

Abstract
The design of multi‐target ligands has become an innovative approach for the identification of effective therapeutic treatments against complex diseases, such as cancer. Recent studies have demonstrated that the combined inhibition of Hsp90 and B‐Raf provides synergistic effects against several types of cancers. Moreover, it has been reported that PDHK1, which presents an ATP‐binding pocket similar to that of Hsp90, plays an important role in tumor initiation, maintenance and progression, participating also to the senescence process induced by B‐Raf oncogenic proteins. Based on these premises, the simultaneous inhibition of these targets may provide several benefits for the treatment of cancer. In this work, we set up a design strategy including the assembly and integration of molecular fragments known to be important for binding to the Hsp90, PDHK1 and B‐Raf targets, aided by molecular docking for the selection of a set of compounds potentially able to exert Hsp90‐B‐Raf‐PDHK1 multi‐target activities. The designed compounds were synthesized and experimentally validated in vitro. According to the in vitro assays, compounds 4 a, 4 d and 4 e potently inhibited Hsp90 and moderately inhibited the PDHK1 kinase. Finally, molecular dynamics simulations were performed to provide further insights into the structural basis of their multi‐target activity.
Keywords: docking, in vitro assays, molecular dynamics, multi-target ligands, polypharmacology
Multitasking ligands: A set of ad hoc ligands has been designed by integrating molecular fragments of known inhibitors of anticancer drug targets. The compounds were docked into the respective targets, and six derivatives were synthesized to be in vitro tested. Molecular dynamics studies were finally performed to provide further insights into the structural basis of the experimentally observed activities.

1. Introduction
Multi‐target inhibitors, that is small molecules able to simultaneously hit multiple targets, have become a major opportunity in drug discovery.[ 1 , 2 , 3 , 4 ] Among their several advantages, they offer the potential of higher efficacy due to synergistic effects, a minor insurgence of drug resistance, the lack of drug‐drug interactions and a better patient compliance. [1] Indeed, multi‐target drugs are recognized to be particularly important for the treatment of complex and multi‐factorial diseases, such as cancer and neurodegeneration,[ 5 , 6 , 7 , 8 ] in which complex networks of multiple and interconnected biological targets need to be simultaneously modulated. For this reason, the rational design of compounds with desired, ad hoc “polypharmacological” profiles has become increasingly important to discover better drug candidates. [9]
Being responsible for the assembly and regulation of a large number of signal transduction and regulatory client proteins, the molecular chaperone Heat shock protein 90 (Hsp90) is a key node in many biological processes, [10] hence it represents an excellent candidate for drug polypharmacology.[ 11 , 12 , 13 , 14 ] Hsp90 refolds, stabilizes and regulates the trafficking of many proteins involved in uncontrolled proliferation and apoptotic resistance, including, but not limited to, multiple protein kinases of the so‐called Hsp90 interactome. Importantly, Hsp90 regulates a number of cellular and disease‐related processes, many of which involve mutant and overexpressed oncoproteins whose activity and regulation depend on Hsp90 activity. [15] Of note, while recent studies showed that administration of a single Hsp90 inhibitor suffers from non‐optimal safety profiles or lack of efficacy, [16] promising drug combinations based on Hsp90 and B‐Raf inhibitors are currently under clinical evaluation against mutated melanoma with promising results (clinicaltrials.gov ID: NCT02721459, NCT01657591). Hsp90 inhibition contributes to the degradation of wild type and V600E mutant B‐Raf kinases. [17] Moreover, such combinations have demonstrated to provide significant synergistic effects against several types of cancers,[ 18 , 19 , 20 , 21 , 22 ] and inhibition of Hsp90 proved to be effective in patients with intrinsic or acquired resistance to Raf inhibitors.[ 15 , 16 , 17 , 18 , 19 , 20 , 21 ] In the context of tumorigenesis, another relevant target is Pyruvate dehydrogenase kinase 1 (PDHK1), an enzyme that inhibits the pyruvate dehydrogenase complex preventing conversion of pyruvate to acetyl‐CoA, the substrate for the Krebs’ cycle. [23] Activation of PDHK1 results in the uncoupling of glycolysis to glucose oxidation. Moreover, PDHK1 is overexpressed in melanoma patients, this effect being associated with the expression of the mTOR pathway effectors and independent of the B‐Raf mutational status. [24] Notably, PDHK1 is not only required for tumor initiation, but also for tumor maintenance and progression, suggesting that targeting PDHK1 may be beneficial for therapeutic intervention in melanoma. [25] In particular, PDHK1 plays an important role in oncogene‐induced senescence in B‐RafV600E melanoma, [25] and the kinase itself is an Hsp90 client adopting a similar fold to that of Hsp90. [26]
In the light of all these considerations, we reasoned that an Hsp90 inhibitor with B‐Raf and PDHK1 kinases multi‐target activity could be of particular interest for cancer treatment. In this study, we describe the design, synthesis and biological evaluation of a small set of potential multi‐target ligands assembled by integrating Hsp90, B‐Raf and PDHK1 key molecular fragments. In particular, the design included an analysis of available crystal structures of ligands in complex with the three targets, as well as docking simulations into selected conformations of the B‐Raf, PDHK1 and Hsp90 binding sites. Although potent inhibitors of Hsp90 were obtained, the resulting compounds did not inhibit B‐RafWT and B‐RafV600E. However, they showed moderate inhibitory activity against PDHK1. The binding mode of the more interesting compounds was further investigated by means of molecular dynamics simulations, in order to shed light into the observed activity profile.
2. Results and Discussion
2.1. Molecular Design
The multi‐target compounds were designed by assembling and integrating scaffolds known to be important for binding to Hsp90, PDHK1 and B‐Raf (Figure 1).
Figure 1.

Design of Hsp90 inhibitors 4 a–4 f with putative multi‐target activity. The compounds were obtained by firstly assembling selected molecular core scaffolds of known Hsp90 inhibitors (i. e., substituted resorcinol, 4‐phenyl‐1,2,3‐triazole molecules), [19] with those selected from reported PDHK1 ligands (based on substituted resorcinol, 4‐phenyl‐(1,2‐oxazole)), [28] and B‐Raf ligands (based on substituted 4‐(1H‐pyrazol‐3‐yl)pyridine) (panel a).[ 29 , 30 , 31 ] Further similarity estimations performed on selected inhibitors of these proteins allowed to identify chemical substituents conferring high potency and selectivity towards B‐Raf protein kinases,[ 30 , 31 ] which are expected to be well accepted also by Hsp90 and PDHK1. Panel b reports few among the most similar Hsp90 and PDHK1 amide substituents, identified with respect to unsubstituted (R1, R2=H) and substituted (R1, R2=F) phenyl‐3‐sulfonamides or phenyl‐3‐sulfamides chemical moieties present in the selected B‐Raf compounds. The assembled molecular core scaffold and selected substituents were finally integrated into chemical entities with the structural details potentially required to achieve efficient binding to Hsp90, B‐Raf and PDHK1 (panel c).
In particular, an analysis of potent Hsp90, PDHK1 and B‐Raf inhibitors reported in the literature was performed, allowing the selection of the resorcinol‐substituted 1,2,3‐triazole Hsp90 ligands reported by Taddei et al., [27] the 4,5‐diarylisoxazole PDHK1 modulators designed by Meng et al. [28] and the pyrazole‐pyridine B‐Raf compounds developed by Hansen et al., [29] as suitable initial structural templates for the design of multi‐target ligands (Figure 1, panel a). The selected resorcinol‐substituted 1,2,3‐triazole Hsp90 inhibitors share significant structural similarity with the 4,5‐diarylisoxazole PDHK1 ligands, the highest similarities evaluated between molecules of these targets being almost 1.0 in terms Tanimoto coefficient for both MACCS and ECFP4 molecular fingerprints (see the “Chemoinformatic analyses” section in the Supporting Information). Slightly different considerations could be drawn for the selected B‐Raf compounds, which share with the diaryl‐triazole Hsp90 ligands and the diaryl‐isoxazole PDHK1 molecules a system composed by three differently substituted aromatic rings, resulting in significant similarity in terms of structural connectivity (see the Supporting Information). Visual inspection of reported Hsp90, PDHK1 and B‐Raf crystal structures highlighted different structural requirements at the three aromatic rings of the selected ligands. Therefore, the phenyl‐triazole and phenyl‐isoxazole chemical fragments were selected as starting points for the design of the desired multi‐target ligands. Notably, replacing the phenyl ring of selected phenyl‐triazole Hsp90 and phenyl‐isoxazole PDHK1 inhibitors with a pyridine ring allowed us to introduce the nitrogen atom required for the binding to the backbone of the Cys532 hinge residue in B‐Raf, as evidenced from the crystallographic structure reported by Hansen et al. (PDB code 3D4Q). [29] Moreover, the performed analyses allowed also to identify significant similarity between some of the more sterically hindered amide substituents of the selected Hsp90 and PHDK1 inhibitors with the phenyl‐3‐sulfonamide and phenyl‐3‐sulfamide chemical moieties of B‐Raf ligands reported by Mathieu et al. [30] and Wenglowsky et al. [31] (Figure 1, panel b), which are engaged in hydrogen bonds to the DFG motif and occupy a small lipophilic pocket of the kinase (see the Supporting Information).
