

Abstract
NRF2 is a basic leucine zipper (bZip) transcription factor that is the master regulator of redox homeostasis. Under basal conditions, the cellular level of NRF2 is low due to a posttranslational regulation by the ubiquitin proteasome system (UPS). But, when an organism is challenged with oxidative or xenobiotic stress, the NRF2 pathway is activated by inhibition of the E3 ubiquitin ligase complex that normally marks NRF2 for destruction. For several decades, researchers have searched for molecules that can intentionally activate NRF2, as this was shown to be a means to prevent certain diseases, at least in animal models. In the present era, there are many compounds known to activate the NRF2 pathway including natural products and synthetic compounds, covalent and non-covalent compounds, and others. However, it was also revealed that like many protective pathways, the NRF2 pathway has a dark side. Just as NRF2 can protect normal cells from damage, it can protect malignant cells from damage. As cells transform, they are exposed to many stressors and aberrant upregulation of NRF2 can facilitate transformation and it can help cancer cells to grow, to spread, and to resist treatment. For this reason, researchers are also interested in the discovery and development of NRF2 inhibitors. In the present review, we will begin with a general discussion of NRF2 structure and function, we will discuss the latest in NRF2 non-covalent activators, and we will discuss the current state of NRF2 inhibitors.
Keywords: Cancer, NRF2, chemoprevention, transcription factor
1. Introduction
Designed chemical modulation of nonreceptor transcription factors remains a seemingly intractable problem in drug discovery, but certainly worth the pursuit, as many of the infamous oncogenes fall into this class (1). Many of the most desirable targets of the oncogene transcription factor class are considered undruggable as they are often intrinsically disordered or largely disordered proteins(2). Intrinsically disordered proteins lack a traditional binding pocket and instead form order only upon binding to another biomolecule (3). Although there have been reports of compounds that bind to these targets (4–7), there are no compounds that are in clinical trials. On the reverse side, in the case of tumor suppressors, the desired outcome is activation of the repressor, again a challenging problem that requires identification of the molecular details of gene suppression and compounds that can reverse this effect (8–11).
The transcription factor NRF2 regulates the antioxidant response, among other actions (discussed below), to protect cells from environmental and endogenous stress. Like many of the oncogenes discussed above (and NRF2 has recently been referred to as an oncogene (12)) the protective function of NRF2 can also lead to protection of cancer cells, facilitating transformation, growth, metastasis, and chemoresistance (13–18). Both protective functions have been the subject of efforts to regulate NRF2 function for chemoprevention (activation of NRF2) or cancer therapy (inhibition of NRF2). Traditionally, NRF2 was the centerpiece of the chemoprevention field where researchers searched for compounds that activated the NRF2 pathway to prevent various diseases related mainly to oxidative damage. There have been many compounds, natural and otherwise, that have been discovered that activate NRF2 and protect cells or animals from insult. More recently, there have been efforts to discover inhibitors of NRF2, but as with other non-receptor transcription factors, this has proven a very challenging task. In the present review we will not extensively discuss all NRF2 activators and inhibitors as this has been recently reviewed by several groups and readers are directed to these reviews (19–24). Instead, we will begin with a discussion of NRF2 structure and function and then discuss work on non-covalent NRF2 activators, followed by a discussion of the dark side of NRF2 and the search for NRF2 inhibitors. Presently, there are no compounds that have been verified to be direct targeting NRF2 inhibitors, but there are a couple of pathway inhibitors, especially the quassinoid brusatol, that have been critical to dissecting the role of NRF2 in the dark side of disease.
2. NRF2 structure and function
When cells are challenged by environmental insults, most prominently oxidants or electrophiles, the NRF2-KEAP1-ARE axis protects the cells by increasing the reductive and nucleophilic power of the cells (25–27). Under basal, non-stressed conditions, the level of NRF2 is low due to constant post-translational control by the ubiquitin proteasome system (UPS) (Figure 1A) (28–32). Two KEAP1 proteins, a CUL3-RBX1 E3 ubiquitin ligase substrate recruiting factor, bind to NRF2. One KEAP1 binds the ETGE motif and the other binds the DLG motif (33, 34). Between the ETGE and DLG are seven lysines that can be ubiquitylated(28). This ubiquitylation recruits the p97-UFD1/NPL4-UBXN7 complex to extract NRF2 from the CUL3 complex and send it to the proteasome for degradation (35). This is the normal state for cells, but when challenged by oxidative stress or electrophilic compounds, KEAP1, which contains a series of sensor cysteine residues, with Cys151 being the most active, becomes modified (30, 32, 36–38). This Cys151 adduction blocks further ubiquitylation of NRF2 either by releasing KEAP1 from the CUL3 complex or by releasing the low affinity DLG motif from one of the KEAP1s (Figure 1B) (33, 34, 39–42). Either or both actions are possible, and both have the effect of blocking the action of the UPS on NRF2. This leads to increasing levels of NRF2, which then translocates to the nucleus where it binds to an antioxidant response element (ARE). To date, over 300 proteins have been shown to contain an ARE including phase I, II and III metabolism, primary metabolism, protein quality control, and antioxidants (26, 43).
Figure 1.
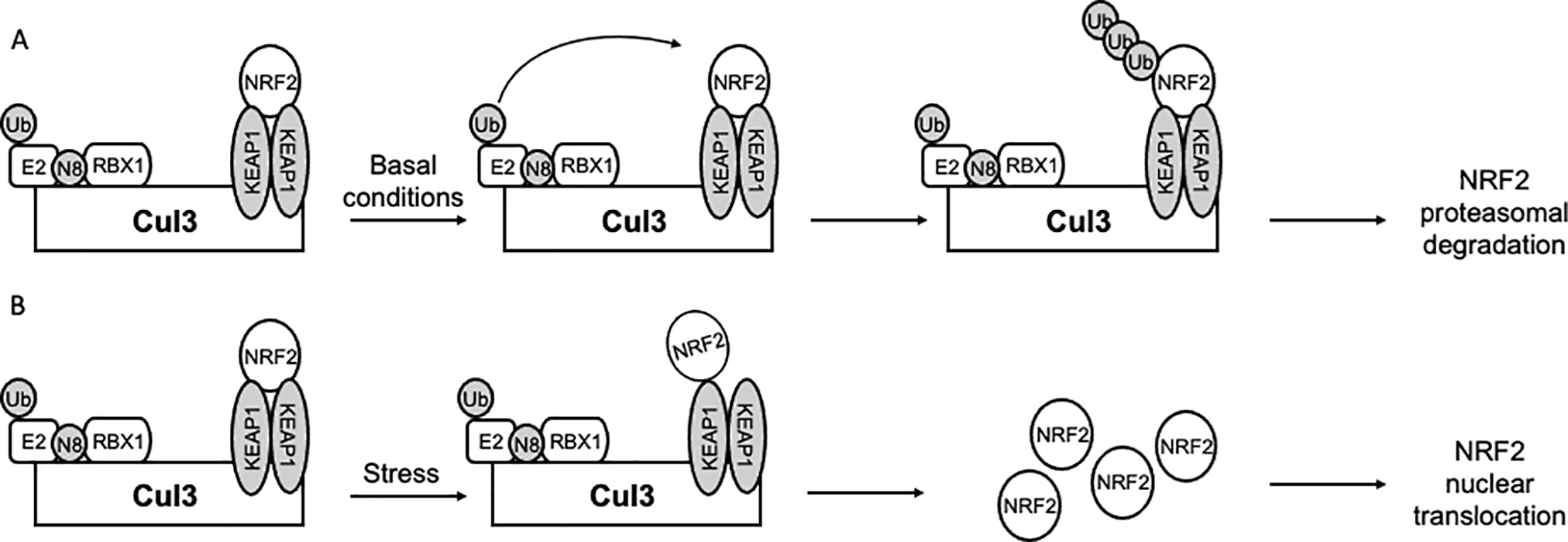
Cellular abundance of NRF2 is primarily regulated by its interaction with KEAP1. (A) NRF2 is rapidly degraded under basal conditions through interaction with a KEAP1 homodimer, which recruits NRF2 to the CUL3-RBX1 E3 ubiquitin ligase leading to proteasomal degradation. (B) Oxidative or electrophilic stress capable of modifying one of the critical cysteines (predominantly Cys151) of KEAP1 prevents UPS-mediated degradation of NRF2 by dissociating the DLG motif from KEAP 1 (shown) or causing KEAP1 dissociation from CUL3 (not shown). This leads to accumulation of NRF2, which translocates to the nucleus, binds sMAF, and transcribes ARE-regulated genes.
NRF2 is a basic leucine zipper (bZip) transcription factor from the Cap ‘n’ Collar (CNC) family (25, 44, 45). It is comprised of seven functional domains defined by homology and functional mapping, but not confirmed to be independent structural elements. In fact, given the disordered nature of NRF2, it is likely that most or all the seven domains are largely unstructured until engaged with their respective binding partners. The organization of the seven NRF2 domains and their assigned functions are shown in Figure 2. Structurally, there are very little data on any of the domains of NRF2. There is one NMR structure of the Neh1 domain that shows a lot of disorder with a couple of small α-helices. Based on the crystal structures of other bZip transcription factors, it is unlikely this is the structure of Neh1 when it is bound to sMAF and engaged with DNA (46–49). The only other structural data are small peptides from the Neh2 domain that bind to KEAP1 (34, 50, 51). These data were important for the discovery of non-covalent NRF2 activators as discussed below.
Figure 2.
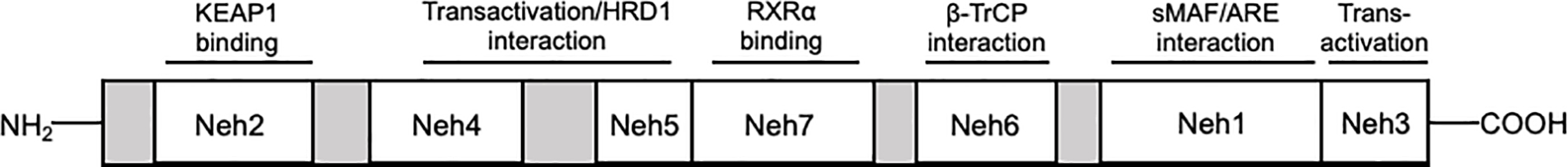
NRF2 domains are assigned based upon function rather than structural independence. From N to C terminus, KEAP1 interacts with NRF2 Neh2 domain, Neh4/5 serve as transactivation domains and a degron that interacts with HRD1, Neh7 serves as the RXRα binding domain, NEH6 is a phosphodegron that interacts with β–TrCP, Neh1 is the bZip domain that interacts with small MAFs and AREs, and Neh3 serves as an additional transactivation domain.
The Neh1 domain is the basic leucine zipper domain. The Neh1 domain forms a heterodimer with sMAF proteins, allowing binding to DNA at the antioxidant response elements. It also contains the nuclear localization sequence and the nuclear export sequence (43, 45, 52–58). The Neh2 domain contains the ETGE and DLG motifs that mediate binding to the KEAP1 homodimer and seven lysine residues between the ETGE and DLG that mediate ubiquitylation (34, 50, 51). The Neh3 domain is one of the transactivation domains that interacts with chromo-ATPase/helicase DNA-binding protein (CHD6) (59). The Neh4 and 5 domains seem to work in concert as both transactivation domains binding CREB-binding protein (CBP) and Brahma-related gene 1(BRG1) (60–62) and as another NRF2 degron that is recognized by the ER localized E3 ubiquitin ligase, HRD1(63). The Neh6 domain is another degron that gets phosphorylated by GSK-3, which recruits the β-TrCP E3 ubiquitin ligase (33, 64–67). Finally, the Neh7 domain is a repressive domain, binding the RXRα repressor (68).
3. Non-covalent NRF2 activators
Discovery of the NRF2-KEAP1-ARE pathway largely came to pass from early studies on compounds that activated what was called the phase 2 response (not to be confused with phase II metabolism) (69–90). Although there are 10 classes of activators, most compounds that have been studied in detail are electrophilic, often Michael acceptor, canonical NRF2 activators that work by modifying Cys151 of KEAP1 (see above for discussion) (91–93). Of this class of compounds, sulforaphane, bardoxolone, and dimethyl fumarate (DMF) have been in clinical trials for a variety of indications including cancers, diabetes, and autoimmune diseases. However, to date only DMF (Tecfidera) has been approved for the treatment of psoriasis in Europe and for relapsing multiple sclerosis. There is also controversy about if the primary mode of action of DMF is through activation of NRF2 (94–98). Bardoxolone and sulforaphane have also been shown to modify many other targets, leading to the possibility of dose limiting toxicity due to off target liabilities and raising some controversy about primary mode of action in the case of bardoxolone (99, 100). For this reason, researchers have explored the possibility of developing compounds (Figure 3) that disrupt the interaction between the ETGE motif of NRF2 and KEAP1 (Figure 4).
Figure 3.
