

Keywords: epithelial repair, HOPX, ischemia, large animal models, stem cell
Abstract
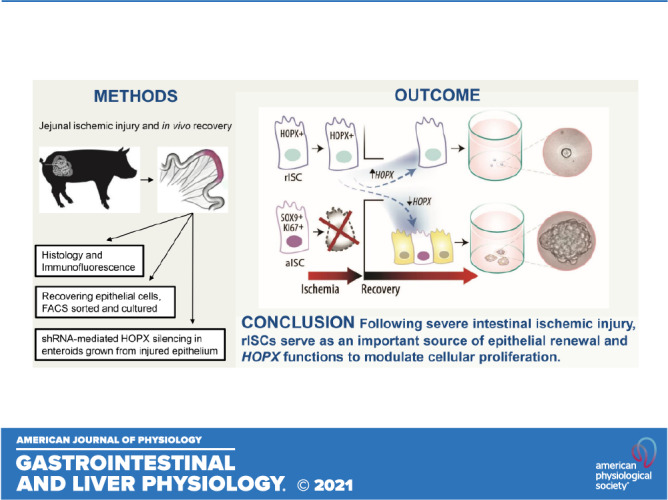
Intestinal ischemia is a life-threatening emergency with mortality rates of 50%–80% due to epithelial cell death and resultant barrier loss. Loss of the epithelial barrier occurs in conditions including intestinal volvulus and neonatal necrotizing enterocolitis. Survival depends on effective epithelial repair; crypt-based intestinal epithelial stem cells (ISCs) are the source of epithelial renewal in homeostasis and after injury. Two ISC populations have been described: 1) active ISC [aISC; highly proliferative; leucine-rich-repeat-containing G protein-coupled receptor 5 (LGR5+)-positive or sex-determining region Y-box 9 -antigen Ki67-positive (SOX9+Ki67+)] and 2) reserve ISC [rISC; less proliferative; homeodomain-only protein X positive (HOPX+)]. The contributions of these ISCs have been evaluated both in vivo and in vitro using a porcine model of mesenteric vascular occlusion to understand mechanisms that modulate ISC recovery responses following ischemic injury. In our previously published work, we observed that rISC conversion to an activated state was associated with decreased HOPX expression during in vitro recovery. In the present study, we wanted to evaluate the direct role of HOPX on cellular proliferation during recovery after injury. Our data demonstrated that during early in vivo recovery, injury-resistant HOPX+ cells maintain quiescence. Subsequent early regeneration within the intestinal crypt occurs around 2 days after injury, a period in which HOPX expression decreased. When HOPX was silenced in vitro, cellular proliferation of injured cells was promoted during recovery. This suggests that HOPX may serve a functional role in ISC-mediated regeneration after injury and could be a target to control ISC proliferation.
NEW & NOTEWORTHY This paper supports that rISCs are resistant to ischemic injury and likely an important source of cellular renewal following near-complete epithelial loss. Furthermore, we have evidence that HOPX controls ISC activity state and may be a critical signaling pathway during ISC-mediated repair. Finally, we use multiple novel methods to evaluate ISCs in a translationally relevant large animal model of severe intestinal injury and provide evidence for the potential role of rISCs as therapeutic targets.
INTRODUCTION
Intestinal ischemia is a life-threatening condition with mortality rates of >50% due to difficulties in early diagnosis, leading to delays in treatment and ultimate intestinal epithelial loss that results in sepsis and remote organ failure (1–4). The intestinal epithelium, made up of a single layer of cells, serves as a barrier to selectively allow for absorption of nutrients while playing a key role in the defense against intraluminal toxins and pathogens (5). Disruption of this barrier occurs during both intestinal ischemia and subsequent tissue reperfusion injury (6). As acute intestinal ischemia is rarely preventable, research efforts must be focused on the development of new therapeutics targeting the postischemic period (7) and more importantly, repair of the damaged mucosa and epithelial barrier. Intestinal epithelial stem cells (ISCs) are responsible for epithelial cell renewal during both homeostasis and after injury. Therefore, ISCs serve as a promising target to improve healing after intestinal injury (8–11).
Published literature supports two pools of intestinal crypt-based ISCs during homeostasis: 1) an actively cycling population (aISC), identified by leucine-rich repeat-containing G protein-coupled receptor 5 (LGR5) gene expression (12) or sex-determining region Y-box 9 and antigen Ki67 (SOX9+Ki67+) protein coexpression (13) that are responsible for self-renewal and daily cellular turnover (13, 14) and 2) a quiescent, slow cycling population (rISCs) that express HOPX (homeodomain-only protein homeobox), BMI1 (B-cell-specific moloney murine leukemia virus integration site-1), or TERT (telomerase reverse transcriptase) that can become activated after intestinal injury (15–18). The ISC compartment has been shown, in mouse models, to respond and to regenerate the intestinal epithelium following a variety of injuries including ionizing radiation, chemotherapeutics, and intestinal resection (19–22). More recent studies have highlighted plasticity within the crypt compartment and the ability of absorptive and secretory progenitor cells to revert to a stem-like state and also contribute to repair (23–26). At this time, the porcine models required to investigate crypt plasticity remain unavailable; however, as both progenitor cells and aISCs are lost following prolonged ischemic injury, it is likely that crypt regeneration depends on remaining rISCs (19). Despite this near complete epithelial loss after severe ischemia, we have previously shown that the small intestinal crypt compartment can regenerate in vivo by 3 days postinjury (DPI). In addition, HOPX+ ISCs are injury resistant and the likely source of new epithelium (13). Our in vitro recovery of crypt epithelium postischemic injury, which included the HOPX+ ISCs, further showed that resultant spheroids initially had increased HOPX gene expression and diminished growth when compared with uninjured cells (13). Subsequent increases in spheroid area corresponded to decreases in HOPX expression, suggesting that HOPX may serve as a functional, molecular switch to control proliferation of recovering ISCs.
HOPX, the smallest homeodomain protein, has been identified as a critical transcription factor in a variety of tissues (27). Direct modulation of HOPX expression has been shown to influence cellular proliferation and differentiation in several cell types including trophoblasts, keratinocytes, T cells, lung alveolar cells, and myocytes (28–32). In addition, HOPX has been labeled a tumor suppressor gene in multiple forms of cancer including both gastric and esophageal (33–35). In colorectal cancer, decreased HOPX expression has also been associated with increased cellular proliferation (36). These works highlight the influence of HOPX expression on cellular proliferation within numerous cell types. To date, HOPX has been frequently used as a rISC biomarker; however, its mechanistic role within these cells, particularly after severe ischemic injury, has yet to be evaluated.
In the present study, we utilized an established model of porcine mesenteric vascular occlusion (13, 37, 38) to evaluate epithelial recovery both in vivo and in vitro immediately following prolonged ischemic injury to both define how ISCs contribute to barrier repair and to understand ISC mechanisms that modulate the recovery response. Based on our previous work, we hypothesized that HOPX+ ISCs contribute actively to the repair process and that peak activation of these cells occurs within 2 days after injury. In addition, we wanted to evaluate whether HOPX expression levels played a direct role on cellular proliferation during recovery after injury; we hypothesized that HOPX silencing would result in increased enteroid growth and cellular proliferation. Our results supported our hypothesis that peak ISC activation occurs by 2 days after injury and further demonstrated that HOPX silencing led to increased cellular proliferation during recovery after ischemic injury. This suggests that HOPX may serve a more functional role in regeneration after injury and may be a potential target to control proliferation.
MATERIALS AND METHODS
Experimental Animals
All animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) of North Carolina State University. Yorkshire crossbred pigs (8- to 10-wk old) of either sex weighing between 15 and 25 kg were used.
Recombinant Adenovirus Construction and Generation
Adenoviral vectors engineered to coexpress a short hairpin RNA (shRNA) and enhanced green fluorescent protein (eGFP) reporter were created using gBlock Gene Fragments (IDT, Inc.) and the recently described Gateway-based (Invitrogen, Inc) pMVP system (39). In brief, gBlocks (IDT, Inc.) containing the human H1 RNA promoter, and one of two shRNA sequences (porcine HOPX-specific GGTCATTCCGATTCATAAA; NM_213792.2) or a scrambled control (Sc) sequence GCCTTACGACCAGTCGTATT), flanked by 5′ and 3′ attL1 and attR5 sequences, and plasmids containing the cytomegalovirus (CMV) promoter (attL5-CMV-attL4 plasmid IH401), the eGFP open reading frame (attR4-eGFP-attR3 plasmid IO001), and the human growth hormone polyadenylation signal (attL3-hGH-attL2 plasmid HB901) were recombined into the Gateway adenoviral acceptor plasmid pAd-PL-DEST, according to the manufacturer’s instructions (Invitrogen, Inc.). The Gateway recombination reactions were transformed into Escherichia coli stbl3 cells (Gibco) and grown overnight at 37°C on LBcarb plates. Colonies were screened in a QuantStudio6 real-time PCR machine (ABI, Inc) using SYBR green primers specific for the H1 RNA promoter (oligos TB1025/TB1026), CMV promoter (TB1507/TB1508), eGFP (TB916/TB917), and Ad5 hexon (TB10145/TB1046). Positive clones were grown in 5 mL of LBcarb for 15 h at 37°C, 225 rpm. Clones were then sequenced with CMV forward oligo TB1013 (GeneWiz). To produce recombinant, replication-deficient adenoviruses, the recombinant adenovirus genomes containing either HOPX- or Sc-shRNA sequences were transfected into HEK293 cells (Invitrogen) for adenovirus generation and propagation (Ad-siHOPX and Ad-Sc). Cells were collected just before lysis in a small volume of media, pelleted, and lysed for an approximate viral titer of 1010 pfu/mL for both Ad-siHOPX and Ad-Sc.
