

Abstract
Systemic infections, especially in patients with chronic diseases, may result in sepsis: an explosive, uncoordinated immune response that can lead to multisystem organ failure with a high mortality rate. Patients with similar clinical phenotypes or sepsis biomarker expression upon diagnosis may have different outcomes, suggesting that the dynamics of sepsis is critical in disease progression. A within-subject study of patients with Gram-negative bacterial sepsis with surviving and fatal outcomes was designed and single-cell transcriptomic analyses of peripheral blood mononuclear cells (PBMC) collected during the critical period between sepsis diagnosis and 6 h were performed. The single-cell observations in the study are consistent with trends from public datasets but also identify dynamic effects in individual cell subsets that change within hours. It is shown that platelet and erythroid precursor responses are drivers of fatal sepsis, with transcriptional signatures that are shared with severe COVID-19 disease. It is also shown that hypoxic stress is a driving factor in immune and metabolic dysfunction of monocytes and erythroid precursors. Last, the data support CD52 as a prognostic biomarker and therapeutic target for sepsis as its expression dynamically increases in lymphocytes and correlates with improved sepsis outcomes. In conclusion, this study describes the first single-cell study that analyzed short-term temporal changes in the immune cell populations and their characteristics in surviving or fatal sepsis. Tracking temporal expression changes in specific cell types could lead to more accurate predictions of sepsis outcomes and identify molecular biomarkers and pathways that could be therapeutically controlled to improve the sepsis trajectory toward better outcomes.
Keywords: CD52, Gram-negative bacteria, inflammation, platelet, sepsis
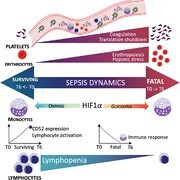
Graphical Abstract
Single cell transcriptomics of peripheral blood mononuclear cells in surviving and fatal sepsis reveal inflammatory and metabolic pathways that change within hours of sepsis recognition.
Graphical Abstract.
INTRODUCTION
Sepsis is an inflammatory syndrome caused by a systemic infection that can lead to multisystem organ failure and death. Sepsis is responsible for a significant percentage of in-hospital healthcare costs both in the United States and worldwide, and it is associated with a high mortality rate.1,2 Despite many efforts, no targeted therapeutics against sepsis have been developed in the last decades. One acknowledged challenge is the complexity of the disease involving the competing interplay between rampant inflammation (cytokine storm) and, paradoxically, the almost simultaneous shutdown of the immune system (immunoparalysis).3,4 Another sepsis challenge is that some patients with nearly identical clinical phenotypes quantified by qSOFA and APACHE scores die at every stage of the disease while others survive.5 This supports the need to understand the molecular level host response to sepsis, which has been studied in blood and peripheral blood mononuclear cell (PBMC) profiling studies by gene expression or proteomics methods.6 These studies identify several prognostic biomarkers, such as lactate, procalcitonin, C-reactive protein (CRP), ferritin, and erythrocyte sedimentation rate (ESR), which along with clinical scores, are standardly utilized to evaluate sepsis patients and determine their care.5
However, connecting these high-level observations to accurate clinical outcomes presents an unresolved challenge, likely due to the complexity and heterogeneity of this disease. Many studies have been conducted to identify a potential sepsis molecular signature to gain molecular insights into this heterogeneity, which could aid in diagnosis or treatment.7 Recently, the first single-cell analysis of the status of immune cells in sepsis was reported, which identified abnormal monocyte states associated with immune dysregulation.8 Here, we apply the same approach to focus on the additional question of immune cell trajectory immediately after diagnosis in sepsis survivor and non-survivor outcomes. We performed single-cell transcriptomics analyses in fatal or surviving sepsis using a within-subject study design of PBMC collected from septic patients in the intensive care unit (ICU) at 0 and 6 h post sepsis diagnosis. There is clinical utility in choosing a 6-h time point, as sepsis resuscitation bundles (both in the United States and internationally) have been modeled after landmark studies9 that demonstrated a significant reduction in mortality with aggressive resuscitation in the first six hours after presentation. Additionally, there is robust data10 that early administration of intravenous antibiotics in the first 60 min after the recognition of septic shock significantly improves mortality. While subsequent trials (PROMISE, ARISE, PROCESS) showed no difference in clinician-driven versus protocol-driven resuscitation at 6 h, the Centers for Medicare and Medicaid Services (CMS) and the Surviving Sepsis Campaign continue to advocate for hospitals and clinicians to use the 6-h time point from initial Emergency Department presentation as a benchmark for resuscitation.11–13 Thus, this time point was chosen to assess molecular changes in the patients after they had received their initial resuscitation (including intravenous antibiotics, intravenous fluids, and vasopressor support).
Our timed analyses revealed the emergence and continuous changes in abnormal immune cells, including new types of cells unique to sepsis and classical cell-types present in both sepsis and healthy controls, but with abnormal gene expression profiles and changes in population ratios. Specifically, we observed that fatal sepsis is associated with the expansion of platelets and erythroid precursors and the immunosuppressive trend of monocytes. Additionally, we identified CD52 expression in lymphocytes as a potential biomarker and therapeutic target for sepsis, where it correlated with increased lymphocyte activation and survival outcomes. At the cellular level, we also observed a switch in the metabolic state from oxidative phosphorylation in survivors to glycolysis in nonsurvivors. Last, we observed that fatal sepsis shared many gene signatures with severe COVID-19 patients, indicating convergent molecular pathways in severe disease. These included genes associated with increased platelet activity, elevated erythroid precursors, and chemokine expression in monocytes.
Overall, this study, which focused on within-subject analyses of PBMC over time, offers a unique perspective on the dynamic changes in immune cells in fatal sepsis. Specifically, we identify abnormal immune cell subsets, changes in functional pathways, and molecular signatures at the single-cell resolution associated with fatal or surviving outcomes in sepsis. This study provides foundation data and identifies specific cell subsets and molecular pathways that can be further explored to better predict and possibly modify sepsis outcomes.
RESULTS
Subject characteristics
To gain a molecular understanding of the immune state in surviving or non-surviving sepsis outcomes, we performed retrospectively single-cell RNA sequencing on PBMCs from 5 hospitalized patients with Gram-negative bacterial sepsis at 0 and 6 h post diagnosis. Three patients survived (Survivor, S) and were discharged from the ICU; two patients had fatal disease courses (Nonsurvivor, NS). Clinical parameters (qSOFA and APACHE scores) were high and could not distinguish between sepsis survivors and nonsurvivors, and all sepsis patients had plasma cytokine levels that were dramatically elevated compared to baseline nonsepsis volunteers (Table 1). These results are consistent with the known phenomenon of the sepsis-induced cytokine storm.4 In contrast, re-stimulation of PBMC from the same sepsis patients with LPS led to reduced TNF-α secretion as compared to PBMC from nonsepsis controls (Table 1), suggesting monocytic deactivation that has been reported in sepsis immunoparalysis.14 Flow cytometric analysis of PBMC was performed according to previously published gating strategies,15–17 and revealed different immune subset distribution with sepsis patients, including increased neutrophils but reduced T cell subsets, especially in the nonsurvivors (Fig. 1A and B). We also observed the emergence of cell subsets that we were unable to define with common PBMC surface antibodies (Fig. 1C, “other”). Together, these data characterize clinical and peripheral immune profiles in sepsis. However, more detailed subsetting of specific immune cells and insights into how temporal changes in their gene expression relate to sepsis outcome were lacking, which we addressed by single-cell RNA sequencing.
TABLE 1.
Characteristics of enrolled non-sepsis volunteers and sepsis patients at sepsis recognition (T0). Clinical parameters, cytokine levels in the plasma, and supernatant following LPS stimulation (10 ng/mL) of PBMCs
| Nonsepsis control (n = 2) | Sepsis nonsurvivor (n = 2) | Sepsis survivor (n = 3) | |||||
|---|---|---|---|---|---|---|---|
| Gender | Male | Female | Male | Female | Male | Female | Female |
| Age range | 35-40 | 45-50 | 90-95 | 65-70 | 45-50 | 65-70 | 70-75 |
| Sepsis etiology | n/a | n/a | E.coli bacteremia | E.coli bacteremia | |||
| APACHEII | n/a | n/a | 18 | 38 | 31 | 41 | 19 |
| SOFA | n/a | n/a | 11 | 16 | 11 | 15 | 7 |
| Time of death (days post enrollment) | n/a | n/a | <30 | 1 | n/a | n/a | n/a |
| Plasma cytokines (ng/mL) | |||||||
| Resistin | 22.5 | 36.7 | 202 | 135.9 | 147 | 281 | 92 |
| IL-6 | N.D | 0.002 | 30.2 | 142.3 | 133 | 2.48 | 0.31 |
| IL-8 | 0.03 | 0.026 | 6.65 | 27.2 | 41.7 | 0.61 | 0.4 |
| IL-10 | N.D. | N.D. | 0.13 | 9.71 | 0.52 | 0.15 | 0.39 |
| LPS-induced TNFα (ng/mL) | |||||||
| TNF-α | 0.656 | 0.979 | 0.45 | 0.0046 | n/a | 0.047 | 0.1 |
Abbreviations: APACHE II, acute physiology and chronic health evaluation II; SOFA, sequential organ failure assessment; N.D., Not detected; n/a, not applicable.
