

Abstract
Cells have the ability to adapt to stressful environments as a part of their evolution. Physical exercise induces an increase of a demand for energy that must be met by mitochondria as the main (ATP) provider. However, this process leads to the increase of free radicals and the so-called reactive oxygen species (ROS), which are necessary for the maintenance of cell signaling and homeostasis. In addition, mitochondrial biogenesis is influenced by exercise in continuous crosstalk between the mitochondria and the nuclear genome. Excessive workloads may induce severe mitochondrial stress, resulting in oxidative damage. In this regard, the objective of this work was to provide a general overview of the molecular mechanisms involved in mitochondrial adaptation during exercise and to understand if some nutrients such as antioxidants may be implicated in blunt adaptation and/or an impact on the performance of exercise by different means.
Keywords: oxidative stress, ROS/RNS, exercise, mitochondrial adaptation, oxidative damage, antioxidants, exercise performance
1. Introduction
The high-oxygen atmospheric concentrations of a million years ago induced metabolic changes, resulting in the majority of living organisms utilizing more oxygen (O2), thus promoting the development of aerobic systems. The basis of aerobic metabolism is supported by the biotransformation of molecular O2 to meet energy demands, homeostasis and to ensure the support of life. In turn, highly reactive molecules result from these cellular processes, such as the reactive oxygen species (ROS), which are considered radical and non-radical derivatives of O2. ROS are formed from one-electron transfers from a redox donor to molecular O2. Further, the term reactive nitrogen species (RNS) refers to reactive molecules derived from nitrogen. Free radicals (FR) are considered an atom/molecule that contains one or more unpaired electrons, which renders them unstable and very reactive [1].
The anion superoxide (O2•−) is the main ROS formed in cells by the one-electron reduction of molecular O2; it is negatively charged and impermeable to the cell membrane as compared with other FR and has a relatively long half-life. The dismutation of O2•− leads to the formation of hydrogen peroxide (H2O2), a permeable non-FR cell membrane, relatively stable, weak in oxidation, and possessing the ability to diffuse within and outside of the cell [2]. Because H2O2 has a long half-life, it can reach high cellular concentrations, inducing cytotoxicity and cell signaling. H2O2, together with O2•− are transformed into hydroxyl radicals (•OH) that are highly reactive molecules with powerful oxidizing potential, but that are not membrane-permeable; thus, they directly react with molecules in close proximity to their production site, causing structural cell damage [1,2].
In addition, among RNS, the most important free radical is oxide nitric (NO), which is synthesized from the amino acid L-arginine by nitric oxide synthase (NOS). NO can also react with O2•− to form peroxynitrite (ONOO−), which is the main cause of the nitration of cellular proteins and the depletion of thiol groups due to its rapid formation. All of these previously mentioned reactive molecules might contribute to oxidative stress (OS), defined as “an imbalance between oxidants and antioxidants in favor of the oxidants, leading to a disruption of redox signaling and control, and/or molecular damage” [3]. Fortunately, cellular systems have developed potent cytoprotector systems as a defense against damage caused by the high levels of ROS and RNS. For instance, endogenous antioxidant systems (EnAS) are activated through the nuclear factor erythroid 2-related factor 2 (Nrf2), a redox-sensitive transcriptional factor that coordinates the antioxidant response and that is also related to mitochondrial biogenesis [4].
Despite the imbalance that ROS and RNS contribute to oxidative stress, it is known that certain levels of these molecules are needed to trigger cell signaling, adaptation, and homeostasis. During exercise, physical work increases energy demands, the O2 uptake, and consequently the formation of ROS [5]. In this manner, mitochondria are susceptible to being attacked in high intense or long sports, which might represent a risk for the detriment in muscle force, contraction, and thus performance. Therefore, the purpose of this review was to provide a general overview of mitochondrial function and the molecular mechanisms implicated in adaptation to stress induced by exercise. Moreover, nutritional insights in terms of the growing evidence with regard to the use of antioxidants and mitochondrial protection are discussed.
2. Oxidative Stress in Exercise
During exertion, muscles are the main source of ROS production within the active muscle fibers from different sites such as the mitochondria, T-tubules, sarcoplasmic reticulum, and sarcolemma. Among these, mitochondria are thought to be the major ROS producer, predominantly in endurance training. Once exercise starts, energy demands rise 10–20 fold in oxygen consumption above that of the resting state [2]. Thus, the augmented oxidative phosphorylation increases O2•− in contracting muscle fibers. However, some recent evidence indicates that mitochondria may not be fully responsible for the abundance of ROS because it is possible to produce more O2•− during basal state-4 respiration compared with active stage-3 respiration [6].
There are other ROS producers, which include nicotinamide, adenine dinucleotide phosphate oxidase (NADPH oxidase), phospholipase A2, (PLA2), xanthine oxidase monoamine oxidases (MAO’s), some dehydrogenases, and some immune-system cells, such as macrophages, eosinophils, monocytes, and neutrophils [2]. For instance, the NADPH oxidase isoform NOX2, located within T-tubes and sarcolemma, is the primary ROS producer in contracting muscle fibers, whereas the other isoform, NOX4, localized in mitochondria and the sarcoplasmic reticulum, contributes to ROS synthesis under baseline conditions [7]. At the same time, PLA2 can activate NADPH oxidases, the calcium-dependent isoform promotes the release of ROS during muscle contraction [8], and the independent-isoform manages oxidation under resting conditions [9]. PLA2 is involved in ROS generation during muscle contraction when the calcium uptake in mitochondria is increased; then, PLA2 is activated within the mitochondria, promoting the interaction of arachidonic acid synthesis with the electron transport chain (ETC). The formation of arachidonic acid elicits an eicosanoid substrate that could also modulate ROS production. In fact, the arachidonic acid released by PLA2 is a substrate of lipoxygenases, a ROS-generating enzyme system deriving from membrane phospholipids [9].
2.1. Impact of Oxidative Stress on Muscle Contraction
The impairment of modulating exercise-induced OS is implicated in overtraining syndrome (OTS), which is characterized by extended periods of fatigue-induced from intense training/competitions and inappropriate recovery post-exercise/competition episodes. In consequence, excessive ROS and OTS are closely involved in performance weakening [10]. Skeletal-muscle fatigue is considered as a decline of maximal force production in response to contractile activity [11]. The production of skeletal-muscle force depends on the contractile mechanism and metabolic factors, including lactate, inorganic phosphate (Pi), hydrogen (H+) ions, ROS, and heat shock protein (HSP), among others, that can enhance muscle fatigue [11].
Some evidence supports the hypothesis that increased fatigue and force depression are the result of the oxidizing state of muscle fibers due to continued exposure to ROS, basically to H2O2. Contrariwise, it has been identified that unfatigued muscle remains predominantly reduced. Also, the sustained intramuscular imbalance of the redox state enhanced chronic inflammation, exerting an impact upon force production and athletic performance [12]. Indeed, the endogenous productions of ROS/RNS play a crucial role in submaximal force, due to that the continuous state of reduced maximal force is induced by Ca2+ myofibrillar sensitivity and/or the decrease of reticulum sarcoplasmic Ca2+ release; these processes are ROS/RNS-dependent. Meanwhile, in rested myofibers, the acute and mild exposure of ROS/RNS raises submaximal force, but extended exposure impacts negatively [13]. Other ROS/RNS-mediated mechanisms associated with skeletal-muscle fatigue include a decrease in cellular-membrane excitability, impairment of the interaction between voltage sensors and Ca2+ ion channels, due to the excitation/contraction-coupling failure and the blocking of the action potential. Furthermore, Ca2+ release interferes in actin-myosin cross-bridge formation and in the reduction of myofibrillar sensitivity [14]. Additionally, there is evidence that ROS also exert a modulatory effect on Na+/K+ pump activity, but that excessive ROS production could impair Na+/K+ pump activity and more proteins implied in excitation-contraction-coupling, affecting muscle force during prolonged endurance exercise [6].
The underlying beneficial or deleterious effects of ROS/RNS on muscle contraction are demonstrated in several studies. Different factors, such as location, time of exposure, type of ROS/RNS, and the combination of endogenous/exogenous antioxidant systems, will determine effective skeletal-muscle function.
2.2. Mitochondrial ROS Production and Antioxidant Defense
Under normal conditions, electron flow along the ETC to complex IV results in their final accumulation in molecular oxygen to produce H2O. Through exercise, predominantly in prolonged endurance activities, ATP production requires reduced equivalents of NADH and FADH2, free ADP, Pi, and O2, and the presence of these elements activates the ETC in mitochondria [15]. Along the ATP synthesis process, mitochondrial ROS (mtROS) is produced for the reaction of electrons with O2 at different sites to form superoxide/hydrogen peroxide fundamentally in complex I and in complex III of ETC, which can leak 1–3% of electron flux as O2•− in state 4 or in the resting state [16,17]. Some evidence indicates that complex II and other mitochondrial enzymes also participate in mtROS formation [18]. It has been found that around 11 different mitochondrial sites leak electrons to O2 to produce the superoxide or hydroperoxide associated with oxidative phosphorylation (OxPho) and substrate oxidation. These sites are complexes I and III, oxoacid dehydrogenase complexes, and complex II, which use ubiquinone as an acceptor. Mitochondrial glycerol 3-phosphate dehydrogenase and complex III produce O2•− to the matrix and the external side of the mitochondrial inner membrane, while the other complexes generate O2•− and H2O2 only in the matrix [19]. Approximately 0.1–0.2% of the O2 consumed by mitochondria is transformed into O2•−. The release of O2•− from mitochondria is in the form of H2O2 or OONO− when superoxide anions react with •NO; this is because the high proton environment in the internal mitochondrial membrane makes it difficult for O2•− to cross the latter. Mitochondria are being found to be major ROS producers; however, inactive contracting muscles, a better-coupled electron flow is observed, and there is minor production of superoxide anions in the ETC. It is noteworthy that unfatigued and healthy muscle fibers could produce less ROS [20,21].
With the purpose of modulating the redox signaling of ROS/RNS, there is a complex defense system constituted of internal and external elements known as the endogenous antioxidant system (EnAS). This system is constituted of mean of enzymatic and non-enzymatic constituents and the exogenous antioxidant system (ExAS), which derives from dietary or supplementary sources such as antioxidant vitamins (vitamins C, E, and A), oligoelements (selenium, copper, and zinc), nucleic acids, and phytochemicals, among others. The latter will be discussed further ahead in this review. The EnAS acts to maintain reactive species within the homeostatic range in order to enhance cell signaling and prevent oxidative damage. Nevertheless, the overproduction of ROS/RNS may surpass the antioxidant defense, generating OS and damage. The dismutation of O2•− is performed by a family of superoxide dismutases (SOD) to produce H2O2 and O2. There are three isoforms as follows: SOD1, located in the cytosol and mitochondrial intermembrane space; SOD2, found in the mitochondrial matrix, and SOD3, situated outside the cell. SOD1 requires copper and zinc as cofactors (Cu/ZnSOD), whereas SOD2 is manganese-dependent (MnSOD) [22]. SOD is highly active in type-I oxidative fibers compared with type-IIX fibers with less oxidative activity [23,24].
H2O2 and peroxides are removed by enzymatic and non-enzymatic elements of the EnAS, such the family of glutathione peroxidases (GPX) in the presence of reduced glutathione (GSH), glutathione reductase (GR), catalases (CAT), thioredoxins (Trx), peroxiredoxins (Prx), and glutaredoxins (Grx) [25]. There are eight isoforms of GPX in mammals (GPX1–GPX8) [26]; all of these reduce H2O2 to form H2O and alcohol, but the different isoforms are related to cell location. GPX1 is active in cytosol and mitochondria is selenium-dependent, GPX2 is exclusively located in cytosol, GPX3 is active in cytosol and the extracellular space, and GPX4 resides in the phospholipid environment to remove lipid peroxides [26]. GPX5 is expressed in mammalian epididymis and protects sperm from ROS and lipid peroxidation damage [27]. GPX6 has been found in epithelial cells [28]. GPX7 and GPX8 are similar in structure, but GPX7 works as a stress signal sensor and transmitter by shuttling disulfide bones to its interacting proteins, which are implicated in diverse signaling pathways to keep redox homeostasis [29]. GPX8 isoform is essentially located in the endoplasmic reticulum, is considered as a primary regulator of tumor aggressiveness in several types of cancer [30]. The reducing power of GPX is found in the use of GSH as electron donor. At the same time, this reaction produces glutathione disulfide (GSSH), the oxidated form of glutathione, which can be reduced by glutathione reductases (GR). In mammalian skeletal muscle and cardiac tissue, the resynthesis of GSH through GR needs NADPH produced by the NADPH-specific isocitrate dehydrogenase and by glyceraldehyde-3-phosphate dehydrogenase [25].
