

Abstract
Septins are a family of multimeric GTP-binding proteins, which are abnormally expressed in cancer. Septin 9 (SEPT9) is an essential and ubiquitously expressed septin with multiple isoforms, which have differential expression patterns and effects in breast cancer cells. It is unknown, however, if SEPT9 isoforms associate with different molecular networks and functions. Here, we performed a proteomic screen in MCF-7 breast cancer cells to identify the interactome of GFP-SEPT9 isoforms 1, 4 and 5, which vary significantly in their N-terminal extensions. While all three isoforms associated with SEPT2 and SEPT7, the truncated SEPT9_i4 and SEPT9_i5 interacted with septins of the SEPT6 group more promiscuously than SEPT9_i1, which bound predominately SEPT8. Spatial mapping and functional clustering of non-septin partners showed isoform-specific differences in interactions with proteins of distinct subcellular organelles (e.g., nuclei, centrosomes, cilia) and functions such as cell signaling and ubiquitination. The interactome of the full length SEPT9_i1 was more enriched in cytoskeletal regulators, while the truncated SEPT9_i4 and SEPT9_i5 exhibited preferential and isoform-specific interactions with nuclear, signaling and ubiquitinating proteins. These data provide evidence for isoform-specific interactions, which arise from truncations in the N-terminal extensions of SEPT9, and point to novel roles in the pathogenesis of breast cancer.
Keywords: septins, SEPT9 isoforms, breast cancer, septin interactome, septin proteomics
1. Introduction
Septins are a large family of GTP-binding proteins that control the intracellular localization of cytoskeletal, membrane and cytosolic proteins [1–5]. Septins interact with one another via their GTP-binding domains (G-domains), forming homomeric and heteromeric complexes which assemble into higher-order filamentous structures [6, 7]. Septins associate with the actin and microtubule cytoskeleton as well as cell membranes, and function in a variety of cellular processes including cell division and migration, intracellular membrane trafficking, cell proliferation and apoptosis [4, 8, 9].
Based on sequence similarity, mammalian septins are classified into four groups named after SEPT2 (SEPT1, SEPT2, SEPT4, SEPT5), SEPT6 (SEPT6, SEPT8, SEPT10, SEPT11, SEPT14), SEPT7 and SEPT3 (SEPT3, SEPT9, SEPT12) [10–12]. Septin paralogs from each of these four groups form complexes of various sizes and composition such as the palindromic hexamers and octamers of SEPT2/6/7 and SEPT2/6/7/9, respectively, which are considered as the minimal units of septin filaments [13–15]. While the precise identity and diversity of septin complexes eludes our knowledge, septin-septin interactions are influenced by GTP binding and hydrolysis, the relative abundance of septin paralogs and isoforms, which can vary between cell types, and the presence of domains (e.g., N-terminal extensions) that may interfere with the G-domain binding interface [9, 16–19]. Importantly, the localization and function of septin complexes appear to depend on the properties and binding partners of their individual subunits [20–22].
Septin 9 (SEPT9) is a ubiquitously expressed septin, which is essential for embryonic development; SEPT9 knock-out mice die early in gestation [23]. A salient feature of SEPT9 is the presence of a long N-terminal extension (NTE), which is unique among septins. Owing to alternative translation start and splice sites, there are at least five different SEPT9 isoforms (SEPT9_i1 to _i5) with NTEs that vary in length and amino acid sequence [24]. The NTE of SEPT9 is structurally disordered and its full-length amino acid sequence comprises a domain (1-148) of basic isolectric point, which contains microtubule- and actin-binding sites, and an acidic proline-rich domain (aa 148-256) [25, 26]. SEPT9 is positioned in the center of the mammalian hetero-octameric complex (SEPT2-SEPT6-SEPT7-SEPT9-SEPT9-SEPT7-SEPT6-SEPT2) interacting with SEPT7 [27, 28], and may influence septin assembly into heteromers in an isoform-dependent manner [15, 19, 20]. Interestingly, in some cell types and processes, SEPT9 proteins appear to localize and function independently of SEPT2/6/7 [19, 29–31].
SEPT9 was among the first septins implicated in cancer as the SEPT9 gene was mapped to the loss of heterozygosity region of chromosome 17 (17q25), which is frequently deleted in breast and ovarian cancers [32, 33]. The SEPT9 gene was also found as a fusion partner of mixed lineage leukemia (MLL) gene and a preferred site of integration for a murine retrovirus that causes T-cell lymphomas [34, 35]. In breast carcinomas and mouse models of breast cancer, the SEPT9 gene is amplified (multiple gene copies) and the expression levels of certain SEPT9 variants are markedly increased [36, 37]. Over-expression of SEPT9 isoforms occurs in ~30% of human breast cancer cases and correlates with poor prognosis and resistance to anti-cancer agents that target microtubules [37–41]. Notably, changes in the methylation status of the SEPT9 gene have been utilized for early detection of colorectal cancer [42, 43]. Several studies have shown that over-expression of SEPT9 isoforms has differential effects on the migratory properties of breast cancer cells and have linked SEPT9_i1 to mechanisms of angiogenesis and cell proliferation [44–48]. Despite these findings, we have a very poor understanding of the roles that different SEPT9 isoforms play in the development and metastasis of breast cancer.
Unbiased shotgun proteomics, which is based on liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS), has been an effective approach in elucidating the molecular networks and complexes of cellular proteins [49, 50]. Combined with bioinformatics, discovery-based proteomics can provide a comprehensive map of the cellular components, pathways and functions of a protein and its binding partners [51]. Yeast hybrid screens have used mammalian septin paralogs as baits to identify potential binding partners [52, 53], but there is a dearth of proteomic data on septin complexes isolated from mammalian cells. Owing to the lack of isoform-specific antibodies, comparative proteomics for septin isoforms are challenging and septin isoform-specific interactomes are underexplored. This is a key challenge for SEPT9, which has a multitude of isoforms that may have differential roles in breast cancer. Here, we used discovery-based bottom-up shotgun proteomics in MCF-7 breast cancer cells, which were stably transfected with GFP-tagged SEPT9 isoforms 1, 4 and 5. We chose these isoforms because their amino acid sequences have the least overlap among all SEPT9 isoforms, and therefore, may have divergent interactomes and functions. Proteomic profiling revealed isoform-specific differences in the interaction with septin paralogs, and non-septin proteins of distinct subcellular localization (e.g., nucleus, centrosome, cilia) and functions including cell signaling and ubiquitin-mediated degradation. Collectively, these data suggest a functional specialization for isoforms of SEPT9, which arises from differences in the length and sequence of the N-terminal extensions of their GTP-binding domains.
2. Materials and Methods
2.1. Cell culture
MCF-7 cells stably expressing GFP-SEPT9_i1, GFP-SEPT9_i4 and GFP-SEPT9_i5 [44] were cultured in Dulbecco’s Modified Eagle’s Medium with high glucose (4500 mg/L), L-glutamine and sodium pyruvate plus 10% fetal bovine serum and the following antibiotics: kanamycin sulfate, penicillin G and streptomycin sulfate. Cells were grown to confluency in 10 cm dishes at 37°C supplemented with 5% CO2. MDCKII/G cells were maintained in Dulbecco’s Modified Eagle’s medium with 1 g/l NaHCO3 and 10% FBS.
2.2. Fluorescence microscopy
MCF-7 cells expressing GFP-tagged isoforms of SEPT9 were fixed with warm PHEM buffer (60 mM Pipes-KOH, pH 6.9, 25 mM Hepes, 10 mM EGTA and 1 mM MgCl2) containing 4% PFA (EM Sciences) and 0.1% Triton X-100 and stained with a mouse antibody to α-tubulin (DM1α; SIGMA) and a secondary donkey DyLight 594-conjugated F(ab’)2 mouse IgGs (Jackson ImmunoResearch Laboratories, Inc) and Rhodamine-phalloidin (Cytoskeleton). Samples were mounted in Vectashield (Vector Laboratories) and imaged with an Olympus Fluoview FV1000 confocal laser scanning microscope with a Plan Apochromat 60X/1.35 NA objective and 488 nm, 543 nm and 635 nm laser lines. Three-dimensional stacks were collected at 0.2 μm-step size. Images were exported to the Slidebook 4.2 software and maximum intensity projections of a selected range of optical planes was generated before importing into Adobe Photoshop, where images were adjusted to 300 dpi before cropping for inclusion in the manuscript’s figures, which were made in Adobe Illustrator.
2.3. Immunoprecipitation (IP)
Cells were washed twice with 2mL PBS at 4°C. 400 μL of IP lysis buffer (0.025M Tris, 0.15M NaCl, 0.001M EDTA, 1% NP-40, 5% glycerol; pH 7.4) supplemented with 2X Halt™ Protease Inhibitor Cocktail (ThermoFisher Scientific) was added to each plate and incubated on a rocker for 10 minutes at 4°C. Cells were scraped and lysate was transferred to a microcentrifuge collection tube and incubated end-over-end for 20 minutes at 4°C. Microcentrifuge collection tubes were then centrifuged at 16,000 xg for 10 minutes and supernatant was collected and frozen at −70°C until use.
Immunoprecipitations (IPs) were performed using Pierce Co-IP Kit (ThermoFisher Scientific) according to manufacturer’s instructions. Briefly, goat polyclonal anti-GFP antibody (Abcam ab6673) was coupled to AminoLink Plus Coupling Resin (10 μg/50 μL) using sodium cyanoborohydride. Antibody-coupled resin was incubated with analyte-containing lysate overnight with gentle rocking at 4°C. Five independent experiments for each isoform were performed. In each experiment, three 10 cm plates of equivalent confluency were individually lysed and IPs were performed in each of three lysates. The eluted complexes from all thee IPs were pooled into a single vial and dried at ambient temperature overnight using the SPD1010 SpeedVac system (Thermo Scientific).
2.4. Western Blots
Western blotting of immunoprecipitates for GFP, GAPDH, ANK3 and RacGAP were performed by transferring SDS-PAGE gels (4-12% 1.5 mM Bis-Tris gradient mini protein gel; NuPage) to 0.45 μm nitrocellulose membranes (Amersham GE Healthcare) at 4°C for 2 h (250 mAmps) or overnight under constant voltage (30V) in Tris-Glycine transfer buffer. Membranes were blocked with 5% non-fat dry milk and 1% BSA in PBS for 1 h at room temperature. Membranes were washed with PBS-T (PBS/0.1% Tween 20) and incubated for 2 h or overnight with primary antibodies against GFP (ProteinTech Group 50430-2-AP and 660002-1-Ig; 1:1,000-2,000), GAPDH (6C5, Novus Biologicals NB600-502, 1:5000), ANK3 (Novus Biologicals NBP2-59310; 1:800) and RacGAP1 (Invitrogen PA5-68992; 1:500). All primary antibodies were diluted PBS-T buffer containing 2% BSA and membranes were washed three times with PBS-T with gentle shaking, and incubated with secondary antibodies IRDye 680RD donkey anti-rabbit IgG (LI-COR 926-68073; 1:10,000) and IRDye 800CW donkey anti-mouse IgG (LI-COR 926-32212; 1:10,000) for 1 h at room temperature with gentle shaking before scanning with an Odyssey infrared imaging system (Odyssey; LICOR).
Western blotting of immunoprecipitates for APC and myomegalin were performed by transferring SDS-PAGE gels to a 0.2 μm nitrocellulose membrane (iBlot2 Transfer Stack) at room temperature using a 9 minute transfer protocol (25V) on the iBlot2 system (ThermoFisher). Membranes were blocked with 5% non-fat dry milk and 1% BSA in PBS for 1 h at room temperature. Membranes were washed with PBS-T and incubated with primary antibodies against APC (Abcam 15270; 1:1,000) and myomegalin (Invitrogen PA5-52969; 1:500) in PBS-T buffer containing 2% BSA for 2 h with gentle shaking at room temperature or overnight at 4°C. Subsequently, membranes were washed three times with PBS-T and incubated for 1 h at room temperature with peroxidase-conjugated AffiniPure goat anti-rabbit IgG (Jackson ImmunoResearch 111-035-144), which was diluted in PBS-T with 2% BSA. Blots were developed for enhanced chemiluminescent detection using SuperSignal™ West Pico PLUS Chemiluminescent Substrate (ThermoFisher, Cat # 34580) before scanning on the myECL Imager (ThermoFisher).
2.5. In-solution Protein Digestion
Vacuum-dried protein pellets were reconstituted in 0.1% (w/v) RapiGest SF Surfactant (Waters) in 50 mM ammonium bicarbonate. Samples were reduced with 5mM dithiothreitol (DTT; ThermoFisher Scientific; cat #: 20291) at 60°C for 30 minutes. Samples were then cooled to room temperature and free cysteines were alkylated with 15 mM iodoacetamide (ThermoFisher Scientific) in the dark at room temperature for 30 minutes. Samples were digested at 37°C overnight in 10 μg MS-grade Trypsin Gold (Promega). Sequencing grade trifluoroacetic acid (TFA; ThermoFisher Scientific) was added to the digested proteins to a final 0.5% concentration and incubated at 37°C for 45 minutes. Samples were centrifuged at 13,000 RPM for 10 minutes after which the supernatant was transferred to an HPLC low volume sample vial.
