

Abstract
Although abiotic catalysts are capable of promoting numerous new-to-nature reactions, only a small subset has so far been successfully integrated into living systems. Research in intracellular catalysis requires an interdisciplinary approach that takes advantage of both chemical and biological tools as well as state-of-the-art instrumentations. In this perspective, we will focus on the techniques that have made studying metal-catalyzed reactions in cells possible using representative examples from the literature. Although the lack of quantitative data in vitro and in vivo has somewhat limited progress in the catalyst development process, recent advances in characterization methods should help overcome some of these deficiencies. Given its tremendous potential, we believe that intracellular catalysis will play a more prominent role in the development of future biotechnologies and therapeutics.
Keywords: intracellular, bioorthogonal, biocompatible, microscopy, flow cytometry, mass spectrometry, bioassays
Graphical Abstract
1. INTRODUCTION
Biological catalysts (e.g., enzymes) have likely existed since the beginning of life,1 which is believed to have occurred billions of years ago.2,3 In comparison, the deliberate use of synthetic catalysts to accelerate chemical reactions was first documented around the 1800s.4 Chemical catalysis today has attained a high level of sophistication, as exemplified by the large number of reactions that have been discovered and our growing insight into mechanism and catalytic species.5–7 An emerging frontier in chemical catalysis is the integration of synthetic metal catalysts with living systems.8–13 Fundamentally, these efforts are driven by intellectual curiosity. Given that nature required millions of years to develop the chemistry of life through natural evolution, can chemists introduce new intracellular transformations on a much shorter timescale? The ability to orchestrate chemical processes inside living systems also has many practical benefits. For example, synthetic catalysts could be used to replace dysfunctional enzymes, label biomolecules,14–16 activate prodrugs,17,18 or degrade toxins.19
Researchers have taken two parallel approaches to introduce new-to-nature reactions inside biological hosts,9 either by using artificial enzymes or synthetic metal catalysts. Artificial enzymes could be engineered using directed evolution20,21 or by combining known protein scaffolds with inorganic complexes.22–25 Synthetic metal catalysts, on the other hand, are not supported by biological scaffolds and have metal active sites that are typically exposed to the external environment.8–13 These complexes can either be homogeneous (e.g., SIMCats = small molecule intracellular metal catalysts8) or heterogeneous (e.g., nanozymes26,27). The advantages of synthetic metal catalysts are that they are cell permeable, synthetically tunable, and easily produced on gram scales or larger. The first demonstration of using metal catalysts to promote intracellular reactions was reported in 198528 and since then, an increasing number of reports has appeared in the literature.29–31 To date, metal catalysts have been used to carry out diverse intracellular reactions, including azide-alkyne cycloaddition,32–36 amide coupling,37 azide reduction,38,39 C–C bond cross-coupling,40,41 olefin metathesis,42 protecting group cleavage,43–49 ring formation,50,51 and transfer hydrogenation52,53 (Scheme 1). Although these examples are remarkable, they represent only a small percentage of what synthetic catalysts is capable of achieving.5 Unfortunately, translating chemistry developed in the reaction flask for applications inside cells or organisms is not trivial.
Scheme 1.
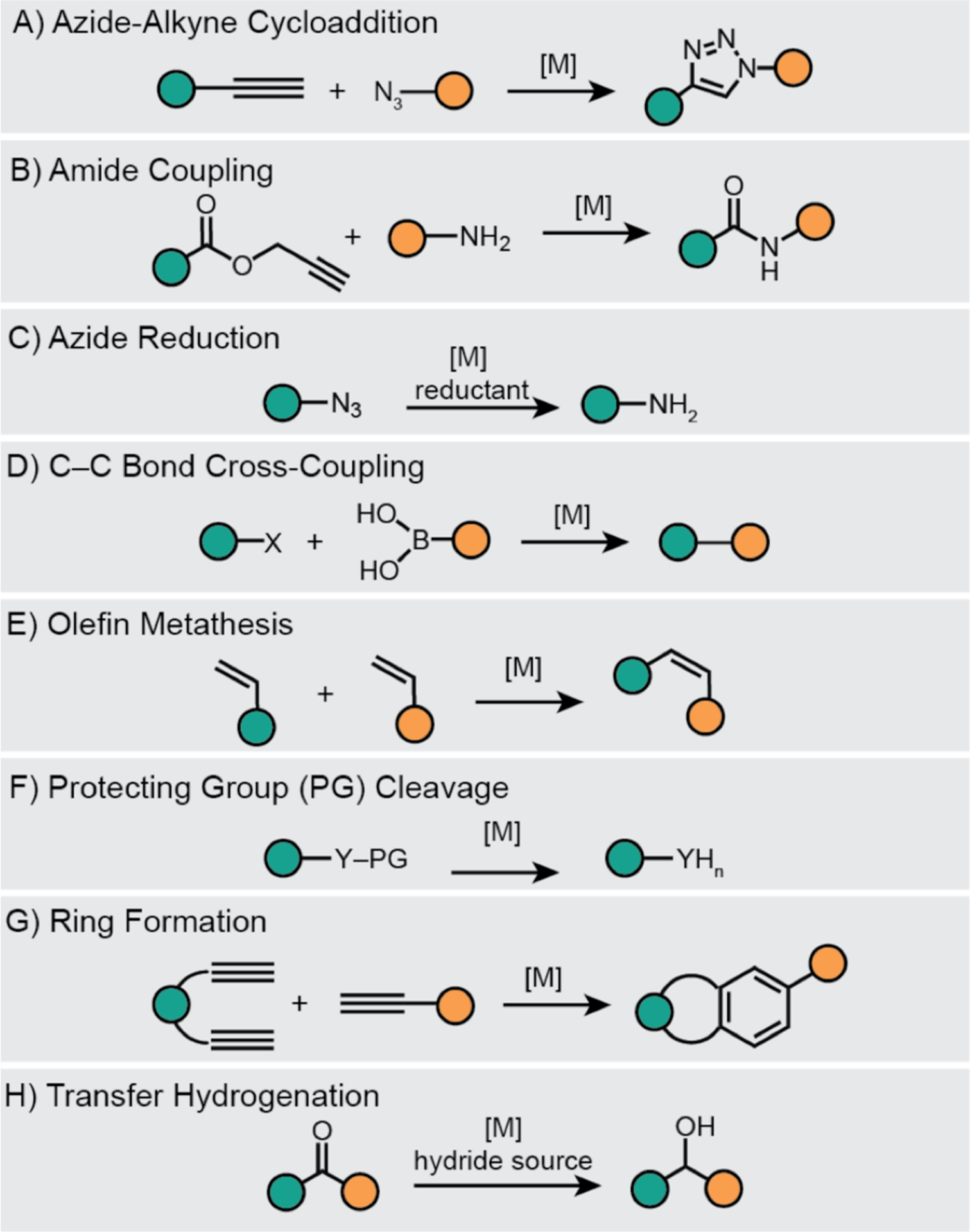
Synthetic metal-catalyzed reactions performed inside living cells and organisms. The teal and orange circles represent different organic substituents. [M] = synthetic metal catalyst, X = halide, Y = O or N.
Although there is no standard approach for developing biocompatible metal catalysts,8–13 most studies contain some, if not all, of the elements shown in Scheme 2. In a typical workflow, the first step is to identify a target reaction. The ideal reaction should not interfere with native biochemical processes (i.e., is bioorthogonal)54 and is novel to the biological host. In step 2, a variety of catalyst candidates should be selected based not only on their reactivity but also on their likely tolerance toward dioxygen (except when working with anaerobes), water, and biological additives. At this stage a robust protocol should be established to screen the activity of these complexes under physiologically relevant conditions (e.g., in phosphate buffered saline (PBS) at 37 °C in air). Next, in step 3, the most promising candidates should be evaluated for their biocompatibility with one or more cell lines. Some useful properties to measure include their cellular uptake and 50% growth inhibition concentration (IC50). In step 4, the catalytic activity of the complexes should be tested in cells, keeping in mind that the catalyst loading should be kept far below its IC50 value to minimize cellular toxicity. For intracellular reaction studies, pro-fluorogenic substrates are commonly used so that upon conversion to product the fluorescence generated could be monitored.43 It is important that neither the starting agents nor products adversely affect cell viability since the ultimate goal is to create new biocompatible chemistry, unless the goal is to kill cells such as in anticancer chemotherapy. Finally, in step 5, the most active catalysts should be subjected to structure-activity relationship (SAR) studies to identify how they could be improved.45,55 The lessons learned from each development cycle will help inform future design efforts.
Scheme 2.
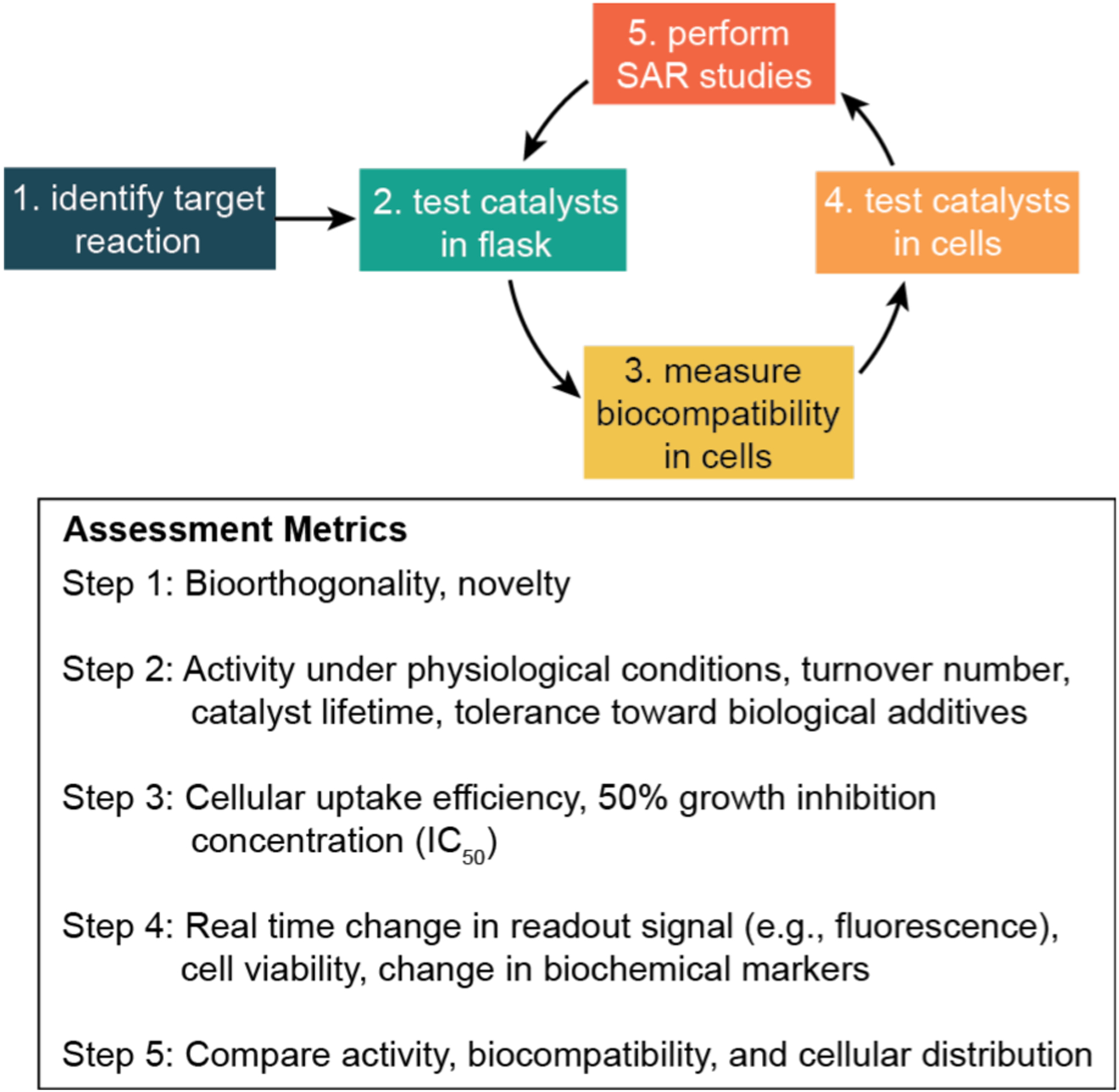
Typical workflow for the discovery and evaluation of biocompatible metal catalysts. SAR = structure-activity relationship.
Studying catalysis inside living systems can be significantly more challenging than in solution. First, living environments are heterogeneous so the behavior of catalysts in cells can vary depending on a variety of factors, such as local pH, presence of biological nucleophiles, or solution viscosity. Thus, any catalytic properties measured in the reaction flask might not necessarily be the same as inside biological cells. In fact, it has been demonstrated that the kinetic parameters Km and kcat for enzymes can differ by several orders of magnitude in living systems vs. in solution.56,57 Second, the species of interest (e.g., small molecules or proteins) are typically present in very low concentrations in cells, often well below micromolar ranges, so extremely sensitive instruments are needed to detect such analytes. In addition, because studies of dynamic catalyst behavior must be conducted with intact cells, non-invasive techniques are usually preferred over invasive ones. Lastly, it is often difficult to determine whether reactions take place intracellularly or extracellularly. To minimize unwanted reactions outside cells, researchers commonly wash cells with fresh media after they are pre-treated with catalyst or substrate (the order of addition usually depends on the cytotoxicity and reactivity of the various agents). However, unless these chemical species could be tagged and monitored directly in real time, the possibility that small molecules could be readily transported in and out of cells make it challenging to ascertain precisely where and when reactions take place. Thus, techniques that give high spatial and temporal resolution are needed to address this ambiguity.
This perspective article will provide a critical overview of the tools available to probe the chemical and biological properties of metal catalysts in cells, including discussions of where new technologies could enhance our investigative power. The sections below are divided into major techniques, specifically fluorescence microscopy, flow cytometry, mass spectrometry, and biological assays (Table 1). We have selected representative examples from the literature to illustrate how these methods are used for studying various aspects of intracellular catalysis (Scheme 1). Most researchers employ a combination of methods in their work but in the interest of clarity, our discussions will focus primarily on the techniques relevant to a particular section. The reader is referred to the original sources for additional details. Because the conditions and environment under which a catalyst is studied can critically influence its behavior, we will make the following distinctions: “in vivo” will be used to describe studies in intact living organisms (e.g., inside zebrafish),56 “in vitro” will be used to describe studies in living models (e.g., in cells grown in petri dish),56 and “in solution” will be used to describe studies in the reaction flask without living specimen. Although researchers sometimes use the term in vivo to refer to biological cells outside their native environment,9,33 we will adhere to the stricter interpretation of the Latin translation.
Table 1.
Summary of Techniques Employed in Biological Cell Studies
| Technique | Typea | Advantagesb | Disadvantagesb | Used to Study Synthetic Catalysis in Cells? |
|---|---|---|---|---|
| Fluorescence microscopy | Non-invasive | Highly sensitive, many fluorophores are available | Requires fluorescent probes with suitable photophysical properties | Yes; most studies are qualitative not quantitative |
| Flow cytometry | Non-invasive | Measurements are automated and fast, has high sensitivity when coupled to fluorescence detection | Lacks spatial resolution inside cells | Yes; only provides ensemble averaged data |
| Mass spectrometry | Invasive | Highly sensitive, particularly useful for quantifying non-native metal concentration | Cells must be lysed prior to analysis, lack spatial resolution | Yes; only provides ensemble averaged data |
| Biological assays | Non-invasive or invasive | Many commercial assays are available to interrogate different biological changes (e.g., cell viability, reactive oxygen species concentrations) | Limited assays available to obtain chemical information about in cell catalysis | Yes; typically used to measure biological response, not chemical properties of the catalyst |
Invasive techniques require destruction of the biological sample whereas non-invasive techniques allow study of specimens in their native form.
The advantages and disadvantages listed are meant to highlight specific features of the technique and are not comprehensive.
The description “living system” will be employed as a generic term for all biological cells and organisms, regardless of their origin. This article will focus primarily on studies in cells due to their less complex nature in comparison to whole organisms. We hope the information contained in this article will be useful to a broad scientific audience, from synthetic chemists to life science researchers.
2. FLUORESCENCE MICROSCOPY
2.1. General Background.
A fluorescence microscope is an instrument that can detect fluorescent signals emanating from light-emitting specimens. In cell biology, fluorescence microscopy provides a way for researchers to not only visualize biological samples at subcellular levels, but also analyze and quantify complex physiological processes in real-time.58 This versatile and non-invasive technique has enabled studies of many intracellular processes, such as protein localization and interactions, DNA transcription, ion transport, and metabolism.59,60 During the past few decades, advances in fluorescence microscopy have allowed scientists to acquire images with deeper penetration, higher resolution, and in less time, making this technique one of the most powerful for life science research.61
When designing fluorescence microscopy experiments, two basic considerations should be taken into account. First, it is important to understand the pros and cons of various imaging modalities available so that the most appropriate could be selected for a particular application.62 The most basic form is widefield fluorescence microscopy, which employs a fluorescent light source to excite samples of interest and an eyepiece or a camera to observe the emitted light (Chart 1A). Light filters are commonly used to select for specific excitation and emission wavelengths. Widefield fluorescence microscopy (WFM) is advantageous because of its fast acquisition time (ms/frame), ease of use, and relatively low cost. Its need for lower power light source (usually μW) is less likely to damage living samples and minimizes photobleaching of fluorophores (Table 2). On the other hand, WFM is diffraction limited (~200 nm) and has poor imaging depth so it is not the most ideal for imaging thick samples such as live cells and tissues.63 Laser scanning confocal microscopy (LSCM) can overcome some of the limitations of WFM.62,64 A laser scanning confocal microscope uses advanced optics to focus laser light to a diffraction-limited spot in the sample and the emitted light is directed to a pinhole that allows optical sectioning (Chart 1B), which gives axial resolution between 0.6–1.0 μm. Although LSCM provides better z spatial resolution than WFM, it has slower acquisition time (s/frame), requires special training to use, can cause phototoxicity65 and photobleaching, and is more expensive. However, because of its high z spatial resolution, LSCM is the preferred choice by many researchers studying metal-catalyzed reactions in live cells.
Chart 1.
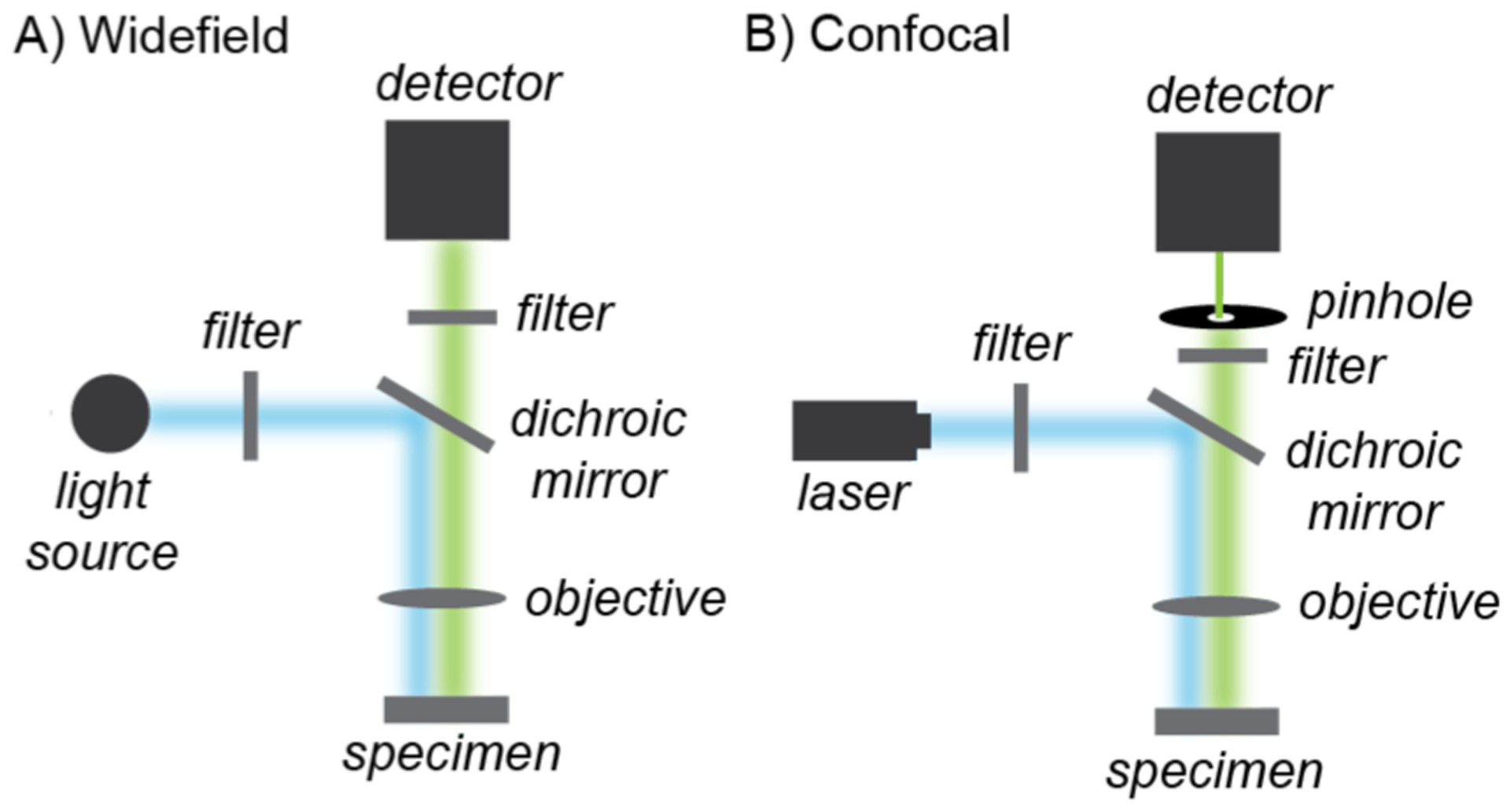
Simplified schematic of widefield (A) and confocal (B) fluorescence microscopy.
Table 2.
