

Abstract
Next-generation sequencing (NGS) has recently been introduced to efficiently and simultaneously detect genetic variations in acute myeloid leukemia (AML). However, its implementation in the clinical routine raises new challenges focused on the diversity of assays and variant reporting criteria. In order to overcome this challenge, the PETHEMA group established a nationwide network of reference laboratories aimed to deliver molecular results in the clinics. We report the technical cross-validation results for NGS panel genes during the standardization process and the clinical validation in 823 samples of 751 patients with newly diagnosed or refractory/relapse AML. Two cross-validation rounds were performed in seven nationwide reference laboratories in order to reach a consensus regarding quality metrics criteria and variant reporting. In the pre-standardization cross-validation round, an overall concordance of 60.98% was obtained with a great variability in selected genes and conditions across laboratories. After consensus of relevant genes and optimization of quality parameters the overall concordance rose to 85.57% in the second cross-validation round. We show that a diagnostic network with harmonized NGS analysis and reporting in seven experienced laboratories is feasible in the context of a scientific group. This cooperative nationwide strategy provides advanced molecular diagnostic for AML patients of the PETHEMA group (clinicaltrials gov. Identifier: NCT03311815).
Introduction
Acute myeloid leukemia (AML) is a heterogeneous disease characterized by a wide spectrum of molecular alterations that lead malignant transformation of normal hematopoietic cells.1 The relevance of chromosomal alterations and gene variants for diagnosis, risk stratification and choice of targeted therapies (i.e, FLT3 and IDH1/2 inhibitors) has remarkably increased the complexity of routine molecular diagnostic strategies.2–5 Next-generation sequencing (NGS) has been established as a new molecular diagnostic tool rapidly adopted by clinical laboratories, being able to simultaneously assess different genetic alterations such as rearrangements, single nucleotide variants (SNV), insertions-deletions (indels) and copy number variations (CNV) in a wide variety of genes.6 NGS gene panels have been preferentially adopted rather than whole genome or exome sequencing due to an easier results interpretation, lower cost and less time consumption, as well as higher read deep needed for low frequency variant detection. Compared to NGS, conventional single-gene approaches by polymerase chain reaction (PCR)7 are laborious, time-consuming and less efficient to detect minor clones, but they are still needed as rapid-screening tests for druggable variants.8
The new scenario for AML molecular diagnosis, requiring rapid screening by conventional PCR and comprehensive characterization by NGS, is a great challenge for molecular biology laboratories. For this purpose, the PETHEMA (Programa Español de Tratamientos en Hematología) group established a nationwide network involving seven central laboratories aimed to deliver molecular results to clinics for newly diagnosed and relapsed/refractory AML patients. The first step was to ensure appropriate logistic support, including geographical localization of highly skilled central laboratories strategically distributed according to population density and distance. The second step was to harmonize NGS and PCR techniques methodology and result reporting across the seven central laboratories, establishing consensus panel genes, quality metrics cutoffs and variant reporting criteria.
In this work, we performed the first analysis of a NGSAML study (clinicaltrials gov. Identifier: NCT03311815), reporting the technical cross-validation results for NGS panel genes during the standardization process and the clinical validation in 823 samples of 751 patients with newly diagnosed or refractory/relapse AML.
Methods
Study design and reference laboratories
This was a prospective, multi-center, non-interventional study, performed in seven Spanish PETHEMA central laboratories: Hospital Universitario La Fe (HULF, Valencia), Hospital Universitario de Salamanca (HUS, Salamanca), Hospital Universitario 12 de Octubre (H12O, Madrid), Hospital Universitario Virgen del Rocío (HUVR, Sevilla), Hospital Universitario Reina Sofía (HURS, Córdoba), Hospital Universitario de Gran Canaria Dr. Negrín (HUDN, Las Palmas de Gran Canaria) and CIMA LAB Diagnostics (UNAV, Pamplona) (see the Online Supplementary Appendix for further details).
Inclusion criteria
All adult patients (≥18 years) with newly diagnosed or relapsed/refractory AML (excluding acute promyelocytic leukemia) according to the World Health Organization criteria (2008), regardless of the treatment received, were eligible for the NGS-AML study. The Institutional Ethics Committee for Clinical Research of each institution approved this study. Written informed consent in accordance with the recommendations of the Declaration of Human Rights, the Conference of Helsinki, and institutional regulations were obtained from all patients.
Cross-validation
The first cross-validation round was developed to evaluate the starting situation of reference laboratories (see the Online Supplementary Appendix). For this purpose, four samples harboring 24 variants were distributed from HULF (coordinator center) and each laboratory carried out NGS analysis according to their already implemented protocols. Reports were sent to the coordinator center to analyze the results.
