

Abstract
Methods that functionalize the periphery of azacylic scaffolds have garnered increasing interest in recent years. Herein, we investigate the selectivity of a solid-state Norrish–Yang cyclization (NYC) and subsequent C–C cleavage/cross-coupling reaction of a strained cyclopropane-fused azacyclic system. Surprisingly, the NYC primarily furnished a single lactam constitutional and diastereo-isomer. The regioselectivity of the C–C cleavage of the α-hydroxy-β-lactam moiety could be varied by altering the ligand set used in the coupling chemistry. Experimental and computational observations are discussed.
Graphical Abstract
Saturated cyclic amines (azacycles) are ubiquitous in natural products, agrochemicals, and pharmaceuticals.1 Among drugs that are prescribed in the United States, piperidines are the most common azacycle, occurring in >10% of the 640 heterocycle-containing drugs on the market in 2014.2 As such, synthetic methods that diversify azacycle-containing compounds to access novel scaffolds are of great importance.3
In recent years, various transition metal-mediated methodologies for the peripheral functionalization of C(sp3)–H rich azacycles (Figure 1A) have been reported. For example, Sames and co-workers demonstrated in a single case that the α-arylation of piperidines bearing an N-amidine directing group could be achieved using Ru catalysis with boronate esters as coupling partners.4 Maes and co-workers disclosed a broader scope for this type of α-arylation using N-pyridyl piperidines under similar conditions, albeit resulting in competing mono- and difunctionalization.5 More recently, Yu and co-workers,6,7 Gong and co-workers,8 and Glorius and co-workers9 demonstrated enantioselective α-functionalization of a range of azacycles, including piperidines, using thioamide or amidoxime directing groups.
Figure 1.
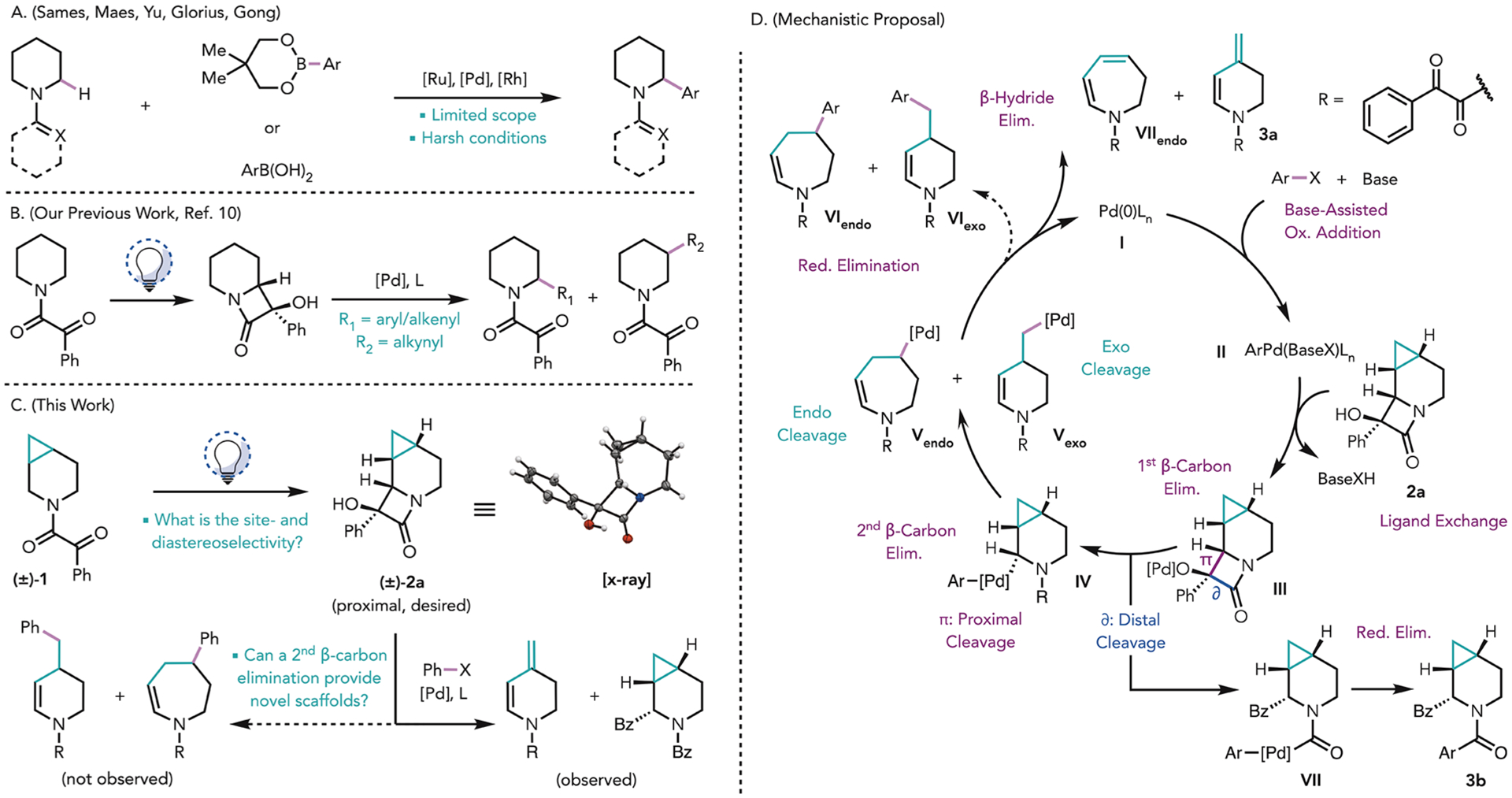
(A) Previous methodologies for the α-functionalization of saturated azacycles. (B) Our recent methodology for the α-functionalization of saturated azacycles. (C) Investigations of the selectivity of the Norrish–Yang cyclization (NYC) and palladium-mediated cleavage/cross-coupling of a cyclopropane-fused piperidine. (D) Mechanistic proposal for the C–C cleavage cross-coupling reaction.
