

Summary
Interfacial inhibitors exert their biological effects through co-association with two macromolecules. The pateamine A (PatA) class of molecules function by stabilizing eukaryotic initiation factor (eIF) 4A RNA helicase onto RNA, resulting in translation initiation inhibition. Here, we present the first crystal structure of an eIF4A1:RNA complex bound to an analogue of the marine sponge-derived natural product PatA, C5-desmethyl pateamine A (DMPatA). One end of this small molecule wedges itself between two RNA bases while the other end is cradled by several protein residues. Strikingly, DMPatA interacts with the eIF4A1:RNA complex in an almost identical fashion as rocaglamide A (RocA), despite being completely unrelated from a structural standpoint. The structural data rationalizes the ability of PatA analogues to target a wider range of RNA substrates compared to RocA. We define the molecular basis of how DMPatA is able to clamp eIF4A1 onto RNA, imparting potent inhibitory properties to this molecule.
Introduction
Eukaryotic initiation factor (eIF) 4A is the prototypical RNA helicase of the DEAD-box family, a group of related ATPases characterized by the presence of two RecA-like domains connected by a short, flexible linker. Clefts between these domains serve as docking sites for RNA, as well as ATP, whose binding and hydrolysis interconverts the protein between open and closed conformations to facilitate RNA unwinding (Andreou and Klostermeier, 2012). eIF4A is required for recruiting the 43S preinitiation complex (PIC: 40S ribosomes and associated initiation factors) to mRNA templates. The ribosome recruitment phase of translation initiation is catalyzed by eIF4F, a complex of three proteins: (i) eIF4E, a cap-binding protein required to bind to m7GpppN cap structures, (ii) eIF4G, a large multi-domain scaffold, and (iii) eIF4A (Pelletier and Sonenberg, 2019). Not all mRNAs display equivalent dependencies towards eIF4F and one feature that increases this reliance is elevated secondary structure within the mRNA 5’ leader region (Pelletier and Sonenberg, 2019). There are three paralogs of eIF4A, with two being implicated in translation initiation (eIF4A1 [DDX2A] and eIF4A2 [DDX2B]), and the third being critical for regulation of gene expression by nonsense-mediated decay (eIF4A3 [DDX48]). eIF4A1 is essential and is the more abundantly expressed homolog, whereas eIF4A2 is not essential and its role in translation is less well-characterized (Pelletier and Sonenberg, 2019). Among the mRNAs that show a higher dependence on eIF4F for initiation, several fuel hallmarks of cancer, making eIF4F and eIF4A targets for drug development (Bhat et al., 2015).
Two distinct natural products, pateamine A (PatA) (Northcote et al., 1991) and the rocaglates, are inhibitors of translation initiation that exert their effects by stabilizing eIF4A onto mRNA (Bordeleau et al., 2006; Bordeleau et al., 2005; Bordeleau et al., 2008; Chu et al., 2020; Iwasaki et al., 2016; Iwasaki et al., 2019; Low et al., 2005). Structural insight into the mechanism of action of rocaglates revealed that rocaglamide A (RocA) simultaneously interacts with both eIF4A1 and two adjacent purine RNA bases to stabilize eIF4A1:RNA complexes (Iwasaki et al., 2019). The critical role played by eIF4A1 residue F163 in mediating interaction with rocalgates was revealed by genetic experiments describing rocaglate-resistant alleles harboring an F163L mutation in yeast and mammalian cells (Chu et al., 2016; Iwasaki et al., 2019; Sadlish et al., 2013).
Structural simplification of PatA by removing the C3-amino and C5-methyl groups led to the synthesis of desmethyl desamino pateamine A (Fig 1a; DMDAPatA), a compound with in vivo anticancer activity in several xenograft models (Kuznetsov et al., 2009). There is currently a void in our understanding of the underlying molecular basis of how PatA or its analogs are able to stabilize eIF4A onto RNA. Herein, we report the crystal structure to 2.9 Å resolution of a quaternary complex consisting of human eIF4A1, AMPPNP, poly (AG)5 RNA, and a PatA analog, C5-desmethyl pateamine A (DMPatA), devoid of the C5-methyl group (Fig 1a).
Figure 1.
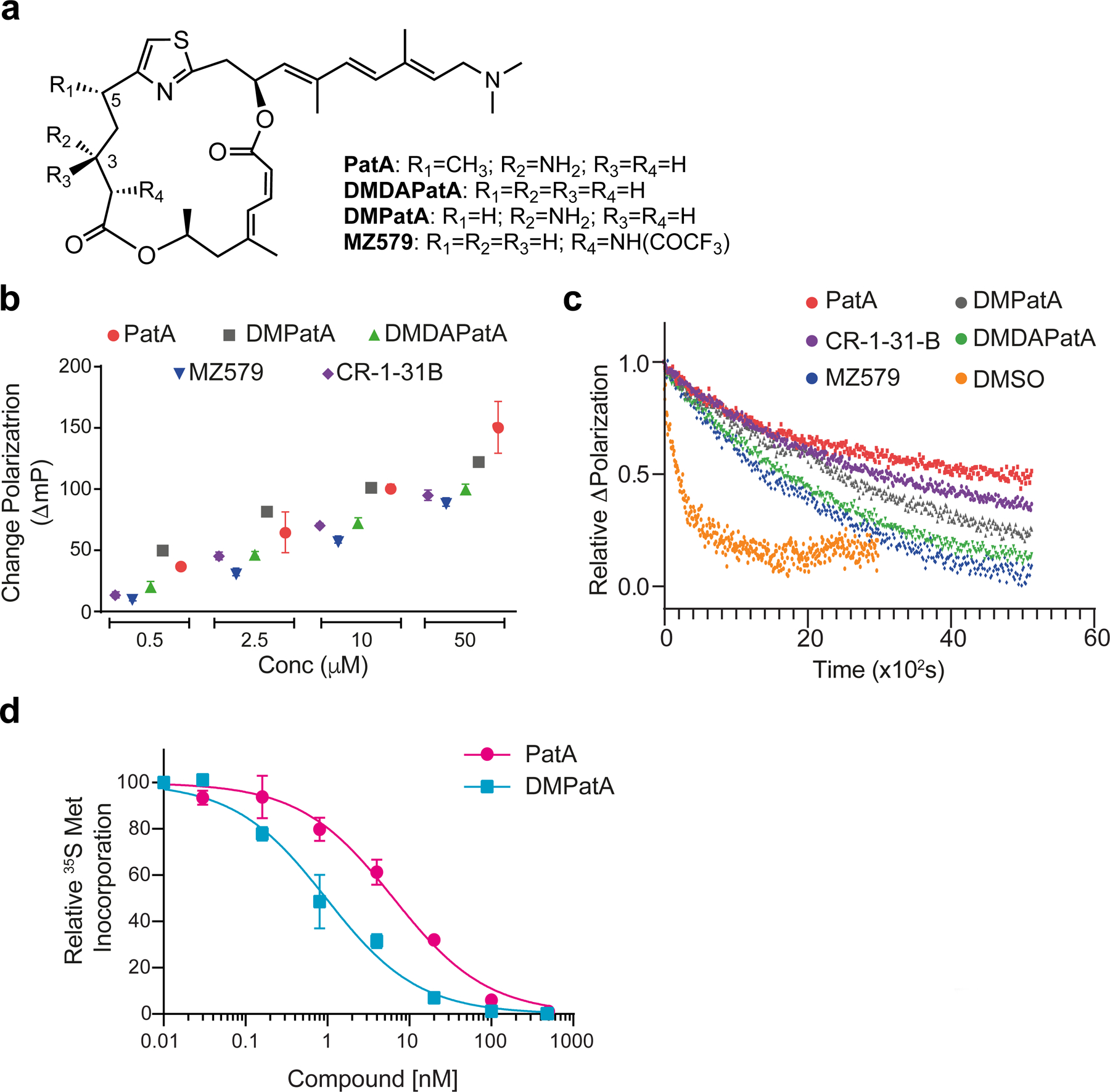
The PatA analogue, DMPatA, is a potent inhibitor of translation. a. Chemical structures of PatA analogs used in this study. b. Assessing compound induced clamping of eIF4A1 to poly (AG)8 RNA. The ΔmP obtained with eIF4A1:poly (AG)8 RNA was measured for the indicate compounds at 0.5, 2.5, 10, and 50 μM. The ΔmP obtained relative to DMSO is shown. n = 3 ± SEM. c. Relative dissociation of pre-formed eIF4A1•ATP•PatA analogs•FAM-poly (AG)8 complexes measured as a function of time in the presence of excess poly (AG)8 RNA. DMSO, t1/2 ~3 ± 0.15 min; PatA (2.5 μM), t1/2 ~65 ± 1.3 min; DMPatA (2.5 μM), t1/2 ~40 ± 0.3 min; DMDAPatA (10 μM), t1/2 ~ 28 ± 0.2 min; MZ579 (10μM), t1/2 ~ 23 ± 0.25 min; CR-1–31-B (10 μM), t1/2 ~ 50 ± 0.5 min. Error values were calculated from the 95% confidence intervals. d. DMPatA is a potent inhibitor of translation. 293T cells were incubated in the presence of the indicated concentrations of compound for 1h. During the last 15 min of incubation, 35S-Met was added to the cells before harvesting, followed by TCA precipitation and quantitation of 35S-Met incorporation into protein. n=2 ± SEM. See also Figure S1.
