

ABSTRACT
The architectural complexity and heterogeneity of the tumor microenvironment (TME) remains a substantial obstacle in the successful treatment of cancer. Hypoxia, caused by insufficient oxygen supply, and acidosis, resulting from the expulsion of acidic metabolites, are prominent features of the TME. To mitigate the consequences of the hostile TME, cancer cells metabolically rewire themselves and express a series of specific transporters and enzymes instrumental to this adaptation. One of these proteins is carbonic anhydrase (CA)IX, a zinc-containing extracellular membrane bound enzyme that has been shown to play a critical role in the maintenance of a neutral intracellular pH (pHi), allowing tumor cells to survive and thrive in these harsh conditions. Although CAIX has been considered a promising cancer target, only two antibody-based therapeutics have been clinically tested so far. To fill this gap, we generated a series of novel monoclonal antibodies (mAbs) that specifically recognize the extracellular domain (ECD) of human CAIX. Here we describe the biophysical and functional properties of a set of antibodies against the CAIX ECD domain and their applicability as: 1) suitable for development as an antibody-drug-conjugate, 2) an inhibitor of CAIX enzyme activity, or 3) an imaging/detection antibody. The results presented here demonstrate the potential of these specific hCAIX mAbs for further development as novel cancer therapeutic and/or diagnostic tools.
KEYWORDS: Carbonic anhydrase (CA)-IX, Antibody-drug-conjugate (ADC), enzyme inhibition, PET/SPECT, hypoxia, in vitro, in vivo
Introduction
The carbonic anhydrases (CA) are a large family of 15 distinct, but related metalloenzymes that can be found in many human organs where they play important roles in the maintenance of a neutral intracellular pH (pHi) and the secretion of electrolytes.1,2 Eight of the CAs are expressed intracellularly (CAI, II, III, VII, VIII, X, XI, and XIII), two in the mitochondria (CAVA and VB) and one is secreted (CAVI), while CAIV, IX, XII and XIV are all extracellular-facing and membrane bound.3,4 Both CAIX and CAXII have been associated with cancer progression,5,6 however the low expression levels in normal tissues of especially CAIX (mostly restricted to the gastro-intestinal tract7,8) makes this protein a very attractive therapeutic target.4,9
CAIX (aka MN or renal cell carcinoma (RCC)-associated protein G250) is a dimeric transmembrane protein consisting of two identical monomers held together by a single intermolecular disulfide bond, several hydrogen bonds and numerous van der Waals interactions.10,11 The protein has a short C-terminal intracellular tail that interacts with several intracellular proteins and functions as a cell-surface signal transducer.12,13 The extracellular domain (ECD) of CAIX consists of a catalytic domain and a N-terminally located proteoglycan (PG)-like domain that is unique for CAIX.14 The catalytic domain of CAIX regulates the reversible hydration of carbon dioxide (CO2) to bicarbonate and protons (H+), while its PG-like domain, which is rich in acidic amino acids, has been suggested to act as an intrinsic proton buffer,15 and is thought to be instrumental in tumor progression.16,17
CAIX can be found on tumor cells located in hypoxic and peri-necrotic regions, where its expression is promoted through induction of hypoxia-inducible factor 1α (HIF1α), but also by the acidifying tumor microenvironment (TME) via HIF1α-independent mechanisms.18,19 An exception are the renal tumors where homogeneous expression of CAIX can be found in >80% of primary renal cell carcinomas (RCCs)20 due to a mutation in the von Hippel Lindau tumor suppressor gene, which renders HIF1α constitutively active leading to the ubiquitous expression of CAIX in the absence of hypoxia.
The tumor-specific expression pattern of CAIX and its virtual absence in normal tissues thus makes it a very attractive therapeutic target, especially for renal cancers4,9 and hypoxic tumors. Several small-molecule inhibitors (SMI), aiming to inhibit enzymatic activity of CAIX, have been developed.5,21,22 Many of these lack CAIX specificity, although SLC-0111, which is highly selective for tumor-associated CAIX and CAXII,23 has successfully completed Phase 1 clinical trials.24 In addition, several monoclonal antibodies (mAbs) targeting CAIX have also been generated. Two of the most prominent anti-CAIX mAbs are M7525 and the G250,26 which bind to the CAIX PG-like and catalytic domain, respectively. M75 has mostly been used for CAIX detection,27 whereas the murine/human chimeric version of G250 (cG250, girentuximab), whose modus operandi relies on an antibody-dependent cellular cytotoxicity (ADCC) response,28 was clinically pursued. Although native cG250 failed to show anti-tumor efficacy in clinical trials,29 it has been used successfully as a carrier for radioisotopes.30 Furthermore, Xu et al.31 isolated a series of single-chain antibody fragments of which one (BAY 79–4620) was further developed as a monomethyl auristatin E (MMAE) antibody-drug-conjugate (ADC).32 Although Phase 1 clinical trials demonstrated potent antitumor efficacy, further development was halted due to adverse events during this trial.
Given the potential of CAIX as a promising therapeutic target, we generated a novel set of 51 mAbs directed against the hCAIX ECD. After biophysical and functional screening, 12 mAbs were selected based on their functionality and sequenced. Of these, six mAbs were recloned in either in a human IgG (c2C7, c2D7, c11H9, c12H8) or mouse IgG2a (m4A2, m9B6) framework, and recombinantly produced and purified. Further functional evaluation showed that these six mAbs can be classified as a: 1) promising ADC candidate, 2) CAIX enzyme inhibitor, and 3) imaging/detection antibody.
Results
Generation and characterization of hybridoma-derived anti-hCAIX antibodies
Mouse immunization
Although several anti-CAIX antibodies have been developed, none of these have reached the clinic as an anti-cancer treatment. We thus generated a novel set of antibodies that specifically target hCAIX and screened for those suitable as: 1) an ADC, 2) a specific hCAIX enzyme inhibitor, or 3) an imaging/detection antibody with good biophysical characteristics. To this end, we expressed a recombinant (r) version of the ECD of human (h)CAIX (Figure 1(a)) consisting of the PG-like (a.a. 38–112) and catalytic domain (a.a. 113–414), and N-terminally flanked by its natural signal peptide (a.a. 1–37). A C-terminal His tag was added to facilitate purification by immobilized metal affinity chromatography (IMAC) of the rhCAIX protein (Suppl. Fig. S1A). Mice immunized with this rhCAIX protein generated a total of 51 mAb-producing clones. Hybridoma cultures were expanded (15 mL) and mAbs were affinity-purified using Protein G for functional screening (Figure 1(b)).
Figure 1.
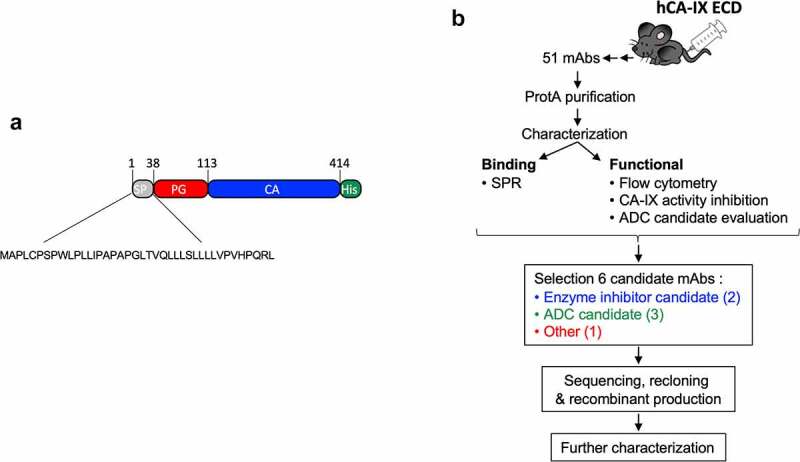
Schematic representation of the workflow used in this study to generate anti-CAIX mAbs. a) Cartoon of the rhCAIX ECD sequence (NP_001207.2) used: SP, signal peptide (gray); PG, proteoglycan-like domain (red); CA, catalytic domain (blue); His, penta-histidine tag (green) used for IMAC purification. b) Workflow of mAb production, characterization and selection process; for details see text
Antibody screening and selection
Purified mAbs were biophysically and functionally assessed (Suppl. Table 1) to identify candidates with suitable (therapeutic) characteristics. Binding studies (at 10 µg/mL) were performed using the hCAIX-positive human clear cell renal carcinoma (CCRC) SK-RC-52 cells.33 Most mAbs bind to these cells albeit with very different affinities, as is illustrated by their varying signal mean fluorescent intensities (MFI; Suppl. Fig. S2A). We also tested the binding of the mAbs using the CAIX-negative SK-RC-59 cells (data not shown) but were unable to detect signals exceeding that of the second mAb alone, indicating the hCAIX specificity of these antibodies. Using the hCAIX-positive SK-RC-52 cells, we then screened the mAbs at a fixed concentration (10 nM) for their ADC potential in a surrogate ADC assay. This assay uses a secondary anti-mouse antibody conjugated to the Saporin toxin (MAP-ZAP), a 30 kDa ribosome-inhibitor protein that is unable to enter the cell by itself and can only evoke cellular toxicity when conjugated to an internalizing antibody. Most of our antibodies induce a certain level of cell death (compared to secondary MAP-ZAP antibody alone). To select our best ADC candidates, we used the M75 mAb9 cellular response as a cutoff, and as such selected the 11H9, 12H8 and 2C7 mAbs as potential ADC candidates (Suppl. Fig. S2B; indicated in green).
CAIX also has a unique extracellular enzyme function that is critical for maintaining a neutral pHi in tumor cells residing in a hypoxic and acidic TME. Using the fluorescent methylumbelliferylacetate (4-MUA) substrate we thus assessed the enzyme activity of rhCAIX (0.5 μM) in the presence of 1 µM of the mAbs and identified 9B6 and 4A2 (Suppl. Fig. S2C; indicated in blue) as inhibitors of CAIX enzyme activity (42.59% and 61.94% enzyme inhibition compared hCAIX alone, respectively). Overall, our screening efforts allowed us to select six mAbs with unique biophysical and functional characteristics that can be used for further in-depth evaluation.
Characterization of recombinant anti-hCAIX antibodies
Evaluation of mAb binding characteristics
The complementarity-determining regions (CDRs) (Suppl. Table S2) of the ADC candidates (2C7, 11H9, and 12H8), enzyme inhibitors (4A2 and 9B6) and an imaging/detection mAb (2D7) were then grafted on either a human IgG1 (11H9, 12H8, 2D7 and 2C7) or murine IgG2a (4A2 and 9B6) backbone. Human chimeric (c) and murine (m) recombinant antibodies were expressed in Chinese hamster ovary (CHO) cells, affinity purified (Protein A) under low-endotoxin conditions. All preparations were subjected to quality control (data not shown) by sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and ultra performance liquid chromatography-size exclusion chromatography (UPLC-SEC; acceptance criteria: ≥ 95% purity) and endotoxin removal (< 0.1 EU/mg protein) prior to further use.
These antibodies were generated by immunizing mice with a mixture of monomeric and dimeric rhCAIX, which thus begs the question whether these mAbs showed any prevalence for binding to the hCAIX monomer or dimer. We thus purified the rhCAIX by size exclusion chromatography (SEC) to obtain monomeric (M) and dimeric (D) rhCAIX (Suppl. Fig. S1A-C) and used surface plasmon resonance (SPR) to evaluate binding of the antibodies to either form of rhCAIX. Antibodies were captured by immobilized hIgG1 (for c11H9, c12H8, c2D7, c2C7 and cG250) or mIgG2a (for m4A2, m9B6), after which purified rhCAIX-M or -D were flowed. Representative sensorgrams for the immobilized mAbs (Figure 2(a)) were used to calculate ka, kd and KD and apparent ka, kd and KD values, when flowing the rhCAIX-M or -D, respectively (Table 1). Significant differences in the kinetic values (ka, kd, KD and Rmax) were observed when using rhCAIX-M or rhCAIX-D (Figure 2(b)). As expected, most mAbs have much slower off-rates (kd) and a smaller KD when rhCAIX-D is flowed, except for m4A2 whose kd and KD remain relatively unchanged. m9B6 stands out in this analysis, showing a very poor binding profile. Despite these poor kinetics m9B6 does show relatively strong hCAIX enzyme inhibiting qualities (Suppl. Fig. S2C). Additional analysis (data not shown) using reverse-phase protein arrays (RPPA; using 40 non-related proteins) and SPR (using cell surface expressed human CA family members rhCAIV, rhCAXII and rhCAIV) confirmed that: 1) all six antibodies are specific for hCAIX, 2) bind cynomolgus CAIX (except c11H9), and 3) none bind dog or mouse CAIX.
Figure 2.
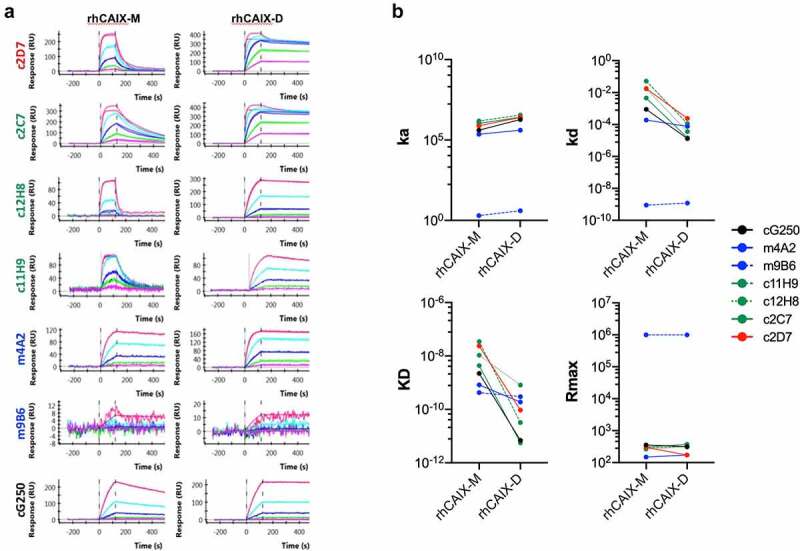
Binding of selected recombinant anti-CAIX antibodies by Surface Plasmon Resonance. Six (6) hybridoma derived mAbs were selected through screening (see Figure 1b, Suppl. Fig. S2A-C), sequenced, recloned in the appropriate IgG framework and recombinantly expressed in CHO cells (for details see text). (a) Purified recombinant antibodies (c11H9, c12H8, c2C7, m4A2, m9B6, c2D7) were captured with the appropriate anti-Fc surface (anti-human Fc: c11H9, c12H8, c2C7, c2D7; anti-mouse Fc: m4A2, m9B6). cG250 was used as a benchmark. Serial dilutions (0.74–60 nM) of rhCAIX monomer (CAIX-M) and dimer (CAIX-D) were then injected, followed by a buffer injection. Sensorgrams were aligned, double-referenced, and fitted to the 1:1 binding model to calculate ka, kd, KD and RUmax when flowing CAIX-M and apparent ka, kd, KD and RUmax when flowing CAIX-D (see Table 1). (b) Graphs depicting changes in the calculated ka, kd, KD and RUmax when flowing rhCAIX-M versus rhCAIX-D over the immobilized antibodies
Table 1.
