

Abstract
From the initial sensing of viral nucleotides by pattern recognition receptors, through the induction of type I and III interferons (IFN), upregulation of antiviral effector proteins, and resolution of the inflammatory response, each step of innate immune signaling is under tight control. Though innate immunity is often associated with broad regulation at the level of gene transcription, RNA-centric post-transcriptional processes have emerged as critical mechanisms for ensuring a proper antiviral response. Here, we explore the diverse RNA regulatory mechanisms that modulate the innate antiviral immune response, with a focus on RNA sensing by RIG-I-like receptors (RLR), interferon (IFN) and IFN signaling pathways, viral pathogenesis, and host genetic variation that contributes to these processes. We address the post-transcriptional interactions with RNA-binding proteins, non-coding RNAs, transcript elements, and modifications that control mRNA stability, as well as alternative splicing events that modulate the innate immune antiviral response.
Keywords: antiviral immunity, interferon, non-coding RNAs, post-transcriptional regulation, RNA biology, RNA-binding proteins
1 |. INTRODUCTION
Innate immune pathways require stringent regulation to promote a robust response to infection while preventing detrimental excess inflammation. The sensing of viral infection activates diverse host antiviral defense programs, but viruses actively counteract these measures to promote their own replication. Broad transcriptional and metabolic changes are key features of both cellular antiviral programs and viral replication. During the rapid timescale of the innate immune response, post-transcriptional regulation of RNA serves as potent mechanism for modulating host antiviral processes.
The function and expression of RNA can be tuned at every stage of its life cycle, beginning with co-transcriptional control of capping, splicing, and polyadenylation; continuing with nuclear export and translation; and terminating with RNA degradation. RNA-binding proteins (RBPs) are inextricably involved in every facet of RNA metabolism. RNA molecules are decorated with RBPs from their inception, and exist exclusively as ribonucleoproteins (RNPs). The critical importance of RNPs is highlighted by the fact that approximately 10% of the human proteome is comprised of RBPs. This percentage continues to grow as additional cellular factors are found to contribute to RNA regulation. Several distinct RBPs bound to a single RNA molecule can act in concert with or against one another. Such interactions between RBPs and RNA lead to transcript-specific molecular outcomes that ultimately shape the cellular transcriptome.
RNA-RBP pairing is mediated by RNA-binding domains within RBPs that interact with specific sequences and structures within RNA molecules.1 For example, the RBP HuR, which contains three globular RNA recognition motifs, interacts with U-rich and AU-rich sequences within introns and 3′ UTRs, influencing the splicing of target mRNAs and promoting RNA stability.2,3 On the other hand, the RBP Staufen1 interacts with structured RNA helices through its double-stranded (dsRNA)-binding domains in a largely sequence-independent fashion.4,5 RBPs may also contain helicase domains that bind highly structured RNAs and unwind them, thereby potentiating additional RNA-RBP interactions.6 In addition to such ordered RNA-binding domains, intrinsically disordered domains can also mediate specific and non-specific RNA-protein interactions and promote phase transition of RNPs into membrane-less organelles where many RNA regulatory processes occur.7–10
Chemical RNA base modifications also engage RBPs to alter RNA fate and function. A 7-methylguanosine (m7G) RNA cap protects the 5′ terminus of mRNAs from exoribonucleolytic activity. This cap is also required for ribosome engagement and translation initiation through the cap-binding protein eIF4E.11 Recently, internal modifications in mRNAs have been discovered and recognized as an additional “epitranscriptomic” regulatory layer of gene regulation.12 For example, N6-methyladenosine (m6A) recruits “reader” RBPs, which contain domains that specifically interact with the methyl group of the modified adenosine.13–18 m6A can also modulate local RNA structure and alter the binding of structure-dependent RBPs such as hnRNPC, hnRNPG, and ADAR1.19–21 Other RNA modifications including pseudouridine, 5-methylcytidine, and N4-acetylcytidine may also modulate RNA function through similar mechanisms.12,22–25
Non-coding RNAs play key roles in RNA regulatory processes through RNA protein as well as RNA-RNA interactions. Although only 2% of the human genome encodes proteins, more than 85% is transcribed into non-coding RNAs, including microRNAs (miRNAs) and long non-coding RNAs (lncRNAs).26,27 miRNAs are single-stranded RNAs that are 21–23 nucleotides in length. After extensive processing of primary miRNA transcripts, miRNAs are loaded into the RNA-induced silencing complex (RISC), where they bind to the 3′ UTRs of target mRNAs through their “seed sequence,” which spans nucleotides 2–8, causing degradation or translational suppression of target mRNAs.28–30 At least 30% of human mRNAs have the potential for regulation via miRNA binding.28–30 LncRNAs, thousands of which are encoded in the human genome, are defined as non-coding RNAs greater than 200 nucleotides in length and are often differentially expressed during viral infection and immune stimulation.31,32 Until recently, lncRNAs were considered transcriptional noise due to their often-low levels of expression. However, lncRNAs are increasingly implicated as diverse regulators of transcription, protein modulation, and RNA activity, most often serving as molecular decoys, guides, or scaffolds.33–37 The biological roles of the vast majority of lncRNAs are still undetermined and there is great need for further investigation of their functions.
Given their importance in controlling biological processes, it is not surprising that RNA-centric mechanisms regulate the immune response to viral infection. Antiviral immune pathways are activated through cellular sensing of viral nucleic acid motifs by a group of sentinel proteins called pattern recognition receptors (PRRs). Viral RNA motifs are sensed by the RIG-I-Like receptors (RLRs) RIG-I and MDA5, as well as by the Toll-Like Receptors (TLRs) TLR3, TLR7, and TLR8.38,39 Viral DNA is detected by TLR9, cGAS, and IFI16.40 Although these sensors reside in diverse subcellular compartments and signal through various adaptors, their detection of viral nucleic acids culminates in the same downstream activation of the interferon regulatory factor (IRF) family of transcription factors and the subsequent production of type I and III interferons (IFNs). IFNs are cytokines that act in both autocrine and paracrine fashions to induce hundreds of interferon-stimulated genes (ISGs), which are potent cellular defense factors that establish an antiviral state and counteract infection.41
In this review, we will highlight examples of RNA-centric processes that modulate the landscape of gene expression during the innate immune response to viruses, thereby shaping the outcome of infection. Because of their critical function in host protection from RNA virus infection, we will focus specifically on the RNA regulatory controls of RLR signaling pathways, type I and type III IFNs.
2 |. RNA REGULATION OF VIRAL SENSING BY RLRS
When the RLRs RIG-I or MDA5 interact with viral “non-self” or endogenous “self” RNA motifs, they undergo conformational changes, oligomerize through their caspase activation and recruitment domains (CARDs), and translocate to the vicinity of the adaptor protein MAVS.38 MAVS is anchored to both mitochondria and contact sites between mitochondria and ER, as well as to peroxisomes. MAVS itself encodes a CARD, and as such, activated RIG-I or MDA5 oligomers trigger the aggregation of MAVS through CARD-CARD interactions, building a platform for downstream signaling. While a diverse array of proteins is involved in MAVS signalosome function, the kinases IKKε and TBK1, which phosphorylate the transcription factors IRF3/IRF7, are especially critical.38,42 Phosphorylated homodimers of IRF3 or IRF7 translocate into the nucleus and induce type I and III IFNs. RLRs and the MAVS signalosome also activate NF-κB, augmenting robust IFN induction. In this section, we highlight RNA-centric mechanisms that control RLR signaling (Figure 1).
FIGURE 1.
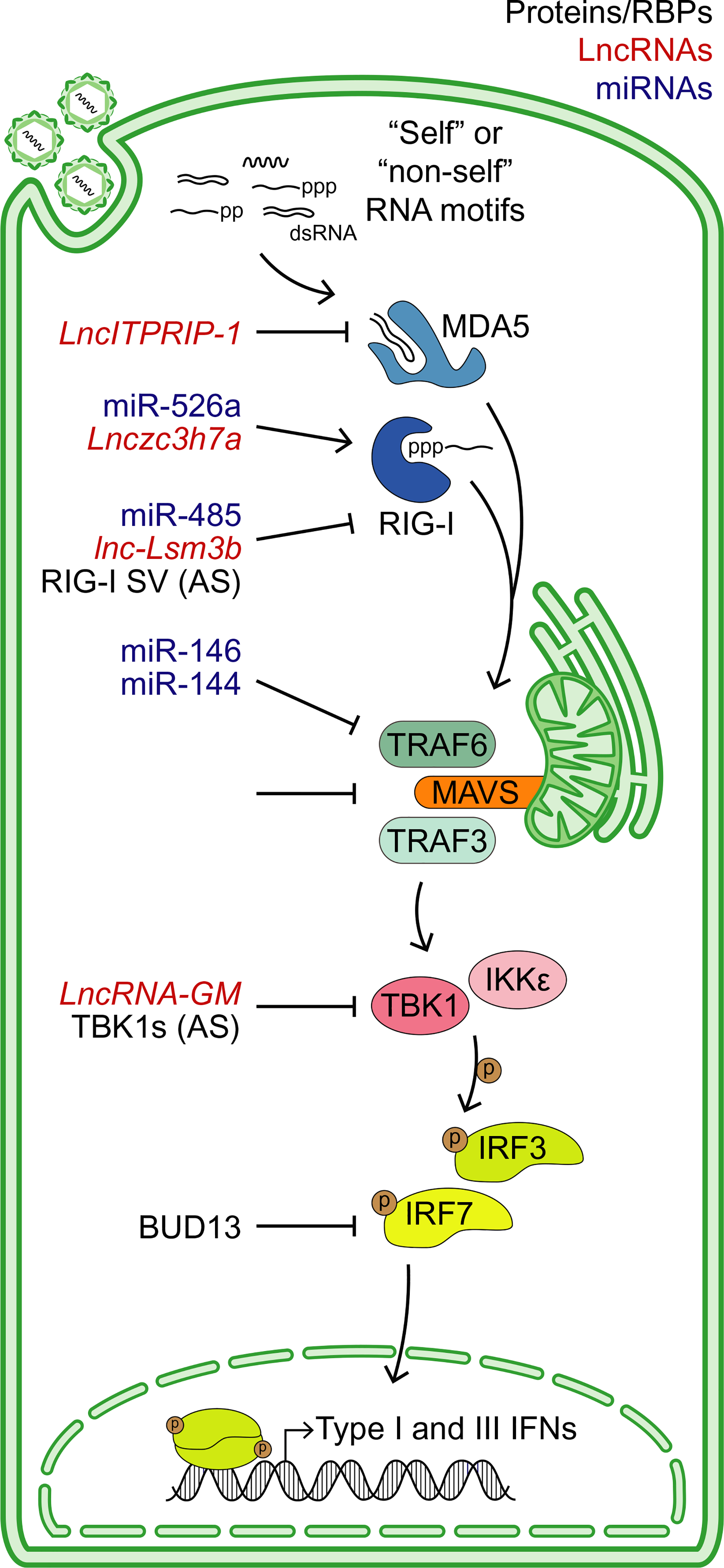
Summary of RNA regulatory mechanisms that control RIG-I-like receptor (RLR) activation. Sensing of viral (“non-self”) or cellular (“self”) RNA motifs by the RLRs RIG-I or MDA5 triggers a signaling cascade centered around the adaptor protein MAVS, ultimately leading to the phosphorylation and nucleation of IRF3 or IRF7, and the induction of type I and III IFNs. These pathways can be regulated by diverse RNA regulatory mechanisms. RNA-binding proteins (RBPs) or alternatively spliced (AS) proteins, lncRNAs, and miRNAs that modulate this pathway are labeled in black, red, and blue, respectively
2.1 |. “Self” and “Non-Self” RNA sensing by RLRs
The composition and structure of “non-self” viral RNA determine how it is sensed by RLRs. Both RIG-I and MDA5 can recognize double-stranded RNA (dsRNA). MDA5 senses long dsRNA intermediates of viral replication during infection by enteroviruses and coronaviruses.43–47 On the other hand, RIG-I is activated by short dsRNA containing either a 5′ triphosphate (5′-ppp) or diphosphate (5′-pp), but lacking an m7G cap.48–51 RIG-I (and potentially MDA5) can also recognize RNA lacking 2′O-methylation at its 5′ end.52–54 Both the m7G cap and terminal 2′O-methylation, found on “self” mRNAs, prevent RIG-I from recognizing endogenous mRNAs and aberrantly activating an IFN response. The antiviral effector IFIT1, an ISG upregulated downstream of RIG-I and MDA5 signaling, senses the lack of 2′O-methylation on viral transcripts and acts to limit the translation of viral RNA.55 Emulating the 5′ cap structure is an efficient strategy for viruses to avoid detection by RIG-I and to engage cap-dependent translation machinery. NS5, the RNA-dependent RNA polymerase of flaviviruses, which are positive-sense, single-stranded RNA viruses, encodes both m7G- and 2′O-methyltransferase activity.56–58 Mutation of NS5 2′O-methyltransferase activity in yellow fever virus and West Nile virus (WNV) results in increased IFN production, demonstrating that flaviviruses use this strategy to evade recognition by RIG-I.52,59 Similarly, the NSP16 protein of coronaviruses has cap 2′O-methyltransferase activity.54,60,61 Ablation of NSP16 cap 2′O-methyltransferase activity attenuates the diverse coronavirus species in a sensing- and IFN-dependent manner. In addition to the 5′ cap structure, several families of viruses contain m6A and other RNA modifications within their transcripts.62 In vitro transcribed RNAs containing modified nucleobases like m6A can suppress detection by RLRs and TLRs.63,64 Inhibition of RLR activation through m6A modifications has been demonstrated to occur during hepatitis C virus (HCV), hepatitis B virus, and human metapneumovirus infection.65,66 Thus, viruses may co-opt cellular RNA modification processes to appear more like endogenous transcripts, thereby shielding their genomes from detection by PRRs.