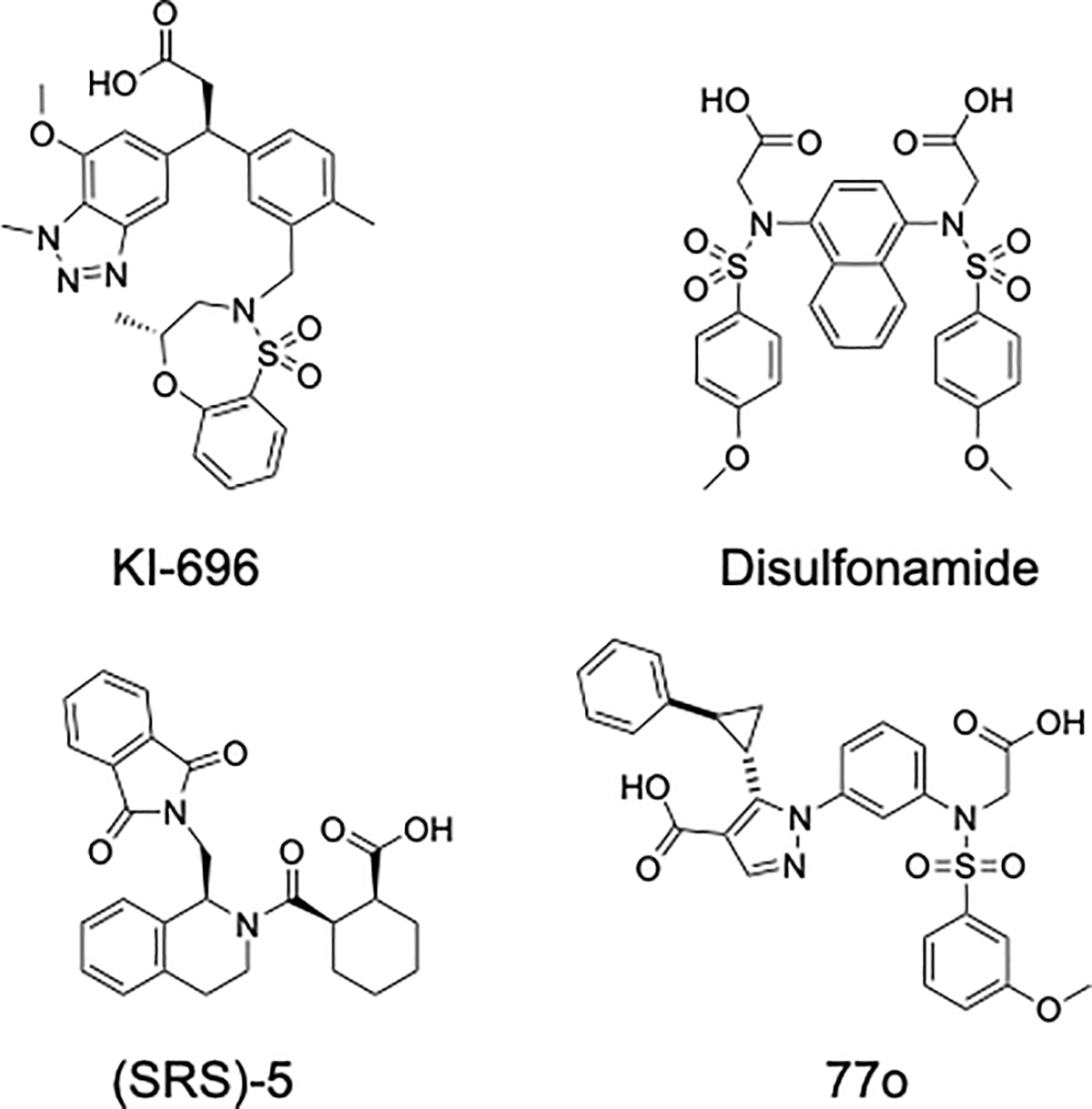
Structures of NRF2-activating molecules which disrupt the NRF2-KEAP1 Kelch domain interaction and promote nuclear accumulation of NRF2 and activation of the NRF2-ARE signaling axis.
Figure 4.

The NRF2 ETGE (and DLG) motifs interact with the KEAP1-Kelch domain. (A) KEAP1-Kelch domain (PDB 5FNR) with the subpockets highlighted; P1 (magenta), P2 (blue), P3 (orange), P4 (teal), and P5 (forest green). (B) KEAP1-Kelch domain (PDB 5FNR) subpocket surface charges; positive charge (blue), negative charge (red), non-polar residues (white). (C) ETGE (green) interacts with all five KEAP1-Kelch domain (PDB 5FNR) subpockets (P1-P5). (D) ETGE (green) orientation within the KEAP1-Kelch domain (PDB 7K2M) with sidechains highlighted using the same colors as subpockets in 4A and 4C.
Mutational studies performed by the Yamamoto group identified the NRF2 mutation E79A reduced the affinity of NRF2 an 9mer peptide containing the ETGE motif by 2–3 orders of magnitude for the KEAP1-Kelch domain. NRF2 E82 was discovered to be the second most energetically significant interaction for KEAP1-Kelch domain binding. Therefore, the E79/E82 interaction in the P1/P2 polar subpockets accounts for the majority of the binding energy due to electrostatic and hydrogen bonding interactions made within these regions of the KEAP1 Kelch domain. (34, 101) The DLG domain binds in a similar manner, but with approximately two orders of magnitude lower affinity (33, 34, 102). These structural and biophysical studies also facilitated many of the efforts to find non-covalent NRF2-ARE activating compounds that block the ETGE (or DLG)-KEAP1-Kelch domain protein-protein interaction and several of these compounds will be discussed in detail below.
We recently published a paper comparing a covalent natural product activator of the NRF2 pathway and a non-covalent semi-synthetic variant. In this study, we showed less potent activation with the non-covalent variant, but that the activation was longer lived as the compound was not susceptible to modifications by phase 2 metabolic processes that are increased due to NRF2 activation. We also demonstrated this compound to be KEAP1 dependent but Cys151 independent, however we did not resolve the precise mechanism of action (103). This will not be discussed in depth in the present review, but it illustrates some of the advantages associated with a non-covalent approach. Instead, we will focus on some of the most important, designed protein-protein interaction inhibitors discovered.
3.1. (SRS)-5
The first characterized non-covalent NRF2 activator (NRF2-KEAP1 protein-protein interaction (PPI) disruptor) was discovered by the Munoz group after screening the 300,000+ compound NIH Molecular Libraries Probe Production Centers Network (MLPCN) library using fluorescence polarization (FP) as the primary screening assay (104). Successful hits from this screen displace a fluorescent NRF2 9mer peptide (containing the ETGE motif) from the KEAP1 Kelch domain (Kd 20–65 nM as determined by surface plasmon resonance (SPR) and FP). This screen acts as a surrogate for displacement of full-length NRF2 from KEAP1, ultimately leading to NRF2 accumulation and increased transcription of NRF2-target genes (105). The strength of this panning approach is the removal of bias from the discovery of covalent NRF2 activators which not only modify critical KEAP1 cysteines outside of the Kelch domain, but also modify other proteins, resulting in systemic side effects (23, 30, 104).
Nearly 500 hits (defined as greater than 12% inhibition of the NRF2 9mer peptide/Kelch domain interaction) were identified and 460 were tested in an 8-point dose response after eliminating fluorescent hit molecules which interfere with the assay readout. In all, eight molecules were confirmed as hits that demonstrated clear dose-dependent inhibition of the NRF2-KEAP1 Kelch domain protein-protein interaction. Hit LH601, with an IC50 of 3 μM in the FP assay and Kd of 1.9 μM by SPR, was explored as the lead compound. Resynthesis of hit molecule LH601 revealed one of its four stereoisomers, named (SRS)-5 (Figure 3), was responsible for most of the activity of the original hit. (SRS)-5 was tested in HepG2 ARE-induction and U2OS NRF2 nuclear translocation assays and was found to have EC50s of 18 and 12 μM, respectively, compared to maximum induction by a standard NRF2 activating compound, tert-butylhydroquinone (tBHQ). IC50 values of 1.4 μM and 0.75 μM were obtained by time-resolved Förster resonance energy transfer (TR-FRET) and SPR, respectively. In addition, chemical shift perturbation analysis by nuclear magnetic resonance (NMR), further supported direct interaction between (SRS)-5 and the KEAP1-Kelch domain (104, 106–108). (SRS)-5 is predicted to interact with at least four of the five KEAP1-Kelch domain subpockets (NRF2 ETGE 9mer peptide interacts with all five (Figure 4)), including the P1 and P2 polar pockets (Figure 5A) (104, 106, 109). It does not appear, however, that the molecule contacts the same residues within the KEAP1 Kelch domain P1 and P2 pockets as the NRF2 ETGE 9mer peptide and may be the basis for the lower affinity of the molecule for the target relative to the peptide.
Figure 5.

NRF2-activator molecule binding poses within the KEAP1-Kelch domain. (Top) (SRS)-5 (A), disulfonamide molecule (B), and KI-696 (C) orientations within the KEAP1-Kelch domain with surface residue charges; positive charge (blue), negative charge (red), hydrophobic (white). (Middle) (SRS)-5 (A) interacts with four of five KEAP1-Kelch domain subpockets, disulfonamide molecule (B) interacts with KEAP1-Kelch domain P3-P5 subpockets, KI-696 (C) interacts with KEAP1-Kelch domain subpockets P1 and P3-P4, P1 (magenta), P2 (blue), P3 (orange), P4 (teal), and P5 (forest green). (Bottom) (SRS)-5 (A), disulfonamide molecule (B), and KI-696 (C) orientations within the KEAP1-Kelch domain with sidechains highlighted with the same colors as the subpockets. (A) PDB 4L7B, (B) PDB 4IQK, (C) PDB 5FNU.
Using MDCK-MDR1 cells in vitro and Mdr1a/1b/BCRP knockout mice as models to test blood-brain barrier (BBB) penetrance, (SRS)-5 was found to undergo significant efflux, and thus low predicted molecule accumulation in the central nervous system (CNS). Attempted bioisosteric replacement of critical (SRS)-5 functional groups significantly enhanced molecule accumulation within the CNS (unbound brain/unbound plasma ratio of 0.9) but decreased binding affinity 100-fold by NRF2 peptide/KEAP1 Kelch domain FP assay (110). Although first in its class as a non-covalent NRF2-KEAP1 interaction inhibitor, (SRS)-5 and its derivatives have proved problematic to modify with coupled retention of biochemical potency and in vitro/in vivo efficacy.
3.2. Benzene-disulfonamides
A second non-covalent NRF2 activator compound class was discovered in a 260,000+ compound screen using FP aided by two-dimensional fluorescence intensity distribution analysis (2D-FIDA). 2D-FIDA is a technique that uses confocal microscopy for greater description of molecule binding including detection of multiple molecule binding modes, aggregation, etc. (111, 112). A fluorescent NRF2 peptide was found to have low nM affinity for the KEAP1-Kelch domain in this assay and hit molecules were carried forth in two counter screens which included testing hit molecules in the FP assay with NRF2 peptide labeled with a different fluorophore as well as running the assays without KEAP-1 to rule out hit-related fluorescence interruption and autofluorescence, respectively. Multiple N-phenyl-benzenesulfonamide hits induced ARE driven expression in a DLD1 (colorectal adenocarcinoma) ARE-luciferase cellular assay. Addition of compound to DLD1 ARE-luciferase cells pretreated with siNRF2 abolished ARE-mediated induction of luciferase, indicating on target effect. A cocrystal structure of a N-phenyl-benzenesulfonamide hit molecule revealed interactions with the Kelch P3, P4, and P5 subpockets (Figure 5B). Despite these interactions, the molecule achieved only low micromolar potency against the target, however interactions with KEAP1-Kelch domain polar pockets 1 and 2 were absent, likely explaining the modest potency (111).
Several groups have developed derivatives of the N-phenyl-benzenesulfonamide scaffold to improve potency (106, 113–117), some of which are predicted to have preferable CNS penetration properties (118). Based upon the crystallography data generated by Marcotte et al., one of the original benzene-disulfonamide hit molecules was modified to mimic the NRF2 Neh2 ETGE E79/E82 interactions with the KEAP1-Kelch domain P1 and P2 subpockets respectively (116). Carboxyl groups added to the benzene-disulfonamide structure (Figure 3), now projecting into and interacting with P1 and P2 subpocket residues within the KEAP1 Kelch domain resulted in a binding affinity 1000-fold greater than the originally discovered molecule as measured by biolayer interferometry (BLI) (116, 119). Jiang et al. demonstrated this optimized compound increased ARE-luciferase activity as well as the expression of NRF2 target gene protein products HO-1, NQO1, and GCLM in a dose and time dependent manner. To obtain a 15-fold increase in ARE-luciferase induction, a compound concentration of more than 1000-fold greater than the compound-KEAP1 Kelch domain BLI Kd was required (116). Although a highly potent binder of the KEAP1 Kelch domain, the modifications made to this molecule to enhance interaction with P1 and P2 subpockets significantly decreased cellular penetrance and potency, likely due to the presence of acidic carboxylic acid groups.