The performed analyses allowed us to design an initial structural core, integrating different structural details of the selected Hsp90, PDHK1 and B‐Raf ligands (Figure 1, panel c), and thus proposing six derivates (4 a–4 f) potentially able to interact with the hot spot residues of the selected targets, as indicated by the corresponding crystal structure complexes. The chemical structures of the 4a–4 f designed compounds are shown in Figure S1 in the Supporting Information. The computationally designed ligands were then docked into representative crystal structures of the three targets, to evaluate whether they are able to provide favorable docking scores and a binding mode consistent with the reported crystal structure information.
2.2. Molecular Docking
Compounds 4 a–4 f were docked into the 2VCI crystal structure of Hsp90 (see experimental methods for details), [32] showing a binding mode fully consistent with that of the 2GJ crystallographic ligand. In particular, the hydroxyl groups of the resorcinol moiety hydrogen bonded to Asp93 and three buried, highly conserved water molecules, the amide nitrogen established an H‐bond interaction with the backbone carbonyl of Gly97, while the pyridine and the aryl sulfonamides or sulfamides were directed outwards. Interestingly, pKa predictions made with Epik (Schrödinger Suite 2020–1)[ 33 , 34 ] showed that the 2,6‐difluoro substituted sulfonamides 4 b–4 d and sulfamides 4 e–4 f were ∼1 and ∼1.5 pKa units more acidic with respect to the unsubstituted compound 4 a (Table S1 in the Supporting Information), suggesting that the former derivatives could bind the protein in their anionic form. This observation is consistent with the electronic effect of an ortho/para di‐fluoro substitution on the acidity of an aryl sulfonamide. Therefore, 4 b–4 f were modelled in their anionic form. Of note, a deprotonated form would potentially favor an interaction with mutant B‐RafV600E, as previously discussed by Tsai et al. [35] Docking scores are reported in Table S2 in the Supporting Information.
Docking calculations predicted that 4 a–4 f are accommodated into the ATP binding site of B‐Raf (PDB code: 3D4Q), [29] with the pyridine nitrogen hydrogen bonded to the Cys532 residue of the hinge, in agreement with the location of the pyridine of ligand SM5 in the experimental complex. [29] The aryl sulfonamide and sulfamide moieties of the ligands interacted with the DFG motif of the kinase, with the small lipophilic tails filling a narrow pocket formed by an outward shift of the αC‐helix, similarly to experimentally observed complexes of RI8 (PDB code: 4EHG), RI9 (PDB code: 4EHE), and BR2 PDB code: 3SKC) crystallographic compounds reported by Mathieu et al. [30] and Wenglowsky et al. [31] Docking scores of the compounds into B‐Raf are reported in Table S2 in the Supporting Information.
Finally, docking of 4 a–4 f into the ATP site of PDHK1 (PDB code: 2Q8G) [26] provided binding modes fully comparable to those obtained in Hsp90. Interestingly, the observed binding poses are consistent, to some extent, with the fact that PDHK1 and Hsp90 have similar folds and share a certain degree of residue conservation of their ATP sites, and that the binding mode of the resorcinol inhibitor radicicol is very similar in the two proteins.[ 26 , 36 ] The docking scores observed for 4 a–4 f in the PDHK1 binding site are reported in Table S2 in the Supporting Information.
2.3. Synthesis of Compounds 4 a–4 f
In line with our previous experience on triazole compounds,[ 37 , 38 , 39 ] the synthetic construction of inhibitors 4 a–4 f was accomplished by a multistep procedure. The preparation of triazole 1 (Scheme 1) is based on Huisgen cycloaddition between azide C and ethyl 3‐(pyridin‐4‐yl)‐propiolate and subsequent base induced ester hydrolysis.
Scheme 1.
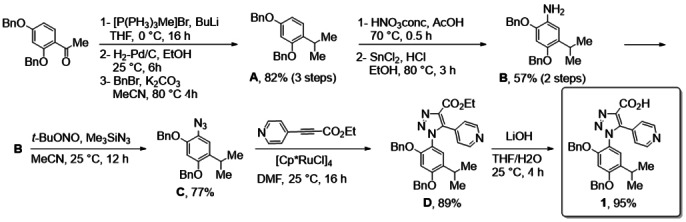
Synthesis of scaffold 1.
Azide C was prepared according to a six‐step procedure. [27] Dibenzyl 4‐acetylresorcinol was used as starting material and the acetyl group was converted into the i‐propyl group by Wittig reaction and hydrogenation. Benzylation reaction was necessary to reintroduce the benzyl groups. The azide group was then introduced by conventional procedure, that is the nitration of compound B, followed by reduction and subsequent diazotization‐azidation reaction. Triazole D was isolated in high yield as a single regioisomer by 1,3‐cycloaddition reaction catalyzed by (Cp*RuCl)4 under classical reaction conditions as reported by Weinreb et al. [40]
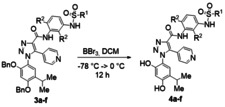
Compound 2 a is commercially available. Compounds 2 b–f were synthetized following literature protocols. [41] Condensation of the common scaffold 1 with various substituted anilines 2 a–f in the presence of HATU as condensing agent (see experimental methods for details) afforded the desired protected compounds 3 a–f with satisfactory yields (Table 1). It is worth of note that the presence of fluorine atoms on the aniline aromatic ring (R2=F, 2 b–f) resulted in a lower reactivity of 2 b–f in the condensation reaction. The final cleavage of the benzyl protecting groups in the presence of BBr3 afforded compounds 4 a–f in good yield after purification (Table 2).
Table 1.
Synthesis of compounds 3 a–f.
|
| |||
|---|---|---|---|
|
Compound |
R1 |
R2 |
Yield [%] |
|
3 a |
−CH3 |
H |
85 |
|
3 b |
−CH3 |
F |
47 |
|
3 c |
−CH2−CH3 |
F |
61 |
|
3 d |
−(CH2)2−CH3 |
F |
49 |
|
3 e |
−N(CH2)4 |
F |
54 |
|
3 f |
−N(CH3)2 |
F |
57 |
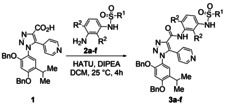
Table 2.
Deprotection of 3 a‐f to give compounds 4 a–f.
|
| |||
|---|---|---|---|
|
Compound |
R1 |
R2 |
Yield [%] |
|
4 a |
−CH3 |
H |
78 |
|
4 b |
−CH3 |
F |
89 |
|
4 c |
−CH2−CH3 |
F |
95 |
|
4 d |
−(CH2)2−CH3 |
F |
98 |
|
4 e |
−N(CH2)4 |
F |
84 |
|
4 f |
−N(CH3)2 |
F |
85 |
2.4. Enzyme Inhibitory Activities
Compounds 4 a–4 e were tested in vitro to evaluate their inhibitory activity on Hsp90, B‐Rafwt, B‐RafV600E and PDHK1 recombinant, purified enzymes (see experimental section for details). The IC50 values of compounds 4 a–4 f along with the IC50 values of the control inhibitors used in the enzymatic assay are reported in Table 3, while dose‐response curves of the active compounds are reported in Figure S2 in the Supporting Information. Compounds 4 a, 4 d and 4 e turned out to be potent nanomolar inhibitors of Hsp90, their IC50 values being 7 nm, 30 nm and 8 nm. The remaining derivatives, that is 4 b, 4 c and 4 f were active in the low micromolar range. All compounds were also tested for the inhibition of the PDHK1 and B‐Raf proteins. None of the tested compounds were active against B‐RafWT or B‐RafV600E. However, 4 a, 4 d and 4 e inhibited PDHK1 with IC50 values of 28.7, 38.7 and 29.4 μm, the compounds being only four‐fold less active than the control inhibitor GW5074 (Table 3).
Table 3.