Operative Technique and Tissue Collection
Before surgery, pigs were fasted for 16–18 h. General anesthesia was induced using xylazine (1.5 mg/kg im) and ketamine (11–20 mg/kg im). Orotracheal intubation was performed, and pigs were maintained under general anesthesia with isoflurane (2%–5%) vaporized in 100% O2 until recovery. Before induction of intestinal ischemia, pigs were placed on heating pads and their temperature monitored. Lactated Ringer solution was administered intravenously through the auricular vein at a maintenance rate of 15 mL/kg/h. Anesthetic monitoring included pulse oximetry, electrocardiography, and indirect blood pressure measurements. Pigs were placed in dorsal recumbency and the abdomen was accessed through a 12-cm ventral midline incision centered at the umbilicus. The jejunum was identified 40 cm oral to the ileocecal junction. In terminal surgeries of ischemia without recovery, 10-cm-long loops of jejunum were delineated by circumferentially ligating the bowel with suture. Mesenteric vasculature to the loop was occluded by suture ligation for 3 and 4 h, and animals were subsequently euthanized. In animals recovered after ischemia, intestinal loops were atraumatically delineated using longitudinal doyen intestinal forceps. The local mesenteric vasculature was clamped using Hopkin’s bulldog clamps for 3 and 4 h after which time the clamps were removed. An additional segment was identified at the start of surgery to serve as an internal normal control. To decrease adhesion formation in recovery animals, carboxymethylcellulose (CMC) was instilled into the abdomen after clamp placement and again before closure (40, 41). After clamp removal was completed, an abdominal lavage was performed using sterile saline. The abdomen was then closed in three layers and animals were recovered for 1 (18–20 h), 2 (40–44 h), or 3 (66–70 h) days after injury (DPI). Postoperatively, pigs were maintained on a soft, canned food diet (Hills Pet Nutrition, Topeka, KS) and were administered intramuscular buprenorphine (0.02–0.05 mg/kg) every 8–12 h as needed for pain. Before being euthanized, animals were reanesthetized using xylazine and ketamine. After animals were euthanized with pentobarbital (85–100 mg/kg iv), intestinal tissue was immediately collected. Tissues were rinsed with 1X phosphate-buffered saline (PBS) and either opened longitudinally along the antimesenteric boarder for histomorphology or sectioned into smaller pieces for epithelial crypt dissociation and enteroid culture.
Tissue Histomorphometric Evaluation
For histomorphological analysis, tissue was fixed in 10% neutral buffered formalin, embedded in paraffin, and sectioned (∼5–8 μm thickness). Slides were stained with hematoxylin and eosin to visualize tissue architecture. A small subset of slides at each time point after injury were examined unblinded by a boarded pathologist (JL) to define each time point of recovery and create an objective scoring system. The different categories and criteria for assessment are shown in Table 1. Slides were then blinded and evaluated by the pathologist. Peracute injury was defined by loss of surface epithelium and contracted villi, but relatively normal crypts characterized by well-differentiated epithelium with tall columnar cells, presence of brush border, basal polarity, rare mitotic figures, and presence of goblet cell differentiation (Fig. 1A). Acute injury was defined by loss of surface epithelium in >75% of the section, absent villi, and abnormal crypts characterized by crypt dilation, loss of crypt epithelium in >75% of the section with remaining cells vacuolated or attenuated, and <10% of crypt epithelium showing signs of early regeneration (Fig. 1A). Early regeneration was defined by loss of surface epithelium in >60% of the section, loss of villi, and early regenerative crypt epithelium characterized by cytoplasmic basophilia, cuboidal to low columnar epithelium, lack of basal polarity, ill-defined brush border, and prominent mitoses. In early regeneration, less than 10% of the crypt epithelium had characteristics of differentiation (Fig. 1A). Late regeneration was defined by an intact surface epithelium over 75% of the section, blunted to absent villi, and >75% of crypts lined by epithelium with characteristics of early to late differentiation. Differentiation within the crypt epithelium was characterized by crypt elongation, low to tall columnar epithelial cells, presence of brush border, restoration of basal polarity, and goblet cell differentiation (Fig. 1A).
Table 1.
Criteria for histological scoring of intestinal biopsies after ischemic injury
| Evidence Regeneration Crypt Epithelium |
Evidence Differentiation Crypt Epithelium |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Classification | Surface Epithelium (% covered) | Villi (Contracted/Absent/Normal) | Percentage of Crypts Lined by Epithelium | Normal Crypts Epithelial Cells | Basophilic Cells | Loss Polarity | Cuboidal | Mitoses | Loss of Basophilia | Basal Polarity | Tall Columnar |
| Normal | 90%–100% | Normal | 95%–100% | Yes | |||||||
| Peracute | <25% | Contracted | >75% | Yes | |||||||
| Acute | <25% | Absent | <25% | No | No | No | No | No | No | No | No |
| Early regeneration | <40% | Absent | 25%–80% | No | Yes | Yes | Yes | Prominent | No/rare | No/rare | No/rare |
| Late regeneration | >25% | Absent/blunted | >75% | No | No/rare | No/rare | No/rare | Rare | Yes | Yes | Yes |
Figure 1.

Histomorphometric analysis of recovering small intestine after 3 h of ischemic injury demonstrates that early epithelial regeneration begins 2 DPI. A: immediately after injury, there is significant surface epithelial loss and contraction of villi to their base (0 DPI). By 1 DPI, the villi are absent and there is loss of crypt epithelial cells. By 2 DPI, the crypts are lined by plump, cuboidal epithelial cells with prominent mitoses. Epithelial differentiation is seen by 3 DPI with tall columnar cells lining crypts, basal polarity, and evidence of crypt elongation. Scale bar inset 20 µm. Scale bar panel 100 µm. B: stack graph defining the histological categories of epithelial recovery and demonstrating that early regeneration occurs at 2 DPI; n = 3–6 pigs. DPI, days postinjury.
Fluorescent Multiplex Immunohistochemistry with Tyramide Signal Amplification
For immunofluorescence analysis, rinsed tissue was fixed in 4% paraformaldehyde (PFA) solution for 16–18 h at 4°C. The tissue was transferred to 30% sucrose solution for at least 24 h at 4°C, embedded in optimal cutting temperature (OCT) media, then frozen and subsequently sectioned at 5–8 μm thickness using a cryotome. Before being stained, sections were washed with PBS to remove OCT. When necessary, heat-induced epitope retrieval (HIER) was performed by placing slides into reveal decloaker solution (Biocare Medical, Concord, CA) for 30 s at 120°C and then 90°C for 10 s, in a pressure cooker. Tissue permeabilization was performed with PBS-0.3% Triton X-100. Antibodies were diluted in antibody diluent (SignalStain, Cell Signaling Technology, Danvers, MA), applied to tissue and incubated for 1 h at room temperature in a humidified chamber. Dilutions for functional antibodies were as follows: αß-catenin (mouse, 1:200, Cell Signaling Technology), αSOX9 (rabbit, 1:2,000, Chemicon/Millipore, Temecula, CA), αKi67 (mouse, 1:1,600, Dako, Santa Clara, CA), and αHOPX (rabbit, 1:500, Santa Cruz Biotechnology, Santa Cruz, CA). After incubation with the first antibody, slides were washed in TBS-T and boosted with a HRP Signal Stain Boost (host specific, rabbit or mouse) for 30 min. A specific fluorophore-conjugated tyramide signal amplification color reagent (Cyanine 3 for HOPX/SOX9, Fluorescein green (GFP) for Ki67, Cyanine 5 for β-catenin; PerkinElmer) was then applied for 10 min. To utilize multiple antibodies raised in the same host, slides were boiled in 10 mM sodium citrate buffer (SignalStain Citrate Unmasking Solution, Cell Signaling Technology), held at a subboiling temperature (95°) for 10 min and then cooled. After this stripping procedure, a different primary antibody and subsequent color reaction was applied. After all reactions, slides were counterstained with bisBenzimide H 33258 nuclear stain (1:1,000, Sigma-Aldrich, St. Louis, MO) and coverslipped (Hydromount; National Diagnostics). Background staining was negligible as determined by nonspecific IgG staining.
For each slide evaluated, well-oriented crypts were used to obtain cell counts. A well-oriented crypt was identified as one with a crypt base in close apposition to the muscularis mucosa and that extended and opened fully into the gut lumen. At least 10 well-oriented crypts for each protein biomarker were imaged, positive cells per crypt counted, and the results averaged for each time point (13, 20, 42).
Image Acquisition
Images were captured on an inverted fluorescence microscope (Olympus IX83, Tokyo, Japan) fitted with a monochrome digital camera (ORCA-flash 4.0, Hamamatsu, Japan) and color camera (DP26, Olympus). The objective lenses used were ×10, ×20, and ×40 with numerical apertures of 0.3, 0.45 and 0.6, respectively (LUC Plan FLN, Olympus).
Intestinal Epithelial Crypt Isolation, Adenovirus Transduction, and Culture
The excised jejunum was washed in cold phosphate-buffered saline solution (PBS) and opened longitudinally. Tissue was cut into small 0.5-cm pieces and incubated for 30 min in a 50-mL conical tube with PBS containing 30 mM ethylenediaminetetraacetic acid (EDTA), 10 mM Y-27632 (Selleck Chemicals, Houston, TX), 1 mM DTT (Sigma-Aldrich), 1X antibiotic-antimycotic (Life Technologies Corporation, Carlsbad, CA), and Primocin (InvivoGen, San Diego, CA). The conical tube was shaken every 5–10 min and kept on ice. Tissue pieces were transferred into a 37°C prewarmed PBS solution containing 30 mM EDTA and 10 mM Y-27632. The tissue was incubated in this solution at 37°C for 10 min and shaken to help mobilize the crypt/villi units. After this step, tissues were placed in ice-cold PBS wash and shaken for 1–2 min. Tissue was transferred into additional washes and shaken until crypt units were seen with minimal background debris. After the final washes, the remnant intestine was removed from the solution. Crypts were aliquoted into RNAse-free microtubes for viral transduction and culture.
