

Abstract
Optical tweezers have become the method of choice in single-molecule manipulation studies. In this Primer, we first review the physical principles of optical tweezers and the characteristics that make them a powerful tool to investigate single molecules. We then introduce the modifications of the method to extend the measurement of forces and displacements to torques and angles, and to develop optical tweezers with single-molecule fluorescence detection capabilities. We discuss force and torque calibration of these instruments, their various modes of operation and most common experimental geometries. We describe the type of data obtained in each experimental design and their analyses. This description is followed by a survey of applications of these methods to the studies of protein–nucleic acid interactions, protein/RNA folding and molecular motors. We also discuss data reproducibility, the factors that lead to the data variability among different laboratories and the need to develop field standards. We cover the current limitations of the methods and possible ways to optimize instrument operation, data extraction and analysis, before suggesting likely areas of future growth.
Single-molecule methods were born out of the desire to follow the dynamics of molecular processes in real time and the impossibility of synchronizing the behaviour of molecules in an ensemble. Most biomolecular processes are thermally activated and subjected to the stochastic nature of molecular collisions from the thermal bath. As a result, attempts to follow the time course of a molecular process necessarily contain the asynchronous contributions of all molecules in the ensemble, which yield at best an unphysical ‘average’ of their behaviour. By contrast, monitoring the time evolution of single molecules as they undergo chemical or biochemical reactions makes it possible to characterize their ‘molecular trajectories’, at the price of obtaining data often dominated by noise from which the signals must be extracted. Once this is done, however, it is frequently possible to gain a greater degree of mechanistic insight into the underlying molecular process than from the average molecular behaviour derived from bulk or ensemble measurements.
Optical tweezers are a method to exert forces or torques on individual molecules and/or to directly measure the forces or torques generated in the course of their biochemical reactions. In 1970, Arthur Ashkin exploited the fact that photons carry momentum to entrain and transport micron-sized latex spheres suspended in water using laser beams1. In 1986, Ashkin et al. demonstrated for the first time the single-beam ‘optical trap’ or ‘optical tweezers’ when they showed that light focused tightly can be used to hold and maintain microscopic particles stably in all three dimensions2. One year later, Ashkin et al. showed that it was possible to use optical tweezers to trap and manipulate bacteria and red blood cells, as well as organelles inside cells3. Optical tweezers also yield the changes in displacement that accompany the application (or generation) of forces and torques. Accordingly, these experiments permit direct access to the work done on (or by) the system of interest. Other methods have been used to accomplish these same tasks, such as atomic force microscopy and magnetic tweezers4. Atomic force microscopy affords higher forces (>100 pN), whereas magnetic tweezers are better suited for low-force applications (sub-piconewtons). However, in some single-molecule applications, atomic force microscopy can be limited by its force resolution; magnetic tweezers, although affording the simultaneous manipulation of many molecules on a surface, have lower spatial and temporal resolution when compared with optical tweezers4. Therefore, because of their force (or torque), spatial and temporal resolution, and versatility and dynamic range, optical tweezers are the method of choice in many biophysical applications.
Photons carry energy, as well as linear and angular momentum. When an object encounters a beam of light, there are two forces exerted by the light on the object: the gradient force and the scattering force. We first define the gradient force Fgrad. For objects much smaller than the wavelength of light, the object behaves as a Rayleigh scatterer whose response to an electric field is characterized by a polarizability α (assumed here to be real). The electric field of light at position where the object is located, and at time t, (where is the amplitude of the electric field) induces an electric dipole moment in the object. The energy (U) of the induced dipole in the electric field is then: . The intensity of a laser beam is proportional to the square of the amplitude of its electric field. Accordingly, the energy of the dipole attains a minimum at the place where the intensity of the beam is a maximum. Therefore, if the field is not homogeneous, as is the case with a Gaussian laser beam, the dipole will experience a force attracting it towards the higher intensities of the beam; this force is proportional to the gradient of the light intensity (I) and tends to minimize the induced dipole’s energy5:
| (1) |
where 〈〉 denotes the time average, c the speed of light in vacuum, nm the index of refraction of the particle and ε0 the permittivity of vacuum. Here, we have used the fact that to write the intensity of the beam as . This is the ‘gradient force’. For particles with positive polarizability (for example, when the polarizability of the particle is greater than that of the surrounding medium), this force attracts the particle towards the higher intensities of the light.
We now define the scattering force Fscat. Particles will also experience a force due to the absorption, and the scattering (refraction or reflection) of light. The scattering force for a Rayleigh particle can be written as:
| (2) |
where σext is the extinction cross-section of the particle with an absorption (σabs) and a scattering (σscat) contribution, and is the time-averaged Poynting vector of the beam, which points in the direction of the propagation of the light and has units of energy per unit area, per unit time. As shown in FIG. 1a, for a Rayleigh particle present in a focused laser beam, the gradient force tends to attract the particle towards the axis of the beam and towards the focus, thus counteracting the effect of the scattering force that tends to push the particle in the direction of the incident light.
Fig. 1 |. Principles of optical tweezers.
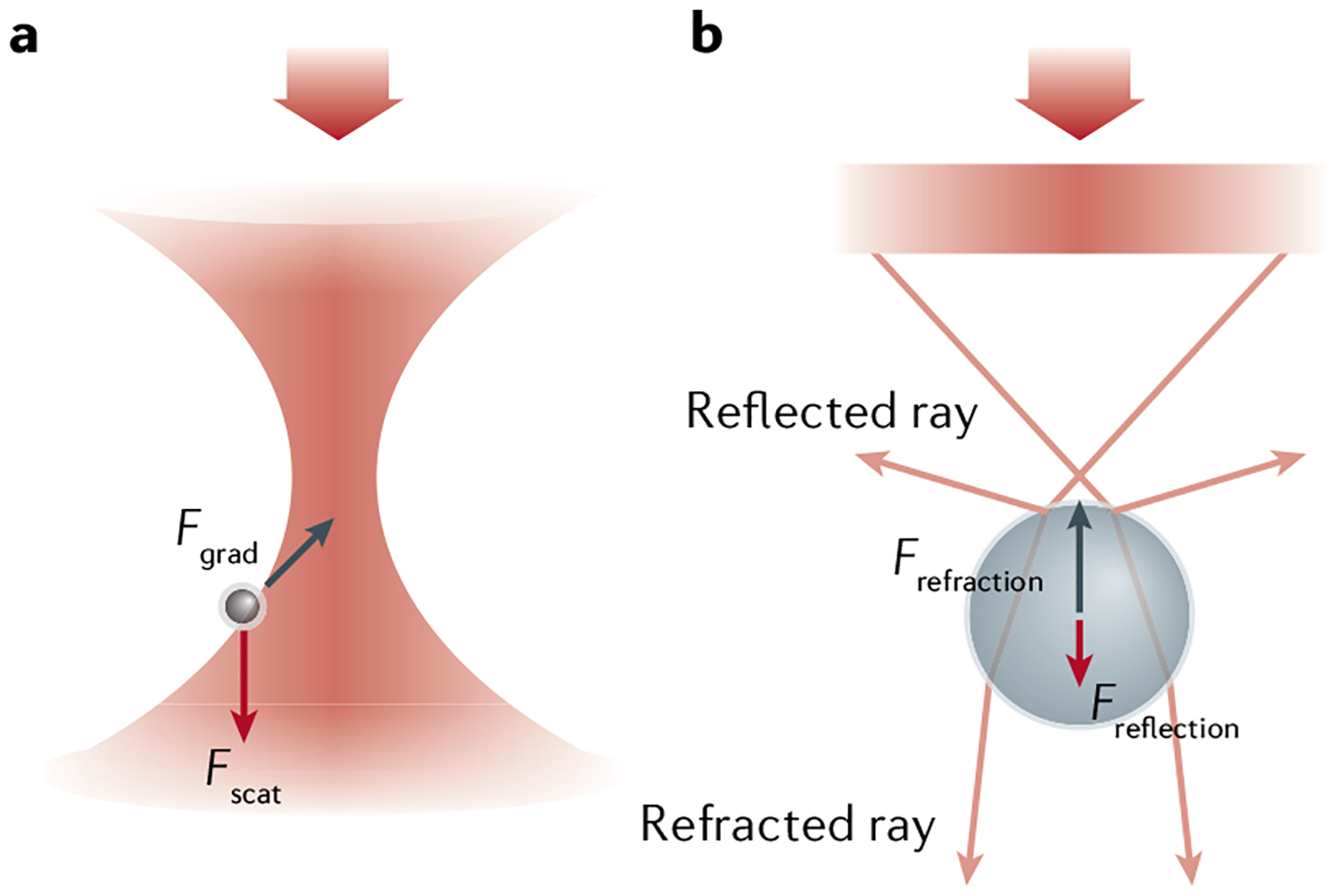
Forces acting on a dielectric sphere interacting with light, with the incident light beam focused by a high-numerical aperture (NA) lens. a | A Rayleigh particle smaller than the wavelength of light experiences a scattering force (Fscat, red arrow) that pushes the particle along the direction of propagation of the light and a gradient force (Fgrad, black arrow) that attracts it towards the focus. b | A dielectric sphere larger than the wavelength of light either reflects or refracts light (pink arrows) focused by a high-NA lens. The change in direction of each ray corresponds to a change in momentum of the light and an equal and opposite change in bead momentum. Reflected rays of light lose forward momentum that is gained by the bead, leading to a net force (Freflection, red arrow) pushing the bead along the direction of propagation of the light. Refracted rays are deflected forward because of the high incidence angle of the light, which generates momentum change and reactive force (Frefraction, black arrow) that pulls the bead towards the focus.
For particles larger than the wavelength of light, such as the beads used in most optical tweezers experiments, the particle acts as a refractive object. Here, the beam of light impinging on the object can be treated as a collection of rays and the force acting on the particle can be described using geometric or ray optics. Newton’s second law defines force as the rate of change of momentum , where is the momentum of the object. Because the momentum of light changes either by being absorbed, reflected or scattered by the particle, conservation of linear momentum dictates that the rate of change of momentum of the light must be accompanied by an identical rate of change of momentum of the object (or force) of opposite sign acting on that object. FIGURE 1b illustrates this limit for a bead in a focused beam. In FIG. 1b, the incident light gains forward momentum by being refracted by the particle (forward pink arrows), which generates a recoil force Frefraction that pulls the bead towards the focus (black arrow). Similarly, the light reflected by the beam (backward pink arrows) loses forward momentum, thus pushing the bead forward with a force Freflection (red arrow). Stable trapping is obtained when these two forces balance each other. A similar argument can be used to show that the bead will be pulled towards the beam axis if the beam intensity is higher there than in the periphery.
In practice, optical tweezers use a beam of laser light, focused through a microscope objective lens, to trap, move and apply calibrated forces to microscopic refractive objects. The most important component of an optical trap is the objective lens, which focuses the trapping beam into the sample chamber. In most optical tweezers experiments, the sample of interest (a protein, RNA molecule and so on) is too small to interact significantly with the trapping light. Instead, the system must be tethered to a micron-sized bead (typically made of polystyrene or silica 0.2–5 μm in diameter6). In order to tether the molecule of interest to the beads, linker molecules are often used; the most common linkers are double-stranded DNA (dsDNA) fragments, referred to as ‘molecular handles’7. If such a molecule attached to a surface is also tethered via a DNA handle to a bead held in an optical trap, then by moving the trap relative to the surface, the trapped bead will follow the beam and a force will be exerted on the molecule through the DNA handle. This force acting on the molecule is equal and opposite to the force sensed by the bead in the trap. Because this force represents a rate of change of momentum of the bead, by conservation of linear momentum, the beam in the optical trap must also experience a rate of change of momentum (or force) of the opposite sign, which can be measured directly from the deflection of the trapping beam using a position-sensitive photo-detector. Thus, optical tweezers not only make it possible to exert forces on molecules but also to measure the forces being exerted.
Single-molecule optical tweezers assays also require a measurable change in a spatial coordinate of the system (for example, conformational change, displacement of the centre of mass, force-induced unfolding of a protein) whose magnitude is well within the spatial resolution of the instrument. In some cases, the change in the spatial coordinate is not large or discrete enough to be directly detected. In such cases, combined optical tweezers and single-molecule fluorescence detection may provide a way to monitor these changes while retaining the ability to act mechanically on the system via the tweezers. In general, in most optical tweezers experiments, the system must be studied one molecule at a time. Therefore, systems that can be repeatedly manipulated (as in protein or RNA mechanical unfolding/refolding studies) or whose operation is cyclical, as in the case of enzyme reactions or molecular motors, make it possible to obtain good statistics on the system. We emphasize that optical tweezers-based single-molecule experiments should not be attempted if a robust bulk biochemistry assay of the system does not already exist, typically arrived at from bulk approaches.
This Primer is not an exhaustive or comprehensive review of all of the main applications of optical tweezers in biophysics; we have limited this review to single-molecule studies. As such, we have not covered the use of optical tweezers to manipulate cells or organelles. Here, we concentrate on the physical foundations of the method and on the best approaches to interpret the data. We describe the instrumentation and experimental designs used in most single-molecule optical tweezers assays (Experimentation), present representative examples of optical tweezers data, data correction and data analysis (Results), and describe the type of information that can be derived from the use of this method to study systems of great biophysical interest, including DNA elasticity, protein and RNA folding, and the dynamics of molecular motors (Applications). We also discuss data reproducibility given the inherent stochastic behaviour of individual molecules, suggest community practices to standardize the results from different laboratories (Reproducibility and data deposition), discuss the current limitations in both instrument performance and the data analysis, and suggest ways to overcome these limitations (Limitations and optimizations). Finally, we explore areas of likely future growth and development of the method and its applications (Outlook). Several reviews are now available that can provide further details on some of the topics addressed here8–13.
Experimentation
In this section, we provide the basic information needed to set up an optical trap experiment. We describe the components of the instrument and its layout, how the instrument is calibrated, the various experimental geometries used and modes of operation, and how samples are prepared. Although commercial systems are becoming increasingly popular, to date a majority of optical tweezers continue to be custom-built instruments and, therefore, designs and operation procedures can vary widely. For the sake of brevity, we do not describe here the variety of instrument layouts in exhaustive detail but, rather, focus on key design features and highlight important differences between set-ups. This section is organized around three main categories of instruments: standard optical tweezers (FIG. 2a) used to measure and exert forces and displacements, optical tweezers supplemented with fluorescence detection and imaging (‘fleezers’) (FIG. 2b) and angular optical tweezers (AOT) (FIG. 2c) that permit one to measure and exert torques and rotations.
Fig. 2 |. Basic designs of optical traps.
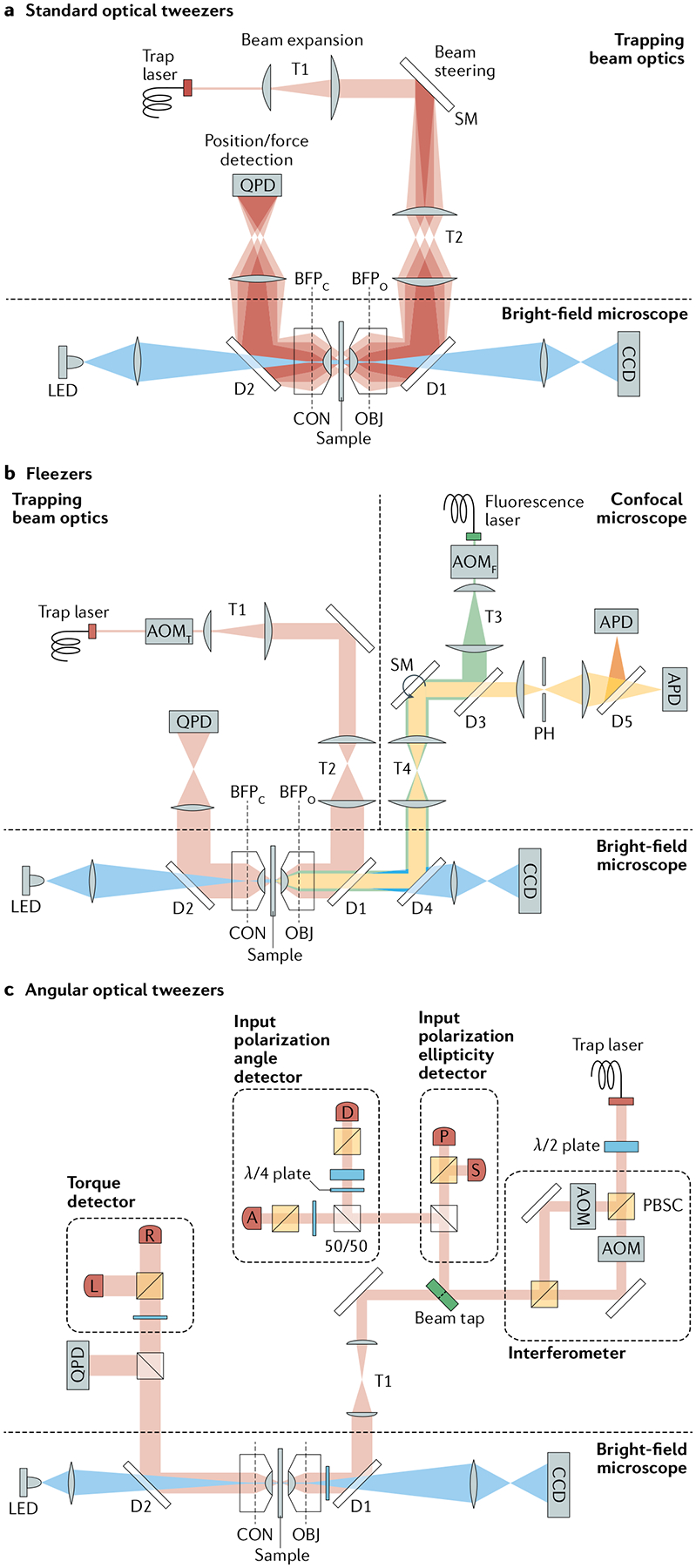
a | Optical layout of a standard single-beam optical trap. A high-power laser generates the trapping beam (pink), which is expanded by telescope T1. Beam-steering optics (here, a steerable mirror (SM)) control the tilt in the beam axis. A high-numerical aperture objective (OBJ) focuses the trapping beam into the sample. T2 images the steering plane (at SM) onto the objective back focal plane (BFPO), so that tilting the beam displaces the trap in the sample plane. A condenser (CON) collects the light scattered by the trapped particle. A lens images the light at the condenser back focal plane (BFPC) onto a position-sensitive quadrant photodetector (QPD) for position/force detection. Two dichroic mirrors (D1 and D2) reflect the trapping beam and transmit visible light (blue) for bright-field illumination (light-emitting diode (LED)) and imaging (charge-coupled device (CCD)) of the sample plane. b | Optical layout for a representative fleezers set-up (dual traps with a confocal microscope)43. A fluorescence excitation beam (green) is expanded (T3) and directed (D3, SM, T4, D4) into the trapping OBJ. The OBJ focuses the beam to a diffraction-limited spot on the sample plane and collects light emitted within the spot. The excitation spot is displaced in the sample plane by a SM. The emitted light (yellow) travels back along the emission path, passing through a dichroic mirror (D3) and into a pinhole aperture (PH) to reject out-of-focus light. Emission light is detected by an avalanche photodiode (APD) (or by two APDs for Förster resonance energy transfer measurements). The trapping and fluorescence excitation beams are interlaced by two out-of-phase acousto-optic modulators (AOMT and AOMF, respectively). The trap layout is similar to that shown in part a, with dual traps generated by time-sharing using AOMT. c | Representative optical layout of angular optical tweezers101. The trapping laser is linearly polarized and split equally into two orthogonally polarized beams at a polarization beam splitter cube (PBSC). Each beam then passes through an AOM, and the two beams are recombined at another PBSC. Prior to the objective, the ellipticity of the laser is measured by the ‘input polarization ellipticity detector’ via photodetectors P and S, while the polarization angle is measured by the ‘input polarization angle detector’ via photodetectors A and D. After the laser interacts with a trapped cylinder in the sample plane, the transmitted laser becomes elliptically polarized, and the optical torque is measured by the ‘torque detector’ via photodetectors R and L. The force on the cylinder is measured by a QPD.
Standard optical tweezers
A standard layout of optical tweezers is shown in FIG. 2a. A high-power laser generates the beam of light used to create the trap. The beam is expanded by a telescope and then passed into a high numerical aperture objective lens (which can be water or oil immersion) that focuses it into a diffraction-limited spot — the optical trap — inside a sample chamber. A condenser lens collects the transmitted light, which is then imaged onto a position-sensitive detector used to measure the displacements of the trapped particle and the force exerted on it. In many designs, the angle of the beam entering the objective is actively steered (for example, with a motorized mirror or with an acousto-optic or electro-optic deflector) to control the trap position inside the sample chamber. In addition, the sample chamber is often mounted on an actuated stage to control its position in all three directions. A camera and bright-field illumination are used to image the sample chamber and traps. Although the set-up is often integrated into a commercial microscope body, this is not essential and many designs are built entirely from individual parts.
The trapping laser is one critical component of the instrument. As the maximum attainable force and the trap stiffness are both proportional to the laser power, a high-power (>1 W) laser is desirable, with high stability to maintain a constant stiffness. As a general rule, a maximum force of ~10–20 pN and a trap stiffness of 0.1–0.3 pN/nm can be achieved per 100 mW of trap power at the specimen plane for a micron-sized bead6,14,15, although the specifics of the instrument design and the material, size and shape of the trapped particle will affect these numbers. The emission wavelength of the laser is another important parameter. In standard designs, near-infrared (750–1,200 nm) lasers are used owing to the availability of high-power (>1 W) lasers emitting in this wavelength range6 and owing to several advantages of this spectral window with respect to biological samples: the relative transparency of aqueous buffers14 and the mitigation of optical damage16.
The most important component of the instrument is the objective lens as it forms the trap. A strongly converging objective with a high numerical aperture (typically 1.2–1.4) is needed to generate the large gradients in light intensity for trapping6. In most designs, stable trapping also requires expanding the trapping beam to ‘overfill’ the back aperture of the objective (FIG. 2a). The reason for this is because the marginal light rays of the beam that are farthest from the optical axis contribute most to the gradient forces when focused by the objective, in contrast to the central rays that contribute mainly to scattering forces. In alternative designs, the trap is formed by two oppositely travelling beams focused at the same point by two objectives to cancel the scattering forces17, removing the need to overfill the objectives.
For detection, most optical tweezers use a technique called back focal plane interferometry18–21. This technique can be implemented using the same light that forms the trap, or using a separate, low-power laser source solely for detecting the particle position (a ‘detection beam’)6. The condenser lens collects this light, and a lens images it onto a position-sensitive photodetector (FIG. 2a). This interferometric method for detecting trapped particle displacements provides a wide linear range of ~100 nm (REF.22) combined with extremely high position sensitivity and temporal resolution. It is possible to detect 1-Å displacements of the trapped particle over a 0.1-ms measurement time, limited only by the detector background noise23, and measurement bandwidths can reach into hundreds of kilohertz. The sensitivity of back focal plane interferometry is a major factor in the high spatial and temporal resolution of optical tweezers.
Calibration.
When reading the position of a trapped particle, the photodetectors output raw signals measured in volts. For quantitative measurements to be possible, these raw signals must be converted into physical displacements (in nanometres). Moreover, to obtain the force exerted on the particle (in piconewtons), the trap stiffness (in piconewtons per nanometre) must be determined. Although some instruments are designed to measure optical forces directly from the change in linear momentum of the light17, most designs require calibration procedures to determine these parameters.