Taking into account the obtained results, the collaborative group established a set of relevant AML genes and minimum quality metrics criteria. Then, a second cross-validation round was designed to strengthen the established quality parameters, the consensus recommendations, and variant reporting for NGS analysis among the seven reference laboratories. Variant detection, variant allele frequency (VAF), dispersion among centers and variant reporting (clinically and non-clinically relevant variants) were assessed in six samples with 30 variants (five with a lower VAF than 5%). Reports were sent to the coordinator center to analyze the results.
Consensus genes establishment
Thrirty genes were established as key genes for AML pathogenesis: ABL1, ASXL1, BRAF, CALR, CBL, CEBPA, CSF3R, DNMT3A, ETV6, EZH2, FLT3, GATA2, HRAS, IDH1, IDH2, JAK2, KIT, KRAS, MPL, NPM1, NRAS, PTPN11, RUNX1, SETBP1, SF3B1, SRSF2, TET2, TP53, U2AF1 and WT1. ASXL1, CEBPA, FLT3, IDH1, IDH2, NPM1, RUNX1, and TP53 were mandatory for their implication in clinical guidelines, targeted therapy and risk stratification. The remaining genes were recommended for NGS panels, according to laboratory features and sequencing panel options.
Sequencing platforms and panels
The sequencing platform and panel were selected by each laboratory using the following criteria: i) to include all eight mandatory genes and, ii) to include the maximum number of other 22 relevant genes (sequencing platforms and panels data are shown in the Online Supplementary Appendix and Online Supplementary Table S1).
Clinical validation
NGS was performed according to already implemented protocols and the consensus parameters established in both cross-validation rounds. Samples meeting the quality metrics criteria established in previous standardization rounds were considered in the clinical validation.
Statistical analyses
All statistics were performed using SPSS version 22 (IBM, Armonk, NY, USA) and GraphPad Prism 4 (GraphPad, La Jolla, CA, USA) software programs. A P-value (P) <0.05 was considered statistically significant (see the Online Supplementary Appendix).
Results
Cross-validation
In the first cross-validation round (pre-standardization), we observed a great diversity in the included genes in each NGS panel. Some AML relevant genes such as NPM1 and CEBPA were not studied, while other non-relevant genes for AML pathogenesis were included. The global error rate was 39.02% with a higher error rate in variants showing a VAF lower than 10% (77.04 ± 6.98% vs. 18.56 ± 29.24%, P<0.001) (Table 1).
In the second round (post-standardization), the mean read depth was 4,032 (range, 1,463-4,532) with a median uniformity of 98.34%. The error rate for all variants was reduced to 14.43% (Table 2). In this round, the error rate was significantly higher in variants with a VAF lower than 5% (28.57 ± 14.28% vs. 12.27 ± 14.39%, P=0.031) (Figure 1). All centers provided a correct clinical classification of the detected variants. No differences in the error rate were observed between indels and SNV. Regarding the accuracy of VAF determination among the different centers, VAF standard deviation (SD) was higher in indel variants than SNV (5.44 ± 2.80 vs. 2.15 ± 2.03, P=0.001).
After cross-validation results, uniformity (>85%) and mean read depth of 1,000X were established as quality control parameters for a valid NGS assay. Synonymous, intronic and polymorphic variants (minor allele frequency [MAF] ≥1% and/or included in the dbSNP database) were filtered out. VAF ≥5% was established as a cutoff value for variant reporting with the exception of pathogenic variants with strong clinical evidence which were reported with a VAF ≥1% (e.g, TP53 or FLT3). Variants accomplishing all these requirements were considered.
Platform performance
The performance of the NGS platforms (Ion Torrent vs. Illumina) regarding the error rate and the VAF SD was assessed after the results of the standardization rounds were obtained. No significant differences were observed when analyzing all the included variants or in any of the sub groups (Indel, SNV, variants with VAF ≤5% and variants with VAF >5%) (Online Supplementary Table S2).
Clinical validation
From October 2017 to October 2019 a total of 823 samples from 751 AML patients were sent to the laboratory network. Disease status at sample collection was: newly diagnosis (DX) (n=639), refractoriness (RS) (n=82), and relapse (RP) (n=102). Patient characteristics are summarized in Table 3. NGS was performed according to already implemented protocols and the consensus parameters established in both cross-validation rounds.
Mutation distribution
A total of 2,052 variants were reported in the 823 samples, with 90.81% of patients showing at least one mutated gene (Online Supplementary Figure S1A). The mean number of variants per sample was 2.49 (range, 0-8). Most patients had three variants (24.37%), followed by patients with two (21.04%) and one (20.77%) variants, respectively (Online Supplementary Figure S1B).
A high frequency of variants in genes involved in signal transduction and epigenetic regulation was observed. FLT3 (24.06%: FLT3 internal tandem duplications [ITD] 16.52%, FLT3-point mutations [PM] 8.87%) was the most prevalent mutated gene followed by IDH (22.60%: IDH1 9.11%, IDH2 13.85%), DNMT3A (21.63%) and NPM1 (21.51%) (Online Supplementary Figure S2).