Catalase (CAT), which also participates in the removal of H2O2 to form H2O and O2, is located in different compartments in the cell, basically in cytosol, peroxisomes and mitochondria [31] and, similar to SOD, it is more active in type-I than in type-IIX oxidative fibers. In contrast to GPX, CAT does not require an electron donor, nor does it eliminate other hydroperoxides [2]. Thioredoxins (Trx) and thioredoxin reductases comprise a group of antioxidant enzymes that maintain proteins in a reduced state by the formation of a disulfide bond with substrate proteins and they are able to transfer two of the electrons that they possess to the target protein. Consequently, Trx is oxidated, but then it can be reduced by Trx reductase in the presence of NADPH as an electron donor. Trx is the main disulfide reductase within cells and there are two isoforms: Trx1, located in the cytosol, and the Trx2 site in mitochondria [32,33]. These isoforms also participate in the removal of hydroperoxides, cell signaling, and vitamin-C recycling [34]. Peroxiredoxins (Prx) are enzymes involved in the reduction of H2O2, alkyl peroxides, and peroxynitrite using electrons of Trx. Six isoforms have been identified: Prx1, 2, and 4 are situated in cytosol, Prx3 is located in mitochondria, Prx5 is found in both mitochondria and cytosol, and Prx6 is localized in the extracellular space. These isoforms are also thought to be involved in cell signaling and circadian-cycle regulation [35,36]. Finally, glutaredoxins (Grx) are small-thiodisulfide oxidoreductases that protect protein and non-protein thiols through the transfer of electrons to the disulfide substrates deriving from NADPH. Grx participates in hydroperoxide elimination, and there are three isoforms: Grx1 is within the cytosol and Grx2 and 3 is situated in the mitochondria [37].
3. Mitochondrial Function and Adaptation
3.1. Mitochondrial Adaptation to Exercise in the Presence of ROS
The practice of exercise is a powerful stimulus that leads to metabolic and physiological adaptations and that eventually allows the body to tolerate heavier workloads, inducing changes in muscle-fiber remodeling and plasticity, which are the result of a complex relationship or diverse regulator factors and signaling processes [38]. During the last two decades, numerous groups of researchers have elucidated a vast list of signals triggered by ROS within muscle contraction and their implications in molecular events related to physiological responses and subsequent cell adaptation. Currently, among the main adaptations mediated by ROS are found the following: mitochondrial biogenesis, increased antioxidant defenses, and myofiber hypertrophy. The role that ROS plays in the previously mentioned adaptations is based on the idea that they affect protein function, enzyme activity, and gene transcription as well they modify membrane and genome integrity [39].
With regard to mitochondrial adaptations, the protagonism must be mentioned of peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α), a transcriptional cofactor that interacts with other transcriptional factors and that owns the ability to induce transcription without binding directly with DNA. This binding to specific transcriptional factors promotes the genomic expression of genes associated with mitochondrial biogenesis, energy metabolism, thermogenesis, and the regulation of uncoupling [40]. PGC-1α coordinates different redox-sensitive programs; for instance, PGC-1α interacts with the estrogen-related receptor alpha (ERRα), Nrf1, and Nrf2 in mitochondrial biogenesis [41]. Thus, PGC-1α is a key element for enhancing adaptative responses in endurance activities [42]. The increase of ROS, particularly H2O2 during prolonged aerobic resistance sports, exacts the activation of PGC-1α and the alternative promoter PGC-1α2 in muscle fibers via the regulation of the upstream stimulatory factor 1 (USF-1), which also depends on beta adrenergic signaling, in turn, activates the angiogenic vascular endothelial growth factor (VEGF), also mediating the orphan nuclear receptor ERRα. Moreover, the adrenergic stimulation of exercise-induced PGC-1α/ERRα/VEGF promotes angiogenesis [43]. PGC-1α is also involved in type-IIX myosin-fiber heavy chains when it interacts with the myocyte enhancer factor-2 (MEF2) [44].
Indeed, the practice of exercise has a potent effect on the activation of the EnAS that, to a great extent, is regulated by the nuclear factor erythroid 2-related factor (Nrf2), a nuclear transcription factor that coordinates more than 250 genes associated with the cytoprotective and antioxidant response. Under homeostatic conditions, Nrf2 is attached to its negative regulator Keap1, a redox-regulator substrate adaptor for the Cullin (Cul)3-RING-box protein (Rbx)1 ubiquitin ligase, which directs Nrf2 toward its degradation by ubiquitination [45]. However, when the cellular concentration of ROS rises, Keap1 cysteine-rich proteins are oxidized, inducing conformational changes. Subsequently, Nrf2 is released from Keap1 to translocate into the nucleus, where it has the ability to heterodimerize with musculoaponeurotic fibrosarcoma proteins (MAF proteins), binding to the antioxidant response element (ARE), a specific DNA sequence (ARE, 50-TGACNNNGC-30); that is the latter is responsible for the activation of a wide-ranging group of genes associated with the antioxidant and protective response, such as the enzymes involves in the biosynthesis and metabolism of glutathione, Trx, phase-II detoxifying enzymes, and NADPH-producing enzymes [46,47]. Nrf2 interacts with PGC-1α in a close relationship to mediate the antioxidant response, together with other signaling pathways. In addition, Nrf2 is involved in the regulation of mitochondrial enzymes that are associated with the activation of PGC-1α. In mitochondrial biogenesis, Nrf2 interacts with PGC-1α and other transcriptional factors such the mitochondrial transcription factor A (TFAM), which encodes active enzymes of the mitochondrial matrix and also stabilizes mtDNA via the synthesis of nucleoids and controls the amount of mtDNA, which is correlated with the amount of TFAM but not necessarily with the transcription level [48]. Nrf2 promotes mitochondrial biogenesis by maintaining levels of nuclear respiratory factor 1 (Nrf1) and promoting purine nucleotide biosynthesis [4]. The mitophagic process is also influenced in the presence of Nrf2: dysfunctional mitochondria are engulfed by autophagosomes that are prepared for their degradation and recycling in lysosomes, preserving mitochondrial homeostasis [4].
Autophagy as a physiological regulator helps to maintain the balance between organelle biogenesis, protein synthesis and degradation. Autophagy preserves cellular homeostasis where damaged organelles and biomolecules are engulfed to be subsequently degraded upon fusion with lysosomes. It can be activated as a pro-survival response to stress; it has been found that autophagy is a redox-sensitive process and ROS/RNS elicits the crosstalk between cell signaling and protein damage [49]. Mitophagy is known as a form of selective autophagy regard to mitochondria turnover is greatly induced by oxidative stress to remove dysfunctional mitochondria [50]. The relation between mitophagy and the complex Nrf2/Keap1 has been elucidated; one way described is via the transcriptional upregulation of p62/sequestosome-1 (p62/SQSTM1) involved in Keap1 autophagic degradation and the activation of Nrf2 promoting the antioxidant response [51]. Recently, Yamada et al. [52] reported that p62/SQSTM1 translocates Keap1 to the mitochondria for the ubiquitination and degradation promoting mitophagy. Another form of the relationship between Nrf2 and mitophagy is through Keap1 inhibitors such as phosphatase and tensin homologue (PTEN)-induced putative kinase 1 (PINK1), which is one of the main regulators of mitophagy [53].
Although the mechanisms underlying exercise-induced mitophagy have not been fully understood, it is being established that mitophagy initiates after 6 h of acute treadmill running in mice observed by increased phosphorylation of AMP-activated protein kinase (Ampk) at tyrosine 172 and of unc-51 like autophagy activating kinase 1 (Ulk1) at serine 555. The mentioned research highlights the effect of exercise on mitochondrial biogenesis and mitochondrial removal, driving mitophagy thru Ampk-Ulk1 signaling in skeletal muscle [54]. In rat myocardium, the elevation of OS after acute heavy exercise triggers inflammatory response via NLRP3 inflammasome activation, promoting mitophagy to compensate for counteracting myocardial damage [55]. The underlying effect of exercise on the activation of EnAS counteracts the elevated level of ROS and their potential effect on destabilizing membrane lipids, structural proteins, or DNA. Abundant evidence indicates that exercise effectively activates Nrf2, the antioxidant response, and mitochondrial biogenesis. It should be noted that an optimal antioxidant response depends largely on the constant feedback between the endogenous and exogenous defense systems (EnAS/ExAS), so the lack of antioxidant dietary intake may directly affect the effectiveness of the cytoprotective response.
3.2. Mitohormesis and Exercise
In 2006, Ji et al. [56] defined hormesis as the beneficial effect conferred by a low dose of an agent or phenomenon that would cause injury or toxicity at high levels. Exercise induces a certain level of stress depending on the intensity, duration, and frequency of the exercise that is necessary for adaptation to physical work: exercise-induced ROS elevation is required for adaptation at physiological levels to permit energy metabolism, stress regulation, protein turnover, immunomodulation, muscle growth, and the regulation of blood flow [57]. Hence, exercise preconditioning/hormesis can be conceived of as the mild OS necessary to increase protection against mechanical/metabolic stressors, extreme temperatures (heat/cold), hypoxic conditions, ischemia, and other factors. In skeletal muscle, exercise preconditioning could be the result of the synergy exerted by different elements, including transcriptional factors, antioxidant defense, the expression of heat shock proteins (HSP), and ROS-producing sources [58].
Nuclear factor (NF) kappaΒ and mitogen-activated protein kinases (MAPK) comprise one of the most important redox-sensitive signal pathways. Transcription factor NF-κΒ remains inactive when is conjugated thru inhibitory I-κΒ proteins, within the cytoplasm are activated by different stimulants such as proinflammatory cytokines including TNF-α, IL-1, and IL-6, lipopolysaccharides (LPS), and toxins, and the presence of H2O2 activates the signal. Then, NF-κB dissociates from I-κΒ by the activation of NFκB-inducing kinase (NIK) phosphorylating serine residues of I-κΒα (ser-32 and ser-36) or I-κΒβ (ser-19 and ser-23), releasing p50/p65 subunits. After this, it translocates into the nucleus and binds to the specific target genes involved in a wide variety of biological functions, such as the antioxidant and the inflammatory response, immunity, apoptosis, and adaptation [59,60]. The target genes are SOD2, vascular cell adhesion molecule-1 (VCAM-1), cyclooxygenase-2 (COX-2), inducible NOS (iNOS), and GCS [61]. On the other hand, MAPK signaling is implicated in transcription, translation, growth, and cell remodeling. MAPK is activated by growth factors, the inflammatory cytokines TNF-α and IL-1, LPS phorbol esters, and ROS [62,63]. The MAPK signal pathway integrates the extracellular signal-regulated kinase (ERK), the c-Jun amino-terminal kinase (JNK), and the p38MAPK, which are controlled by their respective upstream kinases (MEK/MKK) [62,64]. Inactive skeletal muscle, ROS play a role as signal messengers that trigger adaptative responses via the interrelationship of NF-κΒ and MAPK pathways, boosting exercise preconditioning [56].
With respect to the mitochondria, the term mitohormesis refers to exposure to a sublethal dose of stress that could enhance mitochondrial function and that contribute to adaptative hormetic response [65]. In aerobic exercise OS, mechanical, unfolded/misfolded protein, and energy stress can induce several mitohormetic effects, such as antioxidant enzymes expression, myofibrillar protein synthesis, mitochondrial biogenesis, and protein folding (Figure 1). The result in terms of functionality is the increase of muscle mass and strength, improvement in mitochondrial function, oxidative capacity, and antioxidant response, a reduction in ROS production, and the enhancement of proteostasis [66]. Mitohormesis results from the interaction of several transcriptional factors, such as PPARα, EERα, Sp1, PGC-1α, PGC-1β, and Nrf2. In a study with 3- and 12-month-old Nrf2 Wild-Type (WT) and Nrf2 Knock Out (KO) mice submitted to a training protocol of voluntary wheel running, it was demonstrated that Nrf2/KO mice experienced failure in mitochondrial respiration of the intramyocellular fraction (IMC) in 40% of skeletal muscle and that the emission of ROS increased significantly. The aerobic resistance capacity of Nrf2/KO was reduced, along with the highest degree of fatigue associated with the elevated ROS found in mitochondrial samples, which probably affected mitochondrial integrity. Additionally, no significance in mitochondrial content was found; however, the activity of Cytochrome C Oxidase (COX) and of the COXIV subunit increased in Nrf2/WT trained animals. At the same time, Nrf2/KO animals experienced a reduction of NQO1. The study suggested that Nrf2 transcription encouraged by physical exercise is essential for protein stoichiometric adaptations, the enzymatic activity of ETC, and energy metabolism [67].
Figure 1.

Exercise-induced oxidative stress and the development of the mithormetic response. Created with BioRender.com (accessed on 10 November 2021). NADH: reduced nicotinamide adenine dinucleotide; NAD: nicotinamide adenine dinucleotide; FADH: reduced flavin adenine dinucleotide; FAD: flavin adenine dinucleotide; CoQ: coenzyme Q; Cyt C: cytochrome C: PGC1-α: peroxisome proliferator-activated receptor gamma coactivator-1 alpha; Nrf2: nuclear factor erythroid 2-related factor; NF-κB: nuclear factor-kappa beta.
