

ABSTRACT
Plasmodium falciparum is a deadly human pathogen responsible for the devastating disease called malaria. In this study, we measured the differential accumulation of RNA secondary structures in coding and non-coding transcripts from the asexual developmental cycle in P. falciparum in human red blood cells. Our comprehensive analysis that combined high-throughput nuclease mapping of RNA structures by duplex RNA-seq, SHAPE-directed RNA structure validation, immunoaffinity purification and characterization of antisense RNAs collectively measured differentially base-paired RNA regions throughout the parasite’s asexual RBC cycle. Our mapping data not only aligned to a diverse pool of RNAs with known structures but also enabled us to identify new structural RNA regions in the malaria genome. On average, approximately 71% of the genes with secondary structures are found to be protein coding mRNAs. The mapping pattern of these base-paired RNAs corresponded to all regions of mRNAs, including the 5ʹ UTR, CDS and 3ʹ UTR as well as the start and stop codons. Histone family genes which are known to form secondary structures in their mRNAs and transcripts from genes which are important for transcriptional and post-transcriptional control, such as the unique plant-like transcription factor family, ApiAP2, DNA-/RNA-binding protein, Alba3 and proteins important for RBC invasion and malaria cytoadherence also showed strong accumulation of duplex RNA reads in various asexual stages in P. falciparum. Intriguingly, our study determined stage-specific, dynamic relationships between mRNA structural contents and translation efficiency in P. falciparum asexual blood stages, suggesting an essential role of RNA structural changes in malaria gene expression programs.
Abbreviations: CDS: Coding Sequence; DNA: Deoxyribonucleic Acid; dsRNA: double-stranded RNA; IDC: Intra-erythrocytic Developmental Cycle (IDC); m6A: N6-methyladenosine; mRNA: Messenger RNA; ncRNA: Non-coding RNA; RBC: Red Blood cells; RBP: RNA-Binding Protein; REC: Relative Expression Counts; RNA-seq: RNA-sequencing; RNA: Ribonucleic Acid; RNP: Ribonucleoprotein; RPKM: Reads Per Kilobase of transcript Per Million; rRNA: Ribosomal RNA 16. RUFs: RNAs of Unknown Function; SHAPE: Selective 2’-hydroxyl acylation analysed by primer extension; snoRNA: Small Nucleolar RNA; snRNA: Small Nuclear RNA; SRP-RNA: Signal Recognition Particle RNA; ssRNA: (Single-stranded RNA); TE: Translation Efficiency; tRNA: transfer RNA; UTR: Untranslated Region
KEYWORDS: Genome-wide, RNA folding, mRNA, translation, RNA secondary structure, RNA structurome, RNA-shape, plasmodium falciparum, malaria
Graphical abstract
Introduction
RNA is innately structured. The dynamic intramolecular and intermolecular interactions of RNA impact almost all steps of gene expression programs, from transcriptional activation to RNA processing and translation [1]. Changes in RNA folding affect the biological functions in a cell in a measurable fashion and those changes are facilitated by several factors, such as temperature, ion homoeostasis, internal modifications and interactions with proteins. For example, melting of RNA base pairs in response to temperature changes can lead to alterations in RNA structure [2], which can be directly linked to translational regulation as a result of start codon accessibility in mRNAs and ribosomal recruitment for translation initiation process [3–5]. Likewise, drug or metabolite sensing of RNA structures is known to affect mRNA stability or translation efficiency in both prokaryotes and eukaryotes [6–10]. Furthermore, sequence-specific recognition of RNA by RNA-binding proteins (RBPs) [11,12] and chemical modification of RNA nucleotides, such as N6-methyladenosine (m6A) modification [13,14] provides an added layer of regulation which can significantly impact gene function [12,15]. Recent developments in high throughput sequencing technologies and their coupling with RNA structure-probing approaches now provide a comprehensive platform to map the secondary structure of the whole transcriptome of yeasts, plants, metazoans and mammalian cells, and functionally relevant RNA motifs [16–23]. However, our knowledge of RNA structural motifs and their influences in gene regulatory processes found in lower eukaryotes, specifically protists, many of which cause deadly human diseases, is limited. This is due in part to a lack of enough records on genome-wide abundance of RNA secondary structures.
Plasmodium falciparum is a parasitic protist, which is responsible for the deadly disease, malaria. More than 3 billion people live in areas that are at high risk for malaria transmission [24]. Malaria, a febrile illness, is often characterized by cyclical changes in body temperature [25], which can lead to increased adhesive properties of P. falciparum infected red blood cells (iRBCs) [26]. The adhesive properties contribute greatly to the pathology of malaria [27,28]. Recently, biophysical studies have highlighted that the temperature dependence of receptor–ligand interactions in P. falciparum is an important factor in its pathogenesis [29]. Since temperature fluctuations can greatly affect RNA structures and folding energies, which can ultimately influence RNA stability, translation [30,31] and potentially affect virulence properties of pathogenic organisms [32–34], it is crucial to get a clearer view of how RNA folding can affect gene function in a lethal disease like malaria. Ever since the malaria genome was sequenced, numerous studies have shown that transcription in malaria parasite P. falciparum is developmentally regulated [35–38]. Transcriptome-wide studies revealed that the P. falciparum asexual stages, which occur in human RBCs, have a cyclic pattern of steady‐state mRNA expression, with more than 75% of the genes achieving high abundance of mRNAs during only one time‐point within their 48 hours life cycle [39–41]. Additionally, widespread translational delay was reported in malaria [42]. However, gaps remain in our current understanding of how this steady state, timely mRNA expression relates to delay in translation, considering that transcription and translation of malaria genes appear to be tightly linked processes [43]. Therefore, transcriptome-wide control of gene expression and translation remains an active area of exploration in malaria research. In addition, several studies have indicated that post-transcriptional control mechanisms are major means of gene expression regulation in malaria parasites in both sexual and asexual stages. Global ribosome profiling, mechanistic studies on RNA-binding proteins (RBPs) [43–50] and differential gene expression studies have shown that a wide array of genes, including virulence genes in malaria parasites, are regulated at both post-transcriptional and translational level [51–53]. RNA-binding proteins (RBPs) are abundant in P. falciparum and could particularly influence post-transcriptional genetic control cascades in malaria [42,54]. A recent survey of m6A modifications, which affect translation by resolving mRNA secondary structures [55], suggests that these modifications can dynamically calibrate transcriptional and post-transcriptional processes in P. falciparum asexual cycle [56]. Malaria parasites also express antisense RNAs [57–59] that are considered gene regulatory in nature [51,60,61] and several other non-coding RNAs [62–65], many of which fold into characteristic secondary structures [63] that could account for their diverse functional activities. Therefore, like human, yeast and plants [66–69], the structural components of RNA could act as a major force of post-transcriptional and translational regulation in malaria. Indeed, early evidence of differences in the structural content of large subunit ribosomal RNAs (rRNAs) found between asexual and sexual developmental stages indicated that dynamic changes in these rRNAs are related to stage-specific ribosome function in P. falciparum [36,70].
Given the plethora of observations on the functional importance of the RNA secondary structures and on RNA-mediated genetic regulation in malaria parasites, we sought to globally monitor changes in RNA structures in distinct asexual developmental stages of P. falciparum by combining nuclease mapping with high throughput sequencing. Our RNA mapping data not only aligns with known non-coding RNAs that have defined cellular function, it also provides the first genome-wide view of the structural landscape of messenger RNAs (mRNAs) and novel non-coding transcripts which could play important roles in the biology and pathogenesis of malaria. We were also able to purify biologically relevant antisense RNAs from total RNA by immunoprecipitation with antibodies that recognize the double-stranded nature of these RNA molecules. Thus, our findings provide an extensive collection of RNA secondary structures in P. falciparum as well as a valuable new resource for the analysis of RNA structure-function in this organism.
Materials and methods
Plasmodium falciparum culture, synchronization and treatments
P. falciparum laboratory adapted parasite line 3D7 (ATCC, MRA-102) was cultured at 2% haematocrit in purified human erythrocytes/RBCs using standard culturing techniques [71,72] using RPMI 1640 medium (supplemented with 37.5 mM HEPES, 7 mM D-glucose, 6 mM NaOH, 25 µg of gentamicin sulphate/mL, 2 mM L-glutamine, and 10% Albumax II (all reagents from Fisher Scientific/ThermoFisher/Life Technologies) at a pH of 7.2 in a 3% carbon dioxide environment. The asexual blood-stage parasite density was calculated routinely using thick blood films. Baseline RBC count was used to calculate the parasitaemia (parasites/µl). P.falciparum Infected Red Blood Cells (iRBCs) were treated with a pre-warm aliquot of 5% D-sorbitol in RPMI at 37°C to attain synchronized parasite cultures in order to isolate homogeneous stages of parasites. Multiple sorbitol treatments were administered to achieve >90% synchrony of parasites. Parasites were collected from the three different developmental stages (ring, trophozoite and schizont) after stringent synchronizations.
RNA extraction, rRNA depletion and nuclease mapping
Synchronized P. falciparum parasites from the Ring stage (2–12 hours), Trophozoite stage (18–26 hours) and Schizont stage (30–42 hours) were isolated after saponin lysis and washed with incomplete medium (without Albumax). The RNA from each stage (three samples were collected and then pooled for each stage) was isolated by the Trizol (Thermofisher) method followed by Turbo DNAse (Thermofisher) treatment. Total RNA was then subjected to two rounds of rRNA depletion with custom biotinylated oligos designed against P.falciparum’s rRNAs (Suppl. Table 1). Briefly, 500 pmol of biotinylated capture probes was used in a binding buffer containing 10 mM Tris-Cl pH 7.4, 3 mM MgCl2, 300 mM NaCl and 0.5% Igepal-CA 630 (Sigma–Aldrich, 18,896). The hybridization reaction was prepared on ice in the presence of 50 ug of total RNA in each tube and 2 ul of RNase inhibitor. The oligo probe and total RNA mixture was heated at 70°C for 5 minutes followed by gradually reducing the heat until the oligonucleotides have reached room temperature. Finally, rRNA bound oligos were separated by Streptavidin conjugated magnetic Dynabeads M-280 (Invitrogen/Thermofisher, 11,206) followed by ethanol precipitation of rRNA depleted total RNAs.
Next, RNA samples were treated with a single-strand specific ribonuclease, RNase One (Promega, M4261) (5 units per ug of RNA), according to the manufacturer’s instructions. After the nuclease treatment, each RNA sample was fragmented with Fragmentation Reagents (Ambion/Thermofisher, AM8740) and then end corrected with T4PNK (Thermofisher). The RNA went through phenol-chloroform extraction and ethanol precipitation before using them for RNA-seq library preparation.
Library preparation and high-throughput sequencing of duplex RNAs (duplex RNA-seq)
P. falciparum duplex RNA sequencing libraries were prepared from the three developmental stages: ring, trophozoite and schizont (see results section for details). Two independent sets of libraries were prepared (two replicates for each developmental stage). The sequencing library was constructed using the Small RNA Sequencing Kit v3 (Illumina, San Diego, CA) as per manufacturer’s instructions. Library quality was checked by Bioanalyzer and then pair-end sequencing was done on two different Illumina high-throughput sequencing platforms, HiSeq2500 for RNA-seq-1 (Otogenetics Corp., Atlanta, GA) and NextSeq500 for RNA-seq-2 (ACGT Inc., Germantown, MD) (Table-1).
Bioinformatics analysis of duplex RNA-seq data
The paired end reads from the duplex RNA-Seq data were first trimmed to remove any adapter sequences using fastp v0.20.0 [73]. Then, STAR v2.5.2 [74] was used to align the trimmed data to the P. falciparum genome (v34) with the parameters – genomeSAindexNbases 11 and – alignIntronMax 500. The maximum splice junction length was set to a conservative threshold of 500 bp since a larger value of this parameter yielded many false splice junctions and thus, noisy structural peaks. This can be attributed to the short insert sizes in the libraries (Suppl. Table 2) and the relatively shorter length of the trimmed reads post adapter trimming (average length 84nt and 68nt for replicates 1 and replicates 2). A custom filter was then used to remove low mapping quality (mapping quality = 0) and secondary or supplementary alignments. Finally, featurecounts v2.0.0 [75] was used to perform gene counting using the latest gene annotations from [76]. All scripts that were used to perform data analysis have been made available to readers through our ‘Github’ repository at https://github.com/skadri01/duplexRNASeq. Reads that mapped to mitochondrial or apicoplast transcripts were removed from this gene expression analysis. Normalization of gene counts (as seen in Fig. 3A, Suppl. Fig 4) and differential gene expression were performed using DESeq2 [77] v1.26.0 in R v3.6.2 (see more details below). Expression count calculations to estimate the expressed genes are described below.
