

SUMMARY
Group 3 innate lymphoid cells (ILC3s) critically regulate host-microbe interactions in the gastrointestinal tract, but their role in the airway remains poorly understood. Here, we demonstrate that lymphoid-tissue-inducer (LTi)-like ILC3s are enriched in the lung-draining lymph nodes of healthy mice and humans. These ILC3s abundantly express major histocompatibility complex class II (MHC class II) and functionally restrict the expansion of allergen-specific CD4+ T cells upon experimental airway challenge. In a mouse model of house-dust-mite-induced allergic airway inflammation, MHC class II+ ILC3s limit T helper type 2 (Th2) cell responses, eosinophilia, and airway hyperresponsiveness. Furthermore, MHC class II+ ILC3s limit a concomitant Th17 cell response and airway neutrophilia. This exacerbated Th17 cell response requires exposure of the lung to microbial stimuli, which can be found associated with house dust mites. These findings demonstrate a critical role for antigen-presenting ILC3s in orchestrating immune tolerance in the airway by restricting pro-inflammatory T cell responses to both allergens and microbes.
Graphical Abstract
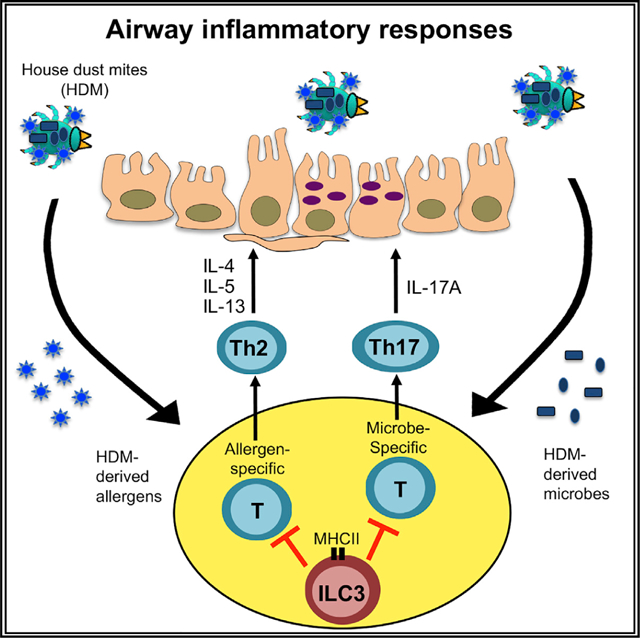
In brief
In this study, Teng et al. demonstrate that an innate immune cell type, ILC3, is enriched in the lung draining lymph node of healthy humans and mice and functions to limit airway inflammation through antigen presentation and control of T cell responses directed against allergens or microbes.
INTRODUCTION
Immune responses in the mammalian airways must be tightly regulated in order to protect from invading pathogens, while also preventing over-active inflammatory responses to innocuous environmental stimuli, including allergens. Indeed, hyper-activated immune responses following repeat exposure with common environmental allergens, such as animal dander, grass pollen, or house dust mite (HDM), underlie the pathogenesis of allergic airway inflammation and asthma (Bosteels et al., 2020; Hammad and Lambrecht, 2021; Hewitt and Lloyd, 2021; Lambrecht et al., 2019; Lloyd and Marsland, 2017). T helper type 2 (Th2) cells are well-defined to be major orchestrators of allergic inflammation in the lung. Through production of interleukin-4 (IL-4), IL-5, and IL-13, Th2 cells promote eosinophilia, goblet cell hyperplasia, smooth muscle contraction, and airway hyperresponsiveness (AHR) (Hammad and Lambrecht, 2021; Hewitt and Lloyd, 2021; Lambrecht et al., 2019; Lloyd and Marsland, 2017). In addition, emerging evidence has demonstrated considerable heterogeneity of the immune response among individuals with asthma, implicating Th17 cell responses and neutrophilic infiltration as key factors driving allergic airway disease (Alcorn et al., 2010; Newcomb and Peebles, 2013; Wang and Wills-Karp, 2011). Elevated Th17 cell responses in asthma patients correlate with severity of disease and resistance to steroid therapies (Alcorn et al., 2010; Newcomb and Peebles, 2013; Wang and Wills-Karp, 2011). Similarly, in murine models of asthma, Th17 cells exacerbate allergic airway inflammation by promoting the influx of neutrophils to the inflamed tissue (Alcorn et al., 2010). Therefore, there is an urgent need to better understand the pathways that control Th2 and Th17 cell responses in the context of lung inflammation.
Innate lymphoid cells (ILCs) are a heterogeneous family of innate lymphocytes that are enriched at mucosal barrier sites of the body, are predominantly tissue resident, and dramatically impact immunity, inflammation, and tolerance (Artis and Spits, 2015; Sonnenberg and Hepworth, 2019; Spits et al., 2013; Vivier et al., 2018). Often referred to as innate counterparts to populations of Th cells, ILCs are classified into three distinct groups based upon shared transcription factor expression and effector cytokine production (Spits et al., 2013). The group 2 ILC (ILC2) subset exhibits phenotypic and functional similarities to Th2 cells, is abundantly present in the airways of humans and mice, and has been robustly studied in the context of asthma (Bartemes and Kita, 2021; Diefenbach et al., 2014; McKenzie et al., 2014; Vivier et al., 2018). ILC2s directly promote allergic airway inflammation through the production of IL-5 and IL-13 and support Th2 cell responses through direct cellular interactions (Bartemes and Kita, 2021; Diefenbach et al., 2014; Halim et al., 2012, 2014; McKenzie et al., 2014; Mirchandani et al., 2014; Oliphant et al., 2014; Vivier et al., 2018). In contrast, group 3 ILCs (ILC3s) resemble Th17 cells and are well-studied in the gastrointestinal tract. At this mucosal site, ILC3s are major regulators of host-microbe interactions through the production of IL-17 or IL-22 (Sonnenberg and Artis, 2015). These cytokines drive antimicrobial responses that shape the microbiota and protect from invading pathogens (Ouyang et al., 2008; Sonnenberg et al., 2011; Zheng et al., 2008). Furthermore, ILC3s are major regulators of immunological tolerance to the microbiota and dietary antigens in the intestine, in part through the production of granulocyte-macrophage colony-stimulating factor (GM-CSF) and/or IL-2 that subsequently support regulatory T cell (Treg) homeostasis (Mortha et al., 2014; Zhou et al., 2019) or through direct regulation of CD4+ T cells by antigen presentation on major histocompatibility complex class II (MHC class II) (Goc et al., 2021; Hepworth et al., 2013, 2015; Melo-Gonzalez et al., 2019; Sonnenberg and Hepworth, 2019).
Despite this knowledge, the role of ILC3s in the airway is poorly understood and it is unclear whether these cells contribute to immune tolerance at this site. This is in part because ILC3s are largely absent from the healthy airway of mice housed in specific pathogen free (SPF) conditions. However, several studies demonstrated that ILC3s are enriched in the airway during neonatal periods or in the context of obesity and tuberculosis infection, in which their production of effector cytokines contributes to host defense and airway inflammation (Ardain et al., 2019; Kim et al., 2014; Oherle et al., 2020; Xiong et al., 2016). Here, we unexpectedly identify a role for ILC3s in allergic airway inflammation. Specifically, we determine that antigen-presenting ILC3s are enriched in the lung draining lymphoid tissues of healthy mice and humans, functionally restrict allergen-specific CD4+ T cell responses, and limit type-2-mediated allergic inflammation. Furthermore, we find that MHC class II+ ILC3s limit the expansion of microbe-reactive Th17 cells in the airway and define the presence of various compositions of microbes directly associated with HDM extracts. These results advance our understanding of the role of ILC3s in the airway, as well as define a pathway controlling pro-inflammatory Th2 and Th17 cell responses in the inflamed lung.
RESULTS
ILC3s are enriched in the lung mediastinal lymph nodes and express high levels of MHC class II
To better define the role of ILC3s in the airway, we examined their presence in the lung and associated lymphoid tissues. Consistent with prior reports (Bartemes and Kita, 2021; Diefenbach et al., 2014; McKenzie et al., 2014; Vivier et al., 2018), ILC2s are the dominant subset within the lung parenchyma of healthy SPF mice and only a small population of ILC3s can be found in this tissue (Figures 1A and 1B). In contrast, we identify that ILC3s are the dominant subset within the lung-draining mediastinal lymph nodes (MedLNs) (Figures 1A, 1B, and S1A). ILC3s in the MedLNs exhibit dimmer CD45, lacked the ILC2 marker KLRG1, expressed higher levels of CD127 and CD25 than ILC2, and lacked markers associated with myeloid cells such as CD11b and CD11c (Figure 1C). Furthermore, these ILC3s exhibit high levels of CCR6, heterogeneous staining for CD4, and absent staining for T-bet and NKp46 (Figure 1C), indicating that they resemble CCR6+ LTi-like ILC3s. The presence of ILC3s in the MedLNs suggests that these populations could impact the adaptive immune response. Consistent with this idea, ILC3s within the mouse MedLNs exhibit robust staining for MHC class II but lack staining for the auto-immune regulator (Aire) (Figure 1C), indicating they are distinct from the recently described Aire+ RORγt+ CD127− immune cell that appears to also present antigens (Yamano et al., 2019). We next examined whether ILC3s could be found in the human lung or associated lymphoid tissues. ILC3s are present at significantly higher frequencies than ILC2s in the MedLNs of healthy human donors, and ILC3s are also present within the lung parenchyma (Figure 1D). Human ILC3s at both tissue sites exhibit dimmer staining for CD45, lack the activation marker NKp44, and highly stain positive for MHC class II (histocompatibility leukocyte antigen-DR [HLA-DR]) relative to ILC2s (Figures 1E and S1B). These data demonstrate that LTi-like ILC3s are present in the airway-associated lymphoid tissues of both mice and humans, and the robust expression of MHC class II indicates the potential to directly modulate antigen-specific CD4+ T cell responses.
Figure 1. ILC3s are enriched in lung-draining mediastinal lymph nodes (MedLNs) and have high expression of MHC class II.