Co-mutations
FLT3, NPM1 and DNMT3A were significantly co-mutated for all combinations (P<0.001). PTPN11 variants also associated with these genes (P<0.01 for NPM1 and P<0.05 for DNMT3A and FLT3). IDH variants associated with NPM1 (P=0.01 for IDH1 and P<0.01 for IDH2) as well as DNMT3A (P<0.01 for IDH1 and P<0.05 for IDH2). SRSF2 was strongly co-mutated with IDH2 and TET2 (P<0.01) (Figure 2).
Table 1.
First cross-validation round results.

Table 2.
Second cross-validation round results.

Exclusion analysis provided 17 MEGS which are defined as lists of exclusive mutated genes. NPM1, TP53, RUNX1 and KIT set was defined as the most significant mutually exclusive gen set (p-nominal: 2,25E-24). TP53 was present in all MEGS being the most exclusive gene. RUNX1 was highly exclusive with NPM1. CEBPA and NRAS were included in several MEGS of three blocks suggesting their exclusive nature (Online Supplementary Figure S3; Online Supplementary Table S3).
Variant allele frequency analysis
Variants in genes involved in signaling pathways (NRAS, FLT3, KIT, KRAS, and PTPN11) showed lower median VAF, whereas genes involved in clonal hematopoiesis of indeterminate potential (CHIP) (ASXL1, DNMT3A and TET2) showed a VAF around 50%. TP53 variants showed the highest median VAF value (Figure 3A). According to the functional categories, epigenetically related genes and tumor suppressor genes were characterized by a high VAF while genes related to signaling pathways genes showed the lowest VAF (Figure 3B).
Mutational landscape according to disease stage
NPM1 and PTPN11 variants were more frequent at diagnosis (NPM1 23.16% vs. 9.76%, P=0.004, and PTPN11 6.62% vs. 1.10%, P=0.048). RUNX1 and SF3B1 variants were more frequent in refractory as compared to diagnosis (RUNX1 30.49% vs. 16.43%, P=0.003 and SF3B1 9.76% vs. 4.23%, P=0.049). IDH1, IDH2 and WT1 variants were more frequent at relapse as compared to diagnosis (IDH1 14.71% vs. 8.14%, P=0.040; IDH2 21.57% vs. 12.52%, P=0.020, and WT1: 7.84% vs. 3.13%, P=0.043). KRAS and PTPN11 variants were more frequent in refractory as compared to relapse stage (KRAS 9.76% vs. 1.96%, P=0.025, and PTPN11 8.54% vs. 1.10%, P=0.028) (Figure 4A and B).
Figure 1.

Variant allele frequency of samples included in the second cross validation round. (A) Variant allele frequency (VAF) >5% variants and (B) VAF ≤5% variants. Black dots indicate VAF reported for each center. Red dots mean not detected variant. Mean VAF is represented by a horizontal bar and whiskers represent the standard deviation.
Age-related mutations
At diagnosis, patients aged ≥65 years had more variants than younger (<65 years) patients (2.74 ± 0.81 vs. 2.18 ± 0.74 variants per patient, P<0.001). The following genes were more frequently mutated in patients aged ≥65 years vs. <65 years: ASXL1, EZH2, IDH2, JAK2, SF3B1, SRSF2, TET2, TP53, and U2AF1. FLT3-ITD and NPM1 mutations were more frequent in younger AML patients (Figure 5).
In relapsed AML, ASXL1 (20.00% vs. 3.57%, P=0.011) and IDH variants (46.70% vs. 25%, P=0.035) were associated with patients aged ≥65 years (Online Supplementary Table S4).
Mutational stability in paired samples
Paired samples at DX-RP (n=14) and DX-RS (n=20) were obtained to assess clonal evolution. The following were stable variants: NPM1 (100%, as no patients acquired or lost variants), TP53, IDH2 (one acquisition at RS and one at RP in each gene), DNMT3A (100% stable at RS and one acquisition at RP), and RUNX1 (stable at RP and one loss at RS). The following variants were unstable: activating signaling pathways genes such as FLT3, NRAS, KRAS, BRAF, KIT and PTPN11 (Online Supplementary Figure S4). Interestingly for targeted therapy, 26.47% of patients changed the mutational status of FLT3 at RS or RP (FLT3- ITD: two gains and three losses; FLT3-PM: one gain and three losses). In all cases, the loss of function mutation of FLT3-PM was located on the Asp835 codon.
Clinically relevant mutations
Overall, 72.30% of patients harbored at least one clinically relevant variant included in the AML clinical guidelines, clinical trials inclusion criteria or as a risk stratification biomarker (ASXL1, CEBPA, FLT3, IDH1/2, NPM1, RUNX1 and TP53) (Online Supplementary Figure S5). Moreover, druggable mutations were present in a significant proportion of patients (FLT3 in 21.14% and IDH1/2 in 22.60%).
