C. Inhibition of SARS pseudovirus cell entry by lactoferrin binding to heparan sulfate proteoglycans. PLoS One. 2011;6(8) doi: 10.1371/journal.pone.0023710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Mousavi S., Moradi M., Khorshidahmad T., Motamedi M. Anti-inflammatory effects of heparin and its derivatives: a systematic review. Adv. Pharmacol. Sci. 2015;2015 doi: 10.1155/2015/507151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Piran S., Schulman S. Treatment of bleeding complications in patients on anticoagulant therapy. Blood. 2019;133(5):425–435. doi: 10.1182/blood-2018-06-820746. [DOI] [PubMed] [Google Scholar]
- 203.Hippensteel J.A., LaRiviere W.B., Colbert J.F., Langouet-Astrie C.J., Schmidt E.P. Heparin as a therapy for COVID-19: current evidence and future possibilities. Am. J. Physiol. Lung C. 2020;319(2):L211–L217. doi: 10.1152/ajplung.00199.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]