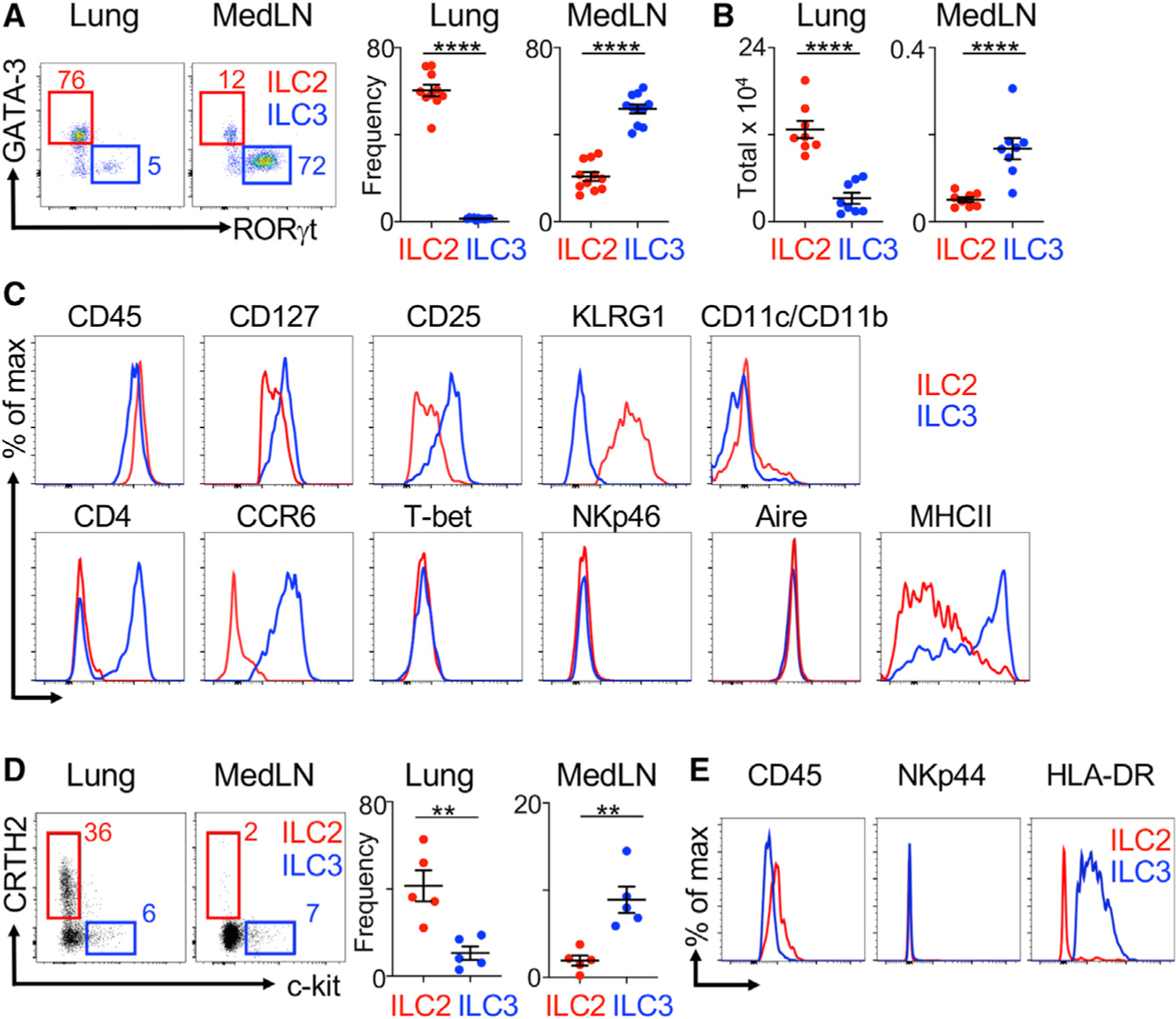
(A and B) Representative plots and quantitative graphs of frequencies (A) or total numbers of ILC2s and ILC3s (B) in mouse lung parenchyma and MedLNs (pooled from 2 independent assays, n = 8–10). Cells are gated on live CD45+ Lin− CD127+ (lineage markers: CD3e, CD5, CD8a, NK1.1, CD11c, CD11b, B220, and F4/80) as in Figure S1A.
(C) Representative histograms of specific markers on gated ILC2s and ILC3s in mouse MedLNs (representative of 6 mice).
(D) Representative plots and quantitative graphs of ratios of ILC2s and ILC3s in human lung and MedLN tissues. Cells were gated on live CD45+ Lin− (CD3, CD4, CD5, CD11b, CD11c, CD14, CD19, FcεRI) CD127+ populations from matched lung and MedLN tissues pooled from 5 donors.
(E) Representative histograms of specific markers on ILC2s and ILC3s from human MedLN tissues (representative from 5 donors). Data are represented as mean ± SEM. Statistics are calculated by Student’s t test. See also Figure S1.
MHC class II+ ILC3s restrict antigen-specific CD4+ T cell responses to allergens in the airway
We have previously defined that MHC class II+ ILC3s in the gastrointestinal tract process and present microbial antigens and that this pathway is essential to promote tolerance by limiting antigen-specific T cell responses to the microbiota (Hepworth et al., 2013, 2015; Melo-Gonzalez et al., 2019). However, whether MHC class II+ ILC3s play a similar role in control of adaptive immunity and airway inflammation is unknown. To address this question, we used HDM to study a clinically relevant model of allergic airway inflammation. Consistent with a prior report (Mackley et al., 2015), we observed co-localization between ILC3s and T cells at interfollicular regions in the MedLNs of healthy mice (Figures S1C and S1D), and despite a striking increase in the size of the MedLN following induction of HDM-induced airway inflammation, co-localization of ILC3s and T cells was still observed at interfollicular regions (Figures 2A and S1C). ILC3s also expanded in the lung parenchyma following HDM-induced airway inflammation and stained positive for CCR7 relative to effector T cells in the airway and MedLN (Figures S1E–S1G). This finding indicates that the limited ILC3s in the lung parenchyma have the ability to migrate to the airway draining lymphoid tissues by the lymphatics, as previously reported for CCR7+ ILC3s in the intestine (Mackley et al., 2015).
Figure 2. ILC3s limit CD4+ T cell responses to allergens in the airway.
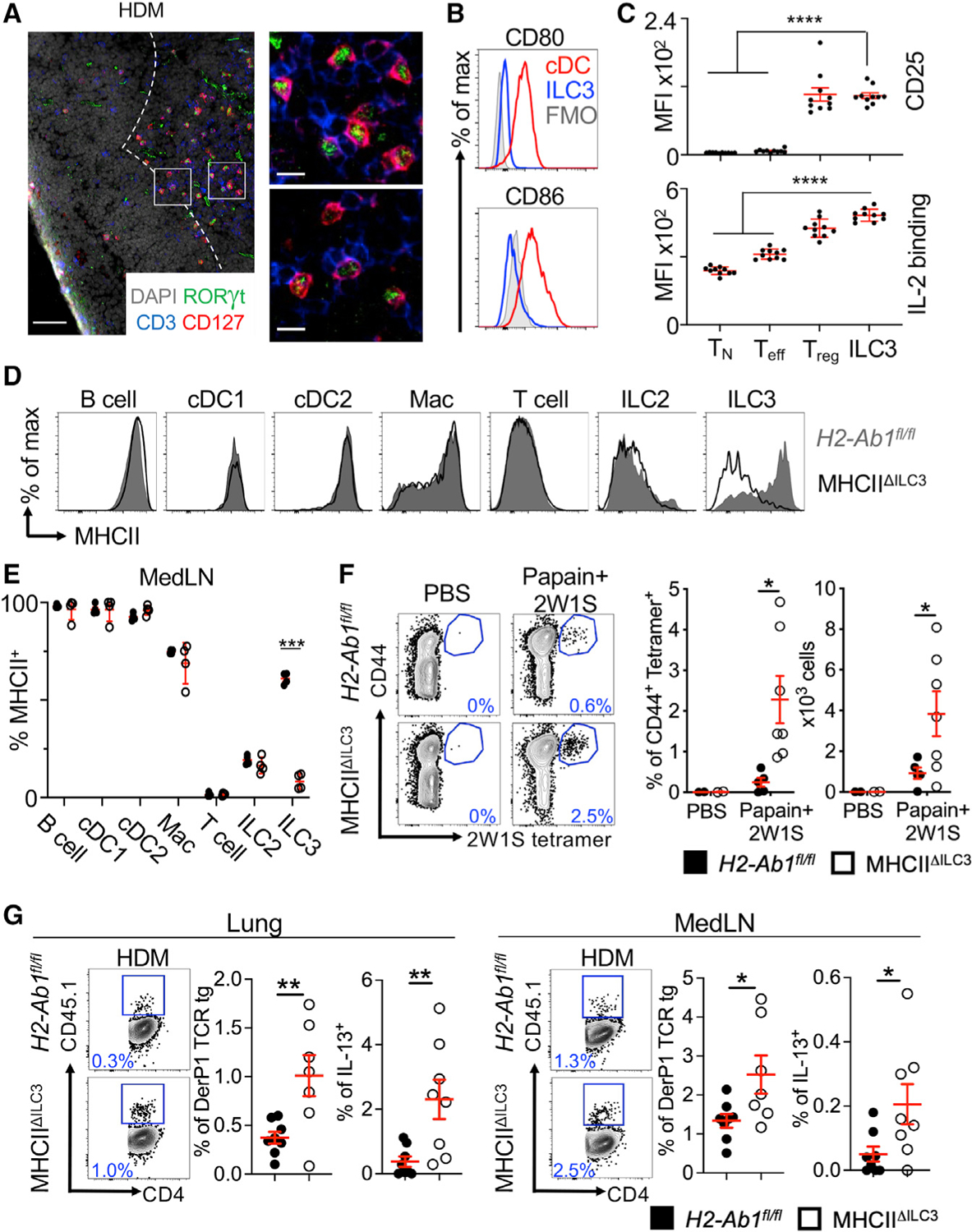
(A) Representative immunofluorescence staining for ILC3s and T cells in the MedLNs of HDM-exposed mice (representative of 2 independent experiments and n = 3 mice per group). Images taken at 20× magnification; scale bars, 50 μm (left panel) and 10 μm (inserts; right panel). Dashed line indicates the B cell follicle border.
(B) Histograms demonstrating CD80 and CD86 expression in MedLN cells following HDM exposure (representative of 2 independent assays containing 8 mice each). FMO, fluorescence minus one.
(C) Graphs quantifying CD25 staining or IL-2 binding in gated MedLN cells following HDM exposure (pooled from 2 independent assays, n = 10). Naive T cells were gated as CD44− CD62L+, effector T cells were gated as CD44+CD62L−, and Tregs were gated as FoxP3+.
(D and E) Representative plots of MHC class II expression in designated immune cells (D) and quantification of MHC class II staining on those cell types in the MedLN following HDM exposure (E) (representative of 2 independent assays, n = 4).
(F) Representative plots and quantitative graphs of the frequency and numbers of 2W1S-specific CD4+ T cells in lung parenchyma from H2-Ab1fl/fl control mice and MHC class IIΔILC3 mice after exposure to papain plus 2W1S peptide or PBS control. Cells were gated on live CD45+ CD19− TCRβ+ CD4+ (data pooled from 2 independent assays).
(G) Representative plots and quantitative graphs of the frequency of total or IL-13+ CD45.1+ donor DerP1-specific TCR transgenic CD4+ T cells in lung parenchyma and MedLNs from recipient H2-Ab1fl/fl and MHC class IIΔILC3 mice after exposure to HDM. Cells were gated on live CD45+ CD19− TCRβ+ CD4+ (data pooled from 2 independent assays, n = 7–8). Data are represented as mean ± SEM. Statistics are calculated by two-way ANOVA (C, E, and F) and Student’s t test (G). See also Figure S2.