Figure 3.
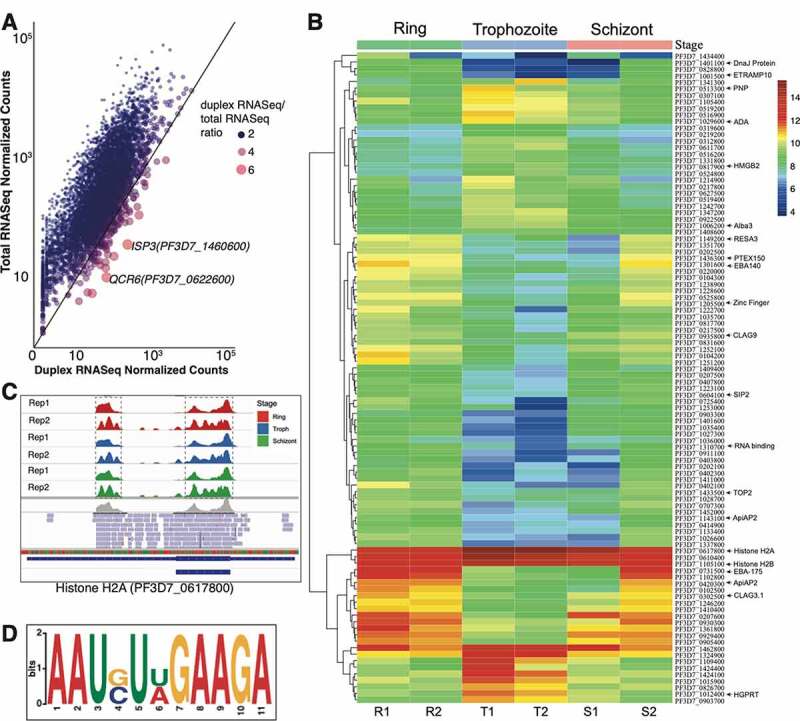
(A) Gene expression of P. falciparum duplex RNA-seq as compared to total RNA-seq data [47]. Representative scatter plot showing correlation between normalized gene counts from total RNA-seq and duplex RNA-seq datasets for ring stage. Trophozoite and schizont stages are shown in Suppl. Fig. 4A and 4B. A pseudocount of 1.0 is added to the normalized counts for both duplex and total RNA-seq data prior to plotting. The axes are in logarithmic scale. The colour and size of the points show the ratio of the duplex RNA-seq normalized counts to the total RNA-seq normalized counts. Larger red points represent genes with higher expression in duplex RNA-seq compared to total RNA-seq as shown in the figure label. (B) Heatmap of the 100 top differentially expressed genes across the three developmental stages. The normalized gene counts from DESeq2 are log transformed in the heatmap. The highlighted genes on the right are referenced across the text. (C) Snapshot of duplex RNA enrichments for all six samples (corresponding to Ring, Trophozoite and Schizont replicates) for Histone H2A visualized by IGV browser. The replicates are indicated as Rep1 and Rep2. The gene contains two sets of peaks in the duplex RNA-seq data, highlighted by dashed boxes. The second replicate show a higher resolution of peaks due to shorter insert sizes (as discussed before) (D) A highly represented sequence-structure motif identified from duplex RNA reads of P. falciparum mRNAs
Figure 4.
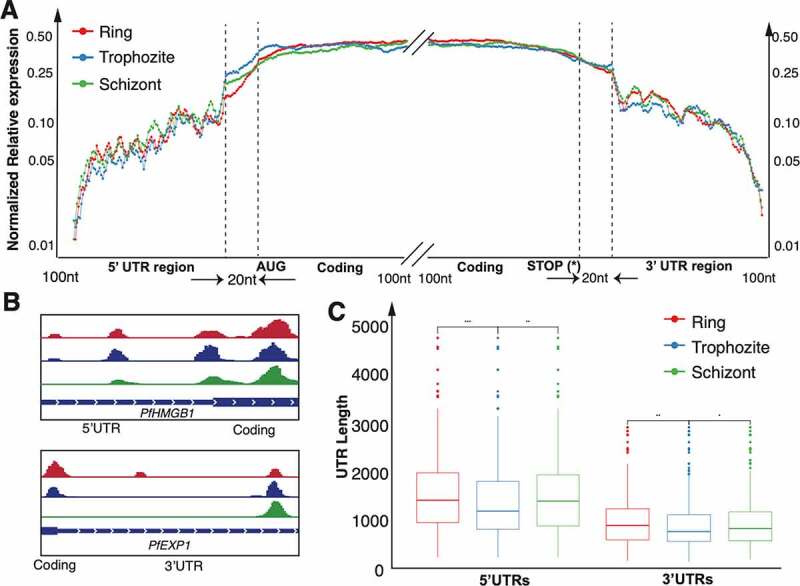
Distribution of duplex RNA-seq data across the top 5% (~200) mRNAs with secondary structures in (A) 5ʹ UTRs and 3ʹUTRs and 100nt near the start and stop codons. The y-axis shows the average normalized relative expression, which is normalized against the highest duplex RNA-seq expression in the region being investigated. This ratio represents the relative distribution of enrichment of base-paired RNAs (See Methods for details). (A) The non-coding UTR regions are binned into 100 bins (UTRs shorter than 100nt are expanded to 100 bins) whereas only 100nt of the coding regions are plotted. Regions that are 20 nt upstream of the start codon (left) and 20 nt downstream of the stop codon (right) are examined to find structures near the start and stop codons respectively. We focus on 100nt of the coding regions downstream of the start codon and upstream of the stop codon for the 5ʹUTR and 3ʹUTR respectively. For each stage the values of the top 5% genes with 5ʹUTR/3ʹUTR structures are averaged. (B) Examples of genes with structures in the 5ʹUTR (PfHMGB1) and 3ʹ UTR (PfEXP1) as visualized in IGV for each of the three stages. (C) Box plots illustrating the lengths of the UTRs of the 5% most structured UTRs (~200 genes each) (See Materials and Methods) across the three time point collections of P. falciparum. The asterisks indicate a significant difference (two-sided Wilcoxon test, p < 0.001)
Expression count calculations to flag potential stable secondary structures
The raw counts from ‘featurecounts’ described above, including all known coding and non-coding genes were normalized by the relative sequencing depth to calculate the ‘Relative Expression Counts’ (REC) using the number of uniquely mapped reads. Only genes with ‘REC’ values greater than 10.0 across 2 or more samples were considered for this analysis. This was done so that outlier noise in single samples would be removed from the analysis. Finally, from the filtered genes, all genes with ‘REC’ values >10.0 filter are considered genes with potential stable structures for each sample. A threshold of 10.0 was selected based on empirical observations of peak data and manual inspection of the data in Integrative Genomics Viewer (IGV) [142] because genes with lower values were typically due to either non-specific noise or low-resolution and unreliable structures. We did not normalize data by gene length (similar to the popular RPKM, i.e. reads per kilobase per million values used in total RNA-seq) because structural data is not as quantitative as total RNA-Seq and does not necessarily contain reads across the entire gene. A longer gene might contain the same number of reads as a similarly expressed shorter gene due to its structural similarities and thus, using RPKM would penalize the longer genes. We used the ‘REC’ values to get a global picture about the genes that form duplex RNA structures across the three developmental stages.
UTR and splice junction analysis
For the top 5% most structured 5ʹUTR and 3ʹUTRs in the P. falciparum genome, we calculated the counts for the duplex RNA-seq at each position in the 5ʹUTR and 3ʹUTR regions as well as 100 nt downstream of the start codon and 100 nt upstream of the stop codon. Within the UTR regions, we separated out 20 nt upstream of the start codon and 20 nt downstream of the stop codons to study the RNA structure data around the start and stop codons, respectively. Since the UTRs vary drastically in size [76], we normalized the UTR lengths into 100nt bins by summing up the counts. UTRs that are shorter than 100nt are normalized into 100nt bins by expanding the counts of the UTR positions into 100 windows. We then calculated the normalized relative expression for each bin or position by normalizing the duplex RNA-seq counts against the maximum duplex RNA-seq expression of the individual regions. This was done to study the relative distribution of duplex RNA-seq reads at the 5ʹ and 3ʹ gene ends. For each stage, the normalized expression for each bin was averaged across the two replicates per P. falciparum stage for the final analysis. Since low gene expression can make the trends noisy due to their stochastic distributions, we only considered the top 5% genes with UTR expression (~200 genes in each of the six samples). A similar analysis was done for the splice junctions by studying the 50nt flanking the top 5% structured splice junctions.
Differential expression analysis (GO/KEGG)
Count data was generated for all duplex RNA-seq datasets using the annotated transcripts from [47] using featurecounts as described above. Differential gene expression was performed using DESeq2 v1.26.0 in R v3.6.2, using default settings and accounting for batch effects. The differentially expressed genes across all three stages (adjusted p-value<0.05) were used for GO analysis (194 genes), which was performed using GO terms from PlasmoDB [78] and topGO v 2.38.1 package in R. The ‘Molecular Function’ ontology was used and the top 20 nodes were studied. We use the Kolmogorov–Smirnov test [79] for the enrichment analysis using the elim method [143] Further filters included removing GO categories with more than 500 genes and collapsing categories with completely overlapping significant gene lists. The resulting GO categories and genes are shown in Fig. 6A.
Figure 6.
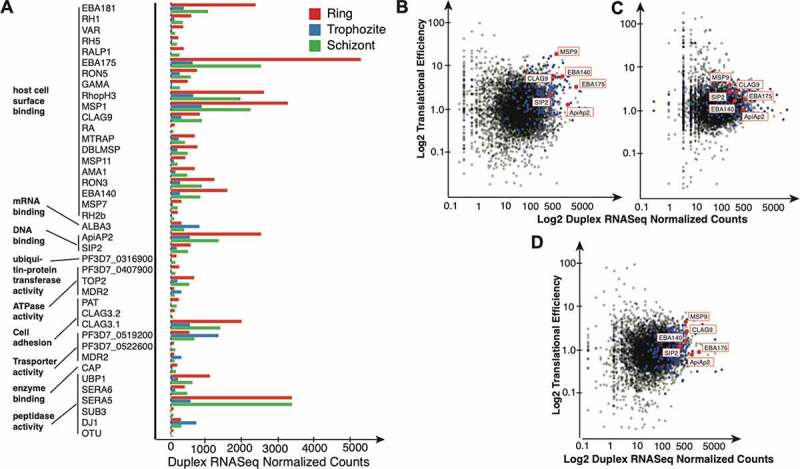
The relationship between mRNA secondary structures and molecular function. (A) Barplot of the normalized gene counts and Gene Ontology (GO) analysis of the top differentially expressed genes (see text for details). Ring vs Trophozoite and Trophozoite vs Schizont, by stage. red: Ring; blue: Trophozoite; green: Schizont. [B-D) Translation efficiency correlation with duplex RNA-seq structures. Scatter plot of normalized duplex RNA-seq counts plotted against translation efficiency calculated using data from 43,for the (B] Ring stage (C) Trophozoite, and (D) Schizont stages (p < 0.001). The 194 differentially expressed genes used for the GO analysis are highlighted in blue. Out of these, 5 selected genes addressed in the text are highlighted in red
P. falciparum gene expression (RNA-seq) data
Fastq files of mRNA-seq data were downloaded from a previously published [47] dataset. The data were run through the similar bioinformatics processing as described for the duplex RNA-seq, which included adapter trimming, read alignment and filtration followed by gene expression counting. The resulting gene counts were normalized using DESeq2 [77] v1.26.0 in R v3.6.2 and used for comparison.
