

ABSTRACT
Type-I interferon (IFN-I) signaling is critical to maintaining antigen-presenting cell function for anti-tumor immunity. However, recent studies have suggested that IFN-I signaling may also contribute to more aggressive phenotypes, raising the possibility that IFN-I downstream signaling in cancer and myeloid cells may exert dichotomous functions.We analyzed the clinicopathologic correlation of cancer-specific IFN-I activation in 195 head and neck squamous cell carcinoma patients. We also characterized the immune impact of IFN-I receptor (IFNAR1)-deficiency in syngeneic tumor models using biochemistry, flow cytometry, and single-cell RNA-Seq. We stained HNSCC tissue microarrays with a sensitive IFN-I downstream signaling activation marker, MX1, and quantitated cancer cell-specific MX1 staining. Kaplan-Meier analysis revealed that MX1-high tumors exhibited worse survival, a phenotype that depends on the number of CD8+ intratumoral T-cells. We found that cancer-specific IFNAR1 engagement promotes cancer stemness and higher expression levels of suppressive immune checkpoint receptor ligands in cancer-derived exosomes. Notably, mice bearing Ifnar1-deficient tumors exhibited lower tumor burden, increased T-cell infiltration, reduced exhausted CD4+PD1high T-cells, and increased effector population CD8+IFN-γ+ T-cells. Then, we performed single-cell RNA-sequencing and discovered that cancer-specific IFN-I signaling not only restricts effector cells expansion but also dampens their functional fitness.The beneficial role of IFN-I activation is largely dependent on the myeloid compartment. Cancer-specific IFN-I receptor engagement promotes cancer stemness and the release of cancer-derived exosomes with high expression levels of immune checkpoint receptor ligands. Cancer-specific IFN-I activation is associated with poor immunogenicity and worse clinical outcomes in HNSCC.
KEYWORDS: Type-I interferon, head and neck cancer, sting1, ifnar1, stemness
Introduction:
As immune checkpoint blockade (ICB) is being moved into the first-line setting for cancer treatment, co-targeting resistance mechanisms have become central to the outcomes of immunotherapies. Conceptually, there are two broad resistance mechanisms. The first mechanism is that tumor and myeloid cells upregulate the expression of the inhibitory immune checkpoint receptor ligands, such as programmed cell death ligand 1 (PD-L1), to dampen the activation of effector T-cells. The second class of resistance mechanisms involves the lack of immune priming signals with minimal inflammatory infiltration in the tumor stroma. The first class of mechanisms can be efficiently targeted by leveraging combinatorial ICBs. However, most tumors show features that fall into the second category. The induction of type-I interferon (IFN-I) signaling has been shown as a powerful priming approach to enhance antigen-presenting cells maturation and effector expansion, which sensitizes hypoimmunogenic cold tumors to ICB in several preclinical cancer models.1–3
According to data from the Centers for Disease Control and Prevention, human papillomavirus (HPV)+ Head and Neck Squamous Cell Carcinoma (HNSCC) was one of the only five cancer types that increased from 1975 to 2009; and the increase is predicted to continue until at least the year 2060.4,5 Notably, in contrast to the generally more favorable response profiles to chemotherapy among patients with HPV+ tumors, the Hazard Ratios for nivolumab were almost identical between the HPV+ and HPV− groups despite the presence of viral epitopes.6,7 Similarly, ICB showed little efficacy over standard-of-care in patients with p16+ tumors.8 Our recent study found that HPV potently suppresses IFN-I production from HNSCC cells and encourages cancer immune escape.9 In preclinical models, we and others engineered IFN-I-inducing vaccines targeting the HPV16 E6/E7 epitopes and showed that IFN-I is essential to condition the tumor microenvironment for a better response of ICB-induced tumor antigen-specific cytotoxic T-cell immunity.1,10,11
Not surprisingly, IFN-I-inducing agents have entered clinical trials, including agonists for a central adaptor molecule stimulator of interferon response cGAMP interactor 1 (STING1), which was pivotal for radiotherapy-mediated priming benefits.3 There are three known human STING1 alleles, which showed different sensitivities to its physiological ligand cGAMP. Novel cyclic di-nucleotides have been engineered to activate all known STING1 alleles. However, a substantial subset of patients remains resistant to even these broadly activating cyclic di-nucleotides.12 In addition, discrepancies are present in studies assessing the role of IFN-I in cancer outcomes. For example, genome instability creates micronuclei whose rupture activates the STING1-IFN-I pathway. This encourages cancer metastasis and genetic suppression of chromosomal instability delays metastasis even in highly aneuploid tumor mouse models.13 Higher expression levels of the Bloom Syndrome RecQ-like Helicase (BLM) and exonuclease 1 (EXO1), which facilitate the generation of STING1-inducing DNA fragments, are also associated with poor prognosis in response to radiotherapy.14
Although the activation of the IFN-I pathway is essential for myeloid M1-like polarization and cross-priming of T-cells, it is equally important to understand the mechanisms underpinning the discrepancies in these different studies. Unlike the type II and type III interferons, whose production is restricted to a small collection of cell types, IFN-I is highly evolutionarily conserved, and almost all normal cell types express IFN-I induction machinery and its receptor IFNAR1. The activation of a spectrum of pattern recognition receptors, including the Toll-like receptors (TLRs), RNA sensors such as DExD/H-Box Helicase 58 (DDX58, aka Retinoic Acid-inducible Gene I or RIG-I), Interferon Induced with Helicase C Domain 1 (IFIH1, aka Melanoma Differentiation-Associated Protein 5 or MDA5), or DNA sensor cyclic GMP-AMP Synthase (cGAS), leads to the phosphorylation of IRF3 and NF-κB. Nuclear phospho-IRF3 and phospho-p65 form an enhanceosome to drive the generation of IFN-I, which includes 13 subtypes of IFN-α, IFN-β, IFN-ω, IFN-ϵ, and IFN-κ. IFN-I functions in an autocrine or paracrine fashion to engage its receptor constituted by IFNAR1 and IFNAR2 on the plasma membrane. Upon activation, tyrosine kinases Tyrosine Kinase 2 (TYK2) and Janus Kinase 1 (JAK1) are phosphorylated, which leads to the phosphorylation of the Signal Transducers and Activators of Transcription (STAT)1 and STAT2. Within the IFNAR1 transcriptional program, MX Dynamin Like GTPase 1 (MX1) is a transgene that is specifically induced by IFN-I and used as a sensitive surrogate marker for IFNAR1 signaling.
Thus, the generation of IFN-I and its downstream signaling upon binding to IFNAR1 are two separate events for cancer cells. The secreted IFN-I may function in a paracrine fashion to stimulate the myeloid compartment, which drives most of the observed benefits. Notably, we and others have shown that multiple solid tumors including HPV− and HPV+ HNSCC show deficiencies in the IFN-I induction pathway to evade innate immune detection. However, their IFN-I downstream signaling pathways are often intact, which can be also activated through the Epidermal Growth Factor Receptor (EGFR) pathway.15 Although IFNAR1 downstream signaling activation in myeloid cells enhances its antigen processing efficiency and cross-priming of CD8+ T-cells, the role of cancer cell-specific IFNAR1 downstream signaling in regulating the tumor immune microenvironment remains unclear. MX1 staining on formalin fixed paraffin-embedded tissues has been well established as a sensitive marker for IFNAR1 signaling activation.16,17 In this study, MX1 is used as a surrogate marker for IFNAR1 signaling activation. MX1 expression is stimulated specifically by type-I interferon signaling and does not respond to other cytokines such as IL-1 or TNF-α.18,19 The mechanism by which IFN-I signaling leads to MX1 induction is also well characterized, involving transcriptional regulation through the JAK/STAT pathway, as with other interferon-stimulated genes.20
In this study, we performed a cohort analysis of 195 HNSCC patients to assess the prognostic significance of IFNAR1 signaling activation in cancer cells. We also characterized the impact of the suppression of the IFNAR1 pathway in cancer cells upon cancer stemness and intra-tumoral immune infiltration using IFNAR1-deficient murine HNSCC models.
Materials and methods
Clinical samples and tissue microarray
Patients with previously untreated HNSCC were recruited by the University of Michigan Head and Neck Cancer Specialized Program of Research Excellence (SPORE) between 2008 to 2012 for a longitudinal study. The patient demographic information and clinical characteristics are summarized in Table 1. The tumor tissues from 218 patients, with a median follow-up of 60 months, were included in tissue microarrays (TMAs). For each tumor, 3 representative 5-μm cores, identified by a head and neck pathologist, were included, each of which contained tumor tissue and surrounding stroma. TMAs were stained with anti-MX1 antibody (1:600; Cell Signaling Technology, catalog #37849S). The secondary antibody was biotinylated goat anti-rabbit antibody (1:400, Vectastain ABC HRP Kit, PK-4001, Vector Laboratories). Immunohistochemical (IHC) staining intensity was quantitated in tumor tissue for each core by selecting a region of interest using Aperio ImageScope, followed by quantification. IHC scores were averaged from the 3 cores, and those that were missing or had insufficient tumor parenchyma were excluded from subsequent analysis. In total, MX1 IHC scores were available for 195 patients. The investigators who scored the TMAs were blinded from the clinical information. In experiments involving multivariate Cox regression modeling to control for CD4+ and CD8+ T-cells, immunohistochemistry was performed for CD4 and CD8 followed by an enumeration of intratumoral CD4+ and CD8+ cell numbers for each TMA, in triplicate, by trained observers blinded to the clinical information. The CD4 and CD8 staining of the TMA has been reported.21
Table 1.
