

Abstract
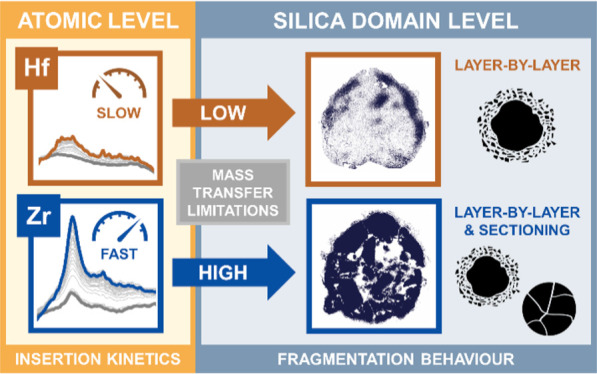
Kinetics-based differences in the early stage fragmentation of two structurally analogous silica-supported hafnocene- and zirconocene-based catalysts were observed during gas-phase ethylene polymerization at low pressures. A combination of focused ion beam-scanning electron microscopy (FIB-SEM) and nanoscale infrared photoinduced force microscopy (IR PiFM) revealed notable differences in the distribution of the support, polymer, and composite phases between the two catalyst materials. By means of time-resolved probe molecule infrared spectroscopy, correlations between this divergence in morphology and the kinetic behavior of the catalysts’ active sites were established. The rate of polymer formation, a property that is inherently related to a catalyst’s kinetics and the applied reaction conditions, ultimately governs mass transfer and thus the degree of homogeneity achieved during support fragmentation. In the absence of strong mass transfer limitations, a layer-by-layer mechanism dominates at the level of the individual catalyst support domains under the given experimental conditions.
Keywords: FIB-SEM, fragmentation, kinetics, mass transfer limitations, metallocenes, PiFM, polymerization, probe molecules
Introduction
In supported olefin polymerization catalysts, the process of support fragmentation is essential for maintaining a high catalyst activity, controlling the morphology of the polymer particles, and achieving a homogeneous distribution of catalyst residues throughout the polymer matrix.1,2 During catalyst particle fragmentation, mechanical forces, which arise from the formation of solid polymer at the active sites of the catalyst, cause the support to disintegrate into smaller fragments. As both polymerization and fragmentation proceed, new active sites are continuously exposed to the reaction environment. Ultimately, mass transfer limitations, due to the buildup of polymer and pore blocking, can be partially overcome and the catalyst activity can be sustained or even increased. Since the early reaction stages are critical in defining the morphology of the final polymer particles, several studies have been dedicated toward understanding the process mechanistically. Two simplified models, namely, the layer-by-layer and sectioning models, are often used to describe experimentally observed fragmentation pathways during early reaction stages.3−9 While the layer-by-layer model involves the progressive fragmentation of the support from its external surface to the interior, the sectioning model is described by a more pronounced and coarser crack formation through the entire catalyst particle, thereby cleaving it into multiple larger fragments. Generally speaking, the morphology, porosity, and mechanical rigidity of the catalyst support all critically affect fragmentation and hence the final polymer morphology and properties.1,3,10−12 The process also strongly depends on the inherent properties of the catalyst’s active sites, the applied reaction conditions, and heat and mass transfer limitations.6,8,9,11,13,14 Thus, with the aim of obtaining a more comprehensive understanding of fragmentation and the key factors behind it, characterization approaches covering both the scale of the single polymerizing particle and the atomic scale of the active sites must be adopted.
In this work, we introduce a multiscale approach that links the morphological evolution of individual catalyst particles to the kinetic behavior of their active sites. Two structurally analogous silica-supported bridged bis-indenyl metallocene catalysts, which were preactivated with methylaluminoxane (MAO) as a cocatalyst (SiO2/MAO/M, M = Hf/Zr) during the catalyst synthesis, were examined. Despite the structural similarity of the hafnocene and zirconocene precursors, the two catalysts yielded substantially different productivities when tested in gas-phase ethylene polymerization, with SiO2/MAO/Zr displaying a more than 40× higher productivity than SiO2/MAO/Hf. The lower activity of hafnocene complexes compared to zirconocene complexes has long been attributed to an inherently lower chain propagation rate, which results from a stronger metal–carbon bond.15−18 More recently, a correlation with the predominantly ionic character of the Hf–C bond was established.19 MAO is also known to be a poor activator for hafnocene complexes as it forms rather stable heterodinuclear compounds with “free” trimethylaluminum (TMA) in equilibrium with its oligomeric part.20,21 The structurally analogous SiO2/MAO/Hf and SiO2/MAO/Zr catalysts, representing two extremes in terms of activity, are thus expected to behave differently during the early stages of the reaction and are therefore ideal candidates for testing our multiscale approach.
The conceptual approach of our work is illustrated in Figure 1. A combination of focused ion beam-scanning electron microscopy (FIB-SEM) and infrared photoinduced force microscopy (IR PiFM) was employed to assess the morphology of individual catalyst particle cross-sections (i.e., the spatial distribution of the support and polymer phases as well as macropores) during the early stages of gas-phase ethylene polymerization (Figure 1A). At the same time, IR spectroscopy in the presence of d-acetonitrile as a probe molecule was used to evaluate the fraction of accessible metal sites and their ability to insert electron-rich molecules into the M–CH3 bond. The latter represents an elementary step in olefin polymerization catalysis (Figure 1B). Finally, by comparing the morphological data obtained at the single-particle level to the compositional and kinetic data collected at the atomic scale, correlations between the catalyst performance and the properties of the active sites were established.
Figure 1.
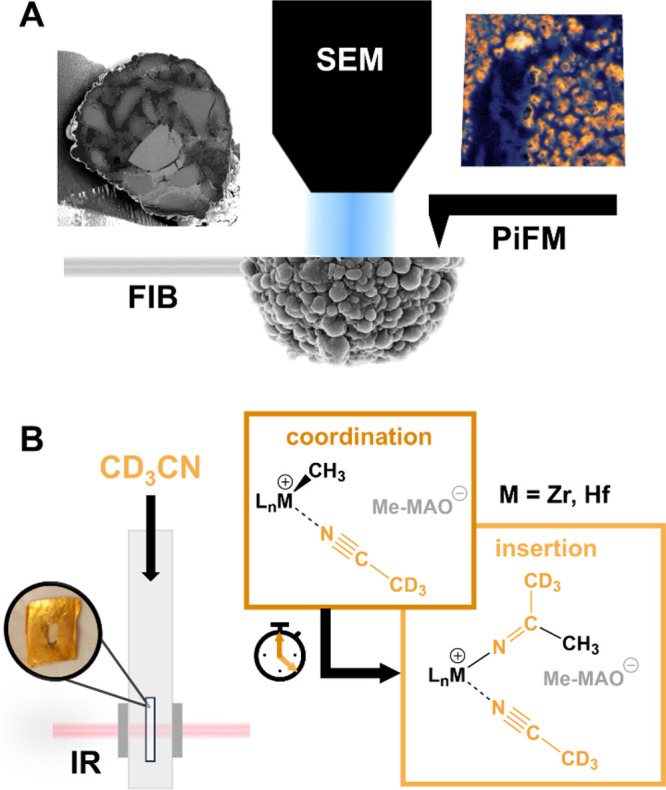
Schematic overview of the multiscale characterization approach applied to the SiO2/MAO/M (M = Hf, Zr) ethylene polymerization catalysts and the information provided by each technique. (A) Correlated focused ion beam-scanning electron microscopy (FIB-SEM) and infrared photoinduced force microscopy (IR PiFM). (B) Time-resolved IR spectroscopy in the presence of d-acetonitrile (d-ACN) as a probe molecule.
Results and Discussion
Assessment of the Catalyst Performance
The SiO2/MAO/Hf and SiO2/MAO/Zr catalysts were prepared following a two-step procedure. First, the 2,2′-biphenylene-bis-2-indenyl MCl2 complexes were suspended in dried toluene and contacted with MAO at an Al:M ratio of 150. Then, polymer-grade SiO2 (D50 = 25.0 μm, precalcined at 600 °C) was added to the solution to form a slurry, followed by solvent removal at room temperature to produce a free-flowing yellow powder. More experimental details are provided in Section S1 of the Supporting Information. It must be noted that the active species (ion-pairs or heterodinuclear complexes with free TMA) were already formed in solution and then impregnated onto the silica support. The final catalysts do not require any additional activation.
The two catalysts were then tested in the gas-phase polymerization of ethylene using pressurized reactors. Under the adopted experimental conditions (T = 87 °C, PC2H4 = 15 bar, and triisobutylaluminum (TiBA) as scavenger; Section S2 and Table S1), SiO2/MAO/Zr displayed a productivity of about 6300 gPE/(gcat·h), while the SiO2/MAO/Hf system displayed a productivity of 150 gPE/(gcat·h) (Table S1). The high temperatures and pressures that were employed during these initial catalytic tests resulted in fast reaction kinetics and high polymer yields (Table S1, see ethylene uptake profiles in Figure S1), thereby impeding any studies on the early stage fragmentation of the two catalyst systems under industrial conditions.
