

Abstract
Brain aging is a complex process that affects everything from the subcellular to the organ level, begins early in life, and accelerates with age. Morphologically, brain aging is primarily characterized by brain volume loss, cortical thinning, white matter degradation, loss of gyrification, and ventricular enlargement. Pathophysiologically, brain aging is associated with neuron cell shrinking, dendritic degeneration, demyelination, small vessel disease, metabolic slowing, microglial activation, and the formation of white matter lesions. In recent years, the mechanics community has demonstrated increasing interest in modeling the brain’s (bio)mechanical behavior and uses constitutive modeling to predict shape changes of anatomically accurate finite element brain models in health and disease. Here, we pursue two objectives. First, we review existing imaging-based data on white and gray matter atrophy rates and organ-level aging patterns. This data is required to calibrate and validate constitutive brain models. Second, we review the most critical cell- and tissue-level aging mechanisms that drive white and gray matter changes. We focuse on aging mechanisms that ultimately manifest as organ-level shape changes based on the idea that the integration of imaging and mechanical modeling may help identify the tipping point when normal aging ends and pathological neurodegeneration begins.
Keywords: Brain aging mechanisms, Cerebral atrophy, Morphological changes, Gray and white matter changes, Vascular changes
1. Introduction
Aging and pathology are associated with significant changes of the brain’s intricate microstructure and results in cognitive decline (Rodrigue and Kennedy, 2011). Brain morphology, i.e., brain shape and anatomy, evolves with age and most commonly undergoes significant atrophy, i.e., cerebral tissue volume loss (Nyberg and Wåhlin, 2020). These changes are accompanied, if not directly the cause, for cognitive deficits that include memory loss (Murman, 2015; Fjell et al., 2016), reduced motor performance (Seidler et al., 2010), and behavior (Park and Reuter-Lorenz, 2009). Due to extensive efforts in large population medical imaging studies, such as the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (Jack et al., 2008b) or the Rotterdam Study (Vinke et al., 2018), structural features of healthy and accelerated brain aging have been identified (Lockhart and DeCarli, 2014; Fox and Schott, 2004). Cross-sectional studies with healthy, or cognitively normal, elderly and subjects with mild cognitive impairment or Alzheimer’s disease (AD), have shown that neurodegenerative diseases accelerate aging mechanisms and lead to more pronounced structural changes of the brain (Coupé et al., 2019). The trajectories of cerebral atrophy in healthy aging and dementia are subject to extensive investigation because a clear distinction between healthy and abnormal changes remains challenging (Fjell et al., 2014b) and early detection of abnormal shape changes may serve as a sensitive biomarker for accelerated aging due to the onset of neurodegeneration (Pini et al., 2016). A major limitation for early detection, however, is that each brain looks different such that it is challenging to identify subtle changes or abnormal developments from a single or even longitudinal magnetic resonance images (MRI) (Scahill et al., 2003; Fjell et al., 2009c, 2013).
Thus far, age- and disease-related brain shape changes have been extensively studied by the neuroimaging community (Oschwald et al., 2020; Fjell and Walhovd, 2010). The development of widely accepted brain atlases and the corresponding tools to register any brain scan to such atlases have enabled direct comparison of shape features from cross-sectional data (Fischl et al., 2002). These features include volume fractions of individual brain regions (Ge et al., 2002), cortical thickness (Madan and Kensinger, 2016), sulcal depth (Rettmann et al., 2006), loss of gyrification (Madan and Kensinger, 2016; Rettmann et al., 2006), and ventricular volume (Yue et al., 1997; Coffey et al., 2001b). While cross-sectional data allows to detect pervasive dominant trends in respective cohorts, the data on personalized brain shape changes remains understudied (Fjell and Walhovd, 2010; Oschwald et al., 2020). Longitudinal image data requires registration of images from consecutive brain scans and physics-based models for interpretation of respective output data (Reuter et al., 2012; Storsve et al., 2014; Wang et al., 2021). While several such registration methods exist (Fjell et al., 2009c; Fjell and Walhovd, 2010; Resnick et al., 2003), it remains challenging to adequately evaluate a subject’s state of health from the subtle changes detected in scans taken during a typically 3–7 year time frame (Reuter et al., 2012).
The progressive microstructural degeneration of white matter (WM) and gray matter (GM) tissue leads to tissue softening (Murphy et al., 2011; Kalra et al., 2019; Hiscox et al., 2021), tissue shrinking (Resnick et al., 2003), and tissue damage associated with such mechanisms as small vessel disease (Wardlaw et al., 2013; Makedonov et al., 2013), demyelination (Liu et al., 2017; Peters, 2002), and leakage of functional barriers such as the ventricular wall (Jiménez et al., 2014). Initially, these aging mechanisms occur predominantly on the cell level due to slowing metabolic activity and ischemia (Sikora et al., 2021), but then gradually manifest as tissue and ultimately organ-level alterations of brain shape (Fox and Schott, 2004; Apostolova et al., 2012). The field of mechanics is suitable to provide a constitutive modeling framework to describe and predict the spatiotemporal evolution of atrophy patterns due to microstructural changes during aging. As such, multiphysics modeling with the finite element method, i.e., the coupling of biology-driven aging mechanisms and structural changes, would allow to simulate healthy and pathological brain shape changes in order to identify abnormal atrophy patterns (Blinkouskaya and Weickenmeier, 2021). To date, however, only few studies have aimed at modeling the mechanical, or mechanobiological, response of the aging brain; most frequently, a continuum approach is used to describe tissue-level changes such as volume loss or the coupling between neurodegeneration, observed in Alzheimer’s disease for example, and cerebral atrophy (Harris et al., 2019; Schäfer et al., 2019; Budday and Kuhl, 2020; Weickenmeier et al., 2018; Blinkouskaya and Weickenmeier, 2021). Multiphysics models will provide new insight into the multiscale mechanisms of brain aging and reveal critical features, i.e., atrophy patterns, discerning accumulation of waste products, advanced cell- and tissue-level damage, that separate normal from accelerated brain aging. Moreover, the calibration and validation of such computer models and biomarkers will be made possible by increasingly available longitudinal imaging data and significantly improve the predictive capabilities of personalized brain aging simulations.
The present review is motivated by the development of advanced constitutive multiphysics models of age-related organ-level brain changes that relate cell- and tissue-level aging mechanisms to organ-level brain shape changes visible in medical imaging. We, therefore, split this review between two major objectives:
The first objective is to provide an overview of the most prominent structural features associated with cerebral atrophy and how they change with age. In Section 3 we summarize quantitative changes reported in literature and differentiate between global brain volume changes, gray matter shrinking, cortical thinning, white matter volume changes, ventricular enlargement, and brain folding changes.
The second objective is to present an overview of the most frequently observed cell- and tissue-level brain aging mechanisms that ultimately manifest as the previously discussed organ-level shape changes. To that end, we differentiate between gray matter mechanisms (Section 4), white matter mechanisms (Section 5), ventricular mechanisms (Section 6), and vascular changes (Section 7).
We begin with Section 2 by reviewing the most frequently used neuroimaging tools to visualize and quantify structural changes of the brain.
2. Common neuroimaging tools and imaging sequences used to quantify aging-related brain changes
The development of neuroimaging tools, such as various MRI sequences and atlas-based medical image analysis techniques, has transformed our ability to study the brain by providing quantitative methods for in vivo visualization of both brain structure and function (Fjell et al., 2009a). Especially with respect to exploring age-related brain changes, the ability to perform repeated measurements holds the promise to uncover subtle morphological changes from longitudinal imaging data (Resnick et al., 2003; Lockhart and DeCarli, 2014). MRI is the most commonly used imaging technique to visualize brain structure at increasingly high resolution. Most cross-sectional studies are based on T1-weighted MRI which provides enhanced contrast between GM, WM, and cerebrospinal fluid (CSF) (Lockhart and DeCarli, 2014). This image type allows to reliably evaluate volume, cortical thickness, degree of folding, and other shape features for the whole brain (Fischl and Dale, 2000; Gautam et al., 2015; Lemieux et al., 1999). Another frequently used MRI sequence is T2-weighted fluid attenuated inversion recovery, or FLAIR (Lockhart and DeCarli, 2014). In FLAIR, CSF signal is suppressed such that cerebral WM abnormalities appear as bright hyperintensities, or also referred to as white matter hyperintensities (WMHs) (Bhagat and Beaulieu, 2004; Kim, 1995). Diffusion tensor imaging (DTI) is used to analyze WM axon fibers, tissue anisotropy, and diffusivity by detecting the amount and direction of myelin water movement in extracellular and intracellular WM spaces (Sullivan and Pfefferbaum, 2006). In extracellular spaces fluid diffuses between fibers and in intracellular spaces fluid diffuses in the axoplasm (Sullivan and Pfefferbaum, 2006). DTI can detect deviations in these two compartments which would indicate abnormal WM degradation (Sullivan and Pfefferbaum, 2006). Functional MRI (fMRI) uses a specific T2-weighted sequence that is sensitive to the level of hemoglobin oxygenation in cerebral blood (Jezzard and Ramsey, 2003). This imaging sequence allows to detect neuronal activity during resting state or task performance, and is the primary technique to analyze cognitive performance (Jezzard and Ramsey, 2003). Lastly, positron emission tomography (PET) is a functional imaging technique that visualizes and measures cerebral blood flow, metabolism, regional chemical composition, absorption, and the presence of targeted disease biomarkers (Villemagne et al., 2021). Following the injection of a protein-specific tracer, PET is used to quantify the standardized uptake value which measures the concentration of the tracer that has adhered to its target (Phelps and Mazziotta, 1985). The quantification of biomarker concentrations, such as amyloid beta and tau, the two most prominent proteins in aging and neurodegenerative diseases, allows to uncover spatial and temporal brain changes and their impact on cognitive decline (Lowe et al., 2018).
Structural MRI, functional MRI, and PET imaging inform about anatomy, physiology, and biochemistry of the brain, respectively. Integration of these individual imaging sequences enables a multiphysics approach to characterizing brain changes in aging and help uncover spatiotemporal progression patterns of primary aging mechanisms. In one such effort, Fagan et al. combined PET imaging of brain amyloid load with a CSF measurement of beta-amyloid to investigate the potential use of those measures as a biomarker for pre-clinical AD (Fagan et al., 2006). Jack et al. investigated how biochemical changes, such as amyloid deposition, affect brain atrophy using both PET and MRI (Jack et al., 2008a). They concluded that both imaging sequences provided complementary information and deliver better predictive outcomes than using either method in isolation. In conclusion, neuroimaging tools open multiple avenues to analyze the aging brain. The combination of longitudinal imaging and computational modeling of aging will deliver reliable biomarkers for early diagnosis of abnormal aging patterns.
3. Morphological changes associated with healthy brain aging
Aging changes brain morphology in both healthy and pathological, or accelerated, aging. Despite each brain’s unique morphology, cross sectional studies have identified hallmark features of age-related changes that follow a persistent trend. Fig. 1 shows the most prevalent features of an aged brain which are GM and WM volume loss (Ge et al., 2002), cortical thinning (Madan and Kensinger, 2016), sulcal widening (Kochunov et al., 2005; Liu et al., 2013b), loss of gyrification (Madan and Kensinger, 2016; Rettmann et al., 2006), increased sulcal depth (Rettmann et al., 2006), and ventricular enlargement (Yue et al., 1997; Coffey et al., 2001b). The effect of aging on the brain’s morphology is highly heterogeneous and exhibits significant spatial and temporal variation (Fjell and Walhovd, 2010). In the following sections we will discuss GM volume changes, cortical thinning, brain folding changes, WM volume loss, and ventricular enlargement. We review quantitative changes presented in literature but would like to point out that these values vary significantly due to patient age range, state of health, MRI settings, and analysis approach. As such, a direct comparison is practically impossible; instead, we understand the following overview of values as a reference point for the comparison and calibration of future constitutive cerebral atrophy models (Blinkouskaya and Weickenmeier, 2021).
Fig. 1.
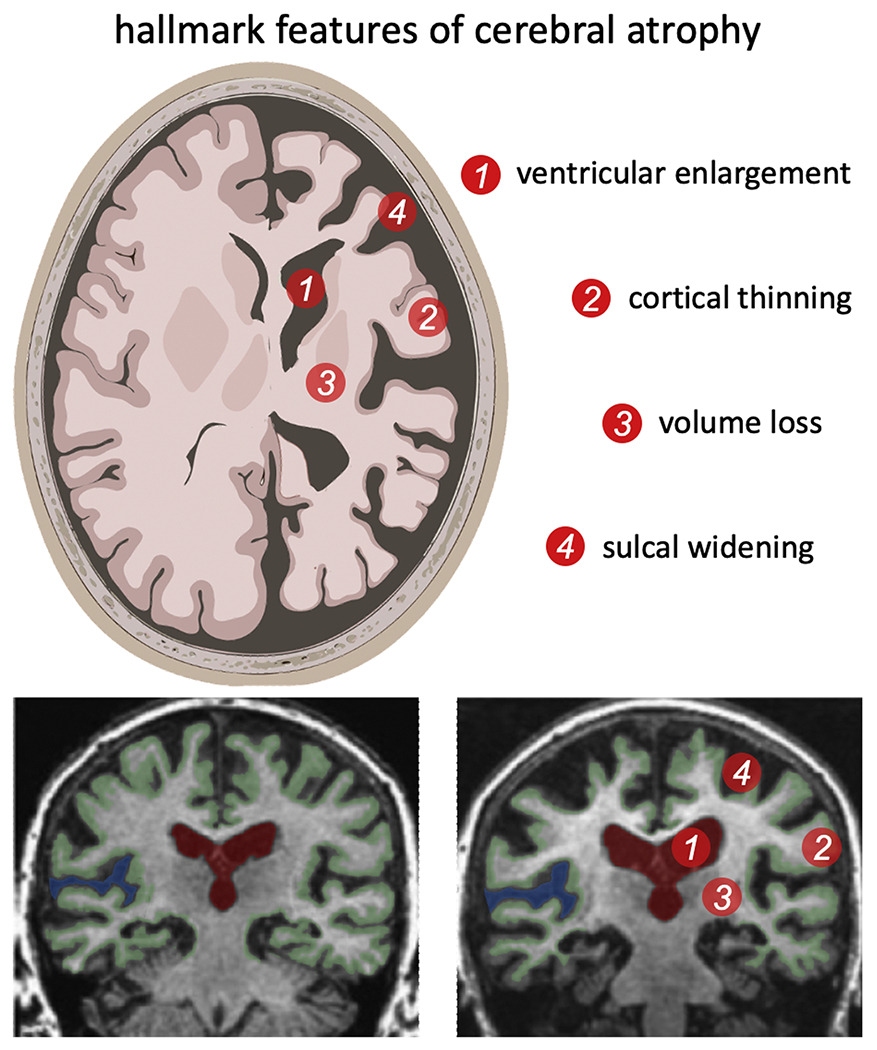
Cerebral atrophy is the most prominent morphological change in the aging brain and includes white and gray matter volume loss, cortical thinning, sulcal widening, and ventricular enlargement. Cross-sectional medical imaging studies have shown that these features gradually intensify in subjects aged 50 years and older (Fjell and Walhovd, 2010; Coupé et al., 2019). Longitudinal imaging data, i.e., two or more scans of the same subject taken at least a couple months apart, are increasingly used to determine the rate of change for individual subjects with the goal to differentiate between healthy and pathological aging processes (Resnick et al., 2003).
3.1. Global volume changes
The review of 56 longitudinal studies of healthy brain volume changes across the lifespan revealed an annual growth of 1% at nine years of age which gradually turns into atrophy at 13 years of age; early adulthood is described as a period of insignificant volume growth or no change in volume; after 35 years of age annual total brain volume loss is found to be 0.2%, which further accelerates and increases to 0.5% by the age of 60, showing a steady volume loss of more than 0.5%/year after that age (Hedman et al., 2012). Cerebral atrophy rates in the elderly are frequently reported on the order of 0.2–0.5%/year (Scahill et al., 2003; Fotenos et al., 2005; Enzinger et al., 2005; Fjell et al., 2009c; Coupé et al., 2019). As such, Fotenos et al., reported longitudinal global brain volume decline in non-demented individuals to be around 0.45%/year (Fotenos et al., 2005). For the cross-sectional group in the same study this rate is reported to be from 0.31% to 0.46% for the 65–95 age range. Jack et al. compared annual rates of volume decline for healthy individuals and individuals with AD (Jack et al., 2004). They reported a median total brain volume loss of 0.4%/year in clinically healthy subjects and substantially higher rates in subjects with AD with an atrophy rate of 0.6–1.4%/year depending on disease severity. It is important to note, however, that values reported in literature vary substantially. Henneman et al., for example, observed total brain atrophy rates of 0.6 ± 0.6%/year in healthy subjects and 1.9 ± 0.9%/year in AD (Henneman et al., 2009); Josephs et al. measured total brain atrophy rates of 0.2%/year for healthy subjects and 1.4%/year for subjects with dementia (Josephs et al., 2008); and Fotenos et al. reported a strong linear, moderate quadratic pattern of total brain volume decline across the adult lifespan with later onset of WM than GM volume loss (Fotenos et al., 2005). In their cohort aged 60–95 healthy subjects observed a mean total brain volume atrophy rate of 0.45%/year; the same rate for individuals with mild AD was twice as high at about 0.98%/year (Fotenos et al., 2005).