3.3. KI-696
Davies et al. employed a fragment-based drug discovery approach to generate a potent, non-covalent NRF2 activator with pharmacokinetic (PK) properties suitable for use in animal studies (120–122). Three common binding sites within the KEAP1 Kelch domain were revealed after screening 330 fragments by x-ray crystallography (122). The most critical molecule/residue interactions discovered were with R483 (electrostatic) in the polar P1 pocket, S602 (H-bonding) in the P3 pocket, and Y525 (π-stacking) in the P4 pocket. Fragments found to make contacts within these critical regions (although individually their affinity for the KEAP1 Kelch domain was greater than 1 mM as determined by NRF2 peptide/Kelch domain FP) were linked and optimized, resulting in an IC50 of less than 15 nM as measured by NRF2 peptide/Kelch domain FP and a 13 nM Kd measured by isothermal calorimetry (ITC) (122, 123). The final lead compound, KI-696 (Figure 3), was found to interact with residues within the KEAP1-Kelch domain subpockets P1, P3, and P4, similarly to the individual fragments (Figure 5C). The authors also showed that 1 μM KI-696 increased NQO1 and GCLM mRNA and protein levels to a similar extent as bardoxolone (although 10-fold less bardoxolone was needed) in normal human epithelial cells, however these effects were abolished in the presence of siNrf2, indicating NRF2 dependence. Pretreatment with KI-696 reversed oxidative stress as measured by cellular GSH levels and furthermore, NRF2 nuclear accumulation increased in a KI-696 dose-dependent manner. Intravenous administration of KI-696 increased transcription of NRF2 target genes and decreased markers of ozone-induced lung inflammation in Han Wistar rats (122). Altogether, KI-696 was demonstrated to be a potent, non-covalent inhibitor of the NRF2-KEAP1 protein-protein interaction with improved cellular penetrance over previously discovered molecules.
3.4. 77o
Most recently, the Bach group implemented a molecule deconstruction/reconstruction strategy using six non-covalent NRF2 activator compound scaffolds to generate a potent lead molecule (124). Seventy-seven fragments, many with desirable physiochemical properties adhering to the rule of three, were generated for initial screening by FP, thermal shift assay (TSA), saturation transfer difference nuclear magnetic resonance (STD NMR), and surface plasmon resonance (SPR) (125–128). Hit rates of 17%, 25%, 64%, and 47% for FP, TSA, STD NMR, and SPR, respectively were observed (124). Fragment hits were selected based upon activity in multiple assays with hits validated by dose response in SPR given the most weight. Validated fragments were then carried forth to x-ray crystallography for examination of binding pose within the KEAP1 Kelch domain binding pocket. Six of the 17 validated hit fragments were found to crystallize and diffract with sufficient resolution to demonstrate molecule binding within the Kelch domain. Two fragments were found to have an overlapping pose in the P3 subpocket; one of the fragments was found to interact with the P1 and P3 subpockets and the other fragment with the P3 and P5 subpockets. After these fragments were linked together, a 380-fold increase in potency was observed relative to either fragment alone. Modifications to these linked fragments further increased potency nearly 100-fold (Ki 40 nM); binding interactions with the P1, P2, P3, and P5 subpockets were also supported by x-ray crystallography. Potent derivatives were stable against metabolism in liver microsome and blood serum assays, however cell penetrance by parallel artificial membrane permeation assay (PAMPA) was very poor (less than 2% of the compound crossed the membrane) (124). Without further optimization of cellular penetration properties, 77o and derivative molecules are useful as biochemical probes, but have little utility in cellular systems or in vivo studies.
4. The dark side of NRF2
Since the discovery of the NRF2-KEAP1-ARE signaling pathway in 1999, NRF2 has been viewed as a good transcription factor that protects against oxidative stress-related diseases, including cancer. Studies have demonstrated that Nrf2−/− mice are more susceptible than wild type mice to chemical-induced tumorigenesis, and controlled NRF2 activation by dietary phytochemicals prevents tumor initiation induced by chemical carcinogens (129, 130). However, in 2008, the Zhang lab demonstrated that NRF2 can promote cancer. This was called the “dark-side” of NRF2 (13), a concept that has been further supported by recent studies from many labs, demonstrating that in tumors, high levels of NRF2 promote tumor progression, metastasis, and chemoresistance. In addition, patients with high NRF2 levels in their tumor tissues have higher risk of recurrence and an overall poor prognosis (13, 16, 17, 131–138). Dysregulation of NRF2, resulting in high NRF2 expression, is common in many human cancers. Somatic NRF2/KEAP1 mutations that disrupt the NRF2-KEAP1 interaction and constitutively activate NRF2 are frequent in certain cancer types, particularly in lung cancer, where these mutations are present in up to one third of patients (139–143). In fact, a recent genome-wide somatic point mutation saturation analysis of 21 tumor types, found that while only a few well-known cancer genes are significantly mutated across different tumor types, KEAP1 is significantly mutated in multiple cancer types, including lung, head and neck, and bladder cancer (as a reference, classical examples of cancer genes such as TP53, KRAS, BRAF and NRAS are significantly mutated across four or more tumor types) (142). Furthermore, in lung adenocarcinoma, KEAP1 is as frequently mutated (>30%) as the tumor suppressor gene TP53 (142). Somatic mutation in NRF2 or KEAP1 in cancer cells, resulting in uncontrolled NRF2 expression, makes them addicted to NRF2 for survival since cancer cells are constantly under increased oxidative, proteotoxic, and metabolic stress (144). Therefore NRF2-addicted cancer cells are far more vulnerable to NRF2 inhibitors than other cells with low NRF2 expression, which has been shown experimentally, as knockdown or deletion of NRF2 in prostate cancer cell lines with high NRF2 expression impedes their ability to grow tumors in vivo (145). More recently, the Zhang lab reported findings suggesting that activation of NRF2 accelerates metastasis of existing tumors in mice (138). The prevalence of constitutive NRF2 upregulation (high expression) in lung tumors (>30%), coupled with genetic and chemical biological data, argues that attenuating NRF2 signaling in NRF2-addicted tumors could be used to slow down cancer progression, block metastasis, and sensitize cancer cells to chemotherapeutic drugs. Furthermore, specific NRF2 inhibition preferentially kills NRF2-addicted cancer cells without harming normal cells, because NRF2 expression is very low in normal cells under unstressed conditions and NRF2 knockout cells and mice develop normally, indicating NRF2 is not essential for normal development and growth. For this reason, researchers have been searching for NRF2 inhibitors (Figure 6), but as indicated in the introduction, this is a daunting task.
Figure 6.

Structures of NRF2 pathway inhibitor molecules which disrupt NRF2 signaling and decrease NRF2-target gene transcription.
Below, we review what we consider to be the most important advances in the discovery of NRF2 inhibitors. Although a direct binding NRF2 inhibitor has been reported, it has not been validated by other research groups and so at this point, there are some pathway inhibitors that have been described, but no bona fide NRF2 inhibitor.
5. NRF2 inhibitors
5.1. All trans retinoic acid (ATRA)
Retinoic acid receptor alpha (RARα) agonists, including all-trans retinoic acid (ATRA), have been identified as antagonists of NRF2-mediated gene transcription (146). RARα agonists relieve co-repression of the nuclear receptor RAR-retinoid x receptor (RXR) heterodimer, subsequently permitting the RAR-RXR heterodimer to interact with transcription factors, ultimately modulating transcriptional output (transrepression vs. transactivation) (147–149). In AREc32 cells (MCF7 cells stably transfected with a plasmid encoding an ARE-driven luciferase reporter), ATRA (Figure 6) treatment decreased induction of tBHQ-mediated ARE-luciferase activity (150, 151). ATRA treatment in these cells also decreased NRF2 target gene AKR1C1/C2 mRNA and protein levels, indicating a partial blockade of NRF2-mediated target gene transcription. NRF2 nuclear localization post tBHQ treatment was not affected by ATRA cotreatment. AREc32 cells treated with four different RAR agonists showed a dose dependent decrease in ARE luciferase activity. Conversely, treatment with RAR antagonists or RAR siRNA increased NRF2-target gene expression after tBHQ exposure. Immunoprecipitation experiments indicated direct interaction between RARα and NRF2 (Figure 7), however tBHQ and ATRA treatment reduced NRF2 binding to ARE by electrophoretic mobility shift assay (EMSA). Vitamin A deficiency (VAD) in mice increased NRF2-target gene expression compared to control mice, whereas VAD mice treated with ATRA abrogated increases in NRF2 target gene expression. Conversely, NRF2-target gene expression was not modulated by vitamin A deficiency or ATRA in NRF2 null mice, indicating agonist-induced activation of RARα is responsible for changes in the NRF2 signaling pathway (151).
Figure 7.
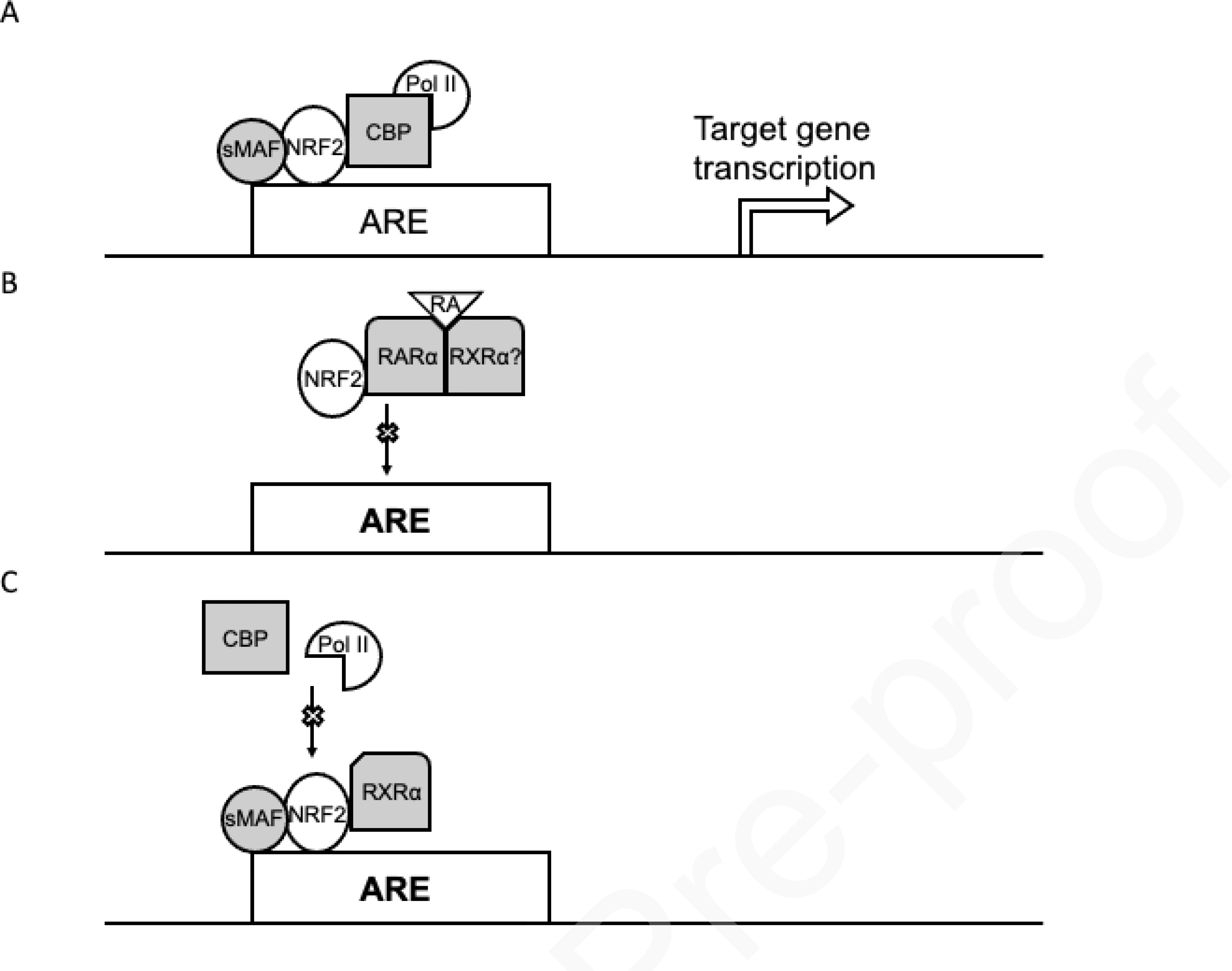
RARα-retinoid agonist and RXRα interact with NRF2 and decrease NRF2-target gene transcription. (A) Recruitment of transactivation machinery after NRF2/sMAF binding to ARE leads to transcription of NRF2-target genes. (B) NRF2-RARα-retinoid agonist interaction decreases NRF2 binding to AREs and reduces NRF2-target gene transcription. (C) NRF2-RXRα interaction does not prevent binding to AREs, but decreases BCP/RNA polymerase II loading, ultimately diminishing transactivation and decreasing NRF2-target gene transcription.
