

SUMMARY
The anti-apoptotic myeloid cell leukemia 1 (MCL1) protein belongs to the pro-survival BCL2 family and is frequently amplified or elevated in human cancers. MCL1 is highly unstable, with its stability being regulated by phosphorylation and ubiquitination. Here, we identify acetylation as another critical post-translational modification regulating MCL1 protein stability. We demonstrate that the lysine acetyltransferase p300 targets MCL1 at K40 for acetylation, which is counteracted by the deacetylase sirtuin 3 (SIRT3). Mechanistically, acetylation enhances MCL1 interaction with USP9X, resulting in deubiquitination and subsequent MCL1 stabilization. Therefore, ectopic expression of acetylation-mimetic MCL1 promotes apoptosis evasion of cancer cells, enhances colony formation potential, and facilitates xenografted tumor progression. We further demonstrate that elevated MCL1 acetylation sensitizes multiple cancer cells to pharmacological inhibition of USP9X. These findings reveal that acetylation of MCL1 is a critical post-translational modification enhancing its oncogenic function and provide a rationale for developing innovative therapeutic strategies for MCL1-dependent tumors.
Graphical Abstract
In brief
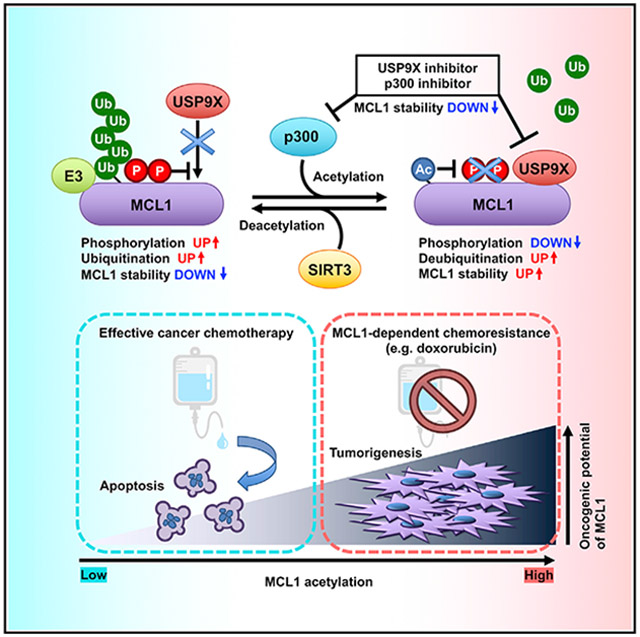
MCL1, an anti-apoptotic BCL2 family protein, is frequently overexpressed in a variety of cancers, and its oncogenic function is finely regulated by post-translational modifications such as phosphorylation and ubiquitination. Shimizu et al. dissect the molecular mechanism of acetylation-mediated MCL1 stability control, providing insights into potential therapeutic intervention targeting the MCL1 protein.
INTRODUCTION
Myeloid cell leukemia 1 (MCL1), an anti-apoptotic BCL2 family member, is an essential pro-survival factor that plays a key role in suppressing apoptosis largely through constraining the activity of the pro-apoptotic BCL2 family of proteins or pro-apoptotic BH-3-only proteins (Brunelle and Letai, 2009; Cory et al., 2016; Merino et al., 2018; Singh et al., 2019). Overexpression or amplification of MCL1 is observed frequently in multiple cancer types (Beroukhim et al., 2010; Campbell et al., 2018; Glaser et al., 2012; Sieghart et al., 2006; Wuillème-Toumi et al., 2005; Xiang et al., 2010) and therefore is regarded as one of the most relevant oncoproteins. Because of its critical pro-survival activity, MCL1 upregulation largely contributes to developing chemoresistance (Konopleva et al., 2006). Thus, understanding the precise molecular mechanisms of regulation of MCL1 oncogenic activity is of great importance to find efficient anti-tumorigenic therapeutic agents and increasing chemosensitivity.
MCL1 protein abundance is tightly controlled by transcriptional, post-transcriptional, translational, and post-translational mechanisms. Increasing evidence has revealed that multiple growth factors and cytokines induce MCL1 gene expression (Thomas et al., 2010), multiple microRNAs (miRNAs) and long non-coding RNAs (lncRNAs) control the stability of MCL1 mRNA (Cui and Placzek, 2018; Senichkin et al., 2020), and mTORC1 promotes MCL1 protein translation (Mills et al., 2008). As a unique property, the MCL1 protein is remarkably unstable compared with other anti-apoptotic BCL2 family members. Thus, rapid MCL1 degradation likely provides a mechanism for cells to promptly commit to apoptosis in response to various intrinsic or extracellular cues (Cuconati et al., 2003; Nijhawan et al., 2003). Mechanistically, MCL1 has a unique extended N-terminal region containing a proline/glutamate/serine/threonine (PEST)-rich sequence, a common motif among unstable proteins with short protein half-lives (Kozopas et al., 1993). Previous studies have indicated that the MCL1 PEST sequence contains multiple phosphorylation sites that affect MCL1 ubiquitination (Mojsa et al., 2014; Senichkin et al., 2020). In support of these observations, MCL1 protein stability is tightly controlled by ubiquitination through the activity of multiple upstream E3 ubiquitin ligases (e.g., c-Mule, SCFFBW7, SCFβ-TRCP, APC/CCdc20, TRIM17, and Parkin) (Carroll et al., 2014; Ding et al., 2007; Harley et al., 2010; Inuzuka et al., 2011; Magiera et al., 2013; Wertz et al., 2011; Zhong et al., 2005) and deubiquitinases (DUBs) (e.g., USP9X, USP13, DUB3, and JOSD1) (Schwickart et al., 2010; Wu et al., 2019, 2020; Zhang et al., 2018). Of these reported E3s and DUBs, SCFFBW7, SCFβ-TRCP, and USP9X require phosphorylation at Ser159 and Thr163 of MCL1 for regulation of their interactions (Ding et al., 2007; Inuzuka et al., 2011; Schwickart et al., 2010; Wertz et al., 2011). These findings imply that dysregulated MCL1 post-translational modifications may cause aberrant MCL1 stabilization, resulting in elevated cell survival and chemoresistance in multiple cancers (Ertel et al., 2013).
Acetylation is an essential type of lysine modification that controls a variety of biological processes, including protein-protein interaction, transcription, subcellular localization, and enzymatic activity of numerous proteins. Additionally, a number of non-histone proteins targeted by acetylation are oncoproteins or tumor suppressors and are directly involved in tumorigenesis, tumor progression, and metastasis (Barneda-Zahonero and Parra, 2012; Buchwald et al., 2009; Spange et al., 2009). Previous studies indicate that acetylation contributes to protein stabilization and degradation in part by interplay with ubiquitination, another type of lysine modification (Caron et al., 2005). For instance, acetylation is found to compete with ubiquitination at the same acceptor lysine residue, leading to stabilization of individual target proteins (Grönroos et al., 2002; Li et al., 2002,2012; Liu et al., 2014; Wang et al., 2017). In addition, acetylation may promote complex formation or dissociation with E3s/DUBs, another potential mechanism of acetylation-mediated protein degradation or stabilization (Caron et al., 2005). Thus, we reasoned that acetylation might be involved in regulating MCL1 ubiquitination and protein stability.
In this study, we demonstrated that MCL1 is acetylated by p300, leading to a decrease in MCL1 ubiquitination and subsequent MCL1 protein stabilization, which is counteracted by sirtuin 3 (SIRT3). Enhanced interaction of USP9X with acetylated MCL1 is likely the underlying molecular mechanism of acetylation-dependent MCL1 stabilization. We further identified that MCL1 acetylation influences apoptotic evasion of cancer cells and facilitates tumor progression in xenograft mouse models. These findings suggest a crucial role of MCL1 acetylation in cell survival and acquired chemoresistance in cancer, which provides insights into targeting acetylated MCL1 as a potential therapeutic intervention.
RESULTS
MCL1 interacts with p300 and is acetylated primarily at the K40 residue
The N-terminal domain of MCL1 is highly modified by post-translational modifications such as ubiquitination and phosphorylation to control its protein stability (Mojsa et al., 2014; Senichkin et al., 2020). Therefore, we hypothesized that acetylation of the N-terminal domain of MCL1 might serve as another layer of control to rapidly modulate MCL1 ubiquitination and, consequently, increase MCL1 protein stability. To assess whether MCL1 is acetylated in cells, we first immunoprecipitated endogenous MCL1 protein and detected its acetylation using the anti-acetylated lysine antibody (Figure 1A). Next, to identify an upstream enzyme responsible for MCL1 acetylation, we ectopically co-expressed MCL1 with various lysine acetyltransferases. Among the five acetyltransferases we tested, p300 and, to a lesser extent, p300/CBP-associated factor (PCAF) and CREB-binding protein (CBP), induced acetylation of MCL1 (Figure 1B). Furthermore, we detected an interaction between MCL1 and p300 at the endogenous level in HeLa cells using co-immunoprecipitation (coIP) (Figures 1C and 1D). In keeping with p300 as the primary enzyme capable of promoting MCL1 acetylation in this experimental setting, we also found that the binding specificity of p300 with MCL1 was relatively higher than that of GCN5, PCAF, and CBP (Figure S1A). Moreover, knockdown of p300 or treatment with the selective p300/CBP inhibitors A-485 and C646 (Bowers et al., 2010; Lasko et al., 2017) resulted in a decrease in MCL1 acetylation (Figures 1E, 1F, and S1B).
Figure 1. p300 binds and acetylates MCL1 at K40.
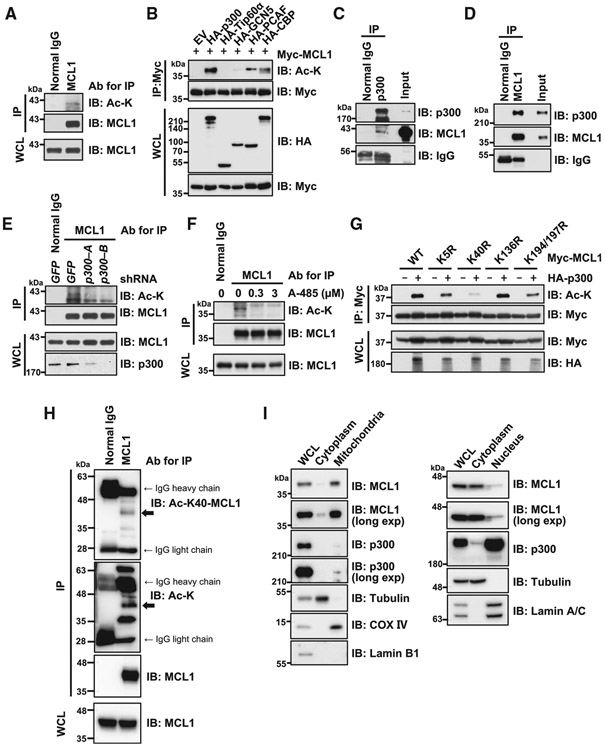
(A) Endogenous MCL1 is targeted by acetylation in HeLa cells. Shown is immunoblot (IB) analysis of whole-cell lysates (WCLs) and anti-MCL1 immunoprecipitates derived from HeLa cells.
(B) p300 promotes MCL1 acetylation in cells. Shown is IB analysis of WCLs and anti-Myc tag immunoprecipitates derived from 293T cells transfected with Myc-MCL1 along with the indicated hemagglutinin (HA)-tagged lysine acetyltransferase constructs.
(C and D) Endogenous interaction between MCL1 and p300. Shown is IB analysis of WCLs (input) and anti-p300 immunoprecipitates (C) or anti-MCL1 immunoprecipitates (D) derived from HeLa cells.
(E) Knockdown of p300 reduces MCL1 acetylation. Shown is IB analysis of WCLs and anti-MCL1 immunoprecipitates derived from HeLa cells stably expressing the lentiviral short hairpin RNA (shRNA) specific for GFP or p300. Cells were treated with MG132 (10 μM) for 6 h before harvesting to stabilize and accumulate MCL1 protein.
(F) Treatment with the p300/CBP inhibitor A-485 reduces MCL1 acetylation. Shown is IB analysis of WCLs and anti-MCL1 immunoprecipitates derived from HeLa cells. Cells were co-treated overnight with A-485 at the indicated concentration and MG132 (5 μM) before harvesting.
(G) Acetylation-deficient K40R markedly diminishes p300-mediated MCL1 acetylation. Shown is IB analysis of WCLs and anti-Myc tag immunoprecipitates derived from 293T cells transfected with the indicated Myc-MCL1 constructs.