Internal Morphology of the Catalyst Particles
To obtain low polymer yield samples that are representative of the initial stages of ethylene polymerization (i.e., the prepolymerization regime), the two catalysts were prepolymerized in gas phase under mild conditions (i.e., 1.6 bar C2H4 and room temperature; Section S3, Table S2, and Figure S2). Remarkably, the two catalysts displayed very similar productivities under the given prepolymerization conditions after 60 min of ethylene polymerization, contrary to the previously mentioned gas-phase polymerization experiments (SiO2/MAO/Hf, 6.7 g of polyethylene (PE) per gram of catalyst; SiO2/MAO/Zr, 5.8 g of PE per gram of catalyst; Table S2). By means of FIB cutting according to a procedure from the literature,22 the cross-sections of randomly selected catalyst particles were accessed and consequently imaged at a nanometer-scale resolution using SEM and PiFM. The SEM images were segmented for the improved visualization of the fragmentation process (Figures 2, 3, S2, and S3; see the Supporting Information for further details).
Figure 2.

Scanning electron microscopy (SEM) images displaying the cross-sections of a pristine (A and A′), 10 (B and B′) min, 30 (C and C′) min, and 60 min (D and D′) prepolymerized particle of the SiO2/MAO/Hf catalyst material. Images were acquired in backscattered electron mode (BSE; silica support, light gray; polyethylene, dark gray; pores, predominantly black). The close-up images (B′, C′, and D′) were segmented (B″, C″, and D″) to clearly illustrate fragmentation events and the morphology of the particles (the silica support is represented in dark blue, and layer-by-layer and sectioning pathways are indicated by orange and turquoise arrows, respectively). Vertical lines, which originate from the focused ion beam (FIB) cutting procedure, are visible in certain SEM images.
Figure 3.

Scanning electron microscopy (SEM) images displaying the cross-sections of a pristine (A and A′), 10 (B and B′) min, 30 (C and C′) min, and 60 min (D, D′) prepolymerized particle of the SiO2/MAO/Zr catalyst material. Images were acquired in backscattered electron mode (BSE; silica support, light gray; polyethylene, dark gray; pores, predominantly black). The close-up images (B′, C′, and D′) were segmented (B″, C″, and D″) to clearly illustrate fragmentation events and the morphology of the particles (the silica support is represented in dark blue, and layer-by-layer and sectioning pathways are indicated by orange and turquoise arrows, respectively). Vertical lines, which originate from the focused ion beam (FIB) cutting procedure, are visible in certain SEM images.
Figures 2 and 3 show representative cross-sectional SEM images of the SiO2/MAO/Hf and SiO2/MAO/Zr catalyst materials, respectively, at multiple reaction stages (i.e., pristine and after 10, 30, and 60 min of ethylene polymerization). As can be seen in the SEM images of the pristine catalyst particles (Figures 2A and A′ and 3A and A′), the spherical support of the particles is constituted by several larger domains of silica that are bound to each other by a phase with a different density, presumably alkali silicate.3 Furthermore, the catalyst particles feature numerous macropores that are postulated to be partially connected (Figures S3 and S4). This interconnected pore network is highly beneficial for ethylene diffusion into the particle interior, especially in the initial reaction stages.
The segmented SEM images of the 10 min prepolymerized SiO2/MAO/Hf and SiO2/MAO/Zr particle cross-sections (Figures 2B–B″ and 3B–B″) do indeed indicate polymerization activity in the particle interior. In the case of the SiO2/MAO/Hf system, most of the larger constituent silica domains (light gray in the backscattered electron mode, BSE) display mild fragmentation in their outer sphere after 10 min of polymerization (Figures 2B–B″). The smaller silica domains, on the other hand, feature a more advanced degree of fragmentation that penetrates the entire silica domain. Up to this reaction stage, we assume that gas-phase ethylene can freely diffuse into the internal pore space of the particle and polymerize on all exposed external and internal surface areas of the catalyst particle. In general, only a limited amount of PE (dark gray in BSE mode) was formed, and large macropores were still clearly visible in the particle. Under the given reaction conditions, the individual silica domains seem to fragment according to a layer-by-layer mechanism (indicated by an orange arrow in Figure 2B″), provided that ethylene gas can access the inner volume of the particle.
Further polymerization evidently leads to a significant decrease in the macropore volume and more pronounced fragmentation (Figure S3). While the 30 min polymerized catalyst particle (Figures 2C–C″) still features larger isolated fragments of the support in the size range of micrometers, the 60 min polymerized particle (Figures 2D–D″) is characterized by a high degree of homogeneous support fragmentation. In fact, the cross-section shows nanometer-sized fragments that are finely dispersed in the PE matrix (Figure 2D″). Remnants of the support in the outer sphere of the catalyst point to the active involvement of the particle interior in the polymerization process, presumably due to the absence of strong mass transfer limitations (Figure 2D″). While the catalyst predominantly fragmented according to a layer-by-layer mechanism (Figures 2B″, C″, and D″; indicated by orange arrows), local diffusion limitations and stress build-up may, to a limited extent, induce the formation of larger cracks in the support matrix at higher PE yields (i.e., sectioning at the support granulate level, indicated by turquoise arrows in Figure 2C″; also refer to Figure S5).
When comparing the SiO2/MAO/Zr catalyst system to the SiO2/MAO/Hf system, no pronounced differences in catalyst support fragmentation are apparent after 10 min of ethylene polymerization (Figures 3B–B″). Similar to the SiO2/MAO/Hf catalyst material, accessible silica domains begin to fragment according to a layer-by-layer mechanism upon exposure to ethylene (Figure 3B″, indicated by an orange arrow). Indications of a more heterogeneous fragmentation pathway are, however, visible in the later reaction stages (Figure 3C–C″ and D–D″). In contrast to the SiO2/MAO/Hf system, large unfragmented support domains are still visible after 30 min for the SiO2/MAO/Zr system (Figures 3C–C″). Interestingly, the cross-section of the 60 min prepolymerized particle also features sizable pristine support domains along with a thick polymer layer that covers the outer surface of the catalyst particles (Figures 3D–D″ and Figure S4). While the layer-by-layer mechanism dominates the fragmentation in the early reaction stages at both the silica domain and the particle level (indicated by orange arrows in Figure 3), the sectioning mechanism contributes more strongly under the imposed mass transfer limitations at later reaction stages (indicated by turquoise arrows in Figure 3). With the particle practically enveloped in a growing layer of PE yet still active in certain domains, the significant accumulation of stress in the particle becomes inevitable. This pressure buildup induces ruptures and more pronounced crack formation,23 resulting in the cleavage of larger support fragments (indicated by turquoise arrows in Figures 3C″ and D″). It must be noted that the outer layers of the catalyst particles in both catalyst systems seem to fragment gradually according to a layer-by-layer mechanism (as indicated by an orange arrow for SiO2/MAO/Zr in Figure 3D″).
To verify our observations, additional particles of the two 60 min prepolymerized catalysts were investigated (Figure S5). While most of the particles within a particular batch demonstrated the expected fragmentation behavior, a certain degree of interparticle heterogeneity was nevertheless apparent. A strong correlation between the catalyst particle fragmentation and the particle size24−26 as well as that between the dimensions, accessibility, and connectivity of the macropore network is expected. This can account for differences in the fragmentation degree and, possibly, the involvement of a particular fragmentation mechanism.
Chemical Composition at a Sub-20 nm Resolution
In principle, the analysis of the SEM images of both catalysts (Figures 2 and 3) via segmentation into their constituent phases (i.e., PE, silica, and pore space; Figures S3 and S4), should yield the relative composition of each cross-section. However, the analysis is hampered by (i) the limited number of particles that are assessed per reaction stage; (ii) the 2D nature of the data; (iii) the degree of polymerization, which varies between particles (Figure S5); and (iv) the detection limit (resolution) of the applied SEM technique. Indeed, at high degrees of polymerization and fragmentation, such as those in the 30 and 60 min prepolymerized SiO2/MAO/Hf cross-sections, a substantial amount of silica fragments remains undetected due to their high dispersion and limited size (nanometer regime).
A nanoscale imaging technique that is arguably better suited to uncovering structural and compositional heterogeneities in our challenging composite materials is PiFM. In the field of polymer science and related disciplines, PiFM27,28 has been extensively used together with AFM-IR29−33 to characterize multicomponent polymer materials at high spatial resolutions. Inspired by this, we used PiFM to characterize the cross-sections of a single 30 min prepolymerized SiO2/MAO/Hf particle at multiple locations and depths (Figures 4, S6, S7, S8, all cross-section 2; and S9, cross-section 1). Nanoscale infrared and topographic imaging27,28,34−40 yielded information on both the chemical composition and morphology of the sample at a sub-20 nm resolution.
Figure 4.