A large cross-sectional study of 2200 participants aged 34–97 years provided one of the earliest comparisons of lobar brain volume loss during aging (DeCarli et al., 2005b). The frontal lobe shrunk most with about 12% volume loss across the cohort. The temporal lobe atrophied by about 9%. The occipital and parietal lobes were observed to undergo a non-significant age-related volume change (DeCarli et al., 2005b) In a longitudinal study with a mean subject age of 75.6 (59.8–90.2) years and two one year follow up studies, Fjell et al. observed an annual decline of about 0.5% in prefrontal cortices and the temporal lobe after year one, while atrophy exceeded 1.0% for most regions (Fjell et al., 2009c).
3.2. Gray matter volume changes
Life-long GM volume changes strongly correlate with total brain volume decline and aging. GM atrophy rates have shown potential sex differences; while Fotenos et al. report no difference (Fotenos et al., 2005), Taki et al. observed an annual GM volume decline of 0.424 ± 0.017% in men and 0.298 ± 0.013% in women (Taki et al., 2011). Gunning-Dixon et al. measured a GM volume decrease by 14% in persons aged 30–90 years (Gunning-Dixon et al., 2009). Anderson et al. proposed GM atrophy rate as a biomarker to differentiate between healthy aging and disease progression in AD (Anderson et al., 2012). In a one year longitudinal follow up study, the observed mean annual GM atrophy rates were reported to be 0.49 ± 1.19% for healthy controls and 2.76 ± 1.64% for AD patients (Anderson et al., 2012).
The hippocampus is of particular interest in assessing age-related changes in brain because of its role in memory and learning. Moreover, the hippocampus has been shown to be particularly vulnerable to aging and neurodegeneration (Geinisman et al., 1995). A number of works compared annual atrophy rates in hippocampus for healthy elderly individuals with patients at different stages of dementia. In a large cross-sectional cohort study of healthy elderly by Raz et al., an annual hippocampus volume loss of 0.35% was observed; strikingly, an accompanying study of longitudinal changes within the same cohort revealed a two-times higher atrophy rate of 0.79% (Raz et al., 2005). Henneman et al. reported atrophy rates of 2.2 ± 1.4%/year in healthy subjects, 3.8 ± 1.2%/year in mild cognitive impairment, and 4.0 ± 1.2%/year in AD (Henneman et al., 2009). In a review article on hippocampus volume changes, Barnes et al. reported an annualized hippocampal atrophy rate of 1.41%/year in healthy adults and 4.66%/year in AD patients (Barnes et al., 2009). Similarly to the hippocampus, atrophy of the entorhinal cortex is considered a valuable indicator for neurodegenerative diseases (Raz et al., 2005); as such, the entorhinal cortex shrinks by 0.11%/year based on cross-sectional data and by 0.32%/year based on longitudinal image data. Other studies reported an annual atrophy of the entorhinal cortex to be in the range of 0.3–2.4% (Du et al., 2003; Coupé et al., 2019). Narvacan et al. observed that substructures of the limbic system decline faster, e.g., 0.4%/year for the hippocampus and 0.7%/year for the thalamus, than basal ganglia structures, e.g., 0.2%/year in the caudate (Narvacan et al., 2017). Their accompanying analysis of longitudinal data, showed that global volume loss ranged from 0.5%/year to 1.5%/year; in subjects aged 46–68 year the hippocampus shrunk by 0.6%/year and the nucleus accumbens, responsible for cognitive processing of motor function, by 0.9%/year; and between the ages 69–83, the hippocampus and amygdala showed an accelerated rate of decline of 1.5%/year and 1.2%/year, respectively (Narvacan et al., 2017).
3.3. Cortical thinning
Cortical thinning and the decrease of surface area are two hallmark features of brain aging. Consistently across many cross-sectional studies, the cortex gradually thins with age across all brain regions and is more correlated with age than surface area changes (Lemaitre et al., 2012; van Velsen et al., 2013; Fjell et al., 2014a). Thinning has also been shown to correlate with cognitive decline (Alegret et al., 2010) and disturbance in memory function (Fjell and Walhovd, 2010), such that it holds the promise to serve as a key biomarker for age-related cognitive decline. Cortical thickness is most frequently calculated from MRI using Free-Surfer (Fischl and Dale, 2000). This technique is well established and has facilitated a large body of work on comparative morphology. However, results on trajectories of cortical thickness changes throughout lifetime are contradictory as highlighted recently in the review by Walhovd et al. (Walhovd et al., 2016). A minor limitation is that today’s spatial resolution of MRI is insufficient to visualize the microstructural changes causing cortical thinning.
Total brain surface area decreases by 3.68 cm2/year and average cortical thickness decreases by 0.004 mm/year (Lemaitre et al., 2012). Another longitudinal study of 297 healthy individuals aged 23–87, reported a mean annual decrease of 0.19%/year for cortical area, a mean cortical volume loss of 0.51%/year, and a mean cortical thickness decrease of 0.35 mm/year (Storsve et al., 2014). Region-wise cortical thickness is the most vulnerable to age-related atrophy in the prefrontal cortex (Lemaitre et al., 2012) and remains nearly constant in the entorhinal and temporal regions until 60 years of age (Hasan et al., 2016). Cortical thickness of the anterior cingulate cortex has an attenuated “U”-shape relationship with age (Sowell et al., 2007) and has been shown to have an extensive impact on superior and inferior frontal areas, medial and superior temporal areas, and supramarginal cortices based on a study of 684 subjects aged 18–94 (Fjell et al., 2009b; Westlye et al., 2010).
3.4. White matter volume changes
Following an initial focus of the neuroimaging community on the study of cortical and subcortical GM changes, we know today that WM integrity plays a fundamental role in age-related cognitive decline (Gunning-Dixon and Raz, 2000). WM mainly consists of myelinated and unmyelinated long distance axonal projections from neurons that are embedded in a dense microglial scaffold. These microglial cells, which include astrocytes, myelin depositing oligodendrocytes, and progenitor cells, regulate neural activity and nutrient supply (Béchade et al., 2013; Gibson et al., 2014). Over the course of life, WM volume changes very differently than GM volume. More specifically, WM volume increases well into adulthood and peaks at about 40–50 years of age (Liu et al., 2017); it then rapidly decreases in the later stages of life (Courchesne et al., 2000; Walhovd et al., 2005; Westlye et al., 2010; Salat et al., 2009). Despite its delayed onset in comparison to GM, WM volume loss typically exceeds GM volume loss in the elderly and shows a volume reduction of 26% (Gunning-Dixon et al., 2009). It is estimated that WM volume reduction at 70 years ranges from 5.6% to 6.4% and at 80 years rapidly decrease to anywhere between 21.6% and 25.0% (Allen et al., 2005). Using atlas-based registration for cross-sectional comparison of individual regions of interest, annual WM volume loss in elderly was found to range from 0.77% in subjects with mean age 70.6 ± 6.1 years (Driscoll et al., 2009) to 0.88% in subjects aged 71.4 ± 0.9 years (Thompson et al., 2003). These WM volume changes are not homogeneous across the brain, but are most prominent in the frontal lobe (Salat et al., 2009). In a cohort aged 59–85 years, the annual atrophy rate was 2.1% in the frontal, 1.1% in the parietal, 1.0% in the temporal, and 0.8% in the occipital lobe (Resnick et al., 2003).
Cross-sectional DTI-based studies have shown that WM axon integrity is compromised in the aged brain which leads to a decreasing fractional anisotropy (FA), i.e., the deterioration of myelin sheaths that are wrapped around axons, thus increasing the diffusivity of free myelin water through the tissue. In the first four decades of life, FA and diffusivity tend to increase due to continued deposition of myelin (Lebel and Beaulieu, 2011). At later stages, however, this trend reverses. Frontal areas appear to be affected the most while temporal and posterior WM regions undergo less drastic changes (Abe et al., 2008; Salat et al., 2005; Lebel et al., 2012). This anterior-posterior gradient has also been observed in the corpus callosum (Lebel et al., 2012). It has also been reported that low FA or high diffusivity correlate with poor cognitive and motor performance in healthy aging (Sullivan et al., 2010). In a cross-sectional study of 430 healthy subjects ranging age 8–85 years, Westlye et al. assessed both WM volume loss and DTI parameters and observed inverted “U”-shape trajectories for WM volume loss and FA (Westlye et al., 2010). WM volume peaked at 50 years and FA peaked at 30 years of age; both measures then declined slowly into late adulthood followed by an accelerating decrease in senescence (Westlye et al., 2010).
3.5. Ventricular enlargement
The ventricular brain system is a network of communicating cavities that encloses and circulates CSF (Shook et al., 2014). The normal aging process can induce alterations in CSF circulation and, thus, impacting neuronal performance (Redzic et al., 2005; Johanson et al., 2008). Excessive accumulation of CSF in the ventricles in elderly individuals without neurological issues leads to ventricular enlargement and, consequently, induces compression of the brain parenchyma (Creasey and Rapoport, 1985; Drayer, 1988; Ambarki et al., 2010; Todd et al., 2018; Meunier et al., 2020). It has been shown that ventricular total intracranial volume fraction may increase by a factor five over the course of life which potentially correlates with a doubling in ventricular volume (Coupé et al., 2019). This leads to the compression of blood vessels, glial activation, and stretching and destruction of periventricular axons that lead to functional and neuropsychological abnormalities (Attier-Zmudka et al., 2019; Meunier et al., 2020). Through the analysis of T2-weighted MRI scans of non-demented, stroke-free subjects aged 65–84 years, Breteler et al. observed that increased ventricular volume correlates with significantly decreased performance on neuropsychological tests (Breteler et al., 1994). Vascular changes, the degeneration of cortical neurons, the subsequent degeneration of axons, and the loss of cerebral WM directly drives age-related ventricular enlargement (Breteler et al., 1994). Quantitative MRI morphometry has uncovered the relationship between cognitive aging and age-related changes in the size of the cerebral hemispheres and CSF spaces in elderly volunteers (ages 66–90 years) (Carmichael et al., 2007). Although cerebral atrophy accelerates at age 50 and above, the actual starting age may vary from subject to subject (Nagata et al., 1987). In later stages of life, CSF volume expansion is one of the most visible changes in MRI (Coffey, 2000).
Size of the lateral and third ventricles are related to poorer performance on visual delayed memory, attention, and psychomotor speed (Coffey et al., 2001a). Common neurodegenerative diseases, such as Parkinson’s disease, Alzheimer’s disease, vascular dementia, progressive supranuclear palsy, and idiopathic chronic hydrocephalus, have been linked to accelerated ventricular enlargement (Missori and Currà, 2015; Apostolova et al., 2012). Ventricle size may therefore serve as a biomarker for early abnormal brain changes (Ott et al., 2010). Ventricular volume is typically assessed via morphometry (Penn et al., 1978; Gado et al., 1982; Nagata et al., 1987; Breteler et al., 1994). Nagata et al. observed that the CSF volume remains nearly constant until the age of 50 years and then gradually increases afterwards (Nagata et al., 1987). More recently, Irimia et al. reported averaged CSF volumes of 159 cm3 at age 20, 266 cm3 at age 45, and 365 cm3 at age 70 (Irimia, 2021). Strikingly, ventricular CSF volume increases by 0.3 mL/year whereas cortical CSF increases by 0.6 mL/year (Pfefferbaum et al., 1994). Ventricular CSF volume starts to increase around age 30 as a result of early GM atrophy associated with cell shrinkage and cellular compacting (Pfefferbaum et al., 1994). Resnick et al. observed from a cross-sectional and a longitudinal neuroimaging study of non-demented participants aged 59–85 years, that ventricular volumes is significantly larger in men in comparison to women, and that GM and WM volumes were significantly smaller in men in comparison to women (Resnick et al., 2000). Their longitudinal analysis also revealed a ventricular volume increase of on average 1.5 cm3/year while the cross-sectional findings provided an increase of 1.3cm3/year (Resnick et al., 2000). Similarly, Scahill et al. observed an average ventricular volume expansion rate of 0.65 cm3/year, which they link to brain atrophy, changes in CSF dynamics, and the distribution of CSF in the ventricular and sulcal spaces (Scahill et al., 2003). While most studies report total ventricular volume changes, Lundervold et al. observed a left-right ventricle expansion asymmetry and reported an annual 2.9% increase of the left and 3.1% of the right ventricular volume (Lundervold et al., 2019).
3.6. Brain folding changes
The brain’s highly folded morphology is one of its many fascinating features. Organ-level properties of these folds, such as position, orientation, and growth during development, are very consistent among individuals (Welker, 1990; Borrell and Reillo, 2012; Wang et al., 2021) and those similarities greatly exceed inter-individual variations (White et al., 1997). There are two frequently used ways to describe brain folding: the gyrification index and the depth and spacing between sulci (Madan, 2021). These measures can be derived from MRI using Free-Surfer (Fischl, 2012).
3.6.1. Gyrification index
Gyrification is the process of brain folding. Quantification of gyrification was first developed by Elias and Schwartz using a stereological approach (Elias and Schwartz, 1969). Zilles et al. suggested the use of the gyrification index (GI), i.e., the ratio between the concise outline of the pial surface and the smoothed outer contour of the cortical layer (Zilles et al., 1988). This measure is often reported for individual coronal sections (Madan, 2021) or as a local or global measure (Schaer et al., 2008; Lamballais et al., 2020). A small GI corresponds to a smoother brain with a less profound folding pattern, while a large GI corresponds to a highly folded section. The consistency of lobes and folds across individuals suggests that gyrification is not simply a random mechanical process (Ronan and Fletcher, 2015). Theories of brain gyrification are mostly concentrated on brain development at young age and can be split into two main theories: folding caused by growth processes in cortical development and folding based on mechanical tension in axons (Zilles et al., 2013; Wang et al., 2020). Examples of that could be theories based on a tension aroused from restricted space in the skull (Mota and Herculano-Houzel, 2012), mechanical tension exerted by fiber tracts (Van Essen, 1997), axonal pushing (Nie et al., 2012), differential growth caused by stress (Bayly et al., 2013), protein regulated radial and tangential expansion (Stahl et al., 2013), and minimization of effective free energy associated with cortical shape (Mota and Herculano-Houzel, 2015).
The degree of gyrification varies across the brain with highest GI values measured in the temporal and parietal lobes (Zilles et al., 1988; Hogstrom et al., 2013). Post-mortem studies (Armstrong et al., 1995; Zilles et al., 2013) and longitudinal imaging studies (Li et al., 2014) have shown that gyrification increases after birth up to adulthood and linearly decreases subsequently (Magnotta et al., 1999; Hogstrom et al., 2013). In a cross-sectional study on lifespan changes from age 4–83 years, Cao et al. confirmed these findings and showed that the GI trajectory follows a logarithmic function of age with a decrease of the GI starting as early as four years old (Cao et al., 2017). Other studies suggest that gyrification continues to increase into adolescence (Blanton et al., 2001) and into the thirties for the entorhinal cortex (Hasan et al., 2016). Lamballais et al. studied late life gyrification with participants mean age of 63.5 years (age range: 45.7–97.9 years) and found a mean annual decline of the GI of about 0.0021 (Lamballais et al., 2020). Madan and Kensinger evaluated structural MRI of adults aged 20–86 and observed a global gyrification decrease of around 0.035 per decade (Madan and Kensinger, 2016). They also reported, however, that their cross-sectional data exhibited a large amount of age-unrelated variability, indicating that longitudinal datasets are prudent for estimating age-related GI changes. In a recent longitudinal study with at least two MRI from 280 healthy adults aged 45–92, Madan observed a decreasing slope of 0.04291 per decade (Madan, 2020). They also showed that there is no consistent anterior-posterior gradient with respect to changes of the GI, but much rather reported a uniform decrease of the GI across all regions with a maximum decline observed in parietal lobe.
3.6.2. Sulcal morphology
Sulcal morphology is automatically estimated from MRI in a similar manner to GI. A number of studies have revealed its relation with age (Kochunov et al., 2005; Rettmann et al., 2006; Liu et al., 2010, 2013a; Li et al., 2011; Shen et al., 2018). A cross-sectional study with participants aged 20–82 years showed that the average sulcal width increases at an approximate rate of 0.7 mm/decade, while the average sulcal depth decreases at an approximate rate of 0.4 mm/decade (Kochunov et al., 2005). A study with two groups of participants consisting of middle aged adults and older healthy individuals found that sulci were, on average, 17.3% wider in the elderly with the largest difference in the left superior frontal sulcus (Jin et al., 2018). Liu et al. observed the biggest increase in superior frontal sulcus in a group of older non-demented individuals (Liu et al., 2010). A longitudinal study of 35 subjects aged 59–84 years reported the decrease in surface area and sulcal depth within a four-year period between scans (Rettmann et al., 2006). A larger longitudinal study with 132 participants followed over a seven year period found the largest rate of increase in fold opening in the superior frontal sulcus with 0.131 mm/year (Shen et al., 2018). Furthermore, this rate appeared to be accelerated after age 80 years. Superior temporal sulci had comparatively low rates of fold opening with 0.035 mm/year for the left hemisphere and 0.085 mm/year for the right hemisphere. It has been shown, that sulcal width and depth mediated the effects of age on GI and accounted for 49.9% of its variability (Madan, 2020). Most of it was connected to sulcal depth (36.2%) and sulcal width explained less mediation (7.2%) with the remaining part associated with direct effect of age (10.6%). Overall, this study showed that sulci morphology is more strongly associated with age than GI (Madan, 2020).
4. Gray matter aging mechanisms
GM atrophy manifests as volume loss and cortical thinning. These changes are driven by microstructural changes on the molecular-, cell-, and tissue-level that are specific to the cortex. The GM layer consists of neuronal cell bodies and blood vessels which account for 16% of the cortex, axons and dendrites each making up around 29%, and glial cells and extracellular space occupying the rest of the GM layer (Braitenberg and Schüz, 2013). Here, we differentiate between aging mechanisms on the cellular and the molecular level.