Comparison Between Widefield and Laser Scanning Confocal Fluorescence Microscopy
| Characteristic | Widefield | Laser Scanning Confocal |
|---|---|---|
| Spatial resolution (xy) | Diffraction limited | Diffraction limited |
| Spatial resolution (z) | Poor | Good |
| Temporal resolution | Fast (ms/frame) | Slow (s/frame) |
| Imaging depth | Poor | Good |
| Ease of use | Simple | Complex |
| Cost | Lower | Higher |
| Phototoxicity/photobleaching | Usually low | Can be high |
A second consideration in the design of fluorescence microscopy experiments is the selection of fluorophores. These light-emitting units can comprise macromolecules,66,67 small-molecules,68,69 or semiconductor nanocrystals70 and should have high quantum yield as well as absorption and emission wavelengths within a desired optical window. Although protein-based fluorophores such as green-fluorescent proteins66,67 are commonly used in biological imaging studies, their major disadvantages are that their large size may interfere with the localization or function of the species of interest and certain emission wavelengths are not easily obtainable. In contrast, synthetic small molecule fluorophores are smaller in size, have more variety in color, can cross cellular membranes, and can be attached to molecular substrates or catalysts.68,71–73 They must also be nontoxic to be biologically useful. Due to their many advantages, synthetic fluorophores are commonly employed in studies of metal-catalyzed reactions in cells.
A fundamental question to ask in intracellular catalysis studies is do the reactions take place inside the living host (e.g., Step 4 in Scheme 2)? To answer this question, researchers have devised creative reaction schemes in which successful catalytic events lead to fluorescence labeling of biomolecules or generation of light-emitting species (Scheme 3). These strategies are highly effective because they offer a straightforward way to monitor reactions in real-time without damaging living specimens. A few other relevant questions to probe in these investigations include: What is the catalytic efficiency? Is catalysis localized in specific cellular compartments? Does catalysis occur inside the cell, outside the cell, or on the cell surface? As we explore the literature examples below, we will examine how some of these inquiries may or may not be possible to answer via fluorescence microscopy.
Scheme 3.
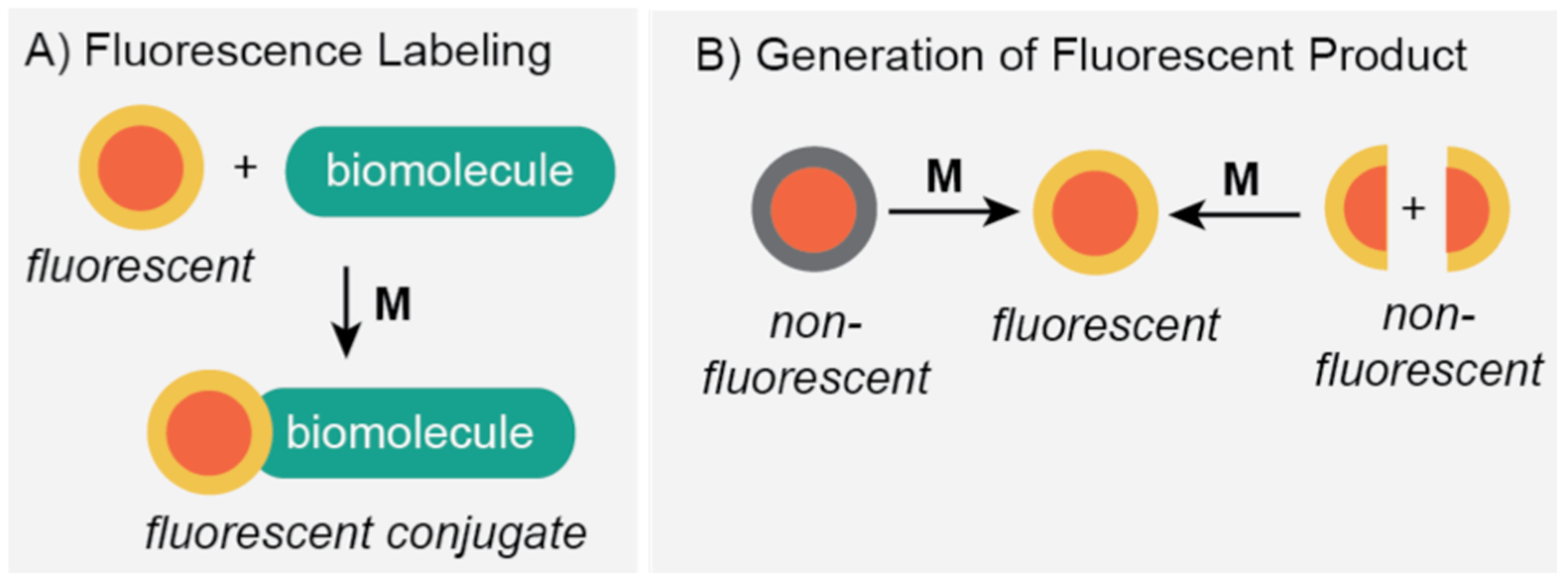
Strategies employed to study metal-catalyzed reactions in living systems by fluorescent microscopy: A) fluorescence labeling; and B) generation of fluorescent product. M = synthetic metal catalyst.
2.2. Studies of Intracellular Metal-Catalyzed Reactions Using Fluorescence Microscopy.
To circumvent the need for genetic manipulations to introduce fluorescent reporters to proteins, Tirrell and coworkers (2005) took advantage of copper-catalyzed azide-alkyne cycloaddition in live cells (Scheme 4).14 The investigators expressed the model protein barstar in E. coli by using either homopropargylglycine (Hpg) as a methionine surrogate or ethynylphenylalanine (Eth) as a phenylalanine surrogate. The bacterial cultures were then treated with CuBr, a tripodal ligand, and 3-azido-7-hydroxycoumarin and allowed to incubate for 14–15 h at 4 °C. After washing twice with fresh buffer, the cells were imaged by fluorescence microscopy. It was observed that cells containing barstar with either Hpg (Scheme 4A) or Eth (Scheme 4B) were strongly emissive, whereas those in the control groups that lacked recombinant barstar were not. Although these results suggested the coumarin fluorophores reacted with the alkyne groups in live cells, it is not clear what percentage of Hpg or Eth in barstar were converted to their corresponding triazole products. Estimation of catalytic efficiency would also require measurement of the intracellular copper concentration, which was not reported. The authors noted that the microscope images showed punctate fluorescence originating from inclusion bodies. However, it is uncertain whether catalysis occurred within these units or if the inclusion bodies formed due to barstar-coumarin aggregation.
Scheme 4.
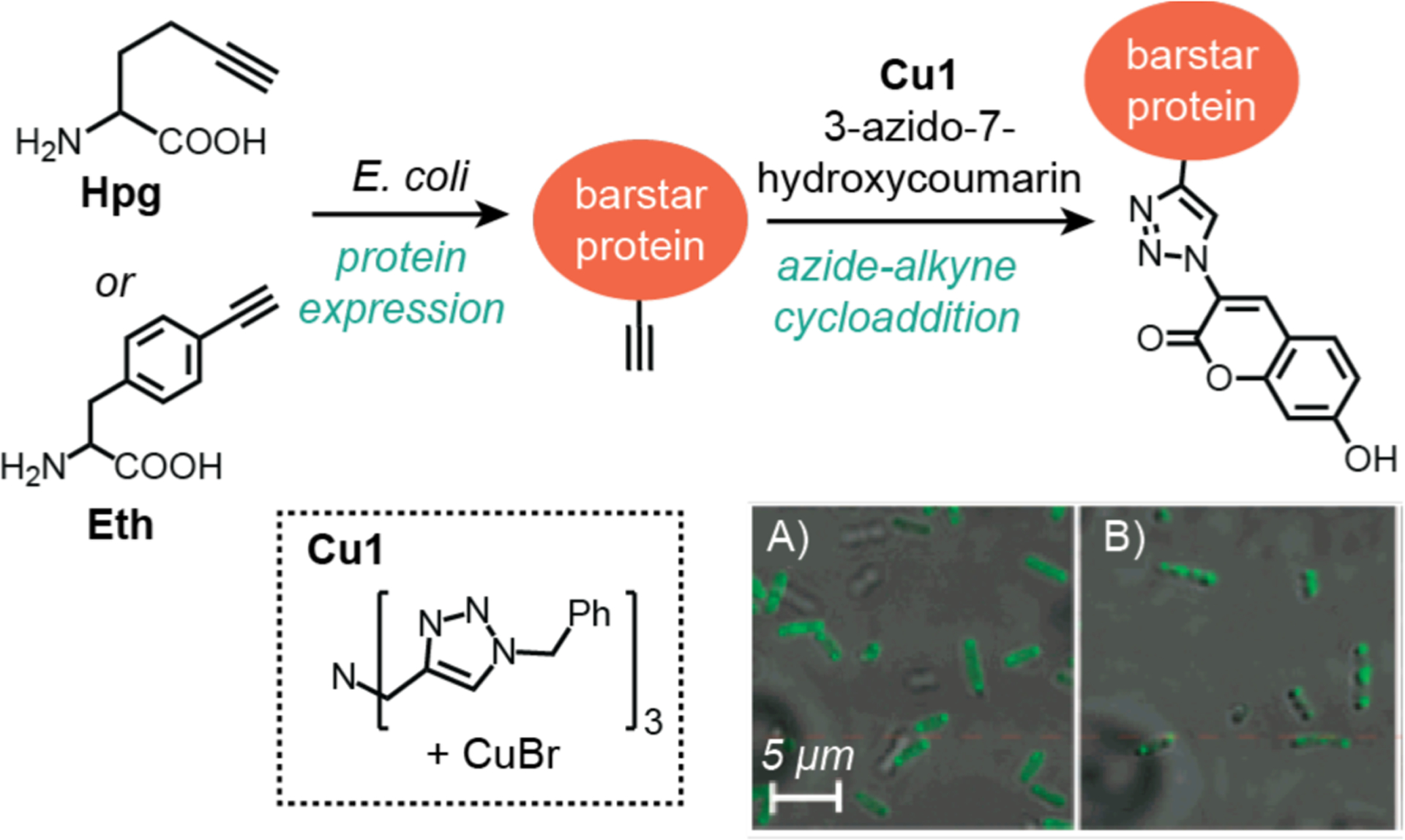
Studies by Tirrell and coworkers demonstrating the use of copper catalysts to promote azide-alkyne cycloaddition in E. coli. The fluorescence microscope images show cells containing barstar with Hpg (A) or Eth (B) after treatment with CuBr and 3-azido-7-hydroxycoumarin. Cells were treated with substrate and catalyst for 14–15 h and then washed prior to imaging. Microscope images adapted with permission from ref 14. Copyright 2005 American Chemical Society.
In 2006, Meggers and coworkers demonstrated that deprotection of pro-fluorogenic substrates is a convenient strategy to study intracellular catalysis (Scheme 5).43 In this work, the researchers employed an organometallic ruthenium catalyst Ru1 and thiophenol to promote the cleavage of allyl carbamate groups to primary or secondary amines. In solution studies, they showed that Ru1 was capable of converting non-fluorescent 1 into the well-known fluorophore rhodamine 110 (compound 2) with up to 80% yield in the presence of glutathione in cell extract. In biological studies, HeLa cells were first incubated with compound 1 and the membrane dye carbocyanine DilC18, washed with PBS to remove excess substrate, and then treated with Ru1 and thiophenol. Real-time imaging by LSCM showed strong emission from the cell interior since the green fluorescence from 2 superimposed quite well with the red fluorescence from the membrane dye (Scheme 5, bottom). It was determined that the fluorescence intensity increased by about 10-fold (presumably relative to background) over 10 min. Unfortunately, this relative change in fluorescence does not provide information about the exact concentration of starting materials or products so reactions yields could not be determined. Based on the cell images provided, it does not appear that the fluorescence signals from 2 were localized within specific regions of the cell. In subsequent studies, Meggers’ group identified other ruthenium complexes that showed significantly greater catalytic activity than Ru1,45,74 indicating that the catalysts are highly tunable. Recent work by Waymouth, Wender, and coworkers suggest that for at least one Ru catalyst variant,75 it is either extracellular or is readily removed from inside cells by washing. However, these studies were performed using 4T1 breast tumor cells with a different probe than that used by Meggers and coworkers.43
Scheme 5.
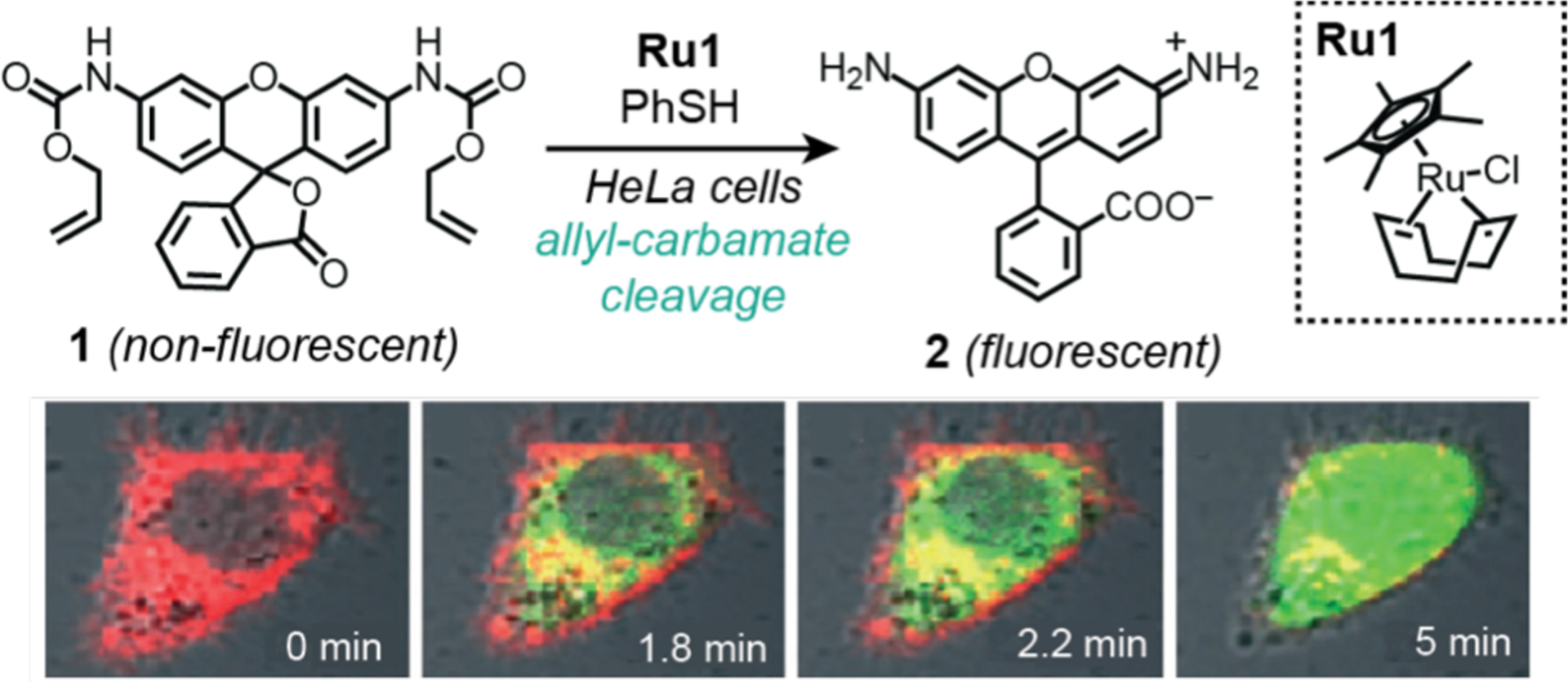
Studies by Meggers and coworkers demonstrating the use of ruthenium catalysts to promote allyl carbamate cleavage in HeLa cells. The confocal fluorescence microscope images show the formation of increasing amounts of rhodamine 110 (2) after reaction of 1 with Ru1 and PhSH. Cells were treated with substrate for 30 min, washed, and then treated with catalyst and thiophenol prior to imaging. Red = membrane dye, green = 2. Microscope images adapted with permission from ref 43. Copyright 2006 John Wiley and Sons.
The protecting group cleavage strategy was expanded to include removal of other chemical functionalities. In 2010, Shin, Ahn, and coworkers developed a propargyl ether deprotection scheme as a way to detect palladium in living organisms (Scheme 6).76 They found that the non-fluorescent propargyl ether compound 3 showed selective turn-on in the presence of various palladium sources (Pd(PPh3)4, PdCl2, Na2PdCl4) due to formation of a fluorescein derivative (4). Remarkably, the investigators demonstrated that this reaction could occur inside live zebrafish. Using five-day-old zebrafish, compound 3 and various concentrations of PdCl2 (0–20 μM) were added to the fish media for 30 min at 28 °C. Visualization by WFM revealed increasing fluorescence inside the living specimen as a function of increasing palladium concentrations. In separate experiments, 3-month-old zebrafish were similarly treated with 3 and PdCl2 for 30 min and then dissected to isolate their organs and tissues. Fluorescence from 3 was detected by microscopy mostly in the brain, eye, and fin. Unfortunately, the fluorescence integrations in these images were not reported so the relative distribution of the probe in these studies could not be determined. It is also not clear whether the location of the probe is indicative of where the palladium was distributed inside the zebrafish since it is possible that the activated probe molecules could have migrated between different organs.
Scheme 6.
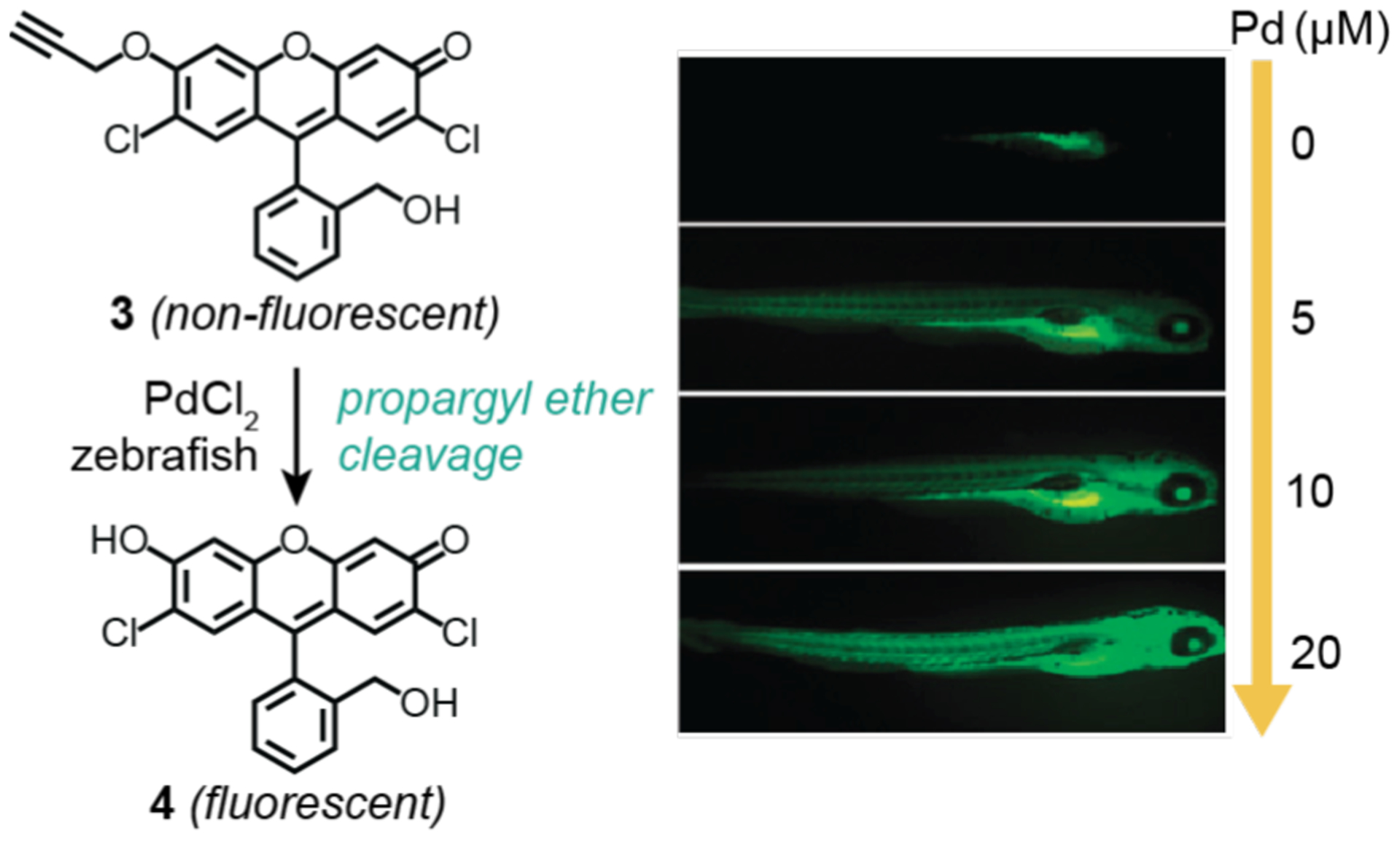
Studies by Shin, Ahn, and coworkers demonstrating the use of palladium salt to promote propargyl ether cleavage in zebrafish. The widefield fluorescence microscope images (right) show five-day old zebrafish incubated with 4 and different concentrations of PdCl2 (0–20 μM). The zebrafish were treated with substrate for 30 min, washed, and then treated with catalyst for 30 min prior to imaging. Microscope images adapted with permission from ref 76. Copyright 2010 The Royal Society of Chemistry.
Rather than using an uncaging approach to generate active fluorophores, Bradley and coworkers sought to synthesize fluorescent products in vitro from non-fluorescent precursors. In 2011, the researchers used palladium nanoparticles to promote Suzuki-Miyaura C–C bond cross-coupling in live cells (Scheme 7).41 In these experiments, HeLa cells were incubated with fluorescently labeled palladium microspheres (Pd1) for 24 h and then thoroughly washed with fresh growth media. The mono-triflate 5 and boronic ester 6 were then added and allowed to incubate for 48 h before the cells were fixed and imaged by LSCM. The presence of green fluorescence indicated that 4 was successfully generated in vitro. Furthermore, 4 appeared to colocalize with Mitotracker Deep Red, which suggests it preferentially targets mitochondria. However, the extent of overlap between 4 and the mitochondrial stain is unclear since no correlation analysis was reported (e.g., Pearson’s or Manders’ coefficients).77–79 A disadvantage of fixing cells prior to imaging is that reaction changes over time cannot be observed in these studies. Furthermore, although the cellular structures were preserved due to fixation, the possibility that 7 could still diffuse in or out of cells leads to some uncertainty about whether it had formed intracellularly.
Scheme 7.