RIG-I-like receptors can also sense “self” RNAs which become unmasked during infection or accumulate due to loss of proper RNA metabolism. Sensing of such transcripts may enhance antiviral signaling. During infection by the DNA viruses herpes simplex virus (HSV)-1 and Epstein-Barr virus (EBV), as well as by the negative-stranded RNA virus influenza A virus (IAV), RIG-I is activated by the mis-localized host 5S ribosomal RNA pseudogene transcript RNA5SP141, which accumulates aberrantly in the cytosol. RNA5SP141 contains dsRNA elements plus a 5′-ppp moiety and is therefore a potent RIG-I substrate. Recognition of RNA5SP141 by RIG-I has been shown to be critical for the induction of IFN and restriction of viral replication during HSV-1 infection.67 Similarly, the reactivation of the DNA virus Kaposi’s sarcoma herpes virus (KSHV) leads to the accumulation of 5′-ppp containing vault RNAs, a class of poorly understood small RNAs, which act as RIG-I agonists. At homeostasis, 5′-ppp on vault RNA is reduced by the cellular triphosphatase DUSP11; however, KSHV reactivation inhibits the transcription of DUSP11, increasing the immunostimulatory potential of these small RNA species.68 During viral infection, the activation of the OAS-RNase L antiviral system also generates “self” ligands that trigger RLR activation. The 2′−5′ oligoadenylate synthetase (OAS) family of ISGs sense viral dsRNA and catalyze the production of the second messenger 2′−5′ adenylate (2′−5′A). 2′−5′A then activates the latent endoribonucleolytic activity of RNase L, which cleaves both viral and host single-stranded RNA.69,70 Although RNase L-mediated RNA cleavage primarily denies a permissive cellular environment for viral replication, RNase L cleavage products can also form small duplex RNA ligands that activate both RIG-I and MDA5 to amplify the antiviral response.70
“Self” RLR activation can promote antiviral immunity through counteracting viral mechanisms that hide viral RNA from cellular pathogen sensors. However, RLR signaling in the absence of infection is detrimental and may lead to inflammatory and autoimmune diseases.71,72 As such, the host employs multiple strategies to restrict sterile activation of RLR pathways during homeostasis. As mentioned, the triphosphatase DUSP11 restricts RIG-I-activating 5′-ppp moieties in certain “self” non-coding RNAs.68,73 Keeping aberrant MDA5 activation in check, however, involves the suppression of endogenous dsRNA. For example, an RNA degradosome comprised of the helicase SUV3 and the polynucleotide phosphorylase PNPT1 is responsible for rapid turnover of dsRNA intermediates that result from mitochondrial transcription. Inhibition of SUV3 and PNPT1 leads to accumulation of mitochondrial dsRNA, which enters the cytoplasm and engages MDA5.74 Pairs of Alu elements, ~300 nt long retrotransposons abundantly dispersed throughout primate genomes, can form long dsRNA regions in cellular RNAs and also act as a potent substrate for MDA5 activation.75–78 However, ADAR edits structured Alu elements, changing adenosine bases to inosine, thereby reducing base-pairing potential and inhibiting MDA5 filament assembly plus downstream signaling.77,78 Gain-of-function mutations in MDA5 that promote MDA5-RNA binding and loss-of-function mutations in ADAR1 that inhibit RNA editing both cause autoimmune interferonopathies such as Aicardi-Guitières syndrome.77,79–81
Differential splicing of cellular RNAs may alter RLR signaling. This facet of RNA regulation is best exemplified in tumor cell suppression of IFN activation. Transcriptomic analyses of tumor samples reveal dramatic alterations to RNA splicing.82 The splicing factor hnRNPC, upregulated in many cancers, suppresses the retention of Alu element-containing introns during mRNA processing.83–85 Depletion of hnRNPC in breast cancer cell lines causes tumor suppression through heightened sensing of Alu elements in endogenous dsRNA ligands, which subsequently induces type I IFN.86 Furthermore, spliceosome-targeted therapies (STTs) which aim to repress key splicing factors, have shown efficacy in cancer models. In a recent report, STTs resulted in the production of dsRNA species via intron retention, thus activating the RLR pathway.87 In murine breast cancer models, STTs that activated the RLR pathway resulted in increased antiviral immune signaling, tumor cell death, and increased adaptive immune responses. Similarly, breast cancer patients with increased intron retention exhibited improved disease-free survival compared to those with lower intron retention.
Sensing of immunostimulatory RNA danger-associated molecular patterns (DAMPs) in cancer cells may also lead to increased resistance to radiotherapy or chemotherapy. In breast cancer models, stromal fibroblasts co-cultured with tumor cells produce RNA containing exosomes, which activate STAT1 in a RIG-I-dependent manner. Activation of STAT1 and ISG induction via exosome RNA (exoRNA) and NOTCH3 signaling in breast cancer cells cooperatively render these cancer cells refractory to therapy, likely by promoting DNA damage resistance.88 Interestingly, these exoRNAs consist mainly of 7SL RNA transcripts such as RN7SL1, which normally nucleate the signal recognition particle (SRP), a highly conserved RNP essential for protein membrane localization and secretion.89,90 RN7SL1 contains a terminal triphosphate moiety within its 5′ Alu-like RNA sequence. In the cytoplasm of homeostatic cells, the heterodimeric SRP proteins SRP9 and SRP14 interact with the Alu domain of RN7SL1 to shield the 5′-ppp from RIG-I. However, reduced SRP9/14 incorporation into exosomes exposes the 5′-ppp of RN7SL1, which can then be sensed by RIG-I in tumor cells.89 Taken together, these findings reveal that both “self” and “non-self” RNAs are important in regulating IFN production through the RLR pathway. While RLR recognition of “non-self” RNA is largely understood, future studies with undoubtedly reveal further cellular factors and mechanisms that either promote or prevent the detection of “self” RNA motifs to control IFN activation.
2.2 |. Post-transcriptional regulation of the RLR pathway
2.2.1 |. Alternative splicing and open reading frames
Nearly 85% of all transcripts undergo alternative splicing.91 Through alternative exon usage, intron retention, or splice site selection, alternative splicing serves to diversify the proteome and generate proteins with distinct properties. Further, alternative splicing can also contribute to post-transcriptional mRNA regulation through nonsense-mediated decay, nuclear retention of transcripts, or modulation of translation efficiency.92 All cellular factors involved in antiviral signaling have the potential to be affected by splicing, and indeed, modulation of both host- and pathogen-driven splicing is abundant during viral infection.91,92 In fact, RIG-I itself undergoes IFN-inducible alternative splicing, generating a truncated isoform termed RIG-I SV. RIG-I SV cannot trigger downstream signaling as it lacks the ability to interact with TRIM25, an E3 ubiquitin ligase required for activating RIG-I through K63-linked ubiquitination. Furthermore, RIG-I SV serves as a dominant-negative signaling inhibitor by forming a complex with full-length RIG-I and preventing its interaction with MAVS.93 An alternate splicing isoform of TBK1 also reduces antiviral signaling. Sendai virus infection or IFN-β treatment induces a truncated isoform TBK1s that lacks the kinase domain required to phosphorylate the transcription factor IRF3. Instead, TBK1s binds directly to RIG-I and disrupts RIG-I/MAVS interactions.94 These findings highlight the potential for alternative splicing to generate dominant-negative isoforms of antiviral signaling molecules as a feedback mechanism to downregulate antiviral signaling.
Conversely, splicing regulatory mechanisms may also promote antiviral signaling, as seen in the case of Irf7, a key transcription factor of IFN in myeloid cells. The fourth intron of Irf7 contains a weak 5′ splice junction site that hampers efficient splicing and limits IRF7 expression. However, the splicing factor BUD13 binds this weak splice site and promotes more efficient splicing. Depletion of BUD13 results in increased retention of intron 4, decreased expression of IRF7 and subsequently diminished type I IFN, causing increased susceptibility to Indiana vesiculovirus (VSV) infection.95 “Bottleneck introns” like intron 4 of Irf7 may constitute a widespread mechanism for proper temporal control of immune-related transcripts.96 More broadly, specific splicing factors have been shown to control the proper splicing of immunity-related regulons.97,98 However, the precise roles of various RBPs in alternative splicing of antiviral signaling elements remains an open area of investigation.
Alternative translation initiation is yet another mechanism that may control RLR signaling. Proper translation initiation at methionine codons depends on their being situated in the context of a strong Kozak nucleotide sequence.99 Leaky ribosome scanning across initiation sites with weaker Kozak context can cause protein translation to begin from downstream methionine codons. For example, the MAVS transcript translation may initiate from at least two distinct methionine codons.100 Translation initiation from the canonical start codon leads to the production of full-length, signaling-competent MAVS protein. However, upstream open reading frames in the MAVS 5′ UTR sometimes mask the canonical start site.100,101 In such scenarios, translation will initiate from a downstream, in-frame methionine codon at amino acid 142, producing a truncated protein called miniMAVS. As miniMAVS lacks the N-terminal CARD, it cannot oligomerize to form a signaling complex and therefore cannot transduce RLR signaling. MiniMAVS negatively regulates IFN production, likely by competing with full-length MAVS for binding of other key signaling platform components like TRAF2 and TRAF6. Recently, other truncated variants of MAVS protein that prevent full-length MAVS oligomerization have been identified.102 These variants are smaller than miniMAVS and are likely translated from methionine codons even further toward the 3′ end of MAVS mRNA than Met142. Ribosome profiling experiments in a human monocytic cell line indicated that mRNAs of other RLR pathway components like MDA5 and TRIM25, as well as downstream antiviral effectors like MX2 and IFITM2, may also be regulated by alternative translation initiation, but this remains to be validated. Much like alternative splicing, alternative translation initiation and cis-regulatory upstream ORFs have profound effects on antiviral signaling pathways. Better understanding the mechanisms by which they regulate RLRs and the IFN response will yield further implications for infection, autoimmunity, and cancer.
2.2.2 |. RNA-binding proteins and RNA modifications
In addition to splicing, RBPs regulate antiviral sensing and signaling via control of nuclear export of mRNA. DEAD-box (DDX) RNA helicases, encoded in domains of both RIG-I and MDA5, have been shown to play diverse roles in antiviral immunity, primarily through modulating export of host mRNA transcripts central to viral RNA sensing pathways.103 For example, DDX46 negatively regulates the production of type I IFN during viral infection by retaining MAVS, TRAF3, and TRAF6 mRNAs in the nucleus.104 These transcripts all contain the RNA modification m6A, which is known to promote nuclear export.105 However, during viral infection, DDX46 recruits the m6A “eraser” ALKBH5 to demethylate these transcripts, resulting in nuclear retention and reduced protein expression. The splicing factor DDX39A also inhibits nuclear export of MAVS, TRAF3, and TRAF6 during viral infection.106 The addition of the small ubiquitin-like modifier protein SUMO to DDX39A inhibits its ability to bind RNA. However, viral infection reduces SUMOylation of DDX39A by downregulating its E3-ligase, RanBP2. In the absence of SUMOylation, DDX39A binds MAVS, TRAF3, and TRAF6 mRNAs and sequesters them in the nucleus. Whether DDX39A and DDX46 co-operate in nuclear retention of the same set of transcripts is unknown.
2.2.3 |. miRNAs
Several miRNAs are known to modulate antiviral immune responses through interactions with various components and regulators of the RLR signaling pathway. miR-485, induced after viral sensing, targets the 3′ UTR of DDX58, which encodes RIG-I. Ectopic expression of miR-485 is sufficient to decrease RIG-I levels, inhibit type I and III IFN production, and promote viral infection. Interestingly, miR-485 also targets the PB1 RNA segment of IAV H5N1. During high dose infection with IAV H5N1, ectopically expressed miR-485 targets PB1 significantly more than RIG-I and thus decreases IAV replication.107 Such examples of bimodal regulation of both host and viral gene expression add complexity to the role of miRNAs in antiviral immunity. In this context, miR-485 negatively regulates the antiviral immune response, but switches to a viral target and protects the host when exposed to high levels of IAV viral RNA. miR-526a, also induced during viral infection in an IRF3- and IRF7-dependent manner, promotes the antiviral response by targeting CYLD, a negative regulator of RIG-I signaling.108 CYLD downregulates RIG-I signaling by removing K63-linked ubiquitin modifications required for critical conformational changes that occur during RIG-I recognition of viral RNA motifs. By downregulating CYLD expression, miR-526a enhances RIG-I ubiquitination and upregulates IFN production. Accordingly, inhibition of miR-526a results in stabilized CYLD, decreased immune responses to virus, and higher viral burden.
Both miR-155 and miR-146 were initially discovered as miRNAs differentially regulated in response to diverse immune stimuli including TLR signaling, and type I IFN, and viral infection,.109–112 Intriguingly, miR-155 and its complementary miR-155* (also called miR-155–3p), encoded from the same primary miRNA locus, affect IFN production in opposite ways. In plasmacytoid dendritic cells (pDCs), major producers of type I IFN, TLR7 activation induces miR-155* at early stages and miR-155 at later times after stimulation, likely due to differences in pri-miR155 processing mediated by the RBP KSRP. miR-155* augments type I IFN production by inhibiting the negative regulator IRAK3, while miR-155 restricts type I IFN by targeting the positive regulator TAB2. Thus, miR-155 and miR-155* help to ensure robust, early induction of IFN and inhibit prolonged, deleterious expression of this inflammatory cytokine.112
miR-146a, in contrast with miR-155, consistently downregulates type I IFN production and allows for increased viral replication during infection. In myeloid cells, VSV infection induces miR-146a in a RIG-I-dependent manner. Treatment with miR-146a mimics quenches IFNB1 expression, while miR-146a inhibitors enhance IFN production; as would be expected, these treatments promote and restrict VSV infection, respectively. Mechanistically, miR-146 targets IRAK1, IRAK2, and TRAF6 mRNAs, all of which encode key signaling molecules positioned at the nexus of IRF and NF-kB activation.109,113 During infection with enterovirus 71, miR-146a blocks TRAF6 and IRAK4 signaling, limiting type I IFN induction.114 Interestingly, like miR-146a, miR-144 also targets TRAF6 to suppress type I IFN production, exemplifying redundant miRNA regulation of a single transcript.115 In human PBMCs, miR-146a overexpression significantly dampens type I IFN and ISG expression, potentially through modulating IRF5 and STAT1 expression.116 This axis of modulation was discovered upon observing lower miR-146a expression in immune cells of individuals with systemic lupus erythematous (SLE), an autoimmune disease characterized by aberrantly high levels of type I IFN production. Though it may seem odd that such a highly induced miRNA actively inhibits inflammation in context of viral infection, the downregulation of miR-146a seen in SLE patients frames this miRNA as an important factor in resolving the type I IFN response to avoid excess inflammatory pathology following viral infection.
2.2.4 |. LncRNAs
Host long lncRNAs have emerged as regulators of several biological processes, including innate immunity to RNA viruses. Viral infection and sterile immune stimulation cause transcriptional changes in hundreds of lncRNAs; however, very few have been functionally dissected and most play unknown roles in the innate immune response to viruses.31,32 The highly abundant nuclear transcript NEAT1 enhances the innate immune response to Hantaan virus. NEAT1 preferentially interacts with the RNA/DNA-binding protein SFPQ and sequesters it in paraspeckles, thereby inhibiting its function as a transcriptional repressor of RIG-I and DDX60.117 NEAT1 also induces expression of the pro-inflammatory cytokine IL-8 through a similar mechanism, releasing transcriptional repression mediated by SFPQ.118 Lastly, NEAT1 is implicated in IFN production following foreign DNA sensing by cGAS through interactions with cGAS, DNA-PK, and the RBP HEXIM1.119 These studies demonstrate pleiotropic roles for NEAT1 in antiviral immune responses.
Cytoplasmic-localized lncRNAs regulate antiviral immune signaling through interactions with diverse proteins in the RLR pathway. Infection-induced LncITPRIP-1 boosts IFN production and restricts HCV replication by acting as a cofactor for MDA5, binding its C-terminus and promoting its oligomerization around target RNAs.120 In mouse models, Lnczc3h7a interacts with TRIM25, an E3 ubiquitin ligase required for robust RIG-I signaling. Deletion of Lnczc3h7a decreases production of type I IFN and increases VSV infection in vitro and in vivo. Lnczc3h7a is proposed to act as a scaffold for TRIM25 and RIG-I, promoting K63-linked ubiquitination of RIG-I and augmenting IFN production.121 Though no human paralog of Lnczc3h7a is known, whether an alternative human lncRNA functions similarly remains an interesting question. Recently, mechanistic investigation of LncRNA-GM, which promotes type I IFN production, revealed a hitherto unknown post-translational modification that inhibits TBK1 function: S-glutathionylation. LncRNA-GM sequesters the glutathione-S-transferase GSTM1 away from TBK1, thus promoting sustained IFN production.122 Intriguingly, infection by several viruses reduces LncRNA-GM expression, enhancing viral replication. This suggests that modulation of lncRNA expression is a useful strategy for viral evasion of host immunity. LncRNAs are not always antiviral and can actively hinder viral RNA sensing. In infected mice, the cytosolic lnc-Lsm3b directly inhibits antiviral signaling by interacting with the sensor RIG-I and competing with its binding of viral RNA.123 lnc-Lsm3b interacts with stem-loop structures to stabilize the CARD-helicase interaction and prevent the release of RIG-I autoinhibition. Consistent with this finding, loss of lnc-Lsm3b both in vitro and in vivo results in heightened antiviral signaling and reduced VSV viral burden.