(H) K40 of endogenous MCL1 is targeted by acetylation in SKBR3 breast cancer cells. Shown is IB analysis of WCLs and anti-MCL1 immunoprecipitates derived from SKBR3 cells.
(I) MCL1 and p300 are present together in cytoplasmic and nuclear compartments. Shown is IB analysis of WCLs and cytoplasmic, mitochondrial, and nuclear fractions derived from BT-20 cells. Data are representative of at least two independent experiments. See also Figure S1.
K5, K40, K136, K194, and K197 are previously reported MCL1 ubiquitination sites (Zhong et al., 2005; Figure S1C), indicating that, for MCL1, the overlap of ubiquitination and acetylation sites may also serve as a regulatory mechanism for lysine modification (Caron et al., 2005; Liu et al., 2014). In keeping with this notion, we performed mutagenesis of the reported ubiquitination sites and determined that substitution of K to R at the K40 residue largely abrogated p300-mediated acetylation of MCL1 in cells (Figure 1G). Furthermore, we performed mass spectrometry following p300 ectopic expression and identified K40 as one of the targeted lysine residues for acetylation (Figure S1D). To confirm the K40 acetylation event, we developed an anti-acetyl-K40-MCL1 antibody (Figures S1E and S1F). Using this generated antibody, we detected K40 acetylation of endogenous MCL1 in SKBR3 breast cancer cells (Figures 1H and S1G). Furthermore, immunofluorescence analysis demonstrated that K40-acetylated MCL1 protein appears to colocalize with p300 in the nucleus and cytoplasm in BT-20 (Figures S1H and S1I). Moreover, in support of the immunofluorescence results, cellular fractionation experiments confirmed that MCL1 and p300 are present in multiple cellular components, including the cytoplasm, mitochondria, and nucleus, in BT-20 and HeLa cells (Figures 1I and S1J). These data indicate that MCL1 is likely subject to acetylation primarily at K40 through the lysine acetyltransferase p300.
p300-mediated acetylation leads to MCL1 stabilization
Several studies have demonstrated that acetylation of oncogenic proteins often increases their protein stability, resulting in elevated oncogenic activities (Buchwald et al., 2009; Inuzuka et al., 2012; Nihira et al., 2017). Therefore, we sought to assess the critical role of p300-mediated acetylation of MCL1 in controlling MCL1 ubiquitination and degradation. We found that p300 depletion resulted in a decrease in MCL1 protein abundance (Figure 2A). Consistent with this finding, p300 knockdown and p300/CBP inhibition with A-485 and C646 shortened MCL1 protein half-life (Figures 2B-2E, S2A, and S2B). Importantly, p300 depletion increased MCL1 poly-ubiquitination in cells (Figure 2F), suggesting that p300 enzymatic activity positively regulates MCL1 protein stability in part by escaping from ubiquitination-mediated proteasomal degradation. Next, to determine the critical role of MCL1 acetylation at K40 on MCL1 protein turnover, we ectopically expressed wild-type (WT) MCL1, an acetylation-mimetic K40Q, or an acetylation-deficient K40R mutant in HeLa cells and performed cellular ubiquitination assays and cycloheximide (CHX) chase experiments. Cellular ubiquitination assays demonstrated a marked impairment in the ubiquitination status of the acetylation-mimetic K40Q mutant in comparison with the WT and the K40R form of MCL1 (Figure 2G). In addition, ectopically expressed p300 reduced ubiquitination of WT MCL1 but not the acetylation-deficient K40R mutant (Figure 2H). In contrast, the p300/CBP inhibitor A-485 enhanced MCL1 ubiquitination in the WT but not K40R mutant (Figure 2I). In support of these observations, CHX chase experiments revealed that the protein half-life of MCL1 was prolonged when an acetylation-mimetic K40Q substitution was introduced (Figures 2J and 2K). To exclude the possibility that the half-life extension was due to an artifactual effect of introducing the K-to-Q substitution, we tested whether p300-mediated MCL1 acetylation modulates MCL1 protein turnover. The half-life of MCL1 WT was prolonged following p300 ectopic co-expression, whereas the acetylation-deficient K40R mutant showed no significant change in its protein turnover when co-expressed with p300 (Figures 2L and 2M). These results indicated that p300-mediated acetylation of MCL1 at K40 likely results in reduced poly-ubiquitination events and increased stabilization of the MCL1 protein.
Figure 2. p300-mediated MCL1 acetylation leads to MCL1 stabilization through decreasing ubiquitination.

(A) p300 knockdown decreases MCL1 protein abundance. Shown is IB analysis of WCLs derived from HeLa cells infected with the indicated lentiviral shRNA specific for GFP or p300.
(B) p300 knockdown shortens MCL1 protein half-life. Shown is IB analysis of WCLs derived from HeLa cells stably expressing the indicated lentiviral shRNA. The cells were treated with the protein synthesis inhibitor cycloheximide (CHX; 100 μg/mL) for the indicated periods before harvesting.
(C) Quantification of the MCL1 band intensities of IB replicates in (B). Data are presented as mean ± SD; n = 3 independent experiments, *p < 0.05.
(D) Treatment with the p300/CBP inhibitor A-485 shortens MCL1 protein half-life. Shown is IB analysis of WCLs derived from HeLa cells. Cells were pretreated with A-485 (3 μM) overnight and then treated with 100 μg/mL CHX for the indicated periods before harvesting.
(E) Quantification of the MCL1 band intensities of IB replicates in (D). Data are presented as mean ± SD; n = 3 independent experiments, **p < 0.01.
(F) HeLa cells stably expressing the indicated lentiviral shRNA specific for GFP or p300 were transfected with His-tagged ubiquitin (His-Ub) and Myc-MCL1. 36 h after transfection, the cells were treated overnight with MG132 (10 μM) before harvesting. His-Ub-conjugated proteins were captured with Ni(2+)-nitrilotriacetic acid (Ni-NTA) agarose beads and subjected to IB analysis.
(G) Acetylation-mimetic K40Q substitution results in decreased poly-ubiquitination of MCL1 in cells. 293T cells were transfected with the indicated Myc-MCL1 and His-Ub constructs. 36 h after transfection, cells were treated overnight with MG132 (10 μM) before harvesting.
(H) Ectopic p300 expression decreases poly-ubiquitination of WT MCL1 but not K40R. 293T cells were transfected with the indicated constructs. 36 h after transfection, cells were treated with MG132 (20 μM) for 5 h and harvested for the Ni-NTA pull-down.
(I) Treatment with the p300 inhibitor A-485 induces poly-ubiquitination of the WT but not the K40R mutant form of MCL1. 293T cells were transfected with the indicated constructs. 24 h after transfection, cells were treated with or without A-485 (3 μM) for 24 h and MG132 (10 μM) for 10 h before harvesting.
(J) Acetylation-mimetic K40Q extends MCL1 protein half-life. Shown is IB analysis of WCLs derived from HeLa cells transfected with the indicated Myc-MCL1 constructs. 48 h after transfection, cells were treated with 100 μg/mL CHX for the indicated periods before harvesting.
(K) Quantification of the Myc band intensities of IB replicates in (J). Data are presented as mean ± SD; n = 3 independent experiments, **p < 0.01.
(L) Ectopic p300 expression extends the protein half-life of the WT but not the K40R form of MCL1. Shown is IB analysis of WCLs derived from 293T cells transfected with Myc-MCL1 and HA-p300 constructs as indicated. 48 h after transfection, cells were treated with 100 μg/mL CHX for the indicated periods before harvesting.
(M) Quantification of the Myc band intensities of IB replicates in (L). Data are presented as mean ± SD; n = 3 independent experiments, **p < 0.01; NS, not significant.
Data in (A) and (F)–(I) are representative of at least two independent experiments. See also Figure S2.
Acetylation of MCL1 inhibits doxorubicin-induced apoptosis and promotes tumorigenicity
Given that MCL1 is eliminated rapidly from damaged cells in response to DNA damage (Cuconati et al., 2003), we evaluated the biological consequences of MCL1 acetylation under doxorubicin-induced genotoxic stress conditions. Overexpression of WT MCL1 protected cells from doxorubicin-induced apoptosis (Figure S3A). MCL1 K40Q exerted a noticeable anti-apoptotic function against dose- and time-dependent effects of doxorubicin compared with the WT and K40R, likely because of acetylation-dependent MCL1 stabilization (Figures S3B and S3C). To further dissect DNA damage-mediated regulation of MCL1, we generated HeLa and HCT116 cells stably expressing MCL1 WT, K40Q, or K40R, in which endogenous MCL1 was eliminated by the CRISPR-Cas9 system. The acetylation-mimetic K40Q mutant was largely protected from doxorubicin-induced downregulation of MCL1 protein abundance (Figures 3A and 3B). Activation of the apoptotic cascade was correlated inversely with MCL1 protein levels. Consistent with this finding, MCL1 K40Q-expressing cells treated with doxorubicin were relatively more resistant to a dose-dependent decrease in cell viability (Figure 3C) and showed enhanced colony formation potential when cells were grown on plastic (Figures 3D and S3D) and in soft agar (Figure 3E). Furthermore, cell lines reconstituted with MCL1 were inoculated subcutaneously into nude mice to evaluate how MCL1 acetylation contributes to tumor progression. This xenograft model showed that the acetylation-mimetic K40Q form of MCL1 accelerates tumor growth more efficiently than the WT and the acetylation-deficient K40R mutant (Figures 3F-3H). These data suggest that stabilization of MCL1 by acetylation likely plays critical roles in acquiring chemotherapeutic resistance and enhancing tumorigenicity in vivo.
Figure 3. Acetylation-mimetic MCL1 K40Q displays enhanced anti-apoptotic function and oncogenicity.

(A and B) Ectopic expression of the acetylation-mimetic MCL1 K40Q mutant confers resistance to doxorubicin-induced downregulation. Shown is IB analysis of WCLs derived from HeLa cells (A) and HCT116 cells (B), which stably express WT MCL1, K40Q, or K40R at a level comparable to that where endogenous MCL1 is eliminated by the CRISPR-Cas9 system. The resulting cells were treated with doxorubicin (1 μg/mL) for 24 h before harvesting.
(C) Acetylation-mimetic MCL1 K40Q enhances its anti-apoptotic function. The HCT116 cells presented in (B) were treated with the indicated concentrations of doxorubicin for 24 h and then subjected to cell viability assays. Data are presented as mean ± SD; n = 3 biological replicates; **p < 0.01, ***p < 0.001.
(D and E) Acetylation-mimetic MCL1 K40Q enhances the tumorigenic activity of MCL1. A colony formation assay was conducted using HeLa cells
(D) presented in (A) and a soft agar assay using HCT116 cells (E) presented in (B). These cells were pretreated overnight with doxorubicin (0.02 μ g/mL) before plating for the assays. Data are presented as mean ± SD; n = 3 biological replicates; *p < 0.05, ***p < 0.001.
(F–H) Acetylation-mimetic MCL1 K40Q promotes tumor growth in the mouse xenograft model. HeLa cells presented in (A) were injected subcutaneously into nude mice (n = 6 for each group). Tumor growth was monitored over the indicated periods (F) and the weight of the dissected tumors (G) and images of the dissected tumors (H) are presented. Data are presented as mean ± SEM; n = 6; *p < 0.05, **p < 0.01, ***p < 0.001.
Data in (A) and (B) are representative of at least two independent experiments. See also Figure S3.
MCL1 acetylation decreases MCL1 ubiquitination via increased interaction with the DUB USP9X
We next explored the molecular mechanisms underlying acetylation-dependent MCL1 stabilization. Given that the MCL1 protein level is controlled tightly by ubiquitination, we hypothesized that the observed MCL1 stabilization is due to dissociation of an E3 ligase or DUB recruitment. We first performed coIP assays using WT MCL1 and K40Q alongside the E3 ligases FBW7, β-TRCP1, and TRIM17. We observed no major differences in the interaction among MCL1 constructs with any of the E3 ligases we assessed (Figures S4A-S4C). On the other hand, of the previously reported MCL1 DUBs we tested, USP9X, but not USP13 and DUB3, bound more strongly to the acetylation-mimetic MCL1 K40Q compared with the WT or K40R (Figures 4A, S4D, and S4E). To investigate the physiological consequence of USP9X recruitment in controlling MCL1 ubiquitination status, we conducted cellular ubiquitination assays and found that depletion of USP9X mediated by two independent shRNAs reversed the decreased poly-ubiquitination level of MCL1 K40Q (Figure 4B). Likewise, the USP9X inhibitor WP1130 (Kapuria et al., 2010) was able to induce ubiquitination of MCL1 K40Q at levels similar to that of the WT and the K40R form of MCL1 (Figure 4C). In line with these findings, no significant difference in shortened protein half-life under the condition of USP9X depletion was observed between the WT and the K40Q form of MCL1 (Figures 4D and 4E). These results indicate that binding of USP9X to MCL1 is enhanced by MCL1 acetylation, likely promoting deubiquitination and stabilization of acetylated MCL1.