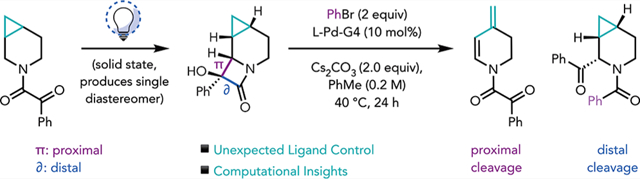
Previously, we described a methodology for the mild functionalization of a variety of saturated azacycles (Figure 1B).10 Our approach exploited a Norrish–Yang cyclization (NYC) to annulate a highly strained α-hydroxy-β-lactam moiety onto azacyclic scaffolds, setting the stage for a subsequent C–C cleavage/cross-coupling to access both α-and β-functionalized azacyclic products. On the basis of this methodology, we questioned whether azacycles fused to highly strained cyclopropanes would engender an additional β-carbon elimination, providing access to additional novel, value-added, scaffolds (Figure 1C). Furthermore, this study could provide support for our recent work11 centered on elucidating the factors that control the regioselectivity of C–C cleavage [i.e., proximal vs distal (see Figure 1D)] using biarylphosphine–Pd complexes. Previously, we found that a crucial donor–acceptor interaction between the ligand and metal controlled the site of C–C cleavage in the α-hydroxy-β-lactam, through what was termed steric enabled electronic effects from the ligand (SEEEL).11
Herein, we report studies of the stereo-, chemo-, and diastereoselectivity of the NYC of a cyclopropane-fused piperidinyl ketoamide [1 (see Figure 1C)]. A high level of site-selectivity, diastereoselectivity, and chemoselectivity was achieved using optimized reaction conditions, leading to the preferential formation of the desired lactam. The Pd-mediated C–C cleavage/cross-coupling reactions of this unique substrate were explored, providing access to novel scaffolds depending on the choice of ligand. Interestingly, a reactivity profile that is complementary to our previous results was observed. Specifically, using APhos as a ligand, cleavage of the distal C–C bond of the α-hydroxy-β-lactam occurred preferentially to furnish α-benzoyl piperidinyl amide 3b (Figure 1D). Using our previously established conditions, which used RuPhos as ligand, proximal C–C bond cleavage was observed. However, under the latter conditions, following C–C bond cleavage, cross-coupling [requiring reductive elimination (RE)] was not observed and only β-hydride elimination occurred under the various conditions that were screened to give 3a. Density functional theory (DFT) calculations corroborated this finding and showed that in this system, proximal C–C bond cleavage is kinetically favored, whereas the downstream RE has a significantly high barrier. In contrast, when APhos was used as a ligand, a switch in selectivity, also supported by computations, was observed. Specifically, in switching from RuPhos to APhos, we calculated a significant decrease in the ΔΔG‡ associated with selectivity for proximal versus distal C–C cleavage, representing a switch in selectivity to distal cleavage under kinetic control. This work extends our previous understanding of the factors that control reactivity and selectivity in these Pd-catalyzed cross-couplings.
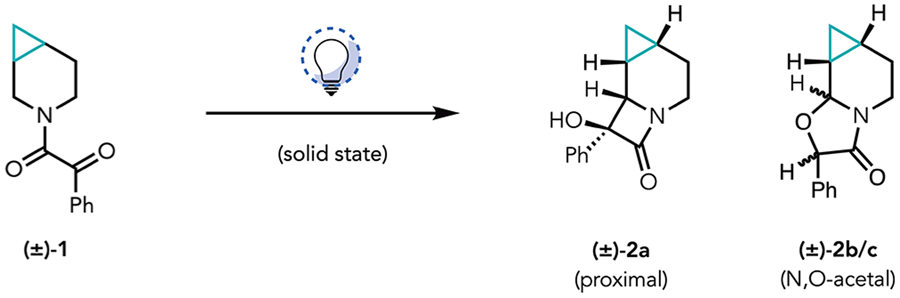
At the outset of this work, we recognized that for our proposed cyclopropane cleavage event to proceed, the NYC had to site-selectively functionalize the cyclopropylcarbinyl methylene group. In this way, the α-Pd intermediate that would be generated following an initial β-carbon elimination (see IV in Figure 1D) would be poised for cyclopropane cleavage through a subsequent β-carbon elimination. The NYC could also lead to numerous stereoisomers (eight possible lactams). Moreover, as had been observed in our previous studies, there was the potential that an undesired N,O-acetal product [2b/c (Table 1), for example] could form instead of the desired lactam, leading to a total of 16 possible isomeric products.
Table 1.
Optimization of the Production of Lactam 2a
| |||||
|---|---|---|---|---|---|
| entry | scale (mg) | time (h) | temp | methoda | 2a:2b/cb |
| 1 | 200 | 16 | rt | vial | 1:0.95 |
| 2 | <5 | 1 | rt | vial | 1:1.1 |
| 3 | 23 | 0.75 | rt | slide | 1:0.56 |
| 4 | 100 | 4 | ~−78 °C | slide with N2 stream | 1:0.22 |
| 5 | 200 | 5 | ~−78 °C | slide with N2 stream | 1:0.24 |
Vial reactions run under a N2 atmosphere.
Ratios determined by 1H NMR analysis of the crude reaction mixture.
We therefore sought to determine the selectivity of the NYC of cyclopropane-fused piperidinyl ketoamide 1 (see the Supporting Information for its preparation). Upon irradiation of the starting ketoamide (1) using our previously established conditions (Table 1, entry 1), we observed full consumption of 1 by 1H NMR. We observed a high site-selectivity for the cyclopropylcarbinyl position, and a single lactam diastereomer (2a) in 18% yield, the relative configuration of which was unambiguously determined by single-crystal X-ray diffraction (XRD) analysis (Figure 1C). In addition to 2a, we observed a pair of N,O-acetal diastereomers (2b and 2c) in roughly a 1:1 ratio (combined yield of 17%). The remainder of the mass balance was ascribed to an intractable mixture of decomposition products and/or unidentifiable constitutional isomers. During our optimization studies, we observed that the reaction had progressed to near completion after 1 h on a 0.02 mmol (5 mg) scale (entry 2). On this basis, we sought to maximize the surface area for this solid phase reaction setup by using a glass slide as the reaction vessel, which proved to be crucial to the reaction rate (entry 3). Following literature precedent,13 we found that cooling the reaction setup (see the Supporting Information for pictures and details of the reaction setup) pushed the product distribution toward the desired lactam (entry 4). Ultimately, we observed full consumption of the starting material, as well as an improved product ratio of 1:0.24, after 5 h (35% isolated yield of lactam) on a 0.9 mmol (200 mg) scale at approximately −78 °C.
Previous studies of solid-state Norrish type II processes have shown that factors such as equilibrium geometries of the diradical intermediate14,15 or the presence of intermolecular hydrogen bonds16 can dictate the stereochemical outcome. We propose, in alignment with the topochemical postulate,17 that the observed stereoselectivity of this solid-state transformation of 1 likely arises from the tendency of crystals to minimize overall reorganization, thus maintaining the greatest level of order in the bulk state. Therefore, it is likely that the equilibrium geometry of the crystal lattice of ketoamide 1 leads to one methylene H atom being accessible with minimal molecular motion. Ultimately, this scenario leads to a stereoselective hydrogen atom transfer (HAT), furnishing a stereogenic radical center, which, upon recombination, provides the observed product with high diastereoselectivity. Under this mechanistic scenario, more than one N,O-acetal diastereomer is likely, as the C–O radical recombination requires additional molecular motion, which would allow access to other modes of reactivity. This is supported by the observed temperature dependence of the product distribution, wherein entropic factors may play a role in the formation of 2b or 2c at increased temperatures, and the fact that the crystal lattice is disrupted upon product formation, leading to melting point depression with reaction progress (the crystalline solid will become an oil if not kept cool as the reaction progresses). Additionally, this hypothesis is supported by single-crystal XRD analysis of the starting ketoamide 1 (Table 1), which shows a clear proximity differential between the ketone moiety and the neighboring methylene groups, favoring the observed positional isomers. Moreover, a large difference in proximity between two of the methylene protons at the cyclopropylcarbinyl position (see the Supporting Information for details) provides an explanation for the high level of diastereoselectivity observed in the lactam product. In comparison to archetypical radical clocks, such as the cyclopropyl methyl radical, the fact that the recombination event is competitive with cyclopropane ring-opening places the rate of recombination on the order of 107 s−1 or faster.12
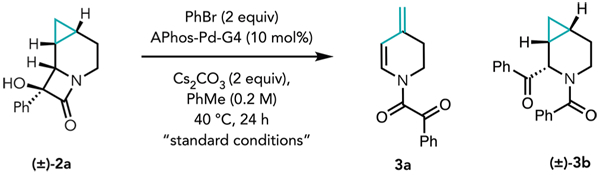
With 2a in hand, we set out to investigate the C–C bond cleavage/coupling chemistry. Using our previously established cross-coupling conditions, starting lactam 2a was fully consumed to form two major products, which were identified as 3a, resulting from sequential C–C cleavage followed by β-hydride elimination, in 60% isolated yield, and α-benzoylated 3b in 12% isolated yield, resulting from distal C–C cleavage of the α-hydroxy-β-lactam (Table 2, entry 2). The observed products are consistent with the structural constraints inherent in starting lactam 2a. Given that β-carbon elimination must occur via a four-membered transition state (TS), the forming and breaking bonds must be syn disposed. However, on the basis of the relative configuration of the α-hydroxy-β-lactam in 2a, only the exo cyclopropyl C–C bond can engage in this process, furnishing a 4-(1-methyl)-palladated intermediate (Figure 1D, Vexo), setting the stage for the observed β-hydride elimination. This is in contrast to an endo cyclopropyl C–C cleavage that would afford the corresponding azepine derivative (Figure 1C, exo vs endo cleavage).
