

SUMMARY
Frontotemporal dementia (FTD) because of MAPT mutation causes pathological accumulation of tau and glutamatergic cortical neuronal death by unknown mechanisms. We used human induced pluripotent stem cell (iPSC)-derived cerebral organoids expressing tau-V337M and isogenic corrected controls to discover early alterations because of the mutation that precede neurodegeneration. At 2 months, mutant organoids show upregulated expression of MAPT, glutamatergic signaling pathways, and regulators, including the RNA-binding protein ELAVL4, and increased stress granules. Over the following 4 months, mutant organoids accumulate splicing changes, disruption of autophagy function, and build-up of tau and P-tau-S396. By 6 months, tau-V337M organoids show specific loss of glutamatergic neurons as seen in individuals with FTD. Mutant neurons are susceptible to glutamate toxicity, which can be rescued pharmacologically by the PIKFYVE kinase inhibitor apilimod. Our results demonstrate a sequence of events that precede neurodegeneration, revealing molecular pathways associated with glutamate signaling as potential targets for therapeutic intervention in FTD.
Graphical abstract
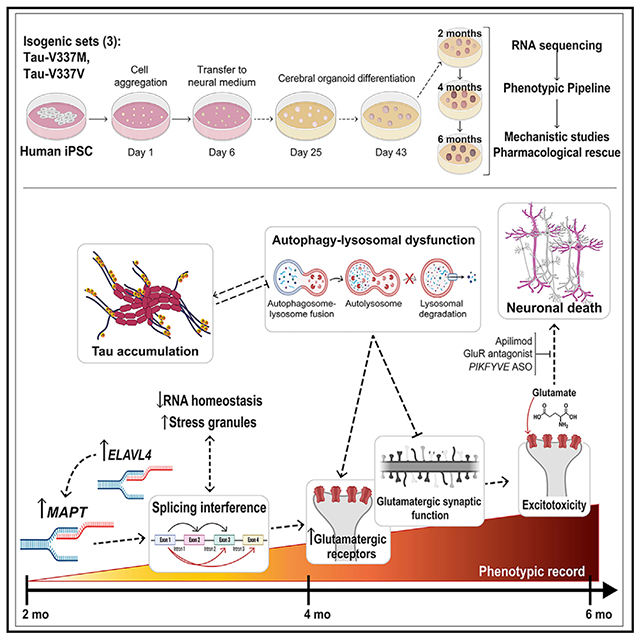
In brief
Characterization of iPSC-derived cerebral organoids with the tau-V337M mutation, which causes frontotemporal dementia, reveals changes preceding neuron death as potential targets for therapeutic intervention, as demonstrated by rescue of susceptibility to glutamatergic toxicity by the PIKFYVE inhibitor apilimod.
INTRODUCTION
Frontotemporal dementia (FTD) encompasses a spectrum of disorders accounting for 5%–6% of all dementias and 20% of cases under the age of 65 (Bird et al., 2003; Olszewska et al., 2016). FTD is heritable, with around 50% of cases having a family history, 10%–20% of which show autosomal dominant inheritance because of mutations in the microtubule-associated protein tau (MAPT) gene (FTD-tau) (Benussi et al., 2015; Rohrer et al., 2009). Pathologic studies reveal accumulation of filamentous, hyperphosphorylated tau protein (P-tau) in cerebral cortical and hippocampal neurons and glia, and tau redistribution from axons to the somatodendritic compartment. These changes correlate with degeneration of synapses and neuronal loss in frontal and temporal cortices (Bodea et al., 2016; Clare et al., 2010). There is great need to identify molecular changes and disease biomarkers that precede cell death when therapeutic intervention will likely have the most effect.
MAPT is alternatively spliced, resulting in two major isoforms, 3R and 4R tau, defined by exclusion or inclusion of exon 10, respectively. To date, 111 unique MAPT variants have been identified (https://www.alzforum.org/mutations/mapt), of which 35 pathogenic mutations are outside exon 10. This is important because little 4R tau is expressed in induced pluripotent stem cell (iPSC)-derived models without a splice-site mutation (lovino et al., 2015; Sato et al., 2018; Sposito et al., 2015; Verheyen et al., 2018). Despite this limitation, iPSC-based models derived from individuals with a MAPT mutation indicate molecular mechanisms underlying tauopathies (Gonzalez et al., 2018; lovino et al., 2015; Jiang et al., 2018; Nakamura et al., 2019; Silva et al., 2016, 2020; Wray, 2017). The tau-V337M mutation selected for this study is located in MAPT exon 12, expressed in iPSC-derived neurons, and associated with FTD with abundant neurofibrillary tangles in frontal and temporal cortices and the cingulate gyrus and atrophy throughout the hippocampus and amygdala (Spillantini et al., 1996; Spina et al., 2017; Sumi et al., 1992). In iPSC-derived induced neurons, tau-V337M disrupts neuronal activity regulation and excitability (Sohn et al., 2019), spurring further studies in more complex models.
Brain organoids derived from iPSCs develop in culture along a trajectory mimicking key features of fetal brain development and can be patterned toward specific brain regions affected in tauopathy (Camp et al., 2015; Lancaster et al., 2017; Paşca et al., 2015; Velasco et al., 2019; Yoon et al., 2019). Increasing evidence showing that early alterations in brain development contribute to later manifestation of neurodegenerative diseases (Faravelli et al., 2020) further encourages use of iPSC-based organoid systems to study FTD-tau. Here we report characterization of over 6,000 cerebral organoids derived from three tau-V337M mutation carriers and respective isogenic CRISPR-corrected lines (Karch et al., 2019; Table S1; Figure 1A). We show that tau-V337M organoids exhibit earlier expression of glutamatergic signaling pathways and synaptic genes, progressive accumulation of total and phosphorylated tau (P-tau S396), alteration of autophagy and lysosomal proteins, formation of stress granules, and widespread changes in splicing with later selective death of glutamatergic neurons. Finally, we demonstrate that glutamate-induced cell death in these organoids can be pharmacologically blocked by apilimod treatment, predicted to target autophagy-lysosomal function and glutamate receptor recycling through PIKFYVE kinase inhibition (Cai et al., 2016; Shi et al., 2018).
Figure 1. Cerebral organoids exhibit similar differentiation patterns as developing fetal brains.
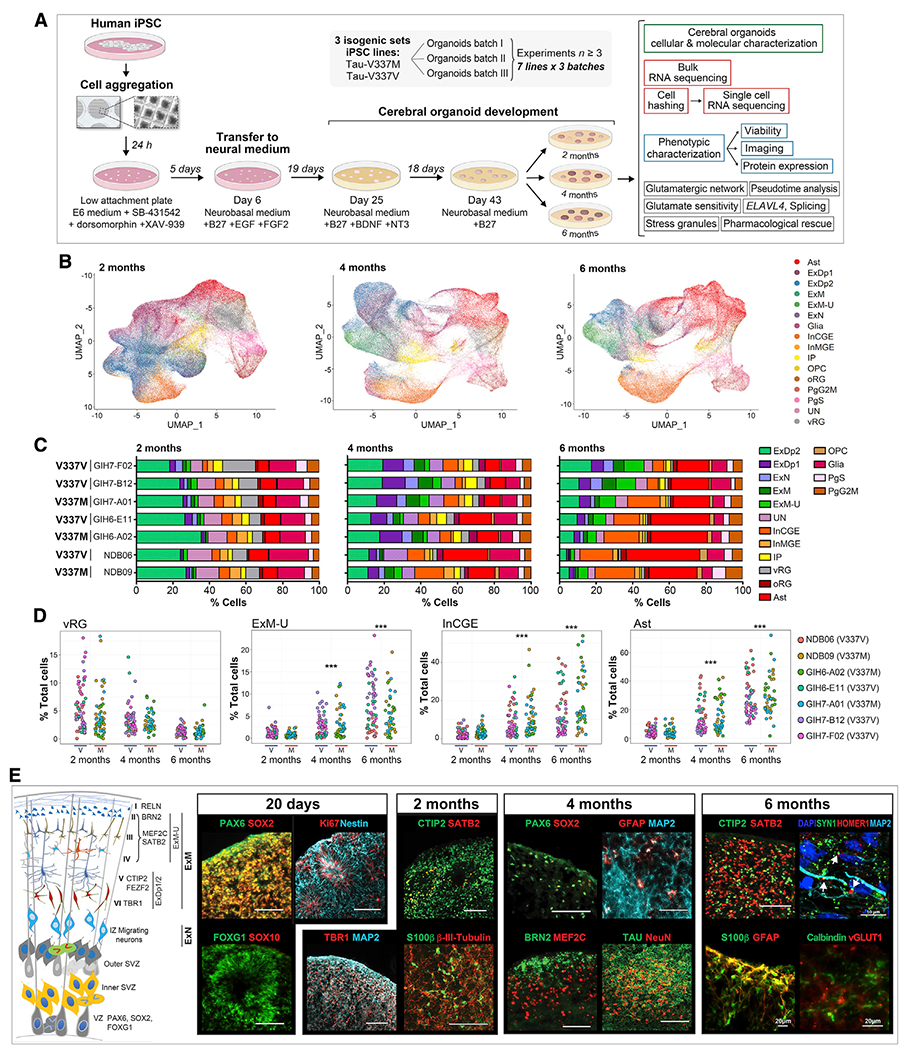
(A) Experiment summary schematic.