Our previous findings demonstrated that tolerance in the intestinal tract was induced in part by ILC3-mediated antigen presentation in the absence of classical co-stimulation and the presence of elevated surface expression of receptors that sequester pro-survival signals, such as IL-2, from effector T cells (Hepworth et al., 2015). Sequestration of IL-2 is also a pathway used by Tregs to control effector T cell responses (Pandiyan et al., 2007). We explored whether similar pathways are employed in MHC class II+ ILC3s from the MedLN, and observed that this population exhibited low staining for the co-stimulatory molecules CD80 and CD86 relative to conventional dendritic cells (cDCs) during both homeostasis (Figure S1H) and following HDM-induced airway inflammation (Figures 2B). Furthermore, following HDM-induced airway inflammation, MedLN ILC3s exhibited significantly higher CD25 and a more efficient ability to bind IL-2 in an in vitro assay, relative to naive and effector T cells, but not Tregs (Figures 2C and S1I). These collective datasets demonstrate that ILC3s in the MedLNs exhibit high levels of MHC class II, an absence of co-stimulatory molecules CD80 and CD86, and an ability to sequester IL-2 from effector T cells. Importantly, this occurs during both homeostasis and airway inflammation, indicating that MedLN ILC3s contribute to immune tolerance.
To more directly examine how ILC3s in the MedLNs functionally impact adaptive immunity, we used a mouse model in which ILC3s selectively lack MHC class II by crossing Rorccre mice and mice with floxed alleles of H2-Ab1 (Goc et al., 2021; Hepworth et al., 2013, 2015; Melo-Gonzalez et al., 2019). Following induction of HDM-induced airway inflammation, we identify that this mouse line significantly and selectively targets for MHC class II deletion only in ILC3s, and does not impact MHC class II levels on B cells, cDC subsets, macrophages (Macs), T cells, or ILC2s in the MedLN and lung parenchyma relative to littermate controls (Figures 2D, 2E, S2A, and S2B). To quantify the impact of MHC class II+ ILC3s on antigen-specific T cell responses in the inflamed airway, we also used the protease allergen papain with the peptide 2W1S as previously described (Halim et al., 2014). Relative to littermate controls, mice lacking ILC3-specific MHC class II exhibit significantly increased frequencies and numbers of 2W1S-specific CD4+ T cells in the lung parenchyma (Figure 2F). In addition, we used HDM-induced airway inflammation in conjunction with pre-transfer of T cell receptor (TCR) transgenic T cells that are specific for the immunodominant HDM allergen DerP1 (Plantinga et al., 2013). In this context, mice lacking ILC3-specific MHC class II exhibit significantly higher frequencies, but not cell numbers, of allergen-specific CD4+ T cells in the lung parenchyma and MedLNs than littermate controls (Figures 2G and S2C). The allergen-specific T cells that developed in the absence of ILC3-specific MHC class II produced significantly more IL-13 following a brief re-stimulation and comparable levels of IL-17 (Figures 2G and S2D). These collective results demonstrate that antigen-presenting ILC3s critically limit antigen-specific adaptive immune responses directed against allergens in the airway.
MHC class II+ ILC3s limit type 2 allergic airway inflammation
Allergic airway inflammation is orchestrated in part by aberrant Th2 cell responses, associated with eosinophilia and airway hyperreactivity (Bosteels et al., 2020; Hammad and Lambrecht, 2021; Hewitt and Lloyd, 2021; Lambrecht et al., 2019; Lloyd and Marsland, 2017). As we observed expanded allergen-specific T cells and enhanced production of IL-13 in the absence of ILC3-specific MHC class II, we next examined whether MHC class II+ ILC3s control type 2 immune responses and airway inflammation. This examination was accomplished by exposing mice lacking ILC3-specific MHC class II to sensitization and challenge with HDM, which resulted in a significant increase in Th2 cell responses in the lung parenchyma and MedLNs relative to littermate controls (Figure 3A). This increase occurred without altered total CD4+ T cell numbers or Treg numbers or changes in chemokine homing receptors on T cell subsets (Figures S3A–S3C). Furthermore, ILC3-specific MHC class II deletion did not impact ILC2 cell numbers or the ability of MedLN ILC3s to produce IL-2 or GM-CSF following HDM-induced airway inflammation (Figures S3D–S3F), indicating that this pathway of tolerance is distinct from these two previously described effector cytokines in ILC3s (Mortha et al., 2014; Zhou et al., 2019). Finally, MHC class II+ ILC3s also were not impacted in the MedLNs by the absence of the metabolite-sensing receptor FFAR2 in this context (Figure S3G), which can modulate ILC3 responses in the gut (Chun et al., 2019; Fachi et al., 2020).
Figure 3. MHC class II+ ILC3s limit Th2 cell responses and allergic airway inflammation.
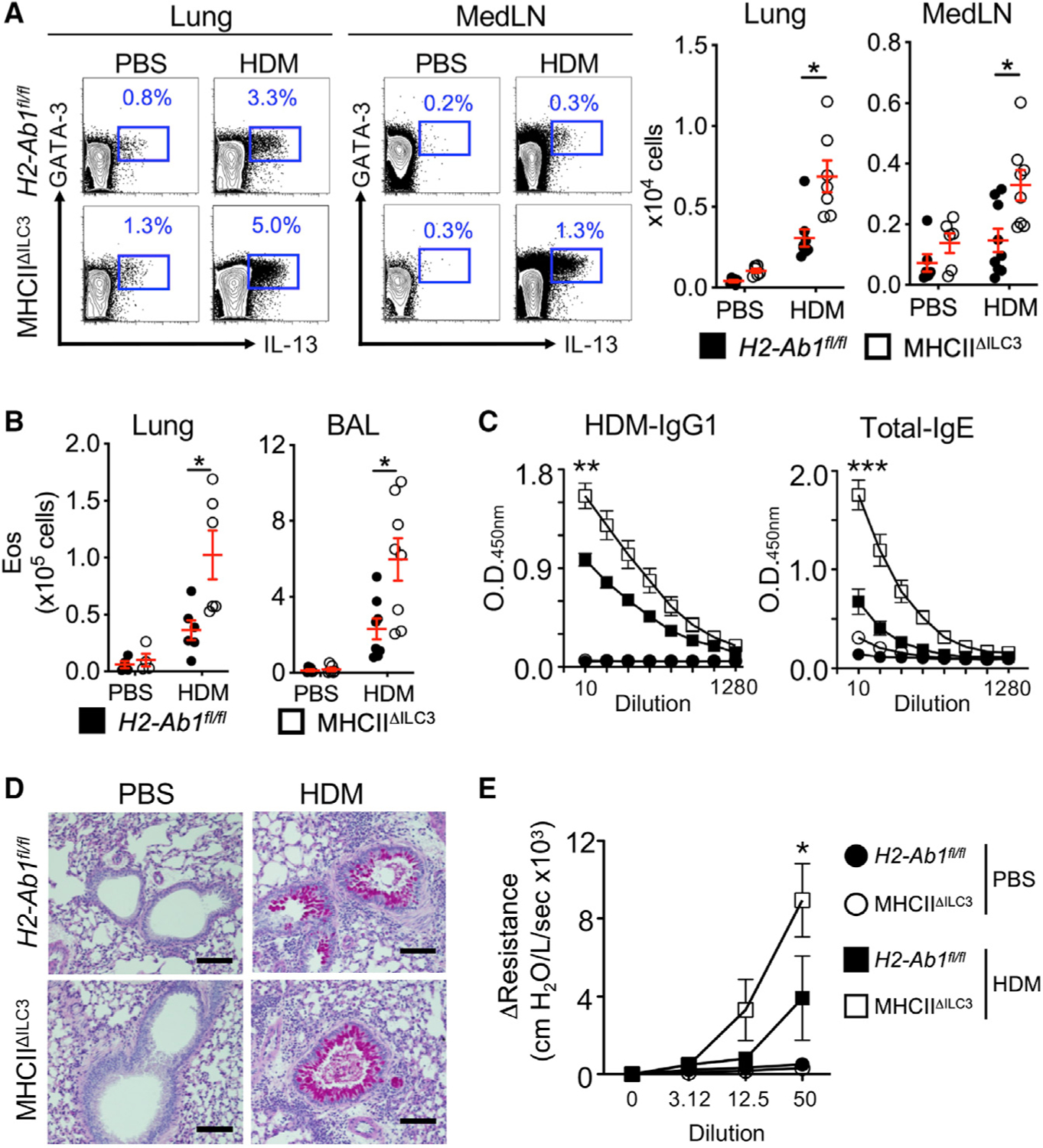
(A) Representative plots and quantitative graphs of the frequency and number of Th2 cells in lung parenchyma and MedLN from H2-Ab1fl/fl control mice and MHC class IIΔILC3 mice after exposure to HDM or PBS. Cells were gated on live CD45+ CD19− CD3/CD5+ CD4+ (data pooled from 4 independent assays).
(B) Quantitative graphs of the numbers of eosinophils in lung parenchyma and BAL from mice in (A). Eosinophils were identified as live CD45+ SSC-Ahi, Siglec-F+ (data pooled from 4 independent assays, n = 8).
(C) ELISA analysis of sera from mice in (A) for HDM-specific IgG1 and total IgE (pooled from 2 independent assays, n = 4).
(D) Representative periodic acid-Schiff staining sections from lung of mice in (A). The scale bars represent 100 μm.
(E) Analysis of airway resistance on mice from (A) (pooled from 2 independent assays, n = 8). Data are represented as mean ± SEM. Statistics are calculated by two-way ANOVA (A and B) and AUC followed by one-way ANOVA (C and E). See also Figure S3.
In line with elevated Th2 responses, mice lacking ILC3-specific MHC class II exhibited significantly elevated levels of eosinophils in the lung parenchyma and bronchoalveolar lavage fluid (BAL) relative to controls, as well as significantly higher titers of HDM-specific IgG1 and total IgE (Figures 3B and 3C). Consistent with this robust type 2 immune response, the absence of ILC3-specific MHC class II results in robust mucus production and significantly increased AHR relative to littermate controls (Figures 3D and 3E). These collective results reveal a critical role for MHC class II+ ILC3s in controlling aberrant Th2 cell responses and allergic type 2 inflammation in the airway.
MHC class II+ ILC3s control microbe-reactive Th17 cells in the inflamed airway
Emerging evidence indicates the presence of robust Th17 cell responses and neutrophilic infiltration as key factors driving allergic airway disease in humans and mouse models (Alcorn et al., 2010; Newcomb and Peebles, 2013; Wang and Wills-Karp, 2011). Therefore, we next examined for the development of Th17 cell responses in our mouse models. Following exposure to HDM, mice lacking ILC3-specific MHC class II exhibit significant increases in frequencies and total numbers of Th17 cells within the lung parenchyma and MedLNs relative to littermate controls (Figure 4A). Consistent with this finding, mice lacking ILC3-specific MHC class II uniquely develop elevated airway neutrophilia (Figure 4B), although the ratio of eosinophils to neutrophils remains unchanged, as both increase in this model system (Figure S3H). These results are surprising, as our earlier results revealed enhanced expansion of allergen-specific Th2 cell responses, but not allergen-specific Th17 cell responses, when mice lacked ILC3-specific MHC class II (Figures 2G and S2D), provoking fundamental questions on the antigen-reactivity of the elevated Th17 cell responses following exposure to HDM.
