

Abstract
We present the results for CAPRI Round 50, the 4th joint CASP-CAPRI protein assembly prediction challenge. The Round comprised a total of 12 targets, including 6 dimers, 3 trimers, and 3 higher-order oligomers. Four of these were easy targets, for which good structural templates were available either for the full assembly, or for the main interfaces (of the higher-order oligomers). Eight were difficult targets for which only distantly related templates were found for the individual subunits. Twenty-five CAPRI groups including 8 automatic servers submitted ~1250 models per target. Twenty groups including 6 servers participated in the CAPRI scoring challenge submitted ~190 models per target. The accuracy of the predicted models was evaluated using the classical CAPRI criteria. The prediction performance was measured by a weighted scoring scheme that takes into account the number of models of acceptable quality or higher submitted by each group as part of their 5 top-ranking models. Compared to the previous CASP-CAPRI challenge, top performing groups submitted such models for a larger fraction (70–75%) of the targets in this Round, but fewer of these models were of high accuracy. Scorer groups achieved stronger performance with more groups submitting correct models for 70–80% of the targets or achieving high accuracy predictions. Servers performed less well in general, except for the MDOCKPP and LZERD servers, who performed on par with human groups. In addition to these results, major advances in methodology are discussed, providing an informative overview of where the prediction of protein assemblies currently stands.
Keywords: CAPRI, CASP, oligomeric state, blind prediction, protein-protein interaction, protein complexes, protein assemblies, template-based modeling, docking, protein docking
INTRODUCTION
Large protein assemblies and complexes of proteins with other proteins and macromolecular components such as DNA or RNA, carry out critical functions in many cellular processes. Their disruption or dysregulation often causes disease 1,2. Characterizing the three-dimensional structure and function of these interactions, at both the molecular and cellular levels, and elucidating the underlying physical principles remains an important goal of biology and medicine.
Much of our current understanding of protein complexes has been derived from the high-resolution three-dimensional structures of protein complexes determined by experimental methods3–6 and deposited in the PDB (Protein Data Bank) 7. But unfortunately, little or no structural information is available for the majority of the protein complexes forming in the cell that can be characterized by modern proteomics and other methods.
The recent spectacular advances in single-molecule cryo-EM techniques, specifically geared at determining the structure of large macromolecular assemblies at atomic resolution 8,9 should enable to narrow the gap, but valuable help is also expected from steady progress in computational procedures.
Thanks to the continued success of structural biology in enriching the structural repertoire of individual proteins, which form the building blocks of larger assemblies, and the recent explosion of the number of available protein sequences, computational approaches are now capable of modeling the 3D structure of individual proteins with increased accuracy from sequence information alone. This is most commonly done by using structures of related proteins deposited in the PDB as templates for the modeling task 10–12. The ability to predict the 3D structure of proteins from sequence in absence of available templates, commonly referred to as ab-initio modeling, has also significantly improved, thanks to computational methods that exploit multiple sequence alignments of related proteins to predict residue-residues contact crucial to defining the protein fold13–15. Further substantial improvements in the performance of 3D protein structure predictions by both template-based and ab-initio approaches, have been achieved by recent Artificial Intelligence (AI)- Deep Learning (DL) techniques16,17, that afford more efficient means of leveraging and integrating information across the known landscape of protein structures and sequences 18–21.
The advantages afforded by these techniques were already highlighted in the previous CASP challenge (CASP13) 22,23 and dramatically confirmed by the superb structure prediction performance of AlphaFold2 from the Google DeepMind team in CASP14 [AlphaFold2, this volume], whose submitted models rivaled in accuracy with high-resolution crystal structures. Strikingly furthermore, AlphaFold2 seemed to very accurately predict the bound conformation of individual subunits of homomeric assemblies (some of which are highly nonglobular) or individual domains of larger proteins [ab-initio structure predictions, this volume], that could not possibly adopt this conformation in isolation. This is particularly relevant for the prediction of protein assemblies, because it suggests that the AlphaFold2 DL-based procedure (of which not much has been revealed at the time of writing this paper) is picking up evolutionary signals that integrate the stable native state of the multi-domain protein or the multi-subunit assembly, where the latter involves preferentially homomeric associations, which tend to be more highly conserved across evolution 24,25.
Computational approaches have also played an important role in the efforts to populate the uncharted landscape of protein assemblies, a role that will hopefully be further bolstered by more closely integrating AI-based techniques with the development of a sufficiently large body of structural data on protein assemblies and their conformational diversity, which currently is still lacking. So far however, the problem of accurately predicting the 3D structure of protein complexes remains a challenging undertaking, which very much depends on the protein system at hand and may therefore be considered as the next frontier in the quest of modeling the functionally relevant states of proteins.
A classical approach to modelling the 3D structures of a protein complex starts form the 3D structures of the individual protein components and uses the so-called ‘docking’ algorithms, and the associated energetic criteria to single out stable binding modes 26–28. CAPRI (Critical Assessment of PRedicted Interactions) (https://www.ebi.ac.uk/pdbe/complex-pred/capri/; http://www.capri-docking.org/) is a community-wide initiative inspired by CASP (Critical Assessment of protein Structure Prediction). Established in 2001, it has offered computational biologists the opportunity to test their algorithms in blind predictions of experimentally determined 3D structures of protein complexes, the ‘targets’, provided to CAPRI prior to publication. Just as CASP has been very instrumental in stimulating the field of protein structure prediction, CAPRI has contributed to advancing the field of modeling protein assemblies. Initially focusing on testing procedures for predicting protein-protein complexes, CAPRI is now also dealing with protein-peptide, protein-nucleic acids, and protein-oligosaccharide complexes. In addition, CAPRI has organized challenges to evaluate computational methods for estimating binding affinity of protein-protein complexes 29–31 and predicting the positions of water molecules at the interfaces of protein complexes 32.
Thanks to the growing ease with which structural templates can be found in the PDB, docking calculations have evolved to routinely take as input homology-built models of the individual components of a complex with an increasing degree of success. It is furthermore not uncommon to find templates for the entire protein assembly. Such cases occur most often for assemblies of identical subunits (homodimers, or higher order homo-oligomers), because their binding modes (oligomeric states) tend to be conserved in related proteins 24,25. In such instances, classical docking calculations may no longer be required because the protein assembly can be modeled directly from the template, a task also called ‘template-based docking’ 10,33,34.
In a significant number of cases however, the modeling task remains challenging because the template structure may differ significantly from the structure of the protein to be modeled, or adequate templates cannot be identified. Overcoming these important roadblocks has called for a much closer integration of methods for predicting the 3D structure of individual protein subunits and those for modeling protein assemblies and developing means for improving the accuracy of the resulting multi-subunit models. This has been the motivation for establishing closer ties between the CASP and CAPRI communities by running joint CASP-CAPRI assembly prediction experiments. Three such experiments were conducted in the summers of 2014, 2016, and 2018, respectively, with results presented at the CASP11, CASP12 and CASP13 meetings in Cancún, Mexico, and Gaeta, Italy, and published in 3 special issues of Proteins 35–38.
Here we present an evaluation of the results obtained in the CASP14-CAPRI challenge, the 4th joint assembly prediction experiment with CASP, representing Round 50 of CAPRI. This prediction Round was held in the summer of 2020 as part of the CASP14 prediction season. Like other CAPRI Rounds, Round 50 also included scoring experiments, uniquely offered by CAPRI, where participants are invited to identify the correct association modes from an ensemble of anonymized predicted complexes generated during the assembly prediction experiment 39,40. In addition, we also evaluate submitted models in terms of their ability to correctly recapitulate the protein-protein interface of the targets36,41, i.e. contain the amino acids residues part of the recognition surfaces of each protein component of the target complex. These evaluations are carried out using criteria and evaluation protocols agreed upon by the CAPRI community. A separate evaluation of the CASP14 assembly prediction performance, reported at the CASP14 meeting and in this Special Issue [Ozden & Karaca, this issue], was performed by the CASP assembly assessment team in collaboration with the CASP prediction center. We wish to highlight the very fruitful collaboration that took place between the CASP teams and the CAPRI assessment in defining the prediction problem for complex targets, discussing evaluation strategies and comparing assessment results.
CAPRI Round 50 comprised a total of 16 targets, a lower number than in some of the previous joint challenges. Experimental structures for 4 of these were not available for evaluation, reducing to 12 the number of targets for which predictions have been evaluated. The 12 targets included 6 dimers (5 homodimers and 1 heterodimer), 3 trimers (2 homotrimers and 1 heterotrimer) and 3 large multi-protein assemblies solved by cryo-EM comprising: the 27 subunits (representing 4 distinct protein chains) of the T5 phase tail distal complex, the 20 subunits homo-oligomeric assembly of a bacterial arginine decarboxylase, and the full viral capsid of the duck hepatitis B virus, (with T=4 icosahedral symmetry, totaling 240 subunits). The targets of Round 50 were hand-picked by the CAPRI management as representing tractable modeling problems for the CAPRI community. A target was considered as tractable, when templates could be identified, for at least a portion of the components of the target complex, using available tools such as HHblits/HHpred 42,43 and applying very lenient thresholds for sequence coverage and divergence. Targets for which such templates could not be identified, were considered as difficult ab-initio fold prediction problems, since both the 3D structures of the subunits and their association modes need to be predicted simultaneously. Although the CASP14 challenge demonstrated that the 3D structures of individual proteins chains may in a good number of cases be predicted to high accuracy by groups such as Google DeepMind, the corresponding models were not available to groups participating in the assembly prediction Round, and CAPRI groups mostly lack the expertise to generate such models. As in previous Rounds, such targets where therefore not included in CAPRI Round 50.
This may change in the future prediction challenges, as DL methods are more closely integrated with assembly prediction and docking procedures, or when groups such as DeepMind automate their prediction method sufficiently to make their accurately modeled 3D structures of individual subunits available to docking experts during the prediction Round. Using such accurate 3D models, which often faithfully represent the bound conformation of the proteins, as input to the docking calculations would be a game changer, particularly in the prediction of homomeric assemblies. We know indeed from earlier CAPRI Rounds and from various benchmark studies that docking calculations performed starting from the bound conformation of the individual subunits, achieve much superior performance 39.
THE TARGETS
The 12 targets of the CASP14-CAPRI assembly prediction experiment, which is henceforth denoted as Round 50, are listed in Table 1, and illustrated in Figure 1. The targets are designated by their CAPRI target ID followed by their corresponding CASP target ID.
Table 1 –
CASP14-CAPRI assembly targets. The columns present respectively the CAPRI and CASP target ID, stoichiometry of the assembly, the number of interfaces, the surface area (or range) of the interfaces, the number of residues per monomer, the PDB code (if available) and a textual description of the target. For target structures not yet deposited in the PDB (N/A in column 7) structural details could not be revealed here. Dimeric and trimeric targets are listed with Easy targets first and then Difficult targets. Difficulty of all targets is indicated by superscript ‘e’ (Easy) or ‘d’ (Difficult) in the CAPRI target ID column. (*) Target T170/H1060 comprises a total of 9 interfaces, with buried surface areas of 1800/1650/1650/950/680/680/550/1200/750, for interfaces 1–9, respectively. (*) T165 shows the area for the A/HL interface.
| Dimers | ||||||||
|
| ||||||||
| Target | ID | Stoich. | #Int. | Area (Å2) | #Res. | PDB | Description | |
|
| ||||||||
| T164e | T1032 | A2 | 1 | 1585 | 284 | N/A | SMCHD1 (human) residues 1616–1899 | |
| T166e | H1045 | A/B | 1 | 765 | 157/173 | N/A | PEX4/PEX22 complex from Arabidopsis thaliana | |
|
| ||||||||
| T169d | T1054 | A2 | 1 | 1530 | 190 | N/A | Outer-membrane lipoprotein from Acinetobacter baumannii | |
|
| ||||||||
| T176d | T1078 | A2 | 1 | 1480 | 138 | N/A | Tsp1 from Trichoderma virens, small secreted cysteine rich protein (SSCRP) | |
|
| ||||||||
| T178d | T1083 | A2 | 1 | 1650 | 98 | 6nq1 | helical segment from Nitrosococcus oceani | |
|
| ||||||||
| T179d | T1087 | A2 | 1 | 1620 | 93 | N/A | helical segment Methylobacter tundripaludum | |
|
| ||||||||
| Trimers | ||||||||
|
| ||||||||
| Target | ID | Stoich. | #Int. | Area (Å2) | #Res. | PDB | Description | |
|
| ||||||||
| T168e | T1052 | A3 | 1 | 2600 | 832 | N/A | Tail fiber of the Salmonella virus epsilon15 | |
|
| ||||||||
| T165d | H1036 | A3H3L3 | 1 | 850* | 931/128/106 | 6vn1 | MC Ab 93k bound to varicella-zoster virus glycoprotein gB | |
|
| ||||||||
| T174d | T1070 | A3 | 1 | 3000 | 335§ | N/A | Protein of attachment region to phage tail | |
|
| ||||||||
| Large assemblies | ||||||||
|
| ||||||||
| Target | ID | Stoich. | #Int. | Area (Å2) | #Res. | PDB | Description | |
|
| ||||||||
| T170d | H1060 | A6/B3/C12/D6 | 9 | 550–1800* | 464/298/140/142 | N/A | Component of the T5 phage tail distal complex. | |
|
| ||||||||
| T177e | T1081 | A20 | 3 | 5000/1250/300 | 758 | 2vyc | Arginine decarboxylase/Bacteria | |
|
| ||||||||
| T180d | T1099 | A240 | 8 (4) | 1110/1970 | 262 | 6ygh | Capsid of duck hepatitis B virus | |
Figure 1: The Targets of Round 50.
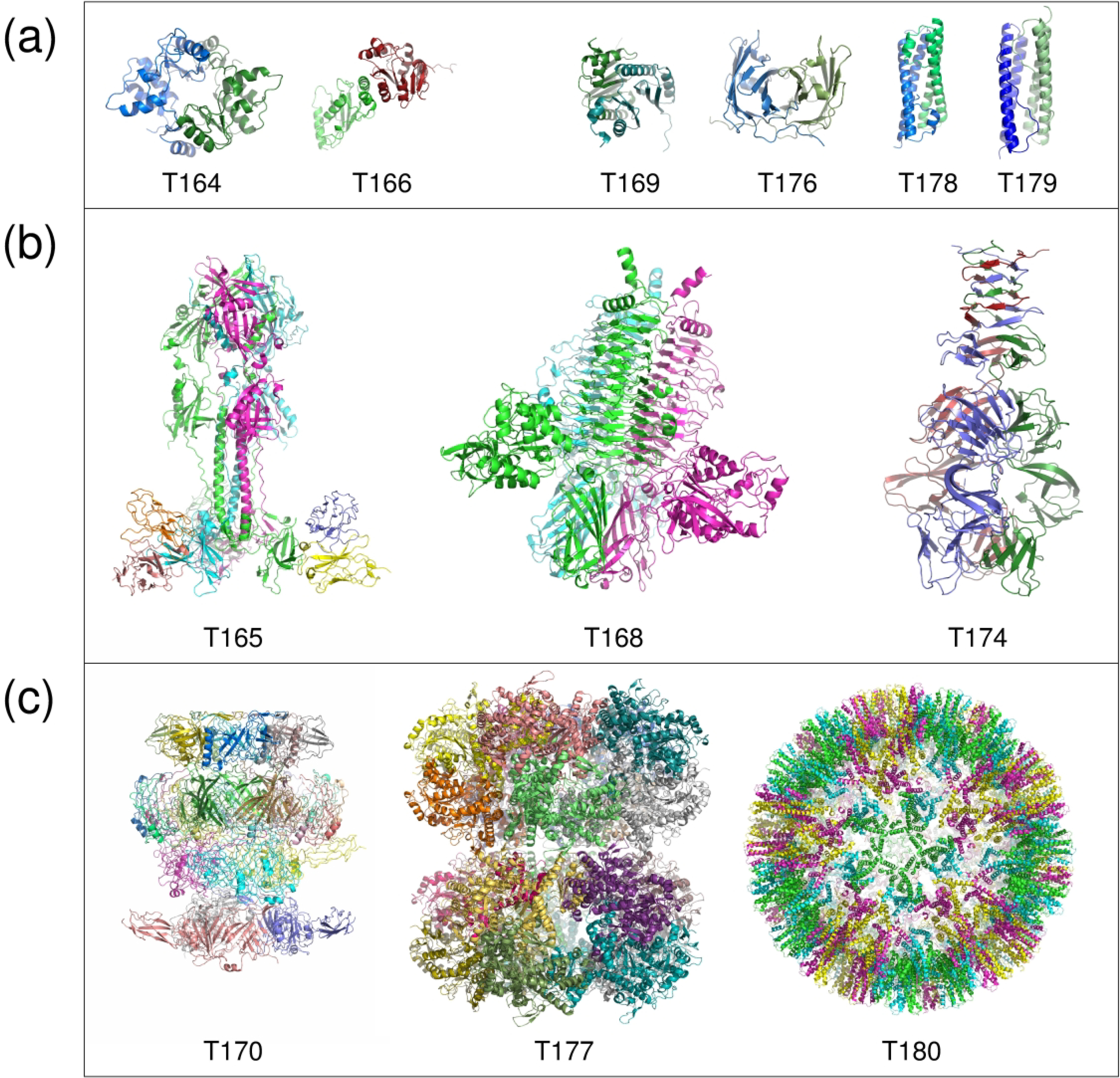
(a) Dimeric targets, (b) trimeric targets, (c) large assemblies. The dimeric targets are divided into Easy (T164/T1032, T166/H1045) and Difficult (T169/T1054, T176/T1078, T178/T1083, T179/T1087) targets. The trimeric targets T165/H1036 and T174/T1070 were Difficult, whereas T168/T1052 was easy. The large assembly target T177/T1081 was an easy target. The remaining targets T170/H1060 and T180/T0199 featured both Easy and Difficult to predict interfaces.
