

Abstract
The increasingly appreciated prevalence of complicated stressor-to-phenotype associations in human disease requires a greater understanding of how specific stressors affect systems or interactome properties. Many currently untreatable diseases arise due to variations in, and through a combination of, multiple stressors of genetic, epigenetic, and environmental nature. Unfortunately, how such stressors lead to a specific disease phenotype or inflict a vulnerability to some cells and tissues but not others remains largely unknown and unsatisfactorily addressed. Analysis of cell- and tissue-specific interactome networks may shed light on organization of biological systems and subsequently to disease vulnerabilities. However, deriving human interactomes across different cell and disease contexts remains a challenge. To this end, this opinion article links stressor-induced protein interactome network perturbations to the formation of pathologic scaffolds termed epichaperomes, revealing a viable and reproducible experimental solution to obtaining rigorous context-dependent interactomes. This article presents our views on how a specialized ‘omics platform called epichaperomics may complement and enhance the currently available conventional approaches and aid the scientific community in defining, understanding, and ultimately controlling interactome networks of complex diseases such as Alzheimer’s disease. Ultimately, this approach may aid the transition from a limited single-alteration perspective in disease to a comprehensive network-based mindset, which we posit will result in precision medicine paradigms for disease diagnosis and treatment.
Keywords: Alzheimer’s disease, complex diseases, edgetic perturbations in disease, epichaperome, epichaperomics, interactome network dysfunctions, precision medicine, protein connectivity dysfunctions, protein–protein interactions, tissue-specific interactome
Introduction
One of the vexing unanswered questions in disease biology is characterizing the genotype-to-phenotype relationship, namely how encoded traits and/or vulnerabilities lead to observable disease manifestations within individuals (e.g., how cause leads to effect). This is not a straightforward and linear relationship. Even in Mendelian diseases presumably controlled by a single gene, the “one-gene/one-protein/one-function” paradigm cannot fully explain genotype-to-phenotype relationships [1]. For example, a specific genetic lesion may not impact all carriers similarly. Moreover, these inherited diseases may be transmitted through unaffected parents. This phenomenon known as reduced or incomplete penetrance is not uncommon. There are many known examples of disease-causing mutations either failing to cause fulminant disease in at least a proportion of the individuals who carry them or manifesting through a spectrum of intensities across affected individuals. Further adding to the paradox is that clinically healthy individuals who do not express features of a disease may carry disease-associated variants that disrupt a protein-coding gene either by leading to a loss of function (e.g., a nonsense or frameshift variant) or by altering an amino acid in the encoded protein (missense variants) [2,3]. A typical healthy individual may have 80 genes severely damaged or inactivated in both copies (i.e., homozygous). The lack of phenotypic effects in such individuals cannot be explained by a normal allele masking the deleterious effects of the disease-causing allele [2,3].
In addition to manifesting differently from patient-to-patient, inherited genetic lesions, albeit present throughout the body and thus common to all cells harboring an affected individual’s genome, also tend to affect specific organs, tissues, and cells differently. Only a minority of genes and proteins are tissue-specific, and the subset of protein-coding genes causal for hereditary diseases is not more tissue specific than protein-coding genes in general. Yet selective vulnerability in Mendelian diseases appears to be ubiquitous with an analysis of over 1200 such diseases showing they manifest in a tissue-specific manner [4]. The genotype-to-phenotype relationship becomes exponentially more challenging to address in complex diseases, where stressors of epigenetic, proteotoxic, and environmental nature combine with genetic defects to shape disease manifestation. Even in diseases such as cancer where genetic lesions have historically provided a main strategy for patient treatment and treatment selection, how somatic mutations affect certain tissues but not others remains an important open question [5]. The relationship between damage to a gene and protein and damage to an individual’s health is therefore far more complex than can be explained by a linear model of a genotype–phenotype relationship [6].
One key means to adequately address complicated stressor-to-phenotype associations in human disease is the understanding of how stressors affect systems or interactome properties [6–10]. In this context, it is the severity of perturbation to the complex network of molecular interactions in the cell that may more closely capture the potential to generate a phenotype. Here, we provide a brief overview of current approaches to address mechanisms underlying disease through evaluating interactomes which result in a disease state and introduce a complementary approach to study interactome dysfunction in a tissue- and cell-specific manner through a novel ‘omics platform we term epichaperomics.
Interactome networks as a gateway to disease
Genes and encoded products function not in isolation but as components of intricate networks of macromolecules linked through interactions, represented as ‘interactomes’ [11–13]. Encoded by the human genome, transcriptional regulation, protein expression, and their biochemical interactions in complex protein machineries as well as their interplay in complex macromolecular interactome networks give rise to cell type- and cell state-specific networks of macromolecules. These include protein–protein or protein–DNA interactions that ultimately result in phenotypic manifestations within cells, tissues, organs, and entire organisms. Disease states represent perturbations of the underlying tissue- and cell-specific molecular networks arising from both internal perturbations (e.g., genetic mutations, proteotoxic stress, hormonal changes, and/or aging) and external stressors (chemical or other environmental exposures, and/or lifestyle choices). The disease interactome is therefore a map of how individual stressors or a combination thereof alter interaction networks and perturb the system as a whole [14]. Interactome structure and biological function are therefore intimately connected, with the interactome providing a direct gateway into understanding disease and potentially uncovering therapeutic opportunities.
Whereas interactomes encompass several biomolecules, as mentioned above, here we will refer specifically to protein–protein interaction (PPI) networks wherein “nodes” represent proteins and “edges” indicate physical interactions between proteins. Interactions among proteins may be through direct or indirect contact (i.e., as part of a protein complex) or functional (i.e., as part of the same protein pathway). Currently available PPI maps are based on several sources [15–17]. One is the available scientific literature mined to catalogue experimentally detected interactions, typically derived from small-scale experiments often performed with differing methodologies. Another source is interactions detected in systematic, proteome-scale mapping efforts, where two main techniques are used—yeast two hybrids for binary interactions and binding affinity purifications coupled to mass spectrometry (MS) for cocomplexes [17–19]. Through these efforts, the known and predicted physical interaction map between human proteins catalogues over 500 000 interactions among approximately 20 000 proteins. The map is continuously expanding through such experimental efforts, as well as through computational predictions, for example, based on protein structure [16]. Several databases have been created that integrate known and predicted interaction data across multiple organisms, and these data repositories were reviewed in recent articles [15,17,20,21]. A study evaluated several human interaction databases, concluding assessing connectivity in the interactome and identifying specific human disease or molecular pathways of interest, and larger networks such as STRING [22] tend to achieve better overall performance, with the caveat that large databases may harbor a significant numbers of false-positive interactions compared with smaller, carefully curated and annotated databases [23]. A compilation of PPI databases can be found in http://www.pathguide.org/. The majority of PPI predictions available in databases to date are unique [16,21], indicating their information is complementary and urging the necessity for the assembly and use of a resource that combines most of them.
The resulting interactome network, of all known potential and predicted PPIs, has become a key framework for studying genotype-to-phenotype relationships [24–26]. However, this network also has limitations. For example, protein interactome networks are derived either by expressing and testing interacting proteins in an in vitro or cell-heterologous system or by exogenously expressing and affinity-purifying a bait protein with its partners from a cell line [15–17]. It is estimated the use of yeast two-hybrid or coprecipitation assays may miss, or be unable to detect interactions, for 50% of the tested proteins in cases where PPIs require additional partners to stabilize the interaction, the protein’s interaction is mediated by specific post-translational modifications (PTMs) not available in the model system, and/or when PPIs are of low interaction strength or executed through dynamic, short-lived complexes [19]. Affinity purification methods may enrich in highly abundant proteins [27], whereas networks generated from literature mining are susceptible to study bias and may be overrepresented in proteins that are widely studied independent of abundance or importance to the network [28,29]. Such technical challenges render protein interaction networks far from complete and in need of careful curation. Whereas it is difficult to predict the constellation of all possible PPIs, some estimate that probably < 20% of all direct PPIs are known to date. Thus, a considerable number of disease-relevant interactions remain to be discovered [19]. In addition to being incompletely curated and annotated, and unlike, for example, transcriptional regulatory interactions mapped across tissues and cell types for several transcription factors, PPIs have often been measured in model systems and lack cell, organ, tissue, and disease contexts [4]. By representing the sum of all observed interactions, such interactomes are inherently generic, thus limiting their utility in understanding disease-specific alterations. Despite these limitations, several elegant concepts and network-based approaches have been developed and applied to these datasets to provide first valuable insights into the complexities of disease interactomes. A number of review articles provide an excellent overview of these efforts [13,14,30–32]. A notable limitation is translating aggregated PPI data into biological insights require sophisticated computational inferences, limiting overall access to the biomedical community. To facilitate these analyses, bioinformatic tools have been developed to represent and explore PPI networks. For example, those associated with Cytoscape (http://www.cytoscape.org/) an open-source bioinformatics software platform can be used for visualizing molecular interaction networks and biological pathways and for integrating these networks with annotations and other types of data [33]. There are several Cytoscape plug-in modules that can be used to download and explore PPIs, and to visualize and analyze human-curated pathway datasets such as WikiPathways [34], Reactome [35], and KEGG [36]. A list of 375 PPI resources has been compiled through extensive literature search (http://startbioinfo.org). Therefore, disease-based interactome network analyses are in their infancy and await further development and use in biological systems.
MS-based methods for PPI mapping
Several techniques coupled with MS have been developed to understand the interactome of a given protein. These include affinity purification mass spectrometry (AP-MS) and proximity labeling, among others [37,38]. For example, AP-MS makes use of epitope tagging, whereby short peptide or protein tags (e.g., FLAG-, TAP-, Strep-Tag, or myc) are fused to a “bait” protein or proteins of interest—either in the context of an exogenous expression construct or under the gene’s endogenous promoter using gene editing technologies such as CRISPR-Cas9 [39–42]. The bait protein functions as an affinity capture probe for interacting, or “prey” proteins, which are then captured by epitope-recognizing matrices and the isolated proteins identified by MS. Several excellent reviews and research articles provide an overview of applications using AP-MS [39–42].
In proximity labeling, proteins proximal to a protein of interest are identified and monitored by expressing in cells a bait protein fused to a promiscuous labeling enzyme [43]. The addition of a small molecule substrate, such as biotin, allows the covalent tagging of endogenous proteins within a 10–20 nm range, capturing the protein’s surrounding environment, including potential interactors. After selective enrichment of biotinylated proteins, commonly through streptavidin binding, interacting proteins are identified by MS. Various proximity labeling methodologies have been developed such as BioID and APEX [44]. For example, BioID utilizes BirA, a biotin ligase with specific mutations rendering the enzyme promiscuous [45]. BirA catalyzes the transformation of biotin to a more reactive form, and the resultant biotin reacts with primary amines of proteins in its vicinity, covalently biotinylating them. APEX uses engineered ascorbic acid peroxidase, which, following addition of H2O2, oxidizes phenol derivatives to biotin-phenoxyl radicals that covalently react with electron-rich amino acids [46]. Important biological information from the use of these methods has been derived, and excellent review articles provide an overview of such achievements [42,43,47,48].