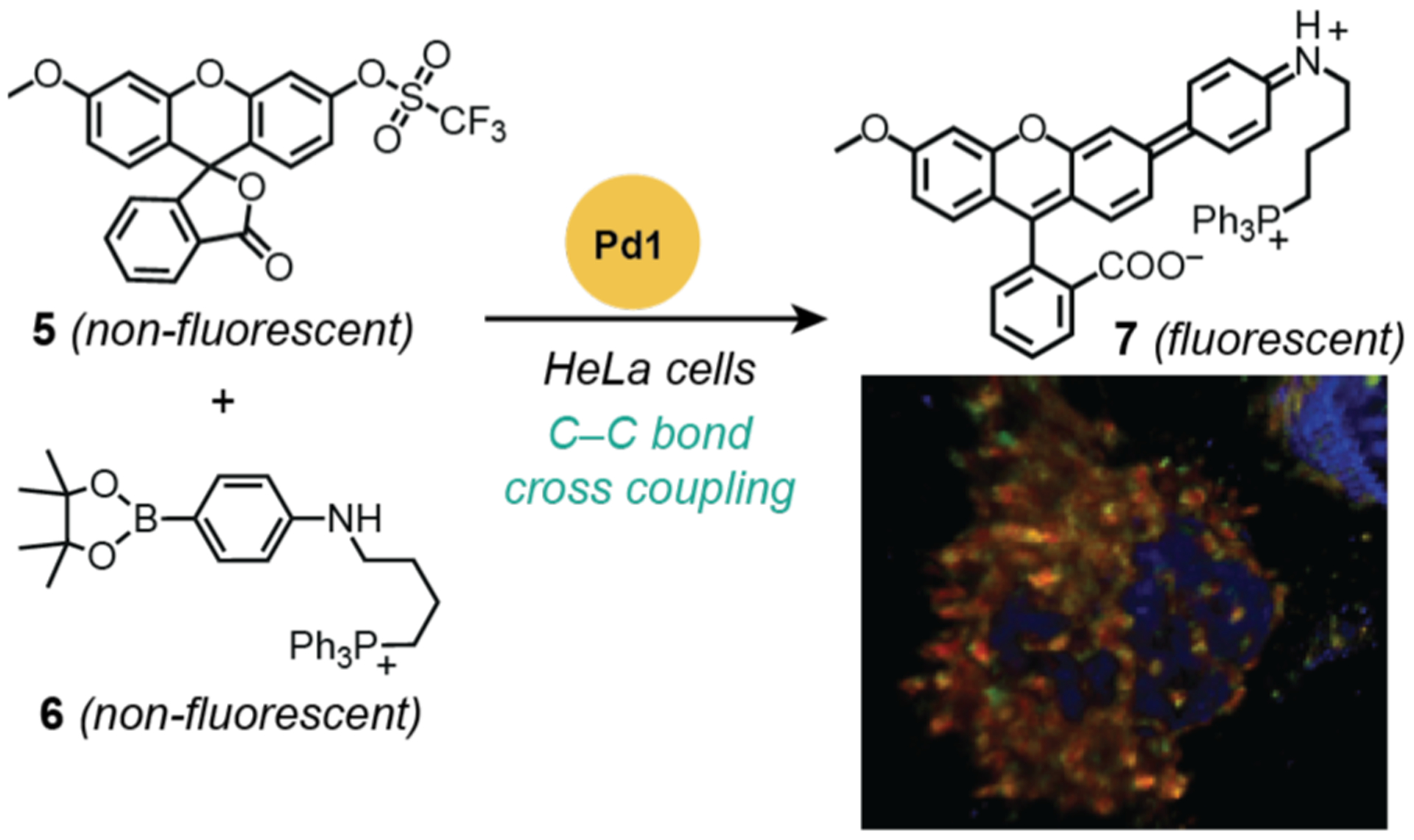
Studies by Bradley and coworkers demonstrating the use of palladium nanoparticles to promote C–C bond cross coupling in HeLa cells. The fluorescence microscope image (bottom right) obtained from merging the red (mitochondrial stain MitoTracker Deep Red), blue (nuclear stain Hoechst 33342), and green (compound 7) channels in fixed cells; orange indicates co-localization between the mitochondrial stain and 7. Cells were treated with catalyst for 24 h, washed, treated with substrates for 48 h, washed, and then fixed with paraformaldehyde for 30 min prior to imaging. Pd1 = palladium(0) nanoparticles prepared from amino-functionalized polystyrene, Pd(OAc)2, and hydrazine. Microscope image adapted with permission from ref 41. Copyright 2011 Springer Nature.
Another method to produce fluorescent products from non-fluorescent (or weakly fluorescent) starting materials is through chemical reduction. Our research group in 2017 disclosed our efforts to develop reductase enzyme mimics using organometallic iridium complexes to catalyze transfer hydrogenation reactions in cells (Scheme 8).52 After testing a variety of probe candidates, we found that the weakly emissive BODIPY-aldehyde 8 converted to a strongly fluorescent compound 9 upon reduction using Ir1 and a hydride source. This reaction was tested in NIH-3T3 mouse fibroblast cells by first treatment with 8 for 4 h, washed with fresh media, and then exposed to Ir1. Imaging by LSCM revealed that these cells showed gradual increase in fluorescence, suggesting that successful transfer hydrogenation had occurred to form 9. Integration of the fluorescence images using the program ImageJ indicated that cells containing 8/Ir1 yielded 1.6-fold higher emission intensity than that of controls containing 8 only, 8/IrCl3, or 8/Ir1/pyruvate (Scheme 8, bottom). The addition of pyruvate suppressed formation of reduced nicotinamide adenine dinucleotide (NADH), which is presumed to be the endogenous hydride source required for transfer hydrogenation in these studies. Although quantification of the microcopy data allowed comparison between the different treatment groups, it does not provide information about the iridium catalyst efficiency or its cellular distribution. In recent work, we have created improved variants of Ir1 but their activity in vitro has not yet been evaluated.19,55
Scheme 8.
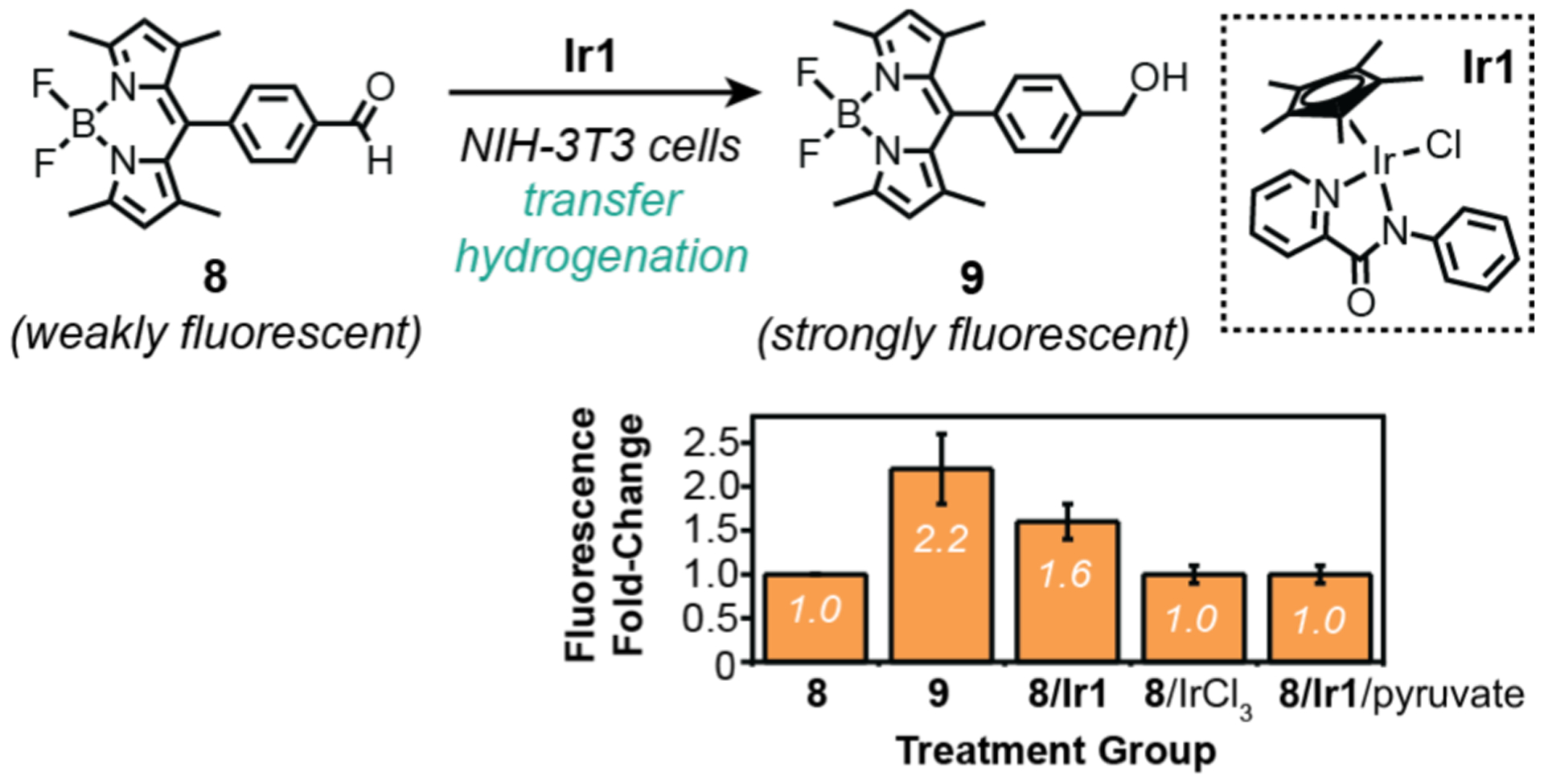
Studies by Do and coworkers demonstrating the use of organometallic iridium catalysts to promote transfer hydrogenation in NIH-3T3 cells. The plot shows change in integrated fluorescence from confocal images of cells after different treatments, relative to that observed from treatment with only aldehyde 8. Cells were treated with substrate for 4 h, washed, and then treated with catalyst (and pyruvate in one treatment group) for 2 h prior to imaging.
2.3. Challenges and Opportunities.
Fluorescence microscopy is a highly sensitive technique capable of providing a tremendous amount of information simultaneously (Table 1), such as the subcellular location of light-emitting analytes, dynamic changes of reactants, and morphology of biological specimens. Although the examples above illustrate why it is an indispensable tool in the investigation of metal-catalyzed reactions in living systems, there are some limitations. First, conventional fluorescence microscopy, including both widefield and confocal (Table 2), has a maximum spatial resolution of ~200 nm due to the diffraction limit of light used.80 The fluorescence images acquired in such studies are ensemble averaged since individual macromolecules (~10 nm) or small-molecules (~1 nm) cannot be resolved. Thus, conventional fluorescence microscopy is unable to probe differences in individual catalyst behavior. Because the living environment is heterogeneous, it is possible that important chemical information is lost through ensemble averaging of the fluorescence data. Second, the integrated fluorescence detected in cells is not necessarily proportional to the total number of light emitters produced. For example, some fluorophores could be photobleached by high intensity light or consumed by subsequent reactions with cellular components.62 Unfortunately, this means that estimation of the amount of substrates, products, or catalysts present in cells is usually made qualitatively rather than quantitatively. Third, prolonged exposure of living specimens to high-power light sources can cause phototoxicity (e.g., continuous illumination using lasers).81 This possibility is important to consider in biocompatibility studies since cell viability measurements are not typically performed under imaging conditions (Scheme 2).
The discovery of super-resolution fluorescence microscopy (SFM) has allowed imaging beyond the diffraction limit,80 which was a milestone achievement recognized by the Nobel Prize to Betzig, Hell, and Moerner in 2014.82 This advance made it possible to visualize biological processes on the single molecule level,83,84 such as protein interactions,85 gene expression,86,87 and enzyme catalysis.88,89 Although SFM has not yet been applied to studies of synthetic metal-catalyzed reactions in cells, we believe it could reveal intracellular behavior not observable by other methods. For example, we could use SFM to monitor individual catalytic events in living cells using pro-fluorogenic substrates and fluorescently labeled catalysts. These data would allow calculation of the reaction kinetics of different catalyst molecules at various cellular locations. In conventional studies, the amount of product generated (or starting material consumed) and the quantity of catalyst present in the cell must be measured separately in order to determine the ensemble averaged catalyst activity. Using SFM, however, there is no need for such measurements since the reaction kinetics of each catalyst molecule are provided directly from the imaging data. It is worth noting that in SFM experiments, photobleaching of fluorophores is actually a desirable process.90 In addition, imaging by SFM will help resolve ambiguity regarding whether catalysis occurs inside cells, outside cells, or on cell surfaces because it can pinpoint exactly when and where individual reaction events take place. In the ensemble-averaged studies described in the preceding section, even if rigorous cell washing procedures were used, there is always the possibility that substrates and catalysts can diffuse outside of the cell where they can react and then subsequently get transported back inside.
To address concerns regarding phototoxicity, there are a variety of options to mitigate the destructive effects of light on biological samples.91 For example, two-photon excitation, temporal separation of light exposure,81 or addition of antioxidants92 have been shown to be successful strategies. Improvements in both hardware and software can also help lessen the impact of photodamage to living speciemens.91
3. FLOW CYTOMETRY
3.1. General Background.
Flow cytometry is a versatile technique that allows rapid multi-parametric analysis of cell populations using optical cameras or detectors.93,94 Modern instruments employ microfluidic systems that align suspended cells into single file as they pass through a laser source for analysis. The forward scattered (FSC) light provides information about cell size whereas the side scattered (SSC) light provides information about cell granularity. In fluorescence flow cytometry (FFC) (Chart 2), cells are pre-treated with different fluorescent dyes, which can report on a wide variety of characteristics (e.g., cell viability, nucleic acid content, oxidative stress, gene expression, etc.). Analysis of the light emitted in different fluorescence channels allows profiling of individual cells. Multi-laser systems can detect up to 20 parameters at a rate of ~25,000 cells per second.95 Although other flow cytometry detection methods are available (e.g., imaging96 or mass spectrometric95,97),98 only the use of fluorescence has been reported so far in investigations of synthetic metal-catalyzed reactions in cells.
Chart 2.
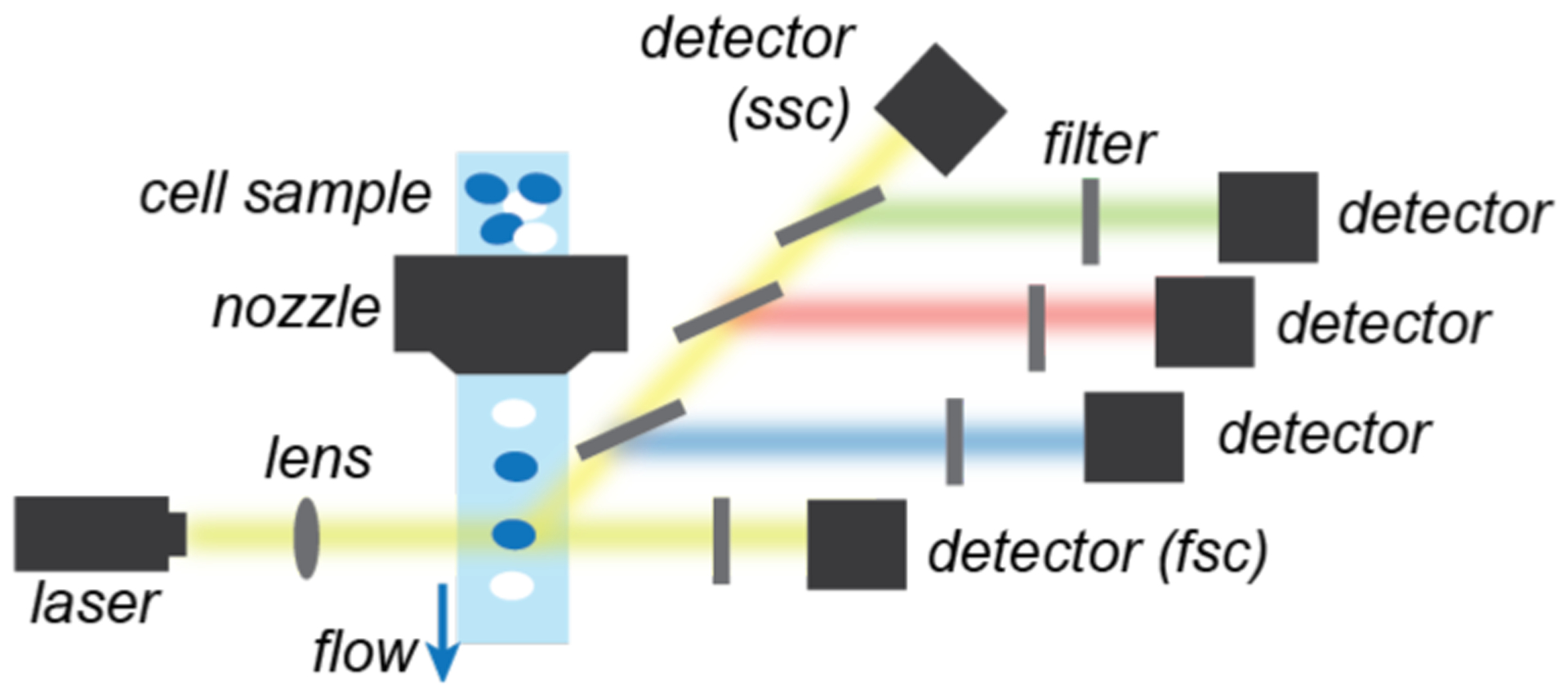
Simplified schematic of typical fluorescence flow cytometers.
Abbreviations: fsc = forward scatter, ssc = side scatter.
When designing FFC experiments, one of the most important factors to consider is the spectral properties of the fluorophores used. For example, it may be desirable to detect the presence of substrates, products, and/or catalysts simultaneously as a way to monitor the progress of intracellular reactions. Each of these chemical species need to be tagged with fluorophores that do not exhibit significant emission overlap so that they could be distinguished from one another using different optical channels. Fluorophores that have high quantum yield and good photostability are the most ideal for this application.
Because FFC enables analysis of individual live cells, it is a useful tool for biocompatible catalyst development investigations (Scheme 2). FFC probes some of the same features as fluorescence microscopy, such as whether catalysis occurred intracellularly, except that it can provide information on much larger cell populations in significantly less time due to its high throughput capability.
3.2. Studies of Intracellular Metal-Catalyzed Reactions Using Flow Cytometry.
The application of flow cytometry to investigate synthetic catalysis on cells was reported by Tirrell and coworkers in 2003 (Scheme 9).32 In this work, the researchers were interested in developing bioorthogonal methods to selectively label cell surfaces. They found that the methionine surrogate azidohomoalanine (10) could be metabolically incorporated into the outer membrane protein C (OmpC) in E. coli to yield OmpC-azide, which placed azido groups on the cell exterior. Mutant cells were also engineered to increase the methionine density of OmpC by six residues relative to that in wild type cells. It was reported that treatment of E. coli containing OmpC-azide with alkyne-biotin compound 11 in the presence of Cu2 (CuSO4/tripodal ligand) and tris(carboxyethyl)phosphine (TCEP) led to successful azide-alkyne cycloaddition. To visualize the cell surface conjugation product, cells were stained with avidin Alexa Fluor 488 and then subjected to flow cytometric analysis. Mutant cells containing OmpC-azide (Scheme 9C) showed about 10-fold increased mean fluorescence in comparison to that of mutant cells containing OmpC-met that lacked clickable azide groups (Scheme 9A). The authors proposed that although wild-type cells bearing OmpC-azide likely reacted with 11, their biotin sites were too sterically hindered to bind avidin for staining (Scheme 9B); thus, these samples gave cytograms similar to those obtained from mutant cells containing OmpC-met (Scheme 9A). Finally, mixing cells featuring OmpC-azide with those featuring OmpC(−) (without added methionine during protein expression) revealed two different cell populations (Scheme 9D) as expected since only cells containing azide groups could be labeled.
Scheme 9.
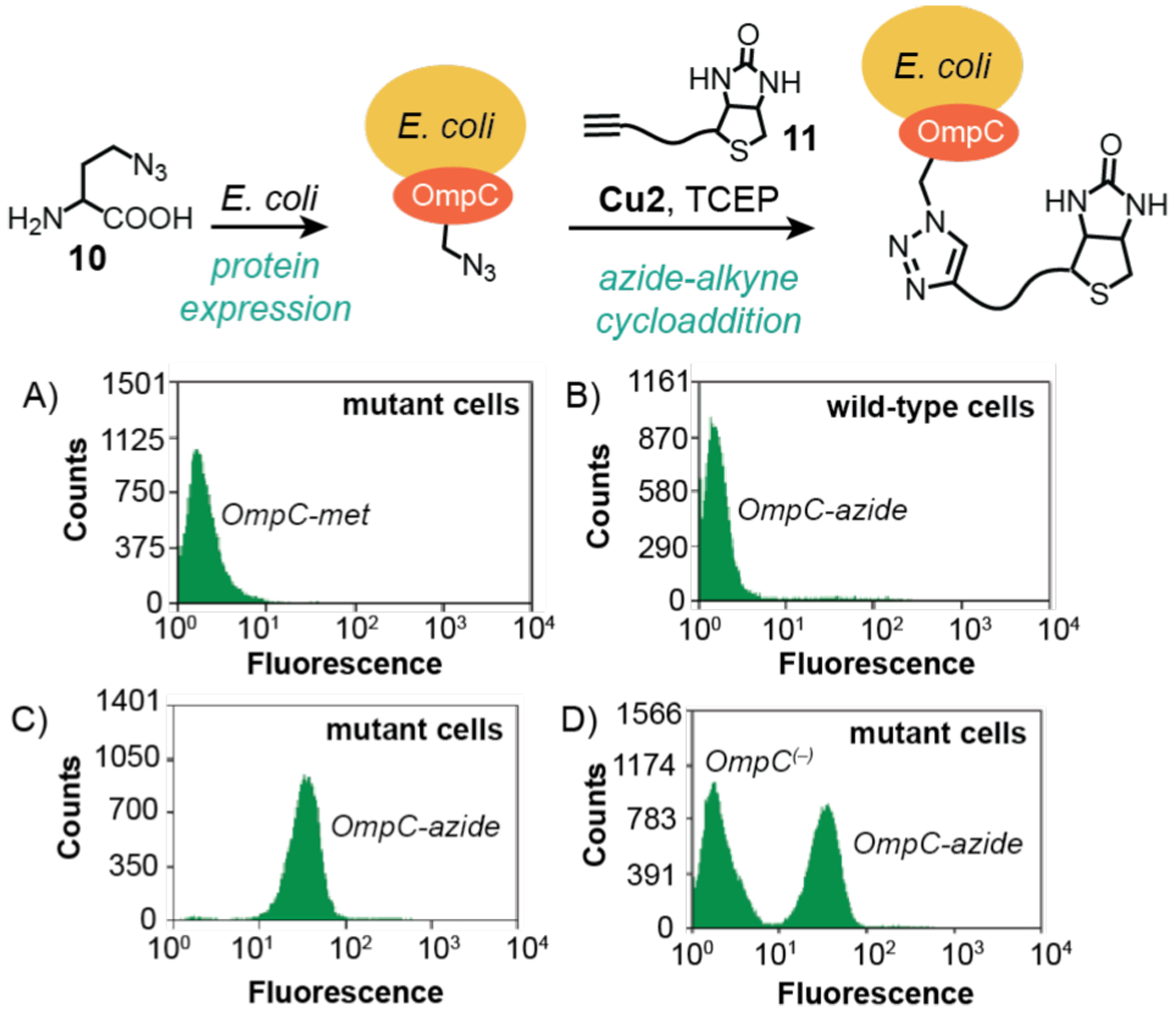
Studies by Tirrell and coworkers demonstrating the use of copper catalysts to promote azide-alkyne cycloaddition on E. coli cell surfaces. The plots show flow cytometry data from: A) mutant cells containing OmpC-met; B) wild-type cells containing OmpC-azide; C) mutant cells containing OmpC-azide; and D) a mix population of mutant cells containing OmpC-azide and cells containing OmpC(–) (without added Met). Cu2 = CuSO4/tripodal ligand. Flow cytograms adapted with permission from ref 32. Copyright 2003 American Chemical Society.