For several decades, neuron cell death was considered to be the leading cause of cortical atrophy in healthy aging (Pakkenberg and Gundersen, 1997; Šimić et al., 1997; Fjell and Walhovd, 2010). This notion has been increasingly challenged due to growing evidence that GM volume loss is caused by cell shrinkage and a degeneration of the dendritic network (Haug et al., 1984; Gómez-Isla et al., 1996; Pakkenberg et al., 2003; Pelvig et al., 2008). In fact, the total number of neurons is estimated to decrease by 2–4% which may account for only up to 10% of overall GM volume loss (von Bartheld, 2018). Instead, a decrease in neuronal body size, degeneration of neuropil, and de-arborization of the dendritic network, and resulting synaptic loss are the driving forces behind cortical changes in the aging brain (Esiri, 2007; Fjell and Walhovd, 2010). Especially neurons with particularly large cell bodies experience substantial volume loss due to gradual metabolic slowing and mitochondrial dysfunction (Castelli et al., 2019). Dendritic arborization plays a central role in neuronal connectivity since spines are the principal sites of signal transmission between neurons (Dickstein et al., 2013). Loss of spines or decay of the dendritic network are likely to affect synaptic events and lead to cognitive decline (Dickstein et al., 2007; Peters et al., 2008). In the healthy brain dendritic spines have an average length of 0.5–2 μm and contain excitatory synapses. Their density varies across the brain and within the dendritic tree itself. Their average density ranges from 1 to 10 spines per micrometer dendritic length (Sorra and Harris, 2000). It is estimated that the average number of dendritic spines in human brain exceeds 1013 (Nimchinsky et al., 2002). In aging primates, Duan et al. observed a loss of cortical neuronal dendritic spines of up to 43% on the apical surface and 27% on the basal surface (Duan et al., 2003). Regression in dendrites was also found in pyramidal neurons located in the prefrontal, pre-central, and superior temporal cortical regions in humans (De Brabander et al., 1998). A post-mortem study by Jacobs et al. revealed a significant decline of dendritic neuropil in older brains when compared to younger subjects. In the aged human brain, dendritic length decreased by 9–11% and spine density decreased nearly 50% (Jacobs et al., 1997). These dendritic changes are very location specific; as such, the spine density of cerebellar Purkinje cells decreases with aging by 17% (Rogers et al., 1984); the caudate of the striatum loses around 50% of its spine density (Levine et al., 1986); and the substantia nigra undergoes severe spine loss in aged subjects (Cruz-Sanchez et al., 1995).
4.1. Cell level
On the cell level, glial cells, such as astrocytes and microglia, play a major role in aging mechanisms. Astrocytes fulfill a wide range of critical functions to maintain homeostasis through nutrient support and ion transport, mitigate neuroinflammation, and preserve the blood brain barrier (BBB) (Liddelow et al., 2017). In the aged brain, however, astrocytes lose their ability to carry out their normal functions and release a toxic factor which kills neurons and oligodendrocytes (Clarke et al., 2018). These two processes directly impact the integrity of GM tissue and contribute to the structural decline in vulnerable brain regions in normal aging.
Microglia, i.e., cells that act as macrophages, form the brain’s immune defense. The brain’s gradual metabolic slowing, clearance of waste products, and potential ischemia leads to a neuroinflamatory response (Norden and Godbout, 2013). It has been shown that a modest increase in the level of microglia activation may already impact cognition (Norden and Godbout, 2013) and exacerbate neurodegeneration in the aging brain (Lull and Block, 2010). Strikingly, pharmacological suppression of a microglial response demonstrated a neuroprotective response (Mattson and Arumugam, 2018; Colonna and Butovsky, 2017).
4.2. Molecule level
Mitochondria play a crucial role in cellular energy metabolism, Ca2+ homeostasis, and apoptosis, i.e., the programmed cell death that occurs in both healthy and pathological conditions (Mattson and Arumugam, 2018; Yun and Finkel, 2014; Mattson, 2000). Studies on the influence of aging on mitochondria revealed a number of age-related changes such as mitochondrial enlargement or fragmentation and increased numbers of mitochondria with depolarized membranes (Grimm and Eckert, 2017), as well as impaired Ca2+ handling (Morozov et al., 2017; Lores-Arnaiz et al., 2016; Pandya et al., 2015). Dysregulation of Ca2+ in hippocampal mitochondria is a common observation in AD and other neurodegenerative diseases which leads to the generation of reactive oxygen species and activation of apoptosis (Calvo-Rodriguez and Bacskai, 2020). Accompanying oxidative stress in neurons as well as the progressive accumulation of misfolded amyloid beta plaques and neurofibrillary tangles impair healthy energy metabolism and ultimately disrupt protein function that controls subcellular calcium dynamics (Camandola and Mattson, 2011).
Aging typically causes an increasing inability to clear oxidatively damaged molecules and an overall low antioxidant protection level are common comorbidities during aging (Paul et al., 2007). For neuronal cells to maintain their structural and functional integrity throughout life, removal of dysfunctional molecules and processes such autophagy and the proteasomal degradation of proteins are vital (Mattson and Arumugam, 2018). It is commonly understood, however, that these mechanisms are compromised in the aging brain (Nixon, 2013). Keller et al., for example, showed that vulnerability to proteasome dysfunction during aging is region-specific because proteasome activity was significantly decreased in hippocampus and cerebral cortex in comparison to the cerebellum and brainstem (Keller et al., 2000).
Aging neurons are characterized by the accumulation of dysfunctional and misfolded proteins (Mattson and Arumugam, 2018). Once misfolded, these proteins turn toxic and initiate a cascade that leads to their progressive recruitment of healthy neighboring proteins and the spreading of plaques and tangles (Jack and Holtzman, 2013). Amyloid beta primarily aggregates in the form of plaques in the extracellular space and hyperphosphorylated tau forms neurofibrillary tangles that accumulate intracellularly (Villemagne et al., 2018; Jucker and Walker, 2013). Amyloid beta and tau are characterized by complex interactions that occur in both healthy and diseased aging brains (Bloom, 2014; Jack et al., 2017; Knopman et al., 2021; Vogt et al., 2021b). It appears that an elevated presence of amyloid beta likely causes the onset of tauopathy which in turn accelerates amyloid beta plaque formation (Bloom, 2014; Knopman et al., 2021). Using PET imaging, it has been shown in cognitively unimpaired subjects with normal amyloid beta loads that aging leads to a minimal accumulation of tau in the temporal lobe; cognitively impaired subjects exhibit progressive tau accumulation in the temporal, parietal, and the frontal lobes with age and amyloid beta load (Bloom, 2014). While elevated amyloid beta is an important precursor for tau accumulation, cognitive decline appears to correlate with tau infiltration (Hanseeuw et al., 2019; Malpetti et al., 2020; Raj et al., 2012). Hence, significant effort has gone into the identification of spatiotemporal trajectories of tau spread (Lowe et al., 2018; Harrison et al., 2019). Given the predominant intracellular accumulation of tau, it has been shown that tau spreads along the axonal network and is able to appear in non-proximal regions of the brain (Braak and Braak, 1991; Raj et al., 2012; Vinke et al., 2018). Vogel et al. recently identified four distinct trajectories that are associated with clinically observed AD progression patterns (Vogel et al., 2021a). These trajectories are characterized by repeatedly observed spatial pathways which include two frequently observed limbic-predominant and medial temporal lobe-sparing patterns as well as a posterior and lateral temporal pattern linked to atypical clinical variants of AD. By adapting initial seeding locations and incorporating secondary seeding locations, Vogel et al. demonstrated that spreading along the corticolimbic network follows various pathways that reproduce PET-based patterns (Vogel et al., 2021a). Our understanding of AD biomarker progression will continue to improve as more longitudinal imaging data becomes available.
5. White matter aging mechanisms
WM changes in the aging brain are characterized by atrophy (Lemaître et al., 2005; Liu et al., 2017), WM tract disruption (Shenkin et al., 2005; Coelho et al., 2021), demyelination (Marner et al., 2003; Faizy et al., 2018), vascular impairment (Kalaria, 2010), and increased inflammation (Raj et al., 2017). Fig. 2 shows a schematic summary of the most prevalent WM aging mechanisms that lead to the tissue-level morphological changes discussed in Section 3.4. The majority of the WM volume is occupied by myelinated axons (~60%), while the extracellular space accounts for about 20%, blood vessels for less than 3%, and the rest of the volume is filled by glial cells (Duval et al., 2016). Age-related WM degeneration has been linked to behavioral changes and cognitive impairment (Madden et al., 2008; Bennett and Madden, 2014). In the following, we will review WM aging mechanisms related to axonal architecture, demyelination, and WMHs. While structural imaging methods are used to quantify organ-level morphological changes, DTI shows increased sensitivity for detecting age-related microstructural alternations in WM (Maillard et al., 2019; Xie et al., 2016; Salami et al., 2012).
Fig. 2.
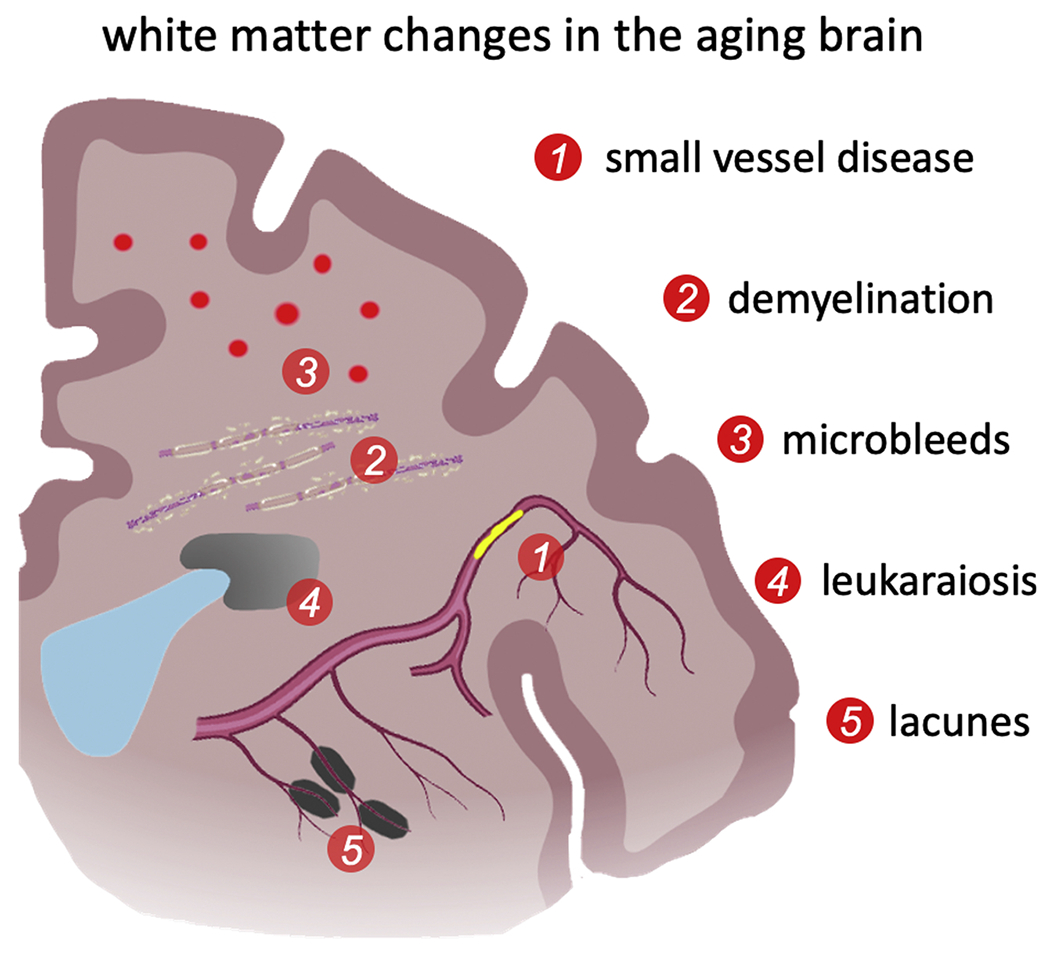
White matter changes are a major source for brain atrophy and loss of brain function. The resulting cognitive decline is linked to neurodegeneration, but is just as impacted by the deterioration of the axonal network (Xiong and Mok, 2011; Chen et al., 2020b). White matter aging is characterized by demyelination, axon degeneration, white matter lesions, and small vessel disease which is associated with microbleeds, lacunes, and ministrokes. White matter lesions drive neuroinflammation and disturb the intricate homeostasis in the brain. Lastly, ischemia is a common driver of gradual cell death through the brain and causes the progressive degeneration of white matter tissue (Mattson and Arumugam, 2018; Liu et al., 2017).
5.1. Alterations in white matter axonal architecture
DTI is the most prominent imaging technique to quantify the degree of anisotropy in WM tissue and reconstruct axonal bundles and WM tracts (González-Reimers et al., 2019). There are four primary diffusion-based measurements of WM structural architecture: fractional anisotropy (FA), mean diffusivity (MD), axial diffusivity (AD), and radial diffusivity (RD) (Basser and Pierpaoli, 2011). To measure these values the eigenvalues of the diffusion tensor are calculated by diagonalization; AD is the first eigenvalue of the diffusion tensor and is associated with diffusion parallel to fiber tracts. A decline in AD may reflect injuries to axons, reduced axonal diameter, or progressively less coherent axonal orientation (Sullivan and Pfefferbaum, 2006). RD is the average of the second and third eigenvalues and is associated with diffusion perpendicular to fiber tracts. RD is sensitive to the local properties of myelin, however high RD may also indicate the presence of axonal loss, loss of myelin, or low axonal packing density (Solowij et al., 2017). Changes in WM diffusivity are driven by tissue degeneration, demyelination, and age-related WM lesion formation (Minati et al., 2007; Liu et al., 2017). FA measures the degree of anisotropy with respect to the axonal network based on the diffusivity of water molecules inside myelinated axons and the extracellular space; in a cohort with 282 subjects aged between 20.2 and 84.2 years, the annual FA percentage change decreased by 0.3 ± 0.8% globally, 0.5 ± 0.9% in the frontal, 0.2 ± 0.8% in the parietal, 0.2±0.8% in the occipital, and increased by 0.1 ± 1.2% in the temporal lobe (Sexton et al., 2014). In myelin-rich axon tracts diffusion is highly anisotropic although axonal diameter, fiber density, and myelin structure will vary spatially (González-Reimers et al., 2019). MD, which is derived from radial and axial diffusivity, represents the overall magnitude of water diffusion (Basser and Pierpaoli, 2011). An increase in MD reflects impaired WM integrity due to local axon and myelin degeneration. Sexton et al. observed an annual MD percentage change of 0.3 ± 0.7% globally, 0.5 ± 0.8% in the frontal, 0.7 ± 0.7% in the parietal, 0.1 ± 0.7% in the occipital, and 0.0 ± 0.9% in the temporal lobe (Sexton et al., 2014). Cross-sectional studies have repeatedly confirmed that FA and MD change at increasing rates in older subjects in comparison to younger adults (Sexton et al., 2014; Pfefferbaum and Sullivan, 2003; Salat et al., 2005; Sullivan and Pfefferbaum, 2006; Minati et al., 2007; Giorgio et al., 2010).
On DTI images, multiple brain areas, especially in mid-hippocampal and posterior thalamic areas, show increased AD and RD values with age resulting from progressive deterioration of axons and supporting myelin (Kumar et al., 2013). Between the age of 20 and 80 years, the length of myelinated fibers reduces by almost 10%/decade, or a total reduction of 45% (Marner et al., 2003). Age-related axon degeneration predominantly affects axons with small diameters with slender myelin sheaths in comparison to thicker axons (Stahon et al., 2016). Additional factors linked to changes in imaging measures are changes in cell density, orientation, size and numbers, as well as volume of axonal fibers (Faizy et al., 2020). A decrease in axonal packing density, e.g., due to a greater loss of myelin or axons in aging, may result in a global increase of extracellular water and the formation of WM lesions (Faizy et al., 2020; Yu et al., 2021). The accumulation of extracellular fluid is frequently associated with the build-up of harmful substances such as plasma proteins that originate from a leaky BBB (Yu et al., 2021). These proteins can be toxic to surrounding WM microstructures including myelin and axons. BBB dysfunction is linked to increased glial cell activation and contributes to age-related neuroinflammation (Norden and Godbout, 2013) and WM lesions (Simpson et al., 2010; Farrall and Wardlaw, 2009). Microglial senecences is characterized by morphological changes in the form of fewer and shorter processes, increased soma volume, and formation of spheroid swellings (Von Bernhardi et al., 2015). These dystrophic microglia co-localize with degenerating neurons and show clumping and accumulation of phagocytic inclusions (Von Bernhardi et al., 2015). Resulting loss of microglial plasticity and immune surveillance is associated with cognitive decline (Norden and Godbout, 2013). Another common factor driving WM change is linked to stiffening of vessels (Maillard et al., 2017). While concise pathological pathways are still being investigated, wall stiffening changes pressure gradients in capillaries and results in increased extravascular free water content due to the accumulation of solutes in the interstitial space. Moreover, age-related hypertension in cerebral vasculature contributes to BBB dysfunction and changes to the composition of the interstitial fluid within WM (Xu et al., 2017).
5.2. Demyelination
Oligodendrocytes form multiple extensions to neighboring axons to deposit and maintain myelin sheaths that wrap around axon segments (Salzer and Zalc, 2016). Myelin not only accelerates signal transduction along axons by up to a factor of 10 (Baumann and Pham-Dinh, 2001), but also provides mechanical integrity and increases WM stiffness (Weickenmeier et al., 2016). Demyelination of WM axon bundles is a common feature in healthy brain aging (Callaghan et al., 2014).