As in previous CASP-CAPRI challenges the majority of the targets (9 out of 12) were homo-oligomers. The remaining 3 targets were hetero complexes. A majority were proteins from bacteria and viruses, with the size of individual subunits spanning a very wide range (93–931 residues). Most targets (7) had their structure determined at high-resolution by X-ray diffraction. The remaining 5 targets, T165/H1036, T168/T1052, T170/H1060, T177/H1081, and T180/T1099, were larger multi-protein assemblies determined by cryo-EM. Most of the targets had annotated biological function and the majority had an author-assigned oligomeric state of the protein.
The 12 targets of Round 50 were grouped into 2 categories: easy targets (4 in total) for which good structural templates were available either for the full assembly, or for the main interfaces (of the higher-order oligomers), and 8 difficult (to model) targets (Table 1). Targets of both categories included dimers, trimers, and large assemblies.
The easy targets were the human SMCHD1 homo dimer (T164/T1032), the PEX4/PEX22 complex from Arabidopsis thaliana (T166/H1045), the homo trimer of the Salmonella virus e15 tail fiber (T168/T1052), and the 20 subunits assembly of the bacterial arginine decarboxylase (T177/T0181) arranged as 2 stacked decameric rings, each adopting D5 symmetry (Figures 1 and 2). The latter assembly target was categorized as easy, because an excellent template was available for the decameric rings.
Figure 2: Evaluated interfaces of the bacterial Arginine decarboxylate (T177/T1081).
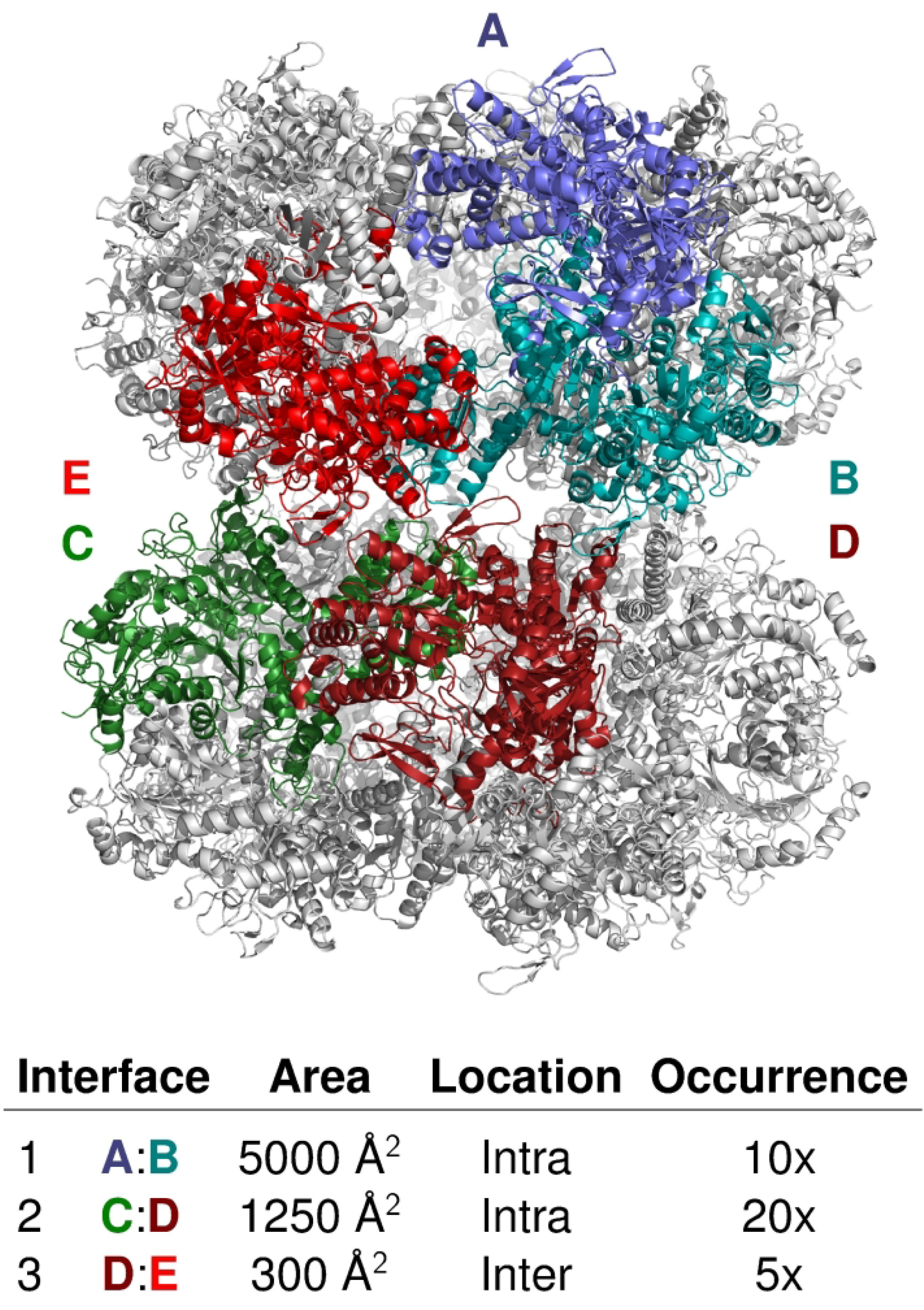
The two primary interfaces are within each decameric ring, the third interface lies between the two rings. Individual subunits illustrating the intra- and inter-decamer interfaces are colored.
The 8 difficult targets include 4 homodimers, 2 trimers, and 2 large assemblies (Table 1). The 4 homodimers comprise two globular bacterial proteins (T169/T1054 and T176/T1078), and two bacterial helical dimers (T178/T1083 and T179/T1087). For all of these, distant templates were available only for the individual subunits of each complex. The 2 difficult trimers include a hetero complex of the varicella-zoster virus glycoprotein gB trimer, bound to a specific monoclonal Ab (T165/T1036), where the main challenge was to predict the Ab binding interface, and the phage tail attachment regions protein (T174/T1070). Of the two difficult to model large assemblies, the first is a multi-protein component of the T5 phage distal complex (T170/H1060), composed of 4 different chains with stoichiometry A6;B3;C12;D6, totaling 27 subunits, arranged in 4 rings stacked on top of one another, one of which is in fact composed of 2 concentric rings the inner B3 ring and the outer C12 ring (see Figure 3a for details and nomenclature used). The cryo-EM structure of the full complex, included 2 additional rings, composed of subunits with significantly disordered regions, for which adequate templates were not available. These additional rings were not part of the prediction challenge.
Figure 3: Subunit arrangement and interfaces of the T5 phage tail distal complex (T170/H1060).
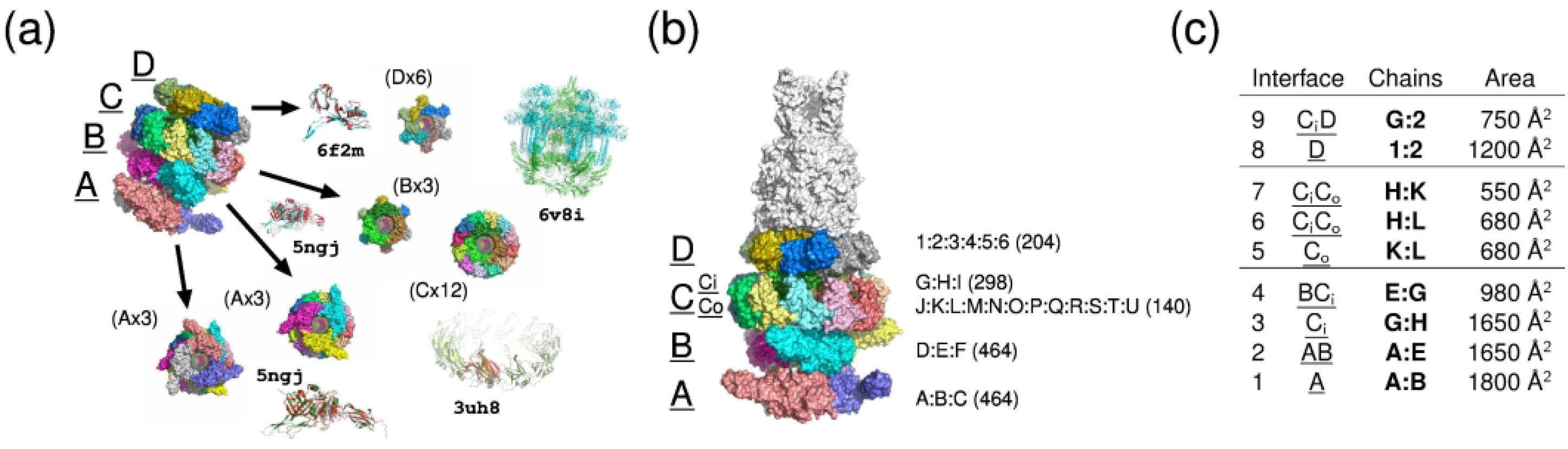
(a) The rings A and B (rings are underlined) consist of 3 identical copies of protein A (proteins are not underlined); ring C contains an inner Ci (3 copies of B) and outer Co (12 copies of C) ring; ring D contains 6 copies of protein D. The best templates for each protein are shown in the image. (b) Shows the organization of the 5 rings in the larger assembly as it was resolved by cryo-EM. To the right of the rings are listed the chain identifiers, with the number of residues in each chain in parentheses. (c) Shows the 9 different interfaces, the rings in or between which they occur, two exemplary chains of the interface and the buried area between the two chains.
The second target of this category (T180/T1099) was the capsid of the of the duck hepatitis B virus, adopting a T=4 icosahedral symmetry with a total of 240 subunits (Figure 4). A template corresponding to a distantly related hepatitis B virus capsid was available, but the corresponding capsid core protein was lacking an insertion exhibited by the target protein, which contributes to the major capsid interface as will be further detailed in our analysis.
Figure 4: Subunit interactions and quasi symmetry of the duck hepatitis B virus capsid (T180/T1099).
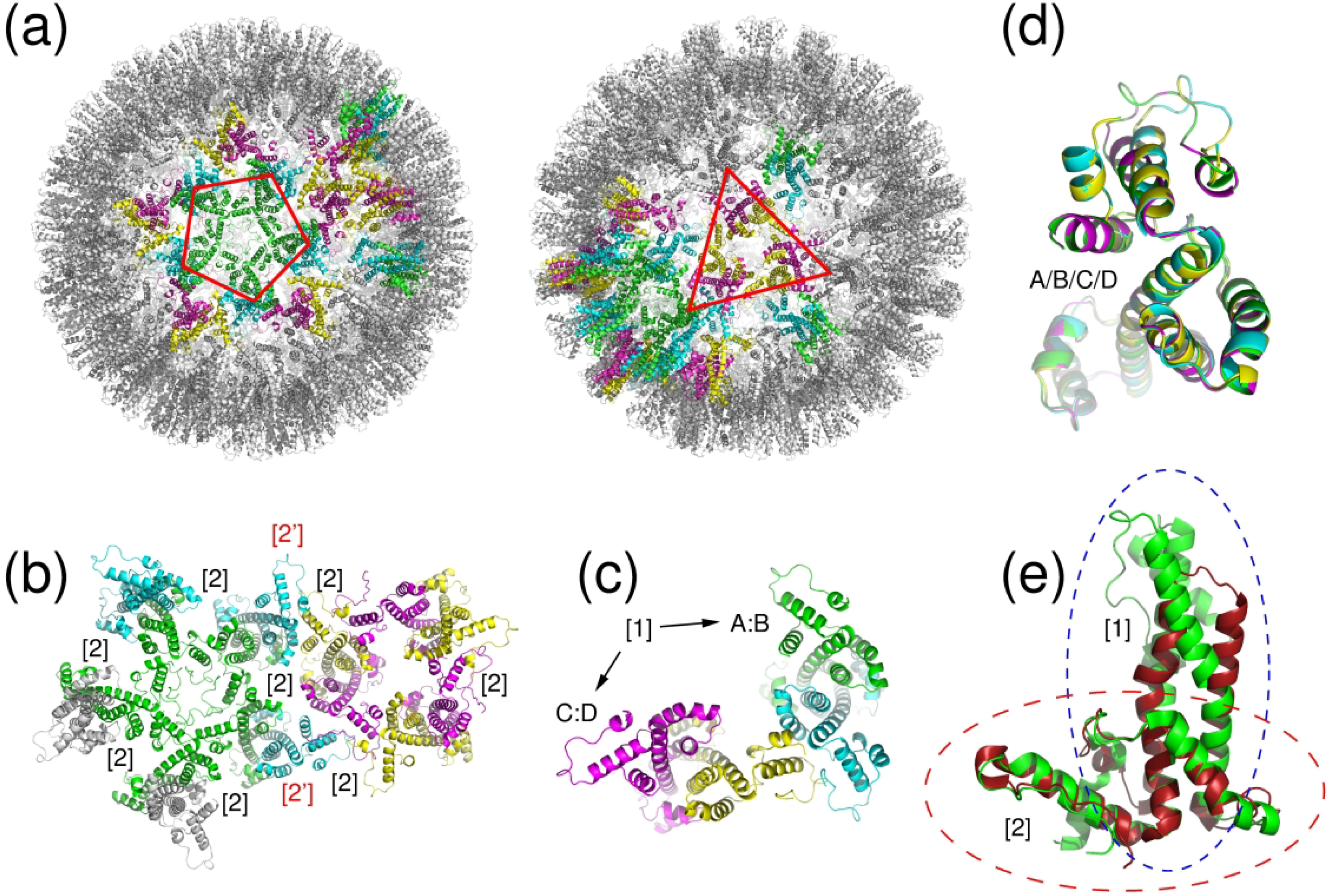
(a) shows the entire capsid, highlighting the five-fold and three-fold symmetry also shown in (b) that is exhibited by the assembly. The capsid contains 60 copies of the four-chain asymmetric unit shown in (c), in which the chain pairs A:B and C:D form the tight, primary interface. The secondary interface, shown in (b), is formed by interactions between chains A (green, forming the pentagon) and chains C (magenta, forming the triangle) of neighboring units. (d) A difference in backbone conformation of chains A/C vs B/D (backbone rmsd 0.6 Å) results in a quasi-identical interface connecting the pentagon and triangle together through interface [2’] of (b). (e) shows the overlap of chain A of the target to its analogue in the template 3j2v, highlighting the regions that needed to be modeled correctly for an accurate prediction of both interfaces.
OVERVIEW OF THE PREDICTION EXPERIMENT
As in previous CASP-CAPRI challenges and in standard CAPRI Rounds, predictor groups were provided with the amino-acid sequence or sequences of the target proteins, usually those of the constructs used to determine the structures. In addition, predictors were given information on the biologically relevant oligomeric state of the protein, provided by the author for most targets, the stoichiometry of the complex and occasionally, some additional relevant details about the protein.
Following the common practice in CAPRI, predictors were invited to submit 100 models for each target, to be used for the scoring challenge (see below). It was stipulated however, that only the 5 top-ranking models would be evaluated. To continue monitoring the ability of predictors to reliably rank their models, we also report the performance of groups on the basis of their single top-ranking models.