FIGURE 1.
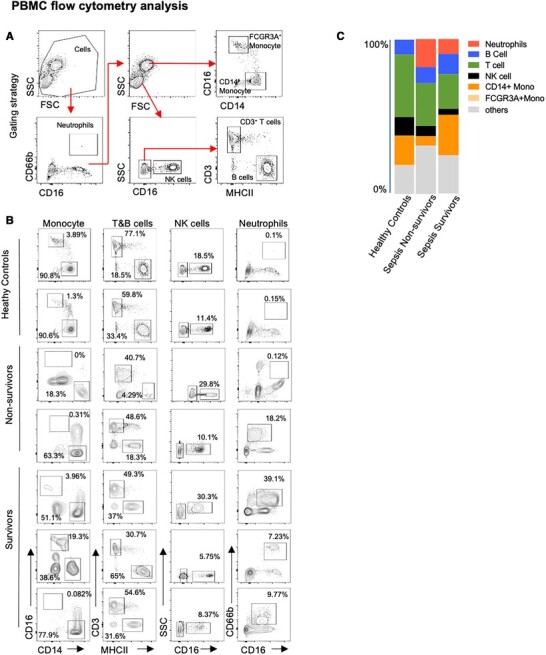
Flow cytometric analysis of PBMC from healthy control (HC), non-survivor (NS), and survivor (S) sepsis patients at first blood collection (T0). (A) Gating strategy. Neutrophils (CD66b+CD16+), FCGR3A+Monocytes (CD66b−SSCHiCD16+CD14low), CD14+Monocytes (CD66b−SSCHiCD16−CD14hi), NK cells (CD66b−SSClowCD16+), T cells (SSClowCD3+), and B cells (SSClowMHCII+). (B) Frequency of immune cell subsets in PBMC. (C) Immune cell proportions in PBMC
Single-cell transcriptomics identify immune cell subsets associated with sepsis severity
Single-cell RNA-seq was performed on a 10× Genomics platform. Consensus-based assignment of cell-types from all subjects revealed 11 cell-types: CD4+ T cells (24%), CD8+ T cells (8%), B cells (20%), Natural killer (NK) cells (9%), CD14+ monocytes (16%), FCGR3A+ monocytes (11%), dendritic cells (DC) (3%), erythroid precursor cells (2%), platelets (6%), neutrophils (1%), and common myeloid progenitor cells (CMP) (< 1%) (Fig. 2A; Supplementary Table S1). Comparison between the samples indicated variability between the individuals and no striking changes between cell subsets within 6 h (Fig. 2B and C; Supplementary Table S1). Among the sepsis patients, the female nonsurvivor (P50) showed an immune profile that was distinct from the ones in the male nonsurvivor (P34) and survivor samples. This individual showed advanced sepsis disease with fatality within 24 h, while the other nonsurvivor passed away within 30 days and had cell distributions more similar to that of the survivors (Fig. 2C; Supplementary Table S1). Therefore, for subsequent analysis, P50 was designated nonsurvivor, late-stage sepsis (NS LS) while P34 was labeled nonsurvivor, early-stage sepsis (NS ES). Sepsis fatality, even within 24 h, was not unexpected in these studies, as these were patients admitted to the ICU with sepsis/septic shock, multisystem organ failure, and high qSOFA scores.
FIGURE 2.
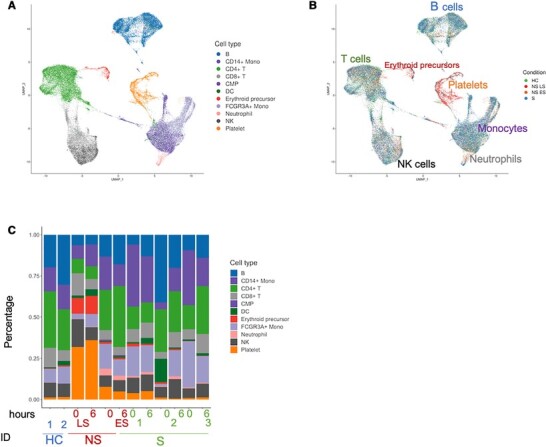
Single-cell transcriptional profiling of PBMC from healthy controls and gram-negative sepsis patients. (A) Cell type UMAP representation of all merged samples. A total of 11 cell types were identified by the consensus method. In total, 57,133 cells are depicted. (B) Sample of origin UMAP representation of all merged samples. Cells were colored by the condition. (C) Bar plots showing the fraction of each sample
Analysis of immune subsets in all the sepsis patients indicated B cell depletion that followed disease severity: 25% in healthy controls (HC), 21% in S, 16% in NS ES, and 6% in NS LS (Fig. 2C; Supplementary Table S1). CD4+ T cell lymphopenia was observed in NS LS (8% compared to 20% in HC, S, and NS ES). We noted an increased proportion of platelets in NS samples, especially in NS LS (34%). The proportion of erythroid precursor cells was also increased with sepsis severity, from 1% in HC, 2% in S, to 6% in NS ES, and 10% in NS LS (Table S1). We investigated potential gender effects among single-cell transcriptomics and found that there were no batch effects related to gender differences with this small sample size (Supplementary Fig. S1). Together, this immune subsetting data by scRNA-seq indicate that lymphocyte subsets are reduced in sepsis, especially in fatal outcomes, and identify the emergence of platelet and erythroid precursors in late-stage fatal sepsis. However, no striking changes in immune subsets were observed within 6 h, prompting us to investigate transcriptional changes within the individual cell types.
Platelet responses are a hallmark of fatal sepsis with similar transcriptional pathways to severe COVID-19 disease
The role of platelets in the development of sepsis pathophysiology is increasingly recognized. Recent studies show that platelets are altered in sepsis and that transcriptional and translational changes in platelets are related to mortality.18 However, timed analysis during the critical early timepoints post sepsis diagnosis has not been performed. Analysis of pathway module scores revealed that platelets in sepsis patients presented with coagulation abnormalities (GO term coagulation, GO:0050817) that was exacerbated over time, especially in fatal disease (Fig. 3A). We also found increased platelet activation (GO:0030168; Fig. 3B) and ATP production modules, which included oxidative phosphorylation (OXPHOS) genes and glycolysis genes19 (Fig. 3C and D). These results are consistent with previous studies that have shown that platelet aggregation is fueled primarily by glycolysis, and that reticulated platelets are more prothrombotic and hyperreactive than mature platelets.20 MHC class I-related genes such as HLA-A, HLA-B, HLA-C, HLA-E, and HLA-F also followed the same trend according to sepsis disease severity (Fig. 3E), indicating CD8 T cell dysregulation by platelets.21,22 Translation initiation modules (GO:0006413) were also changed in sepsis with the lowest score in nonsurvivors, especially in the NS LS, indicating a halt in protein translation as a result of disease (Fig. 3F). Conversely, interferon response modules, including response to type I IFN (GO:0034340), IFN-γ (GO:0034341), and IFN-β (GO:0035456) exhibited the opposite trend with increased scores in sepsis, especially in the nonsurvivors (Fig. 3G–I). This phenomenon might also reflect a general suppression of the protein synthetic apparatus by type I IFN.23
FIGURE 3.