Highly modulated, location-specific expression of lncRNAs during infection implicates these biomolecules as important for fine tuning the innate immune response to viruses. With a greater understanding of the molecular functions of lncRNAs in immunity, these molecules may emerge as viable therapeutic targets for infection or autoimmune disorders driven by antiviral signaling.
3 |. RNA REGULATION OF INTERFERON AND INTERFERON-STIMULATED GENES
Interferons are indispensable for a robust antiviral response. Type I IFNs, comprised of the fourteen subtypes of IFNα plus the singular IFN-β, are arguably the most critical molecules that drive antiviral immunity. Once secreted from infected cells, type I IFNs bind a heterodimeric receptor composed of IFNAR1 and IFNAR2 (referred to as IFNAR) on the surface of the same, neighboring, or distal cells. Signaling downstream of IFNAR proceeds through the JAK and STAT families of signal transducers.124 The four subtypes of type III IFNs act in a similar fashion at epithelial surfaces, albeit through IFNLR1/IL10R2 receptor complex. Through these receptors, both type I and III IFNs activate a common transcription factor complex of composed of STAT1, STAT2, and IRF9 which induces hundreds of ISGs.124,125 Many ISGs code for direct antiviral effectors, as well as proteins involved in feedback loops that amplify or inhibit antiviral pathways.41,126 In this section, we will discuss RNA regulatory mechanisms that directly control IFN and ISG transcripts (Figure 2).
FIGURE 2.
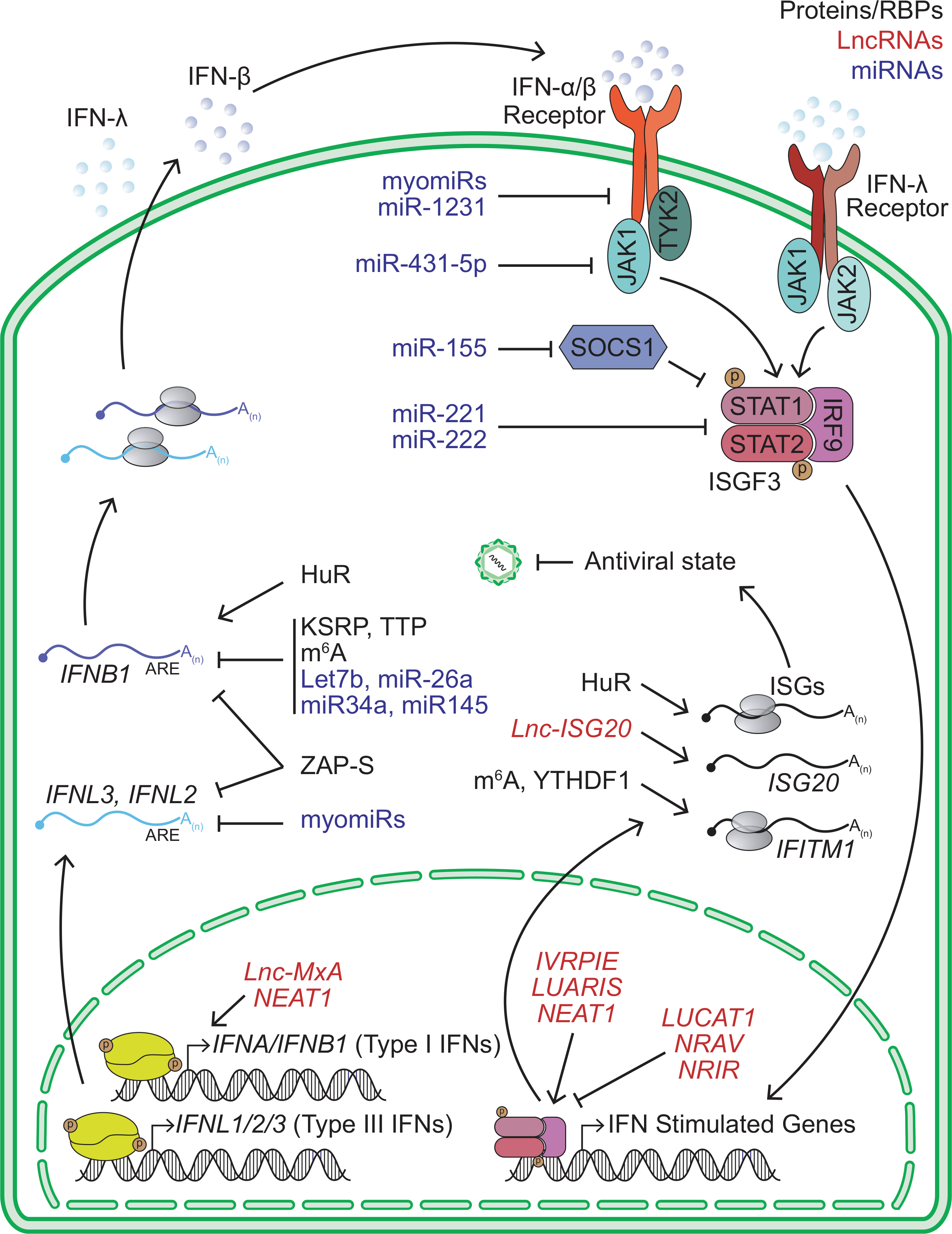
Summary of RNA regulatory mechanisms that control IFN and ISGs expression. Type I and III IFN signaling leads to the activation of the ISGF3 transcription factor complex and the expression of antiviral ISGs. RNA-binding proteins (RBPs) or alternatively spliced (AS) proteins, lncRNAs, and miRNAs that modulate this pathway are labeled in black, red, and blue, respectively
3.1 |. mRNA stability elements and RBPs that control IFNs
While the sensing of viral infection leads to a robust transcriptional induction of antiviral IFNs, type I IFN mRNAs are stringently regulated through several post-transcriptional mechanisms. The 3′ UTRs of all IFN mRNAs, like other transcripts encoding pro-inflammatory cytokines, contain AU-rich elements (AREs).127,128 AREs are cis-regulatory features composed of one or more repeats of the sequence “UUAUUUAUU” or a more minimal sequence of “AUUUA”.129–131 3′ UTR AREs tend to destabilize transcripts through specific AU-rich element-binding proteins (ARE-BPs) which recruit the deadenylase complex to shorten the polyA tail, ultimately leading to mRNA decay.132,133 Beyond controlling RNA stability, 3′ UTR AREs can also positively or negatively influence translation of mRNAs depending on which ARE-BPs they recruit.134,135
The ARE-BP KSRP has been shown to inhibit type I IFN production. Deletion of KSRP in murine cells leads to increased RNA and protein expression of IFN-β and IFN-α4 following treatment with a TLR3 agonist. KSRP interacts directly with Ifnb1 and Ifna4 transcripts and reduces the half-life of these transcripts.136 However, KSRP deletion fails to enhance the basal expression of these type I IFN transcripts in the absence of immune stimulation. It is possible that other ARE-BPs like TTP (encoded by ZPF36) play redundant roles and destabilize these mRNAs, limiting their expression.137 TTP is responsible for the degradation of other pro-inflammatory cytokine transcripts in an ARE-dependent manner, including TNF, IL2, and IFNG.128,138,139 To date, however, a direct role for TTP in the control of type I or III IFN transcripts has not been described. On the other hand, ARE-mediated destabilization of transcripts can be countered by HuR (encoded by ELAVL1) which typically competes with negative regulators like TTP or KSRP for ARE-binding.2,140–143 Consistent with this, HuR interaction with AREs in the 3′ UTR promotes the stability of IFNB1. Both depletion and inhibition of dimerization, required for HuR function, result in a significant reduction of IFNB1 mRNA levels, promoting viral replication.144 Furthermore, HuR was recently shown to have an altered binding pattern across the transcriptome during innate immune stimulus by a dsRNA analog.145 Activating the RLR pathway decreased HuR binding in intronic sequences and increased its interaction within 3′ UTRs. In this context, HuR bound to the 3′ UTRs of several ISGs or ISG regulators and promoted their mRNA stability. Thus, it is clear that AREs and ARE-BPs like HuR, KSRP, and TTP work in concert and antagonistically to modulate antiviral and inflammatory responses.
Recently, our laboratory has discovered that IFN-inducible alternative polyadenylation of the antiviral RBP ZAP (encoded by ZC3HAV1) functionalizes ZAP to act through AREs for regulation of IFN production.146 The alternative polyadenylation of ZAP is driven by the cleavage factor CSTF2 which leads to the expression of two distinct isoforms. The large isoform ZAP-L contains a C-terminal CaaX (cysteine-aliphatic-aliphatic-any residue) amino acid motif. This CaaX motif directs the post-translational prenylation of ZAP-L and targets this isoform to endosomal and lysosomal membranes, where it binds to and degrades viral RNA localized in these compartments. On the other hand, ZAP-S, preferentially induced in response to IFN, does not encode a terminal CaaX motif, and is thus localized diffusely in the cytosol. ZAP-S, but not ZAP-L, binds type I and III IFN mRNAs and destabilizes them. Thus, ZAP-S acts as part of a negative feedback mechanism to prevent prolonged IFN production. AREs in IFN transcripts appear to aid in ZAP binding as mutating AREs in the IFNL3 3′ UTR reduces the interaction of ZAP-S with this transcript.146 However, it is unlikely that ZAP can directly interact with AREs. Although ZAP can bind UA dinucleotide-rich artificial RNAs, it optimally interacts with CG-dinucleotide containing RNA motifs in viral RNA.147–149 Thus, how AREs mediate ZAP binding and control of IFN mRNAs still requires further investigation.
While alternative polyadenylation and divergent subcellular localization direct the ability of ZAP isoforms to interact with different RNA species, what regulates altered RBP interactions with specific mRNAs during immune activation is incompletely understood. miRNA binding to 3′ UTRs has the potential to influence local RBP interactions and vice versa.150 Post-translational modifications are also likely to influence the RBP RNA-binding process.151 Phosphorylation and SUMOylation are post-transcriptional modifications emerging as key mechanisms that regulate the ability of RBPs to bind RNA.98,106 RBP specificity and function are therefore important considerations when studying the immune response to viral infection and thus warrant further investigation.
3.2 |. RNA modifications in the IFN response
The RNA modification m6A affects the host response to viral infection in a virus- and cell-type dependent fashion.62 During infection of fibroblasts by the DNA virus human cytomegalovirus (HCMV), enhanced expression of the m6A methyltransferase METTL3 induces m6A deposition in the 3′ UTR of the IFNB1 transcript near the stop codon. m6A in IFNB1 transcript reduces its half-life, inhibits IFN-β expression, and thus promotes viral infection.152,153 This mechanism of regulation is likely cell-type specific, as neither m6A modification of IFNB1 nor a role for the cellular m6A machinery in IFNB1 expression is observed in hepatocyte or lung epithelial cell lines.154–156 However, during infection of hepatocytes by viruses in the Flaviviridae family, activation of RLR signaling and ER stress pathways alters m6A modification on both pro- and antiviral transcripts.154 Many ISGs are also sensitive to m6A-mediated regulation; indeed, the transcripts of >50 ISGs are modified by m6A. m6A promotes the translation of a subset of these ISGs by facilitating transcript interaction with the m6A-binding protein YTHDF1.155 Currently, the role of m6A in other m6A-modified ISG transcripts remains unknown, but may involve modulation of mRNA processing or half-life.
3.3 |. miRNAs in the IFN response
Beyond regulating RLRs and sensing pathways that lead to IFN induction, miRNAs can directly degrade type I and III IFN transcripts. Several miRNAs target the 3′ UTR of IFNB1, including Let7b, miR-26a, miR-34a, and miR145.157 These miRNAs are induced during both viral infection and type I IFN treatment and inhibit the expression of IFNB1 in human and non-human primates, serving as a miRNA-regulated negative feedback mechanism for type I IFN production. Work from our laboratory has shown that Hepatitis C virus (HCV) can induce the expression of the miRNAs miR-208b and miR-499 in human hepatocytes.158,159 These miRNAs are encoded within the introns of the myosin genes MYH7 and MYH7B. These genes and miRNAs are typically restricted in expression to cardiac and skeletal muscle cells and are therefore called myomiRs. Only HCV infection–not infection by other Flaviviridae family members dengue virus (DENV) or WNV nor stimulus with synthetic immunostimulatory ligands–upregulates myomiRs in hepatocytes, suggesting an HCV-specific mechanism of induction. Further, though type I IFN amplifies the expression of these myomiRs during HCV infection, type III IFN does not. These myomiRs target the 3′ UTRs of the type III IFN mRNAs IFNL2 and IFNL3 as well as the 3′ UTR of IFNAR1, leading to destabilization and downregulation. Our studies demonstrated a mechanism by which HCV can suppress host IFN responses by using host RNA machinery to shut down expression of antiviral mediators.
As demonstrated with myomiRs that target IFNAR1, miRNAs may regulate type I IFN signaling at or downstream of the IFN receptor. Ligation of IFNAR1/2 by IFN-α/β induces the phosphorylation of STAT1 and STAT2, which then associate with IRF9 to form the ISGF3 complex. This transcription factor complex then recognizes IFN-sensitive response elements (ISREs) in the genome and induces expression of hundreds of ISGs. miR-155, discussed previously, upregulates IFN signaling downstream of IFNAR through repression of SOCS1, an ISG that limits IFNAR signal transduction. In mice, miR-155 targets the Socs1 3′ UTR, enhancing STAT1 phosphorylation and ISG expression while reducing viral replication.111 Alternatively, miR-221 and miR-222 negatively regulate IFN signaling. Depleting miR-221/222 increases expression and phosphorylation of STAT1 and STAT2 and heightens induction of ISGs. However, as miR-221/222 do not share seed-sequence complementarity with STAT1 and STAT2 transcripts, it is likely that they influence IFN signaling through an indirect mechanism. With the examples of myomiRs, mir-155 and miR-221/22, and several others, it is evident that miRNAs modulate the expression of type I and III IFNs as well their downstream signaling pathways, thereby fine tuning the immune response to viral infection.