2.6. LC-MS/MS Protein Identification
Each sample was analyzed using online liquid chromatography (Accela pump and autosampler, Thermo, Inc.) coupled to an LTQ-XL linear ion trap mass spectrometer. Samples were loaded onto an UPLC Peptide CSH C18 Column, 130 Å, 1.7 μm, 1 mm x 100 mm (Waters) maintained at 40°C. Solvent A and B were 0.1% formic acid in water and 0.1% formic acid in acetonitrile, respectively (gradient: 98%A/2%B for 59.5 minutes, 50%A/50%B for 0.5 minutes, 98%A/2%B for 10 minutes). The flow rate was set to 100 μL/min and the injection volumes were 10 μL/sample. The samples were ionized using ESI and fragmented using CID. Data were acquired in DDA mode. The mass spectrometer was run in positive ion mode using a source voltage of 3.50 kV, a capillary voltage of 40 V and a capillary temperature of 325°C.
Mass spectra extraction and deconvolution were performed using PEAKS Studio (Bioinformatics Solutions Inc. v8.5). Mass spectra were searched against the Homo sapiens database (UniprotKB) using a precursor mass 1.0 Da (monoisotopic mass) and a fragment ion mass 0.5 Da. Non-specific cleavages were allowed at both ends of the peptide and the number of allowed missed cleavages was set to 5. Specified variable modifications included: carbamidomethylation, deamidation (NQ) and oxidation (M). False positives were eliminated through a systematic four-tier process, which excluded proteins based on the following criteria: i) proteins that did not satisfy a P-value (probability of false identification) score of −10lgP ≥ 15 in peptide-to-spectrum and peptide-to-protein sequence matching by the PEAKS Bioinformatics software (see Supplementary Information, Table S1 for all protein hits with −10lgP ≥ 15); ii) proteins identified in IPs from non-transfected (control) cells; iii) proteins that were not identified in at least two out of five independent IPs; iv) proteins with a score of >150 in the contaminant repository for affinity purification (CRAPome; www.crapome.org) [54]. Based on known caveats of the “two peptide” rule which often results in increased false discovery rates [55], protein hits with single peptide matches were retained if they met a peptide-to-spectrum score of −10lgP ≥ 15 and peptide-to-protein matches were unique to a protein group, which was further verified by manual blasting of peptide sequences against the UniProt database. For all single peptide hits, annotated MS/MS spectra with precursor mass, charge and mass error, retention times, number of spectral matches and −10lgP scores are provided in Supplementary Information, Table S2.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [56] partner repository with the dataset identified PXD012977 and 10.6019/PXD012977 (Username: reviewer71846@ebi.ac.uk, Password: 34TWLIq5).
2.7. Bioinformatic analyses & data graphing
Protein hits were cross-checked against the Biological General Repository for Interaction Datasets (BioGRID; https://thebiogrid.org), septin interactomes [52, 57] and reviews of septin interactions [22], which were used for in litero verification of interactions (asterisks in Figure 2) and to generate Table I. Non-septin protein hits were spatially mapped by assigning each individual protein into one or more subcellular organelles and structures based on knowledge of protein localization in databases such as the human protein atlas (www.proteinatlas.org), GeneCards (www.genecards.org), UniProt (www.uniprot.org), COMPARTMENTS (https://compartments.jensenlab.org) and the Gene Ontology Consortium (www.geneontology.org). Subcellular localizations of protein were further corroborated and researched with pubmed (https://www.ncbi.nlm.nih.gov/pubmed/) queries. A combination of these databases and pubmed searches were used for clustering protein hits into functional categories.
Figure 2. SEPT9 isoforms with shorter N-terminal extensions interact with multiple paralogs of the SEPT6 group.
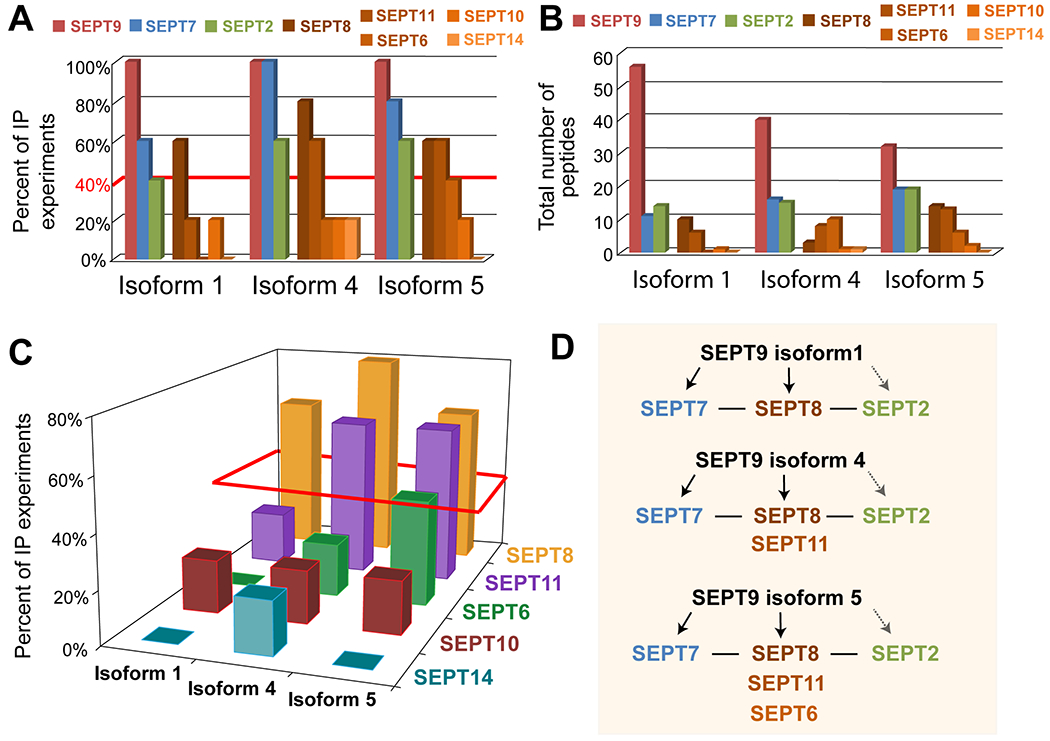
(A) Percent of IP experiments that resulted in the detection of septin paralogs (SEPT9, SEPT7, SEPT2, SEPT8, SEPT11, SEPT6, SEPT10, SEPT14) for each SEPT9 isoform. Red line indicates the cut-off (≥40% of experiments) for bona fide interactions. (B) Bar graph shows the cumulative number of septin peptides that were obtained from five independent immunoprecipitations of GFP-SEPT9 isoforms 1, 4 and 5. (C) Three-dimensional graph of the SEPT6 group paralogs (SEPT6, SEPT8, SEPT10, SEPT11, SEPT14) that co-IPed with SEPT9 isoforms 1, 4 and 5. The z axis indicates the percentage of experiments that resulted in detection of each SEPT6 group septin. The red parallelogram outlines the septin paralogs that were detected in ≥40% of the IP experiments. (D) Schematic summarizing the interactions of each SEPT9 isoform with septin paralogs. Based on previous evidence, solid arrows reflect direct interactions of SEPT9 with SEPT7 and potentially SEPT8, while the dashed opaque arrow indicates a potential indirect interaction with SEPT2 (via septins of the SEPT6 group). Solid lines denote direct interactions between SEPT7, SEPT6 group septins and SEPT2.
Table I:
Septin-interacting proteins that were pulled down but did not meet threshold criteria
| Hits in this study | Interactions in BioGRID & Published Literature | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Proteins | Isoform 1 | Isoform 4 | Isoform 5 | SEPT9 | SEPT7 | SEPT2 | SEPT8 | SEPT11 | SEPT6 |
| OBSL1 (Obscurin Like-1) | OBSL1 | OBSL1 | OBSL1 | ||||||
| BAX (BCL2 Associated X) | BAX | BAX | |||||||
| ESF1, nucleolar pre-rRNA processing protein | ESF1 | ESF1 | |||||||
| FN1 (fibronectin 1) | FN1 | FN1 | FN1 | ||||||
| ANKRD12 (Ankyrin repeat domain 12) | ANKRD12 | ANKRD12 | |||||||
| KMT2B (lysine methyltransferase 2B) | KMT2B | KMT2B | |||||||
| Cyclin-Dependent Kinases | CDK4 | CDK15 CDK4 |
CDK1 | CDK5 | CDK14 | CDK5 | |||
| Component of Oligomeric Golgi Complex | COG6 | COG4 | |||||||
| DEAH-Box Helicase | DHX38 DHX9 |
DHX36 | |||||||
| Eukaryotic translation Elongation Factor 1A | EEF1A2 | EEF1A1 | |||||||
| Heat Shock Protein Family A | HSPA1B HSPA2 HSPA9 |
HSPA5 | HSPA5 HSPA8 |
HSPA5 | HSPA5 | ||||
| Histone Demethylase | KDM3A | KDM1A | |||||||
| Myosin Heavy Chains | MYH10 MYH14 MYH7B |
MYH10 MYH9 |
|||||||
| NADH Dehydrogenase (Complex 1) | NDUFAB1 | NDUFV2 | NDUF36 | ||||||
| Pleckstrin Homology Domain Containing Protein | PLEKHH2 | PLEKHG2 | PLEKHH2 | PLEKHF2 | PLEKHF2 | ||||
| Pre-mRNA Splicing Factors | PRPF38B | PRPF3 PRPF8 |
PRPF3 PRPF8 |
||||||
| RNA Binding Motif Protein | RBMY1J | RBMX RBMXL1 |
RBMX | RBM25 RBM39 |
RBM39 | ||||
| Regulator of G-protein Signaling | RGS14 | RGS3 | RGS3 | ||||||
| SWI/SNF chromatin remodeling complexes | SMARCC2 | SMARCA5 | SMARCA5 | SMARCC2 | SMARCA2 | ||||
| Spectrin beta chain, non-erythrocytic | SPTBN5 | SPTBN4 | SPTBN1 | ||||||
| Tripartite Motif Family | TRIM67 | TRIM67 | TRIM67 | TRIM14 | TRIM55 | TRIM14 TRIM55 |
TRIM14 TRIM32 |
||
Venn diagrams were made according to the numerical size of interactors using the Venn diagram plotter (https://omics.pnl.gov/software/venn-diagram-plotter) developed by the Pacific Northwest National Laboratory (US Department of Energy). Two- and three-dimensional graphs were generated in Microsoft Excel and imported into Abobe Illustrator, where figures were assembled.
2.8. Quantifications of nuclear and cell size, and cell growth rates
MCF-7 cells (2x105) were seeded on coverslip (No 1.5, VWR), which were coated with collagen (Advanced BioMatrix), and allowed to adhere for 4 h in growth media. Cells were fixed with 4% paraformaldehyde (PFA) for 10 minutes, then washed and rinsed with PBS. Fixed cells were permeabilized with 0.1% Triton X-100 for 15 minutes and blocked with 2% BSA in PBS for 1 h after washing and rinsing with PBS. Following blocking, cells were incubated with DAPI (SIGMA, D9542), which labels nuclei, and wheat germ agglutinin (WGA) Alexa Fluor® 647 conjugate (Invitrogen, W32466), which labels cell membranes, using the protocol (concentration and incubation times) recommended by the vendors. Coverslips were mounted in FluorSave™ aqueous mounting medium (EMD Millipore) and imaged with an inverted Zeiss AxioObserver microscope with a 63X/1.4NA oil objective and the Slidebook 6 software. Morphometric analyses were performed with the Slidebook 6 software using the DAPI and WGA fluorescence to outline the nuclear and cell periphery, respectively, and create masks of their corresponding areas. Surface areas were derived automatically from the labeled masks with the Slidebook 6 software. Nuclear aspect ratios were derived after measuring the long and short radius of each nucleus. Cell complexity measurements were calculated by deriving the ratios of total length of cell perimetry to cell surface area. Mean values, standard errors of the mean (S.E.M) and P values were calculated in the GraphPad Prism software. Statistical significance (P values < 0.05) was derived with a one-way ANOVA test and Dunnett’s test for multiple comparisons.
Growth curves were generated by seeding cells (3.5x105; t = 0 days) in multiple 60 mm cell culture dishes in complete medium. After the lapse of each day (24 hours), three cell culture dishes per each cell line were trypsinized, pelleted and resuspended in equal volumes of PBS for counting with a hemocytometer. Total cell numbers were derived and averaged for each triplicate of plates, and plotted for days 0, 1, 2, 3 and 4 for each MCF-7 cell line.
2.9. Imaging and quantification of primary cilia
MCF-7 were plated on glass coverslips (No 1.5) that were coated with type I collagen. After ~48 h, cells were fixed for 15 minutes with 4% PFA in PHEM buffer pH 6.9 (60 mM PIPES, 25 mM HEPES, 10 mM EGTA, 2 mM MgCl2), washed and rinsed three times with PBS, and permeabilized with 0.1% Triton X-100 in PBS for 15 minutes. Following 1 h of blocking with 2% BSA in PBS, cells were stained with a mouse monoclonal antibody against acetylated tubulin (clone 1-11B-1; Sigma-Aldrich T7451) for 2 h in block buffer. Cells were washed with block buffer three times for 5 minutes each, and incubated with a secondary donkey Alexa Fluor 594 AfiiniPure F(ab’)2 anti-mouse IgG (Jackson ImmunoResearch) and subsequently with DAPI (SIGMA D9542) before mounting on microscope slides with FluorSave™ Aqueous Mounting Medium (EMD Millipore). Microscopy was performed on an inverted Zeiss AxioObserver microscope with a 1.63X/1.4 objective and using the Slidebook 6.0 software to acquire 3D image stacks at 0.25 μm step-size. Quantifications were performed by visually inspecting images for the presence of acetylated tubulin-labeled basal bodies on the apex of DAPI-stained nuclei, and the presence of an axoneme. Statistical significance was assessed by a chi-square test of independence.