Interestingly, genetic silencing of RXRα in A549 cells increased NRF2-target gene expression and protein product levels, whereas these levels were decreased with RXRα overexpression. Overexpression of RXRα was found to sensitize A549 cells to traditional chemotherapies. NRF2 immunoprecipitation revealed interaction between the RXRα DNA-binding domain (DBD) and NRF2 residues 209–338 (Neh7). Contrary to the interaction with RARα, the NRF2-RXRα PPI is ligand independent. tBHQ exposed AREc32 and A549 cells increased NRF2/RXRα recruitment to multiple NRF2 target gene AREs, however NRF2 predepletion abolished RXRα binding to these same AREs. RXRα overexpression decreased HO-1 levels compared to empty vector; RNA polymerase II and CBP loading detected at the HO-1 ARE were also decreased, indicating RXRα does not interfere with NRF2 ARE binding but acts as a transrepressor (146). RXRα interaction with Neh7 (Figure 2) may sterically hinder recruitment of transactivation machinery to the adjacent Neh4/5 domain, leading to suppression of NRF2-target gene transcription (62, 152, 153). Overall, the RAR/RXR system has been shown to modulate NRF2-target gene transcription (146). Adoption of vitamin A analog supplementation to lessen the burden of NRF2 pathway activation and mediated resistance in the treatment of select cancer types is not likely to be adopted due to modest effects seen at physiological doses.
5.2. Brusatol
The discovery of brusatol represents the first intentional discovery of an NRF2 pathway inhibitor driven by the dark side of NRF2 hypothesis. Isolated from Brucea javanica “Macassar kernels” (154), brusatol was found to inhibit ARE-luciferase activity in stably transfected MDA-MB-231 cells (14). NRF2 in A549 cells declined to a near undetectable level two hours after exposure to brusatol but returned to baseline within three days even in the presence of inhibitor. A decrease in NRF2-target gene mRNA and protein product levels were observed after brusatol treatment in multiple cell lines with high basal NRF2 levels, indicating disruption of the NRF2 pathway. Cotreatment of multiple NSCLC cell lines with platinum-based compounds and brusatol led to increased intracellular platinum levels greater than platinum treatment alone. This effect may be explained by impaired platinum efflux due to a decrease in expression of NRF2-mediated multidrug-resistance-associated protein genes (14, 155). The combination of brusatol and cisplatin decreased clonogenic survival as well as tumor size in NSCLC xenograft models. Surprisingly, it was found that restoring functional KEAP1 within A549 cells abrogated any additive effect of brusatol with cisplatin as a cotreatment (14).
It has been since uncovered that brusatol likely does not interact directly with NRF2 and that fluorescently labeled brusatol was found to localize to the endoplasmic reticulum (156, 157). Gene expression changes in cells treated with cycloheximide or brusatol were found to have significant overlap. Additionally, brusatol inhibits cap-dependent and cap-independent translation at working concentrations, depleting NRF2 and other proteins with short half-lives (156–158). Brusatol is a potent, small-molecule inhibitor of the NRF2 pathway, however because of its mechanism of action, is unlikely to be explored further as a selective NRF2-pathway inhibitor, but the use of this probe has revealed many important aspects of NRF2 biology.
5.3. Halofuginone
In A549 ARE-luciferase cells, a high throughput screen was conducted by the Yamamoto group to identify inhibitors of the NRF2 pathway. From the hit molecule febrifugine, a less toxic derivative, halofuginone, was used to explore the extent of NRF2 pathway inhibition as well as the mechanism of action for this compound type. In two NRF2-addicted cell lines, halofuginone decreased NRF2 levels in a time and dose-dependent manner. Additionally, in the above cell lines, NRF2 mRNA levels decreased in a dose-dependent manner when treated with Halofuginone (159). Halofuginone is known to inhibit prolyl-tRNA synthetase and supplementing NRF2-addicted cancer cells with proline after exposure to halofuginone in fact restored basal NRF2 levels (159, 160). Halofuginone sensitized NRF2 addicted cancers to traditional chemotherapeutics, however cancers not addicted to NRF2 did not undergo sensitization. Cotreatment of KYSE70 mouse xenografts with halofuginone and traditional chemotherapeutics decreased mouse tumor volume greater than either agent alone without significant changes in mouse liver or kidney function (159). Overall, halofuginone acts as a general translation inhibitor, affecting most proteins with short half-lives, leading to the depletion of NRF2, similarly to brusatol.
5.4. ML385
A compound which was reported to directly bind NRF2 was discovered in a 350,000+ molecule screen by the Biswal group. Their approach involved using an in vitro NRF2-dependent firefly luciferase (Fluc) assay containing an NRF2-specific ARE enhancer sequence-Fluc reporter construct in stably transfected A549 cells. Hit molecules (measured as a decrease in Fluc activity) were verified in multiple human lung adenocarcinoma cell lines containing the above Fluc reporter system. All three cell lines lacked expression of KEAP1 or harbored mutations in KEAP1, both of which resulted in the accumulation of NRF2, enhanced transcription of NRF2 target genes, and increased Fluc activity. Hits were counter screened against native luciferase and a CMV-promoted Fluc reporter system to help rule out Fluc inhibitors and general transcriptional modulators respectively (161).
ML385, a derivative of a hit from the above screen, was found to reduce NRF2-dependent gene expression (including NRF2 itself) as measured by qRT-PCR in a time, dose, and KEAP1-status dependent manner. Protein product expression, regulated by NRF2 target genes, was also decreased in the presence of ML385. Furthermore, glutathione levels and cellular antioxidation capacity were reduced by ML385 in a dose-dependent manner. Using several NSCLC cell lines with perturbed KEAP1-NRF2 regulation, ML385 demonstrated cytotoxicity as a single agent or an additive effect in combination with platinum-containing compounds and other traditional chemotherapeutics in vitro and in vivo (161).
ML385 was found to disrupt the interaction between NRF2-MAFG and fluorescently labeled ARE-DNA as measured by fluorescence polarization, however it remains unknown whether ML385 prevents NRF2-MAFG heterodimerization or NRF2-MAFG heterodimer interaction with DNA (161). Paradoxically, in A549 and H460 cell lines expressing functional KEAP1 (knock-in) exposed to ML385, enhanced clonogenic survival was noted (161, 162). ML385 linked with biotin was found by pulldown to interact with full length NRF2 and the Neh1 domain of NRF2, but not with NRF2ΔNeh1. At working concentrations of ML385, Renilla luciferase (Rluc) reporter expression (driven from a non-NRF2 ARE promoter) was significantly decreased and may indicate potential cytotoxicity unrelated to the inhibition of the NRF2 pathway (161). As a strict molecular probe, ML385 requires further validation as a potent, selective NRF2 inhibitor.
6. Conclusions and future directions
Like many cellular protective pathways, the NRF2 pathway has a dark and a light side. The light side protects cells from environmental stressors and has been intentionally activated to protect organisms form a variety of maladies. Of particular interest in this arena are compounds that disrupt the protein-protein interaction between the ETGE or DLG domain of NRF2 and KEAP1. This approach has been undertaken due to the promiscuous nature of the canonical electrophilic activators. Although this has yet to produce a clinical compound, it is in its infancy and hope remains. The compounds discussed here offer potential for specific NRF2 activators with good pharmacokinetic parameters that can be used to treat a variety of malignancies. However, given the success of DMF, it has been postulated that the optimal use of an NRF2 activating compound might be in neurological disease and therefore a compound that crosses the blood brain barrier (BBB) is being sought. So far, the compounds of this class reported, despite being quite potent, lack BBB penetrance. KI-696 was evaluated in a variety of cellular models and in vivo, but no analysis of neuro activity has been reported.
The dark side of NRF2 comes from the misregulation of the NRF2 pathway, leading to protection of malignant cells. This has been shown to facilitate cancer development, progression, metastasis, and chemoresistance. It has also been shown that inhibition of NRF2 in normal cells is not lethal, offering a potential therapeutic window when treating NRF2-addicted cancers. For this reason, researchers have been searching for NRF2 inhibitors, but this has, to date, produced no direct targeting NRF2 compounds that have been thoroughly verified. Brusatol remains the most important compound in the field, but the complex architecture of the quassinoids likely prohibits chemical optimization of this class. Halofuginone is more tractable synthetically, which leads to the possibility of chemical optimization, but it remains this is not NRF2 specific and careful evaluation of toxicity would be important if this class of compounds were to be developed. It therefore seems a continued search for direct NRF2 targeting compounds is important for the field. Generally, targeting the non-receptor transcription factors has produced no clinical compounds and many have concluded this class of transcription factors are undruggable. However, there have been some recent glimmers of possibility, especially in the case of MYC. There have also been some important advances in the development of proteolysis targeting chimeras (PROTACs), which can target proteins for degradation by binding the protein of interest and recruiting the UPS machinery (163–165). If a potent NRF2 binder were to obtain, it is certainly possible this strategy could produce the desired result. The NRF2 field has been growing rapidly since its initial discovery with thousands of papers published every year. The importance of this regulatory axis and the potential for important clinical advances argues for the ongoing search of potent, clinically viable NRF2 modulating compounds for cancer therapy and other malignancies.
Acknowledgements
This work was supported by R01 ES031463 to E.C., R01 ES023758 to E.C. and D.D.Z., R35 ES031575 to D.D.Z., and T32 GM008804 to J.S.