The most common calibration method uses the Brownian motion of the trapped particle as a reference signal6,24. A particle held in an optical trap will undergo Brownian motion owing to thermal forces from the surrounding bath, and its position relative to the trap centre will fluctuate over time. For a harmonic trap, where the force on the particle is proportional to its displacement x from the trap centre (that is, Fx = κxx, where κx is the trap stiffness in the x direction), these fluctuations can be modelled precisely. Specifically, one can derive the following expression for the power spectrum Sxx(f) of the particle position x, which measures the distribution of noise power over frequency f:
| (3) |
where kB is the Boltzmann constant. T is the temperature and γ the hydrodynamic drag coefficient of the trapped particle, both usually known parameters (for a spherical bead, the drag coefficient is γ = 3πηd, where η is the viscosity of the fluid and d the bead diameter). fc is the characteristic frequency of the trapped particle, given by:
| (4) |
and represents the fact that drag on the particle sets a limit to how fast it can move in the surrounding fluid.
The power spectrum Sxx(f) is determined experimentally by recording the position of a trapped particle x in the trap over time t and taking the Fourier transform of |x(t)|2. Fitting these data to the predicted power spectrum Sxx(f) in Eqs 3 and 4, one extracts both the volts-to-nanometres conversion factor and the trap stiffness κx along the x direction. This approach can easily be extended to the other dimensions y and z. Although this calibration method is standard, there exist many variations and alternatives, and the interested reader is encouraged to refer to the rich literature on this subject6,24,25.
Trapping geometries.
In a single-molecule optical tweezers measurement, trapped micron-sized polystyrene or silica beads (of diameter range 0.2–5 μm) are used to exert forces on the system of interest. Various approaches can be used to this end. For example, in measurements of cytoskeletal motors such as kinesin or dynein, forces are applied by attaching the motor directly to a trapped bead26,27. In other studies, the track on which a motor moves (for example, actin with myosin) is attached to beads18,28. In applications on nucleic acid-binding proteins and nucleic acid-processing motors, a DNA molecule is tethered to trapped beads29–31. Owing to its well-characterized mechanical properties, dsDNA is often used as ‘handles’ to link beads to nucleic acid structures and proteins in mechanical unfolding studies. The choice of tethering approach is as much dictated by the system of study and the desired measurement as by the creativity of the researcher. However, it must be understood that all tethering molecules have mechanical properties and will thus be affected by applied force, which has consequences on the signals measured and on the measurement resolution.
The geometry of the measurement also depends on whether a single-trap or dual-trap instrument is used. In single-trap measurements, one end of the system of interest is attached to the trapped bead and the other is bound to the sample chamber surface29 (FIG. 3a) or to a second bead held by suction on the end of a micropipette10,32–34 (FIG. 3b). In dual-trap systems, the molecule is instead tethered between two optically trapped beads (FIG. 3c). Although the choice of single-trap versus dual-trap design is often dictated by the system of study, it has an impact on trap performance. Surface-based applications are subject to unwanted motion of the sample stage or ‘drift’, which can be significant (~0.1 nm/s) and can limit measurement resolution9,35. Drift in the laser focus, particularly along the axial (z) direction, can arise from heating of the objective by the trapping laser36. Feedback control over the stage position9,37 and temperature control36 of the objective can be implemented to mitigate this drift. In contrast to the above instrument designs, in dual-trap set-ups the traps are decoupled from the surface and its drift. Furthermore, dual traps are usually formed from the same laser, either by splitting the laser into two orthogonally polarized beams38–40 or by ‘time-sharing’, scanning the laser rapidly between two positions using rapid-switching and rapid-steering optics such as acousto-optic and electro-optic modulators and deflectors28,41–43. As a result, they are less sensitive to pointing drift of the laser as both traps are produced from the same source. Dual-trap set-ups generally provide superior stability, with drift in the separation between the traps maintained below 5 nm/h (REF.38), a performance approaching fundamental noise limits set by Brownian motion40.
Fig. 3 |. Measurement geometries of standard optical traps, fleezers and angular optical traps.
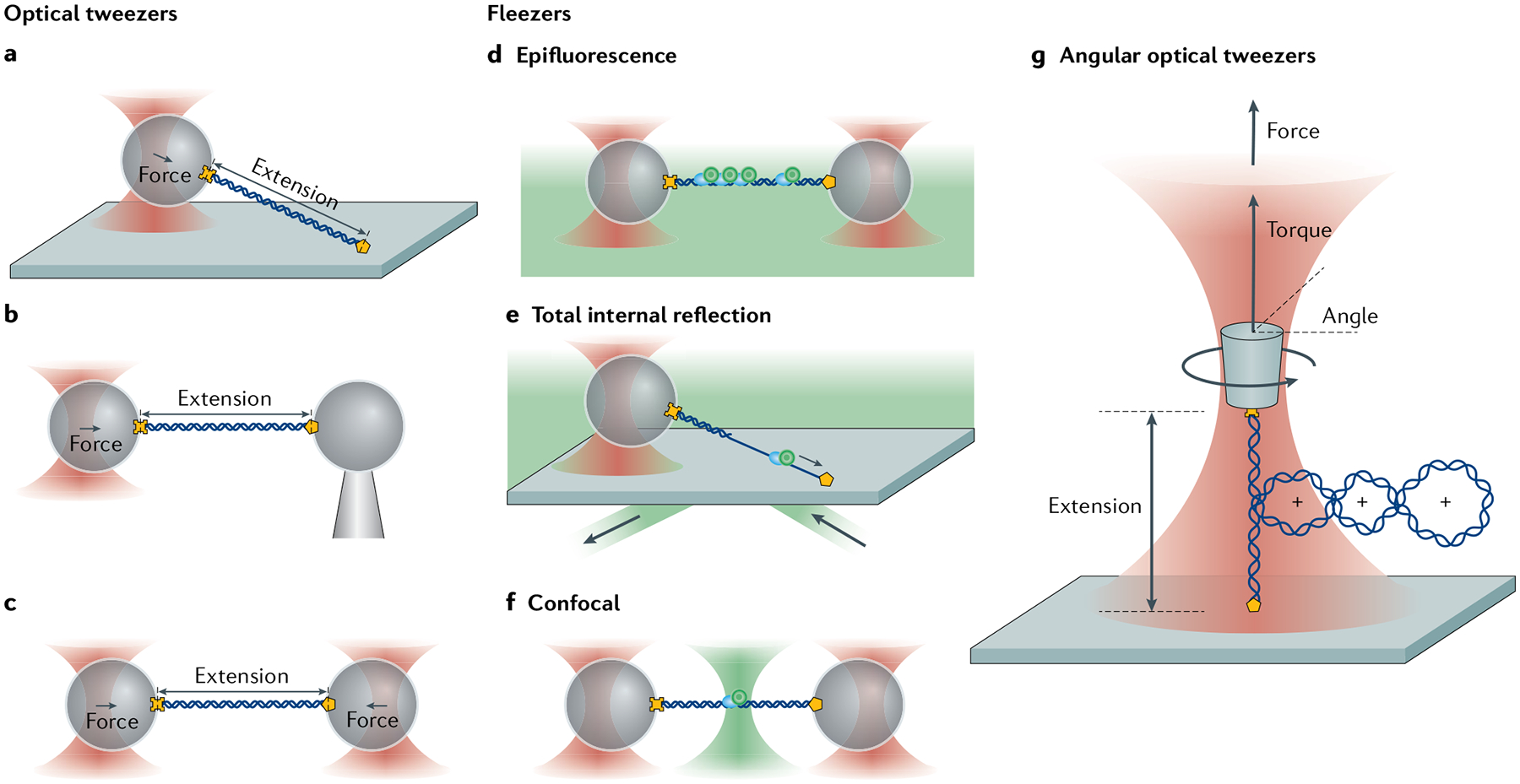
a–c | Common optical trapping geometries11 Single-trap, surface-based geometry where a molecule (here, DNA) functionalized at both ends (yellow cross and pentagon) is tethered between a trapped bead and the sample chamber surface (part a). Single-trap, micropipette-based geometry where the molecule is attached to a trapped bead and a second bead held by suction on the end of a micropipette (part b). Dual-trap geometry where the molecule is tethered between two trapped beads (part c). d–f | Example fleezers configurations11 Dual traps with wide-field, epifluorescence microscopy, where excitation light bathes the specimen plane and fluorescence is collected from dyes (green circles) emitting in the focal plane (part d). Single trap with total internal reflection fluorescence microscopy, where excitation occurs at the chamber surface, in an exponentially decaying evanescent field (typically ~100–200 nm in depth) (part e). Dual traps with confocal fluorescence microscopy, where the excitation light is focused into a diffraction-limited spot inside the sample chamber, and only light emitted from within the spot is collected (part f). g | Basic operational geometry of angular optical tweezers (AOT)91,171. In AOT, a nanofabricated quartz cylinder is trapped. Shown is an example where the AOT are used to investigate the torsional properties of a DNA molecule by twisting the DNA. During this measurement, the AOT exert and measure torque, control rotation and supercoiling, and measure the displacement and force of the trapped cylinder, all at the same time.
Sample preparation.
To carry out an optical trap measurement, the biological system of interest, the beads used to exert forces on the system and the sample chamber in which the measurements are made must all be prepared in advance. As described above, some component of the system must be tethered to trapped beads and/or to the sample chamber surface. Commonly used linkages are streptavidin–biotin and antibody–antigen pairs owing to their strength under force and commercial availability. DNA molecules to be tethered are usually functionalized with biotin and/or digoxigenin — a small-molecule tag that binds to an antibody — via incorporation of modified bases or modified DNA primers, all commercially available. Various approaches have been developed to tether proteins. Protein samples can be attached to beads through a specific antibody or via an affinity peptide tag (for example, 6× histidine). Biotinylation is also possible in recombinant proteins containing the Avi peptide tag using the BirA biotin ligase38,44. Micron-sized beads coated with streptavidin or protein G (to which an antibody can be bound), available from several manufacturers, are then used to bind the molecule of interest. In many protein folding studies, the protein of interest is attached covalently to molecular handles — usually, dsDNA — which separates the protein from the beads and eliminates unwanted interactions. A common strategy is a disulfide bond linkage between a free cysteine in the protein and maleimide-functionalized DNA45–48. Enzyme-based coupling methods have also been used for covalent protein–DNA linkages. In these approaches, a fusion protein (for example, HaloTag) or peptide tag (for example, YbbR tag) engineered into the protein of interest undergoes an enzymatic reaction with a substrate cross-linked to DNA44,49,50.
Beyond their function in applying force to the tethered molecule, the beads play an important but underappreciated role in trap performance, affecting the trap stiffness15 and the maximum force that can be exerted6,14, and the spatial and temporal resolution34,40,51. The beads can also affect the degree to which the traps cause photodamage to the sample; polystyrene beads can act as sensitizers for trap-mediated generation of reactive oxygen species that cause oxidative damage. This effect, however, can be mitigated by adding enzyme-based oxygen scavenging systems to the experimental buffers to remove molecular oxygen or by using beads of a different material such as silica47,52. In sum, bead selection is important in the experimental design.
Optical trap measurements are carried out inside microscope sample chambers, typically consisting of two microscope coverslips bonded together by double-sided tape or melted parafilm, into which buffers and samples are flowed via attached tubing. Measurements are usually carried out at room temperature (or slightly higher, as there can be some residual heating of the sample due to absorption of the laser light by the aqueous buffer53). Temperature control of the sample is possible by heating/cooling the sample chamber with a Peltier device54 or the objective with a water circulation system55, or by laser-based heating56. In surface-based applications (for example, with single-trap instruments; FIG. 3a) where a tether is formed to the chamber surface, streptavidin or antibody is pre-adsorbed to the chamber surface to coat it. Trapping of beads is done in situ and one at a time. Beads are flowed into the sample chamber and the user manually captures one by moving the sample stage or trap position to overlap the bead with the trap focus. Formation of tethers is similarly user-intensive. The tethering molecule is usually coated onto the chamber surface or bead by pre-incubation. Then, the surface and bead (FIG. 3a) or the two beads (FIG. 3b,c) are brought into contact. The presence of a tether is confirmed by retracting the two and observing the appearance of a force on the bead(s). To mitigate the formation of multiple attachments to the same bead, it is advisable to work under conditions where the probability of tether formation per bead is low (<20%).
Operation modes.
Optical tweezers can be operated in various measurement modes depending on the system of interest and the parameters to be extracted from that system. In the simplest measurement mode, the trap or stage position remains fixed (that is, ‘passive’) during instrument operation. Mechanical signals from the tethered molecule — for example, extension changes from folding/unfolding transitions in a molecule or the translocation of a molecular motor — displace the beads out of the centres of the traps, leading to a measured change in the force. As the tether is elastic, it will stretch or relax in response to the increase or decrease in force. Although this mode of operation is simple to implement, it requires knowing the force-dependent compliance of the tether to interpret the force change signal. This problem is overcome by applying a constant force. A ‘force clamp’ can be achieved with passive trap operation in dual-trap systems with unequal traps22: one trap is operated at high power and used to measure force, and the other at low power and used to exert constant force. The low-power trap is configured to hold a bead at the edge of its trapping potential (where the bead is at the cusp of falling out) and where the trap stiffness is effectively zero, leading to a constant force generated as the bead is displaced. This configuration requires a detection beam to monitor the displacements of the bead in the weaker trap. A constant-force operation mode is often used to track the position of a molecular motor over time or to follow transitions between conformational states of a system of interest in real time.
In contrast to passive operation, active control over the trap and/or stage positions allows the application of precisely defined forces over time. Several active modes are commonly used to apply constant forces, force ramps or force jumps to systems of interest during a measurement. To implement a force clamp actively, the tension applied to the tethered molecule is chosen at a preset value and the trap/stage position is adjusted via a negative feedback loop to maintain that tension over time57. In contrast to passive implementations of a force clamp, distance changes in the tethered molecule are obtained from the changes in trap/stage position that maintain the constant tension. There are merits and drawbacks to passive versus active force clamps, with the choice of approach dependent on the application. In the force-ramp operation mode, the traps are moved monotonically (typically at tens of or a few hundred nanometres per second), stretching (or relaxing) the tethered molecule. During stretching, the end-to-end extension of the tethered molecule increases, which in turn leads to a continuous increase of the force acting on it. This operation mode is commonly used to obtain force–extension curves of a molecule and determine its elastic properties, or to obtain the rupture forces at which conformational transitions in the molecule occur. In the force-jump operation mode, the trap/stage position is changed suddenly to attain a preset force58. Usually, the tethered molecule will undergo a conformational transition in response to the force jump. For example, this measurement mode is used in mechanical unfolding applications where the force range at which the folded and the unfolded states coexist is too narrow to permit constant force or passive mode experiments13.
Fleezers
Hybrid instruments combining fluorescence with optical tweezers (fleezers) have become increasingly popular as they enable measurements of mechanical signals and forces simultaneously with fluorescence imaging or Förster resonance energy transfer (FRET) of the same system. A wide variety of fluorescence-trap instrument designs have been implemented integrating both single-trap and dual-trap configurations with wide-field epifluorescence59–61, total internal reflection (TIRF)62–65, and confocal43,66–68 and stimulated emission depletion69 microscopy (discussed below and reviewed in REFS10,11,70,71). In fluorescence microscopy, light at one wavelength is used to excite fluorescent molecules (such as organic dyes or fluorescent proteins) optically, which then emit at a longer wavelength as they relax to the ground state. Thus, only molecules of interest that are fluorescently tagged are imaged, allowing their number, position and dynamics to be tracked in real time. With FRET, an excited ‘donor’ dye transfers its energy to a second ‘acceptor’ dye that emits at a longer wavelength than the first. This energy transfer is extremely sensitive to small (sub-nanometre) changes in dye separation, making it a useful probe for intramolecular and intermolecular dynamics. These fluorescence techniques combined with optical tweezers have been used for single-colour59 and multicolour imaging60, single-molecule fluorescence counting65,72 and tracking65,69,73,74, to monitor chemical processes and displacements simultaneously73, and to measure molecular dynamics by FRET64,66,72,75–77.
Instrument layouts.
Hybrid instruments integrate standard optical trap layouts with fluorescence excitation lasers and detection optics (see, for example, FIG. 2b). Specific excitation lasers are selected to match the absorption wavelengths of the fluorescent dyes to be used. In most cases, the same objective that forms the trap(s) is used both to transmit the fluorescence excitation light into the sample chamber and to collect fluorescence emission. Dichroic mirrors and optical filters, which selectively transmit certain wavelengths and reflect or absorb others, are used to combine the trapping and excitation laser beams into the objective and to separate the trapping and emission light for their respective detection stages. In this way, trapping and fluorescence detection can be done concurrently.
The detailed layout and required optics of the hybrid instrument depend on the type of fluorescence microscopy used. In wide-field microscopy, excitation light bathes the entire sample, which is achieved by focusing the excitation beam onto the back focal plane of the objective59,60 (FIG. 3d). Fluorescence emission is collected from dyes at the sample focal plane. By contrast, with TIRF microscopy, only dyes within a thin layer (~100–200 nm) at the sample surface are excited and their emitted light collected62–65 (FIG. 3e). To achieve TIRF, the excitation beam is positioned at the edge of the objective back aperture, so that it refracts onto the chamber surface at a glancing angle and generates an exponentially decaying light field into the sample. In confocal microscopy, the excitation light is instead focused by the objective to a diffraction-limited spot (~250 nm in diameter) inside the sample, and light emitted only from this spot is collected43,66 (FIG. 3f). The fluorescence emission is focused through a pinhole aperture to reject out-of-focus light (FIG. 2b). Stimulated emission depletion microscopy, one of several super-resolution techniques, breaks the diffraction limit by selective excitation of a smaller area (~50 nm in diameter) than confocal microscopy and requires a more complex instrument set-up. The fluorescence is configured similarly to confocal microscopy, but with two pulsed, co-aligned laser systems, one generating an excitation spot and the other a doughnut-shaped deactivation spot that depletes fluorescence, leaving a small central area capable of fluorescence69.
On the detection end, an electron-multiplying charged-coupled device (EMCCD) camera is typically used with epifluorescence and TIRF microscopy where a wide field is illuminated at once and imaged. EMCCDs have the sensitivity to detect the light from a single fluorophore at each pixel over many pixels. As confocal and stimulated emission depletion microscopy require integrating light emitted from a single spot at one time, a sensitive photodetector such as an avalanche photodiode is a more economical option than an EMCCD and can provide single-photon sensitivity (FIG. 2b).
One technical hurdle in combining fluorescence detection with optical traps is that many fluorophores photobleach rapidly (within 1–2 s) when exposed to the trapping light78,79. The mechanism for photobleaching is a two-photon process involving absorption of a fluorescence excitation photon and an optical trap photon78. In applications in which light from a few dye molecules is collected, rapid photobleaching is a significant limitation. The careful selection of dyes for which absorption at trapping wavelengths is minimal can mitigate this effect64,80, but limits the dyes that can be used. A more common solution is to separate spatially the trap(s) from the dyes to be imaged. In numerous applications (see, for example, REFS60,62,65,66,81,82), the imaging area is spatially separated from the traps by the use of long tethers >10 μm in length (several times the trap diameter). However, this approach comes at a cost in trap spatial resolution, because longer molecules are less stiff mechanically and spatial resolution is proportional to the stiffness of the tethered molecule34,40,51.
An alternative solution is to separate the two light sources temporally, or to interlace them, such that a dye molecule is never exposed to both trapping and fluorescence excitation light simultaneously. In practice, the trapping laser and fluorescence excitation laser(s) are switched on and off out of phase at high rates using rapid-switching optics such as acousto-optic or electro-optic modulators79. High switching rates (typically >10 kHz) prevent the bead from diffusing out of the trapping region in the off period, avoiding the degradation of trap performance79. Interlacing has been implemented in instruments combining a single trap with TIRF microscopy79 and dual traps with confocal microscopy43, allowing the spatial overlap of trap and fluorescence light sources and the use of more compact tethers. A spatial resolution better than 1 nm over a 1-s measurement window with single-molecule fluorescence detection sensitivity was demonstrated with the latter design43. Despite the improved trap performance, this design requires a more complex instrument control/data acquisition architecture to implement the microsecond-level timing accuracy for interlacing the light sources and to synchronize trap and fluorescence data collection within the interlacing cycle43,83.
Apart from the exceptions with interlacing, the data collection and calibration in fleezers are identical to those in standard optical tweezers. Bead displacements from position-sensitive photodetectors are collected in parallel with fluorescence images from EMCCDs or photon counts from avalanche photodiodes. In addition, well-designed hybrid instruments allow the full range of operational modes (for example, force ramp and force clamp) in parallel with fluorescence modalities.
Sample preparation.
Special considerations must be made for sample preparation when incorporating fluorescence measurements. First, the molecule(s) of interest must be labelled fluorescently. DNA stains such as intercalating dyes can be used for imaging the entire molecule, and DNA oligonucleotides chemically modified for labelling with organic dyes are commercially available. Proteins can be labelled by fusion to fluorescent proteins, but these have low fluorescence emission and photobleach rapidly. Thus, proteins are more commonly covalently labelled with organic dyes. Labelling is typically achieved through cysteine-specific chemistry on the protein. Site-directed mutagenesis must often be used to remove native cysteines (whose labelling is undesirable as it can adversely affect protein activity) and to introduce cysteines at the desired positions on the protein. Owing to these difficulties, alternative labelling approaches using genetically encodable tags (such as an aldehyde tag84 and SNAP-tag85) are seeing increased use. Second, all experimental buffers for fluorescent samples must generally be supplemented with oxygen scavengers and quenchers to improve dye stability86–88.
Measurements made near the sample chamber surface (such as TIRF-based assays) also present potential fluorescence background issues due to autofluorescence and non-specific binding of fluorescently labelled molecules to the surface. Fortunately, the single-molecule fluorescence community has largely solved these problems and developed protocols for surface preparation, cleaning and passivation89,90. The trapped beads are another source of fluorescence background due to auto fluorescence and non-specific fluorophore binding. This issue can be significant for applications in which fluorescence must be detected close to the trapped beads, for example when using short tethers. To date, however, less work has been done in developing good practices for bead selection and passivation to reduce this background source.
Angular optical tweezers
In order to investigate and control rotational motion of biomolecules, optical tweezers need to be extended to trap a particle angularly as well as spatially. Stable angular trapping requires confinement of all three angles of the trapped particle orientation. AOT, also termed the optical torque wrench, have allowed stable confinement of all three angles by using birefringent cylinders made of quartz91,92. AOT are capable of direct control and detection of torque and rotation of individual biomolecules, expanding the capability of optical trapping beyond force and extension measurements. There are three core features of AOT8: a birefringent cylinder as the trapping particle, control of the cylinder orientation and detection of the torque on the cylinder. It is worth noting that torque detection has also been demonstrated using the rotary bead assay93,94 and magnetic tweezers95,96.
First, instead of trapping a dielectric microsphere, AOT trap a birefringent cylinder, which is initially nanofabricated out of a quartz wafer92 and is not commercially available (FIG. 3g; see BOX 1 for more details). The cylinder is designed to have its extraordinary axis perpendicular to its cylindrical axis and one of its ends chemically functionalized for attachment to a biological molecule of interest92. Once the cylinder is trapped, its cylindrical axis orients along the direction of light propagation as a result of shape anisotropy, and the cylinder can be rotated about its axis via rotation of the laser polarization.
Box 1 |. Principles of angular optical tweezers.
In angular optical tweezers, the most commonly employed trapping particle thus far is a quartz cylinder. Crystalline quartz has an anisotropic electric susceptibility along the extraordinary axis is greater than χo along the ordinary axes, such that the extraordinary axis of the crystal is more easily polarized than the ordinary axes. The cylinder is designed to have its extraordinary axis perpendicular to its cylindrical axis and one of its ends chemically derivatized for attachment to a biological molecule of interest92. Once the cylinder is trapped, its cylindrical axis orients along the direction of light propagation as a result of shape anisotropy. In the presence of a linearly polarized electric field of the trapping laser , the quartz cylinder develops a polarization , dependent on the electric susceptibility of the cylinder:
| (11) |
where and are unit vectors along the principal axes of the crystal. If the linearly polarized electric field makes an angle θ with the extraordinary axis (FIG. 3g), then a torque is exerted on the cylinder91:
| (12) |
where is the unit vector along the direction of light propagation, and τ0 is the maximum torque that can be exerted on the cylinder where the integration extends over the cylinder’s volume91. Thus, a torque will act on the cylinder until the extraordinary axis is aligned with , at which point . Eq. 12 also shows that the torque varies with θ sinusoidally, with a periodicity of π radians.