Baseline demographic and clinical information corresponding to the TMAs analyzed from an expanded cohort of 195 HNSCC patients (mean follow-up of 60 months)
| Variable | Subgroup | N (%) |
|---|---|---|
| Age Category | <60 | 108 (55%) |
| 60–80 | 76 (39%) | |
| >80 | 11 (6%) | |
| Sex | Male | 147 (75%) |
| Female | 48 (25%) | |
| Stage | 0/1 | 20 (10%) |
| 2 | 20 (10%) | |
| 3 | 29 (15%) | |
| 4 | 126 (65%) | |
| Disease Site | Larynx | 34 (17%) |
| Oral Cavity | 89 (46%) | |
| Oropharynx | 59 (30%) | |
| Hypopharynx/Other | 13 (7%) | |
| ACE Comorbidities Score | none | 58 (30%) |
| mild | 89 (46%) | |
| moderate | 32 (16%) | |
| severe | 15 (7%) | |
| HPV Status | negative | 114 (58%) |
| positive | 60 (31%) | |
| invalid/missing | 21 (11%) | |
| Alcohol consumption | never | 21 (11%) |
| current | 131 (67%) | |
| former (quit >12 months) | 43 (22%) | |
| Smoker (cigarettes) | never | 45 (23%) |
| current | 92 (47%) | |
| former (quit >12 months) | 58 (30%) |
Statistics
Univariate Cox linear regression modeling was used to explore the association between cancer-specific MX1 expression in TMAs and patient clinical variables. The association between MX1 IHC scores and patient survival was conducted using both univariate and multivariate Cox regression models as indicated in Table 2. To dichotomize tumors into MX1-low and MX1-high groups, a threshold score was estimated by the change point in a Cox regression model, generating a change point cutoff score of 41. Statistical significance in survival probability between MX1-low and MX1-high TMAs was evaluated by Kaplan-Meier survival curves and a log-rank test. As a secondary method, we compared overall survival (OS) and recurrence-free time (RFT) in patients exhibiting MX1 scores in the highest quartile (MX1 score ≥ 31.79) vs. patients with lower scores. Statistical analysis between two independent groups was made using unpaired, two-tailed Student’s t-tests. Tumor burden between groups was compared using the generalized estimating equation model. Statistical significance is indicated in all figures according to the following scale: *p < .05; **p < .01; ***p < .001; and ****p < .0001. All graphs are presented as the mean ± SEM. To test the correlation between gene expression levels measured by RNA-seq among 520 TCGA HNSCC patients, we calculated their Spearman correlations and computed corresponding p values using the R package stats.
Table 2.
Association between MX1 score and overall survival or recurrence-free survival using a univariate model or multivariate models controlling for patient age, clinical stage, disease site, comorbidities, HPV status, smoking history, and CD4+ (middle) or CD8+ TILs (bottom)
| MX1 Group | Overall Survival (OS) | Recurrence-Free Survival (RFS) | ||
|---|---|---|---|---|
| Univariate model | HR (95% CI)a | P-value | HR (95% CI) | P-value |
| MX1 score (1 unit increase) | 1.00 (0.99, 1.02) | 0.12 | 1.01 (0.99, 1.02) | 0.35 |
| MX1-high (≥ 41) | 1.97 (1.11, 3.48) | 0.02 | 1.49 (0.73, 3.06) | 0.28 |
| Multivariate model (CD4+) | HR (95% CI) | P-value | HR (95% CI) | P-value |
| MX1 score (1 unit increase) | 1.01 (1.00, 1.03) | 0.14 | 1.00 (0.99, 1.03) | 0.43 |
| MX1-high (≥ 41) | 1.96 (1.02, 3.74) | 0.04 | 1.73 (0.77, 3.87) | 0.18 |
| Multivariate model (CD8+) | HR (95% CI) | P-value | HR (95% CI) | P-value |
| MX1 score (1 unit increase) | 1.00 (0.99, 1.02) | 0.66 | 1.00 (0.98, 1.02) | 0.99 |
| MX1-high (≥ 41) | 1.30 (0.68, 2.48) | 0.42 | 1.25 (0.56, 2.78) | 0.58 |
aAbbreviations: HR: hazard ratio; CI: confidence interval.
Animals
Eight-week-old C57BL/6 J (strain 000664) mice were purchased from The Jackson Laboratory and housed in specific pathogen-free conditions in a temperature- and light-controlled facility. To enhance scientific rigor, and because sex is not a known prognostic variable for HNSCC, both sexes were used. One million empty vector (EV-MOC2-E6/E7) control or shIfnar1-MOC2-E6/E7 cells were implanted subcutaneously in the flank. Seven days after tumor implantation, tumors were measured every 2–3 days and tumor volume calculated according to the formula 1/2 (length × width2). Tumor weight was measured after animals were sacrificed. Mice were euthanized at the time points indicated in the figure legends, followed by tumor, spleen, and TIL processing for the subsequent analyses.
Cell culture
The MOC2-E6/E7 and NOOC1 cell lines were cultured in the following medium: 60% IMDM (SH30228.01, HyClone) with 30% F12 nutrient mix (11764–054, Gibco), 5% FBS, 4 μg/mL puromycin, 5 μg/mL insulin, 40 ng/mL hydrocortisone, 5 ng/mL EGF, 100 U/mL penicillin, and 100 mg/mL streptomycin. Cells were maintained in a humidified incubator at 37°C with 5% CO2. To generate Ifnar1-deficient and control cell lines, MOC2-E6/E7 and NOOC1 cells were transduced with empty vector control (EV) or shIfnar1-expressing lentiviruses followed by puromycin selection. A puromycin killing curve was established using the parental MOC2-E6/E7 cell line. Puromycin at 60 μg/ml was able to kill 100% of the cells and thus was utilized for selection for MOC2-E6/E7, and at 50 µg/ml was used for NOOC1 selection. qPCR on murine Ifnar1, Mx1, Interferon-stimulated genes (Isg)54 and Isg15 was performed to validate the knockdown efficiency. Murine pLKO.1-shIfnar1-puro and pLKO.1–empty vector–puro lentiviral constructs were obtained from Sigma-Aldrich (catalog SHCLNG-NM_010508 and SHC001V, respectively). Lentiviral packaging vectors were provided by Dr. Jenny P.Y. Ting at the University of North Carolina at Chapel Hill (Chapel Hill, North Carolina, USA).
Gene expression qPCR
Total RNA was extracted using QIAshredder and an RNeasy Plus Mini Kit (catalog 79654 and 74134, respectively; Qiagen). A Nanodrop Spectrophotometer (Thermo Fisher Scientific) was used to determine RNA concentrations. RNA was reverse-transcribed into cDNA using High-Capacity cDNA Reverse Transcription Kit and RNase inhibitor (4368814 and N8080119, Applied Biosystems). Primers were obtained from Integrated DNA Technologies and included: Isg54 forward 5′-TCTGGTCACCTGGGGAAACTATG-3′, reverse 5′-TTCTCAATCCTGTAGGGGCTGG-3′; Ifnar1 forward 5′-TCCCCGCAGTATTGATGAGT-3′, reverse 5′-CTGGTCTGTGAGCTGTACTT-3′; Isg15 forward 5′-TGGAAAGGGTAAGACCGTCCT-3′, reverse 5ʹ-GGTGTCCGTGACTAACTCCAT-3′; Mx1 forward 5ʹTCTGAGGAGAGCCAGACGAT −3′, reverse 5ʹ-ACTCTGGTCCCCAATGACAG −3′.
Immunoblotting assay
Whole-cell lysates in each well were collected on ice in RIPA buffer (50 mM Tris-HCl pH 8.0, 1% Triton X-100, 0.05% SDS, 0.25% deoxycholate, 150 mM NaCl, and 50 mM NaF), supplemented with a protease inhibitor cocktail (11836170001, Roche) and Halt Phosphatase inhibitor cocktail (78420, Thermo Fisher Scientific). A total of 10 mg tumor tissue was collected from each tumor-bearing mouse and resuspended in 350 µl RIPA buffer. The mixtures were homogenized and subjected to SDS-PAGE analysis. Antibodies used were: Cd44 (Cell Signaling Technology; #37259; 1:1000 dilution), Bmi-1 (Cell Signaling Technology; #5856; 1:1000 dilution), phospho-Akt (Ser473) (Cell Signaling Technology; #4060; 1:1000 dilution), phospho-P65 (Ser536) (Cell Signaling Technology; #3033; 1:1000 dilution), P65 (Cell Signaling Technology; #8242; 1:1000 dilution), phospho-Irf3 (Ser396) (Cell Signaling Technology; #29047; 1:1000 dilution), Irf3 (Cell Signaling Technology; #4302; 1:1000 dilution), Pd-l1 (Cell Signaling Technology; #64988; 1:1000 dilution), Galectin-9 (Abcam; ab49900; 1:20,000 dilution), β-Actin (Abcam; ab49900; 1:20,000 dilution). The primary antibody was incubated at 4°C overnight. After washing the membranes four times, the secondary antibody was incubated for 1 h at room tempetature. Bands were detected by using an automatic X-ray filmprocessor (Model JP-33, JPI).
Cancer-derived exosome isolation
Cancer-derived exosomes were isolated by using the Total Exosome Isolation Kit (catalog 4478359, invitrogen). The MOC2-E6/E7-EV, MOC2-E6/E7-shIfnar1, NOOC1-EV and NOOC1-shIfnar1 cells were seeded into 10 cm plate with 10 ml culture media. After 48 hours culture, the supernatant was collected and cetrifuged at 2000 × g for 30 minutes to remove the cells and debris. The culture media was mixed with reagent and incubated at 4°C overnight prior to centrifugation at 10,000 × g for 1 hour at 4°C. The pellets at the bottom were resuspended in 120 µl RIPA buffer containing proteinase and phosphatase inhibitors. Equal amounts of exosomes were mixed with 4× NuPAGE LDS Sample Buffer (catalog NP0007, invitrogen) and boiled for 5 minutes at 95°C. The samples were analyzed by SDS-PAGE.
AlamarBlue assay
Empty vector control or shIfnar1 MOC2-E6/E7 cells were seeded at a density of 500 cells/well in flat black bottom 96-well microplates (3904, Corning). Every 24 hours from day 1 to day 4, wells were supplemented with 10% alamarBlue (DAL1025, Invitrogen), and the plate was subsequently incubated at 37°C for 4 hours. The fluorescence intensity (excitation 560 nm, emission 590 nm) of these wells was measured using a Biotek plate reader and Gen5 program (version 2.09), and five replicates per group were simultaneously analyzed.
Flow cytometry
Immune cells from tumors and spleens were purified as we have previously described.10 Briefly, tumors were excised from mice and minced into pieces, followed by dissociation by passing through a 70-μm cell strainer to obtain a single-cell suspension. Spleens were processed by mechanical dissociation, followed by lysis of red blood cells (A10492-01, Gibco). Ficoll-Paque PLUS (17–1440-03, GE Healthcare Life Sciences) was added to the bottom of each tube containing single cell suspensions in RPMI1640, followed by density-gradient centrifugation to purify immune cells. Purified immune cells were stained for multi-fluorophore flow cytometric analysis with the following surface antibodies: anti-CD45 (clone 30-F11, BioLegend), anti-CD3 (clone 17A2, BD Biosciences), anti-TCRβ (clone 56–6.7, BioLegend), anti-CD4 (clone RM4-5, BioLegend), anti-CD8 (clone 53–6.7, BioLegend), anti-CD44 (clone IM7, BioLegend), anti-CD366 [Tim3] (clone RMT3-23, Biolegend), and anti-PD-1 (clone 29F1A12, Biolegend). The Cyto-Fast™ Fix/Perm Buffer Set (Cat # 426803, Biolegend) was used for fixation and permeabilization and anti-mouse IFNγ (clone XMG1.2, BioLegend) antibody was used for intracellular staining. Cell viability was measured using Fixable Viability Dye (FVD) eFluor 780 (65–0865-14, Thermo Fisher Scientific) or Zombie Aqua (423101, BioLegend) diluted 1:1000 in PBS at 4°C for 30 minutes. Acquisition and compensation were conducted on a Beckman Coulter CytoFLEX flow cytometer. FlowJo version 10 software was used for data analysis.