(B) UMAP of scRNA-seq data at 2, 4, and 6 months by cell type. Ast, astrocytes; ExDp1, excitatory deep layer 1; ExDp2, excitatory deep layer 2; ExM, maturing excitatory; ExM-U, maturing excitatory upper enriched; ExN, newborn excitatory; Glia, unspecified glia/non-neuronal cells; InCGE, interneurons caudal ganglionic eminence; InMGE, interneurons medial ganglionic eminence; IP, intermediate progenitors; OPC, oligodendrocyte precursor cells; oRG, outer radial glia; PgG2M, cycling progenitors (G2/M phase); PgS, cycling progenitors (S phase); UN, unspecified neurons; vRG, ventricular radial glia.
(C) Cell type proportions (%) per line at 2, 4, and 6 months.
(D) Cell type proportions (%) for individual organoids over time. Linear model, ***p < 0.001.
(E) Schematic of organoid maturation and neural cell layering (left) and marker visualization (right). Confirmation of dorsal forebrain progenitors at 20 days: PAX6, SOX2, FOXG1, and Nestin; proliferation marker Ki67; absence of SOX10. 2 months: increased MAP2ab, β-III-tubulin neurons; deep-layer glutamatergic neurons TBR1, BCL11B/CTIP2; early glia S100β; few upper layer neurons SATB2+. 4–6 months: few progenitors (SOX2 and PAX6); deep- and upper-layer neurons (BRN2, MEF2C, and SATB2); GFAP+ Ast and Calbindin+ interneurons; robust tau and NeuN; expression of vGLUT1+ and pre- and post-synaptic markers SYN1 and HOMER1, respectively; white arrows indicate adjacent boutons. Scale bars, 100 μm unless otherwise indicated.
See also Figure S1.
RESULTS
Selective loss of glutamatergic neurons in tau-V337M organoids
FTD-tau is associated with glutamatergic neuronal loss in frontal and temporal cortices (Benussi et al., 2019). To determine whether this is recapitulated in cerebral tau-V337M organoids, we assessed changes in the proportions of different neural cell types over 6 months of differentiation. Tau-V337M iPSC lines derived from three donors and corresponding isogenic corrected (V337V) controls, totaling seven lines (Table S1; Karch et al., 2019) were differentiated into cerebral organoids (Yoon et al., 2019) and assessed at 2, 4, and 6 months using multiple phenotypic assays (Figure 1A). We performed bulk RNA sequencing (RNA-seq) on 239 individual organoids and single-cell RNA-seq (scRNA-seq) on 339 individual organoids (>370,000 cells, ~800–1,000 cells per organoid). Oligonucleotide-tagged antibodies against cell surface markers (cell hashing) (Stoeckius et al., 2018) barcoded organoid samples prior to multiplex sequencing to reveal replicable effects.
Following scRNA-seq data normalization, reduction, and clustering, automated annotation with SingleR (Aran et al., 2019), using scRNA-seq expression data from the human neocortex at gestational week 17 (GW17)–GW18 as a reference (Polioudakis et al., 2019), identified 16 different cell types (Figures 1B and 1C). Transcriptional signatures in the human frontal cortex at this age correlate highly with gene expression profiles of organoids (Qian et al., 2016). Organoid development followed the expected temporal changes (Lancaster et al., 2013; Velasco et al., 2019; Watanabe et al., 2017; Yoon et al., 2019; Figures 1B–1D; Figure S1A). At 2 months, all organoids had a similar cell composition with a large proportion of deep-layer excitatory (ExDp2) neurons (Figures 1B and 1C). Overtime, the proportion of upper cortical layer-enriched (ExM-U) neurons increased, as did inhibitory neurons, astrocytes, and oligodendrocytes (all p < 2.2 × 10−16; Figures 1B–1D). Although all lines showed the same overall maturation pattern, variability between lines and individual organoids increased over time (Figures 1C and 1D; Figure S1A), indicating donor and clonal effects on long-term maturation. There was no batch-specific effect on differentiation over three replicates assessed by uniform manifold approximation and projection (UMAP) (Figure S1B) and no clustering because of mutation at 2 or 4 months (Figure S1C). Immunostaining of organoid sections confirmed the progressive maturation of organoids observed by scRNA-seq (Figure 1E; Figures S1D and S1E).
We examined whether tau-V337M affects excitatory neuron survival, as observed in the FTD brain (Benussi et al., 2019). Although there were no differences in neuronal proportions at 2 or 4 months, there was a significant reduction in excitatory neurons in deep-layer (ExDp2, p = 0.004), maturing (ExM, p = 0.003), and newborn (ExN, p = 0.0001) categories in 6-month tau-V337M organoids relative to V337V (Figure 2A). These differences were validated by immunohistochemistry (IHC) analysis, which showed a reduction in neuronal nuclear protein+ (NeuN+) neurons at 4 and 6 months (Figures 2B and 2C) and in MAP2+ neurons at 6 months (Figures S2A and S2B). In contrast, there was no effect on interneuron or astrocyte numbers (Figure S2C). Differential gene expression and Ingenuity Pathway Analysis (IPA) of tau-V337M-enriched astrocyte clusters in scRNA-seq data revealed upregulation of interleukin-6 (IL-6) signaling (Z score = 0.54–1.46, p = 2.26 × 10−1–1.8–10−2), interleukin-8 (IL-8) signaling (Z score = 0.67–1.28, p = 5.73 × 10−4_9.64 × 10−5) and neuroinflammation signaling (Z score = 0.13–3.0, p = 2 × 10−1-2.6 × 10−3) pathways across all three isogenic pairs at 4–6 months, suggesting increased neuroinflammation in mutant organoids (Figures S2D–S2F). These results demonstrate that tau-V337M cerebral organoids recapitulate aspects of the selective excitatory neuron vulnerability and glial inflammatory response observed in the human tauopathy brain (Benussi et al., 2019; Bright et al., 2019).
Figure 2. Tau-V337M organoids exhibit neuronal loss, early autophagy disruption, and progressive tau accumulation.
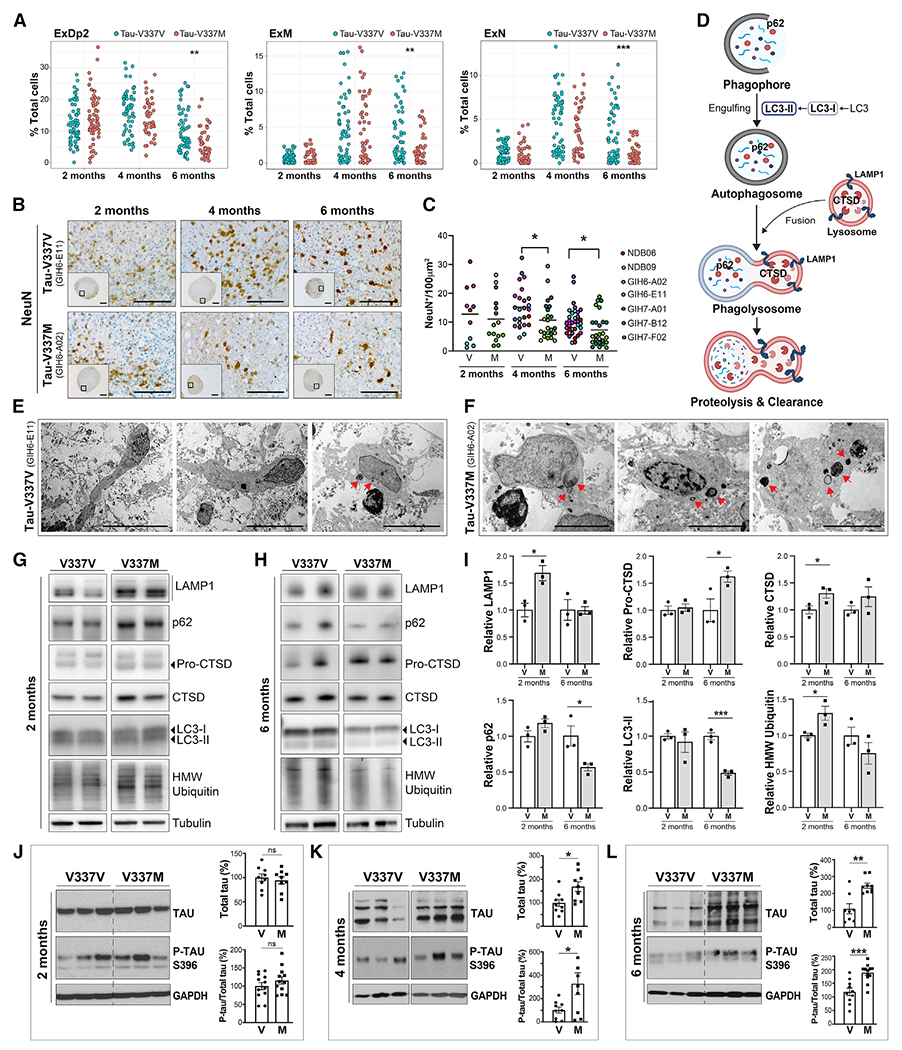
(A) Proportion of glutamatergic neurons (ExDp2, ExM, and ExN) per organoid at 2, 4, and 6 months. Linear model, **p < 0.01, ***p < 0.001.