MDCK cells were plated on collagen-coated coverslips and transfected with GFP, GFP-SEPT9_i1 or GFP-SEPT9_i5 using Lipofectamine 2000 (Invitrogen). After 48 h, cells were stained for acetylated tubulin as described above and mounted on coverslips for immunofluorescence images using the Zeiss AxioObserver microscope with a 1.63X/1.4 objective and the Slidebook 6.0 software. Quantifications of ciliary lengths were performed in the Fiji software using the freehand tool to trace the entire axoneme from its base to the tip. Lengths were derived automatically from the traced line masks. Data distributions were assessed for normality with a D’Agostino & Pearson normality test, and p values were derived with the Mann-Whitney U-test.
3. Results
3.1. The interactome of SEPT9 isoforms i1, i4 and i5 reveal unique and overlapping binding partners
We sought to determine the interactome of SEPT9 isoforms 1 (SEPT9_i1), 4 (SEPT9_i4) and 5 (SEPT9_i5), which share the least overlap in their NTEs from all SEPT9 isoforms (Figure 1A) and therefore, may have differential interactions and functions. MCF-7 cell lines that stably express GFP-SEPT9_i1, GFP-SEPT9_i4 and GFP-SEPT9_i5 were previously established in order to investigate isoform-specific oncogenic properties [44]. Owing to the low copy number of their endogenous SEPT9 gene [44], MCF-7 cells allow to recapitulate the upregulation of SEPT9 isoforms that occurs in breast cancer cells by expressing exogenous GFP-tagged chimeras of SEPT9 isoforms. GFP-SEPT9 isoforms assembled into filaments (Figure 1B) and colocalized with actin filaments and/or microtubules (Supplementary Information, Figure S1), indicating that over-expression was not at levels that would disrupt assembly of SEPT9 isoforms into higher-order filaments and their interaction with the cytoskeleton.
Figure 1. SEPT9 isoforms 1, 4 and 5 have overlapping and distinct interactors in MCF-7 breast cancer cells.
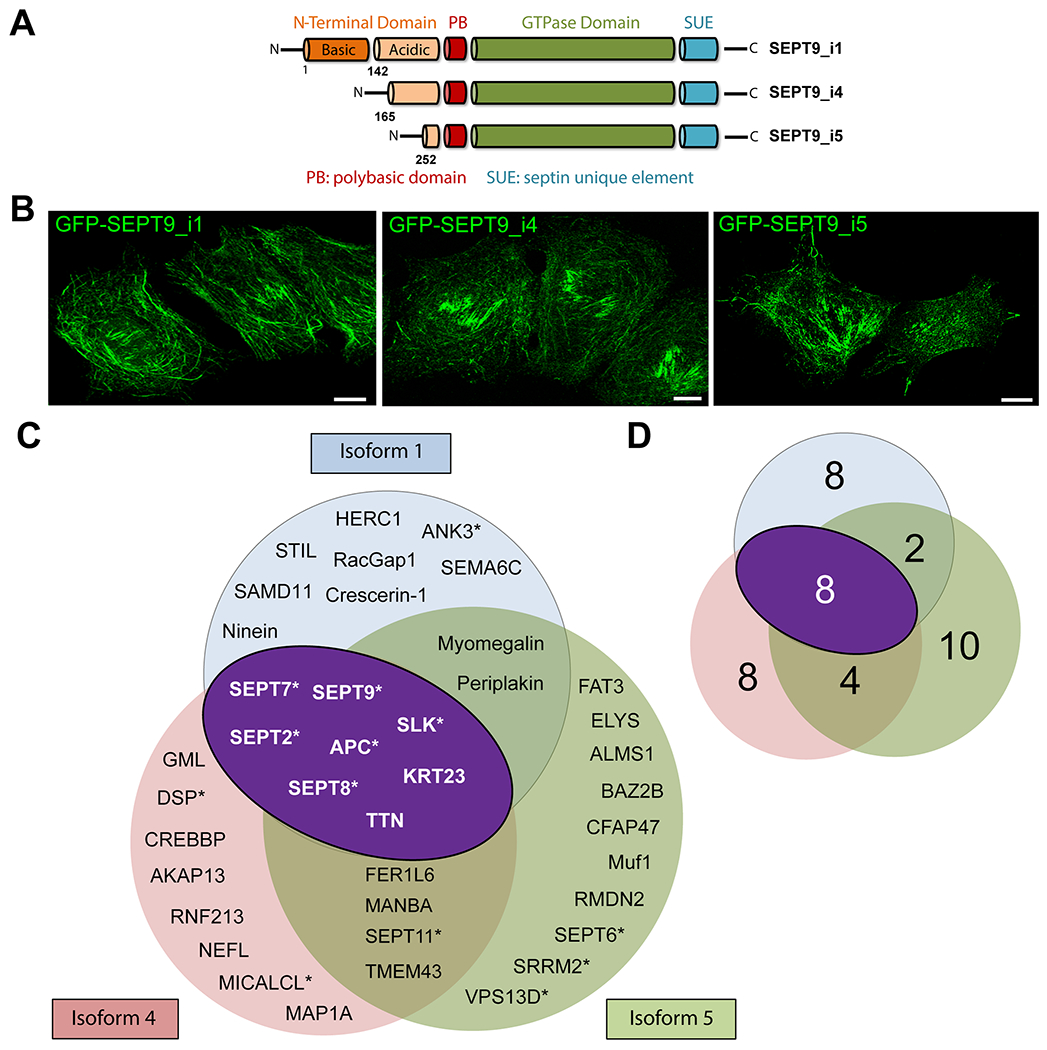
(A) Schematic depicting the lengths and major domains of the human SEPT9_i1, SEPT9_i4 and SEPT9_i5, which vary in the length at their N-terminal extensions. SEPT9_i4 lacks the N-terminal 164 amino acids of SEPT9_i1, and SEPT9_i5 lacks the N-terminal 251 and 87 amino acids of SEPT9_i1 and SEPT9_i4, respectively. (B) Confocal microscopy images of MCF-7 cells that stably express GFP-SEPT9_i1, GFP-SEPT9_i4 and GFP-SEPT9_i5. (C-D) Venn diagrams depicts the names (C) and cumulative numbers (D) of distinct and overlapping interactors of GFP-SEPT9 isoforms 1 (blue), 4 (pink) and 5 (green) in MCF-7 breast cancer cells. The overlapping interactors (white letters) between isoforms 1, 4 and 5 are the same with the overlapping interactors of isoforms 1 and 4, and are shown in the dark purple oval in white text. Asterisks denote previously reported interactions.
To identify the interactome of each SEPT9 isoform, MCF-7 cells were lysed and immunoprecipitations were performed with an antibody against GFP coupled to an amine-reactive resin (Figure S2). Lysates from untransfected MCF-7 cells were used as a negative control for identifying proteins that were precipitated non-specifically by the anti-GFP-coupled resin. Five independent immunoprecipitations were performed with lysates from each MCF-7 SEPT9 isoform-specific cell line. Protein-complexes were eluted from the antibody-bound resin and digested with trypsin prior to injecting into an ultra high-performance liquid chromatography (UHPLC) unit coupled to an electrospray ionization mass spectrometer (ESI-MS). Mass spectra were extracted and deconvoluted with the PEAKS Bioinformatics Studio software, and protein identities were derived by searches against the UniprotKB database for Homo sapiens.
For each isoform, protein hits were subjected to a four-tier stringency for the elimination of false positives. First, only protein hits with P-value (probability of false identification) score of −10lgP ≥ 15 in peptide-to-spectrum and peptide-to-protein matching were retained [58]. Second, proteins that were identified in untransfected (control) cell lysates were removed from the proteome of each SEPT9 isoform. Third, protein hits that were not identified in at least two independent immunoprecipitations were excluded. Fourth, we removed protein hits that are most frequently identified as contaminants per the contaminant repository for affinity purification (CRAPome) database; protein hits that exceeded the score of 150 in the CRAPome database (Table S3).
By applying these stringency criteria, the interactome of SEPT9 isoforms 1, 4 and 5 consisted of 18, 20 and 24 proteins, respectively (Figure 1C and 1D). Approximately, a third of the binding partners of each isoform have been previously reported (Figure 1C; proteins with asterisk), which validates our approach and indicates a high-confidence proteome. Of note, we detected a number of proteins that were previously reported to interact with SEPT9 (Table I, light shaded rows) or septins that form a complex with SEPT9, but did not meet stringency cutoffs; these included interactors that belonged to families of proteins of known association with septins (Table I, dark shaded rows) including myosins, which were reported in a recent proteomic study of SEPT9_i1 [123]. Hence, the interactomes of SEPT9 isoforms 1, 4 and 5 may exclude some bona fide interactions due to high-stringency criteria; a list of all protein hits from each independent experiment is provided in Supplementary Information Table S1.
The majority of the interactome (~75% of total proteins) of each SEPT9 isoform comprised non-septin proteins and nearly half (40-44%) was exclusively isoform-specific; i.e., binding partners were not shared with any other SEPT9 isoform. However, several proteins were common among all three isoforms or between two specific isoforms (Figure 1C). All three isoforms interacted with septins SEPT2, SEPT7 and SEPT8, which is consistent with previous findings of SEPT9 being part of hetero-octameric complex with subunits from the SEPT2, SEPT7 and SEPT6 groups; SEPT8 belongs to the SEPT6 group [14, 15]. In addition to these septins, all three isoforms interacted with the adenomatous polyposis coli (APC) and the STE20-like serine/threonine protein kinase (SLK), which have been previously reported as septin interactors [57, 59], as well as titin (TTN) and keratin 23. Using western-blots, we confirmed the interaction of SEPT9 with APC as well as RacGAP1 and ankyrin-3 (ANK3), which was also recently reported to co-immunoprecipate with septins (SEPT5, SEPT6) in neurons [60] (Figure S3). SEPT9 isoforms 1 and 5 had two additional common binding partners, periplakin and myomegalin, which was validated by immunoblotting (Figure S3). SEPT9 isoforms 4 and 5 shared SEPT11, the transmembrane protein 43 (TMEM43), the FER1-like family member 6 (FER1L6) and β-mannosidase as additional binding partners. Taken together, these data show that SEPT9 isoforms have common and unique interactors, which may arise respectively from shared protein domains (e.g., the GTP-binding domain) and differences in their NTEs.
3.2. SEPT9 isoforms with shorter NTEs exhibit more promiscuous interactions with septins of the SEPT6 group
Septins were the most abundant group of proteins that co-immunoprecipitated with each SEPT9 isoform, which is consistent with their assembly into heteromeric complexes. SEPT9 itself was detected in all five independent immunoprecipitations against each isoform, indicating that SEPT9 was successfully immunoprecipitated in every run (Figure 2A). Collectively, the number of SEPT9 peptides were the most abundant - 56 for GFP-SEPT9_i1, 40 for GFP-SEPT9_i4 and 32 for GFP-SEPT9_i5 – and correlated with the increasingly shorter lengths of each SEPT9 isoform (Figure 2B). Of note, the intensity of surrogate peptides, which is indicative of peptide abundance, was independent of the expression levels of GFP-SEPT9 isoforms. For example, the intensity of peptides (HVDSLSQR, SVQPTSEER), which are shared among all SEPT9 isoforms, was similar in immunoprecipitations of GFP-SEPT9_1 and GFP-SEPT9_i5 which exhibited the highest difference in expression levels from all the three isoforms (Figure S4). Thus, peptide detection and the identified interactors were not impacted by the relative levels of GFP-SEPT9 isoform expression.
Among all septin paralogs that interacted with the SEPT9 isoforms, SEPT7 was most frequently pulled down (Figure 2A–B). Although less frequent than SEPT7, SEPT2 and SEPT8 were also present in the interactome of each SEPT9 isoform (Figure 2A,B). Strikingly, all other septin paralogs that co-immunoprecipitated with SEPT9 belonged to the SEPT6 group and their frequency of detection varied depending on SEPT9 isoform (Figure 2C). For SEPT9_i1, SEPT8 was the predominate paralog of the SEPT6 group; SEPT10 and SEPT11 were pulled down only once out of five immunoprecipitations. In contrast, SEPT9_i4 possessed both SEPT8 and SEPT11 as main binding partners of the SEPT6 group (Figure 2C). In immunoprecipitations of GFP-SEPT9_i5, SEPT6 emerged as a third major binding partner along with SEPT8 and SEPT11 (Figure 2A–C). These results reveal that SEPT9 isoforms with shorter NTEs have more promiscuous interactions with septins of the SEPT6 group (Figure 2D).
3.3. Spatial mapping of the SEPT9 interactome shows isoform-specific differences in binding partners of distinct subcellular localizations.