Appropriate volumes of isolated crypts suspended in PBS from control (uninjured) and 3-h ischemic-injured pigs were separately pelleted by centrifugation (300 g for 5 min at 4°C) to achieve 50 crypts per 50-µL Matrigel (BD Bioscience, San Jose, CA). Similar to the previously described “mix and seed” approach (43), crypt pellets were resuspended in the appropriate volume of ice-chilled Matrigel. Then either Ad-Sc or Ad-siHOPX virus, diluted 1:10 in 1% bovine serum albumin (BSA, Sigma-Aldrich) in PBS, was added to the suspended crypts at 500 multiplicities of infection (MOI) with gentle mixing. Matrigel containing crypts and virus was immediately plated on 24-well plates in 50-µL patties, allowed to set for 30 min at 37°C, then overlayed with 250 µL of Advanced DMEM/F12 containing the supplements 1X N-2 supplement (Life Technologies Corporation, Carlsbad, CA), 1X B-27 supplement minus vitamin A (Life Technologies Corporation), 1X Glutamax (Life Technologies Corporation), 100 mg/mL penicillin-streptomycin, and 1 mM HEPES buffer (Life Technologies Corporation). An additional 250 µL of WRN conditioned media [Wnt-3a (conditioned media, 50% final volume), R-spondin 3 (conditioned medium, 50% final volume), Noggin (conditioned media, 50% final volume)] was also added to each well. Conditioned media were produced from L-WRN cells derived by transfecting L-Wnt3A (ATCC CRL-2647) with an R-spondin 3 and noggin coexpressing vector. Stable clones were selected in medium containing G418 and Hygromycin B. Media were also supplemented with growth factors [50 ng/mL recombinant human EGF (Life Technologies Corporation), 10 mM nicotinamide (Sigma-Aldrich), 10 nM gastrin (Sigma-Aldrich), 10 mM Y-27632, 10 mM SB202190 (Sigma-Aldrich), 500 nM LY2157299 (Selleck Chemicals), and 2.5 μM glycogen synthase kinase 3 inhibitor (GSK3i, CHIR99021)]. Control and 3-h ischemic-injured crypts were also grown without virus and with 1% BSA to serve as negative and vehicle controls, respectively. Growth factors and fresh conditioned media were added 48 h after plating and subsequent 48-h intervals. Successfully transduced resultant spheroid/enteroids, conferred by GFP expression at 24 h in culture, were imaged daily out to 72 h for area measurements. A MOI of 500 resulted in 100% of plated crypts having at least one transduced cell (GFP+, data not shown).
Epithelial Single-Cell Isolation
To isolate epithelial cells (EpCAM+) and ensure collection of ISCs from tissue recovering 1–2 DPI, the entire mucosa was fully dissociated into a single-cell suspension for fluorescence-activated cell sorting (FACS) (44). Full-thickness sections from control and 3-h ischemic-injured jejunum with in vivo recovery were excised and cut into 2-cm square pieces. Each piece was washed in cold 1X Hank’s balanced salt solution wash (HBSS without Ca and Mg, Gibco; 10 mM HEPES; and 0.47% sodium bicarbonate, Corning). For each piece, the serosa was dissected and the remaining mucosa minced, placed in predigest buffer (HBSS wash, 5 mM EDTA, 1.25% BSA, 1 mM DTT, and 10 mM Y-27632), and incubated for 10 min at 37°C, 250 rpm. Tissue pieces were filtered using a 110-µm mesh filter and the cellular filtrate collected. Tissue pieces were then rinsed in HBSS wash with 1.25% BSA, transferred to a gentleMACS C Tube (Miltenyi Biotec) with 0.78 WU Liberase TM (Millipore Sigma), 250 µg DNase (Roche), 3.6 U Dispase (Gibco), and R1.25 (RPMI 1640 + l-glutamine media (Gibco) with 1.25% BSA), and then incubated for 30 min, 37°C, 225 rpm for enzymatic digestion. Then, the C Tube containing tissue was mounted on a gentleMACs dissociator (Miltenyi Biotec) and preset program m_intestine_01 run for further mechanical dissociation. Resultant dissociated single cells were combined with collected cells from predigest A step, filtered, and red blood cells lysed by 1X RBC Lysis Buffer (Invitrogen). Single cells were suspended in flow buffer (PBS with 2% fetal bovine serum and 2 mM EDTA) and counted by trypan blue (Gibco) exclusion in a hemocytometer. Single cells were then incubated with α-CD 326 (EpCAM) (Biolegend, Cat. No. 118211, Alexa Fluor-647 conjugated; 1:200 per 106 cells in 100 µL flow buffer) for 15 min on ice. After being washed, cells were incubated with 7AAD live/dead stain (Novus) and FACS sorted on a Beckman Coulter MoFlo XDP for EpCAM+ live single cells (45–47). EpCAM+ single cells were collected in culture media for three-dimensional (3–D) culture. Approximately 15,000 cells per 50 µL Matrigel were plated on 24-well plates and overlaid with WRN conditioned media, as earlier described. Resultant spheroid/enteroids were imaged and counted daily out to 168 h for area measurements, plating efficiency, and ISC expansion potential (bud count) (48).
Spheroid/Enteroid Area Measurement
Spheroid/enteroid area measurement was obtained after image acquisition using the Freehand Polygon Selection Tool and Measurement Function (NIH ImageJ). Ten to twenty spheroids/enteroids per time point per animal were measured and averaged.
Spheroid/Enteroid Isolation, RNA Extraction, and cDNA Conversion
Adenovirus-transduced spheroids/enteroids were isolated from Matrigel using Corning Cell Recovery Solution (REF 354253), as per manufacturer’s instructions at 24, 48, and 72 h postplating. Spheroids/enteroids were pelleted and snap frozen in liquid nitrogen and stored at −80°C. RNA was extracted using the Ambion PureLink RNA Mini Kit (Thermo Fisher Scientific). Yield and quality control of the extracts were determined by an Agilent 2100 BioAnalyzer performed by the North Carolina State University Genomic Sciences Laboratory Core using Agilent Eukaryote Total Pico Series II chips (Agilent Technologies, Santa Clara, CA). RNA was used if integrity score was >6 and RNA concentration >100 pg/µL. RNA (1 µg) was converted to cDNA using the iScript cDNA synthesis kit (Bio-Rad). Expression of HOPX was determined by quantitative real-time PCR (QuantStudio 6 Flex; Applied Biosystems) on cDNA samples using iTaq Universal SYBR green Supermix (Bio-Rad). The ΔΔCt method was used to measure relative changes in gene expression. Samples were tested in triplicate.
Primer Sequences
Primer sequences are presented in Table 2. Additional primers, including HOPX, ( GGAGGAGACCCAGAAATGGTT and TCTTGGTGGAAGGAAGCAGC) have been previously published (13).
Table 2.
Primer sequences
| Pligo | Forward/Reverse | Target | Sequence |
|---|---|---|---|
| TB916 | For | EGFP | TGACCCTGAAGTTCATCTGCACCA |
| TB917 | Rev | TCTTGTAGTTGCCGTCGTCCTTGA | |
| TB1025 | For | H1 RNA promoter | TCATCAACCCGCTCCAAGGAAT |
| TB1026 | Rev | CCCAGAACACATAGCGACATGCAA | |
| TB1045 | For | Ad5 Hexon | TTGGCGCATCCCATTCTCCAGTAA |
| TB1046 | Rev | ATAAAGAAGGGTGGGCTCGTCCAT | |
| TB1507 | For | CMV promoter (proximal) | CCCACTTGGCAGTACATCAA |
| TB1508 | Rev | CCAAGTAGGAAAGTCCCGTAAG | |
| TB1013 | For | CMV promoter | TTGGCTCATGTCCAATATGACCGC |
| TB1014 | Rev | GGCGGGCCATTTACCGTAAGTTAT |
Spheroid/Enteroid Whole Mount Immunofluorescence
Before fixation, spheroids were incubated with 5-ethynyl-2′-deoxyuridine (EdU Click-iT, Thermo Fisher Scientific) for 1 h before being fixated with 4% PFA. Briefly, spheroids were fixed in 4% PFA at room temperature for 30 min, washed in PBS, and incubated with 0.1% Triton X-100 at room temperature for 30 min to permeabilize. Cells were then incubated in 5% BSA blocking medium at room temperature for 60 min. After a wash, the Click-iT EdU Imaging Kit (Thermo Fisher Scientific) was used to label proliferating cells. Briefly, the cells were incubated in the Click-iT reaction cocktail for 30 min at room temperature. After a 3% BSA wash, cells were stained with 1X Hoescht 33342 for 30 min. At least 15–20 spheroids were counted per pig and evaluated.
Statistics
In general, statistical analyses were performed using Prism 9.0.0 (GraphPad Software, La Jolla, CA) software, unless noted. Outliers were identified using the ROUT method. A Shapiro–Wilk normality test was performed on raw data. For simple comparisons, a Mann–Whitney test was performed. One-way ANOVA was used to evaluate normally distributed data. The Kruskal–Wallis test was performed when any data were either not normally distributed or when sample size was too small to determine normality. A two-way ANOVA was used to evaluate the impact of both animal and recovery time point on number of ISCs during immunofluorescent imaging and the number of buds resultant from single-cell culture. For analyses with significance detected, Tukey’s test, Sidak’s test, or Dunn’s test were utilized for post hoc pairwise multiple comparisons. The α-level for statistical significance was defined as P ≤ 0.05.
To examine the change over time for spheroid/enteroid area from both FACS-sorted EpCAM+ single cells and the virus-transduced crypts, linear mixed models were fit. Model selection used Satterthwaite’s method for degrees of freedom. The full model consisted of the log10-transformed area predicted by the fixed effects of time, treatment, and time squared. Two-way interactions were allowed. Pig was included as a blocking factor and a potential interaction with time. The random effect included was a random intercept for image within pig. After fit, residuals were examined for constant variance, and quantile-quantile plots were generated to check the normality of errors (R version 4.0.2 with lme4, lmerTest, and ggplot2 packages).