Representative kinetic and apparent kinetic values for binding of recombinant anti-CAIX antibodies obtained by SPR analysis and by flowing rhCAIX monomer or dimer, respectively. Antibodies suitable for ADC, CAIX enzyme inhibitor and imaging/detection are listed in green, blue and red, respectively. The cG250 mAb was used as control (black)
| rhCA-IX monomer |
rhCA-IX dimer (Apparent values) |
|||||||
|---|---|---|---|---|---|---|---|---|
| ka (1/Ms) |
kd (1/s) |
KD (M) |
Rmax (RU) | ka (1/Ms) |
kd (1/s) |
KD (M) |
Rmax (RU) | |
| c2C7 | 1.05E+06 | 4.59E-03 | 4.35E-09 | 305.48 | 2.64E+06 | 1.46E-05 | 5.53E-12 | 375.47 |
| c11H9 | 1.19E+06 | 1.72E-02 | 1.07E-08 | 265.23 | 2.18E+06 | 3.59E-05 | 8.18E-10 | 308.54 |
| c12H8 | 1.52E+06 | 5.24E-02 | 3.45E-08 | 286.87 | 3.58E+06 | 1.15E-04 | 3.20E-11 | 323.60 |
| m4A2 | 2.29E+05 | 1.90E-04 | 8.26E-10 | 148.96 | 4.06E+05 | 7.68E-05 | 1.89E-10 | 172.60 |
| m9B6 | 1.97E+00 | 8.93E-10 | 4.26E-10 | 1.00E+06 | 4.02E+00 | 1.20E-09 | 2.99E-10 | 1.00E+06 |
| c2D7 | 7.57E+05 | 1.80E-02 | 2.37E-08 | 300.92 | 2.54E+06 | 2.39E-04 | 9.41E-11 | 172.60 |
| cG250 | 4.05E+05 | 8.94E-04 | 2.21E-09 | 356.64 | 1.88E+06 | 1.30E-05 | 6.92E-12 | 316.37 |
To determine the binding epitope of these antibodies, we applied the PepScan technology (https://www.pepscan.com), using both linear and single-loop conformations of the hCAIX ECD domain. Although this technology does not allow us to assign any definitive-binding epitopes to the antibodies, the results do give some insight into the domain to which these antibodies interact with (Suppl. Fig. S3), i.e., c11H9 and c12H8 likely bind the PG-like domain, whereas m4A2, c2C7 and c2D7 most likely bind the catalytic domain. No signals could be detected when using m9B6, which due to its poor-binding kinetics (Figure 2), is likely washed away during the assay procedure. However, based on the functional characteristics of m9B6 (i.e., hCAIX enzyme inhibition; see Suppl. Fig. S2C), it can be assumed that this antibody binds hCAIX’s catalytic domain. To gain better insight into the binding characteristics and assign binding epitopes to these antibodies, we investigated the catalytic domain-binding antibodies (m4A2, m9B6, c2C7 and c2D7) by Hydrogen deuterium exchange mass spectrometry,34 and those binding the PG-like domain (c11H9 and c12H8) by high resolution nuclear magnetic resonance (data not shown, manuscript in preparation).
Cell surface binding of recombinant anti-hCAIX antibodies
We next sought to further evaluate the cell-binding characteristics of the six antibodies in a plate-based cell binding assay. For this we used SK-RC-52 cells that were incubated with serial antibody dilutions, and used cG250 and M75, two well-characterized hCAIX antibodies, as benchmarks. Figure 3(a) shows that the chimeric antibodies display a variety of binding profiles, which, except for c12H8, are similar to that of cG250, with calculated half-maximal effective concentration (EC50) values ranging from 0.22 nM (c12H8) to 3.483 nM (c11H9) and Bmax values of between 157.1 (c12H8) and 544.4 (c2C7). It should be noted that the reported EC50 value for c11H9 is an estimate, as a binding plateau is not reached. Evaluation of the murine antibodies (Figure 3(b)) confirmed the binding superiority of the M75 antibody (Bmax, 1254; EC50, 1.416). However, while the Bmax value of m4A2 (689.7) is lower than that of M75, its EC50 is very similar (1.156 nM) to that of M75. The cell-binding profile for m9B6 confirmed the poor-binding kinetics observed by SPR (Figure 2(a)), which precluded the calculation of an EC50 for this antibody. A complete overview of the calculated EC50 and Bmax values for the antibodies can be found in Table 2.
Figure 3.
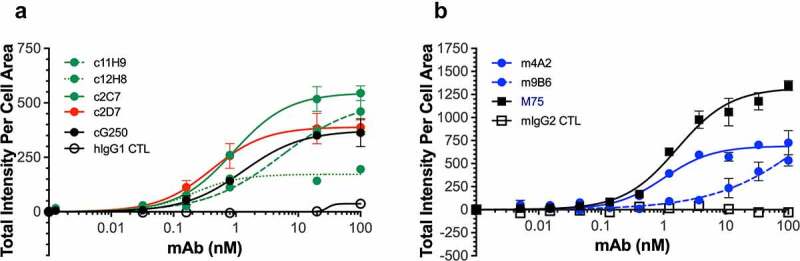
Binding of recombinant antibodies to hCAIX-expressing SK-RC-52 cells. Dose-dependent binding (0–100 nM) to SK-RC-52 cells of recombinantly expressed (a) chimeric antibodies (c11H9, c12H8, c2C7, c2D7) and (b) murine antibodies (m4A2, m9B6), together with the appropriate negative (hIgG1, mIgG2) and positive (cG250, M75) controls. Experiments were carried out in duplicate and repeated twice. Representative KD, Hill slope and Bmax (see Table 2) values were calculated using Graphpad Prism v8
Table 2.
Apparent equilibrium binding constants (EC50) and maximum specific binding (Bmax) values for the recombinant antibodies using SK-RC-52 cells expressing hCA-IX. cG250 and M75(both in black) were used as positive controls; negative controls (not listed) showed no binding (ADC candidates, green; enzyme inhibitors, blue and imaging/detection antibody, red)
| EC50 (nM) | Bmax | |
|---|---|---|
| c2C7 | 0.8234 | 544.4 |
| c11H9 | 3.483 | 479.9 |
| c12H8 | 0.2206 | 157.1 |
| m4A2 | 1.156 | 689.7 |
| m9B6 | 21.78 | ND |
| c2D7 | 0.5195 | 400.9 |
| M75 | 1.416 | 1254 |
| cG250 | 1.410 | 375.6 |
ADCC response
Clinically used cG250 antibody has been described to evoke an ADCC response.35 To evaluate the ADCC response of our antibodies, we used SK-RC-52 target cells (high CAIX) in conjunction with Promega’s human FcγRIIIa (for c11H9, c12H8, c2C7, c2D7; Figure 4(a) and murine FcγRIV (for m4A2, m9B6; Figure 4(b)) in a Jurkat-Luc bioreporter assay. We observed a difference in the ADCC response level between the chimeric and murine antibodies, which is likely a combination of the antibody affinity for hCAIX (e.g., m9B6 does not reach a plateau as was shown in the cell-binding studies) and the human and mouse Fcγ receptor for the respective human and mouse Fc portions of the antibodies. To our surprise, we observed that only antibodies that bind to the catalytic domain of CAIX (i.e., c2C7, c2D7, m4A2, m9B6), but not those that bind the PG-like domain (i.e., c11H9, c12H8), are unable to induce an ADCC response (Figure 4(a,b)). These data thus imply that the nature of an antibody-binding epitope (i.e., structured versus unstructured) may determine whether an ADCC response is induced.
Figure 4.
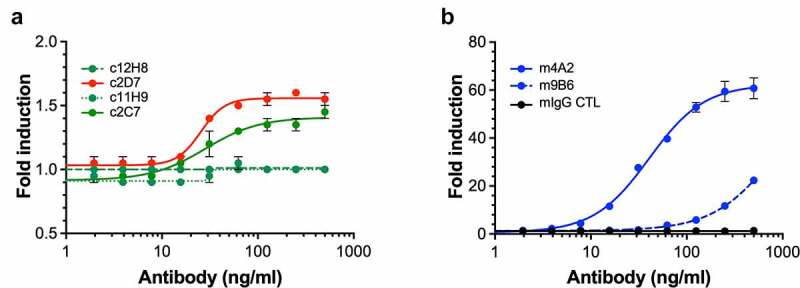
ADCC response of (a) chimeric (human Fc portion) antibodies (c2C7, c11H9, c12H8 and c2D7) and (b) murine antibodies (m4A2 and m9B6) using SK-RC-52 cells in conjunction with Promega’s Promega’s human FcγRIIIa or murine FcγRIV Jurkat-Luc bioreporter assay, respectively. Shown are the results of a representative experiment (± s.e.m.) carried out in triplicate and repeated twice; ADCC response was expressed as fold-induction of non-treated control cells, and data was analyzed using Graphpad Prism v8 software
Inhibition of carbonic anhydrase IX enzyme activity
The expression of enzymatically active CAIX is instrumental for the survival of tumor cells in a hypoxic and/or acidic TME. To further evaluate the m4A2 and m9B6 CAIX enzyme inhibition, we used two approaches. First, we used the same method as was used during our screening, i.e., rhCAIX-D and 4-MUA fluorescent peptide as substrate. Figure 5(a) shows that over time, at a fixed antibody concentration, m9B6, and even more so m4A2, significantly inhibit CAIX’s enzyme activity when compared to the isotype-specific mIgG control. Next, we evaluated the effect of the most potent enzyme inhibiting antibody, m4A2, on the enzymatic activity of rhCAIX in the presence of CO2, the physiologic substrate for CAIX (Figure 5(b,c)). We observed that m4A2 significantly inhibits rhCAIX activity in a dose-dependent manner, with a 5x molar excess of antibody resulting in complete inhibition, relative to the mIgG control. In contrast, similar concentrations of c2D7, which also binds the catalytic domain, did not inhibit CAIX activity (Figure 5(b,c)). Overall, these data show that m4A2, and to a lesser extent m9B6, are potent and specific inhibitors of the catalytic activity of CAIX.
Figure 5.
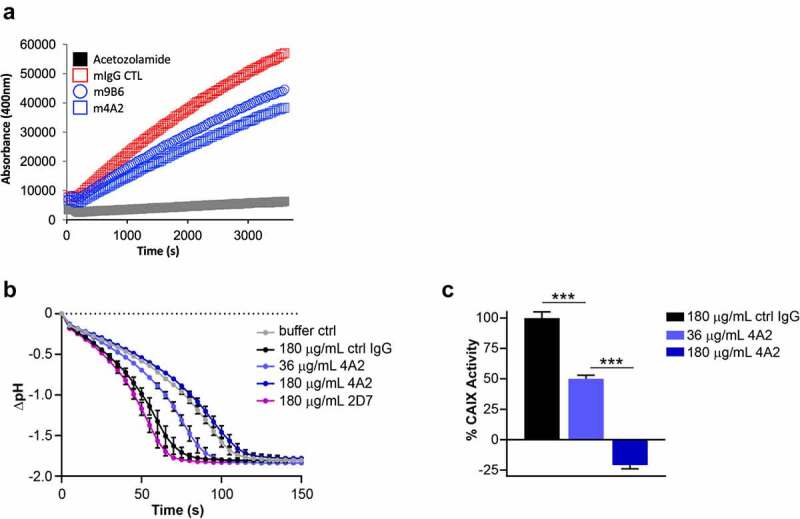
Evaluation of anti-hCAIX antibodies for inhibition of catalytic activity of CAIX. (a) In vitro assay assessing rhCAIX enzyme activity (rhCAIX dimer: 0.5 µM) as a function of 4-MUA (100 µM) substrate-released fluorescence over time in the presence of m4A2 (1 µM, blue open square) or m9B6 (1 µM, blue open circle). Murine IgG CTL mAb (1 µM, red open square) and SMI Acetazolamide (10 µM, black square) were used as negative and positive controls, respectively. (b) Kinetic analysis of the activity of rhCAIX dimer using the physiologic substrate of CAIX, CO2, in response to increasing concentrations of m4A2. Normal mouse IgG and non-function-blocking antibody c2D7 were used as negative controls. The spontaneous change in pH in absence of rhCAIX dimer is shown for reference. Data are presented as the mean ± s.e.m. of technical replicates (n = 3/group) and are representative of 2 independent experiments. (c) Quantification of the relative percent activity of rhCAIX dimer in response to increasing concentrations of m4A2, calculated from the AUC of data in panel B. Data are presented as the mean ± s.e.m. of technical replicates (n = 3/group) and are representative of 2 independent experiments (***P < .001; ANOVA)
Recombinant anti-hCAIX antibodies can be used to detect hCAIX in tissues and in vivo
Initially, we evaluated the six selected CAIX mAbs purified from hybridoma supernatants for the detection of hCAIX in formalin-fixed parafin-embedded (FFPE) tumor tissue sections, using a commercially available CAIX mAb as a benchmark positive control. We focused on human breast tumor xenografts and primary human breast tumor tissues in which we previously demonstrated the presence of strong, hypoxia-induced expression of CAIX.36 We observed that mAb 11H9 showed strong staining of hCAIX in sequential tissue sections from the same MDA-MB-231 LM2-4 orthotopic human breast tumor xenografts (Suppl. Fig. S4A) and human primary breast cancer samples (Suppl. Fig. S4B), with a membrane-specific pattern similar to what is observed for the positive control mAb IgG Ctrl). In addition, mAb 9B6 showed very weak staining in a few regions of the tissues at a concentration of 10 ug/ml, while mAbs 2C7, 12H8, 2D7 and 4A2 did not stain hCAIX in these tissue sections (Suppl. Fig. S4A, B). Next, we stained tumor tissue sections of tumors from trametinib-treated pancreatic ductal adenocarcinoma (PDAC) xenografts, which we have previously demonstrated exhibit high levels of hypoxia-driven hCAIX expression,37 with purified, recombinant c11H9 and m9B6. The M75 anti-CAIX mAb, widely considered a gold standard for immunohistochemistry (IHC)-based detection of CAIX,37,38 was used as a control. We observed robust, membrane-localized staining of hCAIX in PDAC xenografts by both c11H9 and m9B6, with a staining pattern and intensity that mirrored that observed with the M75 mAb (Figure 6(a)). Collectively, these results suggest that c11H9 and m9B6, which recognize epitopes in the PG-like and catalytic domains, respectively, are good candidates for use as diagnostic/biomarker reagents in clinical samples.