(B and C) Imaging and quantification of neuronal density by NeuN+ over time in tau-V337M and isogenic V337V organoids. Mann-Whitney test, *p < 0.05. Scale bars, 250 μm (insets) and 50 μm.
(D) Schematic of the ALP and key markers.
(E and F) Electron photomicrographs of neurons in 2-month-old tau-V337V (E) and V337M (F) organoids. Lamellar bodies are indicated by red arrows. Scale bars, 50 μm (E) and 5 μm (F).
(G–I) Western blot and densitometry quantification of ALP markers in tau-V337M and isogenic organoids at 2 (G) and 6 months (H). Relative densitometry ± SEM. Unpaired t test, *p ≤ 0.01, ***p ≤ 0.0001; n = 3 organoids per group.
(J–L) Western blot and densitometry quantification of total tau and P-tau S396 levels in tau-V337M and isogenic V337V organoids at 2 (J), 4 (K), and 6 (L) months. Bars represent mean total tau densitometry or P-tau/total tau in mutant organoids (%) relative to isogenic controls (100%) ± SEM. Two-tailed unpaired t test, *p ≤ 0.01, **p ≤ 0.001, *p ≤ 0.0001; ns, non-significant; n = 9 per group from 3 independent experiments.
See also Figures S2 and S3.
Early autophagy-lysosomal pathway dysfunction in tau-V337M organoids
Disruption of the autophagy-lysosomal pathway (ALP) is another pathological hallmark in human tauopathy brain (Bain et al., 2019; Piras et al., 2016; Wang et al., 2009). To examine whether the tau-V337M mutation was associated with early disruption of this pathway (Figure 2D), we used electron microscopy (EM) to visualize morphological features of ALP vesicles in 2-month organoids (Figures 2E and 2F). Lysosome-related multi-membrane lamellar bodies were rare in healthy neurons (Figure 2E) but more frequent in tau-V337M neurons, including those showing early signs of apoptosis (Figure 2F), suggesting ALP dysfunction. The key ALP markers LAMP1 and CTSD were increased significantly in tau-V337M organoids compared with isogenic controls at 2 months (Figures 2G and 2I), indicating alterations in lysosomal degradation and proteolytic cleavage of pro-CTSD. High-molecular-weight (HMW) ubiquitinated species were increased significantly in tau-V337M organoids at 2 months, suggesting disrupted degradation and accumulation of poly-ubiquitinated proteins and, thus, increased proteostasis stress in mutant organoids. In contrast, pro-CTSD was increased significantly at 6 months in the absence of increased active CTSD, whereas sequestosome 1 (p62) and microtubule-associated protein 1A/1B-light chain 3 (LC3-II) were reduced significantly at the same time point (Figures 2H and 2I), suggesting defects in proteolytic cleavage and activity.
IHC of 2-month cerebral organoids revealed LAMP1 and p62 primarily in neurons, similar to the staining pattern in the brain from an adult individual, indicating that the predominant ALP dysfunction is neuronal (Figures S3A and S3B), consistent with increased lamellar bodies observed in neurons by EM (Figures 2E and 2F). Notably, we did not observe significant dysregulation of ALP in our transcriptomics data, indicating dysfunction at the protein level. These findings demonstrate that iPSC-tau-V337M organoids phenocopy aspects of ALP dysfunction seen in the FTD brain, which may be triggered by expression of mutant tau protein in neurons early in disease development.
V337M organoids exhibit increased tau and P-tau and degeneration of vulnerable cortical layers
Because tau accumulation is a key hallmark of FTD-tau, we examined this in mutant organoids over time. Tau-V337M organoids showed no genotype-dependent differences at 2 months (Figure 2J), but a significant increase in total tau and the P-tau S396/total tau ratio emerged between 4 and 6 months compared with isogenic controls (Figures 2J–2L). The increased tau burden with age mirrors prior observations in 2D iPSC-derived neuronal models (Ehrlich et al., 2015; Fong et al., 2013; lovino et al., 2015; Silva et al., 2016, 2020; Wren et al., 2015) and in the human V337M brain (Spillantini et al., 1996; Spina et al., 2017). Although expression of 4R tau was low, it increased in tau-V337M and V337V organoids with age (Figures S3C and S3D), with no consistent differences between genotypes over time.
RNA-seq data analysis revealed that MAPT mRNA expression was higher in tau-V337M organoids compared with V337V at 2 months; however, this was no longer observed at 4 months and was reversed at 6 months (Figure S3E), potentially reflecting neuron loss (Figures 2A–2C). Hence, increased tau protein at 6 months (Figure 2L) is likely due to tau accumulation and not increased expression. The scRNA-seq data revealed that MAPT expression was highest in excitatory glutamatergic neurons (Figures 3A–3C; Figure S3F), particularly of cortical layers VI-V (ExDp2; Figure 3C). Moreover, MAPT expression was significantly higher in tau-V337M ExDp2 neurons compared with V337V controls at every time point (2 months, p = 6.49 × 10−23; 4 months, p = 1.88 × 10−18; 6 months, p = 3.27 × 10−08)and in ExDp1 and ExM-U at 4 months (ExDp1, p = 1.16 × 10−07;ExM-U, p = 2.82 × 10−04; Figure 3C). In contrast, there was no difference in MAPT expression in inhibitory interneurons (Figure S3G). Notably, this links high expression of MAPT and mutant tau to later selective vulnerability of the same neuronal subtypes.
Figure 3. Tau-V337M organoids reveal loss of deep- and upper-layer glutamatergic neurons.
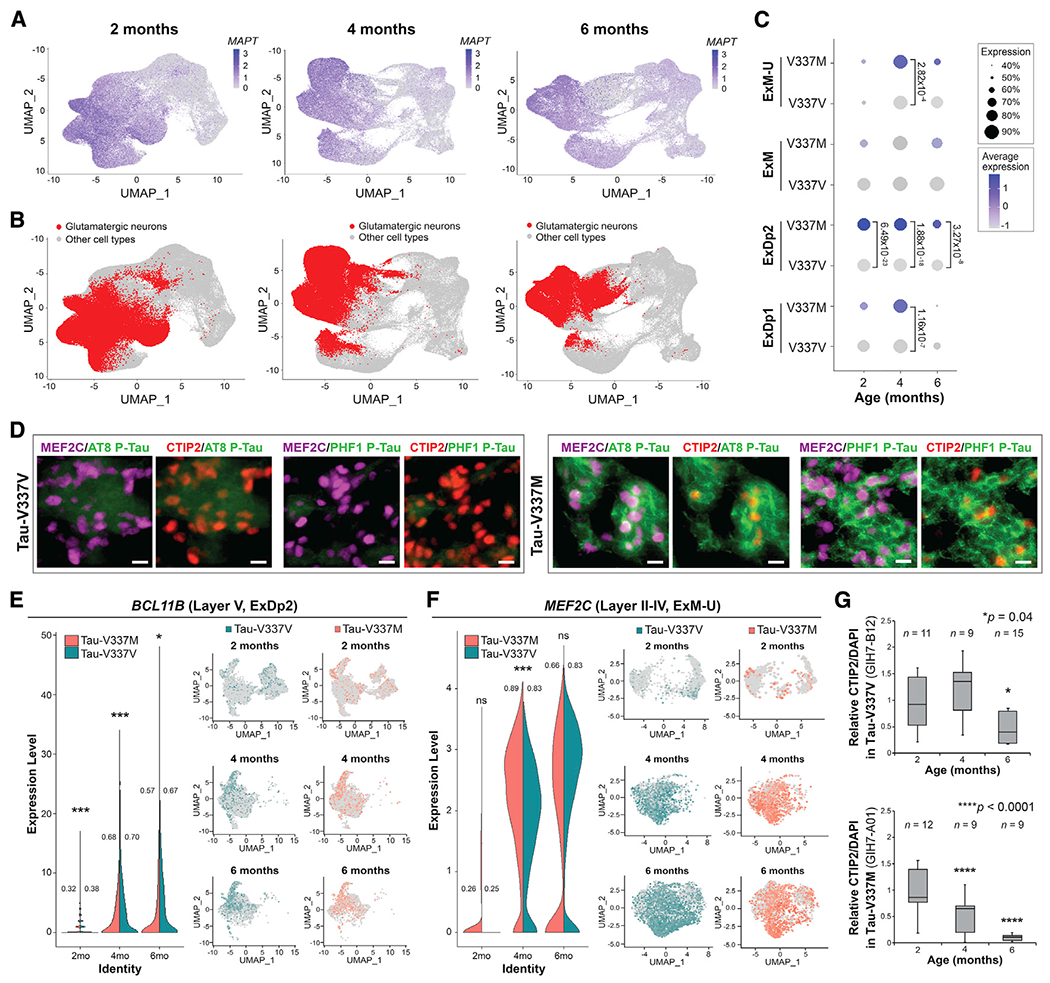
(A and B) Expression of MAPT (A) and glutamatergic neuronal subtypes (ExDp1, ExDp2, ExM, and ExM-U) (B) projected onto scRNA-seq UMAPs at 2, 4, and 6 months.