Inhibitory activities (IC50, μm) of compounds 4 a–4 f against Hsp90α, B‐RafWT, B‐RafV600E and PDHK1. Dose‐response curves of the active compounds are reported in Figure S2 in the Supporting Information.
|
Compound |
IC50 Hsp90α [μm] |
IC50 B‐RafWT [μm] |
IC50 B‐RafV600E [μm] |
IC50 PDHK1 [μm] |
|---|---|---|---|---|
|
controls |
0.009[a] |
0.044[b] |
0.006[b] |
7.1[b] |
|
4 a |
0.007 |
inactive |
inactive |
28.7 |
|
4 b |
1.7 |
inactive |
inactive |
inactive |
|
4 c |
1.1 |
inactive |
inactive |
inactive |
|
4 d |
0.03 |
inactive |
inactive |
38.3 |
|
4 e |
0.008 |
inactive |
inactive |
29.4 |
|
4 f |
0.5 |
inactive |
inactive |
inactive |
Note: [a] Geldanamycin; [b] GW5074;
In silico predictions made with the CLC‐pred webserver (http://www.way2drug.com/cell‐line/, accessed on August 27th, 2021) [42] suggest that the designed compounds could show cytotoxic effects against, for example, non‐small cell lung cancer (NSCLC) and melanoma cancer cells, while not providing significant cytotoxicity on normal human cell lines (see Table S3 in the Supporting Information). These results are particularly interesting, considering that Hsp90 and the other investigated targets have been previously studied for NSCLC treatment.[ 43 , 44 , 45 ]
2.5. Molecular Dynamics
Molecular dynamics simulations were performed to provide further insights into the observed inhibitory activity profiles. Considering that in Hsp90 the di‐fluoro substituted compound 4 b was ∼250‐fold less active than 4 a, but 4 e retained high potency despite bearing a di‐fluoro substitution, compounds 4 a, 4 b and 4 e were considered interesting enough to be selected for further investigation with molecular dynamics (MD). After equilibration, the average root mean square deviations (RMSD) of ligands and binding site residues, along the 100 ns MD production run of each Hsp90‐ligand complex were fairly low. Figure S3 in the Supporting Information shows schematic representations of the hydrogen bond interactions sampled by MD (left panels), along with representative minimized structures (right panels) of each Hsp90‐ligand complex. Table S4 in the Supporting Information reports the percentages of occurrence of hydrogen bond interactions along the simulated MD trajectory (100 ns) and the resulting average hydrogen bond distances in Å. Interestingly, the percentage of occurrence of hydrogen bond interactions between the amide nitrogen of 4 b and the backbone carbonyl of Gly97 was significantly lower compared to that of 4 a (36 % vs 80 %, Table S4 in the Supporting Information). Moreover, the average torsion angle of the 2,6‐diflurophenyl ring of 4 b relative to the amide moiety was nearly orthogonal (80°), while the corresponding angle of the unsubstituted compound 4 a was only 52° out of plane. These results suggest that the 2,6‐difluoro substituent reduced the ability of the amide moiety to hydrogen bond to Gly97, owing to steric effects. In addition, compound 4 a with a neutral sulfonamide hydrogen bonded to Asp102 with 94 % occurrence (Table S4 in the Supporting Information), while such interaction was not observed in the ionized sulfonamide 4 b, which hydrogen bonded to Lys58 and His154 with only 17 % and 7 % occurrences, respectively. Altogether, these findings may explain the lower activity of 4 b with respect to 4 a. Compound 4 e with a pyrrolidine sulfamate resulted in a hydrogen bond network similar to that of 4 b (Table S4 in the Supporting Information), but the additional pyrrolidine ring fitted snugly into a hydrophobic crevice lined by Ile96 (Figure S3 in the Supporting Information), thus explaining the recovered activity of 4 e compared to 4 b.
Although 4 a–4 f docked well into B‐Raf, showing favorable docking scores and binding modes consistent with those experimentally observed in the 3D4Q [29] and 4EHG [30] crystallographic complexes, none of the designed ligands resulted to be active in vitro (Table 2). To shed light into this discrepancy, a MD simulation of the B‐Raf complex with 4 e, which is the compound with the best docking score (Table S2 in the Supporting Information), was performed.
As described in the “molecular docking” section (see above), the pyridine nitrogen atom of 4 e established a H‐bond interaction with the backbone of the Cys532 hinge amino acid, the hydroxyl group hydrogen bonded with His539, and the sulfonamide group hydrogen bonded with the Asp594, Phe595 (of the DFG) and Lys483 (the conserved lysine) residues of the kinase (Figure S4 in the Supporting Information, panels a and b). However, after 20 ns of MD simulation, the pyridine ring moved far away from the hinge and the ligand lost most of the interactions with B‐Raf (Figure S4 in the Supporting Information, panel c). These results were rather unexpected, and contradictory compared to the docking results. We hypothesized that the compound was not able to efficiently accommodate within the B‐Raf binding site due to steric hindrance, which prevented it to stably place the pyridine ring in the hinge and the sulfonamide in the DFG pocket. Future work should consider shorter and less constrained molecules. Altogether, these results show how delicate is the design of multi‐target ligands that bind to targets with different architecture, especially when more than two targets are taken into consideration, as done in the present work. Indeed, the results of this study showed how the integration of different in silico approaches, e. g. docking and molecular dynamics, would provide more robust results in the validation of suitable candidates with respect to the application of a single approach. [46]
Finally, complexes of PDHK1 with compounds 4 a and 4 e were also further investigated by means of MD. As expected, the interactions established by the resorcinol moiety of 4 a and 4 e were very similar to those observed in Hsp90 (Figure S5 and Table S5 in the Supporting Information), the Asp318 residue in PDHK1 corresponding to Asp93 in Hsp90. However, the amide moiety of the two inhibitors gave marginal hydrogen bonds with the backbone carbonyl of Gly322, this residue corresponding to Gly97 in Hsp90. Rather, the amide moiety hydrogen bonded with Gly320, but again with very low percentages of occurrence (Table S5 in the Supporting Information). These findings may explain the lower inhibitory activity of 4 a and 4 e towards PDHK1 compared to Hsp90. Moreover, the hydrophobic crevice formed by Ile96 in Hsp90 is not present in PDHK1, this residue being a glycine in the latter protein, further explaining the lower activity of this compound.
Conclusion
In this study, we have designed and synthesized a set of novel potential multi‐target ligands by integrating molecular fragments selected from known Hsp90, B‐Raf and PDHK1 inhibitors. The study led to the identification of three potent inhibitors of Hsp90 with moderate PDHK1 inhibitory activity. Although activity against B‐Raf could not be obtained, the results herein presented are of significance, considering that three different biological targets were simultaneously addressed. The compounds were designed by following an integrated scaffold approach and exploiting crystal structure information, with the goal of keeping the molecular weight of the resulting compounds to a minimum. Before their synthesis, the designed compounds were validated by means of docking simulations into selected B‐Raf, Hsp90 and PHDHK1 conformations. The binding modes of the newly discovered dual Hsp90/PDHK1 inhibitors were investigated by means of molecular dynamics simulations, providing interesting explanations to the observed activity profiles. The rational design of multi‐target ligands has become a valuable opportunity and a real challenge in medicinal chemistry.[ 1 , 2 , 3 , 4 , 47 , 48 ] The data reported in this study is expected to pave the way to further rational design and optimization of ligands with more balanced multi‐target activity profiles.