Figure 6.
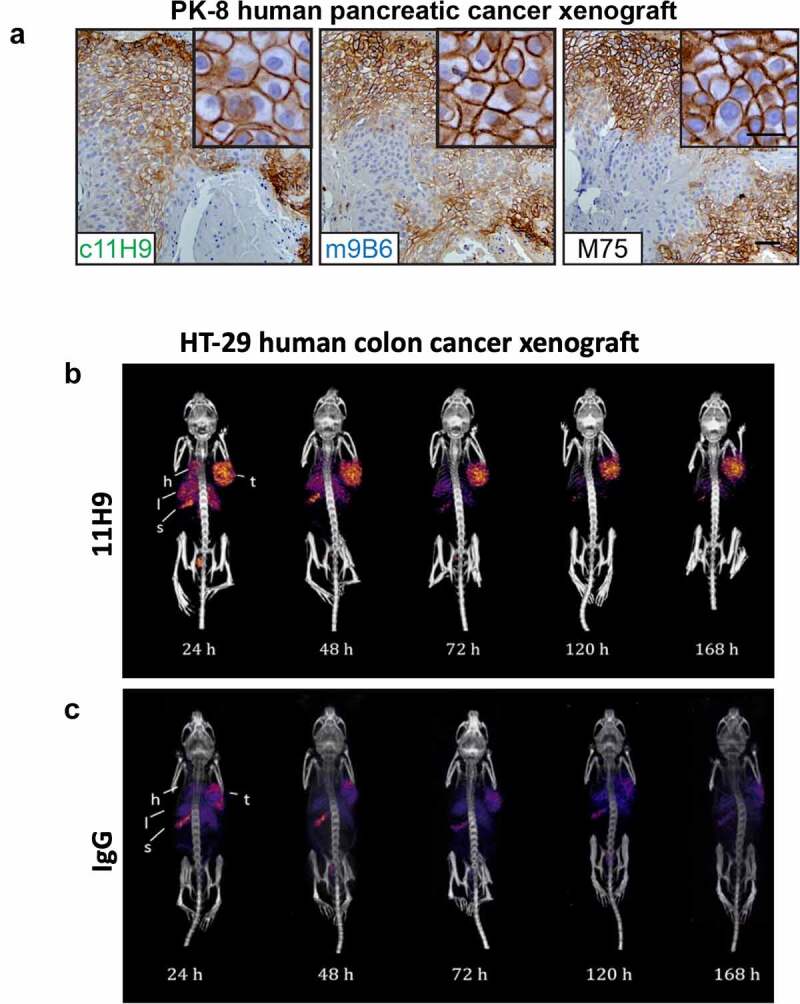
Assessment of recombinant anti-hCAIX antibodies as tools for immunohistochemistry and in vivo imaging applications. (a) Immunohistochemical staining for expression of CAIX in FFPE tissue sections from PK-8 human PDAC xenografts using recombinant antibodies c11H9, m9B6 and c2D7. Commercial anti-CAIX mAb, M75, was used as benchmark-based positive control. Scale bar, 100 μm; inset, 20 μm. Anti-CAIX antibody 11H9 (b) and a (c) control antibody (IgG) were conjugated to the chelator pSCN-Bn-DTPA and radiolabelled with 111In. Conjugates were administered to NODSCID IL2RKO mice bearing subcutaneous hCAIX-positive HT-29 colorectal cancer xenografts (100 mm3). Uptake, accumulation and retention of the radiolabelled 11H9 and IgG control were monitored 24–168 h post-injection by SPECT/CT imaging (B, C; see also Table 3) (t, tumor; l, lung; s, stomach; h, heart)
While both c11H9 and m9B6 mAbs bind effectively to CAIX in FFPE tissues, our data also showed that the binding kinetics of the m9B6 by SPR (Figure 2) and on live cells is very poor compared to that of c11H9 (Figure 3(a)). We therefore used c11H9 to further confirm whether this antibody could also be used to detect hCAIX expressed by tumors in vivo. We thus labeled c11H9 (and c2D7, which was earmarked as suitable for imaging/detection) in addition to an isotype IgG control, with Indium-111 (111In). Radiolabeled antibodies were administered intravenously (i.v.) to mice bearing subcutaneous hCAIX-expressing HT-29 tumors, and their uptake and retention by the tumor and various organs was monitored by biodistribution experiments (Table 3) and single-photon emission computerized tomography/computerized tomography (SPECT/CT) imaging (Figure 6(b,c)). These data not only show that significant amounts of the c11H9 radio-labeled antibodies can be found in the tumor at 24-hrs post-injection (p.i.), but also that 111In-c11H9 is being retained in the tumor over time (Figure 6(b)) compared to the control antibody (Figure 6(c)). Table 3 shows that at 72 h p.i. the mean percent injected dose per gram (%ID/g) of 111In-11H9 was 37.8 ± 4.7%ID/g, which is approximately two-fold higher than c2D7 (19.1 ± 4.8%ID/g). At 168 h, p.i. 35.6%ID/g was detected in the tumor, with little radioactivity in the kidneys and other organs. This contrasts with the 111In-c2D7 and the 111In-IgG, which were almost undetectable at 168 h p.i. These results thus demonstrate the hCAIX-specific binding of c11H9 antibody and its suitability as a tool for the detection of hCAIX in vitro and in vivo.
Table 3.
Biodistribution and tumor/non-target ratios for 111In-SCN-Bn-DTPA-mAbs (ADC candidate, green; detection/imaging antibody, red) 72 h and 168 h post injection (p.i.)
| Organ |
111In-SCN-Bn -DTPA-2D7 |
111In-SCN-Bn -DTPA-11H9 |
111In-SCN-Bn -DTPA-IgG |
|
|---|---|---|---|---|
| 72 h p.i. | 72 h p.i. | 168 h p.i. | 168 h p.i. | |
| (n = 4) | (n = 3) | (n = 1) | (n = 3) | |
| Blood | 6.68 ± 1.41 | 20.62 ± 4.33 | 11.06 | 11.37 ± 2.55 |
| Fat | 1.97 ± 0.69 | 1.88 ± 0.47 | 2.34 | 1.92 ± 0.99 |
| Intestine | 3.05 ± 0.40 | 3.25 ± 0.30 | 1.76 | 2.91 ± 1.57 |
| Stomach | 2.12 ± 0.38 | 2.21 ± 0.44 | 1.98 | 3.08 ± 0.55 |
| Spleen | 78.16 ± 48.53 | 37.84 ± 4.69 | 32.12 | 66.33 ± 22.81 |
| Liver | 18.66 ± 4.42 | 18.68 ± 2.22 | 14.41 | 14.60 ± 2.55 |
| Pancreas | 2.69 ± 0.53 | 2.95 ± 0.59 | 2.09 | 2.42 ± 0.94 |
| Kidney | 10.83 ± 2.78 | 12.86 ± 2.54 | 6.75 | 6.95 ± 1.20 |
| Lungs | 5.88 ± 1.52 | 9.54 ± 0.92 | 9.55 | 4.06 ± 2.92 |
| Heart | 5.49 ± 1.52 | 6.74 ± 1.04 | 3.50 | 2.40 ± 1.81 |
| Muscle | 1.87 ± 0.38 | 2.26 ± 0.29 | 1.35 | 1.48 ± 0.61 |
| Bone | 6.25 ± 2.98 | 4.46 ± 1.62 | 2.83 | 5.55 ± 2.02 |
| Brain | 0.18 ± 0.05 | 0.33 ± 0.04 | 0.20 | 0.25 ± 0.02 |
| Tumor | 19.09 ± 4.81 | 37.84 ± 4.69 | 35.64 | 14.01 ± 0.23 |
| Tumor/muscle | 19.09 ± 4.81 | 15.22 ± 0.64 | 26.38 | 10.47 ± 3.72 |
Values are presented as mean ± SD where possible.
Evaluation of anti-hCAIX mAbs as Antibody-Drug Conjugates in vitro
We next set out to determine the potential of the antibodies to function as ADC candidates. We first assessed the internalization characteristics of our antibodies, which is an important parameter for an ADC. Antibodies were conjugated with pHAb dye, which emits a low fluorescence signal at neutral pH (i.e., when outside the cells), but an increasingly stronger fluorescence signal at lower pH, conditions found in endosomes and lysosomes. SK-RC-52 were incubated with serial dilutions of pHAb-conjugated antibodies (similar avg. dye-to-antibody ratio of 3–4 and >93% monomer content by SEC), at 4°C (to avoid internalization), and after removing unbound antibodies (wash), internalization was initiated by transferring the cells to 37°C. Figure 7(a) shows the internalization and accumulation of c2C7-pHAb (increasing yellow fluorescence), but not the hIgG-pHAb control, by SK-RC-52 cells (nuclei in blue) incubated for up to 24 h. Similar sets of images were obtained for the other antibodies. Figure 7(b) clearly shows that all six antibodies internalize and accumulate in the cell in a dose-dependent and hCAIX-specific manner, as negligible signals are detected for the hIgG-pHAb control. Nonetheless, the cellular accumulation varies significantly amongst the antibodies, reflecting not only their differing intrinsic binding properties (Figure 2(a) and Figure 3(a,b)), but also points toward differences in the cellular routing of the hCAIX-mAb complex. Plotting the fold-change in the fluorescent signal between t = 0 h and 24 h (Figure 7(c)) shows a strong accumulation over time of c2C7, c2D7 and c11H9, while the values for c12H8 and m9B6 are much lower (c2C7> c2D7> c11H9> m4A2> c12H8> m9B6). The cell-binding characteristics of m4A2 are superior to those of c2C7, c2D7 and c11H9 at 10 μg/mL, and yet this antibody accumulates in the cells ~50% less than the aforementioned antibodies. These results thus indicate that affinity (KD) and maximum specific binding (receptor occupancy, Bmax) are important, but not the sole parameters that define internalization and cellular accumulation of an antibody, and that processing and routing of the CAIX-antibody complex play an important role. Nonetheless, these data confirm c2C7 as our best ADC candidate (Suppl. Fig. S2B). To further investigate the ADC efficacy, we conjugated c2C7, and c11H9 and c12H8 (which were also marked as potential ADC candidates; Suppl. Fig. S2B), in addition to an isotype-specific control to the cytotoxic payload mertansine (DM1) and tested these on several hCAIX-expressing cancer cells.
Figure 7.
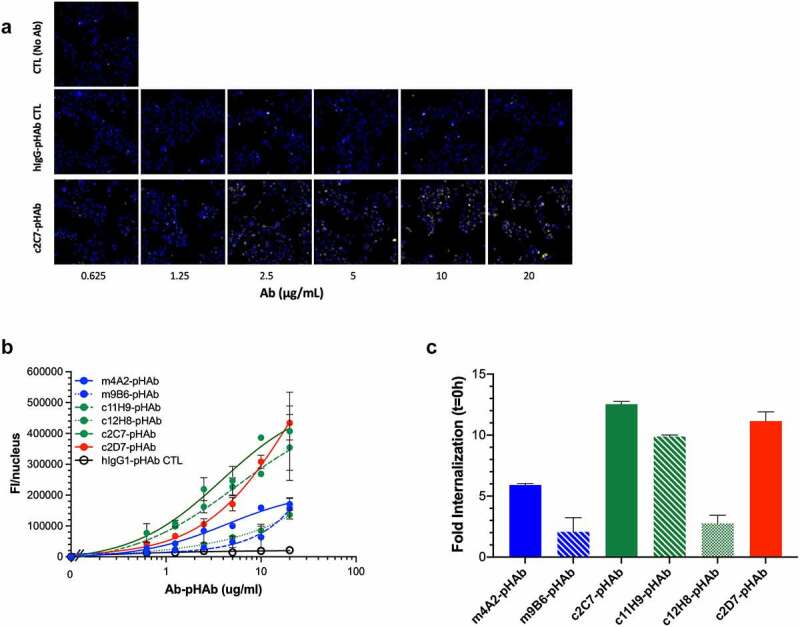
Evaluation of internalization and ADC potential of the CAIX antibodies in vitro (a) Representative processed images of SK-RC-52 cells incubated with serial dilutions of pHAb-labeled c2C7 (bottom) and hIgG-pHAb control (middle). Untreated cells (top) were used as background control (yellow, antibody; blue, nuclei). (b) Internalization dose-response (0–20 µg/mL) of pHAb-labeled mAbs by SK-RC-52 cells that were ‘coated’ with mAbs for 1h at 4°C, washed and then transferred to 37°C for 24 h. Cells were imaged using the ImageXpress Micro (IXM) Widefield High Content Screening System, and images were processed using MetaXpress imaging software and further analyzed using Graphpad Prism v8. (c) Calculation of the total amount of internalized pHAb-labeled mAb (10 μg/mL, at t = 24 h) by SK-RC-52 cells, corrected for nonspecific internalization and expressed as fold change from t = 0 h. For all graphs, ADC candidates (c11H9, c12H8, c2C7), enzyme inhibitors (m4A2, m9B6), the imaging/detection antibody (c2D7), and the control antibody are depicted in green, blue, red, and black respectively
Initially, we examined the ADC response on 67NR mouse breast tumor cells engineered to constitutively express hCAIX, and parental 67NR cells, which do not express hCAIX (Figure 8(a)). We found that all three DM1-conjugated CAIX antibodies robustly reduced viability of CAIX-positive cells in a dose-dependent manner (Figure 8(b); blue curves). In contrast, the ADCs did not appreciably inhibit the viability of the parental 67NR cells (Figure 8(b); black curves), although very modest inhibition was observed at the highest dose of c12H8-DM1. The half-maximal inhibiting concentration (IC50) values calculated from the viability curves demonstrated a spectrum of potency, enabling the ADCs to be ranked according to their IC50 values (Figure 8(b)): c2C7-DM1 (IC50 = 0.6 nM) < c11H9 (IC50 = 1.2 nM) < c12H8 (IC50 = 3.6 nM). Similarly, a DM1-conjugated isotype-specific hIgG control had no effect on the viability of either cell type at all except the highest concentration, demonstrating the target specificity of the DM1-conjugated mAbs. To further investigate the potential of c2C7 as a lead candidate ADC, we performed similar viability assays using MIA PaCa-2 human PDAC cells engineered to constitutively express CAIX (Figure 8(c)). Congruent with the data using the 67NR cell model, c2C7-DM1 potently inhibited the viability of hCAIX-positive MIA PaCa-2 cells with an IC50 of 88pM (Figure 8(d); left panel, blue curves), while the viability of hCAIX-negative control cells was not affected (Figure 8(d); left panel, black curves), demonstrating that c2C7-DM1 functions as a target-specific ADC in cells expressing hCAIX. In addition, non-conjugated c2C7 mAb did not adversely affect the viability of either cell line (Figure 8(d), right panel), further confirming the specificity of the DM1-conjugated antibody.