Second, rapid and flexible control of the input polarization of the trapping laser beam is essential, not only to rotate the trapped cylinder but also to calibrate its angular properties. These capabilities may be achieved via a pair of acousto-optic modulators that independently control the amplitudes and phases of the left (L) and right (R) circular polarizations91. The rotation angle of the polarization Θ is specified by the phase difference ϕ between the two acousto-optic modulators, Θ = ϕ / 2, and polarization may be rotated at an arbitrary rate up to hundreds of kilohertz by modulating this phase difference. Alternatively, an electro-optic modulator may also be used to rotate the laser polarization97. A trapped particle may also be rotated via a circularly polarized light98 or a Laguerre–Gaussian beam99, although their applications to single-molecule studies have yet to be demonstrated.
Third, the optical torque that rotates the cylinder is measured directly as the change in the angular momentum of the trapping laser91 (BOX 1). When the laser exerts a torque on the cylinder, the polarization of the trapping laser will change from linear to elliptical. Thus, the trapping laser that initially carried zero net angular momentum (linearly polarized) leaves the cylinder carrying a net angular momentum (elliptically polarized). This change in angular momentum results in a difference in the power of the left and right-handed circular components (PL and PR) and gives rise to an optical torque91. As shown in FIG. 2c, this optical torque is measured via the torque detector:
| (5) |
where ω0 is the angular frequency of the laser. Because the same laser is used for both torque application and detection, this torque detection method is direct and relies solely on a change in laser angular momentum.
Torque calibration.
In principle, torque may be calculated by change of angular momentum content of the laser (Eq. 5). In practice, the torque detector needs to calibrated to determine a prefactor due to photon losses in the optical system91.
The first step in the torque calibration is to relate the torque signal voltage of the torque detector Vτ, which measures PR − PL, to the cylinder’s angular displacement θ from the trapping laser’s linear polarization direction, using Eqs 5 and 12:
| (6) |
where V0 is the maximum value of the torque signal. This relationship is established via rapid rotation of the polarization (for example, 500 Hz at 10-mW input to the objective). As a trapped cylinder cannot follow such a rapid rotation due to viscous drag, the polarization vector effectively scans a quasi-stationary cylinder. Once V0 is determined, θ is then found via the torque signal Vτ according to Eq. 6.
The second step in the torque calibration is the conversion of the torque signal to physical units of torque91. This step requires the determination of the angular trap stiffness, which is obtained by measuring the Brownian angular fluctuations in θ values of the cylinder. Under a small θ value, the angular trapping potential is effectively harmonic. Using the power spectrum analysis of θ (an angular equivalence of Eq. 3), the angular trap stiffness kθ and the rotational viscous damping coefficient ξ are determined. Both kθ and ξ can be tuned by varying the cylinder size and the birefringent material100. Thus, the torque signal (Vτ) is converted to torque (τ):
| (7) |
Forces exerted on the cylinder.
In addition to torque, AOT can also exert a force on the trapped cylinder. Because the optical torque is exerted axially (Eq. 12), the optimal direction for force application is also the axial direction92. By contrast, conventional optical tweezers typically exert forces transverse to the direction of light propagation (FIG. 3b,c). In an AOT experiment, a molecule of interest (for example, DNA) may be torsionally constrained at one end to the surface of a coverslip, and at the other end to the bottom of a trapped quartz cylinder (FIG. 3g). Thus, the molecule may be twisted, while being stretched axially. This axial stretching may be achieved by moving the sample chamber via a piezoelectric stage to change the distance between the trapped cylinder and the sample chamber surface. For absolute extension measurements, the trap position at which the extension is zero must be established, for example, by finding the piezo position at which the trapped cylinder encounters resistance to further movement as it approaches the surface101.
For an oil immersion objective, the index of refraction mismatch necessitates the use of a factor called the focal shift ratio to convert piezo movement into trap height change. This shift may be determined by using the Fabry–Pérot effect that results from laser beam interference between surfaces of the cylinder and the coverslip6,101,102. Alternatively, it may be obtained by unzipping a DNA molecule of known sequence and using the resulting unzipping signature as a calibration102. Furthermore, both the force trap stiffness and the sensitivity of the z-position detector are functions of trap height. These may also be characterized using the DNA unzipping method102. Once all linear and angular parameters are determined, the AOT are then prepared for precision mechanical studies of a biomolecule.
Results
All variants of optical tweezers, including standard optical tweezers, fleezers and AOT, share the common ability to measure forces and displacements of a trapped particle. In this section, we discuss some typical results from these three types of instrument. We illustrate the use of these instruments using representative examples of operation and discuss alternative ways of operating these instruments.
Optical tweezers
Optical tweezers experiments yield rich information on the mechanical behaviour of tethered molecules, which is manifested as real-time recordings of two measurables: force and extension. Force is calculated as the product of the trap stiffness and the displacement of the trapped bead from the trap centre. Extension is calculated as the distance between the bead centres minus the radii of the two beads. Determination of the absolute extension, however, remains a challenge owing to bead size variation and instrumental uncertainty; the former source is largely dependent on the manufacturer’s quality control, whereas the latter can be mitigated by strategies implemented in the laboratory. Force–extension curves are typically generated from force-ramp experiments. These curves can be used to analyse the elastic properties of the tethered biopolymer by fitting them to theoretical models such as the worm-like-chain model of polymer elasticity103,104 (BOX 2). This type of analysis has been employed to describe the mechanical nature of nucleic acids32,105, peptides106,107, chromatin fibres108,109 and nucleoprotein filaments110.
Box 2 |. Elastic properties of DNA.
The elastic energy of DNA has multiple contributions104,250,255–257, with the following leading terms:
| (13) |
where Ebend, Etwist, Estretch and Etwist–stretch correspond to the bending energy, the twist energy, the stretching energy and the twist–stretch coupling energy, respectively. Theoretical models of DNA elasticity, built upon statistical mechanics principles, have been developed to characterize relationships among four thermodynamic mechanical quantities that are directly measurable: force, extension, rotation and torque104,250,255–257. However, the exact relationship among these quantities is complicated, and analytical solutions are typically unavailable.
The worm-like chain (WLC) model, first formulated by Kratky and Porod258, describes the elastic properties of a semi-flexible polymer in a thermal bath with a single parameter: its persistence length Lp, which can be intuitively understood as the characteristic length scale of a polymer chain beyond which the initial direction of the polymer is randomized by thermal fluctuations. The force–extension relation of double-stranded DNA (dsDNA), under no torsion and in the low force regime (<5 pN), is well described by an interpolating formula of the WLC model103,104. The approximation is given by:
| (14) |
where F is the applied force, x the molecular extension and L0 the contour length (maximum end to end distance). To extend this model to the high-force regime, a modified formula (that is, extensible WLC model) takes into account contributions from the stretch modulus K0, resulting in the following force–extension relation105 (see the figure):
| (15) |
In comparison, elastic properties of single-stranded DNA (ssDNA) are simplified as it can twist freely and does not accumulate torsion, but are complicated by the possible formation of secondary structures. Various models, including the WLC and the alternative freely jointed chain models, have been proposed to describe the force–extension relationship of ssDNA under different conditions32,183,253,259. A thorough review of the elastic properties of single-stranded nucleic acids can be found in REF.184.
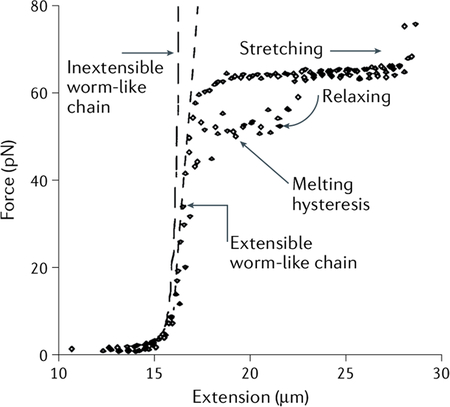
Adapted with permission from REF.32, AAAS.
When the tethered molecule is folded into higher-order structures, the force increases monotonically as the beads are separated until a sudden increase in extension (or ‘rip’), indicating the unfolding of the molecule. Conversely, when the molecule is relaxed by bringing back the beads closer together, the monotonic decrease of the force is interrupted by a sudden contraction in extension (or ‘zip’) that indicates the refolding of the molecule (FIG. 4a). Note that the zips and rips occur each time at different forces (FIG. 4b,c), reflecting the stochastic, thermally induced crossing of the energy barrier. Experiments of this type make it possible to obtain information about the energy landscape over which the molecule transits between the folded and the unfolded states. The unfolding and refolding force distributions obtained at given force-loading rates (dF/dt) can be transformed into force-dependent lifetimes of the folded and unfolded states111,112, whose inverse yields the force-dependent rates of unfolding and refolding of the molecule, respectively. The slope of the logarithm of these rates as a function of force yields the distance to the transition state (FIG. 4d), a key parameter in the description of the energy landscape of the folding process (BOX 3). The unfolding and refolding force distributions often do not coincide, indicating that the molecule is taken out of equilibrium during the process. Accordingly, the area under the rips and zips corresponds to the irreversible work done by the tweezers to unfold the molecule and by the molecule to refold against the tweezers, respectively. Powerful fluctuation theorems113,114 can be used to recuperate the reversible work or free energy of unfolding and refolding from experiments performed out of equilibrium115–117 (BOX 4). This bypasses the requirement for reversible paths and allows the deduction of equilibrium free energies from an ensemble of irreversible force–extension curves. If the zip and rip forces are not too different, then it is possible to find an intermediate range of forces at which the molecule can spend a measurable fraction of time in both its folded and unfolded states. Under these preset forces (with or without active feedback), the molecule is seen to repeatedly ‘hop’ between different states, whose force-dependent lifetimes and their associated unfolding/refolding rates can be directly measured. The folding energy landscape can thus be mapped as described above.
Fig. 4 |. Example optical trapping data.
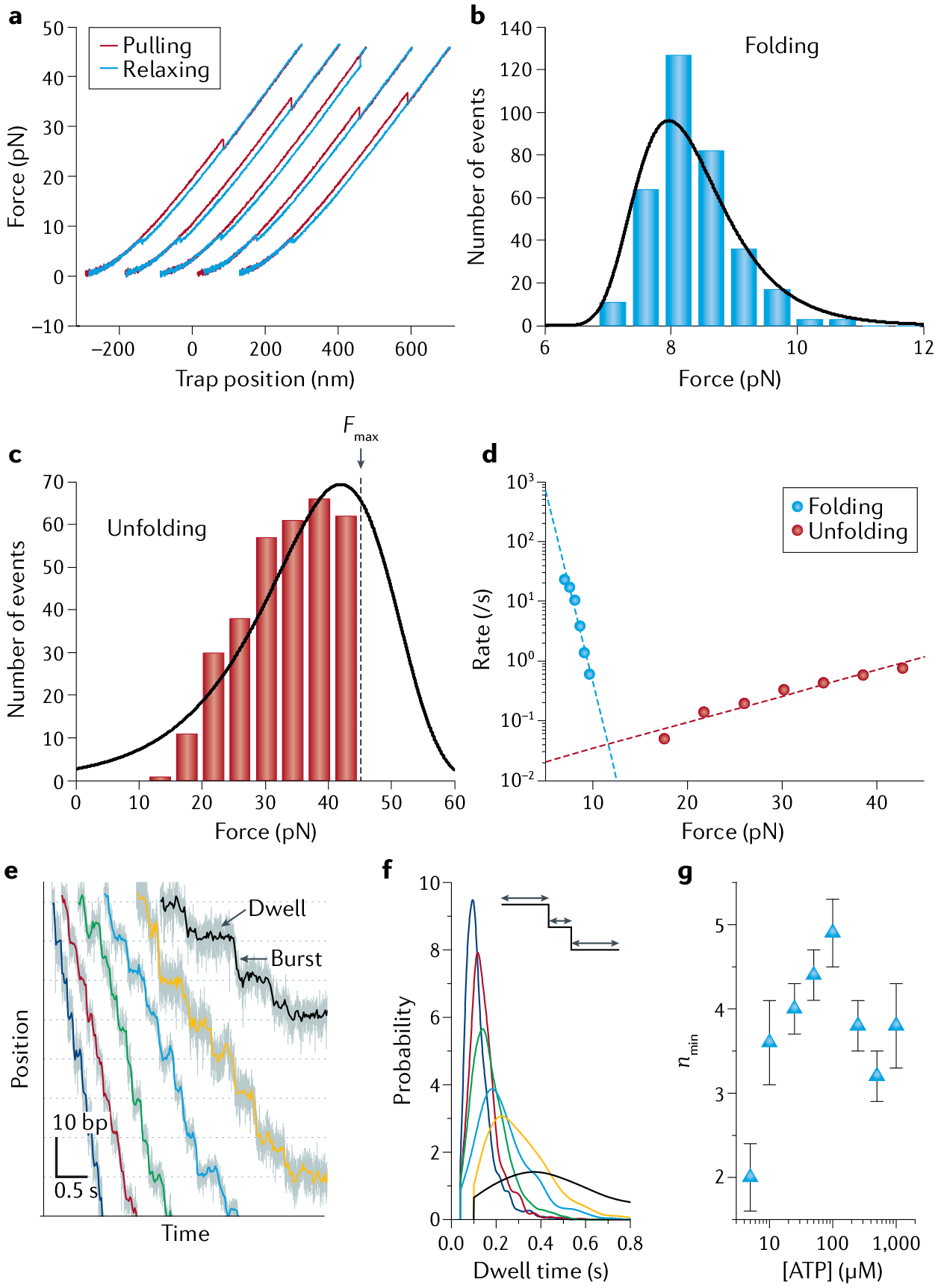
a | Force-ramp cycles of Top7, a de novo designed protein. Successive pulling (red) and relaxing (blue) cycles are offset along the x axis for clarity. b,c | Folding and unfolding force distributions, respectively, of Top7 at a pulling speed of 100 nm/s. Black lines are distributions derived from model fitting in part d. The unfolding distribution in part c is right-censored because the maximum force (Fmax) was set at 45 pN during pulling experiments to avoid tether rupture. d | Force-dependent rates of unfolding (red dots) and refolding (blue dots) extracted from the corresponding force distributions in parts b and c. Dashed lines are fits to Bell’s model254. e | Representative trajectories showing the processive translocation of individual φ29 packaging motors on double-stranded DNA (dsDNA) under a constant force of ~8 pN and different [ATP] (250 μM, 100 μM, 50 μM, 25 μM, 10 μM and 5 μM in blue, red, green, cyan, yellow and black, respectively). Each translocation cycle is composed of a stationary dwell phase and a stepping burst phase. f | Probability distributions of the lifetimes of the dwell phase at different [ATP]. Colour scheme as in part e. g | Values of nmin, the minimum number of rate-limiting kinetic events during the dwell, derived from the dwell time distributions for different [ATP]154. Parts a–d adapted with permission from REF.254, AAAS. Parts e–g adapted from REF.154, Springer Nature Limited.
Box 3 |. Determining the energy landscape of molecular transitions.
Optical tweezers have enabled the characterization of several parameters of the energy landscape for many biomechanical reactions such as RNA/protein folding and molecular motor-driven processes118,211. The application of force F on the molecule amounts to subtracting a term Fx from the free energy surface, where x is the reaction coordinate. The result is to tilt this surface, shifting the equilibrium towards the longer (unfolded) state of the molecule, favouring the spontaneous, thermally induced crossing of the energy barrier between the two states (see the figure).
The effect of force on the equilibrium between the folded and unfolded states is described by211:
| (16) |
where Keq(F) is the force-dependent equilibrium constant, ΔG0 the standard Gibbs free energy (where the superscript denotes the standard state), Δx the difference in extension between the folded and unfolded states at F = 0, and ΔGstretch = Gstretch,2 − Gstretch,1 is the difference in the free energy to stretch the folded (1) and the unfolded (2) states to their respective extensions at force F. Gstretch,i for each state i is calculated by integrating the force over the extension; that is, determining the area under the force–extension curve Fi(x) for that state:
| (17) |
Fi(x) would typically be given by the WLC model in Eq. 14 or 15. At the force at which the molecule spends the same amount of time in the folded state and in the unfolded state F1/2, Keq(F1/2) = 1 and:
| (18) |
Similarly, force increases the unfolding rate by lowering the energy barrier to unfolding. This effect can be approximated by Bell’s model260,261, which reduces the energy to the transition state, ΔG‡, in the standard Arrhenius equation, by the term −Fx‡, where x‡ is the distance to the transition state:
| (19) |
This is the simplest model to describe the dependence of the kinetic rates on force. However, it is phenomenological and does not always accurately describe the experimental data. The observation of a non-linear dependence of ln(k) on F opposite to the linear dependence predicted by Eq. 19 represents the breakdown of Bell’s model262,263. In this case, a more rigorous treatment such as that of Dudko et al. derived using Kramer’s theory263 should be used.
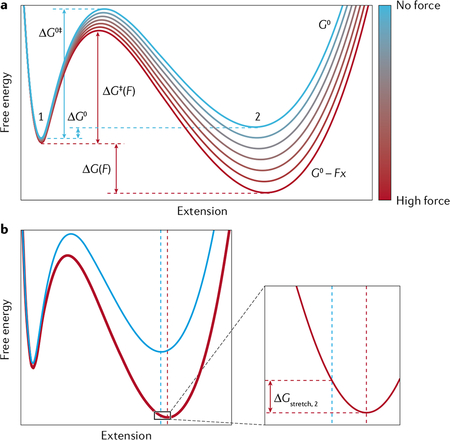
Adapted with permission from REF.13, Annual Reviews.
Box 4 |. Fluctuation theorems.
The second law of thermodynamics states that the mean value of the work (W) done on a system is always larger than or equal to the free energy change (ΔG) of that system depending on whether the process is carried out irreversibly (Wirrev, with dissipation Wdiss) or reversibly (Wrev), respectively:
| (20) |
In 1997, Christopher Jarzynski showed that the Gibbs free energy change for a process (RNA unfolding, for example), could be recovered from the Boltzmann average of the Wirrev values obtained over N realizations performed out of equilibrium, in the limit of large N113,264:
| (21) |
A second result, the Crooks fluctuation theorem (CFT)114,265 relates the probability distribution PF(W) of the work done on a system (W) along a forward (F, unfolding for example) trajectory, and the distribution PR(−W) of the work done by the system (−W) along the reverse (R, refolding) trajectory, with the amount of dissipation in the process (W − ΔG):
| (22) |
The amount of dissipation can be increased or decreased by pulling/relaxing the molecule faster or slower, respectively. Note that when W = ΔG, PF(W) = PR(−W). In other words, the value of W at which both distributions (unfolding and refolding) cross is the free energy change of the process ΔG (see the figure, depicting the test of CFT by unfolding and refolding an RNA hairpin at different loading rates).
Both the Jarzynski and Crooks relationships permit obtaining the free energy of a process from non-equilibrium realizations of that process and have found particularly useful applications in the analysis of single-molecule optical tweezers experiments117,266. These methods work because fluctuations occur around the mean and, although more often than not non-equilibrium trajectories dissipate work, once in a while a fluctuation occurs in which the work done irreversibly is smaller than the reversible work, that is Wirrev < Wrev. This rare situation is reflected in the tails of the distributions on both sides of the value of ΔG (grey dashed line) in the figure. These irreversible work values are smaller and contribute more to the average in Eq. 21. Thus, if enough of them have been recorded (N is large), they can offset the more common, larger irreversible work values, yielding the correct value of the free energy. The larger the amount of dissipation (or hysteresis in the force–extension curves), the greater the number of iterations required for the left-hand side of Eq. 21 to converge to its right-hand side. For dissipations that are not too large (~3–4kBT), ~50 iterations may be necessary, but this number increases fast with the amount of dissipation. In general, the Crooks formulation converges faster than its Jarzynski counterpart, and thus can be used to estimate free energy changes in systems with larger dissipations. Related theorems have been derived during the last few years. A particularly useful one is an extension of the CFT that makes it possible to extract the binding free energy from ligands bound to a macromolecule that is subjected to binding and unbinding via out of equilibrium mechanical unfolding and refolding267.
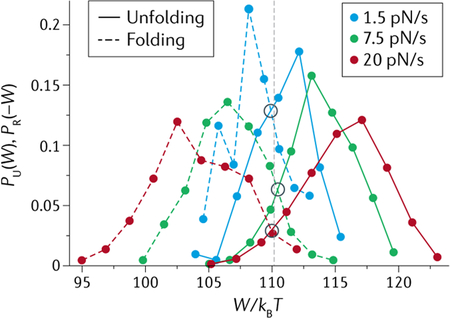
Adapted from REF.117, Springer Nature Limited.
Single-molecule recordings of extension as a function of time generated from constant-force experiments are often used to study dynamic biomolecular processes ranging from reversible interconversions in folding/unfolding of nucleic acids and proteins118 to processive movements of molecular motors119. Various step-finding algorithms, including those based on the residual χ2, the Bayesian information criterion, hidden Markov modelling or machine learning, have been developed to idealize noisy trajectories into discrete stepwise transitions120–124, yielding characteristic step sizes and dwell times (FIG. 4e). The step size (change in extension per transition) can also be inferred from periodogram analysis such as pairwise distance distributions and autocorrelation functions26,125. The dwell time t (duration between steps) harbours all kinetic transitions that precede the mechanical stepping. The shape of the dwell time distribution, which informs on the underlying kinetic mechanism of the biomolecular process, can be formulated by the randomness parameter r or its inverse, nmin (REFS126,127) (FIG. 4f,g):
| (8) |
An r value of 1 corresponds to a single-exponential decay, indicating that a single kinetic step dominates the dwell duration; values of r < 1 correspond to a peaked distribution and indicate at least nmin kinetic steps during the dwell; values of r > 1 typically correspond to a multi-exponential, long-tailed distribution and indicate the existence of parallel pathways or off-pathway states in the kinetic scheme.
Fleezers
Fleezers provide additional and complementary information about the system to that obtained with optical tweezers alone. Increasingly, applications of fleezers have exploited the full, combined power present in simultaneously acquired trap and fluorescence data to carry out correlative analysis of the two (reviewed elsewhere10,11,34,70). A popular example is wide-field epifluorescence or TIRF imaging of DNA and DNA-binding proteins (FIG. 5). The ability to ‘see’ fluorescently labelled proteins allows their location on DNA to be determined as a function of time and applied force59,60. Force is used in combination with microscopy to control the extension of the tethered DNA, which in turn can modulate the location and dynamics of the bound proteins that are imaged59,60. Where fluorescently labelled molecules are sparse and can be localized individually, fluorescence imaging can be used for tracking measurements. The fluorescence intensity profile of an individual dye molecule is well described by a 2D Gaussian function. Thus, the centre-of-mass position of a labelled molecule in each frame of the fluorescence movie is determined by fitting its image to a 2D Gaussian function, and its motion is tracked as a function of force61,63,65,69. A common analysis tool used in imaging applications is the kymograph, in which the fluorescence intensity profiles in one dimension are stacked in a time series to reveal the molecular trajectories of the labelled components. FIGURE 5a–c illustrates this analysis method, where a filament of the fluorescently labelled recombinase RAD51 on DNA is stretched with dual traps and imaged by epifluorescence microscopy81. To create the kymograph, a line scan of RAD51 fluorescence along the stretched tether is obtained for each frame of the fluorescence movie. The line scans are then stacked into a single image, with the position on the tether along one axis (FIG. 5b, y axis) and the frame number (or, equivalently, time) along the other axis (FIG. 5b, x axis). The kymograph thus shows both the location of the bound proteins along the tether and their bound lifetime. In this particular example, the trap is operated in a passive mode, and an increase in force (measured by optical trap) coincides with the DNA contraction triggered by RAD51 dissociation (measured by fluorescence; FIG. 5c).