RESULTS
After Prolonged Ischemic Injury, Crypt-Based Epithelial Cells Recover, with the Earliest Signs of Histologic Regeneration Occurring 2 DPI
Previous studies from our laboratory have shown histological evidence of crypt disruption occurring at durations of acute ischemia beyond 3 h. This disruption is characterized by dramatic loss of both aISCs and transit-amplifying cells but preservation of the rISCs (13). However, the dynamics of crypt epithelial regeneration and the specific contributions of each ISC population to recovery remain unknown. To better define when epithelial regeneration is initiated after prolonged ischemic injury, we first established a histologic time course of recovery using a new scoring system. This system was developed based upon the expected observable histologic response of the intestinal mucosa following ischemic injury (49). Among these expectations, early regenerating crypt epithelium is characterized by epithelial cells with deeply basophilic cytoplasm, loss of regular basal nuclear polarity, low columnar to cuboidal shape with an ill-defined brush border, and high mitotic activity. After early regeneration, crypt epithelial cell differentiation begins, marked by the presence of goblet cells and tall columnar epithelial cells with basal orientation of nuclei and well-defined brush border. Based on our previous work, cell differentiation, crypt expansion, and evidence of epithelial restitution by 3 DPI (13, 37), we postulated that early epithelial regeneration and subsequent stem cell activation occurred on 1 or 2 DPI.
Histological analysis of ischemic (0 DPI) and recovering jejunum revealed that within the first day following the onset of ischemia (1 DPI), injury appeared to be ongoing, evidenced by severe crypt disruption, loss of crypt epithelial cells, and villous contraction followed by complete loss of villi (Fig. 1A). Surviving epithelial cells within the crypts appear flattened and attenuated with loss of the normal brush border. Signs of early epithelial regeneration including presence of plump, cuboidal, deeply basophilic epithelial cells with prominent cellular mitoses were first noted at 2 DPI. The consistency of these early regeneration histological markers at 2 DPI were confirmed by masked evaluation of recovering tissue sections (Fig. 1B). Similar to previous work, crypt expansion, epithelial differentiation, and epithelial restitution, signifying late regeneration, were present by 3 DPI. Once a time course of recovery was established and signs of early epithelial regeneration were identified before 3 DPI, we next wanted to investigate the contribution of ISCs to this early regeneration. Based on our previous work, we postulated that rISCs (HOPX+) were likely responsible for this repair.
Intestinal Stem Cell Quiescence Dominates after Prolonged Ischemic Injury with a Shift to Cellular Proliferation Occurring between 1 and 3 DPI as HOPX+ Cells Steadily Decrease
To investigate ISC population numbers in repair after severe ischemic injury, immunofluorescent protein biomarkers were used to first identify recovering epithelial cells (B-catenin+) and subsequently characterize them as aISCs (SOX9+Ki67+), rISCs (HOPX+), and general proliferative cells (Ki67+) (Fig. 2A). To first identify proliferating cells throughout recovery, total Ki67+ cells per crypt were counted at 0–3 DPI (Fig. 2B). The overall number of proliferating cells significantly increased between 1 and 3 DPI (P = <0.0001), which correlated to the increased crypt expansion seen histologically (Fig. 1).
Figure 2.

After severe ischemic injury, HOPX+ cell numbers are initially enriched and begin to decrease as active intestinal epithelial stem cells (ISCs) recover and proliferate. A: immunofluorescent images of aISC (SOX9+Ki67+, yellow) and rISC (HOPX+, red) cells after severe ischemic injury and during recovery. Epithelium marked by B-catenin (purple). Nuclei were stained with bisbenzimide H 33258 (blue). Dotted line outlines crypts extending down to the muscularis mucosa. Scale bar 20 µm. B: total number of proliferating cells (Ki67+) decrease after ischemic injury and subsequent tissue reperfusion and begin to recover between 1 and 3 DPI. C: total number of active and reserve ISCs postinjury demonstrate that HOPX+ rISCs are injury resistant and numbers are enriched after injury, whereas SOX9+Ki67+ aISCs are lost and recover between 1 and 2 DPI. D: HOPX+ rISCs also colocalize with Ki67 at 1 and 2 DPI. Values are means ± SD. Analysis by two-way ANOVA with post hoc Tukey’s test, *P < 0.05 **P < 0.01 ***P < 0.001 ****P < 0.0001, n = 3–5 pigs per recovery time point, 10 crypts were counted per animal per time point.
We have previously reported an initial loss of SOX9+Ki67+ aISCs due to apoptosis immediately after severe ischemic injury (13). Further investigation into the specific dynamics of aISC and rISC population numbers in recovery confirmed a decrease in aISC numbers at 0 DPI (P = 0.0046) (Fig. 2C). Numbers of aISCs began to recover at 1 DPI but remained significantly decreased compared with control (P = 0.014). In contrast, the relative number of HOPX+ rISCs per intestinal crypt remained unchanged at 0 DPI and then significantly increased to reach a peak 1 DPI (P ≤ 0.0001). A significant number of these cells were also colocalized with Ki67 at 1 and 2 DPI (Fig. 2D). Subsequently, as HOPX+ cell number decreased at 2 DPI, the number of SOX9+Ki67+ cells within the recovering crypt increased, marking a shift in the ISCs contributing to repair.
To further examine epithelial regenerative capacity after injury, we next sought to isolate the epithelial cells that remained from 1 or 2 DPI mucosal tissue, likely enriched in rISCs or aISCs, respectively, to monitor their growth and proliferation potential in vitro. Traditional crypt isolation techniques were not successful in isolating epithelial cells from the actively recovering mucosa; therefore, tissues were chemically and mechanically digested into single cells, stained with a pan-epithelial marker (EpCAM) (44) and fluorescence-activated cell sorted (FACS) for culture.
Epithelial Cells Isolated from 2 DPI Demonstrate Superior Growth When Compared with Cells Isolated from Control and 1 DPI Tissue
Epithelial (EpCAM+) cells were successfully isolated from both control and recovering tissue and grown in culture out to 168 h (Fig. 3, A and B). With a demonstrated loss of villus epithelium and aISCs following severe ischemic injury [Fig. 1A, (13)], the EpCAM+ epithelial cells isolated from 1 DPI tissue were likely enriched in injury-resistant cells including rISCs (Fig. 2). Cells successfully plated from this group (1 DPI) grew 22.0% slower compared with control (P = 0.00639) supporting a quiescent phenotype, whereas overtime, cells isolated from 2 DPI grew 8.6% faster each day than control (P = 0.000122) suggesting increased proliferation at this time. To determine differences between groups at specific time points, the resultant spheroid and/or enteroid log-transformed areas were compared between recovery groups at 24, 48, 96, and 144 h in culture (Fig. 3C). Spheroids are the developmental structures that first form during culture before morphological evidence of more complex enteroid growth such as budding. Compared with uninjured control, spheroids from 1 DPI cells, while initially larger at 24 h (P = 0.0102, 51.9% larger), were no different at 48 h (P = 0.1526). At 96 h in culture, 1 DPI spheroids/enteroids were significantly smaller than control (P = 0.005629, 41.9% smaller). In contrast, spheroids from 2 DPI cells demonstrated significantly increased size; they started larger than control at 24 h and continued to be significantly larger at 48, 96, and 144 h (P = 0.0026, 46.3% larger; P < 0.0001, 106.3% larger; P = 6.411 × 10−11, 159.8% larger; P = 2.198 × 10−8, 204.2% larger, respectively). Furthermore, spheroids/enteroids from 2 DPI cells were also significantly larger than those from 1 DPI at all time points including 48, 96, and 144 h (P = 4.606 × 10−9, P = 8.058 × 1 0–11, and P = 1.485 × 10−6, respectively).
Figure 3.

Two days postinjury (DPI) in vivo recovered epithelial cells grown in culture demonstrate increased size and proliferation compared with control and 1 DPI cells. A: representative gating strategy for FACS-sorted cells isolated from control, 1 DPI, and 2 DPI jejunum based on live/dead (7 AAD) and EpCAM-Alexa647. B: representative time course images of enteroid growth in culture from FACS-sorted EpCAM+ cells from control, 1 DPI, and 2 DPI out to 168 h in culture. Scale bar 20 µm. C: box plots of log transformed daily enteroid area measurements from enteroids in B, showing increased growth of 2 DPI enteroids compared with control and 1 DPI, n = 4 pigs for each recovery day, 15,000 cells plated per 50 µL Matrigel, 15–20 enteroids measured per pig. D: means ± SD of bud counts per enteroid, showing significantly increased proliferative capacity for 2 DPI enteroids at 96 and 144 h in culture compared with control and 1 DPI. Analysis by two-way ANOVA with post hoc Tukey’s test, **P < 0.01, ***P < 0.001, ****P < 0.0001. Data collected from same enteroids in C. E: culture overview images at 168 h, demonstrating increased size and bud count for 2 DPI enteroids. Scale bar 100 µm. FACS, fluorescence-activated cell sorting.
To further quantify enteroid proliferation, the number of bud units per enteroid were counted at 96 and 144 h in culture. Resultant enteroids grown from 2 DPI cells had significantly more buds per enteroid than both control (P = 0.004) and 1 DPI (P < 0.0001) at 96 h (Fig. 3, D and E). By 144 h, 2 DPI enteroids remained more proliferative with increased bud counts when compared with control (P = 0.0005) and 1 DPI (P = 0.0002). One DPI spheroid/enteroids remained small with few buds, suggesting both that signals for cellular activation and proliferation occur between 1 and 2 DPI and that these early cultures are enriched in quiescent rISCs. Despite the marked changes in bud count and area, there were no significant differences in growth efficiency throughout the 7 days (Supplemental Fig. S1, see https://figshare.com/articles/figure/Supplemental_Figure_1/15145038).