Table 2.
Investigation into the Cross-Coupling/Cleavage Reaction of Lactam 2a
| |||
|---|---|---|---|
| entry | deviations from standard conditions | % yield (3a; 3b) | |
| 1 | none | 10; | 52 |
| 2 | RuPhos-Pd-G4 | 60; | 12 |
| 3 | CataCXium A-Pd-G4 | 13; | 49 |
| a4 | QPhos-Pd-G3 | —; | 20 |
| a5 | RuPhos-Pd-G4, p-benzoquinone (10 mol%) | 43; | 9 |
| a6 | w/ p-benzoquinone (10 mol%) | 10; | 50 |
| a7 | K2CO3 instead of Cs2CO3 | returned 2a | |
| a8 | Phl instead of PhBr | 5; | 15 |
| |||
NMR yield based on 1H NMR integration using 1,3,5-trimethoxybenzene as the internal standard. Reactions run on a 0.1 mmol scale.
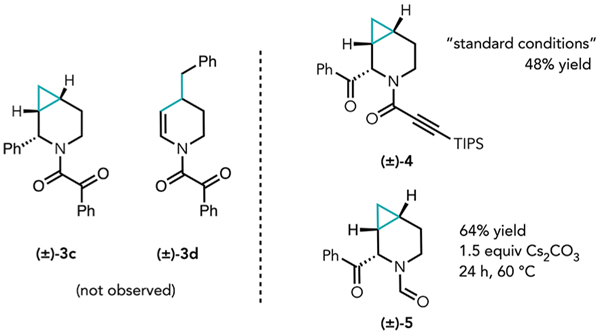
Following these initial observations, we sought conditions to favor RE and therefore cross-coupling. Interestingly, with all of the ligands that were evaluated using microscale high-throughput experimentation (HTE; see the Supporting Information for a list of ligands screened), we observed exclusive proximal C–C cleavage followed by β-carbon and β-hydride elimination to furnish diene product 3a, and/or distal C–C cleavage to furnish α-benzoylated benzamide 3b, with no α-arylated product (3c) or cyclopropyl cleavage cross-coupling product (3d) observed. Ultimately, we found that by using APhos-Pd-G4 (10 mol %) as precatalyst, 3b could be isolated in 52% yield holding all other conditions constant (Table 2, entry 1). The relative configuration of 3b was verified by single-crystal XRD analysis (see the Supporting Information). Similar results were observed using CataCXium A as the ligand (entry 3), and although using QPhos proved to be optimal on an analytical scale, this was not the case on a 0.1 mmol scale (entry 4), which furnished only 20% of 3b as a part of a complex mixture, indicative of many competing side reactions. Attempts to favor RE using RuPhos, for example, by the addition of a π-acid (p-benzoquinone, 10 mol %), decreased the yield of diene 3a (entry 5), likewise leading to a complex mixture. Notably, formation of 3d was still not observed. Finally, addition of π-acids when APhos-Pd-G4 was used as a catalyst did not lead to any improvements in selectivity (entry 6).
In alignment with our previous observations, Cs2CO3 was shown to be the optimal base for this transformation (entry 7), whereas using K2CO3 as a base returned the starting lactam. PhI proved to be significantly less efficient as an aryl source (entry 8), resulting in 35% recovered starting lactam. Interestingly, alkynylated amide 4, the structure of which was confirmed by single-crystal XRD analysis, was isolated in 48% yield under these optimized conditions as a single diastereomer when (bromoethynyl)triisopropylsilane was used as the coupling partner. Finally, when 2a was subjected to our previously established protocol for the conversion of azacycles to the corresponding α-benzoyl-N-formyl derivatives,10 we obtained α-keto aza-bicycloheptanyl formamide 5 in 64% isolated yield as a single diastereomer (verified by single-crystal XRD analysis).
To better understand the role of the ligand in determining the product distribution, we conducted DFT calculations (see the Supporting Information for figures and a full analysis). Using RuPhos, our calculations indicate that cleavage of the proximal C–C bond cleavage is more favorable, with a free energy barrier of 10.60 kcal/mol, whereas the distal cleavage has an activation free energy of 14.05 kcal/mol (see Figure S13). The calculated preference for proximal C–C bond cleavage over distal C–C bond cleavage of the β-lactam using the RuPhos-ligated Pd(II) complex is consistent with our previous computational findings and the experimental observations presented above.10,11,18
In contrast, using monodentate phosphine ligand CataCXium A or APhos, we observed a reversal in the distal versus proximal C–C cleavage selectivity leading to α-benzoylated species 3b as a major product. Specifically, the product ratio reversed by a factor of 26 (Table 2, entries 1 and 2) upon switching from RuPhos to APhos. This experimental observation is in full agreement with our previous findings11 and corresponds to a 2 kcal/mol calculated difference between the free energy barriers of distal and proximal cleavage in comparing RuPhos to APhos or CataCXium A. Calculations for the C–C bond cleavage steps using APhos and CataCXium A as ligands (see Figure S14) revealed ΔΔG‡ values of 0.61 and 0.52 kcal/mol for APhos and CataCXium A, respectively, showing a clear decrease of approximately 3 kcal/mol in selectivity to favor distal cleavage compared to the RuPhos case (ΔΔG⧧ = 3.45 kcal/mol), in full agreement with experiment. Additionally, the distal versus proximal selectivity data presented above for the RuPhos, APhos, and CataCXium A ligated systems are consistent with a distortion interaction analysis,19,20 details of which can be found in the Supporting Information.
In conclusion, for highly strained, cyclopropane-fused piperidinyl ketoamide 1, the NYC to form the corresponding α-hydroxy-β-lactam proceeds in a highly chemo-, diastereo-, and site-selective fashion under the optimized conditions. A C–C bond cleavage/cross-coupling reaction using this substrate provided access to unusual scaffolds such as 3a, 3b, and 4. DFT calculations revealed that in the case of RuPhos, the desired reactivity for the formation of 3c or 3d was hampered by a rapid β-hydride elimination. In contrast, monodentate ligands APhos and CataCXiumA favored distal C–C bond cleavage. The empirical observations of the product distribution in the solid-state photoreaction of 1 are currently being computationally investigated and will be disclosed in due course.