Wang et al. [67] reported that the stimulus of acute exercise promoted the signaling pathway of the redox effector factor 1 (Ref1) and Nrf2 in groups of male ICR/CD-1 mice. The main findings included the role that H2O2 mitochondrial content exerts on the elevation of OS and the activation of Ref1/Nrf2 signaling in a time-dependent manner (45, 90, 120, or 150 min), together with the expression of the antioxidant system: MnSOD and CuZnSOD activities; GSH content; the expression of the Ref1 and Nrf2 genes, and Ref1 and Nrf2 proteins in mouse skeletal muscle (gastrocnemius and quadriceps femoris). This suggested that acute exercise increases H2O2 and OS, enhancing the Ref1/Nrf2 pathway and antioxidant defense, basically GSH and MnSOD (mitochondrial isoform). Merry and Ristow [68] found that the deficiency of Nrf2 in mouse gastrocnemius muscle and muscle cells affects the production of the messenger RNA (mRNA) of Nrf1 and TFAM (mitochondrial biogenesis biomarkers), CAT, SOD1, and SOD2 in acute exercise. Additionally, after a five-week treadmill training protocol, mice deficient in Nrf2 (Nrf/KO) experienced an important decline in mitochondrial mass, SOD activity, and whole-body energy expenditure compared with Nrf2/WT. In isolated muscle-cell C2C12 myoblasts, acute exogenous exposure to H2O2 and diethylenetriamine/NO adduct (NO donor) increased TFAM. These results suggest that ROS and NO are able to regulate the Nrf2 pathway to stimulate mitochondrial biogenesis and the expression of antioxidant protection genes [68].
Mitochondrial ROS production controls a variety of physiological processes within the cell, regulating gene expression and signal transduction. The protein p66Shc is a redox regulator of mitochondrial ROS, acting as H2O2 producer by sequestering electrons from ETC. It modulates H2O2 and proton leak and membrane oxidase activity, thus contributing to mitohormesis in mammalian cells [69,70]. Acute exercise appears to increase the expression of p66Shc and transcription factor FOXO3a in correlation with H2O2 and in a time-dependent manner in gastrocnemius and quadriceps femoris of male ICR/CD-1 mice. Mitochondrial CAT was also affected in a time-dependent manner, but not SOD, which was not significantly modified [71]. FOXO3a is a FOXO family protein implicated in stress modulation and metabolism by interfering in SOD and CAT expression. FOXO3a is also related to the survival of organisms and lifespan extension under energy-nutrient restriction [70]. It is noteworthy that some evidence indicates that there is a more efficient cellular response to stress in trained compared with untrained individuals. Better skeletal-muscle oxidative capacity has been reported with lifelong endurance-exercise training after a single bout of cycling at 75% VO2max in elderly trained individuals (aged 71.3 ± 3.4 years). Different indicators, such as PGC-1α, VEGF mRNA, and AMPK phosphorylation, were significantly higher in trained individuals. These results suggest an improved oxidative muscle capacity and a better ability to respond to acute endurance exercise [72]. It must be noted that single-bout exercise or regular exercise elicits the activation of numerous circuits closely related to mitohormetic adaptations.
3.3. Mitochondrial Uncoupling and Exercise
Mitochondrial uncoupling was initially correlated with mitochondrial dysfunction because it is the dissociation between the generation of membrane potential and the use for ATP synthesis. To a certain degree, uncoupling is necessary to explain diverse biological processes [73]. The lack of coupling could be explained by three possible situations: proton leak, electron leak, and electron slip. Proton leak basically depends on the composition of the inner mitochondrial membrane and can be modulated by a group of selective proteins: the uncoupling proteins (UCP). In addition, the electron leak process in ETC leads to the synthesis of O2•− or of the hydroperoxyl radical (HO2•). Finally, electron slip is the transfer of electrons through the mitochondrial complexes without proton pumping [74,75].
In humans, UCP comprises a protein family constituted of five members (UCP-1–UCP-5). UCP-1, a transmembrane protein that catalyzes proton transport across the mitochondrial membrane, in turn leading to mitochondrial uncoupling, is found in the inner mitochondrial membrane [76]. UCP-1 displays an important role in thermogenesis in brown and beige adipose tissue and the elevation of mitochondrial ROS activates acute thermogenesis. Thermogenic ROS can modify cysteine thiols (cys253) to increase respiration in brown adipose tissue. The sulfenilation of cys253 by mitochondrial ROS supports UCP-1-dependent thermogenesis and the whole-body energy expenditure [77]. Exercise increases uncoupling and mitochondrial biogenesis in skeletal muscle, augmenting the mitochondrial uncoupling-driven thermogenesis and thus energy expenditure [73]. Acute exercise increases the expression of UCP-1 in brown adipose tissue of high fat diet-treated Swiss mice and ICR mice. The thermogenic effect is explained by the enhancement of leptin-induces hypothalamic ERK1/2 phosphorylation, which stimulates thermogenesis in brown adipose tissue [78].
UCP-2 was found to be activated, for instance, is a massive entry of free fatty acids (FFA) into mitochondria [79], similarly to UCP-3, which has been found to be elevated in situations of starvation [80]. The latter is expressed in rat and mouse skeletal muscle; it was demonstrated that the content of UCP-3 in the intermyofibrillar fraction is 1.3-fold higher compared with the subsarcolemmal mitochondrial fraction. In addition, under conditions of stress, in the presence of ROS, UCP-3 promotes the Nrf2 antioxidant response and diminishes superoxide generation by increasing the proton conductance of the internal mitochondrial membrane [81]. UCP-3 is expressed 200–700-fold lower than UCP-1 in brown adipose-tissue mitochondria from warm-adapted hamsters (24–84 µg of UCP-1/mg of mitochondrial protein) [82]. There are some contradictory results found by Nabben et al. [83] in models of acute lipotoxicity–fatty acid-induced oxidative inhibition and fatty acid-induced swelling, in which no evidence was found with regard to UPC-3 protection against OS, ROS regulation, or the protection of acute lipotoxicity in UCP-3 (−/−) mice, backcrossed for 10 generations on a C57Bl/6 background and isolated skeletal-muscle mitochondria.
UCP-3 is expressed in response to exercise and other factors, such as that fat and glucose metabolism modulate its expression during moderate exercise. A rapid biphasic alteration has been observed of UCP-3 gene transcription after 1 h of moderate exercise, and UPC-3 upregulation was sustained for 3 h during the recovery period [84]. In prolonged exercise, that is, 45, 90, 120, and 150 min of a single bout of exercise, UPC-3 expression is rapidly upregulated in rat skeletal muscle, probably due to the gradual increase of intracellular ROS. Due to UCP-3 upregulation during exercise, it is thought that the former potentiates OxPho because it alleviates the proton gradient across the inner mitochondrial membrane, thus reducing further ROS generation via the ETC. The continued physical activity caused a decline in energy-coupling efficiency observed by a rise in respiration rate as a consequence of proton leak and a decrease in the respiratory control index (RCI). In turn, it has been proposed that this is another way of how UCP-3 may act as an antioxidant to protect the mitochondria in muscle cells from exercise-induced OS [85]. In parallel, acute or severe hypobaric hypoxia appears to upregulate UCP-3 and may attenuate the cross-membrane potential (Deltapsi). Through endurance, exercise improves the mitochondrial OxPho by means of better ROS elimination and diminishes hypoxia-induced UPC-3 upregulation [86].
4. Mitochondrial Damage in Exercise Performance
The importance of mitochondrial function is currently being constantly mentioned in terms of maintaining optimal cell conditions. In agreement with hermetic theory, a certain level of stress is required to stimulate or modulate molecular signaling to enhance cell functioning and survival in order to withstand greater insults. Exercise-induced stress is manifested in different forms, including mechanical, metabolic, thermal, and OS, which activates molecular messengers serving as potential boosters of the signaling pathways that regulate adaptative responses to physical training [87]. The big question now appears to be on how to determine the amount of stress necessary to induce cellular adaptations and when the adaptive threshold is exceeded, and cellular damage starts. That is, at what moment are the cytoprotective defenses of the antioxidant system surpassed?
From the bioenergetic perspective, mitochondria comprise the cell core in every single reaction in which ATP is required; thus, a constant oxygen supply to fuel muscle-force production must be achieved. In turn, reducing the OxPho capacity will directly affect the athletic physical performance, contributing to fatigue and the previously mentioned OTS. Mitochondrial-reduced capacity can be the cause of restricted VO2max and the impairment of performing high-intensity sports activities. Very limited studies have been conducted to elucidate the relation between mitochondrial damage indicators and the detriment of athletic performance. The study conducted by Kadaja et al. [88], in a six-week treadmill exercise program to determine changes in energy metabolism in rat myocardium, revealed that chronic exhaustive exercise induces the presence of OTS accompanied by muscle catabolism, body-weight loss, and decreased performance.
It is noteworthy that maximal performance as reflected in physical working capacity was reported in week 3, but in week 6, this decreased remarkably: the animals were unable to maintain that training load for a longer period. Regarding heart–weight changes, a rise was observed to an increased heart-to-body weight ratio, indicating that overtraining induces anabolic responses in the myocardium. Microscopic analysis showed significant myocardium changes, including the disintegration of cardiomyocyte structure, the appearance of peroxisomes, and cellular swelling. Mitochondrial structures appeared to be intact, but had separated from the myofibrils, suggesting that the cytoskeleton surrounding the mitochondria may have disintegrated. Furthermore, the appearance of the peroxisomes, manifested as rounded, electron-dense dark bodies, could be associated with exercise-induced stress. In addition, a decrease was described in OxPhos by means of a reduction in cytochrome c and the impaired coupling of adenylate kinase (AK) to OxPhos. These experiments suggested that overtraining impairs functional coupling between mitochondrial AK2 and ANT protein and produces a drop in the overall capacity of ADP-dependent OxPhos. The study points out that an excessive work volume could result in OTS affecting the myocardium as well.
The reduced ability to perform high-intensity or extended exercises in athletes with high fatigue ratings and OTS has been positively correlated [89]. Reduced mitochondrial respiration may perhaps contribute to the impairment of muscle contraction and, in turn, physical performance (Figure 2a). Vigorous exercise could also lead to mitochondrial DNA deletion and apoptosis [90] and additionally to the mitochondrial structural changes observed as well as the swelling and loss of cristae after a single bout of extraneous exercise [91]. Exhaustive swimming exercise in rats during 10 weeks gave rise to muscle and myocardium damage, reported as the loss of mitochondrial and sarcoplasmic- or endoplasmic-reticulum integrity and the augmented lipid peroxidation of tissue homogenates in exhausted trained and untrained rats [92]. In addition, overtraining might impact muscle metabolism balance by promoting catabolism on contractile protein synthesis (Figure 2b). The overtraining protocol in rats led to a marked quantitative decrease of the slow-twitch oxidative fibers of the soleus muscle and of the fast-twitch glycolytic fibers of plantaris and extensor digitorum longus muscles, body-weight loss, a reduced rate of myosin heavy chain (MHC) synthesis, and an increase of excreted urinary 3-MeHis [93]. On the other hand, after 11 weeks of severe endurance training, male Wistar rats experienced a reduction in performance of 38% in the nonfunctional overreaching group (NFOR) compared with the functional overreaching group (FOR). In this study, remarkably lower citrate-synthase activity was observed, as well as the lower activity of mitochondrial complex IV in the red gastrocnemius muscle of the NFOR group. Cardiomyocyte apoptosis was also described in endurance overtraining as higher in the NFOR than in the FOR group [94].
Figure 2.
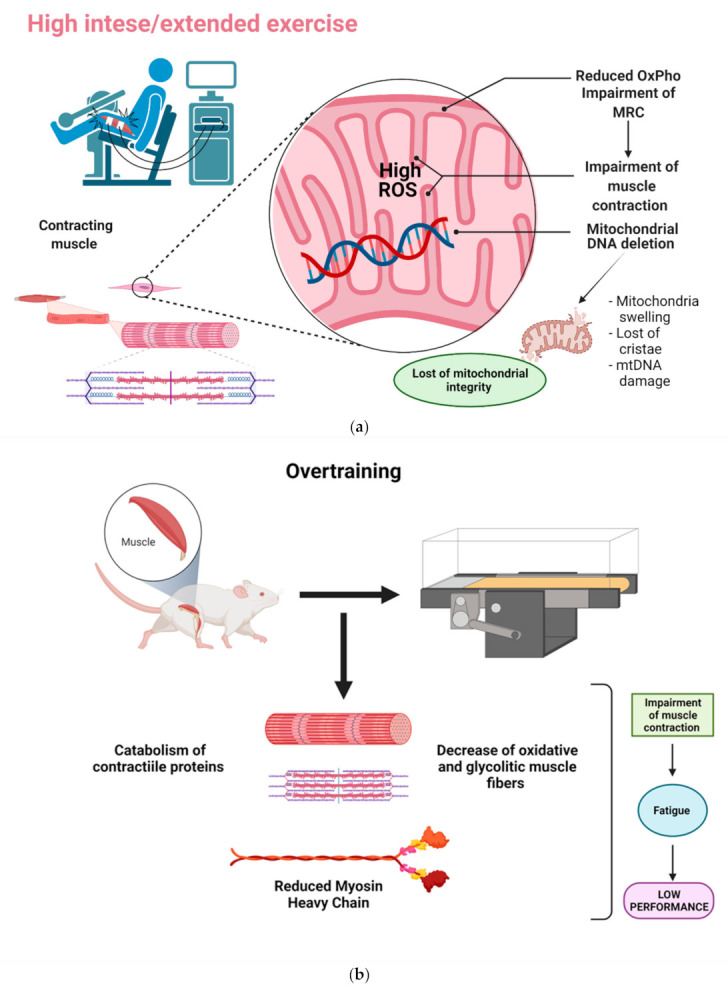
Impact of mitochondrial damage in exercise performance. (a) Graphical representation of mitochondrial reduced function and impairment of muscle contraction in intense or exhausting exercise. (b) Description of overtraining and the impact on muscle catabolism. ROS: reactive oxygen species; OxPho: oxidative phosphorylation; MRC: mitochondrial respiratory chain. Created with BioRednder.com (accessed on 24 October 2021).