In a follow-up study in 2004, Tirrell and coworkers discovered that replacing copper(II) with copper(I) salts led to dramatic improvements in catalytic rates (Scheme 10).99 Using flow cytometry, they were able to show that mutant cells treated with CuSO4/TCEP gave a 20-fold increase in fluorescence relative to background (Scheme 10A), whereas those treated with CuBr gave a 150-fold increase (Scheme 10B). Because FFC can analyze much larger cell populations than fluorescence microscopy in the same amount of time, the former is a more reliable method to obtain aggregate data. An ambiguity in these studies is that since not all surface biotins are sterically accessible by Alexa Fluor 488, it is unclear what percentage of the total cell surface alkynes had reacted with 11, which is a more accurate measure of catalytic efficiency.
Scheme 10.
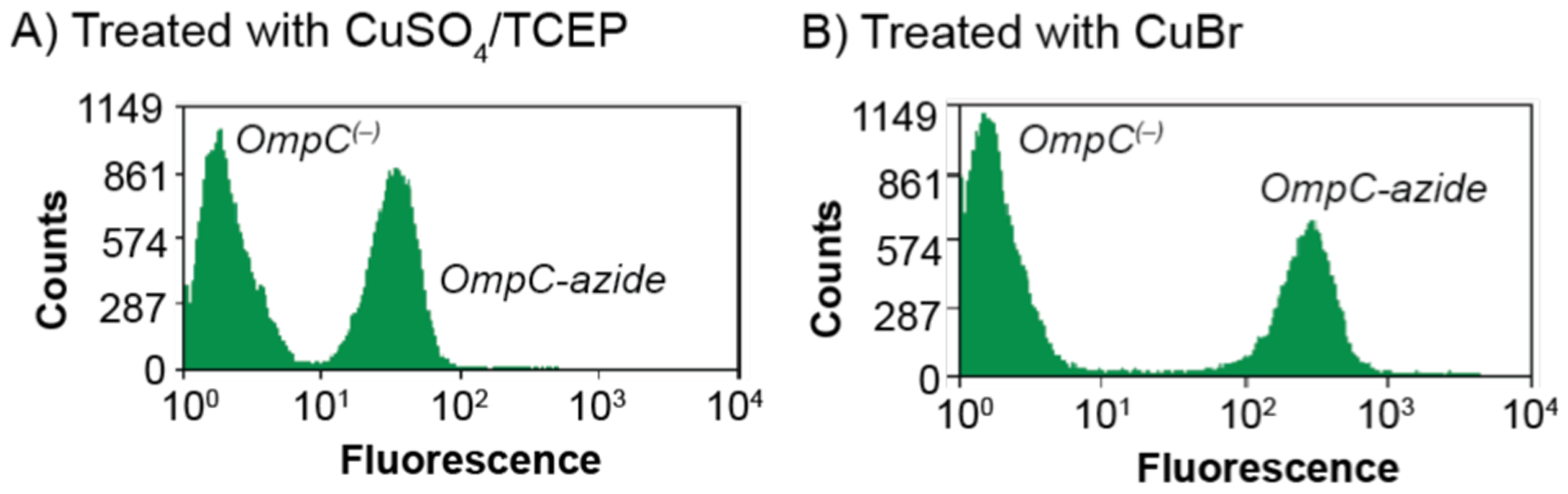
Flow cytograms obtained from studies of mutant E. coli containing either OmpC(−) or OmpC-azide after reaction with 11 using either CuSO4/TCEP (A) or CuBr (B) as the copper source. The cells were stained using avidin Alexa Fluor 488. Flow cytograms adapted with permission from ref 99. Copyright 2004 American Chemical Society.
Recently in 2016, Bradley and coworkers demonstrated that heterogeneous copper catalysts could be used for in situ drug synthesis.35 The authors showed that their entrapped copper nanoparticles (Cu3) were non-toxic and structurally robust (<1% copper leaching after 72 h in H2O at 37 °C). This Cu3 catalyst was used to promote azide-alkyne cycloaddition between 12 and 13 to yield combretastatin A4 (14), which is a tubulin polymerization inhibitor that is highly cytotoxic to a variety of cancer cell lines. The Cu3 catalyst was confirmed to be extracellular based on brightfield microscope images showing that the copper particles were larger than the size of individual cells. Reactions performed in the presence of ovarian adenocarcinoma SKOV-3 cells were evaluated by flow cytometry to interrogate the biological changes induced by in situ formation of 14. Cell proliferation measurements using a commercial Click-iT EdU assay revealed that samples treated with 12, 13, Cu3, and sodium ascorbate (NaAsc) displayed a significantly greater percentage (59%) of cells arrested in the G0/G1 phase compared to those in the control (24%) (Scheme 11A). Generation of the cytotoxic compound 14 was further confirmed using an apoptosis assay, which measures the binding of apoptotic cells to the Annexin V protein and absorption of the DNA intercalator propidium iodide (PI) (Scheme 11B). It was observed that cells grown in combination with drug precursors, reductant, and copper catalyst exhibited 48% apoptosis, which is substantially higher than the 13% in untreated cells (Scheme 11B). The flow cytometry data suggest that 14 generated extracellularly was able to be uptake up by the SKOV-3 cells to induce its cytotoxic effects. However, additional studies are needed to quantify the amount of 14 produced in these in situ reactions.
Scheme 11.
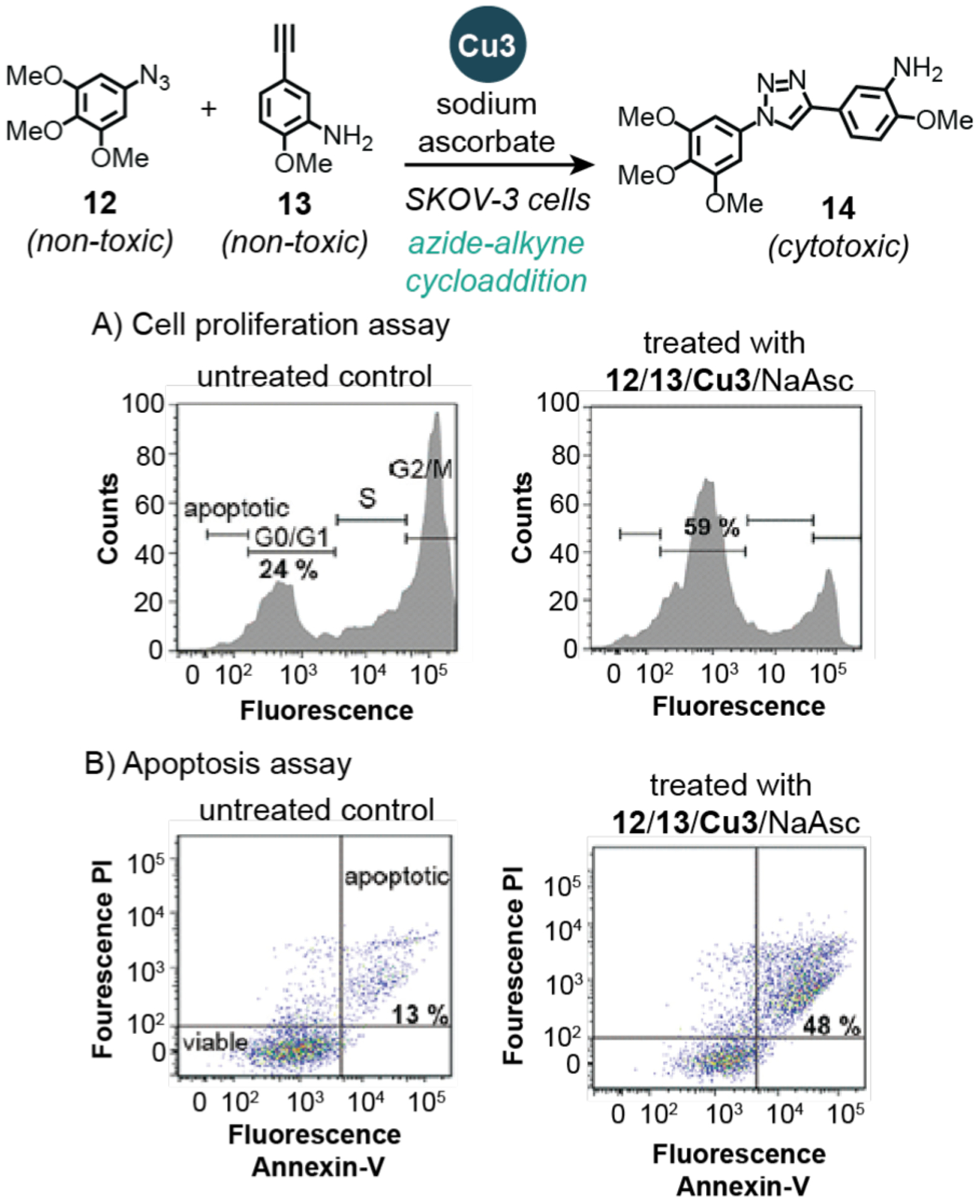
Studies by Bradley and coworkers demonstrating the use of copper nanoparticles to promote azide-alkyne cycloaddition in SKOV-3 cells. The plots show flow cytometry data from: A) cell proliferation assays (cell cycle profiles) and B) apoptosis assays. PI = propidium iodide. Cu3 = copper(0) nanoparticles prepared from amino-functionalized resin, Cu(OAc)2, and hydrazine. Flow cytograms adapted with permission from ref 35. Copyright 2016 John Wiley and Sons.
To overcome the necessity of using large excess of metal salts in intracellular catalysis, Bradley and coworkers reported a simple yet elegant delivery strategy in 2017.100 In this work, the researchers designed a cell-penetrating tris(lysine) peptide containing a pyridine-carbene chelator and sulfonated cyanine Cy5 dye (Scheme 12). Metalation of this peptide with a Pd precursor produced Pd2, which is water soluble and fluorescently traceable. Characterization of a related Pd complex showed it was taken up by cells more readily than Pd(OAc)2. To test the intracellular activity of Pd2, it was incubated with the pro-fluorogenic compound 15 in human prostate PC-3 cells to induce propargyl carbamate cleavage. Because both the product 2 and Pd2 are strongly emissive, the cells could be analyzed by flow cytometry using the green (530/30 nm for 2) and red (660/20 nm for Pd2) band pass filters. As expected, control samples that were either untreated (Scheme 12A) or treated with 15 alone (Scheme 12B) exhibited only background fluorescence. In contrast, cells containing both 15 and Pd2 displayed increased intensity in both green and red fluorescence (Scheme 12C), suggesting that the presence of palladium is necessary for catalysis. This work is an excellent example of how using fluorophores with non-overlapping emission profiles could be useful in studying intracellular reactions. Although not reported by the authors, it may be possible to use their integrated fluorescence data to calculate average turnover numbers (e.g., mole of product per mole of catalyst).
Scheme 12.
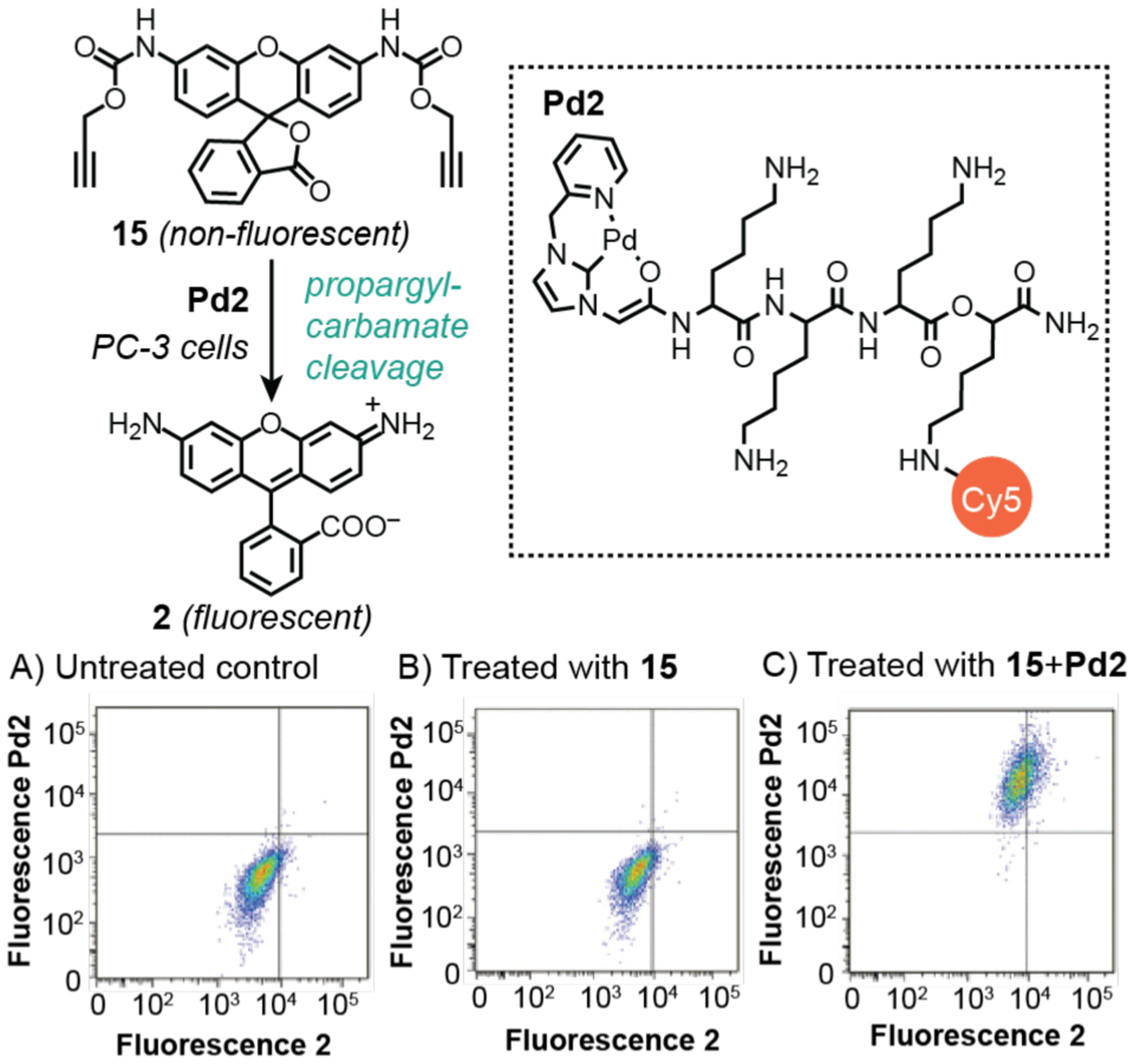
Studies by Bradley and coworkers demonstrating the use of palladium catalysts to promote propargyl carbamate cleavage in PC-3 cells. The plots show flow cytometry data from: A) untreated cells; B) cells treated with 15; and C) cells treated with 15 and Pd2. Cy5 = sulfonated cyanine fluorescent dye. Flow cytograms adapted with permission from ref 100. Copyright 2017 The Royal Society of Chemistry.
3.3. Challenges and Opportunities.
Flow cytometry is arguably unmatched by other live cell analysis techniques in applications requiring fast, accurate, and customizable measurements of large cell populations. Although not as commonly employed as fluorescence microscopy in studies of metal-catalyzed reactions in cells, the examples above demonstrate that it can provide complementary information. However, FFC has a few limitations that users should be aware of. First, FFC detects fluorescence on the single cell level so it cannot differentiate signals originating from different cellular compartments. Thus, studies of nonuniformity in catalyst behavior would require other techniques capable of achieving greater spatial resolution (e.g., fluorescence microscopy). Second, since cells are measured sequentially rather than continuously in flow cytometry, kinetic studies can be complicated.101 A variety of strategies have been introduced to incorporate time as an experimental parameter in studies of enzyme kinetics, such as discontinuous sequential sampling, continuous interrupted sampling, and continuous time recording.102 It has been noted that fast kinetics and heterogeneous populations cannot be resolved.101 Third, FFC is only suitable for studies of cell suspensions so direct measurements of tissues or organisms would require other analytical methods.
In our opinion, the capabilities of FFC have not yet been fully exploited for intracellular catalysis investigations. Current studies typically use only 3–4 parameters, well short of the 20 that are possible.93 We propose that for reaction studies in live cells, simultaneously quantifying markers such as substrate/product/catalyst concentrations, pH, cell viability, glutathione content, and reactive oxygen species would be tremendously helpful. This information would allow us to not only gain insight into the chemistry of interest but also assess any biological changes that might be induced by the xenobiotic components. Of course, implementing such multi-dimensional studies would require that every parameter being assayed has a reporter with distinct spectral signatures to minimize emission overlap.
Technological advances in flow cytometry have expanded its capabilities considerably.98 Mass cytometry (MC) comprises a standard flow system coupled to a mass spectrometer.95,103 While FFC requires the use of fluorophores, MC uses antibodies labeled with rare-earth metal ions. In an MC analysis, individual cells are heated in a plasma torch to ionize all of its elements so that the non-native rare-earth metals from the probes could be detected. Because the mass spectrometer can discriminate between isotopes of heavy metals differing by only one neutron, there is no spillover effect as seen with fluorescence-based reporters. The advantage of MC over traditional FFC is that it can analyze up to 37 parameters. However, it is much slower and is destructive to live cells.
Another alternative to FFC is imaging cytometry (IC).98,104 This method combines high-throughput cytometry with high-resolution optical microscopy. In IC experiments, cells are pre-treated with various fluorescent dyes and then flowed through an imaging platform where their images are captured using one or more microscope objectives. Because the spatial resolution is about 20 nm, subcellular particles could be readily distinguished. However, similar to FFC, a limitation of IC is that the spillover effect of multiple fluorophores overlapping could be problematic.
To the best of our knowledge, neither MC nor IC has been applied for intracellular metal-catalyzed reaction studies. We envision that MC analysis could be very useful in quantifying the total amount of reactants and catalysts present in individual cells, assuming that these species could be tagged with a rare-earth metal. Detecting products using MC would be difficult because the label from the starting material does not change upon conversion to product (i.e., the metal identity would remain the same). Studies using IC could take advantage of its high throughput and high resolution imaging capability to measure where fluorescent products are located within specific regions of the cell. Comparison of results from a large number of cells would reveal to what extent catalytic behavior differs from cell to cell.
4. MASS SPECTROMETRY
4.1. General Background.
Mass spectrometry (MS) is a widely used method for determining the mass-to-charge ratio of ions, which can range from macromolecules to single atoms.105,106 In a typical analysis, a sample is injected into a mass spectrometer where the contents are ionized and the resulting ions are then sorted and detected. Mass spectrometers are classified based on their ionization sources, the most common for biological investigations are electrospray ionization (ESI), matrix-assisted laser desorption ionization (MALDI), and inductively coupled plasma (ICP). They also differ in their mass analyzers (e.g., sector field, quadrupole, or time-of-flight) and can be coupled to different sample separation methods (e.g., gas chromatography (GC), liquid chromatography (LC), or capillary electrophoresis).
The most common MS methods used in intracellular metal-catalyzed reaction studies are ESI for analyzing molecules and ICP for analyzing elements. In ESI-MS, a high voltage is applied to a liquid sample that gets dispersed into a fine spray of charged droplets (Chart 3A).107,108 Through a desolvation process, the ions eventually separate from the droplets and are analyzed by a detector. ESI is considered a “soft” ionization method because it tends to cause minimal fragmentation, which often enables observation of the molecular ions of proteins and small molecules. In ICP-MS, samples are introduced as aerosols that get transferred to an argon plasma, which can reach temperatures as high as 10,000 K (Chart 3B).109 This process atomizes the sample and generates charged ions to be detected. ICP-MS is advantageous over atomic absorption spectroscopy, another common elemental analysis method, because it is more sensitive (can measure samples as low as 0.001 μmol/L) and can detect more than 100 different isotopes simultaneously.110
Chart 3.