(C) Proportion of MAPT-expressing glutamatergic neuronal subtypes over time by mutation. Expression is scaled within each time point. Dot size, proportion of MAPT-expressing cells; color depth, MAPT expression level. Values: differential gene expression p value adjusted by model-based analysis of single-cell transcriptomics (MAST) general linear model comparisons of differential expression.
(D) BCL11B/CTIP2 and MEF2C in tau-V337M (right) and isogenic V337V (left) 6-month organoids colocalized with P-tau S202/T205 (AT8) and P-tau S396/S404 (PHF1) staining. Scale bar, 10 μm.
(E and F) Proportion of V337M and V337V ExDp2 neurons expressing the layer V marker BCL11B/CTIP2 (E) or ExM-U neurons expressing the layer II–IV marker MEF2C (F) at each time point; values: proportion of cells expressing each gene. MAST general linear model, *p < 0.05 **p < 0.01 ***p < 0.001 between V337M and V337V neurons. Shown are UMAPs of gene expression in ExDp2 neurons (E) or ExM-U neurons (F) at each time point.
(G) Time-course image quantitative analysis of BCL11B/CTIP2+ neurons at 2,4, and 6 months normalized to DAPI. n ≥ 3 organoids from 3 separately generated organoid batches (representative image shown in D). n = number of organoids. One-way ANOVA, Tukey post hoc test, *p < 0.05, ****p < 0.0001.
In the tau-V337M human brain, neurofibrillary tangles follow a “tram track” pattern with stronger staining in cortical layers II and V (Spillantini et al., 1996; Spina et al., 2017), which appear to be particularly vulnerable to selective neuronal loss (Ghetti et al., 2015); hence, we investigated this in the organoid model. Immunostaining revealed higher P-Tau S202/T205 (AT8) and P-Tau S396/S404 (PHF1) in V337M organoid neurons expressing MEF2C (predominantly upper layers II-IV; Trevino et al., 2020) or BCL11B/CTIP2 (layer V; Figure 3D), corroborating the tau pathology in V337M human brain tissue (Spillantini et al., 1996; Spina et al., 2017). scRNA-seq data analysis revealed significantly fewer glutamatergic neurons expressing MEF2C and BCL11B/CTIP2 in V337M versus V337V organoids at 6 months (Figures 3E and 3F), supported by immunostaining analysis (Figure 3G). Hence, tau-V337M organoids replicate the cortical layer vulnerability observed in the human FTD brain.
Early neuronal maturation and upregulation of synaptic signaling pathways in mutant glutamatergic neurons
To identify early changes underlying neuronal dysfunction prior to tau aggregation and neurodegeneration, we conducted a gene set enrichment analysis (GSEA) on differentially expressed genes at 2, 4, and 6 months in the RNA-seq data (Figures S4A–S4G and S5A–S5H). In mutant organoids, we observed upregulation of numerous synapse-related pathways compared with isogenic controls at 2 months (Figure S4A, bold text) but downregulation between 2 and 6 months (Figure S4B). Further inspection of these pathways emphasized glutamatergic receptor genes with non-monotonic patterns of early upregulation and late downregulation in mutant compared with control organoids, including GRM5 (p = 1.25 × 10−04, log2 fold change [FC] = 1.08), GRIN1 (p = 3.89 × 10−03, FC = 0.47), and GRM4 (p = 6.9 × 10−03, FC = 0.64) (Figure 4A; Figure S4G).
Figure 4. Tau-V337M organoids exhibit early neuronal maturation and upregulation of synaptic signaling pathways.
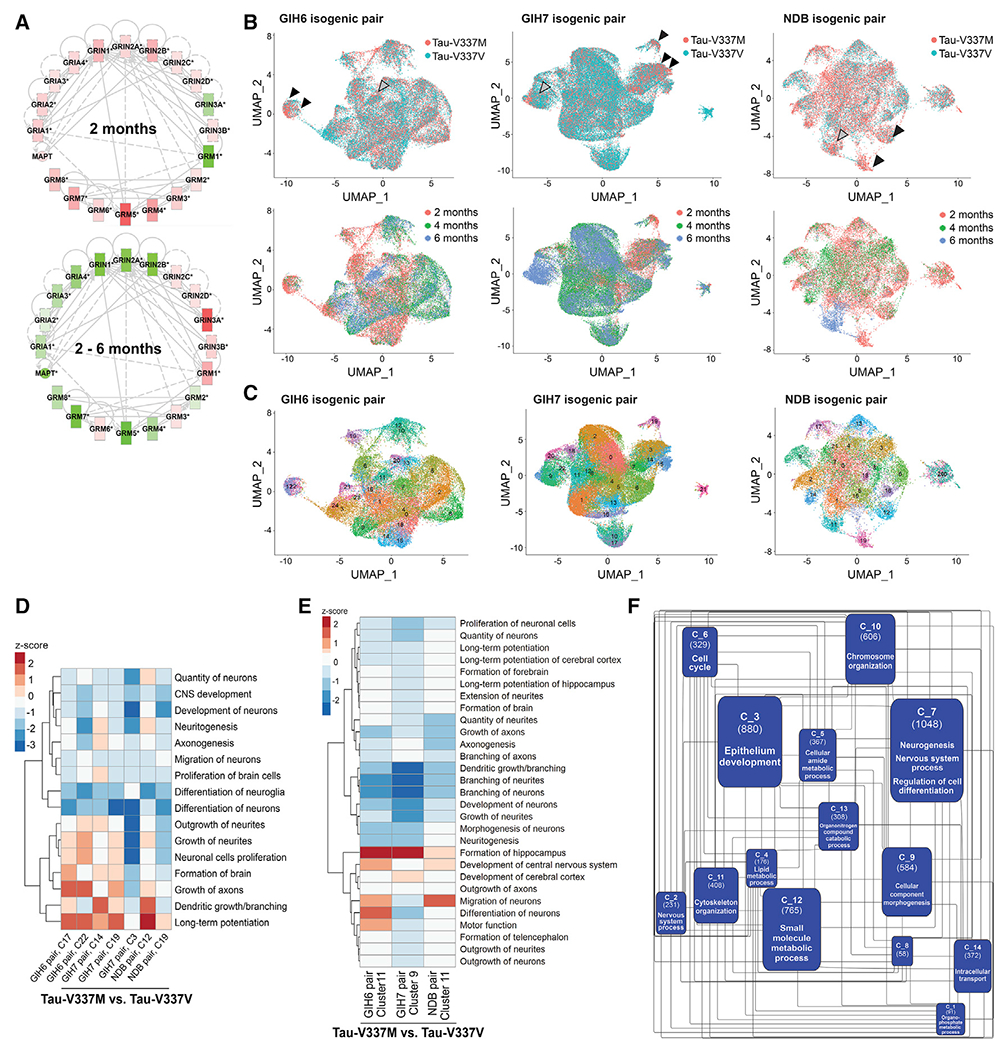
(A) Expression and connectivity of glutamatergic receptor genes and MAPT at 2 months and 2–6 months. Red, upregulation in tau-V337M organoids compared with isogenic V337V; green, downregulation; depth of color, extent of expression fold change.
(B) UMAPs of glutamatergic neurons for each isogenic pair colored by mutation (top) and age (bottom). 2-month V337M-enriched clusters are indicated by black arrowheads, and 6-month V337M-enriched clusters are indicated by gray arrowheads.
(C) UMAPs in (B) colored by Seurat cluster.
(D and E) Z scores for enriched pathways derived from IPA for V337M-enriched 2-month (D) and 6-month (E) glutamatergic neuronal clusters (C) for each isogenic pair.
(F) Network analysis constructed from significantly differentially expressed genes over time between tau-V337V and tau-V337M glutamatergic neurons following pseudobulk analysis of scRNA-seq data.
Communities (C_) are labeled with ID number in bold, with the number of genes in parentheses and the most frequent parent GO term following GO enrichment and semantic similarity analysis. See also Figures S4 and S5.
Following our findings of altered synaptic signaling pathways and loss of glutamatergic neurons over time, we examined excitatory neuronal populations in scRNA-seq data (ExDp2, ExDp1, ExM, and ExM-U). For each isogenic set, excitatory neurons were re-clustered, and V337M-enriched clusters were identified (FC > 1.5; Figures 4B and 4C) and assessed for differential gene expression. Tau-V337M enriched clusters of 2-month neurons showed downregulation of pathways prominent in immature cells, such as differentiation of neuroglia (Z score = −0.274 to −2.175, p = 3.78 × 10−08–4.67 × 10−11) and differentiation of neurons (Z score = −1.131 to −2.606, p = 1.74 × 10−09_1.39 × 10−20) in all three donor lines (Figure 4D). In contrast, and consistent with the bulk data, pathways associated with neuronal maturation and synaptic signaling, such as growth of axons (Z score = 0.239–1.482, p = 9 × 10−06−2.3 × 10−13), dendritic growth/branching (Z score = 0.465–1.608, p = 2.37 × 10−09−5.05 × 10−07), and long-term potentiation (Z score = 0.102–2.775, p = 4.1 × 10−11−6.91 × 10−05) were upregulated at 2 months (Figure 4D) and downregulated at 6 months in V337M-enriched clusters (Figure 4E).