A limitation, however, is these techniques depend on cultured engineered cells (often HEK293T cells) and tagged proteins which restrict the biological information they can provide. Furthermore, transfection introduces cellular stress that could impose artificial interactions among proteins. Importantly, determining the interactome of a given protein cannot be confused with creating a proteome-wide map of the constellation of cellular PPIs. A conclusive understanding of disease interactomes therefore requires that appropriate methods are utilized, and be applicable to, exploring the physiological environment of cells and tissues at the proteome-wide level.
Cellular Thermal Shift Assay (CETSA) coupled with mass spectrometry (MS-CETSA) has recently emerged as a method to study the modulation of protein interaction states in cellular processes and during drug action [49]. CETSA allows quantitative measurement of the stability of proteins within the physiological context of a cell [49]. The method is based on the principle that heat-induced protein unfolding leads to rapid precipitation in the cellular environment. Proteins within complexes are more likely to co-aggregate upon heat denaturation and therefore interacting proteins as part of protein complexes would exhibit similar, nonrandom progressive insolubilities when subjected to an increasing temperature gradient enabling their identification. CETSA, for example, was applied to understand the dynamics of protein complexes during the progression through the steps of cell cycle [50]. It remains to be determined whether most interaction changes within cells will yield measurable CETSA shifts, or whether the method is applicable to complex systems such as human tissues.
Cross-linking mass spectrometry (XLMS) is a method that emerged to fill this gap [51,52]. It allows identification of proximal amino acid pairs—including weak or transient interactions—covalently linked by a chemical cross-linker of a specific length. Protein samples are combined with reagents that form covalent bonds in solution, and upon protein digestion, resulting peptide pairs can be identified by tandem MS. Many chemical and analytical developments have helped apply XLMS at a proteome-wide scale to discover and monitor protein interactions. For example, XLMS has been applied to interrogate the mitochondrial protein interactome [53], synaptic protein interactions [54], and protein conformations and supercomplexes in heart tissue [55]. Despite many advances over the last decade including chemical cross-linkers, analytical methods, and bioinformatic/computational analyses, there are several notable limitations. Cross-linking coverage remains quite variable despite many developments in chemical cross-linking reagents and procedures. Cross-linking relies on a chemical reaction, which can be limited or favored to occur only in stringent buffer conditions. For example, the 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC)-based methods, the most popular zero-length cross-linker for biochemical conjugations, require the protein or complex be stable at pH 6.5 [56,57]. Furthermore, certain reagents need to be introduced into the reaction as acid salts to maintain proper pH. Therefore, the specific system being studied must be tolerant to high ionic strength. Steric shielding or buried surfaces of proteins in protein complexes may render them inaccessible for labeling by the cross-linking reagent. Enrichment of the cross-linked peptide pairs is needed for MS identification [58], requiring strong cation exchange and avidin affinity chromatography, making the method labor intensive. Furthermore, identification of cross-linked peptide spectra is more challenging than a traditional peptide search requiring a more sophisticated computational approach.
Development of new strategies to study changes within interactome networks linking stressors to specific phenotypes remains an unmet need. New approaches must inquire into native biological systems, inform on functionality of interactions, and be unaffected by strength of interactions. Interactome inquiries should anticipate the trajectory and dynamics of highly reconfigurable protein networks and macromolecular complexes, key characteristics of biological systems [59]. They should not rely on sophisticated computational algorithms to extract biological insights which limits access and overall impact to the biomedical community at large.
Cell- and tissue-specific interactome networks
Protein interactions are often context-dependent and context-specific, and a single protein may carry out different functions with different partners in different biological contexts [5,9]. Directly measured context-specific proteome-wide interactome networks are technically challenging using current interrogative approaches and have been approximated by integrating more widely available genomic information [4,60,61]. With technological advances in transcriptomics and proteomics that greatly improved their resolution, accuracy, and content information, cell- and tissue-specific datasets from these ‘omics platforms have started to complement generic interactomes by providing tissue- and cell-specific transcript and/or protein information and, in turn, disease-specific insights [62,63].
Despite tangible advances, success is limited by reliance on ‘omics-derived datasets reflecting an inventory of biomolecules or measure changes in their stoichiometry at a fixed time and condition. Context-dependent interactome maps derived solely from protein stoichiometry inventorying ‘omics analyses may poorly describe the specific molecular interactions that form the basis of all biological processes within a particular system. This challenge is posed by disease biology itself. First, disease-associated proteins tend to interact with each other forming a disease module, a connected subgraph in the interactome that contains the molecular determinants of a disease, or whose perturbation causes the disease [13]. An accurate identification of disease modules is hampered by the incompleteness of a relevant protein list, as determined by current ‘omics platforms, in disease-associated proteins. For example, an analysis of 226 diseases found that disease modules can only be identified for diseases whose number of associated proteins exceeds a critical threshold determined by network incompleteness [64]. The higher the number of disease proteins within the interactome, the higher the biological and functional similarity of corresponding proteins [64]. In other words, incomplete mapping of proteins involved in a disease-specific context makes determinations of molecular functions a difficult challenge. Rather, it is the number of disease module-associated proteins, not the number of proteins identified by a specific method, that is of critical importance for accurate understanding molecular mechanisms underlying disease phenotypes (Fig. 1). Second, global changes in PPIs, which can result from perturbing existing interactions and/or by generating new interactions, are only partly driven by, and dependent on, protein expression alterations [17,65]. A large-scale and high-resolution proteomic study combined with gene copy number analyzed the influence genomic changes on proteomic output, and cellular transformation to find regulation of biological processes and molecular complexes is independent of general copy number changes [65]. Thus, analysis and identification of tissue-specific interactomes may be poorly served by the use of ‘omics approaches limited to detecting, inventorying, and listing changes in the stoichiometry of biomolecules. To expand on these key points, we present a specific example whereby changes in protein connectivity (as determined by epichaperomics, see below) and quantitative bottom-up proteomics (as assayed using isobaric mass tag TMT10plex labeling) were compared (Fig. 1). This study performed on postmortem AD frontal cortex samples {Females (F, n = 8) vs Males (M, n = 6)} reveals how alterations in protein connectivity are independent of protein expression changes. Moreover, changes in expression levels do not necessarily equate to changes in connectivity. For example, both a gain of interaction and a loss of interaction are observed between proteins in the F vs M cohorts (as determined by epichaperomics), despite a trend in lower protein expression overall in F vs M (as determined by proteomics) (Fig. 1A). Notably, epichaperomics is able to detect key changes in connectivity in lowly expressed proteins. Identified proteins in the two datasets influence and determine the ability to make tissue-specific, functional predictions. Determining whether a differentially expressed protein (for proteomics) or differentially connected protein (for epichaperomics) is associated with a certain biological process or molecular function depends on the enrichment of its respective partners in the specific dataset (Fig. 1B).
Fig. 1.
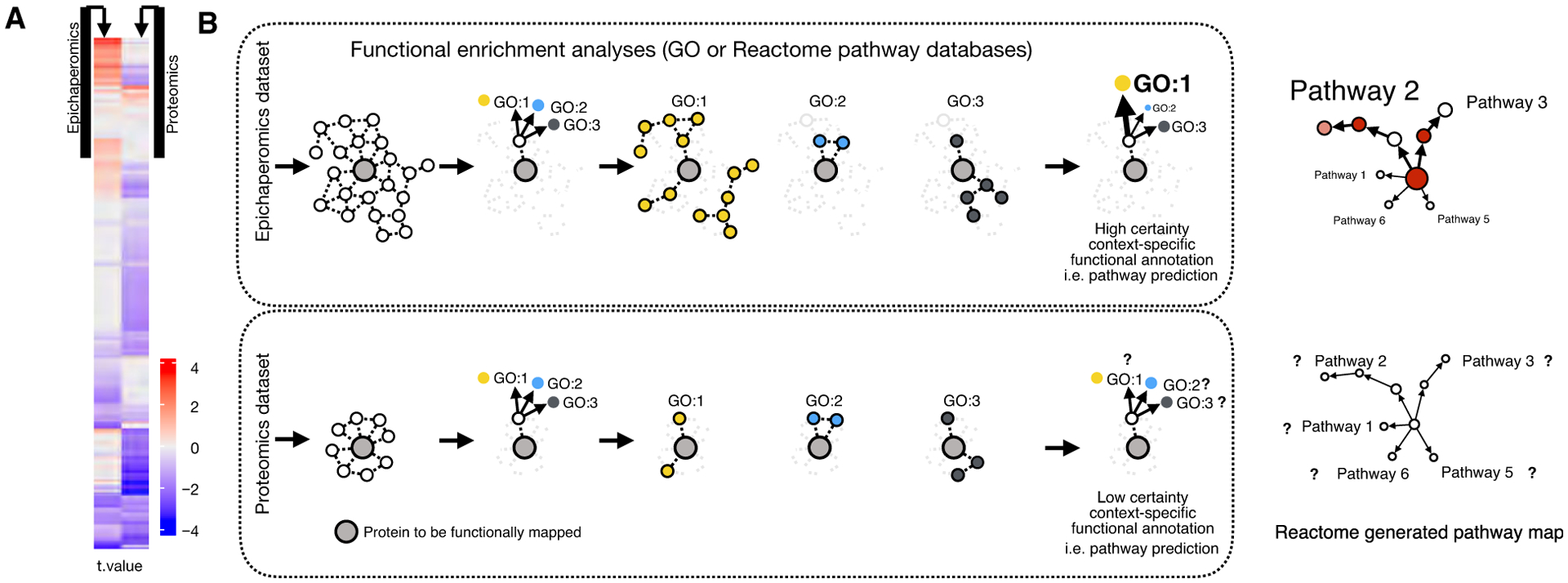
(A) Changes in protein connectivity are independent of protein expression changes, and changes in expression levels do not necessarily equate to changes in connectivity. Epichaperomics and proteomics detected proteins in AD (postmortem AD brain samples {Females (F, n = 8) vs Males (M, n = 6)}). For epichaperomic analysis (left panel), MS-derived files were subjected to Label-Free Quantification analysis using the MaxQuant proteomic data analysis workflow [72]. For proteomic analysis (right panel), quantitative bottom-up proteomics was performed using isobaric mass tag TMT10plex labeling reagents [142]. The calculated differences (t-values) were reordered based on hierarchical clustering. Results indicate connectivity, and expression level changes are independent. (B) Schematic showing the influence of identified proteins in a dataset on the ability to make tissue-specific functional predictions. A single protein may carry out different functions with different partners in different biological contexts. Determining if a differentially expressed protein (for proteomics) or differentially connected protein (for epichaperomics) is associated with a certain biological process or molecular function depends on the enrichment of its partners in the specific dataset. Gene Ontology (GO), which contains standardized annotation of proteins, is commonly used for this purpose. It works by comparing the frequency of individual annotations in the protein list with a reference list. Enrichment of biological pathways supplied by Reactome, WikiPathways, KEGG (Kyoto Encyclopedia of Genes and Genomes), or other pathway analysis resources can be performed in a similar manner.