Figure 4. MCL1 acetylation promotes interaction with USP9X, resulting in MCL1 deubiquitination and stabilization.

(A) Acetylation-mimetic MCL1 K40Q enhances the interaction between MCL1 and USP9X. Shown is IB analysis of WCLs and anti-FLAG immunoprecipitates derived from 293T cells transfected with the indicated constructs.
(B) USP9X depletion reverses the ubiquitination levels of acetylation-mimetic MCL1 K40Q. HeLa cells stably expressing the indicated lentiviral shRNA were transfected with the indicated Myc-MCL1 and His-Ub constructs. 36 h after transfection, cells were treated overnight with MG132 (10 μM) before harvesting.
(C) Treatment with the USP9X inhibitor reverses ubiquitination of acetylation-mimetic MCL1 K40Q. HeLa cells were transfected with the indicated Myc-MCL1 and His-Ub constructs. 36 h after transfection, the cells were treated with MG132 (10 μM) in the presence or absence of WP1130 (5 μM) for 4 h before harvesting. Left: His-Ub-conjugated proteins were captured with Ni-NTA agarose beads. Right: quantification of the Myc-poly-ubiquitination band intensities of IB replicates. Data are presented as mean ± SD, n = 3 independent experiments, *p < 0.05, **p < 0.01.
(D) USP9X depletion abolishes stabilization of MCL1 K40Q. HeLa cells stably expressing the lentiviral shRNA specific for GFP or USP9X were transfected with the indicated Myc-MCL1 constructs. 36 h after transfection, cells were treated with 100 μg/mL CHX for the indicated periods before harvesting.
(E) Quantification of the Myc band intensities of IB replicates in (D). Data are presented as mean ± SD; n = 3 independent experiments, **p < 0.01.
(F and G) Treatment with the USP9X inhibitor WP1130 efficiently downregulates acetylation-mimetic MCL1 K40Q. Shown is IB analysis of WCLs derived from the MCL1-reintroduced CRISPR-Cas9-mediated MCL1 knockout (KO) HeLa (F) and HCT116 (G) cells presented in Figure 3. These cells were treated with WP1130 (10 μM) for the indicated periods before harvesting.
(H and I) Treatment with the USP9X inhibitor WP1130 abrogates the anti-apoptotic effect of acetylation-mimetic MCL1 K40Q. These cells were treated with the indicated concentrations of WP1130 for 24 h and then subjected to cell viability assays. Data are presented as mean ± SD; n = 3 biological replicates.
Data in (A), (B), (F), and (G) are representative of at least two independent experiments. See also Figure S4.
A previous study indicated that MCL1 phosphorylation at S159 and T163 reduces the interaction between USP9X and MCL1 (Schwickart et al., 2010). To establish the precise molecular mechanisms controlling USP9X-MCL1 signaling, we attempted to investigate the potential crosstalk between phosphorylation and acetylation in regulating the interaction between USP9X and MCL1. Our data show that the K40Q mutant exhibited a relatively lower phosphorylation level at T163 and/or S159/T163 (Figures S4F and S4G), which is possibly responsible for suppressing USP9X interaction with MCL1 (Schwickart et al., 2010). To test whether K40 acetylation status-related regulation of S159/T163 phosphorylation limits this MCL1-USP9X interaction, we employed an MCL1 mutant harboring the acetylation-mimetic K40Q and phosphorylationmimetic S159E/T163E substitutions (MCL1 K40Q/S159E/T163E). MCL1 K40Q/S159E/T163E displayed marked impairment of the interaction with USP9X and a consequent increase in poly-ubiquitination (Figures S4H and S4I). On the other hand, phosphorylation-deficient mutations (S159A/T163A) in MCL1 K40R resulted in an increased interaction with USP9X but to a lesser extent. Although further analyses are required to elucidate the detailed underlying molecular mechanisms, our data suggest that the K40 acetylation is likely necessary to impair the phosphorylation status at S159/T163, which, in turn, promotes the association of MCL1 with USP9X (Figure S4J).
Given that the USP9X inhibitor WP1130 can effectively induce ubiquitination of the acetylation-mimetic MCL1 K40Q (Figure 4C), we next sought to verify whether MCL1 K40Q-expressing cells, refractory to doxorubicin treatment (Figures 3A-3E), are relatively more sensitive to WP1130. We treated WT MCL1 and K40Q- and K40R-expressing cell lines with WP1130 and found that the K40Q-dependent anti-apoptotic effect observed in the case of doxorubicin treatment was largely abolished by USP9X inhibition, likely through downregulation of MCL1 K40Q protein abundance (Figures 4F-4I). These results suggest that stabilization of acetylated MCL1 is largely dependent on USP9X and that blockade of USP9X enzymatic activity efficiently induces apoptosis.
SIRT3 antagonizes acetylation-dependent stabilization of MCL1
Acetylation is a reversible post-translational modification that can be removed by specific lysine deacetylases, including the histone deacetylases (HDACs) and SIRT families of proteins. HDAC family members localize in the cytoplasm and nucleus, and SIRT family proteins reside in the nucleus, cytoplasm, and mitochondria (Michishita et al., 2005). Given that MCL1 is localized predominantly in mitochondria, we focused our attention on the SIRT family of proteins and investigated which SIRT activity is required for MCL1 deacetylation. In vitro deacetylation assays showed that SIRT1, SIRT2, and SIRT3 efficiently deacetylated MCL1 (Figure 5A). To identify physiological MCL1 deacetylase, we evaluated endogenous interactions between MCL1 and these SIRT family proteins by coIP assays. We found that MCL1 showed robust interaction with SIRT3, likely because of the mitochondrial localization of both proteins, whereas no and minimal interaction was observed with SIRT1 and SIRT2, respectively (Figure 5B). Consistent with this endogenous interaction, accumulation of MCL1 was observed with depletion of endogenous SIRT3 but not SIRT1 or SIRT2 (Figure 5C), implying possible involvement of SIRT3 in controlling MCL1 acetylation status. In support of this finding, knockdown of SIRT3, but not SIRT1 or SIRT2, exclusively prolonged MCL1 protein half-life (Figures 5D, S5A, and S5B), likely because of a decrease in MCL1 ubiquitination (Figure 5E). Importantly, SIRT3 knockdown resulted in elevation of acetylated MCL1 at K40 (Figure 5F), and ectopic SIRT3 expression enhanced ubiquitination of WT MCL1 but not K40R (Figure 5G, left), implying that SIRT3 expression likely triggers MCL1 deacetylation at K40 and subsequently impairs MCL1-USP9X interaction, resulting in increased MCL1 ubiquitination and shortened protein stability (Figure 5G, right). Next, to investigate the significance of SIRT3-dependent MCL1 deacetylation on a genotoxic stress-induced apoptotic pathway, we treated parental and MCL1 knockout (KO) cells with doxorubicin upon knockdown of SIRT3 and performed a cell viability assay. We found that depletion of SIRT3 increased MCL1 protein abundance and conferred resistance to the doxorubicin-induced decrease in cell viability in WT MCL1 cells but not MCL1 KO cells (Figures 5H, 5I, and S5C). These data indicate that SIRT3-mediated MCL1 deacetylation promotes MCL1 ubiquitination and degradation, sensitizing cancer cells to doxorubicin-elicited apoptosis.
Figure 5. SIRT3 negatively regulates MCL1 stability through deacetylation.

(A) SIRT1, SIRT2, and SIRT3 efficiently deacetylate MCL1 in vitro. Shown is IB analysis of MCL1 acetylation after the in vitro deacetylation reaction and WCLs derived from HeLa cells transfected with the indicated Myc-MCL1 and FLAG-SIRT constructs for MCL1 and SIRT protein purification by FLAG and Myc immunoprecipitates (STAR Methods).
(B) MCL1 interacts with SIRT3 at the endogenous level. Shown is IB analysis of WCLs (input) and anti-MCL1 immunoprecipitates derived from 293T cells.
(C) SIRT3 depletion results in the accumulation of MCL1 protein abundance. Shown is IB analysis of WCLs derived from HeLa cells stably expressing the lentiviral shRNA specific for GFP, SIRT1, SIRT2, or SIRT3.
(D) SIRT3 depletion extends MCL1 protein half-life. HeLa cells stably expressing the lentiviral shRNA specific for GFP or SIRT3 presented in (C) were treated with 100 μg/mL CHX for the indicated period before harvesting.
(E) SIRT3 depletion results in impairment of MCL1 poly-ubiquitination. HeLa cells stably expressing the lentiviral shRNA specific for GFP, SIRT1, SIRT2, or SIRT3 were transfected with Myc-MCL1 and His-Ub constructs. 36 h after transfection, cells were treated with MG132 (10 μM) overnight before harvesting.
(F) SIRT3 depletion accumulates endogenous Ac-K40-MCL1. Shown is IB analysis of WCLs and anti-MCL1 immunoprecipitates derived from HeLa cells stably expressing the indicated lentiviral shRNA specific for GFP or SIRT3.
(G) Ectopic SIRT3 expression increases ubiquitination of WT MCL1 but not K40R. 293T cells were transfected with Myc-MCL1, FLAG-SIRT3, and His-Ub constructs as indicated. 36 h after transfection, the cells were treated with MG132 (20 μM) for 5 h before harvesting. Left: His-Ub-conjugated proteins were captured with Ni-NTA agarose beads. Right: a schematic model of SIRT3-mediated MCL1 ubiquitination through K40 deacetylation followed by USP9X dissociation.
(H and I) SIRT3 depletion confers resistance to doxorubicin-induced apoptosis through MCL1 stabilization. CRISPR-Cas9-mediated MCL1 KO and its parental HeLa cells were infected with the lentiviral shRNA specific for GFP or SIRT3. These cells were treated with the indicated concentrations of doxorubicin for 24 h and then subjected to IB analysis (H) and a cell viability assay (I). Data are presented as mean ± SD; n = 3 biological replicates; **p < 0.01, ***p < 0.001.
Data in (A)–(H) are representative of at least two independent experiments. See also Figure S5.
MCL1 acetylation regulates cancer cell survival in a p300- and USP9X-dependent manner
Having demonstrated that acetylated MCL1 exerts a significant effect on conferring resistance to doxorubicin treatment in HeLa and HCT116 cells (Figure 3), we further explored the biological significance of this acetylation in breast and prostate cancer cells in which MCL1 is a potential prognostic marker and drug target (Arai et al., 2018; Campbell et al., 2018; Dash et al., 2011; Xiao et al., 2015; Young et al., 2016). Elevated levels of MCL1 acetylation were closely correlated with p300 expression in breast (SKBR3 and BT-20) and prostate (C42 and ABL) cancer cell lines (Figure 6A). Furthermore, treatment with shRNA for p300 or a p300/CBP inhibitor, A-485, resulted in decreases in MCL1 protein abundance without decreasing mRNA, at least in cells displaying a high acetylation state of MCL1 (Figures 6B, 6C, S6A, and S6B), implying that MCL1 protein stability is more dependent on p300-mediated acetylation. Importantly, we found a positive correlation between MCL1 and p300 expression levels in breast cancer clinical samples (r = 0.46, n = 49, p < 0.001, Spearman correlation test) (Figure 6D; Table S1). To further assess the molecular basis of acetylation-dependent MCL1 stabilization, we investigated the involvement of USP9X in cancer cells with highly acetylated MCL1 and found that dissociation of USP9X, but not USP13 and DUB3, from MCL1, accompanied by decreased levels of MCL1 acetylation, was caused by the p300/CBP inhibitor A-485 (Figures 6E, 6F, and S6C). In line with these observations, MCL1 protein levels were downregulated by depletion of USP9X but not USP13 (Figure S6D). Given that USP9X inhibition by WP1130 restored poly-ubiquitination of acetylation-mimetic K40Q MCL1 (Figure 4C) and effectively reduced its protein abundance (Figures 4F and 4G), we next evaluated MCL1 levels in breast and prostate cancer cells treated with WP1130. Notably, MCL1 protein levels, but not transcripts, were downregulated by WP1130 treatment in cell lines exhibiting a high acetylation state of MCL1 (Figures 6G, 6H, and S6E). Coupled with the reduction in MCL1 levels, simultaneous activation of the apoptotic cascade was observed in these cells. Indeed, cell viability assays revealed that cells with high MCL1 acetylation were more sensitive to the USP9X inhibitor (Figures 6I and 6J). These data indicate that p300-dependent acetylation of MCL1 is critical for USP9X-mediated MCL1 stabilization, sensitizing cancer cells with relatively higher level of acetylated MCL1 to the USP9X inhibitor WP1130 (Figure 7).