Results
DMPatA is a potent PatA analog that stabilizes eIF4A:RNA complexes.
As a prelude to structural studies, we assessed the activity of PatA and three synthetic PatA analogs in vitro (Fig 1a). Since PatA has been shown to stabilize eIF4A1:RNA complexes (Bordeleau et al., 2006; Bordeleau et al., 2005), we used a fluorescence polarization (FP) assay to measure the ability of PatA and analogs to clamp eIF4A1 onto RNA (Iwasaki et al., 2016). The rocaglate, CR-1–31-B was used as positive control in these experiments and showed RNA clamping to poly (AG)8 RNA comparable to MZ579 and DMDAPatA (Fig 1b). PatA and DMPatA exhibited more robust activity at all four tested concentrations (Fig 1b). To document the stabilizing effects of these compounds on eIF4A1:RNA complexes, we tracked the dissociation of the eIF4A1•RNA•ATP complexes in the presence of 2.5 μM PatA, 2.5 μM DMPatA, 10 μM DMDAPatA, or 10 μM MZ579. Complexes formed with PatA or DMPatA are more stable than those obtained with DMDAPatA or MZ579 (Fig 1c). As well in the absence of ATP, DMPatA (and CR-1–31-B) stimulated eIF4A1:poly (AG)8 complex formation, albeit less efficiently than in its presence, as assessed using the FP assay or a filter binding assay with 32Plabelled poly (AG)8 (Fig S1a). As well, eIF4A1:poly (AG)8 complexes formed in the presence of cold ATP were still active for ATP hydrolysis, as assessed by a chase using γ-32P-ATP (Fig S1b). Hence, DMPatA-clamped eIF4A1 can still hydrolyze ATP (see Discussion). DMPatA, like the natural product, is a 19-membered, thiazole-containing dilactone macrolide with a trienyl amine side-chain but differs from PatA by lacking the C5-methyl group and resultant stereocenter (Fig 1a). Translation was potently inhibited in 293T cells following only a 1 h exposure to DMPatA (IC50 ~1 nM) – more so than what was observed with PatA (IC50 ~6 nM) indicating DMPatA to be a potent translation inhibitor (Fig 1d). Given the similarity in structures and performances of DMPatA and PatA in these assays, and the fact that DMPatA unlike PatA is not limited in supply, we proceeded to undertake crystallization studies with human eIF4A1, DMPatA, and RNA.
Quaternary structure of eIF4A1•AMPPNP •poly (AG)5•DMPatA complexes.
The crystal structure of eIF4A1 bound to poly (AG)5 RNA, AMPPNP, and DMPatA was determined to 2.9 Å resolution (Fig 2a, Table 1). The overall structure of eIF4A1 is quite similar to previously determined complexes of eIF4A1 bound to RocA, polypurine RNA, and AMPPNP (Iwasaki et al., 2019), as well as the Vasa DEAD box helicase bound to ssRNA and AMPPNP (Fig S2a). The protein is in the closed conformation that occurs upon binding both RNA and ATP (Sengoku et al., 2006; Theissen et al., 2008). This results in the RNA helix, bound at the interface between the N-terminal domain (NTD) and C-terminal domain (CTD) being kinked between bases A7 and G8 and creating an exposed cavity on the surface of the complex (Fig S2b). The kinked RNA conformation is thought to play a role in initiating the mechanism of RNA unwinding (Andreou and Klostermeier, 2012). Here, we observed clear differences in electron density for DMPatA wedged within this cavity and spanning the width of the RNA helix (Figs 2a and S2b,c).
Figure 2.
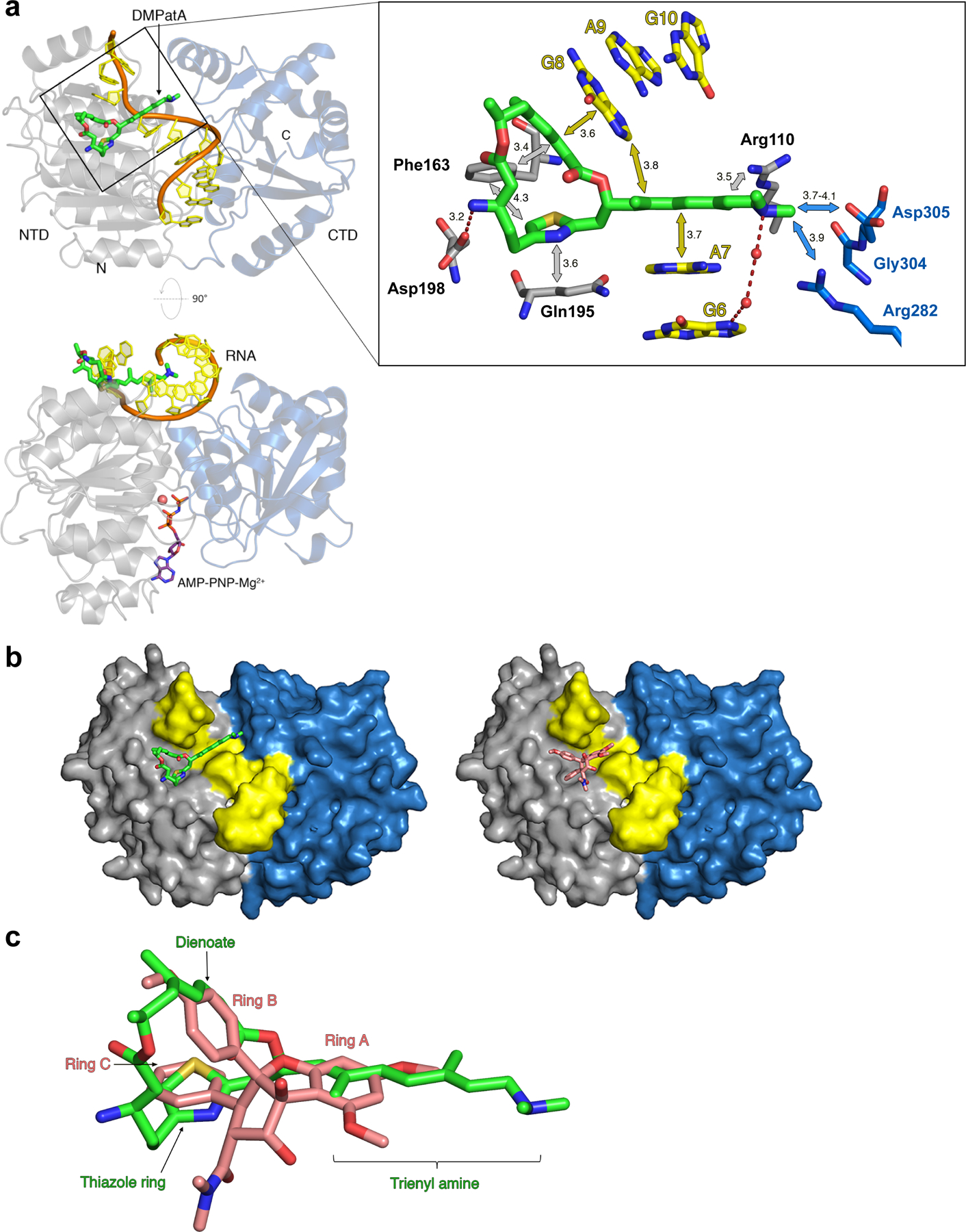
DMPatA interactions with eIF4A1 and poly (AG)5 RNA. a. Structure of DMPatA bound to eIF4A1•poly (AG)5•AMPPNP in two orthogonal views. NTD and CTD are represented as grey and blue ribbons, respectively. The RNA backbone is colored orange and the bases shown in yellow. DMPatA is shown in tube format with carbon, nitrogen, oxygen and sulfur colored green, blue, red and yellow, respectively. AMPPNP (omitted in the top view for clarity) is shown in tube format with pink carbon atoms and a magnesium ion shown as a pink sphere. A close-up of the boxed section in the top view reveals the main interactions between DMPatA and eIF4A1/RNA. NTD and CTD protein residues are shown in tube format with grey and blue carbon atoms, respectively. RNA is shown with yellow carbon atoms. Van der Waals/stacking interactions are indicated with grey/blue and yellow arrows for protein and RNA, respectively, along with corresponding distances in Angstroms. Dashed red lines refer to salt bridges with intervening water molecules represented as red spheres. The trienyl amine arm extends through the space created by the kink between RNA bases A7 and G8, linking the NTD (light grey) to the CTD (light blue). b. Comparison of the DMPatA (left; green) and RocA (right; pink) binding cavities in eIF4A. eIF4A1 is shown as a surface with the NTD, CTD and RNA colored grey, blue and yellow, respectively. c. Overlay of DMPatA (green) on RocA (pink) in the context of their binding to eIF4A1. Key components of each compound are labelled in green and pink for DMPatA and RocA, respectively. See also Figures S2 and S3.