Local catalyst morphology as recorded by scanning electron microscopy (SEM; A, B, C), chemical composition as imaged by infrared photoinduced force microscopy (IR PiFM) (ν(Si–O), 1050–1030 cm–1, A′–C′ and δ(C–H), 1472–1460 cm–1, A″–C″), and relative mechanical properties as determined by phase shift imaging (A‴–C‴). All data were collected on the cross-sections of a single 30 min prepolymerized SiO2/MAO/Hf particle (same particle as in Figure 2C, characterized at a different depth; measurement areas are indicated by circles in the SEM images). Images in panels B–B‴ were recorded in the same area as those in panels A–A‴ but at a higher magnification.
Figure 4 shows correlated SEM images, IR PiFM, and phase shift maps collected at different locations on the above-mentioned particle cross-section (the image in panel B was recorded in the same area as panel A but at higher magnification). The IR PiFM maps were recorded in noncontact mode36 (amplitude ratio set point of 80%, attractive van der Waals force regime; see Table S3 and the Supporting Information for more details) at characteristic wavelengths for the Si–O stretching vibration41,42 (1050–1030 cm–1, ν, Figure 4A′–C′) and the symmetric C–H bending vibration of the methylene group30−32 (1472–1460 cm–1, δ, Figure 4A″–C″). The IR PiFM data stand in excellent agreement with the corresponding SEM images and yield a remarkably clear differentiation between the silica and PE phases. Multiple larger domains of the silica support have partially fragmented following the layer-by-layer mechanism. Silica fragments smaller than 50 nm, infused with polymer that was presumably formed upon exposure of new active sites to ethylene gas, can be observed in the periphery of these domains (Figures 4A′ and B′). Figure 4C–C″ shows a polymer-rich domain in close proximity to a larger network of cracks in the support (Figure S7, cross-section 1). It is conceivable that the emergence of larger amounts of polymer in this particular area resulted in the accumulation of stress in the support, ultimately inducing the formation of cracks. Alternatively, the buildup of stress over the entire particle may have also contributed. Irrespective of the exact formation pathway, the emergence of large and partially unfilled cracks in the pristine support (Figure S7, cross-section 1) will expose new active sites that will consequently contribute to the polymerization and fragmentation of the particle. A similar showcase for PE-induced crack formation is provided in the Supporting Information (Figure S8, cross-section 2).
Interestingly, the phase shift maps in Figure 4 (Figure 4A‴–C‴) display a striking amount of detail, revealing a complex network of overlapping thread-like PE domains in close vicinity to the formed fragments. In general, the phase channel represents a promising complementary imaging tool43,44 due to its correlation with mechanical material properties. It corresponds to the absolute difference in phase between the external excitation (driver) and the tip response and is governed by the dissipation of energy from the tip to the sample.39 This energy dissipation is strongly affected by mechanical material properties such as stiffness, adhesion, and viscoelasticity.45−47 In general, the set point and drive amplitude of the cantilever must be optimized to obtain qualitative insights into the mechanical properties of a given material.48,49 In our case, phase maps were recorded at a 60% set point and 3 nm drive amplitude to enhance the tip–sample contact36 (hard tapping regime, repulsive interactions dominant; Table S3 and Figure S9).
As is evident from Figures 4A‴–C‴, higher phase shift values were obtained for PE, therefore indicating a lower repulsion of the tip from the polymer-rich domains relative to the silica fragments. This is assumed to be a result of the lower mechanical stiffness and higher viscoelasticity of the PE phase. The overall negative phase shift values result from the net repulsive and dissipative force experienced during heavy tapping (Table S3 and Figure S9).
Point spectra recorded of the silica domains, PE/silica composites (Figure 5), and reference materials (Figures S10 and S11) further helped to unambiguously assign the imaged phases. Due to the high degree of intermixing of the silica and PE, it was not possible to acquire point spectra of pure PE on the prepolymerized catalyst cross-section. Furthermore, the PE phase displayed a high sensitivity toward the incident IR laser beam (Figure S12, see Supporting Information for further details). Despite this, PiFM proved to be suitable for the detection and differentiation of the pristine and fragmented support domains, the bulk polymer phase, and the related composite phases at an unparalleled spatial resolution (<20 nm) and also yielded insights into fragmentation events and the associated release of buried active sites.
Figure 5.

Normalized PiFM spectra of the silica support and of PE/silica composites with different amounts of PE.
Probing the Kinetics of Insertion into the M–CH3 Bond
To elucidate the reasons for the different fragmentation behaviors of the two catalysts and their similar productivities under prepolymerization conditions, we designed a series of FT-IR spectroscopy experiments in the presence of acetonitrile as a probe molecule. FT-IR spectroscopy of adsorbed probes is one of the most sensitive methods to retrieve information on the properties of surface sites and has been largely used to characterize heterogeneous catalysts at a molecular level.50,51 This also applies to heterogeneous olefin polymerization catalysts such as the Phillips52,53 and Ziegler-Natta54−57 catalysts. Owing to its mildly basic character, CO is often used as a molecular probe to differentiate between sites based on their acidity. Previous works demonstrate that CO at 100 K is indeed able to probe Lewis acid sites (LAS) belonging to MAO/TMA in SiO2/MAO/metallocene catalyst materials, while it has more difficulty reaching the metallocene cations.58−60 By being inserted into metal–alkyl bonds, CO can also form acyl species.61 Furthermore, CO has traditionally been used to quantify the number of active sites in polymerization catalysts.62−68
In this work, acetonitrile was chosen as a probe molecule over CO due to its comparatively higher basicity.69−74 Hence, it is better suited to probing metal cations. More importantly, acetonitrile can also be inserted into transition metal alkyl bonds to form aza-alkenylidenes, as demonstrated for different cationic titanium and zirconium complexes.75−78 By using acetonitrile as a probe molecule, the acidity and number of the active sites and their ability to insert electron-rich molecules can be assessed simultaneously. All are critical factors in the context of olefin polymerization. It is important to note that the insertion of acetonitrile into the metal–alkyl bond is not affected by diffusion limitations (related to the buildup of polymer at the particle surface). Therefore, the insertion rates evaluated by this method provide direct insights into the inherent insertion ability of the active sites. To the best of our knowledge, there are no similar reports in literature on the use of acetonitrile as a probe or insertion molecule for silica-supported metallocene-based catalyst materials.
The insertion rate of acetonitrile and thus the reactivity of the SiO2/MAO/Hf and SiO2/MAO/Zr catalyst materials was determined by means of time-resolved FT-IR spectroscopy. A blank experiment was also conducted on a SiO2/MAO material for comparison. Deuterated acetonitrile (d-ACN) was used to overcome band doubling due to Fermi resonance effects.70 As a consequence of its interaction with the LAS sites, the ν(C≡N) vibrational mode is expected to increase in energy with respect to the vibrational mode of the free molecule (2265 cm–1). This shift is proportional to the strength of the Lewis acid–base couple. Moreover, the ν(C=N) vibration of the aza-alkenylidene species is expected to decrease in another well-defined spectral region (1720–1600 cm–1) without overlapping with the bands of chemisorbed d-ACN. Figure 6 shows the sequence of FT-IR spectra for SiO2/MAO, SiO2/MAO/Hf, and SiO2/MAO/Zr upon exposure to d-ACN over a period of 3 h in the spectral region of 2400–1300 cm–1. The insets show a magnification of the same spectra in the 1720–1600 cm–1 range to highlight the bands assigned to the Zr- and Hf aza-alkenylidene species (spectra are reported after the subtraction of the spectrum collected before the introduction of d-ACN).
Figure 6.