Footnotes
Conflict of interest
Eli Chapman is a cofounder of BioEL, Inc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Todd R, Wong DT. Oncogenes. Anticancer research. 1999;19(6a):4729–46. Epub 2000/03/04. [PubMed] [Google Scholar]
- 2.Chen A, Koehler AN. Transcription Factor Inhibition: Lessons Learned and Emerging Targets. Trends in molecular medicine. 2020;26(5):508–18. doi: 10.1016/j.molmed.2020.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wright PE, Dyson HJ. Intrinsically disordered proteins in cellular signalling and regulation. Nature Reviews Molecular Cell Biology. 2015;16(1):18–29. doi: 10.1038/nrm3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boike L, Cioffi AG, Majewski FC, Co J, Henning NJ, Jones MD, Liu G, McKenna JM, Tallarico JA, Schirle M, Nomura DK. Discovery of a Functional Covalent Ligand Targeting an Intrinsically Disordered Cysteine within MYC. Cell chemical biology. 2021;28(1):4–13.e7. Epub 2020/09/24. doi: 10.1016/j.chembiol.2020.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou H, Bai L, Xu R, Zhao Y, Chen J, McEachern D, Chinnaswamy K, Wen B, Dai L, Kumar P, Yang CY, Liu Z, Wang M, Liu L, Meagher JL, Yi H, Sun D, Stuckey JA, Wang S. Structure-Based Discovery of SD-36 as a Potent, Selective, and Efficacious PROTAC Degrader of STAT3 Protein. Journal of medicinal chemistry. 2019;62(24):11280–300. Epub 2019/11/21. doi: 10.1021/acs.jmedchem.9b01530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen J, Bai L, Bernard D, Nikolovska-Coleska Z, Gomez C, Zhang J, Yi H, Wang S. Structure-Based Design of Conformationally Constrained, Cell-Permeable STAT3 Inhibitors. ACS medicinal chemistry letters. 2010;1(2):85–9. Epub 2010/07/03. doi: 10.1021/ml100010j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iconaru LI, Ban D, Bharatham K, Ramanathan A, Zhang W, Shelat AA, Zuo J, Kriwacki RW. Discovery of Small Molecules that Inhibit the Disordered Protein, p27(Kip1). Scientific reports. 2015;5:15686. Epub 2015/10/29. doi: 10.1038/srep15686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klein AM, de Queiroz RM, Venkatesh D, Prives C. The roles and regulation of MDM2 and MDMX: it is not just about p53. Genes Dev. 2021. Epub 2021/04/24. doi: 10.1101/gad.347872.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kotelevets L, Scott MGH, Chastre E. Targeting PTEN in Colorectal Cancers. Adv Exp Med Biol. 2018;1110:55–73. Epub 2019/01/10. doi: 10.1007/978-3-030-02771-1_5. [DOI] [PubMed] [Google Scholar]
- 10.Liu Y, Hu X, Han C, Wang L, Zhang X, He X, Lu X. Targeting tumor suppressor genes for cancer therapy. Bioessays. 2015;37(12):1277–86. Epub 2015/10/08. doi: 10.1002/bies.201500093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mazhar S, Taylor SE, Sangodkar J, Narla G. Targeting PP2A in cancer: Combination therapies. Biochim Biophys Acta Mol Cell Res. 2019;1866(1):51–63. Epub 2018/11/08. doi: 10.1016/j.bbamcr.2018.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gañán-Gómez I, Wei Y, Yang H, Boyano-Adánez MC, García-Manero G. Oncogenic functions of the transcription factor Nrf2. Free radical biology & medicine. 2013;65:750–64. Epub 2013/07/04. doi: 10.1016/j.freeradbiomed.2013.06.041. [DOI] [PubMed] [Google Scholar]
- 13.Wang XJ, Sun Z, Villeneuve NF, Zhang S, Zhao F, Li Y, Chen W, Yi X, Zheng W, Wondrak GT, Wong PK, Zhang DD. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis. 2008;29(6):1235–43. Epub 2008/04/17. doi: 10.1093/carcin/bgn095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ren D, Villeneuve NF, Jiang T, Wu T, Lau A, Toppin HA, Zhang DD. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc Natl Acad Sci U S A. 2011;108(4):1433–8. Epub 2011/01/06. doi: 10.1073/pnas.1014275108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Satoh H, Moriguchi T, Takai J, Ebina M, Yamamoto M. Nrf2 Prevents Initiation but Accelerates Progression through the Kras Signaling Pathway during Lung Carcinogenesis. Cancer research. 2013;73(13):4158. doi: 10.1158/0008-5472.CAN-12-4499. [DOI] [PubMed] [Google Scholar]
- 16.Satoh H, Moriguchi T, Saigusa D, Baird L, Yu L, Rokutan H, Igarashi K, Ebina M, Shibata T, Yamamoto M. NRF2 Intensifies Host Defense Systems to Prevent Lung Carcinogenesis, but After Tumor Initiation Accelerates Malignant Cell Growth. Cancer Res. 2016;76(10):3088–96. Epub 2016/03/30. doi: 10.1158/0008-5472.Can-15-1584. [DOI] [PubMed] [Google Scholar]
- 17.Bauer AK, Cho HY, Miller-Degraff L, Walker C, Helms K, Fostel J, Yamamoto M, Kleeberger SR. Targeted deletion of Nrf2 reduces urethane-induced lung tumor development in mice. PLoS One. 2011;6(10):e26590. Epub 2011/11/01. doi: 10.1371/journal.pone.0026590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Wang Y, Zhang Z, Park JY, Guo D, Liao H, Yi X, Zheng Y, Zhang D, Chambers SK, Zheng W. Mechanism of progestin resistance in endometrial precancer/cancer through Nrf2-AKR1C1 pathway. Oncotarget. 2016;7(9):10363–72. Epub 2016/01/30. doi: 10.18632/oncotarget.7004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang DD, Chapman E. The role of natural products in revealing NRF2 function. Natural product reports. 2020;37(6):797–826. doi: 10.1039/C9NP00061E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dodson M, de la Vega MR, Cholanians AB, Schmidlin CJ, Chapman E, Zhang DD. Modulating NRF2 in Disease: Timing Is Everything. Annual review of pharmacology and toxicology. 2019;59:555–75. Epub 2018/09/27. doi: 10.1146/annurev-pharmtox-010818-021856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar H, Kim I-S, More SV, Kim B-W, Choi D-K. Natural product-derived pharmacological modulators of Nrf2/ARE pathway for chronic diseases. Natural product reports. 2014;31(1):109–39. doi: 10.1039/C3NP70065H. [DOI] [PubMed] [Google Scholar]
- 22.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. Epub 2006/09/14. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 23.Cuadrado A, Rojo AI, Wells G, Hayes JD, Cousin SP, Rumsey WL, Attucks OC, Franklin S, Levonen AL, Kensler TW, Dinkova-Kostova AT. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nature reviews Drug discovery. 2019;18(4):295–317. Epub 2019/01/06. doi: 10.1038/s41573-018-0008-x. [DOI] [PubMed] [Google Scholar]
- 24.Zhu J, Wang H, Chen F, Fu J, Xu Y, Hou Y, Kou HH, Zhai C, Nelson MB, Zhang Q, Andersen ME, Pi J. An overview of chemical inhibitors of the Nrf2-ARE signaling pathway and their potential applications in cancer therapy. Free Radic Biol Med. 2016;99:544–56. Epub 2016/10/23. doi: 10.1016/j.freeradbiomed.2016.09.010. [DOI] [PubMed] [Google Scholar]
- 25.Itoh K, Tong KI, Yamamoto M. Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic Biol Med. 2004;36(10):1208–13. Epub 2004/04/28. doi: 10.1016/j.freeradbiomed.2004.02.075. [DOI] [PubMed] [Google Scholar]
- 26.Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci. 2014;39(4):199–218. Epub 2014/03/22. doi: 10.1016/j.tibs.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 27.Harder B, Jiang T, Wu T, Tao S, Rojo de la Vega M, Tian W, Chapman E, Zhang DD. Molecular mechanisms of Nrf2 regulation and how these influence chemical modulation for disease intervention. Biochem Soc Trans. 2015;43(4):680–6. Epub 2015/11/10. doi: 10.1042/bst20150020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol. 2004;24(24):10941–53. Epub 2004/12/02. doi: 10.1128/mcb.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24(16):7130–9. Epub 2004/07/30. doi: 10.1128/mcb.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23(22):8137–51. Epub 2003/10/31. doi: 10.1128/mcb.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Itoh K, Mimura J, Yamamoto M. Discovery of the negative regulator of Nrf2, Keap1: a historical overview. Antioxid Redox Signal. 2010;13(11):1665–78. Epub 2010/05/08. doi: 10.1089/ars.2010.3222. [DOI] [PubMed] [Google Scholar]
- 32.Kansanen E, Kuosmanen SM, Leinonen H, Levonen AL. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013;1(1):45–9. Epub 2013/09/12. doi: 10.1016/j.redox.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J Biol Chem. 2004;279(30):31556–67. Epub 2004/05/15. doi: 10.1074/jbc.M403061200. [DOI] [PubMed] [Google Scholar]
- 34.Tong KI, Padmanabhan B, Kobayashi A, Shang C, Hirotsu Y, Yokoyama S, Yamamoto M. Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol Cell Biol. 2007;27(21):7511–21. Epub 2007/09/06. doi: 10.1128/mcb.00753-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tao S, Liu P, Luo G, Rojo de la Vega M, Chen H, Wu T, Tillotson J, Chapman E, Zhang DD. p97 Negatively Regulates NRF2 by Extracting Ubiquitylated NRF2 from the KEAP1-CUL3 E3 Complex. Molecular and cellular biology. 2017. Epub 2017/01/25. doi: 10.1128/mcb.00660-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suzuki T, Muramatsu A, Saito R, Iso T, Shibata T, Kuwata K, Kawaguchi SI, Iwawaki T, Adachi S, Suda H, Morita M, Uchida K, Baird L, Yamamoto M. Molecular Mechanism of Cellular Oxidative Stress Sensing by Keap1. Cell Rep. 2019;28(3):746–58.e4. Epub 2019/07/18. doi: 10.1016/j.celrep.2019.06.047. [DOI] [PubMed] [Google Scholar]
- 37.Dinkova-Kostova AT, Kostov RV, Canning P. Keap1, the cysteine-based mammalian intracellular sensor for electrophiles and oxidants. Arch Biochem Biophys. 2017;617:84–93. Epub 2016/08/09. doi: 10.1016/j.abb.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McMahon M, Lamont DJ, Beattie KA, Hayes JD. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proc Natl Acad Sci U S A. 2010;107(44):18838–43. Epub 2010/10/20. doi: 10.1073/pnas.1007387107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang M, An C, Gao Y, Leak RK, Chen J, Zhang F. Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog Neurobiol. 2013;100:30–47. Epub 2012/10/03. doi: 10.1016/j.pneurobio.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rachakonda G, Xiong Y, Sekhar KR, Stamer SL, Liebler DC, Freeman ML. Covalent modification at Cys151 dissociates the electrophile sensor Keap1 from the ubiquitin ligase CUL3. Chem Res Toxicol. 2008;21(3):705–10. Epub 2008/02/07. doi: 10.1021/tx700302s. [DOI] [PubMed] [Google Scholar]
- 41.Gao L, Wang J, Sekhar KR, Yin H, Yared NF, Schneider SN, Sasi S, Dalton TP, Anderson ME, Chan JY, Morrow JD, Freeman ML. Novel n-3 fatty acid oxidation products activate Nrf2 by destabilizing the association between Keap1 and Cullin3. J Biol Chem. 2007;282(4):2529–37. Epub 2006/11/28. doi: 10.1074/jbc.M607622200. [DOI] [PubMed] [Google Scholar]
- 42.Kobayashi A, Kang MI, Watai Y, Tong KI, Shibata T, Uchida K, Yamamoto M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol Cell Biol. 2006;26(1):221–9. Epub 2005/12/16. doi: 10.1128/mcb.26.1.221-229.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Malhotra D, Portales-Casamar E, Singh A, Srivastava S, Arenillas D, Happel C, Shyr C, Wakabayashi N, Kensler TW, Wasserman WW, Biswal S. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010;38(17):5718–34. Epub 2010/05/13. doi: 10.1093/nar/gkq212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chan K, Lu R, Chang JC, Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci U S A. 1996;93(24):13943–8. Epub 1996/11/26. doi: 10.1073/pnas.93.24.13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Itoh K, Igarashi K, Hayashi N, Nishizawa M, Yamamoto M. Cloning and characterization of a novel erythroid cell-derived CNC family transcription factor heterodimerizing with the small Maf family proteins. Mol Cell Biol. 1995;15(8):4184–93. Epub 1995/08/01. doi: 10.1128/mcb.15.8.4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fujii Y, Shimizu T, Toda T, Yanagida M, Hakoshima T. Structural basis for the diversity of DNA recognition by bZIP transcription factors. Nature structural biology. 2000;7(10):889–93. doi: 10.1038/82822. [DOI] [PubMed] [Google Scholar]
