

Summary
Soil‐borne microbes can establish compatible relationships with host plants, providing a large variety of nutritive and protective compounds in exchange for photosynthesized sugars. However, the molecular mechanisms mediating the establishment of these beneficial relationships remain unclear. Our previous genetic mapping and whole‐genome resequencing studies identified a gene deletion event of a Populus trichocarpa lectin receptor‐like kinase gene PtLecRLK1 in Populus deltoides that was associated with poor‐root colonization by the ectomycorrhizal fungus Laccaria bicolor. By introducing PtLecRLK1 into a perennial grass known to be a non‐host of L. bicolor, switchgrass (Panicum virgatum L.), we found that L. bicolor colonizes ZmUbipro‐PtLecRLK1 transgenic switchgrass roots, which illustrates that the introduction of PtLecRLK1 has the potential to convert a non‐host to a host of L. bicolor. Furthermore, transcriptomic and proteomic analyses on inoculated‐transgenic switchgrass roots revealed genes/proteins overrepresented in the compatible interaction and underrepresented in the pathogenic defence pathway, consistent with the view that pathogenic defence response is down‐regulated during compatible interaction. Metabolomic profiling revealed that root colonization in the transgenic switchgrass was associated with an increase in N‐containing metabolites and a decrease in organic acids, sugars, and aromatic hydroxycinnamate conjugates, which are often seen in the early steps of establishing compatible interactions. These studies illustrate that PtLecRLK1 is able to render a plant susceptible to colonization by the ectomycorrhizal fungus L. bicolor and shed light on engineering mycorrhizal symbiosis into a non‐host to enhance plant productivity and fitness on marginal lands.
Keywords: PtLecRLK1, ectomycorrhizal symbiosis, Laccaria bicolor, switchgrass, Panicum virgatum
Introduction
In natural ecosystems, ectomycorrhizal soil fungi can form symbiotic relationships with the roots of most temperate forest trees (Horan et al., 1988). During the establishment of ectomycorrhizae (ECM), fungi colonize the plant root with the fungal hyphae penetrating the root from the root cap towards the epidermis and cortical tissue. The hyphal layer sheathes the root tip to form mantle structures, and the internal hyphae grow between the root epidermal and cortical cells to form a network called the Hartig net (Blasius et al., 1986; Horan et al., 1988). Through these physical structures, plants and fungi interact mutualistically; fungi provide water and a large variety of nutrients to host plants in exchange for photosynthesized sugars from the host (Smith and Read, 2010). Ectomycorrhizal fungi, Laccaria bicolor in particular, have attracted considerable attention over the past several years, because they are axenically cultivable, possess plant growth‐promoting characteristics, have a sequenced genome, and contribute several other benefits to their hosts (Felten et al., 2009; Martin et al., 2008; Smith and Read, 2010). For example, a previous report demonstrated that L. bicolor could stimulate lateral root growth in poplar (Populus sp.) and Arabidopsis by regulating auxin signalling (Felten et al., 2009). Furthermore, L. bicolor moderates the effects of phosphorus limitation in poplar by partitioning carbon between carbohydrates and secondary metabolites, whilst sustaining phosphorus uptake and translocation (Shinde et al., 2018). In addition to modulating plant nutrition, ECMs activate stress‐related genes and signalling pathways that confer abiotic stress tolerance in poplar (Luo et al., 2009).
Given that the establishment of ECM requires the interchange of signals between both partners in the symbiosis, identification of the primary factors that regulate the establishment of symbiosis is essential for understanding the role of ECM in plant development and physiology. Previous studies have shown that effector‐type small secreted proteins (SSPs) were present in both L. bicolor and its host plant poplar and are important in establishing the ectomycorrhizal symbiosis (Martin et al., 2008; Pellegrin et al., 2019; Plett et al., 2011, 2017). Further genetic mapping and resequencing in poplar identified a whole gene deletion event of a lectin receptor‐like kinase encoding gene PtLecRLK1 that was associated with a decrease in colonization by L. bicolor (Labbé et al., 2019). Lectin receptor‐like kinases (LecRLKs) are a group of cell‐surface receptors characterized by an extracellular lectin domain, a transmembrane domain, and an intracellular kinase domain (Bouwmeester and Govers, 2009; Herve et al., 1996). LecRLKs are known to be involved in plant innate immunity (Singh and Zimmerli, 2013; Wang and Bouwmeester, 2017). The LecRLK from Arabidopsis, LecRK‐VI.2 was shown to be a mediator of the Arabidopsis pattern‐triggered immunity response (Singh et al., 2012). Arabidopsis LecRK‐a1 was induced during senescence, wounding and in response to oligogalacturonic acid stress (Riou et al., 2002). Arabidopsis LekRK‐I.9 was demonstrated to bind to the integrin ligand present in the oomycete effectors IPI‐O, thereby, contributing to disease resistance and maintaining robust cell wall‐plasma membrane integrations during Phytophthora infestans infection (Bouwmeester et al., 2011). The expression of a poplar LecRLK, PnLPK, was induced by wounding, and the phosphorylation activity of PnLPK was increased in the presence of divalent metal cations (Nishiguchi et al., 2002). Moreover, Nicotiana benthamiana NbLRK1 interacted with Phytophthora infestans INF1 elicitor and mediated INF1‐induced cell death (Kanzaki et al., 2008). In Nicotiana attenuata, LecRK1 was reported to suppress insect‐mediated inhibition of defence responses during Manduca sexta herbivory (Gilardoni et al., 2011).
To date, most studies on RLKs have been performed in herbaceous model plants and focussed on their roles in plant responses to abiotic and biotic stresses, with only a few studies reported on the RLKs in perennial plants such as switchgrass (Panicum virgatum L.). For example, Gill et al. (2018) reported that the expression of a switchgrass RLK gene was increased when using phosphite to inhibit diseases caused by oomycetes. Yongfeng et al. (2018) reported that a gene encoding cysteine‐rich RLK caused delayed flowering time in switchgrass. Unlike annual plants, which need tilling and replanting each year, perennial plants, such as poplar trees and switchgrass plants, retain their aboveground biomass, and their roots exhibit relatively higher soil microbe diversity and long‐lasting relationships with beneficial microbes (Glover et al., 2010). Annual crops can lose five times as much water and 35 times more nitrate than perennial plants (Randall et al., 1997). Therefore, perennial plants can improve soil nutrient and water conservation, creating more sustainable ecosystems (Vukicevich et al., 2016). Due to such traits, perennial plants, including poplar and switchgrass, are being developed as major biomass feedstocks (Ma et al., 2000). However, to improve economic viability, these bioenergy crops are expected to grow on marginal lands so that they do not compete with food crops growing on limited prime agricultural lands. There is a need to improve the fitness of bioenergy crops for growing conditions on marginal lands. A potential way to accomplish this is to engineer beneficial mycorrhization into bioenergy crops, given that increasing evidence has shown that the beneficial mycorrhization can promote plant nutrient acquisition and improve plant performance under abiotic stress. Switchgrass is known to be strongly dependent on arbuscular mycorrhizal fungi (AMF). Sun et al. (2018) reported that the inoculation of AMF increased switchgrass biomass and phosphorus (P) concentration in the cadmium‐contaminated soil. Schroeder‐Moreno et al. (2012) reported that AMF aided switchgrass nitrogen (N) assimilation in a high temperature and high N‐content environment. However, there are only a few studies reporting the relationship of switchgrass with ectomycorrhizal fungi. Ghimire et al. reported that ectomycorrhizal fungus Sebacina vermifera could enhance seed germination and biomass production in switchgrass under drought conditions (Ghimire et al., 2009; Ghimire and Craven, 2011), but the underlying molecular mechanisms remain unclear.
Our previous genomic and genetic studies reported that the lack of colonization of P. deltoides by ectomycorrhizal fungus L. bicolor was associated with a PtLecRLK1 locus deletion (Labbé et al., 2019). PtLecRLK1 introduction into the non‐host, Arabidopsis, enabled the colonization of L. bicolor in the PtLecRLK1 transgenic Arabidopsis root, and was associated with the down‐regulation of plant defence‐related genes and metabolites, thus supporting the role of PtLecRLK1 in mediating plant‐L. bicolor interactions (Labbé et al., 2019). To explore the possibility of greater utilization of switchgrass in marginal environments via ectomycorrhization, and to better understand the role of PtLecRLK1 in mediating plant‐L. bicolor interactions, we hypothesized that introducing PtLecRLK1 into non‐host switchgrass would enable colonization by L. bicolor. By characterizing the fungal colonization and molecular responses of L. bicolor‐inoculated ZmUbipro‐PtLecRLK1 transgenic switchgrass roots, we demonstrated that the introduction of PtLecRLK1 allows the colonization of L. bicolor into switchgrass roots by altering the molecular responses at the transcriptional, translational, and metabolic levels. The initiative to engineer a long‐lasting beneficial relationship between switchgrass and the ectomycorrhizal fungus, L. bicolor, could maximize the utility and productivity of this important bioenergy crop for growth on marginal lands and in stressed environments.
Results and Discussion
In order to promote L. bicolor root colonization in a non‐host plant, we generated transgenic plants by heterologously expressing ZmUbipro‐PtLecRLK1 in switchgrass (a non‐host of L. bicolor). Four switchgrass transgenic lines (i.e. 5007, 5012, 5016 and 5031) expressing ZmUbipro‐PtLecRLK1 were verified by RT‐PCR (Figure S1a) and RNAseq (Figure S1b) analyses and used in the present study.
Ectomycorrhizal development in switchgrass
As a non‐host of L. bicolor, the wild‐type (WT) switchgrass roots had only a thin layer of L. bicolor hyphae loosely attached to the surface with no penetration between the root epidermal cells (Figure 1a,c). In contrast, the ZmUbipro‐PtLecRLK1 transgenic switchgrass roots developed typical ECM structures, including swelling in the root tip, and formation of the hyphal Hartig net between plant cells that extended beyond epidermal cells (Figure 1b,d). These results support our hypothesis that introduction of PtLecRLK1 renders non‐host (switchgrass) receptive to colonization by L. bicolor.
Figure 1.
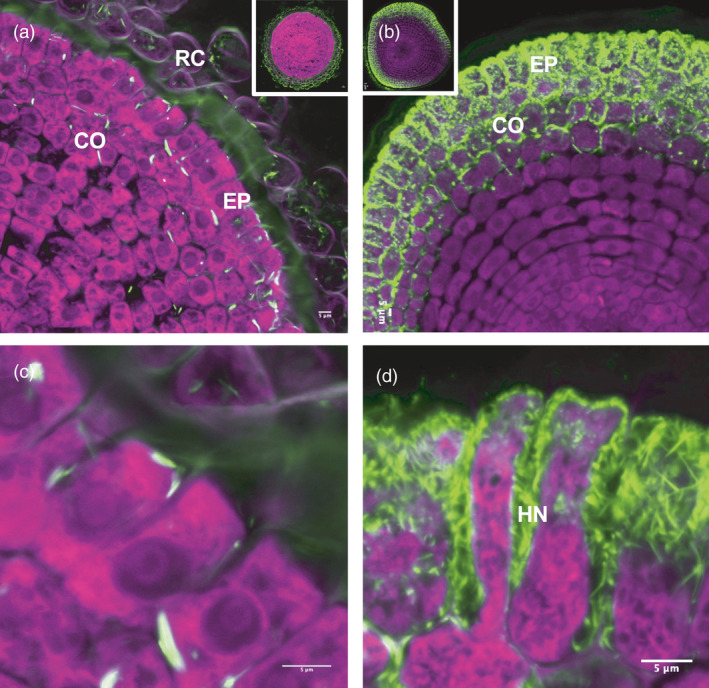
Laccaria bicolor colonization in roots of wild‐type switchgrass (Panicum virgatum L., genotype ‘NFCX01’) and ZmUbipro‐PtLecRLK1 transgenic switchgrass. (a) Dual fluorescence‐stained transverse root section of WT switchgrass. (b) Dual fluorescence‐stained transverse root section of ZmUbipro‐PtLecRLK1 transgenic switchgrass. (c) and (d) Zoom‐ins from (a) and (b). Green colour represents the UVitex stained fungi cell; Purple colour shows the propidium iodide stained root cell. RC, Root cap; Co, Cortex; Ep, epidermis; HN, Hartig net. Images shown are representative images from three experiments. Bars = 5 μm.
Performance of ZmUbipro‐PtLecRLK1 transgenic switchgrass under phosphorus limitation with L. bicolor inoculation
Ray et al. (2020) reported that the AMF Serendipita vermifera ssp. Bescii promoted the growth of winter wheat under P‐limitation. Therefore, we aimed to investigate whether the transgene ZmUbipro‐PtLecRLK1‐induced colonization of L. bicolor would be beneficial to switchgrass growth under nutrient limitation. The plant height, tiller numbers, and tiller dry weight were measured after 2 months of co‐culturing with L. bicolor under the control condition and under the phosphorus (P) limitation condition. As the three‐way ANOVA analysis shows in Table S1, the transgene, L. bicolor inoculation, P‐limitation treatment and the interactions amongst them did not impact plant dry weight. The transgene and P‐limitation treatment interaction caused significant differences in plant height. Under the control‐inoculation condition, WT plants were taller than transgenic plants. However, this difference disappeared under the P‐limitation inoculation condition (Figure S2a). The P‐limitation condition suppressed the height of WT plants, but not the transgenic plants with L. bicolor inoculation. The transgene was also shown to be the main factor causing significantly more tiller numbers in one of the transgenic lines (line 5016) (Figure S2b). Collectively, L. bicolor‐inoculated‐transgenic plants tended to be more tolerant to P‐limitation than the WT plants. A comprehensive evaluation of the influence of L. bicolor inoculation on the performance of the transgenic plants would require field studies with larger sample size and longer inoculation time.
Transcript profiling of switchgrass roots during early interaction with L. bicolor
Given that switchgrass is a non‐host of L. bicolor, we wanted to explore the underlying molecular mechanisms mediating switchgrass‐L. bicolor interaction, and identify molecular processes that are associated with L. bicolor colonization in the ZmUbipro‐PtLecRLK1 transgenic switchgrass. We aimed to uncover the molecular determinants of colonization towards the ultimate goal of mycorrhizal symbiosis engineering. The transcript profiles of WT and ZmUbipro‐PtLecRLK1 transgenic switchgrass roots with L. bicolor inoculation and mock inoculation at 2 months after inoculation (MAI) were generated. This time point corresponded to the penetration of L. bicolor fungal hyphae and formation of the Hartig net between the epidermal cells (Figure 1). Whole roots (containing a mixture of ECM and non‐ECM roots) were collected for RNA extraction. Principal component analysis, where PC1 and PC2 explained 80% of the variance, revealed genes from the different conditions forming distinct clusters (Figure 2a). There was little variation between the two mock conditions in the WT and the transgenic clusters, whereas differences between the inoculated and the mock conditions and between inoculated‐WT and inoculated‐transgenics varied drastically. These differences were also revealed by the following pairwise comparisons amongst the four conditions. Pairwise comparisons between inoculated‐WT and mock‐WT, between inoculated‐transgenic and mock‐transgenic, between mock‐transgenic and mock‐WT, and between inoculated‐transgenic and inoculated‐WT showed 10,130, 6,506, 871, and 6,864 differentially expressed genes (DEGs), respectively, at an absolute fold change ≥2, FDR<0.05 (Figure 2b, Table S2). Laccaria bicolor inoculation in WT switchgrass caused the highest degree of gene regulation (10, 130), whilst the ZmUbipro‐PtLecRLK1 transgene caused the least gene regulation (871). These transcriptomic responses implied that the biotic stimulus (i.e. L. bicolor inoculation) impacted transcriptional regulation more during switchgrass‐microbe interaction than the introduction of an exogenous gene (i.e. PtLecRLK1).
Figure 2.
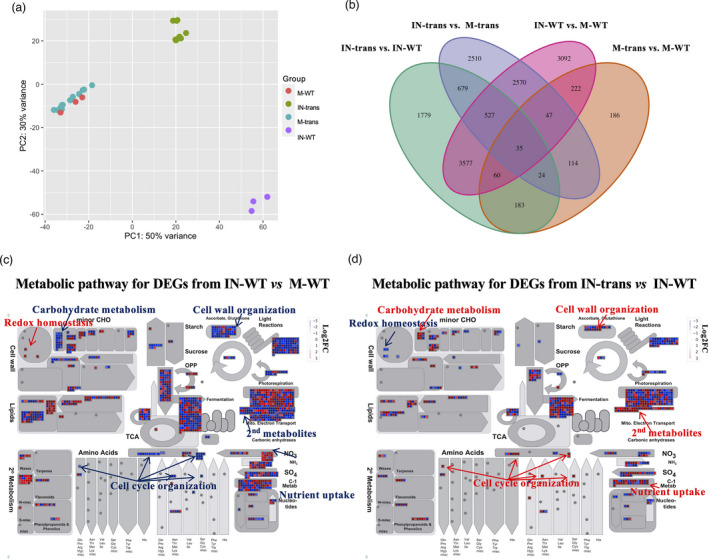
Transcriptomic analysis of switchgrass root samples in response to Laccaria bicolor inoculation. (a) Principal component analysis of gene expression patterns for four conditions of switchgrass samples. (b) Venn diagram showing the number of differentially expressed genes (DEGs) from pair comparisons between different conditions. (c) Overview of the enriched metabolic pathways of DEGs resulting from comparison between IN‐WT and M‐WT. (d) Overview of the enriched metabolic pathways of DEGs resulting from comparison between IN‐trans and IN‐WT. The red and blue colours indicate the log2 fold change of DEGs, showing up‐ or down‐regulated genes, respectively. The metabolic pathways were generated with MapMan software. IN‐trans, M‐trans, IN‐WT, and M‐WT are abbreviations for inoculated‐transgenic, mock‐transgenic, inoculated‐WT, and mock‐WT, respectively.
Through analysing the DEGs from comparison between L. bicolor‐inoculated WT and mock‐WT, we aimed to identify the transcriptional regulation of a typically non‐host plant, switchgrass, in response to the biotic stimulus‐L. bicolor inoculation. There were 5,826 and 4,303 genes down‐ and up‐regulated, respectively. To explore the function of these DEGs, Gene Ontology (GO) enrichment analysis and MapMan analysis were performed to classify these DEGs. The GO enrichment analysis suggested these genes were overrepresented in transcriptional regulation, RNA biosynthesis, and metabolic and ammonium transportation processes, and underrepresented in photosynthesis, phosphorylation, phosphorous, glucan, polysaccharide, and carbohydrate metabolic processes (Figure S3a). The MapMan analysis revealed that up‐regulated genes were involved in redox homeostasis and down‐regulated genes were involved in carbohydrate metabolism, cell wall organization, cell cycle organization, and nutrient uptake process (Figure 2c).
Having established the baseline transcriptomic responses to L. bicolor inoculation in the non‐host switchgrass, we wanted to further examine how introduction of PtLecRLK1 impacted these transcriptomic responses in the inoculated switchgrass transgenic plants. Thus, we compared the inoculated‐transgenic sample with the inoculated‐WT sample. With GO enrichment analysis, we found that 3,896 up‐regulated genes were enriched in reproductive processes, phosphorylation, phosphorus metabolic, and macromolecule modification. Those 2968 down‐regulated genes were overrepresented in response to water, abiotic stimuli, inorganic substance responses, and negative regulation of the mitotic cell cycle (Figure S3b). The following MapMan analysis on these DEGs from the comparison between inoculated‐transgenic and inoculated‐WT indicated a trend contrary to that from the comparison between inoculated‐WT and mock‐WT plants. Redox homeostasis showed a decreasing trend, whereas carbohydrate metabolism, cell wall organization, secondary metabolites, cell cycle regulation, and nutrient uptake processes showed an increasing trend overall (Figure 2d). It is well‐known that the life cycle of mycorrhizal fungi depends on the uptake of carbon from the host plant, and in return, they export nitrogen, phosphorus and minerals to plants (Bonfante and Genre, 2010). Compared to the inoculated‐WT, the genes of inoculated‐transgenic plants associated with nutrient uptake and carbohydrate metabolism were up‐regulated, suggestive of the plausibility of nutrient exchange between the two organisms. Felten et al. (2009) reported that the cell wall related and cell cycle related gene regulations increased in poplar—L. bicolor and Arabidopsis—L. bicolor interactions. The increased cell wall organization and cell cycle organization in inoculated‐transgenic switchgrass supported the feasibility of physical interaction between the two organisms. In summary, the transcriptomic analyses in switchgrass indicated that the introduction of PtLecRLK1 into switchgrass altered its compatibility with L. bicolor.
To better understand the function of PtLecRLK1 in mediating the interaction between switchgrass and L. bicolor, DEGs related to the biotic stress pathway and the nutrient uptake process were analysed in switchgrass responding to L. bicolor‐only (inoculated‐WT vs. mock‐WT), and that responding to L. bicolor + transgene (inoculated‐transgenics vs inoculated‐WT) (Tables S3 and S4). The major difference between DEGs related to biotic stress was the recognition between the two organisms (switchgrass and L. bicolor), which was represented by the categories of cytoskeleton organization, cell wall organization, and external stimuli response (Figure 3a–d). DEGs from plants responding to L. bicolor‐only included 17 up‐ and 45 down‐DEGs belonging to cytoskeleton organization, 39 up‐ and 163 down‐DEGs belonging to cell wall organization, and 0 up‐ and 4 down‐DEGs involved in external stimuli response (Figure 3c). DEGs from plants responding to L. bicolor + transgene included 14 up‐ and 13 down‐DEGs belonging to cytoskeleton organization, 45 up‐ and 30 down‐DEGs belonging to cell wall organization, and 7 up‐ and 1 down‐DEGs involved in external stimuli response (Figure 3d). In terms of nutrient uptake related DEGs, there were 13 up‐ and 7 down‐ DEGs related to nitrogen assimilation, 5 up‐ and 2 down‐ DEGs related to sulphur assimilation, 1 up‐ and 7 down‐ DEGs related to phosphorus assimilation, 14 up‐ and 19 down‐ DEGs related to iron uptake, and 1 up‐ and 1 down‐ DEGs related to copper uptake in response to L. bicolor‐only (Figure 3e). DEGs from plants responding to L. bicolor + transgene showed 4 up‐ and 2 down‐ DEGs related to nitrogen assimilation, 5 up‐ and 2 down‐DEGs related to phosphorus assimilation, 9 up‐ and 6 down‐DEGs related to iron uptake, and 2 up‐ and 1 down‐DEGs related to copper uptake (Figure 3f). There were 15 nutrient uptake related genes regulated in both of the comparisons (inoculated‐WT vs. mock‐WT and inoculated‐transgenics vs. inoculated‐WT), 9 out of 15 genes were up‐regulated in response to L. bicolor + transgene, and 5 genes were up‐regulated in response to L. bicolor‐only (Figure S4). Overall, the data showed higher ratios of up‐ to down‐ DEGs related to biotic stress and nutrient uptake from plants responding to L. bicolor + transgene than those responding to L. bicolor‐only. The data suggest that the introduction of transgene ZmUbipro‐PtLecRLK1 up‐regulated more genes involved in plant response to external stimuli, plant‐microbe recognition (cell wall organization and cytoskeleton organization), and plant nutrient uptake, and presumably allowing the establishment of a compatible interaction between switchgrass and L. bicolor.
Figure 3.
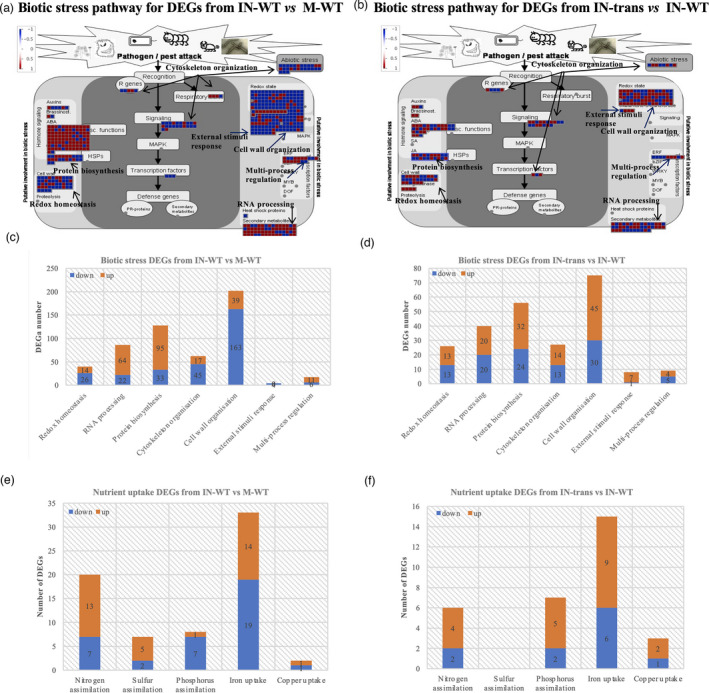
MapMan analyses of differentially expressed genes (DEGs) in biotic stress pathway and nutrient uptake process. (a) Biotic stress pathway analysis for DEGs from comparison between IN‐WT and M‐WT. (b) Biotic stress pathway analysis for DEGs from comparison between IN‐trans and IN‐WT. (c) The number of up‐ and down‐DEGs from comparison between IN‐WT and M‐WT in specific biotic stress categories. (d) The number of up‐ and down‐DEGs from comparison between IN‐trans and IN‐WT in specific biotic stress categories. (e) The number of up‐ and down‐DEGs from comparison between IN‐WT and M‐WT in specific nutrient assimilation categories. (f) The number of up‐ and down‐DEGs from comparison between IN‐trans and IN‐WT in specific nutrient assimilation categories. The red and blue colours indicate the log2 fold change of DEGs, showing up‐ or down‐regulated genes, respectively. The metabolic pathways were generated with MapMan software; IN‐trans, IN‐WT, and M‐WT are abbreviations for inoculated‐transgenic, inoculated‐WT, and mock‐WT, respectively.
It is well known that the RLKs play important roles in plant innate immunity. To understand the role of native LecRLK during the transgenic switchgrass‐L. bicolor interaction, 64 PtLecRLK1 sequence homolog genes (PavirLecRLK) in switchgrass were identified. The phylogenetic relationship amongst those genes is presented in Figure S5a. There were 16 out of 64 members that were differentially regulated in the four conditions (Figure S5b). The regulated PavirLecRLK members included RDA2 that was shown to be involved in transducing immune responses in Arabidopsis (Park et al., 2019), and ARK2, ARK 3 that were shown to be involved in immune‐triggered incompatibility in Arabidopsis (Alcázar et al., 2010). Amongst the 16 PavirLecRLK, there was no PavirLecRLK member that was differentially regulated by the transgene PtLecRLK1 only, but more than 10 were regulated by the L. bicolor infection, and 6 genes were responding to L. bicolor + transgene (Figure S5b). These results suggested that the PtLecRLK1 transgene did not impact endogenous PavirLecRLK expression, but the L. bicolor inoculation did. We also aligned the protein sequences of PtLecRLK1 and the 6 closest switchgrass sequence homologs, and analysed the 4 domains (D‐mannose binding lectin domain, S‐locus binding domain, PAN‐like domain and Protein kinase domain). As shown in Figure S5c–f, except that the protein kinase domain is highly conserved, the other three domains that are predicted to be involved in recognition are not highly conserved, suggestive of the possible distinct recognition mechanisms in poplar and switchgrass. Therefore, we speculate that a functional PtLecRLK1 orthologue, specifically recognizing the ligand secreted by L. bicolor may not be present in switchgrass. It would be interesting to compare the response of transgenic switchgrass to both EM fungi and native AM fungi and/or pathogenic microbes under similar conditions. Such a multiple interaction would more closely mimic the real‐world condition and reflect how the recognition and downstream signal cascade balance/conflict with different microbes to finally establish the symbiotic or defensive relationships.
Co‐expression network analysis highlights transcriptional changes associated with L. bicolor inoculation and PtLecRLK1 introduction
To better reveal the crucial shift of gene expression networks in the establishment of the switchgrass‐L. bicolor interaction, weighted gene co‐expression network analysis (WGCNA) (Langfelder and Horvath, 2008) was used to extract the main patterns of gene co‐expression amongst the four conditions (i.e. mock‐WT, inoculated‐WT, mock‐transgenic, and inoculated‐transgenic). WGCNA resolved the 15, 606 differentially expressed genes into 16 modules, corresponding to branches labelled by colours indicated underneath the tree (Figure 4a, Table S2). Notably, 4 out of the 16 co‐expression modules: turquoise, yellow, black, and green colour modules exhibited condition‐specific (inoculated‐WT, inoculated‐transgenic, mock‐WT, and mock‐transgenic, respectively) expression values (Figure 4b,c). That is, these modules were composed of genes that tended to be overexpressed in a single condition (r > 0.71, P < 2.6e‐92, Figure S6). To compare the trends in gene expression of each group as a function of inoculation + transgenic condition, an overview of expression for each of the 4 groups was presented (Figure 4d). Figure 4e is depicted graphically as the aggregate average expression value for each condition. The two mock groups indicated only a small difference. However, the two inoculated groups were drastically differentiated from each other, and from the two mock groups. In addition, there was an overlapping blue module between the inoculated‐WT and inoculated‐transgenic groups (Figure 4b–d), leading to the similar average expression value amongst those two conditions in Figure 4e. Although the overlap contained fewer genes from the inoculated‐WT in the inoculated‐transgenic module, the average of those overlapped gene expressions was higher than that in the inoculated‐transgenic module. This analysis showed that the gene co‐expression network shifted as the result of the L. bicolor inoculation and the presence of PtLecRLK1.
Figure 4.
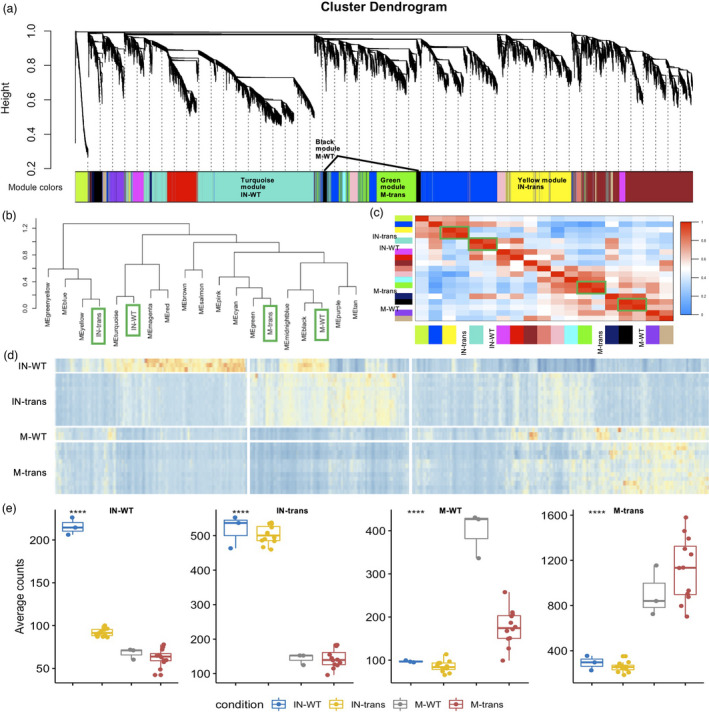
Network analysis of switchgrass root genes. (a) Hierarchical cluster tree showing co‐expression modules identified using WGCNA. Modules correspond to branches and are labelled by colours as indicated underneath the tree. The average gene expression values of all the replicates from the respective condition were used to build the network. (b) Dendrogram tree showing the distance amongst conditions and different coloured‐modules. (c) Heatmap showing the correlation between modules and conditions. The green boxes highlight the correlated conditions and modules. The reported modules and conditions (x‐axis) are often highly correlated with distinct conditions and modules (y‐axis). Colour legend indicates the level of correlation module and condition. (d) Heatmap of co‐expression groups for differentially expressed genes across the four conditions. The vertical axis organizes genes according to replicates in conditions, and the horizontal axis shows individual gene in the conditions labelled on the y‐axis. The colour codes from blue to red indicate the gene expression value from low to high. (e) Boxplots show the distribution of groups expression (mean RPKM of all genes within a given group) for different genotype and inoculation conditions. IN‐WT, inoculated‐WT; IN‐trans, inoculated‐transgenic; M‐WT, mock‐WT; M‐trans, mock‐transgenic; respectively.
The WGCNA showed that PtLecRLK1 and its co‐expressed genes were in the yellow module representing the inoculated‐transgenic samples. We analysed its co‐expressed gene network (Figure S7) and found that there was one gene (Pavir.5KG693600) that was directly connected with the expression of PtLecRLK1. This gene encoded an Integrin‐Linked protein Kinase (ILK) family‐related protein. In mammalian cells, the ILK and its interactors play a key role in mediating the intracellular cytoskeletal and signalling proteins communicating with the extracellular matrix (Wu and Dedhar, 2001). The plant ILK was shown to be coupled with ion transporter function in plant cell development and immune response (Brauer et al., 2016). In addition, the plant ILK with its upstream ILR (Integrin‐like receptor) has been shown to alter the cell wall and plasma membrane via response to multiple signals (Popescu et al., 2017). Therefore, the plant ILK was proposed to be a signal integrator that assembles the transporter and receptor complex to generate the downstream signalling for plant cell growth and immune response (Popescu et al., 2017). In our study, this ILK family‐related protein coding gene, Pavir.5KG693600, was co‐expressed with a hub gene Pavir.5KG551300, encoding a lipid kinase‐phosphatidylinositol 4‐phosphate 5‐kinase MSS4‐like protein, which has been reported to be a cytoskeleton regulator in yeast (Desrivieres et al., 1998) and membrane recycling modulator in Arabidopsis (Sousa et al., 2008). The co‐expression network amongst PtLecRLK1, ILK and phosphatidylinositol 4‐phosphate 5‐kinase MSS4‐like protein suggests a potential role for PtLecRLK1 in switchgrass as an initial integrator for a ligand receptor and related receptor‐like kinase that subsequently triggers the signalling cascade leading to successful establishment of the interaction between switchgrass and L. bicolor. Collectively, our co‐expression network analysis revealed the distinct transcriptional reprogramming for L. bicolor inoculation and the effect of PtLecRLK1, which could be summarized using modules and a co‐expression network with predicted functions.
Functional classification and annotation of differential abundance proteins
On the basis of the transcriptomic evidence showing the potential of PtLecRLK1 to establish a compatible relationship between the transgenic switchgrass and L. bicolor by up‐regulating genes benefiting plant‐microbe recognition and nutrient exchange, and down‐regulating the genes responsible for abiotic stresses, we wanted to extend our analysis to examine the impact of PtLecRLK1 on the translational level regulation. Using the same inoculated‐transgenic and inoculated‐WT plant materials, we generated proteomic profiles to identify proteins that were significantly up‐ or down‐regulated by the introduction of PtLecRLK1 during L. bicolor inoculation.
Principal component analysis on proteins (transcripts were also incorporated into the same analysis) from the two different conditions formed distinct clusters (Figure 5a). Differential abundance proteins were identified with a P < 0.05 and absolute log2 fold change ≥1 between the inoculated‐transgenic and inoculated‐WT. In brief, 1,422 differential abundance proteins (595 high abundance and 827 low abundance) were detected (Table S5). Differential abundance proteins were subjected to enrichment and clustering using functional analysis of GO and MapMan. In the GO function analysis, carbohydrate metabolic, tRNA aminoacylation, asparagine biosynthetic, nitrate assimilation, fatty acid metabolic, and anion transport were overrepresented in the high abundance proteins; for example, nitrate assimilation‐related proteins and nitrate transporter (Pavir.6NG072000, Pavir.6KG067100, Pavir.1NG011219), nitrate reductase (Pavir.6KG411506) and nitrite reductase (Pavir.1KG111021 and Pavir.1NG473400) were at high abundance in the inoculated‐transgenic switchgrass compared with the inoculated‐WT controls. Nitrate transporters are known to participate in nitrogen, phosphorus, and nutrient sensing in plants by regulating plant response to abiotic and biotic stresses (Hu et al., 2019; Krouk et al., 2010), possibly via the phytohormone and receptor‐like kinases (Liu et al., 2020). Nitrogen transportation is known to play a key role in the compatible association during orchid mycorrhizal symbiosis (Dearnaley and Cameron, 2016). Increases in fatty lipid metabolism have also been reported in a compatible interaction between P. trichocarpa and L. bicolor (Tschaplinski et al., 2014). In the low abundance proteins, photosynthesis, oxidation–reduction, nitrogen compound metabolic, phospholipid biosynthesis, and negative regulation of transcription were enriched (Figure 5b). In addition, we observed that pathogen resistance proteins were decreased in the inoculated‐transgenic switchgrass, including WRKY40 (Pavir.1KG087300), WRKY4 (Pavir.3KG465900), LRR, and NB‐ARC domains containing disease resistance proteins: Pavir.6NG062400, Pavir.5NG342200, Pavir.6KG040500, Pavir.6KG045300, Pavir.6KG047205, and Pavir.6KG122900. Furthermore, proteins related to fungal Hartig net formation were altered in the inoculated‐transgenic switchgrass. Plett et al. reported that the application of the ethylene precursor, ACC (1‐aminocyclopropane‐1‐carboxylic acid), in poplar root systems inhibited Hartig net development (Plett et al., 2014). We found that Pavir.5NG258500 and Pavir.5KG296000 encoding ACC oxidases, which synthesize ethylene from ACC, had lower abundance in the inoculated‐transgenic switchgrass than that in the inoculated‐WT control, suggestive of less negative regulation in Hartig net development in the transgenic plants compared to WT. On the other hand, ectomycorrhizal fungi have been reported to produce IAA from tryptophan with the nitrilase enzyme and to excrete IAA to enhance Hartig net formation in the host plant (Plett et al., 2014). We observed that Pavir.1KG395705 and Pavir.1KG319400 encoding nitrilase‐related proteins showed higher abundance in the inoculated‐transgenic switchgrass, potentially increasing the IAA content to enhance Hartig net formation in the transgenic plant roots. To further explore the pathways in which the differential abundance proteins were involved, we analysed these proteins using the MapMan software. The metabolism pathway visualization is shown in Figure 5c. Most proteins involved in light reactions had low abundance. Interestingly, the high abundance proteins were enriched in carbohydrate metabolism, cell cycle organization, nutrient uptake, and phenolics pathways (Figure 5c). Those pathways are similar to those of the up‐regulated DEGs (Figures 2d and 5c). The biotic stress pathway analysis showed a decrease in the abundance of proteins that are related to jasmonic acid and ethylene signalling (Figure 5d), which were reported to act as a negative modulator during the compatible symbiosis between poplar roots and L. bicolor (Plett et al., 2014). Collectively, observation of the proteomic analysis showed that, in the inoculated‐transgenic plants, the high abundance proteins were involved in plant‐microbe compatible interactions, including carbohydrate metabolism, nutrient exchange and Hartig net formation, whereas the low abundance proteins were associated with changes in photosynthesis and plant defence, suggesting that the introduction of an exogenous gene, PtLecRLK1, regulates the switchgrass‐L. bicolor interaction at the translational level, favouring compatible interactions.
Figure 5.
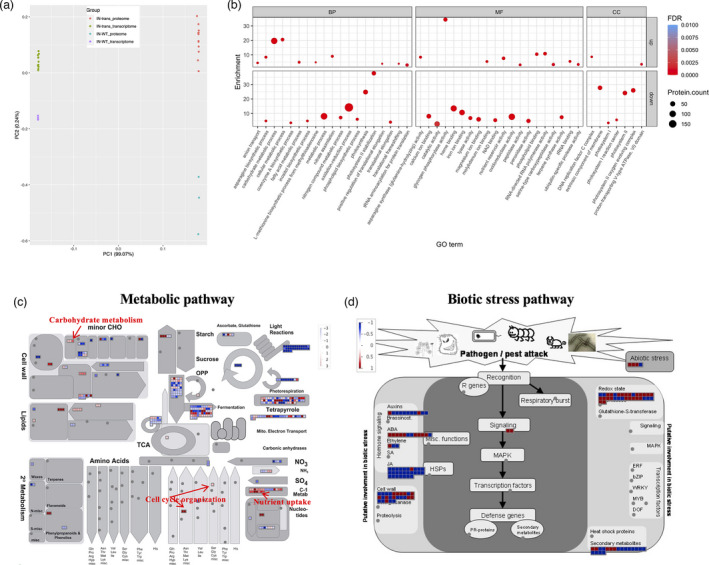
Proteomic analysis of switchgrass root samples in response to Laccaria bicolor inoculation. (a) PCA of inoculated transgenic and WT for transcripts and proteins. (b) GO enrichment analysis for differential abundance proteins from the comparison between inoculated‐transgenic and inoculated‐WT. The dot size reflects the number of genes included in the category. (c) Overview of the metabolic pathways that the differential abundance proteins are involved in. (d) Biotic stress pathway that the differential abundance proteins are involved in. The red and blue colours indicate the log2 fold change of differential abundance proteins, showing high or low abundance proteins, respectively. The metabolic pathways were generated with MapMan software.
Earlier studies reported a weak correlation between transcript level and protein abundance (Greenbaum et al., 2002; Nie et al., 2007; Zhang et al., 2006). Here, we compared 1,422 (high and low abundance: 595 and 827, respectively) differential abundance proteins with 6,864 (up‐ and down‐ regulated: 3,896 and 2968, respectively) DEGs. We found that 105 (up‐ and down‐regulated: 64 and 41, respectively) genes/proteins overlapped (Table S6). Nevertheless, functional enrichment analysis at both the transcriptional and the translational levels showed up‐regulated carbohydrate metabolism, cell cycle organization, and nutrient uptake and down‐regulated photosynthesis processes in the L. bicolor‐inoculated‐transgenic roots. This dichotomy of up‐ and down‐regulated processes suggests concomitant regulation at the transcriptional and translational levels.
Metabolomic profiling of switchgrass roots during early interaction with L. bicolor
From transcriptomic and proteomic analyses, we observed the effect of PtLecRLK1 in switchgrass on up‐regulation of genes and proteins involved in plant‐microbe compatible interactions, such as carbohydrate metabolism, nutrient assimilation, and cell wall organization, and, conversely, down‐regulation of genes and proteins involved in responses to biotic and abiotic stresses. To further elucidate the underlying molecular mechanisms, we generated metabolite profiles of inoculated‐transgenic and WT plants to better understand the PtLecRLK1 regulation of switchgrass responses to L. bicolor inoculation.
Gas chromatography‐mass spectrometry‐based metabolomics were conducted on the same L. bicolor‐inoculated transgenic and WT root samples. PCA analysis demonstrated separation of the WT and transgenic samples (Figure 6a). Figure 6b showed that metabolomic responses of the transgenics were associated with the accumulation of numerous nitrogenous metabolites, such as asparagine, tyrosine, leucine, isoleucine, valine, and other amino acids, and the decline in stearic acid, fumaric acid, malic acid, lactic acid, glucose, mannitol, and aromatic metabolites including hydroxycinnamate conjugates. The enrichment analysis showed that the high abundance metabolites were involved in amino acid biosynthesis and metabolism, and that the low abundance metabolites were involved in the biosynthesis of unsaturated fatty acids and fatty acid biosynthesis (Figure 6c–e and Table S7). Such metabolite changes have been observed in previous studies on plant‐microbe beneficial interactions. For example, the application of the beneficial fungus Trichoderma spp. biocontrol metabolite (6‐pentyl‐2H‐pyran‐2‐one) on tomato plant resulted in increased amino acids, such as tyrosine, valine, glutamine, leucine, arginine and threonine, and phenylalanine, whose high abundance was reported to enable massive enhancement of carbon flux (Mazzei et al., 2016). Increases in asparagine, isoleucine, putrescine, and tryptophan, and decline in glucose were observed in the L. bicolor‐inoculated 35S:PtLecRLK1 transgenic Arabidopsis and the L. bicolor compatible host, P. trichocarpa (Labbé et al., 2019; Tschaplinski et al., 2014). Decreases in glucose, malic and shikimic acid were vital for Panicillium expansum colonization in apple fruit (Žebeljan et al., 2019). Besides sugars, fatty acids are another carbon source that the symbiont can obtain from the plant. For instance, the AM fungi Rhizophagus. irregularis cannot synthesize the fatty acids, and its host plant Medicago can transfer the fatty acid to fungi to build the symbiotic relationship (Jiang et al., 2017; Luginbuehl et al., 2017). A parasitic pathogen fungi, Golovinomyces cichoracearum, can also take up fatty acids from host plants and decrease the fatty acid biosynthesis in plants (Jiang et al., 2017). However, the carbohydrate metabolism and the fatty acid metabolomic pathways were up‐regulated in the inoculated‐transgenic plants in the proteomic analysis. This led us to consider that those increased metabolic activities resulted in providing more available carbon, including fatty acids from transgenic plants to fungi, leading to the decreased fatty acid‐related metabolites in plants. The metabolomic results agreed with the transcriptomic analysis showing increased nitrogen‐containing metabolites associated with the up‐regulated genes enriched in the transmembrane transporter activity, protein metabolic activity, and the tryptophan catabolic process; and decreased carbon‐containing metabolites associated with the down‐regulated genes enriched in photosynthesis. Declines in fatty acids and organic acids were likely caused by increased carbon demands of the microbial symbiont (Larsen et al., 2011). The increased free amino acid concentration in roots was putatively the result of exchanging nutrients for carbon by the fungus. Such metabolites change suggests that the steps including the nutrient exchange in a successful symbiosis occurred between the ZmUbipro‐PtLecRLK1 transgenic switchgrass and L. bicolor.
Figure 6.
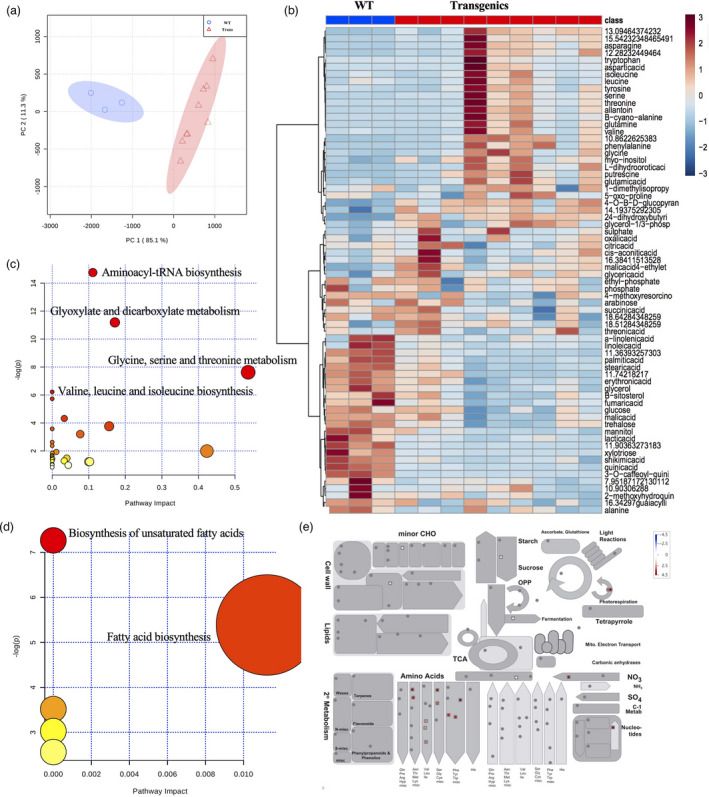
Metabolite response in switchgrass roots inoculated with Laccaria bicolor. (a) PCA separation of switchgrass WT from the transgenic samples. (b) Heatmap showing regulated metabolites in WT and transgenic roots in response to L. bicolor inoculation. (c) High abundance metabolites pathway enrichment analyses. (d) Low abundance metabolites pathway enrichment analyses. The ball size indicates the pathway impact, calculated by the compound number observed divided by the total compound number in the pathway. (e) Overview of the metabolic pasthways that the differential abundance metabolites are involved in. The red and blue colours indicate the log2 fold change of differential abundance metabolites, showing high or low abundance metabolites, respectively. The metabolic pathways were generated with MapMan software.
To gain insight on the L. bicolor transcriptomic response during its colonization in switchgrass roots, we also analysed the fungal genes in the RNAseq dataset. However, only 45 genes out of 23, 125 fungal genes had expression values in at least one of the 15 inoculated root samples. When we performed the pair comparison, we were only able to find one gene down‐regulated in comparison between inoculated‐WT and mock‐WT, and 4 genes up‐regulated in comparison between inoculated‐transgenic and mock‐transgenic plants. The possible reason for detecting a low number of fungal genes was that we focussed on host transcriptomic response and did not enrich fungal RNA for RNAseq, and that the ECM roots in the sample were relative low (as the RNA was extracted from the whole root whilst the ECM root normally occurs in the root tip section). To precisely determine the fungal transcriptomic response, a possible future study would require isolating the ECM root, separating fungal RNA from plant RNA, and measuring the transcriptomic response from both organisms to obtain a complete picture of gene regulation in mutualistic interaction.
Conclusion
The data presented here provide a comprehensive picture of the dynamic changes in transcription, translation, and the concomitant alterations in the level of metabolites that occurred in L. bicolor colonization of switchgrass transgenic plant roots under the regulation of a poplar lectin receptor‐like kinase, PtLecRLK1. Specifically, our data indicated that introduction of PtLecRLK1 into a switchgrass plant alters its susceptibility to fungal colonization by up‐regulating the compatible interaction‐related genes/proteins and metabolites (i.e. nutrient assimilation, carbohydrate metabolism, cell cycle organization, and cell wall organization related molecules, and free amino acids) and by down‐regulating defence‐related genes/proteins and metabolites (i.e. jasmonic acid and ethylene synthesis molecules, organic acids) in response to L. bicolor inoculation. The down‐regulation of defence‐related genes/proteins and metabolites were also observed when poplar trees and PtLecRLK1 transgenic Arabidopsis are colonized by L. bicolor. Despite the down‐regulation, there is no evidence of compromising the plant’s ability to defend against pathogens. It is also worth investigating the influences of PtLecRLK1 and L. bicolor on both the compatible AMF colonization and the pathogenic interaction with switchgrass in future studies. The current results were obtained from greenhouse experiments and analyses of the molecular‐level regulation. It would be interesting to explore the long‐lasting compatible relationship between switchgrass and L. bicolor in field conditions by focussing on the symbiosis‐related physiological traits in the future studies. The present study also provides a basis for further in‐depth investigation of the signal transduction mechanisms underlying PtLecRLK1‐mediated L. bicolor colonization, and builds a foundation for fine‐tuning the engineering of a long‐lasting compatible relationship between the ectomycorrhizal fungus, L. bicolor, and a perennial C4 biofuel crop to improve sustainable switchgrass production on marginal lands.
Experimental procedures
Plant transformation and fungal material growth conditions
ZmUbipro‐PtLecRLK1 transgenic lines of switchgrass were generated by the Noble Research Institute Transformation Core Facility using the protocol published by (Xi et al., 2009). Switchgrass (P. virgatum L., genotype ‘NFCX01’) was used to generate the ZmUbipro‐PtLecRLK1 transgenic lines with the overexpression vector pANIC10A by cloning PtLecRLK1 under the control of the maize UBIQUITIN 1 (ZmUbi1) promoter. The 4 X14″ tall one‐tree pots from Stuewe & Sons, Inc. were used for growing switchgrass in the greenhouse with 16 h photoperiod at 24 °C. The ectomycorrhizal fungus L. bicolor isolate S238N was maintained at 25 °C on potato dextrose agar (PDA 24 g/L) plates.
Switchgrass mycorrhization experiments with L. bicolor in soil
Free‐living mycelia of L. bicolor S238N were grown for 7 days on potato dextrose liquid medium. In parallel, plant material was prepared for inoculation. Switchgrass tillers were pre‐rooted in water for ˜2 weeks. Rooted tillers were then transferred to soil dampened with L. bicolor liquid inoculum as inoculate condition, whilst the tillers transferred to soil dampened with liquid medium only as mock condition. For multi‐omic studies, there were three replicates for each of the transgenic lines and WT plants. The plants were grown in mock condition and/or in inoculate condition for 2 months in the greenhouse under a 16/8 h light/dark photoperiod at 24 °C. All replicates of inoculated and mock root samples from each of the transgenic lines and WT were collected. Plants roots were rinsed, 10–20 root tips (˜1 cm) from middle part of root were randomly cut and fixed with 4% formaldehyde for microscopic assay. The rest of whole roots (including root tips) were collected and snap‐frozen in liquid nitrogen, followed by the homogenization by the TissueLyzer (Invitrogen). The fixed root tips were then processed for imaging, and the ground root samples were used for transcriptomic, proteomic, and metabolomic analyses. For phosphorus (P) limitation assay, two to five individual replicates for each WT and transgenic line (5007, 5012 and 5016) were grown on soil with or without L. bicolor under the control condition or P‐limitation condition. The basal nutrient solution described by (Kovar and Claassen, 2009) was used in this study. The basal solution contained 1.1 mm NO3, 0.5 mm Ca, 0.1 mm NH4, 0.2 mmK, 0.1 mmMg, 0.1 mm S, 0.1 mmMg, 46 μm B, 9.1 μm Mn, 0.8 μm Zn, 0.3 μm Cu, 0.5 μm Mo, and 20 μm Fe. Solution pH was adjusted to 6.4. After 2 weeks of adaptation with the basal solution, P treatment in the form of KH2PO4 was added to the basal nutrient solution to deliver treatment solutions containing 1 μm (P‐limitation) or 100 μm (control). Two months later, the individual plant tiller number was counted, and its height was measured by holding the flexible measure tape close to the ground level, along with the curve of the plant and to the tip of the highest leaf. The plants were then cut at the ground level and dried in a 65 °C oven for 48 h and weighed. The plant tiller number, height, biomass weight were analysed with three‐way ANOVA and Student’s t‐tests, and the criteria for significant difference is P ≤ 0.05.
Microscopy
The fixed root tips were embedded with Tissue‐Tek® O.C.T.TM compound and solidified at −80 °C. Forty micrometre transverse sections were cut using a cryo‐section machine (Leica), followed by double staining with 1% (w/v) UVitex 2B (Polyscience) and 10mM propidium iodide (Sigma‐Aldrich), followed by a 2× rinse with deionized water after each staining. The sections were observed with a Zeiss LSM710 confocal microscope with a 405 nm laser line for UVitex 2B excitation and a 514 nm laser line for Propidium iodide.
Switchgrass LecRLK identification and phylogenetic analysis
BLAST analyses were conducted using PtLecRLK1 as a query against the switchgrass genome. The family members were identified with a stringent cut‐off (e‐value <e − 100). The evolutionary tree was inferred using the Maximum Likelihood method based on the JTT matrix‐based model (Jones et al., 1992) with 1000 bootstraps. Evolutionary analyses were conducted in MEGA X (Kumar et al., 2018; Stecher et al., 2020).
RNA extraction, cDNA synthesis, and quantitative PCR
The total RNA was extracted from roots with hexadecyltrimethylammonium bromide (CTAB) digestion and chloroform/isopropanol purification, followed by RQ1 RNase‐Free DNase (Promega) treatment and RNeasy MinElute cleanup kit (Qiagen). The cDNA synthesis was carried out with the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific Inc.) using the oligo(dT)18 primer. Verification of the expression of PtLecRLK1 in transgenic switchgrass plants was performed with the wild‐type plants as control. Specific primer sets are listed in Table S8. PvEF1a (Gimeno et al., 2014) was used as a reference gene for normalization.
RNAseq analysis
RNA samples were sent to BGI for library construction and sequencing. Sequencing was performed using the DNBseq platform. Filtered and trimmed FASTQ files were mapped to a composite genome of L. bicolor (L. bicolor v2.0) and Switchgrass (P. virgatum v5.1) with STAR v2.6.1b (Dobin et al., 2013). Reads that mapped to the L. bicolor genome were discarded prior to generating raw counts with FeatureCounts v1.6.3 in unstranded mode (Liao et al., 2014). The differential gene expression and gene enrichment analyses were performed in R by DESeq2 v1.22.2 (Love et al., 2014) and topGO (Alexa and Rahnenfuhrer, 2016) packages. For pathway visualization, DEGs (P.adjust <0.05 and >log2FC abs(2)) were annotated with Mercator4 v2.0 and visualized with MapMan 3.5.1R2 (Schwacke et al., 2019). Significantly regulated pathways were determined within MapMan via a Wilcoxon rank‐sum test, and Benjamini–Hochberg adjusted P‐values are displayed.
Construction of co‐expression networks
The gene co‐expression networks related to (i) inoculation, (ii) introduction of PtLecRLK1, or (iii) the interaction of these two factors in switchgrass were constructed with the WGCNA package (Langfelder and Horvath, 2008) within the R environment. The default WGCNA ‘automatic, one‐step network construction’ analysis was used to build clusters of genes displaying similar correlated patterns of transcription (i.e. modules). The parameters used were soft‐power of seven and minimum module size of 30 genes. The adjacency between genes was calculated and a hierarchical clustering tree with the dissimilarity of the topological overlap matrix was constructed. Similar modules were merged by calculating the module eigengenes, clustering them, and assigning a distance threshold. The module eigengene was calculated in each module and then correlated with the treatment (α = 0.05) to identify modules that were significantly associated with L. bicolor inoculation, introduction of PtLecRLK1 or interaction of these two factors in switchgrass. In the weighted gene co‐expression network, gene connectivity was based on the edge weight (ranging from 0 to 1) determined by the topology overlap measure, which reflects the strength of the communication between the two genes. The edge weights above 0.8 were presented in the network and most central and connected genes were considered to be hub genes. Co‐expression patterns and interactions of hub genes were exported to and visualized by Cytoscape (Shannon et al., 2003).
Proteomics sample preparation
Ground samples were solubilized in a lysis buffer and further disrupted by sonication and boiling. After alkylating the samples, proteins were then extracted using methanol/chloroform/water precipitation. Proteins were digested with two separate and sequential aliquots of sequencing‐grade trypsin, then lyophilized/dried in a SpeedVac Concentrator (Thermo Fisher Scientific). Peptide samples were desalted on a desalting spin column, and then resuspended in 0.1% formic acid solution. 15 μg peptide from each sample was allocated for proteomics measurement.
LC‐MS/MS analysis
All samples were analysed on a Q Exactive Plus mass spectrometer (Thermo Scientific) coupled with an RSLC nano UHPLC system (Thermo Scientific). Peptides were separated on a biphasic precolumn (SCX‐RP) coupled to an in‐house‐pulled nanospray emitter of 75 μm inner diameter packed with 30 cm of 1.7 μm of Kinetex C18 resin (Phenomenex). MS data were acquired with the Thermo Xcalibur software using the top 10 data‐dependent acquisition. Other mass spectrometer parameters were set as previously described (Villalobos Solis et al., 2020).
Proteome data analysis and metabolism pathway visualization
All MS/MS spectra collected were processed in Proteome Discoverer v2.4 (Thermo Scientific) using MS Amanda 2.0 (Dorfer et al., 2014) and Percolator (Käll et al., 2007). The following parameters were set up in MS Amanda to derive fully tryptic peptides: MS1 tolerance = 10 ppm; MS2 tolerance = 0.02 Da; missed cleavages = 2; Carbamidomethyl (C, +57.021 Da) as static modification; and oxidation (M, +15.995 Da) as dynamic modifications. The percolator FDR threshold was set to 1% at the PSM and peptide levels. FDR‐controlled peptides were then quantified according to the chromatographic area‐under‐the‐curve and mapped to their respective proteins. Areas were summed to estimate protein‐level abundance. For differential abundance analysis of proteins, the proteins with at least 2 peptide evidence were exported from Proteome Discoverer. Protein abundance values were log2 transformed, LOESS normalized between the biological replicates and mean‐centred across all the conditions using InfernoRDN software (Polpitiya et al., 2008). The differentially abundant proteins were identified by Welch’s T‐test method followed by false discovery rate (FDR) correction using Benjimini Hochberg. Proteins were further filtered using the absolute log2 fold change of 1 (i.e. −2<fold Change>+2). The isoforms for the same protein with the same FDR and fold changes were filtered out. The differential abundance proteins were then plotted in Metabolism overview and biotic stress pathways using MapMan 3.5.1R2 (Schwacke et al., 2019).
Gas chromatography‐mass spectrometry metabolite profiling
Metabolites were extracted from ˜200 mg of fresh‐frozen powdered tissue twice overnight with 2.5 mL 80% ethanol. Sorbitol (75 µL of 1 mg/mL aqueous solution) was added to the first extract as an internal standard to correct for differences in extraction efficiency, subsequent differences in derivatization efficiency and volume changes. The extracts were combined, and a 1 mL aliquot was dried under nitrogen for analysis. Metabolites were silylated to produce trimethylsilyl derivatives by addition of 500 µL silylation‐grade acetonitrile to the dried extract followed by addition of 500 µL of N‐methyl‐N‐trimethylsilyltrifluoroacetamide (MSTFA) with 1% trimethylchlorosilane (TMCS). Samples were heated for 1 h at 70°C and after 2 days, a 1 µL aliquot was injected into an Agilent Technologies 7890A gas chromatograph (GC) coupled to a 5975C inert XL mass spectrometer (MS) configured, as previously described (Abraham et al., 2016). The MS was operated in electron impact (70 eV) ionization mode with a scan range of 50–650 Da. Metabolite peaks were extracted using a key selected ion, characteristic m/z fragment rather than the total ion chromatogram to minimize interference of co‐eluting metabolites, and quantified as previously described (Abraham et al., 2016). The differential abundance metabolites and their enrichment analyses were performed in MetaboAnalyst 4.0 (Chong et al., 2018). Significantly enriched pathways were determined via a Holm–Bonferroni method, and adjusted P‐values and FDR are displayed. The differential abundance metabolites were also visualized with MapMan 3.5.1R2 (Schwacke et al., 2019).
Accession number
Transcriptomic data have been registered with the BioProject database (http://www.ncbi.nlm.nih.gov/bioproject/717062) with the identifier PRJNA717062. Proteomics spectral data have been deposited as a ProteomeXchange dataset (PXD024841) via the MASSIVE repository (https://massive.ucsd.edu/). The data can be reviewed under the username ‘MSV000087067_reviewer’ and password ‘PMI’. Metabolomics data have been deposited to the EMBL‐EBI MetaboLights database (https://doi.org/10.1093/nar/gkz1019, PMID:31691833) with the identifier MTBLS2597. The complete dataset can be accessed via weblink https://www.ebi.ac.uk/metabolights/MTBLS2597.
Conflict of Interest
The authors declare that there is no conflict of interest.
Author contributions
Z.Q., W.M., and J.‐G.C. designed this study. Z.Q. performed experiments, conducted data analysis and wrote the manuscript. T.B.Y., and Y.S. analysed the RNAseq data. T.B.Y., H.K.S., P.E.A., and R.L.H. generated and analysed proteomic data. N.L.E. and T.J.T. generated and analysed metabolomics data. A.F. and Z.‐Y.W. generated switchgrass transgenic lines. J.L. and J.L.M‐F. contributed to imaging analysis. G.A.T., W.M. and J.‐G.C. conceived the study, coordinated research and contributed to experimental design and data interpretation. All authors read and approved the final manuscript.
Supporting information
Figure S1 Verification of ZmUbipro‐PtLecRLK1 expression in the switchgrass transgenic plants.
Figure S2 Phenotypic parameters of switchgrass in different growth conditions.
Figure S3 Gene ontology (GO) enrichment analysis of differentially expressed genes.
Figure S4 Genes related to nutrient assimilation.
Figure S5 PtLecRLK1 sequence homology in switchgrass.
Figure S6 Scatterplots of Gene Significance (GS) for weight vs. Module Membership (MM) in the turquoise, green, yellow, and black modules.
Figure S7 A co‐expression network in the inoculated‐ transgenic module.
Table S1 ANOVA analysis on the plant growth parameters.
Table S2 Switchgrass transcriptome analysis results.
Table S3 Biotic stress‐related DEGs.
Table S4 Nutrient uptake related DEGs.
Table S5 Switchgrass proteome analysis results.
Table S6 DEG and differential abundance proteins overlap.
Table S7 Pathway Enrichment analysis for metabolites.xlsx.
Table S8 Primers used for RT‐PCR.
Acknowledgements
We acknowledge the Noble Research Institute, LLC’s Transformation Core Facility for generating the switchgrass transgenic lines. We thank Mindy M. Clark for growing and maintaining switchgrass plants in ORNL greenhouses. This work was supported by the Plant‐Microbe Interfaces Scientific Focus Area, the Center for Bioenergy Innovation, and the BioEnergy Science Center by the Office of Biological and Environmental Research in the U.S. Department of Energy Office of Science. Oak Ridge National Laboratory is managed by UT‐Battelle, LLC, for the United States Department of Energy under contract DE‐AC05‐00OR22725.
Qiao, Z. , Yates, T. B. , Shrestha, H. K. , Engle, N. L. , Flanagan, A. , Morrell‐Falvey, J. L. , Sun, Y. , Tschaplinski, T. J. , Abraham, P. E. , Labbé, J. , Wang, Z.‐Y. , Hettich, R. L. , Tuskan, G. A. , Muchero, W. and Chen, J.‐G. (2021) Towards engineering ectomycorrhization into switchgrass bioenergy crops via a lectin receptor‐like kinase. Plant Biotechnol. J., 10.1111/pbi.13671
Notice: This manuscript has been authored by UT‐Battelle LLC under Contract No. DE‐AC05‐00OR22725 with the U.S. Department of Energy. The United States Government retains and the publisher by accepting the article for publication, acknowledges that the United States Government retains a non‐exclusive, paid‐up, irrevocable, world‐wide licence to publish or reproduce the published form of this manuscript, or allow others to do so for United States Government purposes. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe‐public‐access‐plan).
Contributor Information
Wellington Muchero, Email: mucherow@ornl.gov.
Jin‐Gui Chen, Email: chenj@ornl.gov.
References
- Abraham, P.E. , Yin, H. , Borland, A.M. , Weighill, D. , Lim, S.D. , De Paoli, H.C. , Engle, N. et al. (2016) Transcript, protein and metabolite temporal dynamics in the CAM plant Agave. Nat. Plants, 2(12), 1–10. [DOI] [PubMed] [Google Scholar]
- Alcázar, R. , García, A.V. , Kronholm, I. , de Meaux, J. , Koornneef, M. , Parker, J.E. and Reymond, M. (2010) Natural variation at Strubbelig receptor kinase 3 drives immune‐triggered incompatibilities between Arabidopsis thaliana accessions. Nat. Genet. 42(12), 1135. [DOI] [PubMed] [Google Scholar]
- Alexa, A. and Rahnenfuhrer, J. (2016) topGO: Enrichment analysis for gene ontology. R package version 2.28. 0. BioConductor Published online.
- Blasius, D. , Feil, W. , Kottke, I. and Oberwinkler, F. (1986) Hartig net structure and formation in fully ensheathed ectomycorrhizas. Nord. J. Bot. 6(6), 837–842. [Google Scholar]
- Bonfante, P. and Genre, A. (2010) Mechanisms underlying beneficial plant–fungus interactions in mycorrhizal symbiosis. Nat. Commun. 1(1), 1–11. [DOI] [PubMed] [Google Scholar]
- Bouwmeester, K. , De Sain, M. , Weide, R. , Gouget, A. , Klamer, S. , Canut, H. and Govers, F. (2011) The lectin receptor kinase LecRK‐I. 9 is a novel Phytophthora resistance component and a potential host target for a RXLR effector. PLoS Pathog. 7(3), e1001327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwmeester, K. and Govers, F. (2009) Arabidopsis L‐type lectin receptor kinases: phylogeny, classification, and expression profiles. J. Exp. Bot. 60(15), 4383–4396. [DOI] [PubMed] [Google Scholar]
- Brauer, E.K. , Ahsan, N. , Dale, R. , Kato, N. , Coluccio, A.E. , Piñeros, M.A. , Kochian, L.V. et al. (2016) The Raf‐like kinase ILK1 and the high affinity K+ transporter HAK5 are required for innate immunity and abiotic stress response. Plant Physiol. 171(2), 1470–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong, J. , Soufan, O. , Li, C. , Caraus, I. , Li, S. , Bourque, G. , Wishart, D.S. et al. (2018) MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 46(W1):W486–W494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dearnaley, J.D. and Cameron, D.D. (2016) Nitrogen transport in the orchid mycorrhizal symbiosis‐further evidence for a mutualistic association. New Phytol. 213(1), 10–12. [DOI] [PubMed] [Google Scholar]
- Desrivieres, S. , Cooke, F.T. , Parker, P.J. and Hall, M.N. (1998) MSS4, a phosphatidylinositol‐4‐phosphate 5‐kinase required for organization of the actin cytoskeleton in Saccharomyces cerevisiae . J. Biol. Chem. 273(25), 15787–15793. [DOI] [PubMed] [Google Scholar]
- Dobin, A. , Davis, C.A. , Schlesinger, F. , Drenkow, J. , Zaleski, C. , Jha, S. , Batut, P. et al. (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics, 29(1), 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorfer, V. , Pichler, P. , Stranzl, T. , Stadlmann, J. , Taus, T. , Winkler, S. and Mechtler, K. (2014) MS Amanda, a universal identification algorithm optimized for high accuracy tandem mass spectra. J. Proteome. Res. 13(8), 3679–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felten, J. , Kohler, A. , Morin, E. , Bhalerao, R.P. , Palme, K. , Martin, F. , Ditengou, F.A. et al. (2009) The ectomycorrhizal fungus Laccaria bicolor stimulates lateral root formation in poplar and Arabidopsis through auxin transport and signaling. Plant. Physiol. 151(4), 1991–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghimire, S.R. , Charlton, N.D. and Craven, K.D. (2009) The mycorrhizal fungus, Sebacina vermifera, enhances seed germination and biomass production in switchgrass (Panicum virgatum L). Bioenergy Res. 2(1–2), 51–58. [Google Scholar]
- Ghimire, S.R. and Craven, K.D. (2011) Enhancement of switchgrass (Panicum virgatum L.) biomass production under drought conditions by the ectomycorrhizal fungus Sebacina vermifera . Appl. Environ. Microbiol. 77(19):7063–7067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilardoni, P.A. , Hettenhausen, C. , Baldwin, I.T. and Bonaventure, G. (2011) Nicotiana attenuata lectin receptor kinase1 suppresses the insect‐mediated inhibition of induced defense responses during Manduca sexta herbivory. Plant Cell, 23(9), 3512–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill, U.S. , Sun, L. , Rustgi, S. , Tang, Y. , von Wettstein, D. and Mysore, K.S. (2018) Transcriptome‐based analyses of phosphite‐mediated suppression of rust pathogens Puccinia emaculata and Phakopsora pachyrhizi and functional characterization of selected fungal target genes. Plant J. 93(5), 894–904. [DOI] [PubMed] [Google Scholar]
- Gimeno, J. , Eattock, N. , Van Deynze, A. and Blumwald, E. (2014) Selection and validation of reference genes for gene expression analysis in switchgrass (Panicum virgatum) using quantitative real‐time RT‐PCR. PLoS One, 9(3), e91474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover, J.D. , Reganold, J.P. , Bell, L.W. , Borevitz, J. , Brummer, E.C. , Buckler, E.S. , Cox, C.M. et al. (2010) Increased food and ecosystem security via perennial grains. Science, 328(5986), 1638–1639. [DOI] [PubMed] [Google Scholar]
- Greenbaum, D. , Jansen, R. and Gerstein, M. (2002) Analysis of mRNA expression and protein abundance data: an approach for the comparison of the enrichment of features in the cellular population of proteins and transcripts. Bioinformatics, 18(4), 585–596. [DOI] [PubMed] [Google Scholar]
- Herve, C. , Dabos, P. , Galaud, J.‐P. , Rougé, P. and Lescure, B. (1996) Characterization of an Arabidopsis thaliana gene that defines a new class of putative plant receptor kinases with an extracellular lectin‐like domain. J. Mol. Biol. 258(5), 778–788. [DOI] [PubMed] [Google Scholar]
- Horan, D. , Chilvers, G. and Lapeyrie, F. (1988) Time sequence of the infection process eucalypt ectomycorrhizas. New Phytol. 109(4), 451–458. [Google Scholar]
- Hu, B. , Jiang, Z. , Wang, W. , Qiu, Y. , Zhang, Z. , Liu, Y. , Li, A. et al. (2019) Nitrate‐NRT1. 1B‐SPX4 cascade integrates nitrogen and phosphorus signalling networks in plants. Nature Plants 5(4), 401. [DOI] [PubMed] [Google Scholar]
- Jiang, Y. , Wang, W. , Xie, Q. , Liu, N. , Liu, L. , Wang, D. , Zhang, X. et al. (2017) Plants transfer lipids to sustain colonization by mutualistic mycorrhizal and parasitic fungi. Science, 356(6343), 1172–1175. 10.1126/science.aam9970 [DOI] [PubMed] [Google Scholar]
- Jones, D.T. , Taylor, W.R. and Thornton, J.M. (1992) The rapid generation of mutation data matrices from protein sequences. Bioinformatics, 8(3), 275–282. [DOI] [PubMed] [Google Scholar]
- Käll, L. , Canterbury, J.D. , Weston, J. , Noble, W.S. and MacCoss, M.J. (2007) Semi‐supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods, 4(11), 923–925. [DOI] [PubMed] [Google Scholar]
- Kanzaki, H. , Saitoh, H. , Takahashi, Y. , Berberich, T. , Ito, A. , Kamoun, S. and Terauchi, R. (2008) NbLRK1, a lectin‐like receptor kinase protein of Nicotiana benthamiana, interacts with Phytophthora infestans INF1 elicitin and mediates INF1‐induced cell death. Planta, 228(6), 977–987. [DOI] [PubMed] [Google Scholar]
- Kovar, J.L. and Claassen, N. (2009) Growth and phosphorus uptake of three riparian grass species. Agron. J. 101(5), 1060–1067. [Google Scholar]
- Krouk, G. , Lacombe, B. , Bielach, A. , Perrine‐Walker, F. , Malinska, K. , Mounier, E. , Hoyerova, K. et al. (2010) Nitrate‐regulated auxin transport by NRT1. 1 defines a mechanism for nutrient sensing in plants. Dev. Cell. 18(6), 927–937. [DOI] [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. , Li, M. , Knyaz, C. and Tamura, K. (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35(6), 1547–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbé, J. , Muchero, W. , Czarnecki, O. , Wang, J. , Wang, X. , Bryan, A.C. , Zheng, K. et al. (2019) Mediation of plant–mycorrhizal interaction by a lectin receptor‐like kinase. Nat. Plants, 5(7), 676–680. [DOI] [PubMed] [Google Scholar]
- Langfelder, P. and Horvath, S. (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinformat. 9(1), 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen, P.E. , Sreedasyam, A. , Trivedi, G. , Podila, G.K. , Cseke, L.J. and Collart, F.R. (2011) Using next generation transcriptome sequencing to predict an ectomycorrhizal metabolome. BMC Syst. Biol. 5(1), 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, Y. , Smyth, G.K. and Shi, W. (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics, 30(7), 923–930. [DOI] [PubMed] [Google Scholar]
- Liu, K.‐H. , Diener, A. , Lin, Z. , Liu, C. and Sheen, J. (2020) Primary nitrate responses mediated by calcium signalling and diverse protein phosphorylation. J. Exp. Bot. 71(15), 4428–4441. 10.1093/jxb/eraa047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love, M.I. , Huber, W. and Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 15(12), 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luginbuehl, L.H. , Menard, G.N. , Kurup, S. , Van Erp, H. , Radhakrishnan, G.V. , Breakspear, A. , Oldroyd, G.E.D. et al. (2017) Fatty acids in arbuscular mycorrhizal fungi are synthesized by the host plant. Science, 356(6343), 1175–1178. 10.1126/science.aan0081. [DOI] [PubMed] [Google Scholar]
- Luo, Z.‐B. , Janz, D. , Jiang, X. , Goebel, C. , Wildhagen, H. , Tan, Y. , Rennenberg, H. et al. (2009) Upgrading root physiology for stress tolerance by ectomycorrhizas: insights from metabolite and transcriptional profiling into reprogramming for stress anticipation. Plant Physiol. 151(4), 1902–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, Z. , Wood, C. and Bransby, D. (2000) Soil management impacts on soil carbon sequestration by switchgrass. Biomass Bioenergy, 18(6), 469–477. [Google Scholar]
- Martin, F. , Aerts, A. , Ahrén, D. , Brun, A. , Danchin, E.G.J. , Duchaussoy, F. , Gibon, J. et al. (2008) The genome of Laccaria bicolor provides insights into mycorrhizal symbiosis. Nature, 452(7183), 88–92. 10.1038/nature06556. [DOI] [PubMed] [Google Scholar]
- Mazzei, P. , Vinale, F. , Woo, S.L. , Pascale, A. , Lorito, M. and Piccolo, A. (2016) Metabolomics by proton high‐resolution magic‐angle‐spinning nuclear magnetic resonance of tomato plants treated with two secondary metabolites isolated from trichoderma. J. Agricult. Food Chem. 64(18), 3538–3545. [DOI] [PubMed] [Google Scholar]
- Nie, L. , Wu, G. , Culley, D.E. , Scholten, J.C. and Zhang, W. (2007) Integrative analysis of transcriptomic and proteomic data: challenges, solutions and applications. Crit. Rev. Biotechnol. 27(2), 63–75. [DOI] [PubMed] [Google Scholar]
- Nishiguchi, M. , Yoshida, K. , Sumizono, T. and Tazaki, K. (2002) A receptor‐like protein kinase with a lectin‐like domain from lombardy poplar: gene expression in response to wounding and characterization of phosphorylation activity. Mol. Genet. Genomics, 267(4), 506–514. [DOI] [PubMed] [Google Scholar]
- Park, J. , Kim, T.‐H. , Takahashi, Y. , Schwab, R. , Dressano, K. , Stephan, A.B. , Ceciliato, P.H.O. et al. (2019) Chemical genetic identification of a lectin receptor kinase that transduces immune responses and interferes with abscisic acid signaling. Plant J. 98(3), 492–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrin, C. , Daguerre, Y. , Ruytinx, J. , Guinet, F. , Kemppainen, M. , Frey, N.F.D. , Puech‐Pagès, V. et al. (2019) Laccaria bicolor MiSSP8 is a small‐secreted protein decisive for the establishment of the ectomycorrhizal symbiosis. Environ. Microbiol. 21(10), 3765–3779. [DOI] [PubMed] [Google Scholar]
- Plett, J.M. , Kemppainen, M. , Kale, S.D. , Kohler, A. , Legué, V. , Brun, A. , Tyler, B.M. et al. (2011) A secreted effector protein of Laccaria bicolor is required for symbiosis development. Curr. Biol. 21(14), 1197–1203. [DOI] [PubMed] [Google Scholar]
- Plett, J.M. , Khachane, A. , Ouassou, M. , Sundberg, B. , Kohler, A. and Martin, F. (2014) Ethylene and jasmonic acid act as negative modulators during mutualistic symbiosis between Laccaria bicolor and Populus roots. New Phytol. 202(1), 270–286. [DOI] [PubMed] [Google Scholar]
- Plett, J.M. , Yin, H. , Mewalal, R. , Hu, R. , Li, T. , Ranjan, P. , Jawdy, S. et al. (2017) Populus trichocarpa encodes small, effector‐like secreted proteins that are highly induced during mutualistic symbiosis. Sci. Rep. 7(1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polpitiya, A.D. , Qian, W.‐J. , Jaitly, N. , Petyuk, V.A. , Adkins, J.N. , Camp, D.G. , Anderson, G.A. et al. (2008) DAnTE: a statistical tool for quantitative analysis of‐omics data. Bioinformatics, 24(13), 1556–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popescu, S.C. , Brauer, E.K. , Dimlioglu, G. and Popescu, G.V. (2017) Insights into the structure, function, and ion‐mediated signaling pathways transduced by plant integrin‐linked kinases. Front. Plant Sci. 8, 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall, G. , Huggins, D. , Russelle, M. , Fuchs, D. , Nelson, W. and Anderson, J. (1997) Nitrate losses through subsurface tile drainage in conservation reserve program, alfalfa, and row crop systems. J. Environ. Qual. 26(5), 1240–1247. [Google Scholar]
- Ray, P. , Guo, Y. , Chi, M.H. , Krom, N. , Saha, M.C. and Craven, K.D. (2020) Serendipita bescii promotes winter wheat growth and modulates the host root transcriptome under phosphorus and nitrogen starvation. Environ. Microbiol. 23(4), 1876–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riou, C. , Hervé, C. , Pacquit, V. , Dabos, P. and Lescure, B. (2002) Expression of an Arabidopsis lectin kinase receptor gene, lecRK‐a1, is induced during senescence, wounding and in response to oligogalacturonic acids. Plant Physiol. Biochem. 40(5), 431–438. [Google Scholar]
- Schroeder‐Moreno, M.S. , Greaver, T.L. , Wang, S. , Hu, S. and Rufty, T.W. (2012) Mycorrhizal‐mediated nitrogen acquisition in switchgrass under elevated temperatures and N enrichment. GCB Bioenergy, 4(3), 266–276. [Google Scholar]
- Schwacke, R. , Ponce‐Soto, G.Y. , Krause, K. , Bolger, A.M. , Arsova, B. , Hallab, A. , Gruden, K. et al. (2019) MapMan4: a refined protein classification and annotation framework applicable to multi‐omics data analysis. Mol. Plant, 12(6), 879–892. [DOI] [PubMed] [Google Scholar]
- Shannon, P. , Markiel, A. , Ozier, O. , Baliga, N.S. , Wang, J.T. , Ramage, D. , Amin, N. et al. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13(11), 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinde, S. , Naik, D. and Cumming, J.R. (2018) Carbon allocation and partitioning in Populus tremuloides are modulated by ectomycorrhizal fungi under phosphorus limitation. Tree Physiol. 38(1), 52–65. [DOI] [PubMed] [Google Scholar]
- Singh, P. , Kuo, Y.‐C. , Mishra, S. , Tsai, C.‐H. , Chien, C.‐C. , Chen, C.‐W. , Desclos‐Theveniau, M. et al. (2012) The lectin receptor kinase‐VI. 2 is required for priming and positively regulates Arabidopsis pattern‐triggered immunity. Plant Cell 24(3), 1256–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, P. and Zimmerli, L.Z. (2013) Lectin receptor kinases in plant innate immunity. Front. Plant. Sci. 4, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, S.E. and Read, D.J. (2010) Mycorrhizal Symbiosis. New York: Academic Press. [Google Scholar]
- Sousa, E. , Kost, B. and Malhó, R. (2008) Arabidopsis phosphatidylinositol‐4‐monophosphate 5‐kinase 4 regulates pollen tube growth and polarity by modulating membrane recycling. Plant Cell, 20(11), 3050–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stecher, G. , Tamura, K. and Kumar, S. (2020) Molecular evolutionary genetics analysis (MEGA) for macOS. Mol. Biol. Evol. 37(4), 1237–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, H. , Xie, Y. , Zheng, Y. , Lin, Y. and Yang, F. (2018) The enhancement by arbuscular mycorrhizal fungi of the Cd remediation ability and bioenergy quality‐related factors of five switchgrass cultivars in Cd‐contaminated soil. PeerJ, 6, e4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschaplinski, T.J. , Plett, J.M. , Engle, N.L. , Deveau, A. , Cushman, K.C. , Martin, M.Z. , Doktycz, M.J. et al. (2014) Populus trichocarpa and Populus deltoides exhibit different metabolomic responses to colonization by the symbiotic fungus Laccaria bicolor . Mol. Plant Microbe. Interact. 27(6), 546–556. [DOI] [PubMed] [Google Scholar]
- Villalobos Solis, M.I. , Poudel, S. , Bonnot, C. , Shrestha, H.K. , Hettich, R.L. , Veneault‐Fourrey, C. , Martin, F. et al. (2020) Integrating state‐of‐the‐art tandem mass spectrometry with de novo‐assisted database peptide sequencing as a viable strategy for the discovery of peptide proteolytic cleavage products (PCPs) in plant‐microbe interactions. Mol. Plant‐Microbe Interact, 33(10), 1177–1188. [DOI] [PubMed] [Google Scholar]
- Vukicevich, E. , Lowery, T. , Bowen, P. , Úrbez‐Torres, J.R. and Hart, M. (2016) Cover crops to increase soil microbial diversity and mitigate decline in perennial agriculture. A review. Agron. Sustain. Dev. 36(3), 48. [Google Scholar]
- Wang, Y. and Bouwmeester, K. (2017) L‐type lectin receptor kinases: new forces in plant immunity. PLoS Pathog. 13(8), e1006433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, C. and Dedhar, S. (2001) Integrin‐linked kinase (ILK) and its interactors: a new paradigm for the coupling of extracellular matrix to actin cytoskeleton and signaling complexes. J. Cell. Biol. 155(4), 505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi, Y. , Fu, C. , Ge, Y. , Nandakumar, R. , Hisano, H. , Bouton, J. and Wang, Z.‐Y. (2009) Agrobacterium‐mediated transformation of switchgrass and inheritance of the transgenes. Bioenergy Res. 2(4), 275–283. [Google Scholar]
- Yongfeng, W. , Aiquan, Z. , Fengli, S. , Mao, L. , Kaijie, X. , Chao, Z. , Shudong, L. et al. (2018) Using transcriptome analysis to identify genes involved in switchgrass flower reversion. Front. Plant Sci. 9, 1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Žebeljan, A. , Vico, I. , Duduk, N. , Žiberna, B. and Krajnc, A.U. (2019) Dynamic changes in common metabolites and antioxidants during Penicillium expansum‐apple fruit interactions. Physiol. Mol. Plant Pathol. 106, 166–174. [Google Scholar]
- Zhang, W. , Culley, D.E. , Scholten, J.C. , Hogan, M. , Vitiritti, L. and Brockman, F.J. (2006) Global transcriptomic analysis of Desulfovibrio vulgaris on different electron donors. Anton. Leeuw. 89(2), 221–237. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Verification of ZmUbipro‐PtLecRLK1 expression in the switchgrass transgenic plants.
Figure S2 Phenotypic parameters of switchgrass in different growth conditions.
Figure S3 Gene ontology (GO) enrichment analysis of differentially expressed genes.
Figure S4 Genes related to nutrient assimilation.
Figure S5 PtLecRLK1 sequence homology in switchgrass.
Figure S6 Scatterplots of Gene Significance (GS) for weight vs. Module Membership (MM) in the turquoise, green, yellow, and black modules.
Figure S7 A co‐expression network in the inoculated‐ transgenic module.
Table S1 ANOVA analysis on the plant growth parameters.
Table S2 Switchgrass transcriptome analysis results.
Table S3 Biotic stress‐related DEGs.
Table S4 Nutrient uptake related DEGs.
Table S5 Switchgrass proteome analysis results.
Table S6 DEG and differential abundance proteins overlap.
Table S7 Pathway Enrichment analysis for metabolites.xlsx.
Table S8 Primers used for RT‐PCR.