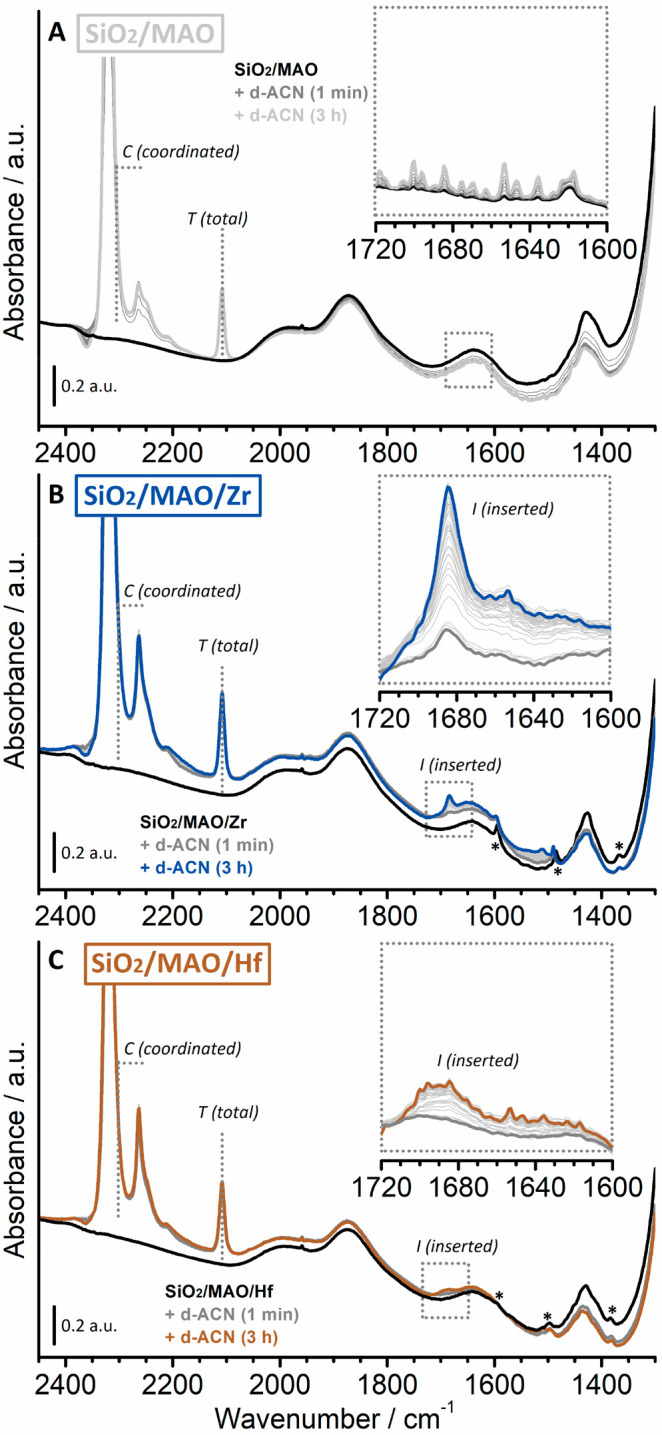
FT-IR spectra recorded of the SiO2/MAO reference material before (black) and after (gray) the interaction with d-ACN at room temperature over a time period of 3 h (light gray) (A). The inset shows the 1720–1600 cm–1 region for the same sequence of FT-IR spectra after the subtraction of the spectrum collected prior to the introduction of d-ACN. Comparable FT-IR spectra for the SiO2/MAO/Zr (B) and SiO2/MAO/Hf (C) catalyst materials. The bands labeled with asterisks (*) are attributed to vibrations of the indenyl ligands in SiO2/MAO/Zr and SiO2/MAO/Hf, respectively.
The initial spectra of the SiO2/MAO, SiO2/MAO/Zr, and SiO2/MAO/Hf catalyst materials (black in Figure 6) are very similar to each other and are dominated by the vibrational features of silica. In addition to these, a limited number of low-intensity bands can be observed. The bands at 1500–1350 cm–1 are assigned to the bending vibrational modes of CH3 groups belonging to MAO (i.e., Al–CH3 species), Si–CH3, or Si–O–Al(CH3)2 species, which might originate from the reaction of MAO/TMA with siloxane bridges at the silica surface. According to Ystenes et al.,79 the δasym(CH3) vibrations of terminal Al–CH3 in MAO are expected to appear at approximately 1435 cm–1, while the corresponding δsym(CH3) vibrational mode is expected to appear around 1300 cm–1 and is hence not detectable due to the broad and intense modes of SiO2, which dominate the spectral region below 1350 cm–1. The δasym(CH3) mode of the −OCH3 species is expected to contribute at around 1470 cm–1 79 but is barely identifiable in our spectra (low intensity and broad band). Finally, the very weak and narrow bands labeled with asterisks are attributed to the vibrations of the indenyl ligands in the activated zirconocene and hafnocene complexes.
In all cases, several intense absorption bands immediately appeared in the 2400–2000 cm–1 region upon introduction of d-ACN, which are discussed as follows:
-
1)
The absorption band at 2108 cm–1 (indicated as T, total in Figure 6) is due to ν(CD3). This band is insensitive to the absorption sites71 and thus not analytically relevant. It will therefore be used as an internal standard to quantify the total amount of d-ACN on the sample, comprising d-ACN coordinated to the LAS sites (belonging to both MAO/TMA and the methylated metallocenes) and physisorbed d-ACN. The concentration of d-ACN in the sample depends on both the equilibrium pressure and the sample mass, the latter being difficult to evaluate due to the complexity of the experimental procedure.
-
2)
In the ν(C≡N) region (2400–2200 cm–1), all spectra are dominated by an intense and symmetric absorption band centered at 2320 cm–1 (i.e., an upward shifted by 55 cm–1 with respect to that of free d-ACN at 2265 cm–1), which is due to the interaction of d-ACN with LAS sites. The band accounts for both the LAS sites of the MAO/TMA species and the Zr or Hf cations (for comparison, d-ACN adsorbed on coordinatively unsaturated Al(III) sites on a triethylaluminum (TEAl)-pretreated SiO2 features a band at 2317 cm–1).80 We expect that the MAO/TMA species are predominantly probed by d-ACN because of their much higher relative content in the two samples (Al/M = 150). Since the associated band rapidly goes out of scale, the total amount of accessible LAS sites was derived from the spectral intensity at 2300 cm–1 for each experiment (indicated as C, coordinated in Figure 6).
-
3)
A second absorption band was observed in the same spectral region at 2265 cm–1 with a shoulder at around 2250 cm–1. The former is ascribed to liquid-like d-ACN and is the only absorption band that decreases in intensity upon degassing (not shown).81 The assignment of the shoulder is more challenging. The low frequency of the ν(C≡N) band suggests a bridging coordination mode, as was observed for other ligands and as suggested for nitriles interacting with two cations in zeolites.82 The appearance of this absorption band indicates the presence of LAS sites that are close enough to each other to be simultaneously complexed by both the nitrogen lone pair and the π-type bonding electrons of the same acetonitrile molecule. A similar absorption band was also observed for d-ACN adsorbed on a Cr(II)/SiO2 Phillips catalyst material activated with TEAl and attributed to a Cr(II)···Al(III) bimetallic species.80
-
4)
In the presence of d-ACN, the SiO2/MAO/Zr (Figure 6B) and SiO2/MAO/Hf (Figure 6C) catalyst materials feature additional weak absorption bands that appear in the 1700–1300 cm–1 spectral region, which slowly grow in intensity over time. In particular, two well-defined absorption bands appear at 1684 and 1690 cm–1 for the SiO2/MAO/Zr and SiO2/MAO/Hf catalyst materials, respectively (labeled as band I, inserted in Figure 6B and C). These absorption bands are attributed to the ν(C=N) of an aza-alkenylidene species that is formed due to the insertion of d-ACN into the Zr–CH3 and Hf–CH3 bonds (Figure 1B). At the same time, all the absorption bands ascribed to the activated metallocene complexes (asterisks) were perturbed. For example, the absorption bands of SiO2/MAO/Zr at 1593 and 1485 cm–1 are upward shifted by a few inverse centimeters, while the absorption band at 1370 cm–1 is lower in intensity. Altogether, this suggests that the coordination modes of the indenyl ligand are changing concomitantly to the coordination of d-ACN to the Zr or Hf cation and its subsequent insertion into the metal–alkyl bond.
In fact, the two absorption bands at 1684 and 1690 cm–1, which are ascribed to the Zr and Hf aza-alkenylidene species respectively, grow at different rates. A significantly faster increase was observed for the SiO2/MAO/Zr system in comparison to that for the SiO2/MAO/Hf system. Interestingly, the rate of insertion of d-ACN in the M–CH3 bond correlates well with the catalyst productivity in ethylene polymerization, as determined by the previously mentioned gas-phase experiments (15 bar ethylene, 87 °C), and not with those determined from the relatively mild prepolymerization experiments. Based on the literature,19−21 our experimental observations may be explained by both: (1) a lower number of active species in the SiO2/MAO/Hf catalyst material due to the formation of stable heterodinuclear compounds with TMA and (2) a different inherent insertion ability of the electron-rich d-ACN in the M–CH3 bonds in the two catalysts.
To clarify the reasons behind the different behaviors of the two structurally analogous catalysts, a series of FT-IR spectroscopy experiments with different d-ACN concentrations, similar to those discussed in Figure 6, were conducted. In total, three experiments were performed for each catalyst (high, medium and low d-ACN concentration). Some of the experiments were repeated to verify the reproducibility of the results. In all experiments, the intensities of the bands T (total amount of d-ACN), C (coordinated d-ACN), and I (inserted d-ACN) were monitored as a function of time (Figure 7, after normalization to the optical pellet thickness and rescaling to the Zr (low) experiment for comparison).
Figure 7.

Evolution of the intensities of the FT-IR absorption bands T (total amount of d-ACN), C (coordinated d-ACN), and I (inserted d-ACN) as a function of time, which were derived from a series of experiments in which SiO2/MAO/Zr (top) and SiO2/MAO/Hf (bottom) were treated with d-ACN at three different concentrations (high, medium, and low). The data were normalized to the optical thickness of the pellets and rescaled with respect to the Zr (Low) experiment for comparison.
For both the SiO2/MAO/Zr system and the SiO2/MAO/Hf system, the T and C bands almost immediately reach their maximum intensity (left and middle in Figure 7, respectively), irrespective of the d-ACN concentration. Based on this, we conclude that there are no intrinsic diffusion limitations. The C bands, on the other hand, slightly decrease in intensity over time due to the gradual insertion of coordinated d-ACN into the M–CH3 bond. The maximum intensities of the T and C bands were determined by extrapolating the linear part at time zero (t = 0) and are reported in Figure S13. In contrast to these, band I (right in Figure 7) grew at an almost constant rate after the first 30 min and did not saturate even after 3 h of reaction. This observation is in agreement with the very slow insertion kinetics of d-ACN in other zirconocene and hafnocene complexes, as reported in the literature.75−78 The insertion rate was derived from the slope of a linear fit that was applied to the curves and can also be found in Figure S13.