Table 1.
Crystallographic data for X-ray structures of the eIF4A1•AMPPNP•poly (AG)5•DMPatA complex.
| Data collection | |
|---|---|
| PDB code | 6XKI |
| Space group | I222 |
| Cell dimensions | |
| a, b, c (Å) | 66.8, 99.9, 153.7 |
| α, β, γ (°) | 90, 90, 90 |
| Wavelength (Å) | 0.98 |
| Resolution (Å) | 50.0 – 2.87 (2.95 – 2.87) |
| Total reflections | 131634 (3533) |
| Unique reflections | 11588 (453) |
| Rmerge | 0.182 (0.864) |
| Rmeas | 0.190 (0.914) |
| Rpim | 0.054 (0.288) |
| CC1/2 | 0.984 (0.811) |
| I / σI | 12.25 (1.4) |
| Completeness (%) | 96.6 (76.6) |
| Redundancy | 11.4 (7.8) |
| Refinement | |
| Resolution (Å) | 45.58 – 2.87 (2.98 – 2.87) |
| No. reflections | 10623 (508) |
| Reflections used for Rfree | 536 (32) |
| Rwork / Rfree | 0.2207 / 0.2614 (0.3173 / 0.3086) |
| No. atoms | |
| Protein | 3244 |
| Ligand/ion | 70 |
| Solvent | 26 |
| Ramachandran favored (%) | 95.48 |
| Ramachandran allowed (%) | 4.52 |
| Ramachandran outliers (%) | 0 |
| Rotamer outliers (%) | 0 |
| Clashscore | 1.38 |
| B-factors | |
| Protein | 62.67 |
| Ligand/ion | 48.87 |
| Solvent | 44.89 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.003 |
| Bond angles (°) | 0.43 |
The macrocyclic ring interacts with both eIF4A1 (mainly the NTD) and RNA through several major interactions (Fig 2a). One edge of the macrocycle, the E,Z-dienoate region, π-stacks with G8 of the RNA segment while the rest of the macrocyclic ring is nestled in a pocket created by three amino acids – (i) Gln195 abuts the thiazole moiety through van der Waals interactions, (ii) Asp198 forms a salt bridge with the C3-primary amine, and (iii) Phe163 interacts with the bottom face of the macrocycle ring through bifurcating π-stacking interactions with the thiazole ring and the E,Zdienoate (Figs. 2a, S3a). Minor van der Waals contacts are also made between the macrocyclic ring and the backbone carbonyl of Pro159 and the side chain of Asn167 (not shown). The linear, conjugated trienyl amine arm extends through the cavity created by the RNA kink and π-stacks with the face of A7 on one side and the edge of G8 on the other through a T-shaped interaction and also makes contacts with Arg110 (Figs 2a, S3a). The tertiary amine at the end of the trienyl arm reaches out to make weak van der Waal interactions with Arg282, Gly304, and Asp305 located in the CTD, as well as water-mediated interactions with the RNA segment (Figs 2a, S3a). Altogether, DMPatA buries 512 Å2 of surface area (222 Å2 RNA and 290 Å2 protein) that spans from the NTD to CTD, locking eIF4A1 in the closed conformation.
DMPatA binding is analogous to eIF4A1•RocA interactions.
Remarkably, although the chemical structures of DMPatA and RocA are unrelated, they occupy the same binding site on eIF4A1 (Figs 2b, S3a). In RocA, all three aromatic rings were identified as important for inserting between the RNA kink and stabilizing the distorted angle between the bases. RocA rings A and B stack with RNA A7 and G8, respectively, whereas ring C is sandwiched between Phe163 and Gln195 (Fig S3b). Superposition of the RocA and DMPatA complexes shows that all three aromatic rings of RocA to a great extent overlap with equivalent conjugated π-systems and the thiazole ring in DMPatA (Fig. 2c). The conformation of the 19-membered dilactone macrolide of DMPatA mimics the angle between rings B and C of RocA with the diene edge overlapping with ring B and the thiazole ring overlapping with ring C. Ring A overlaps with the trienyl amine side chain but does not extend out quite as far. Although the overall interactions made are similar, variation in the substituents of the two inhibitors does impart some differences. For example, the side chain of Gln195 stacks with ring C and also makes a hydrogen bond with the carbonyl group of the C2-dimethylcarboxamide of RocA (Fig S3b), but with DMPatA there is only a stacking interaction (Fig 2a). Asp198 makes an anion-aromatic interaction with RocA and forms a salt bridge to the C3-amino group of DMPatA (Figs 2a, S3). The terminal tertiary amine of the triene extends into the RNA kink to a greater extent and interacts with the eIF4A1 CTD and RNA and this is not observed with RocA, whose interactions are limited to the NTD and RNA. Finally, the C8b-hydroxyl group of RocA hydrogen bonds with G8 in the RNA but no such hydrogen bond exists with DMPatA (Figs 2a, S3).
DMPatA also clamps eIF4A onto polypyrimidine RNA sequences.
Despite similarities in their binding modes, a key differentiating factor between RocA and DMPatA appears to be RNA selectivity. RocA was demonstrated to preferentially interact with, and clamp eIF4A1 onto, polypurine RNA (Iwasaki et al., 2016; Iwasaki et al., 2019). The RocA structure provided a plausible mechanism for this bias as substitution of purine bases at A7 and G8 with pyrimidines is predicted to weaken stacking interactions between ring A and RNA base A7 and disrupt the hydrogen bond between the C8b-hydroxyl group of RocA and G8 (Fig. 3a). However, analogous modelling with DMPatA complexes predicts no such bias – the extended length of the trienyl amine side chain in DMPatA allows it to maintain stacking interactions even with smaller pyrimidine bases and DMPatA does not lose these interactions with the pyrimidine RNA (Fig 3a). Thus, DMPatA is predicted to be able to inhibit a broader repertoire of RNA sequences.
Figure 3.
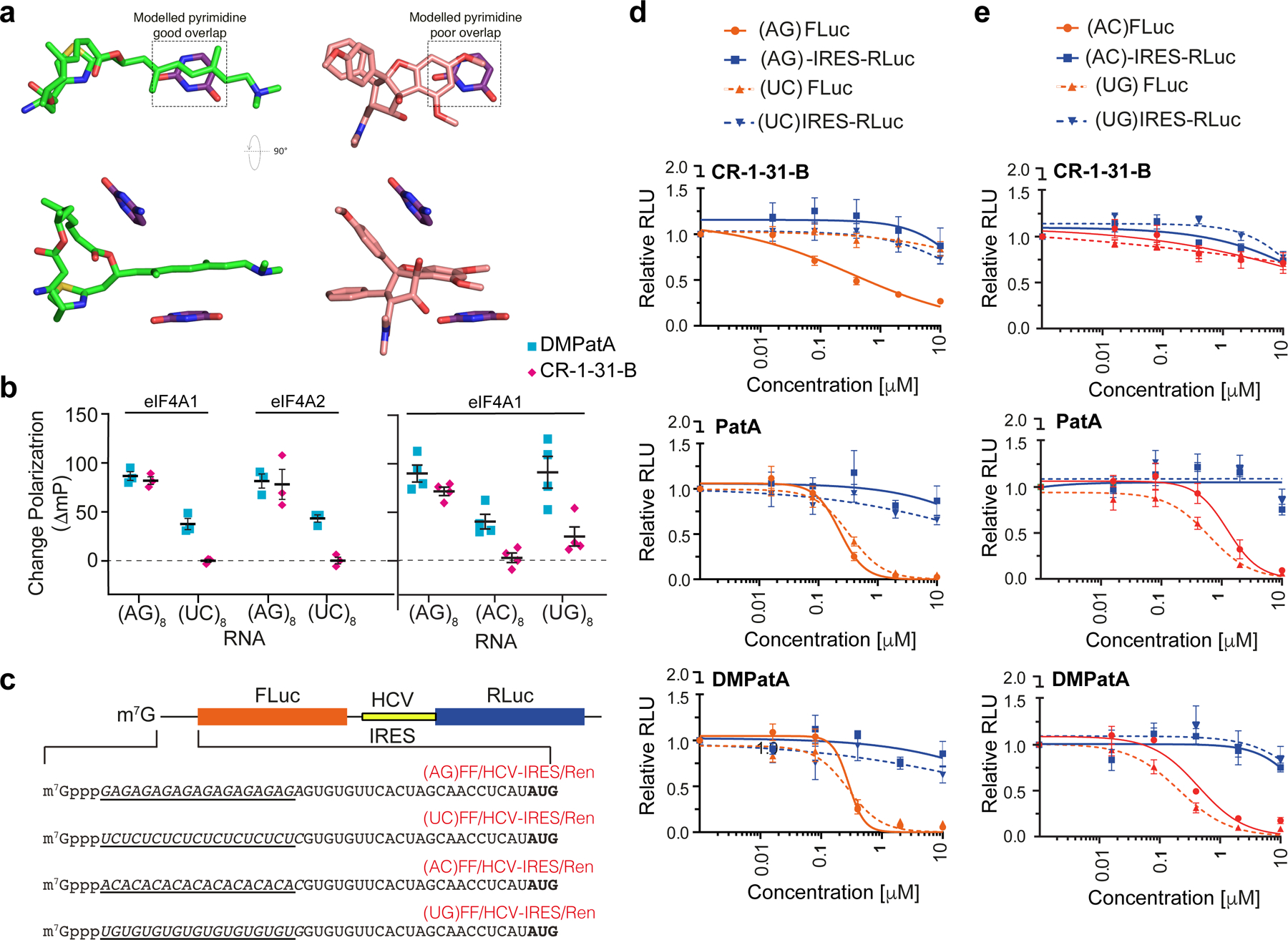