EXPERIMENTAL PROCEDURES
General.
Unless otherwise stated, all reactions were performed in flame-dried or oven-dried glassware under an atmosphere of nitrogen with Teflon-coated stir bars. Photochemical reactions were conducted using a 40 W Kessil A160WE blue LED 400–450 nm fixed wavelength lamp. Mixtures for reactions performed at increased temperatures were heated in an oil bath; room temperature is defined as 23 °C. All commercially available reagents were used without purification. Flash column chromatography was performed with Silicycle SiliaFlash P60 silica gel (230–400 mesh, 40–63 μm particle size). AgNO3-impregnated [10% (w/w)] silica was purchased from Silicycle. Compounds were visualized with UV light (254 nm) and stained with p-anisaldehyde and heat, or potassium permanganate (KMnO4) and heat. TLC and preparative TLC were performed on glass-backed Silicycle SiliaPlate 250 μm thickness, 60 Å porosity F-254 precoated plates. AgNO3-impregnated TLC plates were prepared by running a 10% (w/v) solution of AgNO3 in MeCN to the top of the glass-backed Silicycle SiliaPlate 250 μm thickness, 60 Å porosity F-254 precoated plates and then drying the plates in an oven at 160 °C for 10 min. They were wrapped in foil and stored away from light when not in use. Automated flash chromatography was performed on Yamazen Flash EPCLC W-Prep 2XY (dual channel) automated flash chromatography systems with premium-grade universal columns. Microwave reactions were conducted using a Biotage Initiator and a microwave reactor. All microwave reactions were conducted in sealed microwave vials provided by the manufacturer. Reaction progress was monitored by thin layer chromatography (TLC). NMR spectra were recorded on Bruker AV-300 (NSF Grants CHE-0130862 and CHE-911557), AVQ400 (NSF Grant CHE-0130862), AVB-400 (NSF Grant CHE0130862, NIH Grant S10 RR 03353-01, and NSF Grant CHE8703048), AV-500 (NIH Grant 1S10RR016634-01), and AV600 (NIH Grant SRR023679A) instruments at the University of California Berkeley’s College of Chemistry NMR Facility. Residual solvent was used as internal reference (CDCl3, 7.26 ppm; C6D6, 7.17 ppm). The following abbreviations were used to describe the NMR multiplicities: br, broad; s, singlet; d, doublet; t, triplet; q, quartet; quint, quintet; m, multiplet. Coupling constants J are given in hertz. High-resolution mass spectra (HRMS) were recorded on a PerkinElmer AxION 2 UHPLC-TOF instrument (ESI). Melting point data were collected on an Mel-Temp II melting point apparatus. IR data were collected on a Bruker Vertex 80 FTIR spectrometer. Single-crystal XRD measurements were performed on a Rigaku XtaLAB P200 instrument equipped with a MicroMax 007HF rotating anode and a Pilatus 200K hybrid pixel array detector.
Starting Material Preparation.
N-Boc 3-Azabicyclo[4.1.0]-heptane (S1).
A solution of diethylzinc (1.0 M in hexanes, 40 mL, 40 mmol, 4.0 equiv) was added dropwise via syringe to a solution of CH2Cl2 (50 mL) at 0 °C under a flow of N2. A solution of TFA (3.2 mL, 40 mmol, 4.0 equiv) in CH2Cl2 (10 mL) was then added dropwise via syringe, and the mixture was stirred for 40 min at 0 °C. After this time, a solution of CH2I2 (3.0 mL, 40 mmol, 4.0 equiv) in CH2Cl2 (10 mL) was added dropwise via syringe. Ten milliliters of air then was injected using a syringe. The reaction mixture was stirred for an additional 40 min. A solution of N-Boc-1,2,3,6-tetrahydropyridine (1.8 mL, 10 mmol, 1.0 equiv) in CH2Cl2 (30 mL) was then added dropwise to the reaction mixture via cannula, and the reaction mixture was allowed to warm to room temperature and stirred for 12 h. The reaction mixture was cooled in an ice bath; Et3N (1.7 mL, 12 mmol, 1.2 equiv) and Boc2O (2.4 g, 11 mmol, 1.1 equiv) were added sequentially, and the mixture was stirred at room temperature for 12 h. An additional 1.09 g (5.0 mmol, 0.50 equiv) of Boc2O was then added, and the mixture was stirred for an additional 12 h. After this time, the reaction was quenched with saturated aqueous NH4Cl (40 mL), the organic layer was separated, and the aqueous phase was extracted with CH2Cl2 (3 × 50 mL). The organic layers were combined, dried over NaSO4, and concentrated in vacuo to yield a reddish oil. The crude material was purified in stages using normal phase silica gel column chromatography (0% to 20% EtOAc/hexanes), followed by AgNO3-impregnated silica column chromatography (8 mL of AgNO3–SiO2 per 100 mg of crude material; eluting with 4 column volumes each of 0%, 0.5%, 1.0%, and 1.5% MTBE/pentane followed by an additional 12 column volumes of 1.5% MTBE/pentane), furnishing S1 as a translucent, light-yellow oil with a floral scent (0.97 g, 49% yield). 1H NMR (400 MHz, CDCl3): δ 3.71 (dd, J = 13.3, 1.3 Hz, 1H), 3.52 (dd, J = 13.4, 3.7 Hz, 1H), 3.29 (m, 1H), 2.98 (ddd, J = 13.6, 8.7, 5.4 Hz, 1H), 2.00–1.79 (m, 1H), 1.66 (m, 1H), 1.45 (s, 9H), 0.97 (s, 2H), 0.61 (td, J = 8.6, 4.9 Hz, 1H), 0.13 (q, J = 5.1 Hz, 1H). 13C{1H} NMR (100 MHz, CDCl3): δ 155.2, 79.2, 42.2, 40.3, 28.5, 22.7, 9.6, 8.9, 7.9. IR (neat): 1688 (s) cm−1. HRMS (ESI): calcd for C11H19NO2 [M + Na]+, 220.1308; found, 220.1308.
1-(3-Azabicyclo[4.1.0]heptan-3-yl)-2-phenylethane-1,2-dione (1).
A medium microwave vial was charged with N-Boc 3-azabicyclo[4.1.0]heptane (S1, 269 mg, 1.37 mmol, 1.00 equiv) and anhydrous 1 M Et2O·HCl (7.0 mL, 8.8 mmol, 8.0 equiv). The vial was equipped with a stir bar and fitted with a vial adapter and cap. The mixture was heated in a microwave at 100 °C for 6 min (0 bar additional pressure, absorption level set to “normal”). The pressure of the reaction vessel increased upon heating to the reaction temperature and stabilized at 8 bar for the duration of heating. At the end of the heating period, a fluorescent yellow precipitate had formed that coated the walls of the vial, and a yellow oil had collected at the bottom of the vial. The vial was cooled to room temperature, vented using a needle, and uncapped, and the contents were concentrated under a stream of nitrogen. The residue was dissolved in a minimal amount of CH2Cl2 (<5 mL), and Et2O (~10 mL) was added, which led to the formation of light-yellow, off-white crystals. The solvent was evaporated under a stream of nitrogen, and the resulting crystals were dried in vacuo. This resulting material was used in the following step without further purification.