Several lines of evidence indicate the propensity of many proteins to change connectivity in disease is intrinsically imprinted in their structure. For example, several disease-associated mutations tend to cluster in protein regions that are important for effecting PPIs [66]. Somatic mutations in tumor genomes are overrepresented at protein interaction interfaces [67,68]. Many mutations in cancer are thought to alter the specificity of kinases for their substrates [69]. Many disease mutations affect the motif specificity of transcription factors, with mutations reducing the specificity of DNA binding sites, thereby allowing more promiscuous binding [70]. Formation of splicing variants may also alter protein connectivity, and in cancer, aberrant splicing is more likely to occur at protein domains that mediate interactions [71]. Lastly, bioinformatic analyses suggest physical interactors of major cancer drivers are also frequently mutated in cancer [6].
However, alterations in connectivity in disease cannot be fully explained by defects in the structural integrity of proteins. For example, in the context of Alzheimer’s disease (AD), an analysis of global PPI networks found a large change in protein connectivity between AD and age-matched noncognitively impaired subjects, with approximately 62% of the nodes and 98.5% of the edges significantly altered in AD [72]. New connections were formed in AD (aberrant changes in 35% of nodes and 43% of edges) whereas many normal connections were lost (aberrant changes in 27% of nodes and 55.5% of edges), indicating PPI changes in disease affect a large subset of the context-intrinsic proteome as a whole [72].
In addition to changes in protein partners, protein connectivity changes in disease may be modulated by alterations in the strength of interactions and cellular mislocalization which in turn can be influenced by alterations in PTMs, stabilization of disease-enriched protein conformations, and other protein-modifying mechanisms [59,73–78]. Neither understanding of the genome, its code, expression or genetic alterations, nor profiling of the entire proteome across spatial, temporal, or quantitative dimensions in isolation can singularly explain disease. A curated, interrogative atlas of disease-specific alterations in PPIs is required to understand genotype–phenotype relationships. Accordingly, mechanistic understanding of human disease requires unbiased system-level investigations of interactomes in relevant biological specimens.
Disease-specific interactome alterations via epichaperomics
A solution to gaining insights into context-dependent interactome network changes in disease is provided by nature itself. Discoveries in disease biology link stressor-induced interactome network perturbations to the formation of pathologic scaffolds termed epichaperomes [72,74,79]. Not to be confused with the chaperome, which refers to the assembly of chaperones and cochaperones [80], epichaperomes form when stressors associated with disease alter the connectivity among chaperome proteins by increasing both interaction strength and number of interactions [72,74,81]. Unlike the short-lived chaperone/cochaperone complexes which act in a one-on-one, dynamic cyclic fashion, aiding protein folding, degradation, or disaggregation (Fig. 2A, right) [81], epichaperomes act as scaffolding platforms to mediate how thousands of proteins improperly interact and organize inside cells, aberrantly affecting cellular phenotypes (Fig. 2A, left) [81]. Analogous to scaffold proteins which play central roles in regulating the spatial–temporal organization of many important protein pathways in cells [82], epichaperome scaffolding platforms offer physical platforms to proteins so that transient interactions, such as most functional PPIs are, can be greatly facilitated in the crowded and heterogeneous environment of the cells. Epichaperomes, through their stable oligomeric structure, have an advantage over single-protein scaffolds due to increased quaternary structure diversity, enabling new connection formation to alter function and increase versatility [78,81]. Stabilization of the epichaperome complexes by reducing the dynamic nature of interactions among the constituent proteins may further increase efficiency.
Fig. 2.
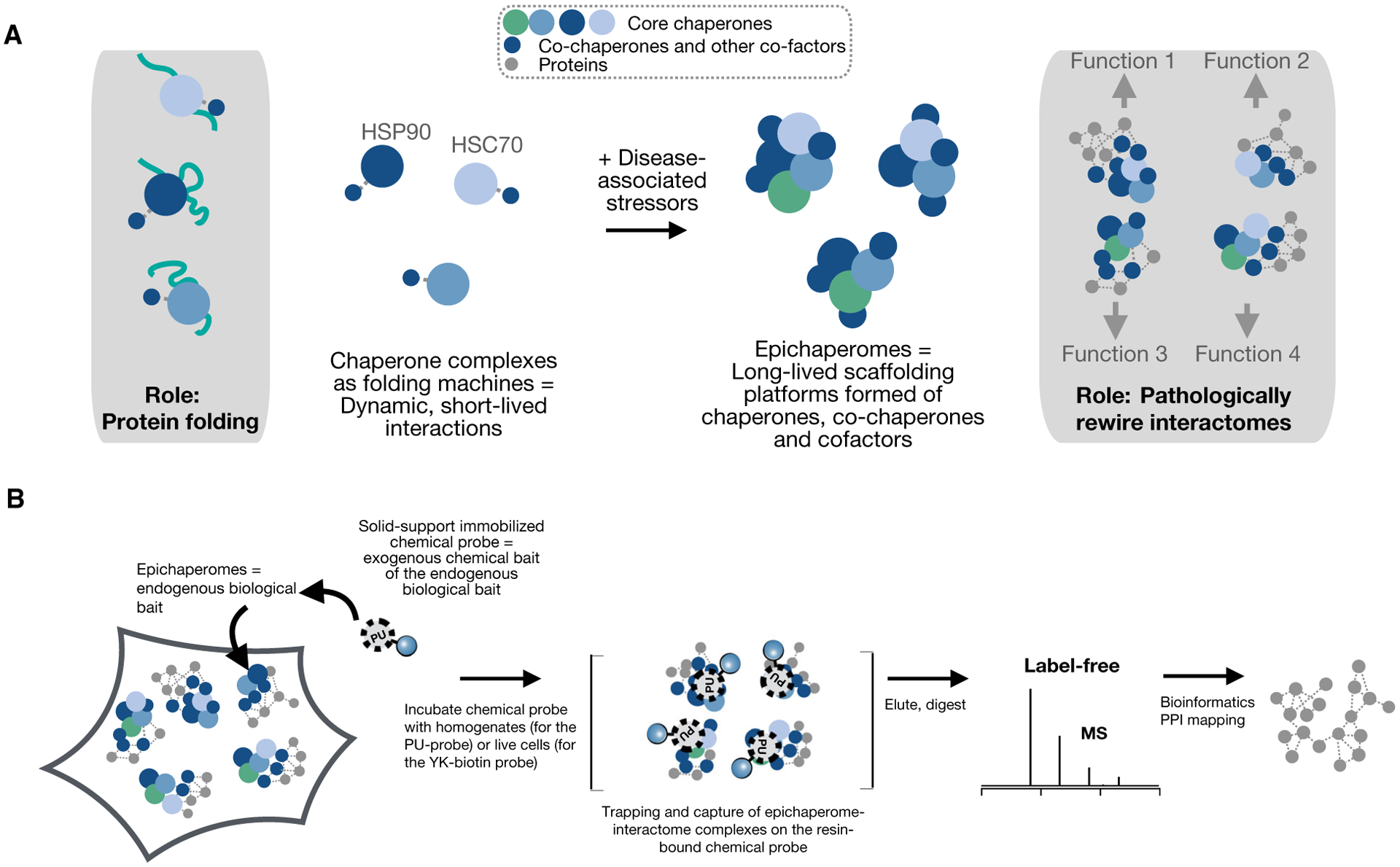
(A) The chaperome is an assembly of chaperones and cochaperones (left panel). Their effects are executed through short-lived chaperome complexes and in a one-to-one, dynamic cyclic fashion, aiding protein folding, degradation, or disaggregation [81]. Stressors associated with disease increase connectivity among chaperome proteins to form stable hetero-oligomeric complexes termed “epichaperomes” (right panel) [72,74,81]. These chaperome pools do not act in protein folding and degradation, but rather as multimolecular scaffolding platforms that pathologically remodel cellular processes [81]. (B) Epichaperomics is an affinity purification technique. Chemical probes that bind key epichaperome components and trap individual epichaperomes bound to their interacting proteins are used to capture and isolate these complexes thus retaining interactions through subsequent isolation steps and enabling their unbiased identification by MS. A bioinformatics pipeline was developed to derive the context-specific interactome maps from the resulting MS datasets [72]. See also Figs 3 and 4. HSP90, heat-shock protein 90 (HSP90α and HSP90β isoforms, encoded by the HSP90AA1 and HSP90AB1 genes, respectively). HSC70, heat-shock cognate 70 protein encoded by the HSPA8 gene.
The chaperones heat-shock protein 90 (HSP90) and heat-shock cognate 70 (HSC70) nucleate (i.e., are at the epicenter of) these epichaperome scaffolds [72,74,78,79]. It should be noted that whereas HSP90 and HSC70 are abundant proteins, averaging each 2–3% of the total protein mass, and found in all cells in the human body, the fraction of HSP90 and HSC70 incorporated into epichaperomes is in contrast minor and localized to diseased cells and tissues [72,80,81,83]. Thus, chaperone proteins safeguard how proteins get made and ensure cellular activities are coordinated properly. Conversely, epichaperomes change how thousands of proteins in the cell interact with each other. In turn, this causes proteins to improperly organize, aberrantly affecting cellular phenotypes. Therefore, HSP90 when acting as a chaperone, folds proteins through dynamic short-lived complexes with cochaperones. Importantly, this role is distinct from HSP90 when part of the epichaperome, whereby HSP90 is stabilized in this context in multiple oligomeric structures with numerous chaperones and cochaperones, and these multimeric structures act as scaffold for remodeling PPIs.
Epichaperomes therefore are pathologic chaperome-containing scaffolding platforms that rewire interactomes in disease. These structures provide a backbone upon which interactome networks become aberrantly reorganized under disease-related stressor conditions, and thus provide handles for direct access to PPI perturbations in native biological systems. In this sense, epichaperomes are not only effectors of PPI changes in disease but also sensors of pathologic alterations [79]. Therefore, capturing epichaperomes and the proteome at large negatively impacted by these critical scaffolds provides direct access to interactome perturbations and to the functional outcome of such changes in native biological systems (Figs 2 and 4).
Fig. 4.