Translational efficiency calculations
Single-end fastq files of ribosomal footprints and the corresponding mRNA-seq from [43] were aligned using STAR v2.5.2 and quantified using feature counts as described above. The count data were normalized using DESeq v1.26.0, and we calculated a ratio of the ribosomal footprint normalized counts and mRNA-seq normalized counts as a representation of translation efficiency in P. falciparum for all protein coding genes in P.falciparum.
Antisense RNA immunoprecipitation with J2 antibody
For immunoprecipitation of total RNA, 50 μg of RNA (rRNA depleted) from asynchronous, mix-stage P. falciparum cultures were incubated overnight at 4°C in a reaction buffer containing 15 mM Tris·HCl (pH 7.5),150 mM KCl, 5 mM MgCl2 and 0.5% Triton X-100 (Sigma–Aldrich) in presence of RNase inhibitor with 10 μg J2 antibody (Scicons). Then, 50 μL of washed (three times in reaction buffer) protein G-Dynabeads magnetic beads were added to the solution with both RNA and J2 antibodies (ThermoFisher) and incubated for 4 hours at 4°C. Complexes were washed four times in reaction buffer and RNA was recovered by phenol-chloroform extraction and ethanol precipitation. RNA integrity, quality and quantity were measured by Qubit 4 fluorometer (ThermoFisher).
Northern blotting
Two micrograms of J2 immunoprecipitated RNA for each sample was resolved on 6% polyacrylamide gel containing 7 M urea, 90 mM Tris-Borate, and 2.5 mM EDTA (TBE) (pH 8.3) after heating at 95°C for 3 min and instantly chilling on ice. RNA from the gel was subsequently transferred onto N+ nylon membranes (Amersham/GE) using a 300 mA constant current for 4 h at 4°C. Then, the RNA was cross-linked onto the membrane by UV light. Northern hybridization was done in 0.5 M sodium phosphate buffer (pH 7.6) with gamma-32P-ATP labelled specific oligodeoxyribonucleotides at ∼5 × 106 cpm/mL. Following hybridization, membranes were washed twice in 2× SSC, 0.1% SDS at hybridization temperature and once at 0.1× SSC, 0.1% SDS at room temperature for 15 min each and exposed to a Phosphorimager.
RNA-SHAPE analysis
For SHAPE modification of P. falciparum U3 RNA, 5ug of in vitro transcribed RNA was denatured at 95°C for 2 min followed by immediate incubation in ice for 3 min. Modification was carried out by incubating the RNA in a 3X SHAPE buffer consisting of 333 mM of NaCl, 333 mM of HEPES and 20 mM of MgCl 2 at 37°C for 30 minutes prior to the addition of 100 mM of 2-methylnicotinic acid imidazolide (NAI; Sigma–Aldrich, 913,839) for 15 min at 37°C. As a negative control, equivalent volumes of DMSO were added to the RNA.
cDNA Synthesis and RNA-SHAPE Analysis by Denaturing Polyacrylamide Gel Electrophoresis (PAGE) was done as follows: 1.6 μg of SHAPE modified RNA was denatured by heating at 95°C for 2 min followed by immediate incubation in ice for 3 min. For primer extension, 5′ cy5 labelled primer for reverse transcription as indicated in Table S3 along with 10 mM dNTPs were mixed with the NAI and DMSO treated RNA and incubated at 65°C for 5 min. Four dideoxy sequencing reactions using ddNTPs were also performed in parallel using the unmodified PfU3 snoRNA in the similar manner as mentioned above. Following incubation, all the reactions were supplemented with 1× SuperScript II (Thermofisher) first-strand buffer (50 mM Tris-HCl, pH 8.3, 75 mM KCl, and 5 mM MgCl2), 10 mM DTT, and 1 μL of RiboLock RNase inhibitor and incubated at 42°C for 2 min. After addition of SuperScript II reverse transcriptase enzyme, the whole reaction was incubated at 42°C for 60 min, followed by inactivation at 70°C for 15 min. The resultant mixtures of cDNA were precipitated by adding isopropanol and then dissolved in water. cDNA generated from primer extension was mixed with 80% formaldehyde, heated at 80°C for 3 min, and then resolved by 12% denaturing PAGE gel. The gel was scanned using Typhoon phosphorimager (GE Life Sciences). Nucleotide position for each band was identified from the dideoxy sequencing lane. Based on the modifications obtained from our denaturing gel experiments, we derived the secondary structure of P. falciparum U3 RNA, based on a previously established model [63] using RNAstructure [80].
Results
Transcript assembly and analysis of duplex RNA-seq from P. falciparum asexual developmental stages
The primary focus of this project was to look into the RNA secondary structures that were formed in the coding and non-coding segments of transcripts in P. falciparum at the genome-wide level. Since the conventional RNA interference (RNAi) machinery that produces double-stranded RNAs (dsRNAs) (via siRNA and miRNA mediated pathways) for gene silencing [81] is absent in malaria pathogen P. falciparum [82], we refer to the double-stranded regions identified in our experiments in P.falciparum as ‘duplex RNAs’ or ‘RNA duplexes’ to avoid any confusion, similar to how they are denoted in previous studies [83,84]. To identify RNA duplexes, i.e. secondary structures that are results of inter- and intra-molecular base pairing interactions by deep sequencing, we optimized a protocol (referred here as ‘duplex RNA-seq’) that was originally developed for double-stranded RNA Sequencing (dsRNA-seq) in plants [19,66,85]. We obtained base-paired RNAs from the asexual blood stage cultures of malaria parasite P. falciparum. Total RNA was obtained from synchronized cultures (>90%) of P. falciparum within its 48 hours asexual RBC cycle and labelled samples as follows: Early or Ring stage collection: 2–12 hours, Mid or Trophozoite stage collection: 18–26 hours and Late or Schizont stage collection: 30–42 hours post-invasion of parasite in human RBCs. Three samples were collected and then pooled for each stage of P. falciparum’s asexual RBC cycle (Figure 1A). Total RNA isolation was followed by two rounds of ribosomal RNA (rRNA) depletion using custom made oligos against P. falciparum rRNAs (see ‘Materials and Methods’ and Suppl. Table-1). Then these RNA samples were treated by RNase One (Promega), a single-stranded RNA (ssRNA) -specific endoribonuclease in order to destroy single-stranded regions of the transcriptome while keeping the base-paired regions intact. Following nuclease treatment, P. falciparum duplex RNAs were purified and stage-specific libraries were subjected to RNA sequencing to obtain the first assembly of P. falciparum strand-specific transcripts, representing duplex RNA abundance from sequencing data. Two biological replicates of each stage-specific sample were sequenced (see ‘Materials and Methods’ for details) and the sequenced reads obtained from duplex RNA-seq were aligned to the 3D7 reference genome (PlasmoDB, Release 34) using STAR aligner [74] (see ‘Materials and Methods’ for details about the analysis). We used two different Illumina sequencing platforms for two separate rounds of duplex RNA-seq, generating the largest number of raw reads of 67,970,256 and 437,153,310 from HiSeq 2500 and NextSeq500 runs respectively (Suppl. Table 2). The read lengths varied from 75bp to 125bp. Correlation analysis between the two replicates at each of the three time points using duplex RNA-seq ‘Relative Expression Counts’ (REC) (see Materials and Methods) reveals high correlation of gene expression of base-paired RNAs across these two platforms (Suppl. Fig 1A). It is worth noting that duplex RNA-seq measured the abundance of base-paired RNA regions among the developmental stages and thus, may not be quantitated in the same way as total RNA-seq data. Despite this and differences in sequencing instruments, we see a high correlation of gene counts between biological replicates of the same stage (Suppl. Fig 1A). When assessing sequencing accuracy, we found on average 83.2% of these total reads showed a Phred quality score of Q ≥ 30. The distribution of GC content in these reads were between 40–50% which is much higher than average GC content of P. falciparum genome (~19%) (Suppl. Table 2). The GC-richness of duplex RNA-seq reads suggests that these reads are aligning mostly to the euchromatic coding sequences or functionally important non-coding regions in the P. falciparum genome which are usually high in GC content [63,86]. In summary, our duplex RNA-seq experiments reliably identified base-paired regions of RNA secondary structures from all three RBC stages of P. falciparum at a global scale.
Figure 1.
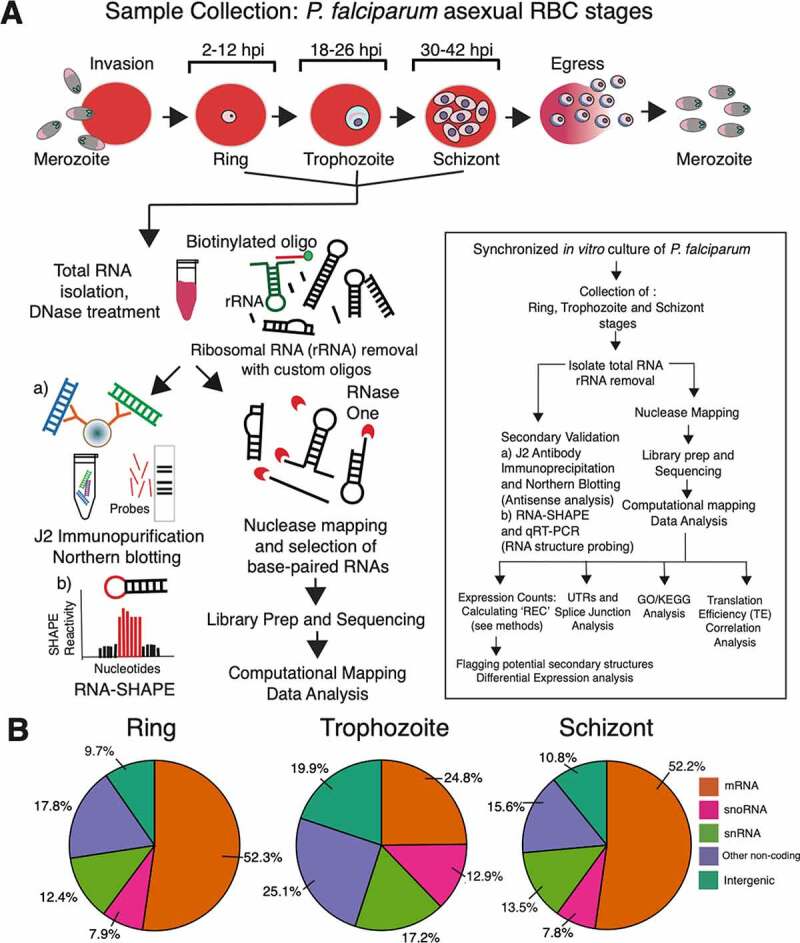
RNA sequencing, analysis pipeline and read distribution profile. A) Total RNA samples collected from three different developmental stages (Ring, Trophozoite and Schizont) of P. falciparum in human RBCs were subjected to nuclease-based structure mapping using RNase One, followed by purification of base-paired RNAs and strand-specific RNA sequencing. Differentially expressed regions of the P. falciparum genome that show significant accumulation of duplex RNA reads were further analysed by (A) immunoprecipitation using anti-dsRNA antibody (J2) for detection of antisense RNAs. Also, notable structural RNAs that were identified by duplex RNA-seq were further validated by (B) mapping to secondary structures following RNA-SHAPE analysis. A detailed flowchart with wet-lab experiments and computational analysis used in this study is shown in the inset box. B) Distribution of paired-end duplex RNA-seq reads that align to different annotated categories of coding and non--coding transcripts in P. falciparum genome (PlasmoDB) [78] using latest gene annotations from [76]. Only reads mapping uniquely to the genome are considered and reads aligning to known rRNAs and tRNAs are removed from this analysis. Abbreviations used are: hpi (hours post infection), REC (relative expression counts), GO (Gene Ontology)/KEGG (Kyoto Encyclopaedia of Genes and Genomes). Additional acronyms are explained in the ‘Abbreviations’ section
Overview of duplex RNA distribution and dynamics in P. falciparum RBC cycle
As anticipated, a large proportion of global coverage of the base-paired RNA reads aligned to genomic regions that are known to produce structural RNAs in P. falciparum [62–64]. Reads that mapped to known tRNAs and residual rRNAs were bioinformatically removed before differential expression analysis of duplex RNAs (see ‘Materials and Methods’). Consequently, a larger proportion of snoRNA, snRNA and known non-coding RNA-specific read accumulations were observed in the trophozoite stage compared to ring and schizont stages of P. falciparum (Figure 1B). In contrast, ring and schizont stages contained >50% reads that correspond to protein-coding transcripts, representing a high abundancy of secondary structures within mRNA molecules in these two stages of the P. falciparum asexual cycle. On average, the ring and schizont stages have ~23% more ‘expressed’ genes, containing duplex RNA reads (see Expression count calculations in the ‘Materials and Methods’ section) than the trophozoite stage. These data corroborate with the stage-specific transcript stabilization that was observed during developmental mRNA dynamics in P. falciparum [87]. This is likely due to the fact that RNA stability plays a critical role in mRNA structure driven translational regulation [88].