Scoring experiments were run for all 12 targets. After the predictor submission deadline, all the submitted models (100 per participating group) were shuffled and made available to all the groups participating in the scoring experiment. The ‘scorer’ groups were in turn invited to evaluate the ensemble of uploaded models using the scoring function of their choice, and to submit their own 5 top-ranking ones. Scorer results based on their top-1 ranking models are also reported. Typical timelines for the prediction and scoring experiments were 3 weeks and 5 days, respectively.
Round 50 participants were invited to submit their models to the CAPRI-EBI management system. In preparation for the CASP14 assembly prediction, the CAPRI management system was updated to generate CASP compliant versions of the 5 top ranking models submitted to CAPRI by predictor and scorer groups, and these compliant versions were automatically forwarded to CASP. With very few exceptions this procedure worked very well, affording a seamless communication between the CASP and CAPRI management teams.
The number of CAPRI groups submitting predictions and the number of models assessed for each target are listed in the Supplementary Material (Table S1). For Round 50 targets, 25 CAPRI groups submitted on average ~1250 models per target of which ~1500 were assessed here. On average 20 scorer groups submitted a total of ~190 models per target, of which a total of ~1200 models were assessed.
ASSESSMENT METRICS AND PROCEDURES
For ready comparison with the results obtained in previous CAPRI Rounds and previous CASP-CAPRI experiments 35,36, models were evaluated using the standard CAPRI assessment protocol. This protocol was complemented with the DockQ score 44, a continuous quality metric that integrates the main quality measures of the standard CAPRI protocol (see details below).
In addition, we evaluated the quality of the predicted protein-protein interfaces in the submitted models, namely the extent to which residues from each of the contacting subunits that make up the binding interface are correctly identified. This is a distinct problem from that of accurately predicting the detailed atomic structure of the binding interface and of the protein complex (or assembly) as a whole. It requires identifying only the residues from each subunit contributing to the interface (as opposed to predicting their contacts) 41 and was therefore assessed separately.
The CAPRI assessment and ranking protocols
The standard CAPRI assessment protocol 39,40 was used to evaluate the quality of the predicted homo- and hetero-complexes. This protocol uses three main parameters, f(nat), L_rms, and i_rms to measure the quality of a predicted model. f(nat) is the fraction of native contacts in the target that is recalled in the model. Atomic contacts below 3 Å are considered clashes and predictions with too many clashes are disqualified (for the definition of native contacts, and the threshold for clashes see reference 39). L_rms is the backbone rmsd (root means square deviation) over the common set of residues (across all submitted models) of the ligand protein, after the receptor protein has been superimposed, and (i_rms) represents the backbone rmsd calculated over the common set of interface residues after these residues have been structurally superimposed. An interface residue is defined as such, when any of its atoms (hydrogen atoms excluded) are located within 10 Å of any of the atoms of the binding partner. On the basis of the values of these 3 parameters models are ranked into 4 categories: high quality, medium quality, acceptable quality and incorrect, as previously described 35.
For targets representing higher order oligomers featuring multiple distinct interfaces, submitted models were evaluated by comparing each pair of interacting subunits in the model to each of the relevant pairs of interacting subunits in the target35. The quality score for the assembly as a whole, ScoreA was computed as a weighted average as follows:
| (1) |
Where nACC, nMED and nHIGH are the number of interfaces of the assembly for which at least 1 acceptable-, medium- and high- quality model respectively, was submitted among the top 5 ranking models. The values of the weights ‘ω’ were taken as ω1 = 1, ω2 = 2 and ω3 = 3. For the purpose of ranking the performance of individual groups across all targets we used the normalized version of Eq. (1): , where K is the number of evaluated interfaces. This was done in order to avoid large assemblies with multiple interfaces weighing too heavily on the global score of individual groups (ScoreG of Eq. (2) below).
The quality of the modeled 3D structure of individual subunits was also evaluated by computing the ‘molecular’ root mean square deviation (M-rms), of backbone atoms of the model versus the target. It was used mainly to gauge the influence of the quality of subunit models on the predicted structure of the assembly. To further evaluate the accuracy of the modelled protein-protein interface we also computed the root mean square deviation of sidechain atoms (S-rms) of residues at the binding interface. This measure uses the backbone rms fit of the i_rms calculation, to compute rms values over side-chain atoms only. It is not used in the classification of models.
The performance of predictor and scorer groups and servers was ranked on the basis of their best-ranking model in the 5-model submission for each target. The final score assigned to a group or a server was expressed as an analogous weighted sum to that of Eq.(1), but considering the performance for individual targets, expressed in each of the three categories (acceptable, medium and high), achieved by that group or server over all targets:
| (2) |
Where NACC, NMED and NHIGH are the number of targets of acceptable-, medium- and high- quality, respectively, and the values of weights ‘ω’ were taken as ω1 = 1, ω2 = 2 and ω3 = 3.
This ranking method was already used in the CASP13-CAPRI challenge 38, and the latest CAPRI assessment 45. It takes into account all models of acceptable quality or higher submitted by a given group. For larger assemblies it takes into account the model quality as defined by the value of <ScoreA> for the assembly, defined above.
Additional assessment measures
To enable a higher-level analysis of the performance across targets, we used a continuous quality metric as formulated by the DockQ score, to evaluate each modeled interface 44 :
| (3) |
With
where f(nat), i_rms, and L_rms are as defined above. The rmsscaled represents the scaled rms deviations corresponding to either L_rms or i_rms and di is a scaling factor, d1 for L_rms and d2 for i_rms,, which was optimized to fit the CAPRI model quality criteria, yielding d1 = 8.5 Å and d2 = 1.5 Å (see ref. 44)
Evaluating predicted interface residues
Models submitted by CAPRI predictor scorer and server groups were also evaluated for the correspondence between residues in the predicted interfaces and those observed in the corresponding structures of the 12 targets of Round 50. A total of 23 distinct protein-protein interfaces, sometimes representing more than one interface for each interacting component, were evaluated. The number of interfaces evaluated for individual targets in both categories (easy and difficult) are listed in Table 1. Interface residues of the receptor (R) and ligand (L) components in both the target and predicted models were defined as those whose solvent accessible surface area (ASA) is reduced (by any amount) in the complex relative to that in the individual components 41. This is a more stringent definition of interfaces residues than the one in the official CAPRI assessment protocol, where residue-residue contacts and backbone conformation are being evaluated. As in the official CAPRI assessment the surface area change was computed from the structures of the individual components in their bound form.
The agreement between the residues in the predicted versus the observed interfaces was evaluated using the two commonly used measures, Recall (sensitivity) and Precision (positive predictive value). Recall is denoted as f(IR), the fraction of the residues in the target interface that are part of the predicted interface. Precision = 1 – f(OP), where f(OP) is the fraction of the residues in the predicted interface that are not part of the target interface, i.e. overpredicted or false positives.
RESULTS AND DISCUSSION
This section is divided into 5 main parts. The first part presents the results of human predictors, servers and scorer groups for the 12 individual CAPRI Round 50 targets for which the prediction and scoring experiments were conducted. In the second part we present the rankings of the same groups established on the basis of their performance across all targets. In the third part we report results of the binding interface predictions obtained by the different categories of participants for all targets. The fourth and final part analyzes methods and factors that may have influenced the prediction performance.
Predictor server and scorer results for individual targets
Detailed results obtained by all groups (predictors, servers, and scorers) for individual targets analyzed in this study can be found in Tables S2 and S3 of the Supplementary Material. Results of the CAPRI evaluation for predictor groups that submitted models only to the CASP prediction center are also included, but will only briefly discussed, since their performance is evaluated in a separate publication (Ozden & Karaca, this issue). Values of all the CAPRI quality assessment measures for individual models submitted by CAPRI participants for the 12 Round 50 targets have been communicated to the participants and will be posted on the CAPRI website (URL: http://pdbe.org/capri). Additional information on the performance of individual groups can be found in the Supplementary Material (Individual Group Summaries).
Easy Dimer targets: T164, T166
The two easy dimer targets were, the homodimer of SMCHD1 (Structural maintenance of chromosomes (SMC) flexible hinge domain-containing protein 1 (T164/ T1032), and the PEX4/PEX22 heterodimer from Arabidopsis thaliana (T166/H1045). The homodimer of T164 featured a sizable interface (1585 Å2 buried area), and several medium quality templates ~30% sequence identity; backbone rmsd values ~2.8 Å), displaying similar interfaces to that of the target, were available. For the hetero dimer (T166), which featured a rather small interface (765 Å2), several good quality templates (21–39% sequence identity; backbone rmsd values 0.5–1.6 Å) were available for each of the subunits, in addition to a good quality template for the complex as a whole.
As expected for this type of targets, models of acceptable quality or higher were submitted by a majority of the CAPRI predictor groups and servers (19/23) for T164. However, only two predictor groups (Gray and Seok) and 1 docking server (MDOCKPP) submitted at least one medium quality model among their top 5 models, whereas none of the groups or servers submitted a high-quality model (Supplementary Table S2). A better performance overall was obtained for T166. A majority of the CAPRI predictor and server groups (18/24) submitted correct models for his target, of which as many as 12 groups (but no server) submitted at least 1 medium quality model and 3 groups (Chang, Venclovas and Takeda-Shitaka) submitted 1 high quality model each (the model of Venclovas featured the highest f(nat) value (0.81), that of Takeda-Shitaka the lowest i-rms (0.74 Å), and the Chang model had the lowest L-rms (2.11 Å). Lastly, 4 servers (GALAXYPPDOCK, MDOCKPP, SWARMDOCK, HDOCK) submitted at least 1 acceptable model each among their top 5 models (Supplementary Table S2).
Of the 8 servers submitting models for T164, 6 submitted correct models, whereas only 1 server (MDOCKPP) submitted a medium quality model for this target. Of the 7 servers submitting models for T166, only the above mentioned 4 servers, each submitted 1 correct model for this homodimer.
Seventeen groups and servers participated in the scoring experiment for T164, and all of those submitted at least one correct model or better among their top 5 ranking models, a rather good performance. Two scorer groups (Bates and Huang), and 3 scorer servers (SWARMDOCK HDOCK and MDOCKPP) submitted medium quality models, whereas the remaining 12 groups and servers submitted acceptable models. Interestingly, the 2 best performing scorer groups and the SWARMDOCK servers (from the Bates group) submitted a medium quality model as their top 1 ranking one, whereas none of the manual predictor groups or servers had such models ranked on top. Scorer groups and servers also performed well for T166. Of the 19 groups participating in the scoring experiment for this target 2 human scorer (Kihara, Takeda-Shitaka) and 1 scoring server (LZERD, by the Kihara group) produced high quality models among their top 5 scoring models, 11 groups and servers produced medium quality models, and 1 group submitted an acceptable model.
Difficult Dimer Targets: T169, T176, T178, T179
These difficult dimer targets included the outer-membrane lipoprotein homodimer from Acinetobacter baumannii (T169/T1054), for which only a distantly related template, adopting a different binding mode from that of the target was available, and as a result no acceptable models were submitted by any of the predictor groups, even though the target dimer features a large buried surface area (1530Å2).
The prediction performance was a little better for T176/T1087, the SSCRP protein, although only very distantly related templates were available for this target (rmsd of 3.9–6.8 Å for the individual subunits; seq-ID of ~11–17%), which furthermore displayed binding modes that differed from that of the target. Yet, 2 predictor groups (Zou and Seok) and the server MDOCKPP submitted acceptable models among their top 5 scoring predictions. On the other hand, nearly half of the participating scorer groups and servers (8/19) were able to identify a correct model in the shuffled set of models offered for scoring, and these included 3 servers (SWARMDOCK, MDOCKPP and HAWKDOCK) in addition to 5 human scorer groups.
Interestingly the AlphaFold2 procedure of Google DeepMind did predict a highly accurate structure for the bound subunit for T176 (with 93% of the Cα atoms of the structure lying with 1 Å of their positions in the target). Had this structure been available to participants for assembly modeling, medium to high quality models would have been obtained, because docking calculations tend to yield more accurate models when using as input the bound structures of the interacting subunits39.
The difficulty with the remaining 2 targets of this category (T178/T1083, T179/T1087) stemmed from the fact that they comprised two very long helical hairpin structures bound to one another, where the main challenge resided in uniquely aligning the helical subunits relative to one another. The very distantly related templates, available for these targets (rmsd 4.7–5.7 Å, seq-ID ~ 7%), were all of higher order helical assemblies, and were therefore of limited relevance.
Nevertheless, of the 26 predictor and server groups submitting prediction for T178/T1083, 12 groups including 3 servers (LZERD, MDOCKPP, HAWKDOCK) submitted an acceptable quality model as one of their top 5 predictions, and only one other group (Venclovas) submitted a medium quality model. The five remaining participating servers submitted only incorrect models. The performance of scorers and scoring server groups was somewhat better than that of predictors. Half of the 19 participating groups submitting at least 1 model of acceptable quality or better (among their top five-ranking models), with however 2 human scorers (Takeda-Shitaka and Chang) and 1 server (HAWKDOCK) submitting medium quality models (Table S2). Interestingly, the scorer group of Venclovas was unable to identify their own medium quality model in the shuffled set and ended up submitting only an acceptable model.
A very similar performance was obtained for T179/T1087. Ten out of the 24 participating human predictor and server groups all submitted only 1 acceptable model for this target. Two of these acceptable models were submitted by the LZERD and MDOCKPP servers, whereas only incorrect models were submitted by the remaining 6 participating servers.
It is again noteworthy that the AlphaFold2 procedure of Google DeepMind did predict a highly accurate structure for the bound subunit of this target (96% of the Cα atoms of the structure lying with 1 Å of their positions in the target). Had this structure been available to participants, most likely more accurate models would have been obtained.
Trimer Targets: T165, T168, T174
These trimer targets include 2 difficult targets, the monoclonal Ab bound to the varicella-zoster virus glycoprotein gB (T165/T1036), and the phage tail attachment region protein (T174/T1070), and one easy target, the tail fiber of the salmonella virus epsilon15 (T168/T1052).
For T165, the main challenge was to predict the binding mode of the monoclonal Ab to the protein trimer, and not to model the viral glycoprotein trimer itself, for which a closely related template was available for the full trimer (backbone rmsd 1.0 Å, seq-id 60%). T174 was a difficult modeling problem, because templates could not be identified even for the individual protein chain, whereas modeling the timer in T168 was an easy problem, given that high quality templates (backbone rmsd 0.76Å, seq-id 42%) were available for the tail fiber viral protein.
For T165/T1036, where we evaluated only the binding mode with the monoclonal Ab, and for T174/T1070, where the full assembly was evaluated, only incorrect models were submitted (see supplementary Table S2). Unsurprisingly in contrast, a very good prediction performance across predictors and servers was obtained for T168/T1051. Of the 24 participating predictor and server groups, 16, including 4 servers (GALAXYPPDOCK, LZERD, MDOCKPP, SWARMDOCK) submitted at least one medium quality model among their top 5 ranking ones, and 2 additional predictor groups submitted 1 acceptable model each. As expected from the good performance of predictors and servers, who contributed many medium quality models to the shuffled set offered for scoring, the scorer performance was very good as well, with all but 2 of the 17 scorer groups submitting at least one medium quality model among their top 5 ranking ones.
Large Assembly Targets: T170, T177, T180
These 3 targets, the component of the T5 phage tail distal complex (T170/H1060), the bacterial arginine decarboxylate from (T177/T1081) and the duck hepatitis B virus capsid (T180/T1099), were all large multi-protein complexes, whose 3D structure was determined by cryo-EM. These large assemblies comprised between 20–240 subunits. They featured different internal symmetries, with protein subunits engaging in several distinct binding modes involving interfaces of varying sizes. Therefore, correctly, not to mention accurately, modelling the 3D structure of the full assembly for each of these targets represented a very challenging prediction problem.
For multi-protein assemblies such as these, predictions were evaluated for individual interfaces of each target, as well as over the full assembly. In the latter case the ScoreA expression of Eq (1) was used. The prediction performance of predictor server and scorer groups for individual interfaces of each target is provided in Table S2 of the Supplementary Material, whereas the performance of the same individual groups for the assembly as a whole, can be found in supplementary Table S3.