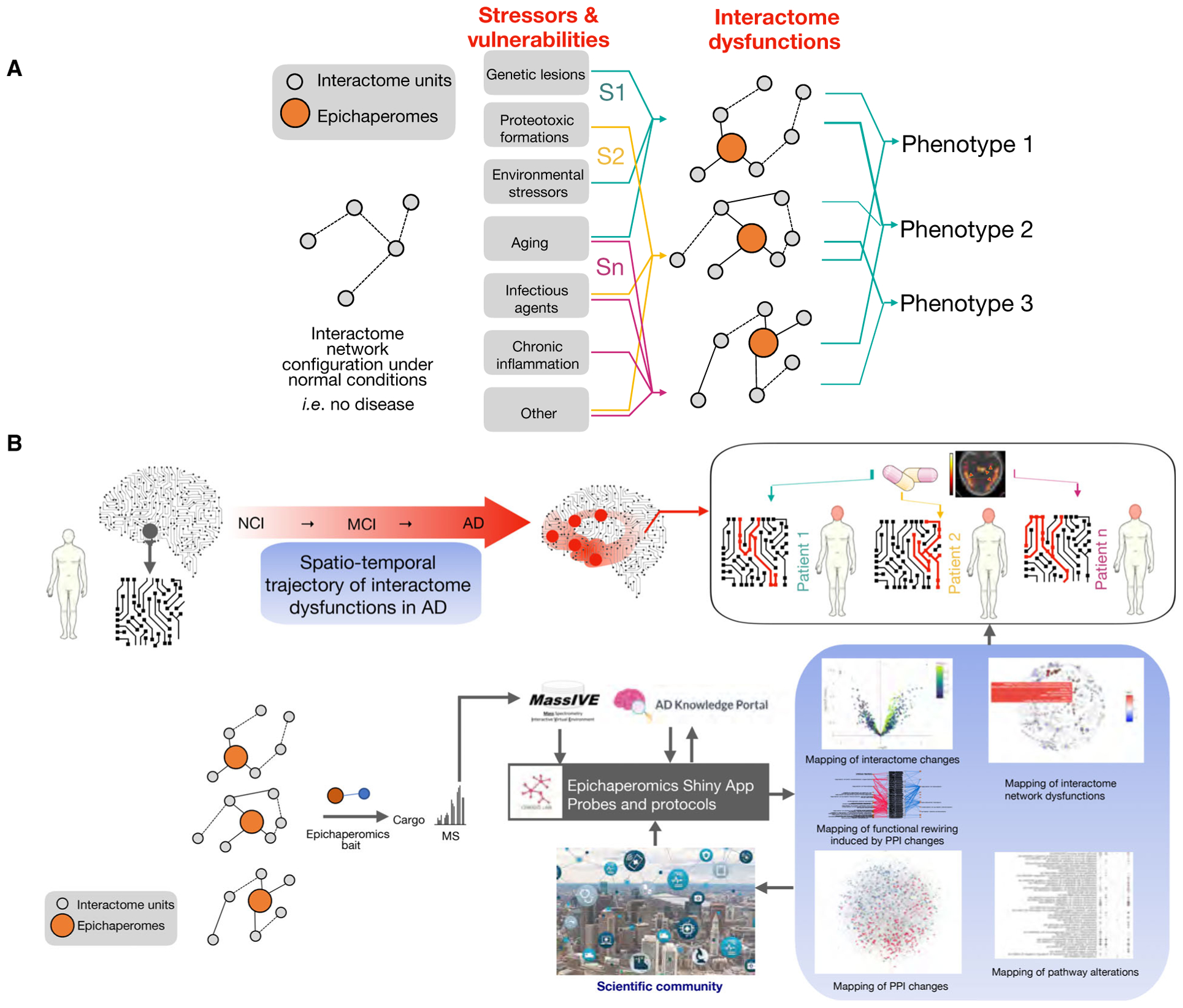
(A) AD stressors induce proteome-wide interactome network dysfunctions through maladaptive epichaperomes. Epichaperomics is positioned to be a new approach to track and study interactome networks across the AD spectrum and provide unprecedented information on molecular mechanisms by which stressors influence phenotypes. (B) Investigating the trajectory of epichaperome-mediated interactome dysfunctions may reveal not only defects within intrinsic neuronal proteins and protein pathways but also connectome disruptions in the intrinsic network connectivity of cells and of brain circuits. By applying epichaperomics to well-characterized brains with available patient-specific genetic, clinical, and pathologic measures, we expect to find clues on specific dysfunctions impacted by these stressors that will lead to novel insights into stressor–phenotype relationships. To make epichaperomics datasets available to and readable by the scientific community, in addition to depositing raw data and analytics into free-access portals such as MassIVE and AD Knowledge Portal Synapse (https://adknowledgeportal.synapse.org/), a web-based user-interface “Epichaperomics shiny app” is envisioned. Its role would be to facilitate data processing and visualization by scientists with or without a bioinformatics background and expertise. We posit a whole new treatment paradigm may open and provide a previously unavailable precision medicine approach to AD by understanding and targeting the interactome.
To enable system-level evaluation of proteome-wide changes in PPIs enabled by epichaperomes, an ‘omics platform was developed called chemical chaperomics or epichaperomics [72] (Fig. 2B). Epichaperomics is an affinity purification method invented by the Chiosis laboratory using epichaperomes as bait to capture the disease promoting complement of proteins and query their disease-specific interactions [72,74,79]. In turn, these “biological” baits are captured by chemical probes that trap epichaperomes bound to their interactome, thus retaining interactions through subsequent isolation steps and enabling their unbiased identification by MS [84–86]. Chemical probes that interact with either HSP90 (e.g., PU beads) or HSC70 (e.g., YK-biotin) preferentially when these chaperones are part of the epichaperomes have been developed [74,80,81,83]. PU beads are used for homogenates [72], whereas YK-biotin, being a cell-permeable probe, can be used for both tissue homogenates and live cells [85]. Because differences in proteins amounts between epichaperomics probes and control probes or between the specimens under analysis have always been large, all epichaperomics applications have used label-free quantification methods [71,73,78]. For experiments that compare many different samples or when sample amounts are small, we recommend a multiplex stable isotope labeling method such as tandem mass tags (TMT) for relative quantitation, or data-independent acquisition to minimize the problem of missing values between samples [87,88].
Epichaperomic profiling in cancer [74], Parkinson’s disease [79], and AD [72] robustly demonstrated this approach provides insights into human disease biology unavailable through other ‘omics platforms and derived datasets, thanks to these important advantages:
Epichaperomics intrinsically detects causal functional changes in the interactome induced by stressors and provides PPI changes likely to be drivers of disease-causing phenotypes [79].
Unlike classical methods that use baits for specific proteins to isolate their interactome, the method is based on the epichaperomes themselves being the “bait” [72,79]. The chaperome reorganizes into epichaperomes under pathologic stress [81]. By isolating these epichaperomes and their interactomes with proven chemical baits, one directly “senses” aberrant changes in protein connectivity at the proteome-wide level [72,74,79,81].
By eliminating the need for exogenous introduction of tagged protein as bait, epichaperomics is applicable for exploring native cellular states [72,74,79,81]. This makes epichaperomics ideal for the analysis of disease from cells to tissues including primary specimens, immune-system associated cell populations, postmortem brain tissues, biopsies, and relevant animal models and cultured cells.
Chemical baits that select, enrich, and trap epichaperome–proteome interactions [78,85,89] enable robust identification of a dynamic range of proteins, increasing the likelihood that both low copy proteins and weak interactions are detected as shown to be the case in postmortem AD and nondemented control samples [72].
Epichaperomics therefore provides an unbiased view of (a) proteome-wide interactome changes (including the number and nature of PPIs) inherent to each phenotype and (b) the functional output of such connections (i.e., how are PPI changes executed and what is their negative functional impact) [72,74]. This platform is complementary to current ‘omics approaches, and its application we believe will aid the scientific community in defining, understanding, and controlling interactome networks of complex diseases. Epichaperomics is also intrinsically a user-friendly ‘omics platform that without complex computational inference, elucidates elusive genotype–phenotype relationships [72]. An open resource may be therefore envisioned whereby a computational pipeline could analyze epichaperomic datasets and associate these observations with additional data from ‘omics platforms as well as clinical data, for example, cognitive and neuropathological data, in the context of AD. Members of the scientific community—with or without computational background—could access freely and without restriction this resource to perform interactome network analyzes of interest.
As with any methods, the technique has its limitations. Dedicated chemical probes are needed and instrumental for epichaperomic studies but so far are limited to a handful of probes [74,78,79,83]. These, such as PU-H71 (the chemical attached to the PU beads) [72,74] and PU-WS13 (the chemical linked to biotin in the PU-WS13-beads) [78], act via binding to HSP90 or its paralog glucose-regulated protein 94 (GRP94), respectively, when these chaperones are incorporated into the epichaperomes. Chaperome composition in epichaperomes is however, context-and stressor-dependent [72,74,78,79]. For example, HSP60 recruitment into HSP90-containing epichaperomes is enhanced under toxic stressors in dopaminergic neurons and in certain cancer cells [74,79], whereas HSC70 recruitment is widespread in cancer and observed also in AD and Parkinson’s disease [72,74,79]. What remains an unmet need is a comprehensive chemical probe toolbox that specifically binds to and traps key chaperones specifically in the context of epichaperomes. Being an affinity purification method, abundant contaminant proteins may constitute a non-negligible background in epichaperomics that could interfere with data interpretation [90]. To overcome these potential shortcomings, control beads, containing an epichaperome-inactive chemical, were also developed [74]. These can be used either to preclear the homogenates of “resin-sticky” proteins or used as a control bait in epichaperomics studies [72,79]. As with any MS associated method, the output of epichaperomics is datasets that include the list of detected proteins and the absolute or relative abundance of the proteins across all samples for each experimental run [72,74,79]. Being identified as interactors of epichaperomes, these proteins are likely to be part of disease modules, thus enabling high confidence disease-specific context determinations of molecular functions (see Fig. 1). This being said, epichaperomics does not indicate which proteins are directly interacting with each other and bioinformatic inference, not direct physical experiments, is used to build the proteome-wide interactome maps. In most cases, it is not important to know the direct binary interactions at the molecular or quantitative level, only which maladaptive pathways are stabilized which can be learned from the interactome data and bioinformatics, with further evidence based on functional assays and outcome assessments, as reported [72,74,79]. If knowing the nature and identity of binary interactions is of interest, epichaperomics could be combined with cross-linking or proximity labeling experiments followed by MS. To date, epichaperomic applications have used label-free quantification methods such as spectral counting or spectrometric signal intensity to measure the protein abundance in individual samples [72,74,79]. It remains to be established whether improvements in sensitivity or MS run time, if needed, can be achieved by the incorporation of TMT labeling or data-independent acquisition techniques.
Interactome network vulnerabilities identified by epichaperomics
To appreciate information derived from epichaperomics, we review a recent study where the epichaperomic platform was applied to postmortem human brains, induced pluripotent stem cell (iPSC)-derived neurons of familial AD, transgenic mouse brains, and cellular models of human tau toxicity [72]. The resulting MS files, a total of 647 raw files and peak files, were deposited in MassIVE (https://massive.ucsd.edu/). Bioinformatic pathway analyses accompanying these studies were deposited in GitHub (https://github.com/chiosislab/Chaperomics_AD_2019) as Cytoscape files. The scientific community has free access to these files, and therefore to the identity of all proteins, pathways and their dysfunctions, as identified under each experimental condition.
A novel layer of information derived from epichaperomics in this study is the full extent of the proteome whose interactions and interaction partners change in AD. Specifically, we analyzed global interactome networks of those diagnosed with AD dementia and age-matched individuals with no cognitive impairment (NCI). We observed approximately 62% of the nodes (in the context of PPI networks, nodes are proteins) and 98.5% of the edges (i.e., an edge is the connection between two proteins) significantly altered in AD compared to the inherent physiological organization of the proteome in NCI brains. New connections were formed in AD whereas many normal connections were lost, whereby 942 proteins lost connection partner(s) and 1191 proteins formed new interactions in the transition from NCI to AD [72]. These analyses performed in sporadic AD brains indicate that despite the intrinsically heterogeneous disease (i.e., pathology, cell composition, underlying disease cause with each patient presenting disease caused by distinct combination of genetic, epigenetic, and environmental factors), these cases have one commonality—they all present global interactome disturbances mediated through epichaperomes [72].