To better understand the RNA structurome of P.falciparum, we decided to split the coverage by chromosome and determine the comparative distribution of the base-paired RNA reads. Thus, we produced a plot of duplex RNA expression from ring, trophozoite and schizont stages on all 14 chromosomes and it revealed that most of the highly expressed protein-coding genes that produce RNA secondary structures in the coding transcripts are residing in chromosomes 6, 11, 13 and 14 (Suppl. Fig. 1B). Interestingly, many of the antigenic gene families in P. falciparum which are involved in host immune evasion and pathogenesis are transcribed from these chromosomes, including multiple copies of var and rif genes [86]. It is noteworthy that the duplex RNA-seq data showed significant enrichment of reads for these mRNAs involved in antigenic variation in P. falciparum (see below).
For known non-coding RNAs, such as snoRNAs, snRNAs and ncRNAs, the transcripts from chromosomes 11, 12 and 14 showed highest accumulation of duplex RNA reads and thus, indicate an abundance of structured regions (Suppl Fig. 2). On the other hand, duplex RNA enrichment on other chromosomes was very minimal. tRNA and rRNA genes were not included in this analysis. As Suppl. Fig 2 shows, this observed trend is not simply a result of a higher number of non-coding genes on these chromosomes (black dots). Rather, the size of non-coding genes also follows a similar pattern as the number of non-coding genes (data not shown), with chromosomes 8 and 11 containing the most areas with non-coding genes (~10kB) and chromosome 14 containing the most enrichment for duplex RNA in the non-coding regions across a length of about 6kB.
Figure 2.
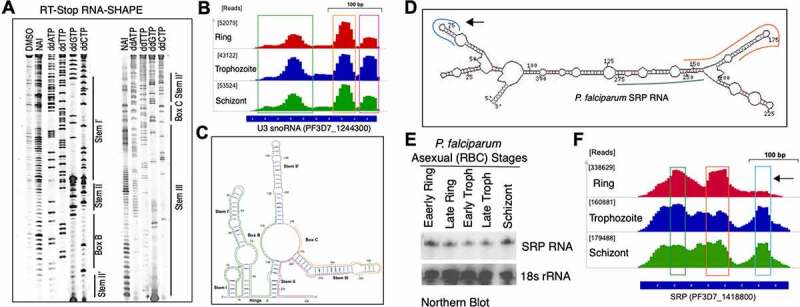
Mapping and validation of duplex RNA-seq data with known RNA secondary structures of non-coding RNAs: U3 RNA (A-C) and SRP RNA (D-F). (A) Two Cy5 labelled denaturing gels for U3 snoRNA domains: Stem I’, Hinge, Box B, Stem II’, Stem II’, Box C and Stem III (Corresponding RNA secondary structure data in panel C). (B) IGV screenshot of the U3 snoRNA peaks representing duplex RNA-seq read enrichments in three different P. falciparum RBC stages. The raw read counts of the highest peak are shown in square brackets ([]) next to each corresponding stage. (C) Secondary structure of the PfU3 snoRNA based on in-gel SHAPE modification of mix stage P. falciparum RNA using NAI (shown in panel A). Unpaired RNA nucleotides are shown in red and orange colour based on their reactivities, where high reactivity is signified by red colour and low reactivity by orange with the SHAPE reagent. Base-paired RNA regions identified from duplex RNA-seq in B are indicated by green, yellow and magenta colour skirting (also shown by matching coloured bars at the bottom of the peaks/reads identified in duplex RNA-seq). (D) Schematic representation of the P. falciparum SRP RNA secondary structure model. Mapped reads from duplex RNA-seq are shown in skirted coloured lines along the structure model. Corresponding peaks of the mapped read regions in P. falciparum genome are shown by three colour matching boxed regions in F. Due to shorter insert sizes in the second set of replicates (replicate – 2) of the data (Suppl. Table 2), the peaks are seen more distinctly and at a higher resolution in these replicates and thus, we selected to display the coverage in these replicates. The remaining three replicates also show the same structures but at a lower resolution. (E) Northern blot analysis of the total RNA from five different time point collections during asexual RBC cycle with SRP RNA-specific probe [63]. (F) IGV screenshot of the SRP RNA duplex RNASeq data in the three P. falciparum RBC stages. Black arrow showing a peak missing from the ring stage samples (also shown by an arrow in structure model in D), indicating absence of domain-specific reads and thus, the ability of the duplex RNA-seq to detect stage specific RNA structural rearrangements during P. falciparum development. Similar to (B), the raw read counts of the highest peak are shown in square brackets ([]) next to each corresponding stage
As a result of the retention of strand information of the molecules from duplex RNA-seq data, we were able to determine the strand bias for different classes of RNAs. DNA replication and transcription can exert a strand bias on the DNA repair process [89,90], particularly the transcription levels which affect the degree of bias [91]. This in turn could affect an organism’s gene expression program. In P. falciparum, a strong enrichment of duplex RNA reads transcribed from the forward (plus) strands was observed for mRNAs and snoRNAs from all three stages (Suppl. Fig. 1C). It was noted that mRNAs and snoRNAs were ~1.5-fold and ~5-fold enriched, respectively, for the forward strand compared to reverse (minus) strand across all chromosomes. Expression of mRNAs with secondary structures was higher in ring and schizont stages compared to trophozoite stages, whereas snoRNAs were mostly expressed in the trophozoite stage, irrespective of strand bias (Suppl. Fig. 1C). On the contrary, snRNAs and other known non-coding RNAs showed a strong reverse strand bias in the duplex RNA-seq data.
Overall, our data analysis not only suggests a stage-dependent enrichment of structured mRNAs in the P. falciparum asexual stages, it also indicates bias for abundance of structured regions in relation to genomic location, gene size and the DNA strand for transcript synthesis.
Genomic distribution of duplex RNA hotspots in P. falciparum
Evidence of highly base-paired regions of RNAs, referred hereby as duplex RNA ‘hotspots’ was identified across the coding and non-coding regions of P. falciparum genome. From previous studies, it is known that structured regions in the mRNA have the potential to act as regulators in cellular processes, such as in RNA stability, turnover and translation in yeast and mammalian cells [88,92,93]. Our duplex RNA-seq data mapped to a total of 3,829 protein coding (mRNAs) and non-coding transcripts with ‘relative expression counts’ (‘REC’) above set thresholds (REC>10) and showed high enrichment (>100 REC) for 969 mRNAs in two or more samples across any of the three RBC stages (See Materials and Methods). This suggests that regions across these 3,829 coding and non-coding transcripts could potentially fold into unique, stable secondary structures. In addition, we identified known structural RNAs as well as novel, structured RNA elements throughout the intronic and intergenic regions of P. falciparum genome.
Structural RNAs
Our duplex RNA-seq data are in agreement with known RNA structures in P. falciparum. We corroborated our duplex RNA-seq data with 73 highly base-paired segments of known, conserved structural RNAs (Suppl. Table 3). To characterize the constituents of duplex RNA-seq reads, we determined the secondary structure of the P. falciparum U3 snoRNA in vitro by in-gel RNA-SHAPE analysis using NAI (Figure 2A) and mapped the duplex RNA-seq data to the U3 secondary structure model. As anticipated, base-paired stem regions originating from intramolecular interactions in PfU3 RNA corresponded to more duplex RNA-seq reads (represented by peaks in Figure 2B) than to the unpaired regions (Figure 2C). Thus, our analysis revealed with confidence that the majority of our data obtained from duplex RNA-seq are related to biologically relevant structures in the malaria genome. Among all these non-coding RNAs, the highest enrichment of reads (>333,000 normalized read counts) mapped to the P. falciparum Signal Recognition Particle RNA (SRP RNA, PF3D7_1418800) [63], which is a conserved RNA universally required for co-translational protein targeting. The three-dimensional structure of mammalian SRP RNA is already known [94] and the secondary structure prediction of P. falciparum SRP RNA was previously reported based on comparative genomics and RNA analysis [63], which corroborates strongly with the duplex RNA-seq data (Figure 2D and 2F). When stage-specific PfSRP expression was compared to the base-paired transcript abundance in our duplex RNA-seq data (Figure 2E and 2F), we observed lower accumulation of SRP RNA domain-specific reads in the ring stage compared to the schizont stage. Although there was lower accumulation of SRP RNA, expression levels were similar in ring and schizont stages as evident from northern analysis shown in Figure 2E (read accumulations are represented by ‘peaks’ in Figure 2F and the missing peak in the ring stage is shown by an arrow in Figure 2F with its corresponding origin in Figure 2D). This suggested that our duplex RNA-seq data were able to capture stage-specific RNA structural rearrangements during P. falciparum development. Among other highly structured classes of RNA molecules that were identified in P. falciparum genome, secondary structure and RNA modification sites for P. falciparum C/D box snoRNA 15 (PF3D7_1222200) was previously established by in vivo DMS probing [63]. The duplex RNA-seq reads accurately mapped to those structured regions of P. falciparum snoR15 (Suppl. Figure 3), suggesting that we are able to reliably capture intramolecular base-pairing interactions by duplex RNA seq and afforded the first genome-wide view of the structural landscape of P. falciparum RNAs, many of which are involved in conserved biological functions. In addition, RNAs of Unknown Function (RUFs) [63], RUF1 (PF3D7_1328700) and RUF 6 (PF3D7_0711800, PF3D7_0712100, PF3D7_0712700), all expressed from chromosome 7, showed enrichment of base-paired RNAs. In addition to previously reported non-coding structural RNAs, we identified potential novel RNA secondary structures in otherwise unannotated genomic regions, which are shown in Suppl. Table 4. Taken together, since a major portion of our duplex RNA-seq reads corresponded to highly conserved, known structured classes of RNA molecules, we deem that the results are reflective of nearly accurate estimation of RNA secondary structures in the malaria genome.
Coding sequence (CDS) of mRNA
Our duplex RNA-seq readout allowed global analysis of RNA secondary structures in the coding regions of P. falciparum mRNAs. As shown in Figure 1B, out of the >50% of dsRNA reads that map to protein-coding genes (after applying the REC>10 threshold to determine ‘expressed’ mRNAs with structures), we found that 3,773 unique protein coding genes have expression indicating structure formation across at least one developmental stage. This 3,773 genes account for 71.4% of all protein coding genes. Thus, we found that nearly 71.4% of the protein-coding genes in P. falciparum align to duplex RNA-seq reads exceeding our set threshold (REC>10), and as a result, define its secondary structure content. Out of the uniquely ‘expressed’ 3773 mRNA regions containing secondary structures, approximately a quarter (26.1%) show high cumulative expression (>100 REC) of transcripts with structured regions in at least one of the three stages analysed in the RBC cycle. Based on the average expression seen across all the dsRNA-Seq time points, we conclude that approximately 71% (3773) of all mRNAs make secondary structures in at least one of the sequenced stages, with expression above set thresholds. When comparing the distribution of expressed genes (REC) between the stages, we found significant difference among ring and trophozoite or trophozoite and schizont stages (two-sided Wilcoxon test, p < 0.001).
In order to study the correlation between the gene expression of protein coding transcripts in P. falciparum total RNA-seq [47] and the duplex RNA-seq data, we normalized each dataset separately using DESeq2 [77] (See Materials and Methods). Then, the average normalized gene expression counts were plotted for each stage across the total RNA-seq and duplex RNA-seq datasets. As expected, the protein coding genes have more expression in the mRNA-seq dataset compared to the duplex RNA-seq as the latter represents a subset of the former, containing expressed structure contents of the protein coding genes (Figure 3A showing ring stage data, Suppl. Figure 4A and 4B showing trophozoite and schizont stage data, respectively). The overall trend in the degree of correlation between the two datasets is moderately high (R2 values are 0.66, 0.65, 0.70 across the three stages).