ALDH assay
This assay was performed using the ALDEFLUOR™ Kit (01700, STEMCELL Technologies). In part, empty vector control or shIFNAR1 MOC2-E6/E7 tumor cells and in vitro lines were resuspended into ALDEFLUOR™ Assay Buffer (01701, STEMCELL Technologies) at a working concentration of 1 × 105. Following ALDEFLUOR staining, cells were stained with anti-CD44, and analyzed on a Beckman Coulter CytoFLEX flow cytometer.
Single-cell immune profiling
Whole tumor lysate was treated with ACK Lysing Buffer (A1049201, ThermoFisher), then processed through an EasySep Dead Cell Removal (Annexin V) Kit (17899, STEMCELL Technologies). Cell suspension was submitted in 1× PBS + 0.04% BSA for 10× Genomics 3ʹ-single cell processing and RNA-Seq at a depth of at least 30,000 reads per cell. Libraries were prepared on 10× Chromium and sequencing conducted on Illumina NovaSeq 6000. We utilized the following criteria to select high-quality transcriptomes: (i) the qualified cells must have a unique feature counts between 200 and 7,500; (ii) the mitochondrial reads must be fewer than 10%; (iii) the transcriptomes must have unique molecular identifiers counts over 500. We integrated 12,907 high-quality transcriptomes for downstream analyses. We utilized the sctransform tool to remove the library size effect and the mutual nearest neighbors algorithm to remove the batch effect for final integration. We selected the top 3,000 genes with the highest cell-to-cell variation as well as features to stabilize the immune population structure. The additional features include Cxcr5, Cd69, Aim2, Irf5, Irf1, Irf3, Lgals9, Ly6e, Nos2, Il6, Tnf, Isg15, Gsdmd, Cd8a, Cd40, Cd80, Cd86, Il10, Tgfb1, Tmem173, Cd274, Cxcl9, Mrc1, Siglec15, Trdc, Cd2, Trac, Cd4, Cd8b1, Foxp3, Trbc1, Trbc2, Gzmb, Eomes, Icos, Cd3d, Cd3e, Ifng, Ncr1, Cd19, Cd79a, Cd79b, Itgam, Itgax, Batf3, Xcr1, Gata3, Ctla4, Rorc, Il17a, Bcl6, Havcr2, Tnfrsf4, Tigit, Cxcl10, Mx1, Ifnb1, Il3ra, Nrp1, Fcer1a, Tbx21, Lag3, Ifnl3, Pdcd1, Cd14, Ifna4, Ly6g, Ly6c1, and Lyz2. We obtained the top 50 principal components from the integrated data set. To characterize the differential gene expression for CD8+ T-cells between conditions, we fitted a generalized linear mixed models on counts using the R package Muscat.22
Results
High MX1 protein expression levels in cancer cells were associated with poor prognosis in HNSCC patients
Separate from the pathways that induce the production of IFN-I, IFN-I downstream signaling is mediated by its binding with the receptor IFNAR1. Although the expression levels of IFNAR1 do not directly correlate with IFN-I signaling activation (Supplementary Figure 1), IFNAR1 engagement launches a large transcriptional program characterized by the induction of MX1 metagene and other interferon-stimulated genes (Figure 1a). To validate MX1 as an IFN-I-induced metagene in HNSCC, we first performed Spearman correlation analysis using 520 specimens available at the TCGA database. We found that the expression levels of MX1 were significantly positively correlated with those of ISG15 (Spearman ρ = 0.76; p < .0001), IFIT2 (Spearman ρ = 0.76; p < .0001), CXCL9 (Spearman ρ = 0.48; p < .0001), CXCL10 (Spearman ρ = 0.64; p < .0001), and OASL (Spearman ρ = 0.71; p < .0001) (Figure 1b-f). Thus, we then stained an internal HNSCC cohort with anti-MX1 antibody. Based on our recent data and assuming the same effect size, we performed a power analysis using the R-package LogRankPower, we found that the power for 200 patients was 83.2% at α = 0.05. Thus, tissue microarrays (TMAs) were generated utilizing specimens from 195 patients with HNSCC. The majority of specimens were collected from patients with advanced-stage disease (15% stage 3; 65% stage 4) and were relatively representative of the natural distribution across disease sites, including the larynx (17%), oral cavity (46%), oropharynx (30%), and hypopharynx/other (7%). Other parameters evaluated in this patient cohort, summarized in Table 1, included age, gender, adult comorbidity evaluation (ACE) score, HPV status, and history or present use of tobacco products and alcohol consumption. To assess the prognostic value of cancer cell-specific IFNAR1 downstream signaling, we analyzed MX1 protein levels by IHC staining of the TMAs. MX1 exhibited a diffuse cytoplasmic staining pattern that was highly variable in density among different tumors. MX1 was also stained in the infiltrating immune cells, as expected. To specifically examine the role of cancer-specific IFNAR1 activation, we manually selected tumor parenchyma for 585 cores, with three cores representing one tumor, prior to quantitation (Figure 1g). Cancer-specific MX1 staining scores were quantitated using Aperio ImageScope, as we previously reported.9 These results supported the heterogeneous nature of MX1 distribution, with descriptive statistics summarized in Supplementary Table 1.
Figure 1.
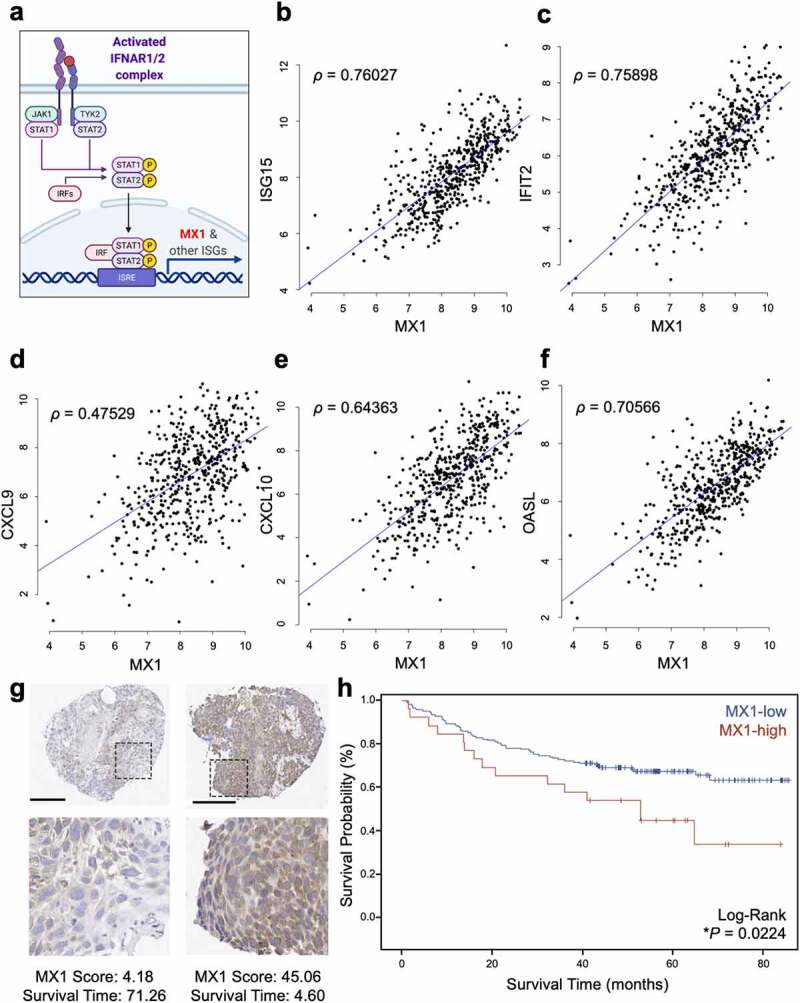
High MX1 protein levels in cancer cells are a negative prognosticator in head and neck squamous cell carcinoma. (a) The signaling schematic shows that the IFNAR1 activation leads to the phosphorylation of STAT1/STAT2, which launches a transcriptional program centering on the ISGs, such as MX1. (b-f) Spearman correlation was performed between the expression levels of MX1 and representative ISGs including ISG15, IFIT2 (ISG54), CXCL9, CXCL10, and OASL using the HNSCC TCGA database (n = 520). (g) MX1 immunohistochemistry in TMAs from HNSCC patients reveals cytoplasmic staining of variable intensity across specimens. Raw MX1 scores and survival time are indicated for each representative TMA. Scale bar: 200 µm. (h) MX1 staining scores were segregated into MX1-low or MX1-high groups and the Kaplan-Meier survival curves of each group were compared using a log-rank test
To analyze the association with relevant clinical variables, MX1 scores were assessed in both continuous form and a dichotomized form wherein tumors were divided into MX1-high and MX1-low groups using the change point threshold in Cox regression modeling, which generated a cutoff score of 41. No significant association was observed between MX1 scores and disease site or nodal metastasis, although a trend existed between the MX1 scores and advanced cancer stage (p = .09) (Supplementary Figure 2a-c). We next examined the impact of cancer cell-specific MX1 protein expression on HNSCC patient overall survival (OS). The Kaplan-Meier survival curves were compared between MX1-high and MX1-low groups using a log-rank test, which revealed that MX1-high staining was significantly correlated with worse overall survival (*p = .02; Figure 1h). Utilizing a univariable Cox regression model where MX1 is the single predictor of outcome, we found that the MX1-high group was correlated with significantly worse OS (*p = .02; Table 2. We also built a multivariate Cox regression model controlling for age, clinical stage, disease site, ACE scores, HPV status, smoking, and CD4+ T-cells among tumor- infiltrating leukocytes (TILs). We found that MX1-high staining remained correlated with significantly worse OS (*p = .04; Table 2). Notably, using a multivariate Cox regression model that controlled for the same aforementioned clinical and social factors but also controlled for CD8+ T-cells instead of CD4+ T-cells among TILs in this cohort of HNSCC specimens, MX1-high tumors were no longer associated with worse prognosis (p = .42; Table 2), suggesting an interaction between high cancer-specific MX1 expression levels and CD8+ T-cells which impacted patient outcomes.