Fig. 5 |. Example fluorescence-trap data.
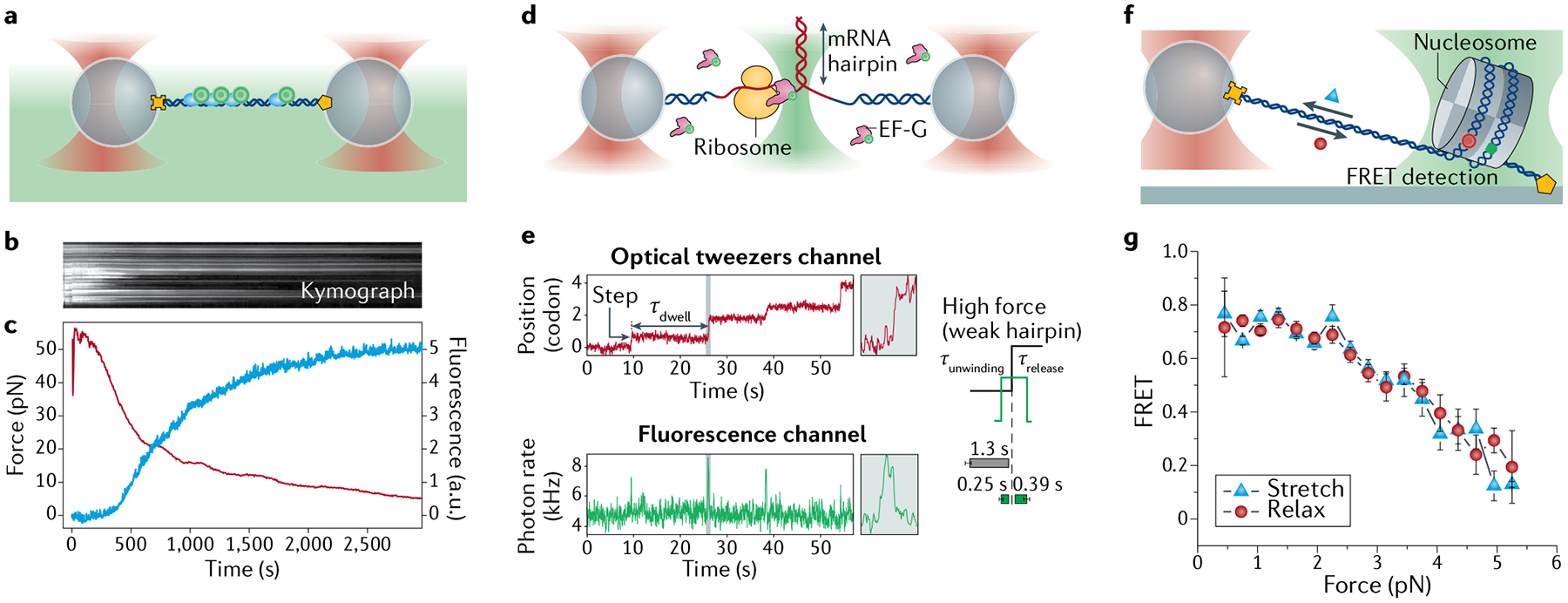
a | Dual-trap plus epifluorescence configuration, showing labelled RAD51 bound to DNA (DNA functionalized at both ends (yellow cross and pentagon) and fluorescence collected from dye (green circles)). b | Kymograph of a RAD51–double-stranded DNA (dsDNA) complex, held at fixed length, triggered to disassemble. c | Fluorescence intensity (red) decreases as tension (blue) increases owing to disassembly-induced DNA contraction81. d | Dual-trap plus confocal configuration, showing a ribosome opening a messenger RNA (mRNA) hairpin and labelled elongation factor G (EF-G) binding. e | Time trace of ribosome translocation in one-codon steps separated by dwells of duration τdwell (red) with simultaneous fluorescence intensity (green) indicating binding and dissociation of EF-G73. Average EF-G residence times before mRNA unwinding (τunwinding) and after unwinding (τrelease) are displayed for a weak hairpin at high force. f | Surface-based, single-trap plus confocal configuration, showing a single nucleosome bound to Förster resonance energy transfer (FRET)-labelled DNA under force. g | Resulting FRET decrease versus force as the nucleosome is unravelled mechanically76. Parts b and c adapted from REF.81, Springer Nature Limited. Parts d and e adapted with permission from REF.73, Elsevier. Parts f and g adapted with permission from REF.76, Elsevier.
The time course of the fluorescence intensity is another result that can be combined with optical trap data. The onset and disappearance of fluorescence signals can determine the bound lifetime of a labelled molecule on a tethered molecule under force62,128. Co-temporal analysis of fluorescence and mechanical signals can further uncover causal relationships between molecular processes, revealing, for example, how molecular motor dynamics correlate with binding of fluorescently labelled substrates or molecular partners62,73. FIGURE 5d,e shows an illustrative example of co-temporal analysis, where ribosome translocation on RNA is monitored with optical traps while binding of fluorescently labelled elongation factor G (EF-G) is detected simultaneously with confocal microscopy73. Analysing the time traces in both channels reveals when EF-G binds and unbinds in relation to the ribosome’s translocation by one codon (three nucleotides) (FIG. 5e). Fluorescence intensity can also be used to count labelled molecules. Over time, fluorescent dyes photobleach, undergoing an irreversible transition into a dark state. This behaviour can be exploited to count the number of labelled molecules at one position, by counting the number of stepwise drops in fluorescence intensity as each dye expires129. Thus, the stoichiometry of a molecular complex may be determined by fluorescence and correlated to its dynamic behaviour as measured by optical trapping. An example of this approach determined how the monomeric versus dimeric forms (counted by fluorescence) of a helicase impact its DNA unwinding activity (measured by an optical trap)65,72. Intensity-based counting approaches are useful in situations where it is impossible to rely on spatial localization to distinguish multiple proteins.
Finally, spectroscopic data obtained from single-molecule FRET86 can be used to probe conformational dynamics in a molecule or complex labelled with a donor–acceptor dye pair. As FRET measures the distance separating the dye pair, it can be combined with optical trapping to monitor, as a function of force, distance changes due to conformational interconversions of nucleic acid structures and nucleoprotein complexes66,75–77,130. An illustrative example of this approach is shown in FIG. 5f,g, in which a single nucleosome is unravelled by force76. Here, the DNA on the histone is labelled with a donor–acceptor dye pair at positions selected such that the two dyes are close when DNA is wrapped (leading to high FRET and acceptor emission) and are far apart when the DNA is unwrapped (leading to low FRET and donor emission). As a force ramp is applied with the optical trap to stretch and relax the tethered DNA, the donor and acceptor dye emission intensities are measured. FIGURE 5g shows how the FRET efficiency — determined from the relation E = IA/(ID + IA), where IA and ID are the acceptor and donor intensities, respectively — decreases as force increases and the nucleosome is unravelled. This approach can be extended to study protein diffusion on DNA by placing one dye of the FRET pair on the protein and the other on the DNA75. As FRET provides measurements of inter-dye distances independent of those obtained from the optical traps, it can be used to track reaction coordinates to which the traps are insensitive. The access to ‘orthogonal’ reaction coordinates enables mapping of multidimensional energy landscapes66,77. As with fluorescence intensity, co-temporal analysis of FRET and mechanical signals can reveal correlations between the conformational states and dynamics of molecular motors72.
Angular optical tweezers
AOT can simultaneously measure multiple physical parameters — torque, angle, force and displacement — and may be operated in different modes depending on the needs of a specific application. The most commonly employed mode is the ‘rotation’ mode, where a trapped cylinder attached to the molecule of interest is twisted via rotation of the laser polarization, as the cylinder closely tracks this polarization92,131,132. This mode is well suited for torsional studies of chromatin fibres and is illustrated with an example in FIG. 6a171. Here, one end of a chromatin fibre was torsionally constrained to the surface of a microscope coverslip and its other end was torsionally constrained to the bottom of a quartz cylinder. The cylinder was first stretched axially and held under a constant force. To introduce turns into DNA, the cylinder was rotated at a constant rate, and the torques exerted on the DNA and the DNA extension were recorded from the change in the polarization of the light and the displacement of the cylinder from the trap centre, respectively (FIG. 6b,c). The measured torque provides information about the torsional modulus of the nucleosomal DNA (FIG. 6c). To date, torques as small as 1 pN·nm have been detected using AOT133.
Fig. 6 |. Example angular optical tweezers data.
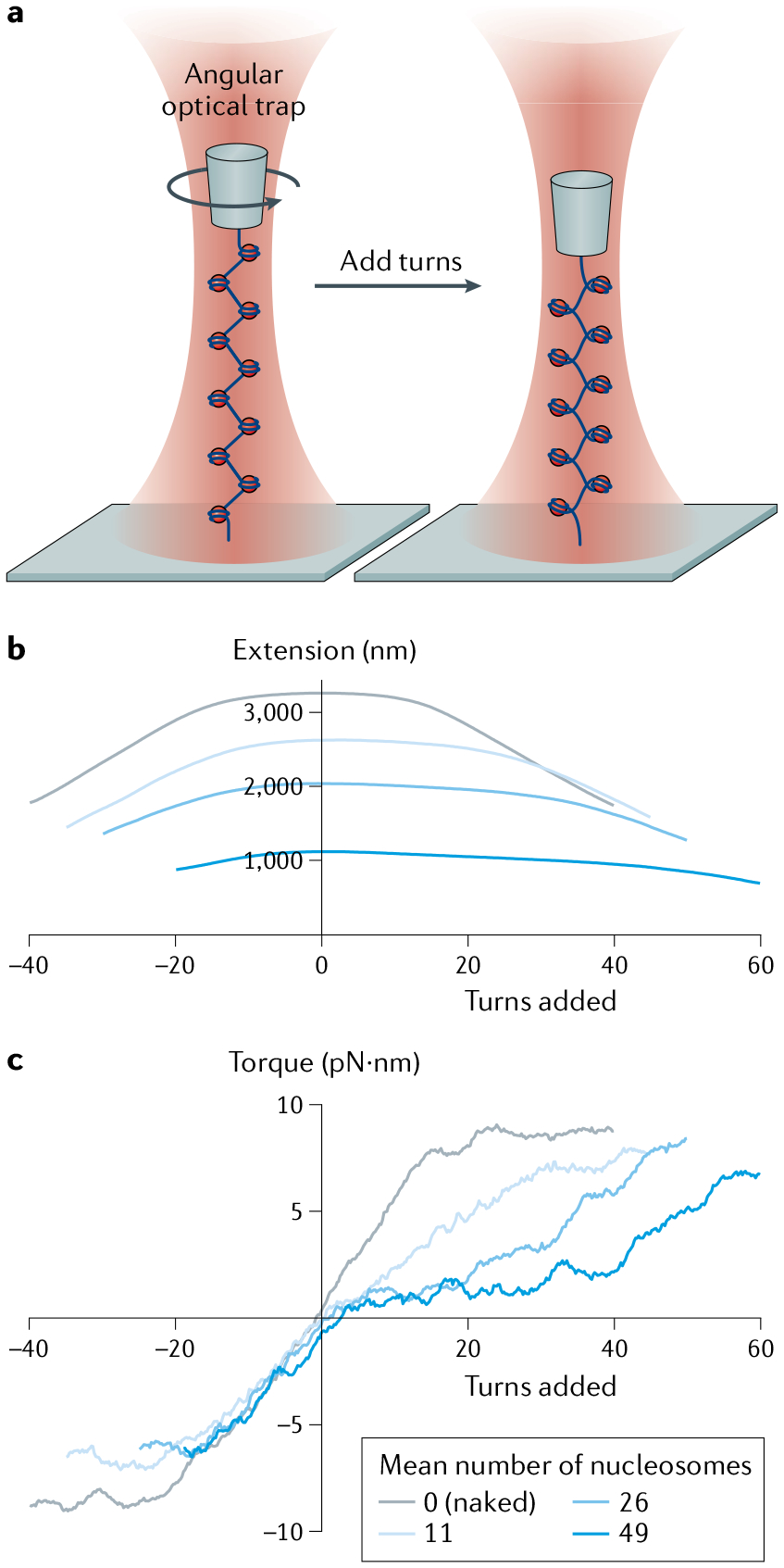
a | Experimental configuration for torsional measurements of a chromatin fibre. Angular optical tweezers are used to twist a chromatin fibre at 4 turns/s, while holding the chromatin fibre under a constant force of 0.5 pN. b | Resulting extension. c | Resulting torque. Measurements are conducted on chromatin fibres containing different numbers of nucleosomes. Extension and torque signals are smoothed by sliding windows of 1 turn and 4 turns, respectively. Torsional properties of each type of substrate may be determined from the torque–turn relation. Adapted with permission from REF.171, Elsevier.
In addition to the rotation mode, AOT can also operate in an active torque wrench mode where a constant torque is maintained on a trapped particle via active feedback on the input polarization angle91. This mode is best suited for applications where the measured torque is much greater than the torque noise. For applications requiring zero or extremely low constant torque, the passive torque wrench mode may be employed134. In this latter mode, a constant optical torque is achieved by a rapid rotation of the laser polarization at a rate much greater than the rotational corner frequency of the cylinder, such that the trapped particle is not able to follow the rotation. This method bears some resemblance to that used during the torque detector calibration. This leads to a constant torque exerted on the particle, with the torque tunable by the polarization rotation rate. In both the active and passive torque wrench modes, torque is directly measured.
Applications
In this section, we discuss several key areas of research where optical tweezers have made revolutionary advances. Although the initial applications of optical tweezers were centred on cytoskeletal motors, the use of optical tweezers rapidly expanded into problems centred around nucleic acids, nucleic-acid based processes and protein folding. In this section, we hope to give the reader a sense of the types of problem that can be uniquely addressed with optical tweezers. Instead of providing an exhaustive list of the applications, below we highlight applications related to nucleic acids, protein–nucleic acid interactions, RNA and protein folding and unfolding, and mechanochemistry of nucleic acid-based motor enzymes.
Protein–nucleic acid interaction studies
DNA is an essential component of protein–DNA interactions, and thus its elastic properties play a direct role in regulating these interactions. In addition, DNA is often used as a molecular handle to study other processes, such as protein folding, and a thorough characterization of the DNA handles is required to subtract their contribution to the overall extension and twist.
Conventional optical tweezers and AOT are excellent tools for the measurement of DNA elastic parameters (BOX 2) such as the persistence lengths of dsDNA and single-stranded DNA (ssDNA)32,105, the stretch moduli of dsDNA and ssDNA32,105, the twist modulus of dsDNA93,131,135 and the twist–stretch coupling modulus of dsDNA136,137 (TABLE 1). The elastic parameters of single-stranded nucleic acids are known to be sequence-dependent138,139, and thus only representative examples are shown in TABLE 1. These studies also showed that when subjected to high forces, dsDNA undergoes an overstretch phase transition32,140–143, and, when subjected to twist, dsDNA transitions to different phases92,131,132,135,137. These behaviours are concisely described in a force–torque diagram8,93,135.
Table 1 |.
Intrinsic elastic parameters of nucleic acids
| Parameter | Value |
|---|---|
| dsDNA persistence lengtha | |
| 43 nm (REF.105) | |
| dsDNA stretch modulus | 1200 pN (REF.105) |
| dsDNA twist persistence lengtha | |
| 102 nm (REFS131,250) | |
| dsDNA twist-stretch coupling modulus | |
| 21 (REF.137) | |
| ssDNA persistence lengtha | |
| 0.71 nm (REF.163) | |
| ssDNA stretch modulus | |
| 420 pN (REF.163) | |
| dsRNA persistence lengtha | 60 nm (REF.251) |
| ssRNA persistence lengtha | 1.3 nm (REF.183) |
| ssRNA stretch modulus | 150 pN (REF.183) |
Measurements were conducted under various physiological buffers. For simplicity, only representative average values are provided. Note that the values of these parameters depend on the ionic composition of the medium183,252,253.
dsDNA, double-stranded DNA; dsRNA, double-stranded RNA; ssDNA, single-stranded DNA; ssRNA, single-stranded RNA.
Multiply by kBT (where kB is the Boltzmann constant, T is the temperature) to obtain the corresponding modulus.
Once the elastic properties of DNA have been well characterized, optical tweezers can then be used in various experimental configurations to investigate DNA processing enzymes and other protein–DNA interactions. Below, we highlight three primary configurations — stretching, unzipping and twisting — each tailored to a specific type of application.
The DNA stretching configuration is the most widely employed configuration and can operate under two primary modes: disruption mode or tracking mode (FIG. 7a). In the disruption mode, a DNA molecule is stretched to disrupt a bound protein, providing information on the disruption force and the length of DNA released from the disruption144–147, which characterize the protein–DNA interactions in terms of the strength of the interaction and the amount of DNA associated with the interaction, respectively. In the tracking mode, a motor protein moving along DNA is anchored to a surface or trapped bead, whereas the upstream or downstream end of the DNA is anchored to another trapped bead or surface. The position of the motor protein on DNA is indirectly inferred from changes in the end-to-end distance between the motor protein and the end of the DNA29,31,148–153. This approach is useful when studying processive motors such as RNA polymerase. For distributive motor proteins, that is, those displaying small processivity such as DNA polymerases, motor movement may also be tracked via the difference in elastic response between ssDNA and dsDNA30. Advanced optical tweezers are capable of detecting motor movement at base pair-scale resolution39,40,124,154–156.
Fig. 7 |. Example applications of optical tweezers to study protein–DNA interactions.
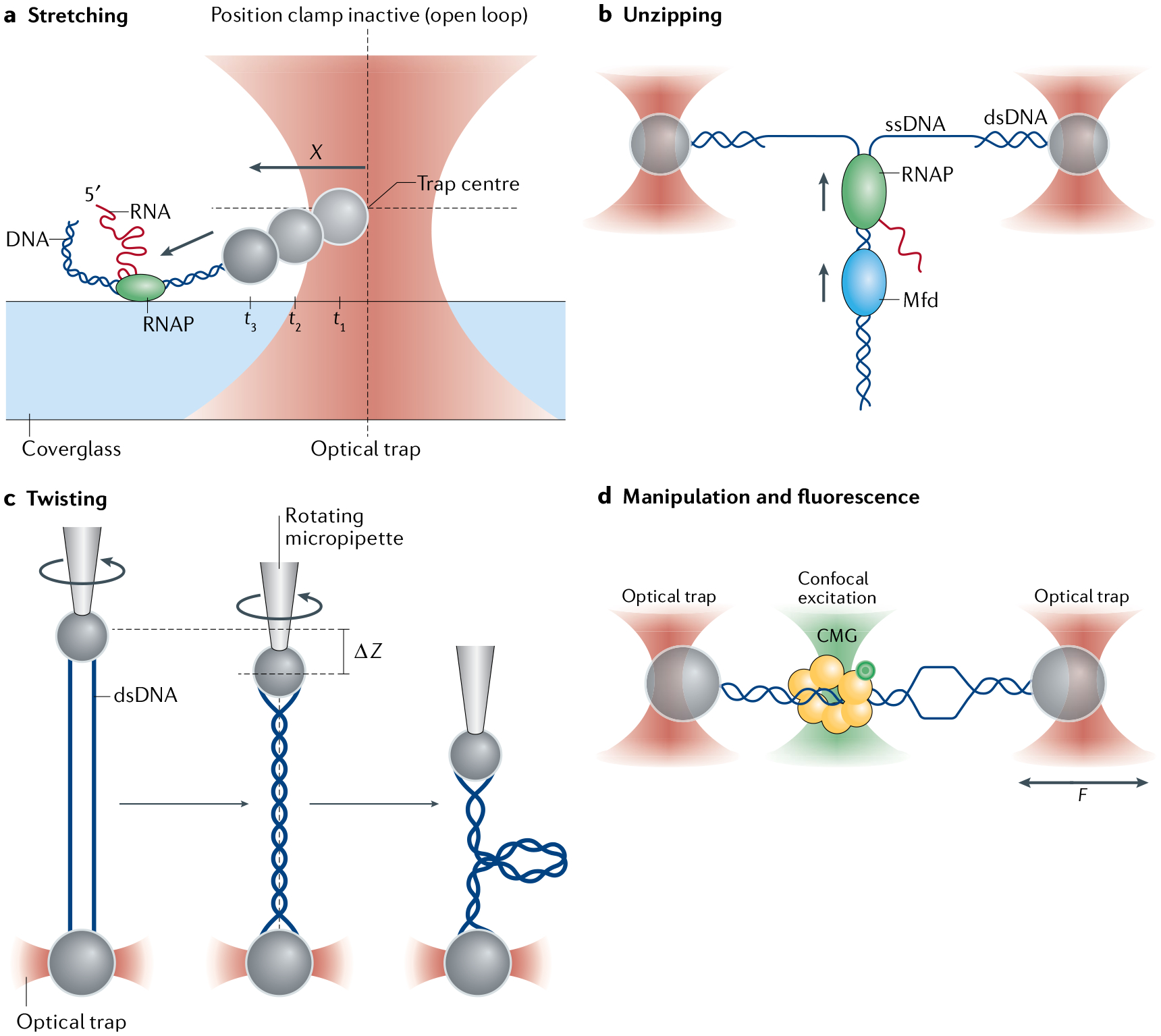
a | By stretching a double-stranded DNA (dsDNA) molecule, RNA polymerase (RNAP) is tracked in real time and stalled under an applied load148. t1, t2 and t3 represent three successive time points as the bead is pulled from the trap centre. b | By unzipping a dsDNA molecule, RNAP, which is trailed by another motor, Mfd, is tracked in real time169. c | By twisting two dsDNA molecules via a combination of optical tweezers and a micropipette, a DNA braid is formed and used to study topoisomerase relaxation174. d | By combining fluorescence detection and force manipulation, the replicative helicase CMG is observed to dynamically transition between single-stranded DNA (ssDNA) and dsDNA178.
The DNA unzipping configuration can employ versatile and flexible modes of operation to probe protein–DNA interactions (FIG. 7b). In the ‘unzipping mapper’ mode, dsDNA is mechanically separated into two single DNA strands to disrupt bound proteins. This approach has been used to characterize the strength of the dsDNA base pairing as a function of sequence157 or to generate a high-resolution map of both the locations and strengths of these protein–DNA interactions158–162. Recent improvements in this technique enable detection of the absolute location at sub-base pair accuracy and precision163. The ‘unzipping tracker’ mode allows detection of motor protein translocation without the need to tag the motor. Unlike the ‘unzipping mapper’ that disrupts a bound protein, this mode is typically operated under a constant force that is high enough to keep the DNA unzipped but low enough not to displace the motor protein off the DNA. The fork position indicates the location of a motor protein interacting with the fork junction via binding to either the ssDNA (as with helicases and DNA polymerases)164–168 or the dsDNA (as with dsDNA translocases)169. If the motor moves towards the fork, the DNA is re-zipped, whereas if the motor moves away from the fork, the DNA is unzipped. A variation of the ‘unzipping tracker’ mode is the ‘unzipping staller’ mode that uses the DNA fork as a barrier to stall a DNA translocase moving towards the fork169.
Finally, the DNA twisting configuration using the AOT (FIG. 7c) allows studies of biological processes under both torsion and force. Some examples include disrupting protein–DNA interactions under torsion170, measuring the torsional stiffness of a substrate (for example, single and braided chromatin fibre)171 and measuring torque generated by motor proteins172,173. In addition, optical tweezers can be combined with other manipulation tools such as a rotary micropipette and a rotor bead, providing alternative means of investigating torque and rotation of DNA-based processes93,136,153,174.