To understand the mRNA structure dynamics of P. falciparum during the asexual stage transitions in human RBCs, we characterized differential expression of structured contents within mRNA coding sequences (CDS), which is depicted as a heatmap in Figure 3B. Results from this parasite’s top 100 most expressed mRNAs with differential secondary structures (that is, out of all the differentially expressed genes) during the 48 hours intra-erythrocytic developmental cycle (IDC) suggest that highly folded RNA coding regions can potentially influence stability and translational outputs of individual transcripts in malaria gene expression programs. Among the highly expressed base-paired mRNA molecules in P. falciparum, histone mRNAs (Histone PfH2A (PF3D7_0617800), PfH2B (PF3D7_1105100) and PfH3 (PF3D7_0610400)) showed highest accumulation (within top 1%) of reads by duplex RNA-seq. Histone mRNAs are one of the most extensively validated functional mRNA structures present not only in metazoans but also in many different protozoa, including alveolates (e.g. P. falciparum) [95]. The histone mRNA forms a stable stem-loop structure at the 3′ UTR structure, which is involved in nucleocytoplasmic transport and regulation of translation efficiency [96]. In addition to strong enrichments of duplex RNA reads at the 3ʹ UTR of P. falciparum histone mRNAs, the read abundance was also observed in the CDS as shown for histone H2A (Figure 3C). The list of highly expressed duplex RNA containing transcripts in P. falciparum also incorporated mRNAs of chaperone proteins HSP70 and HSP90, which are also known to be highly structured [12,97]. These findings substantiated that our duplex RNA-seq identified the desired regions of mRNAs on a genome-wide scale, which are highly enriched in base-pairing interactions.
In regards to P. falciparum gene regulation, one emerging theme is the central role played by DNA and RNA-binding proteins in transcriptional and post-transcriptional control of gene expression [48,87,98]. For example, a plant-like transcription factor family, known as the Apicomplexan AP2 (ApiAP2) protein family, expressed in different stages during malaria parasite development, is critically involved in parasite development and differentiation processes [87,99]. Out of the 27 members, the expression of two members of this gene family, PF3D7_0420300 and PF3D7_1143100 (Figure 3B), were among the top 100 gene candidates to produce high levels of differentially expressed duplex RNAs in their coding regions. They were considered in our study as prominent ‘hotspot’ candidates. Among those, the ApiAP2 (PF3D7_0420300) expressing protein is known to functionally interact with an ACACACAT DNA motif in P. falciparum gene expression regulation [100]. Furthermore, its expression is correlated with a drug resistance transporter, pfCRT, indicating a potential role in resistance evolution to chemotherapeutic molecules in malaria [101]. Similarly, for RNA mediated post-transcriptional gene regulation, RNA-binding proteins (RBPs) in P. falciparum play pivotal role in translational regulation [48,102]. Our duplex RNA-seq data revealed strong accumulation of base-paired reads mapping to DNA-/RNA-binding factor PfAlba3 (PF3D7_1006200) (Figure 3C). The PfAlba3 protein interacts with P. falciparum histone deacetylase, Sir2A [103] and associates with polysome fractions during the P. falciparum IDC [102] and therefore, may play important regulatory roles in transcription as well as in translation. In addition to mRNAs that produce regulatory proteins, transcripts encoding antigens and adhesion molecules involved in host-parasite interaction in P. falciparum are the most highly base-paired classes of RNA molecules. Examples of these antigens and adhesion molecules include the erythrocyte-binding antigen 175 (EBA-175, PF3D7_0731500), which is involved in erythrocyte invasion [104]; SERA5 (PF3D7_0207600), which is important for parasite egress [105] and ring-infected erythrocyte surface antigen; and RESA-1 (PF3D7_0102200) along with RESA-3 (PF3D7_1149200) [106], which may prevent P. falciparum infected erythrocytes from reinfection through interactions with spectrin family proteins [107].
Genetic control of cellular metabolism often involves the sensing of mRNA secondary structures through nutrients and metabolites or interactions with RBPs [108,109]. These mRNA structures are widespread in prokaryotes and are also identified in a handful of eukaryotes. In prokaryotes, these are referred to as RNA genetic switches, which are typically located in non-coding regions of mRNAs (such as UTR regions) of many metabolic enzymes and can modulate gene expression. In eukaryotes, metabolic enzymes can interact with mRNAs to affect mRNA stability or translation efficiency [108]. We have identified base-paired mRNA elements in the coding region of P. falciparum metabolic enzymes. The most enrichment of duplex RNAs was seen in the Hypoxanthine-guanine phosphoribosyltransferase (HGPRT) (PF3D7_1012400) and Adenosylhomocysteinase (SAHH) (PF3D7_0520900). Most parasitic protozoa do not have the de novo purine nucleotide biosynthesis pathway. Instead, they rely mostly on the salvage pathway for purine metabolism, where hypoxanthine is the key precursor, so HGPRT in this pathway makes a promising drug target for P. falciparum infection [110]. However, the cellular interactions and importance of these novel mRNA structural elements in the coding regions of metabolic enzymes remain to be analysed.
We also studied the duplex RNA-seq structures at splice junctions of protein-coding genes (Suppl. Fig. 5) to evaluate differences, if any, between the 5ʹ and 3ʹ exons at these junctions. For the top 5% structured splice junctions in the P. falciparum genome, the 3ʹ exon of the junction was consistently less structured globally than the 5ʹ exons (Suppl. Fig. 5). The differences seen are small and it is difficult to determine whether this consistency is a global trend without additional testing. Next, we surveyed sequence motifs in mRNAs that are vastly appreciated in studying post-transcriptional regulatory processes [111,112], especially because these motifs can act as RNA–protein interface for major types of RBPs. In order to determine whether there were any sequence motifs located within the structured regions of mRNAs across a highly expressed pool of transcripts, we obtained sequences from 50 highly expressed mRNAs that represented a defined structured region (structured regions represented by peaks shown in Figure 3C). We then used the pattern search programme, Improbizer [113], to search through duplex RNA read sequences. We looked for reoccurring short sequence motifs and found six motifs which could be a potential regulatory region or an RBP interaction site on mRNA structures. The highest scoring motif sequence is shown as the sequence logo in Figure 3D.
Figure 5.
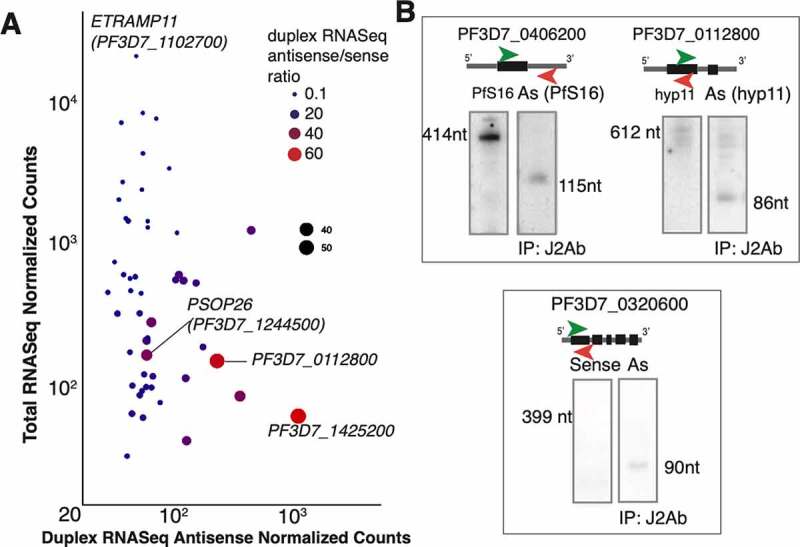
Antisense expression in duplex RNA-seq (A) Antisense duplex RNA-seq counts for the top 50 antisense genes plotted against mRNA-seq counts for ring stage. The other stages are shown in Suppl. Fig 5 C-D. The size and colour of the points indicate the ratio of the duplex RNA-seq antisense to sense counts. Larger red points indicate genes that have more antisense signal than sense signal. Few selected genes with very high and very low ratios are highlighted. A pseudocount of one is added to the normalized counts and axes are in logarithmic scale. (B) Antisense transcripts detected by northern blotting in the J2 antibody immunoprecipitated samples from mix (asynchronous) stages P. falciparum total RNA. Left blot (in all panels) shows sense gene expression and the right blot shows antisense expression from immunoprecipitated samples
In summary, the overall abundance (Figure 1B, Suppl. Fig. 1C) and dynamic changes (Figures 3A, 3B) in mRNA structures during P. falciparum blood-stage development indicate that the extensive stage-specific RNA folding events could be linked to the fine-tuning of protein synthesis in the malaria gene expression programs.
Non-coding regions of mRNAs – 5ʹ and 3ʹ UTRs
To investigate the global changes in P. falciparum mRNA structure during its asexual cycle, especially in the untranslated regions (UTRs), we analysed the top 5% structured regions over development in the UTR regions. Since the UTR lengths have a very broad range in the P. falciparum genome, the UTR regions were analysed as follows: UTR regions were binned into 100 nucleotides (5ʹ and 3ʹ UTRs: defined by [76]; 20 nt upstream of the start codon (for 5ʹUTR) and 20 nt downstream of the stop codon (for 3ʹUTR) to study structures around the start and stop codons; CDS: 100 nt downstream of the start codon (for 5ʹUTR) and 100 nt upstream of the stop codon (for 3ʹUTR)), and then average expression was calculated from ring, trophozoite and schizont stages (see ‘Materials and Methods’ for details about the analysis). Figure 4A and 4B show the distribution of normalized expression ratios of the duplex RNA-seq reads at the 5ʹ and 3ʹ gene ends for the top 5% genes with structures in the UTRs, respectively. The ratio is calculated against the maximum expression of the structures in the region being investigated in order to study the relative enrichment (See Methods). For example, the Schizont structures at the UTRs and splice junctions show lower ratios and thus, lower relative enrichment than the Ring and Schizont stages (Figure 4A-B) (t-test, p < 0.001). Having secondary structure near areas of translation initiation sites or coding regions have been seen to play a role in regulating translation efficiency [93,114,115]. Thus, we studied 20 nt upstream and downstream of the start and stop codons, respectively, and found that the duplex RNA-seq data show structures in these regions. For the non-protein coding regions, that is, 5ʹ UTRs and 3ʹ UTRs of mRNAs molecules, there is a significant enrichment in longer UTRs in the ring and schizont stages compared to the trophozoite stage (Figure 4C) (Wilcoxon test p-value <0.01).