Figure 6. MCL1 acetylation promotes cancer cell survival in a p300- and USP9X-dependent manner.

(A) MCL1 acetylation levels correlate with p300 expression in breast and prostate cancer cell lines. Shown is IB analysis of WCLs and anti-MCL1 immunoprecipitates derived from a panel of breast and prostate cancer cell lines.
(B) p300 depletion impairs MCL1 protein abundance in breast cancer cells with high p300 and acetylated MCL1 levels. Shown is IB analysis of WCLs derived from breast cancer cell lines stably expressing the lentiviral shRNA specific for GFP or p300.
(C) Treatment with the p300/CBP inhibitor A-485 results in decreased MCL1 protein levels in breast and prostate cancer cells with high p300 and acetylated MCL1 levels. Shown is IB analysis of WCLs derived from breast and prostate cancer cell lines treated with A-485 (3 μM) for 48 h before harvesting.
(D) Representative images of MCL1 and p300 expression in breast tumor cells as assessed by immunohistochemistry (IHC). MCL1 and p300 levels were classified as low, moderate, or high, based on the intensities of the IHC staining, and a Spearman correlation test was conducted. Scale bar, 50 μm. See also Table S1.)
(E and F) Treatment with the p300/CBP inhibitor A-485 reduces MCL1 acetylation and promotes dissociation of USP9X from MCL1. Shown is IB analysis of WCLs and anti-MCL1 immunoprecipitates derived from SKBR3 (E) and BT-20 (F) treated overnight with the indicated concentrations of A-485 before harvesting.
(G and H) The USP9X inhibitor WP1130 effectively induces activation of the apoptotic pathway in cells with higher levels of acetylated MCL1. Shown is IB analysis of WCLs derived from the indicated breast cancer (G) and prostate cancer (H) cell lines. These cells were treated with WP1130 (5 μM) for the indicated periods before harvesting.
(I and J) High acetylated MCL1 levels correlate with increased sensitivity to WP1130 in breast and prostate cancer cell lines. Breast cancer (I) and prostate cancer (J) cell lines were treated with the indicated concentrations of WP1130 for 24 h and subjected to cell viability assays. Data are presented as mean ± SD; n = 3 biological replicates; ***p < 0.001. Data in (A)–(C) and (E)–(H) are representative of at least two independent experiments. See also Figure S6.
Figure 7. A schematic diagram of proposed crosstalk among acetylation, phosphorylation, and ubiquitination events in dynamic regulation of MCL1 protein stability.

MCL1 is an unstable protein that is targeted for ubiquitination. p300-directed MCL1 acetylation at K40 leads to enhanced interaction with USP9X, facilitating MCL1 deubiquitination and stabilization. SIRT3 is the potential deacetylase counteracting the acetylation-dependent MCL1 stabilization. Phosphorylation also plays important roles in regulating the interaction with USP9X and certain E3s. Elevated MCL1 acetylation status may correlate with sensitivity to USP9X and p300 inhibitors.
DISCUSSION
MCL1 protein is highly unstable, and several lines of evidence indicate that post-translational modification is one of the major regulatory mechanisms controlling MCL1 biological function (Mojsa et al., 2014; Senichkin et al., 2020). In this study, we report that acetylation is a critical modification for potentiating MCL1 oncogenic activity. We found that the lysine acetyltransferase p300 facilitates MCL1 acetylation at K40, leading to a decrease in MCL1 ubiquitination and subsequent MCL1 protein stabilization. Mechanistically, acetylation of MCL1 resulted in enhanced interaction with USP9X, which, in turn, promotes USP9X-dependent MCL1 deubiquitination. We further identified SIRT3 as the potential MCL1 deacetylase that antagonizes p300-dependent MCL1 acetylation (Figure 7). Although the functions of p300, USP9X, and SIRT3 as drivers of cancer formation or tumor suppressor are cell-type- and context-dependent (Attar and Kurdistani, 2017; Cheng et al., 2019; Wang et al., 2013; Xiong et al., 2016), our data imply that increased p300 abundance may trigger aberrant MCL1 acetylation and subsequently activate the p300-USP9X-MCL1 oncogenic pathway.
Crosstalk between two lysine modifications, acetylation and ubiquitination, affects the stability and activity of several individual cellular proteins (Caron et al., 2005). Although we hypothesized that competition between ubiquitination and acetylation at K40 was the possible mechanism underlying acetylation-dependent MCL1 stabilization, the fact that acetylation-deficient MCL1 K40R was still heavily poly-ubiquitinated at levels comparable with WT MCL1 (Figure 2G) refutes the possibility that competitive inhibition of K40 ubiquitination by K40 acetylation is a primary cause of suppressing overall MCL1 ubiquitination. On the other hand, we demonstrated that USP9X preferred to bind to MCL1 K40Q compared with WT MCL1 or K40R, postulating that acetylation likely acts as a signaling switch regulating MCL1 function, presumably by adjusting the affinity with MCL1-interacting proteins, including USP9X. Furthermore, our data indicate possible cross-talk between acetylation and phosphorylation events. Phosphorylation at T163 and/or S159/T163 was reduced in the acetylation-mimetic form of MCL1 (Figures S4F and S4G). A previous study has demonstrated that phosphorylation of MCL1 at S159 and T163 reduces interaction between MCL1 and USP9X (Schwickart et al., 2010). Consistent with this observation, dephosphorylation of S159/T163 promoted by PP2A has been reported recently to lead to MCL1 stabilization in multiple melanoma cells (Slomp et al., 2021). These results provided a sequential model where K40 acetylation likely promotes the interaction with USP9X by suppressing phosphorylation at S159/T163 (Figure S4J). We previously described the critical role of S159/T163 phosphorylation in inducing proteasomal degradation of MCL1 by establishing a Fbw7 phospho-degron motif (Inuzuka et al., 2011; Wertz et al., 2011). These findings suggest that the integral mechanism between acetylation at K40 and phosphorylation at S159/T163, which affects E3 recruitment and DUB dissociation, cooperatively regulates MCL1 protein stability. Although detailed molecular mechanisms need to be analyzed further, our findings provide a mechanistic explanation for the signal-dependent interaction between MCL1 and USP9X.
MCL1 has been shown to be a potential prognostic marker and drug target in breast and prostate cancer (Attar and Kurdistani, 2017; Campbell et al., 2018; Dash et al., 2011; Xiao et al., 2015; Young et al., 2016). Our results imply that MCL1 acetylation may contribute to conferring cancer cells resistance to doxorubicin treatment (Figures 3A-3E). Given the potential role of MCL1 in acquired chemoresistance, we expect that targeting acetylated MCL1 with genotoxic reagents in combination with an USP9X inhibitor may be an efficient strategy to treat MCL1-over-expressing cancers. Our pharmacological assays using a USP9X inhibitor demonstrates that the viability of cancer cells with high acetylated MCL1 levels is more dependent on USP9X enzymatic activity (Figures 6I and 6J), suggesting that WP1130 is a promising option against tumors with high MCL1 acetylation. On the other hand, cancer cells with low MCL1 acetylation are apt to be richer in other anti-apoptotic BCL2 family proteins, such as Bcl-2 and Bcl-xL (Figure 6A), implying relatively less dependency on MCL1 for cancer cell survival. However, further evaluation is required to clarify the significance of MCL1 acetylation for modulating MCL1 oncogenic potential through an undefined function other than protein stability control. This study characterized acetylation as a key post-translational modification required for MCL1 protein stability control. Given that oncogenic mutations in the MCL1 coding region have not been reported, dysregulation of the upstream degradation signaling pathway is considered a major cause of MCL1 upregulation in a variety of cancers. Here we proposed that aberrant MCL1 acetylation contributes to enhanced cancer cell survival and chemoresistance, promoting tumorigenesis in part through MCL1 protein stabilization. Elucidating the detailed molecular mechanisms underlying MCL1 acetylation may help with development of appropriate diagnostic approaches and provide a potential therapeutic intervention for cancers with elevated p300-USP9X-MCL1 oncogenic signaling.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Wenyi Wei (Department of Pathology, Beth Israel Deaconess Medical Center, Harvard Medical School, 3 Blackfan Circle, Boston, MA 02115; wwei2@bidmc.harvard.edu).
Materials availability
Newly generated materials in this paper are available from the lead contact upon request.
Data and code availability
Mass spectrometry data have been deposited at MassIVE repository and are publicly available as of the date of publication. Accession number is listed in the key resources table. Original western blot images have been deposited at Mendeley Data and are publicly available as of the date of publication. The DOI is listed in the key resources table. Other data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-Mcl-1 (D2W9E) | Cell Signaling Technology | Cat# 94296, RRID: AB_2722740 |
| Rabbit monoclonal anti-Phospho-Mcl-1 (Ser64) | Cell Signaling Technology | Cat# 13297, RRID: AB_2798173 |
| Rabbit polyclonal anti-Phospho-Mcl-1 (Ser159/Thr163) | Cell Signaling Technology | Cat# 4579, RRID: AB_2144100 |
| Rabbit monoclonal anti-Phospho-Mcl-1 (Thr163) (D5M9D) | Cell Signaling Technology | Cat# 14765, RRID: AB_2716686 |
| Rabbit monoclonal anti-Bcl-2 (D55G8) | Cell Signaling Technology | Cat# 4223, RRID: AB_1903909 |
| Rabbit monoclonal anti-Bcl-xL (54H6) | Cell Signaling Technology | Cat# 2764, RRID: AB_2228008 |
| Rabbit polyclonal anti-Acetylated-Lysine | Cell Signaling Technology | Cat# 9441, RRID: AB_331805 |
| Rabbit monoclonal anti-p300 (D8Z4E) | Cell Signaling Technology | Cat# 86377, RRID: AB_2800077 |
| Rabbit monoclonal anti-CBP (D6C5) | Cell Signaling Technology | Cat# 7389, RRID: AB_2616020 |
| Rabbit monoclonal anti-PCAF (C14G9) | Cell Signaling Technology | Cat# 3378, RRID: AB_2128409 |
| Rabbit monoclonal anti-GCN5L2 (C26A10) | Cell Signaling Technology | Cat# 3305, RRID: AB_2128281 |
| Rabbit monoclonal anti-USP9X (D4Y7W) | Cell Signaling Technology | Cat# 14898, RRID: AB_2798640 |
| Rabbit polyclonal anti-SirT1 (1F3) | Cell Signaling Technology | Cat# 8469, RRID: AB_10999470 |
| Rabbit polyclonal anti-SirT2 (D4O5O) | Cell Signaling Technology | Cat# 12650, RRID: AB_2716762 |
| Rabbit monoclonal anti-SirT3 (D22A3) | Cell Signaling Technology | Cat# 5490, RRID: AB_10828246 |
| Rabbit monoclonal anti-Cleaved PARP (Asp214) (D64E10) | Cell Signaling Technology | Cat# 5625, RRID: AB_10699459 |
| Rabbit polyclonal anti-Cleaved Caspase-3 (Asp175) | Cell Signaling Technology | Cat# 9661, RRID: AB_2341188 |