The T177/T1081 assembly of the 2 stacked decamers each adopting D5 symmetry, was undeniably the easiest assembly modelling problem as at least one closely related template (backbone rmsd 0.46Å, Seq-ID, 71%) was available for the entire decameric ring. The assembly features a total of 4 distinct interfaces (I.1-I.4). Three of these are within rings, comprising 2 quite larges interfaces, burying respectively 5000 Å2 (I.1) and 1250 Å2 (I.2), and another very small interface (180 Å2). Only one distinct quite small interface (300 Å2) (I.3), formed diagonally between subunits in different rings and repeated 5 times, affords the inter-ring contacts (Figure 2).
The prediction performance was evaluated for the 2 large interfaces within each ring (I.1, I.2), and the intra-ring interface I.3. Given that a high-quality template was available for the decameric rings, the main challenge for this target was predicting the inter-ring contacts (I.3). Not too surprisingly, given the closely related template for the decameric rings, an excellent prediction performance was obtained for the 2 large intra-ring interfaces I.1, I.2, but a lower performance was achieved for I.3 (Table S2). For example, of the 24 predictor and server groups submitting models for I.1, 17 groups including 5 servers (SWARMDOCK, HDOCK, MDOCKPP, CLUSPRO, LZERD), submitted between 2–5 high quality models among their top 5 ranking models. Another 3 groups (including 1 server: GALAXYPPDOCK) submitted 5 medium quality models.
As expected, an excellent performance for I.1 was also obtained by scorer groups, with 16 out the 18 scorer groups (6 servers included), all submitted between 3–5 high quality models among their top 5 ranking predictions. A very similar tally of high-quality models was obtained across different groups for I.2.
The main challenge posed by T177/T1081, namely, to correctly predict the smaller inter-ring interfaces (I.3), was met by a smaller number of groups and servers, and consequently by scorer groups as well. Among the 24 predictors and server groups submitting model for this inter-ring interface, only one server (MDOCKPP) submitted 1 high quality model (and 4 medium quality ones) as their top 5 ranking ones for this interface. This server thereby surpassed the performance of other groups (Venclovas, Zou, Grudinin, Kozakov/Vajda) and the CLUSPRO server that submitted at best 1–3 medium quality models or only acceptable quality ones (Table S2). The best performing servers were MDOCKPP, SWARMDOCK and CLUSPRO. Somewhat better performance was obtained by scorer groups, with 3 groups (2 servers: SWARMDOCK and HAWKDOCK, and the human scorer Bates), submitting at least 1 high quality model for I.3, and 7 additional groups (including the MDOCKPP, and LZERD server) obtained at least one medium quality model among their top 5 ranking predictions.
Combining the performance across all 3 distinct interfaces of T177/T1081, using the scoring scheme of Eq (1), yields the overall ranking of predictor and scorer groups for the assembly (see Supplementary Table S3). Of the 6 top-ranking predictor groups and servers, submitting models of medium quality or higher for all 3 interfaces, the MDOCKPP ranked first. This server was the only participant submitting high quality models for all three interfaces, including the more challenging inter-ring interfaces (I.3). This top performer is followed by 4 human predictors (Zou, Venclovas, Pierce, Kozakov/Vajda) and the CLUSPRO server, all submitting high quality models for the 2 intra-ring interfaces, and a medium quality model for I.3. Four additional CAPRI groups, and 2 servers (SWARMDOCK, HDOCK), managed only an acceptable model for the I.3, in addition to high quality models for Interfaces I.1 and I.2. Of the 13 CASP predictors, only 5 groups submitted correct models for all 3 interfaces, but only medium and acceptable quality models for interface I.3.
Not surprisingly, the scorer performance was excellent overall (Table S3). Three scorer groups: 2 servers (HAWKDOCK and SWARMDOCK) and the human scorer group of Bates (author of SWARMDOCK), submitted high quality models for all three interfaces of the target. Eight additional groups (including the LZERD and MDOCKPP servers), submitted models of medium quality or better for all three interfaces, and three groups also correctly predicted all three interfaces albeit to lower accuracy.
On the basis of these combined results this assembly can be considered as quite successfully predicted overall. The best model overall was submitted by MDOCKPP and the scoring server HAWKDOCK, as their second-highest ranked model in both cases. It features an average DockQ value of 0.87 ± 0.02, corresponding to f(nat) values of 0.7–0.85, L_rms values of 0.8–1.2 Å and i_rms values of 0.6–0.7 Å for the three interfaces.
Next in terms of the modeling challenge was T180/T1099, the duck hepatitis B virus capsid. This capsid adopts a T=4 icosahedral symmetry with a total of 240 subunits, comprised of identical protein chains. Structurally the subunits assemble into 60 identical copies of an asymmetric unit composed of 4 helical proteins with slightly different conformations (backbone rmsd 0.4–0.74Å). The icosahedral capsid formed by these 60 identical copies engage in a total of 5 distinct interfaces (Figure 4b,c). But the high similarity between the two dimers in the asymmetric unit, and the differences in backbone conformations of the 4 individual subunits of the asymmetric unit, enable the formation of quasi-identical interfaces between the AB dimers in the pentameric face and the CD dimers in the trimeric face of the icosahedron (see Figure 4b,c). As a result, only 2 unique interfaces had to be evaluated for this target: I.1, the larger interfaces between the individual subunits in the AB and CD dimers (1970 Å2), and I.2 the one between subunits B and D between dimers (1100 Å2).
Aware of the high degree of quasi symmetry between the different interfaces forming the capsid of this target, the organizers (of both CASP and CAPRI) invited predictors to submit the minimum number of subunits necessary to include the unique interfaces defining the capsid assembly. As it turned out, many predictor groups were unclear about what this minimum number should be. Only a third of the 125 models submitted by the 25 predictor groups for this target contained 4 subunits (chains), the number of subunits in the asymmetric unit, that were indeed sufficient to define the 2 unique interfaces of this target. A number of other groups submitted assemblies comprising with between 6–20 subunits, and a few groups submitted models with only 2–3 chains.
Several templates of distantly related viral capsids were available for this target. These included the reconstituted hepatitis B viral capsid (3J2V) adopting the same icosahedral symmetry and featuring the most closely similar subunit structure (backbone rmsd 2.0Å). Unfortunately, however, the template protein lacked the crucial insertion (residues 75–125) present in the target protein, which contributes significantly to the target dimer interfaces (I.1) (Figure 4e). This resulted in a very poor prediction performance, with only one predictor group (Seok) submitting a single acceptable model for I.1 among their top 5 submissions, representing a real feat (Table S2), which was achieved with the help of published mutagenesis data on this virus (see the Seok group summary in the Supplementary Material). Of the 18 groups participating in the scoring challenge for this target, only 3 groups (Venclovas, Fernandez-Recio, Huang) and 1 server (Fernandez-Recio’s PYDOCKWEB), were able to identify Seok’s acceptable models for this interface in the shuffled set of models.
It is noteworthy that here too, AlphaFold2 of Google DeepMind predicted a highly accurate model of the individual subunits of the asymmetric unit of the capsid protein (including the extra insertion). Using this model would have certainly enabled more of the participating predictor groups to produce highly accurate models for this interface, and probably for the capsid as a whole, since rather good predictions were obtained for I.2 of this target.
Indeed, a total of 13 groups predictor groups (out of 15) submitted medium quality models for I.2 of T180/T1099, with one group (Venclovas) also submitting 1 high quality model among their 5 top-ranking models, and only 6 groups submitting only incorrect models. Of the 7 participating servers, 3 submitted medium quality models, and 1 server submitted 1 acceptable model. Scorers performed well on this interface, with 11 groups, including 4 servers (LZERD, MDOCKPP, HDOCK, SWARMDOCK) submitting at least one medium quality model, and 6 other groups (including the PYDOCKWEB server) submitting an acceptable model (Table S2). The best performing groups for this interface were the LZERD and MDOCKPP servers, and the group of Zou, but neither was able to identify the high-quality model predicted by Venclovas.
Combining the performance across the two distinct interfaces of T180/T1099, using the scoring scheme of Eq (1), yields the overall ranking of predictor and scorer groups for the assembly (see Supplementary Table S3). The top-ranking groups for this target are Venclovas, who submitted the only high-quality model for I.2, and Seok, with a medium quality model for I.2 in addition to the single acceptable models for the challenging I.1 interface. An additional 12 groups submitted medium quality model (only for I.2), followed by 6 groups who managed only 1 acceptable model for I.2. The best performing prediction servers for this target were LZERD, CLUSPRO and GALAXYPPDOCK. Of the 8 CASP predictor groups for this target (Table S3), 4 groups (Seok-assembly, Kihara-assembly, CoDock and Baker) performed best with 1 medium quality model each, for I.2.
The scorer performance for the assembly was good overall. The best performance was achieved by Huang, the only group submitting correct models for both interfaces: an acceptable model for I.1 and a medium quality model for I.2 (Table S3). Only 3 other groups submitted correct models for both interfaces; all were only of acceptable quality.
By all accounts, the 27-subunit component of the T5 phage tail distal cryo-EM complex (T170/H1060), was the most challenging assembly prediction problem of the entire Round. This component included a total of 4 multi-subunit rings (A-D) stacked on top of one another (Figure 3a). Rings A and B each comprise 3 copies of protein A (464 residues). Ring C comprises 2 concentric rings: an inner ring composed of 3 copies of protein B (298 residues), and an outer ring with 12 copies of protein C (140 residues). Ring D is composed of 6 copies of protein D (204 residues) (bold underlined capital letter are ring identifier; capital letters are protein identifiers).
Closely related templates were available for proteins A and D (monomeric forms), and a rather distantly related templates were available for proteins B and C (see Figure 3b for details).
The 27 subunits of the assembly form a total of 9 unique pairwise interfaces within and between rings. The area buried in these interfaces, the subunits that contribute to each interface (using the chain identifiers provided by the authors) and the total area buried between neighboring rings is listed alongside in Figure 3c.
On the basis of the available templates, and the buried areas between the subunits, the 3 unique interfaces of ring C (interfaces I.5, I.6, I.7), involving proteins B and C, were expected to be the most difficult to predict, whereas the remaining 6 interfaces (I.1-I.4, I.8-I.9) seemed to represent easier prediction problems (see Figure 3c for details). These expectations were partially borne out by the prediction results (Table S2). The best prediction performance was obtained for interfaces I.1 (between subunits within rings A and B), I.5 (between subunits within the outer C ring), and I.8 (between subunits within ring D). For I.1, 13 out of the 22 predictors groups submitted at least 1 acceptable model or better among their 5 top ranking models, among which 2 servers (HDOCK and MDOCKPP), and 3 human predictors (Huang, Shen, Zou) submitted at least one medium quality model. Scorers performed extremely well for this interface, with all 17 scorer groups submitting acceptable models or better, and more than half of these submitting at least 1 medium quality model. For interface I.5, more than half of the predictor groups and one server (CLUSPRO) submitted a model of acceptable quality (7 models) or better (5 medium quality models). Superior performance was achieved by scorers for this interface. The majority of the scorer groups (16/17) submitted models of acceptable quality or better. Ten of these groups, including 2 servers (MDOCKPP, PYDOCKWEB), submitted at least 1 medium quality model among their top 5 ranking ones, with the groups of Shen and Takeda-Shitaka as top performers (Table S2).
A weaker performance was observed for I.8, with only one predictor group (Venclovas) submitting a medium quality model, and 6 groups including 1 server (LZERD) submitting at least one acceptable model among their top 5 ranking ones. Scorer groups performed overall better, with 13 out of the 17 scorer groups (including 3 servers: LZERD, MDOCKPP, PYDOCKWEB) submitting acceptable quality models, of which only the Venclovas scorer group submitting a medium quality model. The only intra-ring interface with a very weak prediction performance was that between the subunit within the inner C ring (I.3), due to the more distant relationship of the B protein to the available template (the latter was more closely related to the A proteins forming the A and B rings) (see Figure 4a,b). For this interface only acceptable models were obtained by 4 predictor groups (Venclovas, Seok, Zou, Shen) and one server (MDOCKPP). Many of these models were identified by a majority of the scorer groups, including 2 servers (MDOCKPP, LZERD) (Table S2).
For the remaining 5 unique interfaces of T170/H1060, the best prediction performance was obtained for I.4 and I.9. For I.4, the interface between ring B and Ci (the C inner ring), 3 predictor groups (Venclovas, Chang, Bates) and one server (HDOCK) submitted at least 1 acceptable model, and scorers did quite well with slightly more than half of the groups submitting at least 1 acceptable model. For I.9 the interface contributing to the contacts between ring D and the inner ring of ring C (Figure 4 c,d), five predictor groups (Huang, Shen, Chang, Kihara, Seok,) and one server (HDOCK) submitted at least 1 acceptable quality model, whereas scorers did quite well with a majority (13 out of 17), submitting at least one acceptable model (Table S2). For the remaining 3 interfaces (I.2, I.6, I.7), all of which are inter-rings, only a single but different group each time, submitted an acceptable model for each of these interfaces, with a commensurate poor performance exhibited by scorer groups (Table S2).
Combining the performance across all 9 distinct interfaces of T170, using our scoring scheme yields the overall ranking of predictor and scorer groups for the assembly (Table S3). The Shen predictor group ranks 1st, with correct models submitted for 6 of the 9 unique interfaces of T170, of which 2 were of medium quality. Venclovas and Chang both correctly predicted 5 of the unique interfaces, of which one (a different one for each group) was of medium quality. These are followed by the groups of Changs, Seok, Kihara, Huang and HDOCK (the best performing server), with acceptable models for 5 interfaces, or correct models for 4 interfaces including a medium quality model for one of those. A further 6 groups (and 2 servers: CLUSPRO, MDOCKPP) submitted correct predictions for only 2 interfaces, including a medium accuracy prediction for interfaces I.1 or I.5. Of the CASP groups, only those of DATE, Baker and Takeda-Shitaka, submitted correct models for 2 interfaces, followed by 2 other groups with only one correctly predicted interface.
Interestingly, scorer groups overall outperformed predictors for the full assembly (Table S3). Two groups (Shen and Zou) correctly predicted 6 of the 9 interfaces of T170, including 2 medium quality models for 2 of these, while the groups of Chang and Kihara, also with 6 correctly predicted interfaces, albeit of lower accuracy. Most of the remaining scorer groups produced correct models of lesser accuracy for between 4–5 interfaces of T170.
Performance of CAPRI predictors servers and scorers across targets
Groups (predictors, servers and scorers) were ranked according to their prediction performance for the 12 assembly targets of Round 50. All the rankings presented here consider, as usual, the best model submitted by each group among the 5 top ranking models for each evaluated interface. For dimer targets or other targets where only one interfaces was evaluated, this amounted to considering the best model submitted for the corresponding target. For higher order assemblies where more than one interface was evaluated, the group score of Table S3 normalized by the number of evaluated interfaces for the target was used (see section on CAPRI assessment and ranking protocols). To avoid bias from the poorer overall performance for T170, the most difficult assembly of this Round with 9 distinct interfaces, this target was sub-divided into 3 sub-targets: T170.1 (assembly defined by interfaces I.1-I.4), T170.2 (assembly defined by interfaces I.5-I.7), and T170.2 (I.8, I.9), with each of the sub-targets evaluated as a distinct assembly target, as outlined above. Taking into account the three sub-targets of T170, the total number of evaluated ‘targets’ amounts to 14. Table 2 presents the ranking of groups that submitted predictions for a total of 10 targets or more out of the 14 targets and sub-targets. The full ranked list can be found in Table S4 of the supplementary material. We did not generate separate ranking across easy and difficult targets this time, given the small number of targets overall, and the fact that they included large assemblies, like the T5 phase tail (T170), which features multiple different subunits and interaction interfaces of varying level of difficulty. Trends among predictor and scorer groups in their ability to tackle more difficult modelling problems, will be discussed in the subsequent sections describing global trends.