Another layer of information gained through epichaperomics is the functional outcome of the large proteome connectivity reorganization executed under specific stressor conditions (Fig. 3). For example, we performed a Reactome pathway analysis on differentially connected (DC) proteins from pairwise comparisons between brains from AD versus NCI subjects (i.e., stressors are complex such as aging, genetics, environmental factors, and others); APP duplication-expressing iPSC-derived neurons versus wild-type APP-expressing iPSC-derived neurons (i.e., the stressor is genetic), PS19 transgenic mouse brains versus non-transgene expressing mice (i.e., stressor is proteotoxic such as produced by mutant tau overexpression), and N2a cells overexpressing human tau versus N2a cells with vector only (i.e., stressor is proteotoxic produced by human tau overexpression). For each stressor condition, we found little overlap between the identified interactomes, which is anticipated for these distinct disease models [72] (Fig. 3, Venn diagram). Conversely, and despite the relatively small sample size, we discovered several interactome network dysfunctions that were commonly shared among all stressor conditions (Fig. 3, Reactome functional mapping). For example, all stressors had a feature in common, in that they all created imbalances in protein pathways important for synaptic plasticity [72]. Therefore, synaptic plasticity is a pervasive interactome vulnerability of brain cells to stressors, with each stressor impacting the connectivity of both overlapping and distinct proteins that map to a specific protein pathway [72].
Fig. 3.
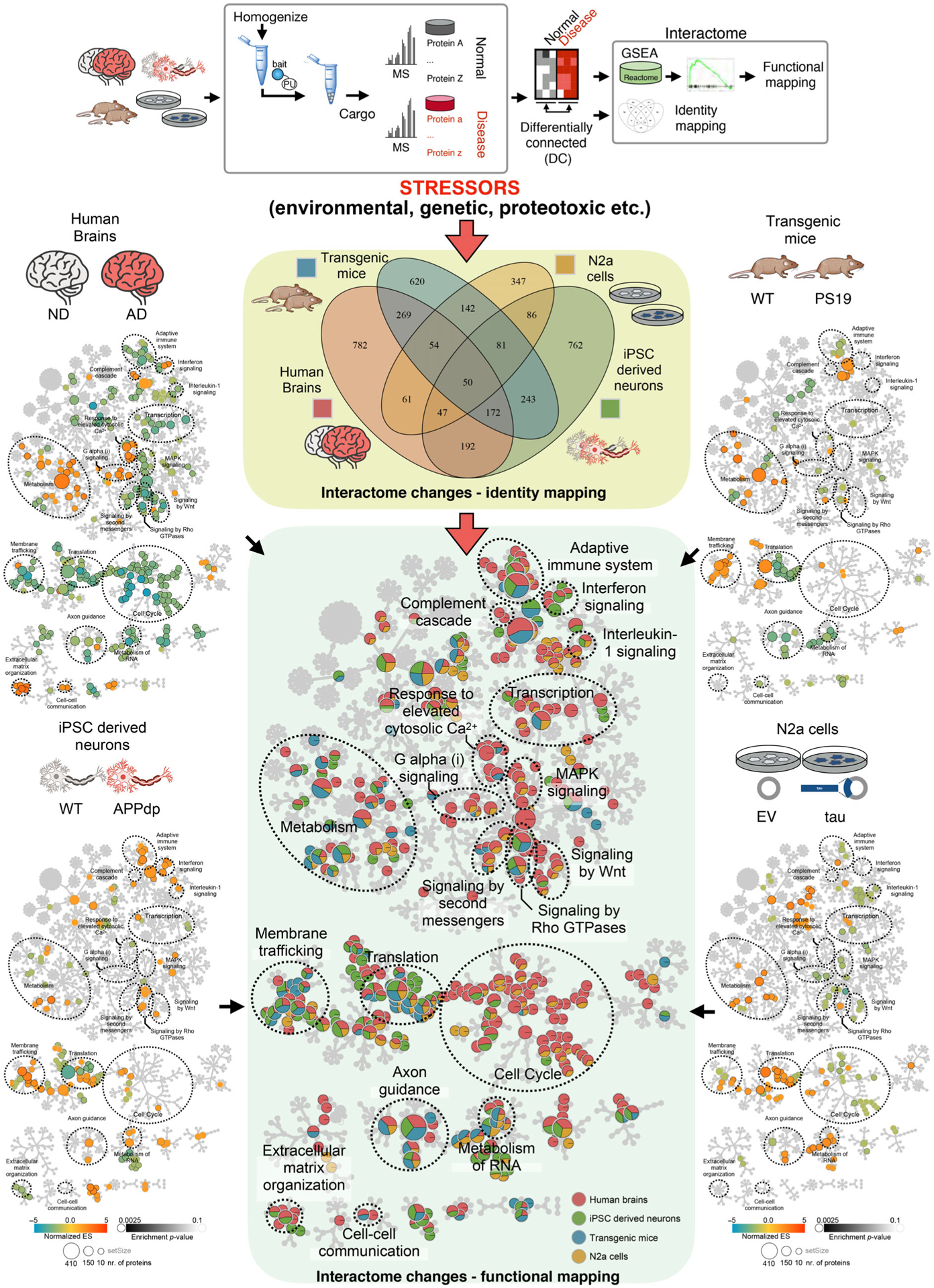
Interactome changes as identified by epichaperomics following individual AD- and AD-relevant model stressor conditions via Venn diagram and Reactome pathway enrichment analyses. In the Venn diagram (center), each circle represents the number of proteins whose connectivity is significantly affected by each stressor. In the Reactome maps, generated in Cytoscape, each circle represents a function (i.e., a protein pathway). The center Reactome map is a merged map depiction of all four individual stressor conditions (left- and right-side panels). If the circle is divided into blue, yellow, red and green segments, it means all four stressors (or stressors characteristic of each condition) mediate imbalances in the select pathway. An exclusively red circle indicates the pathway alteration is AD-specific. Key: human brains (sporadic late-onset AD vs NCI), iPSC-derived neurons (APP duplication vs WT), transgenic mouse brains (PS19 vs WT), and cellular models of human tau toxicity (N2a cells overexpressing human tau vs N2a cells with vector only). Figure adapted from [72].
In humans, epichaperomics found that several synaptic pathways in common to all AD patients were negatively impacted in the transition from NCI to AD dementia including signaling networks such as “signaling by second messenger,” “Galpha(i) signaling,” “signaling by Rho GTPases,” “signaling by Wnt,” “response to elevated cytosolic Ca2+,” “MAPK signaling,” adhesion-regulatory networks such as “extracellular matrix organization” and “cell–cell communication,” as well as protein translation-related networks [72]. Both short-term memories therefore, which rely on post-translational modification of pre-existing synaptic proteins via the above-mentioned signaling networks, and more persistent long-term memories, which require the production of new proteins, are vulnerable to, and negatively affected by global protein connectivity changes.
In addition to synaptic plasticity, metabolic processes, inflammation, and changes in immune response and adaptation, cell cycle re-entry, cell communication processes, and other key processes associated with AD were dysregulated through protein connectivity changes (Fig. 3), as discovered via epichaperomics [72]. Thus, and despite intrinsic heterogeneity in individual cases (i.e., pathology, proteotoxic stress, cell composition, underlying disease cause with each patient presenting with a distinct combination of genetic, epigenetic, and environmental risk factors), these sporadic late-onset AD cases and models have one commonality, notably protein networks altered in AD are among the most vulnerable to interactome changes. These results support epichaperomics is a powerful tool to dissect AD biology, as we propose below.
Opportunities for discovery in AD
AD is an irreversible, age-related neurodegenerative brain disorder responsible for the gradual and insidious failure of cognitive function [91,92]. Once considered a late-life dementia [93,94], AD is now recognized as beginning with brain changes decades prior to clinical symptom onset [92,95–99]. Progressing on a spectrum with three stages—that is, the AD spectrum—from no cognitive impairment (NCI), to mild cognitive impairment (MCI), to dementia over 15–20 years, AD dementia is the severe end of a continuum that begins in midlife [98,100–104]. This conceptual shift suggests early intervention in the prodromal (i.e., preclinical) stage offers the best chance of therapeutic success [99,102,105]. The complex causes behind AD and the multifaceted molecular, cellular, and phenotypic events underlying them greatly hinder development of effective diagnostics and treatments. This sobering reality has led to the realization that AD is not a singular disease but an umbrella of disease states. Many environmental stressors contribute to AD, including diabetes, stroke, high blood pressure, and brain injury [106,107]. Genetic risk factors, gender- and age-related changes, can damage vulnerable cells and brain circuitry over decades [108–111]. Diverse environmental factors may precipitate and modify disease initiation and course in concert with these risk factors [112–114]. Moreover, not every neuronal and non-neuronal cell type displays the same vulnerability and level of molecular, cellular, or functional deficits [115–120], making mechanistic studies difficult in admixed tissues without knowing the contribution of individual cell types to vulnerability. This limits the ability to evaluate and discriminate biomarkers to detect disease prior to AD hallmark pathology {e.g., amyloid-beta (Aβ) and tau} onset and hinders development of therapeutic target(s) to account for the multiple interactome dysfunctions contributing to AD initiation and progression.
We propose that epichaperomics is positioned to be a radical new approach to track and potentially intervene within interactome networks across the AD spectrum (Fig. 4). We propose that investigating the trajectory of epichaperome-mediated interactome dysfunctions will reveal not only defects within intrinsic neuronal proteins and protein pathways but also connectome disruptions in the intrinsic network connectivity of neurons and brain circuits. By applying epichaperomics to well-characterized brains with available patient-specific genetic, clinical, and pathologic measures, we expect to find clues on specific dysfunctions impacted by these stressors that will lead to novel insights into stressor–phenotype relationships complementing current ‘omics efforts [121–127]. We foresee understanding longitudinal changes in intracellular and intercellular networks negatively impacting vulnerable brain regions across the AD spectrum. This is one of the highest priority research topics in AD (e.g., NIA and the National Plan to Address Alzheimer’s Disease; www.nia.nih.gov). Both interactome vulnerabilities imparted by epichaperome formation in specific cell populations and the temporal and regional trajectory of pathologic interactome changes could be discovered and exploited for therapeutic purposes. Success in providing information on even one, if not all proposed cell types, would be highly valuable as the pathways, and molecular mechanisms of cellular activity at the different stages of AD remain either largely unknown or controversial. Interactome network dysfunction trajectory and intensity may also depend on or be influenced by genetics, clinical factors, and/or environmental stressors. Intramolecular and intercellular defects caused by specific stressors alone or in combination may differ in some, but not all, protein pathways, despite functionally converging on a specific dysfunction. We posit epichaperome levels and the extent of interactome dysfunctions, as determined by the epichaperomics platform, may predict interactome network dysfunction severity. This may have clear diagnostic implications as it may be measurable and identifiable through imaging using the PU-AD positron emission tomography (PET) assay [72,128]. For an unequivocal AD diagnosis, there is a need for composite neuroimaging biomarkers combining critical information about molecular alterations, amyloid and tau aggregation, structural and functional alterations, and synaptic and cellular degeneration [129,130]. Together, these may provide precision medicine approaches to better characterize, stage, and classify subtypes of dementias and discriminate AD from more benign age-related changes. Different imaging modalities offer complementary information. The spatial distribution of diverse measurements, including interactome dysfunction via epichaperome PET, can also offer valuable information that can be used for tracking and staging within individuals and groups across the AD spectrum.