3.4 |. LncRNAs in the IFN response
Antiviral signaling leads to widespread changes in lncRNA expression. For example, our laboratory has characterized differentially regulated lncRNAs in plasmacytoid dendritic cells (pDCs), a major source of type I IFN during viral infections. 500 lncRNAs were differentially expressed in pDCs following treatment with either a TLR7/8 agonist which induces IFN or with IFN-β itself.32,160 Several lncRNAs differentially expressed in pDCs during TLR7/8 ligation were also found to be similarly up- or downregulated during IAV infection indicating that these genes also respond to innate immune activation during infection.161 Further, a large proportion of these differentially expressed lncRNA loci were found in proximity on the chromosome to ISGs or other genes involved in immune processes.32 Given that lncRNAs can affect transcription in both cis and trans, lncRNA expression may serve as a means of positive or negative feedback for the cellular antiviral state during infection.31,162,163
Correspondingly, several lncRNAs are implicated in the regulation of the IFN response and viral infection.164 For example, transcription of IFNB1 is inhibited by lnc-MxA, an antisense strand non-coding RNA that originates from the ISG locus MX1.165 When induced via IFN signaling, lnc-MxA interacts directly with the IFNB1 promoter to form an RNA:DNA triplex that inhibits the binding of the transcription factors IRF3 and NF-κB LUCAT1 inhibits ISG expression more broadly through interacting with the transcription factor STAT1.166 The lncRNA NRAV suppresses the expression of several ISGs by altering histone modification and transcriptional availability of their promoters. Specifically, NRAV appears to promote the deposition of the repressive epigenetic mark histone 3 lysine 27 trimethylation (H3K27me3) and impair decoration with the activating mark histone 3 lysine 4 trimethylation (H3K4me3). Intriguingly, NRAV is rapidly degraded during infection through unknown mechanisms, allowing for ISG expression during the host antiviral response.167 NRIR (also called lncRNA-CMPK2) also acts to suppress the expression of proximal and distal ISG loci through epigenetic mechanisms. Unlike NRAV, which is downregulated upon viral infection, NRIR is a bonafide ISG induced by IFN treatment and is thus involved in a negative feedback loop during the IFN response.168 Conversely, the lncRNA IVRPIE promotes ISG expression. Infection-inducible IVRPIE promotes the deposition of the activating histone mark H3K4me3 and blocks that of repressive mark H3K27me3 at IFNB1 and other key ISG loci via interactions with the RBP hnRNPU.169 hnRNPU also stabilizes LUARIS (also called lncRNA#32), which directs the transcription factor ATF2 and promotes transcription of several ISGs.170 While the aforementioned lncRNAs all modulate a broad swathe of antiviral genes, some directly regulate ISGs. For example, Lnc-ISG20 is transcribed in the sense orientation from the ISG20 locus. Despite sharing a significant overlap with the mRNA that encodes the antiviral protein ISG20, Lnc-ISG20 cannot be translated. Instead, this non-coding transcript acts as a sponge for miR-326, which normally targets and degrades ISG20, thereby increasing ISG20 protein levels by incapacitating a direct ISG20 inhibitor.171 Further investigation of lncRNAs differentially expressed during infection or immune stimulus will certainly uncover other roles for these versatile molecules in the antiviral immune response.
4 |. HOST GENETIC VARIATION IN THE RNA REGULATION OF THE ANTIVIRAL RESPONSE
Genetic variation allows for disparate outcomes from infection with the same pathogen in different individuals. Single nucleotide polymorphisms (SNPs) in key genes or regulatory elements may significantly influence the strength of an individual’s antiviral immunity. Genome-wide association studies (GWAS), expression quantitative trait locus (eQTL) mapping, and candidate gene approaches have revealed numerous associations between various polymorphisms and immune processes.172,173,174 However, assigning causality to a particular SNP from a group of polymorphisms in linkage disequilibrium and then following this single SNP’s functional burden proves challenging.175 Despite the difficulty, several single SNP genetic variants have been demonstrated to impact RNA regulatory processes during antiviral immunity.
Splicing regulatory polymorphisms in the IRF5 gene can contribute to systemic lupus erythematosus (SLE), an autoimmune disease characterized by excess production of type I IFN.176 IRF5 is an important component of immune activation downstream of TLR7 and TLR8 signaling.177–179 It directly promotes the transcription of type I IFN, promotes inflammatory pathology, and is also a key regulator of B cell differentiation and function.180–182 Distinct promoters and transcription start sites delineate four potential first exons for the IRF5 gene (exons 1A-D). Exon 1B initiates a constitutively expressed IRF5 transcript. However, this isoform can only be spliced correctly when the T allele at rs2004640 generates a strong 5′ splice donor after exon 1B. Thus, rs2004640T enhances the expression of functional IRF5 transcripts and promotes dysregulated IFN pathways in SLE.183–185 An additional SNP rs10954213A in the 3′ UTR of IRF5 generates a strong polyadenylation sequence (AAT(A/G)A) proximal to the gene’s stop codon and therefore produces IRF5 transcripts with a shorter 3′ UTR.185 This shorter 3′ UTR is associated with extended half-life and higher expression of IRF5, consistent with findings that longer 3′ UTRs promote RNA degradation via facilitating interactions with RBPs and microRNAs.186–189 By influencing differential splicing and polyadenylation of a central player in antiviral pathways, genetic polymorphisms in IRF5 promote type I interferonopathy; how these SNPs affect the outcome of viral infection is still largely unclear.
In addition to functionalizing polyadenylation sequences, polymorphisms in the 3′ UTR of mRNAs can alter sequence or structure-dependent RBP and miRNA interactions, thus impacting transcript metabolism.190,191 One such variant exists in the 3′ UTR of the IFNL3 gene which encodes IFNλ3, a type III IFN crucial for balanced antiviral responses.192,193 Several SNPs at the IFNL3 locus are associated with both response to IFN therapies and with natural clearance of hepatitis C virus (HCV).194–198 We and others have identified the functional mechanism of one SNP in the IFNL3 3′ UTR, rs4803217T/G.158,199 The unfavorable T allele of rs4803217, associated with HCV persistence, inhibits compact RNA structure in the IFNL3 3′ UTR, reducing the translation efficiency of this mRNA.199 The T allele also enhances seed-sequence pairing of the microRNAs miR-208b and miR-499a-5p at the polymorphic site. These microRNAs, together with neighboring AREs, destabilize IFNL3 mRNA harboring the T allele, thereby downregulating cytokine expression and worsening infection outcome.158 As described in the previous section, the myomiRs induced by HCV infection also downregulate IFNL2 and the type I IFN receptor IFNAR1, pointing to a viral strategy that invokes host miRNAs for evasion of type I and type III IFN signaling.158,159
Intriguingly, the unfavorable T allele in IFNL3 described above is strongly linked with the ΔG allele of rs368234815TT/ΔG in IFNL4, a type III IFN gene upstream of the IFNL3 locus.199–201 While the TT allele introduces a frameshift to pseudogenize IFNL4, the ΔG allele can, in principle, lead to functional IFNλ4 expression; thus, IFNλ4 expression may be linked to adverse outcomes of HCV and other viral infections.201 However, our laboratory has shown that while IFNλ4 retains bioactivity comparable to IFNλ3, several RNA regulatory mechanisms suppress IFNλ4 expression.200 IFNL4 mRNA can be alternatively spliced to produce three protein-coding isoforms, but only the largest, IFNλ4p179 (named for the number of amino acids), is secreted, despite each isoform containing identical N-terminal secretion signals. Additional isoforms of IFNL4 harbor retained introns and are thus more likely to be retained in the nucleus or subject to NMD. All IFNL4 transcripts poorly associate with polysomes, indicating inefficient translation. Thus, alternative splicing of IFNL4 may serve to limit functional IFNλ4 protein expression. Lastly, the 3′ UTR of IFNL4 does not encode a canonical polyadenylation sequence, further inhibiting the proper processing and maturation of transcripts from this locus.200 Thus, HCV persistence observed with the rs368234815ΔG phenotype may stem from the inability of functional IFNλ4 to compensate for reduced IFNλ3 expression.
miRNA-mediated mechanisms similar to those described in regulation of IFNL3 during HCV infection may also explain the effect of several polymorphisms in the 3′ UTRs of IFNAR1 (the type I IFN receptor) and JAK1 (a crucial kinase required for signaling downstream of IFNAR1). Polymorphisms in these genes are associated with hepatocellular carcinoma (HCC), a disease strongly linked with viral hepatitis. rs17875871 is a 4 bp indel in the IFNAR1 3′ UTR. Perfect seed-sequence pairing between the deletion allele in IFNAR1 and miR-1231 is disrupted by the 4-nucleotide insertion. Consequently, the hetero- or homozygous deletion allele is associated with reduced IFNAR1 expression and elevated risk of HCC, likely through persistence of viral hepatitis.202 Similarly, rs112395617, a 4 bp indel polymorphism in the 3′ UTR of JAK1 (a kinase crucial for type I and III IFN signaling) alters the binding of miR-431–5p and impacts JAK1 expression.203 While intriguing, more precise functional dissection of these polymorphisms and their interplay with microRNAs is required for full understanding of their effect on antiviral immunity.
Single nucleotide polymorphisms within splice donor and acceptor sites or within cis-regulatory sequences can lead to novel gain-of-function or loss-of-function isoforms of antiviral proteins. These isoforms may contribute to differential control of viral infection and can also lead to autoimmunity. One splicing regulatory polymorphism is found in the OAS1 gene, which encodes an ISG that senses dsRNA and catalyzes the production of the second messenger 2′−5′A. 2′−5′A binding dimerizes and activates the latent endoribonuclease activity of RNase L for degradation of viral RNA.69 The C-terminal of human OAS1 is alternatively spliced to produce several potential isoforms including OAS1 p42, p44, p46, p48, and p52. Of these isoforms, only p42 or p46 are expressed robustly at the protein level due to the instability of the other protein variants and of the RNA isoforms encoding them.203–205 The expression of OAS1 p42 or p46 is controlled by the SNP rs10774671A/G in the splice acceptor site of exon 6. The G allele of this SNP shifts the splice acceptor site by one nucleotide and thus generates p46 with a novel C-terminal amino acid sequence that includes a terminal CaaX motif, subject to post-translational prenylation. Our laboratory has found that prenylation of p46 targets this OAS1 isoform to intracellular organelle membranes such as the Golgi apparatus, whereas unprenylated p42 is localized diffusely in the cytosol. Membrane-localized OAS1 p46 demonstrates enhanced antiviral potential over OAS1 p42 against multiple RNA viruses from distinct families including enteroviruses, flaviviruses, and coronaviruses, all of which use cellular membranes to establish their replication compartments.204 Consequently, by altering the splicing of OAS1, rs10774671G is associated with reduced clinical susceptibility to HCV, WNV, and SARS-CoV-2.204,207–210
As with protein-coding genes, lncRNAs are also subject to regulation by genetic variants which impact viral infection. For example, the SNPs rs1015164 and rs2027820 are polymorphisms in linkage disequilibrium within the CCR5 locus, which encodes a key chemokine and the co-receptor for human immunodeficiency virus-1 (HIV-1).211,212 rs1015164-G and rs2027820-A are associated with decreased expression of an antisense lncRNA CCR5AS from this locus. CCR5AS promotes the expression of CCR5 by sequestering the RBP RALY and inhibiting RALY-mediated destabilization of the CCR5 transcript.212 Therefore, these polymorphisms, which reduce the expression of CCR5AS, also inhibit expression of CCR5 protein levels and therefore promote resistance to HIV-1. However, given that CCR5 deficiency is detrimental to resolution of influenza A virus (IAV) and West Nile virus (WNV) infection, how CCR5AS and its associated genetic variation influences other infections remains in question.213,214
These examples serve to highlight the breadth of mechanisms by which genetic variation can influence the RNA biology that underlies antiviral innate immunity. While functional characterization of most human polymorphisms remains incomplete, most well-validated variants involve changes in protein sequence. However, the vast majority of all pathogenic SNPs identified through GWAS are in non-coding regions of the genome.46,47 Up to 15% of all pathogenic SNPs are intronic, perhaps disrupting cis-regulatory RNA elements or affecting splicing.215 A significant fraction of SNPs (~3.5%) are in untranslated regions.48 Many lie within or regulate lncRNAs.216 Mechanistic investigation of pathogenic SNPs in antiviral genes will enhance our understanding of immune pathways and reveal novel RNA regulatory mechanisms in viral infection.
5 |. RNA REGULATION OF VIRAL PATHOGENESIS AND EVASION OF HOST IMMUNITY
Given the importance of RNA regulation in antiviral immunity, it is not surprising that viruses have evolved mechanisms to co-opt or subvert host RNA metabolism. Viral RNAs themselves may also promote pathogenesis. Viruses often target host splicing to evade the immune response and to enhance their own virluence.92 The ongoing COVID-19 pandemic has led to rapid advances in our knowledge of how SARS-CoV-2 antagonizes host immune responses. It was recently shown that SARS-CoV-2 non-structural protein NSP16 can interfere with cellular mRNA splicing.217 NSP16 interacts with the small nuclear RNAs (snRNAs) U1 and U2, required for canonical intron splicing, at their mRNA splice recognition sites. Infection with SARS-CoV-2 or ectopic expression of NSP16 suppresses global splicing of host mRNAs, including that of antiviral ISGs like RIG-I. DENV disrupts the efficient splicing of cellular genes including RIG-I in a similar manner, as its RNA-dependent RNA polymerase NS5 inhibits host U5 snRNP, another component of the canonical spliceosome.218 While viral RNA from coronaviruses and flaviviruses is not spliced, some RNA viruses like IAV require efficient splicing of their RNA for productive infection.189 During IAV infection, the host splicing factor hnRNPK accumulates in nuclear speckles where viral RNA is spliced, leading to perturbations in hnRNPK-dependent host cellular splicing events.219–221 Reversal of splicing disruptions induced by IAV attenuates viral infection, demonstrating that impeding host splicing is a convergent strategy used by several viruses to generate a permissive environment for their replication.
As obligate intracellular parasites, viruses require the host translational machinery for the production of their proteins. Consequently, they have evolved numerous mechanisms to subvert or shut off host translation while promoting the translation of viral RNA. Conversely, restricting translation is also a host strategy for combatting viral replication. Thus, access to efficient translation is a key zone of evolutionary conflict between virus and host. Translation initiation in eukaryotes typically starts in a cap-dependent manner, where initiation factors recognize the 5′ m7G mRNA cap and recruit the 40S ribosomal subunit.222 However, many viruses including poliovirus and HCV translate their RNA through cap-independent mechanisms involving exquisitely structured RNA sequences known as internal ribosomal entry sites (IRES), which engage translation initiation factors or the 40S ribosomal subunit directly.223 The presence of an IRES in poliovirus RNA allows for its translation despite viral cleavage of the initiation factor eIF4G, which is required for the cap-dependent translation of cellular RNAs.224 On the other hand, the HCV IRES reduces the sensitivity of viral RNA translation to eiF2α phosphorylation. Phosphorylation of the initiation factor eiF2α by kinases such as the dsRNA sensor PKR is a cellular strategy to limit the translation of viral and cellular RNA during infection and stress responses. As the HCV IRES directly recruits the 40S subunit and bypasses eiF2α, eiF2α phosphorylation only cripples antiviral ISG protein production.222,225 5′ terminal RNA structures can also rescue viral RNA from translation inhibition by the antiviral effector IFIT1, which binds to viral RNAs lacking 2′O-methylation in the cap structure. In the case of alphaviruses, defined stem-loop structures in the 5′ UTR prevent the ability of IFIT1 to bind to the unmethylated cap of these viruses and thus allow for efficient translation of viral RNA.55,226
SARS-CoV-2 blocks cellular translation through its NSP1 protein, which interacts with 18S ribosomal RNA and impedes the mRNA entry channel of the ribosome.217 Importantly, the stem-loop 1 (SL1) RNA structure in the viral 5′ UTR allows SARS-CoV-2 mRNAs to bypass this blockage and initiate translation. Given that the proximity of SL1 to the 5′ end of viral RNA is critical in avoiding translational suppression, it is thought that NSP1 likely recognizes SL1 and consequently dissociates from the ribosome, allowing for translation of viral mRNA. NSP1 involvement in host translational shutdown is further supported by cryo-EM structure analysis showing NSP1 interaction with the 40S ribosomal subunit and 18S ribosomal RNA.227,228 In addition to disrupting host translation, SARS-CoV-2 NSP1 can induce degradation of host mRNA transcripts, similar to NSP1 from SARS-CoV.229,230 The precise interplay between cellular translational inhibition and host mRNA degradation mediated by NSP1 during SARS-CoV-2 infection remains unknown.