Table 2–
Overall group performance. Ranking is determined on the combined score (ScoreG of Eq (2)) of the top-5 submission, but performance for top-1 is also listed. The number of targets that a particular group participated in is listed in the column Participation. Ranking is divided between CAPRI predictors, servers, and scorers and scoring servers. The performance of CASP-only predictors is listed but they are not ranked. Their score can however be directly compared to those of the CAPRI predictor groups. Only groups participating in 10 targets or more are shown; the full Table is given as Supplementary Table S4.
| Rank | Predictors | Participation | Top-1 | Top-5 | Score |
|
| |||||
| 1 | Seok | 14 | 8/2** | 9/4** | 13 |
| Venclovas | 14 | 7/2** | 8/1***/3** | 13 | |
| 3 | Chang | 14 | 7/2** | 8/3** | 11 |
| Zou | 14 | 5/3** | 8/3** | 11 | |
| 5 | Kihara | 14 | 5/3** | 7/3** | 10 |
| Pierce | 13 | 6/3** | 7/3** | 10 | |
| 7 | Huang | 14 | 5/3** | 5/3** | 8 |
| Bates, Kozakov/Vajda | 14 | 4/3** | 5/3** | 8 | |
| Fernandez-Recio | 14 | 3/2** | 5/3** | 8 | |
| 11 | Shen | 14 | 3/1** | 6/1** | 7 |
| Vakser | 14 | 3** | 4/3** | 7 | |
| 13 | Nakamura | 11 | 2/1** | 3/2** | 5 |
| 14 | Liwo | 12 | 2 | 3 | 3 |
| Czaplewski | 13 | 2/1** | 2/1** | 3 | |
| Grudinin | 13 | 1 | 1 | 1 | |
|
| |||||
| CASP-only Predictors | Participation | Top-1 | Top-5 | Score | |
|
| |||||
| Baker | 14 | 7/4** | 8/1***/3** | 13 | |
| CoDock | 10 | 5/1** | 6/2** | 8 | |
| Takeda-Shitaka | 14 | 2/1** | 4/1***/1** | 7 | |
| Seok-assembly | 14 | 5/1** | 5/1** | 6 | |
| Kiharalab-assembly | 13 | 3/1** | 5/1** | 6 | |
| Lamoureux | 11 | 3** | 3** | 6 | |
| UNRES | 13 | 2 | 3 | 3 | |
| DATE | 11 | 1 | 2/1** | 3 | |
| Risoluto | 14 | 2 | 2 | 2 | |
| Elofsson | 13 | 1 | 2 | 2 | |
| VoroCNN-select | 13 | 1 | 1** | 2 | |
| Ornate-select | 10 | 1 | 1** | 2 | |
| SBROD | 11 | 0 | 1 | 1 | |
|
| |||||
| Rank | Servers | Participation | Top-1 | Top-5 | Score |
|
| |||||
| 1 | MDOCKPP | 14 | 4/2** | 7/1***/2** | 11 |
| 2 | LZERD | 14 | 4/2** | 6/2** | 8 |
| 3 5 |
GALAXYPPDOCK SWARMDOCK HDOCK, CLUSPRO |
14 14 14 |
5/1** 3/2** 2/1** |
5/1** 4/2** 3/1** |
6 6 4 |
|
| |||||
| Rank | Scorers and Scoring Servers | Participation | Top-1 | Top-5 | Score |
|
| |||||
| 1 | Zou | 14 | 5/3** | 10/3** | 13 |
| Chang | 14 | 6/3** | 9/4** | 13 | |
| MDOCKPP | 14 | 5/3** | 9/4** | 13 | |
| Takeda-Shitaka | 14 | 5/1***/2** | 8/1***/3** | 13 | |
| 5 | Shen | 14 | 5/3** | 9/3** | 12 |
| LZERD | 14 | 7/1***/2** | 8/1***/2** | 12 | |
| 7 | Huang | 14 | 5/4** | 7/4** | 11 |
| 8 | Oliva | 14 | 6/3** | 7/3** | 10 |
| Fernandez-Recio | 14 | 5/2** | 7/3** | 10 | |
| PYDOCKWEB | 14 | 5/1** | 7/3** | 10 | |
| Kihara | 14 | 5/1***/1** | 7/1***/1** | 10 | |
| Bates, SWARMDOCK | 14 | 4/3** | 6/1***/2** | 10 | |
| HAWKDOCK | 10 | 3/2** | 6/1***/2** | 10 | |
| 15 | Venclovas | 13 | 6/2** | 7/2** | 9 |
| HDOCK | 14 | 5/3** | 5/4** | 9 | |
| 17 | Grudinin | 14 | 1 | 5/1** | 6 |
| Bonvin | 14 | 3/2** | 4/2** | 6 | |
Predictor performance
The 4 top ranking predictor groups submitted correct models or better for at least 8 out of the 14 targets, as defined here. These include the group of Seok, with a total of 9 correctly predicted target, of which 4 were predicted to medium accuracy. Next in rank is the group of Venclovas, with 8 correctly predicted targets, including 3 predicted to medium accuracy and 1 to high accuracy, and finally those of Chang and Zou, with 8 correctly predicted targets including 3 medium quality ones. Immediately following are the MDOCKKPP server and the groups of Kihara and Pierce, with 7 correctly predicted targets, including at least 3 targets of medium quality or better. Of the predictor groups who submitted models only to CASP, Baker ranked equal to the best CAPRI predictors, with 8 correctly targets of which 4 were predicted at medium accuracy or higher, and CoDock ranked somewhat lower with 6 correctly predicted targets of which 2 were predicted to medium accuracy.
Server performance.
A total of 8 automatic servers participated Round 50. The ranked performance of 6 of these (each submitting predictions for 14 targets and sub-targets) is listed in Table 2. The best performing server is MDOCKPP, with 7 correctly predicted targets, of which 3 were predicted to medium accuracy or better. The LZERD server follows closely with 6 correctly predicted targets, of which 2 were predicted at medium accuracy. These servers outperform HDOCK and CLUSPRO, two servers that performed particularly well in the CASP13-CAPRI challenge. However, in general, the performance of servers was inferior to that of human predictors, as also highlighted in the individual contributions of participants (see Supplementary Material).
Scorer performance
The scorer performance was overall rather good, and stronger than the performance of predictors and prediction servers. The 7 best performing scorer groups (with score >10 in Table 2) include the MDOCKPP server as top performer, followed by the groups of Zou, Chang, Takeda-Shitalka, the LZERD server, and the groups of Shen and Huang. These scorer groups submitted correct models for at least 7 (Huang) and 10 (Zou) targets, including 2–4 targets predicted at medium accuracy, and 2 groups (the LZERD server and the groups of Takeda-Shitaka), with 1 target predicted at high accuracy.
Lastly it is noteworthy, that the data on the global group ranking of Table 2, and those of Tables S2 and S3, indicate that most predictor groups have improved their ability to rank models. The number of targets for which these groups have a model of acceptable quality or higher ranked on top (top1) is often only slightly lower than when their top-5 ranking models are considered. Prediction servers, and even more so, scorers and scoring servers, are less consistently successful in having their best quality models ranked on top.
Prediction of binding interfaces
Interface predictions were evaluated for 23 binary association modes in the top 5 scoring models submitted for the 12 targets by CAPRI predictors groups (human and servers), as well by CAPRI scorer groups (human and server). The correspondence between the residues defining the interfaces of the individual protein components of each binary association mode in the predicted models and those in the target structure was evaluated using the Recall and Precision measures (see section on Assessment Criteria and Procedures, for further detail).
Global trends
Figure 5 presents scatter plots of the recall and precision values of predicted interfaces for components (receptor and ligand) of the top 5 models submitted for each of the 23 evaluated association modes by predictor and scorer groups. Individual points represent values averaged separately over interfaces of association modes in each of the four categories (incorrect, acceptable, medium, and high) submitted by a given group for a given target.
Figure 5: Global landscape of the interface prediction performance.
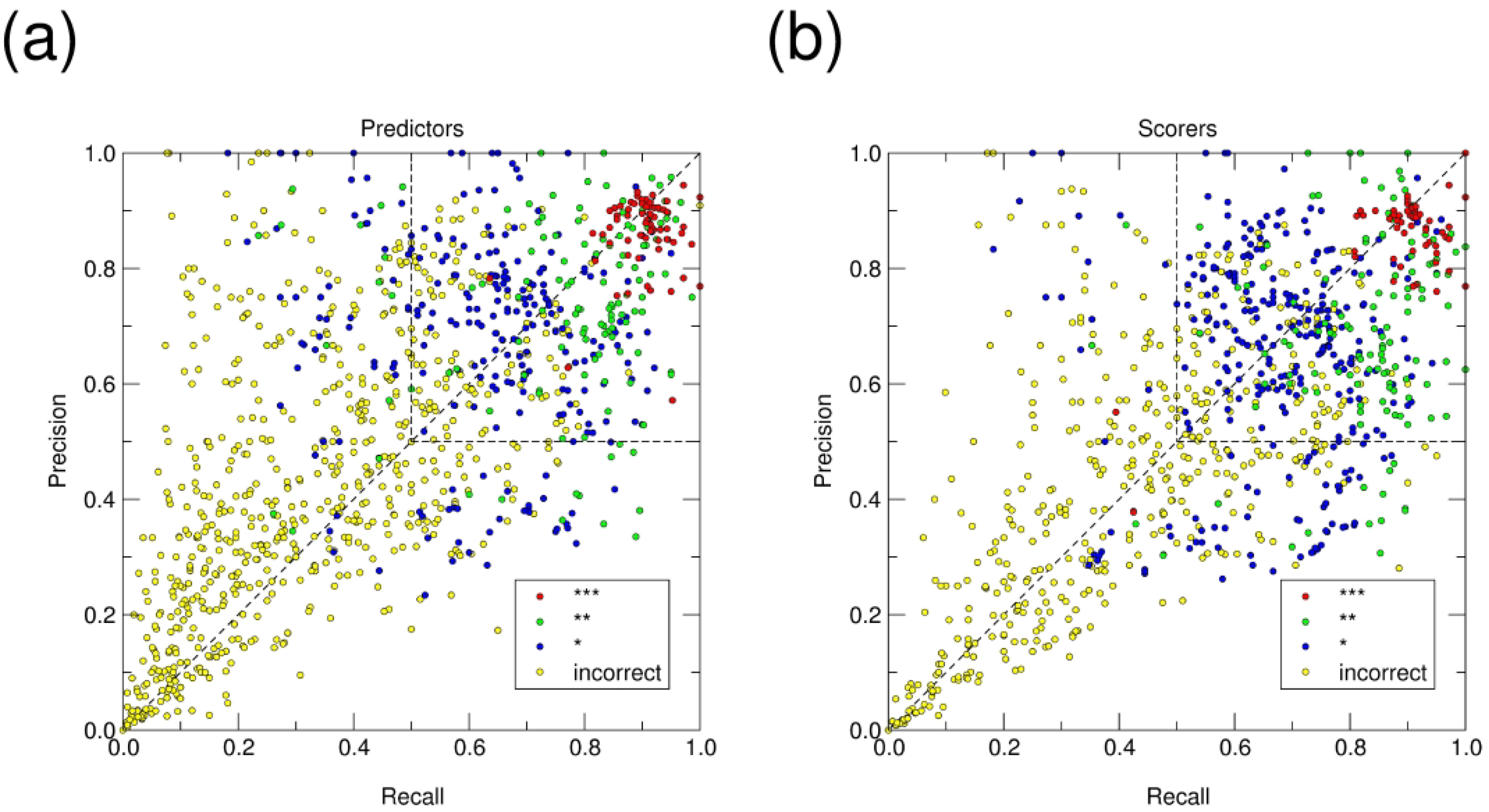
Scatter plot showing the average Recall and Precision values (see main text for definition) of the interfaces in models submitted by all predictors (a) and scorers (b) for the 12 targets of Round 50. Each point represents the average Recall and Precision values for the interfaces of the individual protein components (i.e. the receptor and ligand proteins, respectively) in the 5 models submitted by each participant for one binary association mode. Averaging was performed separately over models in the 4 CAPRI accuracy categories (incorrect, acceptable, medium, and high). For example, for a participant submitting 5 models or which 2 were incorrect, 2 of medium quality and 1 of high quality, average Recall and Precision values were computed for the 2 incorrect models, and the 2 medium-quality ones, respectively, whereas those for the single high-quality models were used as such. Individual points are color-coded by the CAPRI model quality category (as indicated in the legend displayed in the upper left corner of each graph). The upper right-hand quadrant of the graph, with Recall and Precision values above 0.5, contains all points corresponding to “correct” interface predictions.
The 2 salient outlier green points in (a) correspond to the medium accuracy models with high f(non-nat) values submitted by Kozakov/CLUSPRO for the T170.5 interface. The 2 salient outlier red points in (b), correspond to the high accuracy models with however high f(non-nat) values submitted by the group of Zou for the T177.2 interface.
Inspection of the scatter plots reveals that predicted interfaces in the models submitted by both predictors (Figure 5a) and scorers (Figure 5b) span a wide range of recall and precision values. Confirming our previous reports 36,41 we observe that a sizable fraction of the points corresponding to interfaces of incorrect models cluster loosely along the diagonal at very low values, whereas the vast majority of acceptable and higher quality models feature interfaces with recall and precision values ≥ 50% (upper-right quadrant of the scatter plots in Figure 5), which we consider here as the threshold for correct interface predictions. At the same time, a sizable fraction of the points in Figure 5 is spread widely above and below the diagonal. In addition, we see that the fraction of models with higher Recall than Precision values submitted by predictors is smaller (36%) (Figure 5a) than for models submitted by scorers (53%) (Figure 5b). This difference is more pronounced for the incorrect models, and for a fraction of the acceptable models, but becomes much less pronounced for models in the upper right quadrants for points representing models with both Precision and Recall ≥ 50%. Higher precision than recall values correspond to predicted interfaces of smaller size that capture only a fraction of the native interfaces, while including only a few additional residues, and may hence be of predictive value. Interfaces with lower precision than recall values, corresponding to points located below the diagonal, and more particularly the points in the lower left quadrant of the plots in Figure 5 are problematic, and with a few exceptions correspond to incorrect models.
We confirm previous findings that, a) a fraction of incorrect models features in fact correctly predicted interfaces and b) a fraction of correctly predicted interfaces corresponds to incorrect models 36,41. We find indeed that in Round 50, 15.25% the incorrect models submitted by predictors and servers have recall and precision values above 0.5, hence representing correctly predicted interfaces as defined here. For models submitted by scorers this fraction is nearly twice as high (26.35%). Both values are roughly in the range observed earlier: in the CASP13-CAPRI challenge the values ranged between ~11–12% for models of predictors and scorers38, they were 16% in the CASP12-CAPRI challenges 34 and 24% in the initial CAPRI evaluation in 2010 39. At the same time, the fraction of incorrect assembly models in the submissions with correctly predicted interfaces is 29 %, compared to 19%, and ~27% in the CASP13-CAPRI and CASP12-CAPRI challenges, respectively.
The fractions of acceptable and higher quality models featuring correctly predicted interfaces are now 68% and 87%, respectively (reaching 100% for only high-quality models), essentially the same as in the CASP13-CAPRI challenge, and lower than earlier values: 87% and 98% (CASP12) 34, and 92% and 100% respectively, in 2010 39. We also see that medium quality models tend to have higher recall than precision values (although both values are mostly above 0.5), whereas the opposite trend is displayed by acceptable models which are of lower accuracy.
Performance of predictor server and scorer groups
The ranking of groups by their interface prediction performance is listed in the supplementary Table S5. Group performance was ranked on the basis of the fraction of correctly predicted interfaces (interfaces with both recall and precision ≥ 0.5), in the top 5 submitted models for each target.
Nine CAPRI human predictors (Huang, Liwo, Czaplewski, Venclovas, Kozakov/Vajda, Shen,Zou, Bates Grudinin), 7 CASP ones (Risoluto, Elofsson, Seok-assembly, UNRES, Kihara-assembly, Ornate-select, Lamoureux), and 4 prediction servers (MULTICOM-CLUSTER, HDOCK, GALAXYPPDOCK, LZERD) submitted correct predictions for at least 20% of the interfaces. The best performing CAPRI predictor groups were Huang, Liwo and Czaplewski with correct predictions for 27% of the evaluate interfaces, followed by Venclovas who correctly predicted 24% of the interfaces but to a higher accuracy as judged by the corresponding average recall and precision values (Table S5), which remained unmatched by the top 7 CASP predictors, or the 4 CAPRI prediction servers. Like in the CASP13-CAPRI evaluation, some of the human scorers and scoring servers outperformed human predictors and servers, albeit to a more limited extent. Eight human scorer groups had correct prediction for at least 20% of the interfaces, with Bonvin (30% of correct interfaces), followed by Zou (24%), whose models achieved higher average recall and precision values. Only 2 scoring servers (MDOCKPP and HDOCK) submitted correct predictions for at least 20% of the interfaces, achieving average recall and precision values of 50–57%.