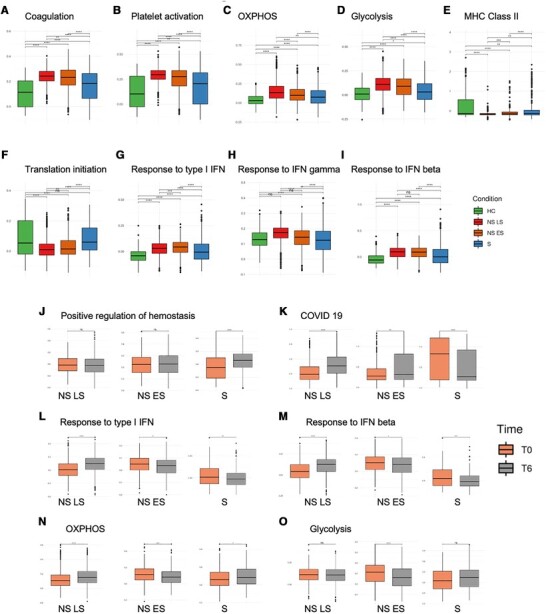
Platelet transcriptional changes over 6 h are associated with sepsis severity. (A– I) Comparisons of pathway module scores across four conditions in platelets. The included modules contain genes related to (A) Coagulation, (B) Platelet activation, (C) OXPHOS, (D) Glycolysis, (E) MHC Class II, (F) Translation initiation, (G) Response to type I IFN, (H) Response to IFN gamma, (I) Response to IFN beta. (J–O) Pathway module scores comparison between T0 vs. T6 in platelets. The included modules contain genes related to (J) positive regulation of hemostasis, (K) COVID-19, (L) response to type I IFN, (M) response to IFN-β, (N) OXPHOS, and (O) glycolysis. The differences in scores associated with adjusted P-values below 0.05, 0.01, 0.001, and 0.0001 are indicated as *, **, ***, and ****, respectively and “ns” – not significant. The significance analysis was performed using Wilcoxon tests
We next focused on dynamic changes in the platelets within 6 -h. Interestingly, analysis of 6-h trajectory transcriptional changes in individual sepsis patients' platelets shared pathways induced in platelets in severe COVID-19 infections, suggesting that platelet transcriptional changes are predictors of severe disease regardless of infection etiology. The most distinguishing pathway, as identified by VENN diagram, which was down-regulated in NS T0→T6 while up-regulated in S T0→T6, was hemostasis (Supplementary Fig. S2A). We analyzed the GO term “positive regulation of hemostasis” (GO:1900048; Fig. 3J) and confirmed that only sepsis survivors exhibited an upward trend, suggesting improved platelet function. Conversely, the most shared pathways that were up-regulated in NS T0→T6 but down-regulated in S T0→T6 included translation initiation, ribosome, and COVID-19 (Supplementary Fig. S2B). To further investigate shared pathways in COVID-19 and sepsis, we investigated genes from Kyoto Encyclopedia of Genes and Genomes Coronavirus disease - COVID-19 (hsa05171) and found that the module scores were significantly increased in NS T6 platelets and decreased in S T6 platelets (Fig. 3K). The COVID-19 megakaryocyte (MK) cell trajectory study reported dysregulated IFN responses in MK cells from patients with severe COVID-19 severe patients, including increased metabolic activity of MKs along the disease trajectory.24 We investigated if IFN response modules changed within the 6-h timeframe and observed that the IFN responses, including type I IFN and IFN-β (Fig. 3L and M), were significantly increased at T6 in NS LS but decreased at T6 in NS ES and S patients. Changes in metabolic activity within 6 h included significantly increased OXPHOS scores in NS LS at T6, while NS ES and S patients had significantly decreased OXPHOS scores (Fig. 3N). The glycolysis score in NS ES was also significantly decreased at T6 (Fig. 3O). Another COVID-19 study reported that circulating platelet-neutrophil, -monocyte, and -T-cell aggregates were elevated in COVID-19 patients compared to healthy donors.25 We used the ligand and receptor database from iTalk26 to score these interactions by calculating the product of average receptor expression and average ligand expression in the respective cell types (see Materials and Methods). Platelet-monocyte interaction scores were significantly elevated in NS LS (Supplementary Fig. S2C). The increased aggregation score to monocytes may explain the unexpected appearance of the platelets in the PBMC fraction. This aggregation was specific to monocytes, as platelet-neutrophil and -T-cell scores were decreased (Supplementary Fig. S2D and E).
Together, our study confirms the theory that platelet coagulation, activation, and energy consumption are functionally linked to sepsis disease severity and identifies shared pathways with COVID-19 disease progression. Further, our timed analysis reveals that these platelet responses are dynamic, changing within a 6-h window, especially in the late stages of fatal sepsis. These data implicate platelet dysfunction as prognostic for disease progression in many infections and suggest that targeting these cell types may be important to prevent fatal outcomes.
Hypoxic stress is a driving factor for erythropoiesis in sepsis with shared pathways in COVID-19 infection
Based on the immune profiling results, which revealed the emergence of erythroid precursors especially in NS LS (see Fig. 2B and C), we investigated transcriptional changes in these cells. Only traces of these cell types are typically present following PBMC isolation by gradient centrifugation, therefore their high levels may suggest abnormal expansion and activation in the oxygen-limiting sepsis environment. Indeed, erythrocyte precursors can be generated through stress erythropoiesis27 as a response to hypoxic conditions.28,29 To test whether these cells were responding to hypoxia, we analyzed the average HIF1A expression in each cell. To avoid the drop-out events known as artifacts in single-cell studies, we removed cells with HIF1A “zero” expression, which left us with 32,185 cells. The HIF1A had the highest expression in NS LS, followed by NS ES, HC then S (Fig. 4A). We investigated dynamic transcriptional changes in erythroid precursors by identifying differentially expressed genes (DEGs) in T0 versus T6. Only the NS LS patient exhibited statistically significant DEG, with the inflammatory protein S100A9 being significantly increased at T6 (adj. P-value < 0.001, log2FC = 0.34).
FIGURE 4.
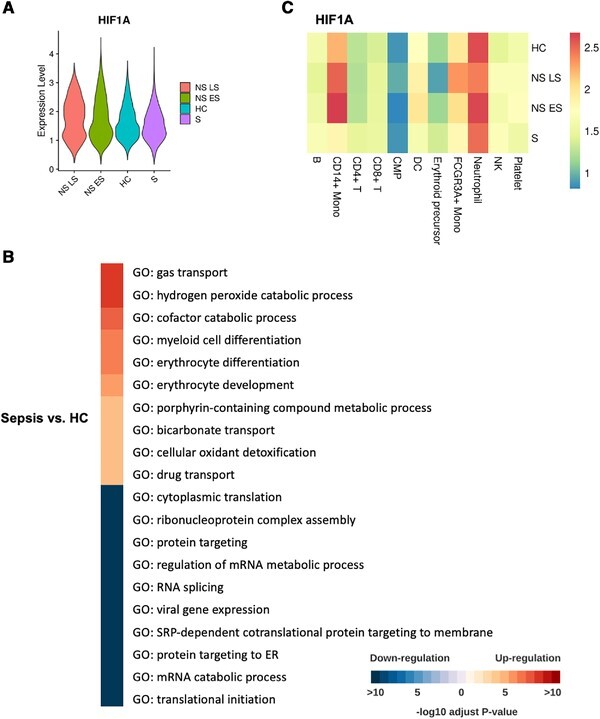
Elevated erythroid precursor cells are associated with hypoxic stress. (A) The expression of the HIF1A gene in erythroid precursors across four conditions. Violin plots are ordered according to the decreasing average value of HIF1A expression. (B) Pathway enrichment when comparing erythroid precursors in sepsis vs. HC. All the GO terms are aligned to representative ones by Revigo70 with a similarity of 0.4. The top 10 -log10 adjust P-values were selected shown in the heatmap. Color red are up-regulated pathways in sepsis patients. The color blue is downregulated pathways in sepsis patients. (C) The comparison of expression of HIF1A in the four conditions. Heatmap coloring represents log-normalized mean gene expression counts averaged across all cells
We next examined gene expression differences in the erythroid precursor cells of sepsis patients and healthy controls (Fig. 4B). Erythroid precursors in sepsis expressed genes related to hypoxic stress (hydrogen peroxide catabolic process, erythrocyte differentiation, cofactor catabolic process, and cellular oxidant detoxification). The down-regulated pathways in sepsis versus HC included cytoplasmic translation, ribonucleoprotein complex assembly, and RNA splicing, suggesting that erythroid precursors in sepsis underwent a halt in protein translation. Our results suggest a strong association between erythropoiesis and fatal sepsis outcomes. This association was also found in a study on COVID-19 infections,24 which proposed that erythroid cells are pivotal components of an unfavorable course of COVID-19. We also investigated if other immune cells were responding to hypoxia and found that monocytes from the sepsis nonsurvivors had the highest HIF1A expression compared to the other patients and other cell types (Fig. 4C). Combined, our findings indicate that sepsis drives hypoxic stress that is associated with disease severity as well as dysfunctional erythropoiesis, which is a shared mechanism in many disease etiologies, including COVID-19 infection.