Fluorescence allows visualization of the locations of molecules, such as proteins and regions of dsDNA and ssDNA60,63,75,81,175–178, as well as detection of conformational dynamics72,77,179 (FIG. 7d). More complex DNA geometries can be used to enable investigation of correspondingly more complex processes. For example, proteins mediating the bridging of two DNA molecules are detected whereas the two DNA molecules are manipulated using multiple optical traps180,181. A DNA ‘Y-structure’ technique, mimicking a DNA replication fork, allows a bound protein to be visualized while the DNA is simultaneously stretched, unzipped and twisted182.
Optical tweezers have also been employed to determine single-stranded RNA and double-stranded RNA elastic parameters183,184 (TABLE 1). These RNA elastic properties have enabled the investigation of RNA-interacting proteins such as RNA helicases that unwind double-stranded RNA164,185 and the ribosome that translocates on single-stranded RNA and invades double-stranded RNA structures73,186–188.
Protein folding studies
There are several reasons for using controlled force to study molecular folding. First, the application of the force establishes both a well-defined reaction coordinate and a potential energy surface over which the molecule’s folding/unfolding transitions take place. Second, unlike its thermal and chemical counterparts, which act globally, force is a selective denaturant that can be applied to one part of the molecule without acting on another. Finally, force may be the more biologically relevant denaturing agent: proteins targeted for destruction in the cell are processed by specialized molecular machines that use the energy of ATP hydrolysis to generate force and mechanically unfold their client proteins44,49.
During folding, proteins often populate intermediate states. Although it is difficult to determine whether these intermediates are on-pathway or off-pathway to the native form using bulk studies, optical tweezers folding studies have made it possible to characterize the nature of these intermediates. Cecconi et al. studied RNaseH and found that the molecule unfolded at 19 pN and refolded at ~5 pN, populating an intermediate that was shown to be on-pathway or obligatory to the native state45, as attaining the native state occurred invariably from the previously formed, transient intermediate. Further characterization of this intermediate revealed that it was a molten globule state.
Using passive-mode optical tweezers, Stigler et al. characterized the folding pathway of calmodulin, a eukaryotic Ca2+-binding protein made up of amino and carboxy-terminal domains, each of which is formed by two EF-hand motifs47 (FIG. 8a). Full and truncation fragments of the protein were used to uncover up to six states in the folding pathway using an ultra-stable instrument and applying a hidden Markov model analysis (FIG. 8b). Transitions between different states but involving similar changes in extension were distinguished by the lifetimes of the initial and final states or by their different connectivities to other states. The authors determined the complete native folding and misfolding pathways.
Fig. 8 |. Example applications of optical tweezers to study protein folding.
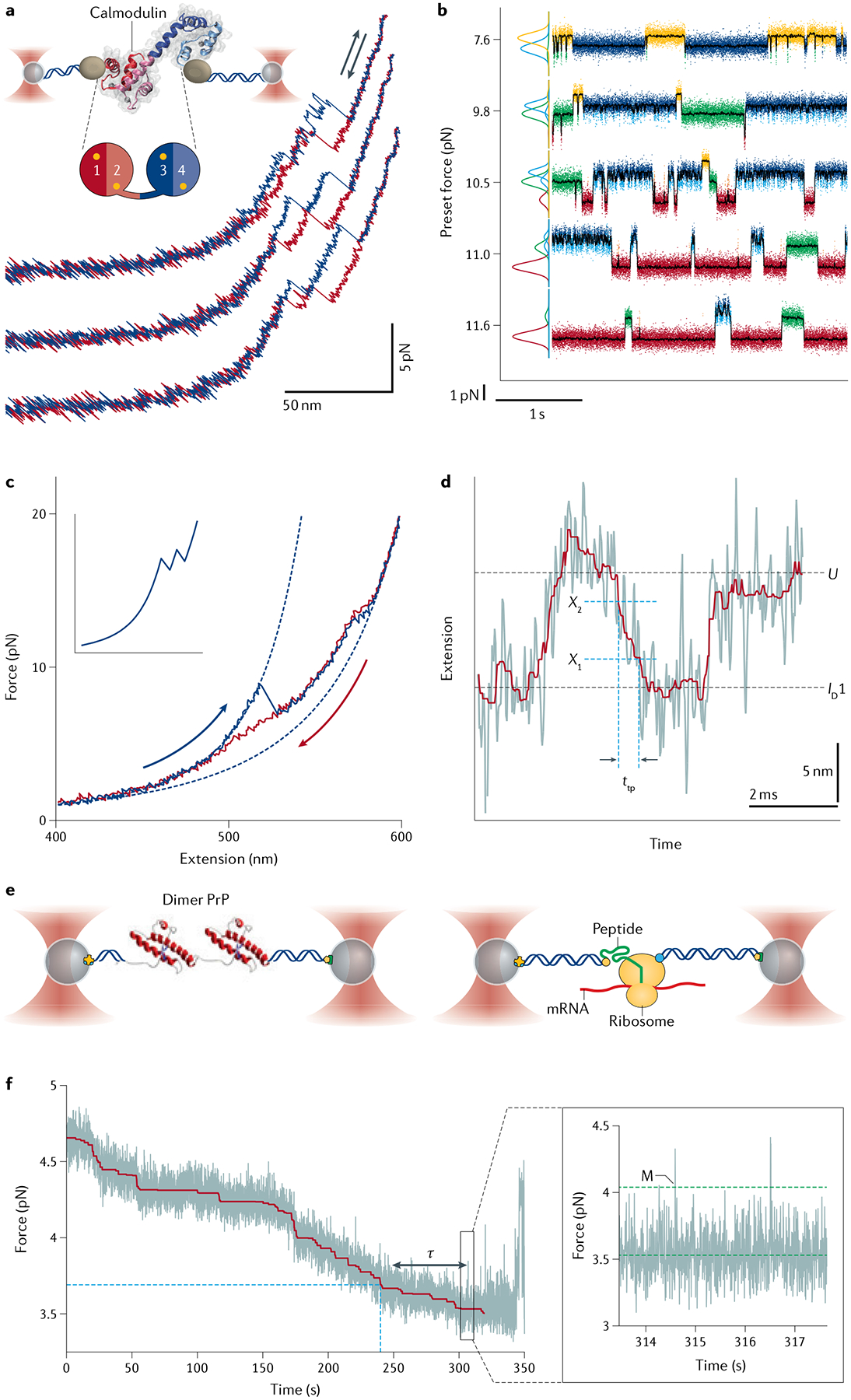
a | Experimental geometry and representative force–extension curves for studying the folding and unfolding of single calmodulin molecules. Stretch and relax cycles are shown in blue and red, respectively. b | Traces of wild-type calmodulin showing folding/unfolding transitions at different preset forces. Data taken in a passive mode. As the preset force increases, the equilibrium is tilted from the native folded state (yellow, all four EF hands folded, F1234) to the unfolded state (red). Four intermediates (blue, F123 folded; cyan, F12 or amino-terminal domain folded; green, F34 or carboxy-terminal domain folded; orange, F23 folded) are also populated in a force-dependent manner. Gaussian fits to histograms of each state are shown on the left47. c | Force–extension curves for studying the misfolding of prion protein (PrP) dimers. Unfolding (blue) and refolding (red) curves show the formation of stable non-native structures, in contrast to the expected sequential unfolding of independently folded native domains (inset). d | Transition path time (ttp) for misfolding measured from constant-force trajectories. Barrier region defined by boundaries X1 and X2 (REF.193). e | Left: experimental geometry for studying the PrP dimers. Right: experimental geometry for studying co-translational folding. DNA handles are attached to the N terminus of the nascent chain and the ribosome itself. f | Example trajectory of real-time translation elongation. The force decreases as the tether gets longer. Blue dashed line corresponds to the codon position where folding would begin under equilibrium conditions. Inset: zoom image showing the folding transitions. ID1, first dimer intermediate state; M, misfolded state; mRNA, messenger RNA; τ, non-equilibrium delay prior to folding210; U, unfolded state. Parts a and b adapted with permission from REF.47, AAAS. Parts c, d and e (left panel) adapted with permission from REF.193, PNAS. Parts e (right panel) and f adapted from REF.210, Springer Nature Limited.
The question of whether individual domains within a protein fold or unfold cooperatively has been investigated using optical tweezers. Shank et al. investigated the folding/unfolding cooperativity of the N-terminal and C-terminal subdomains of T4 lysozyme46. This protein is organized in a re-entrant topology in which the first 12 residues of the N terminus fold as a helix with the C-terminal domain in the tertiary structure. When pulled across both domains, the protein showed a single ‘rip’ (~50 pN) corresponding to the concerted unfolding of both domains. When relaxed, the protein displayed a single ‘zip’ (at ~5 pN) reflecting the simultaneous refolding of both domains. When the force was only applied to the N terminus of the full protein, a single shorter unfolding rip was observed at even higher forces, suggesting that the process also involved the unfolding of the C terminus. Because the unfolding and refolding occurred out of equilibrium, the Crooks fluctuation theorem (BOX 4) was used to obtain the free energy of unfolding of the N-terminal domain. The value was 12.3 kcal/mol, very close to the 14.1 kcal/mol determined for the unfolding of the whole protein by bulk methods, indicating that unfolding of the N-terminal domain led to the unfolding of the C-terminal domain, demonstrating strong coupling and cooperativity in the folding/unfolding transitions of both domains. A circular permutant version of the protein that eliminates its re-entrant topology displayed two unfolding/refolding transitions, indicating that its topology was responsible for the folding cooperativity of the wild type. The authors speculate that evolution may have favoured re-entrant topologies to ensure coordinated folding of domains.
Protein misfolding is likely favoured by the ruggedness of their folding energy landscapes, leading to aggregation. Prion proteins (PrPs) are known to misfold and produce self-propagating aggregated states. Yu et al. showed that the PrP transiently populates three different misfolded states not detected by bulk methods189 (FIG. 8c–e). All three states only formed from the unfolded state. By contrast, folding to the native state occurred as a simple two-state process. Optical tweezers have also been used to resolve some of the complexities arising from intra-domain and inter-domain misfolding in large, multidomain proteins. Jahn et al. dissected the folding of HSP90, a molecular chaperone that functions as a homodimer, with each monomer made of N-terminal, middle and C-terminal domains48,190. Isolated, the N-terminal and middle domains exhibit very fast misfolding. Interestingly, the folding rates for these isolated N-terminal and middle domains are maximal at approximately 3.5 pN and lower at smaller forces, indicating that the potential energy surface bifurcates into a folding and a misfolding path, and that tension suppresses the latter over the former48. Likewise, force also appears to reduce inter-domain interference and misfolding, as the probability of correct folding is higher at 1.8 pN than at 0.6 pN.
Guinn et al. combined a chemical denaturant, mechanical force and site-directed mutagenesis to investigate the two-state folding of the SRC SH3 domain191. These authors showed that the SRC SH3 domain unfolds through a single pathway when pulled in an unzipping geometry, but through two alternative pathways, in a force-dependent manner, when unfolded through a shearing geometry. The existence of these two trajectories was further established using a ϕ–value analysis that describes the effect of a single amino acid mutation on the stability of the transition state. To better characterize the folding intermediates in T4 lysozyme, Motlagh et al. used small molecules that stabilize the folded state in a manner proportional to the change in accessible surface area upon folding (osmolytes)192. The response of an on-pathway intermediate to two different osmolytes, characterized by optical tweezers, allowed these authors to identify it as the C-terminal domain.
The time spent by a molecule crossing the folding barrier (the transition path time) is intrinsically stochastic, thus precluding direct observation by bulk or ensemble experiments. Single-molecule methods are uniquely suited for transition path time measurements. The time resolution of optical tweezers is determined by the corner or roll-off frequency of the instrument. The corner frequency is equal to the ratio of the trap stiffness to the drag coefficient of the trapped bead, both a function of bead size. Smaller beads lead to weaker traps (lower trap stiffness), whereas the drag coefficient of the beads scales linearly with the bead radius. For reference, for a trap with a stiffness of 1 pN/nm and a bead diameter of 1 μm, the time resolution is ~10 μs. Yu et al. determined a transition path time for PrP from the diffusion constant of the protein over its energy landscape, obtaining a value of 2 μs from the unfolded to the native transition193. The transition path time from the unfolded to a misfolded state was 500 μs, which the authors interpreted as reflecting a significantly more rugged landscape (FIG. 8d). They speculate that proteins may have evolved to avoid non-productive interactions, yielding minimally frustrated and smooth energy surfaces. Neupane et al. have determined the transition path time directly using stiff optical tweezers with high temporal resolution194.
Despite decades of research, a central question remains in protein folding: how the native fold is arrived at in such a way as to avoid Levinthal’s paradox195. According to the energy landscape model196–198, proteins attain their native state through many alternative folding pathways over a funnel-shaped energy landscape. In this search, involving bond rotations of each individual residue, the molecule is somehow guided to attain increased native as opposed to non-native interactions by details encoded in the energy landscape. The alternative view, supported by some experimental observations199–201, is the defined-pathway model, which posits that proteins fold in discrete steps through units of 20–30 residues or foldons. In this view, the complexity of the search for the native fold is greatly reduced by the ‘modular’ character lent by larger multi-residue folding units (foldons), whose sequence encodes the necessary intra-foldon interactions that drives them to fold cooperatively, and possibly sequentially, through inter-foldon interactions, to bring about the native state. Single-molecule folding studies can possibly throw some light onto the discussion. For example, proteins containing multiple domains, which could be seen as larger cooperative folding elements or ‘super foldons’, are seen more often than not to attain their native form in a well-defined order often requiring some degree of inter-domain coupling166.
The analysis of barrier crossing of a PrP into its first intermediate173 reveals diverse molecular trajectories, which have been interpreted as confirming the multiple pathways predicted by the energy landscape model202. However, these same authors found that the distribution of transition path times are exponentially distributed, which is to be expected from the dynamics of a well-defined path203. These authors point out that the transition through this path is by no means deterministic and small stochastic deviations in the bond rotational configurations at the amino acid residue level may give rise to the heterogeneity of the molecular trajectories observed. Although the controversy at this point has not been resolved, it is likely that the ability to observe the folding trajectories of individual molecules may provide the crucial discrimination between the two models.
The effects of molecular chaperones on protein folding have been investigated using optical tweezers. Bechtluft et al. showed that chaperone SecB prevents the folding of the periplasmic-resident maltose-bonding protein (MBP), thus favouring its insertion into the membrane in the unfolded state204. A similar effect on the folding of MBP was determined by trigger factor, a chaperone known to interact with fully synthesized and nascent chains on the surface of the ribosome205. Unlike SecB, which prevents the folding of the protein, trigger factor seems to locally interact through different sites with the client protein, allowing it only to fold partially. Interestingly, both of these chaperones prevent the misfolded states resulting from the interaction among tandem repeat copies of MBP, suggesting that they may play such a role in preventing undesirable inter-domain misfolding204,205. During EF-G biosynthesis, the ribosome destabilizes domain II of this GTPase, favouring its interaction with the emerging G domain of the nascent protein and leading to a misfolded state between the two domains. Liu et al.206 showed that trigger factor prevents this interaction. The HSP70 chaperone DnaK binds client proteins transiently in its ATP-bound state, but tightly upon nucleotide hydrolysis. Using optical tweezers, Mashaghi et al.207 found that in addition to its known function of preventing non-native contacts in proteins by binding to their unfolded regions, DnaK stabilizes the core domain of MBP against mechanical unfolding, a conclusion supported by a similar effect of this chaperone observed in bulk on its true client protein RepE.
Optical tweezers were used to characterize the folding of nascent chains on the ribosome surface. Kaiser et al. showed that the refolding of T4 lysozyme tethered by a linker to a ribosome is slowed down >100 times compared with the protein in solution50. The slowing down was greater with shorter linker length, and the effect was shown to be electrostatic in nature. In solution, a truncated T4 lysozyme shortened by 15 amino acids from its C terminus misfolded, whereas the same polypeptide on the surface of the ribosome did not fold. Thus, ribosome–nascent chain interactions might delay premature misfolding until sufficient sequence has been synthesized to enable productive folding. A similar effect was observed for the N-terminal G domain of EF-G206,208. Nascent chain–ribosome interactions also prevent misfolding with other parts of the nascent polypeptide. Misfolding between the two N-terminal domains (G domain and domain II) was markedly reduced on the surface of the ribosome relative to the polypeptide in solution, yielding faster productive folding206.
Finally, optical tweezers have been used to follow the co-translational folding of polypeptides in real time using in vitro reconstituted translation systems. In these experiments, a ribosome is tethered to a bead via a DNA handle and the N terminus of the nascent chain is tethered to another bead via another DNA handle (FIG. 8e). The growth of the polypeptide appears as a lengthening of the end to end distance of the tether, whereas folding transitions are seen as sudden shortening events. Wruck et al. described gradual hydrophobic collapse and partial folding during synthesis209. Using ultra-stable high-resolution optical tweezers, Alexander et al. followed translation of calerythrin in real time210, where a misfolding event that occurs readily with polypeptide fragments in vitro, and more slowly but still rapidly in stalled ribosome–nascent chain complexes, was delayed during active translation by an average time of 63 s, presumably to give time for synthesis to complete or for a chaperone to bind (FIG. 8f). Thus, folding on the surface of the ribosome can be a non-equilibrium process. Stalled ribosome–nascent chain complexes do not show delayed misfolding because the nascent chain reaches conformational equilibrium during the preparation of the sample for the optical tweezers, a process that typically takes tens of minutes.
Mechanochemistry of molecular motor operation
Many essential cellular processes take place in a directional manner and are catalysed by nanometre-scale enzymes known as molecular motors211,212. Ever since the first motor proteins (kinesin and myosin) were discovered213,214, optical tweezers have been instrumental in characterizing these energy-consuming, force-generating microscopic machines18,26,27,105,148. The high spatio-temporal resolution of optical tweezers (ångstrom-scale movement at sub-second bandwidths34) makes it possible to directly visualize the step size of a motor, one of its fundamental properties. For example, the Escherichia coli RNA polymerase (RNAP) was observed to translocate on the dsDNA template in 1-bp (or 3.4-Å) increments39,124 (FIG. 9a–c). Base pair stepping was also observed for other motors such as the HCV NS3 (REF.185) and XPD156 helicases. A hairpin-unwinding geometry is commonly used for the study of DNA/RNA helicases including the SARS-CoV-2 replicative helicase nsp13 (REF.215), in which each base pair of the hairpin substrate opened by the helicase results in an increase in tether extension by two nucleotides. The same geometry was employed to follow the translational dynamics of the ribosome (FIG. 5d), which normally translocates on messenger RNA (mRNA) one codon at a time186,187. Moreover, the ribosome can also take sub-codon steps in the presence of a strong barrier such as a stable mRNA hairpin73, or larger step sizes at a slippery sequence (a stretch of mRNA where the ribosome tends to change its reading frame)188. The detection of a motor’s step size provides critical insight into its internal coordination216. For example, the finding that the bacteriophage φ29 DNA packaging motor took non-integer 2.5-bp steps — which make up 10-bp translocation cycles — suggested that four, rather than five, subunits of this homopentameric ring ATPase participate in DNA translocation154 (FIG. 9d–f). This insight led to a ‘division of labour’ model in which the fifth subunit adopts a distinct regulatory role by making specific contacts with DNA to initiate each cycle155,217,218, which informs the internal coordination among the subunits of the motor.
Fig. 9 |. Example applications of optical tweezers to study molecular motors.
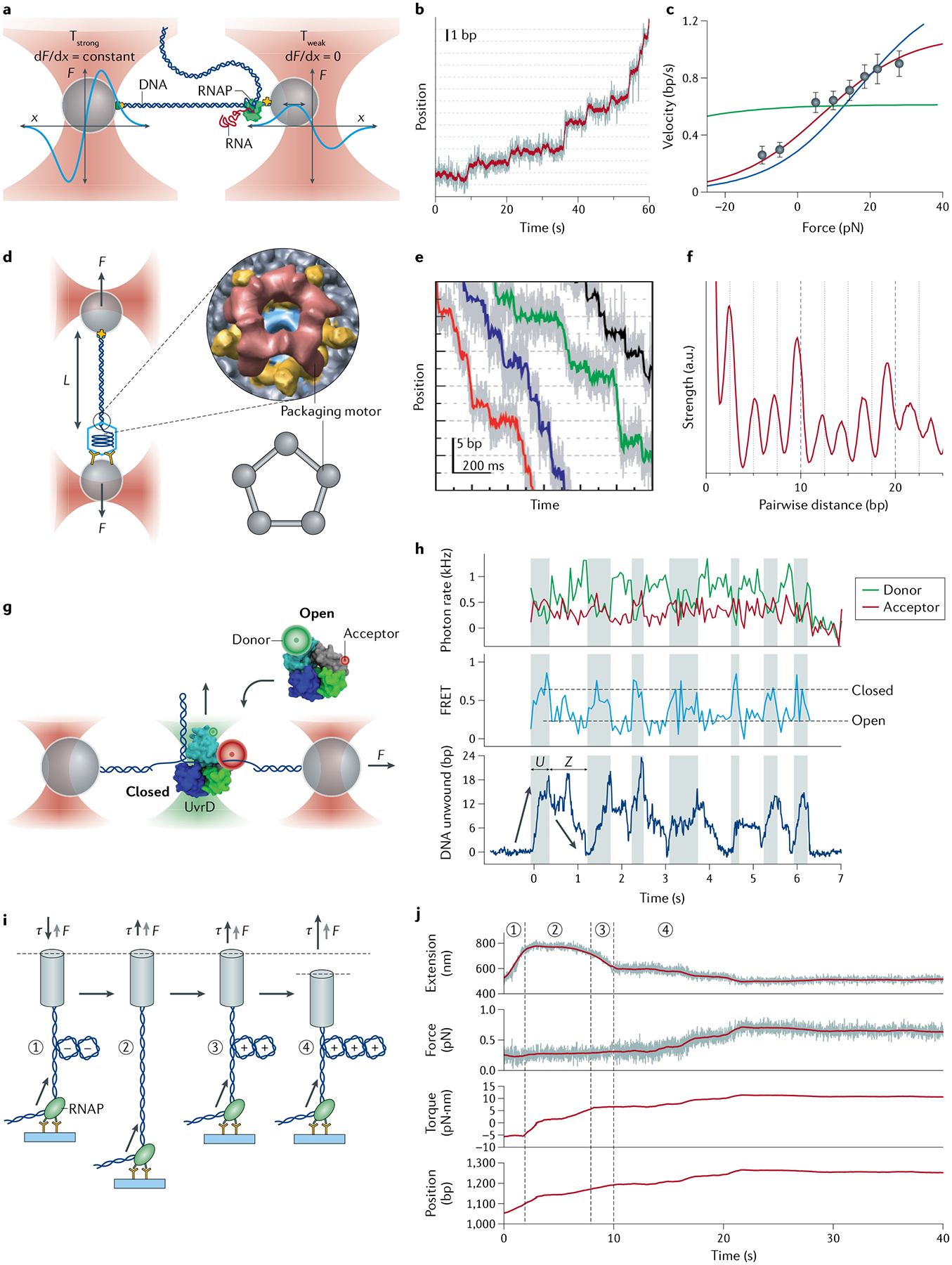
a | Dual-trap optical tweezers set-up for observing the stepping behaviour of Escherichia coli RNA polymerase (RNAP). Schematic force (F) versus position (x) profiles for both traps are shown in cyan. A passive force clamp is maintained in the weak trap (Tweak) on the right. b | Representative trajectory showing a single RNAP translocating on its DNA template in discrete steps. Dotted lines are spaced at 3.4-Å (1-bp) intervals. c | Force–velocity relationship for RNAP at [NTP]eq = 10 μM GTP, 10 μM UTP, 5 μM ATP and 2.5 μM CTP. Fits to a power stroke model (green), a linear Brownian ratchet model (blue) and a branched Brownian ratchet model (red) are shown39. d | Dual-trap optical tweezers set-up for studying DNA packaging by the bacteriophage φ29 packaging motor, a pentameric ring ATPase. The DNA tether length (L) is monitored as a function of time. e | Representative single-molecule packaging trajectories collected with 250 μM ATP and an external force of ~40 pN. Dotted lines are spaced at 2.5-bp intervals. f | Pairwise distance distribution (PWD) averaged from many packaging trajectories indicates the motor’s step size154. g | Fleezers set-up for studying the conformational dynamics and DNA unwinding behaviour of the E. coli UvrD helicase. h | Simultaneous measurements of donor/acceptor fluorescence, Förster resonance energy transfer (FRET) efficiency and number of DNA base pairs unwound enable direct correlation of the conformational states of UvrD (‘Open’ and ‘Closed’) with the motor’s helicase activity (rezipping ‘Z’ and unwinding ‘U’)72. i | Angular optical tweezers (AOT) set-up for studying the transcriptional dynamics of the E. coli RNAP under torsion. j | Representative data of simultaneous force (F), torque (τ) and extension measurements. Under a low-force clamp, RNAP translocation first neutralizes the preformed (−) plectoneme and then induces the formation of (+) plectoneme. After the force clamp is turned off, RNAP continues to translocate along the DNA template, resulting in an increase in force and the corresponding torque until reaching a stall172. Parts a–c adapted from REF.39, Springer Nature Limited. Parts d–f adapted with permission from REF.73, Elsevier. Parts g and h adapted with permission from REF.72, AAAS. Parts i and j adapted with permission from REF.172, AAAS.