After an eight-week training protocol of acute exhaustive exercise in rats conducted by Fang et al. [95], mitochondrial ROS production significantly increased, and the activity of respiratory complex V and ATP content in samples of skeletal muscle and brain was reduced. Acute exhaustive training decreased the expression of prohibitin-1 (PHB1) protein in mitochondria, OxPho was reduced, and the energy metabolism in general mitochondrial function was affected. The PHB comprises a phosphoprotein complex required for mitochondrial homeostasis and survival identified in human T-cells; PHB participates in cell activation, differentiation, function, and viability [96]. Different evidence reveals that mitochondria as the main supplier of ROS throughout exercise might be a central target of OS damage and the consequent undergoing of structural and functional disruptions. The analysis of mitochondrial fractions has demonstrated a proportional relation of the amount of damaged mitochondria with the extension of training bouts [97].
Taken together, this information clearly outlines that these mitochondrial responses to exercise could take two different paths: (1) mitochondrial adaptations in relation to low/moderate exercise, or (2) mitochondrial damage in response to excessive exercise workloads. In this manner, mitochondrial responses will depend to a great degree on the duration, intensity, and the frequency of exercise bouts. Low/moderate exercise promotes mitochondrial biogenesis and with no noticeable mitochondrial impairments such as swelling or disruption of cristae [98,99]. In contrast, exhaustive exercise is associated with cell damage, and with morphological alterations observed as flocculent mitochondria, swelling, incomplete myofibrils, and irregular sarcomeres [100]. Mitochondrial integrity must be a targeted issue in the search for optimal performance beyond adaptations to exercise.
5. Nutrition Insights into Mitochondrial Protection
5.1. General View of Antioxidants, Oxidative Stress, and Exercise Performance
For many years, there has been a constant debate on whether the use of antioxidants truly benefits exercise performance/recovery or impairs biological adaptations to heavy training [101,102,103]. Current available data point out that antioxidant requirements should be covered by a balanced and individualized diet, with the individual consuming the recommended dietary allowance (RDA) doses. Nonetheless, under special circumstances such as high-intensity or prolonged exhaustive sports, antioxidant supplementation can be an optimal way to prevent or reduce the consequences of damage, such as unchaining the previously mentioned fatigue and/or OTS [104,105].
Frequently, antioxidant supplementation is addressed in terms of improving athletic performance, but the evidence is contradictory. It should take into consideration that reducing OS is not necessarily associated with the enhancement of exercise performance. In a double-blind controlled assay, supplementation of vitamin C (500 mg/d) and E (400 IU/d) for 15 days in football players did not modify muscle damage or muscle soreness after acute exercise-induced OS. The reported findings did not exhibit any ergogenic effect; nevertheless, OS was attenuated [106]. Similarly, supplementation with vitamin C and E in improved indices of OS was associated with repetitive loading exercise by increasing the activity of the antioxidant enzyme MnSOD in young animals. CuZnSOD and CAT activity also increased in young and aged animals and improved the positive work output of muscles in aged rodents. These data suggest that more regulatory factors of OS are implicated, such as age and exercise type, which indirectly influence physical performance [107].
On the other hand, four-week antioxidant supplementation with vitamin C (2 × 500 mg/day) and E (400 IU/day) did not exert any effect on skeletal-muscle oxidative OS indicators or in the expression of mitochondrial biogenesis genes (PGC-1α, TFAM, or PGC-related factor). No changes in VO2max, citrate-synthase activity, and mRNA cytochrome oxidase subunit IV (COX IV) in skeletal muscle were observed. In contrast, antioxidant supplementation attenuated skeletal-muscle adaptation biomarkers in SOD activity and the protein abundance of TFAM and SOD 2 [108]. Earlier, it was informed that vitamin C and E supplementation blunts adaptations to endurance training in human trials [109].
Polyphenols are a huge group of molecules that have been largely investigated for their multiple health benefits [110], but their tentative impact on OS and exercise performance remains unclear [111]. Grape polyphenols from beverages, extracts, or supplements have exhibited very good results for the reduction of damage in the intense exercise of diverse sports activities [112]. Lately, some other phytochemicals appear to be potential compounds that could exert an impact on exercise performance and the lowering of OS in some way by their attributed antioxidant activity. For instance, curcumin at doses of 500 mg/day for 8 weeks significantly improved total antioxidant capacity (TAC), malonaldehyde, serum C-reactive protein (CRP), and VO2max in physically active women [113].
Our research group found that silymarin, the flavonolignan extract from Milk thistle, has been demonstrated to increase physical performance and to promote muscle hypertrophy and recovery at doses of 100 mg/kg after eight weeks of regular exercise in a rodent model [114]. Aside, the pretreatment with sulforaphane (SFN) from broccoli in 13-week-age Nrf2+/+ and Nrf2−/− male mice upregulated Nrf2 signaling and downstream genes, improved the endurance capacity associated with a lower OS marker, and provided protection against muscle fatigue during exhaustive exercise. SFN may enhance exercise performance via upregulation of the Nrf2 pathway after an exhaustive treadmill test (progressive-continuous all-out [115]. To date, very few investigations have been conducted on the benefits of SFN in exercise; however, SFN is thought to be a promising molecule on providing an ergogenic effect on exercise performance because preceding literature supports SFN effectiveness in prompting the Nrf2/Keap1/ARE signaling pathway and in suppressing NF-κΒ signaling. Therefore, SFN has great potential for offering protection against OS, inflammation, and fatigue [116]. Of note, silymarin and curcumin have also been reported to be effective bioactivators of Nrf2/Keap1/ARE in a variety of in-vitro and in-vivo studies [117,118,119].
5.2. Mitochondria as Targets for Protection from Oxidative Damage
At present, substantial evidence indicates that DNA damage appears immediately after acute aerobic exercise but that the potential DNA renewal begins after day 3. The relationship concerning what supports the explanation of exercise-induced DNA damage must be understood beyond the hormesis theory, which refers to ROS/RNS as key actors of genomic damage surrounding the breaking of the balance between physical inactivity and overtraining. A multi-dimensional overview has been recently proposed to build a better approach for the complex relationship between DNA integrity and exercise [120]. According to the authors, the multi-dimensional model includes the whole perception of multiple factors regarding the extension of DNA oxidative damage that the exercise-induced. Eight main factors are listed as follows: (1) type of ROS/RNS, from the least to the most reactive; (2) frequency of attacks or episodes of exposure to ROS/RNS; (3) type and extension of DNA damage (single/double-strand breaks); (4) magnitude of DNA oxidation by ROS/RNS; (5) exercise intensity/distance; (6) frequency of training bouts; (7) DNA repair enzymes, and (8) degree of individualization, which comprises further issues, such as age, sex, training level, and the nutrition quantification method [120].
This entire view would be theoretically applicable to the analysis of mtDNA damage caused by exercise, considering that there are certain elements that could render mtDNA more susceptible to damage in comparison with nuclear DNA, such as the accumulation of transition metal ions, the secondary subproducts of DNA, lipid oxidation, and finally, the absence of histones and chromatin organization. More studies are required to determine the mechanism implicated in mtDNA damage as a result of exhaustive/intensive exercise.
An understanding of the importance of mitochondrial dynamics in bioenergetics, signaling, and adaptation to exercise, mitochondrial integrity must be an elemental issue in developing target antioxidants. Mitochondrial dysfunction is being associated with several common pathologies, such as metabolic and cardiovascular diseases, ischemia–reperfusion injury, and neurodegeneration; thus, the mitochondrial features have been largely studied as a crucial drug target to treat the previously mentioned disorders [121]. Several therapeutic approaches are emerging in which some compounds have been designed to provide the target of mitochondrial protection.
Although the effectiveness of conventional antioxidant supplementation and exercise-induced OS has been well-described [122,123,124], there is an absence of evidence regarding mitochondrial-targeted antioxidants in exercise. An example of this is the following double-blind, randomized trial. Twelve healthy male participants (age = 28 ± 10 years, height = 177 ± 12 cm, and body mass = 81 ± 15 kg) were assigned to receive either a daily supplement of 1000 mg of α-lipoic acid (2 × 500-mg tablets) for 14 days or to receive no supplement. The participants performed 100 isolated, continuous, maximal knee extensions (minimal force = 200 N, speed of contraction = 60°·s−1). The analysis of blood and muscle biopsy tissue samples revealed that the exercise promoted mtDNA damage by significantly increasing the 8-hydroxy-2-deoxyguanosine (8-OHdG) concentration; in parallel, total antioxidant capacity was significantly reduced in both groups. An augmented total antioxidant capacity was found in blood samples; the comet essay and 8-OHdG analysis confirmed DNA protection and reduced lipid peroxidation and hydroperoxides in the supplemented group. Both groups experienced protein oxidation indistinctly. The findings suggested that short-term α-lipoic acid supplementation may protect nuclear DNA and lipids, but not mtDNA, in high-intensity, isolated, single-leg exercise. The tentative protective role of α-lipoic acid in blood oxidative markers is perhaps independent of muscle OS indicators and shelters DNA integrity selectively. These results may be associated with a different antioxidant baseline ability within the cell compartment [125].
5.3. Target Mitochondrial Antioxidants in Exercise-Induced Oxidative Damage
In this respect, several factors must be considered, such the inherent mitochondrial biological nature and the chemical nature, such as the molecular dynamics of the bioactive compounds when they are intended to be tested as possible mitochondrial target protectors. Mitochondria possess morphological and physiological features that render them difficult to reach (Figure 3). In addition, bioactive compounds should meet different aspects in order to ensure arrival at the target-cell compartment, and these aspects are herein mentioned as follows: (1) the issue of bioavailability; (2) transportation to the target organelle, and (3) the ability to cross mitochondrial barriers in order to be able to deliver it to the site-of-action. These are the possible limitations that general antioxidants may frequently face in the attempt to exert an antioxidant effect within the mitochondria. Therefore, different current forms of target antioxidants have been designed, such as lipophilic cations, liposome enclosure, and target peptides. Examples of these target antioxidants employed in clinical trials are MitoQ [126,127,128], MitoE [129,130], tiron [131], and MitoC [132]. For an integrative overview of target mitochondrial antioxidants in exercise-induced DNA damage, see the recommended review [133].
Figure 3.

Efficacy of non-target versus target mitochondrial antioxidants supplementation and their effect on exercise performance. MCR: mitochondrial respiratory chain; MPM: micropeptide in mitochondria; SFN: sulforaphane. Created with BioRender.com (accessed on 24 October 2021).
The difference between mitochondrial-target antioxidants and untargeted antioxidants is the potential to remove ROS/RNS from the production source due to the ability to cross the mitochondrial phospholipid bilayer [134]. MitoQ is an orally available mitochondrial-target coenzyme Q10 that contains the antioxidant quinone moiety covalently attached to a lipophilic triphenylphosphonium cation. MitoQ has been applied in a variety of in-vivo studies in rodent models and in diverse human trials [135]. MitoQ has the ability to mimic the antioxidant activity of coenzyme Q (CoQ) and simultaneously increase to supraphysiological levels in a mitochondrial-membrane potential-dependent manner to approximately 100–1000 times greater than non-target derivatives. MitoQ is positively charged, possesses a large molecular weight and is highly lipophilic and low water-soluble. These properties allow MitoQ to pass easily through biological membranes, promoting its mitochondrial accumulation to several hundred-fold, encouraged by the mitochondrial membrane potential [136,137,138]. MitoQ can be accumulated in the inner mitochondrial membrane at the matrix-facing side, where it is reduced by complex II to ubiquinol (MitoQH2) [139]. These localization abilities permit MitoQ to donate hydrogen atoms to oxygen, or even α-tocopherol radicals; thus, it can be effective against lipid peroxidation and peroxynitrite, preventing or attenuating oxidative damage in mitochondrial lipids, proteins, or mtDNA [140]. Consequently, it represents a potential way to deliver precise cytoprotection against exercise-induced oxidative damage.
The study of MitoQ in the context of the exercise was first explored by Shill et al. in 2016 [141] and, after three weeks of MitQ supplementation (10 mg/day), the authors demonstrated that skeletal-muscle and whole-body adaptation to endurance training was not affected by MitoQ in young, healthy men by the absence of a significant difference in VO2max oxidative capacity and circulating angiogenic cells. Recently, the novel work of Williamson et al. [142] demonstrated, in a double-blind placebo controlled study, that in a high-intensity intermittent exercise (HIIE), randomized, placebo-controlled design, 24 healthy male participants divided into two groups (placebo; n = 12, MitoQ; n = 12) were subjected to an exercise trial of 4 × 4-min bouts of cycling at 90–95% of maximal heart rate. The participants received an acute treatment of 20 mg-MitoQ or placebo 1-h pre-exercise and a chronic 21-day phase of supplementation. The results indicated that HIIE induced mtDNA damage in lymphocytes and in human-muscle tissue that was accompanied by modifications in lipid hydroperoxides, α-tocopherol, and ascorbyl free radicals. The MitoQ acute treatment was not sufficiently efficient to protect against mtDNA damage, possibly due to the low bioavailability. Notwithstanding, chronic MitoQ supplementation protected nuclear and mtDNA damage; the likely mechanisms implied that MitoQ helps to metabolize ROS, and that it may also safeguard complexes I and IV (cytochrome c complexes) in the ETC from oxidative damage.