To substantiate the prognostic importance of tumor-specific MX1 expression, we also performed clinical analyses using MX1 scores in the highest quartile (MX1 score ≥ 31.79) vs. patients with lower scores. The Kaplan-Meier survival curves were compared between patients in the MX1-highest 25% and MX1-lowest 75% groups using a log-rank test, which revealed that patients in the highest quartile tended to exhibit worse overall survival and had significantly decreased recurrence-free time (Supplementary Figure 3a-b). Then, we controlled patient age, clinical stage, disease site, comorbidities, and T-cell infiltration in a multivariate model and compared the Kaplan-Meier survival curves. We found a similar trend where MX1-highest 25% group showed a worse prognosis (Supplementary Figure 4a-b). To summarize our findings, we first utilized a univariate model and found that patients with MX1 scores in the highest quartile tended to exhibit worse overall survival (p = .07) and decreased recurrence-free survival (p = .056; Table 3). Then, we built a multivariate Cox model controlling for age, clinical stage, disease site, ACE scores, HPV status, smoking, and CD4+ T-cells. We found that high protein levels of MX1 in cancer cells were associated with significantly worse survival. Notably, the prognostic value of tumor-specific MX1 expression was completely lost when we controlled for CD8+ T-cells (Table 3, Supplementary Figure 5a), in agreement with the results from the dichotomous MX1 expression levels grading model. These results suggest that the clinical impact of tumor-specific MX1 expression depends on CD8+ T-cells.
Table 3.
Association between MX1 scores in the highest quartile vs. all other patients in terms of overall survival or recurrence-free survival using a univariate model or a multivariate model controlling, individually, for patient age, clinical stage, disease site, comorbidities, HPV status, smoking history, CD4+ T-cells, or CD8+ T-cells
|
MX1 Group |
Overall Survival (OS) |
Recurrence-Free Survival (RFS) |
||
|---|---|---|---|---|
| Univariate model | HR (95% CI)a | P-value | HR (95% CI) | P-value |
| MX1-upper quartile vs. lower | 1.66 (0.96, 2.90) | 0.072 | 1.90 (0.99, 3.65) | 0.056 |
|
Multivariate model (MX1-upper quartile vs. lower) |
HR (95% CI) | P-value | HR (95% CI) | P-value |
| Controlling for ageb | 1.03 (1.01, 1.05) | 0.014 | 1.01 (0.98, 1.03) | 0.562 |
| Controlling for stagec | 2.40 (1.12, 5.14) | 0.025 | 3.24 (1.09, 9.60) | 0.034 |
| Controlling for sited | 2.55 (1.26, 5.15) | 0.001 | 2.33 (0.94, 5.77) | 0.066 |
| Controlling for ACE scoree | 5.30 (1.99, 14.2) | 0.001 | 3.99 (1.37, 11.6) | 0.011 |
| Controlling for smoking historyf | 3.78 (1.58, 9.06) | 0.003 | 1.79 (0.73, 4.34) | 0.201 |
| Controlling for CD4+ T-cellsg | 0.68 (0.52, 0.90) | 0.006 | 0.74 (0.56, 0.98) | 0.034 |
| Controlling for CD8+ T-cellsh | 0.93 (0.77, 1.13) | 0.472 | 0.98 (0.80, 1.19) | 0.804 |
aAbbreviations: HR: hazard ratio; CI: confidence interval.
bMultivariate model comparing patients with MX1 scores in upper quartile vs. others while controlling for each additional year older.
cMultivariate model comparing patients with MX1 scores in upper quartile vs. others while controlling for advanced stage (stages 3 and 4 vs. stages 1 and 2).
dMultivariate model comparing patients with MX1 scores in upper quartile vs. others while controlling for disease site (oral cavity vs. larynx).
eMultivariate model comparing patients with MX1 scores in upper quartile vs. others while controlling for ACE score (severe vs. none)
fMultivariate model comparing patients with MX1 scores in upper quartile vs. others while controlling for smoking history (current smoker vs never smoked)
gMultivariate model comparing patients with MX1 scores in upper quartile vs. others while controlling for each 10 unit increase in CD4+ T-cells as determined by IHC.
hMultivariate model comparing patients with MX1 scores in upper quartile vs. others while controlling for each 10 unit increase in CD8+ T-cells as determined by IHC.
Loss of IFN-I signaling in HNSCC cells inhibits cancer-derived exosomal checkpoint receptor ligands expression and reduces tumor burden
To model the impact of the loss of tumor-specific IFN-I downstream signaling upon the CD8+ T-cells and the tumor immune microenvironment, we first utilized an established MOC2-E6/E7 syngeneic HNSCC model.10 This model does not produce IFN-I upon transfection with STING1 agonists,9 which allows us to assess tumor response to IFN-I only from the tumor microenvironment. We engineered a derivative model in which the IFN-I receptor Ifnar1 is defective (MOC2-E6/E7-shIfnar1) (Figure 2a). A deficiency of Ifnar1 resulted in abolished Mx1 gene expression in response to recombinant Ifn-β treatment (Figure 2b). In agreement, Ifnar1-deficient MOC2-E6/E7 cells also exhibited significantly reduced expression levels of two representative ISGs, Isg54 and Isg15 (Figure 2c-d). Recently, we generated a new HNSCC murine model that bears over 90% similarity to human tobacco-associated cancers, NOOC1.23 To validate our findings, we generated Ifnar1-deficient NOOC1 cells (Figure 2e). We found that a defect in Ifnar1 expression resulted in compromised expression of Mx1, Isg54, and Isg15 in response to Ifn-β (Figure 2f-h).
Figure 2.
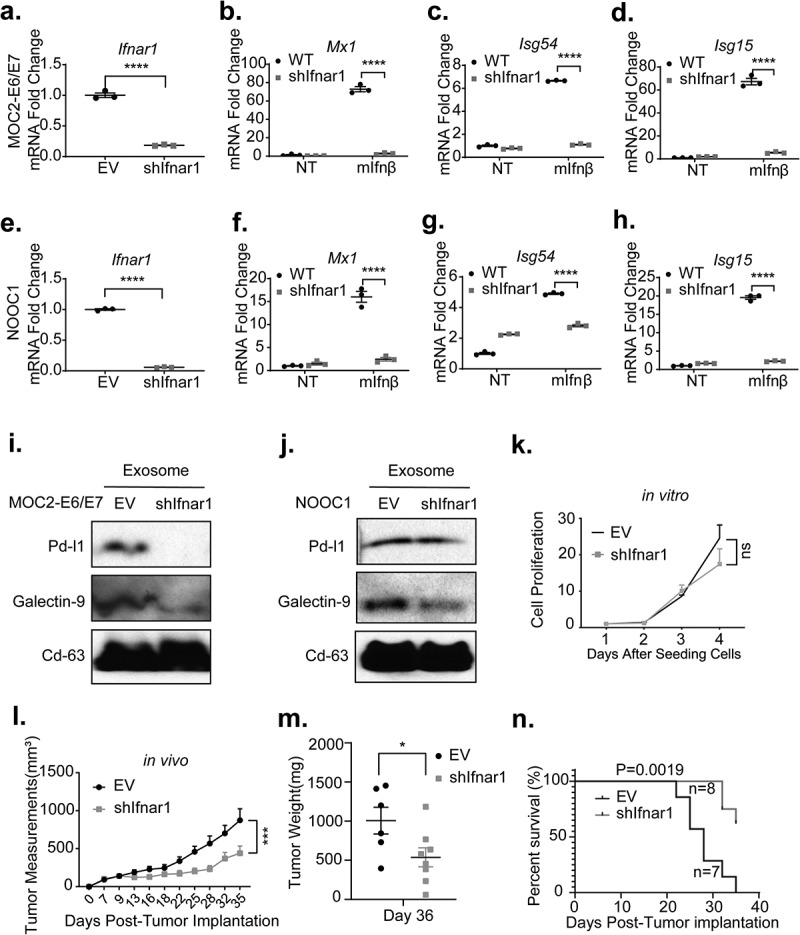
Loss of IFN-I signaling in HNSCC cells inhibits cancer-derived exosomal checkpoint receptor ligands expression and reduces tumor burden. (a) EV-control and Ifnar1-deficient MOC2-E6/E7 cells were generated. The Comparisons between the two groups were made using a two-tailed unpaired t test (****P< .0001). (b-d) The transcription levels of IFN-I target genes Mx1, Isg54, and Isg15 were compared between the EV control and MOC2-E6/E7-shIfnar1 groups with or without a 22-hour incubation with 100 units/ml recombinant murine Ifn-β. (e) EV-control and Ifnar1-deficient NOOC1 cells were validated. (f-h) EV-control and Ifnar1-deficient NOOC1 cells were treated with PBS or 100 units/ml recombinant murine Ifn-β. The expression levels of IFN-I target genes were quantitated using qPCR. Experiments were performed twice. The comparisons were made by two-way ANOVA followed by Sidak’s multiple comparisons test (****P < .0001). Total exosomes were isolated from EV control and shIfnar1 MOC2-E6/E7 cells (f) and shIfnar1 NOOC1 cells (j) culture media and then subjected to immunoblotting for Pd-l1, Galectin-9, and Cd63. (k) The proliferation of EV control and Ifnar1-deficient MOC2-E6/E7 cells was measured via an alamarBlue assay. Each group included 5 replicate wells. Experiments were performed twice. (l-n) One million EV control or IFNAR1-deficient MOC2-E6/E7 cells were implanted subcutaneously in the right flank of C57BL/6 hosts (n = 7 in control group, n = 8 in shIfnar1 group). (l) Tumor measurements were performed every 2–4 days and (m) tumor weight was measured following euthanization. The comparisons were made using a two-tailed unpaired t test (*P< .05, ***P< .001). (n) The Kaplan-Meier survival curves were generated using a cutoff point of 500 mm3 in tumor volume. The comparison was made using a Log-rank (Mantel-Cox) test. The tumor growth in vivo was representative of three repeats
Previous evidence showed that IFN-I promotes the expression of PD-L1 on cancer cell membrane and promotes resistance to anti-PD-1 ICB.24 Exosomal checkpoint ligand-mediated inhibition of effector T-cells correlates with HNSCC progression.25,26 However, it is unclear whether Ifnar1 signaling in cancer cells modulates the content of cancer-derived exosomes. We purified CD63+ cancer-derived exosomes from empty vector control as well as Ifnar1-deficient MOC2-E6/E7 and NOOC1 cells. We found that the loss of Ifnar1 signaling abrogated exosomal expression of immune checkpoint receptor ligands PD-L1 and Galectin-9 in MOC2-E6/E7 cells (Figure 2i). Loss of Ifnar1 signaling did not affect exosomal PD-L1 expression but substantially reduced the exosomal Galectin-9 levels in NOOC1 cells (Figure 2j). Checkpoint ligands represent a critical mechanism driving cancer resistance to immune killing.27 To assess the functional impact of specific disruption of Ifnar1 signaling engagement in cancer cells, we next sought to characterize the immune microenvironment in Ifnar1-deficient tumors. As a control, we showed that impaired downstream IFN-I signaling did not affect cell proliferation in vitro (Figure 2k). Interestingly, when we implanted empty vector control and Ifnar1-deficient MOC2-E6/E7 cells into C57BL/6 mice, we observed a significant reduction in tumor growth and improved survival (**p = .0019) in the Ifnar1-deficient tumor group (Figure 2l-n).
