

ABSTRACT
Autophagosome formation requires PROPPIN/WIPI proteins and monophosphorylated phosphoinositides, such as phosphatidylinositol-3-phosphate (PtdIns3P) or PtdIns5P. This process occurs in association with mammalian endosomes, where the PROPPIN WIPI1 has additional, undefined roles in vesicular traffic. To explore whether these functions are interconnected, we dissected routes and subreactions of endosomal trafficking requiring WIPI1. WIPI1 specifically acts in the formation and fission of tubulo-vesicular endosomal transport carriers. This activity supports the PtdIns(3,5)P2-dependent transport of endosomal cargo toward the plasma membrane, Golgi, and lysosomes, suggesting a general role of WIPI1 in endosomal protein exit. Three features differentiate the endosomal and macroautophagic/autophagic activities of WIPI1: phosphoinositide binding site II, the requirement for PtdIns(3,5)P2, and bilayer deformation through a conserved amphipathic α-helix. Their inactivation preserves autophagy but leads to a strong enlargement of endosomes, which accumulate micrometer-long endosomal membrane tubules carrying cargo proteins. WIPI1 thus supports autophagy and protein exit from endosomes by different modes of action. We propose that the type of phosphoinositides occupying its two lipid binding sites, the most unusual feature of PROPPIN/WIPI family proteins, switches between these effector functions.
Abbreviations: EGF: epidermal growth factorEGFR: epidermal growth factor receptorKD: knockdownKO: knockoutPtdIns3P: phosphatidylinositol-3-phosphatePtdIns5P: phosphatidylinositol-5-phosphatePtdIns(3,5)P2: phosphatidylinositol-3,5-bisphosphateTF: transferrinTFRC: transferrin receptorWT: wildtype
KEYWORDS: Autophagy, autophagosome, EGF receptor, endosome, lysosome, PROPPIN, transferrin receptor, WIPI proteins, vacuole, endosomal transport carrier
Introduction
Autophagy depends on a complex protein machinery that is recruited to phagophores, the sites of autophagosome formation. It allows the growth of a phagophore at this site, recruits cargo into the nascent autophagosome and mediates its closure [1]. The formation of autophagosomes depends on phosphatidylinositol-3-phosphate (PtdIns3P) or phosphatidylinositol-5-phosphate (PtdIns5P) [2–5]. Phosphatidylinositol-3,5-bisphosphate (PI [3,5]P2) appears to be dispensable for formation of autophagosomes but supports their fusion with lysosomes [6–9]. Important binders of these lipids at the phagophore are PROPPIN/WIPI proteins, beta-propellers binding phosphoinositides. They constitute a family of conserved proteins with three members present in yeast (Atg18, Atg21, and Hsv2), and four members in mammals (WIPI1, WIPI2, WDR45B/WIPI3 and WDR45/WIPI4) [10,11]. During autophagy, WIPI1 and WIPI2 localize to autophagic membranes [12,13]. WIPI2 interacts with several factors driving the formation of autophagosomes, such as ATG16L1 and ATG7 [14–17]. Its knockdown inhibits autophagy. WIPI1 interacts with WIPI2 and ATG16L1, but the WIPI1-ATG16L1 interaction appears to be weak and it is was proposed that it might occur indirectly through WIPI2 [17,18]. WIPI1 knockdown reduces virus-induced autophagy, suggesting that it has an important function at the phagophore [19]. WDR45B/WIPI3 and WDR45/WIPI4 were proposed to be part of a signaling complex involved in the induction autophagy [18] and to tether the phagophore membrane to the ER [20–22]. PROPPINs bind the phagophore membrane through two lipid binding sites, which have affinities for PtdIns3P, PtdIns5P and PtdIns(3,5)P2 [3,10,12,23–26]. A major open question is why there are two lipid binding sites, whether they serve as mere membrane anchors, or whether they can confer distinct functions to the WIPI proteins, which might then be defined by the type of phosphoinositide bound.
Aside from their location at phagophores, WIPI1 and WIPI2 also associate with endosomes [27,28]. Here, WIPI2 interacts with the endosomal marker RAB11, a RAB-GTPase that also has an impact on autophagosome formation [29]. Futher molecular links between the autophagic machinery and endosomes were provided by the WIPI1 and WIPI2 interactors ATG16L1 and ATG7, which not only promote autophagy but also influence EGFR (epidermal growth factor receptor) trafficking. This led to the suggestion that autophagy might improve endosomal sorting capacity by removing damaged endosomes [30]. Furthermore, ATG9, a membrane-integral component of the autophagic machinery, was proposed to shuttle between endosomal compartments and the phagophore [28]. Thus, it is possible that endosomal trafficking and autophagosome biogenesis are intimately connected and share some components, among them the WIPI proteins, but it is not known whether these proteins perform equivalent activities in both processes or whether their effects on endosomal sorting are mere consequences of the autophagy of endosomes.
Cells internalize plasma membrane components and extracellular material, such as receptors, transporters, growth factors and solute molecules, by endocytosis. Endocytic vesicles pinch off from the plasma membrane and deliver their cargoes to early endosomes [31]. From there, they can be channeled into several trafficking routes: They can recycle back to the plasma membrane [32–34], move to juxtanuclear endocytic recycling compartments, head for the trans-Golgi network [35], or they can be sorted into multivesicular bodies and degradative lysosomal compartments [36–38]. Arrival of membrane from the cell surface must be balanced by endosomal exit and recycling pathways. These employ tubulo-vesicular endosomal transport carriers and allow cells to control the size and composition of endosomes as well as their signaling properties [39–41].
Important steps in the generation of transport carriers are cargo recruitment, the shaping of the membrane into tubular-vesicular structures, and its constriction and fission. Numerous components that are required for the formation of endosomal transport carriers have been identified, such as the retromer, retriever and CCC complexes, sorting nexins and the WASH complex [42–48]. While structural analyses of retromer have provided us with first impressions of how coats might shape endosomal transport carriers [49,50], their detachment from the endosome has remained very poorly understood. Several factors could contribute. The formation of tubular membranes generates membrane tension, which itself can favor fission [51,52]. In addition, fission can be promoted by the generation of force and friction through the cytoskeleton [43–45,53], and by dedicated fission factors, such as dynamin-like GTPases [54]. During the division of yeast vacuoles, the PROPPIN Atg18 was proposed to drive membrane fission [55,56]. Atg18 carries a hydrophobic CD-loop on blade 6, which must insert into the membrane in order to allow membrane binding [23–26,57,58]. Concomitant folding of this loop into an amphipathic α-helix allows Atg18 to drive membrane fission, e.g. of synthetic giant unilamellar liposomes [55]. Since the potential to form an amphipathic α-helix is conserved among PROPPINs, also other members of this family might carry membrane fission activity. This, however, has not yet been investigated and it has remained unclear whether WIPI proteins provide such an activity on endosomal trafficking pathways.
Inactivation of WIPI1 changes the distribution of M6PR (mannose-6-phosphate receptor, cation dependent) in mammalian cells cell and leads to more elaborated early endosomes, suggesting a function in endosomal membrane traffic [27]. However, trafficking reactions consist of multiple steps, which employ different protein machineries, and it has not been resolved whether WIPI1 participates in the formation, fission, fusion or displacement of endosomal transport carriers. The formation of such carriers depends on PtdIns(3,5)P2, which was proposed to promote their fission [59], but the responsible PtdIns(3,5)P2 effectors have not been identified. WIPI1 is a candidate, because it binds PtdIns3P, PtdIns(3,5)P2, and PtdIns(5)P [3,25,27,60,61]. The lipid binding sites of PROPPINs have often been inactivated by substituting two conserved arginine residues within an FRRG motif. Since this motif participates in both lipid-binding sites [10,12,27,62,63], these substitutions cannot reveal whether the two lipid binding sites provide distinct functions to the protein. Purified PROPPINs distinguish only weakly between PtdIns3P and PtdIns(3,5)P2 at the level of their binding affinities [23–25,57,64]. Since PtdIns3P is much more abundant in cells than PtdIns(3,5)P2 it has thus remained unclear whether WIPI proteins mediate PtdIns(3,5)P2 effects in endosomal membrane traffic and which mechanistic role they might perform.
In this study, we used WIPI1 as a model to dissect the role of PROPPINs in endosomal membrane traffic, which allowed us to differentiate it from that in autophagy. We identified the trafficking routes affected by WIPI1, determined the subreaction of endosomal trafficking requiring it, and tested whether endosomal trafficking utilizes the same molecular features of WIPI1 as the formation of autophagosomes.
Results
Enlargement of endo-lysosomal compartments in WIPI1-deficient cells
We analyzed the effect of WIPI1 on the localization of two compartment markers, the EEA1 (early endosome antigen 1) and the late endosomal and lysosomal LAMP1 (lysosomal associated membrane protein 1). In line with previous findings [27], siRNA-mediated knockdown of endogenous WIPI1 (WIPI1-KD) led to an increase in both number and size of EEA1-positive early endosomal compartments, as judged by immunofluorescence analysis (Figureure. 1A,B). Western blots showed a corresponding 30–40% increase of EEA1 in these cells (Figure. S1 A). WIPI1 depletion increased also the abundance of LAMP1-positive late endosomes and lysosomes, as assessed by immunofluorescence (Figure 1A,C), and led to a 30% increase in LAMP1 signals on immunoblots (Figure. S1 B). The EEA1 and LAMP1 phenotypes of WIPI1-KD cells were rescued by expressing siRNA-resistant WIPI1 in them, excluding off-target effects of the siRNA. Rescue was not observed with WIPI1[FAAG] [27,63,65], a version of WIPI1 in which two arginines that are critical for both lipid-binding sites have been substituted by alanine (Figure 1D,E). HK2 cells from which WIPI1 had been entirely removed through CRISPR-Cas9-mediated knockout (WIPI1 KO) showed phenotypes that were similar to those of WIPI1 KD cells, but slightly stronger (Figure 1A).
Figure 1.

Loss of WIPI1 alters the endo-lysosomal compartment. (A) Representative images of control (CTR), WIPI1-KD and WIPI1 KO HK2 cells immuno-stained with an anti-EEA1 and with anti-LAMP1 antibodies (scale bars: 10 μm). (B-C) Quantification of cells from (A). The average number per cell of enlarged (diameter >2 μm2) EEA1-positive (B) or LAMP1-positive (C) structures, and of all LAMP1- or EEA1-positive structures was determined for CTR and WIPI1 KD cells. The integrated fluorescence intensity per cell was also measured (right panels). Mean values ± s.d. are shown (D) Rescue of WIPI1 knockdown. Experiment as in (A), with cells expressing an siRNA-resistant allele of wild-type (WIPI1) or of a version with substitutions in the lipid binding sites (WIPI1[FAAG]). EEA1 (red), LAMP1 (magenta) and EGFP-WIPI1 (green) are shown. Scale bars: 10 μm. (E) Quantification of EEA1- and LAMP1-fluorescence from (D). Images were acquired and analyzed as in (B). For (B-C) and (E), 180 cells per condition, pooled from 3 independent experiments, were analyzed. Mean values ± s.d are shown
WIPI1 promotes protein exit from early endosomes toward lysosomes, the Golgi and the plasma membrane
We investigated the requirement of WIPI1 for the recycling pathway from early endosomes to the plasma membrane using TFRC (transferrin receptor) [33,66], and for transfer from early to late endosomes and lysosomes using EGFR [67]. In pulse-chase experiments, control and WIPI1 KO cells were incubated with TF (transferrin) for 60 min at 4°C. This treatment loads TFRC at the surface but prevents its internalization, which is confirmed by the fact that abundant TFRC staining appeared in immunofluorescence analysis of non-permeabilized cells, but hardly any staining was detectable after the plasma membrane had been detergent-permeabilized (Figure 2A,B). The surface of WIPI1 KO cells bound less TF than that of control cells, suggesting that the knockouts carry less TFRC at the cell surface. Shifting TF-loaded cells to 37°C triggers internalization of loaded TFRC (Figure 2A). WIPI1 KO cells internalized bound TF at the same rate as control cells, suggesting that endocytosis of TFRC does not depend on WIPI1. Despite the lower initial presence of TFRC at the surface of WIPI1 KO cells, these cells showed more intracellular TFRC than control cells at the later time points (Figure 2A,B). This suggests that TFRC accumulates inside WIPI1 KO cells rather than being efficiently recycled back to the cell surface.
Figure 2.
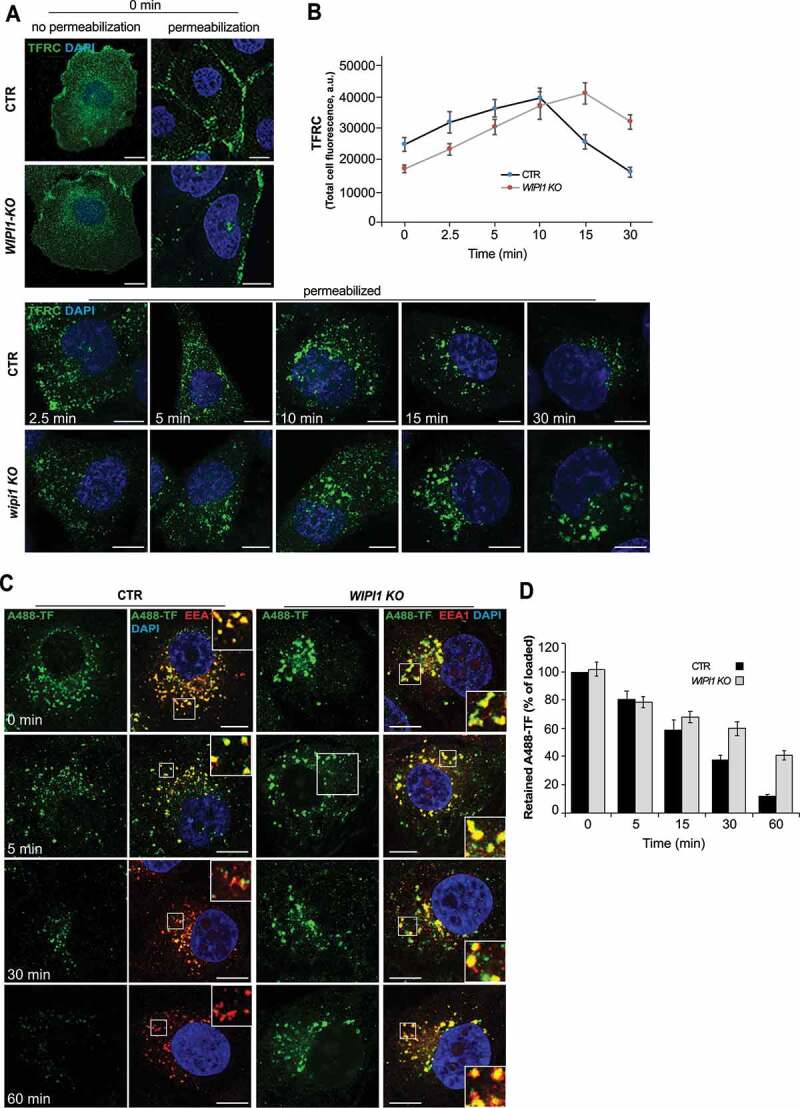
WIPI1 does not affect initial TF uptake, but it is crucial for efficient recycling of TFRC. (A) TFRC surface binding. Control and WIPI1 KO cells were serum-starved for 1 h, washed twice in cold PBS with 1% BSA and incubated for 1 h on ice with 50 μg/ml of transferrin. Cells were transferred to 37°C for the indicated periods of time, acid washed and fixed. Fixed cells were stained with antibodies to TFRC (green) and with DAPI (blue), with detergent permeabilization where indicated. Scale bars: 10 μm. (B) Quantification of TFRC-immunofluorescence in permeabilized cells from (a). Regions of interest (ROIs) corresponding to each cell and in some regions outside the cells (background) were manually defined using ImageJ software. Total cell fluorescence was integrated and corrected for background fluorescence. 195 cells per condition, stemming from 3 independent experiments, were analyzed. Mean values ± s.d. (C) TF recycling. Control and WIPI1 KO cells were serum-starved for 1 h and washed twice in cold PBS with 1% BSA. Cells were then incubated at 37°C for 1 h to load them with 100 μg/ml Alexa-Fluor-488-TF (green), followed by a chase at 37°C in fresh complete HEPES-buffered medium. After the indicated periods of time, cells were acid-washed, fixed and stained with DAPI (blue) and antibodies to EEA1 (red). Scale bars: 10 μm. Insets show enlargements of boxed areas. (D) Quantification. TF-fluorescence in the cells from (C) was integrated as described in (B) and expressed as percentage of the control at 0 min of chase. Data are mean values ± s.d.; (n = 3 independent experiments). 100 cells per condition (CTR and WIPI1 KO) were quantified for each experiment
In order to assess the rate of TFRC recycling from endosomes, cells were incubated with Alexa Fluor 488-conjugated TF at 37°C for 60 min, which allows to load the internal compartments with it (Figure 2C,D). Non-internalized TF was removed by washing and the cells were chased in TF-free medium at 37 °C. While control cells showed a progressive decrease in the TF signal down to 10%, due to recycling of the internalized protein back to the cell surface, from where it dissociates, WIPI1 KO cells retained almost 50% of TF in enlarged EEA1-positive endosomes.