Oligodendrocytes have a unique metabolic demand that includes production and maintenance of myelin sheaths and synthesizing the brain’s cholesterol supply. Consequently, they are vulnerable to a variety of insults including chronic hypoperfusion, toxic products of activated microglia, iron toxicity, and excitotoxicity (Bartzokis, 2004). Age is a deciding factor in the ability of oligodendrocyte progenitor cells to differentiate into myelin-producing oligodendrocytes (Spitzer et al., 2019). These cells are responsible for the long process of intracortical myelination and increase the local cholesterol and iron levels in the process causing increased toxicity to the intracortical environment (Bartzokis, 2004). The loss of myelin in aging WM may be caused by immune-mediated, metabolic, ischaemic, and excitotoxic pathways (Chen et al., 2020b). Immune-mediated myelin injury is mostly driven by activated microglial cells or macrophages which are thus activated, release toxins, and ultimately cause myelin sheath and oligodendrocyte degeneration. Moreover, oligodendrocytes and glial cells are particularly sensitive to ischemia-induced apoptosis (Lassmann, 2001). Hypertensive vascular alterations may gradually obstruct blood flow to WM regions and cause chronic ischemia which leads to progressive loss of myelin and oligodendrocytes (Chen et al., 2020b).
5.3. White matter hyperintensities
WMHs are a common observation in the aging brain and are found in over 90% of WM images of subjects ages 60–64 (Wen and Sachdev, 2004). They show as bright-appearing WM lesions in FLAIR imaging and are associated with vascular degeneration (Fazekas et al., 1987; DeCarli et al., 2005a; Wardlaw et al., 2015). WMHs appear in cerebral WM as a result of hypoperfusion caused by vascular obstruction, reactive gliosis, infarcts, or ministrokes (Fazekas et al., 1998; Salat et al., 2004; Wardlaw et al., 2015). The prevalence of WMHs increases with increasing vascular risk factors, including hypertension, diabetes, and smoking (Habes et al., 2016; Wardlaw et al., 2019). WMHs are typically differentiated into periventricular and deep WM lesions based on their locations (DeCarli et al., 2005a; Kim et al., 2008; Chen et al., 2020a). Both forms are linked to vascular pathology, i.e., small vessel disease (SVD), although periventricular and deep WMHs show some differences with respect to microstructural properties and pathophysiology (Todd et al., 2018; Wardlaw et al., 2015). DTI suggests that periventricular WMHs show significantly lower FA and increased MD, AD, and RD in comparison to deep WMHs (Zhan et al., 2009; Griffanti et al., 2018). Periventricular WMHs have discontinuous ependyma, gliosis, loosening of the WM fibers, and myelin loss around tortuous venules in perivascular spaces; deep WMHs are characterized by less gliosis, but increased axonal loss around perivascular spaces, demyelination, and arteriolosclerosis (Wardlaw et al., 2015; Griffanti et al., 2018).
The severity of WMHs in the brain has been shown to correlate with age and is an indicator for the degree of cognitive decline in subjects and accelerated aging (Schmidt et al., 2005). Cross-sectional imaging data suggests that age-dependent WMH volume growth rates differ for periventricular and deep WMHs with periventricular WMHs increasing by 12%/year and deep WMHs by 7%/year (Nyquist et al., 2015). Periventricular WMHs consistently first appear in the anterior and posterior horns of the ventricles (Fazekas et al., 1987). They are likely associated with a progressive deterioration of the ventricular wall formed by a single layer of ependymal cells leading to CSF leakage into the deeper layers of the ventricular wall and subsequent inflammation (Fernando et al., 2006; Jiménez et al., 2014). On FLAIR images they first show as small linings, then increase to caps, and ultimately penetrate into deep WM (Fazekas et al., 1987). Deep WMHs appear in diffuse locations throughout WM and are linked to ischemic damage from SVD (Fernando et al., 2006; Griffanti et al., 2018). SVD is linked to alterations in the walls of small blood vessels caused by aging, arterial hypertension, and diabetes (Tuladhar et al., 2015). These alterations may cause the consequent narrowing of the lumen and cause small WM infarcts and microbleeds. WM lesions may also be caused by the breakdown of the BBB and macromolecule-leakage, which causes astrocyte activation and gliosis (Wardlaw et al., 2017). It is also known that BBB damage can occur due to traumatic brain injury (TBI); in fact, TBI can lead to oxidative stress and an increase in inflammatory mediators (Song et al., 2020).
6. Ventricular aging mechanisms
Aso et al. have investigated a possible link between venous drainage and ventricular enlargement in both normal and accelerated aging (Aso et al., 2020). They found that changes encountered in cerebral venous drainage stems from venous insufficiency occur in normal aging processes. They motivate ventricular enlargement to result from this change in the venous drainage pattern as well as cerebral atrophy (Aso et al., 2020). This agrees with work by Apostolova et al. that link ventricular enlargement to parenchymal volume loss following the progressive loss of neurons and de-arborization of the dendritic network (Apostolova et al., 2012).
The brain contains three fluid systems: interstitial fluid (ISF), CSF, and vasculature. While the ISF is secreted by endothelial cells, the majority of CSF is produced by the choroid plexus, which is located within the lateral ventricles (Redzic et al., 2005; Brinker et al., 2014). The choroid plexus is a lobulated structure that floats in the CSF space and is formed by a unique and continuous line of epithelial cells that originated from the ependymal wall of the ventricles (Marques et al., 2013). The ependyma is a single layer of ciliated ependymocytes lining the ventricular cavity separating the parenchyma from the CSF. The ependymal cell layer is connected by tight gap and adherens junctions, and contains the water channel protein aquaporin4 (AQP4). These junctions and water channel proteins allow the ependymal lining to act as a bidirectional barrier and a fluid transport system for CSF and ISF, helping to keep the brain free from toxins (Shook et al., 2014). Biochemical changes to the ependymal monolayer affect the efficiency of the bidirectional transport and its clearance mechanisms. Moreover, glial scarring due to age-related ependymal wall failure is accompanied by an increase in AQP4 water channels and inadvertently results in a disruption of CSF/ISF homeostasis and interstitial solute clearance (Shook et al., 2014).
The ventricle’s volume can also be modified by disorders that cause rupture of the BBB’s endothelial cells and epithelial choroidal cells forming the blood–cerebrospinal fluid barrier (Solár et al., 2020). The ventricles and the BBB are responsible for solute clearance of the brain (Shook et al., 2014). It is known that the contraction and expansion of the ventricles involve the interplay of three main factors: changes in brain tissue properties, CSF dynamics, and vascular parameters (Johanson et al., 2008; Meunier et al., 2020). Furthermore, the hemodynamic loading of the ventricles throughout life, causes cyclic ependymal cell stretching and eventually leads to micro tears in the ventricular membrane that cause ependymal cell loss and gliosis (Shook et al., 2014; Todd et al., 2018). Gliosis can lead to glial scar development which replaces ependymal cells with dense astrocytic patches (Acabchuk et al., 2015). In fact, the astrocytic ribbon layer was consistently thicker in regions with decreased ependymal cell coverage (Shook et al., 2014). The deterioration of the ependymal wall compromises periventricular ISF homeostasis and disrupts trans-ependymal bulk flow mechanisms required to clear proteins and metabolites from the brain parenchyma (Shook et al., 2014; Todd et al., 2018). Serot et al. confirmed that aging leads to epithelial atrophy, thickening of the basement membranes, and a decrease in CSF production (Serot et al., 2003). Strikingly, a decrease of epithelial cell height of as low as 10% (15 μm high in newborns and 13.7 μm in elderly) decreases CSF secretion volume by about 50% (Serot et al., 2003). Specifically, CSF secretion volume decreases from 0.41 mL/min at 28 years to 0.19 mL/min at 77 years; CSF turnover drops from six times a day in young adults to 1.7 times a day in elderly subjects (Serot et al., 2003).
CSF circulates from the lateral ventricles through the interventricular foramina into the third ventricle, then through the cerebral aqueduct into the fourth ventricle from where it splits into the central canal of the spinal cord and the cisterns of the subarachnoid space. CSF finally drains into the venous system via the arachnoid villi and cervical lymphatics (Redzic et al., 2005; Brinker et al., 2014). CSF is critical to the central nervous system since it allows exchange of water, small molecules and proteins between the brain parenchyma, and arterial and venous blood by passive diffusion and active transport (Brinker et al., 2014). The ventricular system is important for the movement of nutrient-rich CSF, providing a wide range of vitamins, growth factors, peptides, and nucleosides that are essential to proper brain function (Johanson et al., 2008). Furthermore, CSF is responsible for absorbing mechanical and thermal stress, removing waste products that are formed in the central nervous system, creating an appropriate extracellular molecular composition and transporting humoral mediators and nutrients contributing to brain development, brain homeostasis, and regulation of intracranial pressure and blood supply (Jiménez et al., 2014; Attier-Zmudka et al., 2019; Meunier et al., 2020).
7. Vascular changes
Adequate blood supply to the brain is crucial, and the brain is sensitive to excessive changes in pressure, ischemia, and flow pulsatility changes that result from vascular aging. One of the main characteristics of vascular aging is arterial stiffness and damage to blood vessels (Lin et al., 2017). Other changes include the decrease in capillary density and the increase in BBB permeability (Watanabe et al., 2020). Major limitations of our current understanding of vascular changes are associated with the determination of the most pathological vascular dysfunctions, reversibility of vascular abnormalities, and concise mechanisms of lesion progression (Wardlaw et al., 2019).
The most prominent features of vascular aging are associated with cerebral small vessel disease (CSVD) (Ter Telgte et al., 2018). CSVD is an umbrella term that describes pathologies that affect small arteries, arterioles, and venules (Li et al., 2018; Ter Telgte et al., 2018). Some of these pathologies are intracranial atherosclerosis, cerebral arteriosclerosis, and cerebral amyloid angiopathy (CAA). In cerebral arteriosclerosis, the arterial walls harden and thicken which results in a loss of elasticity (Shim et al., 2015). With aging, the deposition of excess collagen in the walls of veins and venules constricts vessel lumen and causes decreased WM blood flow. It also disturbs the clearing of toxins via the blood stream and the glymphatic system, leading to irreversible WM damage. This pathology is accompanied by a decline in vascular density and impaired autoregulation in the cerebral vascular system which exacerbates WM hypoperfusion (Liu et al., 2017).
Intracranial atherosclerosis is a specific form of arteriosclerosis. It occurs when there is a buildup of cholesterol lipids within blood vessels which eventually leads to stenosis (Wang et al., 2019). This causes blood vessels to harden and lose elasticity resulting in reduced blood flow or stroke (Ritz et al., 2014). Atherosclerotic plaque formation is often associated with the large arteries in the Circle of Willis, but there is a lack of plaque formation in smaller arteries such as the anterior inferior cerebral artery and posterior inferior cerebral artery (Denswil et al., 2016). Intracranial atherosclerosis is often detected using high resolution MRI. However, autopsy studies allow for the direct visualization of atherosclerotic plaques (Suemoto et al., 2018). The gold standard of diagnosis remains to be intra-arterial angiography (Qureshi et al., 2009). The third mechanism, CAA, occurs when amyloid beta begins to build in blood vessels, typically observed in people over the age of 65 years (Ghiso et al., 2010). It can lead to the loss of smooth muscle cells within blood vessels which results in thickening of the vessel wall. This can make the blood vessels extremely brittle and susceptible to rupture. CAA first affects the occipital cortex followed by the frontal, temporal, and parietal cortices (Magaki et al., 2018). Patients with CAA were shown to have pronounced cortical atrophy in the posterior cingulate cortex, precuneus, parietemporal regions, and throughout the medial temporal lobe along with the hippocampus (Kim et al., 2018). The volume of the left hippocampus in CAA patients was found to be approximately 3.5 mm3 whereas the right hippocampal volume was around 3.2 mm3 (Kim et al., 2018). CAA is often associated with stroke and intracerebral haemorrhaging (Weller and Nicoll, 2003). These pathological mechanisms are visible in MRI sequences such as FLAIR and are categorized as microbleeds, lacunes, WMHs, and small subcortical infarcts (Cuadrado-Godia et al., 2018).
7.1. Microbleeds and lacunes
Microbleeds are small circular hypointensities that are commonly seen in the basal ganglia or thalamus. They form upon rupture of a blood vessel caused by sustained hyperintensive vasculopathy or CAA (Martinez-Ramirez et al., 2014). Lobar microbleeds in CAA are typically caused by vessel fragility and rupture following the progressive deposition of amyloid beta within the vessel (Viswanathan and Greenberg, 2011). Hyperintensive vasculopathy describes endothelial dysfunction and arterial remodeling which is often associated with hypertension (Touyz and Montezano, 2015). In hyperintensive vasculopathy, total peripheral vascular resistance increases with age due to a gradual decrease in lumen diameter. Both of these processes severely damage blood vessels and ultimately cause failure.
Lacunes are 3–15 mm CSF-filled cavities in the basal ganglia and WM (Benjamin et al., 2018). They appear following small subcortical infarcts as well as haemorrhages in the vicinity of a perforated arteriole (Mustapha et al., 2019). Small subcortical infarcts are the result of occlusions of small perforating brain arteries, also referred to as atherosclerosis (Nah et al., 2010). Increased levels of cholesterol within blood vessels are a common trigger for the onset of intracranial atherosclerosis. This typically causes hardening of the arterial wall and can lead to progressive stenosis and hypoperfusion (Xu et al., 2017). WM lacunes develop in areas where the perfusion is already compromised, whereas basal ganglia lacunes are more likely to be caused by arterial occlusions due to intracranial atherosclerosis (Gouw et al., 2008). The rupture of an unstable atherosclerotic lesion can lead to platelet activation, thrombus formation, and occlusion of blood vessels (Wang et al., 2019). All these mechanisms are associated with local ischemia that leads to neurodegeneration and progressive cognitive decline.
8. Multiphysics modeling of the brain
Multiphysics modeling allows to formalize the interplay between cell- and tissue-level changes during development, aging, and disease and their gradual manifestation in organ-level structural changes. In the case of brain aging, cellular and subcellular biological processes, many of which were described in previous sections, drive local neurodegeneration. On a constitutive level, it is possible to model the slowing of metabolic activity, the aggregation of waste products, progressive cell and tissue damage, changes in mechanical and structural properties, e. g., FA, diffusivity, and elasticity, or the spreading of neurotoxic proteins and inflammation. The respective measures of these processes can be coupled to local volume change and respective brain deformations that are governed by mechanics.
Many examples of this multiphysics modeling approach exist. For example, brain development, i.e., the early formation of brain folds, has been shown to result from repeated buckling of the cortex due to relative volumetric expansion in the subcortical and cortical layer (Bayly et al., 2014; Budday et al., 2014). Meanwhile, this theory of morphoelastic growth has been extended to incorporate biological processes such as cellular migration from the subventricular zone to the cortical periphery (Verner and Garikipati, 2018; de Rooij and Kuhl, 2018; Zarzor et al., 2021; Wang et al., 2021). These models couple an advection–diffusion equation for cell density changes mimicking cell migration to a mechanical model of volumetric expansion representing the accumulation of neuron cells in the cortical layer. Brain injury is another field of study where mechanics plays a crucial role and aims to provide insight into the relationship between organ-level brain deformations and local damage measures. Work has been done on TBI (Hajiaghamemar et al., 2020; Li et al., 2017; Giordano et al., 2017; Ghazi et al., 2021), chronic traumatic encephalopathy (Noël and Kuhl, 2019; van den Bedem and Kuhl, 2017; Bakhtiarydavijani et al., 2020), and the mechanical-electrophysiological coupling in dislocation injury (Kwong et al., 2019). There are many other examples, including models of neuronal transport (Li et al., 2021), and medical applications such as a decompressive craniectomy (Weickenmeier et al., 2017; Bing et al., 2020), deep brain stimulation (Bikson et al., 2012), and focused ultrasound techniques (Salahshoor et al., 2020). With respect to aging, recent studies modeled cerebral atrophy in the brain during healthy (Harris et al., 2019) and accelerated aging (Weickenmeier et al., 2018; Schäfer et al., 2019; Blinkouskaya and Weickenmeier, 2021). These models couple mechanical shrinking to the spreading of neurotoxic proteins through the brain representative of the biology in neurodegenerative diseases such as Alzheimer’s disease.
Going forward, the combination of multiphysics modeling with cross-sectional and longitudinal imaging data will allow to systematically study the origin of WM and GM changes and uncover the underlying mechanisms driving the onset and progression of neurodegeneration and ultimately cognitive decline.
9. Conclusion
Brain aging is characterized by cell-, tissue-, and organ-level damage and deterioration processes that lead to morphological brain shape changes. Through large scale cross-sectional medical imaging studies, hallmark features of brain aging have been identified and paint a picture of progressive brain volume loss, cortical thinning, ventricular enlargement, and WM deterioration. The brain’s intricate structure-function relationship is significantly impacted by these age-related changes and is commonly accompanied by memory loss, cognitive decline, and behavioral changes. Continuum mechanics theory is particularly suited to model and simulate how cell- and tissue-level damage mechanisms manifest as organ-level morphological changes. Such predictive and personalized brain aging models would prove useful in the differentiation between healthy and accelerated aging. Especially so, because early diagnosis of a particular neurodegenerative disease and immediate personalized intervention have the highest chance of slowing the decline in the quality of life of patients facing long-term disease progression common for AD and related dementias.