Cancer-specific IFNAR1 engagement promotes cancer stemness and effector exhaustion
HNSCC stem cells have been demonstrated to express higher levels of PD-L1 and promote resistance to immune killing.28 Based on our findings that Ifnar1 signaling maintains exosomal immune checkpoint ligands expression and growth advantage in vivo (Figure 2), we next specifically assessed the impact of Ifnar1 signaling on cancer stemness. We observed that Ifnar1 deficiency reduced the fraction of CD44highALDHhigh cells showing features of stemness (Figure 3a-b). To comprehensively profile cancer stemness-associated markers, we employed two high-fidelity models, including MOC2-E6/E7 and NOOC1. In agreement with our flow cytometry findings, loss of Ifnar1 resulted in a substantial reduction of CD44 expression in both models (Figure 3c-d). In addition, we found that Ifnar1 signaling in cancer cells maintained high levels of phosphorylated Akt, which is essential to maintain cancer stemness (Figure 3c-d).29,30 Indeed, we examined a recently established cancer stemness marker for HNSCC, BMI-1.31 We found that Ifnar1 signaling deficiency resulted in a decrease in the levels of BMI-1 in MOC2-E6/E7 and NOOC1 cells (Figure 3c-d). The signaling competency required for the generation of IFN-Is is independent of the Ifnar1 signaling downstream of IFN-Is binding to their receptor. Indeed, the Ifnar1 deficiency only affected the downstream cancer stemness pathway and did not impact the phosphorylation of p65 and IRF3 (Figure 3c-d), which are enhanceosome components for IFN-I genes transcription. To validate these findings in vivo, we homogenized established empty vector control and Ifnar1-deficient MOC2-E6/E7 tumors and immunoblotted CD44. We found that Ifnar1 signaling deficiency resulted in substantial suppression of CD44 (Figure 3e). Then, we examined the immunohistochemical staining of CD44 in the HNSCC TMAs. We identified a significant positive correlation between the scores of CD44 and those of MX1 in oropharyngeal squamous cell carcinomas (Spearman ρ = 0.4, p = .003) (Supplementary Figure 5b).
Figure 3.
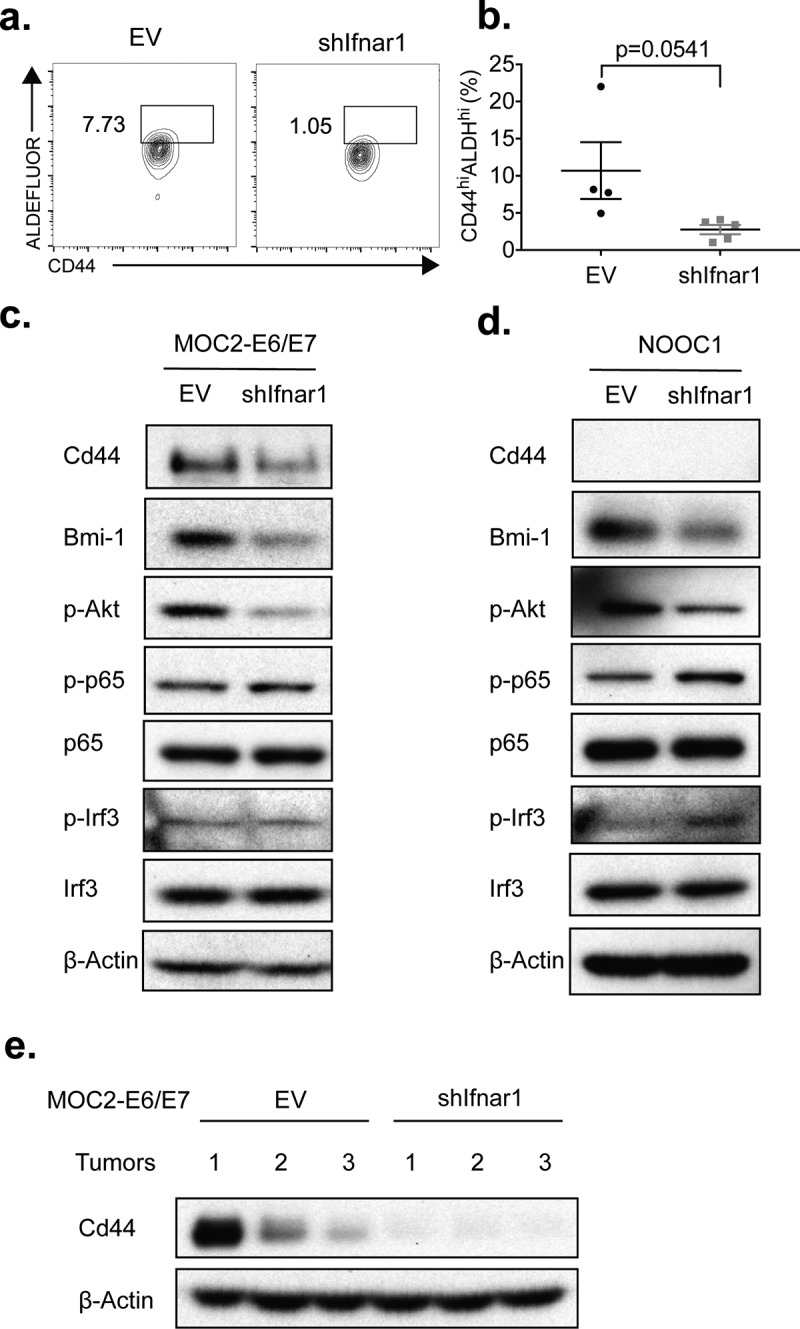
Loss of IFN-I signaling in HNSCC cells promotes cancer stemness. (a-b) EV control and shIfnar1 MOC2-E6/E7 cells were stained using the ALDEFLUOR Assay Kit and analyzed via flow cytometry. Immunoblots were performed to compare the expression levels of the indicated cancer stemness markers in EV control and shIfnar1-MOC2-E6/E7 cells (c) as well as EV control and shIfnar1-NOOC1 cells (d). Experiments were performed twice. (e) 1.0 × 106 EV control and shIfnar1-MOC2-E6/E7 cells were implanted s.c. Upon harvesting, the tumors were homogenized in RIPA buffer and protein extracted from the control and Ifnar1-deficient tumors (n = 3). The tumors were selected randomly. The expression levels of Cd44 were assessed by immunoblotting
Cancer-specific IFN-I downstream signaling impacts the global immune landscape
To directly assess the impact of cancer-specific Ifnar1 signaling activation upon the tumor immune microenvironment, we separated the TILs through Ficoll-Paque gradient centrifugation and identified an enhancement in CD4+ and CD8+ T-cell infiltration in Ifnar1-deficient tumors (Figure 4a-d). Then, we compared the functional status of intratumoral T-cells from empty vector control and Ifnar1-deficient tumors. Notably, we found that a defect in cancer cell-specific IFN-I downstream signaling significantly reduced the terminally exhausted effector T-cell subsets, including CD4+PD1high T-cells (Figure 4e-f).32 To characterize the functional impact of such changes, we next stimulated TILs with PMA/Ionomycin and quantitated intracellular IFN-γ. We found that CD8+ T-cells isolated from Ifnar1-deficient tumors expressed higher levels of IFN-γ (Figure 4g-h), consistent with its reduced tumor burden. Such changes were specific to the tumor microenvironment as no apparent changes were observed from the lymphocytes separated from spleens (Supplementary Fig. 6).
Figure 4.

Cancer-cell-specific IFN-I receptor signaling potentiates effector T-cell exhaustion in the tumor microenvironment. Tumors were harvested 36 days following tumor implantation, and lymphocytes were isolated using a Ficoll-Paque gradient. (a-b) The proportion of CD3+CD4+TILs were assessed by flow cytometry. Gating strategy was as follows: Lymphocytes, single cells, Zombie Aqua negative (viability), CD45-positive, CD3-positive (n = 4 in the control group, n = 5 in the shIfnar1 group). (c-d) The proportion of CD3+CD8+ T-cells in TILs or spleens were assessed by flow cytometry. Gating strategy was as follows: Lymphocytes, single cells, Zombie Aqua negative (viability), CD45-positive, CD3-positive (n = 4 in the control group, n = 5 in the shIfnar1 group). (e-f) The proportion of CD4+PD1highT-cells in TILs were quantitated by flow cytometry. Gating strategy was as follows: Lymphocytes, single cells, Zombie Aqua negative (viability), CD45-positive, TCRβ-positive, CD4-positive (n = 4 in the control group, n = 5 in the shIfnar1 group). (g-h) TILs were stimulated with PMA (1.0 mg/ml), Ionomycin(10.0 mg/ml), and Monensin (1×) for 4 hours and permeabilized for intracellular IFN-γ staining. The proportion of CD8+IFN-γ+ T-cells in TILs was shown. The gating strategy was as follows: Lymphocytes, single cells, Zombie Aqua negative (viability), CD45-positive, TCRβ-positive, CD8-positive (n = 6 in the control group, n = 7 in the shIfnar1 group). The tumor growth in vivo is representative of three repeats. All comparisons between the two groups were made using a two-tailed unpaired t test (*P< .05, *** P.< .001)
To comprehensively characterize the impact of tumor-intrinsic IFN-I signaling on the tumor immune microenvironment, we performed single-cell RNA-sequencing (scRNA-seq) to render the intratumoral immune landscape. We performed Ficoll-Paque purification, followed by CD45+ FACS sorting, to enrich TILs. After filtering, we integrated 12,907 high-quality transcriptomes and identified 20 cell clusters (Figure 5a, Supplementary Table 2, which showed distinct immune lineage segregation and smooth functional transition. The majority of the intratumoral leukocytes consisted of the myeloid cells, including myeloid-derived suppressor cells (MDSCs, clusters 1, 2, 5, 7, 8, 9, and 12), M2-like macrophages (cluster 13), and conventional dendritic cells type 1 (cDC1, cluster 20). The CD4+ T-cells were separated to regulatory T-cells (Treg, cluster 15) and a stem-like cluster with high expression levels of Tcf7 (cluster 3). The effector populations included Tbet+CD8+ T-cells (cluster 6), Tcf7+CD8+ T-cells (cluster 18), natural killer (NK) cells (cluster 17), and γδ T-cells (cluster 16). We also identified Cxcr5+ B-cells (clusters 4, 10, and 11) and a minute population of basophils (cluster 19) (Figure 5a). Global intratumoral immune microenvironment remodeling was noted when tumor cells were deficient in downstream IFN-I signaling (Figure 5b-c). Inhibition of cancer-specific Ifnar1 signaling transduction expanded the frequencies of CD8+ T-cells by 5.4 folds, Tcf7+CD4+ T-cells by 7.3 folds, and γδ T-cells by 3.6 folds. There was an about 50% reduction in MDSCs. Also observed was a 7.6-fold expansion of Cxcr5+ B-cells in the Ifnar1-deficient tumors (Figure 5b-c).