| Rabbit monoclonal anti-COX IV (3E11) | Cell Signaling Technology | Cat# 4850, RRID: AB_2085424 |
| Mouse monoclonal anti-Lamin A/C (4C11) | Cell Signaling Technology | Cat# 4777, RRID: AB_10545756 |
| Mouse monoclonal anti-GFP (4B10) | Cell Signaling Technology | Cat# 2955, RRID: AB_1196614 |
| Rabbit monoclonal anti-Myc-Tag (71D10) | Cell Signaling Technology | Cat# 2278, RRID: AB_490778 |
| Mouse monoclonal anti-Myc-Tag (9B11) | Cell Signaling Technology | Cat# 94296; RRID: AB_2722740 |
| Mouse anti-Myc-Tag immobilized (bead conjugate) | Cell Signaling Technology | Cat# 3400, RRID: AB_10692357 |
| Mouse monoclonal anti-Mcl-1 (B-6) AC | Santa Cruz Biotechnology | Cat# sc-74436 AC, RRID: AB_1126069 |
| Mouse monoclonal anti-USP13 (B-9) | Santa Cruz Biotechnology | Cat# sc-514416 |
| Rabbit polyclonal anti-Vinculin (H-300) | Santa Cruz Biotechnology | Cat# sc-5573, RRID: AB_2214507 |
| Goat polyclonal anti-Lamin B1 (S-20) | Santa Cruz Biotechnology | Cat# sc-30264, RRID: AB_2136305 |
| Rabbit polyclonal anti-HA-probe (Y-11) | Santa Cruz Biotechnology | Cat# sc-805 |
| Mouse monoclonal purified anti-HA.11 Epitope Tag |
BioLegend | Cat# 901503, RRID: AB_2565005 |
| Anti c-Myc antibody beads (10D11) | FUJIFILM Wako | Cat# 016-26503 |
| Mouse monoclonal anti-V5 Tag | Thermo Fisher Scientific | Cat# R960-25, RRID: AB_2556564 |
| Mouse monoclonal anti-DUB3 | Sigma-Aldrich | Cat# WH0377630M1, RRID: AB_1841372 |
| Mouse monoclonal anti-alpha-Tubulin | Sigma-Aldrich | Cat# T5168, RRID: AB_477579 |
| Rabbit polyclonal anti-Flag | Sigma-Aldrich | Cat# F7425, RRID: AB_439687 |
| Mouse monoclonal anti-Flag M2 | Sigma-Aldrich | Cat# F3165, RRID: AB_259529 |
| Anti-Flag M2 Affinity Gel | Sigma-Aldrich | Cat# A2220, RRID: AB_10063035 |
| Mouse monoclonal anti-HA-Agarose antibody | Sigma-Aldrich | Cat# A2095, RRID: AB_257974 |
| Goat polyclonal anti-Mouse IgG (whole molecule), HRP conjugated | Sigma-Aldrich | Cat# A4416, RRID: AB_258167 |
| Goat anti-Rabbit IgG (whole molecule)-Peroxidase antibody | Sigma-Aldrich | Cat# A4914, RRID: AB_258207 |
| Mouse monoclonal anti-acetylated-K40-MCL1 | This paper | N/A |
| Bacterial and virus strains | ||
| XL10-Gold Ultracompetent Cells | Agilent Technologies | Cat# 200315 |
| Chemicals, peptides, and recombinant proteins | ||
| MG132 | Enzo Life Sciences | Cat# BML-PI102-0025 |
| A-485 | MedChemExpress | Cat# HY-107455 |
| C646 | MedChemExpress | Cat# HY-13823 |
| WP1130 | MedChemExpress | Cat# HY-13264 |
| Cycloheximide | Sigma-Aldrich | Cat# C1988 |
| Trichostatin A | Sigma-Aldrich | Cat# T8552 |
| Nicotinamide | Sigma-Aldrich | Cat# 72340 |
| Critical commercial assays | ||
| QuikChange II XL Site-Directed Mutagenesis Kit |
Agilent Technologies | Cat# 200516 |
| NE-PER Nuclear and Cytoplasmic Extraction Reagents |
Thermo Fisher Scientific | Cat# 78833 |
| Cell Fractionation Kit | Abcam | Cat# ab109719 |
| CellTiter-Glo | Promega | Cat# G7571 |
| Deposited data | ||
| Mass spectrometry data | This paper | MassIVE: MSV000088231 |
| Original western blot images | This paper | Mendeley Data: https://doi.org/10.17632/pyfpzd2tv5.1 |
| Experimental models: Cell lines | ||
| HeLa | ATCC | Cat# CCL-2.2, RRID:CVCL_0058 |
| HEK293T | ATCC | Cat# CRL-3216, RRID:CVCL_0063 |
| MCF7 | Dr. Piotr Sicinski lab, DFCI | N/A |
| SKBR3 | Dr. Piotr Sicinski lab, DFCI | N/A |
| MDA-MB-231 | Dr. Alex Toker lab, BIDMC | N/A |
| BT-20 | ATCC | Cat# CRL-7912, RRID:CVCL_0178 |
| C42 | Dr. Steven Balk lab, BIDMC | N/A |
| LNCaP-ABL (ABL) | Dr. Steven Balk lab, BIDMC | N/A |
| PC3 | Dr. Steven Balk lab, BIDMC | N/A |
| DU145 | Dr. Steven Balk lab, BIDMC | N/A |
| Experimental models: Organisms/strains | ||
| Nude Mouse | Charles River | BALB/c-nu/nu |
| Oligonucleotides | ||
| qRT-PCR Primer: Human MCL1 Forward: TGCTTCGGAAACTGGACATCA | This paper | N/A |
| qRT-PCR Primer: Human MCL1 Reverse: TAGCCACAAAGGCACCAAAAG | This paper | N/A |
| qRT-PCR Primer: Human GAPDH Forward: TCCTGCACCACCAACTGCTTA | This paper | N/A |
| qRT-PCR Primer: Human GAPDH Reverse: AGTGATGGCATGGACTGTGGT | This paper | N/A |
| Recombinant DNA | ||
| Myc-MCL1 | Ding et al., 2007 | N/A |
| Myc-MCL1 K5R | Inuzuka et al., 2011 | N/A |
| Myc-MCL1 K40R | Inuzuka et al., 2011 | N/A |
| Myc-MCL1 K40Q | This paper | N/A |
| Myc-MCL1 K136R | Inuzuka et al., 2011 | N/A |
| Myc-MCL1 K194R/197R | Inuzuka et al., 2011 | N/A |
| Myc-MCL1 K40Q/S159E/T163E | This paper | N/A |
| Myc-MCL1 K40R/S159A/T163A | This paper | N/A |
| pLenti-HA-MCL1 | Inuzuka et al., 2011 | N/A |
| pLenti-HA-MCL1 K40R | This paper | N/A |
| pLenti-HA-MCL1 K40Q | This paper | N/A |
| HA-MCL1 | Inuzuka et al., 2011 | N/A |
| HA-p300 | Askew et al., 2010 | Addgene Cat# 89094 |
| Flag-SIRT1 | North et al., 2003 | Addgene Cat# 13812 |
| Flag-SIRT2 | North et al., 2003 | Addgene Cat# 13813 |
| Flag-SIRT3 | North et al., 2003 | Addgene Cat# 13814 |
| Flag-SIRT4 | North et al., 2003 | Addgene Cat# 13815 |
| Flag-SIRT5 | North et al., 2003 | Addgene Cat# 13816 |
| Flag-SIRT6 | North et al., 2003 | Addgene Cat# 13817 |
| Flag-SIRT7 | North et al., 2003 | Addgene Cat# 13818 |
| pLKO-shMCL1 | TRC | N/A |
| pLKO-shp300 | TRC | N/A |
| pLKO-shUSP13 | TRC | N/A |
| HA-GCN5 | This paper | N/A |
| HA-Tip60α | This paper | N/A |
| HA-PCAF | This paper | N/A |
| HA-–-TRCP1 | This paper | N/A |
| Flag-His-USP9X | This paper | N/A |
| pLKO-shUSP9X | A gift from Dr. Qing Zhang, UNC | N/A |
| pLKO-shSIRT1 | TRC | N/A |
| pLKO-shSIRT2 | TRC | N/A |
| pLKO-shSIRT3 | TRC | N/A |
| HA-FBW7 | A gift from Dr. Keiichi Nakayama, Kyushu University, Japan | N/A |
| V5-TRIM17 | A gift from Dr. Tatsuya Sawasaki, Ehime University, Japan | N/A |
| Flag-HA-DUB3 | Sowa et al., 2009 | Addgene Cat# 22593 |
| Myc-Flag-USP13 | A gift from Dr. Lingqiang Zhang, Beijing Institute of Lifeomics, China | N/A |
| Human MCL1 CRISPR/Cas9 KO plasmids | Santa Cruz Biotechnology | Cat# sc-400079 and sc-400079-HDR |
| Software and algorithms | ||
| GraphPad Prism | GraphPad | https://www.graphpad.com |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
HeLa, HEK293T, MCF7, SKBR3, and MDA-MB-231 cells were maintained in DMEM. C42, PC3, LNCaP-ABL (ABL), and PF382 cells were maintained in RPMI 1640. DU145 and BT-20 cells were maintained in MEM. All cells were cultured in a humidified CO2 incubator, 5% CO2 in a temperature at 37°C. Each medium was supplemented with 10% FBS, 100 U of penicillin, and 100 μg/mL streptomycin. For BT-20 cells used in this study, given that some stocks were reported to be misidentified (Nelson-Rees et al., 1981), cell line validation experiments were conducted. The ATCC certification report demonstrated that the BT-20 cells used in this study are not cross-contaminated (Gao et al., 2020). Transfection was performed using polyethylenimine (PEI). Packaging of lentiviruses and subsequent infection of various cell lines were performed according to the protocol described previously (Gao et al., 2011). After viral infection, cells were selected for at least 72 hours in the presence of puromycin (1 μg/mL) or hygromycin (200 μg/mL), depending on the viral vectors used to infect cells. Cycloheximide (CHX) was used at 100 μg/mL for the indicated time periods.
Generation of MCL1-knockout cell lines
HeLa and HCT116 cells were transfected by PEI with human MCL1 CRISPR/Cas9 KO and HDR Plasmids (sc-400079 and sc-400079-HDR, Santa Cruz Biotechnology). At 48 h after transfection, cells were selected in puromycin (1 μg/mL)-containing medium for 3 days. The resulting cells were subjected to clonal isolation by the single cell dilution method in a 96-well plate. The knockout of MCL1 was validated by immunoblot analysis.
Mouse xenograft assays
The MCL1 reintroduced HeLa cells (1 × 106) in 50 μl PBS were injected into the flanks of female nude mice (BALB/cAJcl-nu/nu from CLEA Japan; 5 weeks old). Tumor size was measured every 3 days with a caliper. The tumor volume was determined using the formula L × W2 × 0.52, where L is the longest diameter and W is the shortest diameter. The mice were euthanized at the end of the studies, and in vivo solid tumors were dissected and weighed. All care was taken, and experimental procedures were conducted according to the Tohoku University Institutional Animal Care and Use Committee protocol (2019DnA-019-1).
METHOD DETAILS
Antibodies, plasmids, and materials
Anti-MCL1 (94296), anti-pS64-MCL1 (13297), anti-pS159/T163-MCL1 (4579), anti-pT163-MCL1 (14765), anti-Bcl-2 (4223), anti-Bcl-xL (2764), anti-Acetylated-Lysine (9441), anti-p300 (86377), anti-CBP (7389), anti-PCAF (3378), anti-GCN5L2 (3305), anti-USP9X (14898), anti-SIRT1 (8469), anti-SIRT2 (12650), anti-SIRT3 (5490), anti-Cleaved PARP (5625), anti-Cleaved Caspase-3 (9661), anti-COX IV (4850), anti-Lamin A/C (4777), anti-GFP (2955), polyclonal anti-Myc tag (2278), monoclonal anti-Myc-tag (2276) antibodies, anti-Myc-tag Sepharose beads (3400) were purchased from Cell Signaling Technology. Anti-c-Myc antibody beads (10D11) (016-26503) were purchased from Wako. Anti-MCL1 agarose beads (sc-74436 AC), anti-USP13 (sc-514416), anti-Vinculin (sc-73614), anti-Lamin B1 (sc-30264), and polyclonal anti-HA (sc-805) antibodies were purchased from Santa Cruz Biotechnology. Monoclonal anti-HA antibody (901503) was purchased from Biolegend. Anti-V5 antibody (R960-25) was purchased from Thermo Fisher Scientific. Anti-DUB3 (HPA045642), anti-tubulin (T5168), polyclonal anti-Flag (F7425), and monoclonal anti-Flag (F3165) antibodies, anti-Flag agarose beads (A2220), anti-HA agarose beads (A2095), peroxidase-conjugated anti-mouse secondary antibody (A4416), and peroxidase-conjugated anti-rabbit secondary antibody (A4914) were purchased from Sigma-Aldrich. Monoclonal anti-acetylated-K40-MCL1 antibody was developed in collaboration with mAbProtein. Myc-MCL1, HA-MCL1, HA-p300, HA-FBW7, Flag-SIRTs, pLenti-HA-MCL1, and short hairpin RNAs (shRNAs) specific for p300 and SIRT3 were described previously (Inuzuka et al., 2011, 2012). HA-GCN5, HA-Tip60α, HA-PCAF, and HA-β-TRCP1 were constructed by subcloning the appropriate PCR fragments into pcDNA3-HA plasmid. The open reading frame of USP9X cDNA was amplified by reverse transcription-PCR and cloned into the pcDNA3.1 plasmid. shRNA specific for USP9X is provided by Dr. Qing Zhang. shRNAs specific for SIRT1 and SIRT2 were obtained from Open Biosystems. V5-TRIM17 is provided by Dr. Tatsuya Sawasaki. Myc-Flag-USP13 is a gift from Dr. Lingqiang Zhang. Flag-SIRT1-7 (North et al., 2003), Flag-HA-DUB3 (Sowa et al., 2009), and HA-p300 (Askew et al., 2010) were obtained from Addgene. MCL1 mutants were generated using QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies) according to the manufacturer’s instruction. Proteasome inhibitor MG132 was obtained from Enzo Life Sciences. p300 inhibitor A-485, C646, and USP9X inhibitor WP1130 were obtained from MedChemExpress.