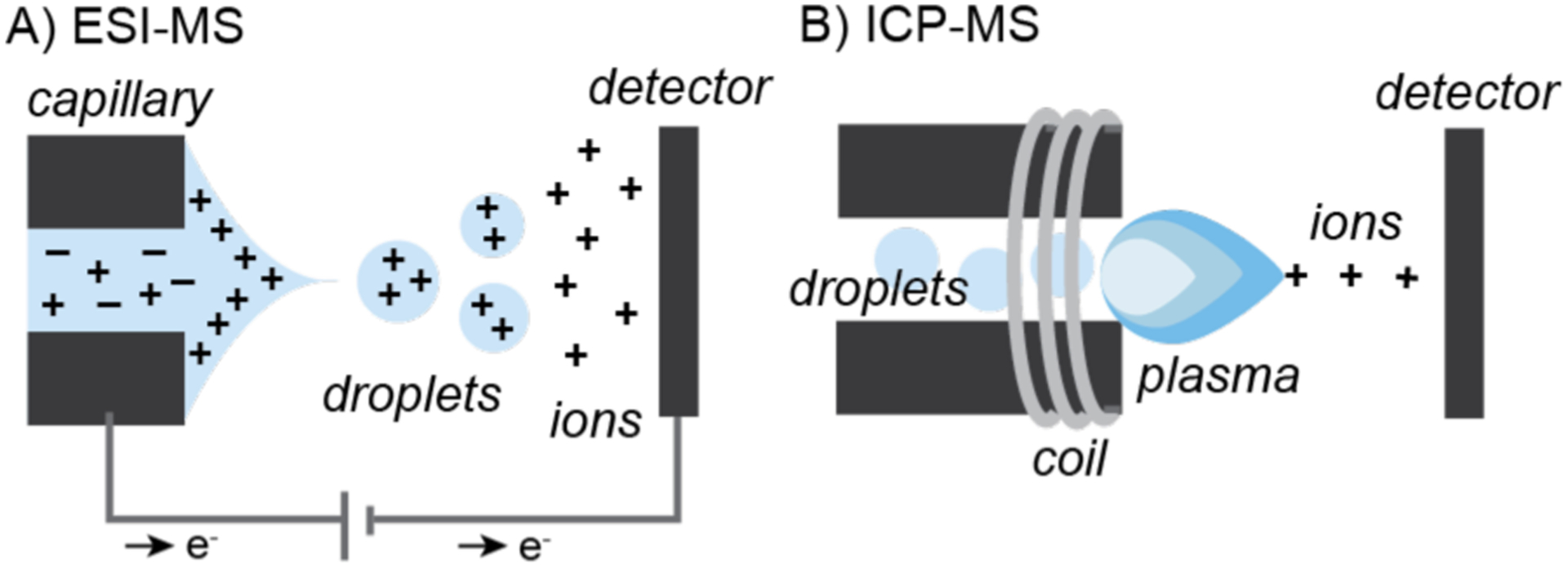
Simplified schematics of A) electrospray-ionization (ESI) and B) inductively-coupled plasma (ICP) mass spectrometry (MS).
When using MS to probe living specimens, it is important to note that this method requires destruction of the biological sample. Although this technique is highly invasive, it is particularly useful in identifying specific molecular species, including those without reporter groups, or quantifying reaction changes.
4.2. Studies of Intracellular Metal-Catalyzed Reactions Using Mass Spectrometry.
In 2014, Chen and coworkers reported a gain-of-function strategy to manipulate proteins under living conditions (Scheme 13).44 In this work, a recombinant green fluorescent protein GFP-Y40-ProcLys containing a N-ε-propargyloxycarbonyllysine group (ProcLys) at Y40 was expressed in HeLa cells for 24 h. These cells were then exposed to Pd2(allyl)2Cl2 for 3 h to induce intracellular propargyl carbamate cleavage, which would generate GFP-Y40-Lys. The authors established that the catalyst was taken up inside cells using ICP-MS (Scheme 13A). Since Pd is not present naturally in cells in appreciable amounts, its presence would indicate successful acquisition by cells from external sources. Analysis of various cell fractions isolated from repeatedly rinsed cells (to remove external Pd) revealed that the catalysts were found mostly in the cytosol and membrane, with a small amount in the nucleus. To measure the depropargylation efficiency, a gel fluorescence assay was developed to quantify the amount of GFP-Y40-Lys produced in cell lysate experiments, giving a yield of ~31%. Liquid chromatography tandem mass spectrometry (MS/MS)111, a technique in which the molecular ion of a specific mass-to-charge ratio is further fragmented and analyzed, was performed on the digested proteins to confirm their identities (e.g., Scheme 13B). Integration of the MS peaks from the cell lysates suggested that the cleavage reaction was about 27% efficient, which was in agreement with their gel fluorescence assay results. The authors hypothesized that the low yields were due to low concentrations of GFP-Y40-ProcLys protein and free Pd species inside cells. This propargyl carbamate cleavage reaction was applied successfully to study intracellular localization of the Erk protein on irreversible dephosphorylation.
Scheme 13.
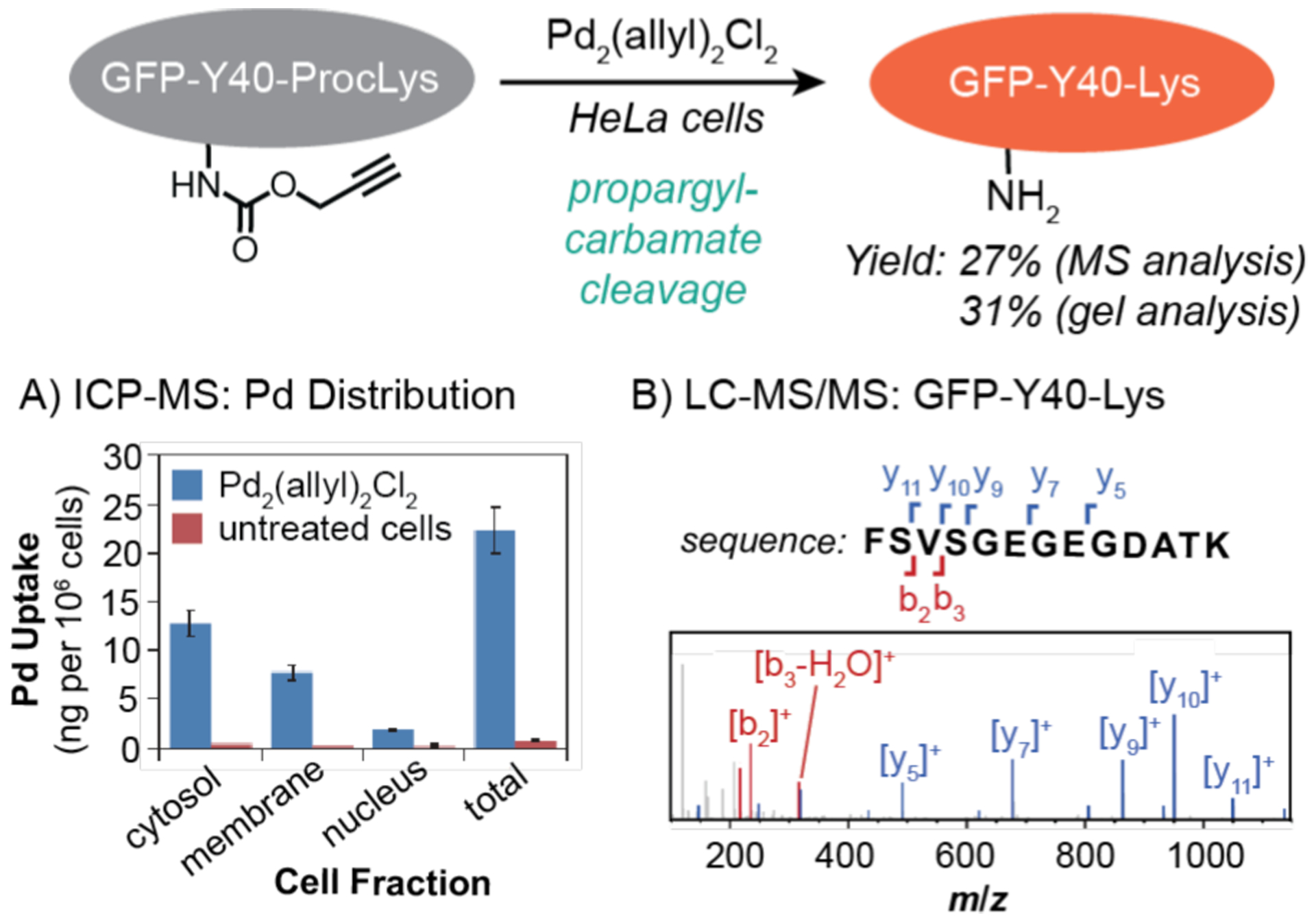
Studies by Chen and coworkers demonstrating the use of palladium catalysts to promote propargyl-carbamate cleavage in HeLa cells. The data are shown for A) distribution of Pd in different cell fractions measured by ICP-MS and B) characterization of the GFPY40-Lys protein measured by LC-MS/MS. The MS data were adapted with permission from ref 44. Copyright 2014 Springer Nature.
Expanding further on the versatility of bioorthogonal palladium catalysis, Chen and coworkers disclosed in 2015 a method for chemical remodeling of cell surface sialic acids.112 Sialic acids are nine-carbon monosaccharides that often reside on the ends of cell surface glycans, such as N-acetylneuramic acid (Neu5Ac) in humans. Although Neu (16), the deacetylated form of Neu5Ac, has been shown to occur in nerve and cancer cells, studying it has been challenging due to its high chemical reactivity in vivo. As a way to generate Neu on cell surfaces, the investigators metabolically incorporated N-propargyloxycarbonylneuramic acid (17) into CHO mammalian cells and then used palladium nanoparticles (Pd3) to catalytically remove the propargyl carbamate groups (Scheme 14). To assess the reaction efficiency, a LC-MS analysis method was developed. After the Pd-catalyzed reactions were complete, the CHO cells were treated with 4-(dimethylamino)-benzoate N-hydroxysuccinimide ester (DMABA-NHS) so that the free amino groups in Neu were protected from further reactions. The cells were then lysed and the precipitated proteins were dissolved in acid to release the sialic acids. The soluble extracts were then analyzed using LC-MS by setting the detector to read only for anion with m/z = 438 [17-H]− (Scheme 14A) and cation with m/z = 415 [18+H]+ (Scheme 14B). Based on the decline in the peak for [17-H]−, it was calculated that the cleavage efficiency was 71%. However, the yield determined from the product peak (i.e., for [18+H]+) was not reported.
Scheme 14.
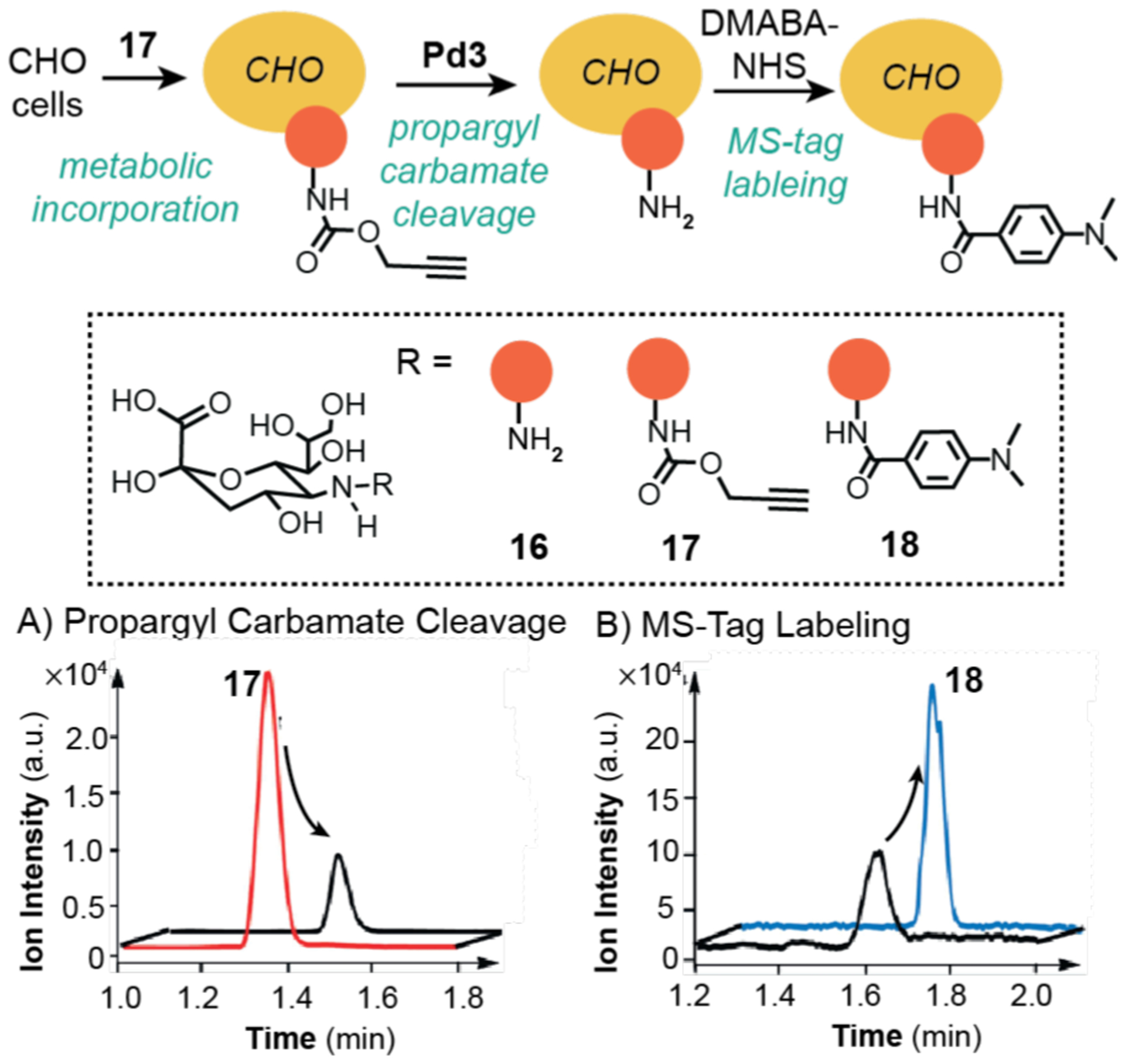
Studies by Chen and coworkers demonstrating the use of palladium catalysts to promote propargyl-carbamate cleavage in CHO cells. The selected ion recording obtained from LC-MS showing the decrease of 17 (A) and increase of 18 (B). Pd3 = palladium(0) nanoparticles prepared from Na2PdCl4 and NaBH4. The MS data were adapted with permission from ref 112. Copyright 2015 John Wiley and Sons.
In 2016, Mascareñas and coworkers used mass spectrometry to establish that synthetic metal catalysts could be targeted to specific cellular organelles (Scheme 15).113 To demonstrate this capability, they used half-sandwich ruthenium complexes as the catalyst platform. The parent complex Ru2 contains a 2-quinolinecarboxylate ligand, whereas its derivatives feature additional phosphonium (Ru3), phosphonium-pyrene (Ru4), and pyrene (Ru5) functionalities. The cationic phosphonium unit is a well-known mitochondria-targeting group and pyrene is a polyaromatic with fluorescent properties. To evaluate the cellular distribution of the ruthenium complexes, HeLa cells were incubated with 50 μM of the different catalysts for 60 min. The cells were then lysed and the resulting contents were separated into mitochondrial and cytosolic fractions. Analysis by ICP-MS revealed that the Ru3 and Ru4 samples gave higher amounts of ruthenium in mitochondria than in the cytosol. In contrast, the Ru2 and Ru5 samples showed the same concentration of ruthenium in both cell fractions, which is expected since complexes that lack phosphonium groups will not likely target mitochondria selectively. Using fluorescence microscopy, the authors demonstrated that Ru3 was capable of catalyzing allyl carbamate cleavage of 1 to 2 inside mitochondria. Interestingly, the targeted catalysts were capable of inducing fast and strong depolarization of the mitochondrial membrane.
Scheme 15.
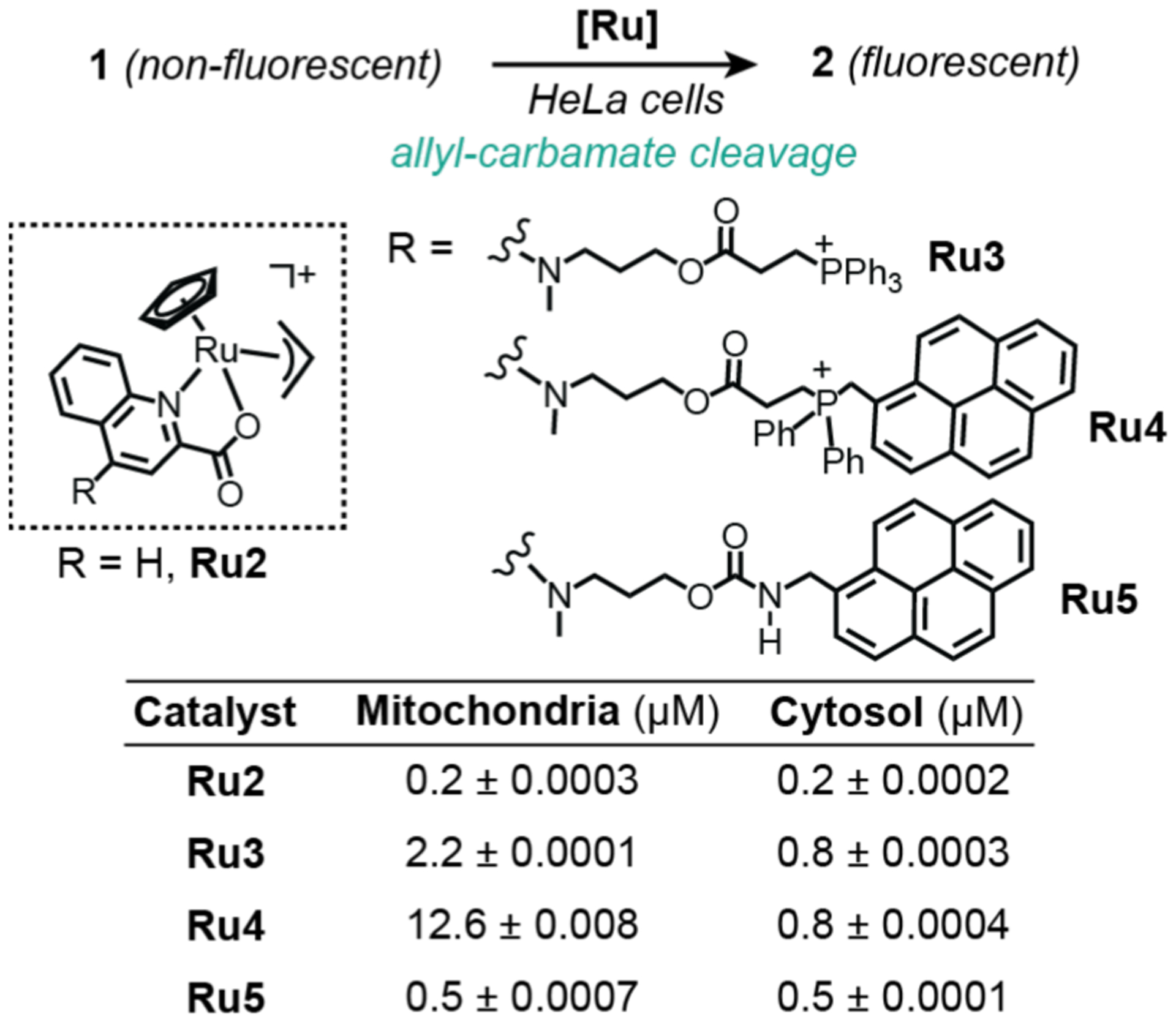
Studies by Mascareñas and coworkers demonstrating the use of ruthenium catalysts to promote allyl-carbamate cleavage in HeLa cells. The ICP-MS data showing the amounts of ruthenium found in the mitochondria and cytosol from cells treated with different catalysts are provided in the bottom table.
Cai and coworkers took up the challenge of quantifying intracellular catalysis in their work on copper-promoted azide-alkyne cycloaddition in 2017 (Scheme 16).114 To maximize the cellular uptake of copper, the investigators tethered a cell-penetrating peptide to a known tripodal ligand. This modified ligand in combination with CuSO4 was found to produce an active catalyst (Cu4). To incorporate alkynyl groups into cellular proteins, the methionine surrogate Hpg was added to human ovarian cancer cells (OVCAR5) during protein expression. These cells were then treated with a biotin-containing azide substrate 19, catalyst Cu4, and sodium ascorbate. After the reaction, the cells were thoroughly washed, lysed, and fractionated into membrane and cytosolic proteins. Complete acid hydrolysis of the biotinylated proteins produced the triazole compound 20. To determine the maximum amount of product obtainable, the total extracted Hpg-containing proteins were denatured using sodium dodecyl sulfate and subjected to copper-catalyzed reaction with 19. Analysis of this sample by LC-MS/MS indicated that the product 20 was present at a concentration of ~49 pg per μg of protein. Based on MS quantification of 20 in the membrane and cytosolic fractions, their corresponding yields were 18 and 0.8%, respectively. The authors proposed that the low yields were due in part to deactivation of the copper catalyst by biological thiols. This study is notable because it is one of the few examples reported in which the reaction yield of a metal-catalyzed reaction was quantified. A complicating factor, however, is that it is unclear what percentage of the alkynyl groups in the folded Hpg-containing protein is chemically accessible, which would affect the calculated yields.
Scheme 16.
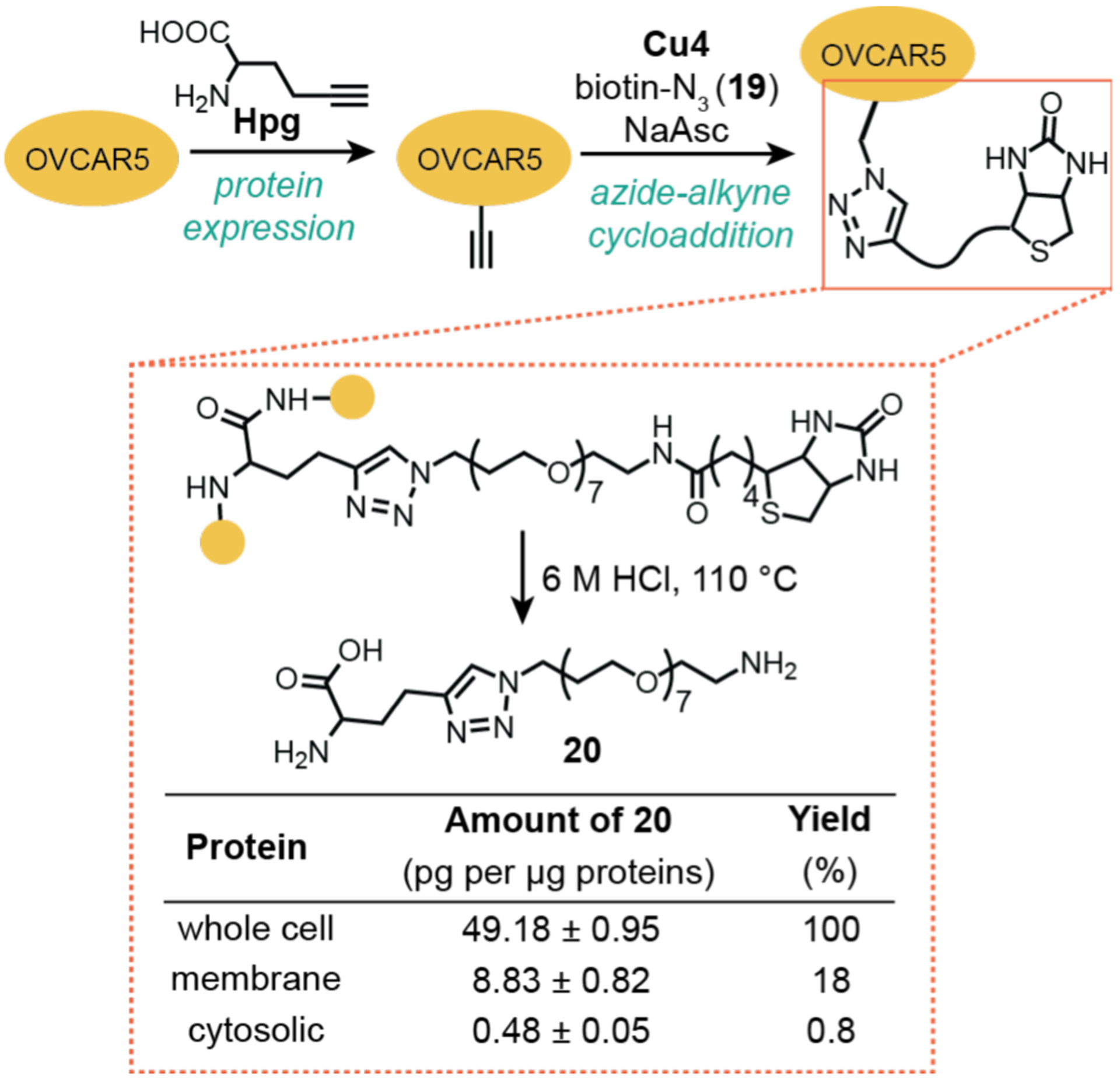
Studies by Cai and coworkers demonstrating the use of copper catalysts to promote azide-alkyne cycloaddition in OVCAR5 cells. The reaction efficiency was determined using LC-MS/MS by quantifying the amount of 20, the triazole product fragment generated via acid hydrolysis. Cu4 = copper(II) complex prepared from a tripodal ligand-peptide conjugate and CuSO4.