The last 4 columns of Table S5 list the average recall and precision values for interfaces of individual models (top 5) submitted by each group, as well as the corresponding standard deviations. It is noteworthy that the average recall and precision values achieved by the best performing groups or servers rarely exceed 50%, compared to 60% in the CASP12-CAPRI challenge 36. With a few exceptions, higher values obtained by some groups correspond to a lower fraction of correctly predicted interfaces overall. The standard deviations are also larger, routinely between 25–30%, and only somewhat lower than in the CASP13-CAPRI challenge. These results indicate that models for individual targets (even those by the best performing groups) tend to vary substantially in terms of the interface prediction accuracy, and that the interface prediction accuracy has in general declined, relative to achievements in previous CAPRI Rounds.
Lastly, we note that most published interface prediction methods reach average recall and precision levels of ~50% and ~25%, respectively, when applied to transient complexes (see reference 46 for review). The best-performing groups of Round 50 achieve somewhat lower recall levels (33–52%) but higher precision (30–56 %) (Supplementary Table S5), for what is most likely a mixture of transient and obligate interfaces of the evaluated targets (especially in the large assemblies with significant multi-valency involving weaker individual association modes). These results support the conclusions that interface prediction methods which model the association modes with the cognate binding partner retain an advantage over interface prediction methods, which do not use such information.
Global overview of the quality of predicted models
A global overview of the quality of models submitted by predictor (and server) groups for the two targets categories is presented in Figure 6. Figure 6a displays the DockQ model scores, color-coded by the CAPRI model quality categories for all the interfaces in individual models submitted by predictors (left column) and scorers (right column) for each of the 23 binary interfaces of the 12 evaluated targets of Round 50. The predictor and scorer DockQ values are compared with those obtained for the best models submitted by respectively, the predictor and scorer versions of the MDOCKPP server (Zou group), the top performing automatic server in this evaluation. Models produced by these servers are used to gauge the baseline performance, analogous to that by the ‘naïve’ predictions 36, or by the best performing HDOCK server38, used in previous evaluations. Figure 6b presents the same data using box plots, illustrating the DockQ score distributions per model quality and target interface.
Figure 6: Global overview of the prediction performance for targets of Round 50.
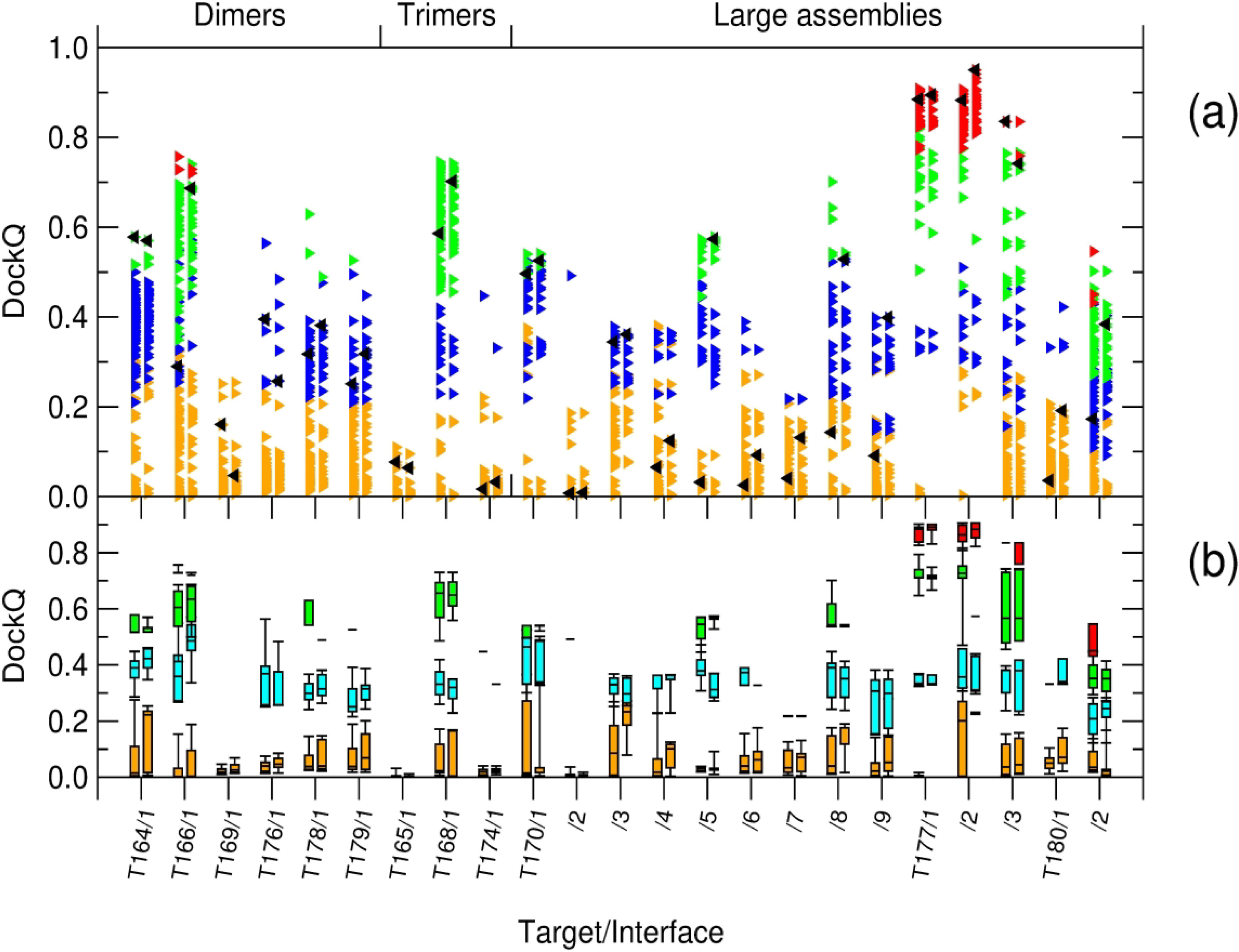
Shown are the distributions of the DockQ values computed for the top-five models submitted by all predictor and scorer groups for individual targets of Round 50. (a) Scatter plots of DockQ values for individual models submitted by predictors (left column) and scorers (right column) for individual targets. The targets are labeled by their CAPRI target number and interface rank. Individual points are color-coded according to the CAPRI model quality category; yellow: incorrect; blue: acceptable; green: medium; red: high. For each target, a baseline-level prediction, represented by the best model of the top-performing automatic server (MDOCKPP; see Table 2), is represented by black triangles. (b) The same information presented as boxplot distributions (whiskers at 9th and 91st percentiles) of models submitted for each target and prediction category; color coding is as for the upper panel, but with a lighter shade of blue for better visibility.
Not too surprisingly, the models produced by predictors and servers for the 2 easy dimer targets (T164, T166) were overall superior to those for the 4 more difficult ones (T169, T176, T178, T179). A good number of medium quality models and 2 high-quality ones were submitted for the heterodimer of T166, but mostly acceptable quality models and only a few medium quality ones were obtained for the T164 homodimer. On the other hand, only incorrect models were obtained for the more difficult T169 dimer, while a small number of acceptable models was generated for T176. For the 2 helical dimers of T178/T179, acceptable quality models were submitted by a good fraction of the groups, whereas a much smaller fraction submitted only medium quality models.
The performance for the 3 trimer targets was mixed. It was poor overall (no correct model) for the 2 difficult targets: for T165, where only the interface with the monoclonal antibody was evaluated, and for T174, the phage tail attachment region protein. But the performance for the easy trimer of T168 was much better, with a majority of the groups (including servers) submitting models of acceptable or medium, although none were of high quality.
The performance across the 14 single interfaces of the large assembly targets was overall above those for the dimer and trimer targets, most likely because adequate templates were available for several of the subcomplexes of these assemblies (e.g. for the ring structures in T170, and T177). A rather good performance was achieved for T177, the arginine decarboxylase, where the main challenge was to correctly predict the inter-ring interface (I.3 or T177/3 in Figure 6), since an excellent template was available for the individual decamer. As expected therefore, the 2 intra-ring interfaces were well predicted by a majority of the groups and servers, with a high fraction of the groups submitting models of medium accuracy or better. The performance was in general lower for the inter-ring interface (I.3, T177/3), with only a single high-quality model submitted by the MDOCKPP server. The global prediction performance for T180, the viral capsid, was disappointing, mainly due to the poor overall performance for interface I.1. The insertion in the target protein was lacking in the available template, resulting in incorrect models being submitted by most groups, except the group of Seok, who submitted the only acceptable quality model for this interface. A much better overall performance was achieved for I.2, for which a large fraction of groups submitted correct models of acceptable quality or better, including the submission of at least 1 high-quality model each, by the groups of Venclovas and Kihara (See Table S2 for detail).
Last but not least, a lower overall performance is observed for T170, the component of the T5 phage tail distal complex, which comprised a total of 9 interfaces. Not unexpectedly, better performance was obtained for the interfaces I.1, I.5, and I.8, all of which are intra-ring (See Figure 4 b,c). These were the only interfaces of T170 for which a fraction of the predictor groups managed to produce medium accuracy models. For 5 of the remaining 6 interfaces, often a smaller fraction of the groups managed to submit at best acceptable models, whereas only incorrect models were submitted for I.2, the interface between the A and B rings (Figure 4 b,c).
We also observe that human predictors produced in general higher quality models than the best performing automatic server (MDOCKPP). This was most prominently the case for interfaces of the large assemblies (interfaces T170/3–9 and T180/1,2), where the server mainly produced incorrect models (Figure 6a). On the other hand, the baseline models produced by the MDOCKPP automatic server were in general on par with those of the best performing manual predictors for the easier-to-model interfaces.
Comparing the quality of models produced by predictors and scorers for the 23 analyzed interfaces, confirms that the best performing scorer groups produce models of similar and sometimes superior quality to those submitted by predictors. This suggests in turn that these scorer groups successfully identify the best models in the shuffled set and often improve their quality through refinement. For about a third of the interfaces, corresponding mainly to the easy-to-model ones, the models of the baseline MDOCKPP scoring server were of similar quality, or better, than those of human scorers (Figure 6a). In addition, the box plots of Figure 6b, which illustrate the DockQ distributions for models in the different CAPRI quality categories, indicate that the distributions for individual categories (incorrect, acceptable, medium and high) tend to be narrower and better separated for the models produced by scorers than those of predictors.
An alternative overview of the quality of the best models submitted by predictors and servers is afforded by plotting the f1 score of the submitted models (a function of the recall, and precision in modeling the residue-residue contact at the binding interface), as a function of the root mean square deviation of the sidechain atoms (S-rms) of interface residues in the model versus the target (see Figure 7 and legend). This plot clearly illustrates that the bulk of the CAPRI ‘acceptable’ models are in fact of rather low quality. Many display low f1 values due mainly to their generously low recall threshold (f(nat) ≥ 0.1) 39, and rather high S-rms values, indicating an overall poor correspondence between the models and target sidechain conformations at the binding interface. A better correspondence with the target interface is displayed by the medium quality models, with most of these models displaying f1 values of 0.4 or higher, and S-rms values < 3.0Å. Nearly all the high-quality models correspond to f1 value > 0.7 with some ranging between 0.8 and 2.5 Å, confirming their high accuracy status. Figure 7 also illustrates the important contribution made by the 3 best human predictors (Seok, Venclovas, Baker), and 2 best servers (MDOCKPP and LZERD), to the more accurate models, and more particularly to the high-quality ones, and that these more accurate models also feature higher residue contact precision and more accurate interface sidechain conformations.
Figure 7: f1 as a function of S-rms.
Each point in the figure represents the best model of a predictor group for each of the 23 interfaces. Individual points are color-coded following the CAPRI model quality as in Figure 6. The results for the best predictors (Baker, Seok, Venclovas) and servers (LZERD, MDOCKPP) are highlighted. See main text for definition of f1 and S-rms. The upper left quadrant features the best models, with S-rms values below 3.5 Å and f1 values above 0.3, corresponding to mostly medium and high-quality models.
Gauging progress
An important goal of community-wide challenges such as CAPRI and CASP, which are repeated over time, is to gauge the progress that is being achieved by the community as a whole in the prediction task that is being evaluated. Assessing progress in predicting the structure of protein-protein complexes and large protein assemblies from blind prediction challenges such as this one is however not straightforward. The problem lies with the small number of targets, in comparison, for example, to the number of targets offered in CASP for the prediction of individual protein chains. This problem is further exacerbated by the substantial variability in the degree of modeling difficulty that these targets represent, leading to significant fluctuations when differences in performance between successive challenges are considered.
Plots quantifying the performance of the 29 top-ranking groups participating in this assembly prediction Round (CASP14-CAPRI) and in the CASP13-CAPRI Round 2-years earlier respectively (Figure 8), illustrate these problems, while at the same time providing useful insights. A clear difference between the 2 challenges is the total number of assembly targets, which as 20 in CASP13 and 12 in this Round. Another is the much larger number of high-quality models (red bars in Figure 8) submitted by the listed groups for many more targets in CASP13, than in CASP14, indicating in turn that most of the targets in the present Round represented more difficult modeling problems.
Figure 8: Gauging progress.
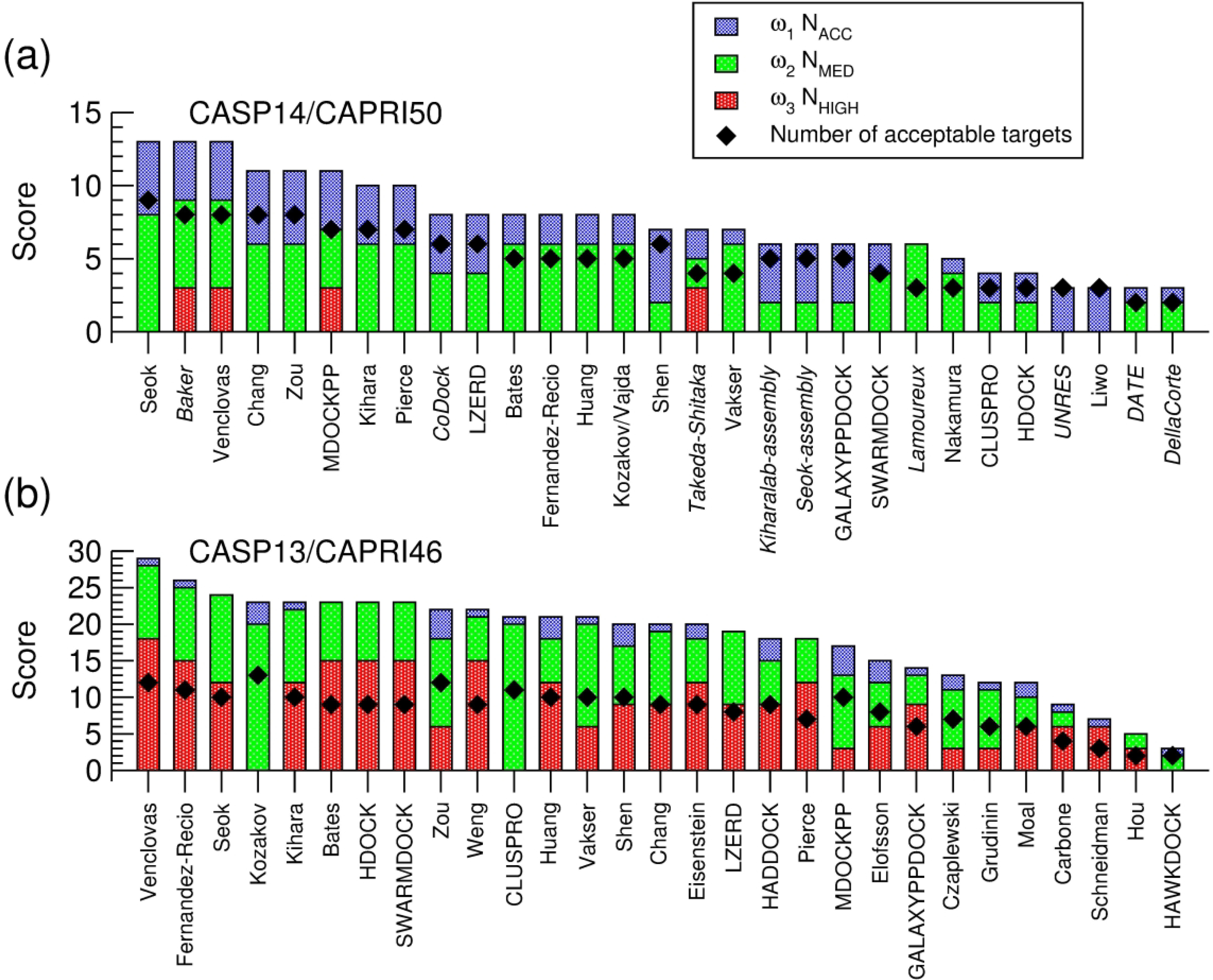
Panel (a) shows the performance score of the top 29 ranking predictor and server groups (both CAPRI and CASP-only groups; server groups are listed in capital letters). The height of the bar is the ScoreG value of Eq. (2), with individual contributions from high, medium, or acceptable-quality models indicated. The total number of targets for which at least an acceptable quality model was produced is indicated in the graph by a diamond. Panel (b) shows the same data from the previous CASP13-CAPRI Round.