Acknowledgements
This work was supported by the National Institute on Aging of the National Institute of Healthunder award R21AG067442 and the National Science Foundation under grant no. 1953323.
References
- Abe O, Yamasue H, Aoki S, Suga M, Yamada H, Kasai K, Masutani Y, Kato N, Kato N, Ohtomo K, 2008. Aging in the cns: comparison of gray/white matter volume and diffusion tensor data. Neurobiol. Aging 29, 102–116. [DOI] [PubMed] [Google Scholar]
- Acabchuk RL, Sun Y, Wolferz R Jr., Eastman MB, Lennington JB, Shook BA, Wu Q, Conover JC, 2015. 3d modeling of the lateral ventricles and histological characterization of periventricular tissue in humans and mouse. JoVE e52328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alegret M, Vinyes-Junqué G, Boada M, Martínez-Lage P, Cuberas G, Espinosa A, Roca I, Hernández I, Valero S, Rosende-Roca M, et al. , 2010. Brain perfusion correlates of visuoperceptual deficits in mild cognitive impairment and mild Alzheimer’s disease. J. Alzheimer’s Dis 21, 557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen JS, Bruss J, Brown CK, Damasio H, 2005. Normal neuroanatomical variation due to age: the major lobes and a parcellation of the temporal region. Neurobiol. Aging 26, 1245–1260. [DOI] [PubMed] [Google Scholar]
- Ambarki K, Israelsson H, Wåhlin A, Birgander R, Eklund A, Malm J, 2010. Brain ventricular size in healthy elderly: comparison between evans index and volume measurement. Neurosurgery 67, 94–99. [DOI] [PubMed] [Google Scholar]
- Anderson VM, Schott JM, Bartlett JW, Leung KK, Miller DH, Fox NC, 2012. Gray matter atrophy rate as a marker of disease progression in ad. Neurobiol. Aging 33, 1194–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolova LG, Green AE, Babakchanian S, Hwang KS, Chou Y-Y, Toga AW, Thompson PM, 2012. Hippocampal atrophy and ventricular enlargement in normal aging, mild cognitive impairment and Alzheimer’s disease. Alzheimer Dis. Assoc. Disord 26, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong E, Schleicher A, Omran H, Curtis M, Zilles K, 1995. The ontogeny of human gyrification. Cereb. Cortex 5, 56–63. [DOI] [PubMed] [Google Scholar]
- Aso T, Sugihara G, Murai T, Ubukata S, Urayama S.-i., Ueno T, Fujimoto G, Thuy DHD, Fukuyama H, Ueda K, 2020. A venous mechanism of ventriculomegaly shared between traumatic brain injury and normal ageing. Brain 143, 1843–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attier-Zmudka J, Sérot J-M, Valluy J, Saffarini M, Macaret A-S, Diouf M, Dao S, Douadi Y, Malinowski KP, Balédent O, 2019. Decreased cerebrospinal fluid flow is associated with cognitive deficit in elderly patients. Front. Aging Neurosci 11, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béchade C, Cantaut-Belarif Y, Bessis A, 2013. Microglial control of neuronal activity. Front. Cell. Neurosci 7, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhtiarydavijani A, Khalid G, Murphy M, Johnson K, Peterson L, Jones M, Horstemeyer M, Dobbins A, Prabhu R, 2020. A mesoscale finite element modeling approach for understanding brain morphology and material heterogeneity effects in chronic traumatic encephalopathy. Comput. Methods Biomech. Biomed. Eng 1–15. [DOI] [PubMed] [Google Scholar]
- Barnes J, Bartlett JW, van de Pol LA, Loy CT, Scahill RI, Frost C, Thompson P, Fox NC, 2009. A meta-analysis of hippocampal atrophy rates in Alzheimer’s disease. Neurobiol. Aging 30, 1711–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartzokis G, 2004. Age-related myelin breakdown: a developmental model of cognitive decline and Alzheimer’s disease. Neurobiol. Aging 25, 5–18. [DOI] [PubMed] [Google Scholar]
- Basser PJ, Pierpaoli C, 2011. Microstructural and physiological features of tissues elucidated by quantitative-diffusion-tensor mri. J. Magn. Reson 213, 560–570. [DOI] [PubMed] [Google Scholar]
- Baumann N, Pham-Dinh D, 2001. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol. Rev [DOI] [PubMed] [Google Scholar]
- Bayly P, Okamoto R, Xu G, Shi Y, Taber L, 2013. A cortical folding model incorporating stress-dependent growth explains gyral wavelengths and stress patterns in the developing brain. Phys. Biol 10, 016005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayly P, Taber L, Kroenke C, 2014. Mechanical forces in cerebral cortical folding: a review of measurements and models. J. Mech. Behav. Biomed. Mater 29, 568–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin P, Trippier S, Lawrence AJ, Lambert C, Zeestraten E, Williams OA, Patel B, Morris RG, Barrick TR, MacKinnon AD, et al. , 2018. Lacunar infarcts, but not perivascular spaces, are predictors of cognitive decline in cerebral small-vessel disease. Stroke 49, 586–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett IJ, Madden DJ, 2014. Disconnected aging: cerebral white matter integrity and age-related differences in cognition. Neuroscience 276, 187–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhagat YA, Beaulieu C, 2004. Diffusion anisotropy in subcortical white matter and cortical gray matter: changes with aging and the role of csf-suppression. J. Magn. Reson. Imaging: Off. J. Int. Soc. Magn. Reson. Med 20, 216–227. [DOI] [PubMed] [Google Scholar]
- Bikson M, Rahman A, Datta A, 2012. Computational models of transcranial direct current stimulation. Clin. EEG Neurosci 43, 176–183. [DOI] [PubMed] [Google Scholar]
- Bing Y, Garcia-Gonzalez D, Voets N, Jérusalem A, 2020. Medical imaging based in silico head model for ischaemic stroke simulation. J. Mech. Behav. Biomed. Mater 101, 103442. [DOI] [PubMed] [Google Scholar]
- Blanton RE, Levitt JG, Thompson PM, Narr KL, Capetillo-Cunliffe L, Nobel A, Singerman JD, McCracken JT, Toga AW, 2001. Mapping cortical asymmetry and complexity patterns in normal children. Psychiatry Res.: Neuroimaging 107, 29–43. [DOI] [PubMed] [Google Scholar]
- Blinkouskaya Y, Weickenmeier J, 2021. Brain shape changes associated with cerebral atrophy in healthy aging and Alzheimer’s disease. Front. Mech. Eng [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom GS, 2014. Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 71, 505–508. [DOI] [PubMed] [Google Scholar]
- Borrell V, Reillo I, 2012. Emerging roles of neural stem cells in cerebral cortex development and evolution. Dev. Neurobiol 72, 955–971. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E, 1991. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. [DOI] [PubMed] [Google Scholar]
- Braitenberg V, Schüz A, 2013. Anatomy of the Cortex: Statistics and Geometry, Vol. 18. Springer Science & Business Media. [Google Scholar]
- Breteler M, van Amerongen NM, van Swieten JC, Claus JJ, Grobbee DE, Van Gijn J, Hofman A, van Harskamp F, 1994. Cognitive correlates of ventricular enlargement and cerebral white matter lesions on magnetic resonance imaging. the Rotterdam study. Stroke 25, 1109–1115. [DOI] [PubMed] [Google Scholar]
- Brinker T, Stopa E, Morrison J, Klinge P, 2014. A new look at cerebrospinal fluid circulation. Fluids Barriers CNS 11, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budday S, Kuhl E, 2020. Modeling the life cycle of the human brain. Curr. Opin. Biomed. Eng 15, 16–25. [Google Scholar]
- Budday S, Steinmann P, Kuhl E, 2014. The role of mechanics during brain development. J. Mech. Phys. Solids 72, 75–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaghan MF, Freund P, Draganski B, Anderson E, Cappelletti M, Chowdhury R, Diedrichsen J, FitzGerald TH, Smittenaar P, Helms G, et al. , 2014. Widespread age-related differences in the human brain microstructure revealed by quantitative magnetic resonance imaging. Neurobiol. Aging 35, 1862–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo-Rodriguez M, Bacskai BJ, 2020. Mitochondria and calcium in Alzheimer’s disease: from cell signaling to neuronal cell death. Trends Neurosci. [DOI] [PubMed] [Google Scholar]
- Camandola S, Mattson MP, 2011. Aberrant subcellular neuronal calcium regulation in aging and Alzheimer’s disease. Biochim. Biophys. Acta (BBA)-Mol. Cell Res 1813, 965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao B, Mwangi B, Passos IC, Wu M-J, Keser Z, Zunta-Soares GB, Xu D, Hasan KM, Soares JC, 2017. Lifespan gyrification trajectories of human brain in healthy individuals and patients with major psychiatric disorders. Sci. Rep 7, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael OT, Kuller LH, Lopez OL, Thompson PM, Dutton RA, Lu A, Lee SE, Lee JY, Aizenstein HJ, Meltzer CC, et al. , 2007. Cerebral ventricular changes associated with transitions between normal cognitive function, mild cognitive impairment, and dementia. Alzheimer Dis. Assoc. Disord 21, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelli V, Benedetti E, Antonosante A, Catanesi M, Pitari G, Ippoliti R, Cimini A, d’Angelo M, 2019. Neuronal cells rearrangement during aging and neurodegenerative disease: metabolism, oxidative stress and organelles dynamic. Front. Mol. Neurosci 12, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Mikheev AV, Yu H, Gruen MD, Rusinek H, Ge Y, Initiative ADN, et al. , 2020a. Bilateral distance partition of periventricular and deep white matter hyperintensities: performance of the method in the aging brain. Acad. Radiol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Huang Y, Shi Z, Li J, Zhang Y, Wang K, Smith AD, Gong Y, Gao Y, 2020b. Demyelinating processes in aging and stroke in the central nervous system and the prospect of treatment strategy. CNS Neurosci. Therapeut 26, 1219–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke LE, Liddelow SA, Chakraborty C, Münch AE, Heiman M, Barres BA, 2018. Normal aging induces a1-like astrocyte reactivity. Proc. Natl. Acad. Sci. U.S.A 115, E1896–E1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho A, Fernandes HM, Magalhães R, Moreira PS, Marques P, Soares JM, Amorim L, Portugal-Nunes C, Castanho T, Santos NC, et al. , 2021. Signatures of white-matter microstructure degradation during aging and its association with cognitive status. Sci. Rep 11, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey CE, Ratcliff G, Saxton JA, Bryan RN, Fried LP, Lucke JF, 2001a. Cognitive correlates of human brain aging: a quantitative magnetic resonance imaging investigation. J. Neuropsychiatry Clin. Neurosci 13, 471–485. [DOI] [PubMed] [Google Scholar]
- Coffey CE, Ratcliff G, Saxton JA, Bryan RN, Fried LP, Lucke JF, 2001b. Cognitive correlates of human brain aging: a quantitative magnetic resonance imaging investigation. J. Neuropsychiatry Clin. Neurosci 13, 471–485. [DOI] [PubMed] [Google Scholar]
- Coffey C, 2000. Anatomic imaging of the aging human brain. Textb. Geriatr. Neuropsychiatry 2, 181–238. [Google Scholar]
- Colonna M, Butovsky O, 2017. Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol 35, 441–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coupé P, Manjón JV, Lanuza E, Catheline G, 2019. Lifespan changes of the human brain in Alzheimer’s disease. Sci. Rep 9, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courchesne E, Chisum HJ, Townsend J, Cowles A, Covington J, Egaas B, Harwood M, Hinds S, Press GA, 2000. Normal brain development and aging: quantitative analysis at in vivo mr imaging in healthy volunteers. Radiology 216, 672–682. [DOI] [PubMed] [Google Scholar]
- Creasey H, Rapoport SI, 1985. The aging human brain. Ann. Neurol.: Off. J. Am. Neurol. Assoc. Child Neurol. Soc 17, 2–10. [DOI] [PubMed] [Google Scholar]
- Cruz-Sanchez F, Cardozo A, Tolosa E, 1995. Neuronal changes in the substantia nigra with aging: a golgi study. J. Neuropathol. Exp. Neurol 54, 74–81. [DOI] [PubMed] [Google Scholar]
- Cuadrado-Godia E, Dwivedi P, Sharma S, Santiago AO, Gonzalez JR, Balcells M, Laird J, Turk M, Suri HS, Nicolaides A, et al. , 2018. Cerebral small vessel disease: a review focusing on pathophysiology, biomarkers, and machine learning strategies. J. Stroke 20, 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Brabander J, Kramers R, Uylings H, 1998. Layer-specific dendritic regression of pyramidal cells with ageing in the human prefrontal cortex. Eur. J. Neurosci [DOI] [PubMed] [Google Scholar]
- de Rooij R, Kuhl E, 2018. A physical multifield model predicts the development of volume and structure in the human brain. J. Mech. Phys. Solids 112, 563–576. [Google Scholar]
- DeCarli C, Fletcher E, Ramey V, Harvey D, Jagust WJ, 2005a. Anatomical mapping of white matter hyperintensities (wmh) exploring the relationships between periventricular wmh, deep wmh, and total wmh burden. Stroke 36, 50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCarli C, Massaro J, Harvey D, Hald J, Tullberg M, Au R, Beiser A, D’Agostino R, Wolf PA, 2005b. Measures of brain morphology and infarction in the framingham heart study: establishing what is normal. Neurobiol. Aging 26, 491–510. [DOI] [PubMed] [Google Scholar]
- Denswil NP, van der Wal AC, Ritz K, de Boer OJ, Aronica E, Troost D, Daemen MJ, 2016. Atherosclerosis in the circle of willis: spatial differences in composition and in distribution of plaques. Atherosclerosis 251, 78–84. [DOI] [PubMed] [Google Scholar]
- Dickstein DL, Kabaso D, Rocher AB, Luebke JI, Wearne SL, Hof PR, 2007. Changes in the structural complexity of the aged brain. Aging Cell 6, 275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickstein DL, Weaver CM, Luebke JI, Hof PR, 2013. Dendritic spine changes associated with normal aging. Neuroscience 251, 21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drayer BP, 1988. Imaging of the aging brain, part i. normal findings. Radiology 166, 785–796. [DOI] [PubMed] [Google Scholar]
- Driscoll I, Davatzikos C, An Y, Wu X, Shen D, Kraut M, Resnick S, 2009. Longitudinal pattern of regional brain volume change differentiates normal aging from mci. Neurology 72, 1906–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du A, Schuff N, Zhu X, Jagust W, Miller B, Reed BR, Kramer J, Mungas D, Yaffe K, Chui H, et al. , 2003. Atrophy rates of entorhinal cortex in ad and normal aging. Neurology 60, 481–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan H, Wearne SL, Rocher AB, Macedo A, Morrison JH, Hof PR, 2003. Age-related dendritic and spine changes in corticocortically projecting neurons in macaque monkeys. Cereb. Cortex 13, 950–961. [DOI] [PubMed] [Google Scholar]
- Duval T, Stikov N, Cohen-Adad J, 2016. Modeling white matter microstructure. Funct. Neurol 31, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias H, Schwartz D, 1969. Surface areas of the cerebral cortex of mammals determined by stereological methods. Science 166, 111–113. [DOI] [PubMed] [Google Scholar]
- Enzinger C, Fazekas F, Matthews P, Ropele S, Schmidt H, Smith S, Schmidt R, 2005. Risk factors for progression of brain atrophy in aging: six-year follow-up of normal subjects. Neurology 64, 1704–1711. [DOI] [PubMed] [Google Scholar]
- Esiri MM, 2007. Ageing and the brain. J. Pathol.: J. Pathol. Soc. Great Britain Ireland 211, 181–187. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Mintun MA, Mach RH, Lee S-Y, Dence CS, Shah AR, LaRossa GN, Spinner ML, Klunk WE, Mathis CA, et al. , 2006. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid aβ42 in humans. Ann. Neurol 59, 512–519. [DOI] [PubMed] [Google Scholar]
- Faizy TD, Kumar D, Broocks G, Thaler C, Flottmann F, Leischner H, Kutzner D, Hewera S, Dotzauer D, Stellmann J-P, et al. , 2018. Age-related measurements of the myelin water fraction derived from 3d multi-echo grase reflect myelin content of the cerebral white matter. Sci. Rep 8, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faizy TD, Thaler C, Broocks G, Flottmann F, Leischner H, Kniep H, Nawabi J, Schön G, Stellmann J-P, Kemmling A, et al. , 2020. The myelin water fraction serves as a marker for age-related myelin alterations in the cerebral white matter-a multiparametric mri aging study. Front. Neurosci 14, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrall AJ, Wardlaw JM, 2009. Blood-brain barrier: ageing and microvascular disease-systematic review and meta-analysis. Neurobiol. Aging 30, 337–352. [DOI] [PubMed] [Google Scholar]
- Fazekas F, Chawluk JB, Alavi A, Hurtig HI, Zimmerman RA, 1987. Mr signal abnormalities at 1.5 t in Alzheimer’s dementia and normal aging. Am. J. Roentgenol 149, 351–356. [DOI] [PubMed] [Google Scholar]
- Fazekas F, Schmidt R, Scheltens P, 1998. Pathophysiologic mechanisms in the development of age-related white matter changes of the brain. Dementia Geriatr. Cogn. Disord 9, 2–5. [DOI] [PubMed] [Google Scholar]
- Fernando MS, Simpson JE, Matthews F, Brayne C, Lewis CE, Barber R, Kalaria RN, Forster G, Esteves F, Wharton SB, et al. , 2006. White matter lesions in an unselected cohort of the elderly: molecular pathology suggests origin from chronic hypoperfusion injury. Stroke 37, 1391–1398. [DOI] [PubMed] [Google Scholar]