- 47.Ellenberger TE, Brandl CJ, Struhl K, Harrison SC. The GCN4 basic region leucine zipper binds DNA as a dimer of uninterrupted α Helices: Crystal structure of the protein-DNA complex. Cell. 1992;71(7):1223–37. doi: 10.1016/S0092-8674(05)80070-4. [DOI] [PubMed] [Google Scholar]
- 48.Yin Z, Machius M, Nestler EJ, Rudenko G. Activator Protein-1: redox switch controlling structure and DNA-binding. Nucleic acids research. 2017;45(19):11425–36. Epub 2017/10/06. doi: 10.1093/nar/gkx795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kurokawa H, Motohashi H, Sueno S, Kimura M, Takagawa H, Kanno Y, Yamamoto M, Tanaka T. Structural basis of alternative DNA recognition by Maf transcription factors. Molecular and cellular biology. 2009;29(23):6232–44. Epub 2009/10/03. doi: 10.1128/mcb.00708-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fukutomi T, Takagi K, Mizushima T, Ohuchi N, Yamamoto M. Kinetic, thermodynamic, and structural characterizations of the association between Nrf2-DLGex degron and Keap1. Mol Cell Biol. 2014;34(5):832–46. Epub 2013/12/25. doi: 10.1128/mcb.01191-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lo SC, Li X, Henzl MT, Beamer LJ, Hannink M. Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. Embo j. 2006;25(15):3605–17. Epub 2006/08/05. doi: 10.1038/sj.emboj.7601243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Venugopal R, Jaiswal AK. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc Natl Acad Sci U S A. 1996;93(25):14960–5. Epub 1996/12/10. doi: 10.1073/pnas.93.25.14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Igarashi K, Kataoka K, Itoh K, Hayashi N, Nishizawa M, Yamamoto M. Regulation of transcription by dimerization of erythroid factor NF-E2 p45 with small Maf proteins. Nature. 1994;367(6463):568–72. Epub 1994/02/10. doi: 10.1038/367568a0. [DOI] [PubMed] [Google Scholar]
- 54.Igarashi K, Itoh K, Motohashi H, Hayashi N, Matuzaki Y, Nakauchi H, Nishizawa M, Yamamoto M. Activity and expression of murine small Maf family protein MafK. J Biol Chem. 1995;270(13):7615–24. Epub 1995/03/31. doi: 10.1074/jbc.270.13.7615. [DOI] [PubMed] [Google Scholar]
- 55.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236(2):313–22. Epub 1997/07/18. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 56.Ellenberger T Getting a grip on DNA recognition: structures of the basic region leucine zipper, and the basic region helix-loop-helix DNA-binding domains. Current Opinion in Structural Biology. 1994;4(1):12–21. doi: 10.1016/S0959-440X(94)90054-X. [DOI] [Google Scholar]
- 57.Amoutzias GD, Robertson DL, Van de Peer Y, Oliver SG. Choose your partners: dimerization in eukaryotic transcription factors. Trends Biochem Sci. 2008;33(5):220–9. Epub 2008/04/15. doi: 10.1016/j.tibs.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 58.Amoutzias GD, Veron AS, Weiner J 3rd, Robinson-Rechavi M, Bornberg-Bauer E, Oliver SG, Robertson DL. One billion years of bZIP transcription factor evolution: conservation and change in dimerization and DNA-binding site specificity. Mol Biol Evol. 2007;24(3):827–35. Epub 2006/12/30. doi: 10.1093/molbev/msl211. [DOI] [PubMed] [Google Scholar]
- 59.Nioi P, Nguyen T, Sherratt PJ, Pickett CB. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol Cell Biol. 2005;25(24):10895–906. Epub 2005/11/30. doi: 10.1128/mcb.25.24.10895-10906.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang J, Hosoya T, Maruyama A, Nishikawa K, Maher JM, Ohta T, Motohashi H, Fukamizu A, Shibahara S, Itoh K, Yamamoto M. Nrf2 Neh5 domain is differentially utilized in the transactivation of cytoprotective genes. Biochem J. 2007;404(3):459–66. Epub 2007/02/23. doi: 10.1042/bj20061611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ki SH, Cho IJ, Choi DW, Kim SG. Glucocorticoid receptor (GR)-associated SMRT binding to C/EBPbeta TAD and Nrf2 Neh4/5: role of SMRT recruited to GR in GSTA2 gene repression. Mol Cell Biol. 2005;25(10):4150–65. Epub 2005/05/05. doi: 10.1128/mcb.25.10.4150-4165.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Katoh Y, Itoh K, Yoshida E, Miyagishi M, Fukamizu A, Yamamoto M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells. 2001;6(10):857–68. Epub 2001/10/31. doi: 10.1046/j.1365-2443.2001.00469.x. [DOI] [PubMed] [Google Scholar]
- 63.Wu T, Zhao F, Gao B, Tan C, Yagishita N, Nakajima T, Wong PK, Chapman E, Fang D, Zhang DD. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 2014;28(7):708–22. Epub 2014/03/19. doi: 10.1101/gad.238246.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rojo AI, Medina-Campos ON, Rada P, Zúñiga-Toalá A, López-Gazcón A, Espada S, Pedraza-Chaverri J, Cuadrado A. Signaling pathways activated by the phytochemical nordihydroguaiaretic acid contribute to a Keap1-independent regulation of Nrf2 stability: Role of glycogen synthase kinase-3. Free Radic Biol Med. 2012;52(2):473–87. Epub 2011/12/07. doi: 10.1016/j.freeradbiomed.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 65.Rada P, Rojo AI, Chowdhry S, McMahon M, Hayes JD, Cuadrado A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol. 2011;31(6):1121–33. Epub 2011/01/20. doi: 10.1128/mcb.01204-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cuadrado A Structural and functional characterization of Nrf2 degradation by glycogen synthase kinase 3/β-TrCP. Free radical biology & medicine. 2015;88(Pt B):147–57. Epub 2015/05/06. doi: 10.1016/j.freeradbiomed.2015.04.029. [DOI] [PubMed] [Google Scholar]
- 67.Chowdhry S, Zhang Y, McMahon M, Sutherland C, Cuadrado A, Hayes JD. Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene. 2013;32(32):3765–81. Epub 2012/09/12. doi: 10.1038/onc.2012.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang H, Liu K, Geng M, Gao P, Wu X, Hai Y, Li Y, Li Y, Luo L, Hayes JD, Wang XJ, Tang X. RXRα inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer research. 2013;73(10):3097–108. Epub 2013/04/25. doi: 10.1158/0008-5472.Can-12-3386. [DOI] [PubMed] [Google Scholar]
- 69.Huggins C, Deuel TF, Fukunishi R. PROTECTION OF ADRENAL CORTEX BY HYDROCARBONS AGAINST INJURY FROM 7,12-DIMETHYLBENZ(A)ANTHRACENE. Biochemische Zeitschrift. 1963;338:106–13. Epub 1963/01/01. [PubMed] [Google Scholar]
- 70.Huggins C, Ford E, Fukunishi R, Jensen EV. AROMATIC-INDUCED PREVENTION OF FETAL TOXICITY OF 7,12-DIMETHYLBENZ(ALPHA)ANTHRACENE. The Journal of experimental medicine. 1964;119:943–54. Epub 1964/01/01. doi: 10.1084/jem.119.6.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huggins C, Fukunishi R. INDUCED PROTECTION OF ADRENAL CORTEX AGAINST 7,12-DIMETHYLBENZ(ALPHA)ANTHRACENE. INFLUENCE OF ETHIONINE. INDUCTION OF MENADIONE REDUCTASE. INCORPORATION OF THYMIDINE-H3. The Journal of experimental medicine. 1964;119:923–42. Epub 1964/01/01. doi: 10.1084/jem.119.6.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huggins C, Fukunishi R. MOLECULAR STRUCTURE OF AROMATICS RELATED TO THEIR ABILITY TO INDUCE ADRENAL PROTECTION. Arzneimittel-Forschung. 1964;14:834–6. Epub 1964/07/01. [PubMed] [Google Scholar]
- 73.Huggins C, Grand L, Fukunishi R. AROMATIC INFLUENCES ON THE YIELDS OF MAMMARY CANCERS FOLLOWING ADMINISTRATION OF 7,12-DIMETHYLBENZ(A)ANTHRACENE. Proc Natl Acad Sci U S A. 1964;51:737–42. Epub 1964/05/01. doi: 10.1073/pnas.51.5.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Loub WD, Wattenberg LW, Davis DW. Aryl hydrocarbon hydroxylase induction in rat tissues by naturally occurring indoles of cruciferous plants. Journal of the National Cancer Institute. 1975;54(4):985–8. Epub 1975/04/01. [PubMed] [Google Scholar]
- 75.Osterberg KA, Wattenberg LW. ENZYME INDUCTION IN IMMATURE GLIA. Proceedings of the Society for Experimental Biology and Medicine Society for Experimental Biology and Medicine (New York, NY). 1965;118:477–9. Epub 1965/02/01. doi: 10.3181/00379727-118-29879. [DOI] [PubMed] [Google Scholar]
- 76.Wattenberg LW. Chemoprophylaxis of carcinogenesis: a review. Cancer research. 1966;26(7):1520–6. Epub 1966/07/01. [PubMed] [Google Scholar]
- 77.Wattenberg LW. Carcinogen-detoxifying mechanisms in the gastrointestinal tract. Gastroenterology. 1966;51(5):932–5. Epub 1966/11/01. [PubMed] [Google Scholar]
- 78.Wattenberg LW. Inhibition of carcinogenic and toxic effects of polycyclic hydrocarbons by phenolic antioxidants and ethoxyquin. Journal of the National Cancer Institute. 1972;48(5):1425–30. Epub 1972/05/01. [PubMed] [Google Scholar]
- 79.Wattenberg LW. Inhibition of chemical carcinogen-induced pulmonary neoplasia by butylated hydroxyanisole. Journal of the National Cancer Institute. 1973;50(6):1541–4. Epub 1973/06/01. doi: 10.1093/jnci/50.6.1541. [DOI] [PubMed] [Google Scholar]
- 80.Wattenberg LW. Inhibition of carcinogenic and toxic effects of polycyclic hydrocarbons by several sulfur-containing compounds. Journal of the National Cancer Institute. 1974;52(5):1583–7. Epub 1974/05/01. doi: 10.1093/jnci/52.5.1583. [DOI] [PubMed] [Google Scholar]
- 81.Wattenberg LW. Potential inhibitors of colon carcinogenesis. The American journal of digestive diseases. 1974;19(10):947–53. Epub 1974/10/01. doi: 10.1007/bf01076221. [DOI] [PubMed] [Google Scholar]
- 82.Wattenberg LW. Inhibition of dimethylhydrazine-induced neoplasia of the large intestine by disulfiram. Journal of the National Cancer Institute. 1975;54(4):1005–6. Epub 1975/04/01. doi: 10.1093/jnci/54.4.1005. [DOI] [PubMed] [Google Scholar]
- 83.Wattenberg LW. Effects of dietary constituents on the metabolism of chemical carcinogens. Cancer research. 1975;35(11 Pt. 2):3326–31. Epub 1975/11/01. [PubMed] [Google Scholar]
- 84.Wattenberg LW. Inhibitors of chemical carcinogenesis. Advances in cancer research. 1978;26:197–226. Epub 1978/01/01. [DOI] [PubMed] [Google Scholar]
- 85.Wattenberg LW, Leong JL. EFFECTS OF PHENOTHIAZINES ON PROTECTIVE SYSTEMS AGAINST POLYCYCLIC HYDROCARBONS. Cancer research. 1965;25:365–70. Epub 1965/04/01. [PubMed] [Google Scholar]
- 86.Benson AM, Batzinger RP, Ou SY, Bueding E, Cha YN, Talalay P. Elevation of hepatic glutathione S-transferase activities and protection against mutagenic metabolites of benzo(a)pyrene by dietary antioxidants. Cancer research. 1978;38(12):4486–95. Epub 1978/12/01. [PubMed] [Google Scholar]
- 87.Benson AM, Cha YN, Bueding E, Heine HS, Talalay P. Elevation of extrahepatic glutathione S-transferase and epoxide hydratase activities by 2(3)-tert-butyl-4-hydroxyanisole. Cancer research. 1979;39(8):2971–7. Epub 1979/08/01. [PubMed] [Google Scholar]
- 88.Pearson WR, Windle JJ, Morrow JF, Benson AM, Talalay P. Increased synthesis of glutathione S-transferases in response to anticarcinogenic antioxidants. Cloning and measurement of messenger RNA. The Journal of biological chemistry. 1983;258(3):2052–62. Epub 1983/02/10. [PubMed] [Google Scholar]
- 89.Talalay P, Batzinger RP, Benson AM, Bueding E, Cha YN. Biochemical studies on the mechanisms by which dietary antioxidants suppress mutagenic activity. Advances in enzyme regulation. 1978;17:23–36. Epub 1978/01/01. [DOI] [PubMed] [Google Scholar]
- 90.Talalay P, Benson AM. Elevation of quinone reductase activity by anticarcinogenic antioxidants. Advances in enzyme regulation. 1982;20:287–300. Epub 1982/01/01. [DOI] [PubMed] [Google Scholar]
- 91.Prestera T, Zhang Y, Spencer SR, Wilczak CA, Talalay P. The electrophile counterattack response: protection against neoplasia and toxicity. Advances in enzyme regulation. 1993;33:281–96. Epub 1993/01/01. doi: 10.1016/0065-2571(93)90024-8. [DOI] [PubMed] [Google Scholar]
- 92.Dinkova-Kostova AT, Holtzclaw WD, Kensler TW. The role of Keap1 in cellular protective responses. Chem Res Toxicol. 2005;18(12):1779–91. Epub 2005/12/20. doi: 10.1021/tx050217c. [DOI] [PubMed] [Google Scholar]