To gain a better insight into the oncogenic pathways and properties of SEPT9 isoforms, we performed spatial and functional clustering of all non-septin binding partners. Based on the bioinformatics databases, we mapped the interactome of each SEPT9 isoform by assigning each protein to a subcellular organelle or structure; note that several proteins associate with more than a single organelle (Table S4). We used this categorization to quantify the relative distribution of the binding partners of SEPT9 isoforms in subcellular locales such as the cytoskeleton (actin filaments, microtubules, intermediate filaments), centrosomes, primary cilia, plasma membrane, nuclei, Golgi, endosomes/lysosomes, mitochondria and cell adhesions.
The spatial distributions of the interactomes of each SEPT9 isoform followed common patterns, but isoform specificities were also found in the organelle enrichment of binding partners (Figure 3A). Approximately half of the interactome of all three SEPT9 isoforms were proteins that localized to the cytoskeleton (20-30%) and the plasma membrane (15-20%). The majority of the cytoskeletal proteins (40-50%) associated with microtubules, while the remaining were linked to actin and intermediate filaments; only a single spectrin-associated protein (ankyrin 3) was found as an isoform 1-specific partner. The rest of the proteome exhibited a fairly equivalent distribution into the nucleus (5-15%), cell adhesions (~10%), the centrosome (5-10%), endolysosomes (3-8%), Golgi (~5%) and primary cilia (0-10%).
Figure 3. Spatial mapping of the SEPT9 interactome reveals isoform-specific interactions with proteins of distinct subcellular localizations.
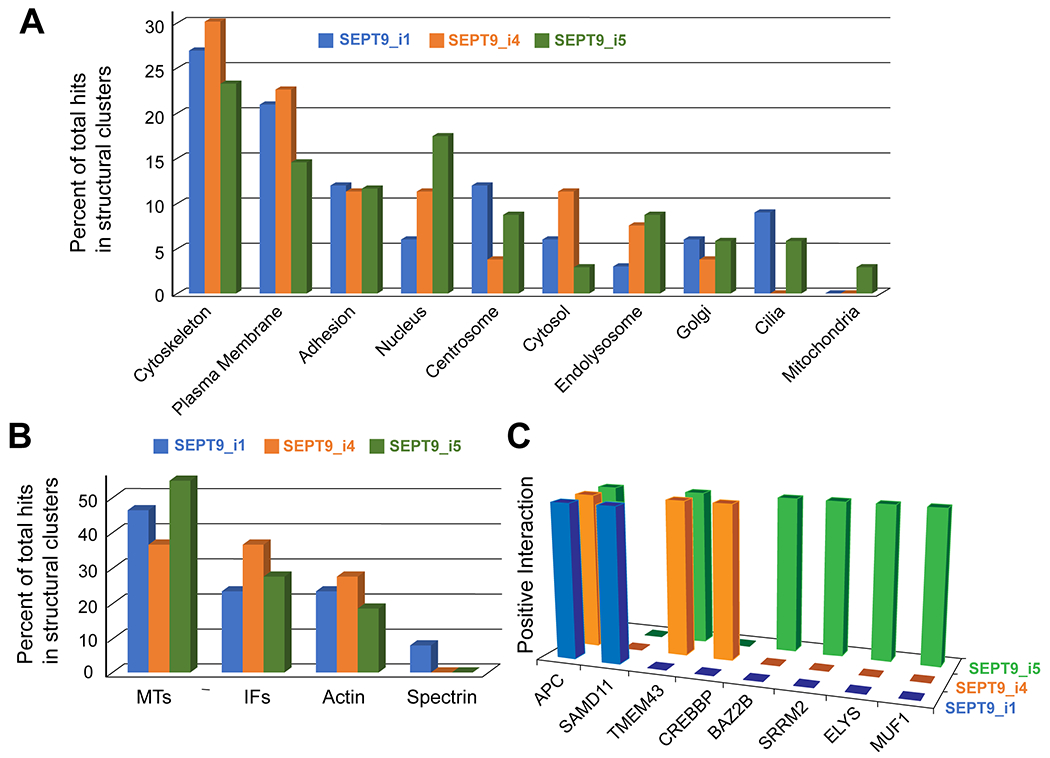
(A) The non-septin binding partners of SEPT9 isoforms 1, 4 and 5 were assigned to one or more subcellular locations based on published findings and bioinformatics databases. The protein number for each subcellular localization was divided by the total number of proteins in all subcellular clusters and plotted as percentage of total protein hits for each SEPT9 isoform. This provided an indication of the relative enrichment of the non-septin interactors of each SEPT9 isoform in distinct subcellular locales. (B) Protein interactors that were components of the MT, IF, actin and spectrin cytoskeletons or associated with these cytoskeletons were further subdivided according to their cytoskeletal identity and affiliation. Relative enrichment in each of the four cytoskeletal networks was quantified by calculating the percentage of total protein interactors assigned to each cytoskeleton. (C) The three-dimensional bar graph shows the identify of protein interactors with nuclear localization for each SEPT9 isoform.
Relative enrichment of SEPT9 binding partners in the cytoskeleton, plasma membrane, Golgi or cell adhesions did not vary significantly across isoforms, but distinct differences were noted for proteins that localize to the nucleus, the centrosome and primary cilia. SEPT9 isoforms with truncated NTEs possessed a greater diversity of interactors with nuclear localization (Figure 3A–C), and several isoform-specific partners were revealed. More strikingly, there was a paucity of centrosomal and ciliary partners for SEPT9_i4, while isoforms 1 and 5 had unique and overlapping proteins that localized to the centrosome and primary cilia (Figure 3A and 4A,B).
Figure 4. SEPT9 isoform-specific interactions with centrosomal and ciliary proteins.
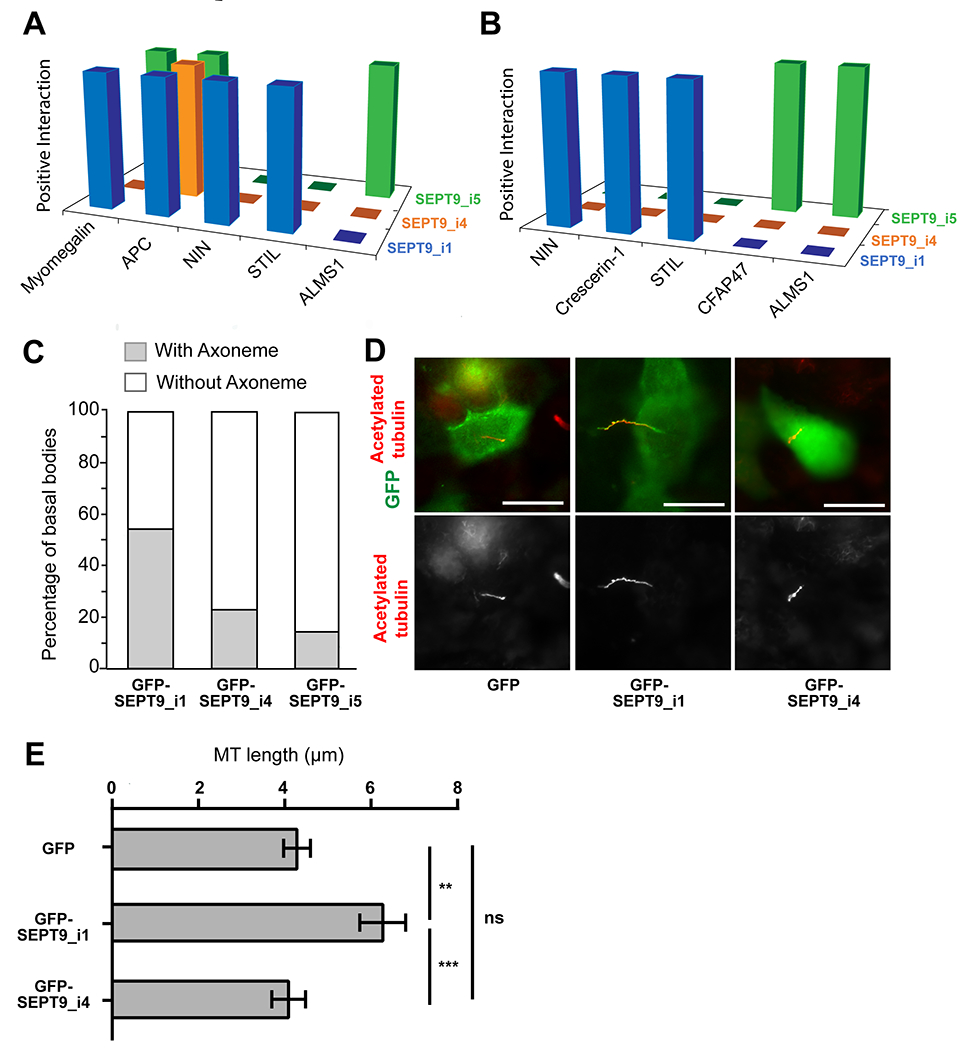
(A-B) The three-dimensional graphs show the centrosomal (A) and ciliary (B) protein hits of SEPT9 isoforms 1, 4 and 5. (C) Quantification shows the percentage of apical basal bodies with and without axonemes in MCF-7 cells that express GFP-SEPT9_i1 (n = 30), GFP-SEPT9_i4 (n = 26) and GFP-SEPT9_i5 (n = 23). The differences in basal bodies with axonemes were statistically significant; chi-square test of independence X 2 (2, N=79) = 12.91, p = .0015. (D) Images of MDCK cells expressing GFP, GFP-SEPT9_i1 and GFP-SEPT9_i4, which were stained for acetylated tubulin. Scale bars, 10 μm. (E) Bar graph shows the mean (±SEM) length of the ciliary axonemes of MDCK cells expressing GFP (n = 44), GFP-SEPT9_i1 (n = 46) and GFP-SEPT9_i4 (n = 49). The Mann-Whitney U test was used to assess statistical significance. **, p < 0.001; ***, p < 0.0001; n.s., non-significant.
3.4. Differential interactions of SEPT9 isoforms with nuclear, centrosomal and ciliary proteins
Several oncogenes and tumor suppressors are nuclear proteins involved in gene transcription and DNA damage repair [61]. Moreover, centrosomes and cilia are linked to cancer pathogenesis through their respective roles in genomic instability and signaling pathways that control cell growth and proliferation (e.g., hedgehog, notch and WNT) [62, 63]. Therefore, SEPT9 isoform-specific interactions with nuclear, centrosomal and ciliary proteins could have implications for the mechanism of tumor progression, depending on the SEPT9 isoform of altered expression.
In the nuclear interactome of SEPT9, the tumor suppressor APC was a shared binding partner among all isoforms and TMEM43/LUMA was found as a common partner of isoforms 4 and 5. The rest of the interactome was isoform specific (Figure 3C). SEPT9_i5 interacted with ELYS (embryonic large molecule derived from yolk sac; also known as AT-hook containing transcription factor 1), a component of nuclear pore complex which together with TMEM43/LUMA have been identified as inner nuclear membrane protein partners of VAPB (vesicle-associated membrane-protein associated protein B) [64, 65]. New studies show that VAPB interacts with VPS13D on ER membranes [66], and interestingly VPS13D was also pulled down as a SEPT9_i5-specific partner (Figure 1C and 5C).
Figure 5. Functional clustering of the non-septin binding partners of SEPT9_i1, SEPT9_i4 and SEPT9_i5, and isoform-specific interactions with signaling and ubiquitinating factors.
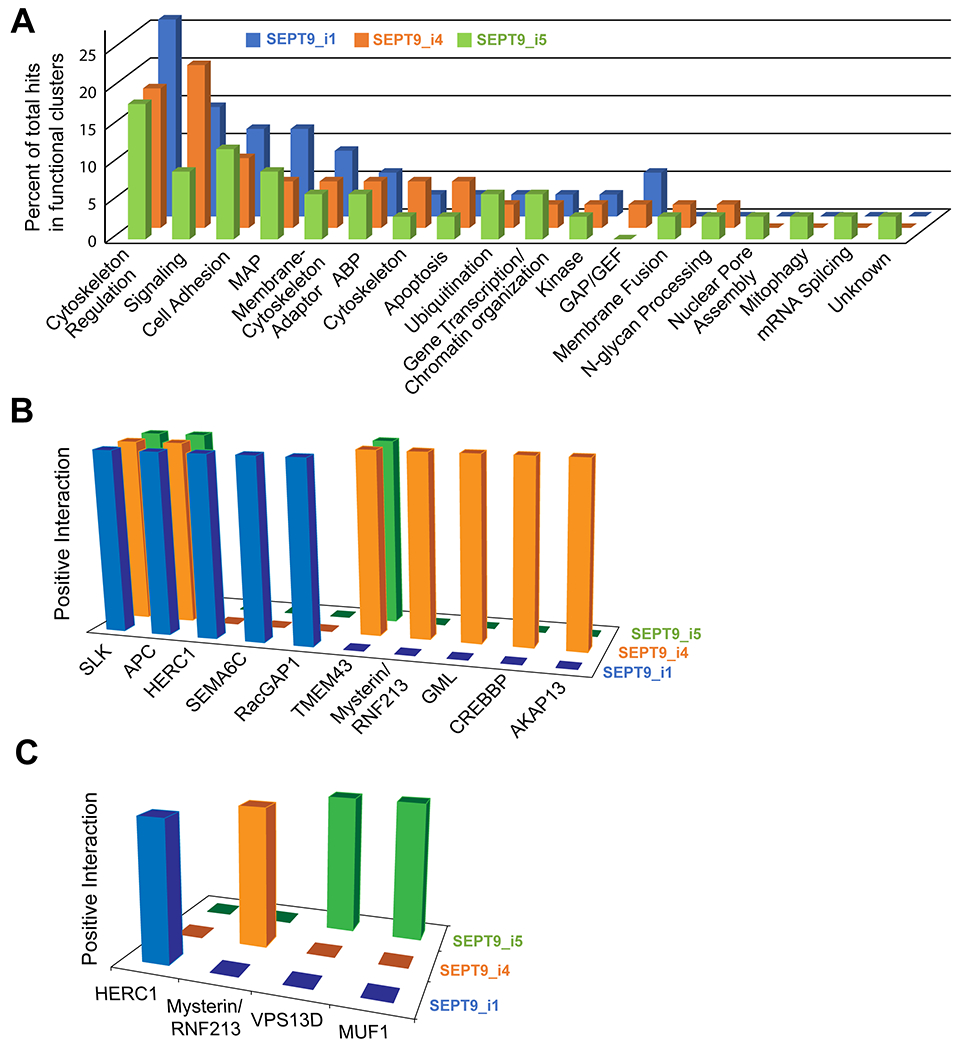
(A) Three-dimensional graph of the distribution of non-septin interactors of SEPT9_i1, SEPT9_i4 and SEPT9_i5 into functional clusters. Proteins of the interactome of each SEPT9 isoform were assigned into biological functions according to bioinformatic databases and published literature. Percent enrichment of each functional cluster was calculated by dividing the number of proteins in each cluster by the total number of proteins in all clusters, and multiplying by 100. (B-C) Three-dimensional graphs show the identify of protein interactors with signaling (B) and ubiquitinating (C) functions.