We used siRNA-mediated knockdowns to test whether TF recycling required also the other three WIPI-proteins as well as Atg2 and Atg16L1, proteins that interact strongly with WIPI2 and weakly or indirectly with WIPI1 [17,18]. The knockdown of all these factors was efficient, reducing their protein amounts by 85–95% (Figure 3A,B). In contrast to the depletion of WIPI1, which reduced recycling of TF to the surface by 50%, depletion of WIPI2, WDR45B/WIPI3, WDR45/WIPI4, ATG2 or ATG16L1 showed only minor effects (Figure 3C,D). These cells recycled 80–90% of TF, close to the value of control cells, which recycled 92% back to the cell surface. Enlargement of endosomal compartments, which is very striking after WIPI1 knockdown, was only weakly evident after WIPI2 knockdown and not observed in the other knockdowns. Thus, WIPI1 promotes the exit of TFRC from early endosomes and its recycling to the plasma membrane. This endosomal activity appears to be largely independent of the other WIPI isoforms and their interactors, such as ATG2 and ATG16L1.
Figure 3.

Influence of the knockdown of WIPI proteins, ATG16L1 and ATG2 on TF recycling. (A) Effects of siRNA treatment on protein levels. HK-2 cells were treated with non-targeting siRNA (CTR) or with siRNA pools to knock down WIPI1, WIPI2, WDR45B/WIPI3, WDR45/WIPI4, ATG2 and ATG16L1 for 72 h. Cell lysates (50 μg/sample) were analyzed by SDS-PAGE and immunoblotted using the indicated antibodies. TUBA/α-tubulin served as a loading control. A representative blot is shown. (B) Quantification of protein levels from 3 independent experiments as in (A). The levels of all proteins were normalized to TUBA/α-tubulin and are expressed as percentage of the CTR signal. (C) TF recycling. Control and cells depleted of the indicated proteins were serum-starved for 1 h and washed twice in cold PBS with 1% BSA. Cells were then incubated at 37°C for 1 h to load them with 100 μg/ml Alexa Fluor 488-TF (green), followed by a chase at 37°C in fresh complete HEPES-buffered medium. After 1 hour, cells were acid-washed, fixed and stained with DAPI (blue) and antibodies to EEA1 (red). Scale bars: 10 μm. (D) Quantification of TF recycling. TF-fluorescence in the cells from (A) was integrated as described in Figure 2D and expressed as percentage of the control at 0 min of chase. Data are mean values ± s.d.; (n = 3 independent experiments). 100 cells per condition were quantified for each experiment
In contrast to these observations, an earlier study proposed that the autophagic machinery, including the WIPI proteins and ATG16L1, promotes endosomal trafficking by autophagic removal of damaged endosomes [30]. This discrepancy probably reflects differences in the approach. Fraser et al. performed all their assays in starving cells, where autophagy is crucial for vitality and survival, whereas we analyzed growing cells in rich media, and in a different cell line. Thus, our observations should not be considered as contradictory to this study.
We next assayed transport from endosomes to the biosynthetic/secretory pathway through Shiga toxin B-subunit. This protein transits from the plasma membrane to the ER via early endosomes and the Golgi apparatus, circumventing the late endocytic pathway [68]. Cy3-labeled Shiga toxin was accumulated in early endosomes by incubating the cells at 19.5°C. Its chase was triggered by shifting the cells to 37°C. These incubations had been performed in the presence of Alexa Fluor488-conjugated TF to label early and recycling endosomes. Initially, TF and Shiga toxin colocalized extensively. But already after 10 min of chase, most of the Shiga toxin in control cells had moved to compartments showing the typical perinuclear morphology of the Golgi [68], while much of the TF had been recycled back to the cell surface. After 30 min, virtually no Shiga toxin remained in small endosomal structures. By contrast, WIPI1 KO cells showed a strong delay in the arrival of Shiga toxin to the Golgi complex and 50% of colocalization between Shiga toxin B-subunit and TF was retained even after 30 min of chase at 37°C (Figure 4A-C). Equivalent experiments, in which cells had been fixed at different time-points of this pulse-chase regime, allowed us to confirm this through immunofluorescence and colocalization with additional markers (Figure. S2 A-C). During the accumulation phase at 19.5°C, Shiga toxin colocalized with the endosomal markers SNX1 (sorting nexin 1) and TFRC (Figure. S2 A, C). After 30 min of chase, Shiga toxin in control cells colocalized almost completely with the Golgi marker beta-1,4-galactosyltransferase (B4GALT1), whereas more than 50% of Shiga toxin had remained outside the Golgi in WIPI1 KO cells (Figure. S2 B-C). This fraction of Shiga toxin had not been rerouted to late endosomes or lysosomes, because it showed virtually no colocalization with LAMP1 (Figure. S2 B-C).
Figure 4.
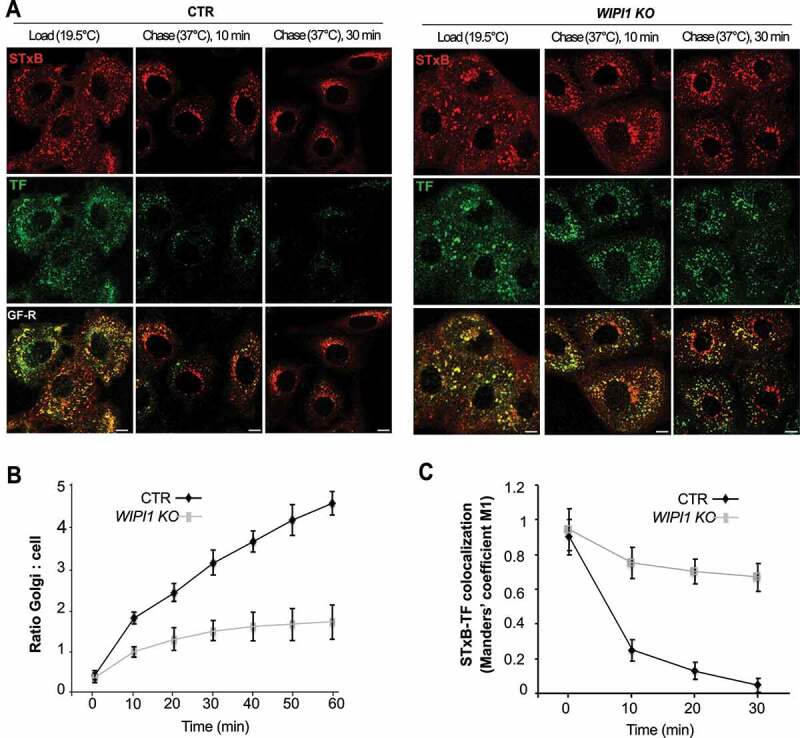
WIPI1 promotes transfer of STxB from early/recycling endosomes to the Golgi. (A) Confocal microscopy of living HK2 cells. Alexa Fluor 488 TF and Cy3-StxB were bound to control and WIPI1 KO cells for 30 min on ice, internalized by incubating cells at 19.5°C for 45 min (LOAD), and finally chased to the Golgi by shifting cells to 37°C for 0–30 min. Scale bars: 10 μm. (B) Quantification of Golgi localized STxB in images from (A). The ratio of average Golgi-associated fluorescence over the average total cell-associated fluorescence is represented as a function of incubation time at 37°C. Data are means ±s.d. from 3 independent experiments. Number of cells quantified: Control = 250; WIPI1 KO = 245 (C) Colocalization of TF and STxB was quantified in images from (A) at different time points after the shift to 37°C. Values are the mean ± s.d. from 3 independent experiments. Number of cells quantified: Control = 240; WIPI1 KO = 220
We analyzed the impact of WIPI1 on cargo transfer from early endosomes to degradative compartments through EGFR. Cells were serum-starved for 24 h, stimulated with EGF and analyzed by immunofluorescence. 15 min after EGF addition, more than 80% of EGFR-positive structures colocalized with the early endosomal marker EEA1 in control cells (Figure 5A,B). Between 30 and 60 min, colocalization with EEA1 gradually decreased, whereas the overlap of EGFR with LAMP1-positive structures increased, indicating the arrival of EGFR at degradative compartments. In WIPI1 KO cells, EGFR reached early endosomes similarly as in control cells within 15 min, but it stayed there and did not colocalize with LAMP1 at later time points. These results were confirmed by western blot analysis. Whereas control cells degraded 80% of EGFR within 60 min after EGF addition, WIPI1 KO cells could not degrade EGFR (Figure 5C-D). Stabilization of EGFR is probably not due to reduced proteolytic capacity of the late endosomal/lysosomal compartments. This is suggested by control experiments with lysotracker, which probes the acidification that is crucial for full proteolytic activity of lysosomes, and with BZiPAR, a probe for lysosomal protease activity in living cells [69]. Staining with LysoTracker and BZiPAR did not reveal significant differences between WIPI1 KO and control cells (Figure. S3 A, B), but the BZiPAR signal was absent when lysosomal hydrolase activity was quenched by the protease inhibitor chymostatin. This suggests that also the transfer of EGFR from early/sorting endosomes to degradative compartments requires WIPI1. WIPI1 is hence involved in multiple protein exit pathways from endosomes.
Figure 5.
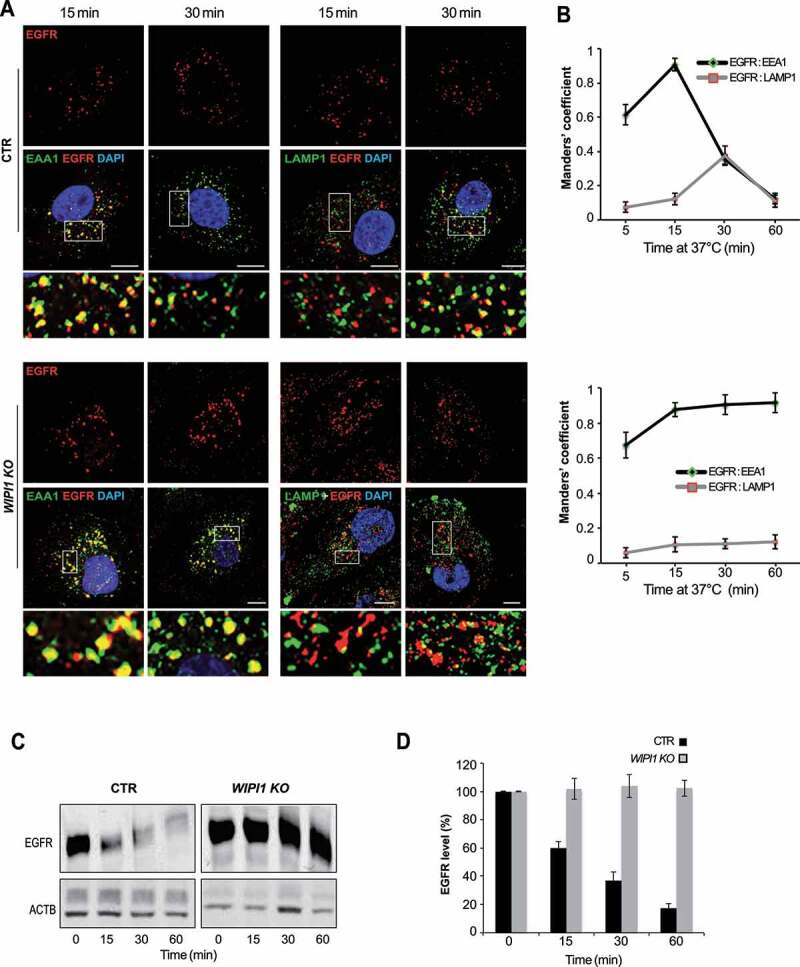
EGFR remains in early endosomes of WIPI1 KO cells and is protected against degradation. (A) CTR and WIPI1 KO cells were serum-starved for 24 h and then supplemented with EGF (100 ng/ml). After the indicated times, cells were fixed, permeabilized, DAPI-labeled (blue) and decorated with antibodies to EGFR (red), EEA1 or LAMP1 (green). Scale bar: 10 μm. Insets show enlargements of the outlined areas. (B) Quantification data from (A). Colocalization of EGFR-EEA1 or EGFR-LAMP1 over time. Manders’ correlation is shown. Values are the mean ± s.d.; (n = 3 independent experiments). Number of cells quantified: CTR = 180; WIPI1 KO = 180. (C) EGFR degradation. CTR and WIPI1 KO cells were stimulated with EGF for different periods of time, lysed and subjected to SDS-PAGE and western analysis for EGFR. ACTB/β-actin served as a loading control. (D) Quantification of EGFR from (C), using the value at time 0 (cells starved for 24 h) as 100% reference. Data are means ±s.d., from three independent experiments
The amphipathic α-helix of WIPI1 is required for formation and fission of endosomal tubules
Protein traffic along the above-mentioned pathways from endosomes to other membranes is mediated by tubulo-vesicular carriers. A defect in these trafficking routes might result from failure to form the carriers, to recruit cargo into them, to transport them or to fuse them with their target compartment. We hence dissected which subreaction is affected by inactivation of WIPI1. Our previous studies had shown that the purified yeast PROPPIN Atg18 can tubulate and fission giant unilamellar liposomes [55]. However, WIPI1 could promote protein exit pathways in other ways (as exemplified through its role in the autophagic pathway, see below), so this is not a given. Therefore, we analyzed whether critical features for liposome fission activity of Atg18 are necessary for the functioning of WIPI1 in endosomal morphology and trafficking in mammalian cells. These include the two lipid binding sites of the protein, its capacity to bind PtdIns(3,5)P2, and the membrane-triggered formation of an amphipathic α-helix, which must insert into the bilayer to deform and fission it.
The role of the amphipathic helix was tested by swapping two pairs of hydrophobic/hydrophilic amino acids on opposite sides of the helix. This generates a scrambled loop of reduced amphipathic character but of identical size and amino acid composition as in the wild-type protein (EGFP-WIPI1[Sloop]; Figure 6A,B). We also designed a variant lacking a significant part of this hydrophobic loop (aa 272–290; EGFP-WIPI1[ΔLoop]; Figure 6B), and a double arginine-to-alanine substitution in the FRRG motif of the lipid binding sites, which completely abolishes phosphoinositide binding by WIPI1 (EGFP-WIPI1[FAAG]). These three variants and a wildtype version of WIPI1 were expressed in control and WIPI1 KO cells at comparable levels, as shown by western blot (Figure. S4 A, B). Fluorescence microscopy revealed that cells transfected with EGFP-WIPI1[Sloop] present extremely long tubular fluorescent structures, which emanate from enlarged globular structures in the perinuclear area and extended into the periphery of the cells (Figure 6C,D). These tubules were produced only when EGFP-WIPI1[Sloop] was expressed on top of the endogenous wildtype alleles, but not upon expression of EGFP-WIPI1[Sloop] in WIPI1 KO cells. Expression of EGFP-WIPI1[ΔLoop] did not show any tubular structures in either background. EGFP-WIPI1[ΔLoop] was recruited to the membranes similarly as WIPI1, indicating that the amphipathic helix is not essential for membrane binding in vivo (Figure 6C). EGFP-WIPI1[FAAG], in agreement with previous studies [27,63,65], did not show any membrane association and did not induce tubules (Figure 6C,D). That tubule formation requires the simultaneous presence of EGFP-WIPI1[Sloop] and of the endogenous wild-type protein suggests that the formation of tubules is a cooperative activity of multiple WIPI1 molecules, at least a fraction of which must carry the amphipathic helix with a sufficient hydrophobic moment.