Assessing DMPatA interactions with different RNA sequences. a. Modelling of pyrimidine bases in place of purines for both DMPatA (left) and RocA (right). Overlapping regions between the compounds and base that may underlie RNA selectivity are boxed. b. The ΔmP (relative to DMSO controls) in the presence of 2.5 μM DMPatA or 10 μM CR-1–31-B using the indicated FAM-labelled RNA with 1.5 μM eIF4A1 or eIF4A2. n=3–4 ± SEM. c. FF/HCV-IRES/Ren mRNA reporters containing cap-proximal poly (AG), (UC), (AC), or (UG) RNA sequences. d. mRNA sensitivity toward CR-1–31-B, PatA and DMPatA with respect to 5’ leader purine or polypyrimidine content. Inhibition of cap-dependent (FLuc) and independent (HCV-IRES/RLuc) translation was measured in response to the indicated compounds in Krebs-2 translation extracts programmed with the noted bicistronic mRNA. IC50’s towards inhibition of FLuc synthesis from (AG)-FF/HCV-IRES/Ren mRNA were: CR-1–31-B, 0.34 ± 0.19 μM; PatA, 0.23 ± 0.04 μM; DMPatA, 0.28 ± 0.09 μM. IC50’s towards inhibition of FLuc synthesis from (UC)-FF/HCV-IRES/Ren mRNA were: CR-1–31-B, >10 μM; PatA, 0.33 ± 0.05 μM; DMPatA, 0.28 ± 0.05 μM; n = 5 ± SEM. e. Translation inhibition of mRNAs harboring mixed purine/pyrimidine 5’ leader region sequences towards PatA and DMPatA in Krebs-2 extracts. IC50’s towards inhibition of FLuc synthesis from (AC)-FF/HCV-IRES/Ren mRNA were: CR-1–31-B, >10 μM; PatA, 1.25 ± 0.29 μM; DMPatA, 0.44 ± 0.08 μM. IC50’s towards inhibition of FLuc synthesis from (UG)-FF/HCV-IRES/Ren mRNA were: CR-1–31-B, >10 μM; PatA, 0.60 ± 0.13 μM; DMPatA, 0.21 ± 0.04 μM. n = 5 ± SEM.
We therefore sought to directly test this prediction using the FP assay. DMPatA was able to induce clamping of both eIF4A1 and eIF4A2 to polypurine and polypyrimidine RNA templates (Fig 3b, left panel). Poly (UC)8 RNA however was not as good a template as poly (AG)8 and this may be a consequence of the smaller size of the pyrimidine bases and reduced buried surface area. The rocaglate, CR-1–31-B, is only able to induce clamping to the polypurine RNA template, as previously reported (Iwasaki et al., 2019). When testing binding to the mixed purine/pyrimidine combinations, poly (AC)8 or poly (UG)8 RNA, we observed that DMPatA but not CR-1–31-B, is also able to mediate significant RNA clamping to eIF4A1 (Fig 3b, right panel). We also assessed compound selectivity in translation assays programmed with mRNA templates harboring cap-proximal polypurine, polypyrimidine, or mixed purine/pyrimidine sequences (Fig 3c). In Krebs-2 in vitro translation extracts, we observed selective inhibition of cap-dependent translation from the (AG)-FF/HCV-IRES/Ren mRNA reporter, but not from (UC)-FF/HCV-IRES/Ren with CR-1–31-B (Fig 3d), as previously documented (Chu et al., 2020). PatA and DMPatA did not show a polypurine bias and blocked expression from both (AG)-FF/HCV-IRES/Ren and (UC)-FF/HCV-IRES/Ren mRNAs. Consistent with the extended base binding specificity of PatA and DMPatA, both molecules also inhibited translation from the mixed purine/pyrimidine reporters, (AC)-FF/HCV-IRES/Ren and (UG)-FF/HCV-IRES/Ren mRNAs, whereas CR-1–31-B did not (Fig 3e).
Assessing the contribution of eIF4A1 residues for DMPatA-induced RNA clamping.
We then probed the involvement of several amino acids implicated in DMPatA binding to assess their relative importance to overall clamping. Since, F163, Q195, and D198 interact with the macrocyclic ring of DMPatA (Fig 2a) a series of single, double, and triple mutants in eIF4A1 at these positions were made. F163 was converted to leucine or serine, Q195 to leucine, and D198 to lysine. Double mutants were also generated that combined F163 substitutions with Q195L or D198K changes. A triple-mutant with F163L/Q195L/D198K was also generated and tested (Fig S4a, left panel). All recombinant proteins maintained the ability to bind to RNA (albeit to slightly different extents), as assessed in the FP assay using poly (AG)8 RNA (Fig S4b). DMPatA stabilized wild-type (wt) eIF4A1 onto FAM-labeled poly (AG)8 and poly (UC)8 RNA probes (Fig 4a). The single mutants, F163L, F163S, Q195L, and D198K, all significantly impaired the ability of DMPatA to bind to eIF4A1 (Fig 4a). All double and triple mutants tested abolished the ability of DMPatA to induce clamping of eIF4A1 to poly (AG)8 and poly (UC)8 RNA probes (Fig 4a). Taken together these results highlight the importance of F163, Q195, and D198 in interacting with the DMPatA macrocyclic ring.
Figure 4.
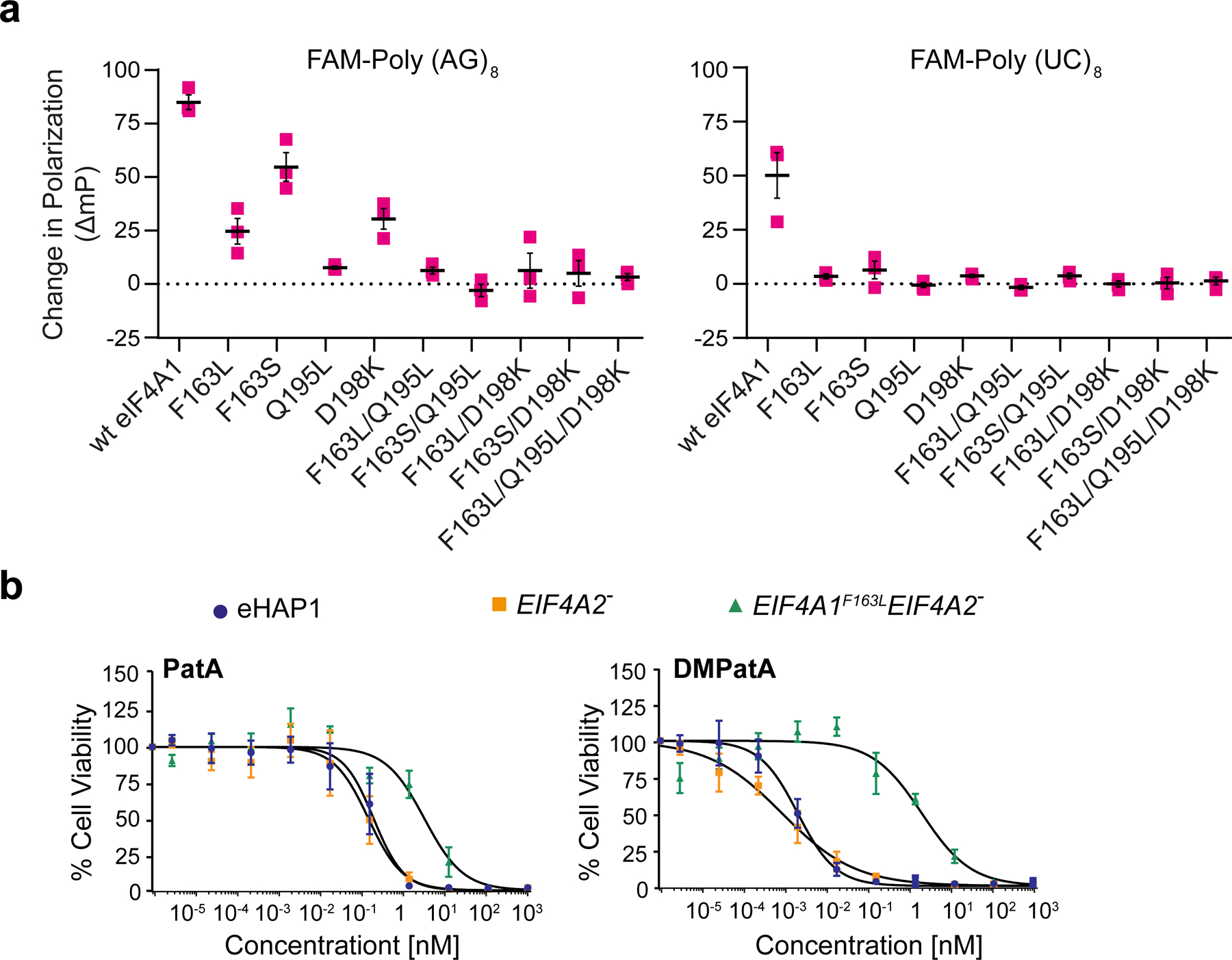
Assessing eIF4A1 amino acid requirement for DMPatA binding. a. Consequence of amino acids substitutions in eIF4A1 on DMPatA-stimulated RNA binding. The ΔmP (relative to DMSO controls) in the presence of 2.5 μM DMPatA using FAM-poly (AG)8 or FAM-poly (UC)8 RNA with the indicated eIF4A1 mutants is presented. n=3 ± SEM. b. Cytotoxicity of PatA and analogs towards wt (blue circles), EIF4A2− (orange squares), and EIF4A1F163L EIF4A2− (green triangles) eHAP1 cells. Cell were exposed to the indicated concentrations of compound for 2 days and viability was measured using the SRB assay. The IC50’s of compounds towards the test cell lines were: PatA/eHAP, 0.2 ± 0.05 nM; PatA/EIF4A2−, 0.1 ± 0.05 nM; PatA/EIF4A1F163LEIF4A2−, 3.2 ± 0.9 nM; DMPatA/eHAP, 1.9 ± 0.4 pM; DMPatA/EIF4A2−, 0.79 ± 0.23 pM; DMPatA/EIF4A1F163LEIF4A2−, 2± 0.69 nM, n = 4 ± SEM. See also Figures S4 and S5.