Figure 4. MHC class II+ ILC3s limit airway Th17 cell responses to microbes upon HDM exposure.
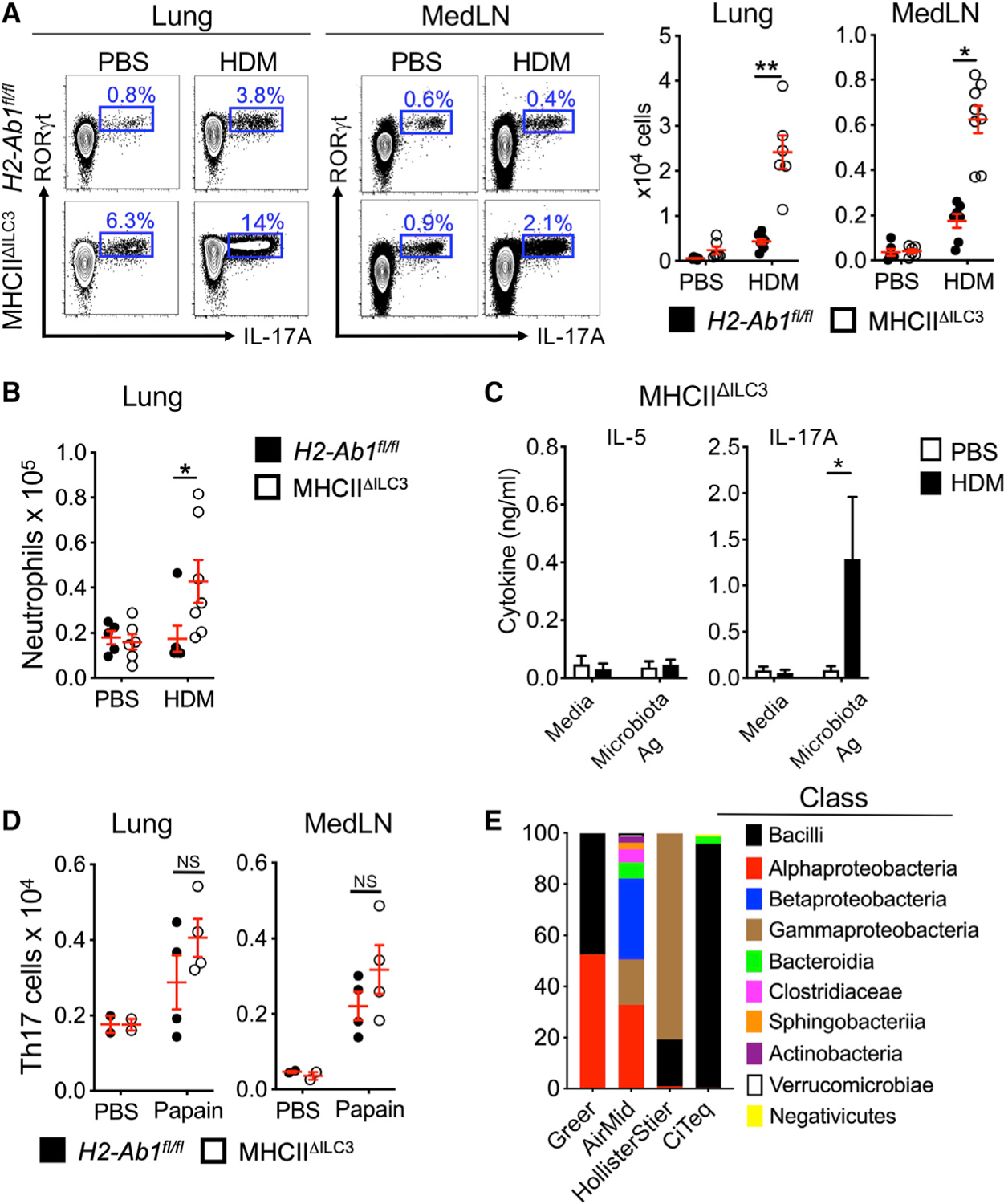
(A) Representative plots and quantitative graphs of the frequency and number of Th17 cells in lung parenchyma and MedLN from H2-Ab1fl/fl control mice and MHC class IIΔILC3 mice after exposure to HDM or PBS. Cells were gated on live CD45+ CD19− CD3/CD5+ CD4+ (data pooled from 4 independent assays, n = 6).
(B) Quantitative graph of the number of neutrophils in lung parenchyma from mice in (A). Neutrophils were identified as live CD45+ CD19− TCRβ− CD11b+ Ly6G+ (data pooled from 4 independent assays, n = 7).
(C) ELISA analysis for IL-5 and IL-17A levels from supernatants of microbiota-extract-restimulated MedLN cells from HDM-exposed MHC class IIΔILC3 mice (data pooled from 2 independent assays).
(D) Quantitative graphs of the number of Th17 cells in lung parenchyma and MedLNs from H2-Ab1fl/fl control mice and MHC class IIΔILC3 mice after exposure to sterile papain (data pooled from 2 independent assays, n = 4).
(E) Bar graphs of major bacterial composition in HDM from four different vendors. Data are represented as mean ± SEM. Statistics are calculated by two-way ANOVA. See also Figure S4.
To explore this result further, we exposed mice to HDM and observed that the elevated IL-13+ and IL-17A+ T cells in the absence of ILC3-specific MHC class II are distinct populations in the lung parenchyma (Figure S3I). Our previous studies demonstrate that ILC3s control microbiota-specific Th17 cells in the intestine (Hepworth et al., 2015). As microbial-driven responses have been implicated in the pathogenesis of asthma, we next tested whether HDM-associated Th17 cells are driven by microbial antigens, as opposed to allergens. Restimulation of MedLNs from HDM-exposed mice that lack ILC3-specific MHC class II revealed the production of IL-17A, but not type 2 cytokines, in response to microbiota-derived extracts (Figure 4C), suggesting that these populations are reactive to microbes. This finding provoked the question as to the source of microbial antigen eliciting this response. In contrast, delivery of a sterile preparation of the protease allergen papain results in comparable Th17 cell responses in the airway of littermate controls and mice lacking ILC3-specific MHC class II (Figure 4D). This result suggest that there are unique microbial stimuli present in HDM extracts that are driving aberrant microbe-reactive Th17 cells in the absence of MHC class II+ ILC3s. It is well appreciated that LPS is present in extract preparations from HDMs, and prior studies have characterized a mite-associated microbiota from various cultures (Chan et al., 2015; Loo et al., 2018); however, the microbial signatures of HDM extracts from commercial vendors have not been described. Therefore, we performed 16S rRNA sequencing on milled raw materials from a Dermatophagoides pteronyssinus culture provided by four independent commercial vendors. Our results identify the presence of microbial sequences from numerous classes of microbes, including Bacilli, Alphaproteobacteria, Betaproteobacteria, Gammaproteobacteria, Bacteroidia, Clostridiaceae, Shingobacteria, Actinobacteria, Verrucomicrobia, and Negativicutes, which varied substantially between commercial vendors (Figure 4E). Furthermore, metagenomic sequencing of milled raw materials of HDM from several of these vendors demonstrate the presence of sequences associated with various bacteria, fungi, and viruses (Figure S4). Our results therefore indicate that antigen-presenting ILC3s critically restrict pro-inflammatory Th17 cell responses to microbes that enter the airway and are associated with a common source of allergens.
DISCUSSION
Defining the pathways that control inappropriate adaptive immune responses at mucosal barrier surfaces is necessary to develop therapeutic approaches for chronic inflammatory diseases such as asthma. Although fundamental research has revealed critical roles for Th2 cells and Th17 cells in the pathogenesis of asthma, the cellular mechanisms that prevent or restrain these T cell responses remain incompletely understood. Our findings suggest that antigen-presenting ILC3s associated with lymphoid tissues that drain the airway critically limit over-active adaptive immune responses. The role of ILC3s in the airway has been vastly understudied. A few reports have highlighted that ILC3s are enriched in the airway during certain contexts and contribute to airway immunity or inflammation through the production of IL-17 and IL-22 (Kim et al., 2014; Oherle et al., 2020; Xiong et al., 2016). However, the study of ILC3s in the airway has been limited by the fact that they are virtually absent from the healthy lungs of mice housed under SPF conditions. Therefore, our finding that ILC3s are enriched in the MedLNs of mice and present within the lung parenchyma of humans is surprising and important. The difference between humans and laboratory mice is likely driven through microbial exposure, as a prior report indicates that ILC3s increase in the airway of laboratory mice when co-housed with mice derived from pet stores (Beura et al., 2016). Combining these observations with the hygiene hypothesis provokes a model whereby early life exposure to microbes could increase the abundance of protective and tolerogenic ILC3s in the airway that subsequently prevent over-active adaptive immune responses in later life.
The ILC3s we describe in the airway and associated lymphoid tissues exhibit an LTi-like phenotype. Based on prior literature, they are likely to be tissue resident or independent of the circulation, even in the context of subsequent inflammatory challenge (Gasteiger et al., 2015; Zhang et al., 2016). There may also be cross-talk between tissue-resident ILC3s and Macs in the MedLNs (Gray et al., 2012) or the potential migration of CCR7+ ILC3s from the lung parenchyma to the MedLNs, as described in other contexts (Mackley et al., 2015). This possibility raises a number of opportunities by which LTi-like ILC3s may acquire antigens and potentially impact adaptive immunity. Additionally, although the microbial metabolite sensor FFAR2 does not impact MHC class II+ ILC3s in the MedLNs, it remains possible that other microbial sensors could impact human or mouse antigen-presenting ILC3s in other contexts. This has certainly been shown in other contexts of ILC3s in the intestine as well as the lung (Beck et al., 2020; Crellin et al., 2010; von Burg et al., 2014), and it will be important to investigate particularly in the context of airway infections.
Furthermore, our results identify a connection by which a type 3 immune response cross-regulates a type 2 immune response. It is surprising that ILC3s can regulate the development of Th2 cells and allergic inflammation in the airway, but these results are in line with a described role for RORγt+ Tregs in controlling type 2 inflammation (Ohnmacht et al., 2015) and expand upon our previous studies that showed that ILC3s limit Th17 cell and T follicular helper cell responses in the intestine (Hepworth et al., 2013, 2015; Melo-Gonzalez et al., 2019). Our data provoke the need to further interrogate a potential regulatory role of ILC3s in multiple allergic diseases. It remains possible that boosting ILC3s could represent a preventative, therapeutic, or curative approach to limit inappropriate type 2 allergic inflammation. This also indicates a functional dichotomy for the ILC family in impacting Th2 cell responses, as prior reports demonstrated that ILC2s play an important role in promoting Th2 cells in the lung through antigen presentation (Mirchandani et al., 2014; Oliphant et al., 2014). Of note, ILC2s express a limited amount of MHC class II in steady state and increase expression upon immune activation, whereas ILC3s abundantly and constitutively express MHC class II during both homeostasis and airway inflammation.