Monocyte transcriptional changes occur within hours of sepsis recognition and reflect immunosuppression
Monocytes are innate immune cells that sense and respond to pathogen invasion by producing inflammatory cytokines and mediating pathogen killing. However, a dysregulated monocyte response can be damaging and fatal. Studies have found that in sepsis, monocytes may produce a flood of inflammatory cytokines triggering a “cytokine storm”, causing widespread inflammation that can lead to a collapse in blood pressure, coagulation abnormalities, and ultimately organ failure and death. In the later stages, patients who survived the cytokine storm may die from immunosuppression, called “immune paralysis” in its extreme form.30 Moreover, the proinflammatory and immunosuppression stages might overlap.31 The stages at which the immune system transits from proinflammatory to suppressive at the cellular and molecular level have not been well studied, mostly because analyses are only performed at a single snapshot in time. To address this question, we analyzed samples from the same patients within 6 h of sepsis diagnosis, providing a picture of the timed trajectory of monocytes during this critical time window. Investigation of the GO Term cytokine activity (GO:0005125) indicated that most cytokines were higher in sepsis nonsurvivors (NS) compared to survivors (S) (Fig. 5A; Supplementary Fig. S3A). However, when investigating changes between T0 and T6, we found that the cytokines were down-regulated in the NS patients from T0 →T6, especially in NS LS (Fig. 5B and C; Supplementary Fig. S3B and C). The observation may indicate that the NS patients had already passed the proinflammatory stage and had instead begun immune shutdown. The proinflammatory cytokines up-regulated in NS compared to S monocytes, but down-regulated at time T6 included CCL2, CCL7, and NAMPT. The chemokines CCL2, CCL7 are vital for the recruitment of CC-chemokine receptor 2-positive (CCR2+) monocytes.32 CCL2 and CCL7 expression were also enriched in the bronchoalveolar fluid from patients with severe COVID-19.33 The only cytokine that was consistently up-regulated in NS T0 versus T6 but down-regulated in S T0 versus T6 was NAMPT (Fig. 5B-D; Supplementary Fig. S3B-D). NAMPT has been reported as a biomarker in sepsis and sepsis-induced acute respiratory distress syndrome (ARDS) in multiple studies.34–37 The NAMPT/TLR4 inflammatory pathway has also been studied as the COVID-19-induced ARDS drug target.38 The cytokines down-regulated over time in NS monocytes, but up-regulated in S monocytes at T6, included TNFSF10/TRAIL, an immunoregulator that mediates leukocyte apoptosis which had been reported to enhance survival in murine polymicrobial sepsis.40, and TNFSF13B/BAFF, a stimulatory factor for B cells41,42 (Fig. 5D; Supplementary Fig. S3D). These data suggest that fatal sepsis is associated with mixed effects on lymphocyte responses, which may be mediated by monocytes.
FIGURE 5.
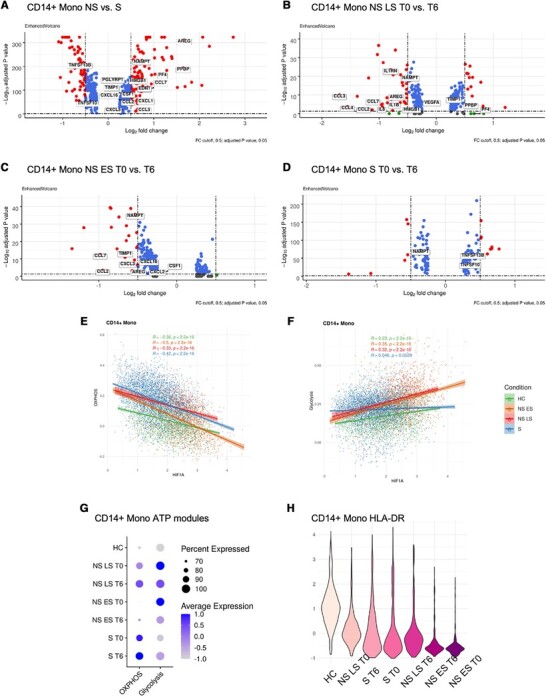
Fatal sepsis patients exhibit immunosuppressive pathways in monocytes. (A-D) Differential expression genes in CD14+ monocytes from (A) NS versus S, (B) NS LS T0 versus T6, (C) NS ES T0 versus T6, (D) S T0 versus T6. Volcano plots were prepared with R package EnhancedVolcano.71 (E and F) The correlations between the HIF1A expression and module score for (E) OXPHOS and (F) glycolysis in CD14+ monocytes across each condition. R‑values from Pearson's correlation, exact 2-sided P-values, and the 95% confidence intervals are shown on each graph. Each dot represents a single cell. Only cells with HIF1A expression ≠ 0 were included in the analysis. Green, orange, red and blue points represent cells from HC, NS ES, NS LS, and S samples, respectively. (G) The percentage of cells with ATP-related pathway modules in CD14+ monocytes across healthy controls and sepsis conditions at T0 and T6. The color saturation indicates the average expression level, and the circle's size indicates the percentage of cells expressing a given module. (H) HLA-DR-related genes expression in CD14+ monocytes across healthy controls and sepsis conditions at T0 and T6. Violin plots are ordered with the decreasing expression average value of HLA-DR-associated genes. The color saturation indicates the average expression level, the darker the color, the lower the average expression level
To profile metabolic changes in monocytes, we investigated the genes that belong to OXPHOS and glycolysis modules, and examined correlation with HIF1A, which was the most highly expressed in monocytes (see Fig. 4C). The OXPHOS modules from all conditions were negatively correlated with HIF1A (Fig. 5E; Supplementary Fig. S3E). On the other hand, the glycolysis modules were positively correlated with HIF1A (Fig. 5F; Supplementary Fig. S3F). Within the group of sepsis survivors, the metabolic activity in CD14+ and FCGR3A+ monocytes was dominated by OXPHOS at both time points. Moreover, the FCGR3A+ monocytes in S had more OXPHOS energy consumption at T6. Overall, monocytes from the sepsis nonsurvivors had shifted energy consumption from OXPHOS to aerobic glycolysis, potentially indicating host defense activation such as production of reactive oxygen species (Fig. 5G). However, within the 6 h timeframe, both CD14+ and FCGR3A+ monocytes from sepsis nonsurvivors exhibited a drop in their glycolysis module scores (Fig. 5G; Supplementary Fig. S3G). One group had demonstrated that defects in the energy metabolism of leukocytes underlie immune paralysis in sepsis, and restoring the ability of immunotolerant leukocytes to mount a glycolytic response might represent a promising novel therapeutic approach to revert the immunotolerant state of sepsis.43 The energy shift to glycolysis, and the decrease in glycolysis consumption at T6 in NS suggest that these monocytes were undergoing immune suppression at the point of sepsis recognition. In contrast, the glycolysis score was increased at T6 in sepsis survivors. We also observed that the HLA-DR module score, as an indicator of monocyte antigen-presenting function,44 was decreased at T6 in the NS LS, indicating immune suppression. However, in the S and NS ES monocytes, the HLA-DR expression was increased at T6, especially in S (Fig. 5H; Supplementary Fig. S3H). Together, these data reveal dynamic transcriptional changes in monocytes within 6 h of sepsis diagnosis, which follow opposite trends in surviving and fatal outcomes. Fatal sepsis is associated with heightened inflammatory and metabolic activity that is down-regulated over time, while improved sepsis outcomes are associated with the restoration of monocyte function within 6 h.
CD52 is a prognostic biomarker for beneficial outcomes in sepsis and is associated with lymphocyte activation
Our preliminary analysis indicated severe lymphopenia in sepsis, especially in the NS LS (see Fig. 2). We further investigated the transcriptional profile of the lymphocytes in sepsis patients and whether it changed over 6 h. Evaluation of activation module scores for CD4+, CD8+ T cell, and B cells (GO terms T cell activation, GO:0042110; B cell activation, GO:0042113) indicated increased activation over 6 h in surviving sepsis patients (Fig. 6A; Supplementary Fig. S4A). In contrast, NS ES had significantly decreased activation scores over time in all lymphocytes (Fig. 6B; Supplementary Fig. S4B). NS LS also exhibited activation scores that decreased significantly, but only in CD4+ T cells, mainly due to the severe CD8+ T cell and B cell lymphopenia (Fig. 6C; Supplementary Fig. S4C).
FIGURE 6.