Conclusions and future directions
Relying on discovering a linear genotype-to-phenotype relationship for each disorder known to mankind is a limited approach to understanding the complex root causes of human disease, especially when evaluating the interplay between internal stressors (e.g., genetic perturbations) and external stressors (e.g., environmental effects). This is especially true considering tissue- and cell-specific vulnerabilities observed in numerous multifactorial diseases, including the well-established selective vulnerability in AD. We posit a realistic approach to understand mechanisms underlying complex disease states is to evaluate cell- and tissue-specific interactome networks. This is an emerging field, and the technology and bioinformatics to yield significant results, especially in the context of deciphering the impact and importance of PPIs, are a moving target. To address this unmet need, we link stressor-derived protein interactome network perturbations to epichaperome formation, which we propose is an innovative method to extract functionally relevant, context-dependent interactomes that can be rigorously employed as a standalone approach. This lead technique can also complement standard ‘omics approaches that are limited to generating lists of presumed candidate genes, encoded proteins, and likely pathways. For proof-of-concept in this opinion piece, we show epichaperomics exposes vulnerabilities used to define protein network dysfunctions across the AD spectrum which were previously unknown or under appreciated. Targeting or controlling the epichaperome for therapeutics is also tenable and may prove to be a transformative strategy for personalized medicine [78,128,131,132], including the context of AD and related dementing illness [72].
A whole new treatment paradigm may open and provide a previously unavailable precision medicine approach to AD [133] by understanding and targeting the interactome [72]. Currently, there is no cure or treatment to delay or stop the onset or progression of AD [92]. To date, the FDA has approved five treatments for AD [92]. Unfortunately, none slow or stop the damage and destruction of neurons or impact AD progression. The major focus of therapeutic targets for AD has been driven by genetic and pathological studies, and directed to the hallmark lesions of amyloid plaques and tau pathology [102,104,134,135]. Treatments targeting Aβ are still being evaluated with largely negative results to date [136–139]. A similar fate may occur for tau immunotherapy [140]. The lack of effective treatment for AD, combined with the projected worldwide increase in AD dementia patients, points to the imminent need for identification and validation of novel therapeutic targets and diagnostics based on core mechanisms of AD. Epichaperomics may identify subgroups of patients that may be considered for early treatment, including epichaperome correction with the PU-AD drug [72], currently in Phase 2 clinical trials [141]. Thus, the combined impact of AD epichaperomics inquiry is of mechanistic, diagnostic, and therapeutic significance. If successful, this platform has the transformative potential to impact the field and individual patient treatment regimens. Epichaperomics could also set a new standard for investigating network dysfunction in neurodegenerative disorders as a means to develop rational therapeutics in the context of a complex multifactorial age-related disorder.
Acknowledgements
This work was supported by NIH grants U01 AG032969, R56 AG061869, R56 AG072599, R01 AG067598, P01 AG014449, P01 AG017617, R01 AG043375, P30 CA008748, R01 NS088627, and R01 NS112144 and the Coins for Alzheimer’s Research, Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research and the Experimental Therapeutics Center of the Memorial Sloan Kettering Cancer Center.
Abbreviations
- AD
Alzheimer’s disease
- AP-MS
affinity purification mass spectrometry
- APP
amyloid-beta precursor protein
- CETSA
cellular thermal shift assay
- DC
differentially connected
- GO
Gene Ontology
- GRP94
glucose-regulated protein 94
- HSC70
heat-shock cognate 70
- HSP90
heat-shock protein 90
- iPSC
induced pluripotent stem cell
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- MS
mass spectrometry
- NCI
no cognitive impairment
- PET
positron emission tomography
- PPI
protein–protein interaction
- PTM
post-translational modification
- TMT
tandem mass tag
- XLMS
cross-linking mass spectrometry
Footnotes
Conflict of interest
Memorial Sloan Kettering Cancer Center holds the intellectual rights to the epichaperome portfolio. G.C. has partial ownership and is a member of the board of directors at Samus Therapeutics Inc, which has licensed this portfolio. G.C. and P.Y. are inventors on the licensed intellectual property. All other authors declare no competing interests.
References
- 1.Cooper DN, Krawczak M, Polychronakos C, Tyler-Smith C & Kehrer-Sawatzki H (2013) Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum Genet 132, 1077–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.MacArthur DG, Balasubramanian S, Frankish A, Huang N, Morris J, Walter K, Jostins L, Habegger L, Pickrell JK, Montgomery SB et al. (2012) A systematic survey of loss-of-function variants in human protein-coding genes. Science 335, 823–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xue Y, Chen Y, Ayub Q, Huang N, Ball EV, Mort M, Phillips AD, Shaw K, Stenson PD, Cooper DN et al. (2012) Deleterious- and disease-allele prevalence in healthy individuals: insights from current predictions, mutation databases, and population-scale resequencing. Am J Hum Genet 91, 1022–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hekselman I & Yeger-Lotem E (2020) Mechanisms of tissue and cell-type specificity in heritable traits and diseases. Nat Rev Genet 21, 137–150. [DOI] [PubMed] [Google Scholar]
- 5.Haigis KM, Cichowski K & Elledge SJ (2019) Tissue-specificity in cancer: the rule, not the exception. Science 363, 1150–1151. [DOI] [PubMed] [Google Scholar]
- 6.Bouhaddou M, Eckhardt M, Chi Naing ZZ, Kim M, Ideker T & Krogan NJ (2019) Mapping the protein-protein and genetic interactions of cancer to guide precision medicine. Curr Opin Genet Dev 54, 110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vidal M, Cusick ME & Barabasi AL (2011) Interactome networks and human disease. Cell 144, 986–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Furlong LI (2013) Human diseases through the lens of network biology. Trends Genet 29, 150–159. [DOI] [PubMed] [Google Scholar]
- 9.Yeger-Lotem E & Sharan R (2015) Human protein interaction networks across tissues and diseases. Front Genet 6, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yadav A, Vidal M & Luck K (2020) Precision medicine - networks to the rescue. Curr Opin Biotechnol 63, 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barabasi AL & Oltvai ZN (2004) Network biology: understanding the cell’s functional organization. Nat Rev Genet 5, 101–113. [DOI] [PubMed] [Google Scholar]
- 12.Bonetta L (2010) Protein-protein interactions: interactome under construction. Nature 468, 851–854. [DOI] [PubMed] [Google Scholar]
- 13.Caldera M, Buphamalai P, Müller F & Menche J (2017) Interactome-based approaches to human disease. Curr Opin Syst Biol 3, 88–94. [Google Scholar]
- 14.Barabasi AL, Gulbahce N & Loscalzo J (2011) Network medicine: a network-based approach to human disease. Nat Rev Genet 12, 56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Las Rivas J & Fontanillo C (2010) Protein-protein interactions essentials: key concepts to building and analyzing interactome networks. PLoS Comput Biol 6, e1000807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garzon JI, Deng L, Murray D, Shapira S, Petrey D & Honig B (2016) A computational interactome and functional annotation for the human proteome. eLife 5, e18715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Capriotti E, Ozturk K & Carter H (2019) Integrating molecular networks with genetic variant interpretation for precision medicine. Wiley Interdiscip Rev Syst Biol Med 11, e1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K et al. (2015) The BioPlex network: a systematic exploration of the human interactome. Cell 162, 425–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luck K, Kim DK, Lambourne L, Spirohn K, Begg BE, Bian W, Brignall R, Cafarelli T, Campos-Laborie FJ, Charloteaux B et al. (2020) A reference map of the human binary protein interactome. Nature 580, 402–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miryala SK, Anbarasu A & Ramaiah S (2018) Discerning molecular interactions: a comprehensive review on biomolecular interaction databases and network analysis tools. Gene 642, 84–94. [DOI] [PubMed] [Google Scholar]
- 21.Bajpai AK, Davuluri S, Tiwary K, Narayanan S, Oguru S, Basavaraju K, Dayalan D, Thirumurugan K & Acharya KK (2019) How helpful are the protein-protein interaction databases and which ones? bioRxiv 566372. [DOI] [PubMed] [Google Scholar]
- 22.Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, Doncheva NT, Legeay M, Fang T, Bork P et al. (2021) The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res 49, D605–D612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang JK, Carlin DE, Yu MK, Zhang W, Kreisberg JF, Tamayo P & Ideker T (2018) Systematic evaluation of molecular networks for discovery of disease genes. Cell Syst 6, 484–495.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cho DY, Kim YA & Przytycka TM (2012) Chapter 5: network biology approach to complex diseases. PLoS Comput Biol 8, e1002820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carter H, Hofree M & Ideker T (2013) Genotype to phenotype via network analysis. Curr Opin Genet Dev 23, 611–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Das J, Fragoza R, Lee HR, Cordero NA, Guo Y, Meyer MJ, Vo TV, Wang X & Yu H (2014) Exploring mechanisms of human disease through structurally resolved protein interactome networks. Mol Biosyst 10, 9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ivanic J, Yu X, Wallqvist A & Reifman J (2009) Influence of protein abundance on high-throughput protein-protein interaction detection. PLoS One 4, e5815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen J, Aronow BJ & Jegga AG (2009) Disease candidate gene identification and prioritization using protein interaction networks. BMC Bioinformatics 10, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rolland T, Tasan M, Charloteaux B, Pevzner SJ, Zhong Q, Sahni N, Yi S, Lemmens I, Fontanillo C, Mosca R et al. (2014) A proteome-scale map of the human interactome network. Cell 159, 1212–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ideker T & Nussinov R (2017) Network approaches and applications in biology. PLoS Comput Biol 13, e1005771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conte F, Fiscon G, Licursi V, Bizzarri D, D’Anto T, Farina L & Paci P (2020) A paradigm shift in medicine: a comprehensive review of network-based approaches. Biochim Biophys Acta Gene Regul Mech 1863, 194416. [DOI] [PubMed] [Google Scholar]
- 32.Faria do Valle Í (2020) Recent advances in network medicine: from disease mechanisms to new treatment strategies. Mult Scler 26, 609–615. [DOI] [PubMed] [Google Scholar]
- 33.Killcoyne S, Carter GW, Smith J & Boyle J (2009) Cytoscape: a community-based framework for network modeling. Methods Mol Biol 563, 219–239. [DOI] [PubMed] [Google Scholar]