To further characterize temporal gene interactions, we used the scRNA-seq data to carry out pseudobulk network analysis on differentially expressed genes between tau-V337M and V337V glutamatergic neurons over time and identified several gene communities (Figure 4F). Gene Ontology (GO) enrichment revealed that the largest community, C_7, was enriched significantly for pathways such as neurogenesis, nervous system process, regulation of cell differentiation, and synaptic signaling (all false discovery rate [FDR] < 0.01) and contained 9 glutamatergic receptor genes, validating that dysregulation of these genes and pathways is a dominant transcriptional effect of tau-V337M expression over time. C_7 interacted with communities related to cellular structure and organization, including cellular component morphogenesis (C_9) and cytoskeleton organization (C_11). Several communities were enriched for metabolic processes, suggesting that mutant tau may impair neuronal metabolic homeostasis over time (Figure 4F).
We next carried out pseudotime analysis (Qiu et al., 2017; Trapnell et al., 2014) to order cells by differentiation state (Figures S6A–S6E). V337M-enriched modules occurred early in pseudotime, encompassing ExDp2 neurons, indicating aberrant gene expression networks early in development (Figure S6F). Of the top 3,000 variable genes across excitatory neurons, 391 had significantly different trajectories in mutant neurons compared with isogenic controls; GO analysis revealed significant enrichment of glutamatergic signaling pathways, including glutamate binding, glutamatergic synapse, clathrin-sculpted glutamate transport vesicle, and glutamate neurotransmitter release cycle (all p < 0.05). We then clustered significantly different gene trajectories to identify those with similar expression patterns (Figure 5A). Of the 27 genes associated with enriched glutamatergic GO pathways, 11 were in cluster 2, including GRIA2 and MAPT (Figure 5A–C). This cluster was characterized by earlier upregulated gene expression in tau-V337M neurons relative to V337V, which converged at later pseudotime points (Figures 5B and 5C). GO enrichment of cluster 2 genes showed enrichment for neuron projection (p = 3.63 × 10−15), synapse (p = 1.34 × 10−13), and axon (p = 2.14 × 10−13) cellular compartments (Figure 5D) as well as nervous system development (p = 5.35 × 10−13) and synapse organization (p = 7.1 × 10−12) molecular functions. We also found significant enrichment of neural differentiation pathways in cluster 1, including forebrain neuron differentiation (p = 8.762 × 10−4), forebrain generation of neurons (p = 1.937 × 10−3), and nervous system development (p = 4.052 × 10−3), with early increased expression similar to cluster 2 (Figure 5A; Figure S6G).
Figure 5. Accelerated glutamatergic gene and ELAVL4 expression precedes aberrant splicing in V337M neurons.
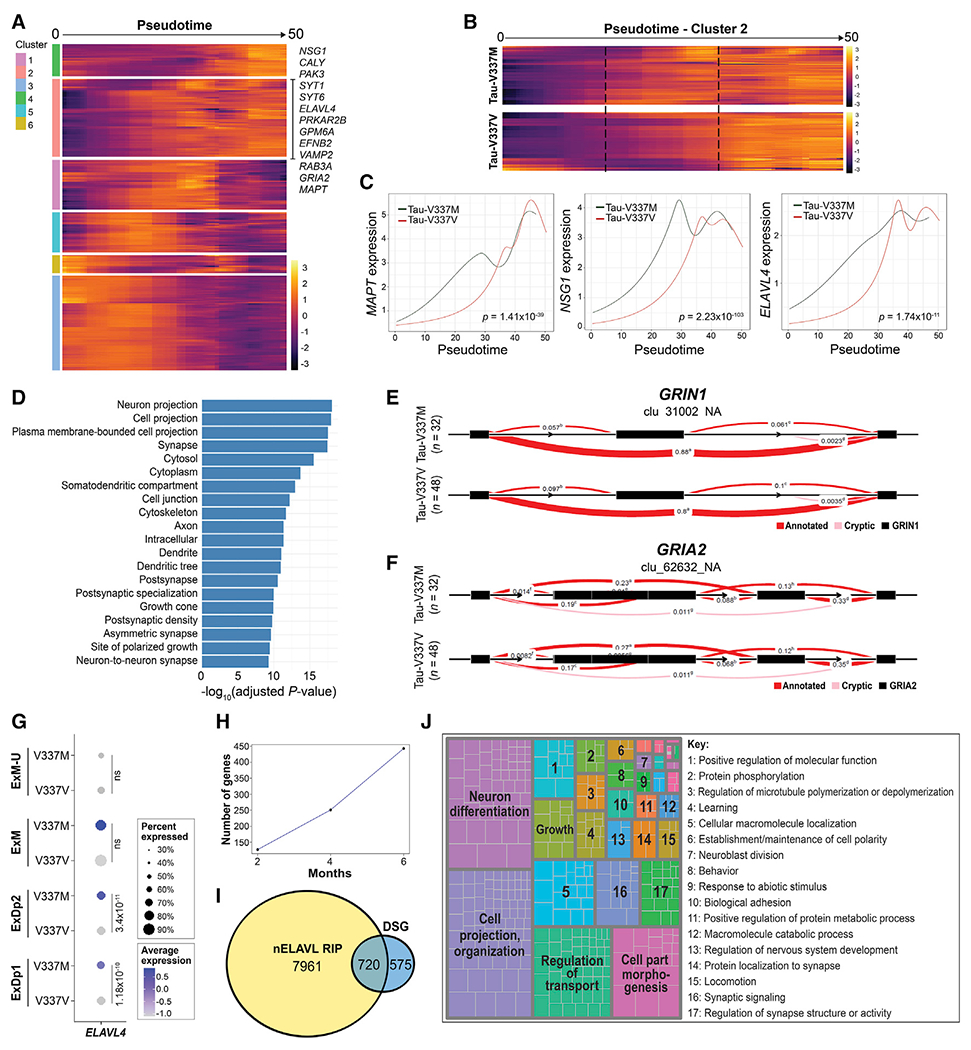
(A) Expression of genes with significantly differential trajectories over pseudotime in tau-V337M versus tau-V337V glutamatergic neurons by spline regression model. Trajectories grouped by unsupervised hierarchical clustering are centered across genes. Genes in significantly enriched glutamatergic signaling pathways are highlighted in cluster 2.
(B) Comparison of cluster 2 pseudotime trajectories in tau-V337M and tau-V337V glutamatergic neurons. Dashed lines highlight the central region of pseudotime, where gene expression differs between mutant and control cells.
(C) Trajectories of average MAPT, glutamatergic pathway gene NSG1, and ELAVL4 expression in tau-V337M and tau-V337V glutamatergic neurons over pseudotime. Adjusted p value for statistical comparison (spline regression) of trajectories is shown in the bottom right corner.
(D) GO pathways enriched for DSGs between 6-month-old tau-V337M and tau-V337V organoids.
(E and F) Leafcutter analysis of differentially spliced intron clusters in the glutamatergic receptor genes GRIN1 (E) and GRIA2 (F). Exons, black boxes. Red band thickness and inserted values represent proportion of spliced exon-exon pairs.
(G) Expression of ELAVL4 in tau-V337M and tau-V337V glutamatergic neurons. Dot size, proportion of cells expressing ELAVL4; depth of color, ELAVL4 expression level. Values: differential gene expression p value adjusted by MAST general linear model comparisons of differential expression.
(H) Number of known nELAVL gene targets by RIP (Scheckel et al., 2016) that are differentially spliced in tau-V337M organoids over time.
(I) Overlap between number of DSGs in tau-V337M organoids and genes in the brain known to be bound by nELAVL by RIP analysis (Scheckel et al., 2016).
(J) Semantic analysis of significant GO pathways enriched for DSGs known to be nELAVL targets (Scheckel et al., 2016).
See also Figure S6.
We noted that a major regulator of glutamatergic cell fate and signaling, ELAVL4, was also present in cluster 2 (Figures 5B and 5C). ELAVL4 encodes a neuronal specific RNA binding protein (RBP) associated with neural development, synaptic plasticity, splicing, glutamate receptor activation and glutamate levels, and multiple neurological diseases (Arendt, 2002; Bolognani et al., 2010; Bronicki and Jasmin, 2013; Ince-Dunn et al., 2012; Popovitchenko et al., 2020; Scheckel et al., 2016; Tiruchinapalli et al., 2008; Zhou et al., 2011; Zhu et al., 2008). Given these functions, we identified ELAVL4 as a potential regulator of accelerated glutamatergic neuron maturation.