- 93.Prestera T, Holtzclaw WD, Zhang Y, Talalay P. Chemical and molecular regulation of enzymes that detoxify carcinogens. Proc Natl Acad Sci U S A. 1993;90(7):2965–9. Epub 1993/04/01. doi: 10.1073/pnas.90.7.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hosseini A, Masjedi A, Baradaran B, Hojjat-Farsangi M, Ghalamfarsa G, Anvari E, Jadidi-Niaragh F. Dimethyl fumarate: Regulatory effects on the immune system in the treatment of multiple sclerosis. Journal of cellular physiology. 2019;234(7):9943–55. Epub 2018/12/12. doi: 10.1002/jcp.27930. [DOI] [PubMed] [Google Scholar]
- 95.Saidu NEB, Kavian N, Leroy K, Jacob C, Nicco C, Batteux F, Alexandre J. Dimethyl fumarate, a two-edged drug: Current status and future directions. Medicinal research reviews. 2019;39(5):1923–52. Epub 2019/02/14. doi: 10.1002/med.21567. [DOI] [PubMed] [Google Scholar]
- 96.Montes Diaz G, Hupperts R, Fraussen J, Somers V. Dimethyl fumarate treatment in multiple sclerosis: Recent advances in clinical and immunological studies. Autoimmunity reviews. 2018;17(12):1240–50. Epub 2018/10/15. doi: 10.1016/j.autrev.2018.07.001. [DOI] [PubMed] [Google Scholar]
- 97.Unni S, Deshmukh P, Krishnappa G, Kommu P, Padmanabhan B. Structural insights into the multiple binding modes of Dimethyl Fumarate (DMF) and its analogs to the Kelch domain of Keap1. The FEBS journal. 2020. Epub 2020/07/17. doi: 10.1111/febs.15485. [DOI] [PubMed] [Google Scholar]
- 98.Schulze-Topphoff U, Varrin-Doyer M, Pekarek K, Spencer CM, Shetty A, Sagan SA, Cree BA, Sobel RA, Wipke BT, Steinman L, Scannevin RH, Zamvil SS. Dimethyl fumarate treatment induces adaptive and innate immune modulation independent of Nrf2. Proceedings of the National Academy of Sciences of the United States of America. 2016;113(17):4777–82. Epub 2016/04/15. doi: 10.1073/pnas.1603907113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yore MM, Kettenbach AN, Sporn MB, Gerber SA, Liby KT. Proteomic analysis shows synthetic oleanane triterpenoid binds to mTOR. PLoS One. 2011;6(7):e22862. Epub 2011/08/06. doi: 10.1371/journal.pone.0022862. with chemical synthesis of new triterpenoids and their application in treatment of cancer, as well as in inflammatory diseases, including human kidney disease (patent 6,974,801 issued 1/1/2004). They strongly adhere to the PLoS ONE policy on sharing of data and materials. The Sporn Laboratory has a long history of being extremely free in sharing of materials, with no strings attached, with other laboratories throughout the world. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang DD. Bardoxolone brings Nrf2-based therapies to light. Antioxidants & redox signaling. 2013;19(5):517–8. Epub 2012/12/12. doi: 10.1089/ars.2012.5118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Padmanabhan B, Tong KI, Ohta T, Nakamura Y, Scharlock M, Ohtsuji M, Kang MI, Kobayashi A, Yokoyama S, Yamamoto M. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Molecular cell. 2006;21(5):689–700. Epub 2006/03/02. doi: 10.1016/j.molcel.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 102.Tong KI, Katoh Y, Kusunoki H, Itoh K, Tanaka T, Yamamoto M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Mol Cell Biol. 2006;26(8):2887–900. Epub 2006/04/04. doi: 10.1128/mcb.26.8.2887-2900.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liu P, Tian W, Tao S, Tillotson J, Wijeratne EMK, Gunatilaka AAL, Zhang DD, Chapman E. Non-covalent NRF2 Activation Confers Greater Cellular Protection than Covalent Activation. Cell chemical biology. 2019. Epub 2019/08/14. doi: 10.1016/j.chembiol.2019.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hu L, Magesh S, Chen L, Wang L, Lewis TA, Chen Y, Khodier C, Inoyama D, Beamer LJ, Emge TJ, Shen J, Kerrigan JE, Kong AN, Dandapani S, Palmer M, Schreiber SL, Munoz B. Discovery of a small-molecule inhibitor and cellular probe of Keap1-Nrf2 protein-protein interaction. Bioorganic & medicinal chemistry letters. 2013;23(10):3039–43. Epub 2013/04/09. doi: 10.1016/j.bmcl.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Inoyama D, Chen Y, Huang X, Beamer LJ, Kong AN, Hu L. Optimization of fluorescently labeled Nrf2 peptide probes and the development of a fluorescence polarization assay for the discovery of inhibitors of Keap1-Nrf2 interaction. Journal of biomolecular screening. 2012;17(4):435–47. Epub 2011/12/14. doi: 10.1177/1087057111430124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bresciani A, Missineo A, Gallo M, Cerretani M, Fezzardi P, Tomei L, Cicero DO, Altamura S, Santoprete A, Ingenito R, Bianchi E, Pacifici R, Dominguez C, Munoz-Sanjuan I, Harper S, Toledo-Sherman L, Park LC. Nuclear factor (erythroid-derived 2)-like 2 (NRF2) drug discovery: Biochemical toolbox to develop NRF2 activators by reversible binding of Kelch-like ECH-associated protein 1 (KEAP1). Archives of biochemistry and biophysics. 2017;631:31–41. Epub 2017/08/13. doi: 10.1016/j.abb.2017.08.003. [DOI] [PubMed] [Google Scholar]
- 107.Ergin E, Dogan A, Parmaksiz M, Elcin AE, Elcin YM. Time-Resolved Fluorescence Resonance Energy Transfer [TR-FRET] Assays for Biochemical Processes. Curr Pharm Biotechnol. 2016;17(14):1222–30. Epub 2016/09/09. doi: 10.2174/1389201017666160809164527. [DOI] [PubMed] [Google Scholar]
- 108.Mureddu L, Vuister GW. Simple high-resolution NMR spectroscopy as a tool in molecular biology. The FEBS journal. 2019;286(11):2035–42. Epub 2019/02/02. doi: 10.1111/febs.14771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Canning P, Sorrell FJ, Bullock AN. Structural basis of Keap1 interactions with Nrf2. Free radical biology & medicine. 2015;88(Pt B):101–7. Epub 2015/06/10. doi: 10.1016/j.freeradbiomed.2015.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jnoff E, Albrecht C, Barker JJ, Barker O, Beaumont E, Bromidge S, Brookfield F, Brooks M, Bubert C, Ceska T, Corden V, Dawson G, Duclos S, Fryatt T, Genicot C, Jigorel E, Kwong J, Maghames R, Mushi I, Pike R, Sands ZA, Smith MA, Stimson CC, Courade JP. Binding mode and structure-activity relationships around direct inhibitors of the Nrf2-Keap1 complex. ChemMedChem. 2014;9(4):699–705. Epub 2014/02/08. doi: 10.1002/cmdc.201300525. [DOI] [PubMed] [Google Scholar]
- 111.Marcotte D, Zeng W, Hus JC, McKenzie A, Hession C, Jin P, Bergeron C, Lugovskoy A, Enyedy I, Cuervo H, Wang D, Atmanene C, Roecklin D, Vecchi M, Vivat V, Kraemer J, Winkler D, Hong V, Chao J, Lukashev M, Silvian L. Small molecules inhibit the interaction of Nrf2 and the Keap1 Kelch domain through a non-covalent mechanism. Bioorg Med Chem. 2013;21(14):4011–9. Epub 2013/05/08. doi: 10.1016/j.bmc.2013.04.019. [DOI] [PubMed] [Google Scholar]
- 112.Kask P, Palo K, Fay N, Brand L, Mets U, Ullmann D, Jungmann J, Pschorr J, Gall K. Two-dimensional fluorescence intensity distribution analysis: theory and applications. Biophysical journal. 2000;78(4):1703–13. Epub 2000/03/29. doi: 10.1016/S0006-3495(00)76722-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhuang C, Narayanapillai S, Zhang W, Sham YY, Xing C. Rapid identification of Keap1-Nrf2 small-molecule inhibitors through structure-based virtual screening and hit-based substructure search. Journal of medicinal chemistry. 2014;57(3):1121–6. Epub 2014/01/15. doi: 10.1021/jm4017174. [DOI] [PubMed] [Google Scholar]
- 114.Yasuda D, Yuasa A, Obata R, Nakajima M, Takahashi K, Ohe T, Ichimura Y, Komatsu M, Yamamoto M, Imamura R, Kojima H, Okabe T, Nagano T, Mashino T. Discovery of benzo[g]indoles as a novel class of non-covalent Keap1-Nrf2 protein-protein interaction inhibitor. Bioorganic & medicinal chemistry letters. 2017;27(22):5006–9. Epub 2017/10/19. doi: 10.1016/j.bmcl.2017.10.008. [DOI] [PubMed] [Google Scholar]
- 115.Winkel AF, Engel CK, Margerie D, Kannt A, Szillat H, Glombik H, Kallus C, Ruf S, Gussregen S, Riedel J, Herling AW, von Knethen A, Weigert A, Brune B, Schmoll D. Characterization of RA839, a Noncovalent Small Molecule Binder to Keap1 and Selective Activator of Nrf2 Signaling. The Journal of biological chemistry. 2015;290(47):28446–55. Epub 2015/10/16. doi: 10.1074/jbc.M115.678136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jiang ZY, Lu MC, Xu LL, Yang TT, Xi MY, Xu XL, Guo XK, Zhang XJ, You QD, Sun HP. Discovery of potent Keap1-Nrf2 protein-protein interaction inhibitor based on molecular binding determinants analysis. Journal of medicinal chemistry. 2014;57(6):2736–45. Epub 2014/02/12. doi: 10.1021/jm5000529. [DOI] [PubMed] [Google Scholar]
- 117.Jain AD, Potteti H, Richardson BG, Kingsley L, Luciano JP, Ryuzoji AF, Lee H, Krunic A, Mesecar AD, Reddy SP, Moore TW. Probing the structural requirements of non-electrophilic naphthalene-based Nrf2 activators. Eur J Med Chem. 2015;103:252–68. Epub 2015/09/13. doi: 10.1016/j.ejmech.2015.08.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tran KT, Pallesen JS, Solbak SMO, Narayanan D, Baig A, Zang J, Aguayo-Orozco A, Carmona RMC, Garcia AD, Bach A. A Comparative Assessment Study of Known Small-Molecule Keap1-Nrf2 Protein-Protein Interaction Inhibitors: Chemical Synthesis, Binding Properties, and Cellular Activity. J Med Chem. 2019;62(17):8028–52. Epub 2019/08/15. doi: 10.1021/acs.jmedchem.9b00723. [DOI] [PubMed] [Google Scholar]
- 119.Ciesielski GL, Hytonen VP, Kaguni LS. Biolayer Interferometry: A Novel Method to Elucidate Protein-Protein and Protein-DNA Interactions in the Mitochondrial DNA Replisome. Methods in molecular biology (Clifton, NJ). 2016;1351:223–31. Epub 2015/11/05. doi: 10.1007/978-1-4939-3040-1_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Heightman TD, Callahan JF, Chiarparin E, Coyle JE, Griffiths-Jones C, Lakdawala AS, McMenamin R, Mortenson PN, Norton D, Peakman TM, Rich SJ, Richardson C, Rumsey WL, Sanchez Y, Saxty G, Willems HMG, Wolfe L 3rd, Woolford AJ, Wu Z, Yan H, Kerns JK, Davies TG. Structure-Activity and Structure-Conformation Relationships of Aryl Propionic Acid Inhibitors of the Kelch-like ECH-Associated Protein 1/Nuclear Factor Erythroid 2-Related Factor 2 (KEAP1/NRF2) Protein-Protein Interaction. Journal of medicinal chemistry. 2019;62(9):4683–702. Epub 2019/04/12. doi: 10.1021/acs.jmedchem.9b00279. [DOI] [PubMed] [Google Scholar]
- 121.Grainger R, Heightman TD, Ley SV, Lima F, Johnson CN. Enabling synthesis in fragment-based drug discovery by reactivity mapping: photoredox-mediated cross-dehydrogenative heteroarylation of cyclic amines. Chem Sci. 2019;10(8):2264–71. Epub 2019/03/19. doi: 10.1039/c8sc04789h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Davies TG, Wixted WE, Coyle JE, Griffiths-Jones C, Hearn K, McMenamin R, Norton D, Rich SJ, Richardson C, Saxty G, Willems HM, Woolford AJ, Cottom JE, Kou JP, Yonchuk JG, Feldser HG, Sanchez Y, Foley JP, Bolognese BJ, Logan G, Podolin PL, Yan H, Callahan JF, Heightman TD, Kerns JK. Monoacidic Inhibitors of the Kelch-like ECH-Associated Protein 1: Nuclear Factor Erythroid 2-Related Factor 2 (KEAP1:NRF2) Protein-Protein Interaction with High Cell Potency Identified by Fragment-Based Discovery. J Med Chem. 2016;59(8):3991–4006. Epub 2016/04/01. doi: 10.1021/acs.jmedchem.6b00228. [DOI] [PubMed] [Google Scholar]
- 123.Archer WR, Schulz MD. Isothermal titration calorimetry: practical approaches and current applications in soft matter. Soft Matter. 2020;16(38):8760–74. Epub 2020/09/19. doi: 10.1039/d0sm01345e. [DOI] [PubMed] [Google Scholar]
- 124.Pallesen JS, Narayanan D, Tran KT, Solbak SMO, Marseglia G, Sorensen LME, Hoj LJ, Munafo F, Carmona RMC, Garcia AD, Desu HL, Brambilla R, Johansen TN, Popowicz GM, Sattler M, Gajhede M, Bach A. Deconstructing Noncovalent Kelch-like ECH-Associated Protein 1 (Keap1) Inhibitors into Fragments to Reconstruct New Potent Compounds. Journal of medicinal chemistry. 2021;64(8):4623–61. Epub 2021/04/06. doi: 10.1021/acs.jmedchem.0c02094. [DOI] [PubMed] [Google Scholar]
- 125.Elgert C, Ruhle A, Sandner P, Behrends S. Thermal shift assay: Strengths and weaknesses of the method to investigate the ligand-induced thermostabilization of soluble guanylyl cyclase. J Pharm Biomed Anal. 2020;181:113065. Epub 2020/02/08. doi: 10.1016/j.jpba.2019.113065. [DOI] [PubMed] [Google Scholar]