D198 appears to interact differently with RocA and DMPatA. Whereas it makes an electrostatic interaction with RocA (Fig S3b) (Iwasaki et al., 2019), it forms a salt bridge with the C3-primary amine of DMPatA (Fig 2a). The D198K mutant showed reduced clamping to poly (AG)8 in the presence of either DMPatA or CR-1–31-B (Fig S4c), indicate that it plays significant roles for both DMPatA and rocaglate binding to eIF4A1.
Unlike what is seen with rocaglates, DMPatA extends to the CTD of eIF4A1 and makes weak interactions with R282 and D305 (Figs 2a,b and S3a). We assessed the role of these in DMPatA binding by generating and purifying R282K and D305A eIF4A1 mutants (Fig S4a, right panel). When tested for DMPatA-induced clamping, there was no significant difference in complex formation, relative to wt eIF4A1, although a slight downward trend was noted for R282K (Fig S4d) indicating that these amino acids make minor contributions to the overall interaction with eIF4A1. These results highlight potential room for improving DMPatA:eIF4A1 interaction through future structure-activity relationship studies at this location.
In cellula validation.
The importance of F163 to DMPatA interaction was also assessed in cellula in haploid eHAP1 cells. To eliminate the possibility that eIF4A2 expression could interfere with interpretation of the results by also being a substrate for DMPatA-induced clamping (Fig 3b), and since eIF4A2 is not essential (Galicia-Vazquez et al., 2015), we generated EIF4A2 null cells using CRISPR/Cas9 (Fig S5a). We then engineered, through homology-directed repair, the F163L mutation into the eIF4A1 locus using an oligonucleotide donor template, thus generating the doubly modified EIF4A1F163LEIF4A2− eHAP1 cell line (Fig S5a). Cells were assessed for their sensitivity towards CR-1–31-B and as previously demonstrated (Chu et al., 2016; Iwasaki et al., 2019), those harboring the EIF4A1F163L allele were significantly more resistant than cells with wild-type EIF4A1 (Fig S5b).
Both wt eHAP1 and EIF4A2− cells showed similar sensitivity to PatA and DMPatA (Fig 4b). Importantly, in both instances the EIF4A1F163LEIF4A2− eHAP1 line showed significant resistance. EIF4A1F163LEIF4A2− cells were 16- and 103-fold more resistant than wt cells to PatA and DMPatA, respectively. These results indicate that F163 is critical to eIF4A:DMPatA interaction in cells and for mediating the cytotoxicity observed for the PatA family of compounds.
Discussion
A number of FDA-approved drugs function as interfacial inhibitors through the stabilization of protein-protein (eg, rapamycin with mTOR/FKBP12) and protein-DNA (eg, etoposide and topoisomerase II/DNA, olaparib and PARP/DNA) complexes (Pommier et al., 2015), but there are no clear examples of drugs acting at the protein-RNA interface. Given the resurgent interest in targeting RNA (Disney et al., 2018), understanding principles governing protein-RNA interfacial interactions is important to defining rules that could enable rational drug design.
The structure of the eIF4A1•AMPPNP•poly (AG)5•DMPatA complex rationalizes how this class of compounds inhibit translation. By interacting with both eIF4A1 and RNA, DMPatA is able to lock eIF4A onto RNA and prevent its release. Drawing from what we know concerning the mode of action of rocaglates, persistence of such stable complexes likely interferes with ribosome scanning when present in mRNA 5’ leader regions (Iwasaki et al., 2016). Although DMPatA-induced clamping of eIF4A1 onto polypyrimidine RNA was not to the same extent as to polypurine RNA (Fig 3b), there was no notable differences in inhibition in in vitro translation assays programmed with (AG)-FF/HCV/Ren or (UC)-FF/HCV/Ren mRNAs. (Fig 3d). We attribute this to the mode of action of DMPatA which induces a gain-of-function property to eIF4A1 and hypothesize that only a fraction of eIF4A1 molecules need to be clamped to inhibit ribosome recruitment or scanning, although this remains to be formally tested. The RNA targeting range of DMPatA is broader than what is observed for rocaglates (RocA or CR-1–31-B) and this may lead to inhibition of a larger spectrum of mRNAs (Fig 3).
Surprisingly, PatA, DMDAPatA, and CR-1–31-B have been reported to stimulate eIF4A1 RNA helicase activity (Bordeleau et al., 2006; Chu et al., 2016; Low et al., 2005), a result that is at odds with the ability of these compounds to “lock” eIF4A1 onto RNA. We hypothesize that shortcomings of the helicase assay used in these studies misguided the interpretations of the results. Given the weak helicase activity of eIF4A1, there is a need to use short duplexes (11 and 12 bp) that are relatively unstable (ΔG ranging from −17.9 to −21.4 kcal/mol) (Rogers et al., 2001). In these assays, one of the two RNA strands has a single-stranded RNA extension and we suspect that eIF4A1-induced clamping by PatA or rocalgate to that extension, and adjacent to the duplex junction, may favor strand displacement during “breathing” or opening at the junction – giving the false impression of increased helicase activity. As well, clamping by eIF4A1 to single-stranded RNA molecules after strand separation (reactions were performed at 35 –37 °C which is near the Tm of the duplexes used) would prevent re-annealing - an anti-association phenomenon that would also give the appearance of increased duplex displacement. Data from our structure is not compatible with an increase in eIF4A1 RNA helicase activity per se.
PatA and DMDAPatA have previously been shown to stimulate eIF4A1 ATPase activity (Bordeleau et al., 2006; Low et al., 2005). The elucidated structure herein shows that AMPPNP binding is compatible with DMPatA binding, since both are in the complex. DMDAPatA significantly increases ATP affinity (reduced KmATP) (Low et al., 2005) and the structure suggests that this is likely because the inhibitor stabilizes the closed conformation of eIF4A whereby the ATP binding site is preformed - enabling ATP to bind with reduced entropy loss. Indeed, we find that DMPatA-clamped complexes can still hydrolyze ATP (Fig S1b).