Finally, besides the substantial amount of evidence linking the gut-lung axis in allergic diseases (Fujimura and Lynch, 2015; Lynch and Boushey, 2016), our findings suggest that impairment of ILC3s is linked to aberrant Th17 cells in the airway that exhibit antigen specificity for the microbes associated with allergens. Seminal studies have defined that the human intestinal microbiota, as well as environmental microbes associated with dust dramatically shape allergic airway inflammation (Ciaccio et al., 2015; Ege et al., 2012; Fujimura et al., 2016; Loo et al., 2018; Stein et al., 2016). Therefore, this cross reactivity of Th17 cells to host- and mite-derived microbiota, and the regulation of this responses by ILC3s, will be important to consider in the broader context of defining how microbes shape allergic airway diseases.
Limitations of the study
Although our study provides a comprehensive understanding of the role of antigen-presenting ILC3s in experimental airway inflammation, several limitations should be mentioned. First, although our in vivo mouse studies demonstrate a regulatory role for MHC class II+ ILC3s in restricting effector T cell responses, we were unable to experimentally examine this further with in vitro co-cultures due to the limited cell numbers available from the MedLNs. Second, our studies suggest that the dysregulated Th17 cells observed in the airway of MHC class IIΔILC3 mice are specific for a mite-associated microbiota. It will be important to definitively demonstrate this in future experiments by generating extract from sterile HDMs depleted of microbiota. Finally, our study does not evaluate the role of antigen-presenting ILC3s in the context of experimental airway infection in mice or in translational samples from human patients with asthma. They will be important future areas of research to build off of this initial study.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Gregory F. Sonnenberg (gfsonnenberg@med.cornell.edu).
Materials availability
This study did not generate new unique reagents. All materials listed in the Key resources table and are available from the Lead Contact.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD45-Brilliant Violet 605 (30-F11) | BioLegend | Cat# 103140, RRID: AB_2562342 |
| CD45-Brilliant Violet 785 (30-F11) | BioLegend | Cat# 103149, RRID: AB_2564590 |
| CD3ε-PerCP-Cy5.5 (145-2C11) | BioLegend | Cat# 100328, RRID: AB_893318 |
| CD3ε-Brilliant Violet 421 (145-2C11) | BioLegend | Cat# 100336, RRID: AB_11203705 |
| CD5-PerCP-Cy5.5 (53-7.3) | BioLegend | Cat# 100624, RRID: AB_2563433 |
| CD8α-PerCP-Cy5.5 (53-6.7) | BioLegend | Cat# 100734, RRID: AB_2075238 |
| NK1.1-PerCP-Cy5.5 (PK136) | BioLegend | Cat# 108728, RRID: AB_2132705 |
| CD11c-APC-Fire750 (N418) | BioLegend | Cat# 117352, RRID: AB_2572124 |
| CD11c-PE-Cy7 (N418) | BioLegend | Cat# 117318, RRID: AB_493568 |
| CD11b-APC-Fire750 (M1/70) | BioLegend | Cat# 101262, RRID: AB_2572122 |
| CD11b-PE-Dazzle 594 (M1/70) | BioLegend | Cat# 101256, RRID: AB_2563648 |
| B220-APC-Fire750 (RA3-6B2) | BioLegend | Cat# 103260, RRID: AB_2572109 |
| CD19-PE-Cy7 (eBio1D3) | ThermoFisher | Cat# 25-0193-82, RRID: AB_657663 |
| CD19-Brilliant Violet 605 (6D5) | BioLegend | Cat# 115540, RRID: AB_2563067 |
| TCRβ-APC-Fire750 (H57-597) | BioLegend | Cat# 109246, RRID: AB_2629697 |
| CD127-PE-Cy5 (A7R34) | BioLegend | Cat# 135016, RRID: AB_1937261 |
| CD90.2-Alexa Fluor 700 (30-H12) | BioLegend | Cat# 105320, RRID: AB_493725 |
| F4/80-APC (BM8) | BioLegend | Cat# 123116, RRID: AB_893481 |
| Ly6G-Alexa Fluor 700 (RB6-8C5) | BioLegend | Cat# 108422, RRID: AB_2137487 |
| Siglec-F-PE (E50-2440) | BD Biosciences | Cat# 552126, RRID: AB_394341 |
| SIRPα-Alexa Fluor 647 (P84) | BioLegend | Cat# 144028, RRID: AB_2721301 |
| XCR1-FITC (ZET) | BioLegend | Cat# 148210, RRID: AB_2564366 |
| CD4-BUV395 (GK1.5) | BD Biosciences | Cat# 563790, RRID: AB_2738426 |
| CD4-Brilliant Violet 650 (RM4-5) | BioLegend | Cat# 100546, RRID: AB_2562098 |
| MHCII-Brilliant Violet 650 (M5/114.15.2) | BioLegend | Cat# 107641, RRID: AB_2565975 |
| CD45.1-Brilliant Violet 421 (A2) | BioLegend | Cat# 110732, RRID: AB_2562563 |
| CD44-APC (IM7) | BioLegend | Cat# 103012, RRID: AB_312963 |
| NKp46-PE (29A1.4) | BioLegend | Cat# 137604 RRID: AB_2235755 |
| CD25-PE-Cy7 (PC61.5) | BioLegend | Cat# 102016, RRID: AB_312865 |
| CCR6-Brilliant Violet 421 (29-2L17) | BioLegend | Cat# 129818, RRID: AB_11219003 |
| KLRG1-APC (2F1) | BioLegend | Cat# 138412 RRID: AB_10641560 |
| CD80-PE-eFluor 610 (16-10A1) | BioLegend | Cat# 104738, RRID: AB_2564175 |
| CD86-BUV395 (GL1) | BD Biosciences | Cat# 564199, RRID: AB_2738664 |
| CCR7-PE (4B12) | BioLegend | Cat# 120106, RRID: AB_389358 |
| CD69-PerCP-Cy5.5 (H1.2F3) | ThermoFisher | Cat# 45-0691-82, RRID: AB_1210703 |
| CCR4-PE (2G12) | BioLegend | Cat# 131204, RRID: AB_1236367 |
| CCR8-FITC (SA214G2) | BioLegend | Cat# 150314, RRID: AB_2629604 |
| CXCR3-Brilliant Violet 605 (CXCR3-173) | BioLegend | Cat # 126523, RRID: AB_2561353 |
| Foxp3-Alexa Fluor 700 (FJK-16 s) | ThermoFisher | Cat# 56-5773-82, RRID: AB_1210557 |
| RORγt-PE (B2D) | ThermoFisher | Cat# 12-6981-82, RRID: AB_10807092 |
| RORγt-PE-eFluor610 (B2D) | ThermoFisher | Cat# 61-6981-82, RRID: AB_2574650 |
| RORγt-Brilliant Violet 650 (Q31-378) | BD Biosciences | Cat# 564722, RRID: AB_2738915 |
| GATA-3-BUV395 (L50-823) | BD Biosciences | Cat# 565448, RRID: AB_2739241 |
| T-bet-Brilliant Violet 421 (4B10) | BD Biosciences | Cat# 644816, RRID: AB_10959653 |
| Aire-eFluor 660 (5H12) | ThermoFisher | Cat# 50-5934-80, RRID: AB_2574256 |
| IL-13-Alexa Fluor 488 (eBio13A) | ThermoFisher | Cat# 53-7133-82, RRID: AB_2016708 |
| IL-2-PE (JES6-5H4) | BioLegend | Cat# 503808, RRID: AB_315302 |
| IL-22-Alexa Fluor 647 (IL22JOP) | ThermoFisher | Cat# 17-7222-82, RRID: AB_10597583 |
| GM-CSF-FITC (MP1-22E9) | BioLegend | Cat# 505404, RRID: AB_315380 |
| IL-17A-BUV395 (TC11-18H10) | BD Biosciences | Cat# 565246, RRID: AB_2722575 |
| CD3-PE-Cy7 (UCHT1) | BioLegend | Cat# 300420, RRID: AB_439781 |
| CD4-PE-Dazzle 594 (OKT4) | BioLegend | Cat# 317448, RRID: AB_2565847 |
| CD5-PE-Cy7 (UCHT2) | BioLegend | Cat# 300622, RRID: AB_2275812 |
| CD11b-Alexa Fluor 700 (M1/70) | BioLegend | Cat# 101222, RRID: AB_493705 |
| CD11c-Alexa Fluor 700 (3.9) | BioLegend | Cat# 301648, RRID: AB_2819923 |
| CD14-Alexa Fluor 700 (HCD14) | BioLegend | Cat# 325614, RRID: AB_830687 |
| CD19-Alexa Fluor 700 (HIB19) | BioLegend | Cat# 302226, RRID: AB_493751 |
| HLA-DR-BUV395 (L203) | BD Biosciences | Cat# 752492, RRID: N/A |
| CRTH2-APC (BM16) | BioLegend | Cat# 350110, RRID: AB_11203707 |
| CD127-APC-eFluor 780 (eBioRDR5) | ThermoFisher | Cat# 47-1278-42, RRID: AB_1548674 |
| FcεRI-PE-Cy7 (AER-37) | BioLegend | Cat# 334620, RRID: AB_10575314 |
| c-kit-PerCP-Cy5.5 (104D2) | BioLegend | Cat# 313214, RRID: AB_2131443 |
| CD45-Brilliant Violet 605 (HI30) | BioLegend | Cat# 304042, RRID: AB_2562106 |
| NKp44-PE (P44-8) | BioLegend | Cat# 325108, RRID: AB_756100 |
| secondary antibodies against murine IgG1-HRP | BD Biosciences | Cat# 559626 |
| secondary antibodies against murine IgE | ThermoFisher | Cat#MA5-16781 |
| Anti-mouse IL-17A, purified | ThermoFisher | Cat# 14-7175-81 |
| Anti-mouse IL-17A, biotin | ThermoFisher | Cat# 13-717781 |
| Anti-mouse IL-13, purified | ThermoFisher | Cat# 14-7133-81 |
| Anti-mouse IL-13, biotin | ThermoFisher | Cat# 13-7135-81 |
| purified anti-mouse RORγt (AFKJS-9) | ThermoFisher | Cat# 14-6988-82, RRID: AB_1834475 |
| biotinylated anti-mouse CD3ε (eBio500A2) | ThermoFisher | Cat# 13-0033-82, RRID: AB_842774 |
| donkey anti-rat-IgG-FITC | Jackson ImmunoResearch | Cat# 712-097-003, RRID: AB_2340655 |
| CD127-eFluor 660 (A7R34) | ThermoFisher | Cat# 50-1271-82, RRID: AB_11219081 |
| donkey anti-rabbit-IgG-Alexa Fluor 488 | ThermoFisher | Cat# A-21206, RRID: AB_2535792 |
| Biological samples | ||
| Human lung and lung mediastinal lymph node tissue samples | LiveOnNY | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Papain | Worthington | Cat#LS003126 |
| 2W1S peptide | GenScript | Peptide sequence: EAWGALANWAVDSA |
| I-A(b) mouse 2W1S tetramer, PE conjugated | NIH Tetramer Core Facility | N/A |
| house dust mite protein extract | Greer Laboratories | Cat# XPB82D3A2.5 |
| Naive CD4+ T Cell Isolation Kit, mouse | Miltenyi Biotec | Cat# 130-104-453 |
| Pentobarbital | American Pharmaceutical Partners | N/A |
| Liberase TL | Sigma | Cat# 5401020001 |
| Collagenase D | Sigma | Cat# 11088882001 |
| ACK lysis buffer | VWR | Cat# 12002-070 |
| RPMI 1640 with L-glutamine | Corning | Cat#10-040-CV |
| trypsin inhibitor | Life Technologies | Cat# 17075-029 |
| DNase I | Sigma | Cat# D5025 |
| Collagenase II | ThermoFisher | Cat#17101015 |
| TMB peroxidase substrate | ThermoFisher | Cat# 00-4201-56 |
| House dust mite milled raw materials | Greer Laboratories | N/A |
| House dust mite milled raw materials | AirMid Healthgroup, | N/A |
| House dust mite milled raw materials | HollisterStier | N/A |