- 126.Jhoti H, Williams G, Rees DC, Murray CW. The ‘rule of three’ for fragment-based drug discovery: where are we now? Nature reviews Drug discovery. 2013;12(8):644–5. Epub 2013/07/13. doi: 10.1038/nrd3926-c1. [DOI] [PubMed] [Google Scholar]
- 127.Tang Y, Zeng X, Liang J. Surface Plasmon Resonance: An Introduction to a Surface Spectroscopy Technique. J Chem Educ. 2010;87(7):742–6. Epub 2011/03/02. doi: 10.1021/ed100186y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Walpole S, Monaco S, Nepravishta R, Angulo J. STD NMR as a Technique for Ligand Screening and Structural Studies. Methods in enzymology. 2019;615:423–51. Epub 2019/01/15. doi: 10.1016/bs.mie.2018.08.018. [DOI] [PubMed] [Google Scholar]
- 129.Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, Kensler TW. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(6):3410–5. Epub 2001/03/15. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Iida K, Itoh K, Kumagai Y, Oyasu R, Hattori K, Kawai K, Shimazui T, Akaza H, Yamamoto M. Nrf2 is essential for the chemopreventive efficacy of oltipraz against urinary bladder carcinogenesis. Cancer research. 2004;64(18):6424–31. Epub 2004/09/18. doi: 10.1158/0008-5472.Can-04-1906. [DOI] [PubMed] [Google Scholar]
- 131.Chio IIC, Jafarnejad SM, Ponz-Sarvise M, Park Y, Rivera K, Palm W, Wilson J, Sangar V, Hao Y, Öhlund D, Wright K, Filippini D, Lee EJ, Da Silva B, Schoepfer C, Wilkinson JE, Buscaglia JM, DeNicola GM, Tiriac H, Hammell M, Crawford HC, Schmidt EE, Thompson CB, Pappin DJ, Sonenberg N, Tuveson DA. NRF2 Promotes Tumor Maintenance by Modulating mRNA Translation in Pancreatic Cancer. Cell. 2016;166(4):963–76. Epub 2016/08/02. doi: 10.1016/j.cell.2016.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, Scrimieri F, Winter JM, Hruban RH, Iacobuzio-Donahue C, Kern SE, Blair IA, Tuveson DA. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475(7354):106–9. Epub 2011/07/08. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jiang T, Chen N, Zhao F, Wang XJ, Kong B, Zheng W, Zhang DD. High levels of Nrf2 determine chemoresistance in type II endometrial cancer. Cancer Res. 2010;70(13):5486–96. Epub 2010/06/10. doi: 10.1158/0008-5472.Can-10-0713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Satoh H, Moriguchi T, Takai J, Ebina M, Yamamoto M. Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer research. 2013;73(13):4158–68. Epub 2013/04/24. doi: 10.1158/0008-5472.Can-12-4499. [DOI] [PubMed] [Google Scholar]
- 135.Solis LM, Behrens C, Dong W, Suraokar M, Ozburn NC, Moran CA, Corvalan AH, Biswal S, Swisher SG, Bekele BN, Minna JD, Stewart DJ, Wistuba II. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin Cancer Res. 2010;16(14):3743–53. Epub 2010/06/11. doi: 10.1158/1078-0432.Ccr-09-3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tao S, Rojo de la Vega M, Chapman E, Ooi A, Zhang DD. The effects of NRF2 modulation on the initiation and progression of chemically and genetically induced lung cancer. Molecular carcinogenesis. 2018;57(2):182–92. Epub 2017/10/05. doi: 10.1002/mc.22745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tao S, Wang S, Moghaddam SJ, Ooi A, Chapman E, Wong PK, Zhang DD. Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer Res. 2014;74(24):7430–41. Epub 2014/10/24. doi: 10.1158/0008-5472.Can-14-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wang H, Liu X, Long M, Huang Y, Zhang L, Zhang R, Zheng Y, Liao X, Wang Y, Liao Q, Li W, Tang Z, Tong Q, Wang X, Fang F, Rojo de la Vega M, Ouyang Q, Zhang DD, Yu S, Zheng H. NRF2 activation by antioxidant antidiabetic agents accelerates tumor metastasis. Sci Transl Med. 2016;8(334):334ra51. Epub 2016/04/15. doi: 10.1126/scitranslmed.aad6095. [DOI] [PubMed] [Google Scholar]
- 139.Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci. 2009;34(4):176–88. Epub 2009/03/27. doi: 10.1016/j.tibs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 140.Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, Herman JG, Baylin SB, Sidransky D, Gabrielson E, Brock MV, Biswal S. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006;3(10):e420. Epub 2006/10/06. doi: 10.1371/journal.pmed.0030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489(7417):519–25. Epub 2012/09/11. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505(7484):495–501. Epub 2014/01/05. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, Yue P, Haverty PM, Bourgon R, Zheng J, Moorhead M, Chaudhuri S, Tomsho LP, Peters BA, Pujara K, Cordes S, Davis DP, Carlton VE, Yuan W, Li L, Wang W, Eigenbrot C, Kaminker JS, Eberhard DA, Waring P, Schuster SC, Modrusan Z, Zhang Z, Stokoe D, de Sauvage FJ, Faham M, Seshagiri S. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466(7308):869–73. Epub 2010/07/30. doi: 10.1038/nature09208. [DOI] [PubMed] [Google Scholar]
- 144.Kitamura H, Motohashi H. NRF2 addiction in cancer cells. Cancer Sci. 2018;109(4):900–11. Epub 2018/02/17. doi: 10.1111/cas.13537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zhang P, Singh A, Yegnasubramanian S, Esopi D, Kombairaju P, Bodas M, Wu H, Bova SG, Biswal S. Loss of Kelch-like ECH-associated protein 1 function in prostate cancer cells causes chemoresistance and radioresistance and promotes tumor growth. Molecular cancer therapeutics. 2010;9(2):336–46. Epub 2010/02/04. doi: 10.1158/1535-7163.Mct-09-0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wang H, Liu K, Geng M, Gao P, Wu X, Hai Y, Li Y, Li Y, Luo L, Hayes JD, Wang XJ, Tang X. RXRalpha inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 2013;73(10):3097–108. Epub 2013/04/25. doi: 10.1158/0008-5472.Can-12-3386. [DOI] [PubMed] [Google Scholar]
- 147.Love JM, Gudas LJ. Vitamin A, differentiation and cancer. Current opinion in cell biology. 1994;6(6):825–31. Epub 1994/12/01. doi: 10.1016/0955-0674(94)90051-5. [DOI] [PubMed] [Google Scholar]
- 148.Pascual G, Glass CK. Nuclear receptors versus inflammation: mechanisms of transrepression. Trends Endocrinol Metab. 2006;17(8):321–7. Epub 2006/09/01. doi: 10.1016/j.tem.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 149.Xu L, Glass CK, Rosenfeld MG. Coactivator and corepressor complexes in nuclear receptor function. Curr Opin Genet Dev. 1999;9(2):140–7. Epub 1999/05/14. doi: 10.1016/S0959-437X(99)80021-5. [DOI] [PubMed] [Google Scholar]
- 150.Wang XJ, Hayes JD, Wolf CR. Generation of a stable antioxidant response element-driven reporter gene cell line and its use to show redox-dependent activation of nrf2 by cancer chemotherapeutic agents. Cancer research. 2006;66(22):10983–94. Epub 2006/11/17. doi: 10.1158/0008-5472.CAN-06-2298. [DOI] [PubMed] [Google Scholar]
- 151.Wang XJ, Hayes JD, Henderson CJ, Wolf CR. Identification of retinoic acid as an inhibitor of transcription factor Nrf2 through activation of retinoic acid receptor alpha. Proc Natl Acad Sci U S A. 2007;104(49):19589–94. Epub 2007/12/01. doi: 10.1073/pnas.0709483104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zhang J, Ohta T, Maruyama A, Hosoya T, Nishikawa K, Maher JM, Shibahara S, Itoh K, Yamamoto M. BRG1 interacts with Nrf2 to selectively mediate HO-1 induction in response to oxidative stress. Molecular and cellular biology. 2006;26(21):7942–52. Epub 2006/08/23. doi: 10.1128/MCB.00700-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Shen G, Hebbar V, Nair S, Xu C, Li W, Lin W, Keum YS, Han J, Gallo MA, Kong AN. Regulation of Nrf2 transactivation domain activity. The differential effects of mitogen-activated protein kinase cascades and synergistic stimulatory effect of Raf and CREB-binding protein. The Journal of biological chemistry. 2004;279(22):23052–60. Epub 2004/03/17. doi: 10.1074/jbc.M401368200. [DOI] [PubMed] [Google Scholar]
- 154.Nadia Bunga Anggraini BE, and Iskandarsyah Iskandarsyah. Antielastase Activity of Macassar Kernels (Rhus javanica) Stem Extract and Skin Elasticity Evaluation of Its Topical Gel Formulation. Advances in Pharmacological and Pharmaceutical Sciences. 2021;2021:11. doi: 10.1155/2021/6690029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ji L, Li H, Gao P, Shang G, Zhang DD, Zhang N, Jiang T. Nrf2 pathway regulates multidrug-resistance-associated protein 1 in small cell lung cancer. PLoS One. 2013;8(5):e63404. Epub 2013/05/15. doi: 10.1371/journal.pone.0063404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Vartanian S, Ma TP, Lee J, Haverty PM, Kirkpatrick DS, Yu K, Stokoe D. Application of Mass Spectrometry Profiling to Establish Brusatol as an Inhibitor of Global Protein Synthesis. Molecular & cellular proteomics : MCP. 2016;15(4):1220–31. Epub 2015/12/30. doi: 10.1074/mcp.M115.055509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Harder B, Tian W, La Clair JJ, Tan AC, Ooi A, Chapman E, Zhang DD. Brusatol overcomes chemoresistance through inhibition of protein translation. Molecular carcinogenesis. 2017;56(5):1493–500. Epub 2016/12/27. doi: 10.1002/mc.22609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Olayanju A, Copple IM, Bryan HK, Edge GT, Sison RL, Wong MW, Lai ZQ, Lin ZX, Dunn K, Sanderson CM, Alghanem AF, Cross MJ, Ellis EC, Ingelman-Sundberg M, Malik HZ, Kitteringham NR, Goldring CE, Park BK. Brusatol provokes a rapid and transient inhibition of Nrf2 signaling and sensitizes mammalian cells to chemical toxicity-implications for therapeutic targeting of Nrf2. Free radical biology & medicine. 2015;78:202–12. Epub 2014/12/03. doi: 10.1016/j.freeradbiomed.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Keller TL, Zocco D, Sundrud MS, Hendrick M, Edenius M, Yum J, Kim YJ, Lee HK, Cortese JF, Wirth DF, Dignam JD, Rao A, Yeo CY, Mazitschek R, Whitman M. Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nature chemical biology. 2012;8(3):311–7. Epub 2012/02/14. doi: 10.1038/nchembio.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Tsuchida K, Tsujita T, Hayashi M, Ojima A, Keleku-Lukwete N, Katsuoka F, Otsuki A, Kikuchi H, Oshima Y, Suzuki M, Yamamoto M. Halofuginone enhances the chemo-sensitivity of cancer cells by suppressing NRF2 accumulation. Free radical biology & medicine. 2017;103:236–47. Epub 2017/01/01. doi: 10.1016/j.freeradbiomed.2016.12.041. [DOI] [PubMed] [Google Scholar]
- 161.Singh A, Venkannagari S, Oh KH, Zhang YQ, Rohde JM, Liu L, Nimmagadda S, Sudini K, Brimacombe KR, Gajghate S, Ma J, Wang A, Xu X, Shahane SA, Xia M, Woo J, Mensah GA, Wang Z, Ferrer M, Gabrielson E, Li Z, Rastinejad F, Shen M, Boxer MB, Biswal S. Small Molecule Inhibitor of NRF2 Selectively Intervenes Therapeutic Resistance in KEAP1-Deficient NSCLC Tumors. ACS chemical biology. 2016;11(11):3214–25. Epub 2016/10/18. doi: 10.1021/acschembio.6b00651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Gong M, Li Y, Ye X, Zhang L, Wang Z, Xu X, Shen Y, Zheng C. Loss-of-function mutations in KEAP1 drive lung cancer progression via KEAP1/NRF2 pathway activation. Cell communication and signaling : CCS. 2020;18(1):98. Epub 2020/06/25. doi: 10.1186/s12964-020-00568-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Jevtić P, Haakonsen DL, Rapé M. An E3 ligase guide to the galaxy of small-molecule-induced protein degradation. Cell chemical biology. 2021. Epub 2021/04/24. doi: 10.1016/j.chembiol.2021.04.002. [DOI] [PubMed] [Google Scholar]
- 164.Kannt A, Đikić I. Expanding the arsenal of E3 ubiquitin ligases for proximity-induced protein degradation. Cell chemical biology. 2021. Epub 2021/05/05. doi: 10.1016/j.chembiol.2021.04.007. [DOI] [PubMed] [Google Scholar]
- 165.Samarasinghe KTG, Crews CM. Targeted protein degradation: a promise for undruggable proteins. Cell chemical biology. 2021. Epub 2021/05/19. doi: 10.1016/j.chembiol.2021.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]