The term mechanochemistry refers to the mechanism by which a molecular motor couples chemical energy (for example, from ATP hydrolysis) to mechanical motion and force219. Similar to its effects on folding, external force — which can be precisely applied by optical tweezers — tilts the free energy landscape along the pulling coordinate that, if designed appropriately, overlaps with the mechanical reaction coordinate of the motor under force. By measuring the dependence of motor velocity on force (F–v relationship), one can distinguish between different classes of mechanisms for force generation and mechanochemical coupling. For example, F–v studies on both bacterial29,39,148 and eukaryotic152,220,221 RNAPs, combined with theoretical modelling222–224, converge on a model in which RNAP operates as a Brownian ratchet (FIG. 9c). In this mechanism, the enzyme oscillates between multiple adjacent positions on the DNA lattice driven by thermal energy; these reversible motions are rectified by the binding of an incoming nucleotide to the active site of RNAP such that net forward translocation is achieved. Similarly, measuring the translation velocity against an applied force by directly tethering the ribosome to a bead, in contrast to the aforementioned hairpin-unwinding geometry, reveals that the ribosome is also a Brownian ratchet motor, generating a maximum force of ~13 pN, just enough to unwind most mRNA secondary structures225. This mechanism contrasts with the power stroke mechanism adopted by motors such as kinesin226 and the φ29 DNA packaging motor151, in which motor movement is directly coupled to a largely irreversible chemical transition227.
By obtaining F–v curves at different substrate (such as ATP) concentrations, one can deduce the Michaelis–Menten kinetic parameters of the motor across force regimes. This information is useful for identifying the location of the force-generating movement step in the motor’s operating cycle57,211. For example, the φ29 DNA packaging motor exhibits force-sensitive maximum velocity Vmax and Michaelis constant KM, but its Vmax/KM ratio is force-insensitive151. This phenotype rules out ATP binding or any chemical transition within the same kinetic block as ATP binding — including ATP tight-binding and ADP release — as the power stroke step, which led to the conclusion that DNA translocation by the φ29 ATPase is powered by the release of inorganic phosphate upon ATP hydrolysis151. Similar analysis of the force dependence of the Michaelis–Menten parameters was employed to dissect the force-generating mechanism of the ClpXP protease228 and the SpoIIIE DNA translocase229. The former uses a constant pulling rate but different gears to translocate polypeptides, whereas the latter displays a biphasic F–v curve that may represent an adaptive mechanism for ultrafast motors such as SpoIIIE to aptly remove DNA-bound roadblocks.
The combination of single-molecule fluorescence spectroscopy with optical tweezers enables multiple aspects of the motor behaviour to be concomitantly measured. For example, fleezers were utilized to follow simultaneously the unwinding activity and the conformational state of UvrD, a DNA repair helicase72 (FIG. 9g,h). It was found that UvrD possesses a conformational switch that controls on which strand of the DNA duplex the helicase translocates. Interconversion between the two conformations leads to intermittent periods of DNA unwinding and rezipping. Such a co-temporal fluorescence detection and force manipulation approach was recently applied to other molecular motors, including the ribosome during protein synthesis73, the polypeptide translocase ClpB61 and the eukaryotic replicative helicase CMG178.
The ability to monitor additional degrees of freedom, such as rotation, facilitates the study of biological motors that generate twist and torque in conjunction with force. For instance, AOT was employed to measure the stall torque and the torque–velocity relationship for the E. coli RNAP172, establishing the polymerase as a potent torsional motor (FIG. 9i,j) that generates DNA supercoiling during transcription. It was subsequently shown that transcription factors regulate the capacity of RNAP to deal with the torsional stress stored in DNA173, which can be further modulated by the chromatin fibre topology in eukaryotes171.
Reproducibility and data deposition
Optical tweezers experiments present numerous challenges in terms of data reproducibility. The first challenge has to do with the low-throughput nature of the experiments and the relatively low statistics derived from them. Although each single-molecule recording affords rich information on the dynamics of that molecule, it is critical that enough replicates are obtained (typically tens to hundreds) so that the full spectrum of behaviours across a population is surveyed. This is particularly important when the population exhibits significant molecular heterogeneity, either from variation between distinct subpopulations (static heterogeneity) or due to interconversion of the same molecule among multiple states over time (dynamic heterogeneity)230. Both types of heterogeneity have been observed in diverse experimental systems150,231–233. They must not be confused with the intrinsic stochastic nature of single-molecule dynamics, which entails that no two molecules — even those with identical kinetic parameters — behave the same. Given these sources of variations, single-molecule researchers should strive to report the full distribution of molecular behaviours observed and disclose the criteria used for excluding any molecules from analysis. Generally, acceptable exclusion includes multiple tethers, functionally inactive molecules and obvious aggregates. It is thus critical to obtain samples as biochemically homogeneous as possible prior to single-molecule experiments.
Although there is no universal standard as to how many molecules constitute a sufficiently large sample size (as this necessarily depends on the extent of heterogeneity and the ease of data collection), one should ensure that the results can be reproduced across multiple days and multiple batches of samples, and, if possible, independently examined by multiple researchers. When presenting results, one should always report errors associated with the measurements and statistical test results when making a comparison between conditions. As the data tend to segregate in classes, it is also important to confirm that the existence of these classes can be corroborated using independent methods and that, when averaged together, the signals recover the behaviour observed in ensemble or bulk experiments. These practices will help minimize implicit biases against undesired results, and to prevent any rare but meaningful behaviour from being overlooked. However, caution must be exercised when extrapolating single-molecule results to bulk measurements, as they often do not measure the same experimental variables.
The other challenge against reproducibility has to do with the various instrumental configurations, experimental settings and data analysis pipelines developed in different laboratories, which make a straightforward comparison between studies difficult. Although not unique to optical tweezers, this problem is exacerbated by the complex and adaptable nature of this technology. Therefore, the methods section of a research article should include detailed technical descriptions such as how the data were filtered, what parameters were chosen for theoretical fitting and which statistical tests were performed.
No standard for data repository and format exists to date. Therefore, the community should converge on a set of practices to standardize the open data format (for example, the HDF5 format) as well as deposit raw data (force, extension, kymograph and so on) and analysis algorithms into open-source repositories, facilitating easy access and re-examination of published results. These practices are already widely adopted with other methodologies in fields such as structural biology and genomics. Moreover, a multi-laboratory endeavour to benchmark different optical tweezers-based protocols (for example, of particular trap stiffnesses and pulling speeds) using a simple and common assay (such as unfolding a particular DNA hairpin or protein), akin to the one recently conducted by the single-molecule FRET community234, may be desirable.
Limitations and optimizations
In this section, we discuss limits to the temporal and spatial resolution, speed of manipulation and throughput of optical tweezers. The temporal resolution of an instrument can be limited by the type of sensors used for detecting bead displacements. Some silicon-based photodetectors exhibit parasitic low-pass filtering above 10 kHz when illuminated with wavelengths >850 nm, above the absorption maximum of silicon235. Photodetector low-pass filtering results in inaccurate measurements of signals at frequencies >10 kHz, including those from bead Brownian motion that can lead to calibration errors. This effect can be mitigated by using non-silicon-based detectors or infrared-enhanced silicon detectors6,83 with which data can be collected at rates exceeding 100 kHz using the appropriate data acquisition card.
Nonetheless, the maximum measurement bandwidth is usually not limited by instrumental factors but, rather, by the trapped beads themselves. Because of the hydrodynamic drag experienced by the beads as they move through a fluid, there is a fundamental limit to how fast they can respond to mechanical signals. A trapped bead has a characteristic frequency fc, above which signals are damped by the viscous drag6. fc depends on the system stiffness and bead size according to Eq. 4. For example, a 1-μm diameter bead and stiffness of 1 pN/nm corresponds to a characteristic frequency of 17 kHz or a temporal resolution of ~10 μs. Temporal resolution can thus be increased by using stiffer systems. In some cases, it can also be increased by decreasing the bead size, although there are practical limits to trapping polystyrene or silica beads smaller than ~200 nm because the polarizability α (see Eq. 1) decreases as the particle volume decreases6. In general, smaller beads require higher laser intensities to attain equivalent forces, increasing the chances of sample damage. Recent work has shown that germanium nanoparticles, whose high refractive index compensates for their small size (as small as ~70 nm), are usable for single-molecule optical trap experiments236. The characteristic frequency fc of the bead can also affect the dynamics of the biological system under investigation. One study has shown that the rates of some DNA-processing enzymes depend inversely on bead size, because the rate of bead thermal motion controls the rate at which transition states are visited by these enzymes237.
The spatial resolution of an optical trap is limited, in part, by instrumental sources of noise. Drift in the sample stage position or objective and fluctuations in trap position or power can all lead to bead displacements that are indistinguishable from signals. Various approaches have been developed to improve instrument stability. In single-trap, surface-based assays, the sample stages can be monitored and stabilized by a feedback loop to better than 1 Å in x, y and z9,37. Drift in the laser focus (particularly along z) due to heating of the objective by absorption of the trap light can be reduced greatly by temperature control of the objective36. Power stability can also be improved by monitoring the trap laser intensity with a photodetector and adjusting the laser power by feedback loop to compensate for intensity fluctuations43,83. Fibre coupling6, in which the laser light is transmitted through an optical fibre, can help improve pointing stability. In dual-trap designs where both traps are formed from the same laser, minimizing differences in the optical paths taken by the two trap-forming beams reduces fluctuations affecting trap separation51,124.
However, even if all sources of instrument noise are eliminated, there remains a fundamental limit to the spatial resolution — the Brownian motion of the trapped beads9,34,51. A useful figure of merit for quantifying spatial resolution is the signal to noise ratio (SNR). The maximum possible SNR is given by the ratio of the bead displacement Δx to the root mean square fluctuation in bead position due to Brownian motion, calculated by integrating Eq. 3 over the measurement bandwidth B. Assuming B ≪ fc, it can be shown that the maximum SNR depends on the tether stiffness κtether and the drag coefficient of the beads γ as34,40:
| (9) |
where Δℓ is the signal of interest, γeff = γ for a bead in a single trap and γeff = γ/2 for identical beads in dual traps. Importantly, neither the trap stiffness nor the operation mode (such as active versus passive mode) impacts the resolution34,40. As a result, it is advantageous to design experiments with small beads, subjected to the limitations mentioned above, and stiff tethers if possible. In applications where the tether is DNA-based, stiff tethers are typically achieved by making them as short as possible and by pulling to high force, although recent work with rigid, self-assembled DNA ‘origami’ beams holds promise for high-resolution measurements238. Assuming reasonable experimental values — for example, beads 1 μm in diameter and a tether stiffness of 0.3 pN/nm (equivalent to that of a 1-μm dsDNA molecule pulled to a force of ~10 pN40 or a single kinesin at its stall force239) — Eq. 9 predicts that a 1-nm signal collected at a 100-Hz bandwidth would be detected with a maximum SNR from 2.5 (for a single trap) to 3.5 (for identical dual traps). Eq. 9 can be used to derive a similar expression for the force resolution of an optical trap:
| (10) |
where ΔF is the force signal. The force resolution is thus also independent of trap stiffness. Finally, the spatial (and force) resolution is intrinsically related to the temporal resolution, as temporal averaging of the data reduces the noise by a factor proportional to the square root of the averaging window (reflected in the dependence on measurement bandwidth B in Eqs 9 and 10). Thus, there is a trade-off between spatial and temporal resolution: under identical conditions, a fourfold increase in temporal resolution leads to a twofold decrease in spatial (and force) resolution.
Manipulation speeds are another important consideration, particularly for constant force measurements, which may require feedback control of the trap position. If the signal of interest changes at a rate faster than the speed at which the sample stage or trap position is controlled, then the instrument cannot maintain a constant force. As a general rule, piezo-actuated linear or mirror stages offer fairly slow scan rates below ~1 kHz (REFS6,51). Other technologies to control the trap position, such as acousto-optic and electro-optic deflectors, which do not rely on moving parts, can achieve much higher scan rates exceeding ~1 MHz (REFS6,83). The choice of technique for controlling the trap position must be made when designing the instrument. An alternative passive force clamp22 method that exploits the edge of the trapping potential can be used to exert constant forces without the need for feedback control of the trap position. This approach can exert constant forces at higher frequencies than possible with piezo-actuated stages, but the distance range over which constant force is maintained is limited to ~50 nm (REF.22). Again, however, the characteristic frequency of the trapped beads sets a fundamental speed limit to any manipulation approach.
In contrast to standard optical tweezers, an AOT instrument is significantly more complex in terms of instrument design and control as well as data acquisition and processing. In particular, the linear polarization of the laser input to the objective needs to be maintained, and deviations from linear polarization need to be carefully monitored and corrected. AOT also require trapping particles with specific anisotropic optical properties (such as quartz cylinders). These particles are not commercially available and must be nanofabricated. Precision torque measurements require smaller cylinders, whereas high force generation requires larger cylinders, so these two considerations must be balanced for a given experiment.
One important limitation of optical tweezers is their low throughput. Although the data produced by optical traps are rich in detail owing to the sensitivity of optical traps, this high information content comes at a cost in throughput. Every bead and tether is individually manipulated, and data are collected one molecule at a time. By contrast, other single-molecule approaches such as magnetic tweezers and TIRF microscopy can interrogate hundreds of molecules in parallel240–243. Nevertheless, this higher throughput is often obtained at the price of reduced spatial and temporal resolution, with the concomitant decrease in information content. Approaches are being explored to parallelize optical traps to increase their throughput.
Outlook
Numerous challenges lie ahead in the application of optical tweezers to the study of biological systems. Some of these will involve incremental, quantitative improvements of the instrument capabilities along the lines discussed above. These developments will likely include the implementation of ever more elaborate methods of controlled microfluidics and improved control of molecular environmental conditions. Likewise, it is clear that the method will witness the increased use of artificial intelligence and machine learning protocols for the control of the experiments and the extraction of the data.
Other developments are, however, more qualitative in nature. Among these we must consider recent efforts aimed at increasing the throughput of optical tweezers. Throughput in optical tweezers experiments is largely limited by the ability to tether the molecular system. ‘Fishing’, as it is commonly referred to, can be automated and computer-controlled, making the task less tedious but not necessarily improving throughput. Once the tether has been formed, the success of the experiment is determined by the robustness of the biological system being studied. Parallelizing optical traps is being explored as a way to increase throughput. By using multiple traps simultaneously, it should be possible to proportionally increase the amount of data obtained from single-molecule experiments. Although time-shared42 or holographic optical traps244 can generate arrays of traps, they suffer from high power requirements that scale linearly with the number of traps. Recent advances in the development of a nanophotonic standing-wave array trap hold promise for precision measurements of biomolecular forces at high throughput on a chip245. In this approach, an array of stable on-chip optical traps is formed at the antinodes of a standing wave in a nanophotonic waveguide. In general, all of these efforts at multiplexing the trapping experiment must solve the challenge of reaching simultaneous and precise control of the experimental molecular geometry in all of the traps. Although throughput is currently a limitation in optical tweezers studies, the amount of mechanistic insight derivable in principle from these single-molecule experiments easily compensates the efforts required to build up statistics.
Another important challenge is the need to fill the gap between in vivo and in vitro studies. This divide is not unique to the optical tweezers field; in fact, it is shared by almost all physical techniques used to study complex biomolecular systems under controlled conditions outside the cell246. In the case of optical tweezers, the use of trapping experiments inside the cell remains limited to date, with the exception of a few applications trapping cytoskeletal cargos after in situ calibration of the traps in the viscoelastic cytoplasm247,248. Because many organelles and vacuoles possess refractive indices slightly above that of the cytoplasm, they are attracted towards the higher intensities of the beam. As a result, it is not possible at present to selectively trap any particular object, even if these are beads specially introduced into the cell by biolistic means. One possible way to address this issue is to develop robust protocols using cell extracts, but this is not a trivial endeavour. In many cases, other components in the extract may bind non-specifically to any part of the system of interest and interfere with the measurement. However, in some cases it may be possible to perform the experiment and use immunoprecipitation to selectively deplete one or another component from the mix, and derive causal relationships from the modified behaviour of the system.
These developments, together with the recent commercial availability of high-resolution optical tweezers, will likely increase both the impact of this powerful technique and its attractiveness among biophysicists, biochemists and quantitative biologists.
Thermal bath.
(Also known as heat bath, or thermal reservoir in thermodynamics and statistical mechanics). A large body held at constant temperature in which the system of interest is immersed and with which it is in thermal equilibrium. It is assumed to possess a large heat capacity and to be essentially an infinite source of thermal energy so that it can exchange energy with the system (as a thermal source or as a heat sink) without altering its temperature.
Torques.
Cross products of the position vector (pointing from the axis of rotation to the point of application of the force) and the force driving the rotation.
Wavelength.
The distance between two maxima of the light’s electric or magnetic field.
Rayleigh scatterer.
An object whose dimensions are much smaller than the wavelength of the light.
Polarizability.
A dipole moment is generated when a distribution of charges experiences a separation of positive and negative charges under the influence of an electric field. The magnitude of the dipole is proportional to the amplitude of the electric field . The constant of proportionality is the polarizability of the charge distribution.
Electric dipole.
A pair of electric charges of equal magnitude but opposite signs separated by a finite distance.
Gaussian laser beam.
A light beam whose intensity can be described with a Gaussian function, maximal at its axis and decreasing towards its periphery.
Permittivity.
A measure of the electric polarizability of the medium.
Absorption.
A process in which the energy carried by light is transferred to the sample.
Refraction.
A change in the direction of the travelling beam by the presence of an object that has a different index of refraction to the surrounding medium, when that beam impinges on it obliquely.
Reflection.
A process by which the light that impinges on a surface of an object bounces back from that surface instead of being absorbed or refracted by the object.
Extinction cross-section.
A measure of the efficiency with which a given object can absorb or scatter the light that impinges on it.
Poynting vector.
A quantity that describes the magnitude and direction of the flow of energy of a propagating electromagnetic wave.
Geometric or ray optics.
A description of light in terms of rays that describe in an approximate manner the paths along which the light travels.
Refractive objects.
Objects that possess a different index of refraction to the medium in which they are immersed and that deflect light that impinges on them obliquely.
High numerical aperture.
The range of angles over which a lens can collect light; the larger the numerical aperture, the larger the angular range.
Condenser lens.
A standard optical microscope component typically used to focus the microscope light into the specimen; in optical traps it is often used to collect scattered trap light.
Back focal plane interferometry.
A technique in which the interference pattern between the light transmitted and the light forward-scattered by the trapped particle is used to determine the position of the particle relative to the trap centre.
Linear range.
The range of input parameters over which the output of the system depends linearly on the input.
Measurement bandwidths.
The rates at which data are collected.
Boltzmann constant.
A fundamental physical constant that relates the kinetic energy of a gas to its absolute temperature. It is closely related to the gas constant R.
Hydrodynamic drag coefficient.
A coefficient characterizing the viscous resistance that a particle moving through a fluid encounters.
Pointing drift.
Unwanted changes in the direction of a beam of light.
Brownian motion.
The random fluctuation of a particle’s position (and orientation) at a given temperature.
Feedback loop.
A self-regulating mechanism in which the output of a system is routed back into an input to the system. In negative feedback, the output is fed back in a fashion that tends to reduce fluctuations in the output.
Fluorophores.
Fluorescent chemical compounds or molecules.
Oxygen scavengers.
Enzymatic systems that remove molecular oxygen detrimental to fluorescence.
Extraordinary axis.
(Also known as the ‘optic axis’). For a uniaxial crystal that has a refractive index of one crystal axis that is different from the other two crystal axes, light propagating parallel to this optic axis experiences the same index of refraction regardless of its polarization.
Laser polarization.
The polarization of the light refers to the direction of the electric field.
Laguerre–Gaussian beam.
A mode of laser that can carry both orbital and spin angular momentum.
Michaelis–Menten kinetic parameters.
Parameters that relate the rate of an enzymatic reaction v to the substrate concentration [s] by the formula v = Vmax[s] / (KM + [s]), where Vmax and KM represent the maximum rate and the substrate concentration at which the reaction rate is half of Vmax, respectively.
Temporal resolution.
The measure of the fastest signal that can be detected accurately.
Biolistic.
A method of introducing beads inside cells by shooting them ballistically inside the cells at supersonic speeds.
Acknowledgements
The authors dedicate this Primer to A. Ashkin (1922–2020), whose ingenious invention of optical tweezers has greatly influenced the field of single-molecule biophysics. This work is supported by the Nanomachines programme (KC1203) funded by the Office of Basic Energy Sciences of the US Department of Energy (DOE) contract no. DE-AC02-05CH11231, National Institutes of Health (NIH) grants R01GM032543 (to C.J.B.), R01GM120353 (to Y.R.C.), DP2HG010510 (to S.L.) and R01GM136894 (to M.D.W.), and National Science Foundation (NSF) grants PHY-1430124 (Physics Frontiers Center (PFC) ‘Center for the Physics of Living Cells’ to Y.R.C.) and MCB-1517764 (to M.D.W.). C.J.B. and M.D.W. are Howard Hughes Medical Institute investigators. S.L. is supported by the Robertson Foundation and thanks M. Wasserman for discussions. M.D.W. thanks J. Inman and G. Xiang for commenting on the manuscript.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Ashkin A Acceleration and trapping of particles by radiation pressure. Phys. Rev. Lett 24, 156 (1970). [DOI] [PubMed] [Google Scholar]
- 2.Ashkin A, Dziedzic JM, Bjorkholm JE & Chu S Observation of a single-beam gradient force optical trap for dielectric particles. Opt. Lett 11, 288 (1986). [DOI] [PubMed] [Google Scholar]; This study provides the first demonstration of a single-beam optical trap that has since been commonly used in single-molecule biophysics.