Experimental Section
Molecular Modeling
The designed ligands were prepared for the structure‐based calculations by using the LigPrep (Schrödinger 2020–1) utility, [49] available in Maestro (Schrödinger 2020–1). [50] pKa predictions were performed using Epik. [33] Structure‐based calculations were performed into the 2VCI, [32] 2Q8G [26] and 3D4Q [29] crystal complexes of Hsp90, PDHK1 and B‐Raf, respectively. In particular, crystal structures were firstly pre‐processed with the Protein Preparation Wizard utility. [51] Atom types and bond connectivity issues were fixed, hydrogen atoms added and H‐bond networks optimized. Ions, water and solvent molecules were also removed from the complexes, except for the three buried water molecules that establish a conserved H‐bond networks with the ligands in Hsp90 and PDHK1.[ 26 , 32 , 52 ] Then, docking of ligands into the binding sites of the investigated targets was performed by using the multistage Induced Fit docking (IFD) protocol available in Maestro of Schrödinger suite 2020–1.[ 53 , 54 ] Re‐docking of the crystallographic compounds into their parent complexes was performed, providing satisfactory results (RMSD<2.0 Å). IFD calculations were performed with default settings, by focusing the calculations at the centroids of the co‐crystallized ligands for Hsp90 and B‐Raf. None of the PDHK1 crystal structures available in the PDB contained a ligand bound in the ATP site. Therefore, in this case the center of the ligand box was determined by superimposing the crystal structure of PDHK1 (PDB code: 2Q8G) with that of the homologous PDHK3 enzyme in complex with radicicol (PDB code: 2Q8I), [26] which shows similar fold and binding site to Hsp90 (PDB code: 4EGK). [36]
Molecular Dynamics
The molecular mechanics (MM) parameters for ligands 4 a, 4 b and 4 e (atom types and atomic charges) were assigned with the Antechamber module of Amber 18. [55] In particular, the ligands were assigned generalized amber force field (GAFF2) [56] atom types and AM1‐BCC [57] atomic charges. Missing force‐field parameters were assigned with the PARMCHECK utility. [55] Starting from the docking complexes, hydrogen atoms were added to the complexes using the internal coordinates of the Amber all‐atom data base. All Lys and Arg residues were positively charged and Glu and Asp residues negatively charged. All calculations in this study were performed with the Amber 18 [55] suite of programs, and the ff14SB force field [58] for the protein and the GAFF2 [56] force field for the ligands. Each protein‐ligand complex was solvated in an octahedral box of TIP3P [59] water molecules centered on the ligand and extending 10 Å outside the protein on all sides. The CUDA version of PMEMD was used to perform all molecular mechanics and molecular dynamics calculations. [55] The simulations employed a residue‐based cut‐off of 10 Å, a time step of 2 fs, and a constraint of bond lengths involving hydrogen atoms using the SHAKE [60] algorithm. The solvated complexes were minimized with 10000 steps of conjugate gradient minimization and equilibrated with MD at 300 K as follows. First, 100 ps MD at constant volume (NVP) with 2 kcal mol−1 Å−2 restraint on the protein and the ligand were performed in order to gradually heat the system from 0 K to 300 K without undesirable drifts of the structures. Then, MD was continued under constant pressure conditions (NPT, 1 atm) for 100 ps with 1 kcal mol−1Å−2 restraint on the same atoms, followed by additional 100+100 ps MD with gradually reduced restraints (0.5 and 0.2 kcal mol−1Å−2). Afterwards, the complexes were equilibrated for 40 ns, without restraints. After equilibration, production runs the length of 100 ns each were performed, and coordinates were collected every 10 ps, resulting in a ten thousand snapshots collected for each trajectory. The coordinates were also averaged every 2 ns for visual inspection. Hydrogen bond and root mean squared deviation analyses on the trajectories were made with the CPPTRAJ module of Amber 18. [55]
Chemical Synthesis
General Procedures
All available chemicals and solvents were purchased from commercial sources and were used without any further purification. Thin layer chromatography (TLC) was performed using 0.25 mm silica gel precoated plates Si 60‐F254 (Merck, Darmstadt, Germany) visualized by UV‐254 light and CAM staining. Purification by flash column chromatography (FCC) was conducted by using silica gel Si 60, 230–400 mesh, 0.040–0.063 mm (Merck). Melting points were determined on a Stuart Scientific SMP3 and are corrected. 1H and 13C NMR spectra were recorded on a Bruker Avance 400 (400 and 101 MHz, respectively) or Bruker Fourier 300 (300 and 75 MHz, respectively); chemical shifts are indicated in parts per million downfield from SiMe4, using the residual proton (CHCl3=7.26 ppm; DMSO=2.54 ppm) and carbon (CDCl3=77.0; ppm; DMSO=40.0 ppm) solvent resonances as internal reference. Protons and carbon assignments were achieved by 13C‐APT, 1H‐1H COSY, and 1H‐13C heteronuclear correlation experiments. Coupling constants values J are given in Hz.
Synthesis and Characterization of New Compounds
Ethyl 1‐[2,4‐bis(benzyloxy)‐5‐isopropylphenyl]‐5‐(pyridin‐4‐yl)‐1H‐1,2,3‐triazole‐4‐carboxylate (D)
[Cp*RuCl]4 (540 mg, 0.48 mmol) was added to a stirred solution of alkyne (ethyl 3‐(pyridin‐4‐yl)propiolate; 1.05 g, 6.0 mmol) and azide C (2.46 g, 6.6 mmol) in anhydrous DMF (8 ml) under nitrogen atmosphere at room temperature. After 16 h, water was added and the reaction was extracted with AcOEt (3×15 mL), washed with brine (1×5 mL) and dried with MgSO4. The solvent was removed under reduced pressure and the residue was purified by FCC – AcOEt/hexane (3 : 7) – on silica gel to afford triazole D as a yellow solid (2.93 g, 89 %). 1H NMR (300 MHz, CDCl3) δ 8.46 (d, 2H, J=5.5), 7.68–7.39 (m, 11H), 6.94 (d, 2H, J=5.30), 6.46 (s, 1H), 5.07 (s, 2H), 4.61 (s, 2H), 4.44–4.42 (m, 2H), 3.36–3.30 (m, 1H), 1.38 (t, 3H, J=7.3) 1.34(s, 3H), 1.22 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 161.5, 158.9, 151.8, 149.7 (2CHAr), 140.6, 137.12, 136.9, 135.8 (2CAr), 129.3, 129.0–128.1 (10CHAr), 125.0 (2CHAr), 124.1, 117.6, 99.4, 72.0, 71.5, 62.9, 27.1, 23.2 (2CH3), 14.8. ESI‐MS Calcd. For C33H32N4O4: 548.24 m/z. Found: [M+H]+549.25 m/z.
1‐[2,4‐bis(benzyloxy)‐5‐isopropylphenyl]‐5‐(pyridin‐4‐yl)‐1H‐1,2,3‐triazole‐4‐carboxylic Acid (1)
LiOH (359 mg, 15.0 mmol) was added to a solution of compound D (2.74 g, 5.0 mmol) in a 1 : 1 mixture of THF:H2O (50.0 mL). The reaction mixture was stirred at room temperature until completion (4 h), then was concentrated and quenched with HCl 1 N (5 mL). After extraction with AcOEt (2×20 mL), the collected organic phases were washed with water (1×10 mL), brine (1×10 mL) and dried over Na2SO4. Evaporation of the solvent in vacuum afforded compound 1 as a brown solid (2.47 g, 95 %). 1H NMR (300 MHz, CDCl3) δ 13.19 (bs, 1H), 8.54 (d, 2H, J=5.2 Hz), 7.42–7.33 (m, 9H), 7.23 (d, 2H, J=6.0), 7.14 (s, 1H), 7.21 (s, 1H), 6.90 (s, 1H), 5.13 (s, 2H), 5.01 (s, 2H), 3.21–3.14 (m, 1H), 1.12 (s, 3H), 1.09 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 166.1, 156.3, 152.1, 149.8 (2CHAr), 140.4, 136.5, 134.9 (2CAr), 129.4, 129.0, 128.4–127.7 (10CHAr), 126.4 (2CHAr), 124.4, 117.7, 99.1, 70.4 (2CH2), 26.7, 23.2 (2CH3). Anal. Calcd. For C31H28N4O4. C, 71.52; H, 5.42; N, 10.76. Found: C, 71.41; H, 5.33; N, 10.88.
General Procedure for the Preparation of Products 3 a–f.
To a stirred solution of carboxylic acid 1 (520 mg, 1.00 mmol) in 15 mL of anhydrous DCM and 0.5 mL of DIPEA were added compound 2 a–f (1.20 mmol) and HATU (570 mg, 1.5 mmol) at room temperature under nitrogen atmosphere. The reaction mixture was magnetically stirred at room temperature for 4 h, then was quenched by HCl 1 M and extracted with DCM (2×10 mL). The collected organic phases were washed with brine (1×10 mL), dried over Na2SO4 and the solvent was evaporated under vacuum (RV). The resulting crude was purified by FCC – AcOEt/hexane (1 : 1) – on silica gel. Yield, physical, spectroscopic and analytical data of products 3 a‐f are as follows.
1‐(2,4‐bis(benzyloxy)‐5‐isopropylphenyl)‐N‐(3‐(methylsulfonamido)phenyl)‐5‐(pyridin‐4‐yl)‐1H‐1,2,3‐triazole‐4‐carboxamide (3 a)
2 a (223 mg). 3 a (585 mg, 85 %). 1H NMR (300 MHz, DMSO‐d6 ) δ 9.33 (bs, 1H), 8.65–8.64 (m, 2H), 8.29 (s, 1H), 7.95 (s, 1H), 7.38–7.16 (m, 14 H), 6.92–6.89 (m, 2H), 6.58 (s, 1H), 4.99 (s, 2H), 4.70 (s, 2H), 3.36–3.27 (m, 1H), 3.12 (s, 3H), 1.21(s, 3H), 1.18(s, 3H); 13C NMR (75 MHz, DMSO‐d6 ) δ 162.6, 156.1, 151.8, 149.9 (2CAr), 149.7, 145.5, 138.8, 138.7, 138.1, 138.0, 131.9, 129.4, 128.2–127.2 (8CAr, 4CHAr), 125.0, 124.6, 122.2, 120.3, 120.0, 115.7, 103.1, 72.9, 72.8, 41.0, 24.5, 21.3 (2 CH3). Anal. Calcd. For C38H36N6O5S: C, 66.26; H, 5.27; N, 12.20. Found: C, 66.13; H, 5.16; N, 12.34.