Increased ELAVL4 expression in mutant organoids and aberrant splicing of synaptic genes
Differential splicing analysis (Li et al., 2018; Figures 5D–5F; Figure S6H) showed a dramatic increase in the number of differentially spliced junctions in tau-V337M compared with tau-V337V organoids, from 190 at 2 months to over 5,000 at 6 months (Figure S6H). GO analysis of differentially spliced genes (DSGs) at 6 months revealed a significant enrichment in several synaptic pathways (Figure 5D). Exon 5 (chromosome 9 [chr9]: 137145902–13749009) of the N-methyl-D-aspartate (NMDA) receptor gene GRIN1 (log likelihood ratio [loglr] = 12.6, p = 2.76 × 10−3) was more frequently excluded in V337M organoids compared with V337V (differential percent spliced in [dPSI] = −0.06; Figure 5E), and exon 14 (chr4: 157360143–157361538) of the β-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor gene GRIA2 (loglr = 27.18, p = 1.96 × 10−6) was more frequently included in V337M organoids (dPSI = 0.038; Figure 5F). Therefore, although the V337M mutation did not affect MAPT splicing, it did affect the regulation of splicing in pathways associated with neuronal and synaptic maturation that may contribute to glutamatergic neuronal vulnerability.
When examining potential splicing regulators, we noted that neuronal ELAVL genes regulate RNA splicing in mouse brain and human neuroblastoma cells and control glutamate levels and neuronal excitability (Ince-Dunn et al., 2012; Scheckel et al., 2016). Expression of ELAVL4 family members is enriched in deep cortical layers in the human brain (Ravanidis et al., 2018), and consistent with this, ELAVL4 is enriched in deep-layer organoid neurons with higher expression in tau V337M compared with V337V (ExDp2 and ExDp1; Figure 5G). ELAVL4 may therefore be relevant to the susceptibility of this neuronal population, and its abnormal expression could contribute to aberrant splicing.
To assess the potential contribution of ELAVL4 expression to altered splicing regulation of synaptic genes in tau-V337M organoids, we determined whether there was an intersection between our DSGs and transcripts bound to neuronal ELAVL proteins (nELAVL) in the human brain detected by RNA immunoprecipitation sequencing (RIP-seq) (Scheckel et al., 2016; Tebaldi et al., 2018). The proportion of DSGs overlapping with nELAVL-bound RNA increased by more than 3-fold with age, revealing over 50% of DSGs as potential ELAVL4 cargo (Figures 5H and 5I). GO enrichment of these potential cargo genes revealed processes associated with RNA splicing (p = 2.4 × 10−08, FDR = 0.2.2 × 10−06) and neuron differentiation (p = 2.1 × 10−24, FDR = 1.4 × 10−20). Semantic analysis of the significant GO categories (FDR < 0.1) showed enrichment of neuronal differentiation, cell projection organization, and other categories, further supporting the concept that aberrant ELAVL4 expression affects several neuronal functions in tau-V337M organoids (Figure 5J).
ELAVL4 binds the MAPT 3′ UTR and co-localizes with tau and stress granules in mutant organoids
Pathological accumulation of tau and RBPs in stress granules in tau-P301S mice (Vanderweyde et al., 2016) contributes to mislocalization of RNAs and splicing components to the cytosol (Lester et al., 2021). Mutant tau may therefore cause cellular changes that impair ELAVL4 function, such as recruitment to stress granules, affecting its regulation of glutamatergic gene expression and splicing (Popovitchenko et al., 2020; Scheckel et al., 2016). Furthermore, studies of murine and HEK293 cells report that ELAVL4 binds and localizes the MAPT transcript and regulates its local translation (Atlas et al., 2007; Behar et al., 1995; Jung and Lee, 2021); therefore, disruption of ELAVL4 function by mutant tau may further promote aberrant MAPT expression and tau mislocalization, observed with disease. Hence, we investigated the interactions between ELAVL4 and the MAPT transcript and tau protein and the incidence of stress granule formation in tau-V337M and tau-V337V organoids over time.
RIP showed that ELAVL4 binds MAPT RNA in tau-V337V and V337M organoids (Figure 6A). scRNA-seq data analysis revealed a significant increase in the stress granule genes TIA1 and G3BP1 in all glutamatergic neuronal subtypes in tau-V337M compared with V337V (Figure 6B). Immunostaining and western blot analyses also showed elevated G3BP1 protein in 2-month tau-V337M organoids (Figures 6C–6F), corroborating the transcriptional analysis and confirming that mutant tau organoids are more susceptible to stress than controls. We found co-localization of G3BP1 with ELAVL4 in tau-V337M neurons (Figure 6G), indicating sequestration of ELAVL4 in granular structures, as reported previously in amyotrophic lateral sclerosis (ALS) models (De Santis et al., 2019). Furthermore, we observed co-localization of tau with ELAVL4 and TIA1 or G3BP1 (Figures 6H and 6I). These findings support a mechanism whereby the mutant tau transcript and protein interact with ELAVL4 to promote stress and expression of stress granule proteins that contribute to ELAVL4 mislocalization and impaired function in glutamatergic neurons.
Figure 6. ELAVL4 binds MAPT RNA and co-localizes with cytosolic stress granules in tau-V337M neurons.
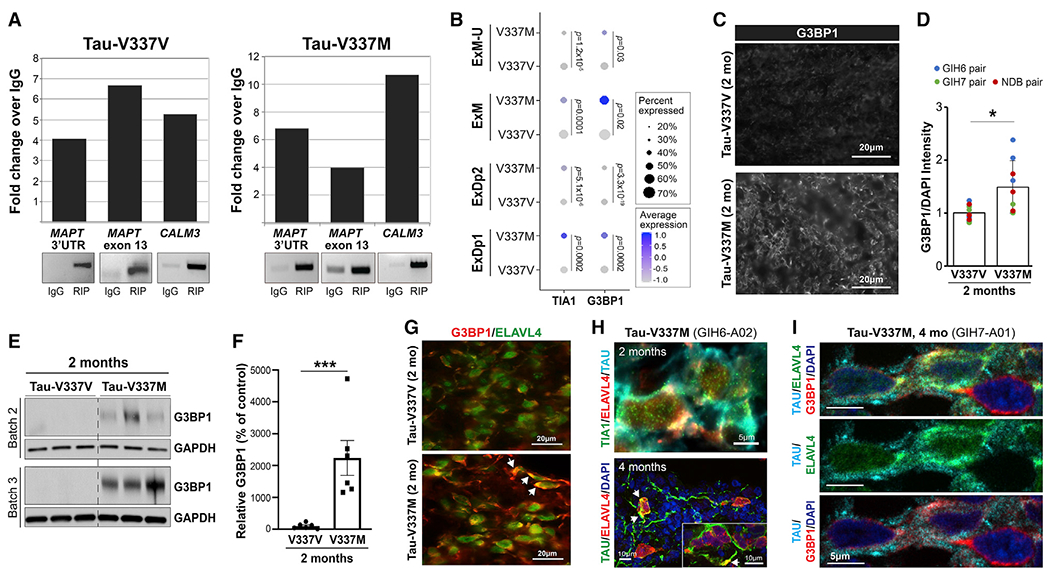
(A) ELAVL4 RNA immunoprecipitation (RIP) detects MAPT 3′ untranslated region (UTR) and exon 13 in tau-V337V (left panel) and tau-V337M organoids (right panel). CALM3, positive control.
(B) Expression of TIA1 and G3BP1 in tau-V337M and tau-V337V glutamatergic neurons. Dot size, proportion of cells expressing a gene; depth of color, expression level. Values: differential gene expression p value adjusted by MAST general linear model.
(C and D) G3BP1 immunostaining (C) and quantification of G3BP1 intensity relative to DAPI (D) in tau-V337V and V337M organoids at 2 months. Bars represent mean intensity ± SD. Unpaired t test, *p ≤ 0.01. n = 4 images per organoid; n = 3 organoids per line for 2 independent experiments.
(E and F) Western blot and densitometry quantification of G3BP1 in tau-V337M and isogenic V337V organoids at 2 months. Bars represent G3BP1 densitometry in mutant organoids (%) relative to isogenic controls ± SEM. Unpaired t test, **p ≤ 0.01; n = 6 per group for 2 independent experiments.
(G) ELAVL4 and G3BP1 colocalization (white arrows) in tau-V337V and V337M organoids at 2 months. Scale bar, 20 μm.
(H) ELAVL4 and TIA1 co-localization with tau at 2 months (scale bar, 5 μm) and ELAVL4 with tau at 4 months (white arrows; scale bar, 10 μm).
(I) Co-localization of tau, ELAVL4, and G3BP1 in tau-V337M organoids at 4 months. Scale bar, 5 μm.
Excitotoxic stress vulnerability and neuronal death in V337M organoids is reversed by apilimod
Because our transcriptomics analyses showed that tau-V337M organoids display early increased expression of glutamatergic signaling components, we examined susceptibility to glutamate stimulation. We used 4- and 12-month organoids to model advancing disease and established a method for longitudinal tracking of individual neurons by transduction with a Synapsin1::GFP lentivirus and daily imaging (Figure 7A; Shi et al., 2019). Viability of labeled cells at the end of the experiment was assessed by co-staining with DRAQ5 (Figure S7A). In the absence of excess glutamate, neurons in control and tau-V337M organoids survived equally well (Figure 7B; Figure S7G). In contrast, repeated treatment with 5 mM glutamate triggered a more rapid and significant loss of tau-V337M neurons relative to isogenic controls (p < 0.0001; Figures 7C and 7D; Figures S7B, S7C, S7H, and S7I). These findings were recapitulated in three isogenic pairs, demonstrating reproducibility of the phenotype (Figure 7E). Importantly, co-treatment of 4-month organoids with selective inhibitors of ionotropic glutamate receptors (3i) significantly rescued survival of isogenic tau-V337V (p = 0.005) and tau-V337M (p < 0.0001) neurons, demonstrating that cell death was driven specifically by excitotoxicity (Figures 7F–7H; Figure S7D). Thus, neurons in tau-V337M organoids were more sensitive to glutamate-induced toxicity than neurons in control organoids.
Figure 7. Tau-V337M susceptibility to glutamate excitotoxicity is reversed by antagonists of excitatory receptors and PIKFYVE inhibition.
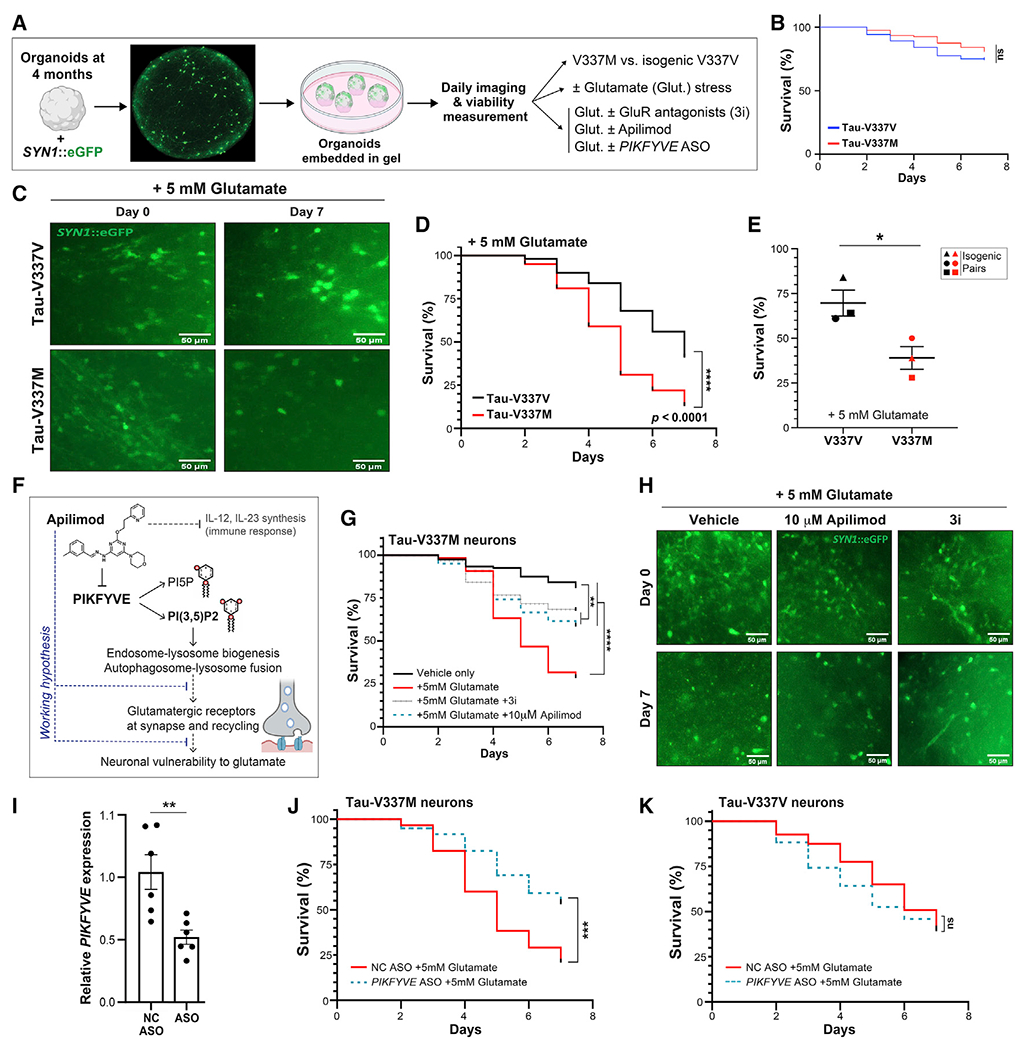
(A) Longitudinal imaging method for tracking neuronal survival in cerebral organoids.
(B) Survival of SYN1::GFP+ neurons in 4-month tau-V337M and isogenic V337V organoids without glutamate treatment. n = 80 neurons from 5 organoids per group. Log rank test. ns, not significant
(C) Images of 4-month tau-V337M and isogenic V337V organoids treated with 5 mM glutamate. Neurons labeled with a lentivirus encoding SYN1::GFP. Scale bars, 50 μm.
(D) Survival of SYN1::GFP+ neurons in tau-V337M and isogenic V337V 4-month organoids with glutamate treatment. n = 100 neurons from 5 individual organoids per group. Log rank test. ****p < 0.0001.
(E) Percentage of surviving neurons following 7 days of glutamate treatment from 3- to 4-month organoids. Each point represents an independent experiment among three isogenic pairs. Two-tailed unpaired t test, *p = 0.0436.
(F) Schematic of the proposed mode of action for apilimod and effect on neuronal vulnerability to excitotoxic stress.
(G) Survival of SYN1::GFP+ neurons in tau-V337M organoids with glutamate treatment and DMSO. 3i, 10 μM CNQX + 10 μM MK-801 + 2 mM nimodipine or 10 μM apilimod. n = 120 neurons from 5 individual organoids per group. Log rank test, **p < 0.01 ****p < 0.0001.
(H) Images of 4-month tau-V337M organoids treated with 5 mM glutamate and DMSO. 3i, 10 μM CNQX + 10 μM MK-801 + 2 μM nimodipine or 10 μM apilimod. Neurons were labeled with SYN1::GFP. Scale bars, 50 μm.
(I) Relative PIKFYVE expression in ND03231 organoids by qRT-PCR following treatment with a PIKFYVE ASO, normalized to 18S expression. Bars represent mean expression ± SEM with n = 6 organoids per group. Two-tailed unpaired t test, **p < 0.01.
(J and K) Survival of SYN1::GFP+ neurons in 4-month tau-V337M (J) and tau-V337V (K) organoids with glutamate and 10 μM negative control (NC) or PIKFYVE ASO treatment. n = 120 neurons from 5 individual organoids per group. Log rank test, ***p = 0.001, ****p < 0.0001.
See also Figure S7.
PIKFYVE is a lipid kinase that regulates endolysosomal trafficking by converting phosphatidylinositol 3-phosphate (PI3P) into phosphatidylinositol-3,5-bisphosphate (PI3,5P2; Marat and Haucke, 2016; Figure 7F). Small-molecule inhibition of PIKFYVE (e.g., apilimod) can prevent recycling of receptors back to post-synaptic densities and protect against NMDA-induced excitotoxicity in vitro and in vivo (Shi et al., 2018; Figure 7F). We hypothesized that PIKFYVE inhibition may rescue glutamate-induced neuronal loss in tau-V337M organoids (Figure 7F). To test this, we performed longitudinal tracking of Synapsin1::GFP-labeled neurons with or without 10 μM apilimod treatment. Apilimod fully rescued the increased cell death in neurons of tau-V337M cortical organoids in response to excess glutamate (p < 0.0001) (Figures 7G and 7H; Figures S7J–S7L). We did not observe any change in total tau or P-tau protein levels following treatment, indicating that the rescue conferred by apilimod was not a result of tau lowering (Figures S7E and S7F). To confirm that the effect was via PIKFYVE, we used an antisense oligonucleotide (ASO) targeting PIKFYVE to directly suppress gene expression (Figures 7I–7K). Compared with the negative control (NC), PIKFYVE ASO treatment significantly improved survival in mutant tau-V337M (p < 0.0001) but not tau-V337V neurons (Figures 7J and 7K). Our results reveal that early deficits in the glutamatergic neuronal network can be detected in cortical organoids derived from iPSCs from individuals with FTD and that vulnerability to excitotoxicity can be blocked pharmacologically.