| House dust mite milled raw materials | CiTeq Biologics | N/A |
| Paraformaldehyde (4% in PBS) | bioWORLD | Cat# 30450002-1 |
| Tissue-Tek OCT compound | Bayer Healthcare | N/A |
| Streptavidin-Alexa Fluor 555 | ThermoFisher | Cat#S21381 |
| DAPI | ThermoFisher | Cat#D1306 |
| ProLong Gold Antifade Mountant | ThermoFisher | Cat# P36934 |
| Critical commercial assays | ||
| Live/Dead Fixable Aqua Dead Cell Stain Kit | Thermo Fisher Scientific | Cat# L34957 |
| Foxp3 transcription factor staining buffer set | Thermo Fisher Scientific | Cat# 00-5523-00 |
| IL-2 fluorokine kit | R&D Systems | Cat# BT202-025 |
| Deposited data | ||
| 16S sequencing of HDM from various vendors | This paper | BioProject ID PRJNA762346 |
| Metagenomic sequencing of HDM from various vendors | This paper | BioProject ID PRJNA762364 |
| Experimental models: Organisms/strains | ||
| C57BL/6J mice | Jackson Laboratory | RRID:IMSR_JAX:000664 |
| Rorccre mice | Dr. Gerard Eberl | N/A |
| H2-Ab1fl/fl mice | Jackson Laboratory | RRID:IMSR_JAX:013181 |
| 1-DEPβ mice | Dr. Bart N. Lambrecht | N/A |
| Ffar2−/− mice | Dr. M van den Brink | N/A |
| Software and algorithms | ||
| Flexivent | Scireq | https://www.scireq.com/flexivent |
| FlowJo V1 0.8.0 | TreeStar | https://www.flowjo.com/solutions/flowjo/downloads |
| FACS Diva | BD Biosciences | https://www.bdbiosciences.com/en-us/products/software/instrument-software/bd-facsdivasoftware#Overview |
| Cutadapt | Caporaso et al., 2011 | https://github.com/marcelm/cutadapt |
| Qiime2 dada2 plugin V 1.10.0 | Caporaso et al., 2011 | https://github.com/qiime2/q2-dada2 |
| GraphPad Prism V 9.2.0 | GraphPad | https://www.graphpad.com |
| ImageJ | NIH | https://imagej.nih.gov/ij/download.html |
Data and code availability
16S sequencing datasets reported in this study are available at NCBI SRA via BioProject ID PRJNA762346. Metagenomic sequencing is available at NCBI SRA via BioProject ID PRJNA762364. The paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Age- and sex matched C57BL/6 wild-type (WT) mice were purchased from Jackson Laboratory (Bar Harbor, ME). All mice were utilized between 8–12 weeks of age for experiments. Both sexes of mice were utilized in experiments and no impact of sex was observed on the reported phenotypes. MHCIIΔILC3 mice and H2-Ab1fl/fl littermate controls were generated as previously described (Hepworth et al., 2013). CD45.1+ β chain TCR transgenic mice specific for DerP1 peptide (1-DEPβ mice) were kindly provided by Dr. B. Lambrecht and were generated as previously described (Plantinga et al., 2013). Ffar2−/− mice were kindly provided by Dr. M van den Brink (Memorial Sloan Kettering Cancer Center). All mice were maintained in specific pathogen-free facilities at Weill Cornell Medicine, New York and where appropriate mutant strains were co-housed with control littermates to ensure equivalent microbiota exposure. All protocols were approved by the Weill Cornell Medicine Institutional Animal Care and Use Committees (IACUC), and all experiments were performed according to the guidelines of the relevant institution. Some experimental replicates were performed under license of the UK Home Office and under approved protocols at the University of Manchester.
Human Tissue Samples
Human tissues were obtained from deceased organ donors at the time of organ acquisition for clinical transplantation through an approved research protocol with LiveOnNY, the organ procurement organization for the New York metropolitan area, as previously described (Carpenter et al., 2018; Thome et al., 2014). All donors were free of chronic disease and cancer, were Hepatitis B-, C-, and HIV-negative. Isolation of tissues from organ donors does not qualify as “human subjects” research, as confirmed by the Columbia University IRB. The age, sex and gender identity are unknown. Tissues were collected after the donor organs were flushed with cold preservation solution and clinical procurement process was completed. Tissue samples were maintained in cold PBS and brought to the laboratory within 4–24 hours of organ procurement.
METHOD DETAILS
Papain and 2W1S Peptide Treatment
Mice were treated intranasally with 10 μg papain (Worthington) and 50 μg 2W1S peptide (peptide sequence: EAWGALANWAVDSA, GenScript) diluted in sterile PBS on Day 0, Day 1 and Day 15, the mice were then euthanized on Day 20 for analysis. The I-A(b) mouse 2W1S tetramer was kindly provided by NIH Tetramer Core Facility. In some assays papain was first diluted in sterile PBS and filtered through 0.22 μm syringe filter unit (Millipore), the sterile papain was then intranasally administered to mice on Days 0, 2, 4, 6, 8, 10 and euthanized on Day 11 for analysis.
House Dust Mite Treatment
Mice were sensitized intranasally with 100 μg house dust mite protein extract (D. pteronyssinus, Greer laboratories) diluted in sterile PBS. One week following sensitization mice were intranasally challenged with 10 μg HDM preparation five times over consecutive days. Mice were euthanized on day 14 post sensitization, unless indicated otherwise. Control mice received an equivalent volume of sterile PBS at same time points.
Adoptive Transfer of TCR Transgenic T Cells
5×106 splenic CD4+ T cells from CD45.1+ 1-DEPβ mice were MACS enriched by CD4+ T cell isolation kit (Miltenyi) and adoptively transferred by intravenous injection to H2-Ab1fl/fl and MHCIIΔILC3 recipient mice. The recipient mice were sensitized intranasally with HDM on the same day of cell transfer, one week following sensitization the recipient mice were intranasally challenged with 10 μg HDM preparation five times over consecutive days. Mice were euthanized on day 14 post sensitization.
Airway Resistance Measurements
After HDM treatment, airway reactivity of MHCIIΔILC3 mice and H2-Ab1fl/fl littermate control mice were assessed. Mice were anaesthetized with pentobarbital (100 mg/kg; American Pharmaceutical Partners), tracheostomized and mechanically ventilated using a computer- controlled animal ventilator (Scireq). Respiratory mechanics were analyzed using the Flexivent software as previously described (Worgall et al., 2013). Broadband forced oscillations were applied to determine Newtonian (airway) resistance (Rn) using a constant phase model. Rn was also assessed following increasing nebulized doses of methacholine (3.125, 12.5 and 50 mg/ml).
Murine Tissue Isolation and Flow Cytometry
Mediastinal lung-draining lymph nodes (MedLN) were harvested and digested for 20 minutes in 0.3 mg/ml Liberase TL (Roche), and passed over a 70 μm filter to generate single-cell suspensions. Bronchoalveolar lavage fluid (BAL) was obtained by flushing lungs twice with 0.7 mL PBS using a tracheal cannula (Terumo, SURFLO, 20G). Lungs were perfused with PBS and minced and digested in 2 mg/ml Collagenase D in PBS for 40 minutes at 37°C. Lung tissue digests were then passed over a 70 μm filter and the remaining lymphocyte-containing pellet was treated with ACK buffer to lyse red blood cells prior to stimulation or staining for flow cytometric analysis.
Fluorochrome-conjugated antibodies were purchased from BioLegend, eBioscience, or BD Biosciences. Cells were fixed and permeabilized utilizing a commercially available kit (eBioscience). For cytokine production, cells were stimulated ex vivo by incubation for 4 hours with 50 ng/mL PMA, 750 ng/mL ionomycin, 10 μg/mL Brefeldin A (all obtained from Sigma-Aldrich) and permeabilized as indicated above. To quantify IL-2 binding, a commercial IL-2 fluorokine kit was utilized following the manufacturer’s instructions (R&D Systems). Dead cells were excluded from analysis using a violet viability stain (Invitrogen). Flow cytometry data collection was performed on an LSR II (BD Biosciences) and cell sorting was performed on an Aria II (BD Biosciences). Data were analyzed using FlowJo software (Tree Star Inc.).
Human Tissue Processing and Flow Cytometry
Lung tissues were minced and incubated at 37°C in enzymatic RPMI media containing 10% FCS, 2 mg/ml Collagenase D (Roche, Nutley, NJ), 1 mg/ml trypsin inhibitor (Life Technologies, Carlsbad, CA) and 0.1 mg/ml DNase I. Digested tissue was passed through a 70μm filter and pelleted via centrifugation. Residual red blood cells were lysed using AKC lysis buffer (Corning) and cells were washed with RPMI media prior to flow cytometric analysis. Lung draining mediastinal lymph node tissues were minced finely and placed in Collagenase II (1 mg/ml) for 1 hour shaking at 37°C. The resulting cell suspension was passed through a 70 μm filter, washed and pelleted via centrifuge. Any residual fat was discarded and red blood cells were lysed using AKC lysis buffer, as above.
Cells were stained with antibodies purchased from BioLegend or eBioscience. Dead cells were excluded from analysis using a viability stain (Invitrogen). After staining the cells were washed and fixed in 2% paraformaldehyede in PBS. Flow cytometry data was collected using a LSR II (BD Biosciences). Data were analyzed using FlowJo software (Tree Star Inc.).