In addition to viral proteins, viral RNA also allows for evasion of host responses and may enhance mechanisms of viral pathogenesis. For instance, flaviviruses subvert host responses through the production of a non-coding RNA species called subgenomic flaviviral RNAs (sfRNAs). sfRNAs are produced by all flaviviruses and contribute to viral pathogenesis.231 sfRNAs are generated from the incomplete digestion of the flavivirus genome by the host 5′−3′ exonuclease XRN1.232 XRN1 colocalizes with the 3′ UTR of WNV in processing bodies, subcellular sites of RNA degradation, and depletion of XRN1 decreases sfRNA generation. Mechanistically, highly structured, pseudoknot reinforced RNA stem loops in the 3′ UTRs called XRN1-resistant RNAs (xrRNAs) work to stall processive XRN1 degradation of the viral genome.233–235 xrRNAs also contribute to sfRNA generation in mosquito cells, as XRN1 is a highly conserved mechanism of RNA decay across species. sfRNAs in insect cells are important determinants of flavivirus transmission.231,236,237 Accordingly, mutations in xrRNAs and associated defects in sfRNA production generate bottlenecks in transmission between insect cells and mammalian cells.238,239
sfRNA-driven flavivirus pathogenesis in human cells occurs through multiple mechanisms. By stalling and sequestering XRN1 at viral RNAs, xrRNA structures and sfRNAs can dysregulate normal cellular mRNA decay, leading to detrimental effects in the host as well as increased viral replication.240 sfRNAs can also dampen the type I IFN response by modulating the function of TRIM25 in RLR signaling.241 TRIM25, an E3 ubiquitin ligase that adds activating K63-linked ubiquitin chains to RIG-I during viral sensing, needs to be deubiquitinated itself to function in this capacity. DENV sfRNAs bind to TRIM25 and inhibit its deubiquitination, thus restricting TRIM25 activation of RIG-I. In line with these findings, infection by DENV strain PR-2B, which maintains a higher ratio of sfRNA to genomic RNA than the strain PR-1, demonstrates reduced TRIM25 deubiquitination and decreased IFN production when compared with PR-1. DENV sfRNAs also disrupt the function of the cellular RBPs G3BP1, G3BP2, and CAPRIN1, which are required for the loading of key antiviral ISG transcripts onto polyribosomes for efficient translation. This body of literature provides strong evidence for the role of sfRNAs in promoting viral replication, antagonizing the translation of ISGs, and impairing the antiviral immune response.
While miRNAs influence the antiviral response in various ways, miR-122 plays a uniquely important role in HCV infection. Expressed abundantly in the liver, two miR-122 transcripts may interact simultaneously with the 5′ UTR of HCV.242 miR-122 is essential for HCV replication; inactivation of miR-122 or mutation of the miR-122 binding site in the HCV 5′ UTR drastically reduces viral RNA and protein levels. miR-122 likely promotes HCV infection through several interconnected mechanisms. First, miR-122 bound to AGO2 serves as an RNA “cap” for the 5′ end of HCV RNA, which lacks a typical m7G cap, thereby increasing the stability of viral RNA.243–245 Second, miR-122 binding to the 5′ UTR promotes the translation of HCV RNA by altering the IRES structure.246–249 Third, miR-122 affects the ratio of viral RNA involved in translation versus replication and thus promotes the synthesis of viral RNA.250 Finally, when miR-122 is bound to viral RNA, it is sequestered from its cellular host targets, the consequential upregulation of which may establish a more permissive cellular environment for HCV replication and persistence.251 miR-122 is so essential to the HCV life cycle that it is considered the primary determinant of the virus’s liver-specific tropism.242,252 Therefore, targeting miR-122 may be a viable host-directed therapeutic strategy to combat HCV infection and is currently being explored in clinical trials.253 Together, these examples clearly indicate that host RNA processes are a key target that viruses subvert or co-opt for their advantage.
6 |. CONCLUDING REMARKS
The examples discussed in this review highlight the diverse roles of RNA regulatory mechanisms in controlling antiviral innate immunity. However, much remains to be discovered. A systematic dissection of post-translational modifications and how they modulate RBP function may shed light on how antiviral signaling events are rapidly integrated into post-transcriptional regulatory circuits. The mechanistic investigation of the thousands of lncRNAs with unknown functions will undoubtedly reveal their widespread involvement in the immune system. We will also likely gain a greater appreciation of RNA-nucleated biomolecular condensates in organizing cellular immune signaling processes. These advances, and others like them, will likely be driven by recently developed technologies. For example, the widespread use of several crosslinking immunoprecipitation techniques as well as affinity purification strategies coupled with mass spectrometry has greatly enhanced our ability to deconvolve RNA-RBP interactomes. Similarly, the advent of long-read sequencing has allowed for the easy identification of alternative splice isoforms with minimal algorithmic processing as compared with traditional short-read sequencing methods. The application of such state-of-the-art techniques to immunological problems will greatly further our understanding of RNA-centric regulation of the immune response to viral pathogens. With RNA molecules increasingly proved as viable druggable targets, manipulating the RNA-associated processes that control antiviral innate immune signaling pathways may be an efficient therapeutic strategy to combat viral infection and immune dysregulation.
ACKNOWLEDGEMENTS
This work was supported by grants from the NIH (AI145974, AI141823, AI147177). NSG was supported by a Helen Hay Whitney Postdoctoral Fellowship. JRS was supported by a NIH T32 training grant (5T32AI106677-08). RDVG was supported by a research training grant from the University of Washington Institute for Translational Health Sciences.
Funding information
Helen Hay Whitney Foundation; University of Washington ITHS, Grant/Award Number: TL1 TR002318; National Institute of Allergy and Infectious Diseases, Grant/Award Number: 5T32AI106677-08, AI141823, AI145974 and AI147177
7 |. DATA AVAILABILITY STATEMENT
No new data were generated for this manuscript.
Footnotes
CONFLICT OF INTEREST
The authors do not declare any conflicts of interest.
REFERENCES
- 1.Corley M, Burns MC, Yeo GW. How RNA-binding proteins interact with RNA: molecules and mechanisms. Mol Cell. 2020;78(1):9–29. 10.1016/j.molcel.2020.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mukherjee N, Corcoran DL, Nusbaum JD, et al. Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol Cell. 2011;43(3):327–339. 10.1016/j.molcel.2011.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nicholson CO, Friedersdorf M, Keene JD. Quantifying RNA binding sites transcriptome-wide using DO-RIP-seq. RNA. 2017;23(1):32–46. 10.1261/rna.058115.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim YK, Furic L, Parisien M, Major F, DesGroseillers L, Maquat LE. Staufen1 regulates diverse classes of mammalian transcripts. EMBO J 2007;26(11):2670–2681. 10.1038/sj.emboj.7601712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marión RM, Fortes P, Beloso A, Dotti C, Ortín J. A human sequence homologue of Staufen is an RNA-binding protein that is associated with polysomes and localizes to the rough endoplasmic reticulum. Mol Cell Biol 1999;19(3):2212–2219. 10.1128/mcb.19.3.2212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bourgeois CF, Mortreux F, Auboeuf D. The multiple functions of RNA helicases as drivers and regulators of gene expression. Nat Rev Mol Cell Biol 2016;17(7):426–438. 10.1038/nrm.2016.50 [DOI] [PubMed] [Google Scholar]
- 7.Balcerak A, Trebinska-Stryjewska A, Konopinski R, Wakula M, Grzybowska EA. RNA-protein interactions: disorder, moonlighting and junk contribute to eukaryotic complexity. Open Biol 2019;9(6):190096. 10.1098/rsob.190096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ottoz DSM, Berchowitz LE. The role of disorder in RNA binding affinity and specificity. Open Biol 2020;10(12):200328. 10.1098/rsob.200328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Uversky VN. Intrinsically disordered proteins in overcrowded milieu: membrane-less organelles, phase separation, and intrinsic disorder. Curr Opin Struct Biol 2017;44:18–30. 10.1016/j.sbi.2016.10.015 [DOI] [PubMed] [Google Scholar]
- 10.Guillén-Boixet J, Kopach A, Holehouse AS, et al. RNA-induced conformational switching and clustering of G3BP drive stress granule assembly by condensation. Cell. 2020;181(2):346–361.e17. 10.1016/j.cell.2020.03.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramanathan A, Robb GB, Chan S-H. mRNA capping: biological functions and applications. Nucleic Acids Res 2016;44(16):7511–7526. 10.1093/nar/gkw551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA modifications in gene expression regulation. Cell. 2017;169(7):1187–1200. 10.1016/j.cell.2017.05.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X, Lu Z, Gomez A, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505(7481):117–120. 10.1038/nature12730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang X, Zhao BS, Roundtree IA, et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161(6):1388–1399. 10.1016/j.cell.2015.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Edupuganti RR, Geiger S, Lindeboom RGH, et al. N6-methyladenosine (m6A) recruits and repels proteins to regulate mRNA homeostasis. Nat Struct Mol Biol 2017;24(10):870–878. 10.1038/nsmb.3462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arguello AE, DeLiberto AN, Kleiner RE. RNA chemical proteomics reveals the N6-methyladenosine (m6A)-regulated protein-RNA interactome. J Am Chem Soc 2017;139(48):17249–17252. 10.1021/jacs.7b09213 [DOI] [PubMed] [Google Scholar]
- 17.Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol 2019;20(10):608–624. 10.1038/s41580-019-0168-5 [DOI] [PubMed] [Google Scholar]
- 18.Patil DP, Pickering BF, Jaffrey SR. Reading m6A in the transcriptome: m6A-binding proteins. Trends Cell Biol 2018;28(2):113–127. 10.1016/j.tcb.2017.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518(7540):560–564. 10.1038/nature14234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu N, Zhou KI, Parisien M, Dai Q, Diatchenko L, Pan T. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res 2017;45(10):6051–6063. 10.1093/nar/gkx141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiang J-F, Yang Q, Liu C-X, Wu M, Chen L-L, Yang L. N6-methyladenosines modulate A-to-I RNA editing. Mol Cell. 2018;69(1):126–135.e6. 10.1016/j.molcel.2017.12.006 [DOI] [PubMed] [Google Scholar]
- 22.Yang Y, Wang L, Han X, et al. RNA 5-methylcytosine facilitates the maternal-to-zygotic transition by preventing maternal mRNA decay. Mol Cell. 2019;75(6):1188–1202.e11. 10.1016/j.molcel.2019.06.033 [DOI] [PubMed] [Google Scholar]
- 23.Yang X, Yang Y, Sun B-F, et al. 5-methylcytosine promotes mRNA export - NSUN2 as the methyltransferase and ALYREF as an m5C reader. Cell Res 2017;27(5):606–625. 10.1038/cr.2017.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arango D, Sturgill D, Alhusaini N, et al. Acetylation of cytidine in mRNA promotes translation efficiency. Cell. 2018;175(7):1872–1886.e24. 10.1016/j.cell.2018.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wiener D, Schwartz S. The epitranscriptome beyond m6A. Nat Rev Genet 2021;22(2):119–131. 10.1038/s41576-020-00295-8 [DOI] [PubMed] [Google Scholar]
- 26.Cech TR, Steitz JA. The noncoding RNA revolution-trashing old rules to forge new ones. Cell. 2014;157(1):77–94. 10.1016/j.cell.2014.03.008 [DOI] [PubMed] [Google Scholar]
- 27.Djebali S, Davis CA, Merkel A, et al. Landscape of transcription in human cells. Nature. 2012;489(7414):101–108. 10.1038/nature11233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mehta A, Baltimore D. MicroRNAs as regulatory elements in immune system logic. Nat Rev Immunol 2016;16(5):279–294. 10.1038/nri.2016.40 [DOI] [PubMed] [Google Scholar]
- 29.Miranda KC, Huynh T, Tay Y, et al. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126(6):1203–1217. 10.1016/j.cell.2006.07.031 [DOI] [PubMed] [Google Scholar]
- 30.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20. 10.1016/j.cell.2004.12.035 [DOI] [PubMed] [Google Scholar]
- 31.Carpenter S, Aiello D, Atianand MK, et al. A long noncoding RNA mediates both activation and repression of immune response genes. Science. 2013;341(6147):789–792. 10.1126/science.1240925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Joslyn RC, Forero A, Green R, Parker SE, Savan R. Long non-coding RNA signatures induced by toll-like receptor 7 and type I interferon signaling in activated human plasmacytoid dendritic cells. J Interferon Cytokine Res 2018;38(9):388–405. 10.1089/jir.2018.0086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robinson EK, Covarrubias S, Carpenter S. The how and why of lncRNA function: an innate immune perspective. Biochim Biophys Acta Gene Regul Mech 2020;1863(4):194419. 10.1016/j.bbagrm.2019.194419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Atianand MK, Caffrey DR, Fitzgerald KA. Immunobiology of long noncoding RNAs. Annu Rev Immunol 2017;35:177–198. 10.1146/annurev-immunol-041015-055459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goff LA, Rinn JL. Linking RNA biology to lncRNAs. Genome Res 2015;25(10):1456–1465. 10.1101/gr.191122.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kopp F, Mendell JT. Functional classification and experimental dissection of long noncoding RNAs. Cell. 2018;172(3):393–407. 10.1016/j.cell.2018.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154(1):26–46. 10.1016/j.cell.2013.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rehwinkel J, Gack MU. RIG-I-like receptors: their regulation and roles in RNA sensing. Nat Rev Immunol 2020;20(9):537–551. 10.1038/s41577-020-0288-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fitzgerald KA, Kagan JC. Toll-like receptors and the control of immunity. Cell. 2020;180(6):1044–1066. 10.1016/j.cell.2020.02.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Briard B, Place DE, Kanneganti T-D. DNA sensing in the innate immune response. Physiology (Bethesda). 2020;35(2):112–124. 10.1152/physiol.00022.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schoggins JW. Interferon-stimulated genes: what do they all do? Annu Rev Virol 2019;6(1):567–584. 10.1146/annurev-virology-092818-015756 [DOI] [PubMed] [Google Scholar]
- 42.Vazquez C, Horner SM. MAVS coordination of antiviral innate immunity. Sullivan CS, ed. J Virol 2015;89(14):6974–6977. 10.1128/JVI.01918-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kato H, Takeuchi O, Mikamo-Satoh E, et al. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med 2008;205(7):1601–1610. 10.1084/jem.20080091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kato H, Takeuchi O, Sato S, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441(7089):101–105. 10.1038/nature04734 [DOI] [PubMed] [Google Scholar]
- 45.Feng Q, Hato SV, Langereis MA, et al. MDA5 detects the double-stranded RNA replicative form in picornavirus-infected cells. Cell Rep 2012;2(5):1187–1196. 10.1016/j.celrep.2012.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roth-Cross JK, Bender SJ, Weiss SR. Murine coronavirus mouse hepatitis virus is recognized by MDA5 and induces type I interferon in brain macrophages/microglia. J Virol 2008;82(20):9829–9838. 10.1128/JVI.01199-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yin X, Riva L, Pu Y, et al. MDA5 governs the innate immune response to SARS-CoV-2 in lung epithelial cells. Cell Rep 2021;34(2):108628. 10.1016/j.celrep.2020.108628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goubau D, Schlee M, Deddouche S, et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature. 2014;514(7522):372–375. 10.1038/nature13590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hornung V, Ellegast J, Kim S, et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314(5801):994–997. 10.1126/science.1132505 [DOI] [PubMed] [Google Scholar]
