

Abstract
The IL-36 cytokines are known to play various roles in mediating the immune response to infection in a tissue- and pathogen-dependent manner. The present study seeks to investigate the role of IL-36R signaling in C57BL/6 mouse corneas in response to Pseudomonas aeruginosa (PA) infection. IL-36α−/−, IL-36γ−/−, and IL-36R−/− mice had significantly more severe keratitis than wild type mice. At 6-hour post-infection (hpi), IL-36α pretreatment augmented PA-induced expression of IL-1Ra, IL36γ, LCN2, and S100A8/A9. At 1-day post-infection (dpi), exogenous IL-36α suppressed, whereas IL-36α deficiency promoted the expression of IL-1β. At 3 dpi, exogenous IL-36α suppressed Th1, but promoted Th2 immune response. IL-36α stimulated the infiltration of IL-22-expressing immune cells and IL-22-neutralization resulted in more severe keratitis. IL36α alone stimulated dendritic cell infiltration in B6 mouse corneas. Taken together, our study suggests that IL-36R signaling plays a protective role in the pathogenesis of PA keratitis by promoting the innate immune defense, Th2 and/or Th22/IL-22 immune responses. Exogenous IL-36α might be a potential therapy for improving the outcome of PA keratitis.
Keywords: Bacterial keratitis, IL-36, IL-22, Th2, Antimicrobial peptides
Introduction
Microbial keratitis is a predominant cause of irreversible visual loss around the world (1). Among all the pathogens in microbial keratitis, Pseudomonas aeruginosa (PA) is the most aggressive and frequently isolated organism, especially in extended contact lens wearers (2). Corneal hypoxia, micro-trauma, decreased tear production, and increased corneal temperature caused by the contact lens allows pathogens to easily adhere to the ocular surface (3, 4). PA keratitis is characterized by its rapid onset and progression, often presenting with strong inflammation and ulceration. Severe complications include anterior chamber hypopyon, descemetocele formation, corneal scarring and perforation (5).
Severe keratitis caused by PA results from both a high virulence of the pathogen and the extreme inflammatory response of the host (6). Various bacterial exotoxins and enzymes help PA digest and degrade the corneal matrix (7). In our experimental model, it takes approximately 18–24 h for PA to cross the corneal epithelial basement membrane and reach the stroma (8). Hence, early recognition and enhanced host defense is essential to suppress bacterial invasion. The innate immunity also has profound effects on adaptive immunity, leading to different responses of the tissues to infection in a tissue- and pathogen-dependent manner. The adaptive immunity response can be activated by antigen presenting cells (dendritic cells and macrophages), residential immune cells in avascular, naïve corneas, that activate Th1, Th2, Th17, and/or Th22 immune responses (9). Conversely, an extensive immune response can also be destructive to the host tissue (10, 11). In the cornea, the TH2 immune response plays a protective role while Th1 and Th17 are detrimental to PA keratitis (12, 13).
IL-36 cytokines are the newest members of the IL-1 superfamily and have been shown to play a role in tissue homeostasis and inflammation (14, 15). The IL-36 family consists of three agonists: IL-36α, IL-36β and IL-36γ. All three share a common heterodimeric receptor IL-36R (16). Binding of IL-36 to IL36R recruits the accessory protein IL-1RAcP, leading to the activation of NF-kb and MAPK pathways, and initiates downstream gene transcription (17). IL36/36R signaling is activated in bacterial, viral, and mycobacterium pneumonia, resulting in the elevation of antimicrobial activity (18–20). As a potent immune stimulator, IL36 signaling has been shown to augment intestine and skin inflammation (21, 22). Like IL-1β, IL-36 cytokines also have a natural antagonist to their receptor, IL-36Ra. IL-36Ra and IL-38 bind IL-36R without recruiting the secondary receptor IL-1RAcP, thus interfering with IL36 signaling (23). Importantly, IL-38 was found to attenuate sepsis by decreasing inflammation and increasing bacterial clearance, suggesting that modulation of IL-36/IL-36R activity might be utilized to control or treat bacterial infection (24). In mouse models of tissue infection, IL-36 agonists exhibited both beneficial and detrimental roles in a tissue- and pathogen-specific manner (18, 20, 25). Using siRNA silencing, we recently showed that IL-36Ra and IL-1Ra played opposing roles in the innate immune response to PA infection, suggesting the potential antagonization of the IL-36/IL36R axis on the IL-1/IL-1R pathway (26). Conventionally, IL-36γ was believed to be the main agonist of IL-36R in mucosal epithelium (27). The knowledge of the role of IL-36α in corneal or mucosal immunity is limited.
In this study, we defined the role of IL-36α in mediating host defense following corneal PA infection. Using a murine model of PA keratitis, we showed an increase in IL-36α expression in response to PA infection. IL-36α protected the cornea by stimulating anti-microbial peptide production, promoting tissue healing by upregulating IL-22 and modulating immune cell infiltration.
Material and Methods
Animals
Specific-pathogen-free, age- and sex-matched C57BL/6 WT mice were purchased from The Jackson Laboratory (Bar Harbor, ME). IL-36R−/− mice on the C57BL/6 genetic background were provided by Amgen (Thousand Oaks, CA) (28). IL-36α−/− and IL-36γ−/− mice bred on a C57BL/6 background were established at the University of Michigan (29). All animal procedures were performed in compliance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Visual Research and were approved by the Institutional Animal Care and Use Committee of Wayne State University.
Mouse model of PA keratitis
Mice were anesthetized with an intraperitoneal injection of ketamine (90 mg/kg) and xylazine (10 mg/kg) before surgical procedures. Mouse corneas were scratched gently with a sterile 26-gauge needle to create three 1-mm incisions to break the epithelial barrier and were inoculated with 1.0 × 104 CFU of ATCC 19660 in 5μl of PBS.
Administration of neutralizing antibody or recombinant protein
To apply neutralizing antibodies or recombinant proteins, mice were subconjunctivally injected with recombinant mouse IL-36α (100ng/5μl; R&D Systems, Minneapolis, MN), anti-IL-22 antibody (400ng/5μl; R&D Systems, Minneapolis, MN), or 0.1% BSA control, 4 h before inoculation with PA.
Isolation of mouse corneal epithelial cells
Mice were euthanized by cervical dislocation. Under the microscope, mouse corneal epithelial cells (CECs) were surgically scraped off from the basement membrane. Cells were collected onto the razor blade. Liquid nitrogen was used to snap freeze the collected cells on the blade, and then the frozen cells were immediately scraped off and transferred into precooled 1.5-ml Eppendorf tubes placed on dry ice. Cells were processed immediately for RNA isolation, protein extraction or were stored at −80°C for later use.
Clinical examination, quantification of PA CFU, and determination of myeloperoxidase units
Corneas were photographed at 1 day or 3-day post-infection (dpi) for the assessment of infection severity. Clinical scores were assigned to the infected corneas in a blinded fashion according to a previously-reported scale (30). Whole corneas were excised and placed in 200 μl sterile PBS. Tissue was homogenized with a TissueLyser II (Qiagen, Valencia, CA). The homogenates were divided into two parts. The first fraction (50μl) was subjected to serial log dilutions for the assessment of viable bacterial numbers. The remaining homogenates were further lysed for myeloperoxidase (MPO) measurement. MPO units were determined according to a previously reported method (31). One MPO unit corresponds to 2.0× 105 PMNs.
Semiquantitative and quantitative PCR
Total RNA was extracted with a RNeasy mini kit (Qiagen) following the manufacturer’s instructions. For semiquantitative PCR, cDNA was amplified with TaqMan technology (Promega, Madison, WI). PCR products were subjected to electrophoresis on 2% agarose gels containing ethidium bromide. For quantitative PCR (qPCR), cDNA was amplified using a StepOnePlus real-time PCR system (Applied Biosystems, University Park, IL) with SYBR Green PCR master mix (Applied Biosystems). Data were analyzed by using the ΔΔ cycle threshold method with β-actin as the internal control. The primers used in this study were listed in Table 1.
Table 1.
Primer sequences used for PCR
| Primers | Forward (5′→3′) | Reverse (5′→3′) |
|---|---|---|
| mβ-actin | GACGGCCAGGTCATCACTATTG | AGGAAGGCTGGAAAAGAGCC |
| mIL-36α | CCAAGAACTGGGGGAAATCT | GGAGGGCTCAGCTTTCTTTT |
| mIL-36γ | CCCATGCAAGTACCCAGAGT | GGGAAAGCCACTGATTCAAA |
| mIL-1β | TGCCACCTTTTGACAGTGATG | AAGGTCCACGGGAAAGACAC |
| mIL-1Ra | GGGACCCTACAGTCACCTAA | GGTCCTTGTAAGTACCCAGCA |
| mIL-22 | CAGGAGGTGGTGCCTTTCCT | TGGTCGTCACCGCTGATGT |
| mIL-23 | AGAAGAAGAGGATGAAGAGAC | GGTGTGAAGTTGGTCCAT |
| mIL-24 | GCTTTCACCAAAGCGACTTC | GCCCAGTAAGGACAATTCCA |
| mIL-17A | TTTAACTCCCTTGGCGCAAAA | CTTTCCCTCCGCATTGACAC |
| mIL-17F | GAGGATAACACTGTGAGAGTTGAC | GAGTTCATGGTGCTGTCTTCC |
| mIL12 | GGTCACACTGGACCAAAGGGACTATG | ATTCTGCTGCCGTGCTTCCAAC |
| mIFN-γ | GTTACTGCCACGGCACAGTCATTG | ACCATCCTTTTGCCAGTTCCTCCAG |
| mIL-4 | TAGTTGTCATCCTGCTCTT | CCAGTACTACGAGTAATCCA |
| mIL-5 | AAAGAGAAGTGTGGCGAGGAGAGAC | CCTTCCATTGCCCACTCTGTACTCATC |
| mLCN2 | CTGAATGGGTGGTGACTGTG | GCTCTCTGGCAACAGGAAAG |
| mS100A8 | TTCGTGACAATGCCGTCTGA | AGGGCATGGTGATTTCCTTGT |
| mS100A9 | TGGGCTTACACTGCTCTTACC | GGTTATGCTGCGCTCCATCT |
m, mouse.
Western blot and ELISA
Mouse corneal samples were lysed with RIPA buffer. The lysates were centrifuged to obtain supernatant. Protein concentration was determined by BCA assay. For Western blot analysis, the protein samples were separated by SDS-PAGE and electrically transferred onto nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA). The membranes were blocked with 5% milk and subsequently incubated with primary and secondary antibodies. Signals were visualized using SuperSignal West Pico chemiluminescent substrate (Thermo Scientific, Pittsburgh, PA). β-actin was used as the loading control. Quantification of protein levels was based on the densitometry of blots by using the software Carestream MI SE (Informer Technologies, Rochester, NY). The antibodies used included: anti-IL-36α, anti-LCN-2 (R&D), and anti-β-actin (Sigma-Aldrich). Enzyme-linked immunosorbent assay (S100A8/9, IL-22; R&D) were performed following manufacturer’s protocols.