Mascareñas and coworkers also took advantage of the analytical capability of LC-MS to quantify intracellular reactions. In 2019, they reported the first example of an allyl alcohol isomerization reaction in cells catalyzed by a ruthenium catalyst (Scheme 17).115 This reaction was demonstrated in HeLa cells using a pro-fluorogenic substrate 21 that converted to 22 upon exposure to Ru6. The reaction products were extracted from cells into methanol and analyzed by LC-MS, which showed that the formation of 22 increased over the course of 6 h (Scheme 17A). Using calibration curves, the total amount of 22 produced was determined. In addition, the amount of ruthenium present inside cells treated with 10 and 25 μM of Ru6 were measured by ICP-MS. Using the equation [concentration of product]/[concentration of catalyst], it was found that the highest turnover number (TON) achieved was 22.4. However, since the quantification of 22 and ruthenium were performed in independent experiments, possible variability between different cell populations could affect the accuracy of the TONs calculated.
Scheme 17.
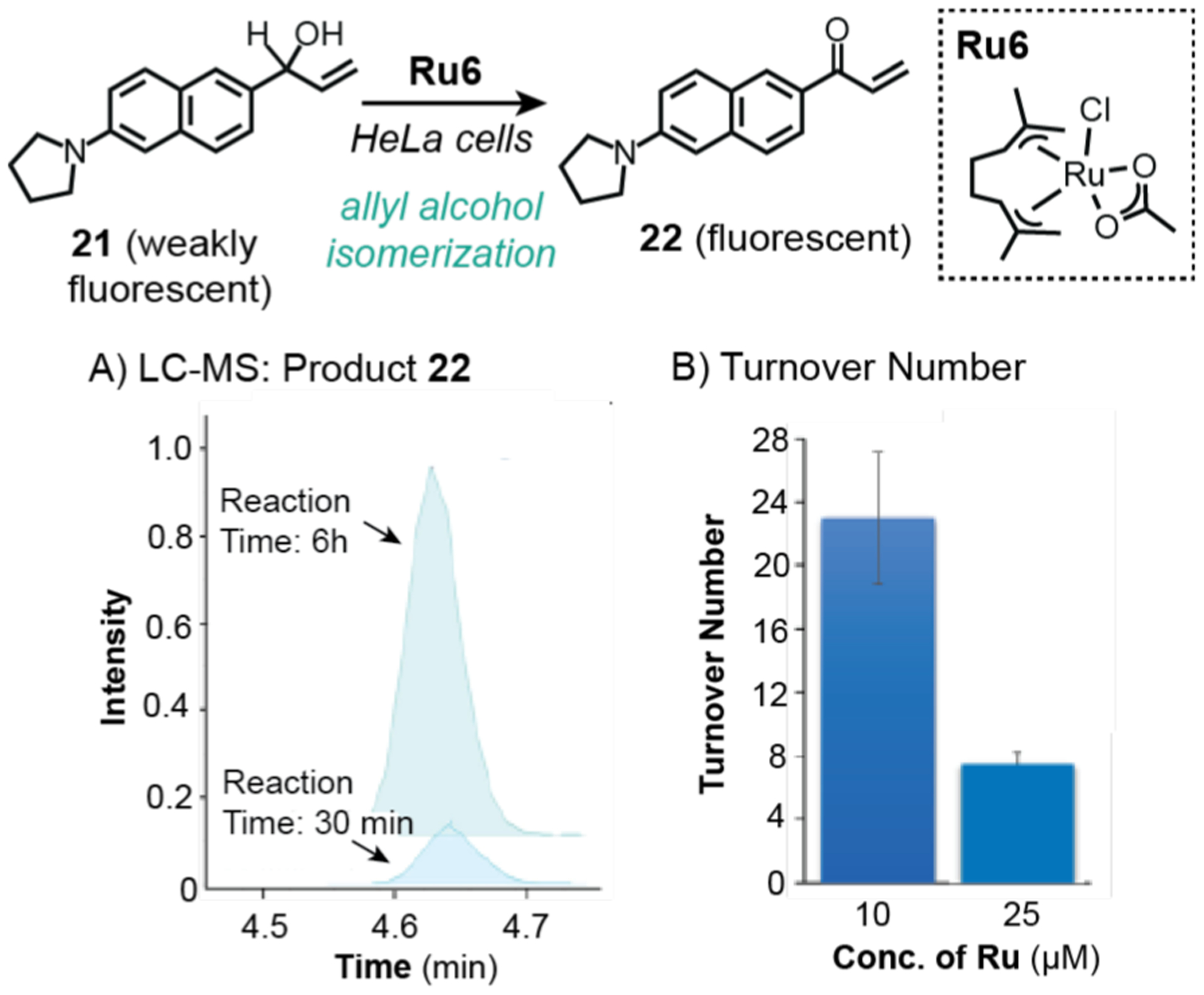
Studies by Mascareñas and coworkers demonstrating the use of ruthenium catalysts to promote allyl alcohol isomerization in HeLa cells. Analysis by LC-MS showed that product 22 increased over time (A). Quantification of 22 and ruthenium concentration in cells allowed determination of turnover number (B). Plots adapted with permission from ref 115. Copyright 2019 American Chemical Society.
4.3. Challenges and Opportunities.
Since the availability of the first commercial mass spectrometer in the 1940s,105 MS is widely used in numerous scientific disciplines. Its high sensitivity and specificity is particularly suited for analyzing biological samples. Although not yet applied routinely to study metal catalyzed reactions in cells, MS offers unique capabilities not attainable by other methods, such as quantifying reaction products without the need for external reporters or identifying unknowns by analyzing mass fragmentations. In the context of intracellular studies, there are several limitations. First, conventional MS methods provide ensemble-averaged data, which means that it cannot give information about different cell populations within a sample. Advanced MS techniques that analyze single cells (e.g., mass cytometry103) can potentially overcome this problem but they do not have subcellular spatial resolution. Second, MS is incapable of reporting on dynamic changes in living cells since the biological specimens must be lysed prior to analysis. Thus, carrying out kinetic studies on a single cell by MS without population averaging would not be possible. To study cells in the living state, non-invasive techniques are needed (e.g., fluorescence microscopy, flow cytometry, etc.).
To improve the spatial resolution of conventional MS techniques, imaging mass spectrometry (IMS) was developed.116,117 This method involves sectioning a sample into a two-dimensional grid and then using a mass spectrometer to ionize the molecules on the sample surface. The mass spectrum of each pixel on the grid is acquired and the enormous amount of data collected is then processed by advanced computational software. Although IMS is commonly used for studying biological tissue sections, new ionization techniques have allowed studies of single cells with spatial resolutions in the low micrometer range. We anticipate that IMS could be a useful tool to map out the cellular distribution of metal catalysts or investigate heterogeneity in intracellular reactions.
5. BIOLOGICAL ASSAYS
5.1. General Background.
Biological assays encompass a wide variety of methods to interrogate living systems, such those probing their viability, mechanism of cell death, function, state, or cellular components (Chart 4).118 They are commonly used in conjunction with analytical instruments, such as microplate readers, fluorometers, cytometers, or gel imagers, to provide either qualitative or quantitative data on one or more parameters being measured. Many biological assays are available as commercial kits, although their costs can range from low to high. Generally, selection of the most appropriates assays should be based on factors such as accuracy, detection limit, convenience, time, safety, and cost.
Chart 4.
Examples of biological assays available to study metal-catalyzed reactions in cells.
Use of biological assays is essential to the intracellular catalyst development process (Scheme 2, Steps 3–5). For example, assessing the biocompatibility of metal complexes or substrates/products requires determining whether they will harm their living hosts.118 One of the most common methods to do so is using colorimetric MTT assays, which determine whether cells are alive based on their ability to convert tetrazolium salts (yellow) to formazan (purple) products. Other alternatives to MTT include MTS, XTT, WST, or SRB, which can differ in their sensitivity or ease of use.119,120 It is important to note that cell viability alone may not be sufficient to ascertain whether a xenobiotic is biocompatible. Measuring additional characteristics such as changes in metabolism121 or redox state122,123 could give a more complete view of overall cell health. Because biological assays are employed routinely to study metal-containing complexes in cells, such as in the development of metallodrugs124,125 and metallodiagnostics,126,127 we will not give an exhaustive review of these methods below. However, the interested reader is encouraged to explore this rich literature further.
Cell-based assays not only provide a wealth of biological information but can also be used to obtain chemical information about intracellular reactions. Commercial assay kits are typically available only for the quantification of naturally occurring compounds, such as NADH128,129 or pyruvate53 as detailed in the next section. Fortunately, synthetic chemists have developed a broad range of molecular probes that could potentially be used for detecting specific chemical analytes or metal ions.130–132
5.2. Studies of Intracellular Metal-Catalyzed Reactions Using Biological Assays.
In 2015, Sadler and coworkers were the first to demonstrate that transfer hydrogenation catalysis could be used for selective killing of cancer cells.128 After screening a series of half-sandwich ruthenium complexes, the researchers found that Ru7 was the most active in A7820 human ovarian cancer cells (Scheme 18). It was proposed that Ru7 was capable of reducing NAD+ to NADH using formate as a hydride source, which led to cell death via induction of reductive stress. This hypothesis was supported by two lines of evidence. First, using an SRB cell viability assay, it was shown that increasing the amount of sodium formate to 2 mM while keeping the concentration of Ru7 constant, resulted in lowering of the ruthenium IC50 value from 13.6 to 1.0 μM (Scheme 18A). Measurements by ICP-MS confirmed that the cells contained equal amounts of ruthenium catalyst in all treatment groups. Furthermore, cells treated with only formate without Ru7 were not adversely affected. Second, the NAD+/NADH ratio decreased upon addition of greater amounts of formate and constant amount of Ru7, which strongly suggested a ruthenium-catalyzed transfer hydrogenation mechanism was operative (Scheme 18B). Further biological studies revealed that the mechanism of cell death was not due to apoptosis, disruption of mitochondrial membrane potential, or necrosis using Annexin V-FITC apoptosis, JC-10 mitochondrial membrane potential, and apoptosis/necrosis assay kits, respectively. Unfortunately, the catalytic efficiency of the ruthenium catalysts inside cells was not addressed in this work.
Scheme 18.
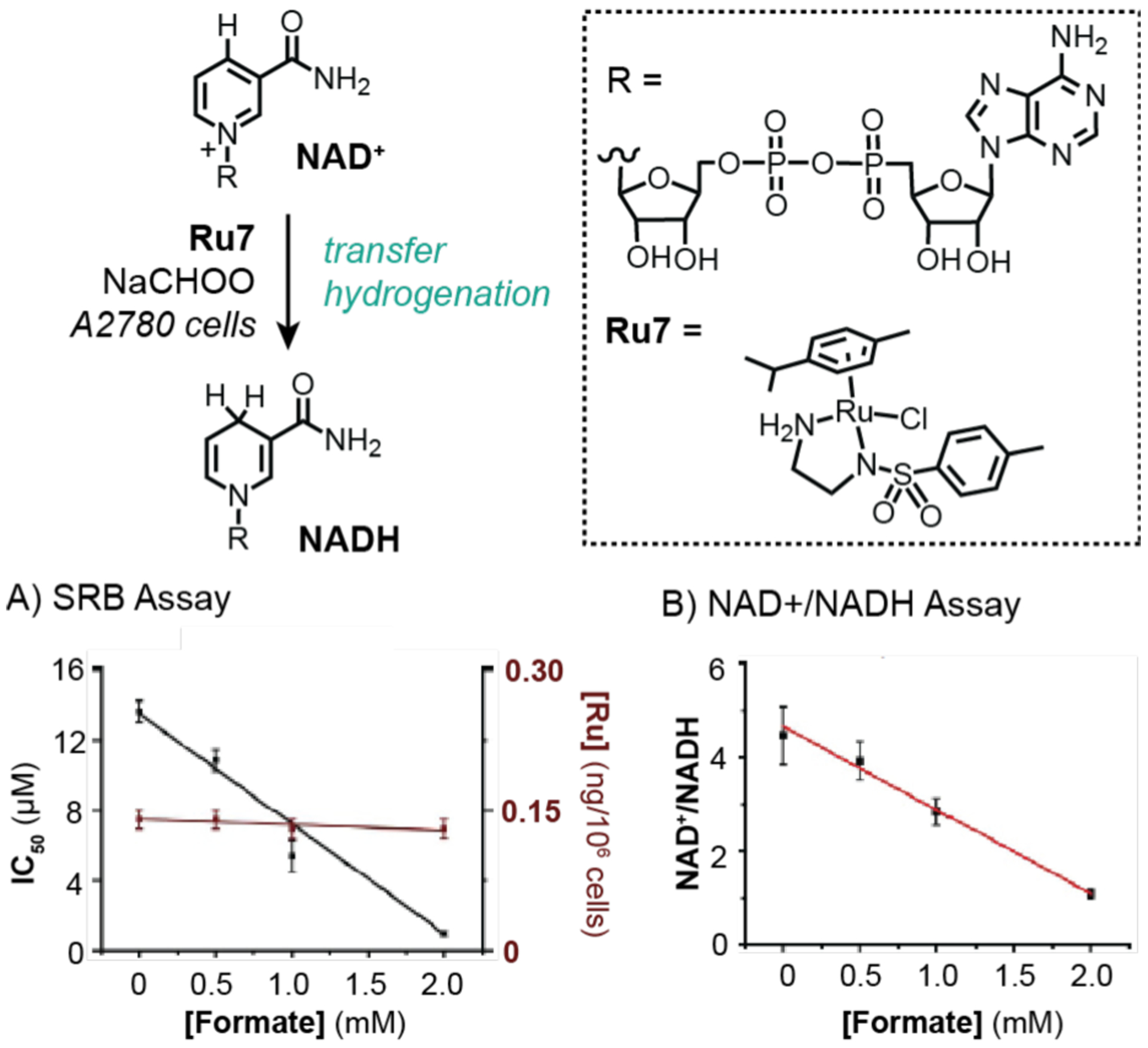
Studies by Sadler and coworkers demonstrating the use of ruthenium catalysts to promote transfer hydrogenation in A2780 cells. An SRB cell viability assay was used to measure IC50 (A) and an NAD+/NADH assay was used to measure redox balance (B). Data were adapted with permission from ref 128. Copyright 2015 Springer Nature.
Sadler and coworkers expanded on their intracellular transfer hydrogenation studies to include osmium complexes in 2018 (Scheme 19).53 The key discovery in this work was that chiral 16-electron osmium complexes were capable of catalyzing enantioselective reduction of pyruvate to lactate in the presence of sodium formate. It was demonstrated that the R,R-Os1 isomer provided D-lactate (83% enantiomeric excess) whereas the S,S-Os1 isomer provided L-lactate (84% enantiomeric excess). Studies in A2780 cells using SRB assays showed that treatment with either S,S-Os1 (Scheme 19A) or R,R-Os1 (data not shown) led to greater cell death upon addition of up to 2 mM of sodium formate. Interestingly, analysis of cells treated with R,R-Os1 and formate using an enantioselective assay detection kit indicated that greater amounts of D-lactate had formed in comparison to that in samples treated with S,S-Os1 or no catalyst (Scheme 19B). However, there does not seem to be a direct correlation between the concentration of D-lactate with cell death. Although the concentrations of lactate products and ruthenium in cells were determined in this work, the turnover numbers were not reported.
Scheme 19.
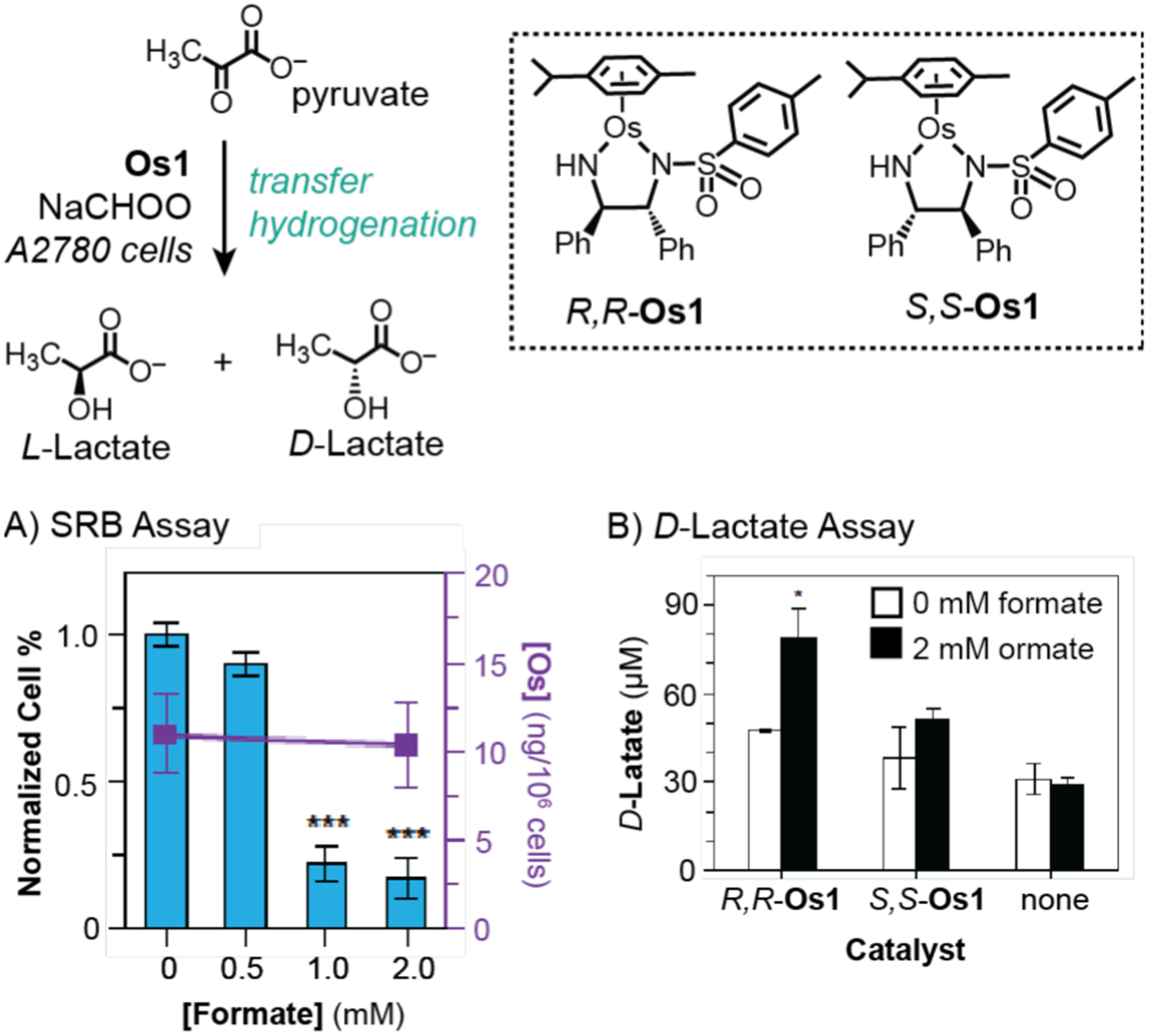
Studies by Sadler and coworkers demonstrating the use of osmium catalysts to promote transfer hydrogenation in A2780 cells. An SRB cell viability assay was used to measure IC50 of S,S-Os1 (A) and a D-lactate assay was used to measure the intracellular concentration of D-lactate (B). Data were adapted with permission from ref 53. Copyright 2018 Springer Nature.
In 2013, Balskus and coworkers took advantage of non-enzymatic chemistry to rescue auxotrophs, which are organisms that are unable to produce key nutrients necessary for their growth and survival (Scheme 20).133 As proof of concept, the researchers obtained three mutant strains of E. coli (ΔpabA, ΔpabB, and ΔaroC) that lack the ability to produce p-aminobenzoic acid (24), an essential biosynthetic precursor to folic acid. To demonstrate that abiotic catalysis could rescue these mutants, a substrate containing an allyl-carbamate group (23) was used in combination with ruthenium catalyst Ru1. It was observed that bacteria inoculated with 23 and Ru1 showed more significant growth compared to that in bacteria treated with 24 directly (Scheme 20A). The optical densities of the different cultures were further monitored over the course of 48 h (Scheme 20B). This simple yet highly effective gain-of-function assay suggested that 24 was generated by the reaction of 23 with Ru1. Since the reaction yields in solution studies were low (only up to 14%), it is likely that the yield in the presence of E. coli was low. The issue of whether catalysis occurred intracellularly or extracellularly was not addressed in this work.
Scheme 20.