- 50.Pichlmair A, Schulz O, Tan CP, et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314(5801):997–1001. 10.1126/science.1132998 [DOI] [PubMed] [Google Scholar]
- 51.Jiang F, Ramanathan A, Miller MT, et al. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature. 2011;479(7373):423–427. 10.1038/nature10537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schuberth-Wagner C, Ludwig J, Bruder AK, et al. A conserved histidine in the RNA sensor RIG-I controls immune tolerance to N1–2′O-methylated self RNA. Immunity. 2015;43(1):41–51. 10.1016/j.immuni.2015.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Devarkar SC, Wang C, Miller MT, et al. Structural basis for m7G recognition and 2′-O-methyl discrimination in capped RNAs by the innate immune receptor RIG-I. Proc Natl Acad Sci USA. 2016;113(3):596–601. 10.1073/pnas.1515152113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Züst R, Cervantes-Barragan L, Habjan M, et al. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat Immunol 2011;12(2):137–143. 10.1038/ni.1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Diamond MS. IFIT1: A dual sensor and effector molecule that detects non-2′-O methylated viral RNA and inhibits its translation. Cytokine Growth Factor Rev 2014;25(5):543–550. 10.1016/j.cytogfr.2014.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ray D, Shah A, Tilgner M, et al. West Nile virus 5′-cap structure is formed by sequential guanine N-7 and ribose 2′-O methylations by nonstructural protein 5. J Virol 2006;80(17):8362–8370. 10.1128/JVI.00814-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou Y, Ray D, Zhao Y, et al. Structure and function of flavivirus NS5 methyltransferase. J Virol 2007;81(8):3891–3903. 10.1128/JVI.02704-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dong H, Chang DC, Xie X, et al. Biochemical and genetic characterization of dengue virus methyltransferase. Virology. 2010;405(2):568–578. 10.1016/j.virol.2010.06.039 [DOI] [PubMed] [Google Scholar]
- 59.Kaiser JA, Luo H, Widen SG, et al. Genotypic and phenotypic characterization of West Nile virus NS5 methyltransferase mutants. Vaccine. 2019;37(48):7155–7164. 10.1016/j.vaccine.2019.09.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Menachery VD, Yount BL, Josset L, et al. Attenuation and restoration of severe acute respiratory syndrome coronavirus mutant lacking 2′-o-methyltransferase activity. J Virol 2014;88(8):4251–4264. 10.1128/JVI.03571-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Menachery VD, Gralinski LE, Mitchell HD, et al. Middle east respiratory syndrome coronavirus nonstructural protein 16 is necessary for interferon resistance and viral pathogenesis. mSphere. 2017;2(6):e00346–17. 10.1128/mSphere.00346-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Williams GD, Gokhale NS, Horner SM. Regulation of viral infection by the RNA modification N6-methyladenosine. Annu Rev Virol 2019;6(1):235–253. 10.1146/annurev-virology-092818-015559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Durbin AF, Wang C, Marcotrigiano J, Gehrke L. RNAs containing modified nucleotides fail to trigger RIG-I conformational changes for innate immune signaling. MBio. 2016;7(5):e00833–16. 10.1128/mBio.00833-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Karikó K, Buckstein M, Ni H, Weissman D. Suppression of RNA recognition by toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23(2):165–175. 10.1016/j.immuni.2005.06.008 [DOI] [PubMed] [Google Scholar]
- 65.Kim G-W, Imam H, Khan M, Siddiqui A. N6-methyladenosine modification of hepatitis B and C viral RNAs attenuates host innate immunity via RIG-I signaling. J Biol Chem 2020;295(37):13123–13133. 10.1074/jbc.RA120.014260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lu M, Zhang Z, Xue M, et al. N6-methyladenosine modification enables viral RNA to escape recognition by RNA sensor RIG-I. Nat Microbiol 2020;5(4):584–598. 10.1038/s41564-019-0653-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chiang JJ, Sparrer KMJ, van Gent M, et al. Viral unmasking of cellular 5S rRNA pseudogene transcripts induces RIG-I-mediated immunity. Nat Immunol 2018;19(1):53–62. 10.1038/s41590-017-0005-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhao Y, Ye X, Dunker W, Song Y, Karijolich J. RIG-I like receptor sensing of host RNAs facilitates the cell-intrinsic immune response to KSHV infection. Nat Commun 2018;9(1):4841. 10.1038/s41467-018-07314-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hornung V, Hartmann R, Ablasser A, Hopfner K-P. OAS proteins and cGAS: unifying concepts in sensing and responding to cytosolic nucleic acids. Nat Rev Immunol 2014;14(8):521–528. 10.1038/nri3719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Malathi K, Dong B, Gale M, Silverman RH. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature. 2007;448(7155):816–819. 10.1038/nature06042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Crow MK, Olferiev M, Kirou KA. Type I interferons in autoimmune disease. Annu Rev Pathol Mech Dis 2019;14(1):369–393. 10.1146/annurev-pathol-020117-043952 [DOI] [PubMed] [Google Scholar]
- 72.Lee-Kirsch MA. The type I interferonopathies. Annu Rev Med 2017;68(1):297–315. 10.1146/annurev-med-050715-104506 [DOI] [PubMed] [Google Scholar]
- 73.Burke JM, Kincaid RP, Nottingham RM, Lambowitz AM, Sullivan CS. DUSP11 activity on triphosphorylated transcripts promotes Argonaute association with noncanonical viral microRNAs and regulates steady-state levels of cellular noncoding RNAs. Genes Dev 2016;30(18):2076–2092. 10.1101/gad.282616.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dhir A, Dhir S, Borowski LS, et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature. 2018;560(7717):238–242. 10.1038/s41586-018-0363-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Deininger P. Alu elements: know the SINEs. Genome Biol 2011;12(12):236. 10.1186/gb-2011-12-12-236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen L-L, DeCerbo JN, Carmichael GG. Alu element-mediated gene silencing. EMBO J 2008;27(12):1694–1705. 10.1038/emboj.2008.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ahmad S, Mu X, Yang F, et al. Breaching self-tolerance to Alu duplex RNA underlies MDA5-mediated inflammation. Cell. 2018;172(4):797–810.e13. 10.1016/j.cell.2017.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chung H, Calis JJA, Wu X, et al. Human ADAR1 prevents endogenous RNA from triggering translational shutdown. Cell. 2018;172(4):811–824.e14. 10.1016/j.cell.2017.12.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rice GI, Del Toro DY, Jenkinson EM, et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet 2014;46(5):503–509. 10.1038/ng.2933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Oda H, Nakagawa K, Abe J, et al. Aicardi-Goutières syndrome is caused by IFIH1 mutations. Am J Hum Genet 2014;95(1):121–125. 10.1016/j.ajhg.2014.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, Stetson DB. Isoforms of RNA-editing enzyme ADAR1 independently control nucleic acid sensor MDA5-driven autoimmunity and multi-organ development. Immunity. 2015;43(5):933–944. 10.1016/j.immuni.2015.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kahles A, Lehmann K-V, Toussaint NC, et al. Comprehensive analysis of alternative splicing across tumors from 8,705 patients. Cancer Cell. 2018;34(2):211–224.e6. 10.1016/j.ccell.2018.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Park YM, Hwang SJ, Masuda K, et al. Heterogeneous nuclear ribonucleoprotein C1/C2 controls the metastatic potential of glioblastoma by regulating PDCD4. Mol Cell Biol 2012;32(20):4237–4244. 10.1128/MCB.00443-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pino I, Pío R, Toledo G, et al. Altered patterns of expression of members of the heterogeneous nuclear ribonucleoprotein (hnRNP) family in lung cancer. Lung Cancer. 2003;41(2):131–143. 10.1016/s0169-5002(03)00193-4 [DOI] [PubMed] [Google Scholar]
- 85.Zarnack K, König J, Tajnik M, et al. Direct competition between hnRNP C and U2AF65 protects the transcriptome from the exonization of Alu elements. Cell. 2013;152(3):453–466. 10.1016/j.cell.2012.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wu Y, Zhao W, Liu Y, et al. Function of HNRNPC in breast cancer cells by controlling the dsRNA-induced interferon response. EMBO J 2018;37(23):e99017. 10.15252/embj.201899017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bowling EA, Wang JH, Gong F, et al. Spliceosome-targeted therapies trigger an antiviral immune response in triple-negative breast cancer. Cell. 2021;184(2):384–403.e21. 10.1016/j.cell.2020.12.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Boelens MC, Wu TJ, Nabet BY, et al. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell. 2014;159(3):499–513. 10.1016/j.cell.2014.09.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nabet BY, Qiu Y, Shabason JE, et al. Exosome RNA unshielding couples stromal activation to pattern recognition receptor signaling in cancer. Cell. 2017;170(2):352–366.e13. 10.1016/j.cell.2017.06.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Akopian D, Shen K, Zhang X, Shan S. Signal recognition particle: an essential protein-targeting machine. Annu Rev Biochem 2013;82:693–721. 10.1146/annurev-biochem-072711-164732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang ET, Sandberg R, Luo S, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456(7221):470–476. 10.1038/nature07509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Boudreault S, Roy P, Lemay G, Bisaillon M. Viral modulation of cellular RNA alternative splicing: a new key player in virus–host interactions? WIREs RNA. 2019;10(5):e1543. 10.1002/wrna.1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gack MU, Kirchhofer A, Shin YC, et al. Roles of RIG-I N-terminal tandem CARD and splice variant in TRIM25-mediated antiviral signal transduction. Proc Natl Acad Sci USA. 2008;105(43):16743–16748. 10.1073/pnas.0804947105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Deng W, Shi M, Han M, et al. Negative regulation of virus-triggered IFN-beta signaling pathway by alternative splicing of TBK1. J Biol Chem 2008;283(51):35590–35597. 10.1074/jbc.M805775200 [DOI] [PubMed] [Google Scholar]
- 95.Frankiw L, Majumdar D, Burns C, et al. BUD13 promotes a type I interferon response by countering intron retention in Irf7. Mol Cell. 2019;73(4):803–814.e6. 10.1016/j.molcel.2018.11.038 [DOI] [PubMed] [Google Scholar]
- 96.Majumdar DS, Frankiw L, Burns CH, Garcia-Flores Y, Baltimore D. Programmed delayed splicing: a mechanism for timed inflammatory gene expression. bioRxiv 2018:443796. 10.1101/443796 [DOI] [Google Scholar]
- 97.Wagner AR, Scott HM, West KO, et al. Global Transcriptomics Uncovers Distinct Contributions From Splicing Regulatory Proteins to the Macrophage Innate Immune Response. Front Immunol 2021;12:656885. 10.3389/fimmu.2021.656885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.West KO, Scott HM, Torres-Odio S, West AP, Patrick KL, Watson RO. The splicing factor hnRNP M is a critical regulator of innate immune gene expression in macrophages. Cell Rep 2019;29(6):1594–1609.e5. 10.1016/j.celrep.2019.09.078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kozak M. Pushing the limits of the scanning mechanism for initiation of translation. Gene. 2002;299(1–2):1–34. 10.1016/s0378-1119(02)01056-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Brubaker SW, Gauthier AE, Mills EW, Ingolia NT, Kagan JC. A bicistronic MAVS transcript highlights a class of truncated variants in antiviral immunity. Cell. 2014;156(4):800–811. 10.1016/j.cell.2014.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shi Y, Wu J, Zhong T, et al. Upstream ORFs prevent MAVS spontaneous aggregation and regulate innate immune homeostasis. iScience. 2020;23(5):101059. 10.1016/j.isci.2020.101059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Qi N, Shi Y, Zhang R, et al. Multiple truncated isoforms of MAVS prevent its spontaneous aggregation in antiviral innate immune signalling. Nat Commun 2017;8:15676. 10.1038/ncomms15676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Perčulija V, Ouyang S. Chapter 9 - Diverse roles of DEAD/DEAH-box helicases in innate immunity and diseases. In: Helicases from All Domains of Life. Elsevier; 2019:141–171. 10.1016/B978-0-12-814685-9.00009-9 [DOI] [Google Scholar]
- 104.Zheng Q, Hou J, Zhou Y, Li Z, Cao X. The RNA helicase DDX46 inhibits innate immunity by entrapping m6A-demethylated antiviral transcripts in the nucleus. Nat Immunol 2017;18(10):1094–1103. 10.1038/ni.3830 [DOI] [PubMed] [Google Scholar]
- 105.Roundtree IA, Luo G-Z, Zhang Z, et al. YTHDC1 mediates nuclear export of N6-methyladenosine methylated mRNAs. eLife. 2017;6:e31311. 10.7554/eLife.31311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shi P, Guo Y, Su Y, et al. SUMOylation of DDX39A alters binding and export of antiviral transcripts to control innate immunity. J Immunol 2020;205(1):168–180. 10.4049/jimmunol.2000053 [DOI] [PubMed] [Google Scholar]
- 107.Ingle H, Kumar S, Raut AA, et al. The microRNA miR-485 targets host and influenza virus transcripts to regulate antiviral immunity and restrict viral replication. Sci Signal 2015;8(406):ra126. 10.1126/scisignal.aab3183 [DOI] [PubMed] [Google Scholar]
- 108.Xu C, He X, Zheng Z, et al. Downregulation of microRNA miR-526a by enterovirus inhibits RIG-I-dependent innate immune response. J Virol 2014;88(19):11356–11368. 10.1128/JVI.01400-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Taganov KD, Boldin MP, Chang K-J, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA. 2006;103(33):12481–12486. 10.1073/pnas.0605298103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci USA. 2007;104(5):1604–1609. 10.1073/pnas.0610731104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang P, Hou J, Lin L, et al. Inducible microRNA-155 feedback promotes type I IFN signaling in antiviral innate immunity by targeting suppressor of cytokine signaling 1. J Immunol 2010;185(10):6226–6233. 10.4049/jimmunol.1000491 [DOI] [PubMed] [Google Scholar]
- 112.Zhou H, Huang X, Cui H, et al. miR-155 and its star-form partner miR-155* cooperatively regulate type I interferon production by human plasmacytoid dendritic cells. Blood. 2010;116(26):5885–5894. 10.1182/blood-2010-04-280156 [DOI] [PubMed] [Google Scholar]
- 113.Hou J, Wang P, Lin L, et al. MicroRNA-146a feedback inhibits RIG-I-dependent Type I IFN production in macrophages by targeting TRAF6, IRAK1, and IRAK2. J Immunol 2009;183(3):2150–2158. 10.4049/jimmunol.0900707 [DOI] [PubMed] [Google Scholar]
- 114.Ho B-C, Yu I-S, Lu L-F, et al. Inhibition of miR-146a prevents enterovirus-induced death by restoring the production of type I interferon. Nat Commun 2014;5:3344. 10.1038/ncomms4344 [DOI] [PubMed] [Google Scholar]
- 115.Rosenberger CM, Podyminogin RL, Diercks AH, et al. miR-144 attenuates the host response to influenza virus by targeting the TRAF6-I RF7 signaling axis. PLoS Pathog 2017;13(4):e1006305. 10.1371/journal.ppat.1006305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tang Y, Luo X, Cui H, et al. MicroRNA-146A contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum 2009;60(4):1065–1075. 10.1002/art.24436 [DOI] [PubMed] [Google Scholar]