ELISA
HDM-specific antibody titers plates were pre-coated with 25 μg/ml HDM extract. Sera was incubated in doubling dilutions, followed by incubation with secondary antibodies against murine IgG1 or IgE (ebioscience). For IL-5 and IL-17A quantifications, 2 × 105 cells from MedLN of HDM exposed MHCIIΔILC3 mice were ex vivo restimulated with 10 μg/ml crude microbiota-derived extracts as previously described (Hepworth et al., 2013), IL-5 and IL-17A levels in the supernatants were measured using commercially available antibody pairs (eBioscience). ELISA plates were developed with TMB peroxidase substrate.
16S-rRNA Analysis
Milled raw materials of D. pteronyssinus HDM were purchased from four different commercially available vendors: Greer, AirMid Healthgroup, HollisterStier and CiTeq Biologics. 200 mg milled raw materials from each vendor were processed and analyzed for 16 s-rRNA sequencing by IDEXX (IDEXX Laboratories). In brief, bacterial 16S rRNA amplicons were constructed via amplification of the V4 region of the 16S rRNA gene with universal primers previously developed against the V4 region, flanked by Illumina standard adaptor sequences (Caporaso et al., 2011; Walters et al., 2011). Dual-indexed forward and reverse primers were used in all reactions. Amplicon pools were combined, thoroughly mixed, and then purified. The final amplicon pool was evaluated using the Advanced Analytical Fragment Analyzer automated electrophoresis system, quantified, and diluted according to Illumina’s standard protocol for sequencing on the MiSeq instrument. Primers were designed to match the 5′ ends of the forward and reverse reads. Cutadapt (version 2.6; https://github.com/marcelm/cutadapt) was used to remove the primer from the 5′ end of the forward read. If found, the reverse complement of the primer to the reverse read was then removed from the forward read as were all bases downstream. Thus, a forward read could be trimmed at both ends if the insert was shorter than the amplicon length. The same approach was used on the reverse read, but with the primers in the opposite roles. Read pairs were rejected if one read or the other did not match a 5′ primer, and an error-rate of 0.1 was allowed. Two passes were made over each read to ensure removal of the second primer. A minimal overlap of 3 with the 3′ end of the primer sequence was required for removal. The Qiime2 dada2 plugin (version 1.10.0) was used to denoise, de-replicate, and count ASVs (amplicon sequence variants), incorporating the following parameters: 1) forward and reverse reads were truncated to 150 bases, 2) forward and reverse reads with number of expected errors higher than 2.0 were discarded, and 3) Chimeras were detected using the “consensus” method and removed. R version 3.5.1 and Biom version 2.1.7 were used in Qiime2. Taxonomies were assigned to final sequences using the Silva.v132 database, using the classify-sklearn procedure.
Metagenomic Analysis
500 mg milled raw materials of HDM from Greer and AirMid were processed and analyzed for metagenomic analysis by CosmosID. In brief, DNA was isolated from dust mite powder samples using the QIAGEN DNeasy PowerLyzer PowerSoil Kit, according to the manufacturer’s protocol. Isolated DNA was quantified by Qubit. DNA libraries were then prepared using the Illumina Nextera XT library preparation kit. Library quantity was assessed with Qubit (ThermoFisher). Libraries were then sequenced on an Illumina HiSeq platform 2×150bp. Unassembled sequencing reads were directly analyzed by CosmosID bioinformatics platform (CosmosID Inc., Rockville, MD) for multi-kingdom microbiome analysis and profiling of antibiotic resistance and virulence genes and quantification of organisms’ relative abundance as previously described (Ponnusamy et al., 2016).
Histology
Lung tissue was fixed in 4% paraformaldehyde, embedded in paraffin and 5 μm sections stained with PAS by IDEXX BioAnalytics.
Immunofluorescence Staining
Staining for RORγt, CD127 and CD3 expression was performed as previously described (Mackley et al., 2015; Melo-Gonzalez et al., 2019). Briefly, MedLN were removed and snap-frozen in Tissue-Tek OCT compound (Bayer Healthcare, Newbury, UK), and 7 μm sections were cut and collected onto Superfrost Plus slides (VWR) and stored at −80°C. Upon thawing slides were fixed in cold acetone at 4°C for 20 min, air-dried for 30 min, and hydrated in PBS for 10 min. Slides were blocked with 10% horse serum in staining buffer (1% BSA in PBS) for 30 min, and subsequently stained with purified anti-mouse RORγt (1:25; clone AFKJS-9; eBioscience) and biotinylated anti-mouse CD3 (1:100; clone eBio500A2; eBioscience) in staining buffer for 1 h at room temperature. Slides were washed with PBS for 10 min and incubated with donkey anti-rat-IgG-FITC (1:100; Jackson ImmunoResearch) secondary antibody for 30 min, for RORγt signal amplification. After washing, slides were blocked with 10% rat serum in staining buffer for 30 min. Subsequently, slides were incubated with rabbit anti-FITC-AF488 (1:100; Life Technologies) tertiary antibody and eFluor 660-conjugated anti-IL-7Ra (1:25; clone A7R34; eBioscience) for 30 min. After washing, slides were stained with donkey anti-rabbit-IgG-AF488 (1:100; Life Technologies) quaternary antibody for RORγt detection and streptavidin Alexa Fluor 555 (1:500; Life Technologies) for CD3 detection. Secondary, tertiary, and quaternary antibodies for anti-RORγt signal amplification were cross-adsorbed for 30min in 10% mouse serum in staining buffer before staining. Sections were counterstained with DAPI (Invitrogen) and mounted using ProLong Gold (Invitrogen). Slides were visualized under a Zeiss Axio Imager.D2 fluorescence microscope or the Olympus BX63 fluorescence slide scanner. Images were processed with ImageJ.
QUANTIFICATION AND STATISTICAL ANALYSIS
Results represent mean + s.e.m. Statistical analyses were performed by Student’s t test or two-way ANOVA unless specified otherwise. For serum ELISA and airway resistance analyses, the area under the curve (AUC) was first calculated for each serial diluted sample followed by one-way ANOVA among groups (Prism 9, Graph-Pad Software). *p < 0.05; **p < 0.01; ***p < 0.001, ****p < 0.0001.
Supplementary Material
Highlights.
ILC3s are enriched in the lung draining lymph node of healthy humans and mice
Antigen presentation by MHC class II+ ILC3s limits allergen-specific T cells in airway
MHC class II+ ILC3s critically control type 2 allergic airway inflammation
MHC class II+ ILC3s also limit airway Th17 cells that respond to mite-associated microbes
ACKNOWLEDGMENTS
We thank members of the Sonnenberg and Hepworth Laboratories for discussions and critical reading of the manuscript. Research in the Sonnenberg Laboratory is supported by the National Institutes of Health (R01AI143842, R01AI123368, R01AI145989, R01AI162936, R21CA249274, and U01AI095608), the NIAID Mucosal Immunology Studies Team (MIST), the Searle Scholars Program, the American Asthma Foundation Scholar Award, an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund, a Wade F.B. Thompson/Cancer Research Institute (CRI) CLIP Investigator grant, the Meyer Cancer Center Collaborative Research Initiative, Linda and Glenn Greenberg, the Dalton Family Foundation, and the Roberts Institute for Research in IBD. G.F.S. is a CRI Lloyd J. Old STAR. Research in the Farber Laboratory is supported by the National Institutes of Health (AI106697). Research in the Hepworth Laboratory is supported by a Sir Henry Dale Fellowship jointly funded by the Wellcome Trust and the Royal Society (grant number 105644/Z/14/Z), a BBSRC responsive mode grant (BB/T014482/1), and a Lister Institute of Preventative Medicine Prize.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.110051.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alcorn JF, Crowe CR, and Kolls JK (2010). TH17 cells in asthma and COPD. Annu. Rev. Physiol 72, 495–516. [DOI] [PubMed] [Google Scholar]
- Ardain A, Domingo-Gonzalez R, Das S, Kazer SW, Howard NC, Singh A, Ahmed M, Nhamoyebonde S, Rangel-Moreno J, Ogongo P, et al. (2019). Group 3 innate lymphoid cells mediate early protective immunity against tuberculosis. Nature 570, 528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artis D, and Spits H (2015). The biology of innate lymphoid cells. Nature 517, 293–301. [DOI] [PubMed] [Google Scholar]
- Bartemes KR, and Kita H (2021). Roles of innate lymphoid cells (ILCs) in allergic diseases: The 10-year anniversary for ILC2s. J. Allergy Clin. Immunol 147, 1531–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck K, Ohno H, and Satoh-Takayama N (2020). Innate Lymphoid Cells: Important Regulators of Host-Bacteria Interaction for Border Defense. Micro-organisms 8, 1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA, Thompson EA, Fraser KA, Rosato PC, Filali-Mouhim A, et al. (2016). Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature 532, 512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosteels C, Neyt K, Vanheerswynghels M, van Helden MJ, Sichien D, Debeuf N, De Prijck S, Bosteels V, Vandamme N, Martens L, et al. (2020). Inflammatory Type 2 cDCs Acquire Features of cDC1s and Macrophages to Orchestrate Immunity to Respiratory Virus Infection. Immunity 52, 1039–1056.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, and Knight R (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 108, 4516–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter DJ, Granot T, Matsuoka N, Senda T, Kumar BV, Thome JJC, Gordon CL, Miron M, Weiner J, Connors T, et al. (2018). Human immunology studies using organ donors: Impact of clinical variations on immune parameters in tissues and circulation. Am. J. Transplant 18, 74–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan TF, Ji KM, Yim AK, Liu XY, Zhou JW, Li RQ, Yang KY, Li J, Li M, Law PT, et al. (2015). The draft genome, transcriptome, and microbiome of Dermatophagoides farinae reveal a broad spectrum of dust mite allergens. J. Allergy Clin. Immunol 135, 539–548. [DOI] [PubMed] [Google Scholar]
- Chun E, Lavoie S, Fonseca-Pereira D, Bae S, Michaud M, Hoveyda HR, Fraser GL, Gallini Comeau CA, Glickman JN, Fuller MH, et al. (2019). Metabolite-Sensing Receptor Ffar2 Regulates Colonic Group 3 Innate Lymphoid Cells and Gut Immunity. Immunity 51, 871–884.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciaccio CE, Barnes C, Kennedy K, Chan M, Portnoy J, and Rosenwasser L (2015). Home dust microbiota is disordered in homes of low-income asthmatic children. J. Asthma 52, 873–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crellin NK, Trifari S, Kaplan CD, Satoh-Takayama N, Di Santo JP, and Spits H (2010). Regulation of cytokine secretion in human CD127(+) LTi-like innate lymphoid cells by Toll-like receptor 2. Immunity 33, 752–764. [DOI] [PubMed] [Google Scholar]