Figure 6.
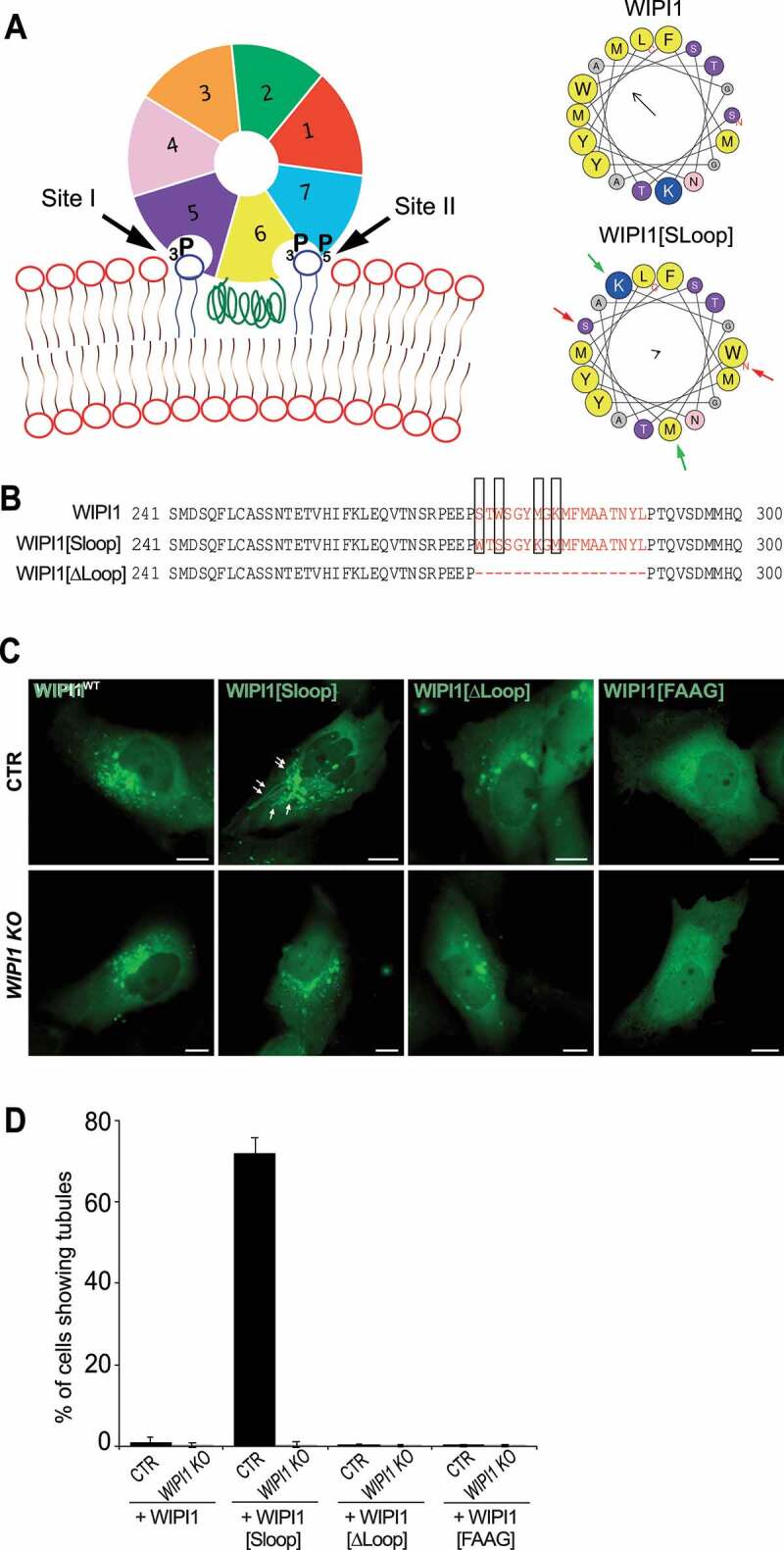
Role of the amphipathic α-helix in blade 6 of WIPI1. (A) Schematic depiction of WIPI1, showing its 7-bladed ß-propeller, the two phosphoinositide binding sites and the hydrophobic CD-loop on blade 6 that folds into an amphipathic helix when in contact with a bilayer [55]. Helical wheel projections show the CD-loop on blade 6 of WIPI1 and WIPI1[Sloop]. Colored arrows indicate the two pairs of amino acids that have been swapped. The magnitude and direction of the hydrophobic moment of the helices was predicted using the online tool Heliquest [118]. It is indicated by the vector in the center of the wheels. (B) Sequences of the hydrophobic loop regions of WIPI1, WIPI1[Sloop] and WIPI1[ΔLoop]. The amino acids removed in WIPI1[ΔLoop] were EEPSTWSGYMGKMFMAATNYL, which comprises the entire sequence shown in the helical wheels. Predicted α-helices analyzed in (A) are plotted in red. The two pairs of hydrophobic/hydrophilic amino acids swapped in WIPI1[Sloop] are highlighted by rectangles. (C) Representative images of CTR and WIPI1 KO cells expressing EGFP-tagged wild-type or the indicated mutant forms of WIPI1. Pictures were taken 18 h after transfection. Tubules are marked by arrows. Scale bar: 10 μm. (D) Quantification of control and WIPI1 KO cells showing tubules after expressing different WIPI1 plasmids for 18 h. The data are mean values ±s.d.; n = 3 independent experiments (20 transfected cells were analyzed per plasmid per experiment in both CTR and WIPI1 KO cells)
WIPI1 tubules stem from a variety of endosomal subtypes
Endosomes form tubular structures, which can give rise to transport carriers that shuttle proteins between these compartments [39,70]. We hence tested the long tubules provoked by WIPI1[Sloop] for the presence of endosomal RAB-GTPases as marker proteins for their origin [71,72]. We co-expressed EGFP-WIPI1[Sloop] with a panel of RAB-GTPases carrying various N-terminal fluorescent protein tags (Figure 7A). The tubules were mostly marked by RAB5, RAB4 and RAB11, markers for early and recycling endosomes, which showed colocalization of 38%, 88% and 76% respectively. Much less colocalization was observed with RAB7 (17%) and RAB9 (4%), markers for late endosomes and lysosomes (Figure 7B). The association of tubules with early and recycling endosomes was confirmed by the enrichment of further markers on them: 58% colocalization with TFRC; 66% with the sorting nexin SNX1; and 52% with the sorting nexin SNX6. By contrast, we found very low colocalization with the late-endosomal marker LAMP1 (15%) and with a subunit of the retriever complex, VPS35L/C16orf62 (6%) (Figure 7C,D). We also observed colocalization between WIPI1[Sloop] and lipids that are enriched in endosomal compartments, as indicated by EGFP-FYVE2 (Figure 8A), a probe with specificity to PtdIns3P [73]. mCherry–ML1N2, a bona fide PtdIns(3,5)P2-labeling probe [74], was mainly enriched on the large, globular structures marked by EGFP-WIPI1[Sloop] in the perinuclear area. On the tubules, it was much less present than EGFP-FYVE2 (Figure 8B,C). These results are consistent with the view that WIPI1-associated tubules originate mainly from early and recycling endosomes.
Figure 7.
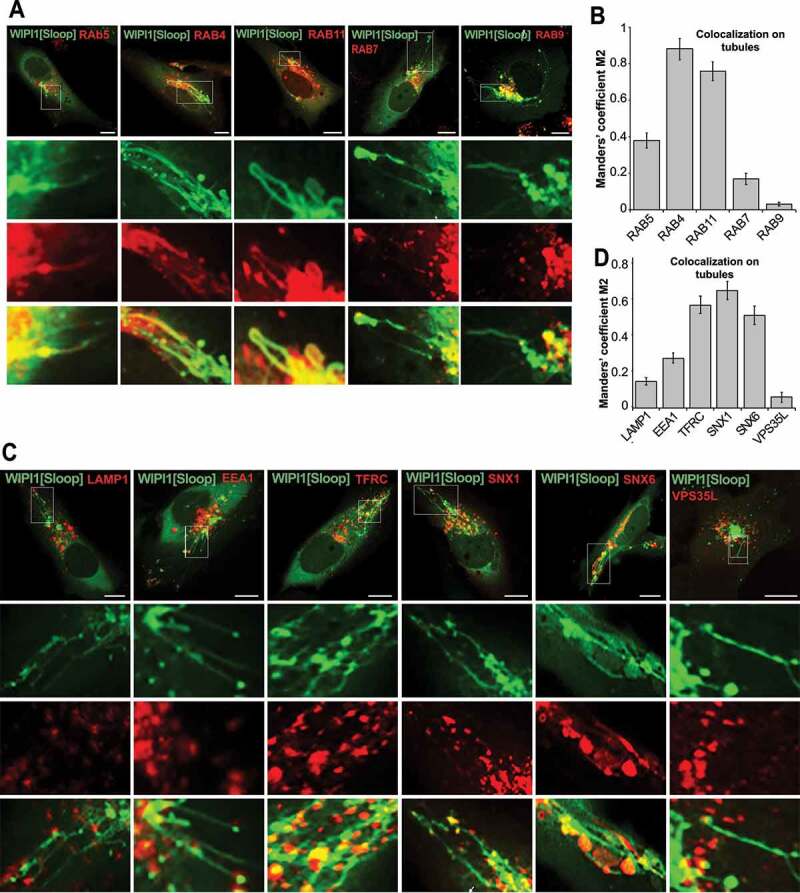
WIPI1[Sloop] is preferentially associated with recycling endosomes. (A) Colocalization with RAB proteins. Live cell images of HK2 cells expressing EGFP-WIPI1[Sloop], mCherry-RAB5, RFP-RAB4, mCherry-RAB7 proteins, DsRed-RAB9 or DsRed-RAB11. Scale bars: 10 μm. Insets show enlargements of the outlined areas. (B) Quantification. Colocalization between WIPI1[Sloop] and different RAB proteins on the tubular structures was assessed using the Manders’ colocalization coefficient M2, calculated in ImageJ. Data are means ±s.d. of n = 150 cells, from three independent experiments. (C) Colocalization with other endo-lysosomal marker proteins. Cells were fixed 18 h after transfection with EGFP-WIPI1[Sloop], stained with the indicated antibodies and imaged by confocal microscopy. Insets show enlargements of the outlined areas. Scale bars: 10 μm. (D) Quantitative colocalization analysis of the images from (C) was carried out for the tubular structures as in (B). Data are means ± s.d. of n = 160 cells from three independent experiments
Figure 8.

WIPI1-associated tubules labeled with probes for PtdIns3P and PtdIns(3,5)P2. HK2 cells expressing (A) EGFP-FYVE2 and mCherry-WIPI1[Sloop], or (B) EGFP-WIPI1[Sloop] and mCherry-ML1N2 were fixed and imaged. Scale bars: 10 μm. The outlined areas are shown at higher magnification. (C) Quantitative colocalization analysis in the entire cell or for the tubular structures was carried out using ImageJ and the pictures from (A) and (B). Data are means ±s.d. of n = 150 cells pooled from three independent experiments
WIPI1-covered tubules carry cargo and are connected to endosomes
The long tubular structures protrude from regions containing enlarged endosomal compartments. We used fluorescence recovery after photobleaching (FRAP) to distinguish whether they are only in the vicinity of these compartments, in which case their membranes should be separated, or whether they actually emanate from them, in which case proteins should move between them. In HK2 cells transiently expressing EGFP-WIPI1[Sloop], tubules were photo-bleached and the recovery of the GFP signal was monitored by confocal microscopy. The tubular structures recovered 40% of the initial fluorescence already within 10 s after bleaching (Figure 9A,B). Fluorescently tagged RAB5, RAB4 and RAB11 proteins on them showed similar recovery, although at a slightly lower rate (Figure 9C-H). Fluorescence recovery along these tubular structures started close to the larger organelle that the tubule was attached to and proceeded from there toward the tip of the tubule (Figure 9C,E,G). This directional recovery suggests that fluorescent proteins repopulate the tubule through diffusion from the larger organelle rather than by direct membrane attachment from a cytosolic pool. Within or next to a tubule, we often found small punctate structures that strongly concentrated TF. A photodepletion approach confirmed that also those structures were connected to the tubules to which they localized, because they exchanged EGFP-WIPI1 with these tubules (Figure 10A,B). These dots might be parts of endosomal compartments that have been pulled out of a larger organelle without separating from it. The tubules thus show properties that we expect from exaggerated forms of endosomal transport carriers that cannot detach from their compartment due to a block in membrane fission.
Figure 9.
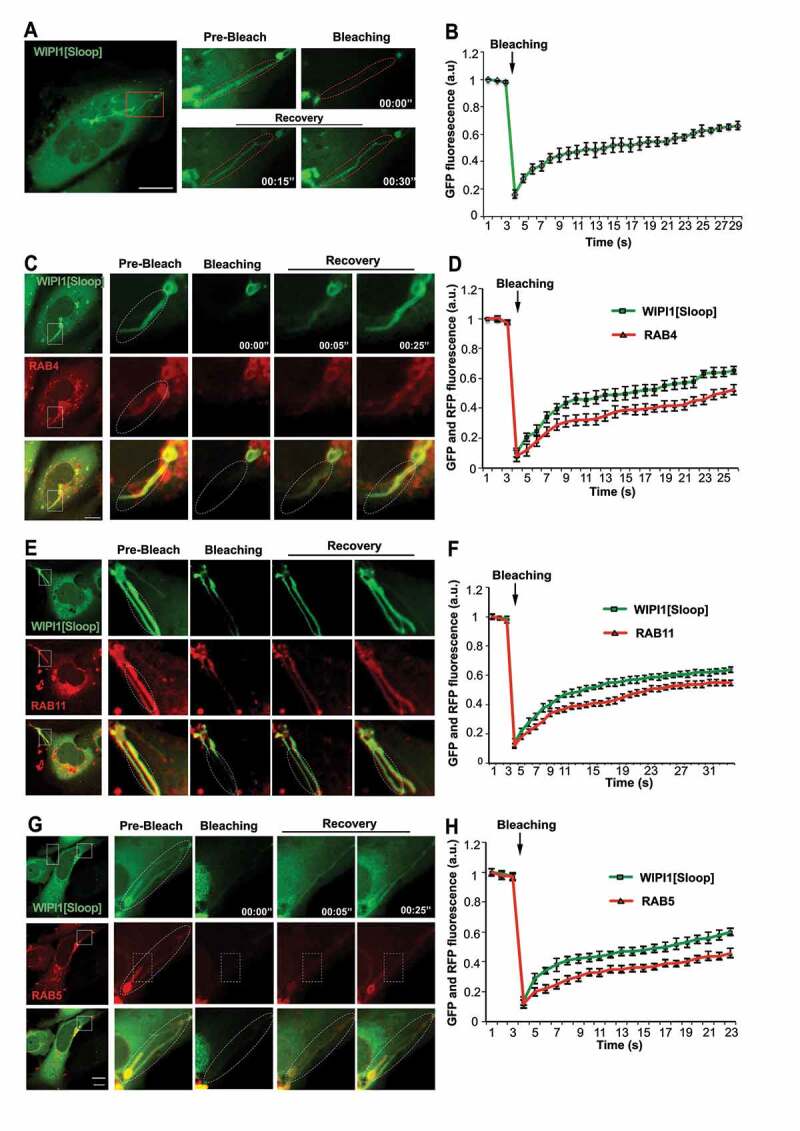
WIPI1-tubules are continuous with endosomes. (A) In HK2 cells expressing EGFP-WIPI1[Sloop], EGFP-labeled tubules (in the boxed area) were photobleached. EGFP fluorescence is shown before and at different times after bleaching. The dashed line indicates the bleached region. The boxed area is shown at higher magnification. Scale bar: 10 μm (B) Quantification of experiments as in a. Means ± s.d. are shown. Data are representative of 3 independent experiments with at least 20 cells analyzed in each experiment. C-H. Experiments as in (A) and (B), respectively, but with cells expressing EGFP-WIPI1[Sloop] plus (C, D) RFP-RAB4, (E, F) DsRed-RAB11 and (G, H) mCherry-RAB5
Figure 10.