MitoQ may have also interacted with proton-motivated forces (uptake reduces proton-motivated force) and favored the coenzyme Q-pool redox state, which possibly affected complex III-mediated superoxide production. In addition, MitoQ could be helping in extramitochondrial activities and in potential participation in metabolizing cytosolic H2O2. This research offers a new understanding with regard to mechanistic insights into mitochondrial redox dynamics and targeted supplementation to safeguard nuclear and mitochondrial genomes following HIIE. MitoQ is the best example of how to target antioxidants elicit benefits in protection against oxidative damage due to heavy training; however, the ability of MitoQ would be opposite to this if mitochondrial concentrations were increased to 0.5–1 µM vs. 2 µM on H2O2 production. It is noteworthy that when the balance between antioxidants and prooxidants is broken, we again have the same concern; that is, when cellular situations of susceptibility, such as unstable lipid membranes, quinone-derived compounds might propagate lipid peroxidation within the mitochondrial membrane [143,144]. This is possible because mitochondria are highly predominant in lipids susceptible to peroxidation, such as anionic cardiolipin, phosphatidylethanolamine, and phosphatidylcholine [145].
Park et al. [146] reported that the acute oral intake of MitoQ (80 mg) improved maximal walking capacity and postponed the onset of limping in individuals with peripheral artery disease. The authors also observed increasing vascular endothelial function and SOD. Although this research was not conducted exclusively in the context of high-intense or extended exercise, it was, to the authors’ knowledge, the first study of mixed exercise-targeted antioxidant supplementation with pathological variables. This highlights that several studies indicating the prophylactic effect attributed to MitoQ are related to the upregulation of the Nrf2/Keap1/ARE signaling pathway and the target antioxidant genes HO-1, NQO, GPx, GR, Trx, CAT, and γ-GCLC [126,128,147], which could also be involved in the complex network of cytoprotection in conjunction with the target mitochondrial antioxidant activity. It is also important to bear in mind the role that Nrf2 signaling plays in mitochondrial biogenesis and adaptation, together with other transcriptional factors, such as PGC-1α, NF-κΒ, PPARα, EERα, TFAM, among others, that we have already discussed. Therefore, it would be very limiting to only address the benefits of MitoQ in terms of a single mode-of-action.
Other interesting molecules appear attractive with respect to the Szeto–Schiller (SS) peptides (SS-19, SS-02, SS-31, and SS-20), which have been analyzed in some trials: these peptides also provide an antioxidant effect attributed to their tyrosine or dimethyl tyrosine residue, which allows them to scavenge ROS easily. These molecules are able to accumulate in the inner mitochondrial membrane at a concentration of 1000–5000-fold by means of hydrophilic and hydrostatic interactions; thus, they are able to cross cellular and mitochondrial membranes because they also possess the alternating aromatic–cationic amino-acid motif [148]. It appears that SS-31 has been mainly efficient according to the data because of its ability to attach to and protect against cardiolipin peroxidation by shifting cytochrome c peroxidase activity [149,150]. In addition, it can scavenge peroxynitrite and hydroxyl radicals due to the addition of dimethyl tyrosine [151]. In 14 days of inactivity-induced muscle atrophy, SS-31 supplementation mitigated mitochondrial ROS, protease activation, and muscle atrophy; to a great extent, skeletal-muscle atrophy is linked to modifications in cytosolic free Ca2+, inducing the activation of caspase-3 and calpain [152].
Peptides also comprise an innovative topic as a goal in mitochondrial protection; micropeptides are small molecules of <100 amino acids with specific functions [153]. Most recently, a novel muscle-enriched micropeptide was identified that was localized to mitochondria and that is denominated a micropeptide in mitochondria (MPM), identified in a ClC2 myoblast. The silencing MPM gene (MPM−/−) in mice permitted the recognition of its implication in myogenic differentiation and in muscle-fiber growth when the expression of Pax7, MyoD, and MyoG expression was impaired in MPM−/−. Furthermore, MPM−/− mice exhibited smaller skeletal-muscle fibers and worse muscle performance, manifested by a reduction in the maximal grip force of limbs, the latency to fall off the rotarod, and the exhausting swimming time. These effects were due to that MPM is involved in mitochondrial respiratory activity and mitochondrial biogenesis simultaneously, thus becoming a suitable target for therapy for muscle dystrophy or perhaps as a therapy for other muscle disorders [154].
Finally, while bioactive compounds from foods or plants, probably similar to those that we have mentioned before, including polyphenols, SFN, curcumin, and silymarin, do not fit into the categorization of target antioxidants, have the potential for very profound exploration in an attempt to elucidate their possible effects on mitochondrial protection and possible interactions within cell signaling, such as activation of the Nrf2 pathway [155,156].
6. Conclusions and Perspectives
The oxidative stress induced by an imbalance between ROS/RNS production and antioxidant systems has been well described in many pathological conditions, such as cancer, diabetes, cardiovascular diseases, immunological, neurodegenerative disorders, and aging. Moreover, as a consequence of increased oxygen consumption and energy demands through physical exercise, more ROS/RNS are produced, unchaining a series of cellular events related to the antioxidant response, molecular signaling linked with adaptation, and other biological responses. It is also known that high-intensity or exhaustive exercise might surpass the EnAS, raising oxidative stress, inflammation, and skeletal-muscle damage that, under chronic exposure, may eventually lead to fatigue and OTS, affecting performance.
Despite the vast literature with respect to exercise-induced muscle damage, few investigations have addressed mitochondrial damage and nutrition-specific strategies. Supplementation with antioxidants received particular attention as an essential tool for preventing or attenuating the damage induced, thus probably exerting a positive impact on sports performance [157]. However, evidence is highly contradictory concerning whether antioxidants truly benefit performance due to their protection or to their impairing adaptations to exercise. Mitochondria comprise one of the major ROS/RNS producers; therefore, they are highly susceptible to being attacked, and EnAS would not be sufficient to counteract mitochondrial damage, leading to a detriment in the performance of exercise and recovery.
Building on the limited body of literature, this review provides new insights into the role of exercise-induced redox perturbations in mitochondria, along with a new perspective on mitochondrial-targeted supplementation, which represents a novel area to explore. At the same time, there are a large number of bioactive compounds that could also offer protection by triggering the signaling involved in cytoprotection, mitochondrial dynamics, mitohormesis and biogenesis. It is noteworthy that the cytoprotector response is the result of constant crosstalk between EnAS and ExAS, and efficacy is notably based on arriving at a systemic equilibrium and an attempt to preserve this balance to the degree possible.
In conclusion, future research should take into consideration that mitochondrial oxidative damage might intervene in cellular adaptation, but that the supplementation of antioxidants might influence exercise performance. To date, it continues to be difficult to reach a conclusion in terms of antioxidant supplementation in athletes. Nonetheless, the use of antioxidants for mitochondrial protection against exercise-induced oxidative damage remains an innovative field to investigate from different angles.
Acknowledgments
Nancy Vargas-Mendoza is a scholarship holder for her postgraduate studies by the Consejo Nacional de Ciencia y Tecnologia (CONACyT) and the BEIFI program, Instituto Politécnico Nacional (IPN), Mexico.
Author Contributions
N.V.-M. and J.A.M.-G. conceived and designed the study; N.V.-M., Á.M.-G., E.O.M.-S., M.M.-M., M.A.-V., E.M.-B., I.Á.-G., J.G.-S., C.E.-C., G.C.-C. and J.M.C.-L. edited and wrote some portions of the paper, compiled the references, and analyzed the data; J.A.M.-G. and N.V.-M. wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Secretaría de Investigación y Posgrado, Instituto Politécnico Nacional: SIP-20210375 and SIP-20210361.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Powers S.K., Ji L.L., Kavazis A.N., Jackson M.J. Reactive oxygen species: Impact on skeletal muscle. Compr. Physiol. 2011;1:941–969. doi: 10.1002/cphy.c100054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Powers S.K., Jackson M.J. Exercise-induced oxidative stress: Cellular mechanisms and impact on muscle force production. Physiol. Rev. 2008;88:1243–1276. doi: 10.1152/physrev.00031.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones D.P. Redefining Oxidative Stress. Antioxid. Redox Signal. 2006;8:1865–1879. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]
- 4.Dinkova-Kostova A.T., Abramov A.Y. The emerging role of Nrf2 in mitochondrial function. Free. Radic. Biol. Med. 2015;88:179–188. doi: 10.1016/j.freeradbiomed.2015.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zorov D.B., Juhaszova M., Sollott S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014;94:909–950. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Powers S.K., Deminice R., Ozdemir M., Yoshihara T., Bomkamp M.P., Hyatt H. Exercise-induced oxidative stress: Friend or foe? J. Sport Health Sci. 2020;9:415–425. doi: 10.1016/j.jshs.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferreira L.F., Laitano O. Regulation of NADPH oxidases in skeletal muscle. Free Radic. Biol. Med. 2016;98:18–28. doi: 10.1016/j.freeradbiomed.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gong M.C., Arbogast S., Guo Z., Mathenia J., Su W., Reid M.B. Calcium-independent phospholipase A2 modulates cytosolic oxidant activity and contractile function in murine skeletal muscle cells. J. Appl. Physiol. 2006;100:399–405. doi: 10.1152/japplphysiol.00873.2005. [DOI] [PubMed] [Google Scholar]
- 9.Nethery D., Stofan D., Callahan L., DiMarco A., Supinski G. Formation of reactive oxygen species by the contracting diaphragm is PLA2dependent. J. Appl. Physiol. 1999;87:792–800. doi: 10.1152/jappl.1999.87.2.792. [DOI] [PubMed] [Google Scholar]
- 10.Cheng A.J., Jude B., Lanner J.T. Intramuscular mechanisms of overtraining. Redox Biol. 2020;35:101480. doi: 10.1016/j.redox.2020.101480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wan J.-J., Qin Z., Wang P.-Y., Sun Y., Liu X. Muscle fatigue: General understanding and treatment. Exp. Mol. Med. 2017;49:e384. doi: 10.1038/emm.2017.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andrade F.H., Reid M.B., Allen D.G., Westerblad H. Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. Pt 2J. Physiol. 1998;509:565–575. doi: 10.1111/j.1469-7793.1998.565bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng A.J., Yamada T., Rassier D.E., Andersson D.C., Westerblad H., Lanner J.T. Reactive oxygen/nitrogen species and contractile function in skeletal muscle during fatigue and recovery. J. Physiol. 2016;594:5149–5160. doi: 10.1113/JP270650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Debold E.P. Potential molecular mechanisms underlying muscle fatigue mediated by reactive oxygen and nitrogen species. Front. Physiol. 2015;6:239. doi: 10.3389/fphys.2015.00239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hargreaves M., Spriet L.L. Skeletal muscle energy metabolism during exercise. Nat. Metab. 2020;2:817–828. doi: 10.1038/s42255-020-0251-4. [DOI] [PubMed] [Google Scholar]