We have previously proposed a “scaffolding” domain, C1-C5 region of the macrocycle, and a “binding” domain (C6-C24) that includes the thiazole, trienyl amine side chain and the E,Z-dienoate of PatA (Romo et al., 2004) with respect to its interaction with eIF4A. This domain analysis of PatA was based on structure-activity relationships (SAR) of PatA and analogues and this early hypothesis is fully consistent with the described structure of the eIF4A1•AMPPNP•poly (AG)5•DMPatA complex. The structural insight also explains the reduced ability of DMDAPatA and MZ579 to clamp eIF4A to RNA, relative to PatA and DMPatA (Fig 1b). The two latter compounds contain a C3-amino group (R2) (Fig 1a) which makes an important salt bridge with D198 (Fig 2a). Since both DMDAPatA and MZ579 lack this functionality, we surmise this reduces their affinity for eIF4A1 revealing that while the C1-C5 region of PatA analogues primarily serves a scaffolding role for the macrocyclic conformation, it can also present functionality (i.e. the C3-amino group) to increase binding interactions with the complex (Low et al., 2014). We have also shown that replacement of the terminal N,N-dimethylamino group of the trienyl side chain with other tertiary amine groups is tolerated (e.g. pyrrolidino) (Low et al., 2014). Modification of the functionality at this site could be used in future studies to modulate binding, physical properties (i.e. aqueous solubility, plasma protein binding and serum stability), cellular specificity, and possibly RNA selectivity.
eIF4A2 shares 90% identity with eIF4A1 and also contains F164 (numbering is offset by 1) and Q196 that can interact with DMPatA, and a conservative amino acid substitution at E199 (D198 in eIF4A1). The longer Glu side chain likely also interacts with the DMPatA C3-amino group and this could explain the similarity in DMPatA-induced clamping to polypurine RNA seen with eIF4A2 and eIF4A1 (Fig 3b). We note that PatA and DMDAPatA have also been documented to inhibit nonsense-mediated decay (NMD) by binding to eIF4A3 (DDX48) (Dang et al., 2009). Indeed, all residues present in eIF4A1 and critical for DMPatA binding (F163, D198, Q195) are conserved in eIF4A3 and provides an explanation for why PatA family members also affect NMD. We were able to validate the critical role that F163 plays in conferring sensitivity to DMPatA in cells using a double mutant EIF4A1F163LEIF4A2− eHAP1 cell line (Fig 4b). EIF4A1F163L alleles confer resistance to rocaglates (Chu et al., 2016; Iwasaki et al., 2019) and we show here that this property also extends to PatA and DMPatA (Fig 4b). DMPatA appears significantly more potent at inhibiting translation and more cytotoxic than PatA, and this bioactivity cannot be rationalized from the structural information and could be the consequence its previously noted lower plasma protein binding (Chen et al., 2019), or other pharmacological properties (Fig 4b).
Our studies have uncovered a striking example of functional mimicry. DMPatA and RocA share very little overall structural similarity, yet both molecules occupy a common region of eIF4A1 and interact with two bases of the nucleic acid substrate. DMPatA is capable of interacting with both polypurine and polypyrimidine sequences, unlike RocA which is restricted to polypurine bases (Fig 3b) (Iwasaki et al., 2016; Iwasaki et al., 2019). The structure predicts that this is likely due to the extended length of the DMPatA trienyl amine side-chain which allows it to maintain sufficient stacking even with smaller pyrimidine bases (Fig 3a). It is truly striking that two structurally unrelated molecules (DMPatA and RocA) exploit a similar binding logic to exert their effects as interfacial inhibitors leading to translation initiation inhibition.
STAR Methods
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to, and will be fulfilled by, the Lead Contact, Jerry Pelletier (jerry.pelletier@mcgill.ca).
Data Availability
The atomic coordinates and structure have been deposited in the Protein Data Bank (PDB) under the accession number 6XKI.
Method Details
General materials.
The 5’-end FAM-labelled poly (AG)8, (UC)8, (AC)8 and UG)8 RNAs, as well as unlabelled poly (AG)5 and (AG)8 RNAs, were obtained from Integrated DNA Technologies (IDT).
Compounds:
The synthesis of pateamine A derivatives has been reported previously (Chen et al., 2019; Low et al., 2014; Romo et al., 2004; Romo et al., 1998). DMDA PatA, DMPatA and MZ579 were prepared in the CPRIT synthesis and drug-lead discovery laboratory at Baylor University. Pateamine A was isolated from the marine sponge Mycale sp. as previously reported (Northcote et al., 1991).
Recombinant DNA constructs.
For crystallization, pET-15b-His6-HRVC-eIF4A1(19–406) was generated and encodes the human eIF4A1 protein [Pro(19) - Ile(406)] with an N-terminal His6 tag and HRV 3C cleavage site. pGEX-GST-HRV 3C protease encodes the human rhinovirus (HRV) 3C protease harboring a GST tag at the NTD. For fluorescence polarization assays, pET-15b-His6-eIF4A1 (wt or mutant) was used to express recombinant proteins. To generate eIF4A1 amino acid substitutions, mutations were introduced into G blocks, subcloned into pET-15b-His6-eIF4A1 and clones verified by Sanger sequencing. Expression plasmids pKS/FF/HCV/Ren, pKS/(AG)-FF/HCV/Ren, and pKS/(UC)-FF/HCV/Ren have been previously described (Chu et al., 2020; Novac et al., 2004). pKS/(AC)-FF/HCV/Ren and pKS/(UG)-FF/HCV/Ren reporters were generated from pKS/(AG)-FF/HCV/Ren by removal of the sequences flanking and containing the polypurine tract using MluI and NdeI and replacing these with annealed oligos containing the sequence of interest.
Cell Viability assays and IC50 determination.
Cell viability assays were performed using sulforhodamine B reagent (SRB). Cells (5000/well) were seeded into 96 well plates and treated with the indicated compounds and drug concentrations for 48 h. To determine viability, cells were washed with PBS and fixed with 50% TCA for 1 h. TCA was washed off and plates were rinsed with water and dried. Fixed cells were stained with 0.5% SRB reagent in 1% acetic acid for 30 mins. Plates were washed with 1% acetic acid four times to remove excess SRB reagent and dried. SRB was resuspended in 100μl 20mM Tris base (pH adjusted to 9). The OD550nm was measured using a microplate reader (SpectraMax M5, Molecular Devices) and relative viability was calculated by normalizing to the DMSO control. Metabolic labelling with 35S-Met was undertaken in 293T cells as previously described (Bordeleau et al., 2005). The IC50 was determined using a non-linear regression model on GraphPad Prism 8.4.0.
Recombinant protein purification.
For FP assays, recombinant full-length His6-tagged eIF4A1 protein was purified as described below. For crystallography studies, pET-15b-His6-HRVC-eIF4A1(19–406) was transformed into BL21 (DE3) competent cells. Starter cultures were grown overnight at 37°C in 20 mL of LB media with ampicillin (100 μg/mL), after which they were transferred to 1 L of LB with ampicillin (100 μg/mL) and grown until the OD600 reached 0.6. Expression was induced with 1 mM IPTG for 4 hours at 30°C. Cells were pelleted at 4000 xg for 20 minutes and resuspended in Buffer A (20 mM Tris pH 7.4, 10% glycerol, 0.1 mM EDTA, 200 mM KCl, 0.1% Triton X-100 and 3.4 mM β-mercaptoethanol) and lysed by sonication. The lysate was clarified by centrifuged (44000 × g for 40 minutes) after which imidazole was added to the supernatant to a final concentration of 20 mM. The supernatant was incubated with Ni2+-NTA beads (2 mL of slurry per 50 mL of supernatant) for one hour on a rotating platform at 4°C. Beads with bound protein were pelleted at 1862 xg and applied to a glass column followed by washing with Buffer 1 (20 mM Tris pH 7.5, 10% glycerol, 0.1 mM EDTA, 800 mM KCl, 20 mM imidazole) and Buffer 2 (Buffer 1 containing 300 mM KCl). The protein was eluted with Buffer E (20 mM Tris pH 7.5, 10% glycerol, 0.1 mM EDTA, 300 mM KCl, 2 mM DTT, and 0.2 M imidazole). PreScission protease was added to cleave the affinity tag and the sample dialyzed overnight in 1L of Dialysis Buffer (20 mM Tris pH 7.5, 10% glycerol, 0.1 mM EDTA, 100 mM KCl) at 4°C for cleavage overnight. Digested protein sample was loaded onto a Q-Sepharose column and eluted using buffer with Dialysis Buffer containing 500 mM KCl. The main peak containing the eluted protein was then incubated with 1 mL of glutathione-agarose beads for one hour to remove the GST-tagged protease. Beads were applied to a glass column and the flow-through was collected. Finally, the sample was applied onto a Superdex 200 gel filtration column in 20 mM Tris pH 7.4, 100 mM KCl.
Complex Formation, crystallization, and data collection.
Conditions for eIF4A1•poly (AG)5•AMPPNP•DMPatA complex formation were similar to those described for obtaining the RocA complex (Iwasaki et al., 2019). Briefly, concentrated eIF4A1 at 20 mg/mL was supplemented with 10-mer (AG)5 RNA (IDT) at a 3:1 molar ratio, AMPPNP at a 16:1 molar ratio, and DMPatA at a 2:1 molar ratio. The complex was then diluted with gel filtration buffer such that the final protein concentration was 3 mg/ml. MgCl2 was added at a concentration of 5 mM.
Crystals were grown by sitting or hanging drop vapor diffusion at room temperature (20 °C). The eIF4A1•poly (AG)5•AMPPNP•DMPatA complex crystallized in 0.1 M MES pH 6.7 and 1.9 M ammonium sulfate. Initial crystals appeared as small plates that were clustered together. These crystals were microseeded with a seed bead kit (Hampton) into pre-equilibrated drops which resulted in larger plates but still clustered. Larger single crystals were pried apart from clusters using a thin hair. Crystals were cryoprotected with 2 M sodium malonate and X-ray diffraction data were collected at 100K on beamline 08ID-1 fitted with a Pilatus3 6M detector at the Canadian Macromolecular Crystallography Facility (CMCF), Canadian Light Source (CLS). Data were processed in HKL2000 with auto-corrections enabled.