Immunoblots and immunoprecipitation
Cells were lysed in EBC buffer (50 mM Tris-HCl pH 7.5, 120 mM NaCl, 0.5% NP-40) supplemented with protease inhibitors (Complete Mini, Roche) and phosphatase inhibitors (PhosSTOP, Roche). The protein concentrations of whole-cell lysates were determined with the Bio-Rad protein assay reagent. Sixty micrograms of whole-cell lysates were resolved by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad). Membranes were blocked with 5% non-fat dry milk in TBST (Tris-buffered saline with 0.05% tween 20, pH 8.0) and probed with indicated antibodies. For immunoprecipitation, one milligram of lysates was incubated with the indicated antibody-conjugated beads for 4 h at 4°C. The immunoprecipitates were washed five times with NETN buffer (20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, and 0.5% NP-40) before being resolved by SDS-PAGE followed by immunoblot analysis with indicated antibodies.
Immunofluorescence
BT-20 (2 × 104) and HeLa (1 × 104) cells were seeded on collagen I-coated coverslips in 24-well plates and cultured for 48h. Cells were then fixed with phosphate buffered 4% paraformaldehyde (PFA) for 15 min at room temperature and then incubated in 100 mM glycine/PBS for 5 min to quench PFA. Membrane permeabilization was performed with 0.1% Triton X-100/PBS for 5 min. For blocking, the cells were incubated in staining buffer (10% FBS/PBS) for 1h. The cells were then incubated with primary antibodies diluted with in staining buffer (anti-Ac-K40-MCL1 at 5 μg/mL; anti-p300 (CST, 86377) at 1:500; anti-COX IV (CST, 4850) at 1:250) for 1 h at room temperature in a humidity box. After washing with PBS (5 min, three times), the cells were stained with secondary antibodies (Alexa Fluor 488 goat anti-mouse IgG, Alexa Fluor 546 goat anti-rabbit IgG, at 1:1000 each) and DAPI for 1h. After washing out the unbound antibodies, the stained cells were mounted onto glass slides with FluorSave (Millipore). Confocal fluorescence images were captured with an LSM700 system (Carl Zeiss) as 16-bit depth with a 63X water-immersion objective lens by scanning each channel four separate times for averaging. Images were analyzed with the accompanying ZEN software to depict the profiles of colocalization plots. The acquired images were processed with Fiji.
Subcellular fractionation
BT-20 and HeLa cells in semiconfluent cultures were harvested with trypsin-EDTA and then washed with PBS. 2 × 106 cells were transferred to a microcentrifuge tube and then centrifuged at 500 × g for 3 min at 4°C to pellet intact cells. Subsequent cytoplasmic and nuclear fractionations were performed with NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific), and separation of cytoplasmic and mitochondrial fractions was performed using Abcam’s Cell Fractionation Kit (standard) (ab109719) according to the manufacturer’s instructions.
Cell viability assays
The indicated cell lines were seeded in 96-well plates (2–3 × 103 cells/well) and cultured for 24 h in 100 μl of medium containing 10% serum. The cells were then treated with various concentrations of doxorubicin or the USP9X inhibitor WP1130 for 24 h. Cell viability was measured using CellTiter-Glo (Promega) according to the manufacturer’s instructions.
In-cell ubiquitination assays
Detection of protein ubiquitination with denaturing Ni-NTA pull-down for mammalian cells was described previously (Tansey, 2006). 293T or HeLa cells were transfected with the indicated constructs along with His-tagged ubiquitin (His-Ub) expression plasmid. At 24-36 h post-transfection, cells were treated with MG132 in the absence or presence of WP1130 or A-485 as described in the figure legends and then lysed with denaturing buffer (6 M guanidine-HCl, 0.1 M Na2HPO4/NaH2PO4,10 mM imidazole pH 8.0) followed by sonication. After 3 h incubation with Ni-NTA agarose beads (QIAGEN), His-ubiquitinated proteins were purified through three washes with the denaturing buffer and TI buffer (20 mM Tris-HCl, 20 mM imidazole pH 6.8) and eluted in SDS sample buffer for subsequent immunoblot analyses.
In vitro deacetylation assay
293T cells were transfected with each of the control vector or Flag-tagged sirtuins (SIRT1-SIRT7) individually (to generate each of the SIRT proteins) or Myc-MCL1 together with HA-p300 (to generate acetylated MCL1), and lysed 48 h after transfection in lysis buffer (50 mM Tris-HCl, pH 7.5, 0.5 mM EDTA, 0.5% NP-40, 150 mM NaCl) in the presence of protease inhibitor cocktail (Complete, Roche). Flag-tagged proteins were immunoprecipitated with anti-Flag M2 agarose affinity gel (Sigma-Aldrich) and Myc-tagged MCL1 was immunoprecipitated with anti-Myc-tag beads (Cell Signaling Technology) for 2 h at 4°C. Immunoprecipitated material was washed three times for 15 min each in lysis buffer and either used for subsequent in vitro activity assays. Immunoprecipitates for Flag-tagged SIRTs and Myc tagged MCL1 were washed two times for 15 min each in SIRT deacetylase buffer (50 mM Tris-HCl, pH 9.0, 4 mM MgCl2, 0.2 mM DTT). Myc-MCL1 immunoprecipitates were resuspended in 800 μL of SIRT deacetylase buffer containing 1 mM NAD+ (Sigma-Aldrich) and mixed to resuspend anti-Myc-agarose beads, and 100 μL was added to each Flag-tagged SIRT immunoprecipitation and incubated for 2 h at 37°C. Reactions were stopped by the addition of 20 μL of 6 × SDS-PAGE buffer. Beads were pelleted by centrifugation at 14,000 rpm for 10 min and 10 μL of each supernatant was separated on 10% SDS-PAGE gels and western blotted as described above.
Detection of MCL1 acetylation site
293T cells were transfected with HA-MCL1 and Myc-p300. Thirty hours post-transfection, 293T cells were treated with 1 mM trichostatin A (TSA) and 5 mM nicotinamide (NAM) for 10 h to block the lysine deacetylases activity. The whole-cell lysates were immunoprecipitated with anti-HA agarose beads (Sigma-Aldrich) in the presence of 2 mM TSA and 10 mM NAM. The immunoprecipitated material was resolved by SDS-PAGE and the MCL1 band was excised. The protein sample were reduced with dithiothreitol, and Cys residues were alkylated with iodoacetamide. The protein was then digested overnight at 37°C using the Trypsin/LysC enzyme. Peptide mixtures were cleaned using a C18 ziptip column and injected on a Thermo EASY-nLC1200 UPLC coupled to a Thermo HF QExactive Orbitrap high resolution mass spectrometer using a 75mm i.d. × 15 cm C18 microcapillary column at a flow rate of 300 nL/min. Data dependent MS/MS acquisitions were performed using the Top 8 method. Tandem mass spectra were extracted by MSConvert version 3.0.9987. All MS/MS samples were analyzed using Mascot (Matrix Science, London, UK; version 2.7.0). Mascot was set up to search the Human_20210616 database (unknown version, 20600 entries) assuming the digestion enzyme trypsin, and acetylation of Lysine was specified as variable modifications.
Colony-formation assays
Cells were seeded in six-well plates (2000 cells per well) and cultured for 8 days until visible colonies formed. The colonies were fixed with 10% acetic acid and 10% methanol for 20 min and then stained with 0.4% crystal violet in 20% ethanol for 20 min. After staining, the plates were washed with distilled water and air-dried. The colonies were counted for statistical analysis.
Soft-agar assays
The anchorage-independent cell-growth assays were performed in six-well plates with the bottom layer containing 0.8% noble agar. Cells (3 × 104 per well) were mixed with noble agar to a final concentration of 0.4% and layered over the bottom agar. The dishes were then cultured in a 37°C incubator for three weeks and 500 μl complete DMEM medium was added to keep the top layer moist. The cells were stained with 1 mg/mL iodonitrotetrazolium chloride (Sigma-Aldrich) for colony visualization and counting.
Real-time RT-PCR analyses
RNA was extracted using QIAGEN RNeasy mini kit, and the reverse-transcription (RT) reaction was performed with ReverTra Ace qPCR RT Master Mix (TOYOBO). The real-time RT-PCR reaction was performed with SYBR Select Master Mix and a 7500 Real-Time PCR System (Applied Biosystems). The relative gene expression was calculated using the ddCt method, and GAPDH was utilized to normalize transcript abundance. All procedures were performed according to the manufacturer’s instructions. The primers used for the PCR reactions are as follows: Human GAPDH, Forward: 5′- TCCTGCACCACCAACTGCTTA 3′, Reverse: 5′- AGT GATGGCATGGACTGTGGT −3′; Human MCL1, Forward: 5′-TGCTTCGGAAACTGGACATCA-3′, Reverse: 5′-TAGCCACAAAGGCACCAAAAG-3′.
Immunohistochemistry
Two serial sections of a tissue microarray containing 49 cases of breast ductal carcinomas (BR10010e, US Biomax, Inc., MD, USA) were examined for expression of p300 and MCL1 by immunohistochemical staining using the avidin-biotin-peroxidase complex (ABC) method. Paraffin sections were deparaffinized and dehydrated through a graded series of ethanol. Antigen retrieval was performed by microwaving at 98°C for 20 min in EDTA-Tris (pH 8.0) for p300 and 0.01 M citrate buffer (pH 6.0) for MCL1, respectively. Endogenous peroxidase activity was blocked with 3% H2O2 in distilled water for 5 min. After blocking non-specific binding with goat serum at 37°C for 30 min, a section was incubated with mouse monoclonal anti-p300 antibody diluted 1:150 (abcam54984, Cambridge, MA, USA) or rabbit polyclonal anti-MCL1 antibody diluted 1:100 (Proteintech) overnight at 4°C. Immunoreactivity was detected using VETASTAIN Elite ABC Kits for rabbit (Rabbit IgG, PK-6101, Vector Laboratories, Burlingame, USA) and mouse (Mouse IgG, PK-6102, Vector Laboratories, Burlingame, USA) and diaminobenzidine. p300 and MCL1 immunoreactivity was scored based on intensity and extent (percentage of positive cancer cells). The intensity was scored on a scale of 0 to 2 (0: no detectable; 1: weak, 2: intense). As all specimens showed less than 5% or more than 50% positive staining of p300 or MCL1 in cancer cells, the extent was scored on a scale of 0 to 1 (0: less than 5%, 1: more than 50%). Specimens final scores were derived from the multiplication of extent by intensity for a score of 0, 1 or 2. Specimen was defined as p300low, p300moderate (p300mod) and p300high when the final score was 0, 1, and 2, respectively. Likewise, the specimen was defined as MCL1low, MCL1moderate (MCL1mod) and MCL1high when the final score was 0, 1, and 2, respectively. Scoring was performed by two pathologists (M. Gi. and S. Suzuki) separately. Discrepant cases were studied together by the two pathologists, and a consensus was reached.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantitative data were presented as mean ± SD or SEM as indicated of at least three independent experiments or biological replicates. Statistical significances were analyzed by Student’s t test for two groups and one-way ANOVA with multiple comparison tests for three or more groups. p < 0.05 was considered as statistically significant. Correlation analysis between MCL1 and p300 expression was evaluated by Spearman correlation test. All the statistical analyses were conducted by Prism 8 (GraphPad Software, Inc., San Diego, CA, USA). Quantification of western blot data was conducted by ImageJ (NIH). All of the statistical details of experiments can be found in the figure legends.
Supplementary Material
Highlights.
p300 acetylates MCL1 at K40, which is counteracted by the deacetylase SIRT3
K40 acetylation recruits USP9X, resulting in MCL1 deubiquitination and stabilization
Acetylation-mimetic MCL1 promotes evasion of apoptosis and facilitates tumorigenesis
Elevated MCL1 acetylation status sensitizes cancer cells to the USP9X inhibitor WP1130
ACKNOWLEDGMENTS
This study was supported by JSPS grants-in-aid for scientific research (18K19282, 20K16146, and CoBiA 16H06277), NIH grants (R35CA197459 and P01 CA120964 to J.M.A. and CA177910 and R35CA253027 to W.W.), an ACS grant, and the Takeda Science Foundation. We thank Dr. Min Yuan for mass spectrometry analysis and Dr. Takeshi Urano, Dr. Yohei Miyagi, and Dr. Yoshiyasu Nakamura for helpful discussions.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109988.