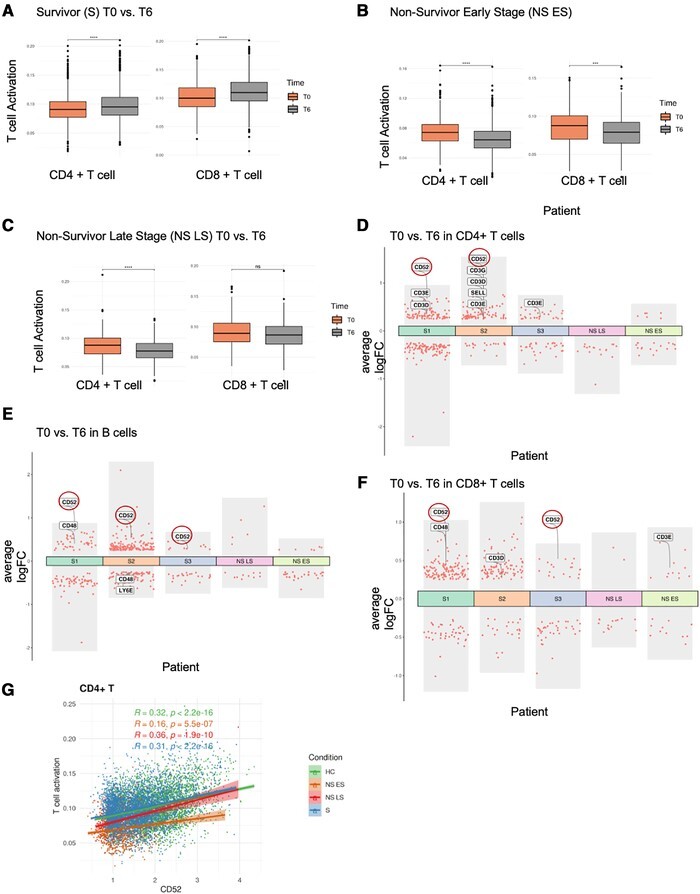
CD52 expression correlates with lymphocyte activation. (A-C) T cell activation pathway module score comparison between T0 and T6 in T cells. (A) Survivors (S), (B) Nonsurvivor early stage (NS ES), and (C) Nonsurvivor late stage (NS LS). The differences in scores associated with adjusted P-values below 0.05, 0.01, 0.001, and 0.0001 are indicated as *, **, ***, and ****, respectively. The significance analysis was performed using Wilcoxon tests. (D-F) Differential gene expression analysis showing up- and down-regulated genes with |log2FC| > 0.25 and adjusted P-value < 0.05 across all 5 sepsis patients between T0 and T6 in (D) CD4+ T cells, (E) B cells, (F) CD8+ T cells. (G) CD52 expression and its correlation with the T cell activation pathway module score in CD4+ T cells across four conditions. R‑values from Pearson's correlation, exact 2-sided P-values, and the 95% confidence intervals are shown on each graph. Each dot represents a single cell. Only cells with CD52 expression ≠ 0 were included in the analysis. Green, orange. red and blue points represent cells from HC, NS ES, NS LS, and S samples, respectively
We next applied DEG analyses between T0 versus T6 in all patients' lymphocytes (Fig. 6D–F, only the genes with |log2FC| > 0.25 and adjusted P-value < 0.05 are shown). To our knowledge, no studies have investigated transcriptional response changes of individual cell types within hours in sepsis patients; however, a study utilizing cecal ligation and puncture as a mouse model for sepsis reported reduced CD4+ T cell activation in the spleen after 6 h, consistent with our current data.45 We therefore investigated the transcripts that were reported in this mouse study: CCR2, CCR6, CD3, CD48, CD52, CD80, ITGB7, SELL, SLAMF6, and Thy1. Of all these genes, only some of them were significantly changed between T0 and T6 in sepsis (Fig. 6D-F). Our analysis identified that CD52, a surface glycoprotein involved in lymphocyte activation, was the most relevant biomarker to predict lymphocyte status and disease outcome. CD52 expression was increased over 6 h in the T and B cells of survivors, but not in sepsis nonsurvivors (Fig. 6D-F). We investigated the other markers, however, did not observe consistent trends associated with protection. To validate that CD52 is correlated with improved lymphocyte function, we plotted CD52 expression against GO term T cell activation and B cell activation module scores. In CD4+ T cells, we observed significant positive correlations in all conditions (Fig. 6G), while in B cells, there were significant positive correlations in the HC, NS LS, and S, but not in the NS ES patient (Supplementary Fig. S4D). Together, our data indicate that increased CD52 expression within hours of sepsis recognition is associated with improved sepsis outcomes. Here, CD52 may act to promote restoration of protective lymphocyte responses, and therefore may serve both as a biomarker for sepsis progression, or as a therapeutic target to promote immune homeostasis.
DISCUSSION
Sepsis is a dysregulated systemic inflammatory response, which results in organ injury with mortality rates of 15–25%.13,46 The molecular level heterogeneity of sepsis makes the study of the dynamics of the individual cell types the ideal tool for understanding sepsis progress and response. However, from over 1000 single-cell transcriptomics studies that have been published to date,47 only 3 have studied sepsis.8,48,49 These studies focused specifically on only two groups of cells: monocytes and myeloid-derived suppressor cells. In contrast, our study used the centrifuge gradient-based approach to isolate PBMC before performing single-cell RNA-seq, which expanded the cell subsets investigated. We additionally collected samples at two different time points from differing outcomes in sepsis, which provided temporal details of the immune response in severe sepsis. These focused analyses were able to identify specific immune cell subsets and gene expression patterns over time that correlated with beneficial or fatal outcomes. Our results are consistent with the previous studies both in single-cell and bulk sepsis transcriptomic studies, but also bring details not seen in other studies, notably molecular changes that occur within hours. Based on this data, future studies evaluating additional time points (such as 24 h and post-discharge) would be relevant to investigate whether the first 6 h are predictive of recovery in surviving sepsis patients. We report in this study that the peripheral blood cell composition of nonsurvivors is more “distant” from healthy controls than the blood cells of survivors, and that severe lymphopenia occurs in fatal sepsis. We also explored cell types that were not previously investigated in sepsis single-cell studies, such as platelets and erythroid precursors, and observed distinct changes in monocytes and lymphocytes within 6 h. Neutrophils are a dominant cell subset in the blood with established antimicrobial but also inflammatory roles in sepsis50,51; however, we were unable to investigate this subset given that most neutrophils are not recovered in the PBMC fraction, and technical issues exist with scRNA-seq of this cell type.
We found that platelets were expanded in sepsis patients, especially in fatal outcomes. Examination of transcriptional changes over time in platelets from the nonsurvivor sepsis patient revealed increased expression of genes related to coagulation, platelet activation, and ATP production modules, including OXPHOS genes and glycolysis genes. These changes were also reported in a COVID-19 MK study,24 suggesting that platelet dysfunction is a shared feature of both diseases, and indicative of clinical severity. This study identified increased metabolic activity of MKs compared to healthy controls. Another study from Holmsen et al.52 demonstrated a correlation between platelet energy demand and aggregation. Consistent with this, we found that the platelets in nonsurvivor sepsis patients had dramatically reduced translation initiation pathways along with the induction of the IFN response pathways, suggesting a general suppression of the protein synthetic apparatus by IFN.23 We also observed that the transcriptional changes in platelets in fatal sepsis were similar to severe COVID-19 patients, including lasting IFN responses, increased metabolic activity, and elevated circulating platelet-monocyte aggregates. Several studies focus on using antiplatelet agents such as aspirin with sepsis,53,54 and a recent COVID-19 study demonstrated that aspirin prescription was associated with decreased mortality rates for COVID-19 positive patients enrolled at the Veterans Health Administration.55 Coagulation disturbances (bleeding and/or clotting) are prominent clinical concerns in sepsis and COVID-19 and deserve further inquiry. Our results support the potential for antiplatelet therapies for the treatment of severe sepsis.
Further investigation of the erythroid precursor subset that was expanded in fatal sepsis revealed the upregulation of genes related to hypoxic stress and apoptosis, reflective of the hypoxic environment in severe sepsis that leads to emergency erythropoiesis. Interestingly, in a longitudinal COVID-19 study,24 erythroid cells were also identified as a hallmark of severe disease with defined molecular signatures linked to a fatal COVID-19 disease outcome. We also observed that erythrocyte expansion and expression of genes related to hypoxic stress were significant predictors of fatal outcomes. Within the erythroid precursor subset, we identified that inflammatory alarmin S100A9 expression dynamically changed in fatal sepsis, with significant increases at T6. S100A9, together with and S100A8 and S100A12, were previously reported as biomarkers for higher risk of death in septic shock patients.56 S100A9 was also identified in a human bone marrow erythropoiesis study,57 which reported its up-regulation at the last stage of maturation of nucleated red blood cell precursors. Together with these studies, our data suggests that S100A9 expression reflects stress erythropoiesis and is associated with rapid fatality in sepsis. S100A9 may serve as a valuable biomarker to stratify sepsis severity, in addition to the clinical scores and other plasma markers.