Among the nuclear protein hits, ELYS and CREBBP (CREP-binding protein), which were identified as isoform 5- and 4-specific interactors, respectively, have been shown to regulate nuclear size [67]. To test whether SEPT9 isoforms 4 and 5 regulate nuclear size, we examined the dimensions of the nuclei in MCF7 cells. Compared to GFP-SEPT9_i1 and _i5, the nuclear surface area of cells that express GFP-SEPT9_i4 was reduced (Figure S5A). However, the nuclear aspect ratio (long-to-short axis ratio) was not altered, indicating that SEPT9_i4 impacts nuclear size without affecting nuclear shape (Figure S5B). This reduction in nuclear size reduction was accompanied by an overall reduction in cell size and the growth rates of MCF-7 cells expressing GFP-SEPT9_i4 (Figure S5C and S5E), while cell morphology was not altered as indicated by quantification of cell complexity (ratio of perimeter length to surface area) (Figure S5D). These data suggest that SEPT9_i4 may regulate nuclear size and cell growth by suppressing CREBBP functions in nuclear size and cell proliferation [67–69].
Similar to the nuclear interactome, several centrosomal and ciliary proteins were identified with specific SEPT9 isoforms (Figure 4A–B). A few of these proteins were common to centrosomes and cilia, which stem from centrioles and thus, have common protein components [70]. All three SEPT9 isoforms interacted with APC, which has been reported to localize and function in centrosomes [71]. The Golgi/centrosome protein myomegalin was a common binding partner of SEPT9_i1 and SEPT9_i5 (Figure 4A). These isoform specific interactions with myomegalin were independently validated by western blots (Figure S3). In contrast to APC and myomegalin, we identified ninein and STIL (centriolar assembly protein) as SEPT9_i1-specific partners, while ALMS1 and CFAP47 (ciliary and flagella protein 47) emerged as a SEPT9_i5-specific interactors (Figure 4B). These interactions are consistent with recent findings of SEPT7 localization and function in the centrosome [72], and SEPT1 interactions with centrosomal proteins (e.g., CEP170, γ-tubulin) on Golgi-membranes [73].
Septin interactions with centrosomal proteins bear significance for the formation of primary cilia, the formation and functions of which involve septins [74, 75]. Our data indicate that SEPT9 isoforms might be critical for the transition of centrosomes to basal bodies and cilia through interactions with components of subdistal appendages, which promote basal body alignment and attachment to the Golgi complex and microtubules of the cell body [76]. Notably, the isoform 5 interactor ALMS1, which localizes to the basal bodies of primary cilia and is required for ciliary biogenesis, regulates the localization of the centrosomal Nek2-associated protein 1 (C-NAP1) [77–79]. C-NAP1 is in turn critical for the recruitment of the subdistal appendage components ninein, a SEPT9_1 hit, and p150Glued and CEP170, which interact SEPT7 and SEPT1, respectively [72, 73, 80]. However, the SEPT9_i1-specific hit crescerin-1, which promotes ciliary elongation by facilitating the addition of tubulin dimers [81], indicates that SEPT9_i1 might be specifically involved in the growth of the ciliary axoneme.
To test if indeed SEPT9_i1 has a unique role in ciliary growth, we examined the primary cilia of MCF-7 cells. Consistent with previous work, which showed that cilia are present at a very low frequency in breast cancer cells [82], MCF-7 cells were largely devoid of cilia with an axoneme that extends from the apical surface. However, a fraction of MCF-7 cells contain apical basal bodies, which are enriched with acetylated tubulin. We found that ~50% of these basal bodies were connected to an axoneme in MCF-7 cells that express GFP-SEPT9_i1 (Figure 4C). Strikingly, this axonemal occurrence was over two-fold higher than in cells that express GFP-SEPT9 isoforms 4 and 5 (Figure 4C). In agreement with a SEPT9_i1-specific role in the growth of the axoneme, expression of GFP-SEPT9_i1 in MDCK cells also increased axonemal lengths (Figure 4D–E). Taken together, these data underscore the known role of septins in ciliary biogenesis, and suggest SEPT9 isoforms have both distinct and overlapping functions in different stages and processes of ciliogenesis.
3.5. Functional clustering of the SEPT9 proteome points to isoform-specific roles in cell signaling and degradative pathways.
Given the isoform-specific interactions with nuclear, centrosomal and ciliary proteins, we probed for isoform-specific functions by sorting the interactome of each SEPT9 isoform according to protein function. Based on published literature and bioinformatic databases, all non-septin binding partners were assigned to one or more functional clusters, which broadly represented the known functions of each protein interactor (Table S4). Cytoskeleton-related proteins were separated into components of cytoskeletal filaments, denoted as “cytoskeleton”, and regulators of the cytoskeletal organization, which were further grouped into microtubule-associated proteins (MAPs), actin-binding proteins (ABPs) and membrane-cytoskeleton adaptors. Functional clusters were also created based on protein involvement in cell signaling and adhesion, gene transcription/regulation, membrane fusion, post-translational modifications such as ubiquitination, phosphorylation (kinases) and N-glycan processing, regulation of small GTPases (GAPs/GEFs), mitophagy, apoptosis, mRNA splicing and nuclear envelope assembly. Each functional cluster contained one or more proteins, which were quantified as percentage of the total number of proteins in all clusters to indicate the relative enrichment of each cluster in the interactome of a SEPT9 isoform.
As predicted by the spatial organization of the interactome, cytoskeleton-related functions were the most enriched for all three isoforms (Figure 5A). The functional cluster of cytoskeletal regulation was highly enriched (~20-25% of the entire interactome), and the MAP, membrane-cytoskeleton adaptors and cell adhesion were also in the top five most-enriched clusters (Figure 5A). Notably, the interactome of the SEPT9 isoform 1 was more enriched in cytoskeletal regulators and MAPs compared to isoforms 4 and 5, which lack the N-terminal basic domain of SEPT9_i1 that interacts with microtubules and actin [25, 26]. Notably, cell signaling was the most enriched cluster for SEPT9_i4 (Figure 5A). The cell signaling cluster of SEPT9_i4 was 1.5- and 2.4-fold more enriched than SEPT9_i1 and SEPT9_5, respectively. The interactomes of isoforms 4 and 5 were more functionally diverse than isoform 1. In particular, isoform 5 exhibited the highest diversity, possessing a number of unique binding partners with functions in mRNA splicing, nuclear pore assembly and mitophagy.
The cell signaling cluster of SEPT9_i4 contains four unique binding partners (Figure 5B): A-kinase anchoring protein 13 (AKAP13), the CREB-binding protein (CREBBP), GML (glycosylphospatidylinositol anchored molecule like) and mysterin/RNF213, all of which are linked to the pathology of breast cancer. AKAP13 is a RhoA GEF that activates Rho signaling and functions as a scaffold for the phosphorylation of the estrogen receptor alpha (ERα) by protein kinase A, which results in resistance to tamoxifen, an estrogen analog that targets ER positive breast tumors [83–85]. Hence, SEPT9_i4 over-expression may impact both the pathogenesis and treatment of breast cancer through AKAP13. Conversely, CREBBP interacts with the BRCA1 tumor suppressor [86], which is the most frequently mutated gene in familial cases of breast cancer. GML is a target of the p53 tumor suppressor and it has been linked to apoptotic pathways that sensitize cancer cells to therapeutic treatments [87–89]. Lastly, mysterin/RNF213 is a AAA ATPase with a ubiquitin ligase domain, which is implicated in signaling pathways involved in vascular development that supports cancer growth and metastasis [90, 91]. Interestingly, mysterin is also involved in the stabilization of lipid droplets, which are regulated by SEPT9 in cells that are infected by the hepatitis C virus [92, 93].
In addition to the signaling cluster, ubiquitination also emerged as a distinct functional cluster with isoform-specific interactors (Figure 5C). SEPT9_i1 associated with HERC1, an E3 ubiqutin ligase that degrades the proto-oncogene serine/threonine-protein kinase c-RAF [94]. The SEPT9_i4 interactome included mysterin/RNF213, which is an E3 ubiquitin ligase [91], and SEPT9_i5 pulled down VPS13D and MUF1. VPS13D has a ubiquitin-associated domain and is involved in mitochondrial clearance (mitophagy) [95], while MUF1 co-assembles with the E3 ubiquitin ligase Cullin/Rbx1 [96]. The interaction with Vps13D is particularly interesting in the context of mitophagy as the mitochondrial SEPT5_i2 interacts with the E3 ubiquitin ligase parkin [97], and septins were recently shown to affect mitochondrial fission [98, 99]. Collectively, these data implicate septins in ubiqutinase-based mechanisms of degradation and may inform future work on understanding how septins are degraded and/or regulate protein degradation. Of note, septins have been shown to inhibit the degradation of several proteins with roles in breast cancer including the EGF receptor, the Erb receptor tyrosine kinase 2 (ErbB2), the c-Jun-N-terminal kinase (JNK) and the hypoxia-inducible factor HIF1α, but the underlying mechanisms are not well understood [48, 100–102].
4. Discussion
The evolutionary expansion of the mammalian family of septins into a multitude of septin paralogs and isoforms suggests a functional specialization, which may arise from distinct interactions and protein-binding properties. However, this has yet to be explored as septin interactomes remain poorly characterized. Proteomic profiling of SEPT9 isoforms, which are over-expressed in breast cancer, revealed isoform-specific differences in interactions with septin paralogs and non-septin proteins of distinct subcellular localizations and functions.
In the absence of isoform-specific antibodies against SEPT9_i4 and SEPT9_i5, our studies were conducted in cell lines that stably express fluorescent SEPT9 isoforms from an exogenous promoter. The localization and filamentous appearance of the over-expressed SEPT9 isoforms were consistent with those of endogenous SEPT9 [44, 46], but septin assembly and interactions may still differ from the native ones. Although this caveat warrants consideration in interpretations of the proteomic results of this work, it may also recapitulate conditions of SEPT9 gene amplification in breast cancer cells where up to 38 copies of the SEPT9 gene have been reported [44]. Given that MCF-7 is a mammary cell line with a low number of SEPT9 gene copies [44], our study may report on the interactome and pathophysiology of endogenously over-expressed SEPT9 isoforms.
Our results suggest that in MCF-7 breast cancer cells, SEPT9 isoforms form complexes with SEPT7, SEPT2 and septin paralogs of the SEPT6 group such as SEPT8, SEPT11 and SEPT6. This is consistent with previous studies showing that i) SEPT7 is a preferred and direct binding partner of SEPT9 [52, 53], and ii) members of the same septin group are exchangeable within the SEPT2/6/7/9 heteromer [13, 15]. It is unclear, however, why there are no other members of SEPT2 group in complex with SEPT9 isoforms and why the truncated SEPT9 isoforms 4 and 5 allow for more flexibility in the exchange of SEPT6 group subunits than the full length SEPT9_i1. The former could be explained by low expression of other SEPT2 group septins in MCF-7 cells [103], while the latter suggests that SEPT9 isoforms possess or enable differential interactions with septin paralogs of the SEPT6 group.