Immunohistochemistry
At the indicated time points, the corneas were enucleated, separated into epithelium and stroma with EDTA; the whole corneas as well as separated epithelial sheet were embedded in Tissue-Tek OCT compound, and frozen in liquid nitrogen. 6-μm thick sections were cut and mounted to poly-L-lysine-coated glass slides, fixed in 4% paraformaldehyde, blocked with PBS containing 2% BSA for 1 h at room temperature. Sections were then incubated with primary Abs: anti-NIMP-R14 (1:50; BD), anti-F4/80 (1:50; BD), anti-IL-22(1:200; R&D) anti-IL-36α (1:50; abcam), followed by the secondary Ab, FITC-conjugated IgG (1:100; Jackson ImmunoResearch Laboratories). Slides were mounted with Vectashield (Vector Laboratories, Burlingame, CA) mounting media containing DAPI. Controls were similarly treated with corresponding IgG from the same animal as the primary antibody.
Flow cytometry analysis
Whole corneas and cervical lymph nodes were digested in 20 μl Liberase TL (2.5 mg/ml; Sigma-Aldrich), followed by incubation at 37°C for 45 minutes. Cell suspensions were passed through a 70 μm filter. Viable cells were then counted using trypan blue dye exclusion. Cells were incubated at 4°C in PBS containing 2% FBS and Fc block. Intracellular staining was performed using Foxp3 fixation/permeabilization kit. The cells were labeled with conjugated CD45(A20), CD4(GK1.5), T-bet(eBio17B7), GATA3(TWAJ), Ly6G(RB6-BC5), F/480(BM8), IL-36α, FITC conjugated goat antirabbit antibody (eBioscence, Thermofisher, Abcam). All samples were washed and reconstituted in PBS. Flow cytometry was performed with FACS system (BD FACSCelesta & Accuri C6), and the data were analyzed using FlowJo software.
Statistical analysis
Data were presented as means ± SD. Statistical differences among three or more groups were identified using one-way analysis of variance (ANOVA), followed by a Bonferroni’s post test to determine statistically significant differences. Analysis of clinical scores was performed by a nonparametric Mann-Whitney U test. Differences were considered statistically significant at p < 0.05.
Results
IL-36/IL-36R signaling plays a critical role in B6 mouse corneal immune defense against PA keratitis
Our previous study showed that the IL-36/IL-36R signaling opposes the role of IL-1β/IL-1R in mediating corneal immune defense against PA infection (26). Our preliminary studies revealed that among the three IL-36R agonists, IL-36α and γ, but not β, were upregulated in response to PA infection in B6 mouse corneas. To further elucidate the role of IL-36R signaling in the pathogenesis of PA keratitis, we used IL-36α, IL-36γ and IL-36R knockout mice with wild type (WT) B6 mice as the controls. These mice were inoculated with 1 ×104 CFU PA in scratch-wounded corneas (Fig. 1). The pathology of PA keratitis was analyzed using clinical scoring, bacterial plate counting, and MPO determination. The WT mice had mild and moderate keratitis at 1 and 3 dpi, respectively. Deficiency of IL-36α and IL-36γ increased the severity of PA keratitis, including higher opacification, significantly elevated clinical scores (9.0±0 vs 10.2±0.4 vs 11.2±0.4), markedly increased bacterial burden (3.52 × 105 vs 5.68 × 106 vs 1.10 × 107 CFU), and patently augmented MPO activity (93.92 vs 141.12 vs 173.69 units), compared to WT mice. IL-36γ−/− mice had more severe keratitis than IL-36α−/− mice while IL-36R−/− mice had the most severe keratitis, with cornea melting as early as 3dpi, suggesting a cumulative and nonoverlapping role of IL-36α and IL-36γ in the immune response to PA keratitis.
Figure 1. IL-36 signaling pathway protects B6 mouse corneas from PA infection.
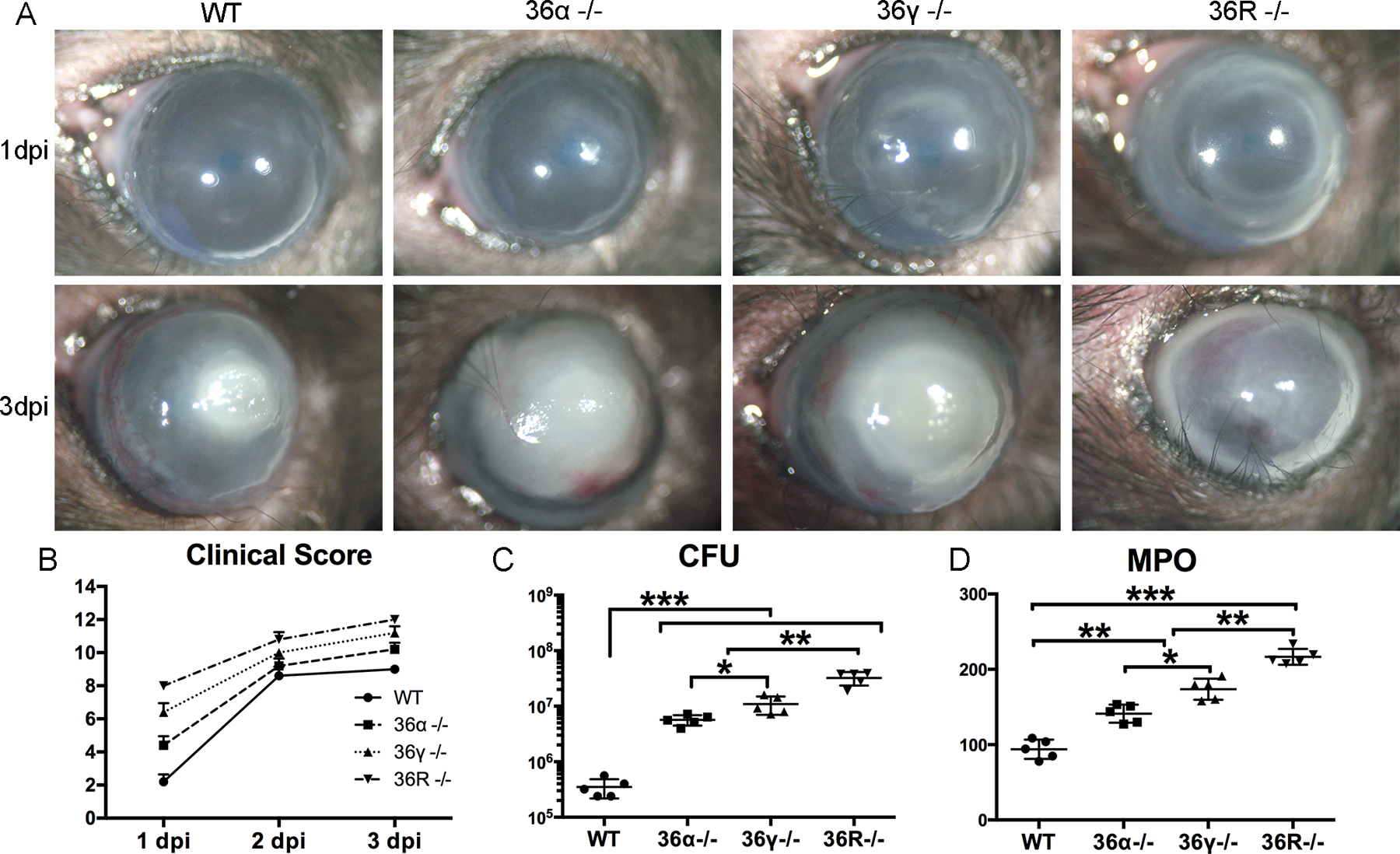
WT, IL-36α−/−, IL-36γ−/−, and IL-36R−/− mouse corneas were gently scratched with a needle to create three 1mm epithelium incisions and inoculated with 1.0×104 CFU PA. (A & B) Eyes were photographed (original magnification × 10) and clinically scored from 1 to 3 dpi. The 1–12 scale clinical scores were graded by an independent blinded observer. All mice were euthanized at 3 dpi. The corneas were excised and subjected to (C) bacterial plate counting presented in log scale and (D) Myeloperoxidase (MPO) determination. The data in (B-D) were presented as median of clinical score (median ± interquartile range), average of CFU or MPO units per cornea (mean ± SD) in dot plots. P values were analyzed with one-way ANOVA, followed by a Bonferroni test. N=5, *P < 0.05, ** P < 0.01, ***P<0.001. The results were representative of three independent experiments.
PA infection increases IL-36α expression in B6 mouse cornea
Our previous study revealed a protective role of IL-36γ in PA keratitis (26). Hence, we focused on the role of IL-36α and investigated its expression in B6 mouse corneas in response to PA infection (Fig. 2). The levels of IL-36 mRNA were increased 8.12-fold in mouse corneas starting from 6 hpi and remaining high up to 18 hpi (16.82-fold) compared to the naïve, control corneas (Fig. 2A). At the protein level, Western-blot analysis showed IL-36α expression in the scraped epithelial cells (Fig. 2B) and an increase in IL-36α protein in the whole corneas starting at 6 hpi, a peak at 9 hpi, and remaining highly elevated up to 18 hpi (Fig. 2C). Immunohistochemistry revealed the corneal epithelium and numerous infiltrating cells are IL-36α positive at 1 dpi (Fig. 2D). The epithelium expression of IL-36α at 1 dpi was further conformed by separating epithelial sheet with the stroma, followed by Western blotting (suppl Fig. 2). Flow cytometry further showed both neutrophils and macrophages produce IL-36α at 1dpi (Fig. 2E). Taken together, Figure 2 reveals that both residential epithelial cells and infiltrated immune cells are the sources of IL-36α in PA-infected corneas at 1 dpi.
Figure 2. PA-infection increases IL-36α expression in B6 mouse cornea.
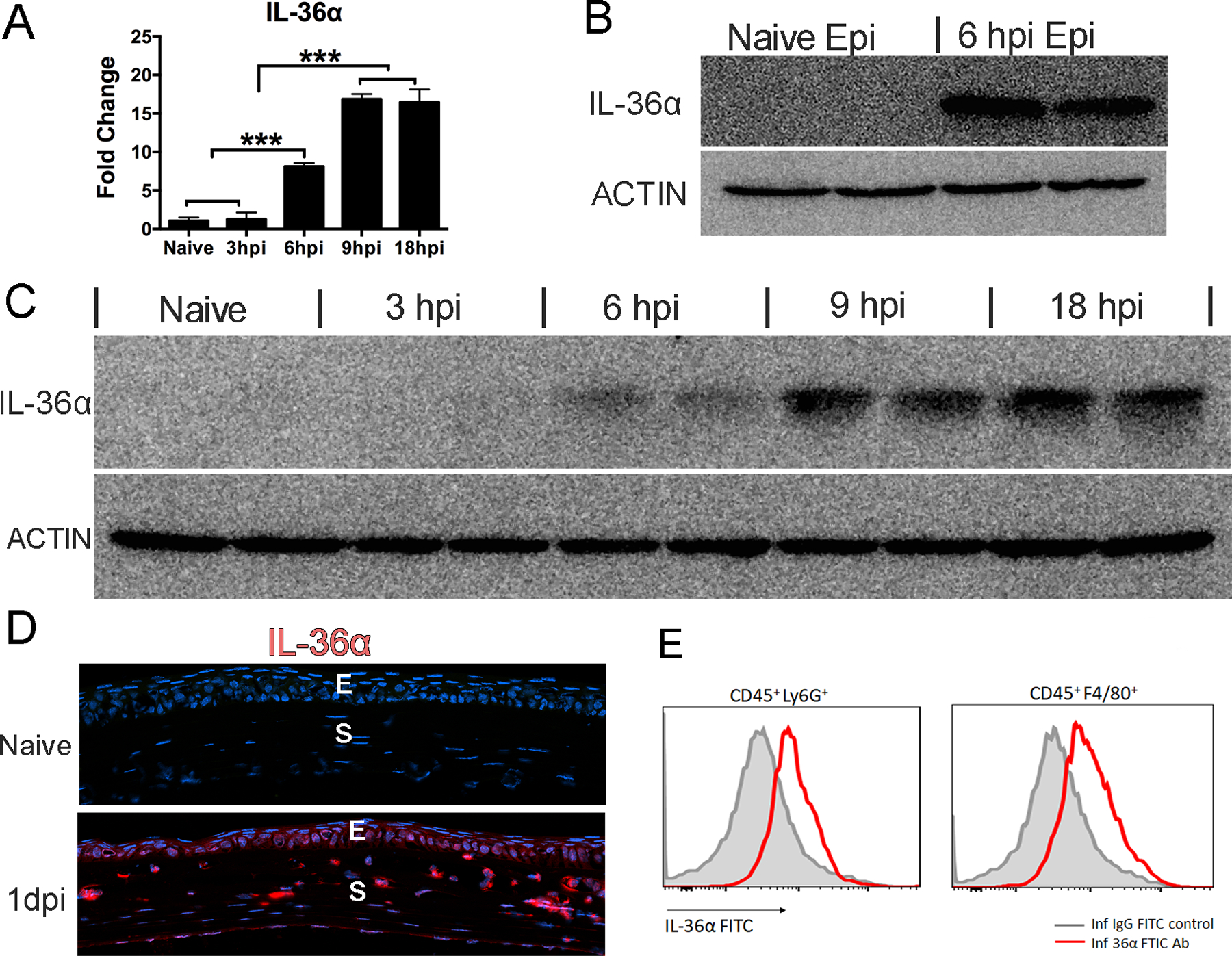
Mouse corneas were gently scratched with a needle to create three 1mm epithelium incisions and inoculated with 1.0×104 CFU PA at 0h. (A) Whole corneas were collected at 3hi, 6hpi, 9hpi 18hpi for q-PCR. P values were analyzed with one-way ANOVA. N=5, *P < 0.05, ** P < 0.01, ***P<0.001. (B) Mouse corneal epithelial cells were collected at 6 hpi for Western-blot analysis of IL-36α. β-actin serves as loading control. (C) Cornea samples representing 5 time points were also collected for Western-blot analysis of IL-36α with β-actin as loading control. Three samples of each time point were used in qPCR and two samples in Western blot. (D) Corneas at 1 dpi were excised and processed for immunohistochemistry analysis. The 6 μm cryostat sections were stained with anti-IL-36α (red), and DAPI (blue) for nuclei. E, epithelium; S, stroma. (E) Flow cytometric analyses of IL-36α, CD45, Ly6G, F4/80 positive immune cells in 1dpi infected corneas. 6 corneas were pooled for each sample. IL36α, CD45+Ly6G+, CD45+F4/80+ cells are shown in the flow cytometric plots. The results were representative of three independent experiments.
IL-36α plays a protective role in PA keratitis in B6 mice
Having identified the increased expression of IL-36α during PA infection in B6 mouse corneas, we next investigated the role of IL-36α in PA keratitis using two complementary approaches: application of recombinant mouse IL-36α (rmIL-36α) prior to PA inoculation or using IL-36α KO mice (Fig. 3). Mouse recombinant protein was subconjunctivally injected 4h before PA inoculation. At 1 dpi and 3 dpi, rmIL-36α treatment resulted in a significant decrease in the severity of PA keratitis, including greatly reduced clinical scores (4.2±0.5 vs 2.0±0.0; 9.2±0.5 vs 6.4±0.9), significantly dampened bacterial burden (5.41 × 104 vs 2.33 × 103 CFU; 2.57 × 106 vs 2.43 × 105 CFU), and decreased MPO activity (13.75 vs 3.30 units; 104.42 vs 44.25 units), compared to the control infected eyes. IL-36α deficiency had opposing effects on PA keratitis outcome. To rule out direct effects of IL-36α on PA, 100 ng recombinant IL-36α in 1 ml with live bacteria exhibited no effects on PA surviving and growth (suppl Fig. 1).
Figure 3. IL-36α plays a protective role in PA keratitis in B6 mice.
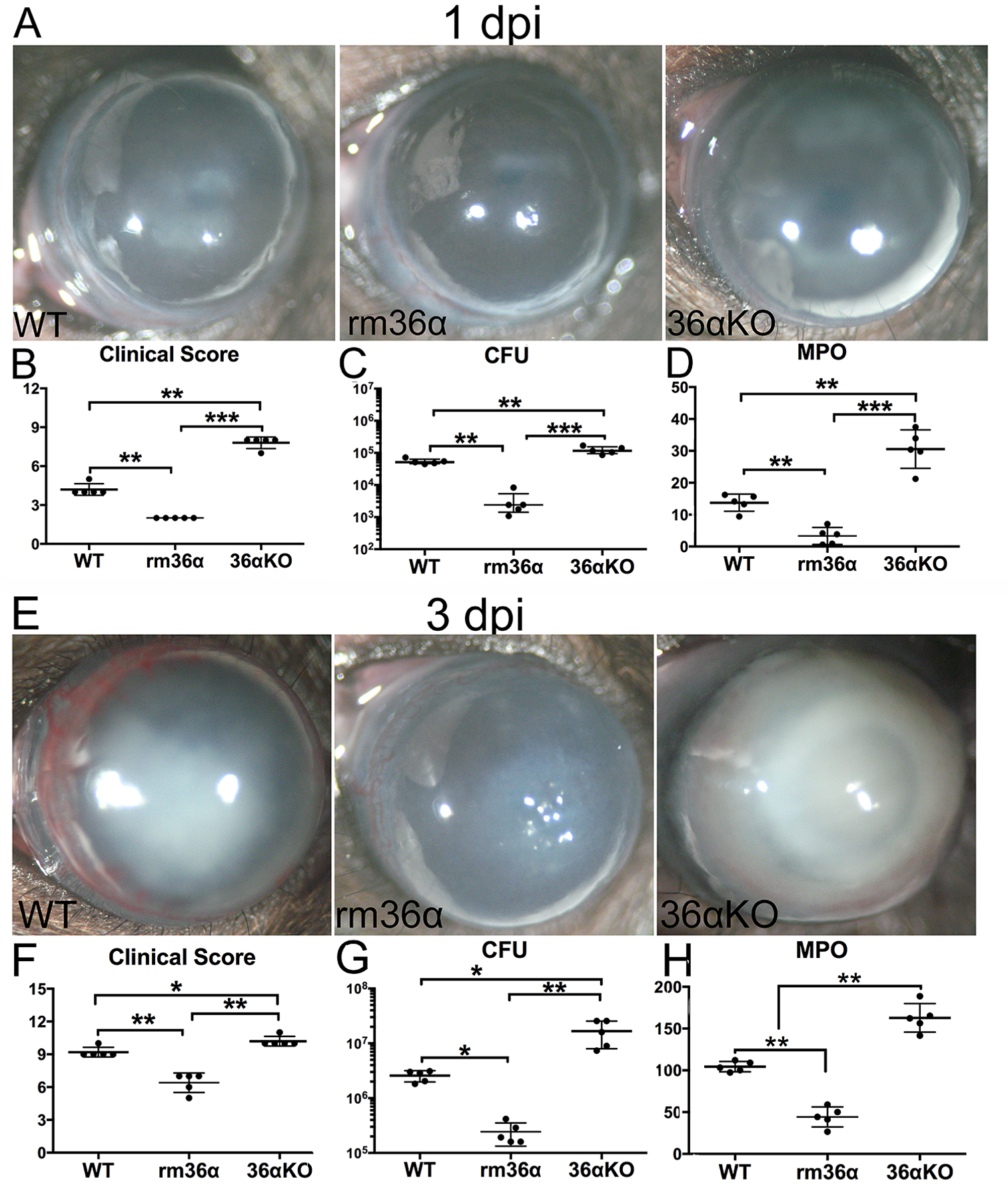
At 4h before PA inoculation, WT mice were subconjunctivally injected with recombinant mouse (rm) IL-36α (100ng/5μl), or 0.1% BSA as control. IL-36α−/− mice were subconjunctivally injected with 0.1% BSA. At 0h, the corneas were gently scratched with a needle to create three 1mm epithelium incisions and inoculated with 1.0×104 CFU PA. (A & E) Mouse corneas were monitored and photographed at 1, 3 dpi, (B & F) and clinical scores were assigned as described in Figure 1. The corneas were excised and subjected to (C & G) bacterial plate counting, the results were presented at log scale; and (D & H) MPO unit determination. The data in (B-D, F-H) were presented as a median of clinical score (median ± interquartile range), average of CFU or MPO units per cornea (mean ± SD). P values were generated by one-way ANOVA, followed by a Bonferroni test. N=5, *P < 0.05, ** P < 0.01, ***P<0.001. The results were representative of three independent experiments.
IL-36α decreases neutrophil and macrophage infiltration in PA infected corneas
Having demonstrated the protective effect of IL-36α in PA keratitis, we next assessed its effect on the infiltration of innate immune cells in PA-infected corneas (Fig. 4). While no neutrophil or macrophage staining was detected in the naive corneas, numerous NIMPR-14- (neutrophil marker) and F4/80- (macrophage marker) positive cells were observed in PA-infected corneal stroma at 1dpi. IL-36α treatment decreased the number of infiltrating neutrophils and macrophages compared with control infected corneas (Fig. 4A). Quantification of neutrophil and macrophage infiltration was evaluated with flow cytometry (Fig. 4B & C). IL-36α treatment significantly reduced the numbers of neutrophils and macrophages cell infiltration compared with control group, which is consistent with immunohistochemistry findings.
Figure 4. IL-36α ameliorates PA-induced inflammation in the B6 mouse corneas.
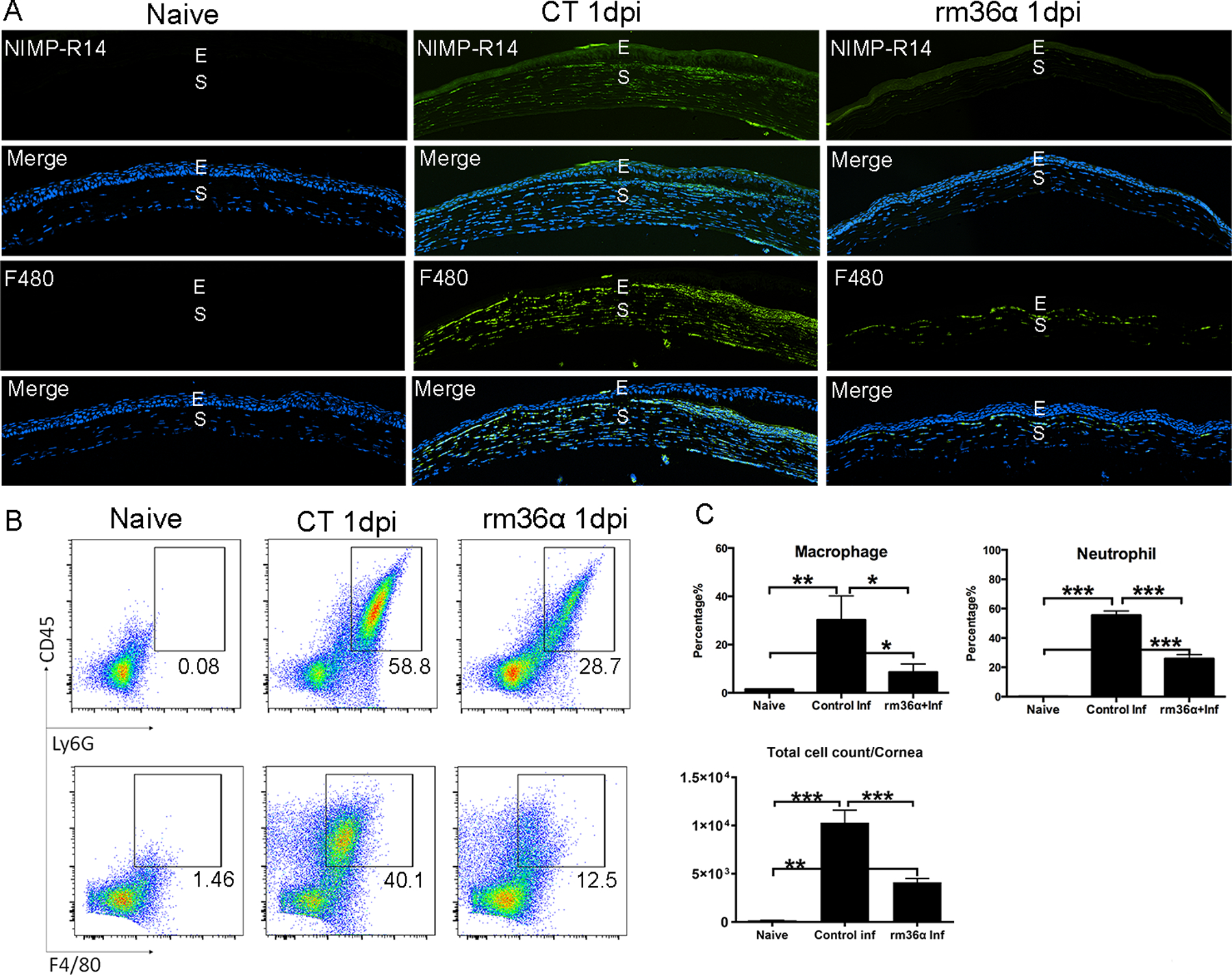
WT mouse corneas were treated with rmIL-36a or 0.1% BSA and inoculated with PA as in figure 3. Naïve corneas were used as negative control. The corneas were collected at 1dpi (A) The corneas were processed for immunohistochemistry analysis. 6μm cryostat sections were stained with NIMP-R14 antibody for neutrophils or F4/80 antibody for macrophages. The images of neutrophils (green), macrophages(green) were merged with DAPI (blue nuclei) staining. E, epithelium; S, stroma. Three independent experiments were performed; 1 representative image for each condition is presented. (B) The corneas were processed for flow cytometry analysis and stained with CD45, Ly6G, F4/80 antibodies. Six corneas were pooled for each sample. (C) Three samples of each groups were used for neutrophil and macrophage cell percentage statistical analysis (mean ± SD). P values were generated by one-way ANOVA, *P < 0.05, ** P < 0.01, ***P<0.001. Three independent experiments were performed; 1 representative image for each condition is presented. E, epithelium; S, stroma.
Exogenous IL-36α alters gene expression in response to PA infection in B6 mouse corneas.
To explore the underlying mechanism of how IL-36α influences the outcome of PA keratitis, we used qPCR to assess the effects of IL-36α on the expression of several innate immune genes that we previously showed to be involved in corneal innate defense (Fig. 5). At 6 hpi, recombinant mouse (rm) IL-36α treatment increased infection-induced expression of factors promoting innate defense such as IL-1Ra (15.00- vs 50.64-fold), IL-36γ (4.04- vs 6.75-fold), and dampened the expression of factors suppressing corneal defense such as IL-24 (319.80- vs 41.71-fold) in B6 mouse corneal epithelial cells (26, 32, 33). Notably, the expression of antimicrobial genes S100A8 (22.73- vs 322.50- fold), S100A9 (3.90- vs 13.34-fold), and LCN2 (6.99- vs 19.29-fold), were also greatly enhanced by rmIL-36α at 6hpi. rmIL-36α exhibited no effects on IL-1β (3.00- vs 3.54-fold) expression and the levels of IL-22 (1.08- vs 1.07-fold) did not change in B6 mouse corneas at 6 hpi (Fig. 5A).
Figure 5. IL-36α alters gene expression in B6 mouse corneas in response to PA-infection.
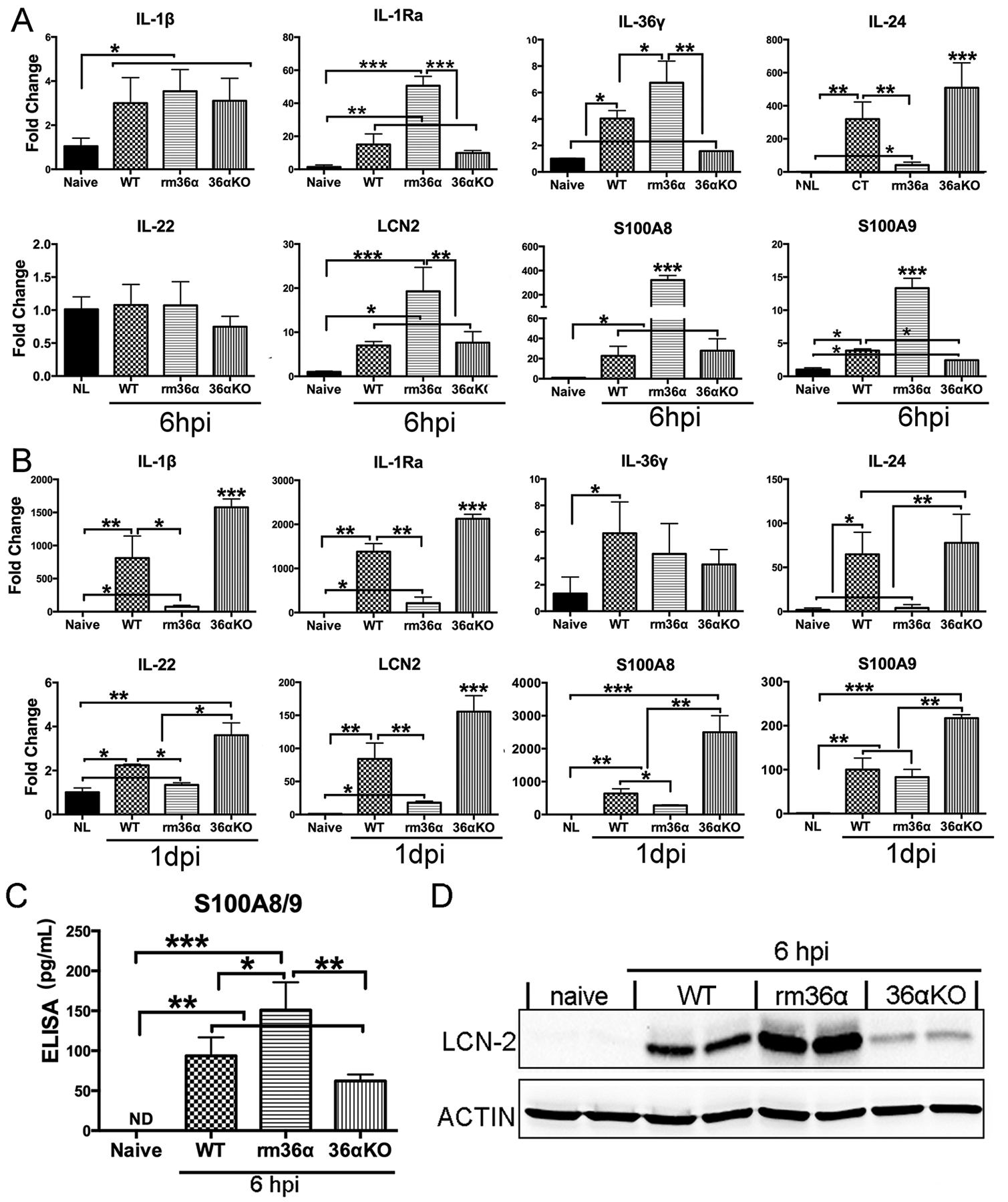
WT, IL-36α−/− mouse corneas were treated with rmIL-36α, or 0.1% BSA and inoculated with PA as in Figure 3. (A) Corneal epithelial cells were collected at 6 hpi and (B) whole cornea samples were collected at 1dpi (immune cell infiltration occurs after 6 hpi) and analyzed by real-time PCR. The results were presented as a relative increase (fold) to those of naive corneas, which were set as a value of 1. Corneal epithelial cells were collected at 6 hpi for (C) ELISA analysis of S100A8/9 dimmer, (D) Western-blot analysis of LCN2. Data were representative of three independent experiments with three corneas per group (mean ± SD). *P<0.05, **P<0.01, ***P<0.001 (One-way ANOVA).
The expression of the aforementioned genes was also assessed in 1 dpi corneas. PA infection-induced levels of IL-1β (811.58- vs 76.42-fold), IL-1Ra (1381.48- vs 214.85-fold), IL-24 (64.83- vs 4.03-fold), Immume cellsIL-22 (2.24- vs 1.35-fold), LCN-2 (83.87- vs 18.02-fold), S100A8 (637.45- vs 277.65-fold) were all decreased in rmIL-36α-treated corneas, consistent with the minimal inflammation in that group. In contrast, IL-36α-deficiency augmented the expression of IL-1β (811.58- vs 1577.10- fold), IL-1Ra (1381.48- vs 2126.75-fold), IL-22 (2.24- vs 3.61-fold), LCN-2 (83.87- vs 155.56-fold), S100A8 (637.45- vs 2498.96-fold) and S100A9 (100.02- vs 217.20- fold). The elevation may due to the severe inflammation in IL-36α−/− mouse corneas (Fig. 5 B).
At the protein level, infection-induced expression of calprotectin (dimmer of S100A8/9) and LCN2 were augmented by the addition of rmIL-36α and dampened in IL-36a−/− corneal epithelia at 6 hpi (Fig. 5 C & D).
IL-36α promotes Th2 immune response in PA-infected B6 mouse corneas
At 1 dpi, corneal defense against PA infection is primarily carried out by the innate immune defense apparatus while adaptive immunity is expected to be initiated, activated, and ultimately participate in the corneal defense at 3 dpi. To determine how IL-36R signaling affects adaptive immune responses, we assessed the expression of two selected cytokines each from Th1 and Th2 pathways (Fig. 6) in 3 dpi corneas. rmIL-36α suppressed Th1 (IFN-γ, 5.65- vs 1.89-fold and IL-12, 3.54- vs 1.96-fold) and enhanced Th2 cytokine (IL-4, 2.47- vs 3.85-fold and IL-5, 1.06- vs 3.95-fold) expression while IL-36R depletion exhibited no apparent effects on the mRNA expression of these genes.
Figure 6. IL-36 signaling regulates adaptive immune gene expression in B6 mouse corneas in response to PA-infection.
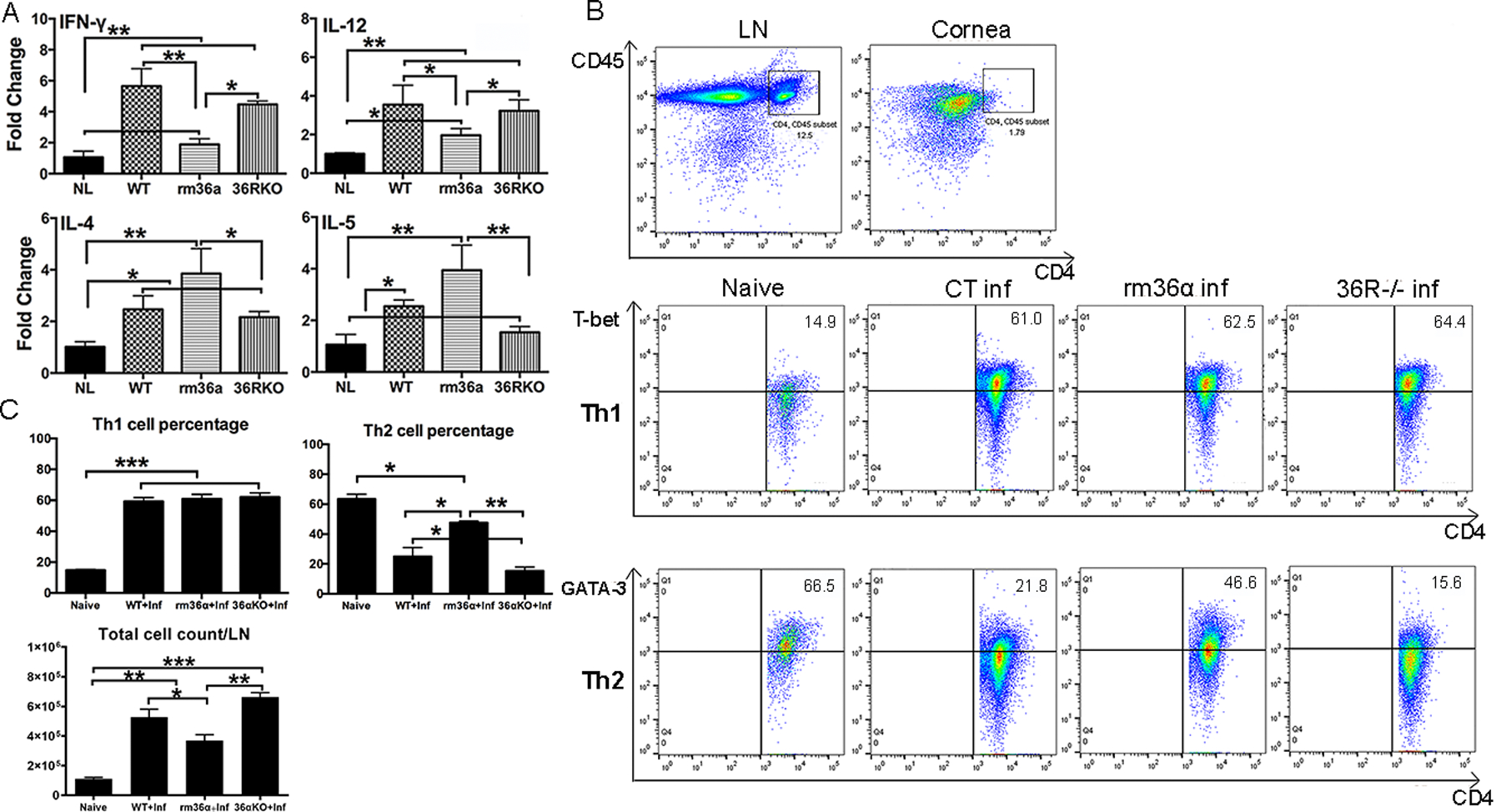
rmIL-36α, or 0.1% BSA treated WT mice, IL-36R−/− mice were scratched and inoculated with PA. (A) At 3 dpi, whole cornea samples were collected and analyzed by real-time PCR. The results were presented as a relative increase (fold) to those of naive corneas, which were set as a value of 1. Three corneas per group (mean ± SD). (B) Six mouse corneas and two cervical lymph nodes were pooled for each sample for flow cytometric analysis. The samples were stained with CD45, CD4, T-bet, GATA3 antibody. Th1 and Th2 cell percentage of mouse lymph nodes were shown in FACS plots. (C)Th1 and Th2 cell percentage out of the total CD4+T cell population, as well as total cell number were analyzed. Data were representative of three independent experiments with three samples per group. *P<0.05, **P<0.01, ***P<0.001 (One-way ANOVA). LN, lymph node.
Flow cytometry revealed that CD45 and CD4 positive T cells were abundant in mouse cervical lymph nodes but barely detectable in the corneas. Further analysis revealed that in 3 dpi mouse cervical lymph nodes, the percentage of Th2 cells was significantly higher in rmIL-36 treated- mice, and lower in IL-36R −/− mice (Fig. 6 B &C). Gating strategy was shown in Supplementary Figure 4. Immunohistochemistry showed that some IL-36α positive cells were also GATA3 positive, suggesting that Th2 cell may also produce IL36α in the cornea at 3 dpi (suppl Fig. 3).
IL-36α induces the infiltration of IL-22 expressing cells and IL-22 neutralization worsens PA keratitis
We recently reported that neutralization of IL-23 regulated Il-17Ra signaling improved the outcome of PA keratitis, suggesting a detrimental role of IL-23-IL-17 axis- in PA keratitis (12). IL-23 is also known to induce IL-22 expression and the IL-23-IL-22 axis was shown to regulate intestinal microbial homeostasis to protect from diet-Induced atherosclerosis (34). To that end, we investigated the relationship of IL-22 expression with IL-36 signaling and the role of IL-22 signaling in immune defense against PA keratitis (Fig. 7). Immunostaining showed that a single dose of rmIL-36α injection to eyes without PA infection resulted in the recruitment of IL-22 positive cells that migrated from the limbus towards the central cornea 1 day after IL-36α treatment, with more staining near the limbus compared to the central cornea. No IL-22 staining was observed in the control BSA-injected corneas. The stromal IL-22 positive cells were co-stained with macrophage marker F4/80 (Fig. 7B).
Figure 7. IL-22 as a downstream cytokine of IL-36α improves the outcome of PA keratitis.
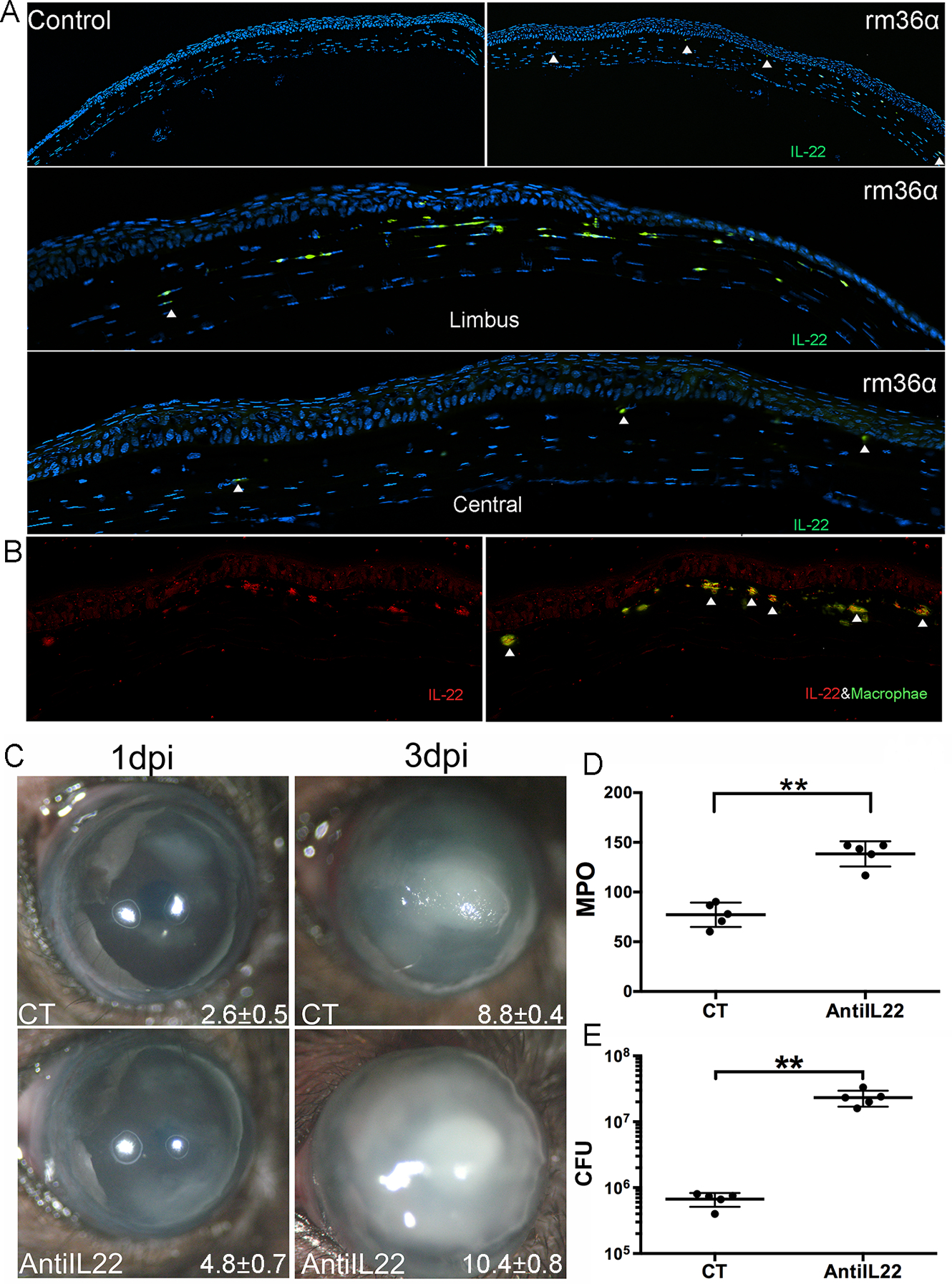
WT mice were subconjunctivally injected with rmIL-36α, or 0.1% BSA as control given 4h prior to PA inoculation. At 1 dpi, corneas were excised and processed for immunohistochemistry analysis. The 6μm cryostat sections were stained with (A) anti-IL22 (green) and DAPI (blue) for nuclei, arrowheads: IL-22 positive cells, or with anti-IL22 (red) and F4/80 (green) antibodies (B), arrowheads: IL-22 and F4/80 positive cells. (C-E) Infected corneas were photographed and scored at 1 and 3 dpi and collected at 3dpi for CFU and MPO determination. (C) The numbers within each eye micrograph are the clinical scores assigned and presented as median plus interquartile range. Data in (D) and (E) were presented as an average of CFU or MPO units per cornea (mean ± SD). N=5, *P<0.05, **P<0.01, ***P<0.001 (paired t-test). Data are representative of three independent experiments. E, epithelium; S, stroma; L, limbus.
The function of IL-22 in PA keratitis was also evaluated using subconjunctival injection of IL-22-neutralizing antibody. Compared to the control corneas injected with IgG, IL-22 neutralization resulted in a marked increase in the severity of PA keratitis (Fig. 7B & C & D) at 3 dpi. There were higher clinical scores (8.8±0.4 vs 10.4±0.8), bacterial burden (6.70 × 105; 2.33 × 107 CFU), and MPO activity (77.17 vs 138.41 units) in anti-IL-22-treated corneas compared to nonspecific IgG-treated corneas.
Exogenous IL-36α induces DC infiltration in B6 mouse corneas.
The aforementioned results indicate that IL-36R signaling induced infiltration of IL-22-expressing cells (TH17 and TH22 cells). Since dendritic cells (DCs) are known to be more sensitive to IL-36 than IL-1β in inducing the production of proinflammatory cytokines IL-12, IL-1β, IL-6, and IL-23 (35), we next assessed whether IL-36α induces DC infiltration in B6 mouse corneas (Fig. 8). At 1 dpi, IL-36α stimulated the migration of numerous CD11c-positive cells from the limbal region towards the central cornea. Most of the recruited CD11c-positive cells were round-shaped rather than dendriform DCs. There were only a few dendriform DCs near the limbal region in control, infected corneas (Fig. 8A). Statistical analysis showed that there were more CD11c positive cells in rm-36α-treated corneas (Fig. 8B).
Figure 8. IL-36α regulates dendritic cell infiltration in B6 mouse corneas.
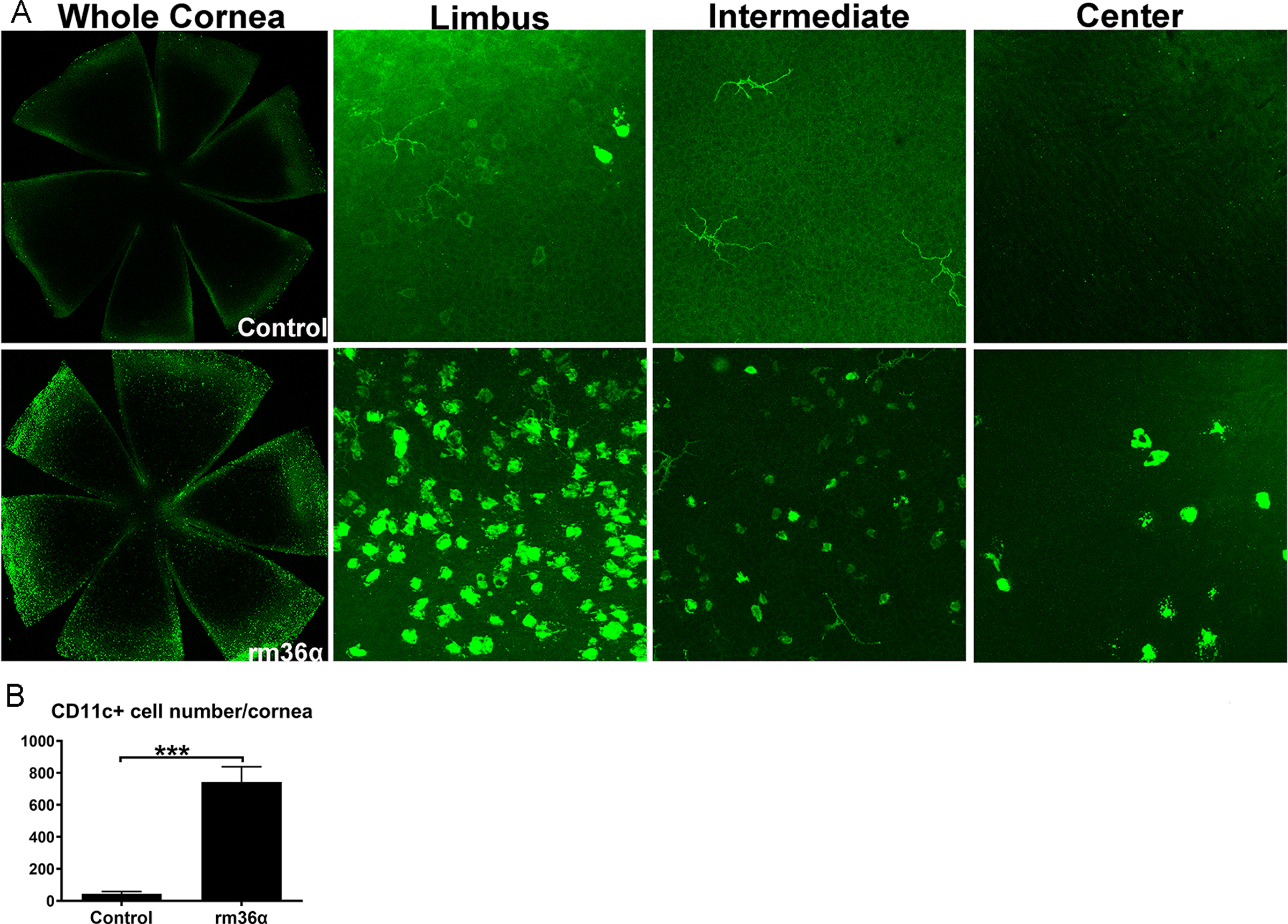
rmIL-36α or 0.1%BSA was subconjunctivally injected into WT mouse corneas. (A) At 1 day post injection, Mouse corneas were collected for whole mount staining with CD11c (green) antibody. (B) The number of CD11c positive cells was quantified and analyzed. Data were representative of three independent experiments with three corneas per group. ***P<0.001 (paired t-test).
Discussion
In this study, we investigated the role of IL-36α and IL-36R signaling in PA keratitis in B6 mice. We demonstrated that IL-36R signaling is required for the proper response of B6 mice to PA infection, and that IL-36α and Il-36γ each contributes to the protective effects of IL-36R signaling. We demonstrated that PA infection induces the upregulation of IL-36α in the cornea and, in addition to the epithelia, certain infiltrated cells also express IL-36α. Functionally, exogenous IL-36α decreases the severity of PA keratitis, promotes bacterial clearance, and ameliorates infection-induced inflammation while IL-36α, IL-36γ, or IL-36R deficiency impairs bacterial clearance and exacerbates PA-induced inflammation. Exogenous IL-36α also dampens the expression of pro-inflammatory cytokines/chemokines and stimulates anti-inflammatory and anti-microbial gene expression. We also found that IL-36α suppressed the mRNA expression of Th1 and promoted Th2 immune responses in PA-infected mouse corneas. Flow cytometry revealed of that IL-36α promoted while IL-36R deficiency suppressed Th2 immune response in mouse cervical lymph nodes. Moreover, IL-36 induced IL-22 expression in mouse corneas and neutralizing IL-22 exacerbated the severity of PA keratitis. IL-36α alone recruited DCs to B6 mouse corneas. Taken together, we conclude that IL-36R signaling mediates corneal immune defense against PA infection through its effects on both innate and adaptive immunity, by regulating inflammatory response, inducing anti-microbial effectors, and modulating Th2 response and/or Th1/Th2 balance in B6 mice.
Our studies show that IL36α mRNA was detected in the naïve corneas, suggesting a basal level of expression. PA infection induces IL-36α upregulation in corneal epithelium as early as 6 hpi. Compared to IL-1β, the expression of which can be detected as early as 1 hpi and increases with time post-PA infection, the upregulation of IL-36 cytokines are at a relatively late stage, detectable in the cornea and more apparent in the corneal epithelia at 6 hpi (26). While IL36 is mostly detected in corneal epithelium at 6 hpi, it was observed in infiltrated cells, neutrophils and macrophages, as well as epithelial cells at 1 dpi. The relatively late upregulation of IL-36α may be related to the fact that, unlike the IL-1 isoforms, IL-36 agonists have a basal expression in naïve corneas. Alternatively, this may be related to the suggested function of the IL-36 family to counteract or antagonize the IL-1 cytokine family after IL-1R activation (26).
Emerging data suggests a significant role of IL-36 in mucosal tissues (36). IL-36γ−/− mice have higher mortality and morbidity in Streptococcus pneumoniae and Klebsiella pneumoniae pulmonary infections, with insufficient type 1 cytokine production (20). However, in the murine Legionella pneumonia model, both IL-36α and IL-36γ were upregulated at mRNA and protein levels in infected lung and only IL-36R−/− (but not IL-36α−/− or IL-36γ−/−) mice exhibited increased mortality, suggesting that IL-36α and IL-36γ have overlapping functions (37). In humans, IL-36α was shown to be highly expressed in the serum and in the salivary glands of primary Sjogren’s syndrome patients (38). Our data in the present study using IL-36α−/−, IL-36γ−/− and IL-36R−/− mice revealed that all three knockout mice have more severe keratitis than WT controls and the severity of keratitis varies, with IL-36R−/− > Il-36γ−/− > IL-36α−/− in terms of decreasing severity. Hence, both IL-36α and IL-36γ contribute to IL-36R signaling-mediated anti-inflammatory and antimicrobial responses and to the host defense against PA keratitis in B6 mouse corneas. We conclude that IL-36α and IL-36γ each play a nonredundant role in mediating corneal immune protection in response to PA infection.
How does IL-36α enhance host defense and protect the cornea from PA infection? Our data show that IL-36α or IL-36R signaling is involved in the innate immunity against PA infection. The epithelium is the first barrier encountered and is the major site for innate immune defense at early stages of infection (39). We showed in this study that activation of IL-36R signaling improved PA keratitis outcomes, and decreased both bacterial load and MPO, indicative of neutrophil infiltration. Neutrophils, which are the first immune cells to respond to PA keratitis, promote bacterial clearance and the production of pro-inflammatory cytokines. As such, one would expect that MPO activity is elevated in IL-36α treated corneas and is attenuated in IL-36R−/− corneas. Our results showed opposing effects. Our previous studies showed that in our model most bacteria were detected within the epithelium, up to 24 h post-PA inoculation. In the present study, we observed that the expression of antimicrobial peptides LCN2 and calprotectin was augmented, while expression of factors exacerbating keratitis, IL-24 and MMP13 (data not shown), was attenuated by IL-36α treatment at 6 hpi. As expected, IL-36R−/− corneas have the opposite effects, providing a molecular mechanism for the protective effects of IL-36 signing at early stages of infection. More importantly, we showed that at the protein level, IL-36α significantly augments, while IL-36R knockout downregulates the expression of calprotectin (S100A8/S100A9 dimer) and LCN-2 at 6 hpi in corneal epithelial cells. Calprotectin can potently kill bacteria and fungi by chelating zinc and manganese (40, 41). LCN2 sequesters bacterial siderophores and interferes with their iron metabolism (42). LCN2 was shown to be required for pulmonary host defense against Klebsiella, an opportunistic extracellular pathogen, and the epithelium-produced LCN2 is important for resistance to dissemination of K pneumoniae (43, 44). Hence, we propose that epithelium-produced LCN2, calprotectin, and potentially other AMPs such as CXCL10 and CRAMP (45, 46), work together to eliminate invading PA within the epithelium at the early stages of infection (6 hpi) resulting in a decrease in the overall inflammatory response. At 1 dpi, the expressions of these factors appeared to be more likely associated with the severity of keratitis. Hence, the innate immune response at 6 hpi, when the number of invading pathogens starts to increase in our model of PA keratitis, is a critical time point for determining the outcome of PA keratitis. Taken together, the epithelium-expressed IL-36α enhances innate host defense by upregulating multiple antimicrobial genes and controls inflammation by inhibiting pro-inflammatory, promoting anti-inflammatory cytokine expression, and suppressing neutrophil and macrophage infiltration.
Our study also suggests a role of IL-36α and IL-36R signaling in mediating the adaptive immune response to PA infection in B6 mouse corneas. Th1 and Th2 are well-characterized adaptive immune responses to pathogens (47). Th1 cells produce IFN-γ to promote cellular immune responses against intracellular microorganisms while Th2 cells produce IL-4, IL-5 and IL-13 to promote humoral immune responses against parasites, extracellular bacteria, and allergens. Consistent with the protective role of IL-36R signaling, our data shows that at the mRNA levels, exogenous IL-36α suppresses Th1 and promotes Th2 response in PA-infected corneas while IL-36R exhibited opposing effects. Early studies have shown that PA infection results in CD4+ T cell infiltration in the cornea and becomes readily apparent as early as 3 dpi with subsequent migration of activated CD4+ T cells by 5 dpi (10, 13, 48–50). To determine if Th1 and/or Th2 cells play a role in keratitis, we performed flow cytometry analysis of infected corneas and local draining lymph nodes. Our data showed only a barely detectable population of CD4 positive T-cells in the infected corneas at 3 dpi. In the cervical lymph nodes, Th2 (GATA3 positive) cell percentage was significantly higher in the exogenous IL-36α treated group, and lower in IL-36R−/− lymph nodes compared with the control infection group. Hence, IL-36R signaling appears to promote Th2 immune responses, consistent with reports that Th2 is protective while Th1 is detrimental for PA keratitis (13). In this case, IL-36 modulates the Th response by decreasing the Th1 to Th2 ratio and thus protecting of the cornea from PA keratitis.
IL-22 is a cytokine in the IL-10 family that is expressed by cells of the innate and adaptive immune system, including Th17 as well as Th22 cells. IL-22 is known to promote host defense, epithelial barrier function, and tissue repair at mucosal surfaces (25, 51, 52). Our results show that at an early stage of corneal infection, 6 hpi, there was no IL-22 induction and at 1 dpi its expression appeared to be correlated to the severity of PA keratitis, consistent with immune cells as the source of IL-22. Indeed, we showed that rmIL-36α alone is sufficient to induce the infiltration of immune cells expressing IL-22 from the limbal region into the corneas assessed at 24 hours after application of the recombinant protein. Neutralization of IL-22 resulted in greatly increased severity of PA keratitis. We propose that IL-36/IL-36R signaling functions as an upstream inducer of Th22 immune response in the cornea, hence promoting bacterial clearance and tissue healing in response to PA-infected corneas. IL-22 is known to induce the production of AMPs, such as S100A8, S100A9, β-defensin 3 and Reg (51, 53, 54), and a recent study identified IL-36/IL-36R signaling as a central upstream driver of the IL-23/IL-22/ AMP pathway during intestinal injury (25). Our study is the first to show the infection-induced and IL-36α-mediated IL-22 expression and potential involvement of Th22 cells in protecting corneas from PA keratitis.
In addition to IL-22 expressing immune cells, we also showed that IL-36α alone stimulated the infiltration of dendritic cells (DCs). Both myeloid and monocyte-derived DCs express IL-36R and respond to IL-36, whereas human T cells or neutrophils do not (55). Myeloid and monocyte-derived DCs may induce both Th1 and Th2 cytokines in naive allogeneic T lymphocytes (56). Our data suggests that IL-36α stimulate DC infiltration and may be a contributing factor for DCs to drive Th2 immune responses in PA-infected corneas.
Taken together, our study demonstrates a vital role of IL-36/IL-36R in the corneal innate immunity against PA infection. IL-36α enhances host defenses by upregulating AMPs, IL-22, and modulates immune cell gene expression and infiltration, suggesting a critical role of IL-36 in the interplay between corneal epithelial cells and immune cells. IL-36α might be used as an adjunctive therapeutic reagent to antibiotics for treating PA and/or other forms of microbial keratitis.
Supplementary Material
Key Points:
IL36 signaling is upregulated and protects mouse corneas from PA infection.
IL-36α modulates inflammation and enhances anti-microbial effectors.
IL-36 promotes IL-22 and Th2 response. IL-22 blocking worsens PA keratitis.
Acknowledgement
The authors declare that there is no duality of interest associated with this manuscript.
The authors declare that there is no conflict of interest associated with this manuscript. We acknowledge support from NIH/NEI R01EY10869, EY17960 (to F.Y.), EY025923 (to P.L.), p30 EY004068 (NEI core to WSU), Research to Prevent Blindness (to Kresge Eye Institute).
Reference
- 1.Al-Mujaini A, Al-Kharusi N, Thakral A, and Wali UK. 2009. Bacterial keratitis: perspective on epidemiology, clinico-pathogenesis, diagnosis and treatment. Sultan Qaboos Univ Med J 9: 184–195. [PMC free article] [PubMed] [Google Scholar]
- 2.Cheng KH, Leung SL, Hoekman HW, Beekhuis WH, Mulder PG, Geerards AJ, and Kijlstra A. 1999. Incidence of contact-lens-associated microbial keratitis and its related morbidity. Lancet 354: 181–185. [DOI] [PubMed] [Google Scholar]
- 3.Stapleton F, and Carnt N. 2012. Contact lens-related microbial keratitis: how have epidemiology and genetics helped us with pathogenesis and prophylaxis. Eye (Lond) 26: 185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Slusher MM, Myrvik QN, Lewis JC, and Gristina AG. 1987. Extended-wear lenses, biofilm, and bacterial adhesion. Arch Ophthalmol 105: 110–115. [DOI] [PubMed] [Google Scholar]
- 5.Guembel HO, and Ohrloff C. 1997. Opportunistic infections of the eye in immunocompromised patients. Ophthalmologica 211 Suppl 1: 53–61. [DOI] [PubMed] [Google Scholar]
- 6.Janeway CA Jr., and Medzhitov R. 2002. Innate immune recognition. Annu Rev Immunol 20: 197–216. [DOI] [PubMed] [Google Scholar]
- 7.Sadikot RT, Blackwell TS, Christman JW, and Prince AS. 2005. Pathogen-host interactions in Pseudomonas aeruginosa pneumonia. Am J Respir Crit Care Med 171: 1209–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hyndiuk RA 1981. Experimental Pseudomonas keratitis. Trans Am Ophthalmol Soc 79: 541–624. [PMC free article] [PubMed] [Google Scholar]
- 9.Iwasaki A, and Medzhitov R. 2015. Control of adaptive immunity by the innate immune system. Nat Immunol 16: 343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hazlett LD 2004. Corneal response to Pseudomonas aeruginosa infection. Prog Retin Eye Res 23: 1–30. [DOI] [PubMed] [Google Scholar]
- 11.Strieter RM, Standiford TJ, Huffnagle GB, Colletti LM, Lukacs NW, and Kunkel SL. 1996. “The good, the bad, and the ugly.” The role of chemokines in models of human disease. J Immunol 156: 3583–3586. [PubMed] [Google Scholar]
- 12.Me R, Gao N, Dai C, and Yu FX. 2020. IL-17 Promotes Pseudomonas aeruginosa Keratitis in C57BL/6 Mouse Corneas. J Immunol 204: 169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hazlett LD, McClellan S, Kwon B, and Barrett R. 2000. Increased severity of Pseudomonas aeruginosa corneal infection in strains of mice designated as Th1 versus Th2 responsive. Invest Ophthalmol Vis Sci 41: 805–810. [PubMed] [Google Scholar]
- 14.Garlanda C, Dinarello CA, and Mantovani A. 2013. The interleukin-1 family: back to the future. Immunity 39: 1003–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walsh PT, and Fallon PG. 2018. The emergence of the IL-36 cytokine family as novel targets for inflammatory diseases. Ann N Y Acad Sci 1417: 23–34. [DOI] [PubMed] [Google Scholar]
- 16.Dinarello C, Arend W, Sims J, Smith D, Blumberg H, O’Neill L, Goldbach-Mansky R, Pizarro T, Hoffman H, Bufler P, Nold M, Ghezzi P, Mantovani A, Garlanda C, Boraschi D, Rubartelli A, Netea M, van der Meer J, Joosten L, Mandrup-Poulsen T, Donath M, Lewis E, Pfeilschifter J, Martin M, Kracht M, Muehl H, Novick D, Lukic M, Conti B, Solinger A, Kelk P, van de Veerdonk F, and Gabel C. 2010. IL-1 family nomenclature. Nat Immunol 11: 973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Towne JE, Garka KE, Renshaw BR, Virca GD, and Sims JE. 2004. Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to NF-kappaB and MAPKs. J Biol Chem 279: 13677–13688. [DOI] [PubMed] [Google Scholar]
- 18.Aoyagi T, Newstead MW, Zeng X, Kunkel SL, Kaku M, and Standiford TJ. 2017. IL-36 receptor deletion attenuates lung injury and decreases mortality in murine influenza pneumonia. Mucosal Immunol 10: 1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Segueni N, Vigne S, Palmer G, Bourigault ML, Olleros ML, Vesin D, Garcia I, Ryffel B, Quesniaux VF, and Gabay C. 2015. Limited Contribution of IL-36 versus IL-1 and TNF Pathways in Host Response to Mycobacterial Infection. PLoS One 10: e0126058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kovach MA, Singer B, Martinez-Colon G, Newstead MW, Zeng X, Mancuso P, Moore TA, Kunkel SL, Peters-Golden M, Moore BB, and Standiford TJ. 2017. IL-36gamma is a crucial proximal component of protective type-1-mediated lung mucosal immunity in Gram-positive and -negative bacterial pneumonia. Mucosal Immunol 10: 1320–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boutet MA, Bart G, Penhoat M, Amiaud J, Brulin B, Charrier C, Morel F, Lecron JC, Rolli-Derkinderen M, Bourreille A, Vigne S, Gabay C, Palmer G, Le Goff B, and Blanchard F. 2016. Distinct expression of interleukin (IL)-36alpha, beta and gamma, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn’s disease. Clin Exp Immunol 184: 159–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kanda T, Nishida A, Takahashi K, Hidaka K, Imaeda H, Inatomi O, Bamba S, Sugimoto M, and Andoh A. 2015. Interleukin(IL)-36alpha and IL-36gamma Induce Proinflammatory Mediators from Human Colonic Subepithelial Myofibroblasts. Front Med (Lausanne) 2: 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Towne JE, Renshaw BR, Douangpanya J, Lipsky BP, Shen M, Gabel CA, and Sims JE. 2011. Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36alpha, IL-36beta, and IL-36gamma) or antagonist (IL-36Ra) activity. J Biol Chem 286: 42594–42602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu F, Lin S, Yan X, Wang C, Tu H, Yin Y, and Cao J. 2018. Interleukin 38 Protects Against Lethal Sepsis. J Infect Dis 218: 1175–1184. [DOI] [PubMed] [Google Scholar]
- 25.Ngo VL, Abo H, Maxim E, Harusato A, Geem D, Medina-Contreras O, Merlin D, Gewirtz AT, Nusrat A, and Denning TL. 2018. A cytokine network involving IL-36gamma, IL-23, and IL-22 promotes antimicrobial defense and recovery from intestinal barrier damage. Proc Natl Acad Sci U S A 115: E5076–E5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao N, Me R, Dai C, Seyoum B, and Yu FX. 2018. Opposing Effects of IL-1Ra and IL-36Ra on Innate Immune Response to Pseudomonas aeruginosa Infection in C57BL/6 Mouse Corneas. J Immunol 201: 688–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heath JE, Scholz GM, Veith PD, and Reynolds EC. 2019. IL-36gamma regulates mediators of tissue homeostasis in epithelial cells. Cytokine 119: 24–31. [DOI] [PubMed] [Google Scholar]
- 28.Blumberg H, Dinh H, Trueblood ES, Pretorius J, Kugler D, Weng N, Kanaly ST, Towne JE, Willis CR, Kuechle MK, Sims JE, and Peschon JJ. 2007. Opposing activities of two novel members of the IL-1 ligand family regulate skin inflammation. J Exp Med 204: 2603–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kovach MA, Singer BH, Newstead MW, Zeng X, Moore TA, White ES, Kunkel SL, Peters-Golden M, and Standiford TJ. 2016. IL-36gamma is secreted in microparticles and exosomes by lung macrophages in response to bacteria and bacterial components. J Leukoc Biol 100: 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu TG, Wilhelmus KR, and Mitchell BM. 2003. Experimental keratomycosis in a mouse model. Invest Ophthalmol Vis Sci 44: 210–216. [DOI] [PubMed] [Google Scholar]
- 31.Williams RN, Paterson CA, Eakins KE, and Bhattacherjee P. 1982. Quantification of ocular inflammation: evaluation of polymorphonuclear leucocyte infiltration by measuring myeloperoxidase activity. Curr Eye Res 2: 465–470. [DOI] [PubMed] [Google Scholar]
- 32.Ross BX, Gao N, Cui X, Standiford TJ, Xu J, and Yu FX. 2017. IL-24 Promotes Pseudomonas aeruginosa Keratitis in C57BL/6 Mouse Corneas. J Immunol 198: 3536–3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao N, Sang Yoon G, Liu X, Mi X, Chen W, Standiford TJ, and Yu FS. 2013. Genome-wide transcriptional analysis of differentially expressed genes in flagellin-pretreated mouse corneal epithelial cells in response to Pseudomonas aeruginosa: involvement of S100A8/A9. Mucosal Immunol 6: 993–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fatkhullina AR, Peshkova IO, Dzutsev A, Aghayev T, McCulloch JA, Thovarai V, Badger JH, Vats R, Sundd P, Tang HY, Kossenkov AV, Hazen SL, Trinchieri G, Grivennikov SI, and Koltsova EK. 2018. An Interleukin-23-Interleukin-22 Axis Regulates Intestinal Microbial Homeostasis to Protect from Diet-Induced Atherosclerosis. Immunity 49: 943–957 e949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vigne S, Palmer G, Lamacchia C, Martin P, Talabot-Ayer D, Rodriguez E, Ronchi F, Sallusto F, Dinh H, Sims JE, and Gabay C. 2011. IL-36R ligands are potent regulators of dendritic and T cells. Blood 118: 5813–5823. [DOI] [PubMed] [Google Scholar]
- 36.Russell SE, Horan RM, Stefanska AM, Carey A, Leon G, Aguilera M, Statovci D, Moran T, Fallon PG, Shanahan F, Brint EK, Melgar S, Hussey S, and Walsh PT. 2016. IL-36alpha expression is elevated in ulcerative colitis and promotes colonic inflammation. Mucosal Immunol. [DOI] [PubMed] [Google Scholar]
- 37.Nanjo Y, Newstead MW, Aoyagi T, Zeng X, Takahashi K, Yu FS, Tateda K, and Standiford TJ. 2019. Overlapping Roles for Interleukin-36 Cytokines in Protective Host Defense against Murine Legionella pneumophila Pneumonia. Infect Immun 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ciccia F, Accardo-Palumbo A, Alessandro R, Alessandri C, Priori R, Guggino G, Raimondo S, Carubbi F, Valesini G, Giacomelli R, Rizzo A, and Triolo G. 2015. Interleukin-36alpha axis is modulated in patients with primary Sjogren’s syndrome. Clin Exp Immunol 181: 230–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao N, Kumar A, and Yu FS. 2015. Matrix Metalloproteinase-13 as a Target for Suppressing Corneal Ulceration Caused by Pseudomonas aeruginosa Infection. J Infect Dis 212: 116–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clark HL, Jhingran A, Sun Y, Vareechon C, de Jesus Carrion S, Skaar EP, Chazin WJ, Calera JA, Hohl TM, and Pearlman E. 2016. Zinc and Manganese Chelation by Neutrophil S100A8/A9 (Calprotectin) Limits Extracellular Aspergillus fumigatus Hyphal Growth and Corneal Infection. J Immunol 196: 336–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Damo SM, Kehl-Fie TE, Sugitani N, Holt ME, Rathi S, Murphy WJ, Zhang Y, Betz C, Hench L, Fritz G, Skaar EP, and Chazin WJ. 2013. Molecular basis for manganese sequestration by calprotectin and roles in the innate immune response to invading bacterial pathogens. Proc Natl Acad Sci U S A 110: 3841–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cassat JE, and Skaar EP. 2013. Iron in infection and immunity. Cell Host Microbe 13: 509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chan YR, Liu JS, Pociask DA, Zheng M, Mietzner TA, Berger T, Mak TW, Clifton MC, Strong RK, Ray P, and Kolls JK. 2009. Lipocalin 2 is required for pulmonary host defense against Klebsiella infection. J Immunol 182: 4947–4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cramer EP, Dahl SL, Rozell B, Knudsen KJ, Thomsen K, Moser C, Cowland JB, and Borregaard N. 2017. Lipocalin-2 from both myeloid cells and the epithelium combats Klebsiella pneumoniae lung infection in mice. Blood 129: 2813–2817. [DOI] [PubMed] [Google Scholar]
- 45.Liu X, Gao N, Dong C, Zhou L, Mi QS, Standiford TJ, and Yu FS. 2014. Flagellin-induced expression of CXCL10 mediates direct fungal killing and recruitment of NK cells to the cornea in response to Candida albicans infection. Eur J Immunol 44: 2667–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kumar A, Gao N, Standiford TJ, Gallo RL, and Yu FS. 2010. Topical flagellin protects the injured corneas from Pseudomonas aeruginosa infection. Microbes Infect 12: 978–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.D’Elios MM, Benagiano M, Della Bella C, and Amedei A. 2011. T-cell response to bacterial agents. J Infect Dev Ctries 5: 640–645. [DOI] [PubMed] [Google Scholar]
- 48.Kwon B, and Hazlett LD. 1997. Association of CD4+ T cell-dependent keratitis with genetic susceptibility to Pseudomonas aeruginosa ocular infection. J Immunol 159: 6283–6290. [PubMed] [Google Scholar]
- 49.Hazlett LD, Huang X, McClellan SA, and Barrett RP. 2003. Further studies on the role of IL-12 in Pseudomonas aeruginosa corneal infection. Eye (Lond) 17: 863–871. [DOI] [PubMed] [Google Scholar]
- 50.Suryawanshi A, Cao Z, Thitiprasert T, Zaidi TS, and Panjwani N. 2013. Galectin-1-mediated suppression of Pseudomonas aeruginosa-induced corneal immunopathology. J Immunol 190: 6397–6409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pociask DA, Scheller EV, Mandalapu S, McHugh KJ, Enelow RI, Fattman CL, Kolls JK, and Alcorn JF. 2013. IL-22 is essential for lung epithelial repair following influenza infection. Am J Pathol 182: 1286–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, and Sabat R. 2004. IL-22 increases the innate immunity of tissues. Immunity 21: 241–254. [DOI] [PubMed] [Google Scholar]
- 53.Eidenschenk C, Rutz S, Liesenfeld O, and Ouyang W. 2014. Role of IL-22 in microbial host defense. Curr Top Microbiol Immunol 380: 213–236. [DOI] [PubMed] [Google Scholar]
- 54.Sonnenberg GF, Fouser LA, and Artis D. 2011. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol 12: 383–390. [DOI] [PubMed] [Google Scholar]
- 55.Foster AM, Baliwag J, Chen CS, Guzman AM, Stoll SW, Gudjonsson JE, Ward NL, and Johnston A. 2014. IL-36 promotes myeloid cell infiltration, activation, and inflammatory activity in skin. J Immunol 192: 6053–6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Segura E, Valladeau-Guilemond J, Donnadieu MH, Sastre-Garau X, Soumelis V, and Amigorena S. 2012. Characterization of resident and migratory dendritic cells in human lymph nodes. J Exp Med 209: 653–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.