- Fischl B, Dale AM, 2000. Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc. Natl. Acad. Sci. U.S.A 97, 11050–11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl B, Salat DH, Busa E, Albert M, Dieterich M, Haselgrove C, Van Der Kouwe A, Killiany R, Kennedy D, Klaveness S, et al. , 2002. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron 33, 341–355. [DOI] [PubMed] [Google Scholar]
- Fischl B, 2012. Freesurfer. Neuroimage 62, 774–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, Walhovd KB, 2010. Structural brain changes in aging: courses, causes and cognitive consequences. Rev. Neurosci 21, 187–221. [DOI] [PubMed] [Google Scholar]
- Fjell AM, Westlye LT, Amlien I, Espeseth T, Reinvang I, Raz N, Agartz I, Salat DH, Greve DN, Fischl B, et al. , 2009a. High consistency of regional cortical thinning in aging across multiple samples. Cereb. Cortex 19, 2001–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, Westlye LT, Amlien I, Espeseth T, Reinvang I, Raz N, Agartz I, Salat DH, Greve DN, Fischl B, et al. , 2009b. Minute effects of sex on the aging brain: a multisample magnetic resonance imaging study of healthy aging and Alzheimer’s disease. J. Neurosci 29, 8774–8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, Walhovd KB, Fennema-Notestine C, McEvoy LK, Hagler DJ, Holland D, Brewer JB, Dale AM, 2009c. One-year brain atrophy evident in healthy aging. J. Neurosci 29, 15223–15231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, McEvoy L, Holland D, Dale AM, Walhovd KB, Initiative ADN, et al. , 2013. Brain changes in older adults at very low risk for Alzheimer’s disease. J. Neurosci 33, 8237–8242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, Westlye LT, Grydeland H, Amlien I, Espeseth T, Reinvang I, Raz N, Dale AM, Walhovd KB, Initiative ADN, 2014a. Accelerating cortical thinning: unique to dementia or universal in aging? Cereb. Cortex 24, 919–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, McEvoy L, Holland D, Dale AM, Walhovd KB, Initiative ADN, et al. , 2014b. What is normal in normal aging? effects of aging, amyloid and Alzheimer’s disease on the cerebral cortex and the hippocampus. Prog. Neurobiol 117, 20–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, Sneve MH, Storsve AB, Grydeland H, Yendiki A, Walhovd KB, 2016. Brain events underlying episodic memory changes in aging: a longitudinal investigation of structural and functional connectivity. Cereb. Cortex 26, 1272–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotenos AF, Snyder A, Girton L, Morris J, Buckner R, 2005. Normative estimates of cross-sectional and longitudinal brain volume decline in aging and ad. Neurology 64, 1032–1039. [DOI] [PubMed] [Google Scholar]
- Fox NC, Schott JM, 2004. Imaging cerebral atrophy: normal ageing to Alzheimer’s disease. Lancet 363, 392–394. [DOI] [PubMed] [Google Scholar]
- Gómez-Isla T, Price JL, McKeel DW Jr., Morris JC, Growdon JH, Hyman BT, 1996. Profound loss of layer ii entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J. Neurosci 16, 4491–4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gado M, Hughes C, Danziger W, Chi D, Jost G, Berg L, 1982. Volumetric measurements of the cerebrospinal fluid spaces in demented subjects and controls. Radiology 144, 535–538. [DOI] [PubMed] [Google Scholar]
- Gautam P, Anstey KJ, Wen W, Sachdev PS, Cherbuin N, 2015. Cortical gyrification and its relationships with cortical volume, cortical thickness, and cognitive performance in healthy mid-life adults. Behav. Brain Res 287, 331–339. [DOI] [PubMed] [Google Scholar]
- Ge Y, Grossman RI, Babb JS, Rabin ML, Mannon LJ, Kolson DL, 2002. Age-related total gray matter and white matter changes in normal adult brain. Part i: volumetric MR imaging analysis. Am. J. Neuroradiol 23, 1327–1333. [PMC free article] [PubMed] [Google Scholar]
- Geinisman Y, Detoledo-Morrell L, Morrell F, Heller RE, 1995. Hippocampal markers of age-related memory dysfunction: behavioral, electrophysiological and morphological perspectives. Prog. Neurobiol 45, 223–252. [DOI] [PubMed] [Google Scholar]
- Ghazi K, Wu S, Zhao W, Ji S, 2021. Instantaneous whole-brain strain estimation in dynamic head impact. J. Neurotrauma 38, 1023–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiso J, Tomidokoro Y, Revesz T, Frangione B, Rostagno A, 2010. Cerebral amyloid angiopathy and Alzheimer’s disease. Hirosaki Igaku [[Hirosaki Med. J.]] 61, S111. [PMC free article] [PubMed] [Google Scholar]
- Gibson EM, Purger D, Mount CW, Goldstein AK, Lin GL, Wood LS, Inema I, Miller SE, Bieri G, Zuchero JB, et al. , 2014. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano C, Zappalà S, Kleiven S, 2017. Anisotropic finite element models for brain injury prediction: the sensitivity of axonal strain to white matter tract inter-subject variability. Biomech. Model. Mechanobiol 16, 1269–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgio A, Santelli L, Tomassini V, Bosnell R, Smith S, De Stefano N, Johansen-Berg H, 2010. Age-related changes in grey and white matter structure throughout adulthood. NeuroImage 51, 943–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Reimers E, Martín-González C, Romero-Acevedo L, Quintero-Platt G, Gonzalez-Arnay E, Santolaria-Fernández F, 2019. Effects of alcohol on the corpus callosum. Neuroscience of Alcohol. Elsevier, pp. 143–152. [Google Scholar]
- Gouw AA, van der Flier WM, Pantoni L, Inzitari D, Erkinjuntti T, Wahlund LO, Waldemar G, Schmidt R, Fazekas F, Scheltens P, et al. , 2008. On the etiology of incident brain lacunes: longitudinal observations from the ladis study. Stroke 39, 3083–3085. [DOI] [PubMed] [Google Scholar]
- Griffanti L, Jenkinson M, Suri S, Zsoldos E, Mahmood A, Filippini N, Sexton CE, Topiwala A, Allan C, Kivimäki M, et al. , 2018. Classification and characterization of periventricular and deep white matter hyperintensities on mri: a study in older adults. NeuroImage 170, 174–181. [DOI] [PubMed] [Google Scholar]
- Grimm A, Eckert A, 2017. Brain aging and neurodegeneration: from a mitochondrial point of view. J. Neurochem 143, 418–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunning-Dixon FM, Raz N, 2000. The cognitive correlates of white matter abnormalities in normal aging: a quantitative review. Neuropsychology 14, 224. [DOI] [PubMed] [Google Scholar]
- Gunning-Dixon FM, Brickman AM, Cheng JC, Alexopoulos GS, 2009. Aging of cerebral white matter: a review of mri findings. Int. J. Geriatr. Psychiatry: J. Psychiatry Late Life Allied Sci 24, 109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habes M, Erus G, Toledo JB, Zhang T, Bryan N, Launer LJ, Rosseel Y, Janowitz D, Doshi J, Van der Auwera S, et al. , 2016. White matter hyperintensities and imaging patterns of brain ageing in the general population. Brain 139, 1164–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajiaghamemar M, Wu T, Panzer MB, Margulies SS, 2020. Embedded axonal fiber tracts improve finite element model predictions of traumatic brain injury. Biomech. Model. Mechanobiol 19, 1109–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanseeuw BJ, Betensky RA, Jacobs HI, Schultz AP, Sepulcre J, Becker JA, Cosio DMO, Farrell M, Quiroz YT, Mormino EC, et al. , 2019. Association of amyloid and tau with cognition in preclinical Alzheimer disease: a longitudinal study. JAMA Neurol. 76, 915–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris TC, de Rooij R, Kuhl E, 2019. The shrinking brain: cerebral atrophy following traumatic brain injury. Ann. Biomed. Eng 47, 1941–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison TM, La Joie R, Maass A, Baker SL, Swinnerton K, Fenton L, Mellinger TJ, Edwards L, Pham J, Miller BL, et al. , 2019. Longitudinal tau accumulation and atrophy in aging and Alzheimer disease. Ann. Neurol 85, 229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan KM, Mwangi B, Cao B, Keser Z, Tustison NJ, Kochunov P, Frye RE, Savatic M, Soares J, 2016. Entorhinal cortex thickness across the human lifespan. J. Neuroimaging 26, 278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haug H, Kühl S, Mecke E, Sass N, Wasner K, 1984. The significance of morphometric procedures in the investigation of age changes in cytoarchitectonic structures of human brain. J. Hirnforsch 25, 353–374. [PubMed] [Google Scholar]
- Hedman AM, van Haren NE, Schnack HG, Kahn RS, Hulshoff Pol HE, 2012. Human brain changes across the life span: a review of 56 longitudinal magnetic resonance imaging studies. Human Brain Mapp. 33, 1987–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henneman W, Sluimer J, Barnes J, Van Der Flier W, Sluimer I, Fox N, Scheltens P, Vrenken H, Barkhof F, 2009. Hippocampal atrophy rates in Alzheimer disease: added value over whole brain volume measures. Neurology 72, 999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiscox LV, Schwarb H, McGarry MD, Johnson CL, 2021. Aging brain mechanics: progress and promise of magnetic resonance elastography. NeuroImage 117889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogstrom LJ, Westlye LT, Walhovd KB, Fjell AM, 2013. The structure of the cerebral cortex across adult life: age-related patterns of surface area, thickness, and gyrification. Cereb. Cortex 23, 2521–2530. [DOI] [PubMed] [Google Scholar]
- Irimia A, 2021. Cross-sectional volumes and trajectories of the human brain, gray matter, white matter and cerebrospinal fluid in 9473 typically aging adults. Neuroinformatics 19, 347–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., Holtzman DM, 2013. Biomarker modeling of Alzheimer’s disease. Neuron 80, 1347–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack C, Shiung M, Gunter J, O’brien P, Weigand S, Knopman D, Boeve B, Ivnik R, Smith G, Cha R, et al. , 2004. Comparison of different mri brain atrophy rate measures with clinical disease progression in ad. Neurology 62, 591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., Lowe VJ, Senjem ML, Weigand SD, Kemp BJ, Shiung MM, Knopman DS, Boeve BF, Klunk WE, Mathis CA, et al. , 2008a. 11c pib and structural mri provide complementary information in imaging of Alzheimer’s disease and amnestic mild cognitive impairment. Brain 131, 665–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., Bernstein MA, Fox NC, Thompson P, Alexander G, Harvey D, Borowski B, Britson PJ, Whitwell JL, Ward C, et al. , 2008b. The Alzheimer’s disease neuroimaging initiative (adni): mri methods. J. Magn. Reson. Imaging: Off. J. Int. Soc. Magn. Reson. Med 27, 685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., Wiste HJ, Weigand SD, Therneau TM, Knopman DS, Lowe V, Vemuri P, Mielke MM, Roberts RO, Machulda MM, et al. , 2017. Age-specific and sex-specific prevalence of cerebral β-amyloidosis, tauopathy, and neurodegeneration in cognitively unimpaired individuals aged 50–95 years: a cross-sectional study. Lancet Neurol. 16, 435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs B, Driscoll L, Schall M, 1997. Life-span dendritic and spine changes in areas 10 and 18 of human cortex: a quantitative golgi study. J. Comp. Neurol 386, 661–680. [PubMed] [Google Scholar]
- Jezzard P, Ramsey NF, 2003. Functional MRI, Quantitative MRI of the Brain: Measuring Changes Caused by Disease, pp. 413–454. [Google Scholar]
- Jiménez AJ, Domínguez-Pinos MD, Guerra MM, Fernández-Llebrez P, Pérez-Fígares JM, 2014. Structure and Function of the Ependymal Barrier and Diseases Associated With Ependyma Disruption. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Zhang T, Shaw M, Sachdev P, Cherbuin N, 2018. Relationship between sulcal characteristics and brain aging. Front. Aging Neurosci 10, 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johanson CE, Duncan JA, Klinge PM, Brinker T, Stopa EG, Silverberg GD, 2008. Multiplicity of cerebrospinal fluid functions: new challenges in health and disease. Cerebrospinal Fluid Res. 5, 1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Whitwell JL, Ahmed Z, Shiung MM, Weigand SD, Knopman DS, Boeve BF, Parisi JE, Petersen RC, Dickson DW, et al. , 2008. β-amyloid burden is not associated with rates of brain atrophy. Ann. Neurol 63, 204–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jucker M, Walker LC, 2013. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501, 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaria RN, 2010. Vascular basis for brain degeneration: faltering controls and risk factors for dementia. Nutr. Rev 68, S74–S87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalra P, Raterman B, Mo X, Kolipaka A, 2019. Magnetic resonance elastography of brain: comparison between anisotropic and isotropic stiffness and its correlation to age. Magn. Reson. Med 82, 671–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller JN, Hanni KB, Markesbery WR, 2000. Possible involvement of proteasome inhibition in aging: implications for oxidative stress. Mech. Ageing Dev 113, 61–70. [DOI] [PubMed] [Google Scholar]
- Kim KW, MacFall JR, Payne ME, 2008. Classification of white matter lesions on magnetic resonance imaging in elderly persons. Biol. Psychiatry 64, 273–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Na HK, Shin J-H, Kim HJ, Seo SW, Seong J-K, Na DL, 2018. Atrophy patterns in cerebral amyloid angiopathy with and without cortical superficial siderosis. Neurology 90, e1751–e1758. [DOI] [PubMed] [Google Scholar]
- Kim S-G, 1995. Quantification of relative cerebral blood flow change by flow-sensitive alternating inversion recovery (fair) technique: application to functional mapping. Magn. Reson. Med 34, 293–301. [DOI] [PubMed] [Google Scholar]
- Knopman DS, Lundt ES, Therneau TM, Albertson SM, Gunter JL, Senjem ML, Schwarz CG, Mielke MM, Machulda MM, Boeve BF, et al. , 2021. Association of initial β-amyloid levels with subsequent flortaucipir positron emission tomography changes in persons without cognitive impairment. JAMA Neurol. 78, 217–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochunov P, Mangin J-F, Coyle T, Lancaster J, Thompson P, Rivière D, Cointepas Y, Régis J, Schlosser A, Royall DR, et al. , 2005. Age-related morphology trends of cortical sulci. Human Brain Mapp. 26, 210–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Chavez AS, Macey PM, Woo MA, Harper RM, 2013. Brain axial and radial diffusivity changes with age and gender in healthy adults. Brain Res. 1512, 22–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong MT, Bianchi F, Malboubi M, García-Grajales JA, Homsi L, Thompson M, Ye H, Noels L, Jírusalem A, 2019. 3d finite element formulation for mechanical-electrophysiological coupling in axonopathy. Comput. Methods Appl. Mech. Eng 346, 1025–1050. [Google Scholar]
- Lamballais S, Vinke EJ, Vernooij MW, Ikram MA, Muetzel RL, 2020. Cortical gyrification in relation to age and cognition in older adults. NeuroImage 212, 116637. [DOI] [PubMed] [Google Scholar]
- Lassmann H, 2001. Classification of demyelinating diseases at the interface between etiology and pathogenesis. Curr. Opin. Neurol 14, 253–258. [DOI] [PubMed] [Google Scholar]
- Lebel C, Beaulieu C, 2011. Longitudinal development of human brain wiring continues from childhood into adulthood. J. Neurosci 31, 10937–10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebel C, Gee M, Camicioli R, Wieler M, Martin W, Beaulieu C, 2012. Diffusion tensor imaging of white matter tract evolution over the lifespan. NeuroImage 60, 340–352. [DOI] [PubMed] [Google Scholar]
- Lemaître H, Crivello F, Grassiot B, Alpérovitch A, Tzourio C, Mazoyer B, 2005. Age-and sex-related effects on the neuroanatomy of healthy elderly. NeuroImage 26, 900–911. [DOI] [PubMed] [Google Scholar]
- Lemaitre H, Goldman AL, Sambataro F, Verchinski BA, Meyer-Lindenberg A, Weinberger DR, Mattay VS, 2012. Normal age-related brain morphometric changes: nonuniformity across cortical thickness, surface area and gray matter volume? Neurobiol. Aging 33, 617–e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux L, Hagemann G, Krakow K, Woermann FG, 1999. Fast, accurate, and reproducible automatic segmentation of the brain in t1-weighted volume MRI data. Magn. Reson. Med.: Off. J. Int. Soc. Magn. Reson. Med 42, 127–135. [DOI] [PubMed] [Google Scholar]
- Levine M, Adinolfi A, Fisher R, Hull C, Buchwald N, McAllister J, 1986. Quantitative morphology of medium-sized caudate spiny neurons in aged cats. Neurobiol. Aging 7, 277–286. [DOI] [PubMed] [Google Scholar]
- Li S, Xia M, Pu F, Li D, Fan Y, Niu H, Pei B, He Y, 2011. Age-related changes in the surface morphology of the central sulcus. NeuroImage 58, 381–390. [DOI] [PubMed] [Google Scholar]