Yeast two-hybrid screens have shown that septins of the SEPT6 group interact directly with SEPT9 [52]. In addition, yeast three-hybrid screens have demonstrated that SEPT9/6/7 complexes can be formed with SEPT9 taking the place of SEPT2 within the canonical SEPT2/6/7 complex [53]. It is therefore plausible that SEPT9 isoforms hetero-dimerize with SEPT6 or form complexes with SEPT6/7 dimers. In this scenario, the full-length NTE of SEPT9_i1 could dictate preference for SEPT8, while the truncated NTEs of isoforms 4 and 5 allow for greater flexibility in interacting with SEPT11 as well as SEPT6. In support of this possibility, the NTE of the yeast septin Cdc3 has been reported to influence binding to the septin paralogs Cdc10 versus Cdc12 through allosteric autoinhibitory interactions with the GTP-binding domain of Cdc3 [17]. Hence, the lengths of the NTEs of SEPT9 isoforms could similarly impact binding to septins of the SEPT6 group, as N-terminal truncations could expose binding sites that allow for more promiscuous interactions; promiscuous septin-septin interactions have been shown to occur via the dimerization interface that involves the core GTP-binding domain [104]. Alternatively, lack of post-translational modifications from the N-termini of shorter SEPT9 isoforms could enhance the diversity of septin and non-septin binding partners (see below).
Spatial and functional clustering of the SEPT9 interactome suggests that the NTEs of SEPT9 isoforms may also bestow a spatial and functional specialization by impacting differentially interactions with non-septin proteins. Our results revealed that SEPT9 isoforms with truncated NTEs have more nuclear binding partners and their non-septin interactome are more functionally diverse. Compared to SEPT9_i1, the nuclear protein cluster was nearly two- and three-fold more enriched for SEPT9_i4 and SEPT9_i5, respectively. Similarly, the signaling protein cluster was markedly more enriched for SEPT9_i4, which appears to downregulate nuclear size and cell growth in an isoform-specific manner. Overall, these isoform-specific differences correlate with protein length- and sequence-dependent differences such as: i) a cytoskeleton-binding domain in the N-terminus of SEPT9_i1, which appears to bias the interactome more toward cytoskeleton-associated proteins; ii) a proline-rich domain in the N-terminal domain of SEPT9_i4, which in the absence of any upstream domains, could bias the interactome toward signaling proteins; proline-rich domains are common binding motifs for signaling molecules with SH3 domains; iii) lack of an NTE in the sequence of SEPT9_i5, which allows for more promiscuous interactions with septin paralogs of the SEPT6 group and consequently, may result in more diverse interactions with non-septin proteins.
In addition to these amino acid sequence differences, post-translational modifications are likely to contribute to isoform-specific interactions [105]. The N-terminus of SEPT9 contains phosphorylatable serines and threonines [106–109], and lysines which are subject to ubiquitylation, sumoylation, acetylation, and methylation [110–115]. Ubiquitylated residues are positioned in the conserved C-terminal sequences of SEPT9 isoforms 1, 4 and 5 [112, 113], but acetylated and methylated sites are in the most N-terminal sequence of SEPT9_i1 and therefore absent from SEPT9_i4 and SEPT9_i5 [111, 114]. In addition, SEPT9_i5 lacks the preponderance of the N-terminal residues, which can be phosphorylated and sumoylated [109, 110, 116, 117]. The absence or presence of isoform-specific post-translational modifications may enable or prohibit certain protein interactions and influence the intracellular localizations of SEPT9.
The increased enrichment of the cell signaling cluster and diversity of the SEPT9_i4 interactome bear significance in breast and ovarian cancers, in which SEPT9_i4 expression increases due to the expression of an mRNA transcript (SEPT9_v4*) with an alternative 5’-UTR sequence that is translated more efficiently [118, 119]. Notably, isoform 4-specific interactions with factors such as CREBBP and GML that interface with the estrogen receptor as well as the BRCA1 and p53 tumor suppressors suggest isoform-specific roles in the pathogenesis and treatment of breast cancer [86, 120]. By downregulating cell proliferation, SEPT9_i4 may impact the response of cancer cells to chemotherapy as observed with alterations in the expression of CREBBP and GML, which also regulate cell proliferation [69, 87]. Of note, SEPT9 isoforms have been implicated in cellular adaptation to anti-cancer drugs [38, 41].
Taken together with previous findings, the proteomic data of this study point to SEPT9 roles in the biogenesis of centrosomes and basal bodies, the formation and organization of the nuclear envelope and mRNA processing. SEPT9 interactions with STIL, ALMS1 and ninein add to recent findings on the regulation of centrosome duplication by SEPT7, and link septins to components of the subdistal appendages of centrioles (e.g., C-NAP1, ninein, p150GLUED) [72, 73, 80]. SEPT9 association with the inner nuclear membrane proteins TMEM43 and ELYS, and VPS13D, all of which partner with VAPB, suggests that SEPT9 may function in the post-mitotic assembly of the nuclear envelope from ER membranes, and promote nuclear membrane attachment to chromatin [64, 66, 121]. Lastly, SEPT9 isoform 5-specific interactions with proteins that regulate the expression of non-coding RNAs, pre-mRNA splicing and nuclear pore-chromatin interactions raises the possibility of isoform-specific effects on gene expression. In support of this possibility, transcriptomic profiling in MCF-7 cells indicates that SEPT9 isoforms have differential effects on gene expression [122], and thereby, may have a global impact on the cellular proteome.
In summary, proteomic profiling of the SEPT9 isoforms 1, 4 and 5 shows isoform-specific roles that have risen from truncations of their N-terminal sequences, which can determine and modulate interactions directly or indirectly through association with other septin paralogs. The interactomes of SEPT9 isoforms 1, 4 and 5 provide rare evidence for septin isoform-specific interactions, implicating SEPT9 isoforms in hitherto unknown mechanisms and pathways, which can benefit future work on the oncogenic functions and therapeutic targeting of SEPT9.
Supplementary Material
Statement of Significance.
Septins are a large family of filamentous GTP-binding proteins that comprise a major component of the cytoskeleton. Septins are abnormally expressed in various diseases including cancer and neurological disorders. Septin 9 (SEPT9) is an essential and ubiquitously expressed septin with multiple isoforms. SEPT9 expression is altered in ~30% of breast cancer cases and SEPT9 isoforms have differential levels of expression and effects on breast cancer cell properties (e.g., migration). However, little is known about the molecular interactions and pathways of SEPT9 isoforms and how they contribute to the development and metastasis of breast cancer. Using comparative shotgun proteomics, we identified and mapped the interactomes of SEPT9 isoforms 1, 4 and 5 into spatial and functional clusters. We found isoform-specific interactions with proteins of distinct subcellular localizations and functions, implicating SEPT9 isoforms in hitherto unknown mechanisms of tumorigenesis. We posit that truncations in the N-terminal sequences of SEPT9 isoforms bestow a functional specialization by biasing toward specific septin and non-septin binding partners. Our results provide rare evidence for septin isoform-specific interactions, which can benefit future work on the oncogenic functions and therapeutic targeting of SEPT9.
Acknowledgments
We thank Drs. Matthew T. Balmer and Arun Arunachalam for their support and Sanofi Pasteur for use of the LCMS instrumentation and bioinformatics software. This work was supported with NIH/NIGMS grants R01 GM097664, R35 GM136337 and a PA Department of Health CURE grant SAP 4100079710 to E.T.S. All microscopy took place in the Cell Imaging Center of Drexel University.
Abbreviations:
- AKAP13
A-kinase anchoring protein 13
- ABP
actin-binding protein
- APC
adenomatous polyposis coli
- BAZ2B
bromodomain adjacent to zinc finger domain 2B
- CREBBP
CREB-binding protein
- CFAP47
ciliary and flagella protein 47
- EGFR
epidermal growth factor receptor
- ELYS
embryonic large molecule derived from yolk sac
- G-domain
GTP-binding domain
- GML
glycosylphospatidylinositol anchored molecule-like protein
- MAP
microtubule-associated protein
- NTE
N-terminal extension
- SAMD11
sterile alpha motif domain containing 11
- SEPT
septin
- SLK
serine/threonine protein kinase
- TMEM43
transmembrane protein 43
Footnotes
The authors have declared no conflict of interest.
6 References
- 1.Mostowy S, and Cossart P (2012). Septins: the fourth component of the cytoskeleton. Nature Reviews Molecular Cell Biology, 13, 183–194. [DOI] [PubMed] [Google Scholar]
- 2.Kinoshita M (2006). Diversity of septin scaffolds. Current Opinion in Cell Biology, 18, 54–60. [DOI] [PubMed] [Google Scholar]
- 3.Gladfelter AS, Pringle JR, and Lew DJ (2001). The septin cortex at the yeast mother-bud neck. Current Opinion in Microbiology, 4, 681–689. [DOI] [PubMed] [Google Scholar]
- 4.Caudron F, and Barral Y (2009). Septins and the lateral compartmentalization of eukaryotic membranes. Developmental Cell, 16, 493–506. [DOI] [PubMed] [Google Scholar]
- 5.Spiliotis ET (2018). Spatial effects - site-specific regulation of actin and microtubule organization by septin GTPases. Journal of Cell Science, 131, jcs207555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sirajuddin M, Farkasovsky M, Hauer F, Kuhlmann D, Macara IG, Weyand M, Stark H, and Wittinghofer A (2007). Structural insight into filament formation by mammalian septins. Nature, 449, 311–315. [DOI] [PubMed] [Google Scholar]
- 7.Valadares NF, d’ Muniz Pereira H, Ulian Araujo AP, and Garratt RC (2017). Septin structure and filament assembly. Biophysical Reviews, 9, 481–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spiliotis ET, and Nakos K (2021). Cellular functions of actin- and microtubule-associated septins. Current Biology 31(10), R651–R666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dolat L, Hu Q, and Spiliotis ET (2014). Septin functions in organ system physiology and pathology. Biological Chemistry, 395, 123–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kinoshita M (2003). The Septins. Genome Biology, 4, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pan F, Malmberg RL, and Momany M (2007). Analysis of septins across kingdoms reveals orthology and new motifs. BMC Evolutionary Biology, 7, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Russell SE, and Hall PA (2011). Septin genomics: a road less travelled. Biological Chemistry, 392, 763–767. [DOI] [PubMed] [Google Scholar]
- 13.Kinoshita M (2003). Assembly of mammalian septins. Journal of Biolochemistry, 134, 491–496. [DOI] [PubMed] [Google Scholar]
- 14.Kim MS, Froese CD, Estey MP, and Trimble WS (2011). SEPT9 occupies the terminal positions in septin octamers and mediates polymerization-dependent functions in abscission. The Journal of Cell Biology, 195, 815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sellin ME, Sandblad L, Stenmark S, and Gullberg M (2011). Deciphering the rules governing assembly order of mammalian septin complexes. Molecular Biology of the Cell, 22, 3152–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abbey M, Gaestel M, and Menon MB (2018). Septins: Active GTPases or just GTP-binding proteins? Cytoskeleton (Hoboken N.J.), 76: 55–62. [DOI] [PubMed] [Google Scholar]
- 17.Weems A, and McMurray M (2017). The step-wise pathway of septin hetero-octamer assembly in budding yeast. Elife, 6, e23689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zent E, and Wittinghofer A (2014). Human septin isoforms and the GDP-GTP cycle. Biological Chemistry, 395, 169–180. [DOI] [PubMed] [Google Scholar]
- 19.Sellin ME, Stenmark S, and Gullberg M (2014). Cell type-specific expression of SEPT3-homology subgroup members controls the subunit number of heteromeric septin complexes. Molecular Biology of the Cell, 25, 1594–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sellin ME, Stenmark S, and Gullberg M (2012). Mammalian SEPT9 isoforms direct microtubule-dependent arrangements of septin core heteromers. Molecular Biology of the Cell, 23, 4242–4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia G 3rd, Finnigan GC, Heasley LR, Sterling SM, Aggarwal A, Pearson CG, Nogales E, McMurray MA, and Thorner J (2016). Assembly, molecular organization, and membrane-binding properties of development-specific septins. The Journal of Cell Biology, 212, 515–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neubauer K, and Zieger B (2017). The Mammalian Septin Interactome. Frontiers in Cell and Developmental Biology, 5, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fuchtbauer A, Lassen LB, Jensen AB, Howard J, Quiroga Ade S, Warming S, Sorensen AB, Pedersen FS, and Fuchtbauer EM (2011). Septin9 is involved in septin filament formation and cellular stability. Biological Chemistry, 392, 769–777. [DOI] [PubMed] [Google Scholar]