For the SiO2/MAO/Zr system, the amount of coordinated d-ACN was found to depend on the total d-ACN concentration (Figures 7 and S12): The higher the concentration or pressure of the electron-rich probe, the higher the fraction of the sites able to coordinate it (comprising both the LAS sites of MAO/TMA and the zirconocene cations). This correlation, however, was not linear. A threefold increase in the total concentration (from T1 to T2) led to twice the amount of coordinated d-ACN (from C1 to C2), while a further twofold increase (from T2 to T3) only resulted in 1.6× the number of coordinated species (from C2 to C3). Interestingly, the d-ACN insertion rates scale perfectly with the fraction of sites able to coordinate d-ACN, i.e., I increases by a factor of 2 from I1 to I2 and by a factor of 1.7 from I2 to I3.
Contrary to the SiO2/MAO/Zr system, the intensities of the three bands T, C, and I in the SiO2/MAO/Hf system rapidly reached a saturation level. Moreover, for comparable d-ACN total concentrations (T), the amount of coordinated d-ACN (C) was lower for SiO2/MAO/Hf than for SiO2/MAO/Zr. This difference became more pronounced at higher d-ACN concentrations. The data stand in agreement with the reported detrimental effect of MAO/TMA on hafnocene complexes relative to comparable zirconocene complexes.20,21 However, this only partly explains the lower insertion rate for the SiO2/MAO/Hf system. A comparison of the experiments performed on the two catalysts at low d-ACN concentrations (T1) revealed that the number of sites coordinating d-ACN (C1) in the SiO2/MAO/Hf system corresponds to 90% of those found in the SiO2/MAO/Zr system, while the insertion rate (I1) is only 75% of that observed for the SiO2/MAO/Zr system. These findings indicate that the insertion kinetics of d-ACN in the SiO2/MAO/Hf system are inherently slower than in the SiO2/MAO/Zr system. As recently suggested,19 this is explained by the more pronounced ionic character of the Hf–C bond compared to the Zr–C bond and, consequently, the larger enthalpic contribution to the activation barrier for Hf–C bond cleavage.
As a final comment, the limited amount of d-ACN that can adsorb on SiO2/MAO/Hf (i.e., T and C signals do not increase significantly when moving to a high d-ACN concentration) might be also explained by a filling of the pore channels that facilitate the diffusion of d-ACN. This hypothesis is compatible with the above-discussed theory on stable heterodinuclear compounds that are formed with TMA.
Correlating Data from Different Length Scales
Based on the time-resolved FT-IR data, which were collected in the presence of d-acetonitrile, we conclude that the insertion kinetics of the SiO2/MAO/Zr catalyst material are notably faster that those of the SiO2/MAO/Hf catalyst material. This is mainly due to the following two reasons: (i) the active sites are more accessible (i.e., less stable heterodinuclear compounds with TMA) in SiO2/MAO/Zr and (ii) the active sites are intrinsically faster. According to the FIB-SEM and IR PiFM data, the faster kinetics of the SiO2/MAO/Zr catalyst material result in the buildup of large amounts of PE at the particle surface as well as in any accessible pores. The imposed diffusion limitations restrict the access of ethylene molecules to the particle interior, thereby limiting both polymerization and concurrent fragmentation under the given experimental conditions. In contrast to this, the kinetically slower SiO2/MAO/Hf catalyst system seems to be less affected by diffusion limitations, which manifests in a more homogeneous fragmentation of the catalyst support. This hypothesis is also reflected by the unexpectedly low PE yield of the kinetically superior SiO2/MAO/Zr catalyst in comparison that of the SiO2/MAO/Hf catalyst under prepolymerization conditions, i.e., 5.8 g of PE per gram of the SiO2/MAO/Zr catalyst vs 6.7 g of PE per gram of the SiO2/MAO/Hf catalyst (Table S2). In fact, low PE yields, which are linked with the buildup of the polymer at the particle surface and thus mass transfer limitations, have also recently been reported by Zanoni et al.9 for a comparable silica-supported zirconocene during gas-phase ethylene polymerization at both 9 and 15 bar.
Conclusions
Our multiscale combined microscopy and spectroscopy approach, which is based on focused ion beam–scanning electron microscopy (FIB-SEM), infrared photoinduced force microscopy (IR PiFM), and time-resolved IR spectroscopy of adsorbed d-acetonitrile (d-ACN), delivered new mechanistic insights into the early stage fragmentation of two structurally analogous metallocene-based catalyst materials with different kinetic profiles during the gas-phase polymerization of ethylene. As summarized in Figure 8, insertion kinetics and associated mass transfer limitations were identified as being critical for the homogeneity of catalyst support fragmentation during the early reaction stages of ethylene polymerization. In the absence of strong mass transfer limitations that are imposed by both the surface buildup of polyethylene and pore filling, the individual support domains disintegrate more uniformly according to a layer-by-layer mechanism, as was observed for the kinetically slower SiO2/MAO/Hf catalyst material. Stronger mass transfer limitations, as evident for the faster SiO2/MAO/Zr catalyst material, significantly inhibit or delay fragmentation during the initial reaction stages of ethylene polymerization under given experimental conditions. This is postulated to induce higher stress accumulation and thus a larger contribution from the sectioning mechanism at the level of the individual silica domains and, possibly, the particle level.
Figure 8.
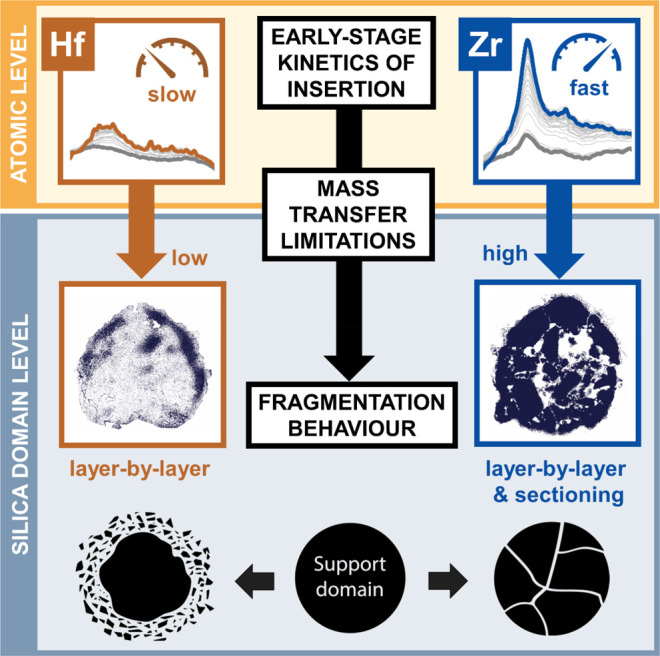
Schematic illustration of the main insights gained from our multiscale microscopy–spectroscopy approach. Insertion kinetics and associated mass transfer limitations are critical for the homogeneity of catalyst support fragmentation during the early stages of ethylene polymerization.
Time-resolved IR spectroscopy in the presence of d-ACN provided an explanation for the different experimentally observed insertion kinetics. Not only are the active sites of SiO2/MAO/Hf less accessible (due to more stable heterodinuclear adducts with TMA), they are also intrinsically slower than the active sites of SiO2/MAO/Zr. It is worth noticing that these conclusions are in agreement with theoretical and experimental data reported in literature. This unprecedented spectroscopic approach can also be applied to similar catalysts to evaluate their accessibility and insertion behavior.
The correlated FIB-SEM-IR PiFM approach, on the other hand, delivered highly resolved morphological information and facilitated the spectroscopic identification of the support, polymer, and composite phases. It represents a novel analytical approach that can also be extended to other industrial-grade catalysts for obtaining information on structure, chemical composition, and mechanical parameters at unparalleled spatial resolutions and, in the case of full catalyst particles, at variable probing depths via FIB cutting.
Acknowledgments
Our appreciation goes to Derek Nowak (Molecular Vista Inc.) for his advice on PiFM.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.1c00324.
Information on catalyst preparation, testing, prepolymerization, and characterization (PDF)
Author Contributions
∇ These authors contributed equally. M.J.W. performed the FIB-SEM experiments, I.C.t.H. and M.J.W. carried out the PiFM experiments, and J.Z. and A.P. conducted the probe molecule FT-IR experiments. C.H. and N.H.F. provided the metallocene-based catalyst samples. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The research was funded by a grant from the Dutch Polymer Institute (DPI, P.O. Box 902, 5600 AX Eindhoven, The Netherlands) and represents a part of the Research Program of DPI project no. 813. Additional funding comes from The Netherlands Center for Multiscale Catalytic Energy Conversion (MCEC), a Dutch Research Council (NWO) Gravitation program funded by the Ministry of Education, Culture and Science of the government of The Netherlands.