Structure determination and refinement.
The structure was solved by molecular replacement using Phaser as implemented in Phenix. The search model was human eIF4A1 bound to RocA (PDB: 5ZC9) using only the protein component with all ligands (RNA, inhibitor, AMPPNP) omitted. Refinement was carried out iteratively in Phenix combined with manual rebuilding in Coot. The molecular replacement solution was initially subjected to torsion-angle simulated annealing (5000 K) restrained to the higher resolution eIF4A1 structure from the RocA complex. Further rounds of refinement included: XYZ coordinates, individual B-factors, Translation-Libration-Screw (TLS) parameters, X-ray/stereochemistry and X-ray/ADP weights optimized. AMPPNP and RNA were placed after confirming their presence in difference maps. DMPatA was built in only after all other components of the complex were refined. Structure figures were produced using Pymol.
Cas9-mediated gene editing of eHAP1 cells.
Targeting of the eIF4A1 and eIF4A2 genes was undertaken using an All-in-One LeGO-based vector expressing Cas9, GFP, and an sgRNA targeting either EIF4A2 exon 5 or EIF4A1 exon 5. First, EIF4A2 was knocked out in eHAP1 to generate EIF4A2− cells. The EIF4A2 targeting sgRNA was: 5’-GTATTGTTGTTGGTACACCC-3’. After 48 h, GFP+ cells were sorted as single cell clones in 96 well plates. Upon expansion, EIF4A2 null cells were identified by Western blotting. Those clones showing no eIF4A2 expression were then selected, RNA was extracted and cDNA prepared. The region spanning exon2 - exon8 of the EIF4A2 cDNA was amplified using primers: eIF4A2ex2fwd (5’-GAACATGGCGGCCCAGAGGGAATG-3’) and eIF4A2ex8rev (5’-CTTCTCAGTCAGCCAGTCCACCTTG-3’), and the PCR product sequenced. An EIF4A2− eHAP1 clone was used for modification of the EIF4A1 locus. The EIF4A1 targeting sgRNA was: 5’-GGTTAAGCATATCAAACACA-3’ and the single-stranded donor oligonucleotide (ssODN) harboring the desired F136L change with a mutated PAM was: 5’-CAACGTGCGTGCTGAGGTGCAGAAACTGCAGATGGAAGCTCCCCACATCATCGTGGGTACCCCTGGACGTGTGCTTGATATGCTTAACCGGAGATACCTGTGTGAGTAATTCGGTTCTCCAATCC-3’. Following transfection into EIF4A2− eHAP1 cells using TurboFectin 8.0 (Origene), cells were given 48 h to recover before being sorted into 96 well plates and undergoing selection with 50 nM CR-1–31-B. A resistant clone, EIF4A1F163LEIF4A2− eHAP1, was expanded and the F163L change confirmed by Sanger sequencing an the exon3-exon10 PCR product, the latter obtained using primers: eIF4A1ex3 fwd (5’-CGCGTATGGTTTTGAGAAGCCCTC-3’) and eIF4A1ex10 rev (5’-CCTGCTGCACATCAATGCCTCTGGC-3’).
Fluorescence polarization assay:
FP assays were performed as previously described (Chu et al., 2020). Briefly, 1.5 μM recombinant wt or mutant eIF4A1 protein was added to 10 nM FAM-labeled RNA in a buffer containing 14.4 mM HEPES-NaOH [pH 8], 108 mM NaCl, 1 mM MgCl2, 14.4% glycerol, 0.1% DMSO, 2 mM DTT and 1mM ATP in the presence or absence of indicated compound in black, low volume 384 well plates (Corning 3820). Binding reactions were allowed to equilibrate for 30 min at room temperature in the dark, prior to measuring polarization values. For the experiments using FAM-labelled poly (AC)8 and poly (UG)8 RNA (Fig 3b) and those in Fig S3c and S3d, FP readings were obtained on a Spectramax M5 unit (Molecular Devices), whereas all other reading were obtained on a Pherastar FS microplate reader (BMG Labtech). For the dissociation experiments, the eIF4A1•FAM-poly (AG)8•compound complexes were pre-assembled with 1.5 μM eIF4A1, 10nM FAM-labelled poly (AG)8 RNA, indicated compound, and 1 mM ATP. Reactions containing only DMSO were performed using 25 μM eIF4A1. Reactions were incubated at room temperature for 30 min in the dark prior to the addition of 1000-fold molar excess of unlabelled poly (AG)8 RNA and polarization measurements were performed. Relative dissociation was measured as a function of time. The half-lives of complexes in Fig 1c were calculated using the “one phase decay” method on Graph Pad. Constraints used for data analysis were (Y0 = 1, Plateau = 0, K > 0). Degrees of freedom were 0.367 for compounds and 0.374 for DMSO and R2 ranged from 0.97–0.84 for compounds and 0.87 for DMSO.
ATPase assays:
ATPase assays were performed as described previously using condition B described by Lorsch and Herschlag (Lorsch and Herschlag, 1998). Briefly, 1.5 μM eIF4A1 was incubated with indicated compound, 5 μM poly (AG)8 RNA, 500 μM cold ATP in the presence of buffer containing 2.5 mM MgCl2, 1mM DTT, 1% glycerol, 20 mM MES-KOH [pH 6.0], 10 mM KOAc for 30 minutes before adding 1 μCi [γ32P]-ATP (3000Ci/mmol). At each time point (0, 30, 60 90, 120 mins), 2 μl of the reaction was put on ice and quenched with a final concentration of 10 mM EDTA. The reactions were then resolved on PEI cellulose TLC plates (Millipore) developed with 1M LiCl, 0.3M NaH2PO4. The extent of ATP hydrolysis was quantitated using a Storm 840 (Molecular dynamics) scanner.
In vitro Transcriptions and Translations:
In vitro translations were performed using 50 ng/ul of reporter mRNA and the indicated compound concentrations in Krebs-2 extracts at 30°C for 1 h, as previously described (Novac et al., 2004). FLuc and RLuc luciferase activities were assessed on a Berthold Lumat LB 9507 luminometer (Berthold Technologies). The IC50’s were determined using a non-linear regression model on GraphPad Prism 8.4.0.
Immunoblotting:
Cells were pelleted, washed in PBS and lysed with protease inhibitor-enriched RIPA buffer (20 mM Tris-HCl [pH 7.6], 100 mM NaCl, 1 mM EDTA,1 mM EGTA, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM PMSF, 4 μg/mL aprotinin, 2 μg/mL leupeptin, 2 μg/mL pepstatin). Cell lysates were collected after centrifuging the samples at 16000 xg for 10 mins. Cell lysates were resolved on a 10% SDS-polyacrylamide gel and transferred to PVDF membrane (Bio-Rad). Antibodies against eIF4A1 (ab31217) and eIF4A2 (ab31218) were obtained from Abcam, whereas anti-eEF2 antibodies (#2332) were from Cell Signaling Technologies.
RNA binding assays:
Body labelled [32P] poly (AG)8 RNA was generated using annealed oligos (T7 promotor (5’-GAAATTAATACGACTCACTATA-3’) and target template (5’-CTCTCTCTCTCTCTCTCTATAGTGAGTCGTATTAATTTC-3’) in in vitro transcription reactions with T7 RNA polymerase, [α32P]-ATP (20 μCi), and supplemented with ATP and GTP to a final concentration of 5 μM and 100 μM, respectively. RNA binding assays (Merrick and Sonenberg, 1997) were performed by incubating eIF4A1, [32P]-poly(AG)8 RNA (100,000 cpm), and the indicated compounds in binding buffer (25 mM Tris [pH 7.5], 1 mM DTT, 100 mM KCl) in the absence or presence of 1 mM ATP/5 mM MgCl2 at 37°C for 10 min. Reactions were terminated by the addition of 1ml STOP buffer ((25 mM Tris [pH 7.5], 100 mM KCl) and passed through nitrocellulose filters (45 μM HA Millipore) which had been preblocked with PBS + 0.1% sodium pyrophosphate. The filters were washed 3 times with 1ml STOP buffer, dried, and the amount of retained [32P]-pol(AG)8 RNA measured by scintillation counting using a scintillation counter (Tri-Carb 2810 TR Perkin Elmer).
Supplementary Material
Significance.
Eukaryotic initiation factor (eIF) 4A is a founding member of the DEAD-box RNA helicase family of proteins, whose members participate in many aspects of RNA biology. Selective targeting of DEAD-box helicases has been difficult to achieve, but is important for uncovering function and potential therapeutic development. Case in point, inhibition of eIF4A1 has been found to exert anti-proliferative effects against several cancer cell lines and in several pre-clinical models, while displaying minimum toxicity towards non-transformed cells. We previously identified a potent, natural product inhibitor of eIF4A, pateamine A (PatA), and showed that this compound stabilizes eIF4A:RNA complexes. To understand the molecular basis of this property, we define the structure of the quaternary complex of eIF4A1, AMPPNP, poly (AG)5 RNA, and a PatA analog, DMPatA. DMPatA behaves as an interfacial inhibitor, making intimate contacts with both eIF4A1 and RNA. Our structural and functional studies rationalize the broad RNA targeting specificity of DMPatA and have the potential to assist in the design of PatA analogs with altered binding affinities or target specificity.