Figure 5.
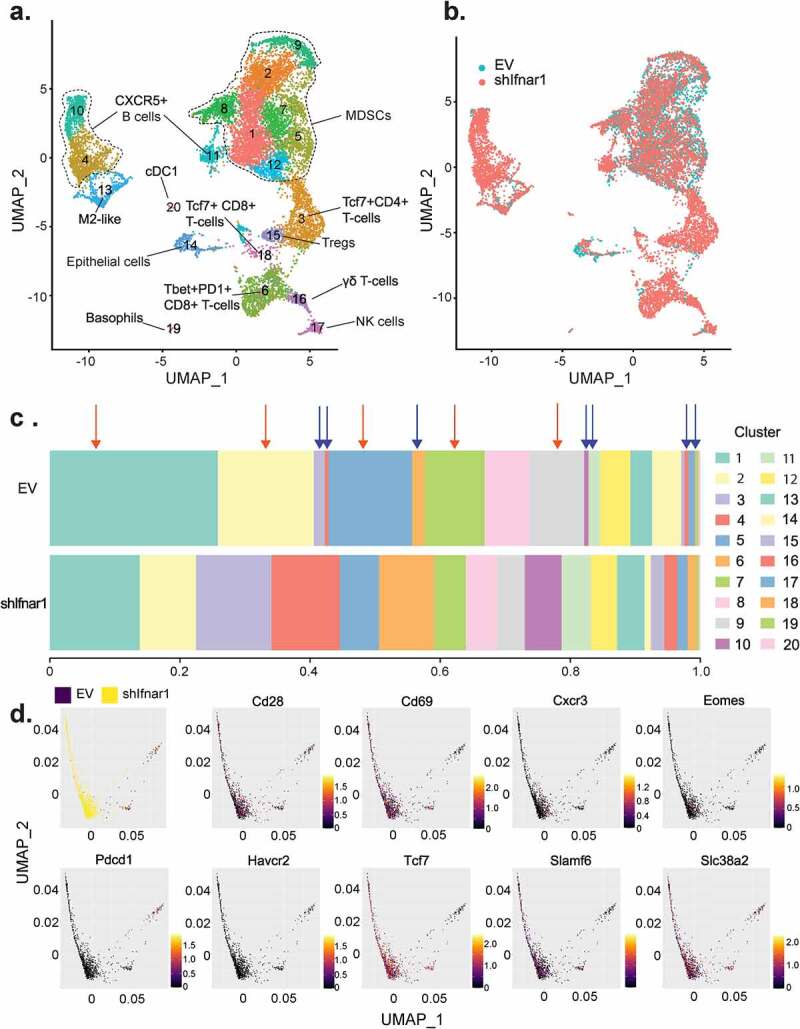
Cancer-cell-specific IFN-I receptor signaling remodels the intratumoral immune landscape. Empty vector (EV) control and Ifnar1-deficient MOC2-E6/E7 tumors were harvested at day 36 after implantation. TILs were purified and pooled from six mice per group and subjected to single-cell RNA-sequencing. (a) After filtering, 12,907 high-quality transcriptomes were integrated for UMAP analysis. Divergent myeloid and lymphoid lineages were defined by top 3,000 genes with the highest cell-to-cell variation and enforced features of 69 immune cell marker genes. (b) UMAP analyses demonstrating the tumor immune landscape in EV control (green) and Ifnar1-deficient (red) tumors are shown. (c) Comparison of the relative proportion of cell types identified in EV control and Ifnar1-deficient tumors. Ifnar1-deficient tumors exhibited a marked decrease in the proportion of several cell types (red arrows), which was particularly pronounced among myeloid-derived suppressor cells (MDSCs, clusters 1, 2, 5, 7, 8, 9, and 12). In Ifnar1-deficient tumors, increased enrichment was observed in the proportion of Tcf7+CD4+ T-cells (cluster 3), CD8+ T-cells (clusters 6 and 18), γδ T-cells (cluster 16), as well as Cxcr5+ B-cells (clusters 4, 10, and 11), as indicated by blue arrows. (d) PHATE analyses were performed to reveal the divergent differentiation trajectories of the CD8+ T-cells (clusters 6 and 18 from Figure 5a) in EV control (black) and Ifnar1-deficient tumors (yellow). The expression patterns of functional markers across the CD8+ T-cell landscape were highlighted in the rest of the panels, with red showing the highest expression levels and black showing the lowest expression levels
Thus, using parallel flow cytometry and single-cell analysis, we found that the activation of downstream IFN-I signaling in cancer cells not only led to lymphoid contraction within the tumors but caused dysfunction of CD8+ T-cells. To delineate the impact of cancer-intrinsic IFN-I activation upon the differentiation of CD8+ T-cells, we separated the CD8+ subsets (clusters 6 and 18) and performed Potential of Heat-diffusion for Affinity-based Transition Embedding (PHATE) analysis, which revealed the functional development pathways of CD8+ T-cells. We found that the CD8+ T-cells in empty vector control and Ifnar1-deficient tumors assumed divergent functional commitments. MOC2 has been established as a hypoimmunogenic HNSCC model that is refractory to ICBs.33,34 In agreement, we found that the CD8+ T-cells in control tumors largely committed to a terminal exhaustion phenotype with high expression levels of Pd-1 and Tim-3. These cells were low on activation markers such as Cd28 and Cd69 as well as stemness markers Tcf7 and Slamf6. In contrast, increased frequency of CD8+ T-cells in Ifnar1-deficient tumors expressed high levels of Cxcr3, which is rapidly induced upon activation of Th1 effectors.35 Notably, most of these CD8+ T-cells in Ifnar1-deficient tumors preserved stemness features, which are critical in maintaining cancer immunogenicity.36,37 Then, we utilized the R-package muscat to identify novel differentially expressed genes between the CD8+ T-cells separated from the two groups (Supplementary Table 3). In agreement with the enhanced effector function in the CD8+ T-cells from Ifnar1-deficient tumors, we observed higher expression levels of Cxcr3, which is expressed by activated Th1 T-cells.35 In addition, the CD8+ T-cells from Ifnar1-deficient tumors also expressed significantly higher levels of Stat5b, which is essential for effector maintenance and proliferation38,39 (Supplementary Table 3). Glutaminolysis has been shown to be activated in Th1 cells to support their effector function.40–42 Interestingly, we found that the terminally exhausted CD8+ T-cells in control tumors largely lost the expression of a glutamine transporter Slc38a2. However, Slc38a2 was highly expressed in CD8+ T-cells that also expressed a stemness marker Tcf7 (Figure 5d).
Discussion
Progress has been made toward understanding cancer resistance to ICB. An optimal response to immunotherapy requires the existance of a pool of tumor-specific T-cells prior to treatment. STING-mediated IFN-I activation underpins the success of multiple innate immune priming strategies that aim to enrich the tumor-specific T-cell pool. Irradiation, inhibition of DNA damage repair, and direct stimulation with cyclic dinucleotides are utilized to trigger DNA-induced STING1 activation. However, the efficacy of this broad class of innate immune priming treatments is challenged by unknown mechanisms that dampen tumor sensitivity to STING1 stimulation. The main focus on the immune remodeling effect of STING1 agonists had been on the M1-like repolarization of myeloid cells. This study aims to understand whether cancer-specific IFN-I downstream signaling contributes to immunosuppression. We first assessed whether cancer intrinsic IFNAR1 signaling is clinically significant for patients with HNSCC. Utilizing univariate and multivariate Cox regression models, we found that high MX1 protein levels in cancer cells were associated with worse overall survival. MX1 is a sensitive surrogate marker for IFNAR1 downstream signaling activation, suggesting that sustained IFNAR1 engagement was an unfavorable prognosticator in HNSCC. Notably, the association between cancer-specific MX1 expression and worse outcomes is dependent on the number of intra-tumoral CD8+ T-cells. A caveat in assessing TILs in TMAs is that the small cores may not represent tumor stromal heterogeneity. To address this potential problem, we sampled three cores for each tumor and also validated the link between cancer-specific IFN-I signaling and intra-tumoral CD8+ T-cells using well-defined preclinical models. Future validation using an independent cohort with other ISGs would also be helpful to characterize further the role of cancer-specific IFN-I activation in patient outcomes.
The current IFN-I agonists were administered to globally activate IFNAR1 signaling in both immune cells and other cell types, including cancer cells. The beneficial effect of STING1-IFN-I activation is dependent on myeloid reprogramming.3,43 Activation of IFNAR1 signaling in immune cells not only enhances antigen processing, antigen presentation, MHC molecule upregulation but also improves trafficking and recruitment of innate and adaptive immune cells to the otherwise T-cell-deficient tumor stroma. However, IFNAR1 activation in cancer cells promotes cancer stemness and potentiates a T-cell exclusion niche. IFN-I stimulation not only makes cancer cells assume stemness features to develop resistance to the immune killing but reprograms the global tumor immune microenvironment. We performed parallel flow cytometry and single-cell RNA-Seq to reveal the divergent myeloid and lymphoid remodeling imposed by cancer-specific IFN-I signaling disruption. We found that Ifnar1-deficient tumors harbor reduced MDSCs and expanded effectors, including CD8+ T-cells, non-Treg CD4+ T-cells, and γδ T-cells. Notably, blocking cancer-specific IFN-I signaling not only expands CD8+ T-cell pool but remodels its differentiation trajectory. We utilized a well-established cold cancer model and indeed found that the majority of the intratumoral CD8+ T-cells have committed to terminal exhaustion with the double expression of Pd-1 and Tim-3. However, Ifnar1-deficient tumors expand Tcf7+ stem-like effector T-cells, which preserve high proliferation potential and responsiveness to ICB. Mechanistically, cancer stem cells in HNSCC are a small yet highly tumorigenic population that selectively express PD-L1 and dampen autologous CD8+ T-cell activation.28 Thus, cancer-intrinsic IFNAR1 signaling activation likely initiates a transcriptional program that is overlapping with the cancer stemness program to dampen the anti-tumor effect of STING1 stimulation.