Consistent with previous studies showing that monocytes play a significant role in the sepsis pathogenesis,30,31 we observed aberrant gene expression and pathway changes in the monocytes of sepsis patients. Overall transcriptional profiles indicated that monocytes were in a hyperinflammatory state in sepsis nonsurvivors. However, focused analyses within the 6-h time window revealed that monocytes from NS patients were undergoing immune suppression, including decreased pro-inflammatory cytokine and HLA-DR expression, and reduced glycolysis energy consumption at T6. Within the 6-h time window, we also identify CD52 as the biomarker for B and T cell activation that correlates with beneficial outcomes. CD52 is a glycoprotein expressed on the surface of mature lymphocytes, monocytes, dendritic cells, and NK cells,58 therefore surface expression could be quantified to predict sepsis progression. Most CD52 targeted therapeutic approaches aim to delete CD52-expressing cells, such as the monoclonal antibody alemtuzumab, which treats chronic lymphocytic leukemia and multiple sclerosis.59 However, our study suggests that promoting CD52 signaling may be beneficial to improving lymphocyte function. In fact, in a study using alemtuzumab, one patient with aggressive multiple sclerosis developed sepsis after treatment.60 Additionally, CD52 had been proposed as a prognostic biomarker in breast cancer, where it is correlated with improved outcomes likely due to increased immune tumoricidal activity.61 CD52 might therefore serve as a biomarker for sepsis prognosis and provides a new therapeutic target for sepsis patients, however, determining its influence on sepsis therapy would require an expanded sample size study.
In conclusion, results from this study indicate that the initial status of the sepsis patient and the dynamic changes in cell behavior during the critical period following diagnosis significantly affect sepsis outcome. Therapeutic intervention to modify these immune trajectories may therefore lead to improved outcomes in these patients that could be identified by biomarkers reported in this study, such as CD52 or S100A9. Future studies that focus on these dysfunctional cell subsets at the individual level, addressing their metabolic dysfunction or how to promote their recovery from exhaustion, may provide therapeutic and prognostic strategies for sepsis, which could be applicable to other fatal diseases such as COVID-19.
MATERIALS AND METHODS
Human blood collection and harvest of PBMCs
Enrollment
Peripheral blood was collected from nonsepsis donors from the Riverside Free Clinic and septic patients with signed informed consent and approval of the University of California, Riverside (UCR, #HS-17-707), and Riverside University Health System (RUHS, #1024190-3) Institutional Review Board. Sepsis patient enrollment was performed according to the following inclusion criteria: (1) Admission to Intensive Care Unit; (2) Age greater than or equal to 18 years old; (3) Suspected or confirmed infection; (4) qSOFA score ≥ 2 (qSOFA variables: altered mentation [GCS ≤ 13], systolic blood pressure < 100 mm Hg and respiratory rate > 22 breaths/min) and/or; (5) Lactate greater than or equal to 2.0 mmol/L and on vasopressor therapy to maintain MAP > 65 mm Hg after 30 mL/kg intravenous fluid bolus.
PBMC analysis
Blood was recovered in Vacutainer glass collection tubes with heparin (BD Biosciences). PBMC were isolated by gradient centrifugation with Histopaque-1077. Plasma was recovered for cytokine quantification by cytokine bead array (BD Biosciences) and resistin ELISA (Peprotech). Cell aliquots were frozen in liquid nitrogen. Following blood draw, PBMC isolation was performed within 24 h through density gradient centrifugation, and cells were stored immediately in liquid nitrogen. Flow cytometry characterization of PBMC involved incubation with Human TruStain FcX™ (Biolegend) and staining with primary Abs: CD14 (HCD14, Biolegend), CD16 (3G8, Biolegend), CD66b (G10F5, eBioscience), CD3 (OKT3, eBioScience). Samples were acquired on a BD LSRII and analyzed on FlowJo (v10).
10X Genomics
For single-cell sequencing, thawed PBMC live cells were recovered by column-based dead cell removal kit (Miltenyi), and viable cells were confirmed by hemocytometer counting (>85% viable). A total of 15,000 cells per sample were loaded onto the 10x genomics platform, and cDNA libraries were prepared according to the manufacturer's instructions (Chromium Next GEM Single Cell V3.1). Samples were sequenced at the UCSD Genomics center on the NovaSeq platform at 250 M reads/sample.
Process and quality control of the single-cell RNA-seq data
The Cell Ranger Software Suite (v.3.1.0) was used to perform sample de-multiplexing, barcode processing, and single-cell 5′ unique molecular identifier (UMI) counting. Specifically, splicing-aware aligner STAR was used in FASTQs alignment. Cell barcodes were then determined based on the distribution of UMI counts automatically. The following criteria were applied to each cell of four sepsis samples and two healthy controls: gene number between 200 and 6000, UMI count > 1000, and mitochondrial gene percentage < 0.2. After filtering, a total of 57,133 cells were left for the following analysis. Finally, all samples' filtered gene-barcode matrix was integrated with Seurat v.362 to remove batch effects across different samples.
Dimensionality reduction, clustering, and consensus-based cell-type annotation
We first analyzed scRNA-seq data from 57,133 cells with 4761 cells on the average per sample. Two-time points were analyzed per sepsis patient. Uniform Manifold Approximation And Projection was used to visualize the cell populations (Fig. 2A and B). The filtered gene barcode matrix was normalized using “LogNormalize” method from Seurat package v.3 with default parameters. In the next step, the vst method implemented in the FindVariableFeatures function of the Seurat package was applied to find the top 2000 most variable genes. It was followed by the principal component analysis (PCA), and the application of the uniform manifold approximation and projection algorithm for cell data visualization performed based on the top 50 principal components. Then the graph-based clustering was performed by applying the FindClusters function of the Seurat package on the PCA-reduced data. With the resolution set to 1.0, 57,133 cells were grouped into 34 clusters. The first method of assignment of cell types to cell clusters was based on their canonical markers: B cells (MS4A1), CD14+ monocytes (CD14 and LYZ), CD4+ T cells (IL7R, CCR7, and CD27), CD8+ T cells (CD8A), DCs (FCER1A, CST3, CD123, and GZMB), erythroid precursors (GYPB and AHSP), FCGR3A+ monocytes (FCGR3A and MS4A7), neutrophils (JAML and SERPINB), NK cells (GNLY and NKG7), and platelets (PPBP). Independently from this initial marker-based cell type assignment, we applied cell-type annotation tools SingleR63 and scCATCH.64 The SingleR program first identifies genes with significant variation between cell types in the reference data set, compares each cell's scRNA-seq data with each sample from the reference data set, and performs iterative fine-tuning to select the most likely cell type of each cell. The microarray dataset from Human Primary Cell Atlas Data with assigned labels was used as the reference. Finally, each cluster was assigned a cell type with the highest percentage of cells assigned to that type by SingleR. The third applied method of cell type assignment was scCATCH, where cell types are assigned using the tissue-specific cellular taxonomy reference databases65–67 and the evidence-based scoring protocol. Our final assignment of cell types to clusters was based on the consensus of the three methods mentioned above as follows: first, each cluster was assigned a cell type selected by most methods if possible. If each method gave a different result, then the priority was given to the assignment based on canonical markers. If the markers-based assignment was inconclusive, the consensus assignment was based on the results from SingleR method.
Differential gene expression analysis and functional annotation of genes
The MAST method68 from the Seurat v.3 package (implemented in FindAllMarkers function) was used with default parameters to perform differential gene expression analysis. A difference in gene expression was considered significant if an adjusted P-value was below 0.05. The false discovery rate (FDR) adjustment was performed by MAST. Only genes with FDR-adjusted P-values < 0.05 were considered in the second step of DEG analysis, where we analyzed differences between the results of the comparisons listed earlier. Pathway enrichment analysis was performed by clusterProfiler69 using database Gene Ontology biological process terms (GO-BP) and Kyoto Encyclopedia of Genes and Genomes pathways. The clusterProfiler program was used for statistical analysis and visualization of functional profiles for DEGs with FDR-adjusted P-value < 0.05.
Comparison of module scores
We used cell module scores to measure the degree to which individual cells expressed certain predefined expression gene sets. The AddModuleScore function from the Seurat v.3 package with default settings was used to perform all calculations and comparisons of module scores. We compared the expression of modules such as T cell activation (GO:0042110), B cell activation (GO:0042113), coagulation (GO:0050817), platelet activation (GO:0030168), OXPHOS, glycolysis, MHC class I, MHC class II, translation initiation (GO:0006413), response to type I IFN (GO:0034340), response to IFN-γ (GO:0034341), response to IFN-β (GO:0035456), Coronavirus disease - COVID-19 (hsa05171), and HLA-DR related genes. The lists of genes defining these modules were prepared based on Gene Ontology and literature. Genes without detectable expression in our data were ignored. The sets of genes defining the modules used in our analysis are listed in Supplementary Table S2.
Statistics
The statistical tools, methods, and significance thresholds for each analysis are described in the Results or Materials and Methods section or in the figure legends.
Supplementary Material
Supplementary methods Calculation of ligand-receptor interaction scores between platelets and other cell types.