- 16.Murphy M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boveris A., Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem. J. 1973;134:707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quinlan C.L., Perevoshchikova I.V., Hey-Mogensen M., Orr A.L., Brand M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013;1:304–312. doi: 10.1016/j.redox.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brand M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016;100:14–31. doi: 10.1016/j.freeradbiomed.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 20.Sakellariou G.K., Jackson M.J., Vasilaki A. Redefining the major contributors to superoxide production in contracting skeletal muscle. The role of NAD(P)H oxidases. Free Radic. Res. 2014;48:12–29. doi: 10.3109/10715762.2013.830718. [DOI] [PubMed] [Google Scholar]
- 21.Sakellariou G.K., Vasilaki A., Palomero J., Kayani A., Zibrik L., McArdle A., Jackson M.J. Studies of mitochondrial and nonmitochondrial sources implicate nicotinamide adenine dinucleotide phosphate oxidase(s) in the increased skeletal muscle superoxide generation that occurs during contractile activity. Antioxid. Redox Signal. 2013;18:603–621. doi: 10.1089/ars.2012.4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Culotta V.C., Yang M., O’Halloran T.V. Activation of superoxide dismutases: Putting the metal to the pedal. Biochim. Biophys. Acta. 2006;1763:747–758. doi: 10.1016/j.bbamcr.2006.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Criswell D., Powers S., Dodd S., Lawler J., Edwards W., Renshler K., Grinton S. High intensity training-induced changes in skeletal muscle antioxidant enzyme activity. Med. Sci. Sports Exerc. 1993;25:1135–1140. doi: 10.1249/00005768-199310000-00009. [DOI] [PubMed] [Google Scholar]
- 24.Powers S.K., Criswell D., Lawler J., Ji L.L., Martin D., Herb R.A., Dudley G. Influence of exercise and fiber type on antioxidant enzyme activity in rat skeletal muscle. Am. J. Physiol. 1994;266:R375–R380. doi: 10.1152/ajpregu.1994.266.2.R375. [DOI] [PubMed] [Google Scholar]
- 25.Deponte M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta. 2013;1830:3217–3266. doi: 10.1016/j.bbagen.2012.09.018. [DOI] [PubMed] [Google Scholar]
- 26.Bermingham E.N., Hesketh J.E., Sinclair B.R., Koolaard J.P., Roy N.C. Selenium-enriched foods are more effective at increasing glutathione peroxidase (GPx) activity compared with selenomethionine: A meta-analysis. Nutrients. 2014;6:4002–4031. doi: 10.3390/nu6104002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li R., Fan X., Zhang T., Song H., Bian X., Nai R., Li J., Zhang J. Expression of selenium-independent glutathione peroxidase 5 (GPx5) in the epididymis of Small Tail Han sheep. Asian-Australas. J. Anim. Sci. 2018;31:1591–1597. doi: 10.5713/ajas.18.0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen J., Zhang Y., Lv Y., Tian M., You J., Chen F., Zhang S., Guan W. Effects of Selenomethionine on Cell Viability, Selenoprotein Expression and Antioxidant Function in Porcine Mammary Epithelial Cells. Front. Nutr. 2021;8:665855. doi: 10.3389/fnut.2021.665855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Y.I., Wei P.C., Hsu J.L., Su F.Y., Lee W.H. NPGPx (GPx7): A novel oxidative stress sensor/transmitter with multiple roles in redox homeostasis. Am. J. Transl. Res. 2016;8:1626–1640. [PMC free article] [PubMed] [Google Scholar]
- 30.Khatib A., Solaimuthu B., Ben Yosef M., Abu Rmaileh A., Tanna M., Oren G., Schlesinger Frisch M., Axelrod J.H., Lichtenstein M., Shaul Y.D. The glutathione peroxidase 8 (GPX8)/IL-6/STAT3 axis is essential in maintaining an aggressive breast cancer phenotype. Proc. Natl. Acad. Sci. USA. 2020;117:21420–21431. doi: 10.1073/pnas.2010275117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schriner S.E., Linford N.J., Martin G.M., Treuting P., Ogburn C.E., Emond M., Coskun P.E., Ladiges W., Wolf N., Van Remmen H., et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 32.Jastrząb A., Skrzydlewska E. Thioredoxin-dependent system. Application of inhibitors. J. Enzym. Inhib. Med. Chem. 2021;36:362–371. doi: 10.1080/14756366.2020.1867121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhong L., Arnér E.S., Holmgren A. Structure and mechanism of mammalian thioredoxin reductase: The active site is a redox-active selenolthiol/selenenylsulfide formed from the conserved cysteine-selenocysteine sequence. Proc. Natl. Acad. Sci. USA. 2000;97:5854–5859. doi: 10.1073/pnas.100114897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arnér E.S., Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000;267:6102–6109. doi: 10.1046/j.1432-1327.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- 35.Rhee S.G., Kil I.S. Multiple Functions and Regulation of Mammalian Peroxiredoxins. Annu. Rev. Biochem. 2017;86:749–775. doi: 10.1146/annurev-biochem-060815-014431. [DOI] [PubMed] [Google Scholar]
- 36.Rhee S.G., Woo H.A., Kang D. The Role of Peroxiredoxins in the Transduction of H(2)O(2) Signals. Antioxid. Redox Signal. 2018;28:537–557. doi: 10.1089/ars.2017.7167. [DOI] [PubMed] [Google Scholar]
- 37.Holmgren A. Antioxidant function of thioredoxin and glutaredoxin systems. Antioxid. Redox Signal. 2000;2:811–820. doi: 10.1089/ars.2000.2.4-811. [DOI] [PubMed] [Google Scholar]
- 38.Ferraro E., Giammarioli A.M., Chiandotto S., Spoletini I., Rosano G. Exercise-induced skeletal muscle remodeling and metabolic adaptation: Redox signaling and role of autophagy. Antioxid. Redox Signal. 2014;21:154–176. doi: 10.1089/ars.2013.5773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gomez-Cabrera M.C., Carretero A., Millan-Domingo F., Garcia-Dominguez E., Correas A.G., Olaso-Gonzalez G., Viña J. Redox-related biomarkers in physical exercise. Redox Biol. 2021;42:101956. doi: 10.1016/j.redox.2021.101956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Puigserver P., Spiegelman B.M. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): Transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003;24:78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- 41.Spiegelman B.M. Transcriptional control of energy homeostasis through the PGC1 coactivators. Novartis Found. Symp. 2007;286 [PubMed] [Google Scholar]
- 42.Baar K. Nutrition and the adaptation to endurance training. Sports Med. 2014;44((Suppl. 1)):S5–S12. doi: 10.1007/s40279-014-0146-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chinsomboon J., Ruas J., Gupta R.K., Thom R., Shoag J., Rowe G.C., Sawada N., Raghuram S., Arany Z. The transcriptional coactivator PGC-1alpha mediates exercise-induced angiogenesis in skeletal muscle. Proc. Natl. Acad. Sci. USA. 2009;106:21401–21406. doi: 10.1073/pnas.0909131106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arany Z., Lebrasseur N., Morris C., Smith E., Yang W., Ma Y., Chin S., Spiegelman B.M. The transcriptional coactivator PGC-1beta drives the formation of oxidative type IIX fibers in skeletal muscle. Cell Metab. 2007;5:35–46. doi: 10.1016/j.cmet.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 45.Eggler A.L., Small E., Hannink M., Mesecar A.D. Cul3-mediated Nrf2 ubiquitination and antioxidant response element (ARE) activation are dependent on the partial molar volume at position 151 of Keap1. Biochem. J. 2009;422:171–180. doi: 10.1042/BJ20090471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bryan H.K., Olayanju A., Goldring C.E., Park B.K. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem. Pharmacol. 2013;85:705–717. doi: 10.1016/j.bcp.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 47.Raghunath A., Sundarraj K., Nagarajan R., Arfuso F., Bian J., Kumar A.P., Sethi G., Perumal E. Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox Biol. 2018;17:297–314. doi: 10.1016/j.redox.2018.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kang D., Kim S.H., Hamasaki N. Mitochondrial transcription factor A (TFAM): Roles in maintenance of mtDNA and cellular functions. Mitochondrion. 2007;7:39–44. doi: 10.1016/j.mito.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 49.Lee J., Giordano S., Zhang J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem. J. 2012;441:523–540. doi: 10.1042/BJ20111451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garza-Lombó C., Pappa A., Panayiotidis M.I., Franco R. Redox homeostasis, oxidative stress and mitophagy. Mitochondrion. 2020;51:105–117. doi: 10.1016/j.mito.2020.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Komatsu M., Kurokawa H., Waguri S., Taguchi K., Kobayashi A., Ichimura Y., Sou Y.S., Ueno I., Sakamoto A., Tong K.I., et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010;12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 52.Yamada T., Murata D., Adachi Y., Itoh K., Kameoka S., Igarashi A., Kato T., Araki Y., Huganir R.L., Dawson T.M., et al. Mitochondrial Stasis Reveals p62-Mediated Ubiquitination in Parkin-Independent Mitophagy and Mitigates Nonalcoholic Fatty Liver Disease. Cell Metab. 2018;28:588–604. doi: 10.1016/j.cmet.2018.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Georgakopoulos N.D., Frison M., Alvarez M.S., Bertrand H., Wells G., Campanella M. Reversible Keap1 inhibitors are preferential pharmacological tools to modulate cellular mitophagy. Sci. Rep. 2017;7:10303. doi: 10.1038/s41598-017-07679-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Laker R.C., Drake J.C., Wilson R.J., Lira V.A., Lewellen B.M., Ryall K.A., Fisher C.C., Zhang M., Saucerman J.J., Goodyear L.J., et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 2017;8:548. doi: 10.1038/s41467-017-00520-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H., Miao W., Ma J., Xv Z., Bo H., Li J., Zhang Y., Ji L.L. Acute Exercise-Induced Mitochondrial Stress Triggers an Inflammatory Response in the Myocardium via NLRP3 Inflammasome Activation with Mitophagy. Oxidative Med. Cell. Longev. 2016;2016:1987149. doi: 10.1155/2016/1987149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ji L.L., Gomez-Cabrera M.C., Vina J. Exercise and hormesis: Activation of cellular antioxidant signaling pathway. Ann. N. Y. Acad. Sci. 2006;1067:425–435. doi: 10.1196/annals.1354.061. [DOI] [PubMed] [Google Scholar]
- 57.Lawler J.M., Rodriguez D.A., Hord J.M. Mitochondria in the middle: Exercise preconditioning protection of striated muscle. J. Physiol. 2016;594:5161–5183. doi: 10.1113/JP270656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alleman R.J., Katunga L.A., Nelson M.A., Brown D.A., Anderson E.J. The “Goldilocks Zone” from a redox perspective-Adaptive vs. deleterious responses to oxidative stress in striated muscle. Front. Physiol. 2014;5:358. doi: 10.3389/fphys.2014.00358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang W.C., Hung M.C. Beyond NF-κB activation: Nuclear functions of IκB kinase α. J. Biomed. Sci. 2013;20:3. doi: 10.1186/1423-0127-20-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li Q., Engelhardt J.F. Interleukin-1beta induction of NFkappaB is partially regulated by H2O2-mediated activation of NFkappaB-inducing kinase. J. Biol. Chem. 2006;281:1495–1505. doi: 10.1074/jbc.M511153200. [DOI] [PubMed] [Google Scholar]
- 61.Ghosh S., Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109((Suppl. 1)):S81–S96. doi: 10.1016/S0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 62.Obata T., Brown G.E., Yaffe M.B. MAP kinase pathways activated by stress: The p38 MAPK pathway. Crit. Care Med. 2000;28:N67–N77. doi: 10.1097/00003246-200004001-00008. [DOI] [PubMed] [Google Scholar]
- 63.Kyriakis J.M., Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 64.Cuschieri J., Maier R.V. Mitogen-activated protein kinase (MAPK) Crit. Care Med. 2005;33:S417–S419. doi: 10.1097/01.CCM.0000191714.39495.A6. [DOI] [PubMed] [Google Scholar]
- 65.Merry T.L., Ristow M. Mitohormesis in exercise training. Free. Radic. Biol. Med. 2016;98:123–130. doi: 10.1016/j.freeradbiomed.2015.11.032. [DOI] [PubMed] [Google Scholar]
- 66.Musci R.V., Hamilton K.L., Linden M.A. Exercise-Induced Mitohormesis for the Maintenance of Skeletal Muscle and Healthspan Extension. Sports. 2019;7:170. doi: 10.3390/sports7070170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang P., Li C.G., Qi Z., Cui D., Ding S. Acute exercise stress promotes Ref1/Nrf2 signalling and increases mitochondrial antioxidant activity in skeletal muscle. Exp. Physiol. 2016;101:410–420. doi: 10.1113/EP085493. [DOI] [PubMed] [Google Scholar]
- 68.Merry T.L., Ristow M. Nuclear factor erythroid-derived 2-like 2 (NFE2L2, Nrf2) mediates exercise-induced mitochondrial biogenesis and the anti-oxidant response in mice. J. Physiol. 2016;594:5195–5207. doi: 10.1113/JP271957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pani G., Galeotti T. Role of MnSOD and p66shc in mitochondrial response to p53. Antioxid. Redox Signal. 2011;15:1715–1727. doi: 10.1089/ars.2010.3499. [DOI] [PubMed] [Google Scholar]