A strong positive link between chronic inflammation and increased cancer risk had been discovered long before the advent of ICB therapy. In fact, a pathologist Rudolf Virchow speculated that chronic inflammation caused cancer in the year 1863.44 NF-κB activation is a key event for IFN-I induction. It is frequently activated in HNSCC,45 and its activation in tumor cells enhances HNSCC invasion and resistance to therapy.46–48 Thus, the duration of immune activation therapy is also likely shaping the evolution of tumor resistance. Most preclinical models for STING1 agonists testing were based on relatively acute administration into implantable subcutaneous tumor models. Chronic STING1 stimulation likely results in sustained NF-κB activation in tumor cells. This pathway encourages transcriptional programs associated with stemness that is associated with a more tolerogenic local immune niche.
To enhance the therapeutic effectiveness and avoid chronic STING1 stimulation, several robust delivery vehicles have been engineered for STING1 agonists. Controlled release of STING1 agonists using a peptide hydrogel significantly extends the survival of tumor-bearing mice compared to treatment with free STING1 agonists treatment alone.11 When STING1 agonists are delivered using nanoparticles in combination with ICB, tumor-specific CD8+ T-cells are significantly expanded.10 Similarly, two other delivery systems, including endosomolytic polymersomes nanoparticles and biodegradable polymer acetylated dextran microparticles also enhance the efficacy of STING1.49,50 Nanoparticles and microparticles are preferably taken up by myeloid cells and protect cargo from rapid degradation. Due to the unfavorable pharmacokinetics property of free cGAMP, the integration of these delivery systems is a highly promising strategy to enhance myeloid response and avoid chronic stimulation of the IFNAR1 signaling in cancer cells.
Conclusions
Overall, to better characterize the mechanisms driving cancer resistance to STING1-inducing immune priming therapies, we separated IFNAR1 signaling in cancer cells from the IFN-I signaling in immune cells. Downstream IFNAR1 signaling activation in cancer cells is associated with poor clinical outcomes. Blocking IFNAR1 signaling in the tumor cells expands stem-like effector T-cells, restricts MDSCs, and reduces tumor burden. Future development of robust IFN-I-inducing formulations is important to rapidly control tumors through myeloid reprograming while avoiding prolonged stimulation of HNSCC cells, which triggers an adaptive mechanism promoting resistance to innate immune priming strategies.
Supplementary Material
Acknowledgments
This research was funded by NIH grants R01 DE026728, U01 DE029255, F31 DE028740, T32 AI007413, and T32 GM008600. The core facilities and bioinformatics analyses are supported, in part, by P30 CA046592 and U24CA232979. We thank the investigators in the University of Michigan Head and Neck SPORE group for their valuable contributions to patient enrollment, tissue procurement, and TMA generation. We also thank the patients who participated in the study.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Funding Statement
This research was funded by NIH grants R01 DE026728, R01 DE030691, U01 DE029255, F31 DE028740, T32 AI007413, and T32 GM008600. The core facilities and bioinformatics analyses are supported, in part, by P30 CA046592 and U24 CA232979.
Disclosure statement
Y.L.L. licensed the NOOC1 model to Kerafast Inc. Y.L.L. is a co-founder and serves on the Scientific Advisory Board for Saros Therapeutics. The other authors declare no conflict of interest relevant to the current study. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Supplementary Materials:
Supplemental data for this article can be accessed on the publisher’s website
Author contributions
Y.L.L. was responsible for the overall conception and direction of this project with intellectual input from C.R.D., J.C.B., S.B.C., J.E.N., H.W., G.T.W., and R.R.J. C.R.D., L.A.D., and E.B. performed the MX1 quantitation and clinicopathologic correlation analyses. C.R.D., B.R.H., W.G., J.W. A.M.N., and Y.X. performed experiments and conducted data analyses. C.R.D. and Y.L.L. wrote the manuscript and all authors edited the manuscript and have read and agreed to its contents.
Ethics statement
All preclinical procedures and procurement of clinical specimens took place at the University of Michigan. Animal procedures were performed in accordance with an active protocol approved by the University of Michigan Institutional Animal Care and Use Committee (PRO00008517 and PRO00010232).The clinical protocol to procure HNSCC specimens was approved by the University of Michigan Institutional Review Board (HUM00113038 and HUM00042189). Informed consent was obtained from all patients enrolled in the study.
References
- 1.Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, Mechette K, Leong JJ, Lauer P, Liu W, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med. 7283: 2015. doi: 10.1126/scitranslmed.aaa4306 [published Online First: 2015/04/17] 283ra52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luo M, Wang H, Wang Z, Cai H, Lu Z, Li Y, Du M, Huang G, Wang C, Chen X, et al. A STING-activating nanovaccine for cancer immunotherapy. Nat Nanotechnol. 2017;12(7):648–16. doi: 10.1038/nnano.2017.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li X-D, Mauceri H, Beckett M, Darga T, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type i interferon-dependent antitumor immunity in immunogenic tumors. Immunity. 2014;41(5):843–852. [published Online First: 2014/12/18]. doi: 10.1016/j.immuni.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jemal A, Simard EP, Dorell C, Noone AM, Markowitz LE, Kohler B, Eheman C, Saraiya M, Bandi P, Saslow D, et al. Annual report to the nation on the status of cancer, 1975-2009, featuring the burden and trends in human papillomavirus(HPV)-associated cancers and HPV vaccination coverage levels. J Natl Cancer Inst. 2013;105(3):175–201. [published Online First: 2013/01/09]. doi: 10.1093/jnci/djs491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gillison ML, Chaturvedi AK, Anderson WF, Fakhry C. Epidemiology of human papillomavirus-positive head and neck squamous cell carcinoma. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology. 2015;33(29):3235–3242. [published Online First: 2015/09/10]. doi: 10.1200/JCO.2015.61.6995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferris RL, Blumenschein G Jr., Fayette J, Guigay J, Colevas AD, Licitra L, Harrington KJ, Kasper S, Vokes EE, Even C, et al. Nivolumab vs investigator’s choice in recurrent or metastatic squamous cell carcinoma of the head and neck: 2-year long-term survival update of CheckMate 141 with analyses by tumor PD-L1 expression. Oral Oncol. 2018;81:45–51. [published Online First: 2018/06/10]. doi: 10.1016/j.oraloncology.2018.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferris RL, Blumenschein G Jr., Fayette J, Guigay J, Colevas AD, Licitra L, Harrington K, Kasper S, Vokes EE, Even C, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375(19):1856–1867. [published Online First: 2016/10/11]. doi: 10.1056/NEJMoa1602252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cohen EEW, Soulieres D, Le Tourneau C, Dinis J, Licitra L, Ahn M-J, Soria A, Machiels J-P, Mach N, Mehra R, et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): a randomised, open-label, phase 3 study. Lancet. 2019;393(10167):156–167. 31999-8 [published Online First: 2018/12/05]. doi: 10.1016/S0140-6736(18)31999-8. [DOI] [PubMed] [Google Scholar]
- 9.Luo X, Donnelly CR, Gong W, Heath BR, Hao Y, Donnelly LA, Moghbeli T, Tan YS, Lin X, Bellile E, et al. HPV16 drives cancer immune escape via NLRX1-mediated degradation of STING. J Clin Invest. 2020;130(4):1635–1652. [published Online First: 2019/12/25]. doi: 10.1172/JCI129497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tan YS, Sansanaphongpricha K, Xie Y, Donnelly CR, Luo X, Heath BR, Zhao X, Bellile E, Hu H, Chen H, et al. Mitigating SOX2-potentiated immune escape of head and neck squamous cell carcinoma with a STING-inducing nanosatellite vaccine. Clin Cancer Res. 2018;24(17):4242–4255. [published Online First: 2018/05/18]. doi: 10.1158/1078-0432.CCR-17-2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leach DG, Dharmaraj N, Piotrowski SL, Lopez-Silva TL, Lei YL, Sikora AG, Young S, Hartgerink JD.. STINGel: controlled release of a cyclic dinucleotide for enhanced cancer immunotherapy. Biomaterials. 2018;163:67–75. [published Online First: 2018/02/18]. doi: 10.1016/j.biomaterials.2018.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flood BA, Higgs EF, Li S, Luke JJ, Gajewski TF. STING pathway agonism as a cancer therapeutic. Immunol Rev. 2019;290(1):24–38. [published Online First: 2019/07/30]. doi: 10.1111/imr.12765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bakhoum SF, Ngo B, Laughney AM, Cavallo J-A, Murphy CJ, Ly P, Shah P, Sriram RK, Watkins TBK, Taunk NK, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature. 2018;553(7689):467–472. [published Online First: 2018/01/18]. doi: 10.1038/nature25432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Erdal E, Haider S, Rehwinkel J, Harris AL, McHugh PJ. A prosurvival DNA damage-induced cytoplasmic interferon response is mediated by end resection factors and is limited by Trex1. Genes Dev. 2017;31(4):353–369. [published Online First: 2017/03/11]. doi: 10.1101/gad.289769.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Concha-Benavente F, Srivastava RM, Trivedi S,Lei Y, Chandran U, Seethala RR, Freeman GJ, Ferris RL. Identification of the cell-intrinsic and -extrinsic pathways downstream of EGFR and IFNgamma that induce PD-L1 expression in head and neck cancer. Cancer Res. 2016;76(5):1031–1043. [published Online First: 2015/12/18]. doi: 10.1158/0008-5472.CAN-15-2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pletneva LM, Haller O, Porter DD, Prince GA, Blanco JCG. Induction of type I interferons and interferon-inducible Mx genes during respiratory syncytial virus infection and reinfection in cotton rats. J Gen Virol. 2008;89(1):261–270. [published Online First: 2007/12/20]. doi: 10.1099/vir.0.83294-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remédios C, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. 2014;20(11):1301–1309. [published Online First: 2014/10/27]. doi: 10.1038/nm.3708. [DOI] [PubMed] [Google Scholar]