- 34.Martens M, Ammar A, Riutta A, Waagmeester A, Slenter DN, Hanspers K, Miller RA, Digles D, Lopes EN, Ehrhart F et al. (2021) WikiPathways: connecting communities. Nucleic Acids Res 49, D613–D621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jassal B, Matthews L, Viteri G, Gong C, Lorente P, Fabregat A, Sidiropoulos K, Cook J, Gillespie M, Haw R et al. (2020) The reactome pathway knowledgebase. Nucleic Acids Res 48, D498–D503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanehisa M, Furumichi M, Sato Y, Ishiguro-Watanabe M & Tanabe M (2021) KEGG: integrating viruses and cellular organisms. Nucleic Acids Res 49, D545–D551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smits AH & Vermeulen M (2016) Characterizing protein-protein interactions using mass spectrometry: challenges and opportunities. Trends Biotechnol 34, 825–834. [DOI] [PubMed] [Google Scholar]
- 38.Greco TM, Kennedy MA & Cristea IM (2020) Proteomic technologies for deciphering local and global protein interactions. Trends Biochem Sci 45, 454–455. [DOI] [PubMed] [Google Scholar]
- 39.Westermarck J, Ivaska J & Corthals GL (2013) Identification of protein interactions involved in cellular signaling. Mol Cell Proteomics 12, 1752–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morris JH, Knudsen GM, Verschueren E, Johnson JR, Cimermancic P, Greninger AL & Pico AR (2014) Affinity purification-mass spectrometry and network analysis to understand protein-protein interactions. Nat Protoc 9, 2539–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Keilhauer EC, Hein MY & Mann M (2015) Accurate protein complex retrieval by affinity enrichment mass spectrometry (AE-MS) rather than affinity purification mass spectrometry (AP-MS). Mol Cell Proteomics 14, 120–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Richards AL, Eckhardt M & Krogan NJ (2021) Mass spectrometry-based protein-protein interaction networks for the study of human diseases. Mol Syst Biol 17, e8792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trinkle-Mulcahy L (2019) Recent advances in proximity-based labeling methods for interactome mapping. F1000Res 8, 135. 10.12688/f1000research.16903.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Samavarchi-Tehrani P, Samson R & Gingras AC (2020) Proximity dependent biotinylation: key enzymes and adaptation to proteomics approaches. Mol Cell Proteomics 19, 757–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roux KJ, Kim DI, Burke B & May DG (2018) BioID: a screen for protein-protein interactions. Curr Protoc Protein Sci 91, 19 23 1–19 23 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hung V, Udeshi ND, Lam SS, Loh KH, Cox KJ, Pedram K, Carr SA & Ting AY (2016) Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2. Nat Protoc 11, 456–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Han S, Li J & Ting AY (2018) Proximity labeling: spatially resolved proteomic mapping for neurobiology. Curr Opin Neurobiol 50, 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu Y, Fan X & Hu Y (2021) In vivo interactome profiling by enzyme-catalyzed proximity labeling. Cell Biosci 11, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prabhu N, Dai L & Nordlund P (2020) CETSA in integrated proteomics studies of cellular processes. Curr Opin Chem Biol 54, 54–62. [DOI] [PubMed] [Google Scholar]
- 50.Dai L, Zhao T, Bisteau X, Sun W, Prabhu N, Lim YT, Sobota RM, Kaldis P & Nordlund P (2018) Modulation of protein-interaction states through the cell cycle. Cell 173, 1481–1494.e13. [DOI] [PubMed] [Google Scholar]
- 51.O’Reilly FJ & Rappsilber J (2018) Cross-linking mass spectrometry: methods and applications in structural, molecular and systems biology. Nat Struct Mol Biol 25, 1000–1008. [DOI] [PubMed] [Google Scholar]
- 52.Yu C & Huang L (2018) Cross-linking mass spectrometry: an emerging technology for interactomics and structural biology. Anal Chem 90, 144–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koshiba T & Kosako H (2020) Mass spectrometry-based methods for analysing the mitochondrial interactome in mammalian cells. J Biochem 167, 225–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gonzalez-Lozano MA, Koopmans F, Sullivan PF, Protze J, Krause G, Verhage M, Li KW, Liu F & Smit AB (2020) Stitching the synapse: cross-linking mass spectrometry into resolving synaptic protein interactions. Sci Adv 6, eaax5783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chavez JD, Lee CF, Caudal A, Keller A, Tian R & Bruce JE (2018) Chemical crosslinking mass spectrometry analysis of protein conformations and supercomplexes in heart tissue. Cell Syst 6, 136–141.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lepvrier E, Doigneaux C, Moullintraffort L, Nazabal A & Garnier C (2014) Optimized protocol for protein macrocomplexes stabilization using the EDC, 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide, zero-length cross-linker. Anal Chem 86, 10524–10530. [DOI] [PubMed] [Google Scholar]
- 57.Wickramathilaka MP & Tao BY (2019) Characterization of covalent crosslinking strategies for synthesizing DNA-based bioconjugates. J Biol Eng 13, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fritzsche R, Ihling CH, Gotze M & Sinz A (2012) Optimizing the enrichment of cross-linked products for mass spectrometric protein analysis. Rapid Commun Mass Spectrom 26, 653–658. [DOI] [PubMed] [Google Scholar]
- 59.Nussinov R, Tsai CJ & Jang H (2019) Protein ensembles link genotype to phenotype. PLoS Comput Biol 15, e1006648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barshir R, Shwartz O, Smoly IY & Yeger-Lotem E (2014) Comparative analysis of human tissue interactomes reveals factors leading to tissue-specific manifestation of hereditary diseases. PLoS Comput Biol 10, e1003632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Greene CS, Krishnan A, Wong AK, Ricciotti E, Zelaya RA, Himmelstein DS, Zhang R, Hartmann BM, Zaslavsky E, Sealfon SC et al. (2015) Understanding multicellular function and disease with human tissue-specific networks. Nat Genet 47, 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sacco F, Silvestri A, Posca D, Pirro S, Gherardini PF, Castagnoli L, Mann M & Cesareni G (2016) Deep proteomics of breast cancer cells reveals that metformin rewires signaling networks away from a pro-growth state. Cell Syst 2, 159–171. [DOI] [PubMed] [Google Scholar]
- 63.Halu A, Wang JG, Iwata H, Mojcher A, Abib AL, Singh SA, Aikawa M & Sharma A (2018) Context-enriched interactome powered by proteomics helps the identification of novel regulators of macrophage activation. eLife 7, e37059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Menche J, Sharma A, Kitsak M, Ghiassian SD, Vidal M, Loscalzo J & Barabasi AL (2015) Disease networks. Uncovering disease-disease relationships through the incomplete interactome. Science 347, 1257601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Geiger T, Cox J & Mann M (2010) Proteomic changes resulting from gene copy number variations in cancer cells. PLoS Genet 6, e1001090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen S, Fragoza R, Klei L, Liu Y, Wang J, Roeder K, Devlin B & Yu H (2018) An interactome perturbation framework prioritizes damaging missense mutations for developmental disorders. Nat Genet 50, 1032–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kamburov A, Lawrence MS, Polak P, Leshchiner I, Lage K, Golub TR, Lander ES & Getz G (2015) Comprehensive assessment of cancer missense mutation clustering in protein structures. Proc Natl Acad Sci USA 112, E5486–E5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Engin HB, Kreisberg JF & Carter H (2016) Structurebased analysis reveals cancer missense mutations target protein interaction interfaces. PLoS One 11, e0152929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Creixell P, Schoof EM, Simpson CD, Longden J, Miller CJ, Lou HJ, Perryman L, Cox TR, Zivanovic N, Palmeri A et al. (2015) Kinome-wide decoding of network-attacking mutations rewiring cancer signaling. Cell 163, 202–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sahni N, Yi S, Taipale M, Fuxman Bass JI, Coulombe-Huntington J, Yang F, Peng J, Weile J, Karras GI, Wang Y et al. (2015) Widespread macromolecular interaction perturbations in human genetic disorders. Cell 161, 647–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Climente-Gonzalez H, Porta-Pardo E, Godzik A & Eyras E (2017) The functional impact of alternative splicing in cancer. Cell Rep 20, 2215–2226. [DOI] [PubMed] [Google Scholar]
- 72.Inda MC, Joshi S, Wang T, Bolaender A, Gandu S, Koren Iii J, Che AY, Taldone T, Yan P, Sun W et al. (2020) The epichaperome is a mediator of toxic hippocampal stress and leads to protein connectivitybased dysfunction. Nat Commun 11, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Harper JW & Bennett EJ (2016) Proteome complexity and the forces that drive proteome imbalance. Nature 537, 328–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rodina A, Wang T, Yan P, Gomes ED, Dunphy MP, Pillarsetty N, Koren J, Gerecitano JF, Taldone T, Zong H et al. (2016) The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature 538, 397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sailer ZR & Harms MJ (2017) Molecular ensembles make evolution unpredictable. Proc Natl Acad Sci USA 114, 11938–11943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Serapian SA & Colombo G (2020) Designing molecular spanners to throw in the protein networks. Chemistry (Easton) 26, 4656–4670. [DOI] [PubMed] [Google Scholar]
- 77.Taggart JC, Zauber H, Selbach M, Li GW & McShane E (2020) Keeping the proportions of protein complex components in check. Cell Syst 10, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yan P, Patel HJ, Sharma S, Corben A, Wang T, Panchal P, Yang C, Sun W, Araujo TL, Rodina A et al. (2020) Molecular stressors engender protein connectivity dysfunction through aberrant N-glycosylation of a chaperone. Cell Rep 31, 107840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kishinevsky S, Wang T, Rodina A, Chung SY, Xu C, Philip J, Taldone T, Joshi S, Alpaugh ML, Bolaender A et al. (2018) HSP90-incorporating chaperome networks as biosensor for disease-related pathways in patient-specific midbrain dopamine neurons. Nat Commun 9, 4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang T, Rodina A, Dunphy MP, Corben A, Modi S, Guzman ML, Gewirth DT & Chiosis G (2019) Chaperome heterogeneity and its implications for cancer study and treatment. J Biol Chem 294, 2162–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Joshi S, Wang T, Araujo TLS, Sharma S, Brodsky JL & Chiosis G (2018) Adapting to stress – chaperome networks in cancer. Nat Rev Cancer 18, 562–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Good MC, Zalatan JG & Lim WA (2011) Scaffold proteins: hubs for controlling the flow of cellular information. Science 332, 680–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Taldone T, Wang T, Rodina A, Pillarsetty NVK, Digwal CS, Sharma S, Yan P, Joshi S, Pagare PP, Bolaender A et al. (2020) A chemical biology approach to the chaperome in cancer-HSP90 and beyond. Cold Spring Harb Perspect Biol 12, a034116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Moulick K, Ahn JH, Zong H, Rodina A, Cerchietti L, Gomes DaGama EM, Caldas-Lopes E, Beebe K, Perna F, Hatzi K et al. (2011) Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat Chem Biol 7, 818–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rodina A, Taldone T, Kang Y, Patel PD, Koren J 3rd, Yan P, DaGama Gomes EM, Yang C, Patel MR, Shrestha L et al. (2014) Affinity purification probes of potential use to investigate the endogenous Hsp70 interactome in cancer. ACS Chem Biol 9, 1698–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shrestha L, Patel HJ & Chiosis G (2016) Chemical tools to investigate mechanisms associated with HSP90 and HSP70 in disease. Cell Chem Biol 23, 158–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Muntel J, Kirkpatrick J, Bruderer R, Huang T, Vitek O, Ori A & Reiter L (2019) Comparison of protein quantification in a complex background by DIA and TMT workflows with fixed instrument time. J Proteome Res 18, 1340–1351. [DOI] [PubMed] [Google Scholar]