- 3.Ashkin A, Dziedzic JM & Yamane T Optical trapping and manipulation of single cells using infrared laser beams. Nature 330, 769–771 (1987). [DOI] [PubMed] [Google Scholar]
- 4.Neuman KC & Nagy A Single-molecule force spectroscopy: optical tweezers, magnetic tweezers and atomic force microscopy. Nat. Methods 5, 491–505 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones PH, Marago OM & Volpe G Optical Tweezers: Principles and Applications (Cambridge Univ. Press, 2015). [Google Scholar]
- 6.Neuman KC & Block SM Optical trapping. Rev. Sci. Instrum 75, 2787–2809 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cecconi C, Shank EA, Dahlquist FW, Marqusee S & Bustamante C Protein–DNA chimeras for single molecule mechanical folding studies with the optical tweezers. Eur. Biophys. J 37, 729–738 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Forth S, Sheinin MY, Inman J & Wang MD Torque measurement at the single-molecule level. Annu. Rev. Biophys 42, 583–604 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perkins TT Angstrom-precision optical traps and applications. Annu. Rev. Biophys 43, 279–302 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Heller I, Hoekstra TP, King GA, Peterman EJ & Wuite GJ Optical tweezers analysis of DNA–protein complexes. Chem. Rev 114, 3087–3119 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Chemla YR High-resolution, hybrid optical trapping methods, and their application to nucleic acid processing proteins. Biopolymers 105, 704–714 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Killian JL, Ye F & Wang MD Optical tweezers: a force to be reckoned with. Cell 175, 1445–1448 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Bustamante C, Alexander L, Maciuba K & Kaiser CM Single-molecule studies of protein folding with optical tweezers. Annu. Rev. Biochem 89, 443–470 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Svoboda K & Block SM Biological applications of optical forces. Annu. Rev. Biophys. Biomol. Struct 23, 247–285 (1994). [DOI] [PubMed] [Google Scholar]
- 15.Rohrbach A Stiffness of optical traps: quantitative agreement between experiment and electromagnetic theory. Phys. Rev. Lett 95, 168102 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Neuman KC, Chadd EH, Liou GF, Bergman K & Block SM Characterization of photodamage to Escherichia coli in optical traps. Biophys. J 77, 2856–2863 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith SB, Cui Y & Bustamante C Optical-trap force transducer that operates by direct measurement of light momentum. Methods Enzymol. 361, 134–162 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Finer JT, Simmons RM & Spudich JA Single myosin molecule mechanics: piconewton forces and nanometre steps. Nature 368, 113–119 (1994). [DOI] [PubMed] [Google Scholar]
- 19.Allersma MW, Gittes F, deCastro MJ, Stewart RJ & Schmidt CF Two-dimensional tracking of ncd motility by back focal plane interferometry. Biophys. J 74, 1074–1085 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gittes F & Schmidt CF Interference model for back-focal-plane displacement detection in optical tweezers. Opt. Lett 23, 7–9 (1998). [DOI] [PubMed] [Google Scholar]
- 21.Pralle A, Prummer M, Florin EL, Stelzer EH & Horber JK Three-dimensional high-resolution particle tracking for optical tweezers by forward scattered light. Microsc. Res. Tech 44, 378–386 (1999). [DOI] [PubMed] [Google Scholar]
- 22.Greenleaf WJ, Woodside MT, Abbondanzieri EA & Block SM Passive all-optical force clamp for high-resolution laser trapping. Phys. Rev. Lett 95, 208102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Denk W & Webb WW Optical measurement of picometer displacements of transparent microscopic objects. Appl. Opt 29, 2382–2391 (1990). [DOI] [PubMed] [Google Scholar]
- 24.Berg-Sørensen K & Flyvbjerg H Power spectrum analysis for optical tweezers. Rev. Sci. Instrum 75, 594 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Tolić-Nørrelykke SF et al. Calibration of optical tweezers with positional detection in the back focal plane. Rev. Sci. Instrum 77, 103101 (2006). [Google Scholar]
- 26.Svoboda K, Schmidt CF, Schnapp BJ & Block SM Direct observation of kinesin stepping by optical trapping interferometry. Nature 365, 721–727 (1993). [DOI] [PubMed] [Google Scholar]; This study presents the observation of individual steps of 8 nm taken by the cytoskeletal motor kinesin, the first direct measurement of the step size of any motor protein.
- 27.Kuo SC & Sheetz MP Force of single kinesin molecules measured with optical tweezers. Science 260, 232–234 (1993). [DOI] [PubMed] [Google Scholar]
- 28.Molloy JE, Burns JE, Kendrick-Jones J, Tregear RT & White DC Movement and force produced by a single myosin head. Nature 378, 209–212 (1995). [DOI] [PubMed] [Google Scholar]
- 29.Yin H et al. Transcription against an applied force. Science 270, 1653–1657 (1995). [DOI] [PubMed] [Google Scholar]
- 30.Wuite GJ, Smith SB, Young M, Keller D & Bustamante C Single-molecule studies of the effect of template tension on T7 DNA polymerase activity. Nature 404, 103–106 (2000). [DOI] [PubMed] [Google Scholar]
- 31.Adelman K et al. Single molecule analysis of RNA polymerase elongation reveals uniform kinetic behavior. Proc. Natl Acad. Sci. USA 99, 13538–13543 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith SB, Cui Y & Bustamante C Overstretching B-DNA: the elastic response of individual double-stranded and single-stranded DNA molecules. Science 271, 795–799 (1996). [DOI] [PubMed] [Google Scholar]; This study reveals that DNA undergoes an overstretching transition at around 65 pN, a feature that has been commonly exploited in numerous subsequent studies.
- 33.Wuite GJ, Davenport RJ, Rappaport A & Bustamante C An integrated laser trap/flow control video microscope for the study of single biomolecules. Biophys. J 79, 1155–1167 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moffitt JR, Chemla YR, Smith SB & Bustamante C Recent advances in optical tweezers. Annu. Rev. Biochem 77, 205–228 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Nugent-Glandorf L & Perkins TT Measuring 0.1-nm motion in 1 ms in an optical microscope with differential back-focal-plane detection. Opt. Lett 29, 2611–2613 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Mahamdeh M & Schaffer E Optical tweezers with millikelvin precision of temperature-controlled objectives and base-pair resolution. Opt. Express 17, 17190–17199 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Carter AR et al. Stabilization of an optical microscope to 0.1 nm in three dimensions. Appl. Opt 46, 421–427 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Shaevitz JW, Abbondanzieri EA, Landick R & Block SM Backtracking by single RNA polymerase molecules observed at near-base-pair resolution. Nature 426, 684–687 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abbondanzieri EA, Greenleaf WJ, Shaevitz JW, Landick R & Block SM Direct observation of base-pair stepping by RNA polymerase. Nature 438, 460–465 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows direct observation of single base-pair (0.34 nm) steps taken by RNA polymerase and represents a milestone in precision measurements of optical trapping.
- 40.Moffitt JR, Chemla YR, Izhaky D & Bustamante C Differential detection of dual traps improves the spatial resolution of optical tweezers. Proc. Natl Acad. Sci. USA 103, 9006–9011 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Visscher K, Brakenhoff GJ & Krol JJ Micromanipulation by multiple optical traps created by a single fast scanning trap integrated with the bilateral confocal scanning laser microscope. Cytometry 14, 105–114 (1993). [DOI] [PubMed] [Google Scholar]
- 42.Visscher K, Gross SP & Block SM Construction of multiple-beam optical traps with nanometer-resolution position sensing. IEEE JSTQE 2, 1066–1076 (1996). [Google Scholar]
- 43.Comstock MJ, Ha T & Chemla YR Ultrahigh-resolution optical trap with single-fluorophore sensitivity. Nat. Methods 8, 335–340 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study presents state-of-the-art high-resolution fleezers that combined a time-shared dual optical trap interlaced with a confocal fluorescence microscope.
- 44.Maillard RA et al. ClpX(P) generates mechanical force to unfold and translocate its protein substrates. Cell 145, 459–469 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cecconi C, Shank EA, Bustamante C & Marqusee S Direct observation of the three-state folding of a single protein molecule. Science 309, 2057–2060 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Shank EA, Cecconi C, Dill JW, Marqusee S & Bustamante C The folding cooperativity of a protein is controlled by its chain topology. Nature 465, 637–640 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stigler J, Ziegler F, Gieseke A, Gebhardt JC & Rief M The complex folding network of single calmodulin molecules. Science 334, 512–516 (2011). [DOI] [PubMed] [Google Scholar]; This study makes direct observations of conformational folding transitions of single calmodulin molecules using dynamic force spectroscopy.
- 48.Jahn M, Buchner J, Hugel T & Rief M Folding and assembly of the large molecular machine Hsp90 studied in single-molecule experiments. Proc. Natl Acad. Sci. USA 113, 1232–1237 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aubin-Tam ME, Olivares AO, Sauer RT, Baker TA & Lang MJ Single-molecule protein unfolding and translocation by an ATP-fueled proteolytic machine. Cell 145, 257–267 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kaiser CM, Goldman DH, Chodera JD, Tinoco I Jr. & Bustamante C The ribosome modulates nascent protein folding. Science 334, 1723–1727 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bustamante C, Chemla YR & Moffitt JR in Single-Molecule Techniques: A Laboratory Manual (eds Selvin PR & Ha T) 297–324 (Cold Spring Harbor Laboratory Press, 2008). [Google Scholar]
- 52.Landry MP, McCall PM, Qi Z & Chemla YR Characterization of photoactivated singlet oxygen damage in single-molecule optical trap experiments. Biophys. J 97, 2128–2136 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peterman EJ, Gittes F & Schmidt CF Laser-induced heating in optical traps. Biophys. J 84, 1308–1316 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Williams MC, Wenner JR, Rouzina I & Bloomfield VA Entropy and heat capacity of DNA melting from temperature dependence of single molecule stretching. Biophys. J 80, 1932–1939 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mao H, Arias-Gonzalez JR, Smith SB, Tinoco I Jr. & Bustamante C Temperature control methods in a laser tweezers system. Biophys. J 89, 1308–1316 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.de Lorenzo S, Ribezzi-Crivellari M, Arias-Gonzalez JR, Smith SB & Ritort F A temperature-jump optical trap for single-molecule manipulation. Biophys. J 108, 2854–2864 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Visscher K, Schnitzer MJ & Block SM Single kinesin molecules studied with a molecular force clamp. Nature 400, 184–189 (1999). [DOI] [PubMed] [Google Scholar]
- 58.Li PT, Collin D, Smith SB, Bustamante C & Tinoco I Jr. Probing the mechanical folding kinetics of TAR RNA by hopping, force-jump, and force-ramp methods. Biophys. J 90, 250–260 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van Mameren J et al. Dissecting elastic heterogeneity along DNA molecules coated partly with Rad51 using concurrent fluorescence microscopy and optical tweezers. Biophys. J 91, L78–L80 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Forget AL & Kowalczykowski SC Single-molecule imaging of DNA pairing by RecA reveals a three-dimensional homology search. Nature 482, 423–427 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Avellaneda MJ et al. Processive extrusion of polypeptide loops by a Hsp100 disaggregase. Nature 578, 317–320 (2020). [DOI] [PubMed] [Google Scholar]
- 62.Ishijima A et al. Simultaneous observation of individual ATPase and mechanical events by a single myosin molecule during interaction with actin. Cell 92, 161–171 (1998). [DOI] [PubMed] [Google Scholar]
- 63.Harada Y et al. Single-molecule imaging of RNA polymerase–DNA interactions in real time. Biophys. J 76, 709–715 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lang MJ, Fordyce PM, Engh AM, Neuman KC & Block SM Simultaneous, coincident optical trapping and single-molecule fluorescence. Nat. Methods 1, 133–139 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee KS, Balci H, Jia H, Lohman TM & Ha T Direct imaging of single UvrD helicase dynamics on long single-stranded DNA. Nat. Commun 4, 1878 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hohng S et al. Fluorescence-force spectroscopy maps two-dimensional reaction landscape of the Holliday junction. Science 318, 279–283 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sirinakis G, Ren Y, Gao Y, Xi Z & Zhang Y Combined versatile high-resolution optical tweezers and single-molecule fluorescence microscopy. Rev. Sci. Instrum 83, 093708 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Duesterberg VK, Fischer-Hwang IT, Perez CF, Hogan DW & Block SM Observation of long-range tertiary interactions during ligand binding by the TPP riboswitch aptamer. eLife 4, e12362 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Heller I et al. STED nanoscopy combined with optical tweezers reveals protein dynamics on densely covered DNA. Nat. Methods 10, 910–916 (2013). [DOI] [PubMed] [Google Scholar]
- 70.van Mameren J, Peterman EJ & Wuite GJ See me, feel me: methods to concurrently visualize and manipulate single DNA molecules and associated proteins. Nucleic Acids Res. 36, 4381–4389 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Candelli A, Wuite GJ & Peterman EJ Combining optical trapping, fluorescence microscopy and microfluidics for single molecule studies of DNA–protein interactions. Phys. Chem. Chem Phys 13, 7263–7272 (2011). [DOI] [PubMed] [Google Scholar]
- 72.Comstock MJ et al. Direct observation of structure–function relationship in a nucleic acid-processing enzyme. Science 348, 352–354 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Desai VP et al. Co-temporal force and fluorescence measurements reveal a ribosomal gear shift mechanism of translation regulation by structured mRNAs. Mol. Cell 75, 1007–1019 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Newton MD et al. DNA stretching induces Cas9 off-target activity. Nat. Struct. Mol. Biol 26, 185–192 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou R et al. SSB functions as a sliding platform that migrates on DNA via reptation. Cell 146, 222–232 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ngo TT, Zhang Q, Zhou R, Yodh JG & Ha T Asymmetric unwrapping of nucleosomes under tension directed by DNA local flexibility. Cell 160, 1135–1144 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Suksombat S, Khafizov R, Kozlov AG, Lohman TM & Chemla YR Structural dynamics of E. coli single-stranded DNA binding protein reveal DNA wrapping and unwrapping pathways. eLife 4, e08193 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.van Dijk MA, Kapitein LC, Mameren J, Schmidt CF & Peterman EJ Combining optical trapping and single-molecule fluorescence spectroscopy: enhanced photobleaching of fluorophores. J. Phys. Chem. B 108, 6479–6484 (2004). [DOI] [PubMed] [Google Scholar]
- 79.Brau RR, Tarsa PB, Ferrer JM, Lee P & Lang MJ Interlaced optical force–fluorescence measurements for single molecule biophysics. Biophys. J 91, 1069–1077 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lang MJ, Fordyce PM & Block SM Combined optical trapping and single-molecule fluorescence. J. Biol 2, 6 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.van Mameren J et al. Counting RAD51 proteins disassembling from nucleoprotein filaments under tension. Nature 457, 745–748 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Candelli A et al. Visualization and quantification of nascent RAD51 filament formation at single-monomer resolution. Proc. Natl Acad. Sci. USA 111, 15090–15095 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Whitley KD, Comstock MJ & Chemla YR High-resolution ‘fleezers’: dual-trap optical tweezers combined with single-molecule fluorescence detection. Methods Mol. Biol 1486, 183–256 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shi X et al. Quantitative fluorescence labeling of aldehyde-tagged proteins for single-molecule imaging. Nat. Methods 9, 499–503 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Keppler A, Pick H, Arrivoli C, Vogel H & Johnsson K Labeling of fusion proteins with synthetic fluorophores in live cells. Proc. Natl Acad. Sci. USA 101, 9955–9959 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ha T Single-molecule fluorescence resonance energy transfer. Methods 25, 78–86 (2001). [DOI] [PubMed] [Google Scholar]
- 87.Rasnik I, McKinney SA & Ha T Nonblinking and long-lasting single-molecule fluorescence imaging. Nat. Methods 3, 891–893 (2006). [DOI] [PubMed] [Google Scholar]
- 88.Swoboda M et al. Enzymatic oxygen scavenging for photostability without pH drop in single-molecule experiments. ACS Nano 6, 6364–6369 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rasnik I, McKinney SA & Ha T Surfaces and orientations: much to FRET about? Acc. Chem. Res 38, 542–548 (2005). [DOI] [PubMed] [Google Scholar]
- 90.Joo C & Ha T in Single-Molecule Techniques: A Laboratory Manual (eds Selvin PR & Ha T) 3–36 (Cold Spring Harbor Laboratory Press, 2008). [Google Scholar]
- 91.La Porta A & Wang MD Optical torque wrench: angular trapping, rotation, and torque detection of quartz microparticles. Phys. Rev. Lett 92, 190801 (2004). [DOI] [PubMed] [Google Scholar]; This study shows the initial implementation of the angular optical trap in which a trapped quartz particle is rotated and the optical torque on the particle is directly measured.
- 92.Deufel C, Forth S, Simmons CR, Dejgosha S & Wang MD Nanofabricated quartz cylinders for angular trapping: DNA supercoiling torque detection. Nat. Methods 4, 223–225 (2007). [DOI] [PubMed] [Google Scholar]
- 93.Bryant Z et al. Structural transitions and elasticity from torque measurements on DNA. Nature 424, 338–341 (2003). [DOI] [PubMed] [Google Scholar]
- 94.Lebel P, Basu A, Oberstrass FC, Tretter EM & Bryant Z Gold rotor bead tracking for high-speed measurements of DNA twist, torque and extension. Nat. Methods 11, 456–462 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Celedon A et al. Magnetic tweezers measurement of single molecule torque. Nano Lett. 9, 1720–1725 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lipfert J, Kerssemakers JW, Jager T & Dekker NH Magnetic torque tweezers: measuring torsional stiffness in DNA and RecA–DNA filaments. Nat. Methods 7, 977–980 (2010). [DOI] [PubMed] [Google Scholar]
- 97.Gutierrez-Medina B, Andreasson JO, Greenleaf WJ, Laporta A & Block SM An optical apparatus for rotation and trapping. Methods Enzymol. 475, 377–404 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Friese ME, Nieminen TA, Heckenberg NR & Rubinsztein-Dunlop H Optical alignment and spinning of laser-trapped microscopic particles. Nature 394, 348–350 (1998). [Google Scholar]
- 99.Paterson L et al. Controlled rotation of optically trapped microscopic particles. Science 292, 912–914 (2001). [DOI] [PubMed] [Google Scholar]
- 100.Ha S et al. Tunable top-down fabrication and functional surface coating of single-crystal titanium dioxide nanostructures and nanoparticles. Nanoscale 8, 10739–10748 (2016). [DOI] [PubMed] [Google Scholar]
- 101.Ma J, Tan C & Wang MD Single-molecule angular optical trapping for studying transcription under torsion. Methods Mol. Biol 1805, 301–332 (2018). [DOI] [PubMed] [Google Scholar]
- 102.Deufel C & Wang MD Detection of forces and displacements along the axial direction in an optical trap. Biophys. J 90, 657–667 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bustamante C, Marko JF, Siggia ED & Smith S Entropic elasticity of λ-phage DNA. Science 265, 1599–1600 (1994). [DOI] [PubMed] [Google Scholar]
- 104.Marko JF & Siggia ED Stretching DNA. Macromolecules 28, 8759–8770 (1995). [Google Scholar]
- 105.Wang MD, Yin H, Landick R, Gelles J & Block SM Stretching DNA with optical tweezers. Biophys. J 72, 1335–1346 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tskhovrebova L, Trinick J, Sleep JA & Simmons RM Elasticity and unfolding of single molecules of the giant muscle protein titin. Nature 387, 308–312 (1997). [DOI] [PubMed] [Google Scholar]
- 107.Kellermayer MS, Smith SB, Granzier HL & Bustamante C Folding–unfolding transitions in single titin molecules characterized with laser tweezers. Science 276, 1112–1116 (1997). [DOI] [PubMed] [Google Scholar]
- 108.Cui Y & Bustamante C Pulling a single chromatin fiber reveals the forces that maintain its higher-order structure. Proc. Natl Acad. Sci. USA 97, 127–132 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bennink ML et al. Unfolding individual nucleosomes by stretching single chromatin fibers with optical tweezers. Nat. Struct. Biol 8, 606–610 (2001). [DOI] [PubMed] [Google Scholar]
- 110.Hegner M, Smith SB & Bustamante C Polymerization and mechanical properties of single RecA–DNA filaments. Proc. Natl Acad. Sci. USA 96, 10109–10114 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Evans E Probing the relation between force–lifetime– and chemistry in single molecular bonds. Annu. Rev. Biophys. Biomol. Struct 30, 105–128 (2001). [DOI] [PubMed] [Google Scholar]
- 112.Dudko OK, Hummer G & Szabo A Theory, analysis, and interpretation of single-molecule force spectroscopy experiments. Proc. Natl Acad. Sci. USA 105, 15755–15760 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study provides the theoretical framework for dynamic force spectroscopy studies by mapping rupture force histograms from constant loading-rate experiments to force-dependent lifetimes in constant-force experiments.