- 70.Pani G., Koch O.R., Galeotti T. The p53-p66shc-Manganese Superoxide Dismutase (MnSOD) network: A mitochondrial intrigue to generate reactive oxygen species. Int. J. Biochem. Cell Biol. 2009;41:1002–1005. doi: 10.1016/j.biocel.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 71.Wang P., Li C.G., Qi Z., Cui D., Ding S. Acute exercise induced mitochondrial H2O2 production in mouse skeletal muscle: Association with p(66Shc) and FOXO3a signaling and antioxidant enzymes. Oxidative Med. Cell. Longev. 2015;2015:536456. doi: 10.1155/2015/536456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Iversen N., Krustrup P., Rasmussen H.N., Rasmussen U.F., Saltin B., Pilegaard H. Mitochondrial biogenesis and angiogenesis in skeletal muscle of the elderly. Exp. Gerontol. 2011;46:670–678. doi: 10.1016/j.exger.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 73.Demine S., Renard P., Arnould T. Mitochondrial Uncoupling: A Key Controller of Biological Processes in Physiology and Diseases. Cells. 2019;8:795. doi: 10.3390/cells8080795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jastroch M., Divakaruni A.S., Mookerjee S., Treberg J.R., Brand M.D. Mitochondrial proton and electron leaks. Essays Biochem. 2010;47:53–67. doi: 10.1042/bse0470053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Murphy M.P. Slip and leak in mitochondrial oxidative phosphorylation. Biochim. Biophys. Acta. 1989;977:123–141. doi: 10.1016/S0005-2728(89)80063-5. [DOI] [PubMed] [Google Scholar]
- 76.Crichton P.G., Lee Y., Kunji E.R. The molecular features of uncoupling protein 1 support a conventional mitochondrial carrier-like mechanism. Biochimie. 2017;134:35–50. doi: 10.1016/j.biochi.2016.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chouchani E.T., Kazak L., Jedrychowski M.P., Lu G.Z., Erickson B.K., Szpyt J., Pierce K.A., Laznik-Bogoslavski D., Vetrivelan R., Clish C.B., et al. Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature. 2016;532:112–116. doi: 10.1038/nature17399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gaspar R.C., Muñoz V.R., Kuga G.K., Nakandakari S., Minuzzi L.G., Botezelli J.D., da Silva A.S.R., Cintra D.E., de Moura L.P., Ropelle E.R., et al. Acute physical exercise increases leptin-induced hypothalamic extracellular signal-regulated kinase1/2 phosphorylation and thermogenesis of obese mice. J. Cell. Biochem. 2019;120:697–704. doi: 10.1002/jcb.27426. [DOI] [PubMed] [Google Scholar]
- 79.Berardi M.J., Chou J.J. Fatty acid flippase activity of UCP2 is essential for its proton transport in mitochondria. Cell Metab. 2014;20:541–552. doi: 10.1016/j.cmet.2014.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cadenas S., Buckingham J.A., Samec S., Seydoux J., Din N., Dulloo A.G., Brand M.D. UCP2 and UCP3 rise in starved rat skeletal muscle but mitochondrial proton conductance is unchanged. FEBS Lett. 1999;462:257–260. doi: 10.1016/S0014-5793(99)01540-9. [DOI] [PubMed] [Google Scholar]
- 81.Anedda A., López-Bernardo E., Acosta-Iborra B., Saadeh Suleiman M., Landázuri M.O., Cadenas S. The transcription factor Nrf2 promotes survival by enhancing the expression of uncoupling protein 3 under conditions of oxidative stress. Free Radic. Biol. Med. 2013;61:395–407. doi: 10.1016/j.freeradbiomed.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 82.Harper J.A., Stuart J.A., Jekabsons M.B., Roussel D., Brindle K.M., Dickinson K., Jones R.B., Brand M.D. Artifactual uncoupling by uncoupling protein 3 in yeast mitochondria at the concentrations found in mouse and rat skeletal-muscle mitochondria. Biochem. J. 2002;361:49–56. doi: 10.1042/bj3610049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nabben M., Shabalina I.G., Moonen-Kornips E., van Beurden D., Cannon B., Schrauwen P., Nedergaard J., Hoeks J. Uncoupled respiration, ROS production, acute lipotoxicity and oxidative damage in isolated skeletal muscle mitochondria from UCP3-ablated mice. Biochim. Biophys. Acta. 2011;1807:1095–1105. doi: 10.1016/j.bbabio.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 84.Kusuhara K., Tobe T., Negoro T., Abe T. A rapid up-regulation in UCP3 transcriptional activity in response to moderate intensity exercise in rat skeletal muscle. J. Sports Sci. Med. 2005;4:170–178. [PMC free article] [PubMed] [Google Scholar]
- 85.Jiang N., Zhang G., Bo H., Qu J., Ma G., Cao D., Wen L., Liu S., Ji L.L., Zhang Y. Upregulation of uncoupling protein-3 in skeletal muscle during exercise: A potential antioxidant function. Free. Radic. Biol. Med. 2009;46:138–145. doi: 10.1016/j.freeradbiomed.2008.09.026. [DOI] [PubMed] [Google Scholar]
- 86.Bo H., Wang Y.H., Li H.Y., Zhao J., Zhang H.Y., Tong C.Q. Endurance training attenuates the bioenergetics alterations of rat skeletal muscle mitochondria submitted to acute hypoxia: Role of ROS and UCP3. Sheng Li Xue Bao [Acta physiologica Sinica] 2008;60:767–776. [PubMed] [Google Scholar]
- 87.Peake J.M., Markworth J.F., Nosaka K., Raastad T., Wadley G.D., Coffey V.G. Modulating exercise-induced hormesis: Does less equal more? J. Appl. Physiol. 2015;119:172–189. doi: 10.1152/japplphysiol.01055.2014. [DOI] [PubMed] [Google Scholar]
- 88.Kadaja L., Eimre M., Paju K., Roosimaa M., Põdramägi T., Kaasik P., Pehme A., Orlova E., Mudist M., Peet N., et al. Impaired oxidative phosphorylation in overtrained rat myocardium. Exp. Clin. Cardiol. 2010;15:e116–e127. [PMC free article] [PubMed] [Google Scholar]
- 89.Urhausen A., Gabriel H.H., Weiler B., Kindermann W. Ergometric and psychological findings during overtraining: A long-term follow-up study in endurance athletes. Int. J. Sports Med. 1998;19:114–120. doi: 10.1055/s-2007-971892. [DOI] [PubMed] [Google Scholar]
- 90.Huang C.C., Lin T.J., Chen C.C., Lin W.T. Endurance training accelerates exhaustive exercise-induced mitochondrial DNA deletion and apoptosis of left ventricle myocardium in rats. Eur. J. Appl. Physiol. 2009;107:697–706. doi: 10.1007/s00421-009-1177-4. [DOI] [PubMed] [Google Scholar]
- 91.King D.W., Gollnick P.D. Ultrastructure of rat heart and liver after exhaustive exercise. Am. J. Physiol. 1970;218:1150–1155. doi: 10.1152/ajplegacy.1970.218.4.1150. [DOI] [PubMed] [Google Scholar]
- 92.Venditti P., Di Meo S. Antioxidants, tissue damage, and endurance in trained and untrained young male rats. Arch. Biochem. Biophys. 1996;331:63–68. doi: 10.1006/abbi.1996.0283. [DOI] [PubMed] [Google Scholar]
- 93.Seene T., Kaasik P., Alev K., Pehme A., Riso E.M. Composition and turnover of contractile proteins in volume-overtrained skeletal muscle. Int. J. Sports Med. 2004;25:438–445. doi: 10.1055/s-2004-820935. [DOI] [PubMed] [Google Scholar]
- 94.Ferraresso R.L., de Oliveira R., Macedo D.V., Nunes L.A., Brenzikofer R., Damas D., Hohl R. Interaction between overtraining and the interindividual variability may (not) trigger muscle oxidative stress and cardiomyocyte apoptosis in rats. Oxidative Med. Cell. Longev. 2012;2012:935483. doi: 10.1155/2012/935483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fang W., Li Z., Liu X.H., Liu Z.M., Dun Y., Feng H. Effects of exhaustive exercise on the expression of PHB1 and the function of mitochondria in rats. Zhongguo Yingyong Shenglixue Zazhi (Chin. J. Appl. Physiol.) 2017;33:544–549. doi: 10.12047/j.cjap.5601.2017.129. [DOI] [PubMed] [Google Scholar]
- 96.Ross J.A., Nagy Z.S., Kirken R.A. The PHB1/2 phosphocomplex is required for mitochondrial homeostasis and survival of human T cells. J. Biol. Chem. 2008;283:4699–4713. doi: 10.1074/jbc.M708232200. [DOI] [PubMed] [Google Scholar]
- 97.Di Meo S., Venditti P. Mitochondria in exercise-induced oxidative stress. Biol. Signals Recept. 2001;10:125–140. doi: 10.1159/000046880. [DOI] [PubMed] [Google Scholar]
- 98.Ljubicic V., Joseph A.M., Saleem A., Uguccioni G., Collu-Marchese M., Lai R.Y., Nguyen L.M., Hood D.A. Transcriptional and post-transcriptional regulation of mitochondrial biogenesis in skeletal muscle: Effects of exercise and aging. Biochim. Biophys. Acta. 2010;1800:223–234. doi: 10.1016/j.bbagen.2009.07.031. [DOI] [PubMed] [Google Scholar]
- 99.Hood D.A. Mechanisms of exercise-induced mitochondrial biogenesis in skeletal muscle. Appl. Physiol. Nutr. Metab. (Physiologie appliquee Nutrition et Metabolisme) 2009;34:465–472. doi: 10.1139/H09-045. [DOI] [PubMed] [Google Scholar]
- 100.Laguens R.P., Gómez-Dumm C.L. Fine structure of myocardial mitochondria in rats after exercise for one-half to two hours. Circ. Res. 1967;21:271–279. doi: 10.1161/01.RES.21.3.271. [DOI] [PubMed] [Google Scholar]
- 101.Merry T.L., Ristow M. Do antioxidant supplements interfere with skeletal muscle adaptation to exercise training? J. Physiol. 2016;594:5135–5147. doi: 10.1113/JP270654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Peternelj T.T., Coombes J.S. Antioxidant supplementation during exercise training: Beneficial or detrimental? Sports Med. 2011;41:1043–1069. doi: 10.2165/11594400-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 103.Slattery K., Bentley D., Coutts A.J. The role of oxidative, inflammatory and neuroendocrinological systems during exercise stress in athletes: Implications of antioxidant supplementation on physiological adaptation during intensified physical training. Sports Med. 2015;45:453–471. doi: 10.1007/s40279-014-0282-7. [DOI] [PubMed] [Google Scholar]
- 104.Antonioni A., Fantini C., Dimauro I., Caporossi D. Redox homeostasis in sport: Do athletes really need antioxidant support? Res. Sports Med. (Print) 2019;27:147–165. doi: 10.1080/15438627.2018.1563899. [DOI] [PubMed] [Google Scholar]
- 105.Margaritis I., Rousseau A.S. Does physical exercise modify antioxidant requirements? Nutr. Res. Rev. 2008;21:3–12. doi: 10.1017/S0954422408018076. [DOI] [PubMed] [Google Scholar]
- 106.de Oliveira D.C.X., Rosa F.T., Simões-Ambrósio L., Jordao A.A., Deminice R. Antioxidant vitamin supplementation prevents oxidative stress but does not enhance performance in young football athletes. Nutrition. 2019;63–64:29–35. doi: 10.1016/j.nut.2019.01.007. [DOI] [PubMed] [Google Scholar]
- 107.Ryan M.J., Dudash H.J., Docherty M., Geronilla K.B., Baker B.A., Haff G.G., Cutlip R.G., Alway S.E. Vitamin E and C supplementation reduces oxidative stress, improves antioxidant enzymes and positive muscle work in chronically loaded muscles of aged rats. Exp. Gerontol. 2010;45:882–895. doi: 10.1016/j.exger.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Morrison D., Hughes J., Della Gatta P.A., Mason S., Lamon S., Russell A.P., Wadley G.D. Vitamin C and E supplementation prevents some of the cellular adaptations to endurance-training in humans. Free. Radic. Biol. Med. 2015;89:852–862. doi: 10.1016/j.freeradbiomed.2015.10.412. [DOI] [PubMed] [Google Scholar]