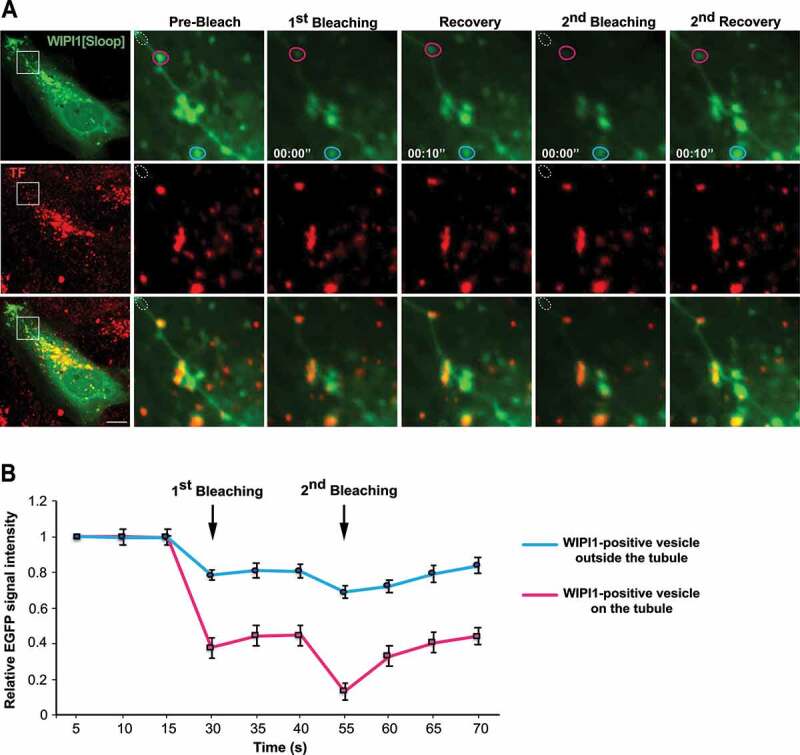
TF-enriched structures on WIPI1 tubules. (A) Photodepletion. HK2 cells were transfected with EGFP-WIPI1[Sloop]. After 18 h, they were loaded with Alexa Fluor 568-TF as in Figure 2 C. A region on a tubule (white dotted circle) was photobleached using a 488-nm laser. The images show concomitant photodepletion of EGFP-fluorescence from a TF-enriched punctate structure, which is outside the bleached area and associated with an EGFP-WIPI1[Sloop]-positive tubule. Two cycles of photodepletion and recovery were recorded. The boxed area of the cell in the image on the left is shown at higher magnification. Scale bar: 10 μm (B) Quantification of experiments as in (a). EGFP-fluorescence intensity was measured over time for EGFP-WIPI1[Sloop]-positive dots localized on a tubule (magenta circle in A), or far away from the tubule (light blue circle in A). Means ±s.d. are shown. 3 independent experiments with 10 cells each were analyzed
PtdIns3P and PtdIns(3,5)P2 are required for different WIPI1 activities
Both PtdIns3P and PtdIns(3,5)P2 are required for membrane recycling events on endosomes. PtdIns(3,5)P2 was proposed to support the fission of endosomal transport carriers [7,59,73,75], but the responsible effector proteins have not yet been identified. We first verified the effects of the PtdIns-3-kinase inhibitor wortmannin and of the PtdIns3P-5-kinase (PIKfyve) inhibitor YM201636 in our cell lines. Block of PtdIns(3,5)P2 production by YM201636 allowed the association of EGFP-WIPI1 with the membranes to persist (Figure. S5). It induced the accumulation of short tubular structures in cells expressing EGFP-WIPI1. In cells expressing EGFP-WIPI1[Sloop], YM201636 further increased the length and the number of the existing tubules. However, it did not induce any tubular structures in cells expressing EGFP-WIPI1[ΔLoop], nor in cells expressing EGFP-WIPI1[FAAG], suggesting that WIPI1 is required for forming the tubules. Wortmannin, which abrogates synthesis of both PtdIns3P and PtdIns(3,5)P2, led to a rapid loss of WIPI1 membrane localization in the cells (Figure. S5) [12,65]. Therefore, we used RFP-RAB4 as alternative marker, since it efficiently marks WIPI1-dependent tubules, as shown above. Wortmannin prevented formation of the tubules that are normally seen in EGFP-WIPI1[Sloop] cells (Figure. S5C). This suggests that PtdIns3P is needed for the generation of WIPI1-dependent tubules. PtdIns(3,5)P2 is dispensable for tubule formation but absence of this lipid provokes their accumulation in an exaggerated, elongated form.
WIPI1 fission activity requires its lipid binding site II
We tested the hypothesis that tubule formation requires WIPI1 and PtdIns3P, whereas WIPI1 in conjunction with PtdIns(3,5)P2 may counteract the accumulation of tubules by supporting their fission. If the two phosphoinositides promote different activities through the same effector protein, substitutions in the two lipid binding sites might differentiate these effector functions. We sought such substitutions by mutating the two lipid binding sites of WIPI1 individually. Work on the yeast PROPPIN Hsv2, a homolog of WIPI1, had suggested that site I preferentially binds PtdIns3P, whereas site II prefers PtdIns(3,5)P2 [23]. The study identified substitutions of conserved residues with differential effects on lipid binding: H179 (site I) impaired binding to PtdIns3P but not to PtdIns(3,5)P2, whereas substitution of H249 (site II) eliminated binding to PtdIns(3,5)P2 and reduced binding to PtdIns3P. These substitutions might hence allow to discriminate between PtdIns3P and PtdIns(3,5)P2-dependent activities. We substituted the two corresponding histidines in WIPI1 (Figure. S6 A) and expressed the following proteins in control and WIPI1 KO cells: WIPI1H185N and WIPI1H185A (affecting site I), and WIPI1H257Q and WIPI1H257A (affecting site II). As negative controls, we substituted single arginines in the FRRG motif (WIPI1R226A and WIPI1R227A), which simultaneously affect both binding sites and strongly reduce membrane association 10,23,63,76. Whereas EGFP-WIPI1R226A and EGFP-WIPI1R227A remained in the cytosol as expected (Figure. S6 B), the proteins with substitutions in site I (WIPI1H185A) and site II (WIPI1H257A) did associate with the membranes. Both substituted proteins showed an increased cytosolic background compared to the wildtype constructs, suggesting reduced affinity for the membrane (Figure. S6 B). This was confirmed by differential centrifugation of extracts from these cells, in which over 80% of the substituted EGFP-WIPI1 remained in the cytosolic supernatant (Figure. S6 C), whereas EGFP-WIPI1 fractionated entirely with the membranes.
In order to circumvent potential effects due to lower membrane binding of WIPI1, we added a prenyl anchor to the C-terminus of WIPI1. Prenyl anchors are used by cells to increase the affinity of a great variety of proteins for membranes. We thus created the corresponding mutants carrying a 15-glycine spacer plus a CVVM motif for prenylation (Figure 11A). The CVVM motif allowed all WIPI1 mutants to be recruited to the membranes as efficiently as the wildtype protein, as judged by microscopy and subcellular fractionation (Figure 11B,C). The endosomal, WIPI1-associated compartments showed strong morphological differences: Substitutions R226A and R227A in the FRRG motif led to fewer and larger compartments, whereas the site I substitution H185A showed little effect. The site II substitution H257A led to an enormous enlargement of the endosomal compartments and to the formation of long tubular structures (Figure 11B, S6 B), which is consistent with perturbed membrane exit from these compartments. Overall, the site II substitution H257A phenocopied the morphological effects of manipulations interfering with fission activity, such as inhibition of PtdIns(3,5)P2 synthesis (Figure. S5) or eliminating the amphipathic character of the hydrophobic loop (Figure 6C). This suggests that PtdIns(3,5)P2 promotes fission through lipid binding site II of WIPI1.
Figure 11.
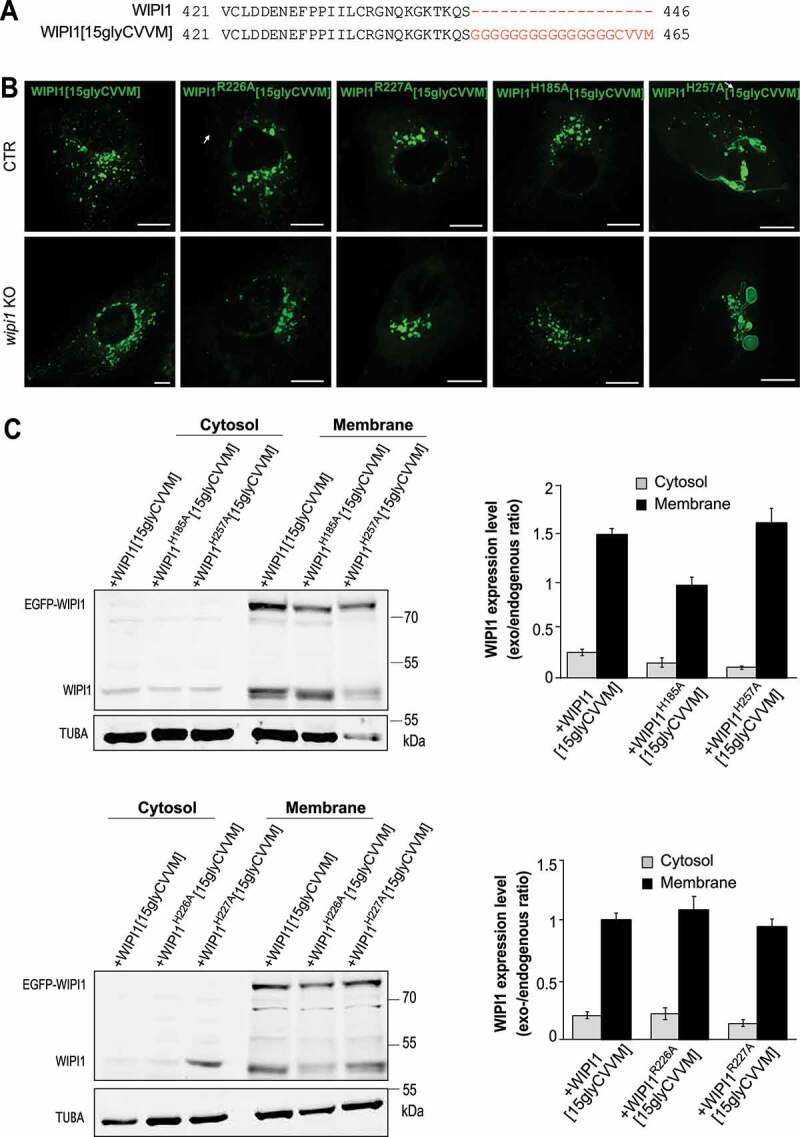
WIPI1 fission activity requires its lipid binding site II. (A) Sequences of WIPI1 and WIPI1[15gly-CVVM] were aligned with the Clustal Omega program. The added prenylation sequence and its spacer are plotted in red. (B) Endosome morphologies. CTR and WIPI1 KO cells were transfected with EGFP-WIPI1[15gly-CVVM] carrying the indicated substitutions and analyzed by live confocal microscopy. Scale bar: 10 μm. (C) Expression levels. CTR cells from (B) were lysed, fractionated by centrifugation into membrane and cytosolic supernatant and subjected to SDS-PAGE and western blotting against WIPI1 and TUBA/α-tubulin as loading control. The quantified signals are displayed as the ratio of the transfected WIPI1 over TUBA as a loading control. All ratios were normalized to that for WIPI1[15gly-CVVM], which had been set to 1. Means ±s.d. are shown; n = 3 independent experiments
Next, we tested whether the WIPI1 versions leading to the accumulation of endosomal tubules (WIPI1[Sloop] and WIPI1H257A[15gly-CVVM]) showed corresponding defects in the recycling of endocytic cargoes and in the sorting from early endosomes to degradative compartments. We used TF and EGFR as model proteins in pulse-chase assays as described above. WIPI1 KO cells transfected with a plasmid expressing EGFP-WIPI1 recovered their TF-recycling and their EGFR degradation (Figure 12). By contrast, EGFP-WIPI1[Sloop] and EGFP-WIPI1H257A[15gly-CVVM] not only failed to induce any rescue in WIPI1 KO cells, but they even exerted a dominant-negative effect on the trafficking of both model proteins, when they were expressed on top of endogenous WIPI1.
Figure 12.
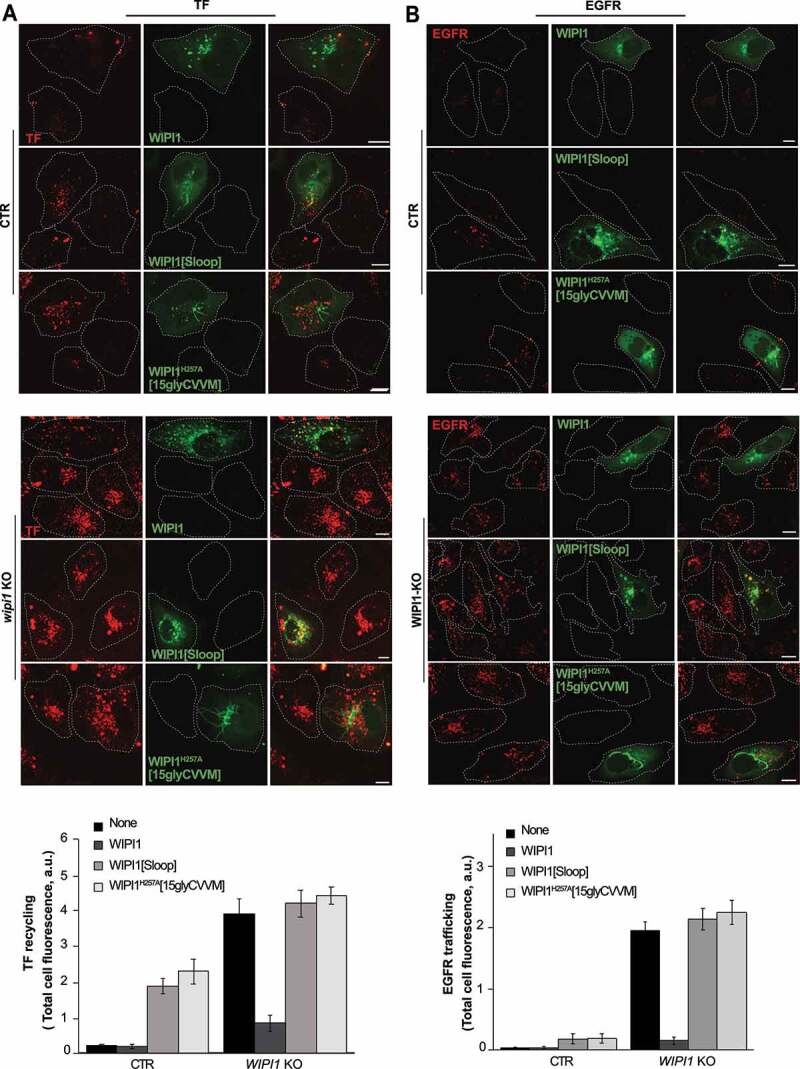
Effect of WIPI1 mutants accumulating endosomal tubules on protein exit from endosomes. (A) TF recycling. Control and WIPI1 KO cells were transfected with EGFP-WIPI1, EGFP-WIPI1[Sloop] or EGFP-WIPI1H257A[15gly-CVVM] for 18 h. Then, they were serum-starved for 60 min, loaded with Alexa Fluor568-conjugated TF and chased at 37°C as in Figure 2 C. Scale bar: 10 μm. The white dashed lines indicate the circumference of the cells. TF-fluorescence was quantified as in Figure. 2B. Mean values ± s.d. are shown. n = 3 independent experiments with a total of 210 cells analyzed per condition. (B) EGFR degradation. CTR and WIPI1 KO cells expressing EGFP-WIPI1, EGFP-WIPI1[Sloop] and EGFP-WIPI1H257A[15gly-CVVM] as in (A) were serum-starved for 24 h and then stimulated with EGF (100 ng/ml) for 60 min. Cells were fixed, permeabilized, and stained with antibody to EGFR (red). Scale bar: 10 μm. EGFR-fluorescence was quantified as in Figure. 2B. 180 cells per condition stemming from 3 independent experiments were analyzed. Mean values ± s.d. are shown
WIPI1 acts in autophagosome formation and in endosomal protein exit through distinct mechanisms
Finally, we asked whether the originally identified function for WIPI proteins in autophagosome formation utilizes the same molecular features as protein exit from endosomes. We first measured the response of our HK2 cell line to 3 hours of starvation in HBSS medium (Figure. S7), using mCherry-LC3 as a marker of autophagosomes in fluorescence microscopy. Starvation triggered a 7-fold increase in the number of punctate structures labeled by mCherry-LC3. In agreement with previous observations in other cell lines [12,13,17], knockdown of WIPI2 reduced the number of puncta in starved cells by 70% (Figure. S7). Knockdown of WIPI1, which occurs with comparable efficiency as the knockdown of WIPI1 (Figure 3A,B), reduced the number of puncta in the starved cells only by 30%.