NPM1 mutations
NPM1 variants were found in 21.51% of samples being 78.53% type A (c.860_863dupTCTG), 6.21% type B (c.863_864insCATG), 6.21% type D (c.863_864insCCTG), and 9.04% had uncommon variants (Online Supplementary Figure S6; Online Supplementary Table S5).
FLT3 mutations
FLT3 was the most frequently mutated gene (24.06%). FLT3 ITD was the most frequent FLT3 aberration (16.52%) followed by D835 and I836 variants (5.71%) and other variants (3.16%) (Online Supplementary Figure S7A). Other variants were mostly SNV (95.18%) located in the tyrosine kinase 1 domain (TKD1; 41.18%), juxtamembrane domain (JMD; 23.53%), tyrosine kinase 2 domain (TKD2; 20.59%), extracellular domain (ED; 11.76%) and kinase insert domain (KID 2.94%). No variants were detected in transmembrane (TMD) and C-terminal domains (CTD). (Online Supplementary Figure S7B). 84.75% of all FLT3 variants were targetable with FLT3 inhibitors and had a direct clinical impact in 21.14% of patients through targeted therapy or clinical trials.
CEBPA mutations
CEBPA variants were found in 5.35% of samples, 3.52% were monoallelic variants and 1.82% were biallelic variants (both CEBPA alleles mutated). Two variants with similar VAF were reported as probably biallelic variants (Online Supplementary Table S6). Although CEBPA variants were distributed along the entire coding sequence, biallelic variants were frequently detected in BZIP (43.33%) and the N-terminal domain (30%). Monoallelic variants, were mostly detected in BZIP (44.83%) and almost equally distributed among TAD1 (13.79%), TAD2 (17.94%) and the N-terminal domain (6.90%).
IDH1 and IDH2 mutations
IDH variants were detected in 22.60% of samples. In mutated IDH1 samples (9.11%), all variants were detected in the Arg132 codon. IDH2 (13.85%) was exclusively mutated in the Arg140 or Arg172 codons (84.20% and 15.79%, respectively). Three patients (1.61%) showed simultaneous variants in IDH1 and IDH2 (Online Supplementary Table S7). All IDH1 and IDH2 variants were targetable mutations by IDH inhibitors as no atypical variants were detected.
ASXL1, RUNX1 and TP53 mutations
Overall, 39.00% of samples showed variants in at least one of them: 18.23% RUNX1, 14.70% TP53 and 12.39% ASXL1. In 26.12% of samples a variant detected in one of these genes was the only clinically relevant variant. Moreover, 28.19% of patients were classified to an unfavorable risk group according to ASXL1, RUNX1 and TP53 mutations, following European LeukemiaNet 2017 recommendations.
Discussion
This study shows that a network platform involving many highly skilled laboratories can successfully deliver robust molecular data for AML patients. This strategy allows for testing NGS in the majority of newly diagnosed and relapsed/refractory AML patients involved in the PETHEMA studies, overcoming the current challenging needs for a high-standard diagnosis in cooperative groups. Our descriptive analysis performed in a large series of reallife patients depicts the complex molecular landscape of AML.
In the last 5 years, NGS has irrupted as a potential routine tool for molecular diagnosis, allowing for precise and simultaneous detection of relevant variants in AML. However, this technique is still non-affordable for many institutions due to: i) remarkable cost as compared to conventional PCR tests, ii) batch of samples, ranging from eight to more than 30, to run the test, and consequently high time consumption, both making it difficult to report results in less than 7-10 days; and iii) the need of expensive machinery and highly- qualified teams for biostatistical and molecular biologists. Moreover, the majority of prognostic or druggable mutations can be rapidly and relatively easily detected by conventional PCR. In fact, from the mandatory NGS panel genes selected by the PETHEMA central laboratories (i.e, ASXL1, CEBPA, FLT3, IDH1, IDH2, NPM1, RUNX1, and TP53), a mutation screening by conventional PCR is still required for FLT3, IDH1, IDH2 and NPM1, as a positive result could allow for rapid implementation of targeted or risk-adapted therapeutic approaches.8 In addition, rapid PCR is also needed for core-binding factor (CBF), PMLRARA and BCR-ABL rearrangements. Under this scenario, our cooperative group designed a nationwide network involving seven central laboratories aimed to deliver homogeneous and comparable molecular results for newly diagnosed and relapsed/refractory AML patients. As far as we know, this is a different strategy as compared to other cooperative groups that usually rely on only one or two central laboratories for molecular diagnostics (e.g, British NCRI).9 Several reasons guided us to make this decision: i) the economic and work burden required to collect samples from the whole group, which covers a wide territory and population, was not affordable for a single laboratory; ii) a minimum referral population is required to permit an efficient diagnosis by studying the appropriate number of samples in every run; iii) the involvement many on-site teams in order to create a true research network; and iv) the need to facilitate rapid delivery of samples while preserving closer and well established relationships between the sample referral institution and the assigned central laboratory. We can affirm that the PETHEMA model for centralized diagnosis has been successful collecting samples from 751 patients in roughly 1 year, enabling the use of this network in routine clinical practice and research.