DECLARATION OF INTERESTS
W.W. is a co-founder and consultant for ReKindle Therapeutics.
REFERENCES
- Arai S, Jonas O, Whitman MA, Corey E, Balk SP, and Chen S (2018). Tyrosine Kinase Inhibitors Increase MCL1 Degradation and in Combination with BCLXL/BCL2 Inhibitors Drive Prostate Cancer Apoptosis. Clin. Cancer Res 24, 5458–5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askew EB, Bai S, Blackwelder AJ, and Wilson EM (2010). Transcriptional synergy between melanoma antigen gene protein-A11 (MAGE-11) and p300 in androgen receptor signaling. J. Biol. Chem 285, 21824–21836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attar N, and Kurdistani SK (2017). Exploitation of EP300 and CREBBP Lysine Acetyltransferases by Cancer. Cold Spring Harb. Perspect. Med 7, a026534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barneda-Zahonero B, and Parra M (2012). Histone deacetylases and cancer. Mol. Oncol 6, 579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. (2010). The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers EM, Yan G, Mukherjee C, Orry A, Wang L, Holbert MA, Crump NT, Hazzalin CA, Liszczak G, Yuan H, et al. (2010). Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem. Biol 17, 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunelle JK, and Letai A (2009). Control of mitochondrial apoptosis by the Bcl-2 family. J. Cell Sci 122, 437–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchwald M, Krämer OH, and Heinzel T (2009). HDACi–targets beyond chromatin. Cancer Lett. 280, 160–167. [DOI] [PubMed] [Google Scholar]
- Campbell KJ, Dhayade S, Ferrari N, Sims AH, Johnson E, Mason SM, Dickson A, Ryan KM, Kalna G, Edwards J, et al. (2018). MCL-1 is a prognostic indicator and drug target in breast cancer. Cell Death Dis. 9, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron C, Boyault C, and Khochbin S (2005). Regulatory cross-talk between lysine acetylation and ubiquitination: role in the control of protein stability. BioEssays 27, 408–415. [DOI] [PubMed] [Google Scholar]
- Carroll RG, Hollville E, and Martin SJ (2014). Parkin sensitizes toward apoptosis induced by mitochondrial depolarization through promoting degradation of Mcl-1. Cell Rep. 9, 1538–1553. [DOI] [PubMed] [Google Scholar]
- Cheng J, Guo J, North BJ, Wang B, Cui CP, Li H, Tao K, Zhang L, and Wei W (2019). Functional analysis of deubiquitylating enzymes in tumorigenesis and development. Biochim. Biophys. Acta Rev. Cancer 1872, 188312. [DOI] [PubMed] [Google Scholar]
- Cory S, Roberts AW, Colman PM, and Adams JM (2016). Targeting BCL-2-like Proteins to Kill Cancer Cells. Trends Cancer 2, 443–460. [DOI] [PubMed] [Google Scholar]
- Cuconati A, Mukherjee C, Perez D, and White E (2003). DNA damage response and MCL-1 destruction initiate apoptosis in adenovirus-infected cells. Genes Dev. 17, 2922–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, and Placzek WJ (2018). Post-Transcriptional Regulation of Anti-Apoptotic BCL2 Family Members. Int. J. Mol. Sci 19, E308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash R, Azab B, Quinn BA, Shen X, Wang XY, Das SK, Rahmani M, Wei J, Hedvat M, Dent P, et al. (2011). Apogossypol derivative BI-97C1 (Sabutoclax) targeting Mcl-1 sensitizes prostate cancer cells to mda-7/IL-24-mediated toxicity. Proc. Natl. Acad. Sci. USA 108, 8785–8790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, He X, Hsu JM, Xia W, Chen CT, Li LY, Lee DF, Liu JC, Zhong Q, Wang X, and Hung MC (2007). Degradation of Mcl-1 by beta-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol. Cell. Biol 27, 4006–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertel F, Nguyen M, Roulston A, and Shore GC (2013). Programming cancer cells for high expression levels of Mcl1. EMBO Rep. 14, 328–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Inuzuka H, Tan MK, Fukushima H, Locasale JW, Liu P, Wan L, Zhai B, Chin YR, Shaik S, et al. (2011). mTOR drives its own activation via SCF(βTrCP)-dependent degradation of the mTOR inhibitor DEPTOR. Mol. Cell 44, 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Nihira NT, Bu X, Chu C, Zhang J, Kolodziejczyk A, Fan Y, Chan NT, Ma L, Liu J, et al. (2020). Acetylation-dependent regulation of PD-L1 nuclear translocation dictates the efficacy of anti-PD-1 immunotherapy. Nat. Cell Biol 22, 1064–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser SP, Lee EF, Trounson E, Bouillet P, Wei A, Fairlie WD, Izon DJ, Zuber J, Rappaport AR, Herold MJ, et al. (2012). Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev. 26, 120–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grönroos E, Hellman U, Heldin CH, and Ericsson J (2002). Control of Smad7 stability by competition between acetylation and ubiquitination. Mol. Cell 10, 483–493. [DOI] [PubMed] [Google Scholar]
- Harley ME, Allan LA, Sanderson HS, and Clarke PR (2010). Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. EMBO J. 29, 2407–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, Zhai B, Wan L, Gutierrez A, Lau AW, et al. (2011). SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature 471, 104–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inuzuka H, Gao D, Finley LW, Yang W, Wan L, Fukushima H, Chin YR, Zhai B, Shaik S, Lau AW, et al. (2012). Acetylation-dependent regulation of Skp2 function. Cell 150, 179–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapuria V, Peterson LF, Fang D, Bornmann WG, Talpaz M, and Donato NJ (2010). Deubiquitinase inhibition by small-molecule WP1130 triggers aggresome formation and tumor cell apoptosis. Cancer Res. 70, 9265–9276. [DOI] [PubMed] [Google Scholar]
- Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, Deng X, Zhai D, Shi YX, Sneed T, et al. (2006). Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 10, 375–388. [DOI] [PubMed] [Google Scholar]
- Kozopas KM, Yang T, Buchan HL, Zhou P, and Craig RW (1993). MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc. Natl. Acad. Sci. USA 90, 3516–3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasko LM, Jakob CG, Edalji RP, Qiu W, Montgomery D, Digiammarino EL, Hansen TM, Risi RM, Frey R, Manaves V, et al. (2017). Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 550, 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Luo J, Brooks CL, and Gu W (2002). Acetylation of p53 inhibits its ubiquitination by Mdm2. J. Biol. Chem 277, 50607–50611. [DOI] [PubMed] [Google Scholar]
- Li H, Wittwer T, Weber A, Schneider H, Moreno R, Maine GN, Kracht M, Schmitz ML, and Burstein E (2012). Regulation of NF-κB activity by competition between RelA acetylation and ubiquitination. Oncogene 31, 611–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Wang Y, Gao T, Pan Z, Cheng H, Yang Q, Cheng Z, Guo A, Ren J, and Xue Y (2014). CPLM: a database of protein lysine modifications. Nucleic Acids Res. 42, D531–D536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magiera MM, Mora S, Mojsa B, Robbins I, Lassot I, and Desagher S (2013). Trim17-mediated ubiquitination and degradation of Mcl-1 initiate apoptosis in neurons. Cell Death Differ. 20, 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino D, Kelly GL, Lessene G, Wei AH, Roberts AW, and Strasser A (2018). BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines. Cancer Cell 34, 879–891. [DOI] [PubMed] [Google Scholar]
- Michishita E, Park JY, Burneskis JM, Barrett JC, and Horikawa I (2005). Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell 16, 4623–4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills JR, Hippo Y, Robert F, Chen SM, Malina A, Lin CJ, Trojahn U, Wendel HG, Charest A, Bronson RT, et al. (2008). mTORC1 promotes survival through translational control of Mcl-1. Proc. Natl. Acad. Sci. USA 105, 10853–10858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojsa B, Lassot I, and Desagher S (2014). Mcl-1 ubiquitination: unique regulation of an essential survival protein. Cells 3, 418–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson-Rees WA, Daniels DW, and Flandermeyer RR (1981). Cross-contamination of cells in culture. Science 212, 446–452. [DOI] [PubMed] [Google Scholar]
- Nihira NT, Ogura K, Shimizu K, North BJ, Zhang J, Gao D, Inuzuka H, and Wei W (2017). Acetylation-dependent regulation of MDM2 E3 ligase activity dictates its oncogenic function. Sci. Signal 10, eaai8026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijhawan D, Fang M, Traer E, Zhong Q, Gao W, Du F, and Wang X (2003). Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 17, 1475–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North BJ, Marshall BL, Borra MT, Denu JM, and Verdin E (2003). The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell 11, 437–444. [DOI] [PubMed] [Google Scholar]
- Schwickart M, Huang X, Lill JR, Liu J, Ferrando R, French DM, Maecker H, O’Rourke K, Bazan F, Eastham-Anderson J, et al. (2010). Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 463, 103–107. [DOI] [PubMed] [Google Scholar]
- Senichkin VV, Streletskaia AY, Gorbunova AS, Zhivotovsky B, and Kopeina GS (2020). Saga of Mcl-1: regulation from transcription to degradation. Cell Death Differ. 27, 405–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieghart W, Losert D, Strommer S, Cejka D, Schmid K, Rasoul-Rockenschaub S, Bodingbauer M, Crevenna R, Monia BP, Peck-Radosavljevic M, and Wacheck V (2006). Mcl-1 overexpression in hepatocellular carcinoma: a potential target for antisense therapy. J. Hepatol 44, 151–157. [DOI] [PubMed] [Google Scholar]
- Singh R, Letai A, and Sarosiek K (2019). Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol 20, 175–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slomp A, Moesbergen LM, Eldering E, Kersten MJ, Minnema MC, and Peperzak V (2021). Phosphatase PP2A enhances MCL-1 protein half-life in multiple myeloma cells. Cell Death Dis. 12, 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowa ME, Bennett EJ, Gygi SP, and Harper JW (2009). Defining the human deubiquitinating enzyme interaction landscape. Cell 138, 389–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spange S, Wagner T, Heinzel T, and Krämer OH (2009). Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int. J. Biochem. Cell Biol 41, 185–198. [DOI] [PubMed] [Google Scholar]
- Tansey WP (2006). Detection of ubiquitylated proteins in mammalian cells. CSH Protoc 2006, pdb.prot4616. [DOI] [PubMed] [Google Scholar]
- Thomas LW, Lam C, and Edwards SW (2010). Mcl-1; the molecular regulation of protein function. FEBS Lett. 584, 2981–2989. [DOI] [PubMed] [Google Scholar]