- 113.Jarzynski C Nonequilibrium equality for free energy differences. Phys. Rev. Lett 78, 2690–2693 (1997). [Google Scholar]
- 114.Crooks GE Entropy production fluctuation theorem and the nonequilibrium work relation for free energy differences. Phys. Rev. E 60, 2721–2726 (1999). [DOI] [PubMed] [Google Scholar]
- 115.Hummer G & Szabo A Free energy reconstruction from nonequilibrium single-molecule pulling experiments. Proc. Natl Acad. Sci. USA 98, 3658–3661 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bustamante C, Liphardt J & Ritort F The nonequilibrium thermodynamics of small systems. Phys. Today 58, 43 (2005). [Google Scholar]
- 117.Collin D et al. Verification of the Crooks fluctuation theorem and recovery of RNA folding free energies. Nature 437, 231–234 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Woodside MT & Block SM Reconstructing folding energy landscapes by single-molecule force spectroscopy. Annu. Rev. Biophys 43, 19–39 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Spudich JA, Rice SE, Rock RS, Purcell TJ & Warrick HM Optical traps to study properties of molecular motors. Cold Spring Harb. Protoc 2011, 1305–1318 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kerssemakers JW et al. Assembly dynamics of microtubules at molecular resolution. Nature 442, 709–712 (2006). [DOI] [PubMed] [Google Scholar]
- 121.Carter BC, Vershinin M & Gross SP A comparison of step-detection methods: how well can you do? Biophys. J 94, 306–319 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kalafut B & Visscher K An objective, model-independent method for detection of non-uniform steps in noisy signals. Comput. Phys. Commun 179, 716–723 (2008). [Google Scholar]
- 123.Syed S, Mullner FE, Selvin PR & Sigworth FJ Improved hidden Markov models for molecular motors, part 2: extensions and application to experimental data. Biophys. J 99, 3696–3703 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Righini M et al. Full molecular trajectories of RNA polymerase at single base-pair resolution. Proc. Natl Acad. Sci. USA 115, 1286–1291 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gelles J, Schnapp BJ & Sheetz MP Tracking kinesin-driven movements with nanometre-scale precision. Nature 331, 450–453 (1988). [DOI] [PubMed] [Google Scholar]
- 126.Schnitzer MJ & Block SM Statistical kinetics of processive enzymes. Cold Spring Harb. Symp. Quant. Biol 60, 793–802 (1995). [DOI] [PubMed] [Google Scholar]
- 127.Moffitt JR, Chemla YR & Bustamante C Methods in statistical kinetics. Methods Enzymol. 475, 221–257 (2010). [DOI] [PubMed] [Google Scholar]
- 128.Whitley KD, Comstock MJ & Chemla YR Elasticity of the transition state for oligonucleotide hybridization. Nucleic Acids Res. 45, 547–555 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Leake MC et al. Stoichiometry and turnover in single, functioning membrane protein complexes. Nature 443, 355–358 (2006). [DOI] [PubMed] [Google Scholar]
- 130.Mitra J et al. Extreme mechanical diversity of human telomeric DNA revealed by fluorescence-force spectroscopy. Proc. Natl Acad. Sci. USA 116, 8350–8359 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Forth S et al. Abrupt buckling transition observed during the plectoneme formation of individual DNA molecules. Phys. Rev. Lett 100, 148301 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sheinin MY, Forth S, Marko JF & Wang MD Underwound DNA under tension: structure, elasticity, and sequence-dependent behaviors. Phys. Rev. Lett 107, 108102 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Forth S, Deufel C, Patel SS & Wang MD Direct measurements of torque during holliday junction migration. Biophys. J 101, L5–L7 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Inman J, Forth S & Wang MD Passive torque wrench and angular position detection using a single-beam optical trap. Opt. Lett 35, 2949–2951 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bustamante C, Bryant Z & Smith SB Ten years of tension: single-molecule DNA mechanics. Nature 421, 423–427 (2003). [DOI] [PubMed] [Google Scholar]
- 136.Gore J et al. DNA overwinds when stretched. Nature 442, 836–839 (2006). [DOI] [PubMed] [Google Scholar]
- 137.Sheinin MY & Wang MD Twist–stretch coupling and phase transition during DNA supercoiling. Phys. Chem. Chem. Phys 11, 4800–4803 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Seol Y, Skinner GM, Visscher K, Buhot A & Halperin A Stretching of homopolymeric RNA reveals single-stranded helices and base-stacking. Phys. Rev. Lett 98, 158103 (2007). [DOI] [PubMed] [Google Scholar]
- 139.McIntosh DB, Duggan G, Gouil Q & Saleh OA Sequence-dependent elasticity and electrostatics of single-stranded DNA: signatures of base-stacking. Biophys. J 106, 659–666 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rouzina I & Bloomfield VA Force-induced melting of the DNA double helix. 2. Effect of solution conditions. Biophys. J 80, 894–900 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bosaeus N et al. Tension induces a base-paired overstretched DNA conformation. Proc. Natl Acad. Sci. USA 109, 15179–15184 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhang X, Chen H, Fu H, Doyle PS & Yan J Two distinct overstretched DNA structures revealed by single-molecule thermodynamics measurements. Proc. Natl Acad. Sci. USA 109, 8103–8108 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.King GA et al. Revealing the competition between peeled ssDNA, melting bubbles, and S-DNA during DNA overstretching using fluorescence microscopy. Proc. Natl Acad. Sci. USA 110, 3859–3864 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Brower-Toland BD et al. Mechanical disruption of individual nucleosomes reveals a reversible multistage release of DNA. Proc. Natl Acad. Sci. USA 99, 1960–1965 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Brower-Toland B et al. Specific contributions of histone tails and their acetylation to the mechanical stability of nucleosomes. J. Mol. Biol 346, 135–146 (2005). [DOI] [PubMed] [Google Scholar]
- 146.Mihardja S, Spakowitz AJ, Zhang Y & Bustamante C Effect of force on mononucleosomal dynamics. Proc. Natl Acad. Sci. USA 103, 15871–15876 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Shundrovsky A, Smith CL, Lis JT, Peterson CL & Wang MD Probing SWI/SNF remodeling of the nucleosome by unzipping single DNA molecules. Nat. Struct. Mol. Biol 13, 549–554 (2006). [DOI] [PubMed] [Google Scholar]
- 148.Wang MD et al. Force and velocity measured for single molecules of RNA polymerase. Science 282, 902–907 (1998). [DOI] [PubMed] [Google Scholar]
- 149.Smith DE et al. The bacteriophage straight φ29 portal motor can package DNA against a large internal force. Nature 413, 748–752 (2001). [DOI] [PubMed] [Google Scholar]
- 150.Perkins TT, Li HW, Dalal RV, Gelles J & Block SM Forward and reverse motion of single RecBCD molecules on DNA. Biophys. J 86, 1640–1648 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Chemla YR et al. Mechanism of force generation of a viral DNA packaging motor. Cell 122, 683–692 (2005). [DOI] [PubMed] [Google Scholar]
- 152.Galburt EA et al. Backtracking determines the force sensitivity of RNAP II in a factor-dependent manner. Nature 446, 820–823 (2007). [DOI] [PubMed] [Google Scholar]
- 153.Liu S et al. A viral packaging motor varies its DNA rotation and step size to preserve subunit coordination as the capsid fills. Cell 157, 702–713 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows direct visualization of the rotational motion of a viral packaging motor, which is a significant application of the rotary bead assay.
- 154.Moffitt JR et al. Intersubunit coordination in a homomeric ring ATPase. Nature 457, 446–450 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Chistol G et al. High degree of coordination and division of labor among subunits in a homomeric ring ATPase. Cell 151, 1017–1028 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Qi Z, Pugh RA, Spies M & Chemla YR Sequence-dependent base pair stepping dynamics in XPD helicase unwinding. eLife 2, e00334 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Huguet JM et al. Single-molecule derivation of salt dependent base-pair free energies in DNA. Proc. Natl Acad. Sci. USA 107, 15431–15436 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Koch SJ, Shundrovsky A, Jantzen BC & Wang MD Probing protein–DNA interactions by unzipping a single DNA double helix. Biophys. J 83, 1098–1105 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Koch SJ & Wang MD Dynamic force spectroscopy of protein–DNA interactions by unzipping DNA. Phys. Rev. Lett 91, 028103 (2003). [DOI] [PubMed] [Google Scholar]
- 160.Hall MA et al. High-resolution dynamic mapping of histone–DNA interactions in a nucleosome. Nat. Struct. Mol. Biol 16, 124–129 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Jin J et al. Synergistic action of RNA polymerases in overcoming the nucleosomal barrier. Nat. Struct. Mol. Biol 17, 745–752 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Chen Z et al. High-resolution and high-accuracy topographic and transcriptional maps of the nucleosome barrier. eLife 8, e48281 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Killian JL, Inman JT & Wang MD High-performance image-based measurements of biological forces and interactions in a dual optical trap. ACS Nano 12, 11963–11974 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Dumont S et al. RNA translocation and unwinding mechanism of HCV NS3 helicase and its coordination by ATP. Nature 439, 105–108 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Johnson DS, Bai L, Smith BY, Patel SS & Wang MD Single-molecule studies reveal dynamics of DNA unwinding by the ring-shaped T7 helicase. Cell 129, 1299–1309 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Sun B et al. ATP-induced helicase slippage reveals highly coordinated subunits. Nature 478, 132–135 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Sun B et al. T7 replisome directly overcomes DNA damage. Nat. Commun 6, 10260 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Sun B et al. Helicase promotes replication re-initiation from an RNA transcript. Nat. Commun 9, 2306 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Le TT et al. Mfd dynamically regulates transcription via a release and catch-up mechanism. Cell 172, 344–357 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Sheinin MY, Li M, Soltani M, Luger K & Wang MD Torque modulates nucleosome stability and facilitates H2A/H2B dimer loss. Nat. Commun 4, 2579 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Le TT et al. Synergistic coordination of chromatin torsional mechanics and topoisomerase activity. Cell 179, 619–631 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ma J, Bai L & Wang MD Transcription under torsion. Science 340, 1580–1583 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study presents the measurement of the torque generated by RNA polymerase during transcription, and is the first application of the angular optical trap to study a motor protein.
- 173.Ma J et al. Transcription factor regulation of RNA polymerase’s torque generation capacity. Proc. Natl Acad. Sci. USA 116, 2583–2588 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Stone MD et al. Chirality sensing by Escherichia coli topoisomerase IV and the mechanism of type II topoisomerases. Proc. Natl Acad. Sci. USA 100, 8654–8659 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Bianco PR et al. Processive translocation and DNA unwinding by individual RecBCD enzyme molecules. Nature 409, 374–378 (2001). [DOI] [PubMed] [Google Scholar]
- 176.Bell JC, Plank JL, Dombrowski CC & Kowalczykowski SC Direct imaging of RecA nucleation and growth on single molecules of SSB-coated ssDNA. Nature 491, 274–278 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lee KS et al. Ultrafast redistribution of E. coli SSB along long single-stranded DNA via intersegment transfer. J. Mol. Biol 426, 2413–2421 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wasserman MR, Schauer GD, O’Donnell ME & Liu S Replication fork activation is enabled by a single-stranded DNA gate in CMG helicase. Cell 178, 600–611 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Arslan S, Khafizov R, Thomas CD, Chemla YR & Ha T Protein structure. Engineering of a superhelicase through conformational control. Science 348, 344–347 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Dame RT, Noom MC & Wuite GJ Bacterial chromatin organization by H-NS protein unravelled using dual DNA manipulation. Nature 444, 387–390 (2006). [DOI] [PubMed] [Google Scholar]
- 181.Brouwer I et al. Sliding sleeves of XRCC4-XLF bridge DNA and connect fragments of broken DNA. Nature 535, 566–569 (2016). [DOI] [PubMed] [Google Scholar]; This study describes the application of a fleezers instrument using dual-and quadruple-trap optical tweezers combined with fluorescence to study the interactions of non-homologous end-joining proteins with their DNA substrates.
- 182.Inman JT et al. DNA Y structure: a versatile, multidimensional single molecule assay. Nano Lett. 14, 6475–6480 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Seol Y, Skinner GM & Visscher K Elastic properties of a single-stranded charged homopolymeric ribonucleotide. Phys. Rev. Lett 93, 118102 (2004). [DOI] [PubMed] [Google Scholar]
- 184.Camunas-Soler J, Ribezzi-Crivellari M & Ritort F Elastic properties of nucleic acids by single-molecule force spectroscopy. Annu. Rev. Biophys 45, 65–84 (2016). [DOI] [PubMed] [Google Scholar]
- 185.Cheng W, Arunajadai SG, Moffitt JR, Tinoco I Jr & Bustamante, C. Single-base pair unwinding and asynchronous RNA release by the hepatitis C virus NS3 helicase. Science 333, 1746–1749 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Wen JD et al. Following translation by single ribosomes one codon at a time. Nature 452, 598–603 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Qu X et al. The ribosome uses two active mechanisms to unwind messenger RNA during translation. Nature 475, 118–121 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Yan S, Wen JD, Bustamante C & Tinoco I Jr. Ribosome excursions during mRNA translocation mediate broad branching of frameshift pathways. Cell 160, 870–881 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Yu H et al. Direct observation of multiple misfolding pathways in a single prion protein molecule. Proc. Natl Acad. Sci. USA 109, 5283–5288 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Jahn M et al. Folding and domain interactions of three orthologs of Hsp90 studied by single-molecule force spectroscopy. Structure 26, 96–105 (2018). [DOI] [PubMed] [Google Scholar]
- 191.Guinn EJ, Jagannathan B & Marqusee S Single-molecule chemo-mechanical unfolding reveals multiple transition state barriers in a small single-domain protein. Nat. Commun 6, 6861 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Motlagh HN, Toptygin D, Kaiser CM & Hilser VJ Single-molecule chemo-mechanical spectroscopy provides structural identity of folding intermediates. Biophys. J 110, 1280–1290 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Yu H et al. Protein misfolding occurs by slow diffusion across multiple barriers in a rough energy landscape. Proc. Natl Acad. Sci. USA 112, 8308–8313 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Neupane K et al. Direct observation of transition paths during the folding of proteins and nucleic acids. Science 352, 239–242 (2016). [DOI] [PubMed] [Google Scholar]
- 195.Levinthal C Are there pathways for protein folding? J. Chim. Phys 65, 44–45 (1968). [Google Scholar]
- 196.Leopold PE, Montal M & Onuchic JN Protein folding funnels: a kinetic approach to the sequence–structure relationship. Proc. Natl Acad. Sci. USA 89, 8721–8725 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Bryngelson JD, Onuchic JN, Socci ND & Wolynes PG Funnels, pathways, and the energy landscape of protein folding: a synthesis. Proteins 21, 167–195 (1995). [DOI] [PubMed] [Google Scholar]
- 198.Dill KA & Chan HS From Levinthal to pathways to funnels. Nat. Struct. Biol 4, 10–19 (1997). [DOI] [PubMed] [Google Scholar]
- 199.Bai Y, Sosnick TR, Mayne L & Englander SW Protein folding intermediates: native-state hydrogen exchange. Science 269, 192–197 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Englander SW & Mayne L The nature of protein folding pathways. Proc. Natl Acad. Sci. USA 111, 15873–15880 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Englander SW, Mayne L, Kan ZY & Hu W Protein folding—how and why: by hydrogen exchange, fragment separation, and mass spectrometry. Annu. Rev. Biophys 45, 135–152 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Wolynes P Biomolecular folding. Moments of excitement. Science 352, 150–151 (2016). [DOI] [PubMed] [Google Scholar]
- 203.Englander SW & Mayne L The case for defined protein folding pathways. Proc. Natl Acad. Sci. USA 114, 8253–8258 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Bechtluft P et al. Direct observation of chaperone-induced changes in a protein folding pathway. Science 318, 1458–1461 (2007). [DOI] [PubMed] [Google Scholar]
- 205.Mashaghi A et al. Reshaping of the conformational search of a protein by the chaperone trigger factor. Nature 500, 98–101 (2013). [DOI] [PubMed] [Google Scholar]
- 206.Liu K, Maciuba K & Kaiser CM The ribosome cooperates with a chaperone to guide multi-domain protein folding. Mol. Cell 74, 310–319 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Mashaghi A et al. Alternative modes of client binding enable functional plasticity of Hsp70. Nature 539, 448–451 (2016). [DOI] [PubMed] [Google Scholar]
- 208.Liu K, Rehfus JE, Mattson E & Kaiser CM The ribosome destabilizes native and non-native structures in a nascent multidomain protein. Protein Sci. 26, 1439–1451 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Wruck F, Katranidis A, Nierhaus KH, Buldt G & Hegner M Translation and folding of single proteins in real time. Proc. Natl Acad. Sci. USA 114, E4399–E4407 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Alexander LM, Goldman DH, Wee LM & Bustamante C Non-equilibrium dynamics of a nascent polypeptide during translation suppress its misfolding. Nat. Commun 10, 2709 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Bustamante C, Chemla YR, Forde NR & Izhaky D Mechanical processes in biochemistry. Annu. Rev. Biochem 73, 705–748 (2004). [DOI] [PubMed] [Google Scholar]
- 212.Kolomeisky AB & Fisher ME Molecular motors: a theorist’s perspective. Annu. Rev. Phys. Chem 58, 675–695 (2007). [DOI] [PubMed] [Google Scholar]
- 213.Sheetz MP & Spudich JA Movement of myosin-coated fluorescent beads on actin cables in vitro. Nature 303, 31–35 (1983). [DOI] [PubMed] [Google Scholar]
- 214.Vale RD, Reese TS & Sheetz MP Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell 42, 39–50 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Mickolajczyk KJ et al. Force-dependent stimulation of RNA unwinding by SARS-CoV-2 nsp13 helicase. Biophys. J 10.1016/j.bpj.2020.11.2276 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Liu S, Chistol G & Bustamante C Mechanical operation and intersubunit coordination of ring-shaped molecular motors: insights from single-molecule studies. Biophys. J 106, 1844–1858 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Aathavan K et al. Substrate interactions and promiscuity in a viral DNA packaging motor. Nature 461, 669–673 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Tafoya S et al. Molecular switch-like regulation enables global subunit coordination in a viral ring ATPase. Proc. Natl Acad. Sci. USA 115, 7961–7966 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Keller D & Bustamante C The mechanochemistry of molecular motors. Biophys. J 78, 541–556 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Larson MH et al. Trigger loop dynamics mediate the balance between the transcriptional fidelity and speed of RNA polymerase II. Proc. Natl Acad. Sci. USA 109, 6555–6560 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Dangkulwanich M et al. Complete dissection of transcription elongation reveals slow translocation of RNA polymerase II in a linear ratchet mechanism. eLife 2, e00971 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Wang HY, Elston T, Mogilner A & Oster G Force generation in RNA polymerase. Biophys. J 74, 1186–1202 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Bai L, Fulbright RM & Wang MD Mechanochemical kinetics of transcription elongation. Phys. Rev. Lett 98, 068103 (2007). [DOI] [PubMed] [Google Scholar]
- 224.D OM, Tadigotla VR, Nudler E & Ruckenstein AE A unified model of transcription elongation: what have we learned from single-molecule experiments? Biophys. J 100, 1157–1166 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Liu T et al. Direct measurement of the mechanical work during translocation by the ribosome. eLife 3, e03406 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Block SM, Asbury CL, Shaevitz JW & Lang MJ Probing the kinesin reaction cycle with a 2D optical force clamp. Proc. Natl Acad. Sci. USA 100, 2351–2356 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Hwang W & Karplus M Structural basis for power stroke vs. Brownian ratchet mechanisms of motor proteins. Proc. Natl Acad. Sci. USA 116, 19777–19785 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Sen M et al. The ClpXP protease unfolds substrates using a constant rate of pulling but different gears. Cell 155, 636–646 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Liu N, Chistol G, Cui Y & Bustamante C Mechanochemical coupling and bi-phasic force-velocity dependence in the ultra-fast ring ATPase SpoIIIE. eLife 7, e32354 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Lu HP, Xun L & Xie XS Single-molecule enzymatic dynamics. Science 282, 1877–1882 (1998). [DOI] [PubMed] [Google Scholar]
- 231.van Oijen AM et al. Single-molecule kinetics of lambda exonuclease reveal base dependence and dynamic disorder. Science 301, 1235–1238 (2003). [DOI] [PubMed] [Google Scholar]
- 232.Fuller DN, Raymer DM, Kottadiel VI, Rao VB & Smith DE Single phage T4 DNA packaging motors exhibit large force generation, high velocity, and dynamic variability. Proc. Natl Acad. Sci. USA 104, 16868–16873 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Liu B, Baskin RJ & Kowalczykowski SC DNA unwinding heterogeneity by RecBCD results from static molecules able to equilibrate. Nature 500, 482–485 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Hellenkamp B et al. Precision and accuracy of single-molecule FRET measurements—a multi-laboratory benchmark study. Nat. Methods 15, 669–676 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Huisstede JH, van Rooijen BD, van der Werf KO, Bennink ML & Subramaniam V Dependence of silicon position-detector bandwidth on wavelength, power, and bias. Opt. Lett 31, 610–612 (2006). [DOI] [PubMed] [Google Scholar]
- 236.Sudhakar S et al. Germanium nanospheres for ultraresolution picotensiometry of kinesin motors. Science 371, eabd9944 (2021). [DOI] [PubMed] [Google Scholar]
- 237.Bai H et al. Remote control of DNA-acting enzymes by varying the Brownian dynamics of a distant DNA end. Proc. Natl Acad. Sci. USA 109, 16546–16551 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Pfitzner E et al. Rigid DNA beams for high-resolution single-molecule mechanics. Angew. Chem. Int. Ed. Engl 52, 7766–7771 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Kojima H, Muto E, Higuchi H & Yanagida T Mechanics of single kinesin molecules measured by optical trapping nanometry. Biophys. J 73, 2012–2022 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Roy R, Hohng S & Ha T A practical guide to single-molecule FRET. Nat. Methods 5, 507–516 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Ribeck N & Saleh OA Multiplexed single-molecule measurements with magnetic tweezers. Rev. Sci. Instrum 79, 094301 (2008). [DOI] [PubMed] [Google Scholar]
- 242.Greene EC, Wind S, Fazio T, Gorman J & Visnapuu ML DNA curtains for high-throughput single-molecule optical imaging. Methods Enzymol. 472, 293–315 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.De Vlaminck I et al. Highly parallel magnetic tweezers by targeted DNA tethering. Nano Lett. 11, 5489–5493 (2011). [DOI] [PubMed] [Google Scholar]
- 244.Farre A, van der Horst A, Blab GA, Downing BP & Forde NR Stretching single DNA molecules to demonstrate high-force capabilities of holographic optical tweezers. J. Biophotonics 3, 224–233 (2010). [DOI] [PubMed] [Google Scholar]
- 245.Soltani M et al. Nanophotonic trapping for precise manipulation of biomolecular arrays. Nat. Nanotechnol 9, 448–452 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Veigel C & Schmidt CF Moving into the cell: single-molecule studies of molecular motors in complex environments. Nat. Rev. Mol. Cell Biol 12, 163–176 (2011). [DOI] [PubMed] [Google Scholar]
- 247.Hendricks AG, Holzbaur EL & Goldman YE Force measurements on cargoes in living cells reveal collective dynamics of microtubule motors. Proc. Natl Acad. Sci. USA 109, 18447–18452 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Blehm BH, Schroer TA, Trybus KM, Chemla YR & Selvin PR In vivo optical trapping indicates kinesin’s stall force is reduced by dynein during intracellular transport. Proc. Natl Acad. Sci. USA 110, 3381–3386 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Smith SB, Finzi L & Bustamante C Direct mechanical measurements of the elasticity of single DNA molecules by using magnetic beads. Science 258, 1122–1126 (1992). [DOI] [PubMed] [Google Scholar]
- 250.Moroz JD & Nelson P Torsional directed walks, entropic elasticity, and DNA twist stiffness. Proc. Natl Acad. Sci. USA 94, 14418–14422 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Herrero-Galan E et al. Mechanical identities of RNA and DNA double helices unveiled at the single-molecule level. J. Am. Chem. Soc 135, 122–131 (2013). [DOI] [PubMed] [Google Scholar]
- 252.Baumann CG, Smith SB, Bloomfield VA & Bustamante C Ionic effects on the elasticity of single DNA molecules. Proc. Natl Acad. Sci. USA 94, 6185–6190 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Saleh OA, McIntosh DB, Pincus P & Ribeck N Nonlinear low-force elasticity of single-stranded DNA molecules. Phys. Rev. Lett 102, 068301 (2009). [DOI] [PubMed] [Google Scholar]
- 254.Goldman DH et al. Ribosome. mechanical force releases nascent chain-mediated ribosome arrest in vitro and in vivo. Science 348, 457–460 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Marko JF & Siggia ED Fluctuations and supercoiling of DNA. Science 265, 506–508 (1994). [DOI] [PubMed] [Google Scholar]
- 256.Odijk T Stiff chains and filaments under tension. Macromolecules 28, 7016–7018 (1995). [Google Scholar]
- 257.Bouchiat C & Mezard M Elasticity model of a supercoiled DNA molecule. Phys. Rev. Lett 80, 1556–1559 (1998). [Google Scholar]
- 258.Kratky O & Porod G Röntgenuntersuchung gelöster fadenmoleküle [German]. Recl. Trav. Chim. Pays-Bas 68, 1106–1123 (1949). [Google Scholar]
- 259.Jacobson DR, McIntosh DB, Stevens MJ, Rubinstein M & Saleh OA Single-stranded nucleic acid elasticity arises from internal electrostatic tension. Proc. Natl Acad. Sci. USA 114, 5095–5100 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Bell GI Models for the specific adhesion of cells to cells. Science 200, 618–627 (1978). [DOI] [PubMed] [Google Scholar]
- 261.Evans E & Ritchie K Dynamic strength of molecular adhesion bonds. Biophys. J 72, 1541–1555 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Hummer G & Szabo A Kinetics from nonequilibrium single-molecule pulling experiments. Biophys. J 85, 5–15 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Dudko OK, Hummer G & Szabo A Intrinsic rates and activation free energies from single-molecule pulling experiments. Phys. Rev. Lett 96, 108101 (2006). [DOI] [PubMed] [Google Scholar]
- 264.Jarzynski C Equilibrium free-energy differences from nonequilibrium measurements: a master-equation approach. Phys. Rev. E 56, 5018–5035 (1997). [Google Scholar]
- 265.Crooks GE Nonequilibrium measurements of free energy differences for microscopically reversible Markovian systems. J. Stat. Phys 90, 1481–1487 (1998). [Google Scholar]
- 266.Liphardt J, Dumont S, Smith SB, Tinoco I Jr. & Bustamante C Equilibrium information from nonequilibrium measurements in an experimental test of Jarzynski’s equality. Science 296, 1832–1835 (2002). [DOI] [PubMed] [Google Scholar]; This study demonstrates the mechanical unfolding and refolding of RNA and is the first experimental validation of Jarzynski’s equality, which relates irreversible work to equilibrium free energy difference.
- 267.Camunas-Soler J, Alemany A & Ritort F Experimental measurement of binding energy, selectivity, and allostery using fluctuation theorems. Science 355, 412–415 (2017). [DOI] [PubMed] [Google Scholar]