Figure 9.

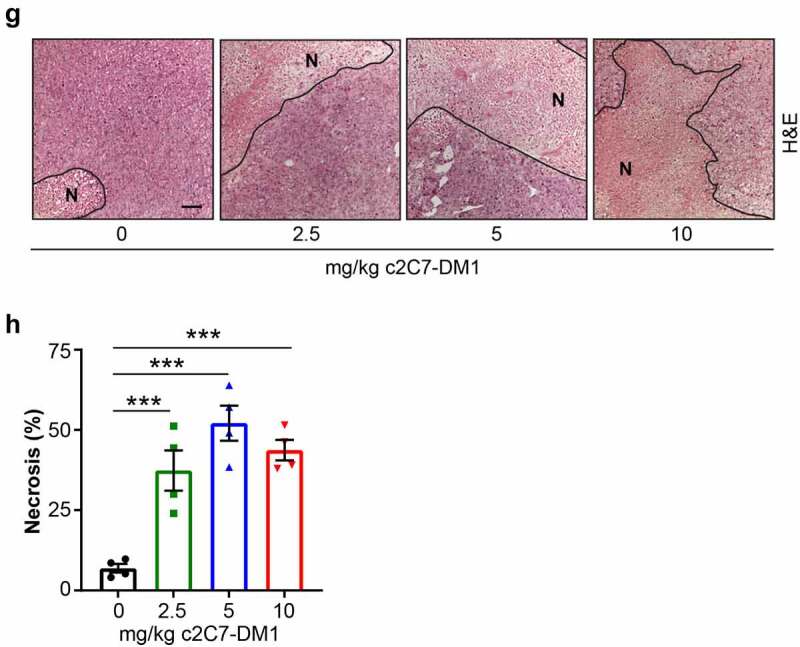
Administration of c2C7-DM1 ADC to mice bearing CAIX-positive MIA PaCa-2 tumors increases tumor cell death. (a) Representative images of tumor tissue sections from CAIX-positive MIA PaCa-2 PDAC xenografts stained for expression of CAIX. Scale bar, 100 μm; inset, 20 μm. (b-d) Mice bearing subcutaneous CAIX-positive MIA PaCa-2 PDAC xenografts were administered increasing doses of c2C7-DM1 (n = 8 mice/group). (b) Study timeline and tumor growth curve. ***P < .001; **P < .01; two-way ANOVA. (c) Tumor growth when control mice harboring tumors and administered vehicle reached the study endpoint. ***P < .001; *P < .05; two-way ANOVA. (d) Survival analysis of tumor-bearing mice administered 10 mg/kg c2C7-DM1, compared to mice given vehicle control. (e) Representative images of tumor tissue sections from CAIX-positive MIA PaCa-2 xenografts administered increasing doses of c2C7-DM1 and stained for cleaved caspase 3, a marker of apoptosis (black arrows). Scale bar, 50 μm; inset, 10 μm. (f) Quantification of cleaved caspase 3 (n = 5, each 5 fields). ***P < .001; *P < .05; two-way ANOVA. (g) Representative images of tumor tissue sections from CAIX-positive MIA PaCa-2 xenografts described in panel A stained with H&E to evaluate regions of necrosis. Boundaries between viable and necrotic tissue () are denoted with a black line. Scale bar, 100 μm. (h) Quantification of necrosis (n = 4, each 5 fields). ***P < .001; *P < .05; two-way ANOVA
Figure 9.
Continued
Figure 8.
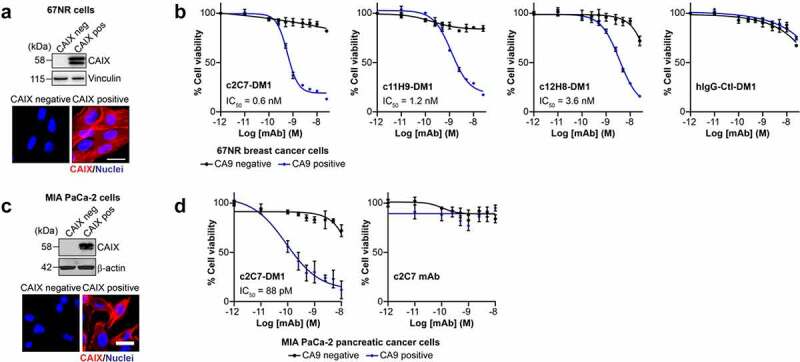
Evaluation of the ADC potential of the CAIX antibodies in vitro. (a) Analysis of expression of hCAIX by 67NR mouse breast cancer cells engineered to constitutively express hCAIX (CAIX-pos), compared to parental, hCAIX-negative (CAIX-neg) cells. Top panel, immunoblot for CAIX. Vinculin was used as a loading control. Bottom panel, immunofluorescence images. Scale bar, 10 μm. (b) Viability of cells described in panel A cultured with increasing concentrations of DM1-conjugated (blue graphs) and non-conjugated (black graphs) CAIX mAbs or a nonspecific IgG CTL antibody. Data show the mean ± s.e.m. of technical replicates (n = 3/group) and are representative of 2 independent experiments. Representative IC50 values were calculated using Graphpad Prism v8. (c) Analysis of expression of hCAIX by MIA PaCa-2 human PDAC cells engineered to constitutively express hCAIX (CAIX-pos), compared to hCAIX-negative (CAIX-neg) control cells. Top panel, immunoblot for CAIX. Vinculin was used as a loading control. Bottom panel, immunofluorescence images. Scale bar, 20 μm. (d) Viability of cells described in panel C cultured with increasing concentrations of c2C7-DM1 (left) or non-conjugated c2C7 mAb (right) (blue graphs, CAIX pos cells; black graphs, CAIX negative cells). Data show the mean ± s.e.m. of technical replicates (n = 3/group) and are representative of 2 independent experiments. Representative IC50 values were calculated using Graphpad Prism v8
Evaluation of c2C7 antibody as an antibody-drug conjugate in vivo
With c2C7 as our confirmed ADC lead, we next set out to investigate the effect of c2C7-DM1 on tumor growth in vivo and implanted the hCAIX-positive MIA PaCa-2 cells subcutaneously into the back of immune-compromised mice. This allowed the tumors, which robustly express CAIX (Figure 9(a)), to establish to a volume of ~100mm3. We then administered increasing doses (0, 2.5 5, 10 mg/kg) of c2C7-DM1 to the mice by i.v. injection twice a week for 3 weeks, which resulted in a significant, dose-dependent inhibition of tumor growth, compared to mice given the vehicle control (Figure 9(b,c)). Furthermore, administration of the ADC at a dose of 10 mg/kg resulted in 100% survival of mice 21 days after initiation of treatment, which is in stark contrast to the vehicle control group of mice (Figure 9(d)). Collectively, these data show that c2C7-DM1 is effective at controlling tumor growth and extending survival of mice harboring CAIX-positive tumors.
Analysis of the tumors at the study endpoint for cleaved caspase 3, a biomarker of apoptosis, showed significantly elevated levels of cleaved caspase 3 in tumors from mice administered c2C7-DM1, compared to vehicle controls (Figure 9(e,f)). Furthermore, significantly higher levels of cleaved caspase 3 were detected at the 10 mg/kg dose, compared to the 2.5 mg/kg dose, demonstrating dose-dependent efficacy of the ADC (Figure 9(f)). Evaluation of the extent of necrosis revealed similar results, with tumors from animals administered the ADC showing significant increases in necrotic tissue area, compared to vehicle controls (Figure 9(g,h)).
Discussion
In the past decade, human mAbs have become an established and well-tolerated treatment option for many different types of cancer. Increasing evidence has indicated that CAIX is a very promising target for both detection and immune-therapy39 in human cancers due to its almost exclusive tumor-specific expression pattern. Here, we present data on a panel of 51 mouse mAbs that were raised against the extracellular domain of hCAIX and that were subjected to a thorough biophysical and functional assessment. Six antibodies with good-binding profiles, hCAIX specificity and functionality (enzyme inhibitor and/or ADC candidate), were selected and sequenced. Three ADC candidates (11H9, 12H8 and 2C7), two enzyme inhibitors (4A2 and 9B6) and one nonfunctional imaging/detection antibody (2D7) were recloned in either a human IgG1 (c11H9, c12H8, c2C7, c2D7) or murine IgG2a (m4A2 and m9B6) framework for large-scale recombinant production and purification. Guided by the screening results, this set of recombinant antibodies was then, both biophysically and functionally, re-assessed in-depth with the aim to identify the best antibodies for further development as novel hCAIX targeting anti-cancer therapeutics.
SPR experiments indicated that the recombinantly produced antibodies, compared to the hybridoma derived mAbs, all retained their binding characteristics. Experiments were carried out on both purified hCAIX monomer and dimer preparations. Interestingly, these results show that the off-rates of the enzyme-inhibiting antibodies (m4A2 and m9B6), in contrast to that of other antibodies, hardly changed when using the hCAIX dimer. This indicates that the binding of the enzyme-inhibiting antibodies, is not affected by avidity. In other words, the location of the binding epitopes of these two antibodies on the CAIX monomer in a dimer configuration does not allow for both arms of a single antibody to bind (avidity), nor does it allow for the binding of two of the same antibody molecules at any given time. In light of this, it should thus be emphasized that the kinetic values obtained for all antibodies, other than m4A2 and m9B6, when flowing dimeric rhCAIX are apparent values, as a dimeric target in combination with a bivalent antibody can introduce avidity. However, we have data showing that one m4A2 and one m9B6 antibody molecule can bind a rhCAIX dimer simultaneously, indicating these antibodies bind to non-competing epitopes.34
One of the mechanisms that can be triggered by antibody therapies, and that is considered to contribute to the killing of tumor cells, is an ADCC response. This immune mechanism is activated through the engagement of the Fc portion of the treatment antibody with the Fcγ receptor-expressing effector cells.40 This is in fact the mechanism of action that has been reported for the clinically tested cG250 antibody.41 Testing our antibodies revealed that, surprisingly, only those antibodies binding the catalytic domain of hCAIX (c2C7, c2D7, m4A2, m9B6) trigger an ADCC response, albeit at very different levels, whereas those binding the PG-like domain (c11H9, c12H8) do not provoke such response. The latter could be related to the ‘floppy’ unstructured character of the PG-like domain, which may prevent the proper cross-linking of the FcγRs expressed by the effector cells and thus prevent an ADCC response. Nonetheless, this intrinsic characteristic may be beneficial for the use of these two antibodies in applications for which an ADCC response is unwanted (i.e., mitigating unwanted toxicity and side effects), without having to re-engineer their Fc portion. On the other hand, the therapeutic effect of antibodies such as m4A2, which functionally inhibits CAIX, may thus benefit from this added intrinsic ADCC response.
When evaluating the binding values of c2C7, c2D7, C11H9 and c12H8 at 10 μg/mL, a correlation can be observed between their binding and their accumulation at that concentration after 24 h of exposure. For example, c2C7, which is a good binder, is also the best internalizing antibody, whereas c12H8 is a relatively poor binder, a characteristic that is reflected in its poor cellular accumulation. However, when comparing the murine m4A2 and m9B6 antibodies to the chimeric antibodies, it could be concluded that avidity does not benefit the internalization process per sé; for example, the binding signal of m4A2 is superior to that of c2C7, nonetheless m4A2 only accumulates in the cells to ~50% of the level of c2C7. These data also demonstrate that for CAIX, whose ECD consists of two very different domains (catalytic and PG-like), binding to the unstructured PG-like domain (i.e., c11H9 and c12H8) or the structured catalytic domain (i.e., m4A2, c2C7, and c2D7), is not predictive of internalization, as the cellular accumulation of c11H9 and c2C7 is rather similar.
A major criterion for the selection of antibodies (and linker-payload combinations) for ADC development is the potential for internalization by target cells following binding to antigen. Our data clearly show that the CAIX antibodies evaluated here for efficacy as ADCs also show robust internalization by tumor cells expressing CAIX. Similar internalization characteristics have been observed for other CAIX mAbs developed for targeted delivery of cytotoxic payloads. Girentuximab (cG250), a clinically validated mAb that targets the CAIX catalytic domain, was observed to be internalized by renal and non-renal cancer cells.42,43 Similarly, antibodies identified through phage display technology that target the CAIX catalytic domain were reported to induce surface CAIX internalization into endosomes of CAIX-positive cancer cells.31 Furthermore, a potent anti-tumor efficacy was observed in preclinical models of lung and colon cancer for BAY79-4620, an ADC composed of MMAE conjugated via an enzyme-cleavable linker to an antibody targeting CAIX that was identified through its capacity to undergo internalization in CAIX-expressing cells.32 Recent studies demonstrated the internalization of anti-CAIX antibody-coated nanoparticles by CAIX-expressing cancer cells44 while doxorubicin-loaded nanoparticles coated with anti-CAIX antibodies for targeted drug delivery were internalized by CAIX-positive 4T1 breast cancer cells. Together with inhibition of tumor growth in vivo,45 this further suggests that internalization is important for therapeutic efficacy when using CAIX-specific antibodies for targeted payload delivery.
In contrast, studies evaluating CAIX-targeted small-molecule drug conjugates (SMDCs) have shown that, when bound to small-molecule ligands, efficient CAIX-mediated internalization does not occur.46 Indeed, recent studies evaluating non-internalizing SMDCs targeting CAIX have demonstrated anti-tumor activity in preclinical models of CAIX-positive renal cancer, alone and in combination with targeted IL-2 therapy47 or immune checkpoint blockade.48 The authors suggest that targeted delivery of linker payload combinations to the extracellular environment may promote efficient, protease-mediated drug release and effective action of drugs on tumor-resident cells and neighboring tumor cells.47 Thus, internalization of CAIX may be most beneficial for therapies involving mAb-based modalities, while successful engineering of SMDCs may involve strategies for abrogating or inhibiting internalization.