- 117.Ma H, Han P, Ye W, et al. The long noncoding RNA NEAT1 exerts antihantaviral effects by acting as positive feedback for RIG-I signaling. J Virol 2017;91(9):e02250–16. 10.1128/JVI.02250-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Imamura K, Imamachi N, Akizuki G, et al. Long noncoding RNA NEAT1-dependent SFPQ relocation from promoter region to paraspeckle mediates IL8 expression upon immune stimuli. Mol Cell. 2014;53(3):393–406. 10.1016/j.molcel.2014.01.009 [DOI] [PubMed] [Google Scholar]
- 119.Morchikh M, Cribier A, Raffel R, et al. HEXIM1 and NEAT1 long non-coding RNA form a multi-subunit complex that regulates DNA-mediated innate immune response. Mol Cell. 2017;67(3):387–399.e5. 10.1016/j.molcel.2017.06.020 [DOI] [PubMed] [Google Scholar]
- 120.Xie Q, Chen S, Tian R, et al. Long noncoding RNA ITPRIP-1 positively regulates the innate immune response through promotion of oligomerization and activation of MDA5. J Virol 2018;92(17):e00507–18. 10.1128/JVI.00507-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lin H, Jiang M, Liu L, et al. The long noncoding RNA Lnczc3h7a promotes a TRIM25-mediated RIG-I antiviral innate immune response. Nat Immunol 2019;20(7):812–823. 10.1038/s41590-019-0379-0 [DOI] [PubMed] [Google Scholar]
- 122.Wang Y, Wang P, Zhang Y, et al. Decreased expression of the host long-noncoding RNA-GM facilitates viral escape by inhibiting the kinase activity TBK1 via S-glutathionylation. Immunity. 2020;53(6):1168–1181.e7. 10.1016/j.immuni.2020.11.010 [DOI] [PubMed] [Google Scholar]
- 123.Jiang M, Zhang S, Yang Z, et al. Self-recognition of an inducible host lncRNA by RIG-I feedback restricts innate immune response. Cell. 2018;173(4):906–919.e13. 10.1016/j.cell.2018.03.064 [DOI] [PubMed] [Google Scholar]
- 124.Lazear HM, Schoggins JW, Diamond MS. Shared and distinct functions of type I and type III interferons. Immunity. 2019;50(4):907–923. 10.1016/j.immuni.2019.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Forero A, Ozarkar S, Li H, et al. Differential activation of the transcription factor IRF1 underlies the distinct immune responses elicited by type I and type III interferons. Immunity. 2019;51(3):451–464.e6. 10.1016/j.immuni.2019.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol 2014;14(1):36–49. 10.1038/nri3581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Pasté M, Huez G, Kruys V. Deadenylation of interferon-beta mRNA is mediated by both the AU-rich element in the 3′-untranslated region and an instability sequence in the coding region. Eur J Biochem 2003;270(7):1590–1597. 10.1046/j.1432-1033.2003.03530.x [DOI] [PubMed] [Google Scholar]
- 128.Stumpo DJ, Lai WS, Blackshear PJ. Inflammation: cytokines and RNA-based regulation. Wiley Interdiscip Rev RNA. 2010;1(1):60–80. 10.1002/wrna.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lagnado CA, Brown CY, Goodall GJ. AUUUA is not sufficient to promote poly(A) shortening and degradation of an mRNA: the functional sequence within AU-rich elements may be UUAUUUA(U/A)(U/A). Mol Cell Biol 1994;14(12):7984–7995. 10.1128/mcb.14.12.7984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zubiaga AM, Belasco JG, Greenberg ME. The nonamer UUAUUUAUU is the key AU-rich sequence motif that mediates mRNA degradation. Mol Cell Biol 1995;15(4):2219–2230. 10.1128/mcb.15.4.2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Barreau C, Paillard L, Osborne HB. AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res 2005;33(22):7138–7150. 10.1093/nar/gki1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gherzi R, Lee K-Y, Briata P, et al. A KH domain RNA binding protein, KSRP, promotes ARE-directed mRNA turnover by recruiting the degradation machinery. Mol Cell. 2004;14(5):571–583. 10.1016/j.molcel.2004.05.002 [DOI] [PubMed] [Google Scholar]
- 133.Chen CY, Gherzi R, Ong SE, et al. AU binding proteins recruit the exosome to degrade ARE-containing mRNAs. Cell. 2001;107(4):451–464. 10.1016/s0092-8674(01)00578-5 [DOI] [PubMed] [Google Scholar]
- 134.Salerno F, Engels S, van den Biggelaar M, et al. Translational repression of pre-formed cytokine-encoding mRNA prevents chronic activation of memory T cells. Nat Immunol 2018;19(8):828–837. 10.1038/s41590-018-0155-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Vasudevan S, Steitz JA. AU-rich-element-mediated upregulation of translation by FXR1 and Argonaute 2. Cell. 2007;128(6):1105–1118. 10.1016/j.cell.2007.01.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lin W-J, Zheng X, Lin C-C, et al. Posttranscriptional control of type I interferon genes by KSRP in the innate immune response against viral infection. Mol Cell Biol 2011;31(16):3196–3 207. 10.1128/MCB.05073-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.King PH, Chen C-Y. Role of KSRP in control of type I interferon and cytokine expression. J Interferon Cytokine Res 2014;34(4):267–274. 10.1089/jir.2013.0143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Carballo E, Lai WS, Blackshear PJ. Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science. 1998;281(5379):1001–1005. 10.1126/science.281.5379.1001 [DOI] [PubMed] [Google Scholar]
- 139.Ogilvie RL, Abelson M, Hau HH, Vlasova I, Blackshear PJ, Bohjanen PR. Tristetraprolin down-regulates IL-2 gene expression through AU-rich element-mediated mRNA decay. J Immunol 2005;174(2):953–961. 10.4049/jimmunol.174.2.953 [DOI] [PubMed] [Google Scholar]
- 140.Tiedje C, Ronkina N, Tehrani M, et al. The p38/MK2-driven exchange between tristetraprolin and HuR regulates AU-rich element-dependent translation. PLoS Genet 2012;8(9):e1002977. 10.1371/journal.pgen.1002977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mukherjee N, Jacobs NC, Hafner M, et al. Global target mRNA specification and regulation by the RNA-binding protein ZFP36. Genome Biol 2014;15(1):R12. 10.1186/gb-2014-15-1-r12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Fan XC, Steitz JA. Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. EMBO J 1998;17(12):3448–3460. 10.1093/emboj/17.12.3448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Brennan CM, Steitz JA. HuR and mRNA stability. Cell Mol Life Sci 2001;58(2):266–277. 10.1007/PL00000854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Herdy B, Karonitsch T, Vladimer GI, et al. The RNA-binding protein HuR/ELAVL1 regulates IFN-β mRNA abundance and the type I IFN response. Eur J Immunol 2015;45(5):1500–1511. 10.1002/eji.201444979 [DOI] [PubMed] [Google Scholar]
- 145.Rothamel K, Arcos S, Kim B, Reasoner C, Lisy S, Mukherjee N, & Ascano, M. ELAVL1 primarily couples MRNA stability with the 3′UTRs of interferon stimulated genes. Mol Biol 2021;35(8):109178. 10.1016/j.celrep.2021.109178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Schwerk J, Soveg FW, Ryan AP, et al. RNA-binding protein isoforms ZAP-S and ZAP-L have distinct antiviral and immune resolution functions. Nat Immunol 2019;20(12):1610–1620. 10.1038/s41590-019-0527-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Odon V, Fros JJ, Goonawardane N, et al. The role of ZAP and OAS3/RNAseL pathways in the attenuation of an RNA virus with elevated frequencies of CpG and UpA dinucleotides. Nucleic Acids Res 2019;47(15):8061–8083. 10.1093/nar/gkz581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Takata MA, Gonçalves-Carneiro D, Zang TM, et al. CG dinucleotide suppression enables antiviral defence targeting non-self RNA. Nature. 2017;550(7674):124–127. 10.1038/nature24039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Luo X, Wang X, Gao Y, et al. Molecular mechanism of RNA recognition by zinc-finger antiviral protein. Cell Rep 2020;30(1):46–52. e4. 10.1016/j.celrep.2019.11.116 [DOI] [PubMed] [Google Scholar]
- 150.Jiang P, Coller H. Functional interactions between microRNAs and RNA binding proteins. Microrna 2012;1(1):70–79. 10.2174/2211536611201010070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Velázquez-Cruz A, Baños-Jaime B, Díaz-Quintana A, De la Rosa MA, Díaz-Moreno I. Post-translational control of RNA-binding proteins and disease-related dysregulation. Front Mol Biosci 2021;8:658852. 10.3389/fmolb.2021.658852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Winkler R, Gillis E, Lasman L, et al. m6A modification controls the innate immune response to infection by targeting type I interferons. Nat Immunol 2019;20(2):173–182. 10.1038/s41590-018-0275-z [DOI] [PubMed] [Google Scholar]
- 153.Rubio RM, Depledge DP, Bianco C, Thompson L, Mohr I. RNA m6 A modification enzymes shape innate responses to DNA by regulating interferon β. Genes Dev 2018;32(23–24):1472–1484. 10.1101/gad.319475.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Gokhale NS, McIntyre ABR, Mattocks MD, et al. Altered m6A modification of specific cellular transcripts affects flaviviridae infection. Mol Cell. 2020;77(3):542–555.e8. 10.1016/j.molcel.2019.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.McFadden MJ, McIntyre ABR, Mourelatos H, et al. Post-transcriptional regulation of antiviral gene expression by N6-methyladenosine. Cell Rep 2021;34(9):108798. 10.1016/j.celrep.2021.108798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Price AM, Hayer KE, McIntyre ABR, et al. Direct RNA sequencing reveals m6A modifications on adenovirus RNA are necessary for efficient splicing. Nat Commun 2020;11(1):6016. 10.1038/s41467-020-19787-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Witwer KW, Sisk JM, Gama L, Clements JE. MicroRNA regulation of IFN-beta protein expression: rapid and sensitive modulation of the innate immune response. J Immunol 2010;184(5):2369–2376. 10.4049/jimmunol.0902712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.McFarland AP, Horner SM, Jarret A, et al. The favorable IFNL3 genotype escapes mRNA decay mediated by AU-rich elements and hepatitis C virus-induced microRNAs. Nat Immunol 2014;15(1):72–79. 10.1038/ni.2758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Jarret A, McFarland AP, Horner SM, et al. Hepatitis-C-virus-induced microRNAs dampen interferon-mediated antiviral signaling. Nat Med 2016;22(12):1475–1481. 10.1038/nm.4211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Colonna M, Krug A, Cella M. Interferon-producing cells: on the front line in immune responses against pathogens. Curr Opin Immunol 2002;14(3):373–379. 10.1016/s0952-7915(02)00349-7 [DOI] [PubMed] [Google Scholar]
- 161.Alculumbre SG, Saint-André V, Di Domizio J, et al. Diversification of human plasmacytoid predendritic cells in response to a single stimulus. Nat Immunol 2018;19(1):63–75. 10.1038/s41590-017-0012-z [DOI] [PubMed] [Google Scholar]
- 162.Fanucchi S, Fok ET, Dalla E, et al. Immune genes are primed for robust transcription by proximal long noncoding RNAs located in nuclear compartments. Nat Genet 2019;51(1):138–150. 10.1038/s41588-018-0298-2 [DOI] [PubMed] [Google Scholar]
- 163.Atianand MK, Hu W, Satpathy AT, et al. A Long Noncoding RNA lincRNA-EPS Acts as a Transcriptional Brake to Restrain Inflammation. Cell. 2016;165(7):1672–1685. 10.1016/j.cell.2016.05.075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Suarez B, Prats-Mari L, Unfried JP, Fortes P. LncRNAs in the type I interferon antiviral response. Int J Mol Sci 2020;21(17):6447. 10.3390/ijms21176447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Li X, Guo G, Lu M, et al. Long noncoding RNA Lnc-MxA inhibits beta interferon transcription by forming RNA-DNA triplexes at its promoter. J Virol 2019;93(21). 10.1128/JVI.00786-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Agarwal S, Vierbuchen T, Ghosh S, et al. The long non-coding RNA LUCAT1 is a negative feedback regulator of interferon responses in humans. Nat Commun 2020;11(1):6348. 10.1038/s41467-020-20165-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ouyang J, Zhu X, Chen Y, et al. NRAV, a long noncoding RNA, modulates antiviral responses through suppression of interferon-stimulated gene transcription. Cell Host Microbe 2014;16(5):616–626. 10.1016/j.chom.2014.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kambara H, Niazi F, Kostadinova L, et al. Negative regulation of the interferon response by an interferon-induced long non-coding RNA. Nucleic Acids Res 2014;42(16):10668–10680. 10.1093/nar/gku713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zhao L, Xia M, Wang K, et al. A long non-coding RNA IVRPIE promotes host antiviral immune responses through regulating interferon β1 and ISG expression. Front Microbiol 2020;11:260. 10.3389/fmicb.2020.00260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Nishitsuji H, Ujino S, Yoshio S, et al. Long noncoding RNA #32 contributes to antiviral responses by controlling interferon-stimulated gene expression. Proc Natl Acad Sci USA. 2016;113(37):10388–10393. 10.1073/pnas.1525022113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Chai W, Li J, Shangguan Q, et al. Lnc-ISG20 inhibits influenza A virus replication by enhancing ISG20 expression. J Virol 2018;92(16):e00539–18. 10.1128/JVI.00539-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Bourdon M, Manet C, Montagutelli X. Host genetic susceptibility to viral infections: the role of type I interferon induction. Genes Immun 2020;21(6–8):365–379. 10.1038/s41435-020-00116-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Forbester JL, Humphreys IR. Genetic influences on viral-induced cytokine responses in the lung. Mucosal Immunol 2021;14(1):14–25. 10.1038/s41385-020-00355-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Hindorff LA, Sethupathy P, Junkins HA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA. 2009;106(23):9362–9367. 10.1073/pnas.0903103106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Tam V, Patel N, Turcotte M, Bossé Y, Paré G, Meyre D. Benefits and limitations of genome-wide association studies. Nat Rev Genet 2019;20(8):467–484. 10.1038/s41576-019-0127-1 [DOI] [PubMed] [Google Scholar]
- 176.Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity. 2006;25(3):383–392. 10.1016/j.immuni.2006.08.010 [DOI] [PubMed] [Google Scholar]
- 177.Negishi H, Taniguchi T, Yanai H. The Interferon (IFN) class of cytokines and the IFN Regulatory Factor (IRF) transcription factor family. Cold Spring Harb Perspect Biol 2018;10(11):a028423. 10.1101/cshperspect.a028423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Yanai H, Negishi H, Taniguchi T. The IRF family of transcription factors: Inception, impact and implications in oncogenesis. Oncoimmunology. 2012;1(8):1376–1386. 10.4161/onci.22475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Schoenemeyer A, Barnes BJ, Mancl ME, et al. The interferon regulatory factor, IRF5, is a central mediator of toll-like receptor 7 signaling. J Biol Chem 2005;280(17):17005–17012. 10.1074/jbc.M412584200 [DOI] [PubMed] [Google Scholar]
- 180.Forbester JL, Clement M, Wellington D, et al. IRF5 promotes influenza virus-induced inflammatory responses in human induced pluripotent stem cell-derived myeloid cells and murine models. J Virol 2020;94(9). e00121–20. 10.1128/JVI.00121-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lazear HM, Lancaster A, Wilkins C, et al. IRF-3, IRF-5, and IRF-7 coordinately regulate the type I IFN response in myeloid dendritic cells downstream of MAVS signaling. PLoS Pathog. 2013;9(1):e1003118. 10.1371/journal.ppat.1003118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Lien C, Fang C-M, Huso D, Livak F, Lu R, Pitha PM. Critical role of IRF-5 in regulation of B-cell differentiation. Proc Natl Acad Sci USA. 2010;107(10):4664–4668. 10.1073/pnas.0911193107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Graham RR, Kozyrev SV, Baechler EC, et al. A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat Genet 2006;38(5):550–555. 10.1038/ng1782 [DOI] [PubMed] [Google Scholar]