- 24.McIlhatton MA, Burrows JF, Donaghy PG, Chanduloy S, Johnston PG, and Russell SE (2001). Genomic organization, complex splicing pattern and expression of a human septin gene on chromosome 17q25.3. Oncogene, 20, 5930–5939. [DOI] [PubMed] [Google Scholar]
- 25.Bai X, Bowen JR, Knox TK, Zhou K, Pendziwiat M, Kuhlenbaumer G, Sindelar CV, and Spiliotis ET (2013). Novel septin 9 repeat motifs altered in neuralgic amyotrophy bind and bundle microtubules. The Journal of Cell Biology, 203, 895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith C, Dolat L, Angelis D, Forgacs E, Spiliotis ET, and Galkin VE (2015). Septin 9 Exhibits Polymorphic Binding to F-Actin and Inhibits Myosin and Cofilin Activity. Journal of Molecular Biology, 427, 3273–3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soroor F, Kim MS, Palander O, Balachandran Y, Collins RF, Benlekbir S, Rubinstein JL, and Trimble WS (2021). Revised subunit order of mammalian septin complexes explains their in vitro polymerization properties. Molecular Biology of the Cell, 32, 289–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mendonca DC, Macedo JN, Guimaraes SL, Barroso da Silva FL, Cassago A, Garratt RC, Portugal RV, and Araujo APU (2019). A revised order of subunits in mammalian septin complexes. Cytoskeleton (Hoboken N.J.), 76, 457–466. [DOI] [PubMed] [Google Scholar]
- 29.Karasmanis EP, Phan CT, Angelis D, Kesisova IA, Hoogenraad CC, McKenney RJ, and Spiliotis ET (2018). Polarity of Neuronal Membrane Traffic Requires Sorting of Kinesin Motor Cargo during Entry into Dendrites by a Microtubule-Associated Septin. Developmental Cell, 46, 204–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Estey MP, Di Ciano-Oliveira C, Froese CD, Bejide MT, and Trimble WS (2010). Distinct roles of septins in cytokinesis: SEPT9 mediates midbody abscission. The Journal of Cell Biology, 191, 741–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Farrugia AJ, Rodriguez J, Orgaz JL, Lucas M, Sanz-Moreno V, and Calvo F (2020). CDC42EP5/BORG3 modulates SEPT9 to promote actomyosin function, migration, and invasion. The Journal of Cell Biology 219(9):e201912159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Russell SE, McIlhatton MA, Burrows JF, Donaghy PG, Chanduloy S, Petty EM, Kalikin LM, Church SW, McIlroy S, Harkin DP, et al. (2000). Isolation and mapping of a human septin gene to a region on chromosome 17q, commonly deleted in sporadic epithelial ovarian tumors. Cancer Research, 60, 4729–4734. [PubMed] [Google Scholar]
- 33.Kalikin LM, Sims HL, and Petty EM (2000). Genomic and expression analyses of alternatively spliced transcripts of the MLL septin-like fusion gene (MSF) that map to a 17q25 region of loss in breast and ovarian tumors. Genomics, 63, 165–172. [DOI] [PubMed] [Google Scholar]
- 34.Osaka M, Rowley JD, and Zeleznik-Le NJ (1999). MSF (MLL septin-like fusion), a fusion partner gene of MLL, in a therapy-related acute myeloid leukemia with a t(11;17)(q23;q25). Proceedings of the National Academy of Sciences of the United States of America, 96, 6428–6433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sorensen AB, Lund AH, Ethelberg S, Copeland NG, Jenkins NA, and Pedersen FS (2000). Sint1, a common integration site in SL3-3-induced T-cell lymphomas, harbors a putative proto-oncogene with homology to the septin gene family. Journal of Virology, 74, 2161–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Montagna C, Lyu MS, Hunter K, Lukes L, Lowther W, Reppert T, Hissong B, Weaver Z, and Ried T (2003). The Septin 9 (MSF) gene is amplified and overexpressed in mouse mammary gland adenocarcinomas and human breast cancer cell lines. Cancer Research, 63, 2179–2187. [PubMed] [Google Scholar]
- 37.Connolly D, Hoang HG, Adler E, Tazearslan C, Simmons N, Bernard VV, Castaldi M, Oktay MH, and Montagna C (2014). Septin 9 amplification and isoform-specific expression in peritumoral and tumor breast tissue. Biological Chemistry, 395, 157–167. [DOI] [PubMed] [Google Scholar]
- 38.Chacko AD, McDade SS, Chanduloy S, Church SW, Kennedy R, Price J, Hall PA, and Russell SE (2012). Expression of the SEPT9_i4 isoform confers resistance to microtubule-interacting drugs. Cellular Oncology (Dordr), 35, 85–93. [DOI] [PubMed] [Google Scholar]
- 39.Froidevaux-Klipfel L, Poirier F, Boursier C, Crepin R, Pous C, Baudin B, and Baillet A (2011). Modulation of septin and molecular motor recruitment in the microtubule environment of the Taxol-resistant human breast cancer cell line MDA-MB-231. Proteomics, 11, 3877–3886. [DOI] [PubMed] [Google Scholar]
- 40.Froidevaux-Klipfel L, Targa B, Cantaloube I, Ahmed-Zaid H, Pous C, and Baillet A (2015). Septin cooperation with tubulin polyglutamylation contributes to cancer cell adaptation to taxanes. Oncotarget, 6, 36063–36080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Amir S, and Mabjeesh NJ (2007). SEPT9_V1 protein expression is associated with human cancer cell resistance to microtubule-disrupting agents. Cancer Biology & Therapy, 6, 1926–1931. [DOI] [PubMed] [Google Scholar]
- 42.Grutzmann R, Molnar B, Pilarsky C, Habermann JK, Schlag PM, Saeger HD, Miehlke S, Stolz T, Model F, Roblick UJ, et al. (2008). Sensitive detection of colorectal cancer in peripheral blood by septin 9 DNA methylation assay. Plos One, 3, e3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Warren JD, Xiong W, Bunker AM, Vaughn CP, Furtado LV, Roberts WL, Fang JC, Samowitz WS, and Heichman KA (2011). Septin 9 methylated DNA is a sensitive and specific blood test for colorectal cancer. BMC Medicine, 9, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Connolly D, Yang Z, Castaldi M, Simmons N, Oktay MH, Coniglio S, Fazzari MJ, Verdier-Pinard P, and Montagna C (2011). Septin 9 isoform expression, localization and epigenetic changes during human and mouse breast cancer progression. Breast Cancer Research, 13, R76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chacko AD, Hyland PL, McDade SS, Hamilton PW, Russell SH, and Hall PA (2005). SEPT9_v4 expression induces morphological change, increased motility and disturbed polarity. The Journal of Pathology, 206, 458–465. [DOI] [PubMed] [Google Scholar]
- 46.Verdier-Pinard P, Salaun D, Bouguenina H, Shimada S, Pophillat M, Audebert S, Agavnian E, Coslet S, Charafe-Jauffret E, Tachibana T, et al. (2017). Septin 9_i2 is downregulated in tumors, impairs cancer cell migration and alters subnuclear actin filaments. Scientific Reports, 7, 44976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gilad R, Meir K, Stein I, German L, Pikarsky E, and Mabjeesh NJ (2015). High SEPT9_i1 protein expression is associated with high-grade prostate cancers. Plos One, 10, e0124251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gonzalez ME, Makarova O, Peterson EA, Privette LM, and Petty EM (2009). Up-regulation of SEPT9_v1 stabilizes c-Jun-N-terminal kinase and contributes to its pro-proliferative activity in mammary epithelial cells. Cellular Signalling, 21, 477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cravatt BF, Simon GM, and Yates JR 3rd (2007). The biological impact of mass-spectrometry-based proteomics. Nature, 450, 991–1000. [DOI] [PubMed] [Google Scholar]
- 50.Walther TC, and Mann M (2010). Mass spectrometry-based proteomics in cell biology. The Journal of Cell Biology, 190, 491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Larance M, and Lamond AI (2015). Multidimensional proteomics for cell biology. Nature reviews. Molecular Cell Biology, 16, 269–280. [DOI] [PubMed] [Google Scholar]
- 52.Nakahira M, Macedo JN, Seraphim TV, Cavalcante N, Souza TA, Damalio JC, Reyes LF, Assmann EM, Alborghetti MR, Garratt RC, et al. (2010). A draft of the human septin interactome. Plos One, 5, e13799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sandrock K, Bartsch I, Blaser S, Busse A, Busse E, and Zieger B (2011). Characterization of human septin interactions. Biological Chemistry, 392, 751–761. [DOI] [PubMed] [Google Scholar]
- 54.Mellacheruvu D, Wright Z, Couzens AL, Lambert JP, St-Denis NA, Li T, Miteva YV, Hauri S, Sardiu ME, Low TY, et al. (2013). The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nature Methods, 10, 730–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gupta N, and Pevzner PA (2009). False discovery rates of protein identifications: a strike against the two-peptide rule. Journal of Proteome Research, 8, 4173–4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, et al. (2019). The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Research, 47, D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vargas-Muniz JM, Renshaw H, Waitt G, Soderblom EJ, Moseley MA, Palmer JM, Juvvadi PR, Keller NP, and Steinbach WJ (2017). Caspofungin exposure alters the core septin AspB interactome of Aspergillus fumigatus. Biochemical Biophysical Research Communications, 485, 221–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ma B, Zhang K, Hendrie C, Liang C, Li M, Doherty-Kirby A, and Lajoie G (2003). PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Communication in Mass Spectrometry, 17, 2337–2342. [DOI] [PubMed] [Google Scholar]
- 59.Bejide MT (2010). Characterization of a novel interaction between septins and the adenomatous polyposis coli tumor suppressor. In Biochemistry, Volume PhD. (Toronto, Canada: University of Toronto; ). [Google Scholar]
- 60.Hamdan H, Lim BC, Torii T, Joshi A, Konning M, Smith C, Palmer DJ, Ng P, Leterrier C, Oses-Prieto JA, et al. (2020). Mapping axon initial segment structure and function by multiplexed proximity biotinylation. Nature Communications, 11, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee EY, and Muller WJ (2010). Oncogenes and tumor suppressor genes. Cold Spring Harbor Perspectives in Biology, 2, a003236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Godinho SA, and Pellman D (2014). Causes and consequences of centrosome abnormalities in cancer. Philosophical Transactions of the Royal Society B: Biological Sciences, 369, 20130467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eguether T, and Hahne M (2018). Mixed signals from the cell’s antennae: primary cilia in cancer. EMBO Reports, 19, e46589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.James C, Muller M, Goldberg MW, Lenz C, Urlaub H, and Kehlenbach RH (2019). Proteomic mapping by rapamycin-dependent targeting of APEX2 identifies binding partners of VAPB at the inner nuclear membrane. Journal of Biological Chemistry, 294, 16241–16254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gillespie PJ, Khoudoli GA, Stewart G, Swedlow JR, and Blow JJ (2007). ELYS/MEL-28 chromatin association coordinates nuclear pore complex assembly and replication licensing. Current Biology, 17, 1657–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guillen-Samander A, Leonzino M, Hanna MG, Tang N, Shen H, and De Camilli P (2021). VPS13D bridges the ER to mitochondria and peroxisomes via Miro. The Journal of Cell Biology, 220, e202010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jevtic P, Schibler AC, Wesley CC, Pegoraro G, Misteli T, and Levy DL (2019). The nucleoporin ELYS regulates nuclear size by controlling NPC number and nuclear import capacity. EMBO Reports, 20, e47283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ait-Si-Ali S, Polesskaya A, Filleur S, Ferreira R, Duquet A, Robin P, Vervish A, Trouche D, Cabon F, and Harel-Bellan A (2000). CBP/p300 histone acetyltransferase activity is important for the G1/S transition. Oncogene, 19, 2430–2437. [DOI] [PubMed] [Google Scholar]
- 69.Gao C, Liu SG, Lu WT, Yue ZX, Zhao XX, Xing TY, Chen ZP, Zheng HY, and Li ZG (2020). Downregulating CREBBP inhibits proliferation and cell cycle progression and induces daunorubicin resistance in leukemia cells. Molecular Medicine Reports, 22, 2905–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kobayashi T, and Dynlacht BD (2011). Regulating the transition from centriole to basal body. The Journal of Cell Biology, 193, 435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lui C, Mills K, Brocardo MG, Sharma M, and Henderson BR (2012). APC as a mobile scaffold: regulation and function at the nucleus, centrosomes, and mitochondria. IUBMB Life, 64, 209–214. [DOI] [PubMed] [Google Scholar]
- 72.Chen TY, Lin TC, Kuo PL, Chen ZR, Cheng HL, Chao YY, Syu JS, Lu FI, and Wang CY (2021). Septin 7 is a centrosomal protein that ensures S phase entry and microtubule nucleation by maintaining the abundance of p150(glued). Journal of Cellular Physiology, 236, 2706–2724. [DOI] [PubMed] [Google Scholar]
- 73.Song K, Gras C, Capin G, Gimber N, Lehmann M, Mohd S, Puchkov D, Rodiger M, Wilhelmi I, Daumke O, et al. (2019). A SEPT1-based scaffold is required for Golgi integrity and function. Journal of Cell Science, 132, jcs225557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mirvis M, Stearns T, and James Nelson W (2018). Cilium structure, assembly, and disassembly regulated by the cytoskeleton. Biochemical Journal, 475, 2329–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Palander O, El-Zeiry M, and Trimble WS (2017). Uncovering the Roles of Septins in Cilia. Frontiers in Cell and Developmental Biology, 5, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hall NA, and Hehnly H (2021). A centriole’s subdistal appendages: contributions to cell division, ciliogenesis and differentiation. Open Biology, 11, 200399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hearn T, Spalluto C, Phillips VJ, Renforth GL, Copin N, Hanley NA, and Wilson DI (2005). Subcellular localization of ALMS1 supports involvement of centrosome and basal body dysfunction in the pathogenesis of obesity, insulin resistance, and type 2 diabetes. Diabetes, 54, 1581–1587. [DOI] [PubMed] [Google Scholar]