It should also be emphasized that both cellular routing and processing of the antigen-antibody complex after internalization, which is dictated by the nature of the epitope–paratope interaction, are critical for cellular accumulation. For example, we have indications (unpublished data) that some of these antibodies display a pH-dependent binding profile, i.e., they bind with higher affinity at low pH, which is likely to affect their routing behavior. Collectively, these data support our initial results using the surrogate ADC, which marked c2C7, c11H9, and to a lesser extent c12H8, as suitable ADC candidates. However, there lies a danger in the screening approach we used, which relied on the response of the M75 antibody as a hard cutoff for the selection of our functional candidates. For example, c2D7, which was shown to internalize, was not marked as an ADC candidate. Even though 2D7’s ADC response did not differ that much from 2C7 in the original screen, this antibody was not superior to that of M75, which was used as cutoff (see Suppl. Fig. S2B). Additional evaluation of both c2C7 and c2D7 as a radio-immuno therapeutic confirmed c2C7 as a potent ADC, but also indicated that c2D7 is nearly as potent (unpublished results). In vitro evaluation of DM1-conjugated c2C7, c12H8 and c11H9, confirmed the superiority of c2C7. Further in vivo testing using a xenograft mouse model of hCAIX-positive MIA-PaCa-2 PDAC cells showed that of c2C7-DM1 inhibits the tumor growth and extends survival in vivo, further confirming its potential as an ADC. However, the relatively modest effect of c2C7-DM1 on tumor volume in this model is likely due to a selective effect on cells located in the core of the tumor and within hypoxic niches, leading to substantial cell death and necrosis in the central areas of the tumor, relative to a thin, more viable layer at the tumor surface that encapsulates the necrotic tissue and leads to a “balloon” effect. Thus, tumor volume likely underestimates the overall efficacy of the ADC in this model. Indeed, treatment with the ADC shows a more robust effect on survival of the mice and these data, together with the high levels of cleaved caspase 3 and necrosis present in the treated tumors, clearly demonstrates proof of principle for the use of this antibody as an effective targeted ADC therapy for CAIX-positive tumors. This concept has been further strengthened by our preliminary data using c2C7 armed with DNA-damaging agents such as PNU-159682 or with α-particle emitting 225Ac. These conjugations were shown to be much more potent in terms of tumor eradication (unpublished data), which thus implies that ADCs armed with microtubule disrupting payloads such as DM1, as shown by Chandran et al.,49 but also DM4 and MMEA (data not shown) are less potent when used for targeting tumor cells that reside in the hypoxic/acidic microenvironment.
The fact that CAIX has an enzyme function that is critical for the survival of tumor cells in the hypoxic and/or acidic tumor TME50 thus opens the door to treatments targeting this functionality. We identified two antibodies (m4A2 and m9B6) that significantly inhibit the hCAIX enzyme function using purified hCAIX. We also tested recombinant m9B6 and m4A2 antigen-binding fragments (Fabs) and found that these were more potent than the full-size antibodies (data not shown), implying that the smaller Fabs may have better access to the active center of hCAIX. Others have reported on enzyme-inhibiting antibodies,31,51 but these and also ours do not achieve the same level of enzyme inhibition as an SMI (e.g., acetozolamide). Nonetheless, there is an advantage to the use of these antibodies, which combine inhibition of CAIX catalysis with target specificity (which SMIs often lack). Furthermore, m4A2, which is being internalized, could be used as a highly selective multifunctional (i.e., by conjugating this antibody to a toxic payload), high affinity therapeutic that could be more efficacious in retarding the growth of hCAIX-expressing tumors by inhibiting CAIX activity combined with an ADC function.
In addition to their ADC and hCAIX enzyme-inhibiting functionality, we also evaluated our antibodies for imaging and detection purposes, both in tissue sections and in vivo. Initial evaluation of the six hybridoma-derived mAbs suggested that only 11H9 was able to robustly detect hCAIX in FFPE human breast cancer xenografts and clinical human primary breast tumor samples. However, further evaluation of the recombinant mAbs in FFPE tumor tissue from PK-8 pancreatic xenografts demonstrated that m9B6, despite its poor-binding kinetics as evaluated by SPR, was also able to detect hCAIX. While the reasons for this are not completely clear, potential differences between the native hybridoma and the recombinant mAb may be, in part, responsible for these results. Importantly, the conditions used for labeling in IHC, especially overnight incubation of the mAb at 4°C, may allow for specific binding in the absence of optimal-binding kinetics evaluated by modalities such as SPR, which are carried out over much shorter time frames. The observation that only c11H9 and m9B6 detected the CAIX epitope in FFPE materials illustrates the fact that not all epitopes are recognizable in fixed, processed tissue. In addition, our findings demonstrate a robust diagnostic mAb set using mAbs that recognize the PG-like and catalytic domains of CAIX.
Due to the suboptimal-binding kinetics observed with m9B6, we chose to use c11H9 as our lead for in vivo imaging, but also included c12H8 and c2D7 which, although unsuitable as immunohistochemistry tools, bind to live cells. Labeled with 111In, these antibodies were evaluated in a HT-29 xenograft in vivo mouse model, which showed that c11H9 and c12H8, but not c2D7, are suitable specific imaging agents for the detection of hCAIX-expressing tumors in vivo. A direct comparison was not performed with the cG250 antibody, and it is known that differences can occur between mouse strains. Compared to published values with the cG250 antibody labeled with 111In in nude mice bearing HT29 xenografts,52 tumor uptake appeared higher with the 11H9 antibody, but blood clearance was slower and the tumor/blood and tumor/muscle ratios were higher with the cG250 antibody. Although cG250 has been used in many instances as an imaging agent,53 the advantage of using c11H9 or c12H8 is that these antibodies add another layer of specificity since hCAIX is the only hCA member with a PG-like domain.21,54 In addition, our results also show that these antibodies do not provoke an ADCC immune response, which may be beneficial for an imaging antibody.
Altogether, the data presented here, which combine biophysical with in vitro and in vivo studies, provides a robust assessment of a novel set of hCAIX-specific antibodies, through which we identified three ADC candidates (c2C7, c11H9, and c12H8; WO2016/199097) and two enzyme inhibiting antibodies (m4A2 and m9B6; WO2019/204939).
In conclusion, the importance of targeting CAIX in tumors has been demonstrated many times, for example Chiche et al.55 showed that silencing of the ca9 gene results in a 40% reduction in tumor growth, while Doyen et al.56 reported that knock-down of CAIX increases tumor sensitivity to radiotherapy. Importantly, Lou et al.36 demonstrated that inhibition of CAIX suppresses breast cancer metastasis in vivo while McDonald et al.38 recently demonstrated that CAIX is a critical, targetable vulnerability in KRAS-driven pancreatic cancer, particularly in combination with chemotherapy. These studies, which extend across tumor types and treatment strategies, suggest that targeting hCAIX with a multifunctional therapeutic antibody may be advantageous, and the antibodies characterized here could thus form the basis for such strategy, by, for example, arming an enzyme-inhibiting antibody such as m4A2 with a radio-active (e.g., 225Ac) or DNA-damaging payload (e.g., PNU-159682), or by using the CDRs in a bispecific T-cell engager or CAR-T format may elicit increased efficacy for inhibiting tumor growth. This type of therapy, that specifically targets CAIX, would thus be able to reach and be effective in the hypoxic and acidic areas in the tumor, which are known to harbor the hard to eradicate tumor stem cells known to express CAIX.57
Materials and methods
Cell lines and reagents
Human clear cell renal carcinoma (CCRC) cell lines SK-RC-52 (hCAIX positive) and SK-RC-59 (hCAIX negative) were kindly provided by Dr G. Ritter (Memorial Sloan-Kettering Cancer Center, USA) and were cultured in Minimum Essential Medium (MEM) supplemented with 10% (v/v) heat-inactivated fetal calf serum and 0.1 mM non-essential amino acids. The MIA PaCa-2 human PDAC cell line was kindly provided by Don Yapp and Sylvia Ng (BC Cancer Research Institute, Canada) and cultured in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies) supplemented with 10% fetal bovine serum (FBS; Life Technologies) as previously described.38 The 67NR murine breast cancer cell line was originally provided by Dr. Fred Miller (Karamanos Cancer Institute, USA) and was cultured as described previously.36 Human colon cancer carcinoma HT-29 cells were obtained from ATCC (HTB-38). To generate of 67NR cells constitutively expressing human CAIX (hCAIX positive), a human wildtype CAIX plasmid (gift from Dr. Jacques Pouyssegur, University of Nice, France) was transfected into 67NR cells using Lipofectmine 2000 as per the manufacturer’s instructions. Transfected cells were selected using Zeocin and then subcloned to select cells with high levels of CAIX expression. The MIA PaCa-2 cells constitutively expressing human CAIX (hCAIX positive) were generated as described below. All cell lines were maintained, subcultured and cell-based experiments were carried out at 37°C in a 5% CO2 containing humidified environment, unless indicated otherwise. All cell lines were routinely tested for mycoplasma contamination using either the LookOut Mycoplasma PCR detection kit (Sigma) or the MycoAlert Mycoplasma detection kit (Lonza). The 67NR and MIA PaCa-2 cell lines have been authenticated using short tandem repeat DNA profiling (DNA fingerprinting) by a commercial testing facility (Genetica). In addition, the cell lines were routinely tested for viability, morphology and expression of CAIX. Mouse monoclonal anti-hCAIX M75 was generously provided by Dr E. Oosterwijk (Radboud University Nijmegen, The Netherlands). Anti-CAIX cG250 antibody was produced in-house using the Chinese Hamster Ovary (CHO) expression platform.58 Mouse monoclonal anti-human CAIX MAB2188 was obtained from R&D Systems (Cat# MAB2188), goat anti-human and goat anti-mouse AlexaFluor(AF)488-labeleled IgG-Fab2 were purchased from Jackson ImmunoResearch (Cat# 109–547-003 and 115–547-003, respectively), recombinant human (rh)CAIV (Cat# 510472-H08S), CAXII (Cat# 10617-H08H) and CAXIV (Cat# 10458H08H) and recombinant murine (rm; Cat# 50660-M08H) and dog (rd; Cat# 70028D08H) CAIX were purchased from Sino Biologics Inc., and cynomolgus CAIX was purchased from ACRO Biosystems (Cat# CA9-C83E6). All reagents were reconstituted according to the manufacturer’s instructions.
Generation of stable CAIX-Positive MIA PaCa-2 cell line
Cloning of pLJM1-CA9-IRES-EGFP: The CA9 ORF was amplified from the pCDNA4-CA9 plasmid (gift from Dr. Jacques Pouyssegur, University of Nice, Nice, France). In parallel the internal ribosome entry site (IRES) from pGIPZ (Thermo Fisher Scientific) was amplified. The two PCR products were mixed and amplified to create the CA9-IRES amplicon. The CA9-IRES amplicon was then cloned into pLJM1-EGFP (gift from David Sabatini; Addgene plasmid # 19319; http://n2t.net/addgene:19319; RRID: Addgene_19319) using NheI and AgeI restriction endonuclease sites to create pLJM1-CA9-IRES-EGFP.
Generation of stable cell lines: HEK239T cells were plated at 5 × 104 cells/cm2 in DMEM + 10% FBS. The following day the cells were transfected with 1 μg pLJM1-CA9-IRES-EGFP, 0.9 μg psPAX2 and 0.1 μg VSV-G plasmids with TransIT-LT1 (MirusBio) according to the manufacturer’s instructions and allowed to express overnight. The following morning, media was removed and 2 mL of DMEM containing 10% FBS and 1% bovine serum albumin (BSA) added. Media was removed 48 hours later and centrifuged at 1500 rpm for 5 min to pellet any cellular debris. Supernatants containing lentivirus were filtered through a 0.45 μm filter and added to MiaPaCa2 cells at a multiplicity of infection of 0.3. The following day, the virus containing media was removed, cells washed twice with PBS and incubated in DMEM containing 10% FBS and 3 μg/mL puromcyin to select for transduced cells. Cell cultures were expanded and screened for CAIX expression by Western blot.
Cloning, expression, and purification of rhCAIX
The hCAIX extracellular domain cDNA (HGNC:1383; a.a. 1–414), was cloned in the PTT5TM expression vector, produced as a C-terminal penta-His-tagged protein in CHO cells,58–60 purified by IMAC affinity chromatography. SDS-PAGE (reducing and non-reducing conditions) indicated the presence of monomeric and dimeric subpopulations that were separated by SEC (Superdex G200, Cytiva Life Sciences). Protein purity and homogeneity of each of the monomer and dimer fractions was assessed by UPLC-SEC and SDS-PAGE under reducing and non-reducing conditions. Monomer and dimer stability were assessed after storage (4 weeks at 4°C and −80°C) by UPLC-SEC.
Recombinant antibody production and purification
Anti-human CAIX mouse mAbs were generated (for details see Suppl. Materials and Methods), functionally screened (Suppl. Figure 2) and sequenced. Full-size heavy and light human IgG chains were ordered from a commercial vendor (Genscript); VH and VL CDR1-3 regions (See Suppl. Table S2) of 11H9, 12H8, 2C7 and 2D7 were grafted on a human IgG1 framework (designated chimeric, ‘c’) whereas those of 4A2 and 9B6 were grafted on a murine IgG2a framework (designated mouse, ‘m’). Antibodies were produced in CHO cells, which were grown to a density of 1.3 × 104 cell/mL F17 medium and then transfected with and 1:1 ratio of heavy and light-chain constructs using PEI MAX™ (Polysciences, Inc.). Cells were kept at 37°C for 24 h, fed with Tryptone N1 at 1% w/v, after which they were maintained at 32°C for another 6 days. Cell culture supernatant was then harvested and analyzed by SDS-PAGE. Antibodies were captured from the supernatant on a MabSelect Sure column (Cytiva Life Sciences), then eluted with acidic buffer and immediately neutralized with 1 M (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES). UPLC-SEC analysis performed after Protein A purification indicated the presence of heavy-chain dimers for certain lots of antibodies. These lots were further purified on a Capto L column (Cytiva Life Sciences) to remove heavy-chain dimers. Purified samples were then subjected to a buffer exchange into PBS and protein purity and homogeneity was assessed by UPLC-SEC and SDS/PAGE. When the percentage of monomer, as determined by UPLC-SEC, was higher than 5%, samples were further purified by SEC (Superdex 200, Cytiva Life Sciences). Finally, purified samples were aliquoted and stored at −80°C.
Antibody conjugations
pHAb dye conjugation: Antibody concentrations were adjusted to 3.1 mg/mL in PBS. pHAb amine reactive dye (Promega, Madison, WI) was dissolved in dimethyl sulfoxide/dH2O (1:1, v/v) at a concentration of 5 mg/mL and then added to a 10 mg/mL antibody solution (molar ratio 14:1), followed by incubation (25°C, 1 h). Excess dye was removed by a ZebaSpin desalting column (Thermo Fisher Scientific) equilibrated in PBS. Samples were centrifuged (20,000 × g, 10 min, 4°C) and supernatant containing the conjugate was retained. Dye-to-antibody ratio was determined using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific) at A280 and A532 nm.
DM1 conjugation: Primary or secondary antibody variants were combined with SMCC-DM1 (Levena Biopharma) in 1x NRC4 buffer (100 mM sodium phosphate, 20 mM NaCl, 2 mM EDTA, pH7.4) and incubated (25°C, 18 h). Polysorbate-20 was added to final concentration of 0.02% w/v. The reaction was passed 3x through a ZebaSpin columns (Thermo Fisher Scientific) and pre-equilibrated with the final formulation buffer (20 mM sodium-succinate, 0.02% polysorbate-20, pH 6.0). When the percentage of monomer, as determined by HPLC-SEC after conjugation, exceeded 3%, conjugated material was further purified by SEC (Superdex 200, Cytiva Life Sciences). Trehalose was added to the final sample at 6% w/v, centrifuged (20000Xg, 10 min) and the drug-to-antibody ratio was determined using a NanoDrop 2000 spectrophotometer (A280 nm and A252 nm) and by size exclusion-high-performance liquid chromatography analysis.