Our previous work demonstrated that resultant spheroid/enteroid cultures derived from 0 DPI ischemic-injured crypts had significantly higher levels of HOPX gene expression; this expression remained increased until 120 h. At that point, a decrease in HOPX gene expression correlated with a significant increase in spheroid size (13). In the current study, we both confirmed preservation of HOPX+ cells at 0 DPI and demonstrated a relative increase in the numbers of HOPX+ cells at 1 DPI (Fig. 2). The same 1 DPI cells plated in culture had decreased growth and proliferative potential (Fig. 3), similar to our previous in vitro recovery findings. However, by 2 DPI, HOPX+ cell numbers decreased (Fig. 2), and growth in culture of 2 DPI cells was significantly increased (Fig. 3). Therefore, given the evidence that decreased HOPX gene and protein expression was associated with increased enteroid growth, we hypothesized that HOPX plays a functional role in the modulation of cellular proliferation. To investigate this hypothesis, an adenovirus coexpressing a HOPX-specific short hairpin RNA (shRNA) and an EGFP reporter (Ad-siHOPX) was developed and used to transduce isolated crypt epithelium.
Decreased HOPX Leads to Increased Spheroid Growth and Cellular Proliferation of Ischemic-Injured Epithelial Crypts
To validate that the adenoviral transduction resulted in silencing of HOPX gene expression, isolated control crypts were first transduced with either Ad-siHOPX or a control adenovirus coexpressing a scrambled shRNA and the EGFP reporter (Ad-Sc), as described. After transduction, resultant cultures were collected at 48 h for RNA isolation and qRT-PCR. HOPX gene expression was significantly decreased in cultures transduced with Ad-siHOPX when compared with Ad-Sc (P = 0.05, Fig. 4A). To determine the functional role of HOPX and assess whether silencing HOPX impacted the growth of ischemic-injured ISCs, crypts isolated immediately after 3 h of ischemic injury (3hrI) were transduced with either Ad-siHOPX or Ad-Sc. Successful transduction was confirmed by the presence of GFP-positive cells within plated crypts at 24 h (Fig. 4B). Area measurements of resultant spheroids in culture were performed every 24 h and the log-transformed values compared between groups (Fig. 4C). At 24 h, spheroids grown from 3hrI crypts transduced with the Ad-siHOPX were significantly larger than both Ad-Sc transduced (36% larger, P = 0.000461) and no virus spheroids (41.6% larger, P = 0.0000533); no virus spheroids were no different from Ad-Sc spheroids (P = 0.498305). At 48 h, Ad-siHOPX spheroids from 3hrI crypts remained larger than both Ad-Sc (29% larger, P = 0.00834) and no virus spheroids (43.9% larger, P = 0.000315); no virus spheroids remained no different from Ad-Sc at 48 h (P = 0.27073). At 72 h, Ad-siHOPX spheroids remained larger than the Ad-Sc group (P = 0.0206) but not the no virus control group (P = 0.2670).
Figure 4.

Ad-siHOPX silences HOPX in culture and leads to increased size of spheroids grown from 3-h ischemic-injured (3hrI) crypts. A: HOPX expression is significantly decreased in control crypts transduced with Ad-siHOPX compared with Ad-Sc at 48 h in culture. Fold changes were calculated relative to Ad-Sc samples and GAPDH as housekeeping gene. Statistics analyzed by two-way ANOVA with *P = 0.05, n = 8. B: representative images of spheroids at 24, 48, and 72 h in culture, grown from no virus, Ad-Sc, and Ad-siHOPX transduced 3hrI crypts. Transduction confirmed by GFP expression (green). Scale bar 20 µm. C: box plots of log transformed daily spheroid area measurements from no virus (green), Ad-Sc (blue), and Ad-siHOPX (red) transduced 3hrI and control crypts, *P < 0.05 **P < 0.01 ***P < 0.001, n = 5 pigs, 15–20 spheroid/enteroids per condition measured.
To determine whether silencing HOPX impacted growth in homeostasis, uninjured control crypts were transduced with Ad-siHOPX and Ad-Sc and evaluated. No significant differences in resultant spheroid area were found between the groups at any time point (Fig. 4D). Although Ad-siHOPX transduction resulted in significant reduction of HOPX expression in uninjured control crypts (Fig. 4A), it is likely that aISC and progenitor cell activity present in all control crypt conditions mitigated any potential spheroid growth effects due to silencing HOPX. Conversely, ischemic injury leads to a loss of crypt-based aISC and progenitor cells. The remaining rISCs transduced with Ad-siHOPX lead to increased spheroid growth rate (Fig. 4C). These data suggest that HOPX is paramount during recovery after injury but not homeostasis.
Based on these results, we postulated that the increased spheroid area of previously injured and HOPX-silenced ISCs was due to increased cellular proliferation. To test this hypothesis, we performed whole mount staining of spheroids grown from 3hrI crypts for DAPI and EdU at 48 h in culture (Fig. 5). Ad-siHOPX spheroids had significantly more DAPI+ cells compared with both Ad-Sc (P = 0.0013) and no virus controls (P = 0.0042), which confirmed that the increased spheroid area (Fig. 4C) was due to an increase in overall cellular number (Fig. 5B). Resultant Ad-siHOPX spheroids were more proliferative when compared with Ad-Sc with significantly more EdU+ cells/spheroid (P = 0.0052, Fig. 5C). There was no difference in the number of EdU+ cells/spheroid between Ad-siHOPX and no virus control (P = 0.2975). Despite an increased total number of EdU+ cells in the Ad-siHOPX group when compared with Ad-Sc, there was no increase in the number of virus-infected, proliferative cells (GFP+EdU+) between groups (P = 0.333, Fig. 5D). It is likely that some of the EdU+ cells seen within the Ad-siHOPX spheroids were replicating daughter cells resulting from the original transduction that would not express GFP. To assess the influence of overall size and total cell count on the number of EdU+ cells/spheroid, the percentage of EdU+ cells relative to the number of total DAPI+ cells/spheroid was calculated and compared between groups (Fig. 5E). Ad-siHOPX spheroids had a higher percentage of EdU+ cells/spheroid when compared with Ad-Sc (P = 0.0369). No virus spheroids had a significantly higher percentage of EdU+ cells relative to DAPI when compared with Ad-Sc (P = 0.0022) but not the Ad-siHOPX group (P = 0.2179, Fig. 5E). Taken together, the data presented in Fig. 5 support that shRNA-mediated suppression of HOPX results in increased cellular proliferation as evidenced by an increased number of total DAPI+ cells/spheroid and EdU+ cells/spheroid when compared with the scrambled shRNA control.
Figure 5.

Silencing HOPX in 3-h ischemic injured (3hrI) crypt epithelium leads to increased overall cell and proliferating cell counts at 48 h in culture. A: representative bright field (BF) and immunofluorescent images of 48 h spheroids from no virus, Ad-Sc, and Ad-siHOPX-transduced 3hrI crypts. Proliferating cells were identified by EdU (red) and Ad-Sc and Ad-siHOPX cells identified by their GFP reporter (green). Nuclei were stained with Hoescht 33342 (blue). Scale bar 20 µm. B: means and standard deviations of total cell count per spheroid, showing significantly increased total cell counts for Ad-siHOPX compared with both Ad-Sc and no virus. C: means and standard deviations of total EdU+ cell counts per spheroid, showing significantly increased EdU+ cells in Ad-siHOPX compared with Ad-Sc. D: means and standard deviations of total EdU+GFP+ colocalized cells per spheroid, showing no difference between Ad-siHOPX and Ad-Sc. No virus spheroids are not included as they did not contain a GFP reporter. E: means and standard deviations of the percentage of EdU+ cells per total cells per spheroid, showing that with Ad-siHOPX leads to significantly increased percentage of proliferating cells. Analysis by two-way ANOVA with post hoc Tukey’s test, *P < 0.05 **P < 0.01 ***P < 0.001, n = 1 or 2 pigs, 15–28 enteroids counted per pig.
DISCUSSION
After prolonged ischemic injury, the epithelium has a remarkable healing capacity with evidence of epithelial restitution and crypt expansion by 3 DPI (Fig. 1). The driving force behind this restitution and repair is the ISC compartment, composed of primarily injury-resistant HOPX+ rISCs. Based on our data shown, HOPX+ rISCs survive after severe ischemia and are enriched within the surviving crypt as the remainder of the epithelium including Ki67+ proliferating cells, SOX9+Ki67+ aISCs and early progenitors are lost (Fig. 2). As the intestine recovers, the population numbers of each ISC pool change, marked by a decrease in HOPX+ rISCs and a subsequent rebound and increase in SOX9+Ki67+ cells. These cells are likely aISCs or early progenitor cells derived from the injury-resistant rISCs. More importantly, with this decline in crypt HOPX+ cells, there is an increase in the number of Ki67+ cells. These findings suggest a role of HOPX to serve as a switch to trigger cellular proliferation.
At this time, the inability to lineage trace porcine epithelial cells in vivo precludes investigations into the specific contributions of each ISC population during recovery. However, by utilizing FACS sorting, we were able to isolate and culture recovering EpCAM+ epithelial cells from 1 and 2 DPI tissue. To the authors’ knowledge, this is the first report of this technique in a porcine model of intestinal injury. When placed in culture, recovering EpCAM+ cells were monitored for 7 days and provided further insight into the dynamics of ISC recovery. Surviving EpCAM+ cells isolated from 1 DPI likely represent injury-resistant rISCs. As shown, cultured cells from this time point survive but grow significantly less than those from control or 2 DPI (Fig. 3). Furthermore, resultant 1 DPI spheroids remain small in culture with minimal growth and budding throughout the 7 days. These data suggest that at 1 DPI, signals within the intestinal crypt support quiescence and dormancy. Immunofluorescent data presented here also showed that in vivo at this time point, there appeared to be a relative increase in the number of HOPX protein-expressing cells (Fig. 2). Interestingly, many of those cells were also positive for the proliferation marker Ki67+ yet remained less proliferative when isolated in culture, supporting that these cells may be noncycling or cycle arrested despite expressing Ki67 protein in immunofluorescence (50). Our previous work (13) also demonstrated that spheroids derived from crypts immediately after ischemic injury that were recovered in vitro had an overall decreased growth and increased levels of HOPX gene expression when compared with uninjured crypts. As HOPX expression decreased over time, the cultures began to grow and proliferate. These experiments support the hypothesis that HOPX serves a more functional role in ISC dynamics during recovery after injury than has been previously reported in the ISC literature.