Benzoylformic acid (254 mg, 1.69 mmol, 1.00 equiv) and CH2Cl2 (30 mL) were added to an oven-dried 100 mL flask equipped with a magnetic stir bar under a flow of N2. The resulting light-yellow solution was stirred for 5 min, at which point oxalyl chloride (0.146 mL, 1.69 mmol, 1.00 equiv) was added. The solution was stirred for 10 min at room temperature, followed by the addition of one drop of anhydrous DMF, at which point the solution grew darker in color. Evolving HCl gas was bubbled through a 1 M solution of NaOH, and the reaction mixture was stirred under N2 until bubbling had ceased (approximately 3 h). The solution was then cooled to 0 °C, and the crystalline material produced in the prior step (182 mg, 1.36 mmol, 0.800 equiv) was added as a solution in CH2Cl2 (5 mL), followed by Et3N (0.354 mL, 2.54 mmol, 1.50 equiv). The reaction mixture was stirred at room temperature for 16 h; then the reaction was quenched with 20 mL of H2O, and the layers were separated. The aqueous phase was extracted with CH2Cl2 (3 × 20 mL), and the organic extracts were combined, dried over Na2SO4, and concentrated in vacuo to give a yellow oil. Purification of the crude material by column chromatography (silica, 0% to 10% EtoAc/hexanes) provided the title compound as a colorless oil, which was crystallized using ice-cooled pentane (250 mg, 80% yield, 1:0.5 mixture of rotamers). Alternatively, addition of a seed crystal and placing the material under a vacuum yields the crystalline material. Single crystals for XRD analysis were grown by slow evaporation of a supersaturated solution of the ketoamide (1, 50 mg) in hot HPLC-grade hexanes (~2 mL). 1H NMR (400 MHz, CDCl3): δ 7.94 (ddd, J = 8.6, 5.4, 1.3 Hz, 1H), 7.65 (dddd, J = 8.7, 5.5, 2.8, 1.4 Hz, 1H), 7.55–7.48 (m, 1H), 4.10 (dd, J = 13.7, 1.6 Hz, 0.5H), 3.86 (dd, J = 13.7, 5.5 Hz, 0.5H), 3.83–3.76 (m, 0.5H), 3.67 (dd, J = 13.5, 4.9 Hz, 0.5H), 3.51 (dd, J = 13.4,2.0 Hz, 0.5H), 3.22 (ddd, J = 13.2, 9.2, 5.6 Hz, 0.5H), 3.11 (m, 1H), 2.13 (ddt, J = 14.2, 7.3, 5.5 Hz, 1H), 1.98 (ddt, J = 11.5, 6.9, 5.7 Hz, 1H), 1.85 (dddd, J = 14.6, 8.9, 5.8, 2.6 Hz, 0.5H), 1.73 (dtd, J = 14.0, 7.1, 6.6, 2.1 Hz, 1H), 1.21–1.06 (m, 1H), 0.99 (dddd, J = 13.7, 8.6, 5.3, 2.1 Hz, 0.5H), 0.80 (td, J = 8.8, 5.2 Hz, 0.5H), 0.71 (td, J = 8.7, 5.2 Hz, 0.5H), 0.32 (dq, J = 8.6, 5.3 Hz, 1H) (eight 1H resonances overlap due to amide rotation). 13C{1H} NMR (126 MHz, CDCl3): δ 191.67, 191.66, 166.3, 166.0, 134.7, 133.2, 133.1, 129.7, 129.0, 128.98, 44.6, 42.3, 40.141, 38.5, 23.5, 22.4, 9.7, 9.6, 9.4, 9.2, 8.1, 7.8 (two 13C resonances overlap due to amide rotation). IR (neat): 1677 (s), 1632 (s) cm−1. Mp: 64–67 °C. HRMS (ESI): calcd for C14H15NO2 [M + Na]+, 252.0995; found, 252.0994.
Low-Temperature Reaction Setup.
On a glass plate (4 cm × 4 cm × 1 mm) was placed 1 (205 mg, 0.890 mmol), previously chopped to a fine consistency with a razor blade, which was secured by placing an additional glass plate of the same size on top, the edges of which were sealed with electrical tape and secured on a ring stand 1 cm above a 40 W Kessil A160WE blue LED 400–450 nm fixed wavelength lamp (Figure S1, top left). A crystallization dish filled with dry ice (omitted from the photos) and iPrOH and covered with aluminum foil was placed on the surface of the glass side, with a nitrogen stream running over the bottom face of the plate (Figure S1, top right). The LED Kessil lamp was turned on full intensity (Figure S1, bottom left), and a bucket was placed over the reaction setup (Figure S1, bottom right) to minimize exposure to the light (see the Supporting Information for details).
(1S,2R,4R,9S)-9-Hydroxy-9-phenyl-7-azatricyclo[5.2.0.02,4]-nonan-8-one (2a).
Compound 2a was prepared according to the procedure outlined in Low-Temperature Reaction Setup (see the Supporting Information for pictures and details). After 5 h, completion of the reaction was determined by NMR analysis, and the material was transferred to a 20 mL scintillation vial using CH2Cl2. Several reaction runs (4 × 205 mg) were combined (total crude mass of 820 mg) and purified using column chromatography (silica, 0% to 60% EtOAc/hexanes). Further purification using column chromatography (silica, 0% to 40% Et2O/hexanes) provided 2a as a white crystalline solid (376 mg, 35% yield). Single crystals for XRD analysis were grown by slow evaporation of a solution of 2a (<3 mg) in HPLC-grade EtOAc (<1 mL). 1H NMR (400 MHz, CDCl3): δ 7.60–7.45 (m, 2H), 7.45–7.31 (m, 3H), 4.23 (d, J = 2.3 Hz, 1H), 3.34 (td, J = 12.6, 6.8 Hz, 1H), 3.23 (ddd, J = 12.6, 7.4, 1.7 Hz, 1H), 2.83 (s, 1H), 2.10 (tdd, J = 12.4, 7.6, 2.5 Hz, 1H), 1.85 (dddd, J = 14.4, 6.8, 3.2, 1.8 Hz, 1H), 1.08–0.88 (m, 2H), 0.17 (q, J = 5.4 Hz, 1H), 0.02 (td, J = 8.2, 6.2 Hz, 1H). 13C{1H} NMR (100 MHz, CDCl3): δ 169.2, 136.5, 128.5, 128.3, 127.6, 88.8, 60.8, 35.4, 17.8, 11.6, 8.8, 2.3. IR (thin film): 3340 (b), 1730 (s) cm−1. Mp: 144–147 °C. HRMS (ESI): calcd for C14H15NO2 [M + H]+, 230.1176; found, 230.1177.
(2S,6aS,7aS,7bS)-2-Phenylhexahydrocyclopropa[c]oxazolo[3,2-a]pyridin-3(2H)-one (2b).