Surface plasmon resonance
All SPR assays were carried out at 25°C using a BioRad ProteOn XPR36 (Bio-Rad Laboratories (Canada) Ltd. (Mississauga, ON)). Sensor chips (GLM and GLC) and coupling reagents (10 mM sodium acetate, pH 4.5; sulfo-N-hydroxysuccinimide (S-NHS); 1-ethyl-3-(3-dimethylaminporpyl)-carbodiamide hydrochloride (EDC) and ethanolamine) were purchased from BioRad Inc. PBS with 0.05% v/v Tween20 was used as running buffer.
GLC sensorchip surfaces were activated by injecting S-NHS/EDC (1:10; 140s, 100 μL/min). Immediately after, 10 ug/mL anti-mouse or anti-human Fc antibody (depending on the anti-CAIX Ab framework) was injected (25 µL/min) in NaOAc (10 mM, pH 4.5) until ~4000 resonance units (RU) were captured. Remaining active groups were quenched by ethanolamine (1 M;140s injection at 100 µL/min); mock-activated inter-spots were used for blank referencing. Anti-CAIX antibodies were then captured (240s, 25 µL/min) onto the appropriate anti-human or anti-mouse Fc antibody (Jackson ImmunoResearch, Cat# 109–005-098 and 115–005-071, respectively) surface (~400 to 600 RUs) after which 60 nM, 20 nM, 6.7 nM, 2.2 nM and 0.74 nM rhCAIX antigen along with a buffer blank was simultaneously injected (120s, 50 μL/min), followed by a buffer injection (300s, 50 μL/min). Antibody surfaces were regenerated by 0.85% phosphoric acid (18s, 100 μL/min). Sensorgrams were aligned and double-referenced using the buffer blank injection, and interspots, analyzed (ProteOn Manager software v3.1) and fitted to the 1:1 binding model to calculate ka, kd, KD and Rmax. A similar approach was used to determine the antibody cross-reactivity for cell surface expressed and secreted rhCAIV, rhCAXII and rhCAXIV, and recombinant murine (rm), dog (rd) and cynomolgus (rc) CAIX. All proteins were reconstituted as recommended by the manufacturer (Sino Biologics) and diluted to 100 nM in running buffer for SPR analysis.
Cell binding experiments
SK-RC-52 cells were seeded in 96-well plates (2000 cells per well) and grown overnight (o/n). Cells were cooled to 4°C the next day, incubated with the indicated anti-CAIX antibodies (0–100 nM) in 100 uL of growth medium (4°C, 2 h), followed by a wash (D-PBS, 4°C). Anti-human (for c11H9, c12H8, c2C7, c2D7 and the hIgG control (Sigma; Cat# 14506–10 MG)) or anti-murine (for m4A2, m9B6 and M75 (Absolute Antibody)) AF488-labeled IgG Fab2 (1:200 v/v dilution; Jackson ImmunoResearch) was then added in the presence of nuclear dye Hoechst 33342 (1:2000 v/v dilution) and CellMask™ Deep Red Plasma Membrane Stain (1:1000 v/v dilution) and incubated for 1 h at 4°C. After a quick wash (D-PBS Ca2+, Mg2+ free) cell images were captured (ImageXpress Micro (IXM) Widefield High Content Imaging System; Molecular Devices), processed (MetaXpress software; Molecular Devices), and further analyzed using GraphPad Prism v8.
ADCC assay
The ADCC assay was performed according to the manufacturer’s instructions (https://bit.ly/2Tk8OWY) with modifications. Briefly, SK-RC-52 target (T) cells were seeded in 96-well plates (12500 cells/well) and grown o/n (37°C, 5% CO2, humidified incubator). hFcγRIIIaV158 (c11H9, c12H8, c2C7, c2D7) or mFcγRIV (m4A2, m9B6) expressing effector (E) Jurkat cells (75000 cells/well) were added to the SK-RC-52 cells (E:T = 6:1). After 6 h of incubation (37°C, 5% CO2, humidified incubator), Bio-GloTM Luciferase assay reagent was added, and luminescence was measured after 5–10 min. Experiments were carried out in triplicate and repeated twice. Final data was analyzed using Graphpad Prism v8.
Internalization studies
SK-RC-52 cells were seeded in 96-well plates (3500 cells/well) and grown o/n. The next day, cells were washed and incubated with serial dilutions (0, 0.63, 1.25, 2.5, 5, 10, 20 µg/mL) of pHAb-labeled mAbs in MEM, containing 1% heat-inactivated FBS, and non-essential amino acids (1 h, 4°C). The cells were washed and imaged (ImageXpress Micro (IXM) Widefield High Content Imaging System; Molecular Devices) at t = 0 h, and then transferred to 37°C for 23.5 h. Cells were briefly removed from the incubator to add the nuclear dye Hoechst 33342 (Thermo Fisher Scientific) and returned to the incubator for an additional 30 min. Internalization was halted by placing the cells on ice, after which they were washed with ice-cold PBS. Cell monolayers were imaged (ImageXpress Micro (IXM) Widefield High Content Imaging System; Molecular Devices) using the following channels and exposure times: Tetramethylrhodamine (1000 ms), 4′,6-diamidino-2-phenylindole (100 ms). Images were processed (Transfluor MetaXpress software module; Molecular Devices) and the internalization data, calculated as the integrated Fluorescent Index normalized to the nuclear Hoechst count, was further analyzed using Graphpad Prism v8.
Cell viability assay
Cell viability was assessed by using cell proliferation and viability assays (Millipore-Sigma) according to the manufacturer’s instructions. Briefly, control and hCAIX-expressing 67NR mouse breast cancer cells or MIA PaCa-2 human pancreatic cancer cells were seeded into 96-well microplates at a density of 2.5 × 103 cells/cm2 in a 90 μL volume and incubated o/n. The following day, non-conjugated mAbs and DM1-conjugated mAbs were serially diluted at 10x final assay concentration in cell culture media, added to the cells (10 μL/well; final concentration 0–25 nM), and cells were incubated for 96 hours. Cells cultured in medium alone were used as a control. Finally, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide labeling reagent (10 μL/well) was added and after 4 h. Formazan crystals were solubilized o/n (37°C) and absorbance was measured using an ELISA reader (570 nm and 660 nm reference). Cell viability was calculated as the percentage of untreated cells; a non-linear regression analyses (log(inhibitor) vs. response – Variable slope (four parameters)) was used to calculate the IC50 values using Graphpad Prism v8. Data are reported as mean ± s.e.m. of three technical replicates/sample; data are representative of at least two independent experiments.
In vitro enzyme inhibition assay
4-MUA substrate: rhCAIX ECD dimer (0.5 μM) was incubated with CAIX mAbs (1 μM, 96-well solid black microplates (Corning), 30 min, RT), and enzymatic reactions were initiated by adding 4-methylumbelliferylacetate (4-MAU; 100 μM). Substrate-released fluorescence was measured (ex: 356 nm; em: 445 nm) as a function of time (0–3600s; EnVision 2104 plate reader, Perkin Elmer). Substrate alone, rhCAIX alone and rhCAIX in the presence of acetazolamide (10 μM, Sigma Aldrich) were used as controls. Substrate hydrolysis rates were determined in the ‘initial’ and ‘end portion’ of each of the curves and corrected for autohydrolysis (substrate alone). Experiments were carried out in triplicate and repeated at least twice.
CO2 substrate: The catalytic activity of CAIX was assessed using a modification of previously described methods.61 For each replicate sample, 1 μl of a 100 μg/ml (2.38 μM) stock solution of recombinant human CAIX (rhCAIX; R&D systems) was incubated with the recombinant anti-CAIX mAbs and PBS in a total volume of 10 μl. Enzyme to mAb molar ratios of 1:1 and 1:5 were evaluated, resulting in antibody concentrations of 36 and 180 μg/ml in the 10 μl incubation volume. Incubation of similar amounts of rhCAIX with normal mouse IgG at the highest mAb concentration served as a control for specificity. Incubations (50 min, room temperature (RT)) were carried out with gentle rotation to allow for binding of the antibodies. Following this incubation, the remainder of the assay was carried out on ice. The enzyme-antibody mixture was added to 790 µl ice cold assay buffer (20 mM Tris, pH8.0) in a 2 ml flat-bottom glass vial equipped with an 8 mm magnetic stir bar. A narrow pH electrode (Accumet) was immersed in the buffer and the sample was equilibrated for 3–5 min on ice with gentle stirring. 200 µl of ice-cold CO2-saturated water was added to initiate the assay and pH readings were recorded at 5 s intervals. Three to 4 technical replicates were evaluated for each sample and data are representative of at least 2 independent experiments. To determine the rate of spontaneous CO2 hydration, measurements were performed on enzyme-free samples (i.e., buffer only). The increase in hydration rate above the spontaneous rate is a measure of CA activity and data were analyzed using Graphpad Prism v8. The relative percent of CAIX activity was calculated from values for the area under the curve (AUC) for the kinetic curves.
Immunohistochemistry and histochemistry
IHC on formalin-fixed, paraffin-embedded tissue was performed as previously described.37,62 Five-micron tissue sections were cut and transferred onto Superfrost-Plus slides (Fisher Scientific). Tissue sections were deparaffinized, rehydrated and antigen retrieval was performed by microwaving in 10 mM citrate buffer, pH 6.0 for 10 minutes. Sections were incubated with 3% H2O2 for 15 minutes to quench endogenous peroxidase activity. Tissue sections were incubated with primary antibody (diluted in 3% skim milk, 1% BSA in PBS) overnight at 4°C. Primary antibodies included anti-human CAIX (M75, Bioscience; 1:200), anti-human CAIX (MAB2188, R&D Systems, 1:200), anti-cleaved caspase 3 (Asp175-5A1E, Cell Signaling, 1:100), and novel anti-CAIX antibodies reported herein, including 11H9, c11H9, 12H8, c12H8, 2C7, c2C7, c2D7, 4A2, m4A2, 9B6 and m9B6 (all diluted to 10 μg/mL). ImmPRESS™ species-specific horseradish peroxidase (HRP) secondary (MP-7405, MP-7500) and HRP substrate (SK-4100) were used according to the manufacturer’s instructions (Vector Laboratories). Tissues labeled with chimeric recombinant CAIX antibodies were incubated with a biotin labeled goat F(ab)2 anti-human IgG-Fc secondary antibody (ab98589, AbCam, 1:1000) for 1 hour at RT and detected using a LSAB+ System Kit (DAKO). Tissues were counterstained with hematoxylin (Leica Biosystems), dehydrated and mounted using Permount. For quantification of cleaved caspase 3 staining, at least 5 randomly selected fields of view (FOV) at 20x magnification were imaged from 1 section/tumor and 5 tumors were analyzed per group. The number of positive cells in each image was counted or the percent area of positive staining was quantified using ImageJ Fiji (National Institutes of Health, USA).
Evaluation of necrotic areas: Five-micron tissue sections prepared as described above with stained with hematoxylin and eosin (H&E) as described previously.37 For quantification of necrotic areas, at least 10 randomly selected FOV at 10x magnification were imaged from 1 section/tumor and 4 tumors were analyzed per group. The areas of necrosis were circumscribed, and the area occupied by necrotic tissue, in pixels, was determined using ImageJ (v1.48, National Institutes of Health, USA). Total tumor area, in pixels, was then determined and the percentage necrosis was calculated as % necrosis = (total area of necrosis in pixels/total tumor area in pixels)*100.
Immunoblotting
Samples were lyzed in RIPA buffer (50 mM Tris-HCl, pH 7.6, 150 mM NaCl, 0.1% (w/v) SDS, 0.5% NaDOC) containing 1 mM Na3VO4, 2 mM NaF and cOmplete™ protease inhibitor cocktail (Sigma), and analyzed by 4–12% gradient SDS PAGE as previously described.37 Briefly, methanol-fixed PVDF Membranes were incubated with primary antibodies in 2% BSA in Tris-buffered saline containing 0.1% Tween® 20 (TBST) overnight at 4°C. Primary antibodies included anti-hCAIX (M75, Bioscience; 1:200) and anti-vinculin (MAB3574, Millipore, 1:2,000), After incubation with primary antibodies, membranes were washed with TBST for 3x10min and then incubated with HRP-conjugated anti-mouse secondary antibodies (Cell Signaling Technologies, 1:5,000) in diluted in 5% skim milk in TBST for 1 hour at RT. After extensive washing with TBST, detection was performed by incubation with Supersignal West Femto chemiluminescence reagents (Life Technologies) and visualization using a Chemidoc XRS+ imaging system (Bio-Rad Laboratories).
Immunofluorescence
Immunofluorescence staining was carried out as previously described13 with modifications. Briefly, cells were grown on glass coverslips, washed 3 times in PBS to remove media, fixed in 4% paraformaldehyde for 10 min, permeabilized with 0.1% Triton X-100 for 10 min and blocked with PBS + 1% horse serum for 30 min. The coverslips were incubated with goat anti-hCAIX (AF2188, R&D Systems, 1:100) primary antibody overnight at 4°C. The coverslips were then washed three times with PBS and incubated with AlexaFluor 594-conjugated secondary antibodies (1:400; Jackson ImmunoResearch Laboratories, Cat# 709–585-149) for 1 h at RT. The coverslips were then washed and mounted in Prolong Diamond anti-fade mounting media with 4′,6-diamidino-2-phenylindole (Thermo Fischer Scientific). Images were acquired using a Zeiss Colibri inverted microscope equipped for epifluorescence (Carl Zeiss Canada Ltd) and processed using ImageJ (v1.48, National Institutes of Health, USA) and Photoshop software (Adobe).
In vivo SPECT/CT imaging
Conjugation of mAb with pSCN-Bn-DTPA: 1.0 mg of antibody was conjugated with pSCN-Bn-DTPA (Macrocyclics) in 500 uL of PBS (buffered to pH 8.9 with 0.1 M Na2CO3). A 5:1 molar ratio of chelator to mAb was used and the reaction was allowed to proceed overnight at RT with gentle agitation. After 24 h incubation, the mixture was purified using a 50 KDa amicon filter (Millipore) to remove any unconjugated chelator. Antibody-DTPA conjugates were washed with PBS and resuspended in 0.15 M ammonium acetate (pH 5.5) and stored as 0.1 mg aliquots at −20°C.