Antisense RNAs
Antisense RNA transcripts, originating from the opposite strand to a protein or RNA-coding strand, have been assigned roles in gene expression regulation by the degradation of the corresponding sense transcripts or by silencing genes at the chromatin level [116]. Therefore, finding pervasive antisense transcription in P. falciparum fuelled the speculation that these RNAs regulate malaria gene expression programs either at the epigenetic or post-transcriptional level [57,59,117,118]. At the transcriptome level, widespread antisense transcription in P. falciparum corresponded to the transcription initiation of an antisense orientation to coding genes [144] [119], transcription start site-associated RNAs (TSS-RNAs) expression [76] as well as antisense transcription in the context of pre-mRNA splicing events [120]. At the gene level, natural antisense transcripts (NATs) that act as antisense long RNAs and that are longer than 200 nt have the potential to regulate P. falciparum virulence gene expression [51,60]. However, the origin and the mechanism of action of these long antisense RNAs transcribed from either ‘silent’ or ‘active’ var genes, remains unclear [121]. To investigate if our duplex RNA reads constitute potential antisense transcripts in the P.falciparum genome, we counted the antisense reads for all coding and non-coding transcripts and normalized these counts by depth of sequencing (REC). The same threshold described earlier (REC>10.0 in at least two samples across any of the three RBC stages of P. falciparum) was applied to the antisense data analysis as this was the empirically set threshold for more reliable duplex RNAs. Since antisense reads are in low abundance (after applying the above stringent cut-off), we found a total of 159 genes (154 mRNAs and 5 non-coding RNAs) with antisense reads above set thresholds. To better understand the relationship between gene expression and the abundance of duplex antisense reads, we looked at the average expression of the top 50 mRNA genes with antisense expression (Figure 5A, Suppl. Fig. 4C-D). As shown in Figure 5A, for example, gene expression from mRNA-seq and duplex antisense expression are not correlated but show an approximately negative relationship (Pearson correlation R2 = −0.11). This means that for certain genes, such as PF3D7_1425200 and PF3D7_0112800, the lower mRNA expression might be explained by antisense transcriptional regulation, although this remains to be validated by functional assays. We found a few interesting types of antisense expression, especially in the 3ʹ UTR regions (36.5% antisense structures were found in the 3ʹUTR). The three types of antisense peaks seen in the 3ʹ UTRs are: (i) antisense 3ʹ UTR structure(s) without any known overlapping genes at the position of the peak(s) (ii) antisense structure(s) in 3ʹUTR with overlapping UTR of another protein coding gene; this could potentially mean that the antisense structures are actually sense structures in the overlapping gene, and (iii) antisense structure(s) in 3ʹUTR from overlapping non-coding gene(s). To validate antisense RNA expression, we immunoprecipitated rRNA depleted P. falciparum total RNA with dsRNA specific J2 monoclonal antibody (Scicons) and probed this RNA with sense and antisense RNA specific complementary oligonucleotides. In this process we confirmed expression of ten (10) sense/antisense RNA pairs from asynchronous, mix stages of RBC cycle. Representative northern blots validating presence of antisense RNAs opposite three (3) genes are shown in Figure 5B. Additional validation were also done by qRT-PCR. Although our deep-sequencing data and Northern blots analysis correspond to the overlapping regions of the RNAs, it remains unclear whether mRNA stability was affected by the expression of these antisense RNAs or by their effect in resolving secondary structures of translatable mRNAs in P. falciparum. Thus, our data revealed several classes of antisense RNAs as components of base-paired regions which could play important role in gene expression regulation in P. falciparum.
Genome-wide relationship between RNA secondary structures and biological processes in P. falciparum
Next, we investigated genome-wide relationships between mRNA structures and the biological functions of the encoded proteins. To accomplish this, we used differential expression data of secondary structures from the three P. falciparum developmental stages within RBCs, that is, ring, trophozoite and schizont stages (Figure 3B). Differential expressions of structured mRNAs can be measured through the read count of the different stages (See Methods). A higher read count in one stage versus another is representative of stage-specific differential accumulation. A GO enrichment analysis (Figure 6A) of the differentially structured transcripts shows that the genes are enriched for molecular functions that are highly relevant for the developmental stages. These processes include host cell surface binding (GO:0046812) (enrichment p-value <1 × 10−3), DNA/RNA binding, which includes mRNA binding (GO:0003729) and DNA binding (GO:0043565) and, lastly, GO terms that belong to processes involved in enzyme activity (GO:0004842, GO:0016887, GO:0019899, GO:0008233) (See Materials and Methods for details). Among these genes, are transcription factors such as plant-like transcription factor ApiAP2 and ALBA3 (PF3D7_1006200) ApiAP2 protein SIP2 (0604100) (P < 0.05 Ring vs Trophozoite stage). In addition to the most significantly enriched molecular functions corresponding to proteins involved in transcription and RNA processing, our duplex RNA-seq data showed GO enrichment for several P. falciparum antigenic genes involved in processes, such as host cell surface binding/red blood cell invasion and host immunity (for example, EBA-181 and EBA-175, P < 0.05, Ring vs Trophozoite stage). Thus, the systematic analysis of mRNA secondary structures in the gene expression and molecular function in P. falciparum has identified substantially enriched candidates for functions relevant to malaria biology and pathogenesis.
Relationship between mRNA secondary structure and translation efficiency in P. falciparum
Dynamics of mRNA unwinding can significantly affect the rate of translation in eukaryotic systems [3]. Previous studies have suggested that the stable secondary structures in the 5ʹUTR or coding regions (CDS) of mRNAs can lead to ribosomal pausing our translational arrests [122,123]. Considering the critical role that mRNA structures can play in translation elongation, we next sought to determine the relationship between the structural contents of coding regions and translation efficiency in P. falciparum. Using existing ribosome profiling data sets [43], we re-analysed the datasets from the original publication and used a ratio of the ribosomal footprint normalized counts and mRNA-seq normalized counts as a representation of translation efficiency in P. falciparum (See Methods). Then, we compared it to the duplex RNA-seq dataset from all three P. falciparum asexual RBC stages. We observed dynamic, stage-specific relationship between mRNA structures and ribosome occupancy (Figure 6B-D, changes in mRNA structures and translational efficiency in individual stages are shown by few examples for genes shown in Figure 6A). Overall, we found stage-dependent, conditionally altered translational efficiency for individual mRNAs, probably reflecting active folding and unfolding events during translation. These data also indicate that factors that determine translational efficiency, such as altered rates of initiation, elongation, or termination, may vary drastically with changes in message (containing secondary structures) abundance and therefore may not reflect a strong overall correlation between mRNA structure and translation. Therefore, a more detailed investigation is required to define the coordinated regulation between mRNA structurome and translatome in P. falciparum.
Discussion
Our analysis of P. falciparum RNA structurome elucidated global, stage-specific RNA structural changes and therefore has a potential to yield significant insights into structure–function relationships in malaria post-transcriptional control. Since the first reports on RNA secondary structures in Plasmodium rRNAs and snRNAs [124,125], much progress has been made on identification and characterization of a variety of non-coding RNAs in P. falciparum [36,57,62–65,76,120]; however, the combined three membrane layers of host and the parasite have posed a challenge for in-cell probing of malaria structure by high-throughput methods. By overcoming this issue through using a robust in vitro assay, we show that reliable detection of structural contents in malaria transcriptome is feasible by duplex RNA-seq. Historically, in vitro characterization of RNA secondary structures, in case of ribozymes and riboswitches, for example, strongly agreed with the folding landscape of RNAs in physiological ensembles [126–128]. Since final native structures of RNAs should have the lowest free energy for the given environment [128–131] and because the experimental conditions we used should favour an energetically favoured folded state of duplex RNA, we anticipate that our data represent individual RNA folding landscapes that are populated by stable RNA structures, akin to ones that occur in the cells. Additionally, our chemical mapping experiments of known, conserved structural RNAs strongly agree with our duplex RNA-seq data that RNA molecules are correctly folded into their secondary structure in given experimental conditions. Although our data provide the first draft of RNA structurome of the P. falciparum asexual blood stages, several new critical advances in both the chemistry of electrophiles (including one from our group) [132–134] as well as probing technologies [22,23,135,136] should allow higher resolution mapping of cellular RNA structures in P. falciparum at a genome-wide scale in near future.
Our comparative analysis with other eukaryotic RNA structuromes has revealed that P. falciparum contains substantially high secondary structures (~71.4%) in protein coding regions as compared to human, yeast and metazoan species. Given that the P. falciparum genome has the highest AT content (~80%) of any eukaryotic organism sequenced so far, it is striking that P. falciparum transcriptome contains substantial stable and dynamic secondary structures in comparison to other organisms with a higher GC content. This also implies that P. falciparum could follow distinct mechanisms akin to one recently suggested for AU-rich mRNAs, which are usually enriched in P-bodies and require specific RNA-binding proteins, particularly ATP-dependent RNA helicases [137] for mRNA stability and translational control process. Recent reports suggest that highly expressed mRNAs usually have extensively structured coding sequences (CDS) [69,88], which could positively correlate with transcript-specific translation efficiency by increasing the mRNA half-life [88]. The ribosome [138] or m6A modifications [139] in CDS or UTRs could be the major remodeller of the mRNA structures for translation. In malaria parasites, mRNA translation facilitates transitions between the developmental stages [87,140], suggesting that malaria disease progression is dependent on stage-specific protein output. Additionally, in vivo measurements of mRNA cellular dynamics by biosynthetic labelling successfully recaptured the ‘just-in-time’ transcriptional programme of P. falciparum mRNAs with variable types of mRNA stabilization profiles [87], which could be due to the differential presence of structure between these mRNA populations. Therefore, it is conceivable that a great proportion of post-transcriptional gene regulatory programme is driven by mRNA secondary structures and RNA–protein interactions in this unique parasitic organism.
Many of these structural regions in coding sequences are phylogenetically conserved at the sequence level and provide an opportunity to study RNA-structure-dependent functions in malaria animal models. Since mRNA molecules are now showing significant promise as vaccine candidates [141], it is important to understand how mRNAs attain structure-dependent stability at greater molecular details. Therefore, thorough interrogations of how these structures in malaria coding regions relate to function should provide mechanistic details for developing future therapeutics that target mRNA structure and stability differently, depending on how they inhibit malaria translation.
Supplementary Material
Acknowledgments
We thank Rania Elbarki, Dipendra Gautam, Arundhati Mohanta and other past and current members of the Chakrabarti laboratory for their help with experiments and data analysis. This work was part of DRA’s undergraduate honors thesis project, as part of UNC Charlotte Honors Program in Biological Sciences. This work was supported by the UNC Charlotte Faculty Development Award and a consortium award by DSF Charitable foundation to KC.
Funding Statement
This work was supported partly by the UNC Charlotte Faculty Development Award and a consortium award by DSF Charitable foundation to KC.
Disclosure of interest
Duplex RNA-seq data for Ring, Trophozoite and Schizont stages of Plasmodium falciparum are publicly available in the NCBI GEO database (GSE163965):
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE163965
Author contributions
Conceived and designed the experiments: DRA, AO, SK, KC. Performed the experiments: DRA, AO, TB, BZ, AD, SB, and KC. Analysed the data: DRA, TB, CL, XS, SK, and KC. Wrote the paper: SK and KC.