The authors declare no competing financial interest.
Supplementary Material
References
- Severn J. R.; Chadwick J. C.; Duchateau R.; Friederichs N. Bound but Not Gagged” - Immobilizing Single-Site α-Olefin Polymerization Catalysts. Chem. Rev. 2005, 105 (11), 4073–4147. 10.1021/cr040670d. [DOI] [PubMed] [Google Scholar]
- Soares J. B. P.; McKenna T. F. L.. Polyolefin Reaction Engineering, 1st ed.; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- Fink G.; Steinmetz B.; Zechlin J.; Przybyla C.; Tesche B. Propene Polymerization with Silica-Supported Metallocene/MAO Catalysts. Chem. Rev. 2000, 100 (4), 1377–1390. 10.1021/cr9804689. [DOI] [PubMed] [Google Scholar]
- Fink G.; Tesche B.; Korber F.; Knoke S. The Particle-Forming Process of SiO2-Supported Metallocene Catalysts. Macromol. Symp. 2001, 173 (1), 77–87. . [DOI] [Google Scholar]
- Horáčková B.; Grof Z.; Kosek J. Dynamics of Fragmentation of Catalyst Carriers in Catalytic Polymerization of Olefins. Chem. Eng. Sci. 2007, 62 (18–20), 5264–5270. 10.1016/j.ces.2007.03.022. [DOI] [Google Scholar]
- Machado F.; Lima E. L.; Pinto J. C.; McKenna T. F. An Experimental Study on the Early Stages of Gas-Phase Olefin Polymerizations Using Supported Ziegler–Natta and Metallocene Catalysts. Polym. Eng. Sci. 2011, 51 (2), 302–310. 10.1002/pen.21830. [DOI] [Google Scholar]
- Bossers K. W.; Valadian R.; Zanoni S.; Smeets R.; Friederichs N.; Garrevoet J.; Meirer F.; Weckhuysen B. M. Correlated X-Ray Ptychography and Fluorescence Nano-Tomography on the Fragmentation Behavior of an Individual Catalyst Particle during the Early Stages of Olefin Polymerization. J. Am. Chem. Soc. 2020, 142 (8), 3691–3695. 10.1021/jacs.9b13485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossers K. W.; Valadian R.; Garrevoet J.; van Malderen S.; Chan R.; Friederichs N.; Severn J.; Wilbers A.; Zanoni S.; Jongkind M. K.; et al. Heterogeneity in the Fragmentation of Ziegler Catalyst Particles during Ethylene Polymerization Quantified by X-Ray Nanotomography. JACS Au 2021, 1 (6), 852–864. 10.1021/jacsau.1c00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanoni S.; Nikolopoulos N.; Welle A.; Vantomme A.; Weckhuysen B. Early-Stage Particle Fragmentation Behavior of a Commercial Silica-Supported Metallocene Catalyst. Catal. Sci. Technol. 2021, 11 (15), 5335–5348. 10.1039/D1CY00930C. [DOI] [Google Scholar]
- Weickert G.; Meier G. B.; Pater J. T. M.; Westerterp K. R. The Particle as Microreactor: Catalytic Propylene Polymerizations with Supported Metallocenes and Ziegler-Natta Catalysts. Chem. Eng. Sci. 1999, 54 (15–16), 3291–3296. 10.1016/S0009-2509(98)00441-2. [DOI] [Google Scholar]
- McKenna T. F.; Soares J. B. P. Single Particle Modelling for Olefin Polymerization on Supported Catalysts: A Review and Proposals for Future Developments. Chem. Eng. Sci. 2001, 56 (13), 3931–3949. 10.1016/S0009-2509(01)00069-0. [DOI] [Google Scholar]
- Böhm L. L. The Ethylene Polymerization with Ziegler Catalysts: Fifty Years after the Discovery. Angew. Chem., Int. Ed. 2003, 42 (41), 5010–5030. 10.1002/anie.200300580. [DOI] [PubMed] [Google Scholar]
- McKenna T.; Mattioli V. Progress in Describing Particle Growth for Polyolefins: A Look at Particle Morphology. Macromol. Symp. 2001, 173, 149–162. . [DOI] [Google Scholar]
- McKenna T. F. L.; Di Martino A.; Weickert G.; Soares J. B. P. Particle Growth during the Polymerisation of Olefins on Supported Catalysts, 1 - Nascent Polymer Structures. Macromol. React. Eng. 2010, 4 (1), 40–64. 10.1002/mren.200900025. [DOI] [Google Scholar]
- Resconi L.; Cavallo L.; Fait A.; Piemontesi F. Selectivity in Propene Polymerization with Metallocene Catalysts. Chem. Rev. 2000, 100 (4), 1253–1345. 10.1021/cr9804691. [DOI] [PubMed] [Google Scholar]
- Ewen J. A.; Haspeslach L.; Atwood J. L.; Zhang H. Crystal Structures and Stereospecific Propylene Polymerizations with Chiral Hafnium Metallocene Catalysts. J. Am. Chem. Soc. 1987, 109 (21), 6544–6545. 10.1021/ja00255a068. [DOI] [Google Scholar]
- Rieger B.; Troll C.; Preuschen J. Ultrahigh Molecular Weight Polypropene Elastomers by High Activity “Dual-Side” Hafnocene Catalysts. Macromolecules 2002, 35 (15), 5742–5743. 10.1021/ma025520o. [DOI] [Google Scholar]
- Laine A.; Linnolahti M.; Pakkanen T. A.; Severn J. R.; Kokko E.; Pakkanen A. Comparative Theoretical Study on Homopolymerization of α-Olefins by Bis(Cyclopentadienyl) Zirconocene and Hafnocene: Elemental Propagation and Termination Reactions between Monomers and Metals. Organometallics 2010, 29 (7), 1541–1550. 10.1021/om900843h. [DOI] [Google Scholar]
- Machat M. R.; Fischer A.; Schmitz D.; Vöst M.; Drees M.; Jandl C.; Pöthig A.; Casati N. P. M.; Scherer W.; Rieger B. Behind the Scenes of Group 4 Metallocene Catalysis: Examination of the Metal-Carbon Bond. Organometallics 2018, 37 (16), 2690–2705. 10.1021/acs.organomet.8b00339. [DOI] [Google Scholar]
- Busico V.; Cipullo R.; Pellecchia R.; Talarico G.; Razavi A. Hafnocenes and MAO: Beware of Trimethylaluminum!. Macromolecules 2009, 42 (6), 1789–1791. 10.1021/ma900066n. [DOI] [Google Scholar]
- Ehm C.; Cipullo R.; Budzelaar P. H. M.; Busico V. Role(s) of TMA in Polymerization. Dalt. Trans. 2016, 45 (16), 6847–6855. 10.1039/C5DT04895H. [DOI] [PubMed] [Google Scholar]
- De Winter D. A. M.; Meirer F.; Weckhuysen B. M. FIB-SEM Tomography Probes the Mesoscale Pore Space of an Individual Catalytic Cracking Particle. ACS Catal. 2016, 6 (5), 3158–3167. 10.1021/acscatal.6b00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grof Z.; Kosek J.; Marek M. Modeling of Morphogenesis of Growing Polyolefin Particles. AIChE J. 2005, 51 (7), 2048–2067. 10.1002/aic.10549. [DOI] [Google Scholar]
- Tisse V. F.; Prades F.; Briquel R.; Boisson C.; McKenna T. F. L. Role of Silica Properties in the Polymerisation of Ethylene Using Supported Metallocene Catalysts. Macromol. Chem. Phys. 2010, 211 (1), 91–102. 10.1002/macp.200900311. [DOI] [Google Scholar]
- Taniike T.; Funako T.; Terano M. Multilateral Characterization for Industrial Ziegler – Natta Catalysts toward Elucidation of Structure – Performance Relationship. J. Catal. 2014, 311, 33–40. 10.1016/j.jcat.2013.10.023. [DOI] [Google Scholar]
- Bashir M. A.; Monteil V.; Boisson C.; McKenna T. F. L. Experimental Proof of the Existence of Mass-Transfer Resistance During Early Stages of Ethylene Polymerization with Silica Supported Metallocene/MAO Catalysts. AIChE J. 2017, 63 (10), 4476–4490. 10.1002/aic.15806. [DOI] [Google Scholar]
- Gu K. L.; Zhou Y.; Morrison W. A.; Park K.; Park S.; Bao Z. Nanoscale Domain Imaging of All-Polymer Organic Solar Cells by Photo-Induced Force Microscopy. ACS Nano 2018, 12 (2), 1473–1481. 10.1021/acsnano.7b07865. [DOI] [PubMed] [Google Scholar]