Compound 2b was isolated as a colorless residue in 5% yield (42 mg) using column chromatography (silica, 0% to 60% EtOAc/hexanes) as a side product of the reaction that yielded 2a. 1H NMR (500 MHz, CDCl3): δ 7.41–7.35 (m, 2H), 7.30–7.27 (m, 2H), 7.25–7.19 (m, 1H), 5.74 (dd, J = 3.4, 2.4 Hz, 1H), 5.19 (d, J = 2.4 Hz, 1H), 3.64 (ddd, J = 13.4, 7.3, 4.9 Hz, 1H), 2.85 (ddd, J = 13.4, 8.7, 6.7 Hz, 1H), 1.90 (dtd, J = 14.4, 6.7, 4.9 Hz, 1H), 1.76–1.68 (m, 1H), 1.37 (tdd, J = 8.6, 5.2, 3.5 Hz, 1H), 1.32–1.25 (m, 1H), 0.74 (td, J = 8.5, 5.6 Hz, 1H), 0.39 (q, J = 5.5 Hz, 1H). 13C{1H} NMR (126 MHz, CDCl3): δ 169.3, 136.9, 128.8, 128.5, 128.5, 126.1, 86.8, 79.4, 37.3, 20.3, 15.7, 11.5, 7.1. IR (thin film): 1707 (s) cm−1. HRMS (ESI): calcd for C14H15NO2 [M + H]+, 230.1176; found, 230.1177.
(2R,6aS,7aS,7bS)-2-Phenylhexahydrocyclopropa[c]oxazolo[3,2-a]pyridin-3(2H)-one (2c).
Compound 2c was isolated in 5% yield (41 mg) as a colorless residue using column chromatography (silica, 0% to 60% EtOAc/hexanes) as a side product of the reaction that yielded 2a. 1H NMR (500 MHz, CDCl3): δ 7.50–7.45 (m, 2H), 7.43–7.36 (m, 2H), 7.35–7.31 (m, 1H), 5.71 (dd, J = 3.5, 2.0 Hz, 1H), 5.20–5.18 (m, 1H), 2.05 (dtd, J = 14.3, 6.6, 5.3 Hz, 1H), 1.79 (dddd, J = 14.3, 8.2, 7.3, 2.5 Hz, 1H), 1.52 (tdd, J = 8.5, 5.2, 3.4 Hz, 1H), 1.45–1.34 (m, 1H), 0.84 (td, J = 8.4, 5.5 Hz, 1H), 0.53 (q, J = 5.5 Hz, 1H). 13C{1H} NMR (126 MHz, CDCl3): δ 169.3, 136.4, 128.6, 128.6, 126.9, 85.7, 79.4, 37.4, 20.5, 15.5, 11.5, 7.1. IR (thin film): 1736 (s) cm−1. HRMS (ESI): calcd for C14H15NO2 [M + H]+, 230.1176; found, 230.1177.
Representative Procedure for C–C Cleavage/Cross-Coupling.
[(1R,2S,6R)-3-Azabicyclo[4.1.0]heptane-2,3-diyl]bis-(phenylmethanone) (3b).
To an oven-dried 1 dram vial charged with a stir bar was added lactam 2a (22.9 mg, 0.100 mmol, 1.00 equiv). The vial was placed in a water- and oxygen-free glovebox, and PhBr (31.4 mg, 0.200 mmol, 2.00 equiv) was added, followed by Cs2CO3 (65.4 mg, 0.200 mmol, 2.00 equiv), APhos-Pd-G4 (6.5 mg, 10 mol %, 0.1 equiv), and toluene (0.5 mL; previously degassed by consecutive freeze–pump–thaw cycles until no bubbles of gas were observed and stored over activated molecular sieves). The vial was sealed with Teflon tape and placed in a heating block at 40 °C for 24 h. After this time, the reaction mixture was cooled to rt and filtered through a glass pipet packed with Celite. The vial and Celite plug were subsequently rinsed with 5% MeOH/CH2Cl2 (3 × 1 mL). The resulting crude material obtained after concentration of the filtrate was purified by PTLC (silica, 20% EtOAc/hexanes) to afford 3b as an opaque white residue (15.9 mg, 52%, 1:0.2 mixture of rotamers). Single crystals for XRD analysis were grown by slow evaporation of a solution of 3b (<3 mg) in HPLC-grade EtOAc (<1 mL). 1H NMR (500 MHz, CDCl3): δ 8.07 (d, J = 7.5 Hz, 2H + 0.2H), 7.58 (q, J = 7.5 Hz, 1H + 0.2H), 7.51 (t, J = 7.6 Hz, 2H + 0.4H), 7.46 (dd, J = 6.7, 3.0 Hz, 2H + 0.2H), 7.41 (dd, J = 5.2, 1.9 Hz, 3H + 0.4H), 6.31 (d, J = 8.8 Hz, 1H), 5.50 (d, J = 9.0 Hz, 0.2H), 4.62 (dd, J = 13.9, 4.1 Hz, 0.2H), 3.59 (ddd, J = 14.2, 5.3, 2.3 Hz, 1H), 3.13 (td, J = 13.7, 3.1 Hz, 1H), 2.86 (td, J = 13.5, 2.5 Hz, 0.2H), 2.03 (tt, J = 13.3, 4.9 Hz, 1H + 0.2H), 1.83 (d, J = 14.1 Hz, 0.2H), 1.75 (dt, J = 13.9, 2.9 Hz, 1H), 1.51 (qd, J = 8.7, 5.2 Hz, 1H + 0.2H), 1.31 (tq, J = 15.4, 10.3, 7.8 Hz, 2H + 0.4H), 0.88 (t, J = 6.9 Hz, 0.2H), 0.49 (td, J = 8.7, 5.6 Hz, 1H), 0.46–0.40 (m, 0.2H), 0.39 (q, J = 5.6 Hz, 1H), 0.34 (q, J = 5.6 Hz, 0.2H) (eight 1H resonances overlap due to amide rotation). 13C{1H} NMR (126 MHz, CDCl3): δ 197.26, 171.16, 135.86, 135.59, 133.12, 129.58, 128.69, 128.63, 128.51, 128.42, 127.83, 127.00, 126.50, 53.01, 40.56, 40.54, 11.11, 11.02, 10.83, 10.81, 5.29, 5.24 (10 13C resonances overlap due to amide rotation). IR (thin film): 1689 (s), 1627 (s) cm−1. Mp: 120–122 °C. HRMS (ESI): calcd for C20H19NO2 [M + Na]+, 328.1308; found, 328.1309.
1-[4-Methylene-3,4-dihydropyridin-1(2H)-yl]-2-phenylethane-1,2-dione (3a).
The title compound was prepared using a procedure analogous to the representative procedure for the formation of 3b using RuPhos-Pd-G4 (8.5 mg, 10 mol %) as the ligand–precatalyst complex. The reaction mixture was filtered over a plug of Celite, eluting with EtOAc to avoid decomposition of the title compound. The crude material was purified by preparative TLC (silica, 20% EtOAc/hexanes) to afford 3a as a yellow residue (13.6 mg, 60%, 1:0.75 mixture of rotamers). Note that the title compound is extremely unstable in the solid state and spontaneously polymerizes upon concentration to dryness. 1H NMR (600 MHz, C6D6): δ 7.97–7.90 (m, 2H + 1.5H), 7.37 (dd, J = 8.2, 1.5 Hz, 0.75H), 7.05 (tt, J = 7.5, 4.1, 1.2 Hz, 1H + 1H), 6.99–6.94 (m, 2H + 1.5H), 6.31 (dd, J = 8.1, 1.5 Hz, 1H), 5.46 (d, J = 8.2 Hz, 0.75H), 5.12 (d, J = 8.1 Hz, 1H), 4.68 (d, J = 1.7 Hz, 0.75H), 4.64 (d, J = 1.5 Hz, 1H), 4.46 (t, J = 1.7 Hz, 1H), 4.43 (p, J = 1.7 Hz, 0.75H), 3.66–3.58 (m, 2H), 3.14–3.07 (m, 1.5H), 2.00 (tt, J = 6.5, 1.6 Hz, 2H), 1.88 (tt, J = 6.5, 1.6 Hz, 1.5H). 13C{1H} NMR (151 MHz, C6D6): δ 190.4, 190.1, 164.8, 163.7, 137.4, 137.3, 134.2, 134.2, 133.5, 129.6, 129.6, 128.8, 128.8, 124.8, 123.5, 113.7, 111.9, 111.0, 110.8, 43.7, 39.6, 29.3, 28.6. IR (thin film): 1705 (s), 1612 (s) cm−1. HRMS (ESI): calcd for C14H14NO2 [M + H]+, 228.0984; found, 228.0987.