It has been demonstrated that HOPX expression modulates cellular proliferation and differentiation in a variety of cell types (28–32, 51); direct upregulation of HOPX has also been shown to decrease cellular proliferation in several cancer cell lines including both esophageal squamous cell carcinoma and colorectal cancer (34, 36, 52, 53). Therefore, we utilized adenoviral transduction to directly influence the recovery of ischemic-injured intestinal crypts in culture by silencing HOPX. Successful transduction of injured crypts led to a significant increase in the growth and proliferation of resultant spheroids in culture (Figs. 4 and 5). This increase in size when compared with the scrambled virus and no virus control was significant up through 72 h further supporting a functional role of HOPX in the modulation of cellular proliferation during severe injury. Effects of the virus, while significant, were short lived, with no differences between groups after 72 h (data not shown). This is likely due to the high MOI used in our model to ensure efficient transduction of whole crypt epithelium. It has been shown that high adenoviral loads can influence cellular behavior in culture leading to growth retardation and apoptosis (54). This may have also contributed to the lack of significant difference between no virus and Ad-siHOPX observed in in vitro proliferation assays after 72 h in culture. In addition, it is possible that other cell types within close proximity to the intestinal stem cells, such as intraepithelial lymphocytes, could be isolated and inadvertently cultured during crypt dissociation thus influencing cellular behavior after a viral infection (55, 56). To minimize the potential impact of neighboring niche cells, future studies should focus on manipulating HOPX expression in isolated epithelial stem cells.
Currently, the mechanisms that control HOPX expression are not completely understood. It has been demonstrated that HOPX gene expression is likely controlled via methylation within the promoter region (36). Hypermethylation of the HOPX promoter results in loss of gene expression and increased cellular proliferation in a variety of body systems (53, 57–59). DNA methylation, a form of epigenetic change, can be influenced by a variety of factors including age, environment, lifestyle, and disease state (60). Interestingly, in hypoxic disease states including colorectal cancer, reactive oxygen species (ROS), and subsequent oxidative stress have been shown to influence epigenetic regulation and gene expression (61, 62). In diseases of ischemia-reperfusion injury where hypoxia-induced ROS production is known to impact tissue damage and recovery following intestinal ischemia-reperfusion injury (7, 63), ROS could also influence gene expression, including HOPX, within injury-resistant rISCs. Additional potential mechanisms to repress HOPX gene expression after ischemia-reperfusion injury include transcription factors. Cellular exposure to hypoxia, as occurs during ischemic injury, results in altered induction of transcription factors to regulate gene expression. Hypoxia-inducible factor (HIF) is a well-described adaptive mechanism by which hypoxia-induced stabilization of HIF then controls expression of cell survival and proliferation genes (64). Another recently described transcription factor in hypoxic injury is Repressor Element 1-Silencing Transcription (REST) factor, which regulates ∼20% of hypoxia-repressed genes. By targeting downstream cell cycle and proliferation genes, REST could play a role in the regulation of HOPX (65). Finally, at the posttranscriptional level, endogenous intracellular mechanisms such as microRNAs play an important role in cellular resistance to injury. Hypoxia-regulated microRNAs have been shown to influence cellular differentiation, proliferation, death, and metabolism via translational repression and mRNA degradation in response to ischemic injury (66, 67). The mechanisms by which HOPX+ injury-resistant rISCs become activated to proliferate would offer further insight into early epithelial repair after ischemic injury.
We have demonstrated the functional impact of HOPX expression on intestinal stem cells following a severe ischemic insult. High levels of HOPX expression correlated to a quiescent state with minimal growth during in vitro recovery (13), whereas silencing of HOPX led to improved growth and increased cellular proliferation (Figs. 4 and 5). Along with the in vivo data demonstrating an increased number of cells positive for HOPX protein expression at 1 DPI (Fig. 2) and a subsequent quiescence of EpCAM+ cells cultured from this time point (Fig. 3), it is likely that a switch occurs around 2 DPI that drives recovering ISCs to become active once again. This switch to proliferation is first demonstrated histologically at 2 DPI, as the earliest signs of epithelial regeneration occur consistently at this time point (Fig. 1). Decreased HOPX protein expression (Fig. 2) and increased growth of EpCAM+ cells cultured at 2 DPI (Fig. 3) further support that additional signals are likely present within the recovering intestine at this time point that drive cellular proliferation and recovery after injury.
Intestinal epithelial stem cells are supported by a variety of other cells and both soluble and cell-associated growth factors crucial in homeostasis and during repair (68). The ISC niche contains subepithelial mesenchymal elements such as myofibroblasts and telocytes as well as hematopoietic cells including lymphocytes and macrophages (69). Niche cells are a known critical source of growth factors for ISCs including Wnt, which promotes epithelial proliferation (70, 71). Additional factors produced by neighboring cells such as T helper cell cytokines may also play a role in the modulation of ISC behavior (72). Future research will investigate the role of supporting factors that signal ISCs during repair with a specific focus on the modulation of cellular proliferation within HOPX+ injury-resistant ISCs. Additional identification of intracellular pathways downstream of HOPX to signal ISC proliferation following intestinal ischemic injury may offer a unique approach to treat ischemic injury by promoting cellular proliferation and improving epithelial repair.
SUPPLEMENTAL DATA
Supplemental Fig. S1: https://figshare.com/articles/figure/Supplemental_Figure_1/15145038.
GRANTS
This study was supported by National Institutes of Health Grants T32 OD011130 (to A. S. Stewart), K01 OD019911-01A1 (to L. M. Gonzalez), R03 OD026598-01 (to L. M. Gonzalez), and National Institute of Diabetes and Digestive and Kidney Diseases Grant P30 DK034987 (to L. M. Gonzalez).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.S.S., C.R.S., and L.M.G. conceived and designed research; A.S.S., C.R.S., J.M.F., T.C.B., S.R.T., and L.M.G. performed experiments; A.S.S., C.R.S., J.B.R., and L.M.G. analyzed data; A.S.S., C.R.S., J.A.L., and L.M.G. interpreted results of experiments; A.S.S., C.R.S., and J.A.L. prepared figures; A.S.S., C.R.S., and L.M.G. drafted manuscript; A.S.S., C.R.S., and L.M.G. edited and revised manuscript; A.S.S., C.R.S., J.A.L., J.M.F., T.C.B., S.R.T., J.B.R., and L.M.G. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Dr. Elsa Ludwig for assistance in performing the porcine surgeries; Dr. Brittany Veerasammy, Dr. Caroline McKinney, Seth Kodikara, Mallory Thomas, and Nichol Henderson for assistance in data acquisition; NCSU-CVM Histology Lab for processing histologic samples and Javid Mohammed (NCSU-CVM Flow Cytometry Core) for patience and assistance with FACS in this project. In addition, we thank all of the technicians and laboratory animal veterinarians of North Carolina State Laboratory Animal Resources for assistance with animal care before, during, and after procedures, as well as for their dedication to this project.