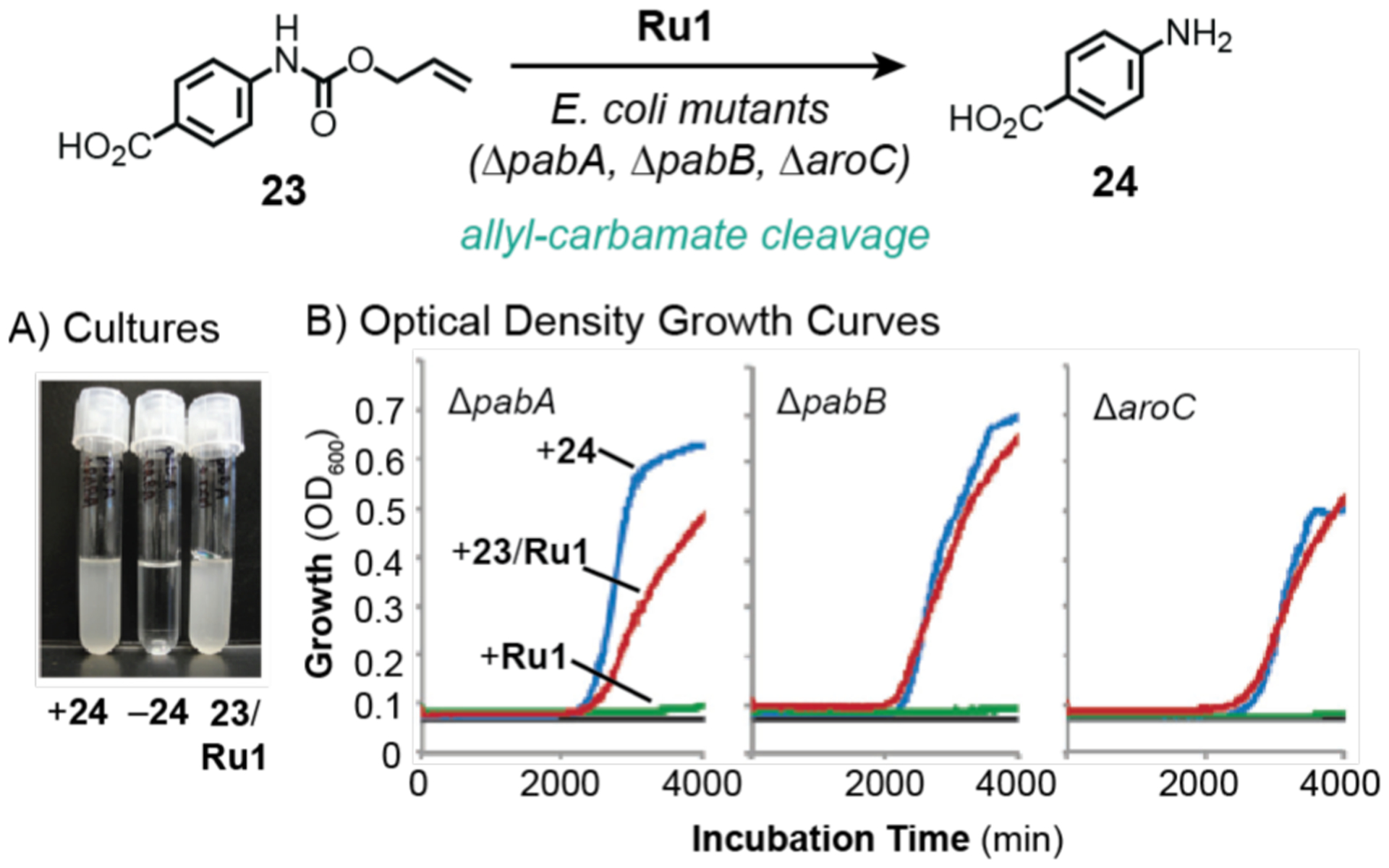
Studies by Balskus and coworkers demonstrating the use of ruthenium catalysts to promote allyl-carbamate cleavage in E. coli mutant cells. Image of bacterial cultures (A) and growth curves based on optical density measurements (B). Data were adapted with permission from ref 133. Copyright 2013 John Wiley and Sons.
5.3. Challenges and Opportunities.
Given the vast number of commercial and non-commercial cell-based assays available for chemical biology research, it is not possible for us to critically assess each one here.119 However, there are several considerations to keep in mind when choosing different assays. First, it is important to understand how a particular assay works so that any incompatibility with a proposed experiment could be avoided. For example, investigations of transfer hydrogenation reactions should not use tetrazolium-based assays because both the assay, which relies on the activity of reductases,120 and synthetic catalysts53 are NADH-dependent. Thus, if the catalyst consumes a significant amount of NAD+ or NADH in an intracellular reaction, the MTT/MTS/WST results for cell viability would be inaccurate. On the other hand, an SRB assay that relies on the binding of sulforhodamine B to proteins would be more appropriate in this cases.119 Second, in fluorescence-based assays, the emission profiles of the assay reporter and fluorescent reaction products should not overlap to prevent obtaining false-positive results. However, since dyes with a wide range of colors are available, this problem should be possible to avoid.
We anticipate that future innovations in technology and analytical methods will lead to even greater assaying capabilities. For example, Waymouth, Wender, and coworkers developed a luciferase reporter system to screen for complexes capable of facilitating allyl carbamate cleavage.75 Mayer and coworkers created a 96-well plate screening platform that measures both catalyst activity and organism fitness.134
6. CONCLUDING REMARKS
The successful integration of synthetic chemistry with living systems is owed largely to the many tools and methods available for carrying out the catalyst development process (Scheme 2). The techniques we discussed, fluorescence microscopy, flow cytometry, mass spectrometry, and biological assays, are complementary to one another and thus, provide different chemical or biological information about the catalytic reactions being studied.
In our view, there are several major barriers to moving the field of intracellular catalysis forward. First, current studies that probe catalytic activity rely on ensemble-averaged measurements that do not take into account possible differences in catalyst behavior inside the cell. It is well established that enzymes often exhibit different catalytic rates in living systems vs. solution due to the heterogeneity of biological environments.56,57 Since synthetic catalysts could be inactivated as a result of binding to biomolecules (e.g., thiols,114,135 nitrogenous bases, sugars, etc.), trapped within membranes, degraded via metabolism, or a variety of other reasons, it is likely that not all catalysts within the cell are active. Knowing what percentage of catalyst molecules are off-target allows researchers to chemically optimize them as needed, which could improve their biocompatibility as well as reduce the need for high catalyst loading. To address this problem, we believe that application of single molecule-based techniques such as SFM might allow interrogation of individual catalysts in their local surroundings.80
Second, it is challenging to determine the catalytic activity or TON of intracellular reactions because these metrics cannot be easily extracted from a single measurement like in synthetic chemistry where the amount of catalyst used is a known quantity. For example, in the work reported in 2019,115 the concentrations of products and catalysts were determined separately by LC-MS and ICP-MS, respectively, to estimate the TON. To complicate matters, this calculated value might not be entirely accurate because it is possible only a small fraction of the metal catalysts inside cells was active (see point above) and the LC-MS and ICP-MS measurements were made using different cell batches. Thus, development of easy-to-use methods that could reliably quantify catalytic performance in vitro or in vivo would be tremendously powerful, particularly in SAR studies where subtle distinctions between catalysts might be important.
Third, although there are numerous techniques available to study intracellular reactions, many of them are not part of the standard synthetic chemistry toolbox, which means that chemists who are unaccustomed to these methods would likely require special training. Furthermore, several of the instruments described above, such as confocal microscopes or flow cytometers, are expensive and might not be available in some research facilities. Some emerging techniques, such as correlative 3D cryo X-ray imaging,136 require use of synchrotron beam lines. Fortunately, issues related to lack of expertise or resources could be overcome by forging collaborations with external researchers who could provide the required knowledge or instrumentations, respectively.
Given that the chemistry of life was developed over the course of billions of years, it is astounding that chemists have achieved so much in such a short amount of time. While we should celebrate these accomplishments, there are still many scientific and technical hurdles to overcome in order to fully harness the power of synthetic chemistry to enhance life. Although the majority of intracellular metal-catalyzed reactions have been studied in cell models, there are promising reports that they could also be carried out in live organisms, such as nematodes,137 zebrafish38,76,138 and mice.48,139–141 Looking ahead even further, we believe that translating this chemistry into clinical practice will be difficult but not unrealistic. Like other candidates under consideration for human trials, synthetic catalysts would need to pass rigorous tests for biological safety and efficacy. As researchers working on metals in medicine have repeatedly demonstrated,124–127,142 inorganic compounds can have unique advantages over their organic counterparts. The fact that a number of metal-based compounds are used clinically or in clinical trials provides the most compelling argument that the possible application of synthetic catalysts in humans is not too far-fetched.
We hope this perspective article will help increase research efficiency by bringing attention to the strengths and weaknesses of various tools available to study living cells. We predict that as new advances are made in analytical hardware and software, the pace of research in intracellular catalysis will accelerate in the coming years. We are excited by the prospect that these efforts will one day lead to a new technological era, in which important problems in health, energy, and the environment could be addressed using creative biosynthetic solutions.
Funding Sources
This work was supported by the Welch Foundation (Grant No. E01894) and the National Institute of General Medical Sciences of the National Institutes of Health (Grant No. R01GM129276).
ABBREVIATIONS
- SIMCat
small-molecule intracellular metal catalyst
- PBS
phosphate buffered saline
- IC50
50% growth inhibition concentration
- SAR
structure-activity relationship
- DNA
deoxyribonucleic acid
- WFM
widefield microscopy
- LSCM
laser scanning confocal microscopy
- E. coli
Escherichia coli
- Hpg
homopropargylglycine
- Eth
ethynylphenylalanine
- NADH
reduced nicotinamide adenine dinucleotide
- NAD+
oxidized nicotinamide adenine dinucleotide
- SFM
super-resolution fluorescence microscopy
- FSC
forward scatter
- SSC
side scatter
- OmpC
outer membrane protein C
- TCEP
tris(carboxyethyl)phosphine
- FFC
fluorescence flow cytometry
- MC
mass cytometry
- IC
imaging cytometry
- MS
mass spectrometry
- ESI
electrospray ionization
- MALDI
matrix-laser desorption ionization
- ICP
inductively coupled plasma
- GC
gas chromatography
- LC
liquid chromatography
- MS/MS
tandem mass spectrometry
- DMABA-NHS
N-hydroxysuccinimide ester
- NaAsc
sodium ascorbate
- IMS
imaging mass spectrometry
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Oparin AI The Origin of Life and the Origin of Enzymes. Adv. Enzymol. Relat. Areas Mol. Biol 1965, 347–380. [DOI] [PubMed] [Google Scholar]
- (2).Schopf JW; Kudryavtsev AB; Czaja AD; Tripathi AB Evidence of Archean Life: Stromatolites and Microfossils. Precambrian Res. 2007, 158, 141–155. [Google Scholar]
- (3).Djokic T; Van Kranendonk MJ; Campbell KA; Walter MR; Ward CR Earliest Signs of Life on Land Preserved in ca. 3.5 Ga Hot Spring Deposits. Nat. Commun 2017, 8, 15263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Robertson AJB The Early History of Catalysis. Platin. Met. Rev 1975, 19, 64–69. [Google Scholar]
- (5).Hartwig JF Organotransition Metal Chemistry; University Science Books: Mill Valley, California, 2010. [Google Scholar]
- (6).Theoretical Aspects of Transition Metal Catalysis; Frenking G, Ed.; Springer: Berlin, Heidelberg, 2005; Vol. 12. [Google Scholar]
- (7).Tsuji J Transition Metal Reagents and Catalysis: Innovations in Organic Synthesis; John Wiley & Sons, Ltd., 2002. [Google Scholar]
- (8).Ngo AH; Bose S; Do LH Intracellular Chemistry: Integrating Molecular Inorganic Catalysts with Living Systems. Chem.-Eur. J 2018, 24, 10584–10594. [DOI] [PubMed] [Google Scholar]
- (9).Rebelein JG; Ward TR In Vivo Catalyzed New-to-Nature Reactions. Curr. Opin. Biotechnol 2018, 53, 106–114. [DOI] [PubMed] [Google Scholar]
- (10).Sasmal PK; Streu CN; Meggers E Metal Complex Catalysis in Living Biological Systems. Chem. Commun 2013, 49, 1581–1587. [DOI] [PubMed] [Google Scholar]
- (11).Soldevila-Barreda JJ; Sadler PJ Approaches to the Design of Catalytic Metallodrugs. Curr. Opin. Chem. Biol 2015, 25, 172–183. [DOI] [PubMed] [Google Scholar]
- (12).Martínez-Calvo M; Mascareñas JL Organometallic Catalysis in Biological Media and Living Settings. Coord. Chem. Rev 2018, 359, 57–79. [Google Scholar]
- (13).Liu Y; Bai Y Design and Engineering of Metal Catalysts for Bioorthogonal Catalysis in Living Systems. ACS Appl. Bio Mater 2020, 3, 4717–4746. [DOI] [PubMed] [Google Scholar]
- (14).Beatty KE; Xie F; Wang Q; Tirrell DA Selective Dye-Labeling of Newly Synthesized Proteins in Bacterial Cells. J. Am. Chem. Soc 2005, 127, 14150–14151. [DOI] [PubMed] [Google Scholar]
- (15).Hong V; Steinmetz NF; Manchester M; Finn MG Labeling Live Cells by Copper-Catalyzed Alkyne-Azide Click Chemistry. Bioconjugate Chem. 2010, 21, 1912–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Li J; Lin S; Wang J; Jia S; Yang M; Hao Z; Zhang X; Chen PR Ligand-Free Palladium-Mediated Site-Specific Protein Labeling Inside Gram-Negative Bacterial Pathogens. J. Am. Chem. Soc 2013, 135, 7330–7338. [DOI] [PubMed] [Google Scholar]
- (17).Weiss JT; Dawson JC; Macleod KG; Rybski W; Fraser C; Torres-Sánchez C; Patton EE; Bradley M; Carragher NO; Unciti-Broceta A Extracellular Palladium-Catalysed Dealkylation of 5-Fluoro-1-Propargyl-Uracil as a Bioorthogonally Activated Prodrug Approach. Nat. Commun 2014, 5, 3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Clavadetscher J; Indrigo E; Chankeshwara SV; Lilienkampf A; Bradley M In-Cell Dual Drug Synthesis by Cancer-Targeting Palladium Catalysts. Angew. Chem. Int. Ed 2017, 56, 6864–6868. [DOI] [PubMed] [Google Scholar]
- (19).Ngo AH; Ibañez M; Do LH Catalytic Hydrogenation of Cytotoxic Aldehydes Using Nicotinamide Adenine Dinucleotide (NADH) in Cell Growth Media. ACS Catal. 2016, 6, 2637–2641. [Google Scholar]
- (20).Qu G; Li A; Acevedo-Rocha CG; Sun Z; Reetz MT The Crucial Role of Methodology Development in Directed Evolution of Selective Enzymes. Angew. Chem. Int. Ed 2020, 59, 13204–13231. [DOI] [PubMed] [Google Scholar]
- (21).Arnold FH Directed Evolution: Bringing New Chemistry to Life. Angew. Chem. Int. Ed 2017, 57, 4143–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Heinisch T; Ward TR Artificial Metalloenzymes Based on the Biotin–Streptavidin Technology: Challenges and Opportunities. Acc. Chem. Res 2016, 49, 1711–1721. [DOI] [PubMed] [Google Scholar]
- (23).Köhler V; Wilson YM; Dürrenberger M; Ghislieri D; Churakova E; Quinto T; Knörr L; Häussinger D; Hollmann F; Turner NJ; Ward TR Synthetic Cascades are Enabled by Combining Biocatalysts with Artificial Metalloenzymes. Nat. Chem 2013, 5, 93–99. [DOI] [PubMed] [Google Scholar]
- (24).Schwizer F; Okamoto Y; Heinisch T; Gu Y; Pellizzoni MM; Lebrun V; Reuter R; Köhler V; Lewis JC; Ward TR Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev 2018, 118, 142–231. [DOI] [PubMed] [Google Scholar]
- (25).Tebo AG; Pecoraro VL Artificial Metalloenzymes Derived From Three-Helix Bundles. Curr. Opin. Chem. Biol 2015, 25, 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wei H; Wang E Nanomaterials with Enzyme-Like Characteristics (Nanozymes): Next-Generation Artificial Enzymes. Chem. Soc. Rev 2013, 42, 6060–6093. [DOI] [PubMed] [Google Scholar]
- (27).Zhou Y; Liu B; Yang R; Liu J Filling in the Gaps Between Nanozymes and Enzymes: Challenges and Opportunities. Bioconjugate Chem. 2017, 28, 2903–2909. [DOI] [PubMed] [Google Scholar]
- (28).Vigh L; Joó F; Cséplő Á Modulation of Membrane Fluidity in Living Protoplasts of Nicotiana plumbaginifolia by Catalytic Hydrogenation. Eur. J. Biochem 1985, 146, 241–244. [DOI] [PubMed] [Google Scholar]
- (29).van de L’Isle MON; Ortega-Liebana MC; Unciti-Broceta A Transition Metal Catalysts for the Bioorthogonal Synthesis of Bioactive Agents. Curr. Opin. Chem. Biol 2021, 61, 32–42. [DOI] [PubMed] [Google Scholar]
- (30).Destito P; Vidal C; López F; Mascareñas JL Transition Metal-Promoted Reactions in Aqueous Media and Biological Settings. Chem.-Eur. J 2021, 27, 4789–4816. [DOI] [PubMed] [Google Scholar]
- (31).Zhang X; Landis RF; Keshri P; Cao-Milán R; Luther DC; Gopalakrishnan S; Liu Y; Huang R; Li G; Malassiné M; Uddin I; Rondon B; Rotello VM Intracellular Activation of Anticancer Therapeutics Using Polymeric Bioorthogonal Nanocatalysts. Adv. Healthc. Mater 2021, 10, 2001627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Link AJ; Tirrell DA Cell Surface Labeling of Escherichia coli via Copper(I)-Catalyzed [3+2] Cycloaddition. J. Am. Chem. Soc 2003, 125, 11164–11165. [DOI] [PubMed] [Google Scholar]
- (33).Speers AE; Adam GC; Cravatt BF Activity-Based Protein Profiling in Vivo Using a Copper(I)-Catalyzed Azide-Alkyne [3 + 2] Cycloaddition. J. Am. Chem. Soc 2003, 125, 4686–4687. [DOI] [PubMed] [Google Scholar]
- (34).Bai Y; Feng X; Xing H; Xu Y; Kim BK; Baig N; Zhou T; Gewirth AA; Lu Y; Oldfield E; Zimmerman SC A Highly Efficient Single-Chain Metal–Organic Nanoparticle Catalyst for Alkyne–Azide “Click” Reactions in Water and in Cells. J. Am. Chem. Soc 2016, 138, 11077–11080. [DOI] [PubMed] [Google Scholar]
- (35).Clavadetscher J; Hoffmann S; Lilienkampf A; Mackay L; Yusop RM; Rider SA; Mullins JJ; Bradley M Copper Catalysis in Living Systems and In Situ Drug Synthesis. Angew. Chem. Int. Ed 2016, 55, 15662–15666. [DOI] [PubMed] [Google Scholar]
- (36).Miguel-Ávila J; Tomás-Gamasa M; Olmos A; Pérez PJ; Mascareñas JL Discrete Cu(I) Complexes for Azide–Alkyne Annulations of Small Molecules Inside Mammalian Cells. Chem. Sci 2018, 9, 1947–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Tsubokura K; Vong KKH; Pradipta AR; Ogura A; Urano S; Tahara T; Nozaki S; Onoe H; Nakao Y; Sibgatullina R; Kurbangalieva A; Watanabe Y; Tanaka K In Vivo Gold Complex Catalysis Wthin Live Mice. Angew. Chem. Int. Ed 2017, 56, 3579–3584. [DOI] [PubMed] [Google Scholar]
- (38).Sasmal PK; Carregal-Romero S; Han AA; Streu CN; Lin Z; Namikawa K; Elliott SL; Köster RW; Parak WJ; Meggers E Catalytic Azide Reduction in Biological Environments. ChemBioChem 2012, 13, 1116–1120. [DOI] [PubMed] [Google Scholar]
- (39).Cao-Milán R; Gopalakrishnan S; He LD; Huang R; Wang L-S; Castellanos L; Luther DC; Landis RF; Makabenta JMV; Li C-H; Zhang X; Scaletti F; Vachet RW; Rotello VM Thermally Gated Bioorthogonal Nanozymes with Supramolecularly Confined Porphyrin Catalysts for Antimicrobial Uses. Chem 2020, 6, 1113–1124. [Google Scholar]
- (40).Li N; Lim RKV; Edwardraja S; Lin Q Copper-Free Sonogashira Cross-Coupling for Functionalization of Alkyne-Encoded Proteins in Aqueous Medium and in Bacterial Cells. J. Am. Chem. Soc 2011, 133, 15316–15319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Yusop RM; Unciti-Broceta A; Johansson EMV; Sánchez-Martín RM; Bradley M Palladium-Mediated Intracellular Chemistry. Nat. Chem 2011, 3, 239–243. [DOI] [PubMed] [Google Scholar]
- (42).Toussaint SNW; Calkins RT; Lee S; Michel BW Olefin Metathesis-Based Fluorescent Probes for the Selective Detection of Ethylene in Live Cells. J. Am. Chem. Soc 2018, 140, 13151–13155. [DOI] [PubMed] [Google Scholar]
- (43).Streu C; Meggers E Ruthenium-Induced Allylcarbamate Cleavage in Living Cells. Angew. Chem. Int. Ed 2006, 45, 5645–5648. [DOI] [PubMed] [Google Scholar]
- (44).Li J; Yu J; Zhao J; Wang J; Zheng S; Lin S; Chen L; Yang M; Jia S; Zhang X; Chen PR Palladium-Triggered Deprotection Chemistry for Protein Activation in Living Cells. Nat. Chem 2014, 6, 352–361. [DOI] [PubMed] [Google Scholar]
- (45).Völker T; Meggers E Chemical Activation in Blood Serum and Human Cell Culture: Improved Ruthenium Complex for Catalytic Uncaging of Alloc-Protected Amines. ChemBioChem 2017, 18, 1083–1086. [DOI] [PubMed] [Google Scholar]
- (46).Learte-Aymamí S; Vidal C; Gutiérrez-González A; Mascareñas JL Intracellular Reactions Promoted by Bis(histidine) Miniproteins Stapled Using Palladium(II) Complexes. Angew. Chem. Int. Ed 2020, 59, 9149–9154. [DOI] [PubMed] [Google Scholar]
- (47).Sancho-Albero M; Rubio-Ruiz B; Pérez-López AM; Sebastián V; Martín-Duque P; Arruebo M; Santamaría J; Unciti-Broceta A Cancer-Derived Exosomes Loaded with Ultrathin Palladium Nanosheets for Targeted Bioorthogonal Catalysis. Nat. Catal 2019, 2, 864–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Miller MA; Askevold B; Mikula H; Kohler RH; Pirovich D; Weissleder R Nano-Palladium is a Cellular Catalyst for in Vivo Chemistry. Nat. Commun 2017, 8, 15906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Wang J; Zheng S; Liu Y; Zhang Z; Lin Z; Li J; Zhang G; Wang X; Li J; Chen PR Palladium-Triggered Chemical Rescue of Intracellular Proteins via Genetically Encoded Allene-Caged Tyrosine. J. Am. Chem. Soc 2016, 138, 15118–15121. [DOI] [PubMed] [Google Scholar]