Interestingly however, despite the increased target difficulty, the best performing group(s) in the present Round produced acceptable or better models for a higher fraction of the targets (70–75%), than the top performers in CASP13 (65%). Seeing this difference roughly maintained across the ranked predictor groups in both challenges, suggests that the lower overall quality of the models submitted in this Round was counter balanced by more targets being predicted less accurately.
The data in Figure 8 also confirm the consistently high relative performance in both CASP13 and CASP14 of several veteran CAPRI predictor groups, such as those of Seok, Venclovas, Zou, and Kihara. It also indicates the progress achieved by servers such as LZERD and MDOCKPP, developed respectively, by the group of Kihara and Zou. It suggests progress in performance by groups such Chang, and Pierce, and reveals new high ranking CASP groups, such as Baker and CoDock, which were not included in the published evaluation of the CASP13-CAPRI challenge. Lastly, some high-ranking servers in CASP13, such as HDOCK, CLUSPRO and SWARMDOCK, or predictor groups such as Kozakov/Vajda and Bates do not maintain their rank in this Round, which probably illustrates the fluctuations associated with this type of limited analyses, and the particular challenges posed by the targets in this Round.
Factors influencing the prediction performance
Round 50 comprised 12 targets that spanned a range of modeling difficulties. These targets included 3 large multiprotein assemblies involving a total of 14 binary protein-protein interfaces. By choice, the majority of the targets had some templates available in the PDB. The majority of the evaluated interfaces were between homomers, or paralogs. For the ‘easy’ targets, for which templates were available for the entire complex (e.g. the dimers of T164, T166; the T168 trimer, or the decameric ring of T177), the prediction task boiled down to template-based modeling of the entire complex and model refinement. For the more difficult targets, where templates, often more distantly related ones, were available only for the individual subunits, the prediction of the complex required modeling the structures of individual subunits, followed by docking calculations and usually some form of model refinement.
Critical factors influencing the prediction performance were therefore 1) the ability to identify templates whose 3D structure and association modes were similar enough to those of the target, to enable building an accurate model of the target assembly, and 2) the extent to which these models were adequately optimized.
Figure 9 displays the backbone rms values of the individual subunits (M-rms) of the submitted models versus those of the experimental structures for all targets of Round 50. Confronting these values with the DockQ scores of models submitted for the corresponding targets (Figure 6), confirms once more the critical impact that model accuracy of the individual subunits has on the prediction performance. For the easy targets such as, T164, T166, T168, or T177, the majority of the M-rms values do not exceed 2.3–3Å. On the other hand, the subunits of poorly predicted complexes such as the T169 dimer, the T174 trimer, or the T180 viral capsid protein, are much less accurately modelled, with M-rms values commonly reaching 10–15Å, because only poor templates (T169, T180), or no templates (T174) could be identified even for the individual subunits.
Figure 9: Model quality of individual protein subunits in assembly models of the 12 targets of Round 50.
Shown are whisker plots (displaying the median, 1st and 3rd quartile, and 9th and 91st percentile) representing the distributions of M-rms values of individual protein subunits in models submitted for each of the targets of Round 50. Targets are labeled by their CAPRI target number; chain identifiers (A, B, etc) are used for the different proteins in the hetero-complexes.
Evidently, identifying the most adequate template is often not straightforward, as multiple templates are often available either for the full complex or for the independent subunits, requiring strategies for optimally exploiting these data. As described in the summaries by the individual CAPRI groups co-authors of this paper (see Supplementary Material), a variety of approaches were used to tackle this crucial step. A number or groups successfully exploited homology models generated by the best performing CASP14 servers, made available during the prediction Rounds, or used publicly available tools such as Modeller 47. Successful approaches involved searching a database of known structures, clustered on the basis of sequence and structure similarity, and relying on various scoring schemes to select the most suitable templates, or a reduced set of templates, for further refinement. Querying the PPI3D web server 48 (by Venclovas), consulting an in-house database of heterodimers (by Seok) for suitable subsets of templates, or running HHblits 42 against a sequence profile database of known structures clustered at 70% sequence identity, as done by many groups, are good examples of such approaches. When no templates could be found for individual subunits, some CAPRI predictors performed structure-based searches against the PDB by submitting CASP server models to the DALI server 49 (by Venclovas), or used 3rd party servers, such as the MULTICOM-CLUSTER, recently updated to include Deep Learning approaches to predict the 3D structure of individual subunits (see Zou- Supplementary Material). New in this Round, some CAPRI groups such as the one of Kihara, used their own recently developed deep learning algorithm to predict ab-initio the structure of individual subunits50.
Further filtering and refining models built from identified templates is likewise important, and here too, different approaches were rather successful (see Individual Group Summaries). The Venclovas group ranked models based on the combination of the VoroMQA scores for the full structure and for the interaction interface 51 whereas the consensus values of several scoring functions were employed by the group of Zou to select top scoring templates. For some targets, close integration of classical template-based modeling with docking calculation (the so-called hybrid docking strategy), carried out by groups like those of Chang, Venclovas, and Seok, was likewise quite effective.
For the more difficult targets (Table 1), the full assembly was predicted using models of the individual subunits, often built on the basis of more distantly related templates and performing ab-initio docking calculations. Interestingly, a number of groups relied on reputable CAPRI docking servers such as CLUSPRO 52, HEX 53,HADDOCK 54 or ZDOCK 55 developed by other CAPRI groups, to generate their docking poses. Some teams like those of Grudinin, and Venclovas exploited the fast-sampling speed of the HEX and SAM 56 docking programs, to perform cross-docking calculations, whereby sets of models are docked to one another, yielding a large set of assembly models that are then scored and optimized. Increasing use was also made of docking algorithms that incorporate symmetry operations (e.g. HSYMDOCK-lite 57, SymDock2 58), or of algorithms that handle multiple chains (e.g. Multi-LZerD 59,60). Promising new developments were also reported on incorporating protein conformational flexibility, by capturing backbone motions of putative interface residues on-the-fly, using replica exchange methods (Gray)61 or normal mode analysis (Shen)62, a lingering challenge that still needs to be effectively addressed
Several of the best-performing CAPRI groups underscored the importance of specialized functions for scoring and ranking protein-protein interfaces for the entire modeled assembly. But the type of functions differed substantially between participants. Examples are the VoroMQA score developed by the Venclovas group63, the combined use of three scoring functions, GOAP64, Dfire65, and ITScore66 by the Kihara group, or the multi-term scoring function of the Vakser group, additionally complemented with sequence-based measures for individual subunits67 and with functional annotations. The quite successful scoring performance of the groups of Chang and Zou/ MDOCKPP relied on an older knowledge-based scoring function for protein-protein recognition68, which the latter group recently augmented by a deep learning model. In addition, several groups (Cheng, Huang), made good use of deep learning methods for predicting inter-subunit residue-residue contacts from multiple sequence alignments.
For further information on factors potentially influencing the performance of individual groups see Supplementary Material (Individual Group Summaries).
As noted in previous assessments 38 the difficult targets, which involved ab-initio docking of homology-built models, gave an advantage to groups with expertise in ab-initio docking and those with more powerful specialized scoring functions. The latter groups clearly had an advantage in the scoring challenge. We also note that the performance of predictors and scorer groups on the set of difficult interfaces weighed more heavily on their ranking for the full set of targets in Round 50, since about 40% of the 23 unique interfaces across the different targets (~9/23) correspond to difficult modeling problems. The impact on the ranking of groups from their performance on the difficult interfaces in the larger assemblies such as that of T170, was however mitigated by applying the normalized weighted scoring scheme of Eq. (1).
CONCLUDING REMARKS
The assessment of the results presented here for the 12 targets of Round 50, the 4th CASP-CAPRI challenge, provides an informative snapshot of the performance of current methods for the prediction of the 3D structure of protein complexes and larger protein assemblies. It shows that a good number of these methods are capable of producing correct to medium accuracy models for homo-oligomers, ranging from dimers to larger assemblies when templates for the full assembly are available. But generating models that accurately reproduce the native interface is still more an exception than the rule, indicating that further efforts are needed to improve model refinement.
Prediction methods are also increasingly successful when closely related templates for individual subunits are available, thanks to better exploitation of data on templates, more efficient integration of docking procedures, and more powerful scoring functions, although, here too, model refinement remains suboptimal.
On the other hand, producing an accurate 3D structure of protein assemblies, for which only distantly related templates are available for the individual components, or where no templates can be found, remains out of reach for modeling tools such as those currently available to the CAPRI community. To tackle the very challenging problem of predicting protein assemblies from sequence information and limited prior information on the structures of the individual subunits, novel approaches are needed. These approaches must integrate more closely the prediction of the 3D structure of individual protein chains with that of their association modes. That this might be within reach in the near future at least for homomeric assemblies, is suggested by the observation that the novel Deep Learning-based approach by AlphaFold2 appears to accurately predict the bound structure of the individual subunits of these assemblies from their amino acid sequences, at least in cases where residue-residue contacts can be predicted from the amino acid sequence data available in public databases. Very preliminary tests performed by the CAPRI predictor groups of Kozakov/Vajda and Seok (see Supplementary Material) suggest that using subunit models produced by AlphaFold2 as input to ab-initio docking calculations, may indeed increase the number of interfaces predicted to acceptable or medium accuracy levels. Additional tests on a larger and more diverse set of targets and, most likely, significant further efforts will be needed to develop Deep Learning methods capable of predicting the structure of protein complexes, including heterocomplexes, to high accuracy. Several CAPRI groups have already started to address the challenge by developing their own Deep-Learning-based methods to directly tackle key bottleneck in the assembly prediction pipeline62,69–71. As the revision of this manuscript was being completed, DeepMind released an open-source version of their successful AlphaFold2 software72 and the teams of Baker & coll. released RoseTTAFold, a new protein structure prediction tool inspired by AlphaFold 2, that also seems to be able to handle the prediction of protein complexes 73. Future CAPRI and CASP prediction Rounds will monitor the impact of these developments.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the CASP Management and in particular Andriy Kryshtafovych, for valuable help and support in running the assembly prediction challenge and acknowledge close collaboration with the CASP assembly assessor team of Ezgi Karaca. We also express gratitude to the structural biologists who provided the targets for this challenge and to the CAPRI management team and predictor groups for stimulating discussion, valuable input and cooperation.
FUNDING STATEMENT
Please see page 2 of the Supplementary Material.
REFERENCES
- 1.Ideker T, Sharan R. Protein networks in disease. Genome Res 2008;18(4):644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barabasi AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet 2011;12(1):56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lo Conte L, Chothia C, Janin J. The atomic structure of protein-protein recognition sites. Journal of molecular biology 1999;285(5):2177–2198. [DOI] [PubMed] [Google Scholar]
- 4.Dey S, Pal A, Chakrabarti P, Janin J. The subunit interfaces of weakly associated homodimeric proteins. Journal of molecular biology 2010;398(1):146–160. [DOI] [PubMed] [Google Scholar]
- 5.Ponstingl H, Kabir T, Gorse D, Thornton JM. Morphological aspects of oligomeric protein structures. Progress in biophysics and molecular biology 2005;89(1):9–35. [DOI] [PubMed] [Google Scholar]
- 6.Nooren IM, Thornton JM. Structural characterisation and functional significance of transient protein-protein interactions. Journal of molecular biology 2003;325(5):991–1018. [DOI] [PubMed] [Google Scholar]
- 7.Berman HM, Battistuz T, Bhat TN, Bluhm WF, Bourne PE, Burkhardt K, Feng Z, Gilliland GL, Iype L, Jain S, Fagan P, Marvin J, Padilla D, Ravichandran V, Schneider B, Thanki N, Weissig H, Westbrook JD, Zardecki C. The Protein Data Bank. Acta crystallographica Section D, Biological crystallography 2002;58(Pt 6 No 1):899–907. [DOI] [PubMed] [Google Scholar]
- 8.Bai XC, McMullan G, Scheres SH. How cryo-EM is revolutionizing structural biology. Trends in biochemical sciences 2015;40(1):49–57. [DOI] [PubMed] [Google Scholar]
- 9.Cheng Y, Glaeser RM, Nogales E. How Cryo-EM Became so Hot. Cell 2017;171(6):1229–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kundrotas PJ, Zhu Z, Janin J, Vakser IA. Templates are available to model nearly all complexes of structurally characterized proteins. Proceedings of the National Academy of Sciences of the United States of America 2012;109(24):9438–9441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berman HM, Coimbatore Narayanan B, Di Costanzo L, Dutta S, Ghosh S, Hudson BP, Lawson CL, Peisach E, Prlic A, Rose PW, Shao C, Yang H, Young J, Zardecki C. Trendspotting in the Protein Data Bank. FEBS letters 2013;587(8):1036–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ovchinnikov S, Park H, Varghese N, Huang PS, Pavlopoulos GA, Kim DE, Kamisetty H, Kyrpides NC, Baker D. Protein structure determination using metagenome sequence data. Science 2017;355(6322):294–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marks DS, Hopf TA, Sander C. Protein structure prediction from sequence variation. Nature biotechnology 2012;30(11):1072–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ovchinnikov S, Kim DE, Wang RY, Liu Y, DiMaio F, Baker D. Improved de novo structure prediction in CASP11 by incorporating coevolution information into Rosetta. Proteins 2016;84 Suppl 1:67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones DT, Singh T, Kosciolek T, Tetchner S. MetaPSICOV: combining coevolution methods for accurate prediction of contacts and long range hydrogen bonding in proteins. Bioinformatics 2015;31(7):999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Min S, Lee B, Yoon S. Deep learning in bioinformatics. Briefings in bioinformatics 2017;18(5):851–869. [DOI] [PubMed] [Google Scholar]