We also assayed autophagy in the knockdowns through a western blot assay of the lipidation of LC3, which coincides with the formation of autophagosomes (Figure. S7 C, D) [76,77]. This modification converts the cytosolic form LC3-I into the autophagosome-associated LC3-II, which accumulates when its degradation in lysosomal compartments is suppressed by protease inhibitors. Inhibition of lysosomal protease activity was confirmed through an assay with BZiPAR (Figure. S8). The LC3-II assay showed a strong effect for the knockdown of WIPI2, but only a weak and statistically non-significant effect of the knockdown of WIPI1. This agrees with earlier WIPI1 knockdown experiments in other cell lines [13]. That WIPI1 knockdown did not lead to a significant reduction of LC3-II in the blot assay, whereas it had a robust effect in the microscopic assay with knockdowns (Figures. S7 A,B, S8) as well as well as in WIPI1 KO cells (see below) may be due to the fact that the blot is an ensemble assay, in which non-transfected cells provide an elevated background. This background of non-transfected cells is excluded in the microscopy assay and not present in a WIPI1 KO.
We used WIPI1 KO cells to express the WIPI1 variants that interfere with fission activity and test their impact on autophagy. These variants include substitutions in site I or site II (EGFP-WIPI1H185A[15gly-CVVM] and EGFP-WIPI1H257A[15gly-CVVM]), the FRRG motif (EGFP-WIPI1R226A[15gly-CVVM] and EGFP-WIPI1R227A[15gly-CVVM]) and the amphipathic helix (EGFP-WIPI1[Sloop]). WIPI1 and all variants increased the formation of mCherry-LC3 puncta (Figure 13) and the formation of LC3-II (Figure 14) to a similar degree, by 50%. Therefore, the amphipathic α-helix and lipid binding site II are not essential for autophagy, whereas they are crucial for receptor exit from endosomes.
Figure 13.
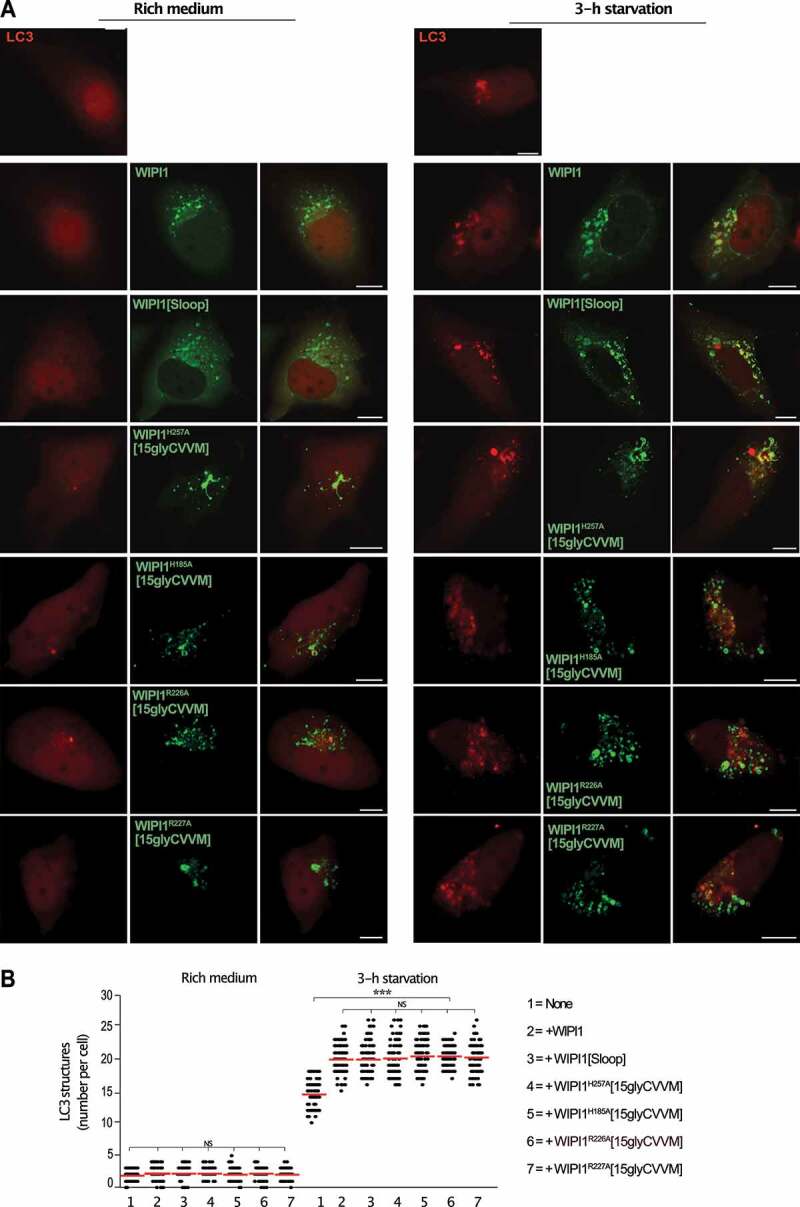
WIPI1 variants hindering protein exit from endosomes promote formation of mCherry-LC3 puncta like WIPI1 (A) WIPI1 KO cells expressing mCherry-LC3 and the indicated variants of EGFP-WIPI1 were incubated in growth medium or in HBSS for 3 h and analyzed by confocal microscopy. Scale bar: 10 µm. (B) Quantification. The number of LC3-positive structures in the cells from A was counted. The red line indicates the mean; n = 150 cells per condition, pooled from three independent experiments. P-values were calculated by one-way ANOVA and t-test t (analysis performed with 99% confidence ***p < 0.0001, NS = not significant)
Figure 14.
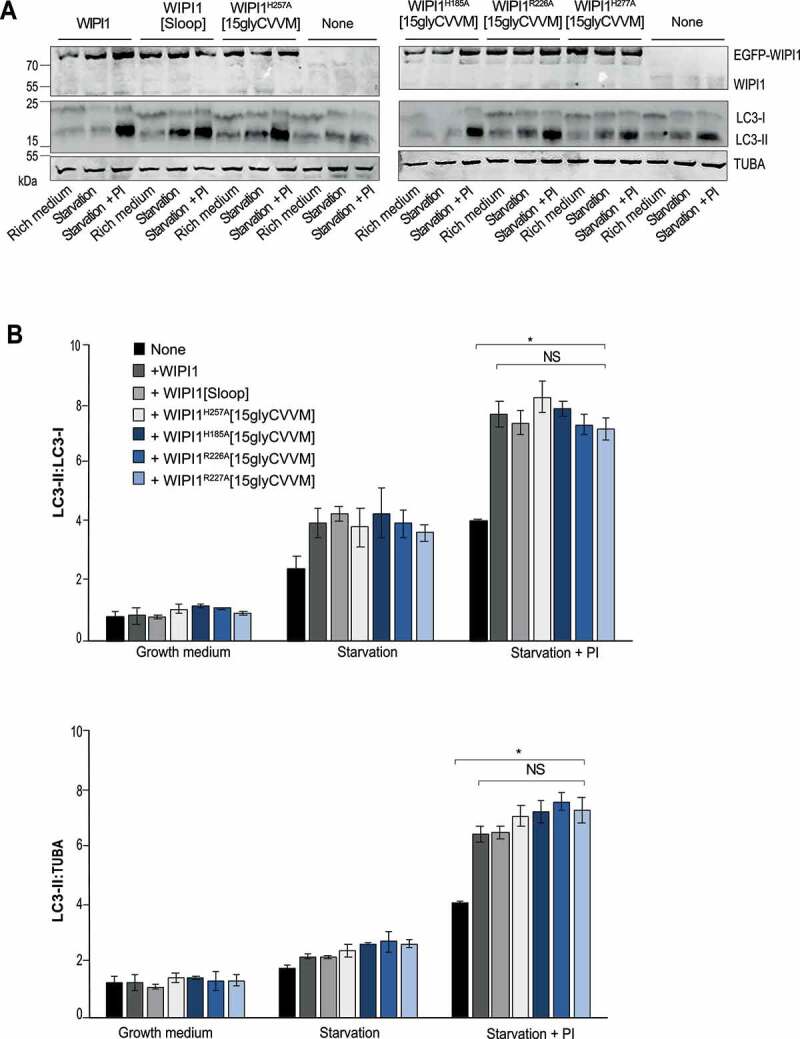
Formation of LC3-II is not affected by WIPI1 mutations hindering protein exit from endosomes. (A) Total cell lysates of the cells from WIPI1 KO cells re-expressing the indicated WIPI1 variants (from Figure. 13 A; 50 μg of protein per sample) were analyzed by SDS–PAGE and western blotting using the indicated antibodies. TUBA/α-tubulin served as a loading control. A representative blot is shown. (B) The ratio LC3-II:LC3-I and LC3-II:TUBA/α-tubulin was quantified by a fluorescence scanner. Data are means ±s.d.; n = 3 independent experiments. P values are indicated and were calculated by t-Test. The analysis was performed with 99% confidence: *p < 0.01; NS = not significant)
By integrating our observations with the known property of the WIPI1 homolog Atg18 to undergo PtdIns(3,5)P2-dependent oligomerization [55,78], we can formulate a working model of WIPI1 function on endo-lysosomal compartments. We propose that WIPI1 in conjunction with PtdIns3P promotes the formation of tubular endosomal transport carriers. This activity requires lipid binding site I, but not PtdIns(3,5)P2. Lipid binding site II and PtdIns(3,5)P2 become important to induce the fission activity of the protein, which depends on the insertion of its amphipathic helix into the membrane. PtdIns(3,5)P2-dependent oligomerization should facilitate fission by locally concentrates these membrane-inserting helices [79]. The role of WIPI1 in autophagy is independent of these fission-related features. Here, the protein exploits its affinity for PtdIns3P, probably in a templating function that recruits other factors to the phagophore, similarly as shown for the WIPI1 homologs Atg18 and WIPI2 [13,16,17,80–82].
Discussion
While the coat proteins involved in the formation of tubulo-vesicular endosomal transport carriers become increasingly well understood [39,70], it is still unclear how these carriers are detached from the donor organelle. In contrast to previous suggestions that, in starving cells, the autophagic machinery promotes endosomal trafficking by autophagic removal of damaged endosomes [30], our observations present evidence for a direct function of specifically WIPI1 in both the formation and fission of these tubules in living mammalian cells. They permit to separate these two functions through their differential requirements for PtdIns3P, PtdIns(3,5)P2 and the two WIPI1 lipid binding sites. They underscore that the two lipid binding sites modify the function of the protein and do not merely serve as membrane anchors. We propose that formation and release of the carriers depend on the membrane-insertion of the amphipathic helix on CD-loop 6 of WIPI1. It is notable that several substitutions in WIPI1, which were identified in melanoma or pancreatic cancer cells [11], map to this loop. This provides a hint on the nature of the underlying cellular defect, because the amphipathic helix is only required for the membrane fission activity of WIPI1, but not for autophagy. Our results hence suggest that the substitutions promote these cancers through perturbation of cellular signaling at the level of the endosome. In support of this argument, we show that inactivation of WIPI1 hinders the degradation of EGFR and leads to an accumulation of the protein in the endo-lysosomal system, from where it can actually continue to signal [83]. Since EGFR activity is necessary for the growth of melanomas and EGFR overactivation can even induce it [84–86], we propose that WIPI1 mutations contribute to these cancers through excessive signaling of accumulated endosomal EGFR and/or other growth factor receptors [87]. This hypothesis will be tested in future studies.
Our observations suggest that WIPI1 acts cooperatively on endosomal membranes in vivo, providing a first indication that oligomerization, which can observed with the purified WIPI1 homolog Atg18 in vitro [55,78], is functionally relevant. Oligomerization can support fission by enhancing the induction of curvature by inserted amphipathic helices [88,89]. Previous studies suggested an involvement of the dynamin-like GTPase Vps1 in the fission of retrograde transport carriers in yeast [90,91]. Yeast Atg18 promotes retrograde trafficking in a so far undefined manner [10]. Its homology to WIPI1 render it very likely that it also promotes fission of transport carriers in this pathway. Then, the formation of these carriers shows remarkable similarity to the fission of yeast vacuoles into smaller vesicles, which also requires both Atg18 and Vps1 [10,55,56,92–96]. For exit from mammalian endosomes, a role of a dynamin-related GTPase has not been firmly established. However, the EHD proteins, which are present on endosomes, share some properties with dynamin-related GTPases and show membrane fission activity in vitro [97–102]. Like dynamin-related GTPases, the EHD proteins self-interact, hydrolyze nucleotides and are potential mechanochemical devices to deform or constrict membrane tubules. This leads us to the working model that membrane fission at endo-lysosomal compartments may generally require a PROPPIN and a mechanochemical nucleoside triphosphatase. They may need to cooperate to first gradually constrain the diameter of a membrane tubule and then sever it. We envision a situation similar to endocytosis, where mechanochemical dynamin-related GTPases collaborate with membrane-bending BAR-domain proteins such as SH3GL/endophilin and AMPH (amphiphysin) to facilitate fission. Depending on the type of endocytic vesicle formed, dynamin is not strictly required and it can be replaced by other mechanical stresses, mediated e.g. through the cytoskeleton and motor proteins [53,103–106]. In the formation of endosomal transport carriers, additional mechanical stress can be provided by the actin-cytoskeleton and the WASH complex [42–44,107]. Therefore, we now undertake systematic analyses of the interactions of PROPPINs with other factors involved in protein exit from endosomes.
Protein sorting from endosomes requires both PtdIns(3,5)P2 and PtdIns3P [61]. Whereas coat complexes such as the retromer, retriever and commander [39,40,108] utilize PtdIns3P in order to bind the membrane, a requirement for PtdIns(3,5)P2 in their functioning has not been reported and the effector for PtdIns(3,5)P2 in protein exit from endosomes has remained unknown. WIPI1 and other PROPPINs are good candidates because of their affinities for both PtdIns3P and PtdIns(3,5)P2 [10,13,23–25,27,65,109]. Our data support WIPI1 as effector of PtdIns(3,5)P2, because substitutions in its lipid binding site II phenocopy the effect of an inhibition of PtdIns(3,5)P2 synthesis. Both manipulations provoke the accumulation of micrometer-long endosomal membrane tubules, suggesting that the PtdIns3P-dependent tubulation of endosomes continues while the PtdIns(3,5)P2-dependent fission of these structures is impaired. When both lipid binding sites are ablated or PtdIns3P formation is inhibited, also tubule formation is blocked. Since both tubulation and fission require WIPI1, but differ in their need for PtdIns3P, PtdIns(3,5)P2 and the second lipid binding site of WIPI1, we propose that the activities of the protein in the formation and fission of carrier vesicles are switched by the phosphoinositides that it binds.