Acknowledgements
We wish to dedicate this paper to the isolation chemists who first discovered pateamine A, Profs. Murray H.G. Munro and John W. Blunt (Univ. of Canterbury, NZ) and their student at the time, Prof. Peter T. Northcote (Victoria University, Wellington, NZ). We thank the staff at the Canadian Macromolecular Crystallography Facility, Canadian Light Source, which is supported by the Natural Sciences and Engineering Research Council of Canada, the National Research Council Canada, the Canadian Institutes of Health Research, the Province of Saskatchewan, Western Economic Diversification Canada, and the University of Saskatchewan. This study was supported by research funding to PTS (Worldwide Cancer Research, 15-1253), DR (National Institutes of Health, National Institute of General Medical Sciences, GM134910, GM052964; Baylor University-CPRIT Drug Lead Discovery Laboratory), BN (Canadian Institutes of Health Research [#MOP-133535]), and JP (Canadian Institutes of Health Research [#FDN-148366]).
Footnotes
Declaration of Interests
The authors declare no competing interests.
References
- Afonine PV, Mustyakimov M, Grosse-Kunstieve RW, Moriarty NW, Langan P, and Adams PD (2010). Joint X-ray and neutron refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr 66, 1153–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreou AZ, and Klostermeier D (2012). The DEAD-box helicase eIF4A: paradigm or the odd one out? RNA Biol 10, 19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat M, Robichaud N, Hulea L, Sonenberg N, Pelletier J, and Topisirovic I (2015). Targeting the translation machinery in cancer. Nat Rev Drug Discov 14, 261–278. [DOI] [PubMed] [Google Scholar]
- Bordeleau ME, Cencic R, Lindqvist L, Oberer M, Northcote P, Wagner G, and Pelletier J (2006). RNA-mediated sequestration of the RNA helicase eIF4A by Pateamine A inhibits translation initiation. Chem Biol 13, 1287–1295. [DOI] [PubMed] [Google Scholar]
- Bordeleau ME, Matthews J, Wojnar JM, Lindqvist L, Novac O, Jankowsky E, Sonenberg N, Northcote P, Teesdale-Spittle P, and Pelletier J (2005). Stimulation of mammalian translation initiation factor eIF4A activity by a small molecule inhibitor of eukaryotic translation. Proc Natl Acad Sci USA 102, 10460–10465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordeleau ME, Robert F, Gerard B, Lindqvist L, Chen SM, Wendel HG, Brem B, Greger H, Lowe SW, Porco JA Jr., et al. (2008). Therapeutic suppression of translation initiation modulates chemosensitivity in a mouse lymphoma model. J Clin Invest 118, 2651–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Zhu M, Chaudhari RR, Robles O, Chen Y, Skillern W, Qin Q, Wierda WG, Zhang S, Hull KG, et al. (2019). Creating novel translation inhibitors to target pro-survival proteins in chronic lymphocytic leukemia. Leukemia 33, 1663–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J, Galicia-Vazquez G, Cencic R, Mills JR, Katigbak A, Porco JA Jr., and Pelletier J (2016). CRISPR-Mediated Drug-Target Validation Reveals Selective Pharmacological Inhibition of the RNA Helicase, eIF4A. Cell Rep 15, 2340–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J, Zhang W, Cencic R, O’Connor PBF, Robert F, Devine WG, Selznick A, Henkel T, Merrick WC, Brown LE, et al. (2020). Rocaglates Induce Gain-of-Function Alterations to eIF4A and eIF4F. Cell Rep 30, 2481–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang Y, Low WK, Xu J, Gehring NH, Dietz HC, Romo D, and Liu JO (2009). Inhibition of nonsense-mediated mRNA decay by the natural product pateamine A through eukaryotic initiation factor 4AIII. J Biol Chem 284, 23613–23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disney MD, Dwyer BG, and Childs-Disney JL (2018). Drugging the RNA World. Cold Spring Harb Perspect Biol 10, a034769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galicia-Vazquez G, Chu J, and Pelletier J (2015). eIF4AII is dispensable for miRNA-mediated gene silencing. RNA 21, 1826–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki S, Floor SN, and Ingolia NT (2016). Rocaglates convert DEAD-box protein eIF4A into a sequence-selective translational repressor. Nature 534, 558–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki S, Iwasaki W, Takahashi M, Sakamoto A, Watanabe C, Shichino Y, Floor SN, Fujiwara K, Mito M, Dodo K, et al. (2019). The Translation Inhibitor Rocaglamide Targets a Bimolecular Cavity between eIF4A and Polypurine RNA. Mol Cell 73, 738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsov G, Xu Q, Rudolph-Owen L, Tendyke K, Liu J, Towle M, Zhao N, Marsh J, Agoulnik S, Twine N, et al. (2009). Potent in vitro and in vivo anticancer activities of des-methyl, des-amino pateamine A, a synthetic analogue of marine natural product pateamine A. Mol Cancer Ther 8, 1250–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, and Swindells MB (2011). LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model 51, 2778–2786. [DOI] [PubMed] [Google Scholar]
- Lorsch JR, and Herschlag D (1998). The DEAD box protein eIF4A. 1. A minimal kinetic and thermodynamic framework reveals coupled binding of RNA and nucleotide. Biochemistry 37, 21802193. [DOI] [PubMed] [Google Scholar]
- Low WK, Dang Y, Schneider-Poetsch T, Shi Z, Choi NS, Merrick WC, Romo D, and Liu JO (2005). Inhibition of eukaryotic translation initiation by the marine natural product pateamine A. Mol Cell 20, 709–722. [DOI] [PubMed] [Google Scholar]
- Low WK, Li J, Zhu M, Kommaraju SS, Shah-Mittal J, Hull K, Liu JO, and Romo D (2014). Second-generation derivatives of the eukaryotic translation initiation inhibitor pateamine A targeting eIF4A as potential anticancer agents. Bioorg Med Chem 22, 116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrick WC, and Sonenberg N (1997). Assays for eukaryotic translation factors that bind mRNA. Methods 11, 333–342. [DOI] [PubMed] [Google Scholar]
- Northcote PT, Blunt JW, and Munro MHG (1991). Pateamine - a Potent Cytotoxin from the New-Zealand Marine Sponge, Mycale Sp. Tetrahedron Letters 32, 6411–6414. [Google Scholar]
- Novac O, Guenier AS, and Pelletier J (2004). Inhibitors of protein synthesis identified by a high throughput multiplexed translation screen. Nucleic Acids Res 32, 902–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier J, and Sonenberg N (2019). The Organizing Principles of Eukaryotic Ribosome Recruitment. Annu Rev Biochem 88, 307–335. [DOI] [PubMed] [Google Scholar]
- Pommier Y, Kiselev E, and Marchand C (2015). Interfacial inhibitors. Bioorg Med Chem Lett 25, 3961–3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigo CM, Cencic R, Roche SP, Pelletier J, and Porco JA (2012). Synthesis of rocaglamide hydroxamates and related compounds as eukaryotic translation inihibitors: synthetic and biological studies. J Med Chem. 55, 558–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers GW Jr., Lima WF, and Merrick WC (2001). Further characterization of the helicase activity of eIF4A. Substrate specificity. J Biol Chem 276, 12598–12608. [DOI] [PubMed] [Google Scholar]
- Romo D, Choi NS, Li S, Buchler I, Shi Z, and Liu JO (2004). Evidence for separate binding and scaffolding domains in the immunosuppressive and antitumor marine natural product, pateamine a: design, synthesis, and activity studies leading to a potent simplified derivative. J Am Chem Soc 126, 10582–10588. [DOI] [PubMed] [Google Scholar]
- Romo D, Rzasa RM, Shea HA, Park K, Langenhan JM, Sun L, Akhiezer A, and Liu JO (1998). Total synthesis and immunosuppressive activity of (-)-pateamine A and related compounds: Implementation of a b-lactam-based macrocyclization. J Am Chem Soc 120, 12237–12254. [Google Scholar]
- Sadlish H, Galicia-Vazquez G, Paris CG, Aust T, Bhullar B, Chang L, Helliwell SB, Hoepfner D, Knapp B, Riedl R, et al. (2013). Evidence for a functionally relevant rocaglamide binding site on the eIF4A-RNA complex. ACS Chem Biol 8, 1519–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengoku T, Nureki O, Nakamura A, Kobayashi S, and Yokoyama S (2006). Structural basis for RNA unwinding by the DEAD-box protein Drosophila Vasa. Cell 125, 287–300. [DOI] [PubMed] [Google Scholar]
- Theissen B, Karow AR, Kohler J, Gubaev A, and Klostermeier D (2008). Cooperative binding of ATP and RNA induces a closed conformation in a DEAD box RNA helicase. Proc Natl Acad Sci USA 105, 548–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The atomic coordinates and structure have been deposited in the Protein Data Bank (PDB) under the accession number 6XKI.