- Li G, Wang L, Shi F, Lyall AE, Lin W, Gilmore JH, Shen D, 2014. Mapping longitudinal development of local cortical gyrification in infants from birth to 2 years of age. J. Neurosci 34, 4228–4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Sandler H, Kleiven S, 2017. The importance of nonlinear tissue modelling in finite element simulations of infant head impacts. Biomech. Model. Mechanobiol 16, 823–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Yang Y, Reis C, Tao T, Li W, Li X, Zhang JH, 2018. Cerebral small vessel disease. Cell Transplant. 27, 1711–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Farimani AB, Zhang YJ, 2021. Deep learning of material transport in complex neurite networks. Sci. Rep 11, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung W-S, Peterson TC, et al. , 2017. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C-H, Cheng H-M, Chuang S-Y, Chen C-H, 2017. Vascular aging and cognitive dysfunction: silent midlife crisis in the brain. Pulse 5, 127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Wen W, Zhu W, Trollor J, Reppermund S, Crawford J, Jin JS, Luo S, Brodaty H, Sachdev P, 2010. The effects of age and sex on cortical sulci in the elderly. NeuroImage 51, 19–27. [DOI] [PubMed] [Google Scholar]
- Liu T, Sachdev PS, Lipnicki DM, Jiang J, Geng G, Zhu W, Reppermund S, Tao D, Trollor JN, Brodaty H, et al. , 2013a. Limited relationships between two-year changes in sulcal morphology and other common neuroimaging indices in the elderly. NeuroImage 83, 12–17. [DOI] [PubMed] [Google Scholar]
- Liu T, Sachdev PS, Lipnicki DM, Jiang J, Cui Y, Kochan NA, Reppermund S, Trollor JN, Brodaty H, Wen W, 2013b. Longitudinal changes in sulcal morphology associated with late-life aging and mci. NeuroImage 74, 337–342. [DOI] [PubMed] [Google Scholar]
- Liu H, Yang Y, Xia Y, Zhu W, Leak RK, Wei Z, Wang J, Hu X, 2017. Aging of cerebral white matter. Ageing Res. Rev 34, 64–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockhart SN, DeCarli C, 2014. Structural imaging measures of brain aging. Neuropsychol. Rev 24, 271–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lores-Arnaiz S, Lombardi P, Karadayian AG, Orgambide F, Cicerchia D, Bustamante J, 2016. Brain cortex mitochondrial bioenergetics in synaptosomes and non-synaptic mitochondria during aging. Neurochem. Res 41, 353–363. [DOI] [PubMed] [Google Scholar]
- Lowe VJ, Wiste HJ, Senjem ML, Weigand SD, Therneau TM, Boeve BF, Josephs KA, Fang P, Pandey MK, Murray ME, et al. , 2018. Widespread brain tau and its association with ageing, Braak stage and Alzheimer’s dementia. Brain 141, 271–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lull ME, Block ML, 2010. Microglial activation and chronic neurodegeneration. Neurotherapeutics 7, 354–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundervold AJ, Vik A, Lundervold A, 2019. Lateral ventricle volume trajectories predict response inhibition in older age – a longitudinal brain imaging and machine learning approach. Plos one 14, e0207967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan CR, Kensinger EA, 2016. Cortical complexity as a measure of age-related brain atrophy. NeuroImage 134, 617–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan CR, 2020. Age-related decrements in cortical gyrification: evidence from an accelerated longitudinal dataset. Eur. J. Neurosci [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan CR, 2021. Age-related decrements in cortical gyrification: evidence from an accelerated longitudinal dataset. Eur. J. Neurosci 53, 1661–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden DJ, Spaniol J, Costello MC, Bucur B, White LE, Cabeza R, Davis SW, Dennis NA, Provenzale JM, Huettel SA, 2008. Cerebral white matter integrity mediates adult age differences in cognitive performance. J. Cogn. Neurosci 21, 289–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magaki S, Tang Z, Tung S, Williams CK, Lo D, Yong WH, Khanlou N, Vinters HV, 2018. The effects of cerebral amyloid angiopathy on integrity of the blood-brain barrier. Neurobiol. Aging 70, 70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnotta VA, Andreasen NC, Schultz SK, Harris G, Cizadlo T, Heckel D, Nopoulos P, Flaum M, 1999. Quantitative in vivo measurement of gyrification in the human brain: changes associated with aging. Cereb. Cortex 9, 151–160. [DOI] [PubMed] [Google Scholar]
- Maillard P, Mitchell GF, Himali JJ, Beiser A, Fletcher E, Tsao CW, Pase MP, Satizabal CL, Vasan RS, Seshadri S, et al. , 2017. Aortic stiffness, increased white matter free water, and altered microstructural integrity: a continuum of injury. Stroke 48, 1567–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maillard P, Fletcher E, Singh B, Martinez O, Johnson DK, Olichney JM, Farias ST, DeCarli C, 2019. Cerebral white matter free water: a sensitive biomarker of cognition and function. Neurology 92, e2221–e2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makedonov I, Black S, MacIntosh B, 2013. Cerebral small vessel disease in aging and Alzheimer’s disease: a comparative study using MRI and SPECT. Eur. J. Neurol 20, 243–250. [DOI] [PubMed] [Google Scholar]
- Malpetti M, Kievit RA, Passamonti L, Jones PS, Tsvetanov KA., Rittman T, Mak E, Nicastro N, Bevan-Jones WR, Su L, et al. , 2020. Microglial activation and tau burden predict cognitive decline in Alzheimer’s disease. Brain 143, 1588–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marner L, Nyengaard JR, Tang Y, Pakkenberg B, 2003. Marked loss of myelinated nerve fibers in the human brain with age. J. Comp. Neurol 462, 144–152. [DOI] [PubMed] [Google Scholar]
- Marques F, Sousa JC, Sousa N, Palha JA, 2013. Blood-brain-barriers in aging and in Alzheimer’s disease. Mol. Neurodegen 8, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Ramirez S, Greenberg SM, Viswanathan A, 2014. Cerebral microbleeds: overview and implications in cognitive impairment. Alzheimer’s Res. Ther 6, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Arumugam TV, 2018. Hallmarks of brain aging: adaptive and pathological modification by metabolic states. Cell Metab. 27, 1176–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, 2000. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol 1, 120–130. [DOI] [PubMed] [Google Scholar]
- Meunier A, Sawamoto K, Spassky N, 2020. Ependyma. Patterning and Cell Type Specification in the Developing CNS and PNS. Elsevier, pp. 1021–1036. [Google Scholar]
- Minati L, Grisoli M, Bruzzone M, 2007. Mr spectroscopy, functional mri, and diffusion-tensor imaging in the aging brain: a conceptual review. J. Geriatr. Psychiatry Neurol 20, 3–21. [DOI] [PubMed] [Google Scholar]
- Missori P, Currà A, 2015. Progressive cognitive impairment evolving to dementia parallels parieto-occipital and temporal enlargement in idiopathic chronic hydrocephalus: a retrospective cohort study. Front. Neurol 6, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozov YM, Datta D, Paspalas CD, Arnsten AF, 2017. Ultrastructural evidence for impaired mitochondrial fission in the aged rhesus monkey dorsolateral prefrontal cortex. Neurobiol. Aging 51, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mota B, Herculano-Houzel S, 2012. How the cortex gets its folds: an inside-out, connectivity-driven model for the scaling of mammalian cortical folding. Front. Neuroanat 6, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mota B, Herculano-Houzel S, 2015. Cortical folding scales universally with surface area and thickness, not number of neurons. Science 349, 74–77. [DOI] [PubMed] [Google Scholar]
- Murman DL, 2015. The impact of age on cognition. Seminars in Hearing, Vol. 36. Thieme Medical Publishers, p. 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MC, Huston J III, Jack CR Jr., Glaser KJ, Manduca A, Felmlee JP, Ehman RL, 2011. Decreased brain stiffness in Alzheimer’s disease determined by magnetic resonance elastography. J. Magn. Reson. Imaging 34, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustapha M, Nassir CMNCM, Aminuddin N, Safri AA, Ghazali MM, 2019. Cerebral small vessel disease (csvd)-lessons from the animal models. Front. Physiol 10, 1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata K, Basugi N, Fukushima T, Tango T, Suzuki I, Kaminuma T, Kurashina S, 1987. A quantitative study of physiological cerebral atrophy with aging. Neuroradiology 29, 327–332. [DOI] [PubMed] [Google Scholar]
- Nah H-W, Kang D-W, Kwon SU, Kim JS, 2010. Diversity of single small subcortical infarctions according to infarct location and parent artery disease: analysis of indicators for small vessel disease and atherosclerosis. Stroke 41, 2822–2827. [DOI] [PubMed] [Google Scholar]
- Narvacan K, Treit S, Camicioli R, Martin W, Beaulieu C, 2017. Evolution of deep gray matter volume across the human lifespan. Human Brain Mapp. 38, 3771–3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie J, Guo L, Li K, Wang Y, Chen G, Li L, Chen H, Deng F, Jiang X, Zhang T, et al. , 2012. Axonal fiber terminations concentrate on gyri. Cereb. Cortex 22, 2831–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimchinsky EA, Sabatini BL, Svoboda K, 2002. Structure and function of dendritic spines. Annu. Rev. Physiol 64, 313–353. [DOI] [PubMed] [Google Scholar]
- Nixon RA, 2013. The role of autophagy in neurodegenerative disease. Nat. Med 19, 983–997. [DOI] [PubMed] [Google Scholar]
- Noël L, Kuhl E, 2019. Modeling neurodegeneration in chronic traumatic encephalopathy using gradient damage models. Comput. Mech 64, 1375–1387. [Google Scholar]
- Norden DM, Godbout JP, 2013. Microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol. Appl. Neurobiol 39, 19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyberg L, Wåhlin A, 2020. Imaging: the many facets of brain aging. Elife 9, e56640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyquist PA, Bilgel M, Gottesman R, Yanek LR, Moy TF, Becker LC, Cuzzocreo JL, Prince J, Wasserman BA, Yousem DM, et al. , 2015. Age differences in periventricular and deep white matter lesions. Neurobiol. Aging 36, 1653–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oschwald J, Guye S, Liem F, Rast P, Willis S, Röcke C, Jäncke L, Martin M, Mérillat S, 2020. Brain structure and cognitive ability in healthy aging: a review on longitudinal correlated change. Rev. Neurosci 31, 1–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott BR, Cohen RA, Gongvatana A, Okonkwo OC, Johanson CE, Stopa EG, Donahue JE, Silverberg GD, 2010. Brain ventricular volume and cerebrospinal fluid biomarkers of Alzheimer’s disease. J. Alzheimer’s Dis 20, 647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakkenberg B, Gundersen HJG, 1997. Neocortical neuron number in humans: effect of sex and age. J. Comp. Neurol 384, 312–320. [PubMed] [Google Scholar]
- Pakkenberg B, Pelvig D, Marner L, Bundgaard MJ, Gundersen HJG, Nyengaard JR, Regeur L, 2003. Aging and the human neocortex. Exp. Gerontol 38, 95–99. [DOI] [PubMed] [Google Scholar]
- Pandya JD, Grondin R, Yonutas HM, Haghnazar H, Gash DM, Zhang Z, Sullivan PG, 2015. Decreased mitochondrial bioenergetics and calcium buffering capacity in the basal ganglia correlates with motor deficits in a nonhuman primate model of aging. Neurobiol. Aging 36, 1903–1913. [DOI] [PubMed] [Google Scholar]
- Park DC, Reuter-Lorenz P, 2009. The adaptive brain: aging and neurocognitive scaffolding. Annu. Rev. Psychol 60, 173–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul A, Belton A, Nag S, Martin I, Grotewiel MS, Duttaroy A, 2007. Reduced mitochondrial sod displays mortality characteristics reminiscent of natural aging. Mech. Ageing Dev 128, 706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelvig DP, Pakkenberg H, Stark AK, Pakkenberg B, 2008. Neocortical glial cell numbers in human brains. Neurobiol. Aging 29, 1754–1762. [DOI] [PubMed] [Google Scholar]
- Penn RD, Belanger MG, Yasnoff WA, 1978. Ventricular volume in man computed from cat scans. Ann. Neurol.: Off. J. Am. Neurol. Assoc. Child Neurol. Soc 3, 216–223. [DOI] [PubMed] [Google Scholar]
- Peters A, Sethares C, Luebke J, 2008. Synapses are lost during aging in the primate prefrontal cortex. Neuroscience 152, 970–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, 2002. The effects of normal aging on myelin and nerve fibers: a review. J. Neurocytol 31, 581–593. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Sullivan EV, 2003. Increased brain white matter diffusivity in normal adult aging: relationship to anisotropy and partial voluming. Magn. Reson. Med.: Off. J. Int. Soc. Magn. Reson. Med 49, 953–961. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Mathalon DH, Sullivan EV, Rawles JM, Zipursky RB, Lim KO, 1994. A quantitative magnetic resonance imaging study of changes in brain morphology from infancy to late adulthood. Arch. Neurol 51, 874–887. [DOI] [PubMed] [Google Scholar]
- Phelps ME, Mazziotta JC, 1985. Positron emission tomography: human brain function and biochemistry. Science 228, 799–809. [DOI] [PubMed] [Google Scholar]
- Pini L, Pievani M, Bocchetta M, Altomare D, Bosco P, Cavedo E, Galluzzi S, Marizzoni M, Frisoni GB, 2016. Brain atrophy in Alzheimer’s disease and aging. Ageing Res. Rev 30, 25–48. [DOI] [PubMed] [Google Scholar]
- Qureshi AI, Feldmann E, Gomez CR, Johnston SC, Kasner SE, Quick DC, Rasmussen PA, Suri MFK, Taylor RA, Zaidat OO, 2009. Intracranial atherosclerotic disease: an update. Ann. Neurol.: Off. J. Am. Neurol. Assoc. Child Neurol. Soc 66, 730–738. [DOI] [PubMed] [Google Scholar]
- Raj A, Kuceyeski A, Weiner M, 2012. A network diffusion model of disease progression in dementia. Neuron 73, 1204–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj D, Yin Z, Breur M, Doorduin J, Holtman IR, Olah M, Mantingh-Otter IJ, Van Dam D, De Deyn PP, den Dunnen W, et al. , 2017. Increased white matter inflammation in aging- and Alzheimer’s disease brain. Front. Mol. Neurosci 10, 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz N, Lindenberger U, Rodrigue KM, Kennedy KM, Head D, Williamson A, Dahle C, Gerstorf D, Acker JD, 2005. Regional brain changes in aging healthy adults: general trends, individual differences and modifiers. Cereb. Cortex 15, 1676–1689. [DOI] [PubMed] [Google Scholar]
- Redzic ZB, Preston JE, Duncan JA, Chodobski A, Szmydynger-Chodobska J, 2005. The choroid plexus-cerebrospinal fluid system: from development to aging. Curr. Top. Dev. Biol 71, 1–52. [DOI] [PubMed] [Google Scholar]
- Resnick SM, Goldszal AF, Davatzikos C, Golski S, Kraut MA, Metter EJ, Bryan RN, Zonderman AB, 2000. One-year age changes in mri brain volumes in older adults. Cereb. Cortex 10, 464–472. [DOI] [PubMed] [Google Scholar]
- Resnick SM, Pham DL, Kraut MA, Zonderman AB, Davatzikos C, 2003. Longitudinal magnetic resonance imaging studies of older adults: a shrinking brain. J. Neurosci 23, 3295–3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettmann ME, Kraut MA, Prince JL, Resnick SM, 2006. Cross-sectional and longitudinal analyses of anatomical sulcal changes associated with aging. Cereb. Cortex 16, 1584–1594. [DOI] [PubMed] [Google Scholar]
- Reuter M, Schmansky NJ, Rosas HD, Fischl B, 2012. Within-subject template estimation for unbiased longitudinal image analysis. NeuroImage 61, 1402–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz K, Denswil NP, Stam OC, van Lieshout JJ, Daemen MJ, 2014. Cause and mechanisms of intracranial atherosclerosis. Circulation 130, 1407–1414. [DOI] [PubMed] [Google Scholar]
- Rodrigue KM, Kennedy KM, 2011. The cognitive consequences of structural changes to the aging brain. Handbook of the Psychology of Aging, pp. 73–91. [Google Scholar]
- Rogers J, Zornetzer SF, Bloom FE, Mervis RE, 1984. Senescent microstructural changes in rat cerebellum. Brain Res. 292, 23–32. [DOI] [PubMed] [Google Scholar]
- Ronan L, Fletcher PC, 2015. From genes to folds: a review of cortical gyrification theory. Brain Struct. Funct 220, 2475–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salahshoor H, Shapiro MG, Ortiz M, 2020. Transcranial focused ultrasound generates skull-conducted shear waves: computational model and implications for neuromodulation. Appl. Phys. Lett 117, 033702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salami A, Eriksson J, Nilsson L-G, Nyberg L, 2012. Age-related white matter microstructural differences partly mediate age-related decline in processing speed but not cognition. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis 1822, 408–415. [DOI] [PubMed] [Google Scholar]
- Salat DH, Buckner RL, Snyder AZ, Greve DN, Desikan RS, Busa E, Morris JC, Dale AM, Fischl B, 2004. Thinning of the cerebral cortex in aging. Cereb. Cortex 14, 721–730. [DOI] [PubMed] [Google Scholar]