- Wang F, Marshall CB, and Ikura M (2013). Transcriptional/epigenetic regulator CBP/p300 in tumorigenesis: structural and functional versatility in target recognition. Cell. Mol. Life Sci 70, 3989–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Li S, Gilbert J, Gritton HJ, Wang Z, Li Z, Han X, Selkoe DJ, and Man HY (2017). Crucial Roles for SIRT2 and AMPA Receptor Acetylation in Synaptic Plasticity and Memory. Cell Rep. 20, 1335–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J, et al. (2011). Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 471, 110–114. [DOI] [PubMed] [Google Scholar]
- Wu X, Luo Q, Zhao P, Chang W, Wang Y, Shu T, Ding F, Li B, and Liu Z (2019). MGMT-activated DUB3 stabilizes MCL1 and drives chemoresistance in ovarian cancer. Proc. Natl. Acad. Sci. USA 116, 2961–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Luo Q, Zhao P, Chang W, Wang Y, Shu T, Ding F, Li B, and Liu Z (2020). JOSD1 inhibits mitochondrial apoptotic signalling to drive acquired chemoresistance in gynaecological cancer by stabilizing MCL1. Cell Death Differ. 27, 55–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuillème-Toumi S, Robillard N, Gomez P, Moreau P, Le Gouill S, Avet-Loiseau H, Harousseau JL, Amiot M, and Bataille R (2005). Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia 19, 1248–1252. [DOI] [PubMed] [Google Scholar]
- Xiang Z, Luo H, Payton JE, Cain J, Ley TJ, Opferman JT, and Tomasson MH (2010). Mcl1 haploinsufficiency protects mice from Myc-induced acute myeloid leukemia. J. Clin. Invest 120, 2109–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Nimmer P, Sheppard GS, Bruncko M, Hessler P, Lu X, Roberts-Rapp L, Pappano WN, Elmore SW, Souers AJ, et al. (2015). MCL-1 Is a Key Determinant of Breast Cancer Cell Survival: Validation of MCL-1 Dependency Utilizing a Highly Selective Small Molecule Inhibitor. Mol. Cancer Ther 14, 1837–1847. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Wang M, Zhao J, Han Y, and Jia L (2016). Sirtuin 3: A Janus face in cancer. Int. J. Oncol 49, 2227–2235. [DOI] [PubMed] [Google Scholar]
- Young AI, Law AM, Castillo L, Chong S, Cullen HD, Koehler M, Herzog S, Brummer T, Lee EF, Fairlie WD, et al. (2016). MCL-1 inhibition provides a new way to suppress breast cancer metastasis and increase sensitivity to dasatinib. Breast Cancer Res. 18, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Zhang M, Jing Y, Yin X, Ma P, Zhang Z, Wang X, Di W, and Zhuang G (2018). Deubiquitinase USP13 dictates MCL1 stability and sensitivity to BH3 mimetic inhibitors. Nat. Commun 9, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Q, Gao W, Du F, and Wang X (2005). Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 121, 1085–1095. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Mass spectrometry data have been deposited at MassIVE repository and are publicly available as of the date of publication. Accession number is listed in the key resources table. Original western blot images have been deposited at Mendeley Data and are publicly available as of the date of publication. The DOI is listed in the key resources table. Other data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-Mcl-1 (D2W9E) | Cell Signaling Technology | Cat# 94296, RRID: AB_2722740 |
| Rabbit monoclonal anti-Phospho-Mcl-1 (Ser64) | Cell Signaling Technology | Cat# 13297, RRID: AB_2798173 |
| Rabbit polyclonal anti-Phospho-Mcl-1 (Ser159/Thr163) | Cell Signaling Technology | Cat# 4579, RRID: AB_2144100 |
| Rabbit monoclonal anti-Phospho-Mcl-1 (Thr163) (D5M9D) | Cell Signaling Technology | Cat# 14765, RRID: AB_2716686 |
| Rabbit monoclonal anti-Bcl-2 (D55G8) | Cell Signaling Technology | Cat# 4223, RRID: AB_1903909 |
| Rabbit monoclonal anti-Bcl-xL (54H6) | Cell Signaling Technology | Cat# 2764, RRID: AB_2228008 |
| Rabbit polyclonal anti-Acetylated-Lysine | Cell Signaling Technology | Cat# 9441, RRID: AB_331805 |
| Rabbit monoclonal anti-p300 (D8Z4E) | Cell Signaling Technology | Cat# 86377, RRID: AB_2800077 |
| Rabbit monoclonal anti-CBP (D6C5) | Cell Signaling Technology | Cat# 7389, RRID: AB_2616020 |
| Rabbit monoclonal anti-PCAF (C14G9) | Cell Signaling Technology | Cat# 3378, RRID: AB_2128409 |
| Rabbit monoclonal anti-GCN5L2 (C26A10) | Cell Signaling Technology | Cat# 3305, RRID: AB_2128281 |
| Rabbit monoclonal anti-USP9X (D4Y7W) | Cell Signaling Technology | Cat# 14898, RRID: AB_2798640 |
| Rabbit polyclonal anti-SirT1 (1F3) | Cell Signaling Technology | Cat# 8469, RRID: AB_10999470 |
| Rabbit polyclonal anti-SirT2 (D4O5O) | Cell Signaling Technology | Cat# 12650, RRID: AB_2716762 |
| Rabbit monoclonal anti-SirT3 (D22A3) | Cell Signaling Technology | Cat# 5490, RRID: AB_10828246 |
| Rabbit monoclonal anti-Cleaved PARP (Asp214) (D64E10) | Cell Signaling Technology | Cat# 5625, RRID: AB_10699459 |
| Rabbit polyclonal anti-Cleaved Caspase-3 (Asp175) | Cell Signaling Technology | Cat# 9661, RRID: AB_2341188 |
| Rabbit monoclonal anti-COX IV (3E11) | Cell Signaling Technology | Cat# 4850, RRID: AB_2085424 |
| Mouse monoclonal anti-Lamin A/C (4C11) | Cell Signaling Technology | Cat# 4777, RRID: AB_10545756 |
| Mouse monoclonal anti-GFP (4B10) | Cell Signaling Technology | Cat# 2955, RRID: AB_1196614 |
| Rabbit monoclonal anti-Myc-Tag (71D10) | Cell Signaling Technology | Cat# 2278, RRID: AB_490778 |
| Mouse monoclonal anti-Myc-Tag (9B11) | Cell Signaling Technology | Cat# 94296; RRID: AB_2722740 |
| Mouse anti-Myc-Tag immobilized (bead conjugate) | Cell Signaling Technology | Cat# 3400, RRID: AB_10692357 |
| Mouse monoclonal anti-Mcl-1 (B-6) AC | Santa Cruz Biotechnology | Cat# sc-74436 AC, RRID: AB_1126069 |
| Mouse monoclonal anti-USP13 (B-9) | Santa Cruz Biotechnology | Cat# sc-514416 |
| Rabbit polyclonal anti-Vinculin (H-300) | Santa Cruz Biotechnology | Cat# sc-5573, RRID: AB_2214507 |
| Goat polyclonal anti-Lamin B1 (S-20) | Santa Cruz Biotechnology | Cat# sc-30264, RRID: AB_2136305 |
| Rabbit polyclonal anti-HA-probe (Y-11) | Santa Cruz Biotechnology | Cat# sc-805 |
| Mouse monoclonal purified anti-HA.11 Epitope Tag |
BioLegend | Cat# 901503, RRID: AB_2565005 |
| Anti c-Myc antibody beads (10D11) | FUJIFILM Wako | Cat# 016-26503 |
| Mouse monoclonal anti-V5 Tag | Thermo Fisher Scientific | Cat# R960-25, RRID: AB_2556564 |
| Mouse monoclonal anti-DUB3 | Sigma-Aldrich | Cat# WH0377630M1, RRID: AB_1841372 |
| Mouse monoclonal anti-alpha-Tubulin | Sigma-Aldrich | Cat# T5168, RRID: AB_477579 |
| Rabbit polyclonal anti-Flag | Sigma-Aldrich | Cat# F7425, RRID: AB_439687 |
| Mouse monoclonal anti-Flag M2 | Sigma-Aldrich | Cat# F3165, RRID: AB_259529 |
| Anti-Flag M2 Affinity Gel | Sigma-Aldrich | Cat# A2220, RRID: AB_10063035 |
| Mouse monoclonal anti-HA-Agarose antibody | Sigma-Aldrich | Cat# A2095, RRID: AB_257974 |
| Goat polyclonal anti-Mouse IgG (whole molecule), HRP conjugated | Sigma-Aldrich | Cat# A4416, RRID: AB_258167 |
| Goat anti-Rabbit IgG (whole molecule)-Peroxidase antibody | Sigma-Aldrich | Cat# A4914, RRID: AB_258207 |
| Mouse monoclonal anti-acetylated-K40-MCL1 | This paper | N/A |
| Bacterial and virus strains | ||
| XL10-Gold Ultracompetent Cells | Agilent Technologies | Cat# 200315 |
| Chemicals, peptides, and recombinant proteins | ||
| MG132 | Enzo Life Sciences | Cat# BML-PI102-0025 |
| A-485 | MedChemExpress | Cat# HY-107455 |
| C646 | MedChemExpress | Cat# HY-13823 |
| WP1130 | MedChemExpress | Cat# HY-13264 |
| Cycloheximide | Sigma-Aldrich | Cat# C1988 |
| Trichostatin A | Sigma-Aldrich | Cat# T8552 |
| Nicotinamide | Sigma-Aldrich | Cat# 72340 |
| Critical commercial assays | ||
| QuikChange II XL Site-Directed Mutagenesis Kit |
Agilent Technologies | Cat# 200516 |
| NE-PER Nuclear and Cytoplasmic Extraction Reagents |
Thermo Fisher Scientific | Cat# 78833 |
| Cell Fractionation Kit | Abcam | Cat# ab109719 |
| CellTiter-Glo | Promega | Cat# G7571 |
| Deposited data | ||
| Mass spectrometry data | This paper | MassIVE: MSV000088231 |
| Original western blot images | This paper | Mendeley Data: https://doi.org/10.17632/pyfpzd2tv5.1 |
| Experimental models: Cell lines | ||
| HeLa | ATCC | Cat# CCL-2.2, RRID:CVCL_0058 |
| HEK293T | ATCC | Cat# CRL-3216, RRID:CVCL_0063 |
| MCF7 | Dr. Piotr Sicinski lab, DFCI | N/A |
| SKBR3 | Dr. Piotr Sicinski lab, DFCI | N/A |
| MDA-MB-231 | Dr. Alex Toker lab, BIDMC | N/A |
| BT-20 | ATCC | Cat# CRL-7912, RRID:CVCL_0178 |
| C42 | Dr. Steven Balk lab, BIDMC | N/A |
| LNCaP-ABL (ABL) | Dr. Steven Balk lab, BIDMC | N/A |
| PC3 | Dr. Steven Balk lab, BIDMC | N/A |
| DU145 | Dr. Steven Balk lab, BIDMC | N/A |
| Experimental models: Organisms/strains | ||
| Nude Mouse | Charles River | BALB/c-nu/nu |
| Oligonucleotides | ||
| qRT-PCR Primer: Human MCL1 Forward: TGCTTCGGAAACTGGACATCA | This paper | N/A |
| qRT-PCR Primer: Human MCL1 Reverse: TAGCCACAAAGGCACCAAAAG | This paper | N/A |
| qRT-PCR Primer: Human GAPDH Forward: TCCTGCACCACCAACTGCTTA | This paper | N/A |
| qRT-PCR Primer: Human GAPDH Reverse: AGTGATGGCATGGACTGTGGT | This paper | N/A |
| Recombinant DNA | ||
| Myc-MCL1 | Ding et al., 2007 | N/A |
| Myc-MCL1 K5R | Inuzuka et al., 2011 | N/A |
| Myc-MCL1 K40R | Inuzuka et al., 2011 | N/A |
| Myc-MCL1 K40Q | This paper | N/A |
| Myc-MCL1 K136R | Inuzuka et al., 2011 | N/A |
| Myc-MCL1 K194R/197R | Inuzuka et al., 2011 | N/A |
| Myc-MCL1 K40Q/S159E/T163E | This paper | N/A |
| Myc-MCL1 K40R/S159A/T163A | This paper | N/A |
| pLenti-HA-MCL1 | Inuzuka et al., 2011 | N/A |
| pLenti-HA-MCL1 K40R | This paper | N/A |
| pLenti-HA-MCL1 K40Q | This paper | N/A |
| HA-MCL1 | Inuzuka et al., 2011 | N/A |
| HA-p300 | Askew et al., 2010 | Addgene Cat# 89094 |
| Flag-SIRT1 | North et al., 2003 | Addgene Cat# 13812 |
| Flag-SIRT2 | North et al., 2003 | Addgene Cat# 13813 |
| Flag-SIRT3 | North et al., 2003 | Addgene Cat# 13814 |
| Flag-SIRT4 | North et al., 2003 | Addgene Cat# 13815 |
| Flag-SIRT5 | North et al., 2003 | Addgene Cat# 13816 |
| Flag-SIRT6 | North et al., 2003 | Addgene Cat# 13817 |
| Flag-SIRT7 | North et al., 2003 | Addgene Cat# 13818 |
| pLKO-shMCL1 | TRC | N/A |
| pLKO-shp300 | TRC | N/A |
| pLKO-shUSP13 | TRC | N/A |
| HA-GCN5 | This paper | N/A |
| HA-Tip60α | This paper | N/A |
| HA-PCAF | This paper | N/A |
| HA-–-TRCP1 | This paper | N/A |
| Flag-His-USP9X | This paper | N/A |
| pLKO-shUSP9X | A gift from Dr. Qing Zhang, UNC | N/A |
| pLKO-shSIRT1 | TRC | N/A |
| pLKO-shSIRT2 | TRC | N/A |
| pLKO-shSIRT3 | TRC | N/A |
| HA-FBW7 | A gift from Dr. Keiichi Nakayama, Kyushu University, Japan | N/A |
| V5-TRIM17 | A gift from Dr. Tatsuya Sawasaki, Ehime University, Japan | N/A |
| Flag-HA-DUB3 | Sowa et al., 2009 | Addgene Cat# 22593 |
| Myc-Flag-USP13 | A gift from Dr. Lingqiang Zhang, Beijing Institute of Lifeomics, China | N/A |
| Human MCL1 CRISPR/Cas9 KO plasmids | Santa Cruz Biotechnology | Cat# sc-400079 and sc-400079-HDR |
| Software and algorithms | ||
| GraphPad Prism | GraphPad | https://www.graphpad.com |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |