

Abstract
A remote C3–H activation of pyridine-containing substrates can be achieved with a directive Ni catalyst. The bifunctional NHC ligand incorporates an Al-binding side-arm that recruits and orients the substrate leading to the assembly of the requisite macrocyclophane transition state through reversible coordination. This assembly not only induces the reactivity of the otherwise unreactive Ni catalyst, but also overrides the intrinsic C2/C4 electronic bias of the Al-bound pyridine substrate, allowing for the first time, the C3 alkenylation of a variety of pyridine and heteroarene substrates as the limiting reagent.
Graphical Abstract
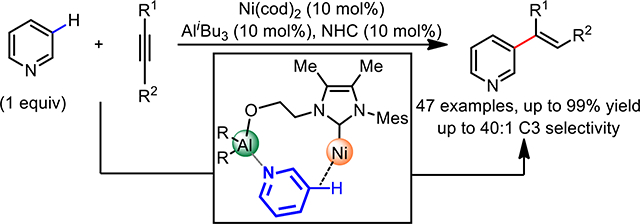
The ubiquity of C–H bonds in organic molecules and their often-marginal chemical differences renders the site-selective activation of C–H bonds an enduring challenge.1 The directing approach bearing a covalent bond between a directing group and a substrate has proved to be a particularly promising strategy for proximate and increasingly for remote C–H bond activations (Type I, Scheme 1a),2 In particular, the importance of distance and geometric considerations for tuning the macrocyclophane transition state has been demonstrated for predictable remote C–H activation.3,4 However, this covalent template strategy faces practical challenges that impede synthetic application, where stoichiometric template use and separate template attachment and removal steps are required. To address these challenges, a reversible template-substrate anchoring strategy has been developed (Type II, Scheme 1a).5 This strategy merges the role of template and ligand into a single bifunctional scaffold, renders the template catalytic and eliminates the extra steps required for template attachment/removal. Though attractive, this strategy has seen limited applications in the ability to catalyze wide-ranging functionalizations; success has so far been restricted to Ir-catalyzed remote C‒H borylation through reversible interactions such as H bonding,6 ion pairing7 and Lewis acid coordination.8 In contrast, direct C‒C bond forming reactions of remote C‒H bonds has been scarcely explored, partly attributed to lower reactivity and harsher conditions required. Addressing this limitation, our group has recently achieved the first example of remote C‒C bond forming reaction via Pd-Pd homobimetallic catalysis, furnishing remote C‒H alkenylation of relatively electron-rich arenes using the azine nitrogen as the template anchoring group (Scheme 1b).9 Despite a step forward, the requirements of high loadings of Pd, template, ligand, and super-stoichiometric amounts of metal oxidants for catalyst turnover offer much scope for practical improvement. Moreover, electron-deficient pyridine and related derivatives—challenging substrates widely encountered in natural products and pharmaceuticals—remain incompatible in such a reaction. Herein, we report a Ni‒Al heterobimetallic catalyst for the C3‒H alkenylation of pyridines with alkynes, providing an atom-economical method for direct C‒C bond formation without the need for external oxidants (Scheme 1c). Importantly, this manifold enables the use of pyridines as the limiting reagent for the first time, allowing the late-stage C3-H alkenylation of pyridine motifs in complex molecules. A bifunctional NHC was identified as the critical ligand, which recruits and positions the substrate via Al anchorage to the vicinity of the Ni catalyst. This directive ligand not only enables catalytic reactivity through substrate and ligand binding, but also reverses the conventional C2/C4 site-selectivity obtained in low-valent Ni-catalyzed C–H activation processes.
Scheme 1.

Remote C–H Activation via Macrocyclophane Transition State
The C3-selective alkenylation of pyridine-containing heterocycles represents a desirable transformation in the realm of medicinal chemistry due to both its prevalence in pharmaceutical agents and its facile entry point to a range of functionalities (Scheme 2a).10,11 However, the strong σ-coordinative ability of pyridines often poison metal catalysts, rendering the development of catalytic processes a formidable challenge. In 2011, we reported a Pd(II)-catalyzed C3–H alkenylation of pyridines,12,13 which gave high C3 selectivity arising from an electrophilic palladation process (Scheme 2b). However, a large excess of pyridine substrate (16 equivalents) was required to achieve reasonable reactivity. This drawback also rendered this reaction incompatible with the late-stage functionalization of complex pyridine-containing substrates, where the large excesses required poses unfavorable resource, cost and solubility issues in a synthetic setting. In pioneering studies by Nakao and Hiyama,14,15 coordination with Al Lewis acids was demonstrated to mask the pyridyl nitrogen, and polarized the pyridine ring to enhance the reactivity of C2/C4 positions towards nucleophilic low-valent Ni C–H oxidative addition. Prompted by this finding, we envisioned that a bifunctional carbene ligand could coordinate to both Ni and Al and form a heterobimetallic catalyst16 directed towards the C3(5) position, thus achieving the Type II template approach (Scheme 1) and reversing the conventional C2/C4 selectivity (Scheme 2c).
Scheme 2.

C3-Alkenylation of Pyridinesa
We selected non-substituted pyridine (1a) and oct-4-yne (2a) as the model substrate and coupling partner to explore the necessary ligands and reaction conditions for this transformation. Preliminary results showed that traditional phosphines and NHC ligands were poorly reactive and, as expected, delivered the alkenylated product at the C2 or C4 positions exclusively (Table S1). As expected, the in situ formation of NHCs from their precursors led to significant decrease in reactivity, attributed to the generation and deleterious coordination of tBuOH to the Al Lewis acid. To our delight, we found that ligand L1 bearing a coordinating alkoxy group provided the desired mono-C3-alkenylated pyridine in moderate yields (33%, Table S1), overruling the intrinsic C2/C4 selectivity of the substrate. Notably, the observed reactivity for L1 could be obtained through the use of the imidazolium halide precursor, obviating the need for carbene pre-generation required in previous reports.15a,b Then we systematically surveyed a range of Al Lewis acids and varied the linker length on the ligand (Table S3 and S6). We found that best results were obtained by using AliBu3 as the anchoring Lewis acid, in conjunction with a two-carbon alkoxy side-arm (L1), improving the combined yield to 61% (Scheme 3). The importance of linker length was affirmed by the use of a homologated side-arm (L3), which resulted in both a reduction in yield and selectivity. Notably, the use of unsubstituted aryl NHC ligand (L4) was ineffective, inferring that the assembly of the putative macrocyclophane intermediate may be facilitated by conformational restriction. This observation was reinforced by the further incorporation of methyl groups onto the imidazolium backbone (L6, Table S6), which elevated the yield to 64% (10:1, C3:others). Additional tuning of the NHC aryl group revealed that the para-methoxy substitution was optimal (L10, 87%, ca. 16:1, C3:others, Scheme 3). Importantly, methylation of the coordinating alkoxy group completely shut down the reaction (L12, Scheme 3),17 demonstrating that Al coordination by the ligand side arm was crucial for both the reactivity and the selectivity of this process. Further mechanistic evidence validating such a three-component assembly process was obtained by 1H NMR studies, which showed that 3-phenylpyridine, AlMe3 and ligand (L10) formed a new complex demonstrated by marked downfield shifts of H2, H3, and H6 of 3-phenylpyridine (Page S13 and S14). In addition, heating the aforementioned three-component complex under the reaction conditions led to the C3-alkenylation product in 34% (Page S15), supporting the productive role this ternary complex plays in this reaction.
Scheme 3. Ligand Optimizationa.

aReaction conditions: 1a (0.20 mmol), 2a (0.6 mmol), toluene (0.5 mL); Ni(cod)2, Ligand, tBuONa and toluene at 80 °C for 30 min, then pyridine, AliBu3 and alkyne substrate at 100 °C under N2 for 12 h; yield of isolated mixed isomers; ratio of isomers determined by 1H NMR. bL10 (10 mol%).
With the optimized conditions in hand, we proceeded to examine the scope of pyridines and other heteroarenes for this reaction (Scheme 4). Electron-donating substituents at C3 positions such as alkyls (3b, 3c, 3d), alkoxyl (3e) and amino groups (3f and 3g) were compatible with the reaction, providing the corresponding products in 49% to 64% yield with the desired C5 selectivity (up to 30:1). It was pleasing to observe that increasing the electron density of the pyridine ring system tends to inhibit oxidative addition of the Ni(0) catalyst with C–H bonds. As expected, electron-withdrawing groups such as F (3h) and CF3 (3i) significantly elevated the yield to 91% (44:1, C5:C4) and 99% (4:1, C5:C4), respectively. Though highly reactive, decreased C5 selectivity was observed for 3i containing the CF3 group, attributed to poorer binding between the pyridyl group and the Al Lewis acid resultant of its strongly electron-withdrawing nature. A wide range of functionalities were well-tolerated in this reaction; 3j and 3k containing ester or amide groups both gave good yields and high C5 selectivity. In addition, aryl groups (3l–3p) bearing a range of functional groups such as methoxy (3n), silyl (3o) and boryl groups (3p) are tolerated, giving the desired products in 72–92% yields and excellent C5 selectivity (16:1 to 33:1). The presence of a C4-phenyl group (3q) led to decreased reactivity and C3 selectivity, presumably owing to both its electron-donating effect as well as heightened steric hindrance. On the other hand, the smaller C4 fluoro group (3r) provided near-quantitative yield and high C5 selectivity (9:1). Consistent with the proposed coordination of pyridine with Al as a crucial mechanistic component, the presence of C2 substituents gave poorer reactivity and selectivity (3s and 3t). Notably, other azaheteroarenes were also compatible (3u to 3x): alkenylation of diazaheteroarenes such as pyridazine (3u) and pyrimidines (3v and 3w) afforded the desired products in 63–86% yields (6:1–25:1, C5:others); quinoline gave the alkenylated product in 72% yield, albeit with a lower selectivity (1:1, 3x) owing to poorer coordination with Al Lewis acid.
Scheme 4. Scope of Pyridinesa.

aReaction conditions: 1 (0.40 mmol), 2a (1.20 mmol), toluene (1.0 mL); Ni(cod)2, L10, tBuONa and toluene at 80 °C for 30 min, then pyridine, AliBu3 and alkyne substrate at 100 °C under N2 for 12 h; yield of isolated mixed isomers; ratio of isomers determined by 1H NMR. bNi(cod)2 (20 mol% ), L10 (20 mol%), AliBu3 (20 mol%), and tBuONa (25 mol%). c140 °C. dAliBu3 instead of AlMe3.
The scope of alkyne coupling partners was next surveyed using 3-fluoropyridine (1h) as a model substrate. Considering the synthetic versatility of the olefin motif, it was pleasing to observe that a broad range of alkylalkynes was well-tolerated, affording the trisubstituted alkenylated products with excellent C5 selectivity (Scheme 5, >40:1 C5:others). Both symmetrical dialkylalkynes (4a to 4e) and non-symmetrical alkylalkynes (4f to 4h) afforded the corresponding products in 82–93% yields. For non-symmetrical alkynes, the regiochemical outcomes were governed by the relative steric hindrance between the two alkyne substituents, with larger size differences giving higher alkene regioselectivity. As well, alkylalkynes bearing potentially acid and Lewis acid sensitive groups such as silanes (4i and 4j) and silyl ethers (4k to 4o) were all compatible substrates in this reaction.
Scheme 5. Scope of Alkynesa.

aReaction conditions: 1h (0.40 mmol), 2 (1.20 mmol), toluene (1.0 mL); Ni(cod)2, L10, tBuONa and toluene at 80 °C for 30 min, then pyridine, AliBu3 and alkyne substrate at 100 °C under N2 for 12 h; yield of isolated products; bRegioisomer ratio of alkenes determined by 1H NMR.
In contrast to previously reported C–H olefination of pyridines,12 this newly-developed catalyst allows for the use of pyridine substrates as the limiting agent, thus opening new avenues for the efficient late-stage modification of heterocycle-containing bioactive molecules (Scheme 6). To demonstrate this, we applied our C–H alkenylation reaction to a range of nicotinic acid-derived complex molecules, such as (−)-menthol (5a), (−)-borneol (5b), diacetonefructose (5c), (−)-Corey lactone diol (5d), and cholesterol (5e). Gratifyingly, the reactions proceeded smoothly, providing the desired products in 43–79% yield and with high C5 selectivity (11:1–32:1). Medicinally relevant compounds were also competent in this process; azabicyclic compound (5f), representing an important class of agent active in the central nervous system, was alkenylated in 60% yield (26:1 C5:others). Abiraterone (5g), an anticancer drug, was alkenylated in 65% yield (34:1 C5:others). In addition, bioactive steroid hormones such as estrone (5h) and estradiol (5i) were also suitable substrates, providing the corresponding alkenylated products in 88% and 89% yield, respectively, both with high C5 selectivity (15:1–30:1).
Scheme 6. Late-Stage Alkenylation of Pyridine-Containing Bioactive Moleculesa.

aReaction conditions: 1 (0.40 mmol), 2a (1.20 mmol), toluene (1.0 mL); Ni(cod)2, L10, tBuONa and toluene at 80 °C for 30 min, then pyridine, AliBu3 and alkyne substrate at 100 °C under N2 for 12 h; yield of isolated mixed isomers; ratio of C5/C6/C4 determined by 1H NMR. bNi(cod)2 (20 mol%), L10 (20 mol%), AliBu3 (20 mol%), tBuONa (25 mol%).
In conclusion, we have developed a bifunctional Ni catalyst that allows, for the first time, the C3(5)-selective C–H alkenylation of pyridine-containing hetereocycles as the limiting reagent. As the alkene functionality could be readily derivatized, the broad scope and synthetic practicality of this reaction could enable the facile access of diverse C3(5)-functionalized motifs bearing a range of carbon oxidation states. We determined that the assembly of a putative macrocyclophane intermediate through reversible Al coordination was crucial to enable both catalyst reactivity and site-selectivity. As a testament to the strength of the directing effect, the ligand allows for the Ni catalyst to override the intrinsic electronic activation of the C2 and C4 and achieve the selective metalation at the C3–H bond of pyridines. This process further validates the applicability of a merged ligand-template strategy in C–H activation, where we anticipate these design principles applied to a wider range of remote functionalization processes.
Supplementary Material
ACKNOWLEDGMENT
We thank the National Natural Science Foundation of China (91856104, 21871145 and 21672107) and “the Fundamental Research Funds for the Central Universities” (63191601) for financial support. We also gratefully acknowledge The Scripps Research Institute, the Lindemann Trust (N.Y.S.L.), and the NIH ((National Institute of General Medical Sciences grant R01GM102265) for financial support.
Footnotes
The authors declare no competing financial interests.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website. Experimental procedures and spectral data for all new compounds (PDF). Metrical parameters for the structure of L10 are available free of charge from the Cambridge Crystallographic Data Centre under reference number CCDC 1434263.
REFERENCES
- (1).(a) For selected reviews on site-selective activation of C–H bonds, see: Neufeldt SR; Sanford MS Controlling Site Selectivity in Palladium-Catalyzed C-H Bond Functionalization. Acc. Chem. Res. 2012, 45, 936–946. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mahatthananchai J; Dumas AM; Bode JW Catalytic Selective Synthesis. Angew. Chem. Int. Ed. 2012, 51, 10954–10990. [DOI] [PubMed] [Google Scholar]; (c) Kuhl N; Hopkinson MN; Wencel-Delord J; Glorius F Beyond Directing Groups: Transition-Metal-Catalyzed C-H Activation of Simple Arenes. Angew. Chem. Int. Ed. 2012, 51, 10236–10254. [DOI] [PubMed] [Google Scholar]; (d) Tobisu M; Chatani N Remote Control by Steric Effects. Science 2014, 343, 850–851. [DOI] [PubMed] [Google Scholar]; (e) Funken N; Zhang Y-Q; Gansäuer A Regiodivergent Catalysis: A Powerful Tool for Selective Catalysis. Chem. Eur. J. 2017, 23, 19–32. [DOI] [PubMed] [Google Scholar]; (f) Ping L; Chung DS; Bouffard J; Lee S Transition metal-catalyzed site- and regio-divergent C–H bond functionalization. Chem. Soc. Rev. 2017, 46, 4299–4328. [DOI] [PubMed] [Google Scholar]
- (2).(a) For selected reviews on chelated proximate C–H bond activations, see: Chen X; Engle KM; Wang D-H; Yu J-Q Palladium(II)-Catalyzed C-H Activation/C-C Cross-Coupling Reactions: Versatility and Practicality. Angew. Chem. Int. Ed. 2009, 48, 5094–5115. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Colby DA; Bergman RG; Ellman JA Rhodium-Catalyzed C−C Bond Formation via Heteroatom-Directed C−H Bond Activation. Chem. Rev. 2010, 110, 624–655. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lyons TW; Sanford MS Palladium-Catalyzed Ligand-Directed C−H Functionalization Reactions. Chem. Rev. 2010, 110, 1147–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ackermann L Carboxylate-Assisted Transition-Metal-Catalyzed C−H Bond Functionalizations: Mechanism and Scope. Chem. Rev. 2011, 111, 1315–1345. [DOI] [PubMed] [Google Scholar]; (e) Colby DA; Bergman RG; Ellman JA Rhodium-Catalyzed C-C Bond Formation via Heteroatom-Directed C-H Bond Activation. Chem. Rev. 2010, 110, 624–655. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Chen Z; Wang B; Zhang J; Yu W; Liu Z; Zhang Y Transition Metal-Catalyzed C-H Bond Functionalizations by the Use of Diverse Directing Groups. Org. Chem. Front. 2015, 2, 1107−1295. [Google Scholar]; (g) Newton CG; Wang S-G; Oliveira CC; Cramer N Catalytic enantioselective transformations involving C-H bond cleavage by transition-metal complexes. Chem. Rev. 2017, 117, 8908−8976. [DOI] [PubMed] [Google Scholar]; (h) Saint-Denis TG; Zhu R-Y; Chen G; Wu Q-F; Yu J-Q Enantioselective C(sp3)-H bond activation by chiral transition metal catalysts. Science 2018. 359, eaao4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) For selected reviews on chelated remote C–H bond activations, see: Li J; De Sarkar S; Ackermann L meta- and para-Selective C−H Functionalization by C−H Activation. Top. Organomet. Chem. 2015, 55, 217–257. [Google Scholar]; (b) Sharma R; Sharma U Remote C-H bond activation/transformations: A continuous growing synthetic tool; Part II. Catal. Rev. 2018, 60, 497–565. [Google Scholar]; (c) Mihai MT; Genov GR; Phipps RJ Access to the meta position of arenes through transition metal catalysed C–H bond functionalisation: a focus on metals other than palladium. Chem. Soc. Rev. 2018, 47, 149–171. [DOI] [PubMed] [Google Scholar]; (d) Dey A; Sinha SK; Achar TK; Maiti D Accessing Remote meta- and para-C(sp2)-H Bonds with Covalently Attached Directing Groups. Angew. Chem., Int. Ed. 2019, 58, 10820–10843. [DOI] [PubMed] [Google Scholar]; (e) Shao Q; Wu K; Zhuang Z; Qian S; Yu J-Q From Pd(OAc)2 to Chiral Catalysts: The Discovery and Development of Bifunctional Mono-N-Protected Amino Acid Ligands for Diverse C-H Functionalization Reactions. Acc. Chem. Res. 2020, 53, 833–851. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Meng G; Lam NYS; Lucas EL; Saint-Denis TG; Verma P; Chekshin N; Yu J-Q Achieving site-selectivity for C-H activation processes based on distance and geometry: A carpenter’s approach. J. Am. Chem. Soc. 2020, 142, 10571–10591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) For selected examples on chelated remote C–H bond activations, see: Leow D; Li G; Mei T-S; Yu J-Q Activation of remote meta-C-H bond assisted by an end-on template. Nature 2012, 486, 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hofmann N; Ackermann L Meta-Selective C-H Bond Alkylation with Secondary Alkyl Halides. J. Am. Chem. Soc. 2013, 135, 5877−5884. [DOI] [PubMed] [Google Scholar]; (c) Wang X-C; Gong W; Fang L-Z; Zhu R-Y; Li S; Engle KM; Yu J-Q Ligand-Enabled meta-C-H Activation Using a Transient Mediator. Nature 2015, 519, 334–338. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Phipps RJ; Gaunt MJ A Meta-Selective Copper-Catalyzed C–H Bond Arylation. Science 2009, 323, 1593–1597. [DOI] [PubMed] [Google Scholar]; (e) Bag S; Patra T; Modak A; Deb A; Maity S; Dutta U; Dey A; Kancherla R; Maji A; Hazra A; Bera M; Maiti D Remote para C-H Functionalization of Arenes by a D-Shaped Biphenyl TemplateBased Assembly. J. Am. Chem. Soc. 2015, 137, 11888−11891. [DOI] [PubMed] [Google Scholar]; (f) Das S; Incarvito CD; Crabtree RH; Brudvig GW Molecular recognition in the selective oxygenation of saturated C-H Bonds by a dimanganese catalyst. Science 2006, 312, 1941–1943. [DOI] [PubMed] [Google Scholar]
- (5).(a) For relevant reviews, see: Gandeepan P; Ackermann L Transient Directing Groups for Transformative C–H Activation by Synergistic Metal Catalysis. Chem 2018, 4, 199–222. [Google Scholar]; (b) Davis HJ; Phipps RJ Harnessing non-covalent interactions to exert control over regioselectivity and site-selectivity in catalytic reactions. Chem. Sci. 2017, 8, 864–877. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kim DS; Park W-J; Jun C-H Metal−Organic Cooperative Catalysis in C−H and C−C Bond Activation. [DOI] [PubMed] [Google Scholar]; (d) Rousseau G; Breit B Removable Directing Groups in Organic Synthesis and Catalysis. Angew. Chem. Int. Ed. 2011, 50, 2450‒2494. [DOI] [PubMed] [Google Scholar]
- (6).(a) For examples on H-bonding interaction, see: Roosen PC; Kallepalli VA; Chattopadhyay B; Singleton DA; Maleczka RE; Smith MR III Outer-Sphere Direction in Iridium C−H Borylation. J. Am. Chem. Soc. 2012, 134, 11350−11353. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Preshlock SM; Plattner DL; Maligres PE; Krska SW; Maleczka RE Jr.; Smith Milton R. III A Traceless Directing Group for C-H Borylation. Angew. Chem. Int. Ed. 2013, 52, 12915–12919. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kuninobu Y; Ida H; Nishi M; Kanai M A meta-selective C-H borylation directed by a secondary interaction between ligand and substrate. Nat. Chem. 2015, 7, 712–717. [DOI] [PubMed] [Google Scholar]; (d) Davis HJ; Genov GR; Phipps RJ Meta-Selective C-H Borylation of Benzylamine, Phenethylamine and Phenylpropylamine-Derived Amides Enabled by a Single Anionic Ligand. Angew. Chem. Int. Ed. 2017, 56, 13351‒13355. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Lu X; Yoshigoe Y; Ida H; Nishi M; Kanai M; Kuninobu Y Hydrogen Bond-Accelerated meta-Selective C-H Borylation of Aromatic Compounds and Expression of Functional Group and Substrate Specificities. ACS Catal. 2019, 9, 1705‒1709. [Google Scholar]; (f) Genov GR; Douthwaite JL; Lahdenperä ASK; Gibson DC; Phipps RJ Enantioselective remote C-H activation directed by a chiral cation. Science 2020, 367, 1246–1251. [DOI] [PubMed] [Google Scholar]
- (7).(a) For examples on ion-pair or electrostatic interaction, see: Davis HJ; Mihai MT; Phipps RJ Ion pair-directed regiocontrol in transition-metal catalysis: A meta-selective C-H borylation of aromatic quaternary ammonium salts. J. Am. Chem. Soc. 2016, 138, 12759–12762. [DOI] [PubMed] [Google Scholar]; (b) Chattopadhyay B; Dannatt JE; Andujar-De Sanctis IL; Gore KA; Maleczka RE; Singleton DA; Smith MR III Ir-Catalyzed ortho-Borylation of Phenols Directed by Substrate-Ligand Electrostatic Interactions: A Combined Experimental/in Silico Strategy for Optimizing Weak Interactions. J. Am. Chem. Soc. 2017, 139, 7864‒7871. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mihai MT; Phipps RJ Ion-Pair-Directed meta-Selective C–H Borylation of Aromatic Quaternary Ammonium Salts. Synlett 2017, 28, 1011‒1017. [DOI] [PubMed] [Google Scholar]
- (8).(a) For examples on Lewis acid coordination, see: Bisht R; Chattopadhyay B Formal Ir-Catalyzed Ligand-Enabled Ortho and Meta Borylation of Aromatic Aldehydes via in Situ-Generated Imines. J. Am. Chem. Soc. 2016, 138, 84–87. [DOI] [PubMed] [Google Scholar]; (b) Li HL; Kuninobu Y; Kanai M Lewis Acid–base interaction-controlled ortho-selective C-H borylation of aryl sulfides. Angew. Chem. Int. Ed. 2017, 56, 1495–1499. [DOI] [PubMed] [Google Scholar]; (c) Hoque ME; Bisht R; Haldar C; Chattopadhyay B Noncovalent interactions in Ir-catalyzed C-H activation: L-shaped ligand for para-selective borylation of aromatic esters. J. Am. Chem. Soc. 2017, 139, 7745–7748. [DOI] [PubMed] [Google Scholar]; (d) Bisht R; Hoque ME; Chattopadhyay B Amide effects in C-H activation: Noncovalent interactions with L-shaped ligand for meta borylation of aromatic amides. Angew. Chem. Int. Ed. 2018, 57, 15762–15766. [DOI] [PubMed] [Google Scholar]; (e) Yang L; Uemura N; Nakao Y meta-Selective C-H borylation of benzamides and pyridines by an iridium−Lewis acid bifunctional catalyst. J. Am. Chem. Soc. 2019, 141, 7972–7979. [DOI] [PubMed] [Google Scholar]
- (9) (a).Zhang Z; Tanaka K; Yu J-Q, Remote site-selective C-H activation directed by a catalytic bifunctional template. Nature 2017, 543, 538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shi H; Lu Y; Weng J; Bay KL; Chen X; Tanaka K; Verma P; Houk KN; Yu J-Q Differentiation and functionalization of remote C–H bonds in adjacent positions. Nat. Chem. 2020, 12, 399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Vitaku E; Smith DT; Njardarson JT Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- (11).(a) For relevant reviews on C-H activation of pyridines, see: Murakami K; Yamada S; Kaneda T; Itami K C−H Functionalization of Azines. Chem. Rev. 2017, 117, 9302–9332. [DOI] [PubMed] [Google Scholar]; (b) Stephens DE; Larionov OV Recent advances in the C−H functionalization of the distal positions in pyridines and quinolines. Tetrahedron 2015, 71, 8683–8716. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Iwai T; Sawamura M Transition-Metal-Catalyzed Site-Selective C-H Functionalization of Quinolines beyond C2 Selectivity. ACS Catal. 2015, 5, 5031–5040. [Google Scholar]; (d) Nakao Y Transition-Metal-Catalyzed C−H Functionalization for the Synthesis of Substituted Pyridines. Synthesis 2011, 20, 3209–3219. [Google Scholar]; (e) Nakao Y Hydroarylation of Alkynes Catalyzed by Nickel. Chem. Rec. 2011, 11, 242–251. [DOI] [PubMed] [Google Scholar]
- (12) (a).Ye M; Gao G-L; Yu J-Q Ligand-promoted C-3 selective C-H olefination of pyridines with Pd catalysts. J. Am. Chem. Soc. 2011. 133, 6964–6967. [DOI] [PubMed] [Google Scholar]; (b) For related work, see: Cong X; Tang H; Wu C; Zeng X Role of mono-N-protected amino acid ligands in palladium(II)-catalyzed dehydrogenative Heck reactions of electron-deficient (hetero)arenes: experimental and computational studies. Organometallics 2013, 32, 6565–6575. [Google Scholar]
- (13) (a).Takagi J; Sato K; Hartwig JF; Ishiyama T; Miyaura N Iridium-catalyzed C–H coupling reaction of heteroaromatic compounds with bis(pinacolato)diboron: regioselective synthesis of heteroarylboronates. Tetrahedron Lett. 2002, 43, 5649−5651. [Google Scholar]; (b) Mkhalid IAI; Conventry DN; Albesa-Jove D; Batsanov AS; Howard JAK; Perutz RN; Marder TB Ir-catalyzed borylation of C-H bonds in N-containing heterocycles: Regioselectivity in the synthesis of heteroaryl boronate esters. Angew. Chem. Int. Ed. 2006, 45, 489–491. [DOI] [PubMed] [Google Scholar]; (c) Murphy JM; Liao X; Hartwig JF Meta Halogenation of 1,3-Disubstituted Arenes via Iridium-Catalyzed Arene Borylation. J. Am. Chem. Soc. 2007, 129, 15434−15435. [DOI] [PubMed] [Google Scholar]; (d) Li B-J; Shi Z-J Ir-catalyzed highly selective addition of pyridyl C–H bonds to aldehydes promoted by triethylsilane. Chem. Sci. 2011, 2, 488−493. [Google Scholar]; (e) Ye M; Gao G-L; Edmunds AJF; Worthington PA; Morris JA; Yu J-Q Ligand-promoted C3-selective arylation of pyridines with Pd catalysts: gram-scale synthesis of (±)-preclamol. J. Am. Chem. Soc. 2011. 133, 19090–19093. [DOI] [PubMed] [Google Scholar]; (f) Dai F; Gui Q; Liu J; Yang Z; Chen X; Guo R; Tan Z Pd-catalyzed C3-selective arylation of pyridines with phenyl tosylates. Chem. Commun. 2013, 49, 4634−4636. [DOI] [PubMed] [Google Scholar]; (g) Larsen MA; Hartwig JF Iridium-Catalyzed C−H Borylation of Heteroarenes: Scope, Regioselectivity, Application to Late-Stage Functionalization, and Mechanism. J. Am. Chem. Soc. 2014, 136, 4287−4299. [DOI] [PubMed] [Google Scholar]; (h) Cheng C; Hartwig JF Iridium-Catalyzed Silylation of Aryl C−H Bonds. J. Am. Chem. Soc. 2015, 137, 592−595. [DOI] [PubMed] [Google Scholar]; (i) Wübbolt S; Oestreich M Catalytic Electrophilic C-H Silylation of Pyridines Enabled by Temporary Dearomatization. Angew. Chem. Int. Ed. 2015, 54, 15876–15879. [DOI] [PubMed] [Google Scholar]; (j) Jiao J; Murakami K; Itami K Palladium-catalyzed C-H Arylation of Pyridines with Aryl Triflates. Chem. Lett. 2016, 45, 529–531. [Google Scholar]; (k) Yamada S; Murakami K; Itami K Regiodivergent Cross-Dehydrogenative Coupling of Pyridines and Benzoxazoles: Discovery of Organic Halides as Regio-Switching Oxidants. Org. Lett. 2016, 18, 2415−2418. [DOI] [PubMed] [Google Scholar]; (l) Yang L; Uemura N; Nakao Y meta-Selective C−H Borylation of Benzamides and Pyridines by an Iridium−Lewis Acid Bifunctional Catalyst. J. Am. Chem. Soc. 2019, 141, 7972−7979. [DOI] [PubMed] [Google Scholar]; (m) Zhang W-B; Yang X-T; Ma J-B; Su Z-M; Shi S-L Regio- and enantioselective C−H cyclization of pyridines with alkenes enabled by a nickel/N-heterocyclic carbene catalysis. J. Am. Chem. Soc. 2019, 141, 5628−5634. [DOI] [PubMed] [Google Scholar]; (n) Xie H; Shao Y; Gui J; Lan J; Liu Z; Ke Z; Deng Y; Jiang H; Zeng W Co(II)-Catalyzed Regioselective Pyridine C-H Coupling with Diazoacetates. Org. Lett. 2019, 21, 3427−3430. [DOI] [PubMed] [Google Scholar]
- (14).Nakao Y; Kanyiva KS; Hiyama T A strategy for C-H activation of pyridines: Direct C-2 selective alkenylation of pyridines by nickel/Lewis acid catalysis. J. Am. Chem. Soc. 2008, 130, 2448–2449. [DOI] [PubMed] [Google Scholar]
- (15).(a) For relevant works, see: Tsai C-C; Shih W-C; Fang C-H; Li C-Y; Ong T-G; Yap GPA Bimetallic nickel aluminun mediated para-selective alkenylation of pyridine: Direct observation of η2, η1-pyridine Ni(0)-Al(III) intermediates prior to C-H Bond Activation. J. Am. Chem. Soc. 2010, 132, 11887–11889. [DOI] [PubMed] [Google Scholar]; (b) Nakao Y; Yamada Y; Kashihara N; Hiyama T Selective C-4 alkylation of pyridine by nickel/Lewis acid catalysis. J. Am. Chem. Soc. 2010, 132, 13666–13668. [DOI] [PubMed] [Google Scholar]; (c) Lee W-C; Chen C-H; Liu C-Y; Yu M-S; Lina Y-H; Ong T-G Nickel-catalysed para-CH activation of pyridine with switchable regioselective hydroheteroarylation of allylarenes. Chem. Commun. 2015, 51, 17104−17107. [DOI] [PubMed] [Google Scholar]; (d) Singh V; Nakao Y; Sakaki S; Deshmukh MM Theoretical Study of Nickel-Catalyzed Selective Alkenylation of Pyridine: Reaction Mechanism and Crucial Roles of Lewis Acid and Ligands in Determining the Selectivity. J. Org. Chem. 2017, 82, 289−301. [DOI] [PubMed] [Google Scholar]
- (16).(a) For relevant reviews on NHC ligands, see: Thongpaen J; Manguin R; Basle O Chiral N-heterocyclic carbene ligands enable asymmetric C-H bond functionalization. Angew. Chem. Int. Ed. 2020, 59, 10242–10251. [DOI] [PubMed] [Google Scholar]; (b) Zhao Q; Meng G; Nolan SP; Szostak M N‑Heterocyclic carbene complexes in C-H activation reactions. Chem. Rev. 2020, 120, 1981–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).As expected, the use of the imidazolium halide precursor to L5 led to the complete shut-down of reactivity in a manner similar to the use of IPr·HCl and IMes·HCl, where the formed tBuOH deleteriously reacts with the Al Lewis acid. This result also signifies the importance of the free hydroxyl group in the directive NHC ligands, where it preferentially reacts with the Al Lewis acid and modulates the reactivity such that further reaction with tBuOH is inhibited. Unfortunately, our attempt at generating and employing the free carbene from L5 was unsuccessful due to the instability of the formed carbene species.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.