- 17.Wang S, Sun S, Li Z, Zhang R, Xu J. Accurate De Novo Prediction of Protein Contact Map by Ultra-Deep Learning Model. PLoS computational biology 2017;13(1):e1005324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kocher JP, Rooman MJ, Wodak SJ. Factors influencing the ability of knowledge-based potentials to identify native sequence-structure matches. Journal of molecular biology 1994;235(5):1598–1613. [DOI] [PubMed] [Google Scholar]
- 19.Rooman MJ, Kocher JP, Wodak SJ. Prediction of protein backbone conformation based on seven structure assignments. Influence of local interactions. Journal of molecular biology 1991;221(3):961–979. [DOI] [PubMed] [Google Scholar]
- 20.Rooman MJ, Kocher JP, Wodak SJ. Extracting information on folding from the amino acid sequence: accurate predictions for protein regions with preferred conformation in the absence of tertiary interactions. Biochemistry 1992;31(42):10226–10238. [DOI] [PubMed] [Google Scholar]
- 21.Rooman M, Wodak SJ. Generating and testing protein folds. Current opinion in structural biology 1993;3(2):247–259. [Google Scholar]
- 22.Senior AW, Evans R, Jumper J, Kirkpatrick J, Sifre L, Green T, Qin C, Zidek A, Nelson AWR, Bridgland A, Penedones H, Petersen S, Simonyan K, Crossan S, Kohli P, Jones DT, Silver D, Kavukcuoglu K, Hassabis D. Protein structure prediction using multiple deep neural networks in the 13th Critical Assessment of Protein Structure Prediction (CASP13). Proteins 2019;87(12):1141–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Senior AW, Evans R, Jumper J, Kirkpatrick J, Sifre L, Green T, Qin C, Zidek A, Nelson AWR, Bridgland A, Penedones H, Petersen S, Simonyan K, Crossan S, Kohli P, Jones DT, Silver D, Kavukcuoglu K, Hassabis D. Improved protein structure prediction using potentials from deep learning. Nature 2020;577(7792):706–710. [DOI] [PubMed] [Google Scholar]
- 24.Xu Q, Canutescu AA, Wang G, Shapovalov M, Obradovic Z, Dunbrack RL Jr. Statistical analysis of interface similarity in crystals of homologous proteins. Journal of molecular biology 2008;381(2):487–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levy ED, Teichmann S. Structural, evolutionary, and assembly principles of protein oligomerization. Progress in molecular biology and translational science 2013;117:25–51. [DOI] [PubMed] [Google Scholar]
- 26.Wodak SJ, Janin J. Structural basis of macromolecular recognition. Advances in protein chemistry 2002;61:9–73. [DOI] [PubMed] [Google Scholar]
- 27.Ritchie DW. Recent progress and future directions in protein-protein docking. Current protein & peptide science 2008;9(1):1–15. [DOI] [PubMed] [Google Scholar]
- 28.Vajda S, Kozakov D. Convergence and combination of methods in protein-protein docking. Current opinion in structural biology 2009;19(2):164–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fleishman SJ, Whitehead TA, Strauch EM, Corn JE, Qin S, Zhou HX, Mitchell JC, Demerdash ON, Takeda-Shitaka M, Terashi G, Moal IH, Li X, Bates PA, Zacharias M, Park H, Ko JS, Lee H, Seok C, Bourquard T, Bernauer J, Poupon A, Aze J, Soner S, Ovali SK, Ozbek P, Tal NB, Haliloglu T, Hwang H, Vreven T, Pierce BG, Weng Z, Perez-Cano L, Pons C, Fernandez-Recio J, Jiang F, Yang F, Gong X, Cao L, Xu X, Liu B, Wang P, Li C, Wang C, Robert CH, Guharoy M, Liu S, Huang Y, Li L, Guo D, Chen Y, Xiao Y, London N, Itzhaki Z, Schueler-Furman O, Inbar Y, Potapov V, Cohen M, Schreiber G, Tsuchiya Y, Kanamori E, Standley DM, Nakamura H, Kinoshita K, Driggers CM, Hall RG, Morgan JL, Hsu VL, Zhan J, Yang Y, Zhou Y, Kastritis PL, Bonvin AM, Zhang W, Camacho CJ, Kilambi KP, Sircar A, Gray JJ, Ohue M, Uchikoga N, Matsuzaki Y, Ishida T, Akiyama Y, Khashan R, Bush S, Fouches D, Tropsha A, Esquivel-Rodriguez J, Kihara D, Stranges PB, Jacak R, Kuhlman B, Huang SY, Zou X, Wodak SJ, Janin J, Baker D. Community-wide assessment of protein-interface modeling suggests improvements to design methodology. Journal of molecular biology 2011;414(2):289–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moretti R, Fleishman SJ, Agius R, Torchala M, Bates PA, Kastritis PL, Rodrigues JP, Trellet M, Bonvin AM, Cui M, Rooman M, Gillis D, Dehouck Y, Moal I, Romero-Durana M, Perez-Cano L, Pallara C, Jimenez B, Fernandez-Recio J, Flores S, Pacella M, Praneeth Kilambi K, Gray JJ, Popov P, Grudinin S, Esquivel-Rodriguez J, Kihara D, Zhao N, Korkin D, Zhu X, Demerdash ON, Mitchell JC, Kanamori E, Tsuchiya Y, Nakamura H, Lee H, Park H, Seok C, Sarmiento J, Liang S, Teraguchi S, Standley DM, Shimoyama H, Terashi G, Takeda-Shitaka M, Iwadate M, Umeyama H, Beglov D, Hall DR, Kozakov D, Vajda S, Pierce BG, Hwang H, Vreven T, Weng Z, Huang Y, Li H, Yang X, Ji X, Liu S, Xiao Y, Zacharias M, Qin S, Zhou HX, Huang SY, Zou X, Velankar S, Janin J, Wodak SJ, Baker D. Community-wide evaluation of methods for predicting the effect of mutations on protein-protein interactions. Proteins 2013;81(11):1980–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lensink MF, Wodak SJ. Docking, scoring, and affinity prediction in CAPRI. Proteins 2013;81(12):2082–2095. [DOI] [PubMed] [Google Scholar]
- 32.Lensink MF, Moal IH, Bates PA, Kastritis PL, Melquiond AS, Karaca E, Schmitz C, van Dijk M, Bonvin AM, Eisenstein M, Jimenez-Garcia B, Grosdidier S, Solernou A, Perez-Cano L, Pallara C, Fernandez-Recio J, Xu J, Muthu P, Praneeth Kilambi K, Gray JJ, Grudinin S, Derevyanko G, Mitchell JC, Wieting J, Kanamori E, Tsuchiya Y, Murakami Y, Sarmiento J, Standley DM, Shirota M, Kinoshita K, Nakamura H, Chavent M, Ritchie DW, Park H, Ko J, Lee H, Seok C, Shen Y, Kozakov D, Vajda S, Kundrotas PJ, Vakser IA, Pierce BG, Hwang H, Vreven T, Weng Z, Buch I, Farkash E, Wolfson HJ, Zacharias M, Qin S, Zhou HX, Huang SY, Zou X, Wojdyla JA, Kleanthous C, Wodak SJ. Blind prediction of interfacial water positions in CAPRI. Proteins 2014;82(4):620–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Negroni J, Mosca R, Aloy P. Assessing the applicability of template-based protein docking in the twilight zone. Structure 2014;22(9):1356–1362. [DOI] [PubMed] [Google Scholar]
- 34.Szilagyi A, Zhang Y. Template-based structure modeling of protein-protein interactions. Current opinion in structural biology 2014;24:10–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lensink MF, Velankar S, Kryshtafovych A, Huang SY, Schneidman-Duhovny D, Sali A, Segura J, Fernandez-Fuentes N, Viswanath S, Elber R, Grudinin S, Popov P, Neveu E, Lee H, Baek M, Park S, Heo L, Rie Lee G, Seok C, Qin S, Zhou HX, Ritchie DW, Maigret B, Devignes MD, Ghoorah A, Torchala M, Chaleil RA, Bates PA, Ben-Zeev E, Eisenstein M, Negi SS, Weng Z, Vreven T, Pierce BG, Borrman TM, Yu J, Ochsenbein F, Guerois R, Vangone A, Rodrigues JP, van Zundert G, Nellen M, Xue L, Karaca E, Melquiond AS, Visscher K, Kastritis PL, Bonvin AM, Xu X, Qiu L, Yan C, Li J, Ma Z, Cheng J, Zou X, Shen Y, Peterson LX, Kim HR, Roy A, Han X, Esquivel-Rodriguez J, Kihara D, Yu X, Bruce NJ, Fuller JC, Wade RC, Anishchenko I, Kundrotas PJ, Vakser IA, Imai K, Yamada K, Oda T, Nakamura T, Tomii K, Pallara C, Romero-Durana M, Jimenez-Garcia B, Moal IH, Fernandez-Recio J, Joung JY, Kim JY, Joo K, Lee J, Kozakov D, Vajda S, Mottarella S, Hall DR, Beglov D, Mamonov A, Xia B, Bohnuud T, Del Carpio CA, Ichiishi E, Marze N, Kuroda D, Roy Burman SS, Gray JJ, Chermak E, Cavallo L, Oliva R, Tovchigrechko A, Wodak SJ. Prediction of homoprotein and heteroprotein complexes by protein docking and template-based modeling: A CASP-CAPRI experiment. Proteins 2016;84 Suppl 1:323–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lensink MF, Velankar S, Baek M, Heo L, Seok C, Wodak SJ. The challenge of modeling protein assemblies: the CASP12-CAPRI experiment. Proteins 2018;86 Suppl 1:257–273. [DOI] [PubMed] [Google Scholar]
- 37.Lafita A, Bliven S, Kryshtafovych A, Bertoni M, Monastyrskyy B, Duarte JM, Schwede T, Capitani G. Assessment of protein assembly prediction in CASP12. Proteins 2018;86 Suppl 1:247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lensink MF, Brysbaert G, Nadzirin N, Velankar S, Chaleil RAG, Gerguri T, Bates PA, Laine E, Carbone A, Grudinin S, Kong R, Liu RR, Xu XM, Shi H, Chang S, Eisenstein M, Karczynska A, Czaplewski C, Lubecka E, Lipska A, Krupa P, Mozolewska M, Golon L, Samsonov S, Liwo A, Crivelli S, Pages G, Karasikov M, Kadukova M, Yan Y, Huang SY, Rosell M, Rodriguez-Lumbreras LA, Romero-Durana M, Diaz-Bueno L, Fernandez-Recio J, Christoffer C, Terashi G, Shin WH, Aderinwale T, Maddhuri Venkata Subraman SR, Kihara D, Kozakov D, Vajda S, Porter K, Padhorny D, Desta I, Beglov D, Ignatov M, Kotelnikov S, Moal IH, Ritchie DW, Chauvot de Beauchene I, Maigret B, Devignes MD, Ruiz Echartea ME, Barradas-Bautista D, Cao Z, Cavallo L, Oliva R, Cao Y, Shen Y, Baek M, Park T, Woo H, Seok C, Braitbard M, Bitton L, Scheidman-Duhovny D, Dapkunas J, Olechnovic K, Venclovas C, Kundrotas PJ, Belkin S, Chakravarty D, Badal VD, Vakser IA, Vreven T, Vangaveti S, Borrman T, Weng Z, Guest JD, Gowthaman R, Pierce BG, Xu X, Duan R, Qiu L, Hou J, Ryan Merideth B, Ma Z, Cheng J, Zou X, Koukos PI, Roel-Touris J, Ambrosetti F, Geng C, Schaarschmidt J, Trellet ME, Melquiond ASJ, Xue L, Jimenez-Garcia B, van Noort CW, Honorato RV, Bonvin A, Wodak SJ. Blind prediction of homo- and hetero-protein complexes: The CASP13-CAPRI experiment. Proteins 2019;87(12):1200–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lensink MF, Mendez R, Wodak SJ. Docking and scoring protein complexes: CAPRI 3rd Edition. Proteins 2007;69(4):704–718. [DOI] [PubMed] [Google Scholar]
- 40.Lensink MF, Wodak SJ. Docking and scoring protein interactions: CAPRI 2009. Proteins 2010;78(15):3073–3084. [DOI] [PubMed] [Google Scholar]
- 41.Lensink MF, Wodak SJ. Blind predictions of protein interfaces by docking calculations in CAPRI. Proteins 2010;78(15):3085–3095. [DOI] [PubMed] [Google Scholar]
- 42.Remmert M, Biegert A, Hauser A, Soding J. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nature methods 2012;9(2):173–175. [DOI] [PubMed] [Google Scholar]
- 43.Zimmermann L, Stephens A, Nam SZ, Rau D, Kubler J, Lozajic M, Gabler F, Soding J, Lupas AN, Alva V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. Journal of molecular biology 2018;430(15):2237–2243. [DOI] [PubMed] [Google Scholar]
- 44.Basu S, Wallner B. DockQ: A Quality Measure for Protein-Protein Docking Models. PloS one 2016;11(8):e0161879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lensink MF, Nadzirin N, Velankar S, Wodak SJ. Modeling protein-protein, protein-peptide, and protein-oligosaccharide complexes: CAPRI 7th edition. Proteins 2020;88(8):916–938. [DOI] [PubMed] [Google Scholar]
- 46.de Vries SJ, Bonvin AM. How proteins get in touch: interface prediction in the study of biomolecular complexes. Current protein & peptide science 2008;9(4):394–406. [DOI] [PubMed] [Google Scholar]
- 47.Webb B, Sali A. Protein Structure Modeling with MODELLER. Methods in molecular biology 2017;1654:39–54. [DOI] [PubMed] [Google Scholar]
- 48.Dapkunas J, Timinskas A, Olechnovic K, Margelevicius M, Diciunas R, Venclovas C. The PPI3D web server for searching, analyzing and modeling protein-protein interactions in the context of 3D structures. Bioinformatics 2017;33(6):935–937. [DOI] [PubMed] [Google Scholar]
- 49.Holm L DALI and the persistence of protein shape. Protein Sci 2020;29(1):128–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jain A, Terashi G, Kagaya Y, Maddhuri Venkata Subramaniya SR, Christoffer C, Kihara D. Analyzing effect of quadruple multiple sequence alignments on deep learning based protein inter-residue distance prediction. Sci Rep 2021;11(1):7574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dapkunas J, Olechnovic K, Venclovas C. Structural modeling of protein complexes: Current capabilities and challenges. Proteins 2019;87(12):1222–1232. [DOI] [PubMed] [Google Scholar]
- 52.Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, Beglov D, Vajda S. The ClusPro web server for protein-protein docking. Nature protocols 2017;12(2):255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ritchie DW, Kemp GJ. Protein docking using spherical polar Fourier correlations. Proteins 2000;39(2):178–194. [PubMed] [Google Scholar]
- 54.van Zundert GCP, Rodrigues J, Trellet M, Schmitz C, Kastritis PL, Karaca E, Melquiond ASJ, van Dijk M, de Vries SJ, Bonvin A. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J Mol Biol 2016;428(4):720–725. [DOI] [PubMed] [Google Scholar]
- 55.Pierce BG, Wiehe K, Hwang H, Kim BH, Vreven T, Weng Z. ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014;30(12):1771–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.El Houasli M, Maigret B, Devignes MD, Ghoorah AW, Grudinin S, Ritchie DW. Modeling and minimizing CAPRI round 30 symmetrical protein complexes from CASP-11 structural models. Proteins 2017;85(3):463–469. [DOI] [PubMed] [Google Scholar]
- 57.Yan Y, Tao H, Huang SY. HSYMDOCK: a docking web server for predicting the structure of protein homo-oligomers with Cn or Dn symmetry. Nucleic acids research 2018;46(W1):W423–W431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roy Burman SS, Yovanno RA, Gray JJ. Flexible Backbone Assembly and Refinement of Symmetrical Homomeric Complexes. Structure 2019;27(6):1041–1051 e1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Esquivel-Rodriguez J, Yang YD, Kihara D. Multi-LZerD: multiple protein docking for asymmetric complexes. Proteins 2012;80(7):1818–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Christoffer C, Chen S, Bharadwaj V, Aderinwale T, Kumar V, Hormati M, Kihara D. LZerD webserver for pairwise and multiple protein-protein docking. Nucleic Acids Res 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Z, Schindler CE, Lange OF, Zacharias M. Application of Enhanced Sampling Monte Carlo Methods for High-Resolution Protein-Protein Docking in Rosetta. PLoS One 2015;10(6):e0125941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cao Y, Shen Y. Bayesian Active Learning for Optimization and Uncertainty Quantification in Protein Docking. J Chem Theory Comput 2020;16(8):5334–5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Olechnovic K, Venclovas C. VoroMQA: Assessment of protein structure quality using interatomic contact areas. Proteins 2017;85(6):1131–1145. [DOI] [PubMed] [Google Scholar]
- 64.Zhou H, Skolnick J. GOAP: a generalized orientation-dependent, all-atom statistical potential for protein structure prediction. Biophysical journal 2011;101(8):2043–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhou H, Zhou Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein science : a publication of the Protein Society 2002;11(11):2714–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huang SY, Zou X. Statistical mechanics-based method to extract atomic distance-dependent potentials from protein structures. Proteins 2011;79(9):2648–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kundrotas PJ, Anishchenko I, Badal VD, Das M, Dauzhenka T, Vakser IA. Modeling CAPRI targets 110–120 by template-based and free docking using contact potential and combined scoring function. Proteins 2018;86 Suppl 1:302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huang SY, Zou X. An iterative knowledge-based scoring function for protein-protein recognition. Proteins 2008;72(2):557–579. [DOI] [PubMed] [Google Scholar]
- 69.Cao Y, Shen Y. Energy-based graph convolutional networks for scoring protein docking models. Proteins 2020;88(8):1091–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Geng C, Jung Y, Renaud N, Honavar V, Bonvin A, Xue LC. iScore: a novel graph kernel-based function for scoring protein-protein docking models. Bioinformatics 2020;36(1):112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang X, Flannery ST, Kihara D. Protein Docking Model Evaluation by Graph Neural Networks. BioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D. Highly accurate protein structure prediction with AlphaFold. Nature 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, Wang J, Cong Q, Kinch LN, Schaeffer RD, Millan C, Park H, Adams C, Glassman CR, DeGiovanni A, Pereira JH, Rodrigues AV, van Dijk AA, Ebrecht AC, Opperman DJ, Sagmeister T, Buhlheller C, Pavkov-Keller T, Rathinaswamy MK, Dalwadi U, Yip CK, Burke JE, Garcia KC, Grishin NV, Adams PD, Read RJ, Baker D. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.