Studies with synthetic liposomes yielded differing results with respect to the specificities of the two lipid binding sites of PROPPINs. Whereas both binding sites interact with PtdIns3P, a moderate preference for PtdIns(3,5)P2 was assigned either to binding site I or II [23–25,57]. Whether this reflects a genuine functional distinction among PROPPIN family members cannot yet be judged, because these studies used different experimental conditions and different PROPPINs. However, the fact that all binding studies have revealed only moderate preferences of the two binding sites for PtdIns3P or PtdIns(3,5)P2 allows two interpretations, which are not mutually exclusive: First, PROPPINs might simply not differentiate between the two phosphoinositides. This hypothesis is inconsistent with the fact that WIPI1 substitutions in two different regions of the protein (the amphipathic helix and lipid binding site II) phenocopy the suppression of PtdIns(3,5)P2 synthesis but not PtdIns3P-dependent functions, such as formation of endosomal tubules and autophagosomes. As a second possibility, the two phosphoinositides might be differentiated through their agonist properties rather than their binding affinity and membrane recruitment. This concept is well established for receptor proteins, where numerous examples have shown that while a variety of ligands may bind a receptor with similar affinities, their agonist properties, i.e. their capacity to change the activity and conformation of the receptor, can grossly differ [110]. Therefore, we took a functional in vivo approach rather than a study of binding affinities to analyze the roles of the lipid binding sites of WIPI1. We substituted a conserved histidine, which is placed at an equivalent location in both lipid binding sites [23,25]. In contrast to the commonly used substitutions in the FRRG motif, which simultaneously affect both lipid binding sites, each of these histidines coordinates a phosphoinositide head group in only a single binding site. Since a point mutation in lipid binding site II entails the same accumulation of endosomal tubules as the ablation of fission through depletion of PtdIns(3,5)P2 or scrambling the hydrophobic loop, fission activity and the effect of PtdIns(3,5)P2 in protein exit from endosomes can be tentatively assigned to site II. This suggests a functional differentiation between the two lipid binding sites in vivo.
Several observations further support the notion that the two lipid binding sites of PROPPINs are not simple membrane anchors but activate specific effector functions. First, when WIPI1 lipid binding sites are compromised or when PtdIns(3,5)P2 synthesis is inhibited, normal membrane recruitment is rescued through addition of a prenyl anchor. This restores the function of the protein in autophagy, but not in endosomal protein exit. Second, PtdIns3P suffices for the WIPI1-dependent formation of endosomal tubules, but PtdIns(3,5)P2 and lipid binding site II are required for fission. Since PtdIns(3,5)P2 is bound in a pocket of the protein, the only conceivable way how it could trigger this specific endosomal effector function is through a conformational change of the protein.
Our results thus suggest that PROPPINs have two fundamentally different functional faces. We propose a working model in which PtdIns3P allows WIPI1 to support the tubulation of the endosomal membrane. PtdIns(3,5)P2 triggers WIPI1 to insert its amphiphilic helix, oligomerize and cooperatively fission the membrane during protein exit form endosomes. In autophagy, this latter feature is not utilized. The protein relies on its membrane attachment through PtdIns3P and may serve as an interaction platform for other proteins that build and expand the phagophore membrane. Different lipids bound to a PROPPIN may thus activate distinct effector functions.
Materials and Methods
Antibodies and reagents
All chemical reagents were from Sigma-Aldrich unless otherwise specified. Primary and secondary antibodies used in this study are listed in Table 1. Other reagents: Opti-MEM (Thermo Fisher, 11,058,021) and Trypsin (Thermo Fisher, 27,250,018); Alexa Fluor® 488 or 568-conjugated Transferrin from Human Serum (ThermoFisher Scientific, T13342); rhodamine110, bis-(CBZ-L-isoleucyl-L-prolyl-L-arginine amide) dihydrochloride (BZiPAR; Thermo Fisher Scientific, R6505); LysoTracker® Blue DND-22 (Thermo Fisher Scientific, L7525; Cy3-STxB was a gift from Prof. Ludger Johannes (Institute Curie, Paris, France). Protease inhibitor (PI) cocktail (final concentrations: 40 μM pefablock SC (Merck, 11,429,876,001), 2.1 μM leupeptin (Merck, 11,529,048,001), 80 μM o-phenantroline (Merck,131,377), 1.5 μM pepstatin A (Merck, 11,524,488,001).
Table 1.
Antibodies used
| ATG2A | ab226931 | Abcam | 1/500 | WB | |
|---|---|---|---|---|---|
| ATG16L1 | 8089 | D6D5 | Cell Signaling Technology | 1/1000 | WB |
| ACTB/β-actin | A2066 | Sigma | 1/10,000 | WB | |
| B4GALT1/β4-GALT | HPA010807 | Sigma | 1/200 | IF | |
| EEA1 | 2411 | Cell Signaling | 1/500; 1/1000 | IF WB | |
| EGFR | sc-03 | 1005 | Santa Cruz Biotechnology | 1/400; 1/1000 | IF WB |
| LAMP1 | H4A3 | H4A3 | USBiological Life Sciences | 1/500; 1/1000 | IF WB |
| MAP1LC3B/LC3 | NB100-2220 | Novus Biologicals | 1/1000 | WB | |
| SNX1 | 611,482 | 51/SNX1 | BD transduction Laboratories | 1/500 | IF |
| SNX6 | HPA049374 | Atlas antibodies | 1/200 | IF | |
| TFRC | 13–6800 | H68.4 | Invitrogen | 1/200 | IF |
| WIPI1 | H00055062-M02 | 3 C1 | Abnova | 1/1000 | WB |
| WIPI1 | W2394 | Sigma | 1/1000 | WB | |
| WIPI2 | HPA019852 | Sigma | 1/500 | WB | |
| WDR45B/WIPI3 | sc-514,194 | B-7 | Santa Cruz | 1/500 | WB |
| WDR45/WIPI4 | ab240905 | Abcam | 1/1000 | WB | |
| Cy3-conjugated AffiniPure donkey anti-mouse IgG (H + L) secondary antibody | 715–165-151 | Jackson Immuno Research | 1/400 | IF | |
| Cy3-conjugated AffiniPure donkey anti-rabbit IgG (H + L) secondary antibody | 711–165,152 | Jackson Immuno Research | 1/400 | IF | |
| Alexa Fluor® 488-conjugated AffiniPure donkey anti-mouse IgG (H + L) secondary antibody | 715–545-151 | Jackson Immuno Research | 1/400 | IF | |
| Alexa Fluor® 488-conjugated AffiniPure donkey anti-rabbit IgG (H + L) secondary antibody | 711–545-152 | Jackson Immuno Research | 1/400 | IF | |
| Alexa Fluor® 647-conjugated AffiniPure donkey anti-rabbit IgG (H + L) secondary antibody | 711–605-152 | Jackson Immuno Research | 1/400 | IF | |
| Alexa Fluor® 647-conjugated AffiniPure donkey anti-mouse IgG (H + L) secondary antibody | 715–605-151 | Jackson Immuno Research | 1/400 | IF | |
| IRDye® 800CW goat anti-mouse IgG (H + L) | 926–32,210 | Li-Cor | 1/10,000 | WB | |
| IRDye® 800CW goat anti-rabbit IgG (H + L) | 926–32,211 | Li-Cor | 1/10,000 | WB | |
| IRDye® 680RD goat anti-mouse IgG (H + L) | 926–68,070 | Li-Cor | 1/10,000 | WB | |
| IRDye® 680LT goat anti-rabbit IgG (H + L) | 926–68,071 | Li-Cor | 1/10,000 | WB |
Complementary DNA (cDNA) constructs
Vectors expressing tagged-RAB proteins were purchased from Addgene: RFP-RAB4 (79,800; deposited by J.D. Johnson); DsRed-RAB11 (12,679; deposited by R. Pagano); mCherry-RAB7 (61,804; deposited by G. Voeltz); mCherry-RAB5 (49,201; deposited by G. Voeltz); DsRed-RAB9 (12,677; deposited by R. Pagano); mCherry-hLC3B-pcDNA3.1 (40,827; deposited by D. Rubinszstein). The following vectors were kindly provided by colleagues: EGFP-WIPI1 (pAR31CD vector) and EGFP-WIPI1[FAAG] (Tassula Proikas-Cezanne, Tübingen, Germany); pEGFP-FYVE2 (Harald Stenmark, Department of Molecular Cell Biology, Institute for Cancer Research, Oslo, Norway); mCherry-ML1N2 (Haoxing Xu, University of Michigan, Ann Arbor, USA).
To generate mCherry-WIPI1, mCherry and WIPI1 fragments were amplified from pFA6a-mCherry-V5-KanMX6 (from Fulvio Reggiori, University Medical Center Groningen, Netherlands) and EGFP-WIPI1 plasmid, respectively, by using the primers listed in Table S1. Then, the two fragments were fused by using overlap extension-PCR and cloned into the pAR31CD vector between Age1 and EcoR1 restriction sites.
To obtain EGFP-WIPI1[15Gly-CVVM] a sequence with 15 glycine and a CVVM motif were added to EGFP-WIPI1 plasmid by PCR using the primers listed in Table S1. The product was sub cloned into the pAR31CD-WIPI1 vector between Xmn1 and EcoR1 restriction sites.
Site-directed mutagenesis
All constructs were verified by DNA sequencing. EGFP-WIPI1 was used as DNA template for site-directed mutagenesis (QuikChange mutagenesis system, Agilent Technologies, 200,524) to generate EGFP-WIPI1[Sloop] and EGFP-WIPI1[ΔLoop]) following the manufacturer’s protocol using primers (Microsynth) listed in Table S1. For EGFP-WIPI1[Sloop] the sequence was STWSGYMGKMFMAATNYL changed into WTSSGYKGMMFMAATNYL (substituted amino acids are underlined and in bold). For EGFP-WIPI1[ΔLoop] a sequence of 18 amino acids was deleted (Figure 5). EGFP-WIPI1 was used as template to generate point mutations in the FRRG motif (R226A and R227A) and in the two histidine (H185A, H185N, H257Q and H257A) by using site-directed mutagenesis system and the primers containing desired mutations are listed in Table S1. Nonmutated template vector was removed from the PCR mixture through digestion by the enzyme Dpn1 for 1 h at 37°C. The product was purified using NucleoSpin PCR Clean-up (Macherey-Nagel, 740,609.50S) and transformed into Escherichia coli. Plasmid DNA was purified and sequenced.
The site-directed mutagenesis system was also used to generate the siRNA-resistant cDNA for WIPI1. Forward and reverse sequences of the primers are listed in Table S1.
Cell culture, transfection and treatments
HK2 cells were grown in DMEM-HAM’s F12 (Thermo Fisher, 11,765,054) supplemented with 5% fetal calf serum (Gibco, 10,270,106), 50 U/mL penicillin/50 mg/mL streptomycin (ThermoFisher, 15,140,148), ITSE (5 μg/mL insulin, 5 μg/mL TF, 5 ng/mL selenium; LuBio Science, 00–101-100ML). Cells were grown at 37°C in 5% CO2 and at 98% humidity.
HK2 cells were transfected with different plasmids using X-tremeGENETM HP DNA transfection reagent (Sigma-Aldrich, 6,366,546,001), unless otherwise specified, according to the manufacturer’s instructions, and incubated for 18–24 h before fixation or live cell imaging. The HK2 cell line was checked for mycoplasma contamination by a PCR-based method. All cell-based experiments were repeated at least three times.
RNA interference
HK2 cells were transfected with siRNA for 72 h using Lipofectamine® RNAiMax (Thermo Fisher Scientific, 13,778,150) according to the manufacturer’s instructions. Control cells (CTR2 in Figure. S1) were treated with identical concentrations of siGENOME Control Pool Non-Targeting from Dharmacon (D-001206-13-05).
siRNAs targeting WIPI1, WIPI2, WDR45B/WIPI3, WDR45/WIPI4 and ATG16L1 were from Dharmacon (siGenome Human WIPI1 D-01820503; ON-TARGETplus Human WIPI2 J-020521; ON-TARGETplus Human WDR45B J-017119; ON-TARGETplus Human WDR45 J-019758; ON-TARGETplus Human ATG16L1 J-021033). siRNA targeting ATG2 was form Sigma (ATG2A 5ʹ- GCAUUCCCAGUUGUUGGAGUUCCUA-3ʹ; ATG2B 5ʹ-AGGUCUCUCUUGUCUGGCAUCUUUA-3ʹ). All siRNAs were used at a final concentration of 20 nM.
CRISPR-Cas9 gene editing
CRISPR sequences targeting the region that precedes the phosphoinositide-binding site of human WIPI1 were designed using the online Cas9 target design tool developed by the F. Zhang laboratory (http://www.genome-engineering.org/) [111,112]. The seed sequences preceding the protospacer adjacent motif (PAM) are the following: WIPI1 oligo 1–5′-CACCGCTTGAAGATGTGTACCGTCT-3′; WIPI1 oligo 2–5′-AAACAGACGGTACACATCTTCAAGC-3′; We used a sequence targeting luciferase as non-relevant single-guide RNA (sgRNA), (CTR1 in Figure. S1): luciferase oligo 1–5′-CACCGCTTCGAAATGTCCGTTCGGT-3′; and luciferase oligo 2–5′-AAACACCGAACGGACATTTCGAAGC-3′. Nucleotides in italics show the overhangs necessary for incorporation into the restriction site for BbsI of the LentiCRISPR-v2 vector expressing Cas9 and sgRNA (Addgene, 52,961; deposited by Feng Zhang). Lentiviruses were obtained from 293 T cells as previously described [113], were kindly provided from Fabio Martinon (University of Lausanne, Switzerland). HK2 cells were infected with lentiCRISPv2 viruses targeting WIPI1 (WIPI1 KO) or luciferase (CTR1). Positive cells were selected with 2 μg/mL puromycin for 15 days. The population was subjected to limiting dilution to obtain individual clones. WIPI1 expression was assessed by immunoblotting of whole-cell extracts (Figure. S1).
To generate WIPI1 cDNA resistant to Cas9 digestion, a silent mutation was inserted in the PAM sequence (NGG) of the Cas9 target sequence (CCGAGACGGTACACATCTTCAAG), generating the sequence CTGAGACGGTACACATCTTCAAG.
Endocytosis assays
TF binding and internalization
Cells were serum-starved for 60 min at 37°C, washed twice in cold PBS (137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.4 mM KH2PO4) with 1% BSA (Sigma-Aldrich A 8806) and incubated for 60 min on ice with 50 μg/ml of TF. The cells were fixed or chased in complete fresh HEPES-buffered medium at 37°C for the indicated time points. At the end of this incubation at 37°C, the cells were quickly acid-washed (150 mM NaCl, 10 mM acetic acid, pH 3.5) and then fixed.
TF recycling
HK2 cells were serum-starved for 60 min at 37°C, washed twice in cold PBS with 1% BSA, and then exposed to 100 μg/ml Alexa Fluor 488-TF for 60 min at 37°C (load). After extensive washing with complete fresh HEPES-buffered serum free-medium, the recycling of TF was followed by incubating the cells in TF-free complete medium (chase) for the indicated periods of time at 37°C. The cells were acid-washed (see above) before fixing.
Shiga toxin B-fragment transport
Transport of Shiga toxin B-fragment was investigated in fixed and living cells as described [68]. For experiments on fixed cells, cells on coverslips were placed on ice with 1 μg/ml Cy3-labeled B-fragment in HEPES-buffered DMEM and incubated for 30 min. Cells were washed three times with ice-cold culture medium and shifted in HEPES-buffered DMEM to either 19.5°C for 45 min or to 37°C in a CO2 incubator. After incubation for different periods of time, cells were washed three times with PBS containing 0.5 mM CaCl2 and 1 mM MgCl2, fixed with 3% PFA for 10 min, permeabilized with saponin, stained with the indicated primary and secondary antibodies, and then mounted. Slides were analyzed by confocal microscopy.