REFERENCES
- 1.Aliosmanoglu I, Gul M, Kapan M, Arikanoglu Z, Taskesen F, Basol O, Aldemir M. Risk factors effecting mortality in acute mesenteric ischemia and mortality rates: a single center experience. Int Surg 98: 76–81, 2013. doi: 10.9738/CC112.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bobadilla JL. Mesenteric ischemia. Surg Clin North Am 93: 925–940, ix, 2013. doi: 10.1016/j.suc.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 3.Heys SD, Brittenden J, Crofts TJ. Acute mesenteric ischaemia: the continuing difficulty in early diagnosis. Postgrad Med J 69: 48–51, 1993. doi: 10.1136/pgmj.69.807.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park WM, Gloviczki P, Cherry KJ Jr, Hallett JW Jr, Bower TC, Panneton JM, Schleck C, Ilstrup D, Harmsen WS, Aa N. Contemporary management of acute mesenteric ischemia: factors associated with survival. J Vasc Surg 35: 445–452, 2002. doi: 10.1067/mva.2002.120373. [DOI] [PubMed] [Google Scholar]
- 5.Groschwitz KR, Hogan SP. Intestinal barrier function: molecular regulation and disease pathogenesis. J Allergy Clin Immunol 124: 3–20, 2009. doi: 10.1016/j.jaci.2009.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grootjans J, Lenaerts K, Derikx JP, Matthijsen RA, de Bruïne AP, van Bijnen AA, van Dam RM, Dejong CH, Buurman WA. Human intestinal ischemia-reperfusion-induced inflammation characterized: experiences from a new translational model. Am J Pathol 176: 2283–2291, 2010. doi: 10.2353/ajpath.2010.091069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gonzalez LM, Moeser AJ, Blikslager AT. Animal models of ischemia-reperfusion-induced intestinal injury: progress and promise for translational research. Am J Physiol Gastrointest Liver Physiol 308: G63–G75, 2015. doi: 10.1152/ajpgi.00112.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Markel TA, Crisostomo PR, Lahm T, Novotny NM, Rescorla FJ, Tector J, Meldrum DR. Stem cells as a potential future treatment of pediatric intestinal disorders. J Pediatr Surg 43: 1953–1963, 2008. doi: 10.1016/j.jpedsurg.2008.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Potten CS, Booth C, Pritchard DM. The intestinal epithelial stem cell: the mucosal governor. Int J Exp Pathol 78: 219–243, 1997. doi: 10.1046/j.1365-2613.1997.280362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yui S, Nakamura T, Sato T, Nemoto Y, Mizutani T, Zheng X, Ichinose S, Nagaishi T, Okamoto R, Tsuchiya K, Clevers H, Watanabe M. Functional engraftment of colon epithelium expanded in vitro from a single adult Lgr5(+) stem cell. Nat Med 18: 618–623, 2012. doi: 10.1038/nm.2695. [DOI] [PubMed] [Google Scholar]
- 11.Fordham RP, Yui S, Hannan NR, Soendergaard C, Madgwick A, Schweiger PJ, Nielsen OH, Vallier L, Pedersen RA, Nakamura T, Watanabe M, Jensen KB. Transplantation of expanded fetal intestinal progenitors contributes to colon regeneration after injury. Cell stem cell 13: 734–744, 2013. doi: 10.1016/j.stem.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, Clevers H. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459: 262–265, 2009. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 13.Gonzalez LM, Stewart AS, Freund J, Kucera CR, Dekaney CM, Magness ST, Blikslager AT. Preservation of reserve intestinal epithelial stem cells following severe ischemic injury. Am J Physiol Gastrointest Liver Physiol 316: G482–G494, 2019. doi: 10.1152/ajpgi.00262.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, Clevers H. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449: 1003–1007, 2007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 15.Takeda N, Jain R, LeBoeuf MR, Wang Q, Lu MM, Epstein JA. Interconversion between intestinal stem cell populations in distinct niches. Science 334: 1420–1424, 2011. doi: 10.1126/science.1213214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henning SJ, von Furstenberg RJ. GI stem cells - new insights into roles in physiology and pathophysiology. J Physiol 594: 4769–4779, 2016. doi: 10.1113/JP271663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richmond CA, Shah MS, Carlone DL, Breault DT. An enduring role for quiescent stem cells. Dev Dyn 245: 718–726, 2016. doi: 10.1002/dvdy.24416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bankaitis ED, Ha A, Kuo CJ, Magness ST. Reserve stem cells in intestinal homeostasis and injury. Gastroenterology 155: 1348–1361, 2018. doi: 10.1053/j.gastro.2018.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Landeghem L, Santoro MA, Krebs AE, Mah AT, Dehmer JJ, Gracz AD, Scull BP, McNaughton K, Magness ST, Lund PK. Activation of two distinct Sox9-EGFP-expressing intestinal stem cell populations during crypt regeneration after irradiation. Am J Physiol Gastrointest Liver Physiol 302: G1111–G1132, 2012. doi: 10.1152/ajpgi.00519.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dekaney CM, Gulati AS, Garrison AP, Helmrath MA, Henning SJ. Regeneration of intestinal stem/progenitor cells following doxorubicin treatment of mice. Am J Physiol Gastrointest Liver Physiol 297: G461–G470, 2009. doi: 10.1152/ajpgi.90446.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Helmrath MA, Fong JJ, Dekaney CM, Henning SJ. Rapid expansion of intestinal secretory lineages following a massive small bowel resection in mice. Am J Physiol Gastrointest Liver Physiol 292: G215–G222, 2007. doi: 10.1152/ajpgi.00188.2006. [DOI] [PubMed] [Google Scholar]
- 22.Kim CK, Yang VW, Bialkowska AB. The role of intestinal stem cells in epithelial regeneration following radiation-induced gut injury. Curr Stem Cell Rep 3: 320–332, 2017. [Erratum in Curr Stem Cell Rep 4: 95, 2018]. doi: 10.1007/s40778-017-0103-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tetteh PW, Basak O, Farin HF, Wiebrands K, Kretzschmar K, Begthel H, van den Born M, Korving J, de Sauvage F, van Es JH, van Oudenaarden A, Clevers H. Replacement of lost Lgr5-positive stem cells through plasticity of their enterocyte-lineage daughters. Cell Stem Cell 18: 203–213, 2016. doi: 10.1016/j.stem.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 24.Ayyaz A, Kumar S, Sangiorgi B, Ghoshal B, Gosio J, Ouladan S, Fink M, Barutcu S, Trcka D, Shen J, Chan K, Wrana JL, Gregorieff A. Single-cell transcriptomes of the regenerating intestine reveal a revival stem cell. Nature 569: 121–125, 2019. doi: 10.1038/s41586-019-1154-y. [DOI] [PubMed] [Google Scholar]
- 25.Murata K, Jadhav U, Madha S, van Es J, Dean J, Cavazza A, Wucherpfennig K, Michor F, Clevers H, Shivdasani RA. Ascl2-dependent cell dedifferentiation drives regeneration of ablated intestinal stem cells. Cell Stem Cell 26: 377–390, 2020. e376. doi: 10.1016/j.stem.2019.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rees WD, Tandun R, Yau E, Zachos NC, Steiner TS. Regenerative intestinal stem cells induced by acute and chronic injury: the saving grace of the epithelium? Front Cell Dev Biol 8: 583919, 2020. doi: 10.3389/fcell.2020.583919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Y, Zhang W. The role of HOPX in normal tissues and tumor progression. Biosci Rep 40, 2020. doi: 10.1042/BSR20191953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheung WK, Zhao M, Liu Z, Stevens LE, Cao PD, Fang JE, Westbrook TF, Nguyen DX. Control of alveolar differentiation by the lineage transcription factors GATA6 and HOPX inhibits lung adenocarcinoma metastasis. Cancer Cell 23: 725–738, 2013. doi: 10.1016/j.ccr.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Asanoma K, Kato H, Yamaguchi S, Shin CH, Liu ZP, Kato K, Inoue T, Miyanari Y, Yoshikawa K, Sonoda K, Fukushima K, Wake N. HOP/NECC1, a novel regulator of mouse trophoblast differentiation. J Biol Chem 282: 24065–24074, 2007. doi: 10.1074/jbc.M701380200. [DOI] [PubMed] [Google Scholar]
- 30.Kee HJ, Kim JR, Nam KI, Park HY, Shin S, Kim JC, Shimono Y, Takahashi M, Jeong MH, Kim N, Kim KK, Kook H. Enhancer of polycomb1, a novel homeodomain only protein-binding partner, induces skeletal muscle differentiation. J Biol Chem 282: 7700–7709, 2007. doi: 10.1074/jbc.M611198200. [DOI] [PubMed] [Google Scholar]
- 31.Obarzanek-Fojt M, Favre B, Kypriotou M, Ryser S, Huber M, Hohl D. Homeodomain-only protein HOP is a novel modulator of late differentiation in keratinocytes. Eur J Cell Biol 90: 279–290, 2011. doi: 10.1016/j.ejcb.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 32.Hawiger D, Wan YY, Eynon EE, Flavell RA. The transcription cofactor Hopx is required for regulatory T cell function in dendritic cell-mediated peripheral T cell unresponsiveness. Nat Immunol 11: 962–968, 2010. doi: 10.1038/ni.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ooki A, Yamashita K, Kikuchi S, Sakuramoto S, Katada N, Kokubo K, Kobayashi H, Kim MS, Sidransky D, Watanabe M. Potential utility of HOP homeobox gene promoter methylation as a marker of tumor aggressiveness in gastric cancer. Oncogene 29: 3263–3275, 2010. doi: 10.1038/onc.2010.76. [DOI] [PubMed] [Google Scholar]
- 34.Yamashita K, Kim MS, Park HL, Tokumaru Y, Osada M, Inoue H, Mori M, Sidransky D. HOP/OB1/NECC1 promoter DNA is frequently hypermethylated and involved in tumorigenic ability in esophageal squamous cell carcinoma. Mol Cancer Res 6: 31–41, 2008. doi: 10.1158/1541-7786.MCR-07-0213. [DOI] [PubMed] [Google Scholar]
- 35.Yap LF, Lai SL, Patmanathan SN, Gokulan R, Robinson CM, White JB, Chai SJ, Rajadurai P, Prepageran N, Liew YT, Lopes V, Wei W, Hollows RJ, Murray PG, Lambert DW, Hunter KD, Paterson IC. HOPX functions as a tumour suppressor in head and neck cancer. Sci Rep 6: 38758, 2016. doi: 10.1038/srep38758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Katoh H, Yamashita K, Waraya M, Margalit O, Ooki A, Tamaki H, Sakagami H, Kokubo K, Sidransky D, Watanabe M. Epigenetic silencing of HOPX promotes cancer progression in colorectal cancer. Neoplasia 14: 559–571, 2012. doi: 10.1593/neo.12330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blikslager AT, Rhoads JM, Bristol DG, Roberts MC, Argenzio RA. Glutamine and transforming growth factor-alpha stimulate extracellular regulated kinases and enhance recovery of villous surface area in porcine ischemic-injured intestine. Surgery 125: 186–194, 1999. [PubMed] [Google Scholar]