- Salat D, Tuch D, Greve D, Van Der Kouwe A, Hevelone N, Zaleta A, Rosen B, Fischl B, Corkin S, Rosas HD, et al. , 2005. Age-related alterations in white matter microstructure measured by diffusion tensor imaging. Neurobiol. Aging 26, 1215–1227. [DOI] [PubMed] [Google Scholar]
- Salat DH, Greve DN, Pacheco JL, Quinn BT, Helmer KG, Buckner RL, Fischl B, 2009. Regional white matter volume differences in nondemented aging and Alzheimer’s disease. NeuroImage 44, 1247–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzer J, Zalc B, 2016. Myelination. Curr. Biol 26, R971–R975. [DOI] [PubMed] [Google Scholar]
- Scahill RI, Frost C, Jenkins R, Whitwell JL, Rossor MN, Fox NC, 2003. A longitudinal study of brain volume changes in normal aging using serial registered magnetic resonance imaging. Arch. Neurol 60, 989–994. [DOI] [PubMed] [Google Scholar]
- Schäfer A, Weickenmeier J, Kuhl E, 2019. The interplay of biochemical and biomechanical degeneration in Alzheimer’s disease. Comput. Methods Appl. Mech. Eng 352, 369–388. [Google Scholar]
- Schaer M, Cuadra MB, Tamarit L, Lazeyras F, Eliez S, Thiran J-P, 2008. A surface-based approach to quantify local cortical gyrification. IEEE Trans. Med. Imaging 27, 161–170. [DOI] [PubMed] [Google Scholar]
- Schmidt R, Ropele S, Enzinger C, Petrovic K, Smith S, Schmidt H, Matthews PM, Fazekas F, 2005. White matter lesion progression, brain atrophy, and cognitive decline: the austrian stroke prevention study. Ann. Neurol.: Off. J. Am. Neurol. Assoc. Child Neurol. Soc 58, 610–616. [DOI] [PubMed] [Google Scholar]
- Seidler RD, Bernard JA, Burutolu TB, Fling BW, Gordon MT, Gwin JT, Kwak Y, Lipps DB, 2010. Motor control and aging: links to age-related brain structural, functional, and biochemical effects. Neurosci. Biobehav. Rev 34, 721–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serot J-M, Béné M-C, Faure GC, 2003. Choroid plexus, aging of the brain, and Alzheimer’s disease. Front. Biosci 8, s515–s521. [DOI] [PubMed] [Google Scholar]
- Sexton CE, Walhovd KB, Storsve AB, Tamnes CK, Westlye LT, Johansen-Berg H, Fjell AM, 2014. Accelerated changes in white matter microstructure during aging: a longitudinal diffusion tensor imaging study. J. Neurosci 34, 15425–15436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Liu T, Tao D, Fan Y, Zhang J, Li S, Jiang J, Zhu W, Wang Y, Wang Y, et al. , 2018. Variation in longitudinal trajectories of cortical sulci in normal elderly. NeuroImage 166, 1–9. [DOI] [PubMed] [Google Scholar]
- Shenkin S, Bastin M, Macgill ivray T, Deary I, Starr J, Rivers C, Wardlaw J, 2005. Cognitive correlates of cerebral white matter lesions and water diffusion tensor parameters in community-dwelling older people. Cerebrovasc. Dis 20, 310–318. [DOI] [PubMed] [Google Scholar]
- Shim YS, Yang D-W, Roe CM, Coats MA, Benzinger TL, Xiong C, Galvin JE, Cairns NJ, Morris JC, 2015. Pathological correlates of white matter hyperintensities on magnetic resonance imaging. Dementia Geriatr. Cogn. Disord 39, 92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shook BA, Lennington JB, Acabchuk RL, Halling M, Sun Y, Peters J, Wu Q, Mahajan A, Fellows DW, Conover JC, 2014. Ventriculomegaly associated with ependymal gliosis and declines in barrier integrity in the aging human and mouse brain. Aging Cell 13, 340–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikora E, Bielak-Zmijewska A, Dudkowska M, Krzystyniak A, Mosieniak G, Wesierska M, Wlodarczyk J, 2021. Cellular senescence in brain aging. Front. Aging Neurosci 13, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šimić G, Kostović I, Winblad B, Bogdanović N, 1997. Volume and number of neurons of the human hippocampal formation in normal aging and Alzheimer’s disease. J. Comp. Neurol 379, 482–494. [DOI] [PubMed] [Google Scholar]
- Simpson JE, Wharton SB, Cooper J, Gelsthorpe C, Baxter L, Forster G, Shaw PJ, Savva G, Matthews FE, Brayne C, et al. , 2010. Alterations of the blood-brain barrier in cerebral white matter lesions in the ageing brain. Neurosci. Lett 486, 246–251. [DOI] [PubMed] [Google Scholar]
- Solár P, Zamani A, Kubíčková L, Dubov‘y P, Joukal M, 2020. Choroid plexus and the blood-cerebrospinal fluid barrier in disease. Fluids Barriers CNS 17, 1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solowij N, Zalesky A, Lorenzetti V, Yücel M, 2017. Chronic cannabis use and axonal fiber connectivity. Handbook of Cannabis and Related Pathologies. Elsevier, pp. 391–400. [Google Scholar]
- Song K, Li Y, Zhang H, An N, Wei Y, Wang L, Tian C, Yuan M, Sun Y, Xing Y, et al. , 2020. Oxidative stress-mediated blood-brain barrier (bbb) disruption in neurological diseases. Oxid. Med. Cell. Longevity 2020. [Google Scholar]
- Sorra KE, Harris KM, 2000. Overview on the structure, composition, function, development, and plasticity of hippocampal dendritic spines. Hippocampus 10, 501–511. [DOI] [PubMed] [Google Scholar]
- Sowell ER, Peterson BS, Kan E, Woods RP, Yoshii J, Bansal R, Xu D, Zhu H, Thompson PM, Toga AW, 2007. Sex differences in cortical thickness mapped in 176 healthy individuals between 7 and 87 years of age. Cereb. Cortex 17, 1550–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer SO, Sitnikov S, Kamen Y, Evans KA, Kronenberg-Versteeg D, Dietmann S, de Faria O Jr., Agathou S, Káradóttir RT, 2019. Oligodendrocyte progenitor cells become regionally diverse and heterogeneous with age. Neuron 101, 459–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl R, Walcher T, Romero CDJ, Pilz GA, Cappello S, Irmler M, Sanz-Aquela JM, Beckers J, Blum R, Borrell V, et al. , 2013. Trnp1 regulates expansion and folding of the mammalian cerebral cortex by control of radial glial fate. Cell 153, 535–549. [DOI] [PubMed] [Google Scholar]
- Stahon KE, Bastian C, Griffith S, Kidd GJ, Brunet S, Baltan S, 2016. Age-related changes in axonal and mitochondrial ultrastructure and function in white matter. J. Neurosci 36, 9990–10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storsve AB, Fjell AM, Tamnes CK, Westlye LT, Overbye K, Aasland HW, Walhovd KB, 2014. Differential longitudinal changes in cortical thickness, surface area and volume across the adult life span: regions of accelerating and decelerating change. J. Neurosci 34, 8488–8498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suemoto CK, Grinberg LT, Leite RE, Ferretti-Rebustini RE, Jacob-Filho W, Yaffe K, Nitrini R, Pasqualucci CA, 2018. Morphometric measurements of extracranial and intracranial atherosclerotic disease: a population-based autopsy study. Atherosclerosis 270, 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan EV, Pfefferbaum A, 2006. Diffusion tensor imaging and aging. Neurosci. Biobehav. Rev 30, 749–761. [DOI] [PubMed] [Google Scholar]
- Sullivan EV, Rohlfing T, Pfefferbaum A, 2010. Longitudinal study of callosal microstructure in the normal adult aging brain using quantitative dti fiber tracking. Dev. Neuropsychol 35, 233–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taki Y, Kinomura S, Sato K, Goto R, Kawashima R, Fukuda H, 2011. A longitudinal study of gray matter volume decline with age and modifying factors. Neurobiol. Aging 32, 907–915. [DOI] [PubMed] [Google Scholar]
- Ter Telgte A, van Leijsen EM, Wiegertjes K, Klijn CJ, Tuladhar AM, de Leeuw F-E, 2018. Cerebral small vessel disease: from a focal to a global perspective. Nat. Rev. Neurol 14, 387–398. [DOI] [PubMed] [Google Scholar]
- Thompson PM, Hayashi KM, De Zubicaray G, Janke AL, Rose SE, Semple J, Herman D, Hong MS, Dittmer SS, Doddrell DM, et al. , 2003. Dynamics of gray matter loss in Alzheimer’s disease. J. Neurosci 23, 994–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd KL, Brighton T, Norton ES, Schick S, Elkins W, Pletnikova O, Fortinsky RH, Troncoso JC, Molfese PJ, Resnick SM, et al. , 2018. Ventricular and periventricular anomalies in the aging and cognitively impaired brain. Front. Aging Neurosci 9, 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touyz RM, Montezano AC, 2015. Hypertensive Vasculopathy. [Google Scholar]
- Tuladhar AM, van Norden AG, de Laat KF, Zwiers MP, van Dijk EJ, Norris DG, de Leeuw F-E, 2015. White matter integrity in small vessel disease is related to cognition. NeuroImage: Clin 7, 518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bedem H, Kuhl E, 2017. Molecular mechanisms of chronic traumatic encephalopathy. Curr. Opin. Biomed. Eng 1, 23–30. [Google Scholar]
- Van Essen DC, 1997. A tension-based theory of morphogenesis and compact wiring in the central nervous system. Nature 385, 313–318. [DOI] [PubMed] [Google Scholar]
- van Velsen EF, Vernooij MW, Vrooman HA, van der Lugt A, Breteler MM, Hofman A, Niessen WJ, Ikram MA, 2013. Brain cortical thickness in the general elderly population: the Rotterdam scan study. Neurosci. Lett 550, 189–194. [DOI] [PubMed] [Google Scholar]
- Verner S, Garikipati K, 2018. A computational study of the mechanisms of growth-driven folding patterns on shells, with application to the developing brain. Extreme Mech. Lett 18, 58–69. [Google Scholar]
- Villemagne VL, Doré V, Burnham SC, Masters CL, Rowe CC, 2018. Imaging tau and amyloid-β proteinopathies in Alzheimer disease and other conditions. Nat. Rev. Neurol 14, 225–236. [DOI] [PubMed] [Google Scholar]
- Villemagne VL, Doré V, Burnham S, Rowe CC, 2021. Aβ imaging in aging, Alzheimer’s disease, and other neurodegenerative conditions. PET and SPECT in Neurology. Springer, pp. 283–343. [Google Scholar]
- Vinke EJ, de Groot M, Venkatraghavan V, Klein S, Niessen WJ, Ikram MA, Vernooij MW, 2018. Trajectories of imaging markers in brain aging: the Rotterdam study. Neurobiol. Aging 71, 32–40. [DOI] [PubMed] [Google Scholar]
- Viswanathan A, Greenberg SM, 2011. Cerebral amyloid angiopathy in the elderly. Ann. Neurol 70, 871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel JW, Young AL, Oxtoby NP, Smith R, Ossenkoppele R, Strandberg OT, La Joie R, Aksman LM, Grothe MJ, Iturria-Medina Y, et al. , 2021a. Four distinct trajectories of tau deposition identified in Alzheimer’s disease. Nat. Med 27, 871–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt NM, Hunt JF, Adluru N, Ma Y, Van Hulle CA Iii, D.C.D., Kecskemeti SR, Chin NA, Carlsson CM, Asthana S, et al. , 2021b. Interaction of amyloid and tau on cortical microstructure in cognitively unimpaired adults. Alzheimer’s Dementia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bartheld CS, 2018. Myths and truths about the cellular composition of the human brain: a review of influential concepts. J. Chem. Neuroanat 93, 2–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Bernhardi R, Eugenín-von Bernhardi L, Eugenín J, 2015. Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci 7, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walhovd KB, Fjell AM, Reinvang I, Lundervold A, Dale AM, Eilertsen DE, Quinn BT, Salat D, Makris N, Fischl B, 2005. Effects of age on volumes of cortex, white matter and subcortical structures. Neurobiol. Aging 26, 1261–1270. [DOI] [PubMed] [Google Scholar]
- Walhovd KB, Fjell AM, Giedd J, Dale AM, Brown TT, 2016. Through thick and thin: a need to reconcile contradictory results on trajectories in human cortical development. Cereb. Cortex 27, bhv301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Meng R, Liu G, Cao C, Chen F, Jin K, Ji X, Cao G, 2019. Intracranial atherosclerotic disease. Neurobiol. Dis 124, 118–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Demirci N, Holland MA, 2020. Numerical investigation of biomechanically coupled growth in cortical folding. Biomech. Model. Mechanobiol 1–13. [DOI] [PubMed] [Google Scholar]
- Wang Z, Martin B, Weickenmeier J, Garikipati K, 2021. An inverse modelling study on the local volume changes during early morphoelastic growth of the fetal human brain. Brain Multiphys. 100023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardlaw JM, Smith EE, Biessels GJ, Cordonnier C, Fazekas F, Frayne R, Lindley RI, O’Brien JT, Barkhof F, Benavente OR, et al. , 2013. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 12, 822–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardlaw JM, Valdés Hernández MC, Muñoz-Maniega S, 2015. What are white matter hyperintensities made of? Relevance to vascular cognitive impairment. J. Am. Heart Assoc 4, e001140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardlaw JM, Makin SJ, Hernández MCV, Armitage PA, Heye AK, Chappell FM, Munoz-Maniega S, Sakka E, Shuler K, Dennis MS, et al. , 2017. Blood-brain barrier failure as a core mechanism in cerebral small vessel disease and dementia: evidence from a cohort study. Alzheimer’s Dementia 13, 634–643. [Google Scholar]
- Wardlaw JM, Smith C, Dichgans M, 2019. Small vessel disease: mechanisms and clinical implications. Lancet Neurol. 18, 684–696. [DOI] [PubMed] [Google Scholar]
- Watanabe C, Imaizumi T, Kawai H, Suda K, Honma Y, Ichihashi M, Ema M, Mizutani K.-i., 2020. Aging of the vascular system and neural diseases. Front. Aging Neurosci 12, 309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weickenmeier J, de Rooij R, Budday S, Steinmann P, Ovaert TC, Kuhl E, 2016. Brain stiffness increases with myelin content. Acta Biomater. 42, 265–272. [DOI] [PubMed] [Google Scholar]
- Weickenmeier J, Butler C, Young P, Goriely A, Kuhl E, 2017. The mechanics of decompressive craniectomy: personalized simulations. Comput. Methods Appl. Mech. Eng 314, 180–195. [Google Scholar]
- Weickenmeier J, Kuhl E, Goriely A, 2018. Multiphysics of prionlike diseases: progression and atrophy. Phys. Rev. Lett 121, 158101. [DOI] [PubMed] [Google Scholar]
- Welker W, 1990. Why does cerebral cortex fissure and fold? Cerebral Cortex. Springer, pp. 3–136. [Google Scholar]
- Weller RO, Nicoll JA, 2003. Cerebral amyloid angiopathy: pathogenesis and effects on the ageing and Alzheimer brain. Neurol. Res 25, 611–616. [DOI] [PubMed] [Google Scholar]
- Wen W, Sachdev P, 2004. The topography of white matter hyperintensities on brain mri in healthy 60-to 64-year-old individuals. NeuroImage 22, 144–154. [DOI] [PubMed] [Google Scholar]
- Westlye LT, Walhovd KB, Dale AM, Bjørnerud A, Due-Tønnessen P, Engvig A, Grydeland H, Tamnes CK, Østby Y, Fjell AM, 2010. Life-span changes of the human brain white matter: diffusion tensor imaging (dti) and volumetry. Cereb. Cortex 20, 2055–2068. [DOI] [PubMed] [Google Scholar]
- White L, Andrews T, Hulette C, Richards A, Groelle M, Paydarfar J, Purves D, 1997. Structure of the human sensorimotor system. I. Morphology and cytoarchitecture of the central sulcus. Cereb. Cortex (New York, NY: 1991) 7, 18–30. [DOI] [PubMed] [Google Scholar]
- Xie S, Zhang Z, Chang F, Wang Y, Zhang Z, Zhou Z, Guo H, 2016. Subcortical white matter changes with normal aging detected by multi-shot high resolution diffusion tensor imaging. PLOS ONE 11, e0157533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong YY, Mok V, 2011. Age-related white matter changes. J. Aging Res 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Wang B, Ren C, Hu J, Greenberg DA, Chen T, Xie L, Jin K, 2017. Age-related impairment of vascular structure and functions. Aging Dis. 8, 590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Yin X, Hong H, Wang S, Jiaerken Y, Zhang F, Pasternak O, Zhang R, Yang L, Lou M, et al. , 2021. Increased extracellular fluid is associated with white matter fiber degeneration in cadasil: in vivo evidence from diffusion magnetic resonance imaging. Fluids Barriers CNS 18, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue NC, Arnold AM, Longstreth WT Jr., Elster AD, Jungreis CA, O’Leary DH, Poirier VC, Bryan RN, 1997. Sulcal, ventricular, and white matter changes at MR imaging in the aging brain: data from the cardiovascular health study. Radiology 202, 33–39. [DOI] [PubMed] [Google Scholar]
- Yun J, Finkel T, 2014. Mitohormesis. Cell Metab. 19, 757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarzor M, Kaessmair S, Steinmann P, Blümcke I, Budday S, 2021. A two-field computational model couples cellular brain development with cortical folding. Brain Multiphys. 2, 100025. [Google Scholar]
- Zhan W, Zhang Y, Mueller SG, Lorenzen P, Hadjidemetriou S, Schuff N, Weiner MW, 2009. Characterization of white matter degeneration in elderly subjects by magnetic resonance diffusion and flair imaging correlation. NeuroImage 47, T58–T65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilles K, Armstrong E, Schleicher A, Kretschmann H-J, 1988. The human pattern of gyrification in the cerebral cortex. Anat. Embryol 179, 173–179. [DOI] [PubMed] [Google Scholar]
- Zilles K, Palomero-Gallagher N, Amunts K, 2013. Development of cortical folding during evolution and ontogeny. Trends Neurosci. 36, 275–284. [DOI] [PubMed] [Google Scholar]