111In radiolabelling of mAb-DTPA conjugates: 111In was purchased from Nordion as [111In]InCl solution. 0.9% NaCl solution was added to 111In solution to create a ~ 5 mCi/100 uL stock solution. 500 uL of 0.1 M HEPES solution (pH 5.5) was added to 100 uL of [111In]InCl solution, 0.4 mg of mAb-DTPA was added, followed by 1 h incubation at RT. This reaction mixture was purified (50 KDa amicon filter, Millipore) to remove unbound radioisotope and radiochemical purity was determined by size-exclusion radio-HPLC (Agilent).
SPECT Imaging studies and Data analysis: Four (4) NODSCID IL2RKO female mice were subcutaneously inoculated with 5 × 106 HT29 cells in 50 μL 50% Matrigel (in PBS). 10 days post-inoculation, mice were injected with ~3.7 MBq (biodistribution) or 37 MBq (imaging) of 111In-11H9, 111In-2D7 or 111In-IgG CTL antibody. Biodistribution experiments were performed by euthanizing groups of animals at 2, 72 and 168 hours, at which time blood and the organs were collected, rinsed, blotted dry, weighed and counted in a gamma counter (PerkinElmer Wizard2). The uptake in biodistribution studies was expressed in percent of the injected dose per gram of tissue (%ID/g). SPECT/CT images were acquired using a U-SPECT-II/CT system (MILabs) at 24 h, 48 h, 72 h, and 168 h post-injection, after which mice were euthanized. Images were prepared using PMOD/ImageJ software.
In vivo evaluation of DM1-conjugated mAbs
All experimental animal procedures were carried out at the BC Cancer Animal Resource Center in accordance with protocol A14-0058 approved by the institutional Animal Care Committee at the University of British Columbia, Vancouver. The studies are compliant with all relevant ethical regulations regarding animal research. All mice were housed in ventilated cages in a pathogen-free, environment-controlled room at 19–21°C. The relative humidity ranged between 40% and 70% and a photoperiod of 12 hours light and 12 hours darkness was provided. Food and water were provided ad libitum.
MIA PaCa-2 pancreatic cancer cells constitutively expressing human CAIX (MIA PaCa-2hCAIX) were injected subcutaneously (5.0x106 cells/animal, suspended in 100 μL sterile PBS) on the back of 8–10-week-old female NOD/SCID IL2Rγ-/- mice obtained through an “in-house” breeding program using breeders from Jackson Laboratories and tumors were allowed to establish. All tumors were measured using digital calipers and tumor volumes were calculated as previously described37,38 using the modified ellipsoidal formula of volume = 0.5(length x width2). When tumors reached approximately 100 mm3, mice were sorted into groups of similar average tumor volume and drug administration was initiated. Anti-CAIX antibodies were administered twice per week by i.v. injection through the tail vein and a total of 6 doses were administered. Anti-CAIX ADC c2C7-DM1 (5.8 mg/ml) was formulated in 20 mM succinate, 0.02% polysorbate-20, 6% trehalose, pH6.0 and was diluted in sterile PBS prior to administration. The doses used were 2.5 mg/kg, 5 mg/kg and 10 mg/kg. All animals were weighed prior to dosing and doses were administered at a volume of 5 μL/g body weight. The vehicle control group was administered the vehicle alone at the 10 mg/kg dose equivalent. For survival analyses, a surrogate threshold was used and survival events occurred once tumors crossed the threshold volume of 800 mm3.
Tumors were harvested and fixed in formalin and embedded in paraffin for downstream analyses by IHC. For assessment of drug toxicity, body weights were measured during the treatment phase of the study. A clinical observation and health monitoring record was maintained for each individual animal throughout the study. The investigators were not blinded to the identity of the study groups and the number of animals per group was chosen based on data from previous studies.36,38,63
Statistical analysis
Statistical significance between two groups was assessed by unpaired Student’s t-test. One-way analysis of variance (ANOVA) was used to analyze more than two groups. Two-way ANOVA was used to analyze quantification of IHC. Analysis of tumor growth curves was performed using TumGrowth software64 (https://kroemerlab.shinyapps.io/TumGrowth). GraphPad Prism v7 and v8 software were used for all other statistical analyses. All cell culture experiments were performed at least two times. Data are presented as mean ± s.e.m. and P values less than 0.05 were considered statistically significant. Further statistical details, including values of n and definitions of what n represents, can be found in the figure legends.
Supplementary Material
Acknowledgments
We would like to thank all colleagues from the National Research Council and the BC Cancer Research Institute for supporting this study, and Dr N. Rohani-Larijani for critical reading and editing of our manuscript.
Funding Statement
This research was supported by the National Research Council of Canada, and, in part, by a grant from the Canadian Institutes of Health Research (S.D.; CIHR grant # FDN-143318), and a program project grant from the Terry Fox Research Institute (F.B.; Grant #1045).
Abbreviations
%ID/g Mean percent injected dose per gram
4-MUA 4-Methylumbelliferylacetate
ADC Antibody-drug-conjugate
ADCC Antibody-dependent cellular cytotoxicity
ANOVA One-way analysis of variance
BSA Bovine serum albumin
CA Carbonic anhydrase
CAIX Carbonic anhydrase IX
CCRC Clear cell renal carcinomaCHOChinese hamster ovary
CO2 Carbon dioxide
D-PBS Dulbecco’s phosphate-buffered saline
DM1 MertansineDMEMDulbecco’s modified Eagle’s medium
EC50 Half-maximal effective concentration
ECDExtracellular domain
EDC 1-ethyl-3-(3-dimethylaminporpyl)-carbodiamide hydrochloride
FBS Fetal bovine serumFFPEFormalin-fixed paraffin-embedded
FOV Fields of view
H+ Proton
HEPES(4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
HIF1α Hypoxia-inducible factor 1α
HRP Horseradish peroxidase
i.v. Intravenous
IC50 Half-maximal inhibiting concentrationIHCImmunohistochemistry
IMAC Immobilized metal affinity chromatography
MAP-ZAP Anti-mouse antibody conjugated to the Saporin toxin
MEM Minimum essential medium
MMAE Monomethyl auristatin E
p.i. Post injection
PDAC Pancreatic ductal adenocarcinoma
PG Proteoglycan
RCC Renal cell carcinoma
RPPA Reverse phase protein array
RU Resonance unit
S-NHS Sulfo-N-hydroxysuccinimide
s.e.m. Standard error of the mean
SDS-PAGE Sodium dodecyl-sulfate polyacrylamide gel electrophoresis
SMI Small molecule inhibitor
SPECT/CT Single-photon emission computerized tomography/computerized tomography
SPR Surface plasmon resonance
TBST Tris-buffered saline containing 0.1% Tween® 20
TME Tumor microenvironment
UPLC-SEC Ultra performance liquid chromatography-size exclusion chromatography
VHL von Hippel Lindau
%ID/g Mean percent injected dose per gram
4-MUA4-Methylumbelliferylacetate
Author contributions
A.E.G.L. conceived the project, outlined the experiments and drafted the original manuscript. D.L., A.P., F.M., Y.D., M.D. and C.G. contributed to the expression of the antibodies and rhCAIX. M.P., H.O., A.D., M.P., M.L., A.A. and N.B-G. contributed to the purification of the antibodies. R.M., M.P., D.L., and S.G. contributed to the purification of rhCAIX. J.B., S.G. and B.P-R. performed the SPR analysis of antibodies. Y.F. and C.W. contributed to the RPPA experiments. P.L., M.G. and P.C.M. performed the enzyme inhibition studies with the antibodies. A.R., C.C., Y.C-D., M.J., S.R., P.S., M.A., and P.C.M. executed the cell-based evaluation of the antibodies. S.C.C, O.N., J.L. and F.B. contributed and performed the in vivo PET/SPECT studies. P.C.M., M.A. contributed to the in vivo ADC studies. P.C.M. carried out the IHC evaluations. P.C.M., M.P., S.D. and J.B. reviewed and edited the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Zatovicova M, Sedlakova O, Svastova E, Ohradanova A, Ciampor F, Arribas J, Pastorek J, Pastorekova S.. 2005. Ectodomain shedding of the hypoxia-induced carbonic anhydrase IX is a metalloprotease-dependent process regulated by TACE/ADAM17. Br J Cancer. 93(11):1267–20. doi: 10.1038/sj.bjc.6602861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thiry A, Supuran CT, Masereel B, Dogne JM. 2008. Recent developments of carbonic anhydrase inhibitors as potential anticancer drugs. J Med Chem. 51(11):3051–56. doi: 10.1021/jm701526d. [DOI] [PubMed] [Google Scholar]
- 3.Thiry A, Dogne JM, Masereel B, Supuran CT. 2006. Targeting tumor-associated carbonic anhydrase IX in cancer therapy. Trends Pharmacol Sci. 27(11):566–73. doi: 10.1016/j.tips.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 4.Pastorekova S, Parkkila S, Pastorek J, Supuran CT. 2004. Carbonic anhydrases: current state of the art, therapeutic applications and future prospects. J Enzyme Inhib Med Chem. 19(3):199–229. doi: 10.1080/14756360410001689540. [DOI] [PubMed] [Google Scholar]
- 5.Neri D, Supuran CT. 2011. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov. 10(10):767–77. doi: 10.1038/nrd3554. [DOI] [PubMed] [Google Scholar]
- 6.Supuran CT, Alterio V, Di Fiore A, Carta F, Monti SM, et al. 2018. Inhibition of carbonic anhydrase IX targets primary tumors, metastases, and cancer stem cells: three for the price of one. Med Res Rev. 38:1799–836. [DOI] [PubMed] [Google Scholar]
- 7.Pastorekova S, Parkkila S, Parkkila AK, Opavsky R, Zelnik V, Saarnio J, Pastorek J. 1997. Carbonic anhydrase IX, MN/CA IX: analysis of stomach complementary DNA sequence and expression in human and rat alimentary tracts. Gastroenterology. 112(2):398–408. doi: 10.1053/gast.1997.v112.pm9024293. [DOI] [PubMed] [Google Scholar]
- 8.Saarnio J, Parkkila S, Parkkila AK, Waheed A, Casey MC, Zhou XY, Pastoreková S, Pastorek J, Karttunen T, Haukipuro K, et al. 1998. Immunohistochemistry of carbonic anhydrase isozyme IX (MN/CA IX) in human gut reveals polarized expression in the epithelial cells with the highest proliferative capacity. J Histochem Cytochem. 46(4):497–504. doi: 10.1177/002215549804600409. [DOI] [PubMed] [Google Scholar]
- 9.Zavada J, Zavadova Z, Pastorek J, Biesova Z, Jezek J, Velek J. 2000. Human tumour-associated cell adhesion protein MN/CA IX: identification of M75 epitope and of the region mediating cell adhesion. Br J Cancer. 82(11):1808–13. doi: 10.1054/bjoc.2000.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alterio V, Hilvo M, Di Fiore A, Supuran CT, Pan P, Parkkila S, Scaloni A, Pastorek J, Pastorekova S, and Pedone C. 2009. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc Natl Acad Sci U S A. 106(38):16233–38. doi: 10.1073/pnas.0908301106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mahon BP, Pinard MA, McKenna R. 2015. Targeting carbonic anhydrase IX activity and expression. Molecules. 20(2):2323–48. doi: 10.3390/molecules20022323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buanne P, Renzone G, Monteleone F, Vitale M, Monti SM, Sandomenico A, Garbi C, Montanaro D, Accardo M, Troncone G, et al. 2013. Characterization of carbonic anhydrase IX interactome reveals proteins assisting its nuclear localization in hypoxic cells. J Proteome Res. 12(1):282–92. doi: 10.1021/pr300565w. [DOI] [PubMed] [Google Scholar]
- 13.Swayampakula M, McDonald PC, Vallejo M, Coyaud E, Chafe SC, Westerback A, Venkateswaran G, Shankar J, Gao G, Laurent EMN, et al. 2017. The interactome of metabolic enzyme carbonic anhydrase IX reveals novel roles in tumor cell migration and invadopodia/MMP14-mediated invasion. Oncogene. 36(45):6244–61. doi: 10.1038/onc.2017.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Opavsky R, Pastorekova S, Zelnik V, Gibadulinova A, Stanbridge EJ, Zavada J, Kettmann R, Pastorek J. 1996. Human MN/CA9 gene, a novel member of the carbonic anhydrase family: structure and exon to protein domain relationships. Genomics. 33(3):480–87. doi: 10.1006/geno.1996.0223. [DOI] [PubMed] [Google Scholar]
- 15.Innocenti A, Pastorekova S, Pastorek J, Scozzafava A, De Simone G, Supuran CT. 2009. The proteoglycan region of the tumor-associated carbonic anhydrase isoform IX acts as anintrinsic buffer optimizing CO2 hydration at acidic pH values characteristic of solid tumors. Bioorg Med Chem Lett. 19(20):5825–28. doi: 10.1016/j.bmcl.2009.08.088. [DOI] [PubMed] [Google Scholar]
- 16.Ames S, Pastorekova S, Becker HM. 2018. The proteoglycan-like domain of carbonic anhydrase IX mediates non-catalytic facilitation of lactate transport in cancer cells. Oncotarget. 9(46):27940–57. doi: 10.18632/oncotarget.25371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jamali S, Klier M, Ames S, Barros LF, McKenna R, Deitmer JW, Becker HM. 2015. Hypoxia-induced carbonic anhydrase IX facilitates lactate flux in human breast cancer cells by non-catalytic function. Sci Rep. 5(1):13605. doi: 10.1038/srep13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andreucci E, Peppicelli S, Carta F, Brisotto G, Biscontin E, Ruzzolini J, Bianchini F, Biagioni A, Supuran CT, Calorini L, et al. 2017. Carbonic anhydrase IX inhibition affects viability of cancer cells adapted to extracellular acidosis. J Mol Med (Berl). 95(12):1341–53. doi: 10.1007/s00109-017-1590-9. [DOI] [PubMed] [Google Scholar]