1‐(2,4‐bis(benzyloxy)‐5‐isopropylphenyl)‐N‐(2,6‐difluoro‐3‐(methylsulfonamido)phenyl)‐5‐(pyridin‐4‐yl)‐1H‐1,2,3‐triazole‐4‐carboxamide (3 b)
2 b (267 mg). 3 b (341 mg, 47 %). 1H‐NMR (300 MHz, DMSO‐d6 ) δ 9.00 (bs, 1H), 8.45 (d, 2H, J=5.4 Hz), 7.37–7.28 (m, 9H), 7.17–7.15 (d, 2H, J=5.5 Hz), 7.03–7.01 (m, 1H), 6.96–6.95 (m, 2H), 6.75 (t, 1H, J=9.2 Hz), 6.70 (s, 1H) 6.57 (bs, 1H), 5.13 (s, 2H), 4.84 (s, 2H), 3.52 (s, 3H), 3.24–3.22 (m, 1H), 1.13 (s, 3H), 1.11 (s, 3H); 13C NMR (75 MHz, DMSO‐d6 ) δ 159.9, 154.2, 153.3–150.0 (m, 2CFAr), 151.5, 151.0, 149.4 (2CAr), 145.9, 138.1, 136.7, 133.1–122.4 (13CAr, 6CHAr), 120.0, 101.7, 73.9, 73.8, 50.3, 25.0, 23.9 (2CH3). Anal. Calcd. For C38H34F2N6O5S: C, 62.97; H, 4.73; N, 11.60. Found: C, 63.11; H, 4.85; N, 11.45.
[2,4‐bis(benzyloxy)‐5‐isopropylphenyl]‐N‐[3‐(ethylsulfonamido)‐2,6‐difluorophenyl]‐5‐(pyridin‐4‐yl)‐1H‐1,2,3‐triazole‐4‐carboxamide (3 c)
2 c (283 mg). 3 c (451 mg, 61 %). 1H NMR (400 MHz, DMSO‐d6 ) δ 8.86 (bs, 1H), 8.55 (d, 2H, J=5.1 Hz), 7.53 (d, 2H, J=5.1 Hz), 7.40–7.28 (m, 9H), 7.23 (d, 2H, J=5.6 Hz), 7.03–7.01 (m, 1H), 6.96–6.94 (m, 2H), 6.45 (s, 1H), 5.00 (s, 2H), 4.74 (s, 2H), 3.39–3.30 (m, 1H), 3.15–3.08 (m, 2H), 1.40 (t, 3H, J=7.5 Hz), 1.23 (s, 3H), 1.21 (s, 3H); 13C NMR (100 MHz, DMSO‐d6 ) δ 162.1, 155.0, 152.6–150.1 (m, 2CFAr), 151.3, 150.1, 149.6 (2CAr), 146.0, 138.5, 137.7, 134.1–122.1 (13CAr, 6CHAr), 120.0–119.9 (m, 1CHAr), 102.3, 74.3, 74.2, 51.1, 24.8, 23.9 (2 CH3), 15.9. Anal. Calcd. For C39H36F2N6O5S: C, 63.40; H, 4.91; N, 11.38. Found: C, 63.64; H, 5.12; N, 11.12.
1‐[2,4‐bis(benzyloxy)‐5‐isopropylphenyl]‐N‐[2,6‐difluoro‐3‐(propylsulfonamido)phenyl]‐5‐(pyridin‐4‐yl)‐1H‐1,2,3‐triazole‐4‐carboxamide (3 d)
2 d (300 mg). 3 d (369 mg, 49 %). 1H NMR (300 MHz, CDCl3) δ 8.82 (bs, 1H), 8.51 (d, 2H, J=5.5 Hz), 7.50 (d, 2H, J=5.5 Hz), 7.37–7.26 (m, 9H), 7.19 (d, 2H, J=5.6 Hz), 7.00 (t, 1H, J=7.2 Hz), 6.92–6.89 (m, 2H), 6.41 (s, 1H), 4.97 (s, 2H), 4.71 (s, 2H), 3.34–3.27 (m, 1H), 3.06–3.01 (m, 2H), 1.89–1.82 (m, 2H), 1.20 (s, 3H), 1.18 (s, 3H), 1.02 (t, 3H, J=7.1 Hz); 13C NMR (75 MHz, CDCl3) δ 164.6, 156.6, 153.8–151.1 (m, 2CFAr), 151.0, 149.6, 149.4 (2CAr), 145.8, 138.1, 187.9, 133.3–122.2 (13CAr, 6CHAr), 119.5–119.3 (m, 1CHAr), 101.4, 72.3, 72.2, 52.9, 25.2, 24.4 (2CH3), 18.6, 11.2. Anal. Calcd. For C40H38F2N6O5S: C, 63.82; H, 5.09; N, 11.16. Found: C, 64.05; H, 5.33; N, 10.95.
1‐[2,4‐bis(benzyloxy)‐5‐isopropylphenyl]‐N‐{3‐[(N,N‐dimethylsulfamoyl)amino]‐2,6‐difluorophenyl}‐5‐(pyridin‐4‐yl)‐1H‐1,2,3‐triazole‐4‐carboxamide (3 e)
2 e (302 mg). 3 e (407 mg, 54 %). 1H NMR (300 MHz, CDCl3) δ 8.89 (bs, 1H), 8.45 (d, 2H, J=5.4 Hz), 7.74 (d, 2H, J=5.4 Hz), 7.39–7.35 (m, 6H), 7.29–7.27 (m, 5H), 7.22–7.20 (m, 1H), 7.04–7.01 (m, 2H), 6.71 (s, 1H), 5.12 (s, 2H), 4.86 (s, 2H), 4.01–3.97 (m, 1H), 3.81 (s, 6H), 1.19 (s, 3H), 1.16(s, 3H); 13C NMR (75 MHz, DMSO‐d6 ) δ 161.9, 155.7, 154.4, 152.7–150.0 (m, 2CFAr), 149.5 (2CHAr), 141.4, 137.0, 136.9, 135.9, 135.0, 131.3, 129.6–128.5 (1CAr, 10CHAr), 126.1 (2CHAr), 125.9, 124.3, 118.2, 118.0, 112.4, 103.3, 72.2, 72.1, 46.1 (2CH3), 27.0, 22.4 (2CH3). Anal. Calcd. For C39H37F2N7O5S: C, 62.14; H, 4.95; N, 13.01. Found: C, C, 61.96; H, 4.81; N, 13.18.
1‐[2,4‐bis(benzyloxy)‐5‐isopropylphenyl]‐N‐[2,6‐difluoro‐3‐(pyrrolidine‐1‐sulfonamido)phenyl]‐5‐(pyridin‐4‐yl)‐1H‐1,2,3‐triazole‐4‐carboxamide (3 f)
2 e (333 mg). 3 e (445 mg, 57 %). 1H NMR (300 MHz, CDCl3) δ 8.91 (bs, 1H), 8.56–8.54 (d, 2H, J=Hz), 7.59–7.57 (m, 2H), 7.44–7.31 (m, 9H), 7.24 (d, 2H, J=5.5 Hz), 7.04–7.02 (m, 3H), 6.65 (s, 1H), 5.06 (s, 2H), 4.82 (s, 2H), 3.49–3.34 (m, 5H), 1.89–1.85 (m, 4H), 1.25(s, 3H), 1.23 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 160.4, 155.9, 153.0, 152.6–149.7 (m, 2CFAr), 150.1 (2CHAr), 142.2, 138.5, 135.6, 135.5, 135.1, 130.9, 130.1–128.5 (1CAr, 10CHAr), 126.7 (2CHAr), 126.0, 124.0, 117.8, 116.9, 111.1, 104.1, 73.2, 73.1, 46.9 (2CH2), 27.3, 23.9 (2CH3), 22.5 (2CH2). Anal. Calcd. For C41H39F2N7O5S: C, 63.15; H, 5.04; N, 12.57. Found: C, 63.43; H, 5.22; N, 12.41.
General Procedure for the Synthesis of Products 4 a–f
To a solution of 3 a‐f (0.3 mmol) in DCM dry (3 mL) was added BBr3 1 M in DCM (0.6 mL, 0.6 mmol) under nitrogen atmosphere at −78 °C. Then, the mixture was stirred at room temperature overnight. The reaction was cooled to 0 °C, methanol was added dropwise and the solvent was removed under vacuo. The residual was dissolved in ethyl acetate and washed with HCl 1 M (1×5 mL), brine (1×5 mL), dried over Na2SO4, and the solvent was removed under vacuo. The crude was purified by FCC – MeOH/DCM (5 : 95) – on silica gel. Yield, physical, spectroscopic and analytical data of products 4 a–f are as follows.
1‐(2,4‐dihydroxy‐5‐isopropylphenyl)‐N‐[3‐(methylsulfonamido)phenyl]‐5‐(pyridin‐4‐yl)‐1H‐1,2,3‐triazole‐4‐carboxamide (4 a)
3 a (207 mg). 4 a (119 mg, 78 %). 1H NMR (400 MHz, DMSO‐d6 ) δ 9.01 (bs, 2H), 8.69 (d, 2H, J=5.3 Hz), 7.81 (d, 2H, J=5.3 Hz), 7.89 (s, 1H), 7.61–7.52 (m, 4H), 6.77 (bs, 1H), 6.49 (s, 1H), 5.79 (bs, 1H), 3.17–3.13 (m, 1H), 3.00 (s, 3H), 1.10 (s, 3H), 1.08 (s, 3H); 13C NMR (75 MHz, DMSO‐d6 ) δ 163.0, 155.8, 152.2, 149.9 (2CHAr), 142.3, 139.1, 138.5, 134.0, 130.9, 129.2, 128.2, 124.9 (2CHAr), 124.0, 120.1, 119.4, 119.3, 114.7, 104.4, 42.1, 26.0, 21.4 (2CH3). Anal. Calcd. For C24H24N6O5S: C, 56.68; H, 4.76; N, 16.53. Found: C, 56.83; H, 4.87; N, 16.44.
N‐[2,6‐difluoro‐3‐(methylsulfonamido)phenyl]‐1‐(2,4‐dihydroxy‐5‐isopropylphenyl)‐5‐(pyridin‐4‐yl)‐1H‐1,2,3‐triazole‐4‐carboxamide (4 b)
3 b (217 mg). 4 b (145 mg, 89 %). 1H NMR (400 MHz, DMSO‐d6 ) δ 9.82 (bs, 3H), 8.63–8.61 (m, 2H), 7.31–7.29 (m, 2H), 7.02 (s, 1H), 6.95–6.93 (m, 1H), 6.72–6.69 (m, 1H), 6.37 (s, 1H), 5.49 (bs, 1H), 3.19 (s, 3H), 3.07–3.01 (m, 1H), 1.06 (s, 3H), 1.04 (s, 3H); 13C NMR (100 MHz, DMSO‐d6 ) δ 161.7, 154.1, 151.7, 152.1–149.5 (m, 2CFAr), 149.8 (2CHAr), 143.4, 140.0, 133.9, 130.5, 129.6, 124.4 (2CHAr), 124.3, 122.2, 119.9–119.8 (m, 1CHAr), 119.7, 119.0–118.8 (m,1CHAr), 104.0, 41.0, 25.8, 22.2 (2CH3). Anal. Calcd. For C24H22F2N6O5S: C, 52.94; H, 4.07; N, 15.43. Found: C, 53.23; H, 4.33; N, 15.16.
1‐(2,4‐dihydroxy‐5‐isopropylphenyl)‐N‐[3‐(ethylsulfonamido)‐2,6‐difluorophenyl]‐5‐(pyridin‐4‐yl)‐1H‐1,2,3‐triazole‐4‐carboxamide (4 c)
3 c (222 mg). 4 c (159 mg, 95 %). 1H NMR (400 MHz, DMSO‐d6 ) δ 9.45 (bs, 4H), 8.57–8.55 (m, 2H), 7.32–7.30 (m, 2H), 7.39 (s, 1H), 7.05–7.01 (m, 1H), 6.91–6.93 (m, 1H), 6.44 (s, 1H), 3.30–3.11 (m, 3H), 1.07 (s, 3H), 1.04–1.01 (m, 6H); 13C NMR (75 MHz, DMSO‐d6 ) δ 159.4, 153.6, 151.9, 151.9–149.5 (m, 2CFAr), 149.9 (2CHAr), 145.2, 141.3, 134.7, 131.0, 129.4, 124.1 (2CHAr), 122.9, 122.8, 120.1–119.8 (m, 1CHAr), 119.0, 117.9–117.8 (m,1CHAr), 105.1, 44.0, 25.8, 23.3 (2CH3), 21.9. Anal. Calcd. For C25H24F2N6O5S: C, 53.76; H, 4.33; N, 15.05. Found: C, 54.01; H, 4.54; N, 14.82.
N‐[2,6‐difluoro‐3‐(propylsulfonamido)phenyl]‐1‐(2,4‐dihydroxy‐5‐isopropylphenyl)‐5‐(pyridin‐4‐yl)‐1H‐1,2,3‐triazole‐4‐carboxamide (4 d)
3 d (226 mg). 4 d (168 mg, 98 %).1H NMR (300 MHz, DMSO‐d6 ) δ 9.69 (bs, 3H), 8.67 (d, 2H, J=5.5 Hz), 7.41 (d, 2H, J=5.5 Hz), 7.36 (s, 1H), 7.00–6.96 (m, 1H), 6.89–6.87 (m, 1H), 6.50 (s, 1H), 4.99 (bs, 1H), 3.21–3.09 (m, 3H), 1.86–1.79 (m, 2H), 1.06 (s, 3H), 1.04–1.00 (m, 6H); 13C NMR (75 MHz, DMSO‐d6 ) δ 163.0, 154.9, 151.1, 151.3–148.8 (m, 2CFAr), 149.9 (2 CHAr), 144.1, 141.0, 134.3, 131.2, 129.4, 124.0 (2CHAr), 123.8, 122.2, 120.0–119.9 (m, 1CHAr), 119.6, 119.2–118.9 (m,1 CHAr), 103.7, 45.8, 27.0, 24.9 (2CH3), 21.4, 17.9. Anal. Calcd. For C26H26F2N6O5S: C, 54.54; H, 4.58; N, 14.68. Found: C, 54.77; H, 4.74; N, 14.88.
1‐(2,4‐dihydroxy‐5‐isopropylphenyl)‐N‐{3‐[(N,N‐dimethylsulfamoyl)amino]‐2,6‐difluorophenyl}‐5‐(pyridin‐4‐yl)‐1H‐1,2,3‐triazole‐4‐carboxamide (4 e)
3 e (226 mg). 4 e (145 mg, 84 %). 1H NMR (400 MHz, DMSO‐d6 ) δ 9.82 (bs, 4H), 8.59 (d, 2H, J=5.4 Hz), 7.35 (d, 2H, J=5.4 Hz), 7.16–7.12 (m, 1H), 7.12 (s, 1H), 7.11–7.10 (m, 1H), 6.38 (s, 1H), 3.78 (s, 6H), 3.081–2.96 (m, 1H), 1.09 (s, 3H), 1.07 (s, 3H); 13C NMR (100 MHz, DMSO‐d6 ) δ 160.9, 153.8, 152.5, 152.0–149.4 (m, 2CFAr), 149.9 (2CHAr), 142.0, 138.8, 130.2, 130.1, 129.9, 124.3, 124.1 (2CHAr), 120.0, 119.9 (m, 1CHAr), 119.5, 119.0 (m, 1CHAr), 105.1, 49.7 (2CH3), 23.9, 21.4 (2CH3). Anal. Calcd. For C25H25F2N7O5S: C, 52.35; H, 4.39; N, 17.09. Found: C, 52.16; H, 4.21; N, 17.25.
N‐[2,6‐difluoro‐3‐(pyrrolidine‐1‐sulfonamido)phenyl]‐1‐(2,4‐dihydroxy‐5‐isopropylphenyl)‐5‐(pyridin‐4‐yl)‐1H‐1,2,3‐triazole‐4‐carboxamide (4 f)
3 f (234 mg). 4 f (153 mg, 85 %). 1H NMR (300 MHz, DMSO‐d6 ) δ 9.01–8.99 (bs, 3H), 8.68 (d, 2H, J=5.5 Hz), 7.85 (d, 2H, J=5.5 Hz), 7.27–7.10 (m, 3H), 6.45 (s, 1H), 6.00 (bs, 1H), 3.75–3.59 (m, 4H), 3.081–2.96 (m, 1H), 1.78–1.69 (m, 4H), 1.11 (s, 3H), 1.08 (s, 3H); 13C NMR (75 MHz, DMSO‐d6 ) δ 162.3, 154.9, 153.6, 151.8–149.7 (m, 2CFAr), 150.2 (2CHAr), 142.5, 138.4, 131.3, 130.6, 129.2, 124.4 (2CHAr), 124.3, 120.2, 120.0 (m, 1CHAr), 119.1, 119.0 (m, 1CHAr), 103.7, 50.1 (2CH2), 25.0, 24.8 (2CH2), 21.4 (2CH3). Anal. Calcd. For C27H27F2N7O5S: C, 54.08; H, 4.54; N, 16.35. Found: C, 54.33; H, 4.76; N, 16.08.
Biological Assays
Hsp90 Assays
The Hsp90 assay is based on the competition of fluorescently labelled geldanamycin (FITC‐GM) for the binding to Hsp90. [61] Because FITC‐GM binds to the ATP binding pocket of Hsp90, the assay is able to detect ATP‐competitive inhibitors. Hsp90 assays were performed at Reaction Biology Corporation using human recombinant Hsp90α at a 30 nm concentration (GenBank Accession No. NM_005348) with His‐tag, MW=90 kDa, expressed in E.coli expression system. The reaction buffer consisted in 20 mM HEPES, pH 7.5, 50 mM NaCl, 10 mM MgCl2, 0.02 % Brij 35, Add fresh: 2 mM DTT, 0.02 mg/ml BSA, 1 % DMSO. The compounds were firstly solubilized at 10 mM in 100 % DMSO, then serially diluted 1 to 3 (10 concentrations) starting from 100 μM. Geldanamycin was used as a positive control. Compounds were delivered into the enzyme mixture by Acoustic technology (Echo550; nanoliter range), and incubated for 30 min. The FITC‐labeled Geldanamycin probe was added at a 5 nm concentration to initiate the reaction and incubated for 3 hr at room temperature with gentle mixing. Fluorescence polarization was then read and IC50 values and curve fits were calculated using Prism (GraphPad Software).
Kinase Assays
Kinase assays (B‐RafWT, B‐RafV600E and PDHK1) were performed at Reaction Biology Corporation using the “HotSpot” assay platform. [62] To this aim, specific kinase/substrate pairs along with required cofactors were prepared in reaction buffer; 20 mm Hepes pH 7.5, 10 mm MgCl2, 1 mM EGTA, 0.01 % Brij35, 0.02 mg/ml BSA, 0.1 mM Na3VO4, 2 mm DTT, 1 % DMSO. The compounds were first solubilized at 10 mm in 100 % DMSO, then serially diluted 1 to 3 (10 concentrations) starting from 100 μM. GW5074 was used as a positive control. Compounds were delivered into the reaction, followed ∼20 min later by addition of a mixture of ATP (Sigma) and 33P‐ATP (PerkinElmer) to a final 10 μm concentration to initiate the reaction. Reactions were carried out at 25 °C for 120 min, followed by spotting of the reactions onto P81 ion exchange filter paper (Whatman). Unbound phosphate was removed by extensive washing of filters in 0.75 % phosphoric acid. After subtraction of background derived from control reactions containing inactive enzyme, kinase activity data were expressed as the percent remaining kinase activity in test samples compared to vehicle (dimethyl sulfoxide) reactions. IC50 values and curve fits were obtained using Prism (GraphPad Software).
Conflict of interest
The authors declare no conflict of interest relevant to the ideas or the contents of this manuscript.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
This work was supported by a grant from the Associazione Italiana per la Ricerca sul Cancro [AIRC IG 23635]. L.P. was supported by a FIRC‐AIRC fellowship [rif. 24096].
L. Pinzi, F. Foschi, M. S. Christodoulou, D. Passarella, G. Rastelli, ChemistryOpen 2021, 10, 1177.
References
- 1. Anighoro A., Bajorath J., Rastelli G., J. Med. Chem. 2014, 57, 7874–7887. [DOI] [PubMed] [Google Scholar]
- 2. Peters J.-U., J. Med. Chem. 2013, 56, 8955–8971. [DOI] [PubMed] [Google Scholar]
- 3. Proschak E., Stark H., Merk D., J. Med. Chem. 2019, 62, 420–444. [DOI] [PubMed] [Google Scholar]
- 4. Bolognesi M. L., ACS Med. Chem. Lett. 2019, 10, 273–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Makhoba X. H., Jr Viegas C., Mosa R. A., Viegas F. P. D., Pooe O. J., Drug Des. Dev. Ther. 2020, 14, 3235–3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yumura M., Nagano T., Nishimura Y., Molecules. 2020, 25(17), 3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang T., Sui X., Yu B., Shen Y., Cong H., Curr. Med. Chem. 2020, 27(28), 4720–4740. [DOI] [PubMed] [Google Scholar]
- 8. Maramai S., Benchekroun M., Gabr M. T., Yahiaoui S., BioMed Res. Int. 2020, 2020, 5120230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rastelli G., Pinzi L., Front. Pharmacol. 2015, 6, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Echeverría P. C., Picard D., in The Molecular Chaperones Interaction Networks in Protein Folding and Degradation, ed. W. A. Houry, Springer-Verlag, New York, 2014, ch. 5, pp. 133–150. [Google Scholar]
- 11. Anighoro A., Stumpfe D., Heikamp K., Beebe K., Neckers L. M., Bajorath J., Rastelli G., J. Chem. Inf. Model. 2015, 55, 676–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lin S., Zhang L., Zhang X., Yu Z., Huang X., Xu J., Liu Y., Chen L., Wu L., Bioorg. Med. Chem. 2020, 28(9), 115434. [DOI] [PubMed] [Google Scholar]
- 13. Yao L., Ohlson S., Dymock B. W., Bioorg. Med. Chem. Lett. 2018, 28, 1357–1362. [DOI] [PubMed] [Google Scholar]
- 14. Antolin A. A., Clarke P. A., Collins I., Workman P., Al-Lazikani B., Cell Chem. Biol. 2021, 27, S2451–9456(21)00221-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li J., Buchner J., Biomed J 2013, 36, 106–117. [DOI] [PubMed] [Google Scholar]
- 16. Neckers L., Workman P., Clin. Cancer Res. 2012, 18, 64–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Grbovic O. M., Basso A. D., Sawai A., Ye Q., Friedlander P., Solit D., Rosen N., Proc. Natl. Acad. Sci. USA 2006, 103, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Solárová Z., Mojžiš J., Solár P., Int. J. Oncol. 2015, 46, 907–926. [DOI] [PubMed] [Google Scholar]
- 19. Acquaviva J., Smith D. L., Jimenez J.-P., Zhang C., Sequeira M., He S., Sang J., Bates R. C., Proia D. A., Mol. Cancer Ther. 2014, 13, 353–363. [DOI] [PubMed] [Google Scholar]
- 20. Zhang G., Frederick D. T., Wu L., Wei Z., Krepler C., Srinivasan S., Chae Y. C., Xu X., Choi H., Dimwamwa E., Ope O., Shannan B., Basu D., Zhang D., Guha M., Xiao M., Randell S., Sproesser K., Xu W., Liu J., Karakousis G. C., Schuchter L. M., Gangadhar T. C., Amaravadi R. K., Gu M., Xu C., Ghosh A., Xu W., Tian T., Zhang J., Zha S., Liu Q., Brafford P., Weeraratna A., Davies M. A., Wargo J. A., Avadhani N. G., Lu Y., Mills G. B., Altieri D. C., Flaherty K. T., Herlyn M., J. Clin. Invest. 2016, 126, 1834–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Suzuki R., Kikuchi S., Harada T., Mimura N., Minami J., Ohguchi H., Yoshida Y., Sagawa M., Gorgun G., Cirstea D., Cottini F., Jakubikova J., Tai Y.-T., Chauhan D., Richardson P. G., Munshi N., Ando K., Utsugi T., Hideshima T., Anderson K. C., PLoS One 2015, 10, e0143847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kryeziu K., Bruun J., Guren T. K., Sveen A., Lothe R. A., Biochim. Biophys. Acta Rev. Cancer. 2019, 1871(2), 240–247. [DOI] [PubMed] [Google Scholar]
- 23. Bowker-Kinley M., Popov K. M., Biochem. J. 1999, 344 Pt 1, 47–53. [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang W., Zhang S.-L., Hu X., Tam K. Y., Int. J. Biol. Sci. 2015, 11, 1390–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kaplon J., Zheng L., Meissl K., Chaneton B., Selivanov V. A., Mackay G., van der Burg S. H., Verdegaal E. M. E., Cascante M., Shlomi T., Gottlieb E., Peeper D. S., Nature 2013, 498, 109–112. [DOI] [PubMed] [Google Scholar]
- 26. Kato M., Li J., Chuang J. L., Chuang D. T., Structure 2007, 15, 992–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Taddei M., Ferrini S., Giannotti L., Corsi M., Manetti F., Giannini G., Vesci L., Milazzo F. M., Alloatti D., Guglielmi M. B., Castorina M., Cervoni M. L., Barbarino M., Foderà R., Carollo V., Pisano C., Armaroli S., Cabri W., J. Med. Chem. 2014, 57, 2258–2274. [DOI] [PubMed] [Google Scholar]
- 28. Meng T., Zhang D., Xie Z., Yu T., Wu S., Wyder L., Regenass U., Hilpert K., Huang M., Geng M., Shen J., J. Med. Chem. 2014, 57, 9832–9843. [DOI] [PubMed] [Google Scholar]
- 29. Hansen J. D., Grina J., Newhouse B., Welch M., Topalov G., Littman N., Callejo M., Gloor S., Martinson M., Laird E., Brandhuber B. J., Vigers G., Morales T., Woessner R., Randolph N., Lyssikatos J., Olivero A., Bioorg. Med. Chem. Lett. 2008, 18, 4692–4695. [DOI] [PubMed] [Google Scholar]
- 30. Mathieu S., Gradl S. N., Ren L., Wen Z., Aliagas I., Gunzner-Toste J., Lee W., Pulk R., Zhao G., Alicke B., Boggs J. W., Buckmelter A. J., Choo E. F., Dinkel V., Gloor S. L., Gould S. E., Hansen J. D., Hastings G., Hatzivassiliou G., Laird E. R., Moreno D., Ran Y., Voegtli W. C., Wenglowsky S., Grina J., Rudolph J., J. Med. Chem. 2012, 55, 2869–2881. [DOI] [PubMed] [Google Scholar]
- 31. Wenglowsky S., Ahrendt K. A., Buckmelter A. J., Feng B., Gloor S. L., Gradl S., Grina J., Hansen J. D., Laird E. R., Lunghofer P., Mathieu S., Moreno D., Newhouse B., Ren L., Risom T., Rudolph J., Seo J., Sturgis H. L., Voegtli W. C., Wen Z., Bioorg. Med. Chem. Lett. 2011, 21, 5533–5537. [DOI] [PubMed] [Google Scholar]
- 32. Brough P. A., Aherne W., Barril X., Borgognoni J., Boxall K., Cansfield J. E., Cheung K.-M. J., Collins I., Davies N. G. M., Drysdale M. J., Dymock B., Eccles S. A., Finch H., Fink A., Hayes A., Howes R., Hubbard R. E., James K., Jordan A. M., Lockie A., Martins V., Massey A., Matthews T. P., McDonald E., Northfield C. J., Pearl L. H., Prodromou C., Ray S., Raynaud F. I., Roughley S. D., Sharp S. Y., Surgenor A., Walmsley D. L., Webb P., Wood M., Workman P., Wright L., J. Med. Chem. 2008, 51, 196–218. [DOI] [PubMed] [Google Scholar]
- 33. Schrödinger Release 2020–1: Epik, Schrödinger, LLC, New York, NY, 2020.