- Diefenbach A, Colonna M, and Koyasu S (2014). Development, differentiation, and diversity of innate lymphoid cells. Immunity 41, 354–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ege MJ, Mayer M, Schwaiger K, Mattes J, Pershagen G, van Hage M, Scheynius A, Bauer J, and von Mutius E (2012). Environmental bacteria and childhood asthma. Allergy 67, 1565–1571. [DOI] [PubMed] [Google Scholar]
- Fachi JL, Sécca C, Rodrigues PB, Mato FCP, Di Luccia B, Felipe JS, Pral LP, Rungue M, Rocha VM, Sato FT, et al. (2020). Acetate coordinates neutrophil and ILC3 responses against C. difficile through FFAR2. J. Exp. Med 217, jem.20190489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimura KE, and Lynch SV (2015). Microbiota in allergy and asthma and the emerging relationship with the gut microbiome. Cell Host Microbe 17, 592–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimura KE, Sitarik AR, Havstad S, Lin DL, Levan S, Fadrosh D, Panzer AR, LaMere B, Rackaityte E, Lukacs NW, et al. (2016). Neonatal gut microbiota associates with childhood multisensitized atopy and T cell differentiation. Nat. Med 22, 1187–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger G, Fan X, Dikiy S, Lee SY, and Rudensky AY (2015). Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science 350, 981–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goc J, Lv M, Bessman NJ, Flamar AL, Sahota S, Suzuki H, Teng F, Putzel GG, Eberl G, Withers DR, et al. ; JRI Live Cell Bank (2021). Dysregulation of ILC3s unleashes progression and immunotherapy resistance in colon cancer. Cell 184, 5015–5030.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray EE, Friend S, Suzuki K, Phan TG, and Cyster JG (2012). Subcapsular sinus macrophage fragmentation and CD169+ bleb acquisition by closely associated IL-17-committed innate-like lymphocytes. PLoS One 7, e38258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halim TY, Krauss RH, Sun AC, and Takei F (2012). Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity 36, 451–463. [DOI] [PubMed] [Google Scholar]
- Halim TY, Steer CA, Mathä L, Gold MJ, Martinez-Gonzalez I, McNagny KM, McKenzie AN, and Takei F (2014). Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 40, 425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammad H, and Lambrecht BN (2021). The basic immunology of asthma. Cell 184, 2521–2522. [DOI] [PubMed] [Google Scholar]
- Hepworth MR, Monticelli LA, Fung TC, Ziegler CG, Grunberg S, Sinha R, Mantegazza AR, Ma HL, Crawford A, Angelosanto JM, et al. (2013). Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature 498, 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepworth MR, Fung TC, Masur SH, Kelsen JR, McConnell FM, Dubrot J, Withers DR, Hugues S, Farrar MA, Reith W, et al. (2015). Immune tolerance. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4+ T cells. Science 348, 1031–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt RJ, and Lloyd CM (2021). Regulation of immune responses by the airway epithelial cell landscape. Nat. Rev. Immunol 21, 347–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HY, Lee HJ, Chang YJ, Pichavant M, Shore SA, Fitzgerald KA, Iwakura Y, Israel E, Bolger K, Faul J, et al. (2014). Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat. Med 20, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrecht BN, Hammad H, and Fahy JV (2019). The Cytokines of Asthma. Immunity 50, 975–991. [DOI] [PubMed] [Google Scholar]
- Lloyd CM, and Marsland BJ (2017). Lung Homeostasis: Influence of Age, Microbes, and the Immune System. Immunity 46, 549–561. [DOI] [PubMed] [Google Scholar]
- Loo EXL, Chew LJM, Zulkifli AB, Ta LDH, Kuo IC, Goh A, Teoh OH, Van Bever H, Gluckman PD, Yap F, et al. (2018). Comparison of microbiota and allergen profile in house dust from homes of allergic and non-allergic subjects- results from the GUSTO study. World Allergy Organ. J 11, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch SV, and Boushey HA (2016). The microbiome and development of allergic disease. Curr. Opin. Allergy Clin. Immunol 16, 165–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackley EC, Houston S, Marriott CL, Halford EE, Lucas B, Cerovic V, Filbey KJ, Maizels RM, Hepworth MR, Sonnenberg GF, et al. (2015). CCR7-dependent trafficking of RORγ+ ILCs creates a unique microenvironment within mucosal draining lymph nodes. Nat. Commun 6, 5862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie ANJ, Spits H, and Eberl G (2014). Innate lymphoid cells in inflammation and immunity. Immunity 41, 366–374. [DOI] [PubMed] [Google Scholar]
- Melo-Gonzalez F, Kammoun H, Evren E, Dutton EE, Papadopoulou M, Bradford BM, Tanes C, Fardus-Reid F, Swann JR, Bittinger K, et al. (2019). Antigen-presenting ILC3 regulate T cell-dependent IgA responses to colonic mucosal bacteria. J. Exp. Med 216, 728–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirchandani AS, Besnard AG, Yip E, Scott C, Bain CC, Cerovic V, Salmond RJ, and Liew FY (2014). Type 2 innate lymphoid cells drive CD4+ Th2 cell responses. J. Immunol 192, 2442–2448. [DOI] [PubMed] [Google Scholar]
- Mortha A, Chudnovskiy A, Hashimoto D, Bogunovic M, Spencer SP, Belkaid Y, and Merad M (2014). Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science 343, 1249288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomb DC, and Peebles RS Jr. (2013). Th17-mediated inflammation in asthma. Curr. Opin. Immunol 25, 755–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oherle K, Acker E, Bonfield M, Wang T, Gray J, Lang I, Bridges J, Lewkowich I, Xu Y, Ahlfeld S, et al. (2020). Insulin-like Growth Factor 1 Supports a Pulmonary Niche that Promotes Type 3 Innate Lymphoid Cell Development in Newborn Lungs. Immunity 52, 275–294.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnmacht C, Park JH, Cording S, Wing JB, Atarashi K, Obata Y, Gaboriau-Routhiau V, Marques R, Dulauroy S, Fedoseeva M, et al. (2015). MUCOSAL IMMUNOLOGY. The microbiota regulates type 2 immunity through RORγt+ T cells. Science 349, 989–993. [DOI] [PubMed] [Google Scholar]
- Oliphant CJ, Hwang YY, Walker JA, Salimi M, Wong SH, Brewer JM, Englezakis A, Barlow JL, Hams E, Scanlon ST, et al. (2014). MHCII-mediated dialog between group 2 innate lymphoid cells and CD4(+) T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity 41, 283–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang W, Kolls JK, and Zheng Y (2008). The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity 28, 454–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandiyan P, Zheng L, Ishihara S, Reed J, and Lenardo MJ (2007). CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat. Immunol 8, 1353–1362. [DOI] [PubMed] [Google Scholar]
- Plantinga M, Guilliams M, Vanheerswynghels M, Deswarte K, Branco-Madeira F, Toussaint W, Vanhoutte L, Neyt K, Killeen N, Malissen B, et al. (2013). Conventional and monocyte-derived CD11b(+) dendritic cells initiate and maintain T helper 2 cell-mediated immunity to house dust mite allergen. Immunity 38, 322–335. [DOI] [PubMed] [Google Scholar]
- Ponnusamy D, Kozlova EV, Sha J, Erova TE, Azar SR, Fitts EC, Kirtley ML, Tiner BL, Andersson JA, Grim CJ, et al. (2016). Cross-talk among flesh-eating Aeromonas hydrophila strains in mixed infection leading to necrotizing fasciitis. Proc. Natl. Acad. Sci. USA 113, 722–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenberg GF, and Artis D (2015). Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat. Med 21, 698–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenberg GF, and Hepworth MR (2019). Functional interactions between innate lymphoid cells and adaptive immunity. Nat. Rev. Immunol 19, 599–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenberg GF, Fouser LA, and Artis D (2011). Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat. Immunol 12, 383–390. [DOI] [PubMed] [Google Scholar]
- Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie AN, Mebius RE, et al. (2013). Innate lymphoid cells–a proposal for uniform nomenclature. Nat. Rev. Immunol 13, 145–149. [DOI] [PubMed] [Google Scholar]
- Stein MM, Hrusch CL, Gozdz J, Igartua C, Pivniouk V, Murray SE, Ledford JG, Marques Dos Santos M, Anderson RL, Metwali N, et al. (2016). Innate Immunity and Asthma Risk in Amish and Hutterite Farm Children. N. Engl. J. Med 375, 411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thome JJ, Yudanin N, Ohmura Y, Kubota M, Grinshpun B, Sathaliyawala T, Kato T, Lerner H, Shen Y, and Farber DL (2014). Spatial map of human T cell compartmentalization and maintenance over decades of life. Cell 159, 814–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie ANJ, Mebius RE, et al. (2018). Innate Lymphoid Cells: 10 Years On. Cell 174, 1054–1066. [DOI] [PubMed] [Google Scholar]
- von Burg N, Chappaz S, Baerenwaldt A, Horvath E, Bose Dasgupta S, Ashok D, Pieters J, Tacchini-Cottier F, Rolink A, Acha-Orbea H, and Finke D (2014). Activated group 3 innate lymphoid cells promote T-cell-mediated immune responses. Proc. Natl. Acad. Sci. USA 111, 12835–12840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters WA, Caporaso JG, Lauber CL, Berg-Lyons D, Fierer N, and Knight R (2011). PrimerProspector: de novo design and taxonomic analysis of barcoded polymerase chain reaction primers. Bioinformatics 27, 1159–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YH, and Wills-Karp M (2011). The potential role of interleukin-17 in severe asthma. Curr. Allergy Asthma Rep 11, 388–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worgall TS, Veerappan A, Sung B, Kim BI, Weiner E, Bholah R, Silver RB, Jiang XC, and Worgall S (2013). Impaired sphingolipid synthesis in the respiratory tract induces airway hyperreactivity. Sci. Transl. Med 5, 186ra67. [DOI] [PubMed] [Google Scholar]
- Xiong H, Keith JW, Samilo DW, Carter RA, Leiner IM, and Pamer EG (2016). Innate Lymphocyte/Ly6C(hi) Monocyte Crosstalk Promotes Klebsiella Pneumoniae Clearance. Cell 165, 679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano T, Dobeš J, Vobořil M, Steinert M, Brabec T, Ziętara N, Dobešová M, Ohnmacht C, Laan M, Peterson P, et al. (2019). Aire-expressing ILC3-like cells in the lymph node display potent APC features. J. Exp. Med 216, 1027–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Roth TL, Gray EE, Chen H, Rodda LB, Liang Y, Ventura P, Villeda S, Crocker PR, and Cyster JG (2016). Migratory and adhesive cues controlling innate-like lymphocyte surveillance of the pathogen-exposed surface of the lymph node. eLife 5, e18156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, and Ouyang W (2008). Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med 14, 282–289. [DOI] [PubMed] [Google Scholar]
- Zhou L, Chu C, Teng F, Bessman NJ, Goc J, Santosa EK, Putzel GG, Kabata H, Kelsen JR, Baldassano RN, et al. (2019). Innate lymphoid cells support regulatory T cells in the intestine through interleukin-2. Nature 568, 405–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
16S sequencing datasets reported in this study are available at NCBI SRA via BioProject ID PRJNA762346. Metagenomic sequencing is available at NCBI SRA via BioProject ID PRJNA762364. The paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.