- Sun C.; Pan F.; Bin H.; Zhang J.; Xue L.; Qiu B.; Wei Z.; Zhang Z. G.; Li Y. A Low Cost and High Performance Polymer Donor Material for Polymer Solar Cells. Nat. Commun. 2018, 9 (1), 1–10. 10.1038/s41467-018-03207-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dazzi A.; Prater C. B.; Hu Q.; Chase D. B.; Rabolt J. F.; Marcott C. AFM – IR : Combining Atomic Force Microscopy and Infrared Spectroscopy for Nanoscale Chemical Characterization. Appl. Spectrosc. 2012, 66 (12), 1365–1384. 10.1366/12-06804. [DOI] [PubMed] [Google Scholar]
- Eby T.; Gundusharma U.; Lo M.; Sahagian K.; Marcott C.; Kjoller K. Reverse Engineering of Polymeric Multilayers Using AFM-Based Nanoscale IR Spectroscopy and Thermal Analysis. Spectrosc. Eur. 2012, 24 (3), 18–21. [Google Scholar]
- Tang F.; Bao P.; Su Z. Analysis of Nanodomain Composition in High-Impact Polypropylene by Atomic Force Microscopy-Infrared. Anal. Chem. 2016, 88, 4926–4930. 10.1021/acs.analchem.6b00798. [DOI] [PubMed] [Google Scholar]
- Kelchtermans M.; Lo M.; Dillon E.; Kjoller K.; Marcott C. Characterization of a Polyethylene-Polyamide Multilayer Film Using Nanoscale Infrared Spectroscopy and Imaging. Vib. Spectrosc. 2016, 82, 10–15. 10.1016/j.vibspec.2015.11.004. [DOI] [Google Scholar]
- Nguyen-Tri P.; Ghassemi P.; Carriere P.; Nanda S.; Assadi A. A.; Nguyen D. D. Recent Applications of Advanced Atomic Force Microscopy in Polymer Science: A Review. Polymers 2020, 12 (5), 1142. 10.3390/polym12051142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak D.; Morrison W.; Wickramasinghe H. K.; Jahng J.; Potma E.; Wan L.; Ruiz R.; Albrecht T. R.; Schmidt K.; Frommer J.; et al. Nanoscale Chemical Imaging by Photoinduced Force Microscopy. Sci. Adv. 2016, 2 (3), e1501571 10.1126/sciadv.1501571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu D.; Park K.; Delen G.; Attila Ö.; Meirer F.; Nowak D.; Park S.; Schmidt J. E.; Weckhuysen B. M. Nanoscale Infrared Imaging of Zeolites Using Photoinduced Force Microscopy. Chem. Commun. 2017, 53 (97), 13012–13014. 10.1039/C7CC06832H. [DOI] [PubMed] [Google Scholar]
- Murdick R. A.; Morrison W.; Nowak D.; Albrecht T. R.; Jahng J.; Park S. Photoinduced Force Microscopy: A Technique for Hyperspectral Nanochemical Mapping. Jpn. J. Appl. Phys. 2017, 56 (8S1), 08LA04 10.7567/JJAP.56.08LA04. [DOI] [Google Scholar]
- Delen G.; Monai M.; Meirer F.; Weckhuysen B. M. In Situ Nanoscale Infrared Spectroscopy of Water Adsorption on Nanoislands of Surface-Anchored Metal-Organic Frameworks. Angew. Chem. 2021, 133 (3), 1644–1648. 10.1002/ange.202011564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Jahng J.; Pang J.; Morrison W.; Li J.; Lee E. S.; Xu J. J.; Chen H. Y.; Xia X. H. Tip-Enhanced Infrared Imaging with Sub-10 nm Resolution and Hypersensitivity. J. Phys. Chem. Lett. 2020, 11 (5), 1697–1701. 10.1021/acs.jpclett.0c00129. [DOI] [PubMed] [Google Scholar]
- Almajhadi M. A.; Uddin S. M. A.; Wickramasinghe H. K. Observation of Nanoscale Opto-Mechanical Molecular Damping as the Origin of Spectroscopic Contrast in Photo Induced Force Microscopy. Nat. Commun. 2020, 11 (1), 1–9. 10.1038/s41467-020-19067-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Have I. C.; Duijndam A. J. A.; Oord R.; Berlo-van den Broek H. J. M.; Vollmer I.; Weckhuysen B. M.; Meirer F. Photoinduced Force Microscopy as an Efficient Method Towards the Detection of Nanoplastics. Chem. Methods 2021, 1 (5), 205–209. 10.1002/cmtd.202100017. [DOI] [Google Scholar]
- Ocaña M.; Fornés V.; Serna C. J. The Variability of the Infrared Powder Spectrum of Amorphous SiO2. J. Non-Cryst. Solids 1989, 107, 187–192. 10.1016/0022-3093(89)90461-4. [DOI] [Google Scholar]
- Almeida R. M. Comment on “Infrared-Reflectance Spectra of Heat-Treated, Sol-Gel-Derived Silica.. Phys. Rev. B: Condens. Matter Mater. Phys. 1996, 53 (21), 14656–14658. 10.1103/PhysRevB.53.14656. [DOI] [PubMed] [Google Scholar]
- Ruddick V. J.; Badyal J. P. S. AFM Study of the Breakup of Catalyst Particles during Ethylene Polymerization. J. Phys. Chem. B 1997, 101 (10), 1791–1793. 10.1021/jp963206o. [DOI] [Google Scholar]
- Pang G. K. H.; Baba-Kishi K. Z.; Patel A. Topographic and Phase-Contrast Imaging in Atomic Force Microscopy. Ultramicroscopy 2000, 81 (2), 35–40. 10.1016/S0304-3991(99)00164-3. [DOI] [PubMed] [Google Scholar]
- Schmitz I.; Schreiner M.; Friedbacher G.; Grasserbauer M. Phase Imaging as an Extension to Tapping Mode AFM for the Identification of Material Properties on Humidity-Sensitive Surfaces. Appl. Surf. Sci. 1997, 115 (2), 190–198. 10.1016/S0169-4332(97)80204-8. [DOI] [Google Scholar]
- Magonov S. N.; Elings V.; Whangbo M. H. Phase Imaging and Stiffness in Tapping-Mode Atomic Force Microscopy. Surf. Sci. 1997, 375 (2–3), L385–L391. 10.1016/S0039-6028(96)01591-9. [DOI] [Google Scholar]
- Scott W. W.; Bhushan B. Use of Phase Imaging in Atomic Force Microscopy for Measurement of Viscoelastic Contrast in Polymer Nanocomposites and Molecularly Thick Lubricant Films. Ultramicroscopy 2003, 97 (1–4), 151–169. 10.1016/S0304-3991(03)00040-8. [DOI] [PubMed] [Google Scholar]
- Bar G.; Thomann Y.; Brandsch R.; Cantow H. J.; Whangbo M. H. Factors Affecting the Height and Phase Images in Tapping Mode Atomic Force Microscopy. Study of Phase-Separated Polymer Blends of Poly(Ethene-Co-Styrene) and Poly(2,6-Dimethyl-1,4-Phenylene Oxide). Langmuir 1997, 13 (14), 3807–3812. 10.1021/la970091m. [DOI] [Google Scholar]
- Wang L. Role of Damping in Phase Imaging in Tapping Mode Atomic Force Microscopy. Surf. Sci. 1999, 429 (1), 178–185. 10.1016/S0039-6028(99)00368-4. [DOI] [Google Scholar]
- Zecchina A.; Areán C. O. Diatomic Molecular Probes for Mid-IR Studies of Zeolites. Chem. Soc. Rev. 1996, 25 (3), 187–197. 10.1039/CS9962500187. [DOI] [Google Scholar]
- Lamberti C.; Zecchina A.; Groppo E.; Bordiga S. Probing the Surfaces of Heterogeneous Catalysts by in Situ IR Spectroscopy. Chem. Soc. Rev. 2010, 39 (12), 4951–5001. 10.1039/c0cs00117a. [DOI] [PubMed] [Google Scholar]
- Groppo E.; Lamberti C.; Bordiga S.; Spoto G.; Zecchina A. The Structure of Active Centers and the Ethylene Polymerization Mechanism on the Cr/SiO2 Catalyst: A Frontier for the Characterization Methods. Chem. Rev. 2005, 105 (1), 115–183. 10.1021/cr040083s. [DOI] [PubMed] [Google Scholar]
- Groppo E.; Martino G. A.; Piovano A.; Barzan C. The Active Sites in the Phillips Catalysts: Origins of a Lively Debate and a Vision for the Future. ACS Catal. 2018, 8 (11), 10846–10863. 10.1021/acscatal.8b02521. [DOI] [Google Scholar]
- Zakharov V. A.; Paukshtis E. A.; Mikenas T. B.; Volodin A. M.; Vitus E. N.; Potapov A. G. Surface Acidic Sites of Highly Disperse Magnesium Chloride: IR and ESR Spectroscopy Studies. Macromol. Symp. 1995, 89 (1), 55–61. 10.1002/masy.19950890108. [DOI] [Google Scholar]
- Thushara K. S.; D’Amore M.; Piovano A.; Bordiga S.; Groppo E. The Influence of Alcohols in Driving the Morphology of Magnesium Chloride Nanocrystals. ChemCatChem 2017, 9 (10), 1782–1787. 10.1002/cctc.201700101. [DOI] [Google Scholar]