- (50).Miguel-Ávila J; Tomás-Gamasa M; Mascareñas JL Intracellular Ruthenium-Promoted (2+2+2) Cycloadditions. Angew. Chem. Int. Ed 2020, 59, 17628–17633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Vidal C; Tomás-Gamasa M; Destito P; López F; Mascareñas JL Concurrent and Orthogonal Gold(I) and Ruthenium(II) Catalysis Inside Living Cells. Nat. Commun 2018, 9, 1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Bose S; Ngo AH; Do LH Intracellular Transfer Hydrogenation Mediated by Unprotected Organoiridium Catalysts. J. Am. Chem. Soc 2017, 139, 8792–8795. [DOI] [PubMed] [Google Scholar]
- (53).Coverdale JPC; Romero-Canelón I; Sanchez-Cano C; Clarkson GJ; Habtemariam A; Wills M; Sadler PJ Asymmetric Transfer Hydrogenation by Synthetic Catalysts in Cancer Cells. Nat. Chem 2018, 10, 347–354. [DOI] [PubMed] [Google Scholar]
- (54).Sletten EM; Bertozzi CR Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew. Chem. Int. Ed 2009, 48, 6974–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Ngo AH; Do LH Structure–Activity Relationship Study of Half-Sandwich Metal Complexes in Aqueous Transfer Hydrogenation Catalysis. Inorg. Chem. Front 2020, 7, 583–591. [Google Scholar]
- (56).Davidi D; Milo R Lessons on Enzyme Kinetics from Quantitative Proteomics. Curr. Opin. Biotechnol 2017, 46, 81–89. [DOI] [PubMed] [Google Scholar]
- (57).Van Noorden CJF; Jonges GN Analysis of Enzyme Reactions In Situ. Histochem. J 1995, 27, 101–118. [DOI] [PubMed] [Google Scholar]
- (58).Masters BR The Development of Fluorescence Microscopy. In Encylcopedia of Life Sciences; John Wiley & Sons: Chichester, 2010. [Google Scholar]
- (59).Bright FV Bioanalytical Applications of Fluorescence Spectroscopy. Anal. Chem 1988, 60, 1031A–1039A. [DOI] [PubMed] [Google Scholar]
- (60).Albani JR Principles and Applications of Fluorescence Spectroscopy; John Wiley & Sons, 2008. [Google Scholar]
- (61).Sanderson MJ; Smith I; Parker I; Bootman MD Fluorescence Microscopy. Cold Spring Harb. Protoc 2014, 2014, 1042–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Combs CA Fluorescence Microscopy: A Concise Guide to Current Imaging Methods. Curr. Protoc. Neurosci 2010, 50, 2.1.1–2.1.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Stephens DJ; Allan VJ Light Microscopy Techniques for Live Cell Imaging. Science 2003, 300, 82–86. [DOI] [PubMed] [Google Scholar]
- (64).Paddock SW Confocal Laser Scanning Microscopy. BioTechniques 1999, 27, 992–1004. [DOI] [PubMed] [Google Scholar]
- (65).Icha J; Weber M; Waters JC; Norden C Phototoxicity in Live Fluorescence Microscopy, and How to Avoid It. Bioessays 2017, 39, 1700003. [DOI] [PubMed] [Google Scholar]
- (66).Tsien RY The Green Fluorescent Protein. Annu. Rev. Biochem 1998, 67, 509–544. [DOI] [PubMed] [Google Scholar]
- (67).Zimmer M Green Fluorescent Protein (GFP): Applications, Structure, and Related Photophysical Behavior. Chem. Rev 2002, 102, 759–782. [DOI] [PubMed] [Google Scholar]
- (68).Wang L; Frei MS; Salim A; Johnsson K Small-Molecule Fluorescent Probes for Live-Cell Super-Resolution Microscopy. J. Am. Chem. Soc 2019, 141, 2770–2781. [DOI] [PubMed] [Google Scholar]
- (69).Alamudi SH; Chang Y-T Advances in the Design of Cell-Permeable Fluorescent Probes for Applications in Live Cell Imaging. Chem. Commun 2018, 54, 13641–13653. [DOI] [PubMed] [Google Scholar]
- (70).Michalet X; Pinaud FF; Bentolila LA; Tsay JM; Doose S; Li JJ; Sundaresan G; Wu AM; Gambhir SS; Weiss S Quantum Dots for Live Cells, in Vivo Imaging, and Diagnostics. Science 2005, 307, 538–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Pak YL; Swamy K; Yoon J Recent Progress in Fluorescent Imaging Probes. Sensors 2015, 15, 24374–24396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Wessendorf MW; Brelje TC Multicolor Fluorescence Microscopy Using the Laser-Scanning Confocal Microscope. Neuroprotocols 1993, 2, 121–140. [Google Scholar]
- (73).Baggaley E; Weinstein JA; Williams JAG Lighting the Way to See Inside the Live Cell with Luminescent Transition Metal Complexes. Coord. Chem. Rev 2012, 256, 1762–1785. [Google Scholar]
- (74).Völker T; Dempwolff F; Graumann PL; Meggers E Progress Towards Bioorthogonal Catalysis with Organometallic Compounds. Angew. Chem. Int. Ed 2014, 53, 10536–10540. [DOI] [PubMed] [Google Scholar]
- (75).Hsu H-T; Trantow BM; Waymouth RM; Wender PA Bioorthogonal Catalysis: A General Method To Evaluate Metal-Catalyzed Reactions in Real Time in Living Systems Using a Cellular Luciferase Reporter System. Bioconjugate Chem. 2016, 27, 376–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Santra M; Ko S-K; Shin I; Ahn KH Fluorescent Detection of Palladium Species with an O-Propargylated Fluorescein. Chem. Commun 2010, 46, 3964–3966. [DOI] [PubMed] [Google Scholar]
- (77).Manders EMM; Verbeek FJ; Aten JA Measurement of Co-localization of Objects in Dual-Colour Confocal Images. J. Micrsoc 1993, 169, 375–382. [DOI] [PubMed] [Google Scholar]
- (78).Aaron JS; Taylor AB; Chew T-L Image Co-localization – Co-occurrence Versus Correlation. J. Cell Sci 2018, 131, jcs211847. [DOI] [PubMed] [Google Scholar]
- (79).Owen D; Ashdown G; Griffié J; Shannon M Co-Localisation and Correlation in Fluorescence Microscopy Data. In Standard and Super- Resolution Bioimaging Data Analysis 2017, p 143–171. [Google Scholar]
- (80).Sauer M; Hofkens J; Enderlein J Handbook of Fluorescence Spectroscopy and Imaging; Wiley-VCH Verlag & Co. KGaA: Weinheim, Germany, 2011. [Google Scholar]
- (81).Hoebe RA; Van Oven CH; Gadella TWJ Jr.; Dhonukshe PB; Van Noorden CJF; Manders EMM Controlled Light-Exposure Microscopy Reduces Photobleaching and Phototoxicity in Fluorescence Live-Cell Imaging. Nat. Biotechnol 2007, 25, 249–253. [DOI] [PubMed] [Google Scholar]
- (82).Lippincott-Schwartz J Profile of Eric Betzig, Stefan Hell, and W. E. Moerner, 2014 Nobel Laureates in Chemistry. Proc. Nat. Acad. Sci 2015, 112, 2630–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Chen P; Andoy NM Single-Molecule Fluorescence Studies from a Bioinorganic Perspective. Inorg. Chim. Acta 2008, 361, 809–819. [Google Scholar]
- (84).Xie XS; Choi PJ; Li G-W; Lee NK; Lia G Single-Molecule Approach to Molecular Biology in Living Bacterial Cells. Annu. Rev. Biophys 2008, 37, 417–444. [DOI] [PubMed] [Google Scholar]
- (85).Chen T-Y; Cheng Y-S; Huang P-S; Chen P Facilitated Unbinding via Multivalency-Enabled Ternary Complexes: New Paradigm for Protein–DNA Interactions. Acc. Chem. Res 2018, 51, 860–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Cai L; Friedman N; Xie XS Stochastic Protein Expression in Individual Cells at the Single Molecule Level. Nature 2006, 440, 358–362. [DOI] [PubMed] [Google Scholar]
- (87).Yu J; Xiao J; Ren X; Lao K; Xie XS Probing Gene Expression in Live Cells, One Protein Molecule at a Time. Science 2006, 311, 1600–1603. [DOI] [PubMed] [Google Scholar]
- (88).Funatsu T; Harada Y; Tokunaga M; Saito K; Yanagida T Imaging of Single Fluorescent Molecules and Individual ATP Turnovers by Single Myosin Molecules in Aqueous Solution. Nature 1995, 374, 555–559. [DOI] [PubMed] [Google Scholar]
- (89).Shi J; Palfey BA; Dertouzos J; Jensen KF; Gafni A; Steel D Multiple States of the Tyr318Leu Mutant of Dihydroorotate Dehydrogenase Revealed by Single-Molecule Kinetics. J. Am. Chem. Soc 2004, 126, 6914–6922. [DOI] [PubMed] [Google Scholar]
- (90).Zhang H; Guo P Single Molecule Photobleaching (SMPB) Technology for Counting of RNA, DNA, Protein and Other Molecules in Nanoparticles and Biological Complexes by TIRF Instrumentation. Methods 2014, 67, 169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Tosheva KL; Yuan Y; Matos PP; Culley S; Henriques R Between Life and Death: Strategies to Reduce Phototoxicity in Super-Resolution Microscopy. J. Phys. D: Appl. Phys 2020, 53, 163001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Stockley JH; Evans K; Matthey M; Volbracht K; Agathou S; Mukanowa J; Burrone J; Káradóttir RT Surpassing Light-Induced Cell Damage in Vitro with Novel Cell Culture Media. Sci. Rep 2017, 7, 849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).McKinnon KM Flow Cytometry: An Overview. Curr. Protoc. Immun 2018, 120, 5.1.1–5.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Chattopadhyay PK; Roederer M Cytometry: Today’s Technology and Tomorrow’s Horizons. Methods 2012, 57, 251–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Bendall SC; Nolan GP; Roederer M; Chattopadhyay PK A Deep Profiler’s Guide to Cytometry. Trends Immunol. 2012, 33, 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Basiji DA; Ortyn WE; Liang L; Venkatachalam V; Morrissey P Cellular Image Analysis and Imaging by Flow Cytometry. Clin. Lab. Med 2007, 27, 653–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Ornatsky O; Bandura D; Baranov V; Nitz M; Winnik MA; Tanner S Highly Multiparametric Analysis by Mass Cytometry. J. Immunol. Methods 2010, 361, 1–20. [DOI] [PubMed] [Google Scholar]
- (98).Doan H; Chinn GM; Jahan-Tigh RR Flow Cytometry II: Mass and Imaging Cytometry. J. Invest. Dermatol 2015, 135, e36. [DOI] [PubMed] [Google Scholar]
- (99).Link AJ; Vink MKS; Tirrell DA Presentation and Detection of Azide Functionality in Bacterial Cell Surface Proteins. J. Am. Chem. Soc 2004, 126, 10598–10602 [DOI] [PubMed] [Google Scholar]
- (100).Indrigo E; Clavadetscher J; Chankeshwara SV; Megia-Fernandez A; Lilienkampf A; Bradley M Intracellular Delivery of a Catalytic Organometallic Complex. Chem. Commun 2017, 53, 6712–6715. [DOI] [PubMed] [Google Scholar]
- (101).Martin JC; Swartzendruber DE Time: A New Parameter for Kinetic Measurements in Flow Cytometry. Science 1980, 207, 199–201. [DOI] [PubMed] [Google Scholar]
- (102).Dive C; Workman P; Marrone BL; Watson JV In Flow Cytometry; Jacquemin-Sablon A, Ed.; Springer Berlin Heidelberg: Berlin, Heidelberg, 1993, p 29–47. [Google Scholar]
- (103).Spitzer MH; Nolan GP Mass Cytometry: Single Cells, Many Features. Cell 2016, 165, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Barteneva NS; Fasler-Kan E; Vorobjev IA Imaging Flow Cytometry: Coping with Heterogeneity in Biological Systems. J. Histochem. Cytochem 2012, 60, 723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Griffiths J A Brief History of Mass Spectrometry. Anal. Chem 2008, 80, 5678–5683. [DOI] [PubMed] [Google Scholar]
- (106).de Hoffmann E; Stroobant V Mass Spectrometry: Principles and Applications; Wiley, 2007. [Google Scholar]
- (107).Fenn JB; Mann M; Meng CK; Wong SF; Whitehouse CM Electrospray Ionization for Mass Spectrometry of Large Biomolecules. Science 1989, 246, 64–71. [DOI] [PubMed] [Google Scholar]
- (108).Konermann L; Ahadi E; Rodriguez AD; Vahidi S Unraveling the Mechanism of Electrospray Ionization. Anal. Chem 2013, 85, 2–9. [DOI] [PubMed] [Google Scholar]
- (109).Wilschefski SC; Baxter MR Inductively Coupled Plasma Mass Spectrometry: Introduction to Analytical Aspects. Clin. Biochem. Rev 2019, 40, 115–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Liu R; Wu P; Yang L; Hou X; Lv Y Inductively Coupled Plasma Mass Spectrometry-Based Immunoassay: A Review. Mass Spectrom. Rev 2014, 33, 373–393. [DOI] [PubMed] [Google Scholar]
- (111).Bantscheff M; Schirle M; Sweetman G; Rick J; Kuster B Quantitative Mass Spectrometry in Proteomics: A Critical Review. Anal. Bioanal. Chem 2007, 389, 1017–1031. [DOI] [PubMed] [Google Scholar]
- (112).Wang J; Cheng B; Li J; Zhang Z; Hong W; Chen X; Chen PR Chemical Remodeling of Cell-Surface Sialic Acids through a Palladium-Triggered Bioorthogonal Elimination Reaction. Angew. Chem. Int. Ed 2015, 54, 5364–5368. [DOI] [PubMed] [Google Scholar]
- (113).Tomás-Gamasa M; Martínez-Calvo M; Couceiro JR; Mascareñas JL Transition Metal Catalysis in the Mitochondria of Living Cells. Nat. Commun 2016, 7, 12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Li S; Wang L; Yu F; Zhu Z; Shobaki D; Chen H; Wang M; Wang J; Qin G; Erasquin UJ; Ren L; Wang Y; Cai C Copper-Catalyzed Click Reaction on/in Live Cells. Chem. Sci 2017, 8, 2107–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Vidal C; Tomás-Gamasa M; Gutiérrez-González A; Mascareñas JL Ruthenium-Catalyzed Redox Isomerizations inside Living Cells. J. Am. Chem. Soc 2019, 141, 5125–5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Buchberger AR; DeLaney K; Johnson J; Li L Mass Spectrometry Imaging: A Review of Emerging Advancements and Future Insights. Anal. Chem 2018, 90, 240–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Weaver EM; Hummon AB Imaging Mass Spectrometry: From Tissue Sections to Cell Cultures. Adv. Drug Deliv. Rev 2013, 65, 1039–1055. [DOI] [PubMed] [Google Scholar]
- (118).Groth T; Falck P; Miethke R-R Cytotoxicity of Biomaterials—Basic Mechanisms and in Vitro Test Methods: A Review. Altern. Lab Anim 1995, 23, 790–799. [Google Scholar]
- (119).Aslantürk ÖS In Vitro Cytotoxicity and Cell Viability Assays: Principles, Advantages, and Disadvantages; InTech, 2018; Vol. 2. [Google Scholar]
- (120).Adan A; Kiraz Y; Baran Y Cell Proliferation and Cytotoxicity Assays. Curr. Pharm. Biotechnol 2016, 17, 1213–1221. [DOI] [PubMed] [Google Scholar]
- (121).Zhang J; Nuebel E; Wisidagama DRR; Setoguchi K; Hong JS; Van Horn CM; Imam SS; Vergnes L; Malone CS; Koehler CM; Teitell MA Measuring Energy Metabolism in Cultured Cells, Including Human Pluripotent Stem Cells and Differentiated Cells. Nat. Protoc 2012, 7, 1068–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Zhang Y; Dai M; Yuan Z Methods for the Detection of Reactive Oxygen Species. Anal. Methods 2018, 10, 4625–4638. [Google Scholar]
- (123).Burns JM; Cooper WJ; Ferry JL; King DW; DiMento BP; McNeill K; Miller CJ; Miller WL; Peake BM; Rusak SA; Rose AL; Waite TD Methods for Reactive Oxygen Species (ROS) Detection in Aqueous Environments. Aquat. Sci 2012, 74, 683–734. [Google Scholar]
- (124).Anthony EJ; Bolitho EM; Bridgewater HE; Carter OWL; Donnelly JM; Imberti C; Lant EC; Lermyte F; Needham RJ; Palau M; Sadler PJ; Shi H; Wang F-X; Zhang W-Y; Zhang Z Metallodrugs Are Unique: Opportunities and Challenges of Discovery and Development. Chem. Sci 2020, 11, 12888–12917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Ott I 3.32 - Biodistribution of Metals and Metallodrugs. In Comprehensive Inorganic Chemistry II (Second Edition); Reedijk J, Poeppelmeier K, Eds.; Elsevier: Amsterdam, 2013, p 933–949. [Google Scholar]
- (126).Sedgwick AC; Brewster JT II; Harvey P; Iovan DA; Smith G; He X-P; Tian H; Sessler JL; James TD Metal-based Imaging Agents: Progress Towards Interrogating Neurodegenerative Disease. Chem. Soc. Rev 2020, 49, 2886–2915. [DOI] [PubMed] [Google Scholar]
- (127).Markwalter CF; Kantor AG; Moore CP; Richardson KA; Wright DW Inorganic Complexes and Metal-Based Nanomaterials for Infectious Disease Diagnostics. Chem. Rev 2019, 119, 1456–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Soldevila-Barreda JJ; Romero-Canelón I; Habtemariam A; Sadler PJ Transfer Hydrogenation Catalysis in Cells as a New Approach to Anticancer Drug Design. Nat. Commun 2015, 6, 6582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Yang L; Bose S; Ngo AH; Do LH Innocent But Deadly: Nontoxic Organoiridium Catalysts Promote Selective Cancer Cell Death. ChemMedChem 2017, 12, 292–299. [DOI] [PubMed] [Google Scholar]
- (130).Szente L; Szemán J Cyclodextrins in Analytical Chemistry: Host–Guest Type Molecular Recognition. Anal. Chem 2013, 85, 8024–8030. [DOI] [PubMed] [Google Scholar]
- (131).Cao D; Liu Z; Verwilst P; Koo S; Jangjili P; Kim JS; Lin W Coumarin-Based Small-Molecule Fluorescent Chemosensors. Chem. Rev 2019, 119, 10403–10519. [DOI] [PubMed] [Google Scholar]
- (132).Bruemmer KJ; Crossley SWM; Chang CJ Activity-Based Sensing: A Synthetic Methods Approach for Selective Molecular Imaging and Beyond. Angew. Chem. Int. Ed 2020, 59, 13734–13762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Lee Y; Umeano A; Balskus EP Rescuing Auxotrophic Microorganisms with Nonenzymatic Chemistry. Angew. Chem. Int. Ed 2013, 52, 11800–11803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Rubini R; Ivanov I; Mayer C A Screening Platform to Identify and Tailor Biocompatible Small-Molecule Catalysts. Chem.-Eur. J 2019, 25, 16017–16021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Wilson YM; Dürrenberger M; Nogueira ES; Ward TR Neutralizing the Detrimental Effect of Glutathione on Precious Metal Catalysts. J. Am. Chem. Soc 2014, 136, 8928–8932. [DOI] [PubMed] [Google Scholar]
- (136).Conesa JJ; Carrasco AC; Rodriguez-Fanjul V; Yang Y; Carrascosa JL; Cloetens P; Pereiro E; Pizarro AM Unambiguous Intracellular Localization and Quantification of a Potent Iridium Anticancer Compound by Correlative 3D Cryo X-Ray Imaging. Angew. Chem. Int. Ed 2020, 59, 1270–1278. [DOI] [PubMed] [Google Scholar]
- (137).You Y; Cao F; Zhao Y; Deng Q; Sang Y; Li Y; Dong K; Ren J; Qu X Near-Infrared Light Dual-Promoted Heterogeneous Copper Nanocatalyst for Highly Efficient Bioorthogonal Chemistry in Vivo. ACS Nano 2020, 14, 4178–4187. [DOI] [PubMed] [Google Scholar]
- (138).Soriano del Amo D; Wang W; Jiang H; Besanceney C; Yan AC; Levy M; Liu Y; Marlow FL; Wu P Biocompatible Copper(I) Catalysts for in Vivo Imaging of Glycans. J. Am. Chem. Soc 2010, 132, 16893–16899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Miller MA; Mikula H; Luthria G; Li R; Kronister S; Prytyskach M; Kohler RH; Mitchison T; Weissleder R Modular Nanoparticulate Prodrug Design Enables Efficient Treatment of Solid Tumors Using Bioorthogonal Activation. ACS Nano 2018, 12, 12814–12826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Li B; Liu P; Wu H; Xie X; Chen Z; Zeng F; Wu S A Bioorthogonal Nanosystem for Imaging and in Vivo Tumor Inhibition. Biomaterials 2017, 138, 57–68. [DOI] [PubMed] [Google Scholar]
- (141).Hoop M; Ribeiro AS; Rösch D; Weinand P; Mendes N; Mushtaq F; Chen X-Z; Shen Y; Pujante CF; Puigmartí-Luis J; Paredes J; Nelson BJ; Pêgo AP; Pané S Mobile Magnetic Nanocatalysts for Bioorthogonal Targeted Cancer Therapy. Adv. Funct. Mater 2018, 28, 1705920. [Google Scholar]
- (142).Johnstone TC; Suntharalingam K; Lippard SJ The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs. Chem. Rev 2016, 116, 3436–3486. [DOI] [PMC free article] [PubMed] [Google Scholar]