For experiments on living cells, cells were plated on 42-mm glass cover slips. Cy3-labeled B-fragment was bound to these cells on ice and then internalized at 19.5°C, as described above, together with 10 μg/ml A488-TF. The cover slip was rapidly transferred to a POC-chamber on the heated stage of a confocal microscope, culture medium at 37°C was added, and image acquisition was started. Fluorescence intensity was measured with ImageJ software (NIH) on z-projections, either from the entire cell, or from the Golgi region, as defined by B4GALT1 labeling. The ratio was calculated as an index of Golgi localization as reported in Figure 3 B. A background correction factor was determined from cells that did not have internalized B-fragment.
EGFR degradation
HK2 cells were serum-starved for 24 h and then stimulated with 100 ng/ml EGF for the indicated periods of time. Immediately after stimulation, the cells were fixed or lysed in lysis buffer (50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 1% [v:v] Triton X-100 (Sigma-Aldrich, 648,462), 5 mM ethylenediaminetetraacetic acid, 200 mM Na3VO4, 50 mM NaF, 40 mM β-glycerophosphate, and protease inhibitors). The cell lysates were analyzed by SDS-PAGE and western blotting.
Immunofluorescence
In general, cells were fixed for 10 min in 4% paraformaldehyde in PBS. However, when preservation of tubular structures was required, cells were fixed for 8 min with 0.2% glutaraldehyde and 2% paraformaldehyde. After fixation, cells were permeabilized in 0.1% (w:v) saponin (Sigma-Aldrich, 558,255), 0.5% (w:v) BSA and 50 mM NH4Cl in PBS (blocking buffer) for 30 min at room temperature. The cells were incubated for 1 h with primary antibodies in blocking buffer, washed three times in PBS, incubated for 1 h with the secondary antibodies (Alexa Fluor-conjugated), washed three times in PBS, mounted with Mowiol (Sigma-Aldrich, 475,904-M) on slides and analyzed by confocal microscopy.
Confocal fluorescence microscopy, image processing, and colocalization analysis
HK2 cells were grown to 70% confluence on glass coverslips and immunofluorescence microscopy was performed as described above. The experiments were repeated at least three times and representative images are shown. The level of colocalization was analyzed by acquiring serial sections from about 80–100 cells per sample. Images were exported in TIFF format and processed as previously described [114]. Images of samples to be compared were acquired using the same settings (i.e., laser power, photomultiplier gain and pinhole size), avoiding pixel saturation. The images were processed in the same way using ImageJ software. Channels from each image were converted into 8-bit format and the ‘Auto Local Threshold’ Plug-in with the “Default” method was used to segment grayscale images, identify the structures of interest and subtract background. The ImageJ “Analyze Particles” Plug-in was then used to identify and count the total number of the structures (with an area above 0.10 μm2), e.g., for EEA1- and LAMP1-positive structures in the Figure 1E.
TFRC, TF, EGF-R and BZiPAR fluorescence levels were quantified in CTR and WIPI1 KO cells using ImageJ. z-stack images were acquired and compressed into a single plane using the “maximum intensity Z-projection” function in ImageJ. Individual cells were selected using the freeform drawing tool to create a ROI (ROI). The “Measure” function provided the area, the mean gray value and integrated intensity of the ROI. The mean background level was obtained by measuring the intensity in three different regions outside the cells, dividing them by the area of the regions measured, and averaging the values obtained. To make a more accurate proportion background noise given off by unadhered dye has to be removed so per each cell the CTCF (corrected total cell fluorescence) was calculated using the formula: CTCF = integrated intensity of cell ROI − (area of ROI × mean fluorescence of background).
To quantify the degree of colocalization, confocal z-stacks were acquired, single channels from each image in 8-bit format were thresholded to subtract background and then the “Just Another Colocalisation Plug-in” (JACOP) was used to measure the overlap coefficient according to Manders. Manders’ coefficient indicates an overlap of the signals and represents the degree of colocalization:
where S1 represents signal intensity of pixels in the channel 1 and S2 represents signal intensity of pixels in the channel 2. The colocalization coefficients M1 and M2 describe contribution of each channel to the pixels. They are not dependent on the intensity of the overlapping pixels, but they are sensitive to background so a threshold has to be set [115]. For example, if the red-green pair of channels is selected and M1 and M2 are 1.0 and 0.2, respectively, this means that all red pixels colocalize with green pixels, but only 20% of green pixels colocalize with red ones. In our case M1 indicates the fraction of red pixels overlapping with green one, while M2 indicates the fraction of green pixels overlapping with the red ones.
Furthermore, to measure the levels of colocalization of RABs and endosomal markers on tubules in cells overexpressing EGFP-WIPI1[Sloop] (Figure 6B,D) we used an approach based on the detection of signal edges by through the “Canny or Sobel filters” Plug-in for ImageJ [116]. The surrounded fields were filled to generate binary mask images. In this way, we were able to select tubules in order to separate signal from background, but also to determine a common region for analyzing both channels (i.e., red for Rabs and green for WIPI1[Sloop]). These mask images were then subjected to thresholding as described above and the levels of colocalization were evaluated with ImageJ JACOP Plug-in by calculating Manders’ coefficient.
Confocal microscopy was performed on an inverted confocal laser microscope (Zeiss LSM 880 with airyscan) with a 63 × 1.4 NA oil immersion lens unless stated otherwise.
FRAP analysis
HK2 cells expressing EGFP-WIPI1[Sloop] and RFP-RAB4, DsRed-RAB11 and mCherry-RAB5 were plated into live-cell-imaging μ-dishes (Ibidi® Cell in Focus), incubated at 37°C in carbonate-free culture medium in a humidified temperature-controlled incubation chamber on a LSM880 confocal microscope. Cells were imaged using a 488 nm or 561 nm laser with a 63x NA 1.4 oil DIC M27 immersion objective (Zeiss). After collecting three pre‐bleaching images (pixel dwell 2.38 μsec), a selected ROI was bleached to < 80% of the original signal by a 488 nm (or 561 nm) laser pulse (100% power laser and pixel dwell 26.17 μsec). Post-bleaching images were collected at 1 s intervals for a total period of 150 s.
At each time point, the mean fluorescence intensity of the bleached ROI was corrected by the background signal (regions from the same field but without any recognizable fluorescent structures) and by the fluorescence decay of a non-bleached area. The values were expressed as a percentage of the maximum fluorescence intensity in that ROI of the last pre-bleach image.
Gel electrophoresis and western blot
Cells were plated into 12-well tissue culture test plates (TPP) until they reached 90% confluency, except for KD-cells, which were cultured until 72 h after transfection with the siRNAs. Cells were then washed three times with ice-cold PBS (phosphate-buffered saline), scraped, and proteins were extracted in ice-cold lysis buffer (150 mM NaCl, 2 mM EDTA, 40 mM HEPES, and 1% Triton X-100 supplemented with phosphatase and protease inhibitor cocktail. Protein extracts were supplemented with 1/4 volume of 5x reducing sample buffer (250 mM Tris‐Cl, pH 6.8, 5% β-mercaptoethanol, 10% SDS, 30% glycerol, 0.02% bromophenol blue) and heated to 95 °C for 5 min. The samples were run on either 8%, 10% or 12.5% SDS-polyacrylamide gels (W x L x H: 8.6 × 6.8 x 0.15 cm). The stacking gels were prepared as follows: 6% acrylamide, 0.16% bis-acrylamide, 0.1 M Tris, pH 6.8, 0.1% SDS, 0.1% TEMED, 0.05% ammonium persulfate. Running gels were: 10% or 12.5% acrylamide, 0.27% or 0.34% bis-acrylamide, 0.38 M Tris, pH 8.8, 0.1% SDS (Applichem, 475,904-M), 0.06% TEMED (Applichem, A1148), 0.06% APS (Applichem, A2941). The gels were run at constant current (20–30 mA). Proteins were blotted onto nitrocellulose membrane by the semi-dry method for 80 min at 400 mA (Trans-Blot® SD Semi-Dry Electrophoretic Transfer Cell, Bio-Rad). After incubation with the primary antibody, signals were detected by secondary antibodies coupled to infrared dyes (LI-COR) and detected on a LI-COR Odyssey Infrared Imager. Images were exported as TIFF files and processed in Adobe Photoshop. Band intensity was quantified using ImageJ band analysis [117].
Membrane-cytosol fractionation
Cell lysis and fractionation were performed on ice using pre-chilled solutions. Centrifugations and incubations were carried out at 4°C. All buffers were supplemented with 1x protease inhibitor solution and 1 mM DTT directly before use. Cells were washed 3 times with PBS and pelleted for 5 min at 300 x g. The cell pellet was resuspended in 1 ml of buffer 1 (50 mM HEPES-KOH, pH 7.4, 100 mM KCl, 1 mM MgCl2) and cell lysis was performed by using a French press. The lysate was centrifuged at 800 x g for 15 min. The supernatant was centrifuged again at 1,000 x g for 15 min. The supernatant was centrifuged for 1 h at 100,000 x g in an ultracentrifuge. This final supernatant was used as the fraction of cytosolic proteins. The pellet of this last centrifugation was solubilized in 0.5 ml of buffer 2 (20 mM Tris HCl, pH 7.4, 0.4 M NaCl, 15% glycerol, 1.5% Triton X-100), shaken for 1 h at 1400 rpm and 4°C, and then centrifuged at 9,000 x g for 30 min. The supernatant was used as the membrane protein fraction.
Statistical analysis
Differences between the means have been evaluated by Student’s t-test unless otherwise specified. They are mentioned as relevant differences only when p < 0.01. Immunofluorescence experiments were repeated independently three times and at least 50 cells were analyzed from a single experiment. Representative images are shown. Western blotting experiments were repeated at least three times and representative blots are shown. Most data are presented as the means ± standard deviation (s.d.) unless otherwise specified.
Supplementary Material
Acknowledgments
We thank Tassula Proikas-Cezanne, Haoxing Xu, Fulvio Reggiori, Ludger Johannes and Harald Stenmark for providing plasmids or proteins; the ACCESS facility of the NCCR Chemical Biology for help with automated microscopy at the beginning of this project; and Navin Gopaldass and Thibault Courtellement for discussions. This work was supported by grants from the SNSF (31003A_179306) and ERC (788442) to AM.
Funding Statement
This work was supported by the European Research Council [788442].
Disclosure statement
All authors declare no conflict of interest.
Supplemental material
Supplemental data for this article can be accessed here.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Ohsumi Y. Historical landmarks of autophagy research. Cell Res. 2014;24:9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Itakura E, Kishi C, Inoue K, et al. Beclin 1 Forms Two Distinct Phosphatidylinositol 3-Kinase Complexes with Mammalian Atg14 and UVRAG. Mol Biol Cell. 2008;19:5360–5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Vicinanza M, Korolchuk VI, Ashkenazi A, et al. PI(5)P regulates autophagosome biogenesis. Mol Cell. 2015;57:219–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kihara A, Noda T, Ishihara N, et al. Two Distinct Vps34 phosphatidylinositol 3–kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol. 2001;152:519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Petiot A, Ogier-Denis E, Blommaart EF, et al. Distinct classes of phosphatidylinositol 3ʹ-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem. 2000;275:992–998. [DOI] [PubMed] [Google Scholar]
- [6].Rusten TE, Vaccari T, Lindmo K, et al. ESCRTs and Fab1 regulate distinct steps of autophagy. Curr Biol. 2007;17:1817–1825. [DOI] [PubMed] [Google Scholar]
- [7].De Lartigue J, Polson H, Feldman M, et al. PIKfyve regulation of endosome-linked pathways. Traffic. 2009;10:883–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ferguson CJ, Lenk GM, Meisler MH.. Defective autophagy in neurons and astrocytes from mice deficient in PI(3,5)P2. Hum Mol Genet. 2009;18:4868–4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sharma G, Guardia CM, Roy A, et al. A family of PIKFYVE inhibitors with therapeutic potential against autophagy-dependent cancer cells disrupt multiple events in lysosome homeostasis. Autophagy. 2019;15:1694–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dove SK, Piper RC, McEwen RK, et al. Svp1p defines a family of phosphatidylinositol 3,5-bisphosphate effectors. Embo J. 2004;23:1922–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Proikas-Cezanne T, Takacs Z, Dönnes P, et al. WIPI proteins: essential PtdIns3P effectors at the nascent autophagosome. J Cell Sci. 2015;128:207–217. [DOI] [PubMed] [Google Scholar]
- [12].Proikas-Cezanne T, Waddell S, Gaugel A, et al. WIPI-1alpha (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy. Oncogene. 2004;23:9314–9325. [DOI] [PubMed] [Google Scholar]
- [13].Polson HEJ, De Lartigue J, Rigden DJ, et al. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy. 2010;6:506–522. [DOI] [PubMed] [Google Scholar]
- [14].Barth H, Meiling-Wesse K, Epple UD, et al. Autophagy and the cytoplasm to vacuole targeting pathway both require Aut10p. FEBS Lett. 2001;508:23–28. [DOI] [PubMed] [Google Scholar]
- [15].Guan J, Stromhaug PE, George MD, et al. Cvt18/Gsa12 is required for cytoplasm-to-vacuole transport, pexophagy, and autophagy in Saccharomyces cerevisiae and Pichia pastoris. Mol Biol Cell. 2001;12:3821–3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Obara K, Sekito T, Niimi K, et al. The Atg18-Atg2 complex is recruited to autophagic membranes via phosphatidylinositol 3-phosphate and exerts an essential function. J Biol Chem. 2008;283:23972–23980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dooley HC, Razi M, Polson HEJ, et al. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol Cell. 2014;55:238–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bakula D, Müller AJ, Zuleger T, et al. WIPI3 and WIPI4 β-propellers are scaffolds for LKB1-AMPK-TSC signalling circuits in the control of autophagy. Nat Commun. 2017;8:15637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Liao -C-C, Ho M-Y, Liang S-M, et al. Recombinant protein rVP1 upregulates BECN1-independent autophagy, MAPK1/3 phosphorylation and MMP9 activity via WIPI1/WIPI2 to promote macrophage migration. Autophagy. 2013;9:5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chowdhury S, Otomo C, Leitner A, et al. Insights into autophagosome biogenesis from structural and biochemical analyses of the ATG2A-WIPI4 complex. Proc Natl Acad Sci USA. 2018;58:201811874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Stanga D, Zhao Q, Milev MP, et al. TRAPPC11 functions in autophagy by recruiting ATG2B-WIPI4/WDR45 to preautophagosomal membranes. Traffic. 2019;20:325–345. [DOI] [PubMed] [Google Scholar]
- [22].Otomo T, Chowdhury S, Lander GC. The rod-shaped ATG2A-WIPI4 complex tethers membranes in vitro. Contact (Thousand Oaks). 2018;1:251525641881993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Baskaran S, Ragusa MJ, Boura E, et al. Two-site recognition of phosphatidylinositol 3-phosphate by PROPPINs in autophagy. Mol Cell. 2012;47:339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Krick R, Busse RA, Scacioc A, et al. Structural and functional characterization of the two phosphoinositide binding sites of PROPPINs, a β-propeller protein family. Proc Natl Acad Sci USA. 2012;109(30):E2042–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liang R, Ren J, Zhang Y, et al. Structural conservation of the two phosphoinositide-binding sites in WIPI proteins. J Mol Biol. 2019;431:1494–1505. [DOI] [PubMed] [Google Scholar]
- [26].Watanabe Y, Kobayashi T, Yamamoto H, et al. Structure-based analyses reveal distinct binding sites for Atg2 and phosphoinositides in Atg18. J Biol Chem. 2012;287:31681–31690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jeffries TR, Dove SK, Michell RH, et al. PtdIns-specific MPR pathway association of a novel WD40 repeat protein, WIPI49. Mol Biol Cell. 2004;15:2652–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Orsi A, Razi M, Dooley HC, et al. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol Biol Cell. 2012;23:1860–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Puri C, Vicinanza M, Ashkenazi A, et al. The RAB11A-positive compartment is a primary platform for autophagosome assembly mediated by WIPI2 recognition of PI3P-RAB11A. Dev Cell. 2018;45:114–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Fraser J, Simpson J, Fontana R, et al. Targeting of early endosomes by autophagy facilitates EGFR recycling and signalling. EMBO Rep. 2019;20:e47734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mellman I. Endocytosis and molecular sorting. Annu Rev Cell Dev Biol. 1996;12:575–625. [DOI] [PubMed] [Google Scholar]