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].Sharp PA. The centrality of RNA. Cell. 2009;136(4):577–580. [DOI] [PubMed] [Google Scholar]
- [2].Nikolova EN, Al-Hashimi HM.. Thermodynamics of RNA melting, one base pair at a time. RNA. 2010;16(9):1687–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kramer MC, Gregory BD. Does RNA secondary structure drive translation or vice versa? Nat Struct Mol Biol. 2018;25(8):641–643. [DOI] [PubMed] [Google Scholar]
- [4].Mustoe AM, Corley M, Laederach A, et al. Messenger RNA Structure Regulates Translation Initiation: a Mechanism Exploited from Bacteria to Humans. Biochemistry. 2018b;57(26):3537–3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Narberhaus F. Translational control of bacterial heat shock and virulence genes by temperature-sensing mRNAs. RNA Biol. 2010;7(1):84–89. [DOI] [PubMed] [Google Scholar]
- [6].Barrick JE, Breaker RR. The distributions, mechanisms, and structures of metabolite-binding riboswitches. Genome Biol. 2007;8(11):R239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Breaker RR. Riboswitches and Translation Control. Cold Spring Harb Perspect Biol. 2018;10(11):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cheah MT, Wachter A, Sudarsan N, et al. Control of alternative RNA splicing and gene expression by eukaryotic riboswitches. Nature. 2007;447(7143):497–500. [DOI] [PubMed] [Google Scholar]
- [9].Henkin TM. Riboswitch RNAs: using RNA to sense cellular metabolism. Genes Dev. 2008;22(24):3383–3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Warner KD, Hajdin CE, Weeks KM. Principles for targeting RNA with drug-like small molecules. Nat Rev Drug Discov. 2018;17(8):547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jones S, Daley DT, Luscombe NM, et al. Protein-RNA interactions: a structural analysis. Nucleic Acids Res. 2001;29(4):943–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sanchez De Groot N, Armaos A, Graña-Montes R, Sanchez de Groot N, Armaos A, Grana-Montes R, Alriquet M, Calloni G, Vabulas RM, Tartaglia GG . RNA structure drives interaction with proteins. Nat Commun. 2019;10(1):3246. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Meyer KD, Jaffrey SR. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat Rev Mol Cell Biol. 2014;15(5):313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Roundtree IA, Evans ME, Pan T, et al. Dynamic RNA Modifications in Gene Expression Regulation. Cell. 2017;169(7):1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Corley M, Burns MC, Yeo GW. How RNA-Binding Proteins Interact with RNA: molecules and Mechanisms. Mol Cell. 2020;78(1):9–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ding Y, Kwok CK, Tang Y, et al. Genome-wide profiling of in vivo RNA structure at single-nucleotide resolution using structure-seq. Nat Protoc. 2015;10(7):1050–1066. [DOI] [PubMed] [Google Scholar]
- [17].Kertesz M, Wan Y, Mazor E, et al. Genome-wide measurement of RNA secondary structure in yeast. Nature. 2010;467(7311):103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kwok CK, Tang Y, Assmann SM, et al. The RNA structurome: transcriptome-wide structure probing with next-generation sequencing. Trends Biochem Sci. 2015;40(4):221–232. [DOI] [PubMed] [Google Scholar]
- [19].Li F, Zheng Q, Ryvkin P, et al. Global analysis of RNA secondary structure in two metazoans. Cell Rep. 2012;1(1):69–82. . [DOI] [PubMed] [Google Scholar]
- [20].McGinnis JL, Liu Q, Lavender CA, et al. In-cell SHAPE reveals that free 30S ribosome subunits are in the inactive state. Proc Natl Acad Sci U S A. 2015;112(8):2425–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Smola MJ, Christy TW, Inoue K, et al. SHAPE reveals transcript-wide interactions, complex structural domains, and protein interactions across the Xist lncRNA in living cells. Proc Natl Acad Sci U S A. 2016;113(37):10322–10327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Smola MJ, Weeks KM. In-cell RNA structure probing with SHAPE-MaP. Nat Protoc. 2018;13(6):1181–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Spitale RC, Crisalli P, Flynn RA, et al. RNA SHAPE analysis in living cells. Nat Chem Biol. 2013;9(1):18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Val F, Costa FT, King L, et al. Tafenoquine for the prophylaxis, treatment and elimination of malaria: eagerness must meet prudence. Future Microbiol. 2019;14(15):1261–1279. [DOI] [PubMed] [Google Scholar]
- [25].Oakley MS, Gerald N, McCutchan TF, et al. Clinical and molecular aspects of malaria fever. Trends Parasitol. 2011;27(10):442–449. [DOI] [PubMed] [Google Scholar]
- [26].Zhang R, Chandramohanadas R, Lim CT, et al. Febrile Temperature Elevates the Expression of Phosphatidylserine on Plasmodium falciparum (FCR3CSA) Infected Red Blood Cell Surface Leading to Increased Cytoadhesion. Sci Rep. 2018;8(1):15022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chen Q, Schlichtherle M, Wahlgren M. Molecular aspects of severe malaria. Clin Microbiol Rev. 2000;13(3):439–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Miller LH, Ackerman HC, Su XZ, et al. Malaria biology and disease pathogenesis: insights for new treatments. Nat Med. 2013;19(2):156–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lim YB, Thingna J, Kong F, et al. Temperature-Induced Catch-Slip to Slip Bond Transit in Plasmodium falciparum-Infected Erythrocytes. Biophys J. 2020;118(1):105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Qi F, Frishman D. Melting temperature highlights functionally important RNA structure and sequence elements in yeast mRNA coding regions. Nucleic Acids Res. 2017;45(10):6109–6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wan Y, Qu K, Ouyang Z, et al. Genome-wide measurement of RNA folding energies. Mol Cell. 2012;48(2):169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Brower-Sinning R, Carter DM, Crevar CJ, et al. The role of RNA folding free energy in the evolution of the polymerase genes of the influenza A virus. Genome Biol. 2009;10(2):R18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Leach MD, Cowen LE. Surviving the heat of the moment: a fungal pathogens perspective. PLoS Pathog. 2013;9(3):e1003163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Meyer S, Carlson PD, Lucks JB. Characterizing the Structure-Function Relationship of a Naturally Occurring RNA Thermometer. Biochemistry. 2017;56(51):6629–6638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Coulson RM, Hall N, Ouzounis CA. Comparative genomics of transcriptional control in the human malaria parasite Plasmodium falciparum. Genome Res. 2004;14(8):1548–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hughes KR, Philip N, Starnes GL, et al. From cradle to grave: RNA biology in malaria parasites. Wiley Interdiscip Rev RNA. 2010;1(2):287–303. [DOI] [PubMed] [Google Scholar]
- [37].Le Roch KG, Johnson JR, Florens L, et al. Global analysis of transcript and protein levels across the Plasmodium falciparum life cycle. Genome Res. 2004;14(11):2308–2318. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Llinas M, DeRisi JL. Pernicious plans revealed: plasmodium falciparum genome wide expression analysis. Curr Opin Microbiol. 2004;7(4):382–387. [DOI] [PubMed] [Google Scholar]
- [39].Bozdech Z, Llinas M, Pulliam BL, et al. The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol. 2003;1(1):E5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Le Roch KG, Zhou Y, Blair PL, et al. Discovery of gene function by expression profiling of the malaria parasite life cycle. Science. 2003;301(5639):1503–1508. . [DOI] [PubMed] [Google Scholar]
- [41].Llinas M, Bozdech Z, Wong ED, et al. Comparative whole genome transcriptome analysis of three Plasmodium falciparum strains. Nucleic Acids Res. 2006;34(4):1166–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bunnik EM, Chung DW, Hamilton M, et al. Polysome profiling reveals translational control of gene expression in the human malaria parasite Plasmodium falciparum. Genome Biol. 2013;14(11):R128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Caro F, Ahyong V, Betegon M, et al. Genome-wide regulatory dynamics of translation in the Plasmodium falciparum asexual blood stages. Elife. 2014;3:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lu XM, Batugedara G, Lee M, et al. Nascent RNA sequencing reveals mechanisms of gene regulation in the human malaria parasite Plasmodium falciparum. Nucleic Acids Res. 2017;45(13):7825–7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Mair GR, Braks JA, Garver LS, et al. Regulation of sexual development of Plasmodium by translational repression. Science. 2006;313(5787):667–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mair GR, Lasonder E, Garver LS, et al. Universal features of post-transcriptional gene regulation are critical for Plasmodium zygote development. PLoS Pathog. 2010;6(2):e1000767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Otto TD, Wilinski D, Assefa S, et al. New insights into the blood-stage transcriptome of Plasmodium falciparum using RNA-Seq. Mol Microbiol. 2010;76(1):12–24. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Rios KT, Lindner SE. Protein-RNA interactions important for Plasmodium transmission. PLoS Pathog. 2019;15(12):e1008095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sims JS, Militello KT, Sims PA, et al. Patterns of gene-specific and total transcriptional activity during the Plasmodium falciparum intraerythrocytic developmental cycle. Eukaryot Cell. 2009;8(3):327–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zhang Q, Siegel TN, Martins RM, et al. Exonuclease-mediated degradation of nascent RNA silences genes linked to severe malaria. Nature. 2014b;513(7518):431–435. . [DOI] [PubMed] [Google Scholar]
- [51].Amit-Avraham I, Pozner G, Eshar S, et al. Antisense long noncoding RNAs regulate vargene activation in the malaria parasite Plasmodium falciparum. Proc Natl Acad Sci U S A. 2015;112(9):E982–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Amulic B, Salanti A, Lavstsen T, et al. An upstream open reading frame controls translation of var2csa, a gene implicated in placental malaria. PLoS Pathog. 2009;5(1):e1000256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Chan S, Frasch A, Mandava CS, et al. Regulation of PfEMP1-VAR2CSA translation by a Plasmodium translation-enhancing factor. Nat Microbiol. 2017;2(7):17068. . [DOI] [PubMed] [Google Scholar]
- [54].Reddy BP, Shrestha S, Hart KJ, et al. A bioinformatic survey of RNA-binding proteins in Plasmodium. BMC Genomics. 2015;16(1):890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Liu N, Dai Q, Zheng G, et al. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518(7540):560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Baumgarten S, Bryant JM, Sinha A, et al. Transcriptome-wide dynamics of extensive m(6)A mRNA methylation during Plasmodium falciparum blood-stage development. Nat Microbiol. 2019;4(12):2246–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Lopez-Barragan MJ, Lemieux J, Quinones M, et al. Directional gene expression and antisense transcripts in sexual and asexual stages of Plasmodium falciparum. BMC Genomics. 2011;12(1):587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Militello KT, Patel V, Chessler AD, et al. RNA polymerase II synthesizes antisense RNA in Plasmodium falciparum. RNA. 2005;11(4):365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Siegel TN, Hon CC, Zhang Q, et al. Strand-specific RNA-Seq reveals widespread and developmentally regulated transcription of natural antisense transcripts in Plasmodium falciparum. BMC Genomics. 2014;15(1):150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Epp C, Li F, Howitt CA, et al. Chromatin associated sense and antisense noncoding RNAs are transcribed from the var gene family of virulence genes of the malaria parasite Plasmodium falciparum. RNA. 2009;15(1):116–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Gardiner DL, Holt DC, Thomas EA, et al. Inhibition of Plasmodium falciparum clag9 gene function by antisense RNA. Mol Biochem Parasitol. 2000;110(1):33–41. [DOI] [PubMed] [Google Scholar]
- [62].Broadbent KM, Broadbent JC, Ribacke U, et al. Strand-specific RNA sequencing in Plasmodium falciparum malaria identifies developmentally regulated long non-coding RNA and circular RNA. BMC Genomics. 2015;16(1):454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Chakrabarti K, Pearson M, Grate L, et al. Structural RNAs of known and unknown function identified in malaria parasites by comparative genomics and RNA analysis. RNA. 2007;13(11):1923–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Mourier T, Carret C, Kyes S, et al. Genome-wide discovery and verification of novel structured RNAs in Plasmodium falciparum. Genome Res. 2008;18(2):281–292. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Vembar SS, Scherf A, Siegel TN. Noncoding RNAs as emerging regulators of Plasmodium falciparum virulence gene expression. Curr Opin Microbiol. 2014;20:153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Foley SW, Vandivier LE, Kuksa PP, et al. Transcriptome-wide measurement of plant RNA secondary structure. Curr Opin Plant Biol. 2015;27:36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Rouskin S, Zubradt M, Washietl S, et al. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature. 2014;505(7485):701–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Wan Y, Kertesz M, Spitale RC, et al. Understanding the transcriptome through RNA structure. Nat Rev Genet. 2011;12(9):641–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Wan Y, Qu K, Zhang QC, et al. Landscape and variation of RNA secondary structure across the human transcriptome. Nature. 2014;505(7485):706–709. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Rogers MJ, Gutell RR, Damberger SH, et al. Structural features of the large subunit rRNA expressed in Plasmodium falciparum sporozoites that distinguish it from the asexually expressed subunit rRNA. RNA. 1996;2(2):134–145. [PMC free article] [PubMed] [Google Scholar]
- [71].Duffy S, Avery VM. Routine In Vitro Culture of Plasmodium falciparum: experimental Consequences? Trends Parasitol. 2018;34(7):564–575. [DOI] [PubMed] [Google Scholar]