Table 3.
Demographic and baseline characteristics of the study population (n=751).
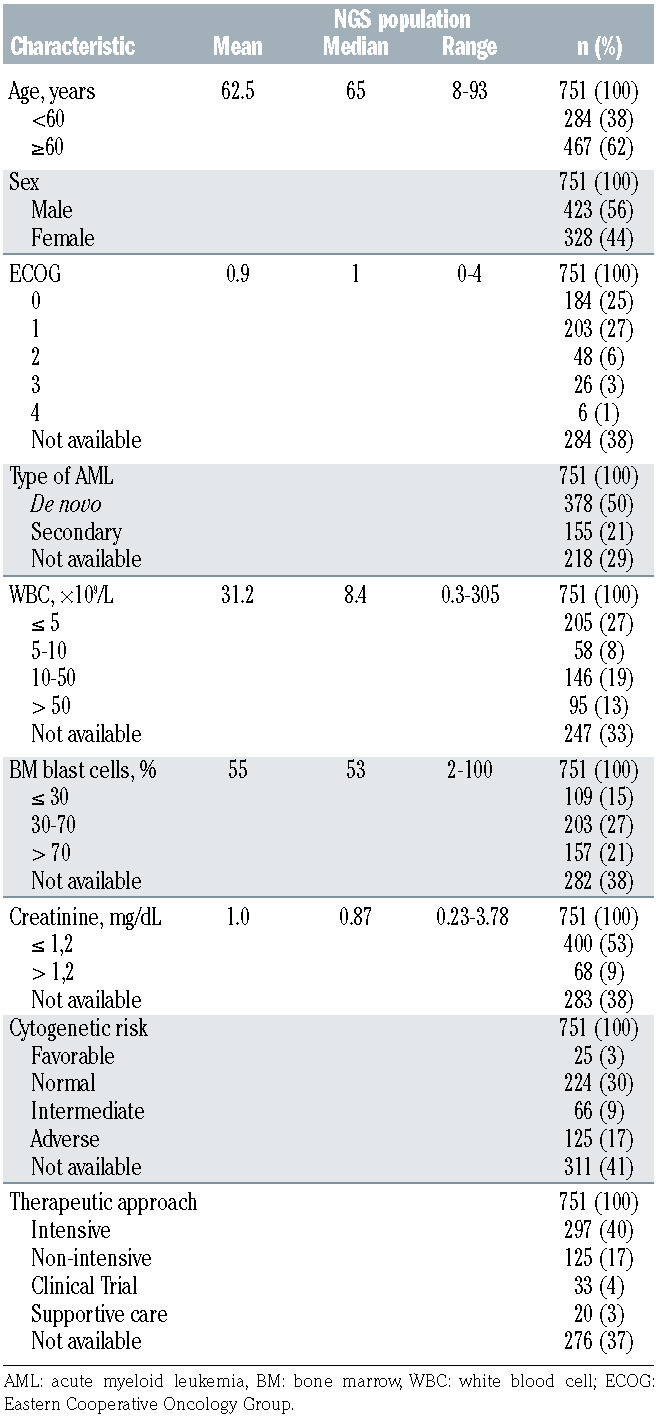
Figure 2.

Circos diagram showing mutation concurrences in our cohort.
Figure 3.

Variant allele frequency analysis. (A) Mean variant allele frequency (VAF) for individual genes and (B) mean VAF according functional categories. Each dot represents one variant, median VAF is represented by a horizontal bar and whiskers represent the interquartile range. ITD: internal tandem duplication; PM: point mutations.
Figure 4.

Distribution of gene alterations in acute myeloid leukemia samples. (A) Mutational landscape in the global cohort. Horizontal green bars: diagnosis, orange: refractory, and red: relapse.Vertical dark blue bars: positive (in global FLT3 row represents internal tandem duplications [ITD]), red: FLT3-D835/I836, green: other FLT3 point mutations (PM), orange: FLT3-ITD and D835/I836, light blue: FLT3-ITD and other FLT3-PM, light grey: negative, dark grey: not tested, yellow: biallelic variants in CEBPA. (B) Mutational prevalence according to disease stage. Diagnosis are represented as blue bars, refractory as red bars and relapse as green bars. *P<0.05, **P<0.01.
Our study demonstrates that harmonized and reliable NGS results can be achieved across several laboratories, even if they are using their own diagnostic platforms. As shown in the pre-standardization cross-validation round, an overall concordance of 60.98% was obtained with a great variability in selected genes and conditions across laboratories. After consensus of AML relevant genes and optimization of quality parameters (uniformity >85%; mean read depth of 1,000X) the overall concordance rose to 85.57% in the second cross-validation round. This was a remarkable achievement for all laboratories taking into account that low VAF (≤5%) variants were included in this second round. To the best of our knowledge, there are no similar studies reported in the literature for AML.
Clinical validation of our AML cohort was consistent with previous reports. Roughly 91% of AML patients had at least one variant, and many harbored three, four and up to eight variants reflecting the heterogeneous AML mutational profile.10 FLT3, IDH1/2, DNMT3A and NPM1 were the most frequently mutated genes,11,12 and we showed that up to 73% of patients had variants with clinical implications for risk stratification or targeted therapy-based approaches (i.e, ASXL1, CEBPA, FLT3, IDH1/2, NPM1, RUNX1 and TP53).13 Moreover, ASXL1, RUNX1 and TP53 variants which are not easily analyzed with conventional molecular techniques,14–16 were the unique clinically relevant alteration detected in up to 28.19% of patients, highlighting that NGS-based mutational profiling seems crucial to categorize AML risk according to the European LeukemiaNet 2017 guidelines.3
As reported by other groups,17–20 elderly patients had a higher number of variants, which were enriched in spliceosome machinery, epigenetic regulators and in DNA repair (i.e, SRSF2, U2AF1, SF3B1, ASXL1, TET2, IDH2 and TP53). In line with previous studies, NPM1 variants were more frequent in younger AML patients, and we noticed a striking decrease of FLT3 variants in older patients.21 We can affirm that a lower number of older patients may benefit from tyrosine kinase inhibitors-based approaches,22 but more from novel IDH-inhibitors.23 In our experience, NGS has efficiently screened FLT3 gene variants, including less frequent variants, which could also be informative for therapeutic decisions.24,25 Furthermore, NGS is a promising tool to assess FLT3-ITD duplicated region, which could have prognostic impact regarding its location and extension.26
We also provide insights on clonal evolution and leukemogenesis: i) variants in signalling pathway genes (FLT3, KIT, RAS) had lower VAF, reflecting their role as late events,27; ii) genes related to CHIP showed higher VAF values, 28 and iii) median VAF in AML patients with TP53 variants was above 50%, indicating the frequent loss of the wild-type TP53 allele. Recent studies suggest that a higher clonal size of TP53 variants, determined by VAF and chromosomal aberrations (del 17p), could discriminate patients with worse prognosis among TP53 mutated AML.29,30
Figure 5.

Age-related mutational profile. Pvalues (P) for statistically significant results are shown. ITD: internal tandem duplication; PM: point mutations.
Our study allowed to observe differences in the mutational profile of relapsed/refractory patients as compared to newly diagnosed/untreated subjects. Interestingly, RUNX1 variants were more frequent among refractory AML patients, which is consistent with previously reported poorer outcomes after intensive chemotherapy and its association with older age.31 The same was observed with SFRS2, depicting poorer outcomes when commutated with IDH232 which was highly associated in our cohort. NPM1 variants were significantly less frequent in refractory patients, as compared to newly diagnosed or relapsed, reflecting the known high complete remission rates in this setting.33,34 Interestingly, IDH variants were the most frequent alteration at relapse, suggesting that they are associated with higher relapse risk,35 but also have more possibilities to obtain an initial response with front-line therapies.18,36
We analyzed 35 patients with paired samples at diagnosis versus relapse/refractory setting, confirming some findings from scarce studies on clonal evolution: i) as a founder variant, NPM1 was very stable,37,38 ii) DNMT3A variants were very stable, probably due to its early acquisition and preleukemic occurrence,39,40 as well as their persistence during remission and disease progression,41 and iii) activating signaling pathways genes were unstable (FLT3, NRAS, KRAS, BRAF, KIT and PTPN11). This is particularly relevant for the management of patients who acquire FLT3 mutations during relapse and refractoriness which may benefit from second generation inhibitors such as gilteritinib, available for the treatment of relapsed/refractory AML with FLT3 mutation.42,43 In line with the study by Kronke et al., we show that IDH2 variants were very stable, contrarily to IDH1, but numbers are low and should be cautiously interpreted. 38
The main limitation of our study is that, apart from age and disease phase, baseline clinical characteristics, treatment patterns, and outcomes, were not available for a minor proportion of patients at the time of this interim analysis of the NGS-AML trial. This is why the results herein presented focused on the overall network building strategy, as well as in the cross-validation of samples and reporting harmonization, showing a mutational landscape of AML consistent with the current knowledge. We should mention that our diagnostic platform has many areas of improvement: i) time to NGS reporting, which is longer than conventional molecular analysis (approximately 2-3 weeks), ii) need to promote larger participation of PETHEMA clinical sites, as a sizable proportion of patients are not yet benefiting from advanced laboratory centralization (especially in the relapse/refractory setting), and ii) budgetary vulnerability.
In conclusion, the PETHEMA cooperative scientific group has adopted the reported nationwide strategy network with centralized NGS analyses. Sample and information exchange allowed us to unify analysis criteria and decrease reporting variability in order to offer reliable and consistent NGS results. This cooperative strategy has also been applied to rapid screening by conventional PCR and quantitive real-time PCR to measure residual disease, and is being expanded to other AML diagnostic areas (e.g., biobanking and multiparametric flow cytometry). Ongoing therapeutic guidelines (NCT01296178) and clinical trials (clinicaltrials gov. Identifier: NCT04230239, NCT04107727, NCT04112589, NCT04090736) by the PETHEMA group are benefiting from this diagnostic network.
Supplementary Material
Acknowledgments
The authors would like to thank María D. García, Carlos Pastorini, Rafael Vianney, and Mar Benlloch for data collection and management; and Data Science, Biostatistics and Bioinformatics Unit from IIS La Fe for its collaboration in statistical analysis.
Funding Statement
Funding: This work was partially supported by a Celgene grant, the Subdirección General de Investigación Sanitaria (Instituto de Salud Carlos III, Spain) Spanish Ministry of Economy and Competitiveness: PI15/01706, PI16/00517 PI16/0665, PI16/01530, PI18/01340, PI18/01946 PI19/00730, PI19/01518, FI19/00059, Fundación Española de Hematología y Hemoterapia (FEHH) grant, CRIS against Cancer foundation 2018/001. CIBERONC-CB16/12/00233 and “Una manera de hacer Europa” (Innocampus; CEI-2010-1-0010), Instituto de Investigación Sanitaria La Fe (Contrato de Investigación postresidentes 2019-052-1)
References
- 1.Saultz J, Garzon R. Acute myeloid leukemia: a concise review. J Clin Med. 2016;5(3):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405. [DOI] [PubMed] [Google Scholar]
- 3.Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424-447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hackl H, Astanina K, Wieser R. Molecular and genetic alterations associated with therapy resistance and relapse of acute myeloid leukemia. J Hematol Oncol. 2017;10(1):51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luppi M, Fabbiano F, Visani G, Martinelli G, Venditti A. Novel agents for acute myeloid leukemia. Cancers (Basel). 2018;10(11):429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Riva L, Luzi L, Pelicci PG. Genomics of acute myeloid leukemia: the next generation. Front Oncol. 2012;2:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuo FC, Mar BG, Lindsley RC, Lindeman NI. The relative utilities of genome-wide, gene panel, and individual gene sequencing in clinical practice. Blood. 2017;130(4):433-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bacher U, Shumilov E, Flach J, et al. Challenges in the introduction of next-generation sequencing (NGS) for diagnostics of myeloid malignancies into clinical routine use. Blood Cancer J. 2018;8(11):113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Macklin PS, Pillay N, Lee JL, et al. CM-Path Molecular Diagnostics Forum—consensus statement on the development and implementation of molecular diagnostic tests in the United Kingdom. Br J Cancer. 2019; 121(9):738-743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ley TJ, Miller C, Ding L, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059-2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tyner JW, Tognon CE, Bottomly D, et al. Functional genomic landscape of acute myeloid leukaemia. Nature. 2018;562(7728):526-531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209-2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castelli G, Pelosi E, Testa U. Targeted therapies in the treatment of adult acute myeloid leukemias: current status and future perspectives. Int J Hematol Oncol. 2016;5(4):143-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gelsi-Boyer V, Brecqueville M, Devillier R, Murati A, Mozziconacci MJ, Birnbaum D. Mutations in ASXL1 are associated with poor prognosis across the spectrum of malignant myeloid diseases. J Hematol Oncol. 2012;5:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kohlmann A, Nadarajah N, Alpermann T, et al. Monitoring of residual disease by next-generation deep-sequencing of RUNX1 mutations can identify acute myeloid leukemia patients with resistant disease. Leukemia. 2014;28(1):129-137. [DOI] [PubMed] [Google Scholar]
- 16.Barbosa K, Li S, Adams PD, Deshpande AJ. The role of TP53 in acute myeloid leukemia: challenges and opportunities. Genes Chromosomes Cancer. 2019;58(12): 875-888. [DOI] [PubMed] [Google Scholar]
- 17.Dinardo CD, Ravandi F, Agresta S, et al. Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am J Hematol. 2015;90(8):732-736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Medeiros BC, Fathi AT, DiNardo CD, Pollyea DA, Chan SM, Swords R. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia. 2017;31(2):272-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prassek VV, Rothenberg-Thurley M, Sauerland MC, et al. Genetics of acute myeloid leukemia in the elderly: Mutation spectrum and clinical impact in intensively treated patients aged 75 years or older. Haematologica. 2018;103(11):1853-1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Metzeler KH, Herold T, Rothenberg-Thurley M, et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood. 2016;128(5):686-698. [DOI] [PubMed] [Google Scholar]
- 21.Schneider F, Hoster E, Schneider S, et al. Age-dependent frequencies of NPM1 mutations and FLT3-ITD in patients with normal karyotype AML (NK-AML). Ann Hematol. 2012;91(1):9-18. [DOI] [PubMed] [Google Scholar]
- 22.Daver N, Schlenk RF, Russell NH, Levis MJ. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia. 2019;33(2):299-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu X, Gong Y. Isocitrate dehydrogenase inhibitors in acute myeloid leukemia. Biomark Res. 2019;7(1):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams AB, Nguyen B, Li L, et al. Mutations of FLT3/ITD confer resistance to multiple tyrosine kinase inhibitors. Leukemia. 2013;27(1):48-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heidel F, Solem FK, Breitenbuecher F, et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood. 2006;107(1):293-300. [DOI] [PubMed] [Google Scholar]
- 26.Liu SB, Dong HJ, Bao XB, et al. Impact of FLT3-ITD length on prognosis of acute myeloid leukemia. Haematologica. 2019; 104(1):e9-e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirsch P, Zhang Y, Tang R, et al. Genetic hierarchy and temporal variegation in the clonal history of acute myeloid leukaemia. Nat Commun. 2016;7(1):1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martignoles JA, Delhommeau F, Hirsch P. Genetic hierarchy of acute myeloid leukemia: from clonal hematopoiesis to molecular residual disease. Int J Mol Sci. 2018;19(12):3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goel S, Hall J, Pradhan K, et al. High prevalence and allele burden-independent prognostic importance of p53 mutations in an inner-city MDS/AML cohort. Leukemia. 2016;30(8):1793-1795. [DOI] [PubMed] [Google Scholar]
- 30.Rücker FG, Schlenk RF, Bullinger L, et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. 2012;119(9):2114-2121. [DOI] [PubMed] [Google Scholar]
- 31.Gaidzik VI, Bullinger L, Schlenk RF, et al. RUNX1 mutations in acute myeloid leukemia: results from a comprehensive genetic and clinical analysis from the AML study group. J Clin Oncol. 2011;29(10): 1364-1372. [DOI] [PubMed] [Google Scholar]
- 32.Yoshimi A, Lin KT, Wiseman DH, et al. Coordinated alterations in RNA splicing and epigenetic regulation drive leukaemogenesis. Nature. 2019;574(7777):273-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jain P, Kantarjian H, Patel K, et al. Mutated NPM1 in patients with acute myeloid leukemia in remission and relapse. Leuk Lymphoma. 2014;55(6):1337-1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schneider F, Hoster E, Unterhalt M, et al. NPM1 but not FLT3-ITD mutations predict early blast cell clearance and CR rate in patients with normal karyotype AML (NKAML) or high-risk myelodysplastic syndrome (MDS). Blood. 2009;113(21):5250-5253. [DOI] [PubMed] [Google Scholar]
- 35.Ok CY, Loghavi S, Sui D, et al. Persistent IDH1/2 mutations in remission can predict relapse in patients with acute myeloid leukemia. Haematologica. 2019;104(2):305-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Megías-Vericat JE, Ballesta-López O, Barragán E, Montesinos P. IDH1-mutated relapsed or refractory AML: current challenges and future prospects. Blood Lymphat Cancer Targets Ther. 2019;9:19-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cocciardi S, Dolnik A, Kapp-Schwoerer S, et al. Clonal evolution patterns in acute myeloid leukemia with NPM1 mutation. Nat Commun. 2019;10(1):2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krönke J, Bullinger L, Teleanu V, et al. Clonal evolution in relapsed NPM1-mutated acute myeloid leukemia. Blood. 2013;122(1):100-108. [DOI] [PubMed] [Google Scholar]
- 39.Bhatnagar B, Eisfeld AK, Nicolet D, et al. Persistence of DNMT3A R882 mutations during remission does not adversely affect outcomes of patients with acute myeloid leukaemia. Br J Haematol. 2016;175(2):226-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hou HA, Kuo YY, Liu CY, et al. DNMT3A mutations in acute myeloid leukemia: stability during disease evolution and clinical implications. Blood. 2012;119(2):559-568. [DOI] [PubMed] [Google Scholar]
- 41.Gaidzik VI, Weber D, Paschka P, et al. DNMT3A mutant transcript levels persist in remission and do not predict outcome in patients with acute myeloid leukemia. Leukemia. 2018;32(1):30-37. [DOI] [PubMed] [Google Scholar]
- 42.Larrosa-Garcia M, Baer MR. FLT3 Inhibitors in acute myeloid leukemia: current status & future directions. Mol Cancer Ther. 2017;16(6):991-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Short NJ, Kantarjian H, Ravandi F, Daver N. Emerging treatment paradigms with FLT3 inhibitors in acute myeloid leukemia. Ther Adv Hematol. 2019;10:2040620719827310. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.