1-[(1R,2S,6R)-2-Benzoyl-3-azabicyclo[4.1.0]heptan-3-yl]-3-(triisopropylsilyl)prop-2-yn-1-one (4).
The title compound was prepared using a procedure analogous to the representative procedure for the formation of 3b but using (bromoethynyl)triisopropylsilane as the cross-coupling partner (52 mg, 0.2 mmol, 2 equiv). The crude material was purified by PTLC (silica, 5% EtOAc/hexanes) to provide 4 as a white residue (19.8 mg, 48% yield, 1:0.3 mixture of rotamers). Single crystals for XRD analysis were grown from slow evaporation of a solution of 4 (<3 mg) in HPLC-grade EtOAc (<1 mL). 1H NMR (500 MHz, CDCl3): δ 8.01 (dt, J = 8.3, 1.2 Hz, 2H + 0.6H), 7.61–7.55 (m, 1H + 0.3H), 7.48 (dd, J = 8.3, 7.0 Hz, 2H + 0.6H), 6.20 (d, J = 9.1 Hz, 0.3H), 6.16 (d, J = 8.6 Hz, 1H), 4.46 (ddd, J = 14.2, 5.6, 2.7 Hz, 0.3H), 4.36 (ddd, J = 14.2, 5.5, 2.2 Hz, 1H), 3.23 (td, J = 13.6, 3.2 Hz, 1H), 2.75 (td, J = 13.2, 3.4 Hz, 0.3H), 2.05 (tq, J = 14.3, 4.7 Hz, 1H), 2.01–1.92 (m, 0.3H), 1.86 (ddt, J = 13.5, 3.8, 2.1 Hz, 1H), 1.78 (dq, J = 13.7, 3.0 Hz, 0.3H), 1.55 (qd, J = 8.8, 5.2 Hz, 0.3H), 1.42 (qd, J = 8.7, 5.2 Hz, 1H), 1.36–1.20 (m, 2H + 0.6H), 1.16–1.06 (m, 18H), 0.97–0.87 (m, 5.5H), 0.49 (dtd, J = 17.6, 8.7, 5.7 Hz, 1H + 0.3H), 0.39 (q, J = 5.6 Hz, 1H), 0.32 (q, J = 5.5 Hz, 0.3H) (six 1H resonances overlap due to amide rotation). 13C{1H} NMR (126 MHz, CDCl3): δ 196.5, 195.6, 153.4, 153.2, 135.4, 134.7, 133.4, 133.2, 128.7, 128.5, 128.5, 98.3, 97.6, 95.2, 94.7, 57.1, 52.5, 40.1, 34.2, 22.6, 21.7, 18.5, 18.41, 18.37, 11.85, 11.77, 11.5, 11.4, 11.1, 11.06, 11.02, 10.97, 10.9, 5.98, 5.94, 5.2, 5.1 (one pair of 13C resonances overlap due to amide rotation. IR (thin film): 1693 (s), 1621 (s) cm−1. Mp: 90–93 °C. HRMS (ESI): calcd for C25H35NO2Si [M + Na]+, 432.2332; found, 432.2333.
(1R,2S,6R)-2-Benzoyl-3-azabicyclo[4.1.0]heptane-3-carbalde-hyde (5).
The title compound was prepared according to a literature procedure.10 The crude material was purified using PTLC (silica, 20% EtOAc/hexanes) to provide 5 as a white residue (14.4 mg, 63% yield, 1:0.1 mixture of rotamers). Single crystals for XRD analysis were grown from slow evaporation of a solution of 5 (<3 mg) in HPLC-grade EtOAc (<1 mL). 1H NMR (500 MHz, CDCl3): δ 8.10 (s, 1H + 0.1H), 8.03–7.99 (m, 2H + 0.2H), 7.58 (tt, J = 6.9, 1.2 Hz, 1H + 0.1H), 7.51–7.46 (m, 2H + 0.2H), 6.04 (d, J = 8.6 Hz, 1H), 5.41 (d, J = 9.0 Hz, 0.1H), 4.30 (ddd, J = 14.1, 5.7, 1.9 Hz, 0.1H), 3.48 (ddd, J = 14.0, 5.5, 2.1 Hz, 1H), 3.21 (td, J = 13.5, 3.6 Hz, 1H), 2.71 (td, J = 13.5, 3.6 Hz, 0.1H), 1.99 (tt, J = 13.1, 5.0 Hz, 1H), 1.90 (ddt, J = 13.4, 3.8, 1.9 Hz, 1H), 1.80–1.74 (m, 0.1H), 1.53 (qd, J = 8.8, 5.2 Hz, 0.1H), 1.43 (qd, J = 8.7, 5.3 Hz, 1H), 1.32 (dqt, J = 11.6, 4.5, 2.2 Hz, 1H), 0.55–0.45 (m, 1H + 0.1H), 0.37 (qd, J = 5.6, 2.2 Hz, 1H), 0.34–0.29 (m, 0.1H) (five 1H resonances overlap due to amide rotation). 13C{1H} NMR (126 MHz, CDCl3): δ 196.3, 161.7, 135.2, 133.3, 128.9, 128.7, 128.5, 128.4, 56.2, 51.5, 39.2, 32.9, 22.8, 21.5, 11.8, 11.7, 11.44, 11.43, 10.6, 10.5, 6.2, 5.0 (two 13C resonances overlap due to amide rotation). IR (thin film): 1693 (s), 1662 (s) cm−1. Mp: 92–95 °C. HRMS (ESI): calcd for C14H15NO2 [M + H], 230.1176; found, 230.1179.
Supplementary Material
ACKNOWLEDGMENTS
C.A.R. thanks the Reed College Marsh Cronyn Fellowship for partial funding. R.L.L. thanks Reed College for startup funds. C.S.Y. thanks the Disruptive Chemistry Fellowship program of Merck & Co., Inc. (Kenilworth, NJ), for support. D.G.M. and L.-P.X. gratefully acknowledge the use of the resources of the Cherry Emerson Center for Scientific Computation at Emory University. D.G.M. thanks the National Science Foundation under the CCI Center for Selective C–H Functionalization (CHE-1700982). L.-P.X. acknowledges the National Natural Science Foundation of China (NSFC 21702126) and the China Scholarship Council for support. R.S. thanks the National Institute of General Medical Sciences (R35 GM130345A) for support of a portion of the work conducted at Berkeley. The authors thank Dr. Hasan Celik and the University of California Berkeley’s NMR facility in the College of Chemistry (CoC-NMR) for spectroscopic assistance. Instruments in CoC-NMR are supported in part by Nationa Institutes of Health Grant S10OD024998. The authors acknowledge the Catalysis Facility of Lawrence Berkeley National Laboratory, which is supported by the Director, Office of Science, of the U.S. Department of Energy under Contract DE-AC02- 05CH11231 for use of resources. The authors thank Dr. Nick Settineri (University of California Berkeley) for assistance with X-ray crystallographic data acquisition.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.1c01466.