- 18.Holzinger D, Jorns C, Stertz S,Boisson-Dupuis S, Thimme R, Weidmann M, Casanova JL, Haller O, Kochs G. Induction of MxA gene expression by influenza A virus requires type I or type III interferon signaling. J Virol. 2007;81(14):7776–7785. [published Online First: 2007/05/12]. doi: 10.1128/JVI.00546-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simon A, Fah J, Haller O,Staeheli P. Interferon-regulated Mx genes are not responsive to interleukin-1, tumor necrosis factor, and other cytokines. J Virol. 1991;65(2):968–971. [published Online First: 1991/02/01]. doi: 10.1128/JVI.65.2.968-971.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zav’yalov VP, Hamalainen-Laanaya H, Korpela TK,Wahlroos T. Interferon-inducible myxovirus resistance proteins: potential biomarkers for differentiating viral from bacterial infections. Clin Chem. 2019;65(6):739–750. [published Online First: 2018/12/30]. doi: 10.1373/clinchem.2018.292391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nguyen N, Bellile E, Thomas D, McHugh J, Rozek L, Virani S, Peterson L, Carey TE, Walline H, Moyer J, et al. Tumor infiltrating lymphocytes and survival in patients with head and neck squamous cell carcinoma. Head Neck. 2016;38(7):1074–1084. [published Online First: 2016/02/18]. doi: 10.1002/hed.24406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crowell HL, Soneson C, Germain PL,Calini D, Collin L, Raposo C, Malhotra D, Robinson MD. Muscat detects subpopulation-specific state transitions from multi-sample multi-condition single-cell transcriptomics data. Nat Commun. 2020;11(1):6077. [published Online First: 2020/12/02]. doi: 10.1038/s41467-020-19894-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun X, Zhang Y, Li J, Park KS, Han K, Zhou X, Xu Y, Nam J, Xu J, Shi X, et al. Amplifying STING activation by cyclic dinucleotide-manganese particles for local and systemic cancer metalloimmunotherapy. Nat Nanotechnol. 2021;[published Online First: 09/ 30/2021]. doi: 10.1038/s41565-021-00962-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacquelot N, Yamazaki T, Roberti MP, Duong CPM, Andrews MC, Verlingue L, Ferrere G, Becharef S, Vétizou M, Daillère R, et al. Sustained Type I interferon signaling as a mechanism of resistance to PD-1 blockade. Cell Res. 2019;29(10):846–861. [published Online First: 2019/09/05]. doi: 10.1038/s41422-019-0224-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Theodoraki MN, Yerneni SS, Hoffmann TK,Gooding WE, Whiteside TL. Clinical significance of PD-L1(+) exosomes in plasma of head and neck cancer patients. Clin Cancer Res. 2018;24(4):896–905. [published Online First: 2017/12/14]. doi: 10.1158/1078-0432.CCR-17-2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Theodoraki MN, Yerneni S, Gooding WE,Ohr J, Clump DA, Bauman JE, Ferris RL, Whiteside TL. Circulating exosomes measure responses to therapy in head and neck cancer patients treated with cetuximab, ipilimumab, and IMRT. Oncoimmunology. 2019;8(7):1593805. [published Online First: 2019/05/31]. doi: 10.1080/2162402X.2019.1593805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poggio M, Hu T, Pai CC, Chu B, Belair CD, Chang A, Montabana E, Lang UE, Fu Q, Fong L, et al. Suppression of exosomal PD-L1 induces systemic anti-tumor immunity and memory. Cell. 2019;177(2):414–27e13. [published Online First: 2019/04/06]. doi: 10.1016/j.cell.2019.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee Y, Shin JH, Longmire M,Wang H, Kohrt HE, Chang HY, Sunwoo JB. CD44+ Cells in head and neck squamous cell carcinoma suppress T-Cell-Mediated immunity by selective constitutive and inducible expression of PD-L1. Clin Cancer Res. 2016;22(14):3571–3581. [published Online First: 2016/02/13]. doi: 10.1158/1078-0432.CCR-15-2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dubrovska A, Kim S, Salamone RJ,Walker JR, Maira SM, García-Echeverría C, Schultz PG, Reddy VA. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proc Natl Acad Sci U S A. 2009;106(1):268–273. [published Online First: 2009/01/01]. doi: 10.1073/pnas.0810956106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoon C, Lu J, Yi BC,Chang KK, Simon MC, Ryeom S, Yoon SS. PI3K/Akt pathway and Nanog maintain cancer stem cells in sarcomas. Oncogenesis. 2021;10(1):12. [published Online First: 2021/01/21]. doi: 10.1038/s41389-020-00300-z. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31.Jia L, Zhang W, Wang CY. BMI1 inhibition eliminates residual cancer stem cells after PD1 blockade and activates antitumor immunity to prevent metastasis and relapse. Cell Stem Cell. 2020;27(2):238–53e6. doi: 10.1016/j.stem.2020.06.022. [published Online First: 2020/07/23] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kansy BA, Concha-Benavente F, Srivastava RM, Jie H-B, Shayan G, Lei Y, Moskovitz J, Moy J, Li J, Brandau S, et al. PD-1 Status in CD8 + T Cells associates with survival and Anti-PD-1 therapeutic outcomes in head and neck cancer. Cancer Res. 2017;77(22):6353–6364. [published Online First: 2017/09/15]. doi: 10.1158/0008-5472.CAN-16-3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou L, Mudianto T, Ma X,Riley R, Uppaluri R. Targeting EZH2 enhances antigen presentation, antitumor immunity, and circumvents Anti-PD-1 resistance in head and neck cancer. Clin Cancer Res. 2020;26(1):290–300. [published Online First: 2019/09/29]. doi: 10.1158/1078-0432.CCR-19-1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moore E, Clavijo PE, Davis R,Cash H, Van Waes C, Kim Y, Allen C. Established T cell-inflamed tumors rejected after adaptive resistance was reversed by combination STING activation and PD-1 pathway blockade. Cancer Immunol Res. 2016;4(12):1061–1071. [published Online First: 2016/11/09]. doi: 10.1158/2326-6066.CIR-16-0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim CH, Rott L, Kunkel EJ,Genovese MC, Andrew DP, Wu L, Butcher EC. Rules of chemokine receptor association with T cell polarization in vivo. J Clin Invest. 2001;108(9):1331–1339. [published Online First: 2001/11/07]. doi: 10.1172/JCI13543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, et al. Intratumoral Tcf1(+)PD-1(+)CD8(+) T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity. 2019;50(1):195–211e10. [published Online First: 2019/01/13]. doi: 10.1016/j.immuni.2018.12.021. [DOI] [PubMed] [Google Scholar]
- 37.Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol. 2019;20(3):326–336. [published Online First: 2019/02/20]. doi: 10.1038/s41590-019-0312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tripathi P, Kurtulus S, Wojciechowski S,Sholl A, Hoebe K, Morris SC, Finkelman FD, Grimes HL, Hildeman DA. STAT5 is critical to maintain effector CD8+ T cell responses. J Immunol. 2010;185(4):2116–2124. [published Online First: 2010/07/21]. doi: 10.4049/jimmunol.1000842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burchill MA, Goetz CA, Prlic M, O’Neil JJ, Harmon IR, Bensinger SJ, Turka LA, Brennan P, Jameson SC, Farrar MA, et al. Distinct Effects of STAT5 Activation on CD4 + and CD8 + T cell homeostasis: development of CD4 + CD25 + Regulatory T Cells versus CD8 + Memory T Cells. J Immunol. 2003;171(11):5853–5864. [published Online First: 2003/11/25]. doi: 10.4049/jimmunol.171.11.5853. [DOI] [PubMed] [Google Scholar]
- 40.Chisolm DA, Savic D, Moore AJ,Ballesteros-Tato A, León B, Crossman DK, Murre C, Myers RM, Weinmann AS. CCCTC-binding factor translates interleukin 2- and alpha-ketoglutarate-sensitive metabolic changes in T Cells into context-dependent gene programs. Immunity. 2017;47(2):251–67e7. [published Online First: 2017/08/17]. doi: 10.1016/j.immuni.2017.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Edwards DN, Ngwa VM, Raybuck AL, Wang S, Hwang Y, Kim LC, Cho SH, Paik Y, Wang Q, Zhang S, et al. Selective glutamine metabolism inhibition in tumor cells improves antitumor T lymphocyte activity in triple-negative breast cancer. J Clin Invest. 2021;131(4). [published Online First: 2020/12/16]. doi: 10.1172/JCI140100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buck MD, O’Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med. 2015;212(9):1345–1360. doi: 10.1084/jem.20151159. [published Online First: 2015/08/12] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hou Y, Liang H, Rao E, Zheng W, Huang X, Deng L, Zhang Y, Yu X, Xu M, Mauceri H, et al. Non-canonical NF-kappaB antagonizes STING sensor-mediated DNA sensing in radiotherapy. Immunity. 2018;49(3):490–503e4. [published Online First: 2018/09/02]. doi: 10.1016/j.immuni.2018.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357(9255):539–545. doi: 10.1016/S0140-6736(00)04046-0. [published Online First: 2001/03/07] [DOI] [PubMed] [Google Scholar]
- 45.Yang X, Cheng H, Chen J, Wang R, Saleh A, Si H, Lee S, Guven-Maiorov E, Keskin O, Gursoy A, et al. Head and neck cancers promote an inflammatory transcriptome through coactivation of classic and alternative NF-kappaB pathways. Cancer Immunol Res. 2019;7(11):1760–1774. [published Online First: 2019/10/19]. doi: 10.1158/2326-6066.CIR-18-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rehman AO, Wang CY. CXCL12/SDF-1 alpha activates NF-kappaB and promotes oral cancer invasion through the Carma3/Bcl10/Malt1 complex. Int J Oral Sci. 2009;1(3):105–118. doi: 10.4248/IJOS.09059. [published Online First: 2010/08/10] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang Z, Liao J, Cullen KJ,Dan H. Inhibition of IKKbeta/NF-kappaB signaling pathway to improve Dasatinib efficacy in suppression of cisplatin-resistant head and neck squamous cell carcinoma. Cell Death Discov. 2020;6(1):36. [published Online First: 2020/05/22]. doi: 10.1038/s41420-020-0270-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li Z, Liao J, Yang Z,Choi EY, Lapidus RG, Liu X, Cullen KJ, Dan H. Co-targeting EGFR and IKKbeta/NF-kappaB signalling pathways in head and neck squamous cell carcinoma: a potential novel therapy for head and neck squamous cell cancer. Br J Cancer. 2019;120(3):306–316. [published Online First: 2018/12/27]. doi: 10.1038/s41416-018-0351-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shae D, Becker KW, Christov P, Yun DS, Lytton-Jean AKR, Sevimli S, Ascano M, Kelley M, Johnson DB, Balko JM, et al. Endosomolytic polymersomes increase the activity of cyclic dinucleotide STING agonists to enhance cancer immunotherapy. Nat Nanotechnol. 2019;14(3):269–278. [published Online First: 2019/01/22]. doi: 10.1038/s41565-018-0342-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Watkins-Schulz R, Tiet P, Gallovic MD,Junkins RD, Batty C, Bachelder EM, Ainslie KM, Ting JPY. A microparticle platform for STING-targeted immunotherapy enhances natural killer cell- and CD8(+) T cell-mediated anti-tumor immunity. Biomaterials. 2019;205:94–105. [published Online First: 2019/03/26]. doi: 10.1016/j.biomaterials.2019.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.