- 78.Li G, Vega R, Nelms K, Gekakis N, Goodnow C, McNamara P, Wu H, Hong NA, and Glynne R (2007). A role for Alstrom syndrome protein, alms1, in kidney ciliogenesis and cellular quiescence. Plos Genetics, 3, e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Knorz VJ, Spalluto C, Lessard M, Purvis TL, Adigun FF, Collin GB, Hanley NA, Wilson DI, and Hearn T (2010). Centriolar association of ALMS1 and likely centrosomal functions of the ALMS motif-containing proteins C10orf90 and KIAA1731. Molecular Biology of the Cell, 21, 3617–3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mazo G, Soplop N, Wang WJ, Uryu K, and Tsou MF (2016). Spatial Control of Primary Ciliogenesis by Subdistal Appendages Alters Sensation-Associated Properties of Cilia. Developmental Cell, 39, 424–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Das A, Dickinson DJ, Wood CC, Goldstein B, and Slep KC (2015). Crescerin uses a TOG domain array to regulate microtubules in the primary cilium. Molecular Biology of the Cell, 26, 4248–4264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yuan K, Frolova N, Xie Y, Wang D, Cook L, Kwon YJ, Steg AD, Serra R, and Frost AR (2010). Primary cilia are decreased in breast cancer: analysis of a collection of human breast cancer cell lines and tissues. Journal of Histochemistry and Cytochemistry, 58, 857–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bentin Toaldo C, Alexi X, Beelen K, Kok M, Hauptmann M, Jansen M, Berns E, Neefjes J, Linn S, Michalides R, et al. (2015). Protein Kinase A-induced tamoxifen resistance is mediated by anchoring protein AKAP13. BMC Cancer, 15, 588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sterpetti P, Hack AA, Bashar MP, Park B, Cheng SD, Knoll JH, Urano T, Feig LA, and Toksoz D (1999). Activation of the Lbc Rho exchange factor proto-oncogene by truncation of an extended C terminus that regulates transformation and targeting. Molecular and Cellular Biology, 19, 1334–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Diviani D, Soderling J, and Scott JD (2001). AKAP-Lbc anchors protein kinase A and nucleates Galpha 12-selective Rho-mediated stress fiber formation. Journal of Biological Chemistry, 276, 44247–44257. [DOI] [PubMed] [Google Scholar]
- 86.Pao GM, Janknecht R, Ruffner H, Hunter T, and Verma IM (2000). CBP/p300 interact with and function as transcriptional coactivators of BRCA1. Proceedings of the National Academy of Sciences of the United States of America, 97, 1020–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kimura Y, Furuhata T, Shiratsuchi T, Nishimori H, Hirata K, Nakamura Y, and Tokino T (1997). GML sensitizes cancer cells to Taxol by induction of apoptosis. Oncogene, 15, 1369–1374. [DOI] [PubMed] [Google Scholar]
- 88.Kagawa K, Inoue T, Tokino T, Nakamura Y, and Akiyama T (1997). Overexpression of GML promotes radiation-induced cell cycle arrest and apoptosis. Biochemical and Biophysical Research Communications, 241, 481–485. [DOI] [PubMed] [Google Scholar]
- 89.Higashiyama M, Miyoshi Y, Kodama K, Yokouchi H, Takami K, Nishijima M, Nakayama T, Kobayashi H, Minamigawa K, and Nakamura Y (2000). p53-regulated GML gene expression in non-small cell lung cancer. a promising relationship to cisplatin chemosensitivity. European Journal of Cancer, 36, 489–495. [DOI] [PubMed] [Google Scholar]
- 90.Kobayashi H, Matsuda Y, Hitomi T, Okuda H, Shioi H, Matsuda T, Imai H, Sone M, Taura D, Harada KH, et al. (2015). Biochemical and Functional Characterization of RNF213 (Mysterin) R4810K, a Susceptibility Mutation of Moyamoya Disease, in Angiogenesis In Vitro and In Vivo. Journal of the American Heart Association, 4, e002146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu W, Morito D, Takashima S, Mineharu Y, Kobayashi H, Hitomi T, Hashikata H, Matsuura N, Yamazaki S, Toyoda A, et al. (2011). Identification of RNF213 as a susceptibility gene for moyamoya disease and its possible role in vascular development. Plos One, 6, e22542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Akil A, Peng J, Omrane M, Gondeau C, Desterke C, Marin M, Tronchere H, Taveneau C, Sar S, Briolotti P, et al. (2016). Septin 9 induces lipid droplets growth by a phosphatidylinositol-5-phosphate and microtubule-dependent mechanism hijacked by HCV. Nature Communications, 7, 12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sugihara M, Morito D, Ainuki S, Hirano Y, Ogino K, Kitamura A, Hirata H, and Nagata K (2019). The AAA+ ATPase/ubiquitin ligase mysterin stabilizes cytoplasmic lipid droplets. The Journal of Cell Biology, 218, 949–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schneider T, Martinez-Martinez A, Cubillos-Rojas M, Bartrons R, Ventura F, and Rosa JL (2018). The E3 ubiquitin ligase HERC1 controls the ERK signaling pathway targeting C-RAF for degradation. Oncotarget, 9, 31531–31548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Anding AL, Wang C, Chang TK, Sliter DA, Powers CM, Hofmann K, Youle RJ, and Baehrecke EH (2018). Vps13D Encodes a Ubiquitin-Binding Protein that Is Required for the Regulation of Mitochondrial Size and Clearance. Current Biology, 28, 287–295 e286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kamura T, Burian D, Yan Q, Schmidt SL, Lane WS, Querido E, Branton PE, Shilatifard A, Conaway RC, and Conaway JW (2001). Muf1, a novel Elongin BC-interacting leucine-rich repeat protein that can assemble with Cul5 and Rbx1 to reconstitute a ubiquitin ligase. Journal of Biological Chemistry, 276, 29748–29753. [DOI] [PubMed] [Google Scholar]
- 97.Choi P, Snyder H, Petrucelli L, Theisler C, Chong M, Zhang Y, Lim K, Chung KK, Kehoe K, D’Adamio L, et al. (2003). SEPT5_v2 is a parkin-binding protein. Molecular Brain Research, 117, 179–189. [DOI] [PubMed] [Google Scholar]
- 98.Pagliuso A, Tham TN, Stevens JK, Lagache T, Persson R, Salles A, Olivo-Marin JC, Oddos S, Spang A, Cossart P, et al. (2016). A role for septin 2 in Drp1-mediated mitochondrial fission. EMBO Reports, 17, 858–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sirianni A, Krokowski S, Lobato-Marquez D, Buranyi S, Pfanzelter J, Galea D, Willis A, Culley S, Henriques R, Larrouy-Maumus G, et al. (2016). Mitochondria mediate septin cage assembly to promote autophagy of Shigella. EMBO Reports, 17, 1029–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Amir S, Wang R, Simons JW, and Mabjeesh NJ (2009). SEPT9_v1 up-regulates hypoxia-inducible factor 1 by preventing its RACK1-mediated degradation. Journal of Biological Chemistry, 284, 11142–11151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Marcus EA, Tokhtaeva E, Turdikulova S, Capri J, Whitelegge JP, Scott DR, Sachs G, Berditchevski F, and Vagin O (2016). Septin oligomerization regulates persistent expression of ErbB2/HER2 in gastric cancer cells. Biochemical Journal, 473, 1703–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Diesenberg K, Beerbaum M, Fink U, Schmieder P, and Krauss M (2015). SEPT9 negatively regulates ubiquitin-dependent downregulation of EGFR. Journal of Cell Science, 128, 397–407. [DOI] [PubMed] [Google Scholar]
- 103.Orre LM, Vesterlund M, Pan Y, Arslan T, Zhu Y, Fernandez Woodbridge A, Frings O, Fredlund E, and Lehtio J (2019). SubCellBarCode: Proteome-wide Mapping of Protein Localization and Relocalization. Molecular Cell, 73, 166–182 e167. [DOI] [PubMed] [Google Scholar]
- 104.Kim MS, Froese CD, Xie H, and Trimble WS (2012). Uncovering principles that control septin-septin interactions. Journal of Biological Chemistry, 287, 30406–30413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hernandez-Rodriguez Y, and Momany M (2012). Posttranslational modifications and assembly of septin heteropolymers and higher-order structures. Current Opinion in Microbiology, 15, 660–668. [DOI] [PubMed] [Google Scholar]
- 106.Estey MP, Di Ciano-Oliveira C, Froese CD, Fung KY, Steels JD, Litchfield DW, and Trimble WS (2013). Mitotic regulation of SEPT9 protein by cyclin-dependent kinase 1 (Cdk1) and Pin1 protein is important for the completion of cytokinesis. Journal of Biological Chemistry, 288, 30075–30086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Scholz R, Imami K, Scott NE, Trimble WS, Foster LJ, and Finlay BB (2015). Novel Host Proteins and Signaling Pathways in Enteropathogenic E. coli Pathogenesis Identified by Global Phosphoproteome Analysis. Molecular and Cellular Proteomics, 14, 1927–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shinde MY, Sidoli S, Kulej K, Mallory MJ, Radens CM, Reicherter AL, Myers RL, Barash Y, Lynch KW, Garcia BA, et al. (2017). Phosphoproteomics reveals that glycogen synthase kinase-3 phosphorylates multiple splicing factors and is associated with alternative splicing. Journal of Biological Chemistry, 292, 18240–18255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Beausoleil SA, Villen J, Gerber SA, Rush J, and Gygi SP (2006). A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nature Biotechnology, 24, 1285–1292. [DOI] [PubMed] [Google Scholar]
- 110.Ribet D, Boscaini S, Cauvin C, Siguier M, Mostowy S, Echard A, and Cossart P (2017). SUMOylation of human septins is critical for septin filament bundling and cytokinesis. The Journal of Cell Biology, 216, 4041–4052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Van Damme P, Lasa M, Polevoda B, Gazquez C, Elosegui-Artola A, Kim DS, De Juan-Pardo E, Demeyer K, Hole K, Larrea E, et al. (2012). N-terminal acetylome analyses and functional insights of the N-terminal acetyltransferase NatB. Proceedings in National Academy of Sciences of the United States of America, 109, 12449–12454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Akimov V, Barrio-Hernandez I, Hansen SVF, Hallenborg P, Pedersen AK, Bekker-Jensen DB, Puglia M, Christensen SDK, Vanselow JT, Nielsen MM, et al. (2018). UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nature Structural & Molecular Biology, 25, 631–640. [DOI] [PubMed] [Google Scholar]
- 113.Udeshi ND, Svinkina T, Mertins P, Kuhn E, Mani DR, Qiao JW, and Carr SA (2013). Refined preparation and use of anti-diglycine remnant (K-epsilon-GG) antibody enables routine quantification of 10,000s of ubiquitination sites in single proteomics experiments. Molecular and Cellular Proteomics, 12(3), 825–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Larsen SC, Sylvestersen KB, Mund A, Lyon D, Mullari M, Madsen MV, Daniel JA, Jensen LJ, and Nielsen ML (2016). Proteome-wide analysis of arginine monomethylation reveals widespread occurrence in human cells. Science Signaling, 9, rs9. [DOI] [PubMed] [Google Scholar]
- 115.Hendriks IA, Lyon D, Young C, Jensen LJ, Vertegaal AC, and Nielsen ML (2017). Site-specific mapping of the human SUMO proteome reveals co-modification with phosphorylation. Nature Structural & Molecular Biology, 24, 325–336. [DOI] [PubMed] [Google Scholar]
- 116.Zhou H, Di Palma S, Preisinger C, Peng M, Polat AN, Heck AJ, and Mohammed S (2013). Toward a comprehensive characterization of a human cancer cell phosphoproteome. Journal of Proteome Research, 12, 260–271. [DOI] [PubMed] [Google Scholar]
- 117.Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, Gnad F, Cox J, Jensen TS, Nigg EA, et al. (2010). Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Science Signaling, 3, ra3. [DOI] [PubMed] [Google Scholar]
- 118.McDade SS, Hall PA, and Russell SE (2007). Translational control of SEPT9 isoforms is perturbed in disease. Human Molecular Genetics, 16, 742–752. [DOI] [PubMed] [Google Scholar]
- 119.Scott M, McCluggage WG, Hillan KJ, Hall PA, and Russell SE (2006). Altered patterns of transcription of the septin gene, SEPT9, in ovarian tumorigenesis. International Journal of Cancer, 118, 1325–1329. [DOI] [PubMed] [Google Scholar]
- 120.Furuhata T, Tokino T, Urano T, and Nakamura Y (1996). Isolation of a novel GPI-anchored gene specifically regulated by p53; correlation between its expression and anti-cancer drug sensitivity. Oncogene, 13, 1965–1970. [PubMed] [Google Scholar]
- 121.Clever M, Funakoshi T, Mimura Y, Takagi M, and Imamoto N (2012). The nucleoporin ELYS/Mel28 regulates nuclear envelope subdomain formation in HeLa cells. Nucleus, 3, 187–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Marcus J, Bejerano-Sagie, Patterson N, Bagchi S, Verkhusha VV, Connolly D, Goldberg GL, Golden A, Sharma VP, Condeelis J, and Montagna C (2019). Septin 9 isoforms promote tumorigenesis in mammary epithelial cells by increasing migration and ECM degradation through metalloproteinase secretion at focal adhesions. Oncogene 38(30):5838–5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hecht M, Rosler R, Wiese S, Johnsson N, Gronemyer T (2019) An interaction network of the human SEPT9 established by quantitative mass spectroscopy. G3 Genes / Genomes / Genetics, 9, 1869–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.