Supplementary Figure 1. UMAP representation of all merged samples.
Supplementary Figure 2. Aberrant platelet function changes observed in fatal sepsis.
Supplementary Figure 3. Immunosuppressive trends in monocytes from sepsis non-survivors.
Supplementary Figure 4. The association between CD52 expression and B cell activation.
Table S1. Absolute cell numbers for each cell type from all samples.
Table S2. Genes used for gene module scoring.
ACKNOWLEDGMENTS
This research was supported by the UCR School of Medicine (to A.G. and M.G.N.), the Dean Innovation Fund (to J.B. and M.G.N.), and the National Institutes of Health (NIAID, R21AI37830, and R01AI153195 to M.G.N.). The data from this study were generated at the UC San Diego IGM Genomics Center utilizing an Illumina NovaSeq 6000 that was purchased with funding from a National Institutes of Health SIG grant (#S10 OD026929). We thank Dr. Hashini Batugedara, Dr. Luqman Nasouf, Mr. Joseph Miller, Mr. Sang Woo, the RUHS-MC sepsis response team, and the Riverside Free Clinic for assistance with participant enrollment, blood collection, and blood processing, and Dr. Karine Le Roch for access to the 10X equipment.
Abbreviations
- ARDS
acute respiratory distress syndrome
- CMP
common myeloid progenitor cell
- CRP
C-reactive protein
- DEG
differentially expressed gene
- ES
early stage
- ESR
erythrocyte sedimentation rate
- FDR
false discovery rate
- HC
healthy control
- ICU
intensive care unit
- LS
late stage
- MK
megakaryocyte
- NS
nonsurvivor
- OXPHOS
oxidative phosphorylation
- PBMC
peripheral blood mononuclear cell
- S
survivor
Contributor Information
Xinru Qiu, Graduate Program in Genetics, Genomics and Bioinformatics, University of California Riverside, Riverside, California, USA.
Jiang Li, Division of Biomedical Sciences, School of Medicine, University of California Riverside, Riverside, California, USA.
Jeff Bonenfant, Division of Pulmonary and Critical Care, Riverside University Health System Medical Center, Riverside, California, USA; Department of Internal Medicine, Division of Pulmonary and Critical Care, Loma Linda University, Loma Linda, California, USA.
Lukasz Jaroszewski, Division of Biomedical Sciences, School of Medicine, University of California Riverside, Riverside, California, USA.
Aarti Mittal, Division of Pulmonary and Critical Care, Riverside University Health System Medical Center, Riverside, California, USA.
Walter Klein, Division of Pulmonary and Critical Care, Riverside University Health System Medical Center, Riverside, California, USA.
Adam Godzik, Division of Biomedical Sciences, School of Medicine, University of California Riverside, Riverside, California, USA.
Meera G Nair, Division of Biomedical Sciences, School of Medicine, University of California Riverside, Riverside, California, USA.
DATA AVAILABILITY STATEMENT
The raw data have been deposited with the Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo) at the GEO accession GSE167363. Other supporting raw data are available from the corresponding author upon request. Source data are provided with this paper.
CODE AVAILABILITY
Experimental protocols and the data analysis pipeline used in our work follow the 10X Genomics and Seurat official websites. The analysis steps, functions, and parameters used are described in detail in the Materials and Methods section. Custom scripts for analyzing data are available upon reasonable request. Source data are provided with this paper.
AUTHORSHIP
M.G.N., A.G., X.Q., J.L., J.B., A.M., and W.K. conceptualized the study. J.L., X.Q., A.M., M.G.N., and A.G. developed the methodology. M.G.N., A.G., X.Q., J.L., and J.B. performed the investigation. M.G.N., A.G., X.Q., J.L., and L.J. provided the formal analysis. X.Q., A.G., L.J., and M.G.N. wrote the article. W.K., J.B., and A.M. edited the manuscript and contributed clinical expertise. M.G.N. and A.G. supervised the study.
Xinru Qiu and Jiang Li contributed equally to this work.
REFERENCES
- 1. Tiru B, Dinino EK, Orenstein A et al. The economic and humanistic burden of severe sepsis. Pharmacoeconomics. 2015;33:925–937. [DOI] [PubMed] [Google Scholar]
- 2. Rudd KE, Johnson SC, Agesa KM et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. Lancet. 2020;395:200–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aziz M, Jacob A, Yang W-L, Matsuda A, Wang P. Current trends in inflammatory and immunomodulatory mediators in sepsis. J Leukoc Biol. 2013;93:329–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Van Der Poll T, Van De Veerdonk FL, Scicluna BP, Netea MG. The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol. 2017;17:407–420. [DOI] [PubMed] [Google Scholar]
- 5. Pierrakos C, Velissaris D, Bisdorff M, Marshall JC, Vincent J-L. Biomarkers of sepsis: time for a reappraisal. Crit Care. 2020;24:287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Davenport EE, Burnham KL, Radhakrishnan J et al. Genomic landscape of the individual host response and outcomes in sepsis: a prospective cohort study. Lancet Respir Med. 2016;4:259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sweeney TE, Perumal TM, Henao R et al. A community approach to mortality prediction in sepsis via gene expression analysis. Nat Commun. 2018;9:694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Reyes M, Filbin MR, Bhattacharyya RP et al. An immune-cell signature of bacterial sepsis. Nat Med. 2020;26:333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rivers E, Nguyen B, Havstad S et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345:1368–1377. [DOI] [PubMed] [Google Scholar]
- 10. Kumar A, Roberts D, Wood KE et al. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit Care Med. 2006;34:1589–1596. [DOI] [PubMed] [Google Scholar]
- 11. Mouncey PR, Osborn TM, Power GS et al. Trial of early, goal-directed resuscitation for septic shock. N Engl J Med. 2015;372:1301–1311. [DOI] [PubMed] [Google Scholar]
- 12. Peake SL, Delaney A, Bailey M et al. Goal-directed resuscitation for patients with early septic shock. N Engl J Med. 2014;371:1496–1506. [DOI] [PubMed] [Google Scholar]
- 13. Pro CI et al. A randomized trial of protocol-based care for early septic shock. N Engl J Med. 2014;370:1683–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kox WJ, Volk T, Kox SN, Volk H-D. Immunomodulatory therapies in sepsis. Intensive Care Med. 2000;26:S124–128. Suppl. [DOI] [PubMed] [Google Scholar]
- 15. Marimuthu R, Francis H, Dervish S, Li SCH, Medbury H, Williams H. Characterization of human monocyte subsets by whole blood flow cytometry analysis. J Vis Exp. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Slyker JA, Lohman-Payne B, John-Stewart GC, Dong T, Mbori-Ngacha D, Tapia K et al. The impact of HIV-1 infection and exposure on natural killer (NK) cell phenotype in Kenyan infants during the first year of life. Front Immunol. 2012;3:399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Medina F, Segundo C, Campos-Caro A, González-García I, Brieva J. The heterogeneity shown by human plasma cells from tonsil, blood, and bone marrow reveals graded stages of increasing maturity, but local profiles of adhesion molecule expression. Blood. 2002;99:2154–2161. [DOI] [PubMed] [Google Scholar]
- 18. Middleton EA, Rowley JW, Campbell RA et al. Sepsis alters the transcriptional and translational landscape of human and murine platelets. Blood. 2019;134:911–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Frederick M, Skinner HD, Kazi SA, Sikora AG, Sandulache VC. High expression of oxidative phosphorylation genes predicts improved survival in squamous cell carcinomas of the head and neck and lung. Sci Rep. 2020;10:6380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. George MJ, Bynum J, Nair P et al. Platelet biomechanics, platelet bioenergetics, and applications to clinical practice and translational research. Platelets. 2018;29:431–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Garraud O, Cognasse F. Are platelets cells? And if yes, are they immune cells. Front Immunol. 2015;6:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chapman LM, Aggrey AA, Field DJ et al. Platelets present antigen in the context of MHC class I. J Immunol. 2012;189:916–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jiang H, Lin JJ, Tao J, Fisher PB. Suppression of human ribosomal protein L23A expression during cell growth inhibition by interferon-beta. Oncogene. 1997;14:473–480. [DOI] [PubMed] [Google Scholar]
- 24. Bernardes JP, Mishra N, Tran F et al. Longitudinal multi-omics analyses identify responses of megakaryocytes, erythroid cells, and plasmablasts as hallmarks of severe COVID-19. Immunity. 2020;53:1296–1314 e1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Manne BK, Denorme F, Middleton EA et al. Platelet gene expression and function in patients with COVID-19. Blood. 2020;136:1317–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang Y et al. iTALK: an R package to characterize and illustrate intercellular communication. bioRxiv. 2019:507871. [Google Scholar]