- 109.Paulsen G., Cumming K.T., Holden G., Hallén J., Rønnestad B.R., Sveen O., Skaug A., Paur I., Bastani N.E., Østgaard H.N., et al. Vitamin C and E supplementation hampers cellular adaptation to endurance training in humans: A double-blind, randomised, controlled trial. J. Physiol. 2014;592:1887–1901. doi: 10.1113/jphysiol.2013.267419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cory H., Passarelli S., Szeto J., Tamez M., Mattei J. The Role of Polyphenols in Human Health and Food Systems: A Mini-Review. Front. Nutr. 2018;5:87. doi: 10.3389/fnut.2018.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Myburgh K.H. Polyphenol supplementation: Benefits for exercise performance or oxidative stress? Sports Med. 2014;44((Suppl. 1)):S57–S70. doi: 10.1007/s40279-014-0151-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Elejalde E., Villarán M.C., Alonso R.M. Grape polyphenols supplementation for exercise-induced oxidative stress. J. Int. Soc. Sports Nutr. 2021;18:3. doi: 10.1186/s12970-020-00395-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Salehi M., Mashhadi N.S., Esfahani P.S., Feizi A., Hadi A., Askari G. The Effects of Curcumin Supplementation on Muscle Damage, Oxidative Stress, and Inflammatory Markers in Healthy Females with Moderate Physical Activity: A Randomized, Double-Blind, Placebo-Controlled Clinical Trial. Int. J. Prev. Med. 2021;12:94. doi: 10.4103/ijpvm.IJPVM_138_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Vargas-Mendoza N., Ángeles-Valencia M., Madrigal-Santillán E.O., Morales-Martínez M., Tirado-Lule J.M., Solano-Urrusquieta A., Madrigal-Bujaidar E., Álvarez-González I., Fregoso-Aguilar T., Morales-González Á., et al. Effect of Silymarin Supplementation on Physical Performance, Muscle and Myocardium Histological Changes, Bodyweight, and Food Consumption in Rats Subjected to Regular Exercise Training. Int. J. Mol. Sci. 2020;21:7724. doi: 10.3390/ijms21207724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Oh S., Komine S., Warabi E., Akiyama K., Ishii A., Ishige K., Mizokami Y., Kuga K., Horie M., Miwa Y., et al. Nuclear factor (erythroid derived 2)-like 2 activation increases exercise endurance capacity via redox modulation in skeletal muscles. Sci. Rep. 2017;7:12902. doi: 10.1038/s41598-017-12926-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ruhee R.T., Suzuki K. The Integrative Role of Sulforaphane in Preventing Inflammation, Oxidative Stress and Fatigue: A Review of a Potential Protective Phytochemical. Antioxidants. 2020;9:521. doi: 10.3390/antiox9060521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Vargas-Mendoza N., Morales-González Á., Morales-Martínez M., Soriano-Ursúa M.A., Delgado-Olivares L., Sandoval-Gallegos E.M., Madrigal-Bujaidar E., Álvarez-González I., Madrigal-Santillán E., Morales-Gonzalez J.A. Flavolignans from Silymarin as Nrf2 Bioactivators and Their Therapeutic Applications. Biomedicines. 2020;8:122. doi: 10.3390/biomedicines8050122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Farzaei M.H., Zobeiri M., Parvizi F., El-Senduny F.F., Marmouzi I., Coy-Barrera E., Naseri R., Nabavi S.M., Rahimi R., Abdollahi M. Curcumin in Liver Diseases: A Systematic Review of the Cellular Mechanisms of Oxidative Stress and Clinical Perspective. Nutrients. 2018;10:855. doi: 10.3390/nu10070855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hassan F.U., Rehman M.S., Khan M.S., Ali M.A., Javed A., Nawaz A., Yang C. Curcumin as an Alternative Epigenetic Modulator: Mechanism of Action and Potential Effects. Front. Genet. 2019;10:514. doi: 10.3389/fgene.2019.00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tryfidou D.V., McClean C., Nikolaidis M.G., Davison G.W. DNA Damage Following Acute Aerobic Exercise: A Systematic Review and Meta-analysis. Sports Med. 2020;50:103–127. doi: 10.1007/s40279-019-01181-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Murphy M.P., Hartley R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018;17:865–886. doi: 10.1038/nrd.2018.174. [DOI] [PubMed] [Google Scholar]
- 122.Yavari A., Javadi M., Mirmiran P., Bahadoran Z. Exercise-induced oxidative stress and dietary antioxidants. Asian J. Sports Med. 2015;6:e24898. doi: 10.5812/asjsm.24898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pingitore A., Lima G.P., Mastorci F., Quinones A., Iervasi G., Vassalle C. Exercise and oxidative stress: Potential effects of antioxidant dietary strategies in sports. Nutrition. 2015;31:916–922. doi: 10.1016/j.nut.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 124.Neubauer O., Yfanti C. Antioxidants in Athlete’s Basic Nutrition: Considerations towards a Guideline for the Intake of Vitamin C and Vitamin E. In: Lamprecht M., editor. Antioxidants in Sport Nutrition. Taylor & Francis Group, LLC.; Boca Raton, FL, USA: 2015. [PubMed] [Google Scholar]
- 125.Fogarty M.C., Devito G., Hughes C.M., Burke G., Brown J.C., McEneny J., Brown D., McClean C., Davison G.W. Effects of α-lipoic acid on mtDNA damage after isolated muscle contractions. Med. Sci. Sports Exerc. 2013;45:1469–1477. doi: 10.1249/MSS.0b013e31828bf31e. [DOI] [PubMed] [Google Scholar]
- 126.Xiao L., Xu X., Zhang F., Wang M., Xu Y., Tang D., Wang J., Qin Y., Liu Y., Tang C., et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017;11:297–311. doi: 10.1016/j.redox.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rossman M.J., Santos-Parker J.R., Steward C.A.C., Bispham N.Z., Cuevas L.M., Rosenberg H.L., Woodward K.A., Chonchol M., Gioscia-Ryan R.A., Murphy M.P., et al. Chronic Supplementation With a Mitochondrial Antioxidant (MitoQ) Improves Vascular Function in Healthy Older Adults. Hypertension. 2018;71:1056–1063. doi: 10.1161/HYPERTENSIONAHA.117.10787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhou J., Wang H., Shen R., Fang J., Yang Y., Dai W., Zhu Y., Zhou M. Mitochondrial-targeted antioxidant MitoQ provides neuroprotection and reduces neuronal apoptosis in experimental traumatic brain injury possibly via the Nrf2-ARE pathway. Am. J. Transl. Res. 2018;10:1887–1899. [PMC free article] [PubMed] [Google Scholar]
- 129.Mao G., Kraus G.A., Kim I., Spurlock M.E., Bailey T.B., Zhang Q., Beitz D.C. A mitochondria-targeted vitamin E derivative decreases hepatic oxidative stress and inhibits fat deposition in mice. J. Nutr. 2010;140:1425–1431. doi: 10.3945/jn.110.121715. [DOI] [PubMed] [Google Scholar]
- 130.Mao G., Kraus G.A., Kim I., Spurlock M.E., Bailey T.B., Beitz D.C. Effect of a mitochondria-targeted vitamin E derivative on mitochondrial alteration and systemic oxidative stress in mice. Br. J. Nutr. 2011;106:87–95. doi: 10.1017/S0007114510005830. [DOI] [PubMed] [Google Scholar]
- 131.Fang Y., Hu X.-H., Jia Z.-G., Xu M.-H., Guo Z.-Y., Gao F.-H. Tiron protects against UVB-induced senescence-like characteristics in human dermal fibroblasts by the inhibition of superoxide anion production and glutathione depletion. Australas. J. Dermatol. 2012;53:172–180. doi: 10.1111/j.1440-0960.2012.00912.x. [DOI] [PubMed] [Google Scholar]
- 132.Finichiu P.G., Larsen D.S., Evans C., Larsen L., Bright T.P., Robb E.L., Trnka J., Prime T.A., James A.M., Smith R.A.J., et al. A mitochondria-targeted derivative of ascorbate: MitoC. Free. Radic. Biol. Med. 2015;89:668–678. doi: 10.1016/j.freeradbiomed.2015.07.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Williamson J., Davison G. Targeted Antioxidants in Exercise-Induced Mitochondrial Oxidative Stress: Emphasis on DNA Damage. Antioxidants. 2020;9:1142. doi: 10.3390/antiox9111142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Oyewole A.O., Birch-Machin M.A. Mitochondria-targeted antioxidants. FASEB J. 2015;29:4766–4771. doi: 10.1096/fj.15-275404. [DOI] [PubMed] [Google Scholar]
- 135.Smith R.A.J., Murphy M.P. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann. N. Y. Acad. Sci. 2010;1201:96–103. doi: 10.1111/j.1749-6632.2010.05627.x. [DOI] [PubMed] [Google Scholar]
- 136.Cochemé H.M., Kelso G.F., James A.M., Ross M.F., Trnka J., Mahendiran T., Asin-Cayuela J., Blaikie F.H., Manas A.R., Porteous C.M., et al. Mitochondrial targeting of quinones: Therapeutic implications. Mitochondrion. 2007;7((Suppl.)):S94–S102. doi: 10.1016/j.mito.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 137.Ross M.F., Kelso G.F., Blaikie F.H., James A.M., Cochemé H.M., Filipovska A., Da Ros T., Hurd T.R., Smith R.A., Murphy M.P. Lipophilic triphenylphosphonium cations as tools in mitochondrial bioenergetics and free radical biology. Biochem. Biokhimiia. 2005;70:222–230. doi: 10.1007/s10541-005-0104-5. [DOI] [PubMed] [Google Scholar]
- 138.Murphy M.P. Targeting lipophilic cations to mitochondria. Biochim. Biophys. Acta. 2008;1777:1028–1031. doi: 10.1016/j.bbabio.2008.03.029. [DOI] [PubMed] [Google Scholar]
- 139.James A.M., Sharpley M.S., Manas A.R., Frerman F.E., Hirst J., Smith R.A., Murphy M.P. Interaction of the mitochondria-targeted antioxidant MitoQ with phospholipid bilayers and ubiquinone oxidoreductases. J. Biol. Chem. 2007;282:14708–14718. doi: 10.1074/jbc.M611463200. [DOI] [PubMed] [Google Scholar]
- 140.James A.M., Cochemé H.M., Smith R.A., Murphy M.P. Interactions of mitochondria-targeted and untargeted ubiquinones with the mitochondrial respiratory chain and reactive oxygen species. Implications for the use of exogenous ubiquinones as therapies and experimental tools. J. Biol. Chem. 2005;280:21295–21312. doi: 10.1074/jbc.M501527200. [DOI] [PubMed] [Google Scholar]
- 141.Shill D.D., Southern W.M., Willingham T.B., Lansford K.A., McCully K.K., Jenkins N.T. Mitochondria-specific antioxidant supplementation does not influence endurance exercise training-induced adaptations in circulating angiogenic cells, skeletal muscle oxidative capacity or maximal oxygen uptake. J. Physiol. 2016;594:7005–7014. doi: 10.1113/JP272491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Williamson J., Hughes C.M., Cobley J.N., Davison G.W. The mitochondria-targeted antioxidant MitoQ, attenuates exercise-induced mitochondrial DNA damage. Redox Biol. 2020;36:101673. doi: 10.1016/j.redox.2020.101673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Murphy M.P. Understanding and preventing mitochondrial oxidative damage. Biochem. Soc. Trans. 2016;44:1219–1226. doi: 10.1042/BST20160108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Antonenko Y.N., Avetisyan A.V., Bakeeva L.E., Chernyak B.V., Chertkov V.A., Domnina L.V., Ivanova O.Y., Izyumov D.S., Khailova L.S., Klishin S.S., et al. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 1. Cationic plastoquinone derivatives: Synthesis and in vitro studies. Biochemistry. 2008;73:1273–1287. doi: 10.1134/S0006297908120018. [DOI] [PubMed] [Google Scholar]
- 145.Schlame M., Greenberg M.L. Biosynthesis, remodeling and turnover of mitochondrial cardiolipin. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids. 2017;1862:3–7. doi: 10.1016/j.bbalip.2016.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Park S.Y., Pekas E.J., Headid R.J., 3rd, Son W.M., Wooden T.K., Song J., Layec G., Yadav S.K., Mishra P.K., Pipinos I.I. Acute mitochondrial antioxidant intake improves endothelial function, antioxidant enzyme activity, and exercise tolerance in patients with peripheral artery disease. Am. J. Physiol. Heart Circ. Physiol. 2020;319:H456–H467. doi: 10.1152/ajpheart.00235.2020. [DOI] [PubMed] [Google Scholar]
- 147.Hu Q., Ren J., Li G., Wu J., Wu X., Wang G., Gu G., Ren H., Hong Z., Li J. The mitochondrially targeted antioxidant MitoQ protects the intestinal barrier by ameliorating mitochondrial DNA damage via the Nrf2/ARE signaling pathway. Cell Death Dis. 2018;9:403. doi: 10.1038/s41419-018-0436-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Boris A.F., Vladimir P.S. Cellular and Molecular Mechanisms of Action of Mitochondria-Targeted Antioxidants. Curr. Aging Sci. 2017;10:41–48. doi: 10.2174/1874609809666160921113706. [DOI] [PubMed] [Google Scholar]
- 149.Birk A.V., Chao W.M., Bracken C., Warren J.D., Szeto H.H. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br. J. Pharmacol. 2014;171:2017–2028. doi: 10.1111/bph.12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Birk A.V., Liu S., Soong Y., Mills W., Singh P., Warren J.D., Seshan S.V., Pardee J.D., Szeto H.H. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J. Am. Soc. Nephrol. JASN. 2013;24:1250–1261. doi: 10.1681/ASN.2012121216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Szeto H.H. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br. J. Pharmacol. 2014;171:2029–2050. doi: 10.1111/bph.12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Min K., Smuder A.J., Kwon O.S., Kavazis A.N., Szeto H.H., Powers S.K. Mitochondrial-targeted antioxidants protect skeletal muscle against immobilization-induced muscle atrophy. J. Appl. Physiol. 2011;111:1459–1466. doi: 10.1152/japplphysiol.00591.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Andrews S.J., Rothnagel J.A. Emerging evidence for functional peptides encoded by short open reading frames. Nat. Rev. Genet. 2014;15:193–204. doi: 10.1038/nrg3520. [DOI] [PubMed] [Google Scholar]
- 154.Lin Y.F., Xiao M.H., Chen H.X., Meng Y., Zhao N., Yang L., Tang H., Wang J.L., Liu X., Zhu Y., et al. A novel mitochondrial micropeptide MPM enhances mitochondrial respiratory activity and promotes myogenic differentiation. Cell Death Dis. 2019;10:528. doi: 10.1038/s41419-019-1767-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Houghton C.A., Fassett R.G., Coombes J.S. Sulforaphane and Other Nutrigenomic Nrf2 Activators: Can the Clinician’s Expectation Be Matched by the Reality? Oxidative Med. Cell. Longev. 2016;2016:7857186. doi: 10.1155/2016/7857186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bhattacharjee S., Dashwood R.H. Epigenetic Regulation of NRF2/KEAP1 by Phytochemicals. Antioxidants. 2020;9:865. doi: 10.3390/antiox9090865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Nocella C., Cammisotto V., Pigozzi F., Borrione P., Fossati C., D’Amico A., Cangemi R., Peruzzi M., Gobbi G., Ettorre E., et al. Impairment between Oxidant and Antioxidant Systems: Short- and Long-term Implications for Athletes’ Health. Nutrients. 2019;11:1353. doi: 10.3390/nu11061353. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.