- 38.Stieler Stewart A, Freund JM, Blikslager AT, Gonzalez LM. Intestinal stem cell isolation and culture in a porcine model of segmental small intestinal ischemia. J Vis Exp 135: 57647, 2018. doi: 10.3791/57647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haldeman JM, Conway AE, Arlotto ME, Slentz DH, Muoio DM, Becker TC, Newgard CB. Creation of versatile cloning platforms for transgene expression and dCas9-based epigenome editing. Nucleic Acids Res 47: e23, 2019. doi: 10.1093/nar/gky1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hay WP, Mueller PO, Harmon B, Amoroso L. One percent sodium carboxymethylcellulose prevents experimentally induced abdominal adhesions in horses. Vet Surg 30: 223–227, 2001. doi: 10.1053/jvet.2001.17849. [DOI] [PubMed] [Google Scholar]
- 41.Reijnen MM, Skrabut EM, Postma VA, Burns JW, van Goor H. Polyanionic polysaccharides reduce intra-abdominal adhesion and abscess formation in a rat peritonitis model. J Surg Res 101: 248–253, 2001. doi: 10.1006/jsre.2001.6288. [DOI] [PubMed] [Google Scholar]
- 42.Pereira-Fantini PM, Thomas SL, Wilson G, Taylor RG, Sourial M, Bines JE. Short- and long-term effects of small bowel resection: a unique histological study in a piglet model of short bowel syndrome. Histochem Cell Biol 135: 195–202, 2011. doi: 10.1007/s00418-011-0778-2. [DOI] [PubMed] [Google Scholar]
- 43.Wang N, Zhang H, Zhang B-Q, Liu W, Zhang Z, Qiao M, Zhang H, Deng F, Wu N, Chen X, Wen S, Zhang J, Liao Z, Zhang Q, Yan Z, Yin L, Ye J, Deng Y, Luu HH, Haydon RC, Liang H, He T-C. Adenovirus-mediated efficient gene transfer into cultured three-dimensional organoids. PloS One 9: e93608, 2014. doi: 10.1371/journal.pone.0093608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gonzalez LM, Williamson I, Piedrahita JA, Blikslager AT, Magness ST. Cell lineage identification and stem cell culture in a porcine model for the study of intestinal epithelial regeneration. PloS One 8: e66465, 2013. doi: 10.1371/journal.pone.0066465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoshida S, Miwa H, Kawachi T, Kume S, Takahashi K. Generation of intestinal organoids derived from human pluripotent stem cells for drug testing. Sci Rep 10: 5989, 2020. doi: 10.1038/s41598-020-63151-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luo H, Zheng J, Chen Y, Wang T, Zhang Z, Shan Y, Xu J, Yue M, Fang W, Li X. Utility Evaluation of porcine enteroids as PDCoV infection model in vitro. Front Microbiol 11: 821, 2020. doi: 10.3389/fmicb.2020.00821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gracz AD, Puthoff BJ, Magness ST. Identification, isolation, and culture of intestinal epithelial stem cells from murine intestine. Methods Mol Biol 879: 89–107, 2012. doi: 10.1007/978-1-61779-815-3_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fuller MK, Faulk DM, Sundaram N, Shroyer NF, Henning SJ, Helmrath MA. Intestinal crypts reproducibly expand in culture. J Surg Res 178: 48–54, 2012. doi: 10.1016/j.jss.2012.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Uzal FA, Brandon P, Hostetter J. Alimentary system; intestinal ischemia and infarction. In: Jubb, Kennedy & Palmer’s Pathology of Domestic Animals, edited by Maxie MG. St. Louis, MO: Elsevier, 2016, p. 81–82. [Google Scholar]
- 50.van Oijen MG, Medema RH, Slootweg PJ, Rijksen G. Positivity of the proliferation marker Ki-67 in noncycling cells. Am J Clin Pathol 110: 24–31, 1998. doi: 10.1093/ajcp/110.1.24. [DOI] [PubMed] [Google Scholar]
- 51.Ota C, Ng-Blichfeldt JP, Korfei M, Alsafadi HN, Lehmann M, Skronska-Wasek W, Mds M, Guenther A, Wagner DE, Königshoff M. Dynamic expression of HOPX in alveolar epithelial cells reflects injury and repair during the progression of pulmonary fibrosis. Sci Rep 8: 12983, 2018. doi: 10.1038/s41598-018-31214-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yamaguchi S, Asanoma K, Takao T, Kato K, Wake N. Homeobox gene HOPX is epigenetically silenced in human uterine endometrial cancer and suppresses estrogen-stimulated proliferation of cancer cells by inhibiting serum response factor. Int J Cancer 124: 2577–2588, 2009. doi: 10.1002/ijc.24217. [DOI] [PubMed] [Google Scholar]
- 53.Waraya M, Yamashita K, Katoh H, Ooki A, Kawamata H, Nishimiya H, Nakamura K, Ema A, Watanabe M. Cancer specific promoter CpG Islands hypermethylation of HOP homeobox (HOPX) gene and its potential tumor suppressive role in pancreatic carcinogenesis. BMC Cancer 12: 397, 2012. doi: 10.1186/1471-2407-12-397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brand K, Klocke R, Possling A, Paul D, Strauss M. Induction of apoptosis and G2/M arrest by infection with replication-deficient adenovirus at high multiplicity of infection. Gene Ther 6: 1054–1063, 1999. doi: 10.1038/sj.gt.3300914. [DOI] [PubMed] [Google Scholar]
- 55.Rogoz A, Reis BS, Karssemeijer RA, Mucida D. A 3-D enteroid-based model to study T-cell and epithelial cell interaction. J Immunol Methods 421: 89–95, 2015. doi: 10.1016/j.jim.2015.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kawabata S, Boyaka PN, Coste M, Fujihashi K, Yamamoto M, McGhee JR, Kiyono H. Intraepithelial lymphocytes from villus tip and crypt portions of the murine small intestine show distinct characteristics. Gastroenterology 115: 866–873, 1998. doi: 10.1016/s0016-5085(98)70258-6. [DOI] [PubMed] [Google Scholar]
- 57.Ren X, Yang X, Cheng B, Chen X, Zhang T, He Q, Li B, Li Y, Tang X, Wen X, Zhong Q, Kang T, Zeng M, Liu N, Ma J. HOPX hypermethylation promotes metastasis via activating SNAIL transcription in nasopharyngeal carcinoma. Nat Commun 8: 14053, 2017. doi: 10.1038/ncomms14053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kikuchi M, Katoh H, Waraya M, Tanaka Y, Ishii S, Tanaka T, Nishizawa N, Yokoi K, Minatani N, Ema A, Kosaka Y, Tanino H, Yamashita K, Watanabe M. Epigenetic silencing of HOPX contributes to cancer aggressiveness in breast cancer. Cancer Lett 384: 70–78, 2017. doi: 10.1016/j.canlet.2016.10.017. [DOI] [PubMed] [Google Scholar]
- 59.Rodrigues MF, Esteves CM, Xavier FC, Nunes FD. Methylation status of homeobox genes in common human cancers. Genomics 108: 185–193, 2016. doi: 10.1016/j.ygeno.2016.11.001. [DOI] [PubMed] [Google Scholar]
- 60.Rodríguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med 17: 330–339, 2011. doi: 10.1038/nm.2305. [DOI] [PubMed] [Google Scholar]
- 61.Zhang R, Kang KA, Kim KC, Na SY, Chang WY, Kim GY, Kim HS, Hyun JW. Oxidative stress causes epigenetic alteration of CDX1 expression in colorectal cancer cells. Gene 524: 214–219, 2013. doi: 10.1016/j.gene.2013.04.024. [DOI] [PubMed] [Google Scholar]
- 62.García-Guede Á, Vera O, Ibáñez-de-Caceres I. When oxidative stress meets epigenetics: implications in cancer development. Antioxidants (Basel) 9, 2020. doi: 10.3390/antiox9060468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sasaki M, Joh T. Oxidative stress and ischemia-reperfusion injury in gastrointestinal tract and antioxidant, protective agents. J Clin Biochem Nutr 40: 1–12, 2007. doi: 10.3164/jcbn.40.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Benita Y, Kikuchi H, Smith AD, Zhang MQ, Chung DC, Xavier RJ. An integrative genomics approach identifies hypoxia inducible factor-1 (HIF-1)-target genes that form the core response to hypoxia. Nucleic Acids Res 37: 4587–4602, 2009. doi: 10.1093/nar/gkp425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cavadas MA, Mesnieres M, Crifo B, Manresa MC, Selfridge AC, Keogh CE, Fabian Z, Scholz CC, Nolan KA, Rocha LM, Tambuwala MM, Brown S, Wdowicz A, Corbett D, Murphy KJ, Godson C, Cummins EP, Taylor CT, Cheong A. REST is a hypoxia-responsive transcriptional repressor. Sci Rep 6: 31355, 2016. doi: 10.1038/srep31355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kulshreshtha R, Davuluri RV, Calin GA, Ivan M. A microRNA component of the hypoxic response. Cell Death Differ 15: 667–671, 2008. doi: 10.1038/sj.cdd.4402310. [DOI] [PubMed] [Google Scholar]
- 67.Liang H, Wang C, Gao K, Li J, Jia R. ΜicroRNA-421 promotes the progression of non-small cell lung cancer by targeting HOPX and regulating the Wnt/β-catenin signaling pathway. Mol Med Rep 20: 151–161, 2019. doi: 10.3892/mmr.2019.10226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sailaja BS, He XC, Li L. The regulatory niche of intestinal stem cells. J Physiol 594: 4827–4836, 2016. doi: 10.1113/JP271931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Santos AJ, Lo YH, Mah AT, Kuo CJ. The intestinal stem cell niche: homeostasis and adaptations. Trends Cell Biol 28: 1062–1078, 2018. doi: 10.1016/j.tcb.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kaestner KH. The intestinal stem cell niche: a central role for Foxl1-expressing subepithelial telocytes. Cell Mol Gastroenterol Hepatol 8: 111–117, 2019. doi: 10.1016/j.jcmgh.2019.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shoshkes-Carmel M, Wang YJ, Wangensteen KJ, Tóth B, Kondo A, Massassa EE, Itzkovitz S, Kaestner KH. Subepithelial telocytes are an important source of Wnts that supports intestinal crypts. Nature 557: 242–246, 2018. doi: 10.1038/s41586-018-0084-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Biton M, Haber AL, Rogel N, Burgin G, Beyaz S, Schnell A, Ashenberg O, Su C, Smillie C, Shekhar K, Chen Z, Wu C, Ordovas-Montanes J, Alvarez D, Herbst RH, Zhang M, Tirosh I, Dionne D, Nguyen LT, Xifaras ME, Shalek AK, von Andrian UH, Graham DB, Rozenblatt-Rosen O, Shi HN, Kuchroo V, Yilmaz OH, Regev A, Xavier RJ. T helper cell cytokines modulate intestinal stem cell renewal and differentiation. Cell 175: 1307–1320, 2018. doi: 10.1016/j.cell.2018.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]