- D’Amore M.; Thushara K. S.; Piovano A.; Causà M.; Bordiga S.; Groppo E. Surface Investigation and Morphological Analysis of Structurally Disordered MgCl2 and MgCl2/TiCl4 Ziegler–Natta Catalysts. ACS Catal. 2016, 6 (9), 5786–5796. 10.1021/acscatal.6b00871. [DOI] [Google Scholar]
- Pletcher P.; Welle A.; Vantomme A.; Weckhuysen B. M. Quality Control for Ziegler-Natta Catalysis via Spectroscopic Fingerprinting. J. Catal. 2018, 363, 128–135. 10.1016/j.jcat.2018.04.012. [DOI] [Google Scholar]
- Velthoen M. E. Z.; Muñoz-Murillo A.; Bouhmadi A.; Cecius M.; Diefenbach S.; Weckhuysen B. M. The Multifaceted Role of Methylaluminoxane in Metallocene-Based Olefin Polymerization Catalysis. Macromolecules 2018, 51 (2), 343–355. 10.1021/acs.macromol.7b02169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchenko V. N.; Danilova I. G.; Zakharov V. A.; Paukshtis E. A. An IR-Spectroscopic Study of the State of Zirconium in Supported Zirconocene Catalysts. Kinet. Catal. 2004, 45 (4), 547–553. 10.1023/B:KICA.0000038083.22397.bf. [DOI] [Google Scholar]
- Marsella J. A.; Curtis C. J.; Bercaw J. E.; Caulton K. G. Low-Temperature Infrared Study of d° Carbonyl Complexes. J. Am. Chem. Soc. 1980, 102 (24), 7244–7246. 10.1021/ja00544a012. [DOI] [Google Scholar]
- Piovano A.; Zarupski J.; Groppo E. Disclosing the Interaction between Carbon Monoxide and Alkylated Ti3+ Species: A Direct Insight into Ziegler-Natta Catalysis. J. Phys. Chem. Lett. 2020, 11 (14), 5632–5637. 10.1021/acs.jpclett.0c01665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desert X.; Carpentier J. F.; Kirillov E. Quantification of Active Sites in Single-Site Group 4 Metal Olefin Polymerization Catalysis. Coord. Chem. Rev. 2019, 386, 50–68. 10.1016/j.ccr.2019.01.025. [DOI] [Google Scholar]
- Doi Y.; Murata M.; Yano K.; Keii T. Gas-Phase Polymerization of Propene with the Supported Ziegler Catalyst: TiCl4/MgCl2/C6H5COOC2H5/Al(C2H5)3. Ind. Eng. Chem. Prod. Res. Dev. 1982, 21 (4), 580–585. 10.1021/i300008a013. [DOI] [Google Scholar]
- Taniike T.; Sano S.; Ikeya M.; Thang V. Q.; Terano M. Development of a Large-Scale Stopped-Flow System for Heterogeneous Olefin Polymerization Kinetics. Macromol. React. Eng. 2012, 6 (6–7), 275–279. 10.1002/mren.201200003. [DOI] [Google Scholar]
- Thakur A.; Wada T.; Chammingkwan P.; Terano M.; Taniike T. Development of Large-Scale Stopped-Flow Technique and Its Application in Elucidation of Initial Ziegler-Natta Olefin Polymerization Kinetics. Polymers 2019, 11 (6), 1012. 10.3390/polym11061012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukatov G.; Goncharov V.; Zakharov V. Interaction of 14CO with Ziegler-type Heterogeneous Catalysts and Effect of Interaction Products on the Determination of the Amount of Active Centers. Makromol. Chem. 1986, 187 (5), 1041–1051. 10.1002/macp.1986.021870501. [DOI] [Google Scholar]
- Shiono T.; Ohgizawa M.; Soga K. Reaction between Carbon Monoxide and a Ti-polyethylene Bond with a MgCl2-supported TiCl4 Catalyst System. Makromol. Chem. 1993, 194 (7), 2075–2085. 10.1002/macp.1993.021940720. [DOI] [Google Scholar]
- Tritto I.; Sacchi M. C.; Locatelli P. On the Insertion Reaction of Carbon Oxides into Metal-carbon Bonds of Ziegler-Natta Catalysts. Makromol. Chem., Rapid Commun. 1983, 4 (9), 623–627. 10.1002/marc.1983.030040908. [DOI] [Google Scholar]
- Knözinger H. Infrared Spectroscopy as a Probe of Surface Acidity. Elem. React. Steps Heterog. Catal. 1993, 267–285. 10.1007/978-94-011-1693-0_16. [DOI] [Google Scholar]
- Knoezinger H.; Krietenbrink H. Infrared Spectroscopic Study of the Adsorption of Nitriles on Aluminium Oxide. Fermi Resonance in Coordinated Acetonitrile. J. Chem. Soc., Faraday Trans. 1 1975, 71, 2421–2430. 10.1039/f19757102421. [DOI] [Google Scholar]
- Morterra C.; Peñarroya Mentruit M.; Cerrato G. Acetonitrile Adsorption as an IR Spectroscopic Probe for Surface Acidity/Basicity of Pure and Modified Zirconias. Phys. Chem. Chem. Phys. 2002, 4 (4), 676–687. 10.1039/b109047j. [DOI] [Google Scholar]
- Morterra C.; Cerrato G.; Novarino E.; Peñarroya Mentruit M. On the Adsorption of Acetonitrile on Pure and Sulfated Tetragonal Zirconia (t-ZrO2). Langmuir 2003, 19 (14), 5708–5721. 10.1021/la0270284. [DOI] [Google Scholar]
- Escalona Platero E.; Peñarroya Mentruit M.; Morterra C. Fourier Transform Infrared Spectroscopy Study of CD3CN Adsorbed on Pure and Doped γ-Alumina. Langmuir 1999, 15 (15), 5079–5087. 10.1021/la981654c. [DOI] [Google Scholar]
- Cerruti M.; Bolis V.; Magnacca G.; Morterra C. Surface Chemical Functionalities in Bioactive Glasses. The Gas/Solid Adsorption of Acetonitrile. Phys. Chem. Chem. Phys. 2004, 6 (9), 2468–2479. 10.1039/b315418a. [DOI] [Google Scholar]
- Bochmann M.; Wilson L. M. Synthesis and Insertion Reactions of Cationic Alkylbis(Cyclopentadienyl) Titanium Complexes. J. Chem. Soc., Chem. Commun. 1986, 0 (21), 1610–1611. 10.1039/c39860001610. [DOI] [Google Scholar]
- Jordan R. F.; Bajgur C. S.; Dasher W. E.; Rheingold A. L. Hydrogenation of Cationic Dicyclopentadienyl Zirconium(IV) Alkyl Complexes. Characterization of Cationic Zirconium(IV) Hydrides. Organometallics 1987, 6 (5), 1041–1051. 10.1021/om00148a023. [DOI] [Google Scholar]
- Bochmann M.; Wilson L. M.; Hursthouse M. B.; Motevalli M. Insertion Reactions of Nitriles in Cationic Alkylbis(Cyclopentadienyl)Titanium Complexes: The Facile Synthesis of Azaalkenylidene Titanium Complexes and the Crystal and Molecular Structure of [(Indenyl)2Ti(NCMePh)(NCPh)]BPh4. Organometallics 1988, 7 (5), 1148–1154. 10.1021/om00095a020. [DOI] [Google Scholar]
- Alelyunas Y. W.; Jordan R. F.; Echols S. F.; Borkowsky S. L.; Bradley P. K. Organometallics 1991, 10 (5), 1406–1416. 10.1021/om00051a036. [DOI] [Google Scholar]
- Ystenes M.; Eilertsen J. L.; Liu J.; Ott M.; Rytter E.; Støvneng J. A. Experimental and Theoretical Investigations of the Structure of Methylaluminoxane (MAO) Cocatalysts for Olefin Polymerization. J. Polym. Sci., Part A: Polym. Chem. 2000, 38 (17), 3106–3127. . [DOI] [Google Scholar]
- Martino G. A.; Piovano A.; Barzan C.; Rabeah J.; Agostini G.; Bruekner A.; Leone G.; Zanchin G.; Monoi T.; Groppo E. Rationalizing the Effect of Triethylaluminum on the Cr/SiO2 Phillips Catalysts. ACS Catal. 2020, 10 (4), 2694–2706. 10.1021/acscatal.9b04726. [DOI] [Google Scholar]
- Zecchina A.; Guglielminotti E.; Coluccia S.; Borello E. Infrared Spectra of Nitriles of Chromia-Silica Catalyst. J. Chem. Soc. A 1969, 2196–2199. 10.1039/J19690002196. [DOI] [Google Scholar]
- Montanari T.; Bevilacqua M.; Busca G. Use of Nitriles as Probe Molecules for the Accessibility of the Active Sites and the Detection of Complex Interactions in Zeolites through IR Spectroscopy. Appl. Catal., A 2006, 307 (1), 21–29. 10.1016/j.apcata.2006.03.003. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.