- [72].Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193(4254):673–675. [DOI] [PubMed] [Google Scholar]
- [73].Chen S, Zhou Y, Chen Y, et al. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–930. [DOI] [PubMed] [Google Scholar]
- [76].Chappell L, Ross P, Orchard L, et al. Refining the transcriptome of the human malaria parasite Plasmodium falciparum using amplification-free RNA-seq. BMC Genomics. 2020;21(1):395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Aurrecoechea C, Brestelli J, Brunk BP, et al. PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res. 2009;37(Database):D539–543. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Alexa A, Rahnenfuhrer J, Lengauer T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics. 2006;22(13):1600–1607. [DOI] [PubMed] [Google Scholar]
- [80].Xu ZZ, Mathews DH. Experiment-Assisted Secondary Structure Prediction with RNAstructure. Methods Mol Biol. 2016;1490:163–176. [DOI] [PubMed] [Google Scholar]
- [81].Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009;136(4):642–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Baum J, Papenfuss AT, Mair GR, et al. Molecular genetics and comparative genomics reveal RNAi is not functional in malaria parasites. Nucleic Acids Res. 2009;37(11):3788–3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Lu Z, Gong J, Zhang QC. PARIS: psoralen Analysis of RNA Interactions and Structures with High Throughput and Resolution. Methods Mol Biol. 2018;1649:59–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Piao M, Sun L, Zhang QC. RNA Regulations and Functions Decoded by Transcriptome-wide RNA Structure Probing. Genomics Proteomics Bioinformatics. 2017;15(5):267–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Zheng Q, Ryvkin P, Li F, et al. Genome-wide double-stranded RNA sequencing reveals the functional significance of base-paired RNAs in Arabidopsis. PLoS Genet. 2010;6(9):e1001141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Gardner MJ, Hall N, Fung E, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419(6906):498–511. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Painter HJ, Chung NC, Sebastian A, et al. Genome-wide real-time in vivo transcriptional dynamics during Plasmodium falciparum blood-stage development. Nat Commun. 2018;9(1):2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Mauger DM, Cabral BJ, Presnyak V, et al. mRNA structure regulates protein expression through changes in functional half-life. Proc Natl Acad Sci U S A. 2019;116(48):24075–24083. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Brachman EE, Kmiec EB. DNA replication and transcription direct a DNA strand bias in the process of targeted gene repair in mammalian cells. J Cell Sci. 2004;117(17):3867–3874. [DOI] [PubMed] [Google Scholar]
- [90].Liu L, Rice MC, Drury M, et al. Strand bias in targeted gene repair is influenced by transcriptional activity. Mol Cell Biol. 2002;22(11):3852–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Strick TR, Savery NJ. Understanding bias in DNA repair. Proc Natl Acad Sci U S A. 2017;114(11):2791–2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Jacobs E, Mills JD, Janitz M. The role of RNA structure in posttranscriptional regulation of gene expression. J Genet Genomics. 2012;39(10):535–543. [DOI] [PubMed] [Google Scholar]
- [93].Leppek K, Das R, Barna M. Functional 5ʹ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat Rev Mol Cell Biol. 2018;19(3):158–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Halic M, Becker T, Pool MR, et al. Structure of the signal recognition particle interacting with the elongation-arrested ribosome. Nature. 2004;427(6977):808–814. [DOI] [PubMed] [Google Scholar]
- [95].Davila Lopez M, Samuelsson T. Early evolution of histone mRNA 3ʹ end processing. RNA. 2008;14(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Zhang J, Tan D, DeRose EF, et al. Molecular mechanisms for the regulation of histone mRNA stem-loop-binding protein by phosphorylation. Proc Natl Acad Sci U S A. 2014a;111(29):E2937–2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Ahmed R, Duncan RF. Translational regulation of Hsp90 mRNA. AUG-proximal 5ʹ-untranslated region elements essential for preferential heat shock translation. J Biol Chem. 2004;279(48):49919–49930. [DOI] [PubMed] [Google Scholar]
- [98].Vembar SS, Droll D, Scherf A. Translational regulation in blood stages of the malaria parasite Plasmodium spp. : systems-wide studies pave the way. Wiley Interdiscip Rev RNA. 2016;7(6):772–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Balaji S, Babu MM, Iyer LM, et al. Discovery of the principal specific transcription factors of Apicomplexa and their implication for the evolution of the AP2-integrase DNA binding domains. Nucleic Acids Res. 2005;33(13):3994–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Toenhake CG, Fraschka SA, Vijayabaskar MS, et al. Chromatin Accessibility-Based Characterization of the Gene Regulatory Network Underlying Plasmodium falciparum Blood-Stage Development. Cell Host Microbe. 2018;23(4):e559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Siwo GH, Tan A, Button-Simons KA, et al. Predicting functional and regulatory divergence of a drug resistance transporter gene in the human malaria parasite. BMC Genomics. 2015;16(1):115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Bunnik EM, Batugedara G, Saraf A, et al. The mRNA-bound proteome of the human malaria parasite Plasmodium falciparum. Genome Biol. 2016;17(1):147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Goyal M, Alam A, Iqbal MS, et al. Identification and molecular characterization of an Alba-family protein from human malaria parasite Plasmodium falciparum. Nucleic Acids Res. 2012;40(3):1174–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Mamillapalli A, Pattnaik P, Sharma M, et al. Sequence polymorphisms in the receptor-binding domain of Plasmodium falciparum EBA-175: implications for malaria vaccine development. Mol Biochem Parasitol. 2006;146(1):120–123. [DOI] [PubMed] [Google Scholar]
- [105].Collins CR, Hackett F, Atid J, et al. The Plasmodium falciparum pseudoprotease SERA5 regulates the kinetics and efficiency of malaria parasite egress from host erythrocytes. PLoS Pathog. 2017;13(7):e1006453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Favaloro JM, Coppel RL, Corcoran LM, et al. Structure of the RESA gene of Pias falciparum. Nucleic Acids Res. 1986;14(21):8265–8277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Pei X, Guo X, Coppel R, et al. The ring-infected erythrocyte surface antigen (RESA) of Plasmodium falciparum stabilizes spectrin tetramers and suppresses further invasion. Blood. 2007;110(3):1036–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Clingman CC, Ryder SP. Metabolite sensing in eukaryotic mRNA biology. Wiley Interdiscip Rev RNA. 2013;4(4):387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Sudarsan N, Barrick JE, Breaker RR. Metabolite-binding RNA domains are present in the genes of eukaryotes. RNA. 2003;9(6):644–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Cheviet T, Lefebvre-Tournier I, Wein S, et al. Plasmodium Purine Metabolism and Its Inhibition by Nucleoside and Nucleotide Analogues. J Med Chem. 2019;62(18):8365–8391. [DOI] [PubMed] [Google Scholar]
- [111].Dominguez D, Freese P, Alexis MS, et al. Sequence, Structure, and Context Preferences of Human RNA Binding Proteins. Mol Cell. 2018;70(5):e859. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Rabani M, Kertesz M, Segal E. Computational prediction of RNA structural motifs involved in posttranscriptional regulatory processes. Proc Natl Acad Sci U S A. 2008;105(39):14885–14890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Ao W, Gaudet J, Kent WJ, et al. Environmentally induced foregut remodeling by PHA-4/FoxA and DAF-12/NHR. Science. 2004;305(5691):1743–1746. [DOI] [PubMed] [Google Scholar]
- [114].Chen C, Zhang H, Broitman SL, et al. Dynamics of translation by single ribosomes through mRNA secondary structures. Nat Struct Mol Biol. 2013;20(5):582–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Faure G, Ogurtsov AY, Shabalina SA, et al. Role of mRNA structure in the control of protein folding. Nucleic Acids Res. 2016;44(22):10898–10911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Faghihi MA, Wahlestedt C. Regulatory roles of natural antisense transcripts. Nat Rev Mol Cell Biol. 2009;10(9):637–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Gunasekera AM, Patankar S, Schug J, et al. Widespread distribution of antisense transcripts in the Plasmodium falciparum genome. Mol Biochem Parasitol. 2004;136(1):35–42. [DOI] [PubMed] [Google Scholar]
- [118].Jiang L, Mu J, Zhang Q, et al. PfSETvs methylation of histone H3K36 represses virulence genes in Plasmodium falciparum. Nature. 2013;499(7457):223–227. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Adjalley SH, Chabbert CD, Klaus B, et al. Landscape and Dynamics of Transcription Initiation in the Malaria Parasite Plasmodium falciparum. Cell Rep. 2016;14(10):2463–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Sorber K, Dimon MT, DeRisi JL. RNA-Seq analysis of splicing in Plasmodium falciparum uncovers new splice junctions, alternative splicing and splicing of antisense transcripts. Nucleic Acids Res. 2011;39(9):3820–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Ralph SA, Bischoff E, Mattei D, et al. Transcriptome analysis of antigenic variation in Plasmodium falciparum–var silencing is not dependent on antisense RNA. Genome Biol. 2005;6(11):R93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Mao Y, Liu H, Liu Y, et al. Deciphering the rules by which dynamics of mRNA secondary structure affect translation efficiency in Saccharomyces cerevisiae. Nucleic Acids Res. 2014;42(8):4813–4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Rodnina MV. The ribosome in action: tuning of translational efficiency and protein folding. Protein Sci. 2016;25(8):1390–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Francoeur AM, Gritzmacher CA, Peebles CL, et al. Synthesis of small nuclear ribonucleoprotein particles by the malarial parasite Plasmodium falciparum. Proc Natl Acad Sci U S A. 1985;82(11):3635–3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Waters AP, White W, McCutchan TF. The structure of the large subunit rRNA expressed in blood stages of Plasmodium falciparum. Mol Biochem Parasitol. 1995;72(1–2):227–237. [DOI] [PubMed] [Google Scholar]
- [126].Andreasson JOL, Savinov A, Block SM, et al. Comprehensive sequence-to-function mapping of cofactor-dependent RNA catalysis in the glmS ribozyme. Nat Commun. 2020;11(1):1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Kato Y, Kuwabara T, Warashina M, et al. Relationships between the activities in vitro and in vivo of various kinds of ribozyme and their intracellular localization in mammalian cells. J Biol Chem. 2001;276(18):15378–15385. [DOI] [PubMed] [Google Scholar]
- [128].Leamy KA, Assmann SM, Mathews DH, et al. Bridging the gap between in vitro and in vivo RNA folding. Q Rev Biophys. 2016;49:e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Cruz JA, Westhof E. The dynamic landscapes of RNA architecture. Cell. 2009;136(4):604–609. [DOI] [PubMed] [Google Scholar]
- [130].Dibrov SM, Parsons J, Hermann T. A model for the study of ligand binding to the ribosomal RNA helix h44. Nucleic Acids Res. 2010;38(13):4458–4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Watters KE, Yu AM, Strobel EJ, et al. Characterizing RNA structures in vitro and in vivo with selective 2ʹ-hydroxyl acylation analyzed by primer extension sequencing (SHAPE-Seq). Methods. 2016;103:34–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Busan S, Weidmann CA, Sengupta A, et al. Guidelines for SHAPE Reagent Choice and Detection Strategy for RNA Structure Probing Studies. Biochemistry. 2019;58(23):2655–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Fessler AB, Dey A, Finis DS, et al. Innately Water-Soluble Isatoic Anhydrides with Modulated Reactivities for RNA SHAPE Analysis. Bioconjug Chem. 2020;31(3):884–888. [DOI] [PubMed] [Google Scholar]
- [134].Lee B, Flynn RA, Kadina A, et al. Comparison of SHAPE reagents for mapping RNA structures inside living cells. RNA. 2017;23(2):169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Mitchell D 3rd, Assmann SM, Bevilacqua PC. Probing RNA structure in vivo. Curr Opin Struct Biol. 2019;59:151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Mustoe AM, Busan S, Rice GM, et al. Pervasive Regulatory Functions of mRNA Structure Revealed by High-Resolution SHAPE Probing. Cell. 2018a;173(1):e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Courel M, Clement Y, Bossevain C, et al. GC content shapes mRNA storage and decay in human cells. Elife. 2019;8:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Beaudoin JD, Novoa EM, Vejnar CE, et al. Analyses of mRNA structure dynamics identify embryonic gene regulatory programs. Nat Struct Mol Biol. 2018;25(8):677–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Mao Y, Dong L, Liu XM, et al. m(6)A in mRNA coding regions promotes translation via the RNA helicase-containing YTHDC2. Nat Commun. 2019;10(1):5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Cui L, Lindner S, Miao J. Translational regulation during stage transitions in malaria parasites. Ann N Y Acad Sci. 2015;1342(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Pardi N, Hogan MJ, Porter FW, et al. mRNA vaccines - a new era in vaccinology. Nat Rev Drug Discov. 2018;17(4):261–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Robinson JT, Thorvaldsdóttir H, Winckler W, et al. Integrative Genomics Viewer. Nat Biotechnol. 2011;29(1):24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Ackermann M, Strimmer K. et al. A general modular framework for gene set enrichment analysis. BMC Bioinformatics. 2009;10:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Adjalley SH, Chabbert CD, Klaus B, Pelechano V, Steinmetz LM.2016. Landscape and Dynamics of Transcription Initiation in the Malaria Parasite Plasmodium falciparum. Cell Rep 14:2463–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.