- [32].Anderson RG, Brown MS, Beisiegel U, et al. Surface distribution and recycling of the low density lipoprotein receptor as visualized with antireceptor antibodies. J Cell Biol. 1982;93:523–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Dautry-Varsat A, Ciechanover A, Lodish HF. pH and the recycling of transferrin during receptor-mediated endocytosis. PNAS. 1983;80:2258–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hopkins CR. Intracellular routing of transferrin and transferrin receptors in epidermoid carcinoma A431 cells. Cell. 1983;35:321–330. [DOI] [PubMed] [Google Scholar]
- [35].Umeda A, Fujita H, Kuronita T, et al. Distribution and trafficking of MPR300 is normal in cells with cholesterol accumulated in late endocytic compartments: evidence for early endosome-to-TGN trafficking of MPR300. J Lipid Res. 2003;44:1821–1832. [DOI] [PubMed] [Google Scholar]
- [36].Katzmann DJ, Babst M, Emr SD. Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell. 2001;106:145–155. [DOI] [PubMed] [Google Scholar]
- [37].Maxfield FR, McGraw TE. Endocytic recycling. Nat Rev Mol Cell Biol. 2004;5:121–132. [DOI] [PubMed] [Google Scholar]
- [38].Luzio JP, Hackmann Y, Dieckmann NMG, et al. The biogenesis of lysosomes and lysosome-related organelles. Cold Spring Harb Perspect Biol. 2014;6:a016840–0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Cullen PJ, Steinberg F. To degrade or not to degrade: mechanisms and significance of endocytic recycling. Nat Rev Mol Cell Biol. 2018;19:679–696. [DOI] [PubMed] [Google Scholar]
- [40].Ma M, Burd CG. Retrograde trafficking and plasma membrane recycling pathways of the budding yeast Saccharomyces cerevisiae. Traffic. 2019;21:45–59:tra.12693. [DOI] [PubMed] [Google Scholar]
- [41].Seaman MNJ. Back from the brink: retrieval of membrane proteins from terminal compartments: unexpected pathways for membrane protein retrieval from vacuoles and endolysosomes. BioEssays. 2019;41:e1800146. [DOI] [PubMed] [Google Scholar]
- [42].Chen K-E, Healy MD, Collins BM. Towards a molecular understanding of endosomal trafficking by Retromer and Retriever. Traffic. 2019;20:tra.12649. [DOI] [PubMed] [Google Scholar]
- [43].Derivery E, Sousa C, Gautier JJ, et al. The Arp2/3 activator WASH controls the fission of endosomes through a large multiprotein complex. Dev Cell. 2009;17:712–723. [DOI] [PubMed] [Google Scholar]
- [44].Gomez TS, Billadeau DD. A FAM21-containing WASH complex regulates retromer-dependent sorting. Dev Cell. 2009;17:699–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Phillips-Krawczak CA, Singla A, Starokadomskyy P, et al. COMMD1 is linked to the WASH complex and regulates endosomal trafficking of the copper transporter ATP7A. Mol Biol Cell. 2015;26:91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Rojas R, Kametaka S, Haft CR, et al. Interchangeable but essential functions of SNX1 and SNX2 in the association of retromer with endosomes and the trafficking of mannose 6-phosphate receptors. Mol Cell Biol. 2007;27:1112–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Temkin P, Lauffer B, Jäger S, et al. SNX27 mediates retromer tubule entry and endosome-to-plasma membrane trafficking of signalling receptors. Nat Cell Biol. 2011;13:715–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lucas M, Gershlick DC, Vidaurrazaga A, et al. Structural Mechanism for Cargo Recognition by the Retromer Complex. Cell. 2016;167:1623–1635.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hierro A, Rojas AL, Rojas R, et al. Functional architecture of the retromer cargo-recognition complex. Nature. 2007;449:1063–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kovtun O, Leneva N, Bykov YS, et al. Structure of the membrane-assembled retromer coat determined by cryo-electron tomography. Nature. 2018;561:561–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Bar-Ziv R, Tlusty T, Moses E, et al. Pearling in cells: a clue to understanding cell shape. PNAS. 1999;96:10140–10145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Markin VS, Tanelian DL, Jersild RA, et al. Biomechanics of stretch-induced beading. Biophys J. 1999;76:2852–2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Simunovic M, Manneville J-B, Renard H-F, et al. Friction mediates scission of tubular membranes scaffolded by BAR proteins. Cell. 2017;170:172–184.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Antonny B, Burd C, De Camilli P, et al. Membrane fission by dynamin: what we know and what we need to know. Embo J. 2016;35:e201694613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Gopaldass N, Fauvet B, Lashuel H, et al. Membrane scission driven by the PROPPIN Atg18. Embo J. 2017;36:3274–3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zieger M, Mayer A. Yeast vacuoles fragment in an asymmetrical two-phase process with distinct protein requirements. Mol Biol Cell. 2012;23:3438–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Busse RA, Scacioc A, Krick R, et al. Characterization of PROPPIN-Phosphoinositide Binding and Role of Loop 6CD in PROPPIN-Membrane Binding. Biophys J. 2015;108:2223–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Tamura N, Oku M, Ito M, et al. Atg18 phosphoregulation controls organellar dynamics by modulating its phosphoinositide-binding activity. J Cell Biol. 2013;202:685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Rutherford AC, Traer C, Wassmer T, et al. The mammalian phosphatidylinositol 3-phosphate 5-kinase (PIKfyve) regulates endosome-to-TGN retrograde transport. J Cell Sci. 2006;119:3944–3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Schink KO, Raiborg C, Stenmark H. Phosphatidylinositol 3-phosphate, a lipid that regulates membrane dynamics, protein sorting and cell signalling. BioEssays. 2013;35:900–912. [DOI] [PubMed] [Google Scholar]
- [61].McCartney AJ, Zhang Y, Weisman LS. Phosphatidylinositol 3,5-bisphosphate: low abundance, high significance. BioEssays. 2014;36:52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Krick R, Tolstrup J, Appelles A, et al. The relevance of the phosphatidylinositolphosphat-binding motif FRRGT of Atg18 and Atg21 for the Cvt pathway and autophagy. FEBS Lett. 2006;580:4632–4638. [DOI] [PubMed] [Google Scholar]
- [63].Gaugel A, Bakula D, Hoffmann A, et al. Defining regulatory and phosphoinositide-binding sites in the human WIPI-1 β-propeller responsible for autophagosomal membrane localization downstream of mTORC1 inhibition. J Mol Signal. 2012;7:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Hasegawa J, Strunk BS, Weisman LS. PI5P and PI(3,5)P2: minor, but Essential Phosphoinositides. Cell Struct Funct. 2017;42:49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Proikas-Cezanne T, Ruckerbauer S, Stierhof Y-D, et al. Human WIPI-1 puncta-formation: a novel assay to assess mammalian autophagy. FEBS Lett. 2007;581:3396–3404. [DOI] [PubMed] [Google Scholar]
- [66].Hopkins CR, Trowbridge IS. Internalization and processing of transferrin and the transferrin receptor in human carcinoma A431 cells. J Cell Biol. 1983;97:508–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Beguinot L, Lyall RM, Willingham MC, et al. Down-regulation of the epidermal growth factor receptor in KB cells is due to receptor internalization and subsequent degradation in lysosomes. PNAS. 1984;81:2384–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Mallard F, Antony C, Tenza D, et al. Direct pathway from early/recycling endosomes to the Golgi apparatus revealed through the study of shiga toxin B-fragment transport. J Cell Biol. 1998;143:973–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Kupcho K, Somberg R, Bulleit B, et al. A homogeneous, nonradioactive high-throughput fluorogenic protein kinase assay. Anal Biochem. 2003;317:210–217. [DOI] [PubMed] [Google Scholar]
- [70].Chi RJ, Harrison MS, Burd CG. Biogenesis of endosome-derived transport carriers. Cell Mol Life Sci. 2015;72:3441–3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Wandinger-Ness A, Zerial M. Rab proteins and the compartmentalization of the endosomal system. Cold Spring Harb Perspect Biol. 2014;6:a022616–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Pfeffer SR. Rab GTPase regulation of membrane identity. Curr Opin Cell Biol. 2013;25:414–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Gillooly DJ, Morrow IC, Lindsay M, et al. Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. Embo J. 2000;19:4577–4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Li X, Wang X, Zhang X, et al. Genetically encoded fluorescent probe to visualize intracellular phosphatidylinositol 3,5-bisphosphate localization and dynamics. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:21165–21170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Wallroth A, Haucke V. Phosphoinositide conversion in endocytosis and the endolysosomal system. J Biol Chem. 2018;293:1526–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000;19:5720–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. 3rd Ed. Autophagy. 2016; Vol. 12, p. 1–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Scacioc A, Schmidt C, Hofmann T, et al. Structure based biophysical characterization of the PROPPIN Atg18 shows Atg18 oligomerization upon membrane binding. Sci Rep. 2017;7:14008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Boucrot E, Pick A, Camdere G, et al. Membrane fission is promoted by insertion of amphipathic helices and is restricted by crescent bar domains. Cell. 2012;149:124–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Velikkakath AKG, Nishimura T, Oita E, et al. Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol Biol Cell. 2012;23:896–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Maeda S, Otomo C, Otomo T. The autophagic membrane tether ATG2A transfers lipids between membranes. elife. 2019;8:E3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Osawa T, Kotani T, Kawaoka T, et al. Atg2 mediates direct lipid transfer between membranes for autophagosome formation. Nat Struct Mol Biol. 2019;26:281–288. [DOI] [PubMed] [Google Scholar]
- [83].Wang Y, Pennock S, Chen X, et al. Endosomal signaling of epidermal growth factor receptor stimulates signal transduction pathways leading to cell survival. Mol Cell Biol. 2002;22:7279–7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Schartl M, Wilde B, Laisney JAGC, et al. A mutated EGFR is sufficient to induce malignant melanoma with genetic background-dependent histopathologies. J Invest Dermatol. 2010;130:249–258. [DOI] [PubMed] [Google Scholar]
- [85].Girotti MR, Pedersen M, Sanchez-Laorden B, et al. Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov. 2013;3:158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Bardeesy N, Kim M, Xu J, et al. Role of epidermal growth factor receptor signaling in RAS-driven melanoma. Mol Cell Biol. 2005;25:4176–4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Cendrowski J, Mamińska A, Miaczynska M. Endocytic regulation of cytokine receptor signaling. Cytokine Growth Factor Rev. 2016;32:63–73. [DOI] [PubMed] [Google Scholar]
- [88].Simunovic M, Voth GA, Callan-Jones A, et al. When physics takes over: BAR proteins and membrane curvature. Trends Cell Biol. 2015;25:780–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Daumke O, Roux A, Haucke V. BAR domain scaffolds in dynamin-mediated membrane fission. Cell. 2014;156:882–892. [DOI] [PubMed] [Google Scholar]
- [90].Chi RJ, Liu J, West M, et al. Fission of SNX-BAR-coated endosomal retrograde transport carriers is promoted by the dynamin-related protein Vps1. J Cell Biol. 2014;204:793–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Arlt H, Reggiori F, Ungermann C. Retromer and the dynamin Vps1 cooperate in the retrieval of transmembrane proteins from vacuoles. J Cell Sci. 2015;128:645–655. [DOI] [PubMed] [Google Scholar]
- [92].Efe JA, Botelho RJ, Emr SD. Atg18 regulates organelle morphology and Fab1 kinase activity independent of its membrane recruitment by phosphatidylinositol 3,5-bisphosphate. Mol Biol Cell. 2007;18:4232–4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Peters C, Baars TL, Buhler S, et al. Mutual control of membrane fission and fusion proteins. Cell. 2004;119:667–678. [DOI] [PubMed] [Google Scholar]
- [94].Alpadi K, Kulkarni A, Namjoshi S, et al. Dynamin-SNARE interactions control trans-SNARE formation in intracellular membrane fusion. Nat Commun. 2013;4:1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Michaillat L, Mayer A. Identification of genes affecting vacuole membrane fragmentation in Saccharomyces cerevisiae. PLoS ONE. 2013;8:e54160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Michaillat L, Baars TL, Mayer A. Cell-free reconstitution of vacuole membrane fragmentation reveals regulation of vacuole size and number by TORC1. Mol Biol Cell. 2012;23:881–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Daumke O, Lundmark R, Vallis Y, et al. Architectural and mechanistic insights into an EHD ATPase involved in membrane remodelling. Nature. 2007;449:923–927. [DOI] [PubMed] [Google Scholar]
- [98].Cai B, Xie S, Caplan S, et al. GRAF1 forms a complex with MICAL-L1 and EHD1 to cooperate in tubular recycling endosome vesiculation. Front Cell Dev Biol. 2014;2:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Deo R, Kushwah MS, Kamerkar SC, et al. ATP-dependent membrane remodeling links EHD1 functions to endocytic recycling. Nat Commun. 2018;9:5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Cai B, Caplan S, Naslavsky N. cPLA2α and EHD1 interact and regulate the vesiculation of cholesterol-rich, GPI-anchored, protein-containing endosomes. Mol Biol Cell. 2012;23:1874–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Zhang J, Reiling C, Reinecke JB, et al. Rabankyrin-5 interacts with EHD1 and Vps26 to regulate endocytic trafficking and retromer function. Traffic. 2012;13:745–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].McKenzie JE, Raisley B, Zhou X, et al. Retromer guides STxB and CD8-M6PR from early to recycling endosomes, EHD1 guides STxB from recycling endosome to Golgi. Traffic. 2012;13:1140–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Boucrot E, Ferreira APA, Almeida-Souza L, et al. Endophilin marks and controls a clathrin-independent endocytic pathway. Nature. 2015;517:460–465. [DOI] [PubMed] [Google Scholar]
- [104].Renard H-F, Simunovic M, Lemière J, et al. Endophilin-A2 functions in membrane scission in clathrin-independent endocytosis. Nature. 2015;517:493–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Riezman H, Munn A, Geli MI, et al. Actin-, myosin- and ubiquitin-dependent endocytosis. Experientia. 1996;52:1033–1041. [DOI] [PubMed] [Google Scholar]
- [106].Römer W, Pontani -L-L, Sorre B, et al. Actin dynamics drive membrane reorganization and scission in clathrin-independent endocytosis. Cell. 2010;140:540–553. [DOI] [PubMed] [Google Scholar]
- [107].Simonetti B, Cullen PJ. Actin-dependent endosomal receptor recycling. Curr Opin Cell Biol. 2019;56:22–33. [DOI] [PubMed] [Google Scholar]
- [108].Suzuki SW, Emr SD. Membrane protein recycling from the vacuole/lysosome membrane. J Cell Biol. 2018;217(5):1623–1632. jcb.201709162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Stromhaug PE, Reggiori F, Guan J, et al. Atg21 is a phosphoinositide binding protein required for efficient lipidation and localization of Atg8 during uptake of aminopeptidase I by selective autophagy. Mol Biol Cell. 2004;15:3553–3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Colquhoun D. Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Br J Pharmacol. 1998;125:924–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Shalem O, Sanjana NE, Hartenian E, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343(6166):84–87. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Bagnis C, Bailly P, Chapel-Fernandes S. Using an EGFPmeter to evaluate the lentiviral vector production: tricks and traps. Methods Mol Biol. 2009;515:151–163. [DOI] [PubMed] [Google Scholar]
- [114].Vicinanza M, Di Campli A, Polishchuk E, et al. OCRL controls trafficking through early endosomes via PtdIns4,5P2-dependent regulation of endosomal actin. Embo J. 2011;30(24):4970–4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Manders EMM, Verbeek FJ, Aten JA. Measurement of co-localization of objects in dual-colour confocal images. J Microsc. 2011;169:375–382. [DOI] [PubMed] [Google Scholar]
- [116].Canny J. A computational approach to edge detection. IEEE Trans Pattern Anal Mach Intell. 1986;8:679–698. [PubMed] [Google Scholar]
- [117].Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Gautier R, Douguet D, Antonny B, et al. HELIQUEST: a web server to screen sequences with specific alpha-helical properties. Bioinformatics. 2008;24:2101–2102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.