- 27. Paulson RF, Shi L, Wu D-C. Stress erythropoiesis: new signals and new stress progenitor cells. Curr Opin Hematol. 2011;18:139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bapat A, Schippel N, Shi X et al. Hypoxia promotes erythroid differentiation through the development of progenitors and proerythroblasts. Exp Hematol. 2021;97:32–46.e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rogers HM, Yu X, Wen J, Smith R, Fibach E, Noguchi CT. Hypoxia alters progression of the erythroid program. Exp Hematol. 2008;36:17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis. 2013;13:260–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gomez HG, Gonzalez SM, Londoño JM et al. Immunological characterization of compensatory anti-inflammatory response syndrome in patients with severe sepsis: a longitudinal study*. Crit Care Med. 2014;42:771–780. [DOI] [PubMed] [Google Scholar]
- 32. Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11:762–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhou Z, Ren L, Zhang Li et al. Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe. 2020;27:883–890.e882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Camp SM, Ceco E, Evenoski CL et al. Unique Toll-like receptor 4 activation by NAMPT/PBEF induces NFκB signaling and inflammatory lung injury. Sci Rep. 2015;5:13135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ye SQ, Simon BA, Maloney JP, Zambelli-Weiner A et al. Pre-B-cell colony-enhancing factor as a potential novel biomarker in acute lung injury. Am J Respir Crit Care Med. 2005;171:361–370. [DOI] [PubMed] [Google Scholar]
- 36. Gesing J, Scheuermann K, Wagner IV et al. NAMPT serum levels are selectively elevated in acute infectious disease and in acute relapse of chronic inflammatory diseases in children. PLoS One. 2017;12:e0183027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Karampela I, Christodoulatos GS, Kandri E et al. Circulating eNampt and resistin as a proinflammatory duet predicting independently mortality in critically ill patients with sepsis: a prospective observational study. Cytokine. 2019;119:62–70. [DOI] [PubMed] [Google Scholar]
- 38. Quijada H, Bermudez T, Kempf CL et al. Endothelial eNAMPT amplifies pre-clinical acute lung injury: efficacy of an eNAMPT-neutralising monoclonal antibody. Eur Respir J. 2021;57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schenck EJ, Ma KC, Price DR et al. Circulating cell death biomarker TRAIL is associated with increased organ dysfunction in sepsis. JCI Insight. 2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cziupka K, Busemann A, Partecke LI et al. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) improves the innate immune response and enhances survival in murine polymicrobial sepsis. Crit Care Med. 2010;38:2169–2174. [DOI] [PubMed] [Google Scholar]
- 41. Yan M, Marsters SA, Grewal IS, Wang H, Ashkenazi A, Dixit VM. Identification of a receptor for BLyS demonstrates a crucial role in humoral immunity. Nat Immunol. 2000;1:37–41. [DOI] [PubMed] [Google Scholar]
- 42. Hadjadj J, Yatim N, Barnabei L et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369:718–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cheng S-C, Scicluna BP, Arts RJW et al. Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat Immunol. 2016;17:406–413. [DOI] [PubMed] [Google Scholar]
- 44. Cheron A, Floccard B, Allaouchiche B et al. Lack of recovery in monocyte human leukocyte antigen-DR expression is independently associated with the development of sepsis after major trauma. Crit Care. 2010;14:R208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mcdunn J, Turnbull I, Polpitiya A et al. Splenic CD4+ T cells have a distinct transcriptional response six hours after the onset of sepsis. J Am Coll Surg. 2006;203:365–375. [DOI] [PubMed] [Google Scholar]
- 46. Hotchkiss RS, Moldawer LL, Opal SM, Reinhart K, Turnbull IR, Vincent J-L. Sepsis and septic shock. Nat Rev Dis Primers. 2016;2:16045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Svensson V, da Veiga Beltrame E, Pachter L. A curated database reveals trends in single-cell transcriptomics. BioRxiv. 2019:742304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jiang Y, Rosborough BR, Chen J et al. Single cell RNA sequencing identifies an early monocyte gene signature in acute respiratory distress syndrome. JCI Insight. 2020;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Darden DB, Bacher R, Brusko MA et al. Single cell RNA-SEQ of human myeloid derived suppressor cells in late sepsis reveals multiple subsets with unique transcriptional responses: a pilot study. Shock. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shen X-F, Cao Ke, Jiang J-P, Guan W-X, Du J-F. Neutrophil dysregulation during sepsis: an overview and update. J Cell Mol Med. 2017;21:1687–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Demaret J, Venet F, Friggeri A et al. Marked alterations of neutrophil functions during sepsis-induced immunosuppression. J Leukoc Biol. 2015;98:1081–1090. [DOI] [PubMed] [Google Scholar]
- 52. Holmsen H, Kaplan KL, Dangelmaier CA. Differential energy requirements for platelet responses. A simultaneous study of aggregation, three secretory processes, arachidonate liberation, phosphatidylinositol breakdown and phosphatidate production. Biochem J. 1982;208:9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Akinosoglou K, Alexopoulos D. Use of antiplatelet agents in sepsis: a glimpse into the future. Thromb Res. 2014;133:131–138. [DOI] [PubMed] [Google Scholar]
- 54. Tsai M-J, Shih C-J, Chen Y-T. Association of prior antiplatelet agents with mortality in sepsis patients. Intensive Care Med. 2016;42:605–607. [DOI] [PubMed] [Google Scholar]
- 55. Osborne TF, Veigulis ZP, Arreola DM, Mahajan SM, Röösli E, Curtin CM. Association of mortality and aspirin prescription for COVID-19 patients at the Veterans Health Administration. PLoS One. 2021;16:e0246825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dubois C, Marcé D, Faivre V et al. High plasma level of S100A8/S100A9 and S100A12 at admission indicates a higher risk of death in septic shock patients. Sci Rep. 2019;9:15660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mello FV, Land MGP, Costa ES et al. Maturation-associated gene expression profiles during normal human bone marrow erythropoiesis. Cell Death Discov. 2019;5:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lifely MR, Treumann A, Schneider P, Ferguson MAJ. Primary structure of CD52. J Biol Chem. 1995;270:6088–6099. [DOI] [PubMed] [Google Scholar]
- 59. Evan JR, Bozkurt SB, Thomas NC, Bagnato F. Alemtuzumab for the treatment of multiple sclerosis. Expert Opin Biol Ther. 2018;18:323–334. [DOI] [PubMed] [Google Scholar]
- 60. Zhang X, Tao Y, Chopra M et al. Differential reconstitution of T cell subsets following immunodepleting treatment with alemtuzumab (anti-CD52 monoclonal antibody) in patients with relapsing-remitting multiple sclerosis. J Immunol. 2013;191:5867–5874. [DOI] [PubMed] [Google Scholar]
- 61. Wang J, Zhang G, Sui Y et al. CD52 is a prognostic biomarker and associated with tumor microenvironment in breast cancer. Front Genet. 2020;11:578002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Stuart T, Butler A, Hoffman P et al. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902 e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Aran D, Looney AP, Liu L et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol. 2019;20:163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shao X, Liao J, Lu X, Xue R, Ai Ni, Fan X. scCATCH: automatic annotation on cell types of clusters from single-cell RNA sequencing data. iScience. 2020;23:100882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhang X, Lan Y, Xu J et al. CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 2019;47:D721–D728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Han X, Wang R, Zhou Y et al. Mapping the mouse cell atlas by Microwell-Seq. Cell. 2018;173:1307. [DOI] [PubMed] [Google Scholar]
- 67. Yuan H, Yan M, Zhang G et al. CancerSEA: a cancer single-cell state atlas. Nucleic Acids Res. 2019;47:D900–D908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Finak G, Mcdavid A, Yajima M et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015;16:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yu G, Wang Li-G, Han Y, He Q-Yu. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Supek F, Bošnjak M, Škunca N, Šmuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One. 2011;6:e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Blighe K, R S, Lewis M. EnhancedVolcano: publication-ready volcano plots with enhanced colouring and labeling. R package version 1100. 2021. https://github.com/kevinblighe/EnhancedVolcano [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary methods Calculation of ligand-receptor interaction scores between platelets and other cell types.
Supplementary Figure 1. UMAP representation of all merged samples.
Supplementary Figure 2. Aberrant platelet function changes observed in fatal sepsis.
Supplementary Figure 3. Immunosuppressive trends in monocytes from sepsis non-survivors.
Supplementary Figure 4. The association between CD52 expression and B cell activation.
Table S1. Absolute cell numbers for each cell type from all samples.
Table S2. Genes used for gene module scoring.
Data Availability Statement
The raw data have been deposited with the Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo) at the GEO accession GSE167363. Other supporting raw data are available from the corresponding author upon request. Source data are provided with this paper.