- 19.Ihnatko R, Kubes M, Takacova M, Sedlakova O, Sedlak J, Pastorek J, KOPACEK J, and PASTOREKOVA S, et al. 2006. Extracellular acidosis elevates carbonic anhydrase IX in human glioblastoma cells via transcriptional modulation that does not depend on hypoxia. INTERNATIONAL JOURNAL OF ONCOLOGY. 29:1025–33. [PubMed] [Google Scholar]
- 20.Oosterwijk-Wakka JC, Boerman OC, Mulders PF, Oosterwijk E. 2013. Application of monoclonal antibody G250 recognizing carbonic anhydrase IX in renal cell carcinoma. Int J Mol Sci. 14(6):11402–23. doi: 10.3390/ijms140611402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Supuran CT. 2008. Carbonic Anhydrases An Overview. Current Pharmaceutical Design. 14(7):603–14. doi: 10.2174/138161208783877884. [DOI] [PubMed] [Google Scholar]
- 22.Supuran CT. 2018. Carbonic anhydrase inhibitors as emerging agents for the treatment and imaging of hypoxic tumors. Expert Opinion on Investigational Drugs. 27(12):963–70. doi: 10.1080/13543784.2018.1548608. [DOI] [PubMed] [Google Scholar]
- 23.Pacchiano F, Carta F, McDonald PC, Lou Y, Vullo D, Scozzafava A, Dedhar S, Supuran CT. 2011. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J Med Chem. 54(6):1896–902. doi: 10.1021/jm101541x. [DOI] [PubMed] [Google Scholar]
- 24.McDonald PC, Chia S, Bedard PL, Chu Q, Lyle M, Tang L, Singh M, Zhang Z, Supuran CT, Renouf DJ, et al. 2020. A Phase 1 study of SLC-0111, a novel inhibitor of carbonic anhydrase IX, in patients with advanced solid tumors. Am J Clin Oncol. 43(7):484–90. doi: 10.1097/COC.0000000000000691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zavada J, Zavadova Z, Pastorekova S, Ciampor F, Pastorek J, and Zelnik V. Expression of MaTu-MN protein in human tumor cultures and in clinical specimens. Int J Cancer . 1993;54:268-274. [DOI] [PubMed] [Google Scholar]
- 26.Oosterwijk E, Ruiter DJ, Hoedemaeker PJ, Ekj P, Jonas U, Zwartendijk J, and Warnaar, SO. 1986. Monoclonal antibody G250 recognizes a determinant normal kidney present in renal-cell carcinoma and absent from normal kidney. Int J Cancer. 38. [DOI] [PubMed] [Google Scholar]
- 27.Zatovicova M, Jelenska L, Hulikova A, Csaderova L, Ditte Z, Ditte P, Goliasova T, Pastorek J, Pastorekova S. 2010. Carbonic anhydrase IX as an anticancer therapy target: preclinical evaluation of internalizing monoclonal antibody directed to catalytic domain. Curr Pharm Des. 16(29):3255–63. doi: 10.2174/138161210793429832. [DOI] [PubMed] [Google Scholar]
- 28.Surfus JE, Hank JA, Oosterwijk E, Welt S, Lindstrom MJ, Albertini MR, Schiller JH, Sondel PM. 1996. Anti-renal-cell carcinoma chimeric antibody G250 facilitates antibody-dependent cellular cytotoxicity with in vitro and in vivo interleukin-2-activated effectors. J Immunother Emphasis Tumor Immunol. 19(3):184–91. doi: 10.1097/00002371-199605000-00003. [DOI] [PubMed] [Google Scholar]
- 29.Chamie K, Donin NM, Klopfer P, Bevan P, Fall B, Wilhelm O, Störkel S, Said J, Gambla M, Hawkins RE, et al. 2017. Adjuvant weekly girentuximab following nephrectomy for high-risk renal cell carcinoma: the ARISER randomized clinical trial. JAMA Oncol. 3(7):913–20. doi: 10.1001/jamaoncol.2016.4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Basaco T, Pektor S, Bermudez JM, Meneses N, Heller M, Galvan JA, Boligán K, Schürch S, Von Gunten S, Türler A, et al. 2018. Evaluation of radiolabeled girentuximab in vitro and in vivo. Pharmaceuticals (Basel). 11(4):132. doi: 10.3390/ph11040132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu C, Lo A, Yammanuru A, Tallarico AS, Brady K, Murakami A, Barteneva N, Zhu Q, Marasco WA, et al. 2010. Unique biological properties of catalytic domain directed human anti-CAIX antibodies discovered through phage-display technology. PLoS One. 5(3):e9625. doi: 10.1371/journal.pone.0009625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petrul HM, Schatz CA, Kopitz CC, Adnane L, McCabe TJ, Trail P, Ha S, Chang YS, Voznesensky A, Ranges G, et al. 2012. Therapeutic mechanism and efficacy of the antibody-drug conjugate BAY 79-4620 targeting human carbonic anhydrase 9. Mol Cancer Ther. 11(2):340–49. doi: 10.1158/1535-7163.MCT-11-0523. [DOI] [PubMed] [Google Scholar]
- 33.Ebert T, Bander NH, Finstad CL, Ramsawak RD, Old LJ. 1990. Establishment and characterization of human renal cancer and normal kidney cell lines. Cancer Res. 50:5531–36. [PubMed] [Google Scholar]
- 34.Sheff JGKJ, Robotham A, Sulea T, Malenfant F, L’Abbé D, Duchesne M, Pelletier A, Lefebvre J, Acel A, et al. 2021. HDX-MS reveals three unique binding responses of mAbs to the catalytic domain of hCAIX. MAbs. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Z, Smyth FE, Renner C, Lee FT, Oosterwijk E, Scott AM. 2002. Anti-renal cell carcinoma chimeric antibody G250: cytokine enhancement of in vitro antibody-dependent cellular cytotoxicity. Cancer Immunol Immunother. 51(3):171–77. doi: 10.1007/s00262-002-0268-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lou Y, McDonald PC, Oloumi A, Chia S, Ostlund C, Ahmadi A, Kyle A, Auf Dem Keller U, Leung S, Huntsman D, et al. 2011. Targeting tumor hypoxia: suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Res. 71(9):3364–76. doi: 10.1158/0008-5472.CAN-10-4261. [DOI] [PubMed] [Google Scholar]
- 37.Brown WS, McDonald PC, Nemirovsky O, Awrey S, Chafe SC, Schaeffer DF, Li J, Renouf DJ, Stanger BZ, Dedhar S, et al. 2020. Overcoming adaptive resistance to KRAS and MEK inhibitors by Co-targeting mTORC1/2 complexes in pancreatic cancer. Cell Rep Med. 1(8):100131. doi: 10.1016/j.xcrm.2020.100131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McDonald PC, Chafe SC, Brown WS, Saberi S, Swayampakula M, Venkateswaran G, Nemirovsky O, Gillespie JA, Karasinska JM, Kalloger SE, et al. 2019. Regulation of pH by carbonic anhydrase 9 mediates survival of pancreatic cancer cells with activated KRAS in response to hypoxia. Gastroenterology. 157(3):823–37. doi: 10.1053/j.gastro.2019.05.004. [DOI] [PubMed] [Google Scholar]
- 39.Pandit-Taskar N, Postow MA. 2020. Immune-directed molecular imaging biomarkers. Semin Nucl Med. 50(6):584–603. doi: 10.1053/j.semnuclmed.2020.06.005. [DOI] [PubMed] [Google Scholar]
- 40.Ochoa MC, Minute L, Rodriguez I, Garasa S, Perez-Ruiz E, Inoges S, Melero I, Berraondo P. 2017. Antibody-dependent cell cytotoxicity: immunotherapy strategies enhancing effector NK cells. Immunol Cell Biol. 95(4):347–55. doi: 10.1038/icb.2017.6. [DOI] [PubMed] [Google Scholar]
- 41.Davis ID, Liu Z, Saunders W, Lee FT, Spirkoska V, Hopkins W, et al. 2007. A pilot study of monoclonal antibody cG250 and low dose subcutaneous IL-2 in patients with advanced renal cell carcinoma. Cancer Immun. 7:14. [PMC free article] [PubMed] [Google Scholar]
- 42.Lawrentschuk N, Lee FT, Jones G, Rigopoulos A, Mountain A, O’Keefe G, Papenfuss AT, Bolton DM, Davis ID, Scott AM, et al. 2011. Investigation of hypoxia and carbonic anhydrase IX expression in a renal cell carcinoma xenograft model with oxygen tension measurements and (1)(2)(4) I-cG250PET/CT. Urol Oncol. 29(4):411–20. doi: 10.1016/j.urolonc.2009.03.028. [DOI] [PubMed] [Google Scholar]
- 43.Zatovicova M, Jelenska L, Hulikova A, Ditte P, Ditte Z, Csaderova L, Svastova E, Schmalix W, Boettger V, Bevan P, et al. 2014. Monoclonal antibody G250 targeting CA: binding specificity, internalization and therapeutic effects in a non-renal cancer model. Int J Oncol. 45(6):2455–67. doi: 10.3892/ijo.2014.2658. [DOI] [PubMed] [Google Scholar]
- 44.Antal I, Koneracka M, Kubovcikova M, Zavisova V, Jurikova A, Khmara I, Omastova M, Micusik M, Barathova M, Jelenska L, et al. 2021. Targeting of carbonic anhydrase IX-positive cancer cells by glycine-coated superparamagnetic nanoparticles. Colloids Surf B Biointerfaces. 205:111893. doi: 10.1016/j.colsurfb.2021.111893. [DOI] [PubMed] [Google Scholar]
- 45.Chen M, Hu J, Wang L, Li Y, Zhu C, Chen C, Shi M, Ju Z, Cao X, Zhang Z, et al. 2020. Targeted and redox-responsive drug delivery systems based on carbonic anhydrase IX-decorated mesoporous silica nanoparticles for cancer therapy. Sci Rep. 10(1):14447. doi: 10.1038/s41598-020-71071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krall N, Pretto F, Decurtins W, Bernardes GJ, Supuran CT, Neri D. 2014. A small-molecule drug conjugate for the treatment of carbonic anhydrase IX expressing tumors. Angew Chem Int Ed Engl. 53(16):4231–35. doi: 10.1002/anie.201310709. [DOI] [PubMed] [Google Scholar]
- 47.Cazzamalli S, Ziffels B, Widmayer F, Murer P, Pellegrini G, Pretto F, Wulhfard S, Neri D. 2018. Enhanced Therapeutic activity of non-internalizing small-molecule-drug conjugates targeting carbonic anhydrase IX in combination with targeted interleukin-2. Clin Cancer Res. 24(15):3656–67. doi: 10.1158/1078-0432.CCR-17-3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Millul J, Krudewig C, Zana A, Dakhel Plaza S, Puca E, Villa A, Neri D, Cazzamalli S. 2021. Immunotherapy with Immunocytokines and PD-1 blockade enhances the anticancer activity of small molecule-drug conjugates targeting carbonic anhydrase IX. Mol Cancer Ther. 20(3):512–22. doi: 10.1158/1535-7163.MCT-20-0361. [DOI] [PubMed] [Google Scholar]
- 49.Indira Chandran V, Mansson AS, Barbachowska M, Cerezo-Magana M, Nodin B, Joshi B, Koppada N, Saad OM, Gluz O, Isaksson K, et al. 2020. Hypoxia attenuates trastuzumab uptake and Trastuzumab-Emtansine (T-DM1) cytotoxicity through redistribution of phosphorylated caveolin-1. Mol Cancer Res. 18(4):644–56. doi: 10.1158/1541-7786.MCR-19-0856. [DOI] [PubMed] [Google Scholar]
- 50.Casey SC, Amedei A, Aquilano K, Azmi AS, Benencia F, Bhakta D, et al. 2015. Cancer prevention and therapy through the modulation of the tumor microenvironment. Semin Cancer Biol. 35(Suppl):S199–S223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murri-Plesko MT, Hulikova A, Oosterwijk E, Scott AM, Zortea A, Harris AL, Ritter G, Old L, Bauer S, Swietach P, et al. 2011. Antibody inhibiting enzymatic activity of tumour-associated carbonic anhydrase isoform IX. Eur J Pharmacol. 657(1–3):173–83. doi: 10.1016/j.ejphar.2011.01.063. [DOI] [PubMed] [Google Scholar]
- 52.Carlin S, Khan N, Ku T, Longo VA, Larson SM, Smith-Jones PM, Boswell A. 2010. Molecular targeting of carbonic anhydrase IX in mice with hypoxic HT29 colorectal tumor xenografts. PLoS One. 5(5):e10857. doi: 10.1371/journal.pone.0010857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lau J, Lin K-S, Benard F. 2017. past, present, and future: development of theranostic agents targeting carbonic anhydrase IX. Theranostics. 7(17):4322–39. doi: 10.7150/thno.21848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Supuran CT. 2016. Structure and function of carbonic anhydrases. Biochem J. 473(14):2023–32. doi: 10.1042/BCJ20160115. [DOI] [PubMed] [Google Scholar]
- 55.Chiche J, Ilc K, Laferriere J, Trottier E, Dayan F, Mazure NM, Brahimi-Horn MC, Pouysségur J. 2009. Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer Res. 69(1):358–68. doi: 10.1158/0008-5472.CAN-08-2470. [DOI] [PubMed] [Google Scholar]
- 56.Doyen J, Parks SK, Marcie S, Pouyssegur J, Chiche J. 2012. Knock-down of hypoxia-induced carbonic anhydrases IX and XII radiosensitizes tumor cells by increasing intracellular acidosis. Front Oncol. 2:199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lock FE, McDonald PC, Lou Y, Serrano I, Chafe SC, Ostlund C, Aparicio S, Winum J-Y, Supuran CT, Dedhar S, et al. 2013. Targeting carbonic anhydrase IX depletes breast cancer stem cells within the hypoxic niche. Oncogene. 32(44):5210–19. doi: 10.1038/onc.2012.550. [DOI] [PubMed] [Google Scholar]
- 58.Durocher Y, Butler M. 2009. Expression systems for therapeutic glycoprotein production. Curr Opin Biotechnol. 20(6):700–07. doi: 10.1016/j.copbio.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 59.Durocher Y, Perret S, Kamen A. 2002. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 30(2):E9. doi: 10.1093/nar/30.2.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tom R, Bisson L, Durocher Y. 2008. Purification of his-tagged proteins using fractogel-cobalt. CSH Protoc. 2008:pdb prot4980. [DOI] [PubMed] [Google Scholar]
- 61.Wilbur KM, Andrson NG. 1948. Electrometric and colorimetric determination of carbonic anhydrase. J Biol Chem. 176(1):147–54. doi: 10.1016/S0021-9258(18)51011-5. [DOI] [PubMed] [Google Scholar]
- 62.Chafe SC, McDonald PC, Saberi S, Nemirovsky O, Venkateswaran G, Burugu S, Gao D, Delaidelli A, Kyle AH, Baker JHE, et al. 2019. Targeting hypoxia-induced carbonic anhydrase IX enhances immune-checkpoint blockade locally and systemically. Cancer Immunol Res. 7(7):1064–78. doi: 10.1158/2326-6066.CIR-18-0657. [DOI] [PubMed] [Google Scholar]
- 63.Brown WS, McDonald PC, Nemirovsky O, Awrey S, Chafe SC, Schaeffer DF, et al. 2020. Overcoming adaptive resistance to KRAS and MEK inhibitors by co-targeting mTORC1/2 complexes in pancreatic cancer. Cell Rep Med. 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Enot DP, Vacchelli E, Jacquelot N, Zitvogel L, Kroemer G. 2018. TumGrowth: an open-access web tool for the statistical analysis of tumor growth curves. Oncoimmunology. 7(9):e1462431. doi: 10.1080/2162402X.2018.1462431. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.