Experimental procedures, computational details, and compound characterization (PDF)
Accession Codes
CCDC 2090469–2090473 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.joc.1c01466
The authors declare no competing financial interest.
Contributor Information
Charis Amber Roberts, Department of Chemistry, University of California, Berkeley, California 94720, United States; Chemistry Department, Reed College, Portland, Oregon 97202, United States.
Bohyun Park, Department of Chemistry, University of California, Berkeley, California 94720, United States; Department of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea; Center for Catalytic Hydrocarbon Functionalizations, Institute for Basic Science (IBS), Daejeon 34141, Republic of Korea;.
Li-Ping Xu, Cherry L. Emerson Center for Scientific Computation and Department of Chemistry, Emory University, Atlanta, Georgia 30322, United States;.
Jose B. Roque, Department of Chemistry, University of California, Berkeley, California 94720, United States;.
Charles S. Yeung, Disruptive Chemistry Fellow, Department of Discovery Chemistry, Merck & Company, Inc., Boston, Massachusetts 02115, United States;.
Djamaladdin G. Musaev, Cherry L. Emerson Center for Scientific Computation and Department of Chemistry, Emory University, Atlanta, Georgia 30322, United States;.
Richmond Sarpong, Department of Chemistry, University of California, Berkeley, California 94720, United States;.
Rebecca Lyn LaLonde, Chemistry Department, Reed College, Portland, Oregon 97202, United States;.
REFERENCES
- (1).Mitchell EA; Peschiulli A; Lefevre N; Meerpoel L; Maes BUW Direct α-Functionalization of Saturated Cyclic Amines. Chem. - Eur. J 2012, 18 (33), 10092–10142. [DOI] [PubMed] [Google Scholar]
- (2).Vitaku E; Smith DT; Njardarson JT Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem 2014, 57 (24), 10257–10274. [DOI] [PubMed] [Google Scholar]
- (3).Drews J Drug Discovery: A Historical Perspective. Science 2000, 287 (5460), 1960–1964. [DOI] [PubMed] [Google Scholar]
- (4).Godula K; Sames D C-H Bond Functionalization in Complex Organic Synthesis. Science 2006, 312 (5770), 67–72. [DOI] [PubMed] [Google Scholar]
- (5).Prokopcová H; Bergman SD; Aelvoet K; Smout V; Herrebout W; Van der Veken B; Meerpoel L; Maes BUW C-2 Arylation of Piperidines through Directed Transition-Metal-Catalyzed sp3 C–H Activation. Chem. - Eur. J 2010, 16 (44), 13063–13067. [DOI] [PubMed] [Google Scholar]
- (6).Jain P; Verma P; Xia G; Yu J-Q Enantioselective Amine α-Functionalization via Palladium-Catalysed C–H Arylation of Thioamides. Nat. Chem 2017, 9 (2), 140–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Verma P; Richter JM; Chekshin N; Qiao JX; Yu J-Q Iridium(I)-Catalyzed α-C(sp3)–H Alkylation of Saturated Azacycles. J. Am. Chem. Soc 2020, 142 (11), 5117–5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Jiang H-J; Zhong X-M; Yu J; Zhang Y; Zhang X; Wu Y-D; Gong L-Z Assembling a Hybrid Pd Catalyst from a Chiral Anionic CoIII Complex and Ligand for Asymmetric C(sp3)–H Functionalization. Angew. Chem., Int. Ed 2019, 58 (6), 1803–1807. [DOI] [PubMed] [Google Scholar]
- (9).Greßies S; Klauck FJR; Kim JH; Daniliuc CG; Glorius F Ligand-Enabled Enantioselective C – H Activation of Tetrahydroquinolines and Saturated Aza-Heterocycles by RhI. Angew. Chem., Int. Ed 2018, 57 (31), 9950–9954. [DOI] [PubMed] [Google Scholar]
- (10).Roque JB; Kuroda Y; Jurczyk J; Xu L-P; Ham JS; Göttemann LT; Roberts CA; Adpressa D; Saurí J; Joyce LA; Musaev DG; Yeung CS; Sarpong R C–C Cleavage Approach to C–H Functionalization of Saturated Aza-Cycles. ACS Catal 2020, 10 (5), 2929–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Xu L-P; Roque JB; Sarpong R; Musaev DG Reactivity and Selectivity Controlling Factors in the Pd/Dialkylbiarylphosphine-Catalyzed C–C Cleavage/Cross-Coupling of an N-Fused Bicyclo α-Hydroxy-β-Lactam. J. Am. Chem. Soc 2020, 142 (50), 21140–21152. [DOI] [PubMed] [Google Scholar]
- (12).Anslyn EV; Dougherty DA Modern Physical Organic Chemistry; University Science Books: Sausalito, CA, 2006. [Google Scholar]
- (13).Aoyama H; Hasegawa T; Omote Y Solid State Photochemistry of N,N-Dialkyl-.Alpha.-Oxoamides. Type II Reactions in the Crystalline State. J. Am. Chem. Soc 1979, 101 (18), 5343–5347. [Google Scholar]
- (14).Oelgemöller M; Hoffmann N Studies in Organic and Physical Photochemistry – an Interdisciplinary Approach. Org. Biomol. Chem 2016, 14 (31), 7392–7442. [DOI] [PubMed] [Google Scholar]
- (15).Griesbeck AG; Heckroth H Stereoselective Synthesis of 2-Aminocyclobutanols via Photocyclization of α-Amido Alkylaryl Ketones: Mechanistic Implications for the Norrish/Yang Reaction. J. Am. Chem. Soc 2002, 124 (3), 396–403. [DOI] [PubMed] [Google Scholar]
- (16).Sinicropi A; Barbosa F; Basosi R; Giese B; Olivucci M Mechanism of the Norrish–Yang Photocyclization Reaction of an Alanine Derivative in the Singlet State: Origin of the Chiral-Memory Effect. Angew. Chem., Int. Ed 2005, 44 (16), 2390–2393. [DOI] [PubMed] [Google Scholar]
- (17).Kohlschütter V; Haenni P Zur Kenntnis des Graphitischen Kohlenstoffs und der Graphitsäure. Z. Für Anorg. Allg. Chem 1919, 105 (1), 121–144. [Google Scholar]
- (18).Ham JS; Park B; Son M; Roque JB; Jurczyk J; Yeung CS; Baik M-H; Sarpong R C–H/C–C Functionalization Approach to N-Fused Heterocycles from Saturated Azacycles. J. Am. Chem. Soc 2020, 142 (30), 13041–13050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Morokuma K Molecular Orbital Studies of Hydrogen Bonds. III. C=O⋯H–O Hydrogen Bond in H2CO⋯H2O and H2CO⋯2H2O. J. Chem. Phys 1971, 55 (3), 1236–1244. [Google Scholar]
- (20).Ziegler T; Rauk A On the Calculation of Bonding Energies by the Hartree Fock Slater Method. Theor. Chim. Acta 1977, 46 (1), 1–10. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.