- 184.Clark DN, Read RD, Mayhew V, et al. Four promoters of IRF5 respond distinctly to stimuli and are affected by autoimmune-risk polymorphisms. Front Immunol 2013;4:360. 10.3389/fimmu.2013.00360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Graham RR, Kyogoku C, Sigurdsson S, et al. Three functional variants of IFN regulatory factor 5 (IRF5) define risk and protective haplotypes for human lupus. Proc Natl Acad Sci USA. 2007;104(16):6758–6763. 10.1073/pnas.0701266104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Mayr C, Bartel DP. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138(4):673–684. 10.1016/j.cell.2009.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science. 2008;320(5883):1643–1647. 10.1126/science.1155390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Pai AA, Baharian G, Pagé Sabourin A, et al. Widespread shortening of 3′ untranslated regions and increased exon inclusion are evolutionarily conserved features of innate immune responses to infection. PLoS Genet 2016;12(9):e1006338. 10.1371/journal.pgen.1006338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Mariella E, Marotta F, Grassi E, Gilotto S, Provero P. The length of the expressed 3′ UTR is an intermediate molecular phenotype linking genetic variants to complex diseases. Front Genet 2019;10:714. 10.3389/fgene.2019.00714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Steri M, Idda ML, Whalen MB, Orrù V. Genetic variants in mRNA untranslated regions. Wiley Interdiscip Rev RNA. 2018;9(4):e1474. 10.1002/wrna.1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Zhang F, Lupski JR. Non-coding genetic variants in human disease. Hum Mol Genet 2015;24(R1):R102–R110. 10.1093/hmg/ddv259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Syedbasha M, Egli A. Interferon lambda: modulating immunity in infectious diseases. Front Immunol 2017;8:119. 10.3389/fimmu.2017.00119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Hemann EA, Gale M, Savan R. Interferon lambda genetics and biology in regulation of viral control. Front Immunol 2017;8:1707. 10.3389/fimmu.2017.01707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461(7262):399–401. 10.1038/nature08309 [DOI] [PubMed] [Google Scholar]
- 195.Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet 2009;41(10):1100–1104. 10.1038/ng.447 [DOI] [PubMed] [Google Scholar]
- 196.Tanaka Y, Nishida N, Sugiyama M, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet 2009;41(10):1105–1109. 10.1038/ng.449 [DOI] [PubMed] [Google Scholar]
- 197.Thomas DL, Thio CL, Martin MP, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature. 2009;461(7265):798–801. 10.1038/nature08463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Sheahan T, Imanaka N, Marukian S, et al. Interferon lambda alleles predict innate antiviral immune responses and hepatitis C virus permissiveness. Cell Host Microbe 2014;15(2):190–202. 10.1016/j.chom.2014.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Lu Y-F, Mauger DM, Goldstein DB, Urban TJ, Weeks KM, Bradrick SS. IFNL3 mRNA structure is remodeled by a functional non-coding polymorphism associated with hepatitis C virus clearance. Sci Rep 2015;5:16037. 10.1038/srep16037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Hong M, Schwerk J, Lim C, et al. Interferon lambda 4 expression is suppressed by the host during viral infection. J Exp Med 2016;213(12):2539–2552. 10.1084/jem.20160437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Prokunina-Olsson L, Muchmore B, Tang W, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet 2013;45(2):164–171. 10.1038/ng.2521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Zhou C, Yu Q, Chen L, Wang J, Zheng S, Zhang J. A miR-1231 binding site polymorphism in the 3′UTR of IFNAR1 is associated with hepatocellular carcinoma susceptibility. Gene. 2012;507(1):95–98. 10.1016/j.gene.2012.06.073 [DOI] [PubMed] [Google Scholar]
- 203.Yu Q, Qian W, Wang J, Wu Y, Zhang J, Chen W. An indel polymorphism in the 3′ untranslated region of JAK1 confers risk for hepatocellular carcinoma possibly by regulating JAK1 transcriptional activity in a Chinese population. Oncol Lett 2018;15(5):8088–8094. 10.3892/ol.2018.8347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Soveg FW, Schwerk J, Gokhale NS, et al. Endomembrane targeting of human OAS1 P46 augments antiviral activity. eLife. 2021;10:e71047. 10.7554/eLife.71047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Li H, Reksten TR, Ice JA, et al. Identification of a Sjögren’s syndrome susceptibility locus at OAS1 that influences isoform switching, protein expression, and responsiveness to type I interferons. PLoS Genet 2017;13(6):e1006820. 10.1371/journal.pgen.1006820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Frankiw L, Mann M, Li G, Joglekar A, Baltimore D. Alternative splicing coupled with transcript degradation modulates OAS1g antiviral activity. RNA. 2020;26(2):126–136. 10.1261/rna.073825.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.El Awady MK, Anany MA, Esmat G, et al. Single nucleotide polymorphism at exon 7 splice acceptor site of OAS1 gene determines response of hepatitis C virus patients to interferon therapy. J Gastroenterol Hepatol 2011;26(5):843–850. 10.1111/j.1440-1746.2010.06605.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Lim JK, Lisco A, McDermott DH, et al. Genetic variation in OAS1 is a risk factor for initial infection with West Nile virus in man. PLoS Pathog 2009;5(2):e1000321. 10.1371/journal.ppat.1000321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Pairo-Castineira E, Clohisey S, Klaric L, et al. Genetic mechanisms of critical illness in COVID-19. Nature. 2021;591(7848):92–98. 10.1038/s41586-020-03065-y [DOI] [PubMed] [Google Scholar]
- 210.Zhou S, Butler-Laporte G, Nakanishi T, et al. A Neanderthal OAS1 isoform protects individuals of European ancestry against COVID-19 susceptibility and severity. Nat Med 2021;27(4):659–667. 10.1038/s41591-021-01281-1 [DOI] [PubMed] [Google Scholar]
- 211.Deng H, Liu R, Ellmeier W, et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996;381(6584):661–666. 10.1038/381661a0 [DOI] [PubMed] [Google Scholar]
- 212.Kulkarni S, Lied A, Kulkarni V, et al. CCR5AS lncRNA variation differentially regulates CCR5, influencing HIV disease outcome. Nat Immunol 2019;20(7):824–834. 10.1038/s41590-019-0406-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Glass WG, McDermott DH, Lim JK, et al. CCR5 deficiency increases risk of symptomatic West Nile virus infection. J Exp Med 2006;203(1):35–40. 10.1084/jem.20051970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Falcon A, Cuevas MT, Rodriguez-Frandsen A, et al. CCR5 deficiency predisposes to fatal outcome in influenza virus infection. J Gen Virol 2015;96(8):2074–2078. 10.1099/vir.0.000165 [DOI] [PubMed] [Google Scholar]
- 215.Xiong HY, Alipanahi B, Lee LJ, et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science. 2015;347(6218):1254806. 10.1126/science.1254806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Kumar V, Westra H-J, Karjalainen J, et al. Human disease-associated genetic variation impacts large intergenic non-coding RNA expression. PLoS Genet 2013;9(1):e1003201. 10.1371/journal.pgen.1003201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Banerjee AK, Blanco MR, Bruce EA, et al. SARS-CoV-2 disrupts splicing, translation, and protein trafficking to suppress host defenses. Cell. 2020;183(5):1325–1339.e21. 10.1016/j.cell.2020.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.De Maio FA, Risso G, Iglesias NG, et al. The dengue virus NS5 protein intrudes in the cellular spliceosome and modulates splicing. Pierson TC, ed. PLoS Pathog 2016;12(8):e1005841. 10.1371/journal.ppat.1005841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Mor A, White A, Zhang K, et al. Influenza virus mRNA trafficking through host nuclear speckles. Nat Microbiol 2016;1(7):16069. 10.1038/nmicrobiol.2016.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Thompson MG, Dittmar M, Mallory MJ, et al. Viral-induced alternative splicing of host genes promotes influenza replication. eLife. 2020;9:e55500. 10.7554/eLife.55500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Thompson MG, Muñoz-Moreno R, Bhat P, et al. Co-regulatory activity of hnRNP K and NS1-BP in influenza and human mRNA splicing. Nat Commun 2018;9(1):2407. 10.1038/s41467-018-04779-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Walsh D, Mohr I. Viral subversion of the host protein synthesis machinery. Nat Rev Microbiol 2011;9(12):860–875. 10.1038/nrmicro2655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Martinez-Salas E, Francisco-Velilla R, Fernandez-Chamorro J, Embarek AM. Insights into structural and mechanistic features of viral IRES elements. Front Microbiol 2018;8:2629. 10.3389/fmicb.2017.02629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Lamphear BJ, Kirchweger R, Skern T, Rhoads RE. Mapping of functional domains in eukaryotic protein synthesis initiation factor 4G (eIF4G) with picornaviral proteases. Implications for cap-dependent and cap-independent translational initiation. J Biol Chem 1995;270(37):21975–21983. 10.1074/jbc.270.37.21975 [DOI] [PubMed] [Google Scholar]
- 225.Garaigorta U, Chisari FV. Hepatitis C virus blocks interferon effector function by inducing protein kinase R phosphorylation. Cell Host Microbe 2009;6(6):513–522. 10.1016/j.chom.2009.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Hyde JL, Gardner CL, Kimura T, et al. A viral RNA structural element alters host recognition of nonself RNA. Science. 2014;343(6172):783–787. 10.1126/science.1248465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Thoms M, Buschauer R, Ameismeier M, et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science. 2020;369(6508):1249–1255. 10.1126/science.abc8665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Schubert K, Karousis ED, Jomaa A, et al. SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat Struct Mol Biol 2020;27(10):959–966. 10.1038/s41594-020-0511-8 [DOI] [PubMed] [Google Scholar]
- 229.Finkel Y, Gluck A, Nachshon A, et al. SARS-CoV-2 uses a multipronged strategy to impede host protein synthesis. Nature. 2021;594(7862):240–245. 10.1038/s41586-021-03610-3 [DOI] [PubMed] [Google Scholar]
- 230.Kamitani W, Narayanan K, Huang C, et al. Severe acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation. Proc Natl Acad Sci USA. 2006;103(34):12885–12890. 10.1073/pnas.0603144103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Slonchak A, Khromykh AA. Subgenomic flaviviral RNAs: what do we know after the first decade of research. Antiviral Res 2018;159:13–25. 10.1016/j.antiviral.2018.09.006 [DOI] [PubMed] [Google Scholar]
- 232.Pijlman GP, Funk A, Kondratieva N, et al. A highly structured, nuclease-resistant, noncoding RNA produced by flaviviruses is required for pathogenicity. Cell Host Microbe 2008;4(6):579–591. 10.1016/j.chom.2008.10.007 [DOI] [PubMed] [Google Scholar]
- 233.Chapman EG, Costantino DA, Rabe JL, et al. The structural basis of pathogenic subgenomic flavivirus RNA (sfRNA) production. Science. 2014;344(6181):307–310. 10.1126/science.1250897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Funk A, Truong K, Nagasaki T, et al. RNA structures required for production of subgenomic flavivirus RNA. J Virol 2010;84(21):11407–11417. 10.1128/JVI.01159-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Akiyama BM, Laurence HM, Massey AR, et al. Zika virus produces noncoding RNAs using a multi-pseudoknot structure that confounds a cellular exonuclease. Science. 2016;354(6316):1148–1152. 10.1126/science.aah3963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Göertz GP, Fros JJ, Miesen P, et al. Noncoding subgenomic flavivirus RNA is processed by the mosquito RNA interference machinery and determines west nile virus transmission by Culex pipiens mosquitoes. J Virol 2016;90(22):10145–10159. 10.1128/JVI.00930-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Göertz GP, van Bree JWM, Hiralal A, et al. Subgenomic flavivirus RNA binds the mosquito DEAD/H-box helicase ME31B and determines Zika virus transmission by Aedes aegypti. Proc Natl Acad Sci USA. 2019;116(38):19136–19144. 10.1073/pnas.1905617116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Filomatori CV, Carballeda JM, Villordo SM, et al. Dengue virus genomic variation associated with mosquito adaptation defines the pattern of viral non-coding RNAs and fitness in human cells. PLoS Pathog 2017;13(3):e1006265. 10.1371/journal.ppat.1006265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Pallarés HM, Costa Navarro GS, Villordo SM, et al. Zika virus subgenomic flavivirus RNA generation requires cooperativity between duplicated RNA structures that are essential for productive infection in human cells. J Virol 2020;94(18):e00343–20. 10.1128/JVI.00343-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Moon SL, Anderson JR, Kumagai Y, et al. A noncoding RNA produced by arthropod-borne flaviviruses inhibits the cellular exoribonuclease XRN1 and alters host mRNA stability. RNA. 2012;18(11):2029–2040. 10.1261/rna.034330.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Manokaran G, Finol E, Wang C, et al. Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science. 2015;350(6257):217–221. 10.1126/science.aab3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science. 2005;309(5740):1577–1581. 10.1126/science.1113329 [DOI] [PubMed] [Google Scholar]
- 243.Machlin ES, Sarnow P, Sagan SM. Masking the 5′ terminal nucleotides of the hepatitis C virus genome by an unconventional microRNA-target RNA complex. Proc Natl Acad Sci USA. 2011;108(8):3193–3198. 10.1073/pnas.1012464108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Shimakami T, Yamane D, Jangra RK, et al. Stabilization of hepatitis C virus RNA by an Ago2-miR-122 complex. Proc Natl Acad Sci USA. 2012;109(3):941–946. 10.1073/pnas.1112263109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Sedano CD, Sarnow P. Hepatitis C virus subverts liver-specific miR-122 to protect the viral genome from exoribonuclease Xrn2. Cell Host Microbe. 2014;16(2):257–264. 10.1016/j.chom.2014.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Jangra RK, Yi M, Lemon SM. Regulation of hepatitis C virus translation and infectious virus production by the microRNA miR-122. J Virol 2010;84(13):6615–6625. 10.1128/JVI.00417-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Henke JI, Goergen D, Zheng J, et al. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J 2008;27(24):3300–3310. 10.1038/emboj.2008.244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Schult P, Roth H, Adams RL, et al. microRNA-122 amplifies hepatitis C virus translation by shaping the structure of the internal ribosomal entry site. Nat Commun 2018;9(1):2613. 10.1038/s41467-018-05053-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Chahal J, Gebert LFR, Gan HH, et al. miR-122 and Ago interactions with the HCV genome alter the structure of the viral 5′ terminus. Nucleic Acids Res 2019;47(10):5307–5324. 10.1093/nar/gkz194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Masaki T, Arend KC, Li Y, et al. miR-122 stimulates hepatitis C virus RNA synthesis by altering the balance of viral RNAs engaged in replication versus translation. Cell Host Microbe 2015;17(2):217–228. 10.1016/j.chom.2014.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Luna JM, Scheel TKH, Danino T, et al. Hepatitis C virus RNA functionally sequesters miR-122. Cell. 2015;160(6):1099–1110. 10.1016/j.cell.2015.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Fukuhara T, Kambara H, Shiokawa M, et al. Expression of microRNA miR-122 facilitates an efficient replication in nonhepatic cells upon infection with hepatitis C virus. J Virol 2012;86(15):7918–7933. 10.1128/JVI.00567-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Janssen HLA, Reesink HW, Lawitz EJ, et al. Treatment of HCV infection by targeting microRNA. N Engl J Med 2013;368(18):1685–1694. 10.1056/NEJMoa1209026 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new data were generated for this manuscript.