- 34. Greenwood J. R., Calkins D., Sullivan A. P., Shelley J. C., J. Comput.-Aided Mol. Des. 2010, 24, 591–604. [DOI] [PubMed] [Google Scholar]
- 35. Tsai J., Lee J. T., Wang W., Zhang J., Cho H., Mamo S., Bremer R., Gillette S., Kong J., Haass N. K., Sproesser K., Li L., Smalley K. S., Fong D., Zhu Y. L., Marimuthu A., Nguyen H., Lam B., Liu J., Cheung I., Rice J., Suzuki Y., Luu C., Settachatgul C., Shellooe R., Cantwell J., Kim S. H., Schlessinger J., Zhang K. Y., West B. L., Powell B., Habets G., Zhang C., Ibrahim P. N., Hirth P., Artis D. R., Herlyn M., Bollag G., Proc. Natl. Acad. Sci. USA. 2008, 105(8), 3041–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Austin C., Pettit S. N., Magnolo S. K., Sanvoisin J., Chen W., Wood S. P., Freeman L. D., Pengelly R. J., Hughes D. E., J. Biomol. Screening 2012, 17, 868–876. [DOI] [PubMed] [Google Scholar]
- 37. Bonandi E., Christodoulou M. S., Fumagalli G., Perdicchia D., Rastelli G., Passarella D., Drug Discovery Today 2017, 22, 1572–1581. [DOI] [PubMed] [Google Scholar]
- 38. Gabba A., Robakiewicz S., Taciak B., Ulewicz K., Broggini G., Rastelli G., Krol M., Murphy P. V., Passarella D., Eur. J. Org. Chem. 2017, 2017, 60–69. [Google Scholar]
- 39. Colombo F., Tintori C., Furlan A., Borrelli S., Christodoulou M. S., Dono R., Maina F., Botta M., Amat M., Bosch J., Passarella D., Bioorg. Med. Chem. Lett. 2012, 22, 4693–4696. [DOI] [PubMed] [Google Scholar]
- 40. Majireck M. M., Weinreb S. M., J. Org. Chem. 2006, 71, 8680–8683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu L., Lee M. R., Kim J. L., Whittington D. A., Bregman H., Hua Z., Lewis R. T., Martin M. W., Nishimura N., Potashman M., Yang K., Yi S., Vaida K. R., Epstein L. F., Babij C., Fernando M., Carnahan J., Norman M. H., Bioorg. Med. Chem. 2016, 24, 2215–2234. [DOI] [PubMed] [Google Scholar]
- 42. Lagunin A. A., Dubovskaja V. I., Rudik A. V., Pogodin P. V., Druzhilovskiy D. S., Gloriozova T. A., Filimonov D. A., Sastry N. G., Poroikov V. V., PLoS One 2018, 13(1), e0191838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Esfahani K., Cohen V., Lung Cancer (Auckl) 2016, 7, 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dyrstad S. E., Lotsberg M. L., Tan T. Z., Pettersen I. K. N., Hjellbrekke S., Tusubira D., Engelsen A. S. T., Daubon T., Mourier A., Thiery J. P., Dahl O., Lorens J. B., Tronstad K. J., Røsland G. V., Cancers 2021, 13(5), 941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Leonetti A., Facchinetti F., Rossi G., Minari R., Conti A., Friboulet L., Tiseo M., Planchard D., Cancer Treat. Rev. 2018, 66, 82–94. [DOI] [PubMed] [Google Scholar]
- 46. Pinzi L., Rastelli G., Int. J. Mol. Sci. 2019, 20(18), 4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Anighoro A., Pinzi L., Marverti G., Bajorath J., Rastelli G., RSC Adv. 2017, 7, 31069–31074. [Google Scholar]
- 48. Pinzi L., Benedetti R., Altucci L., Rastelli G., ACS Omega 2020, 5, 11473–11480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schrödinger Release 2020–1: LigPrep, Schrödinger, LLC, New York, NY, 2020.
- 50. Schrödinger Release 2020–1: Maestro, Schrödinger, LLC, New York, NY, 2020.
- 51. Sastry G. M., Adzhigirey M., Day T., Annabhimoju R., Sherman W., J. Comput.-Aided Mol. Des. 2013, 27(3), 221–234. [DOI] [PubMed] [Google Scholar]
- 52. Sgobba M., Rastelli G., ChemMedChem 2009, 4, 1399–1409. [DOI] [PubMed] [Google Scholar]
- 53. Schrödinger Release 2020–1: Induced Fit Docking protocol; Glide, Schrödinger, LLC, New York, NY, 2020; Prime, Schrödinger, LLC, New York, NY, 2020.
- 54. Sherman W., Day T., Jacobson M. P., Friesner R. A., Farid R., J. Med. Chem. 2006, 49, 534–553. [DOI] [PubMed] [Google Scholar]
- 55. Case D. A., Belfon K., Ben-Shalom I. Y., Brozell S. R., Cerutti D. S., T. E. Cheatham, III , Cruzeiro V. W. D., Darden T. A., Duke R. E., Giambasu G., Gilson M. K., Gohlke H., Goetz A. W., Harris R., Izadi S., Izmailov S. A., Kasavajhala K., Kovalenko A., Krasny R., Kurtzman T., Lee T. S., LeGrand S., Li P., Lin C., Liu J., Luchko T., Luo R., Man V., Merz K. M., Miao Y., Mikhailovskii O., Monard G., Nguyen H., Onufriev A., Pan F., Pantano S., Qi R., Roe D. R., Roitberg A., Sagui C., Schott-Verdugo S., Shen J., Simmerling C. L., Skrynnikov N. R., Smith J., Swails J., Walker R. C., Wang J., Wilson L., Wolf R. M., Wu X., Xiong Y., Xue Y., York D. M., Kollman P. A., AMBER 2018, University Of California, San Francisco, 2018. [Google Scholar]
- 56. He X., Man V. H., Yang W., Lee T.-S., Wang J., J. Chem. Phys. 2020, 153, 114502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jakalian A., Bush B. L., Jack D. B., Bayly C. I., J. Comput. Chem. 2000, 21, 132–146. [Google Scholar]
- 58. Tian C., Kasavajhala K., Belfon K. A. A., Raguette L., Huang H., Migues A. N., Bickel J., Wang Y., Pincay J., Wu Q., Simmerling C., J. Chem. Theory Comput. 2020, 16, 528–552. [DOI] [PubMed] [Google Scholar]
- 59. Åqvist J., J. Phys. Chem. 1990, 94, 8021–8024. [Google Scholar]
- 60. Ryckaert J.-P., Ciccotti G., Berendsen H. J. C., J. Comput. Phys. 1977, 23, 327–341. [Google Scholar]
- 61. Howes R., Barril X., Dymock B. W., Grant K., Northfield C. J., Robertson A. G. S., Surgenor A., Wayne J., Wright L., James K., Matthews T., Cheung K.-M., McDonald E., Workman P., Drysdale M. J., Anal. Biochem. 2006, 350, 202–213. [DOI] [PubMed] [Google Scholar]
- 62. Anastassiadis T., Deacon S. W., Devarajan K., Ma H., Peterson J. R., Nat. Biotechnol. 2011, 29, 1039–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information