- 88.O’Connell JD, Paulo JA, O’Brien JJ & Gygi SP (2018) Proteome-wide evaluation of two common protein quantification methods. J Proteome Res 17, 1934–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Patel PD, Yan P, Seidler PM, Patel HJ, Sun W, Yang C, Que NS, Taldone T, Finotti P, Stephani RA et al. (2013) Paralog-selective Hsp90 inhibitors define tumor-specific regulation of HER2. Nat Chem Biol 9, 677–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mellacheruvu D, Wright Z, Couzens AL, Lambert JP, St-Denis NA, Li T, Miteva YV, Hauri S, Sardiu ME, Low TY et al. (2013) The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat Methods 10, 730–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D & Jones E (2011) Alzheimer’s disease. Lancet 377, 1019–1031. [DOI] [PubMed] [Google Scholar]
- 92.(2020) 2020 Alzheimer’s disease facts and figures. Alzheimers Dement 16, 391–460. [DOI] [PubMed] [Google Scholar]
- 93.Reisberg B, London E, Ferris SH, Anand R & de Leon MJ (1983) Novel pharmacologic approaches to the treatment of senile dementia of the Alzheimer’s type (SDAT). Psychopharmacol Bull 19, 220–225. [PubMed] [Google Scholar]
- 94.Serrano-Pozo A, Frosch MP, Masliah E & Hyman BT (2011) Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 1, a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen RC et al. (2011) The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7, 270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R et al. (2011) The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR Jr, Kaye J, Montine TJ et al. (2011) Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7, 280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Petersen RC (2012) New clinical criteria for the Alzheimer’s disease spectrum. Minn Med 95, 42–45. [PubMed] [Google Scholar]
- 99.Dubois B, Hampel H, Feldman HH, Scheltens P, Aisen P, Andrieu S, Bakardjian H, Benali H, Bertram L, Blennow K et al. (2016) Preclinical Alzheimer’s disease: definition, natural history, and diagnostic criteria. Alzheimers Dement 12, 292–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schneider JA, Arvanitakis Z, Leurgans SE & Bennett DA (2009) The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol 66, 200–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bennett DA & Launer LJ (2012) Longitudinal epidemiologic clinical-pathologic studies of aging and Alzheimer’s disease. Curr Alzheimer Res 9, 617–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mufson EJ, Ikonomovic MD, Counts SE, Perez SE, Malek-Ahmadi M, Scheff SW & Ginsberg SD (2016) Molecular and cellular pathophysiology of preclinical Alzheimer’s disease. Behav Brain Res 311, 54–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS & Schneider JA (2018) Religious orders study and rush memory and aging project. J Alzheimers Dis 64, S161–S189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jellinger KA (2020) Neuropathological assessment of the Alzheimer spectrum. J Neural Transm (Vienna) 127, 1229–1256. [DOI] [PubMed] [Google Scholar]
- 105.Vos SJB & Visser PJ (2018) Preclinical Alzheimer’s disease: implications for refinement of the concept. J Alzheimers Dis 64, S213–S227. [DOI] [PubMed] [Google Scholar]
- 106.Edwards Iii GA, Gamez N, Escobedo G Jr, Calderon O & Moreno-Gonzalez I (2019) Modifiable risk factors for Alzheimer’s disease. Front Aging Neurosci 11, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rochoy M, Rivas V, Chazard E, Decarpentry E, Saudemont G, Hazard PA, Puisieux F, Gautier S & Bordet R (2019) Factors associated with Alzheimer’s disease: an overview of reviews. J Prev Alzheimers Dis 6, 121–134. [DOI] [PubMed] [Google Scholar]
- 108.Altmann A, Tian L, Henderson VW, Greicius MD & Alzheimer’s Disease Neuroimaging Initiative I (2014) Sex modifies the APOE-related risk of developing Alzheimer disease. Ann Neurol 75, 563–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Misra A, Chakrabarti SS & Gambhir IS (2018) New genetic players in late-onset Alzheimer’s disease: findings of genome-wide association studies. Indian J Med Res 148, 135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Oveisgharan S, Arvanitakis Z, Yu L, Farfel J, Schneider JA & Bennett DA (2018) Sex differences in Alzheimer’s disease and common neuropathologies of aging. Acta Neuropathol 136, 887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Toro CA, Zhang L, Cao J & Cai D (2019) Sex differences in Alzheimer’s disease: understanding the molecular impact. Brain Res 1719, 194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Xu W, Tan L, Wang HF, Jiang T, Tan MS, Tan L, Zhao QF, Li JQ, Wang J & Yu JT (2015) Meta-analysis of modifiable risk factors for Alzheimer’s disease. J Neurol Neurosurg Psychiatry 86, 1299–1306. [DOI] [PubMed] [Google Scholar]
- 113.Eid A, Mhatre I & Richardson JR (2019) Gene-environment interactions in Alzheimer’s disease: a potential path to precision medicine. Pharmacol Ther 199, 173–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, Boland A, Vronskaya M, van der Lee SJ, Amlie-Wolf A et al. (2019) Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet 51, 414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ginsberg SD & Che S (2005) Expression profile analysis within the human hippocampus: comparison of CA1 and CA3 pyramidal neurons. J Comp Neurol 487, 107–118. [DOI] [PubMed] [Google Scholar]
- 116.Ginsberg SD, Alldred MJ, Gunnam SM, Schiroli C, Lee SH, Morgello S & Fischer T (2018) Expression profiling suggests microglial impairment in human immunodeficiency virus neuropathogenesis. Ann Neurol 83, 406–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mostafavi S, Gaiteri C, Sullivan SE, White CC, Tasaki S, Xu J, Taga M, Klein HU, Patrick E, Komashko V et al. (2018) A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat Neurosci 21, 811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Canchi S, Raao B, Masliah D, Rosenthal SB, Sasik R, Fisch KM, De Jager PL, Bennett DA & Rissman RA (2019) Integrating gene and protein expression reveals perturbed functional networks in Alzheimer’s disease. Cell Rep 28, 1103–1116.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ginsberg SD, Malek-Ahmadi MH, Alldred MJ, Che S, Elarova I, Chen Y, Jeanneteau F, Kranz TM, Chao MV, Counts SE & et al. (2019) Selective decline of neurotrophin and neurotrophin receptor genes within CA1 pyramidal neurons and hippocampus proper: Correlation with cognitive performance and neuropathology in mild cognitive impairment and Alzheimer’s disease. Hippocampus 29, 422–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang ZT, Zhang C, Wang YJ, Dong Q, Tan L & Yu JT (2020) Selective neuronal vulnerability in Alzheimer’s disease. Ageing Res Rev 62, 101114. [DOI] [PubMed] [Google Scholar]
- 121.De Jager PL, Ma Y, McCabe C, Xu J, Vardarajan BN, Felsky D, Klein HU, White CC, Peters MA, Lodgson B et al. (2018) A multi-omic atlas of the human frontal cortex for aging and Alzheimer’s disease research. Sci Data 5, 180142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sancesario GM & Bernardini S (2018) Alzheimer’s disease in the omics era. Clin Biochem 59, 9–16. [DOI] [PubMed] [Google Scholar]
- 123.Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X et al. (2019) Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Johnson ECB, Dammer EB, Duong DM, Ping L, Zhou M, Yin L, Higginbotham LA, Guajardo A, White B, Troncoso JC et al. (2020) Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat Med 26, 769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nativio R, Lan Y, Donahue G, Sidoli S, Berson A, Srinivasan AR, Shcherbakova O, Amlie-Wolf A, Nie J, Cui X et al. (2020) An integrated multi-omics approach identifies epigenetic alterations associated with Alzheimer’s disease. Nat Genet 52, 1024–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang X, Allen M, Li S, Quicksall ZS, Patel TA, Carnwath TP, Reddy JS, Carrasquillo MM, Lincoln SJ, Nguyen TT et al. (2020) Deciphering cellular transcriptional alterations in Alzheimer’s disease brains. Mol Neurodegener 15, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Neff RA, Wang M, Vatansever S, Guo L, Ming C, Wang Q, Wang E, Horgusluoglu-Moloch E, Song W-M, Li A et al. (2021) Molecular subtyping of Alzheimer’s disease using RNA sequencing data reveals novel mechanisms and targets. Sci Adv 7, eabb5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pillarsetty N, Jhaveri K, Taldone T, Caldas-Lopes E, Punzalan B, Joshi S, Bolaender A, Uddin MM, Rodina A, Yan P et al. (2019) Paradigms for precision medicine in epichaperome cancer therapy. Cancer Cell 36, 559–573.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Marquez F & Yassa MA (2019) Neuroimaging biomarkers for Alzheimer’s disease. Mol Neurodegener 14, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Young PNE, Estarellas M, Coomans E, Srikrishna M, Beaumont H, Maass A, Venkataraman AV, Lissaman R, Jimenez D, Betts MJ et al. (2020) Imaging biomarkers in neurodegeneration: current and future practices. Alzheimers Res Ther 12, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jhaveri KL, Dos Anjos CH, Taldone T, Wang R, Comen E, Fornier M, Bromberg JF, Ma W, Patil S, Rodina A et al. (2020) Measuring tumor epichaperome expression using [(124)I] PU-H71 positron emission tomography as a biomarker of response for PU-H71 plus Nab-Paclitaxel in HER2-negative metastatic breast cancer. JCO Precis Oncol 4, 1414–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sugita M, Wilkes D, Bareja R, Eng K, Nataraj S, Jimenez-Flores R, Yan L, de Leon J, Croyle J, Kaner J et al. (2020) Targeting the epichaperome as an effective precision medicine approach in a novel PML-SYK fusion acute myeloid leukemia. NPJ Precis Oncol 5, 44. 10.1038/s41698-021-00183-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Isaacson RS, Ganzer CA, Hristov H, Hackett K, Caesar E, Cohen R, Kachko R, Melendez-Cabrero J, Rahman A, Scheyer O et al. (2018) The clinical practice of risk reduction for Alzheimer’s disease: a precision medicine approach. Alzheimers Dement 14, 1663–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bennett DA, Yu L & De Jager PL (2014) Building a pipeline to discover and validate novel therapeutic targets and lead compounds for Alzheimer’s disease. Biochem Pharmacol 88, 617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chiang K & Koo EH (2014) Emerging therapeutics for Alzheimer’s disease. Annu Rev Pharmacol Toxicol 54, 381–405. [DOI] [PubMed] [Google Scholar]
- 136.Behl C (2017) Amyloid in Alzheimer’s disease: guilty beyond reasonable doubt? Trends Pharmacol Sci 38, 849–851. [DOI] [PubMed] [Google Scholar]
- 137.Gold M (2017) Phase II clinical trials of anti-amyloid beta antibodies: when is enough, enough? Alzheimers Dement (N Y) 3, 402–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Honig LS, Vellas B, Woodward M, Boada M, Bullock R, Borrie M, Hager K, Andreasen N, Scarpini E, Liu-Seifert H et al. (2018) Trial of solanezumab for mild dementia due to Alzheimer’s disease. N Engl J Med 378, 321–330. [DOI] [PubMed] [Google Scholar]
- 139.Shi J, Sabbagh MN & Vellas B (2020) Alzheimer’s disease beyond amyloid: strategies for future therapeutic interventions. BMJ 371, m3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Mullard A (2021) Failure of first anti-tau antibody in Alzheimer disease highlights risks of history repeating. Nat Rev Drug Discov 20, 3–5. [DOI] [PubMed] [Google Scholar]
- 141.ClinicalTrials.gov Identifier: NCT04311515.
- 142.Huang FK, Zhang G, Lawlor K, Nazarian A, Philip J, Tempst P, Dephoure N & Neubert TA (2017) Deep coverage of global protein expression and phosphorylation in breast tumor cell lines using TMT 10-plex isobaric labeling. J Proteome Res 16, 1121–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]