DISCUSSION
We have characterized a 3D cerebral organoid model of FTD using iPSC lines from three donors with the tau-V337M mutation and isogenic corrected controls to identify temporal changes leading to neurodegeneration. This model exhibits progressive accumulation of total tau and P-tau, glial inflammation, impaired autophagy function, and, later, specific loss of glutamatergic neurons. Upregulation of glutamatergic signaling pathways, increased ALP markers, and splicing changes are seen in 2-month mutant neurons preceding cell death. We showed that ELAVL4 protein directly binds MAPT RNA and co-localizes with tau. Upregulated expression of ELAVL4 at 2 months and mislocalization of ELAVL4 with G3BP1-positive stress granules suggests a mechanism by which mutant tau may lead to aberrant expression and function of this RBP. Additionally, we show that these abnormalities lead to neuronal dysfunction and vulnerability to excitotoxicity. Hence, we uncovered a time course of molecular changes in critical pathways associated with MAPT mutation that result in preferential deep cortical layer glutamatergic neuronal death.
Given that organoid models do not accurately reflect adult aging, it is interesting that we observed neurodegeneration. This may be due to model-associated stress (Bhaduri et al., 2020) on a vulnerable neuronal population contributing to accelerated neuronal death. Indeed, we observed increased stress granules in tau-V337M organoids compared with isogenic controls, also observed in mouse tau-P301L brain (Vanderweyde et al., 2016). Expression of tau-V337M resulted in progressive accumulation of total tau and P-tau S396 as organoids aged, as seen in human FTD brain (Shiarli et al., 2006; Spillantini et al., 1996; Sumi et al., 1992). Furthermore, ALP markers increased at 2 months in tau-V337M organoids, consistent with early impaired function (Wang et al., 2009) and corroborating increased LAMP1 and accumulation of autophagic vesicles in the human brain (Piras et al., 2016) and models of tauopathy (Lin et al., 2003; Nixon et al., 2005; Yu et al., 2004). Interestingly, by 6 months, LC3II and p62 levels were reduced significantly in tau-V337M organoids, suggesting impairments in autophagosome/phagolysosome formation and reduced proteolysis. Differences in ALP markers between 2 and 6 months may reflect proteolytic stress and neuronal dysfunction as well as neuronal loss with time. Because ALP impairment contributes to pathological accumulation of tau (Bain et al., 2019; Bendiske and Bahr, 2003; Wang et al., 2009), it is likely that ALP dysfunction associated with mutant tau further contributes to tau accumulation.
Tau neurofibrillary tangles occur primarily in cortical layers II and V in the tau-V337M brain (Spillantini et al., 1996; Spina et al., 2017), and layer V neuronal subsets preferentially die (Lin et al., 2019). We found that MAPT expression was highest in deep-layer glutamatergic excitatory neurons encompassing layer V and more substantial loss of deep cortical neurons at 6 months in tau-V337M organoids, suggesting that high MAPT expression may underlie selective neuronal vulnerability, but with longer culture periods we may see increased loss of later-born upper-layer cells.
Our studies point to major alterations in glutamatergic signaling pathways associated with MAPT mutation. Glutamate excitotoxicity is the predominant cause of cerebral neurodegeneration (Benussi et al., 2019; Bowie, 2008). Human clinical studies of FTD and tau-V337M animal models suggest impairments in glutamatergic circuits and signaling (Borroni et al., 2017, 2018; Leuzy et al., 2016; Warmus et al., 2014). We show early upregulation of these pathways, consistent with electrophysiological evidence of advanced synaptic maturation and excitability in tau-P301L, N279K, and V337M MAPT mutation iPSC neurons in 2D culture (lovino et al., 2015; Sohn et al., 2019) and with increased seizure susceptibility in individuals with FTD early in disease (Beagle et al., 2017). Premature synaptic maturation and glutamatergic signaling likely predispose neurons to dysfunction and excitotoxicity, resulting in later synaptic loss and neurodegeneration, and may be common across different MAPT mutations.
Alternative splicing is crucial for synaptic maturation (Weyn-Vanhentenryck et al., 2018). We observed splicing dysregulation in tau-V337M organoids in genes converging on synaptic signaling pathways and increased stress granule markers, consistent with aberrant splicing processes (Anderson and Kedersha, 2008; Lester et al., 2021; Vanderweyde et al., 2016). In the tau-P301S mouse, aggregation of RBPs with tau in stress granules and the resulting alterations in splicing converge on synaptic transmission pathways (Apicco et al., 2019), similar to tau-V337M organoids. These mice exhibit increased inclusion of exon 14 of the AMPA receptor subunit GRIA2 (Apicco et al., 2019), also observed in tau-V337M organoids, which can cause an excitatory phenotype by slower desensitization of AMPA receptors (Koike et al., 2000; Pei et al., 2009). We also identified increased exclusion of GRIN1 exon 5 in tau-V337M organoids, associated previously with overproduction of excitatory synapses in layer V pyramidal neurons in adult mice, resulting in seizure susceptibility (Liu et al., 2019), which may contribute to the cortical layer vulnerability observed in our model and in FTD brains.
ELAVL4 is selectively expressed in neuronal populations vulnerable to cell death in tau-V337M organoids, consistent with its reported expression in the brain (Bronicki and Jasmin, 2013; DeBoer et al., 2014). ELAVL4 expression decreases self-renewal of neural progenitor cells and promotes deep-layer glutamatergic neuronal differentiation (Akamatsu et al., 2005; DeBoer et al., 2014; Popovitchenko et al., 2020). Our pathway analysis of mutant-enriched glutamatergic neuron clusters showed downregulation of neuronal differentiation pathways at 2 months and that neuronal differentiation pathways were accelerated in tau-V337M neurons over pseudotime, supporting the assertion that early ELAVL4 expression promotes neuronal maturation. ELAVL4 regulates RNA splicing (Scheckel et al., 2016), glutamate levels, and neuronal excitability (Ince-Dunn et al., 2012; Jung and Lee, 2021) and binds to the glutamatergic receptor GRM5 and other genes within the long-term potentiation (LTP) pathway (Bolognani et al., 2010), which were upregulated in tau-V337M organoids.
Here we demonstrate that ELAVL4 likely binds MAPT RNA at the 3′ UTR and exonic sites. Interestingly, ELAVL4 binds the 3′ UTR of MAPT RNA in rat primary neurons and in mouse P19 cells, where it has been proposed to regulate MAPT stability and translation, contributing to normal tau localization and neuronal polarity (Atlas et al., 2007; Behar et al., 1995; Jung and Lee, 2021). We observed ELAVL4 co-localized with tau protein and the stress granule markers TIA1 and G3BP1. Tau influences stress granule formation in neurons, and tau-TIA1 interaction can lead to formation and stabilization of toxic tau oligomers (Ash et al., 2021).
Given its pleiotropic functions, our observation of perturbed ELAVL4 expression and localization could contribute to several phenotypes observed in tau-V337M organoids, including aberrant MAPT/tau expression, early neuronal maturation, aberrant glutamate signaling, and dysregulated splicing. Additionally, dysfunctional proteostasis may result in increased levels of tau and ELAVL4 because of impaired protein clearance, promoting their cytosolic accumulation. Dysregulation of ELAVL4 may not be restricted to FTD-tau; ELAVL4 is a target of FUS activity, co-localizes with FUS in cytoplasmic speckles and stress granules, and is present in TAR DNA-binding protein 43 (TDP-43) inclusions (Fallini et al., 2012; De Santis et al., 2019). ELAVL4-mediated splicing dysregulation resulting from its mislocalization and sequestration to stress granules may therefore be a shared mechanism unifying FTD-tau with other familial forms of FTD.
We demonstrate that susceptibility to glutamate-induced neuronal death in tau-V337M neurons can be blocked by treatment with the PIKFYVE inhibitor apilimod and through knockdown of PIKFYVE mRNA using an ASO. It has been proposed that apilimod reduces glutamatergic receptor expression and electrophysiological activity by preventing RAS-associated binding protein (RAB)-dependent recycling of the receptors back to the cell surface (Seebohm et al., 2012). In addition, PIKFYVE inhibition increases the number of EEA1-positive endosomes and LAMP1-positive lysosomes and improves proteostasis in neurons (Shi et al., 2018; Staats et al., 2019). Therefore, PIKFYVE inhibition may rescue glutamate-induced neurodegeneration by correcting ALP dysfunction, increasing turnover of misfolded tau, and favoring elimination of internalized NMDA and AMPA receptors over recycling back to the synapse. Importantly, humans and mice haplodeficient for PIKFYVE are normal, and apilimod has shown good tolerability in phase I and II clinical trials (Ikonomov et al., 2011; Sands et al., 2010), indicating potential therapeutic value.
Temporal analysis of human organoid models has revealed early pathological events in neurodegeneration. In our working model, the MAPT mutation results in accumulation of mutant tau, which alters glutamatergic synaptic signaling via two mechanisms: first by altered splicing and homeostasis of glutamatergic signaling genes driven in part by dysregulated ELAVL4 expression and function because of sequestration in stress granules, and second by impaired proteostasis, preventing recycling of receptors away from the synapse and promoting further tau accumulation. Both mechanisms result in increased susceptibility to excitotoxicity. Although these changes are initially accommodated by plasticity, over time they result in glutamatergic neuron death. Our discovery that PIKFYVE inhibition prevents selective glutamatergic cell death encourages further study of the mechanism as a therapeutic strategy in FTD.
Limitations of the study
This study demonstrates the value of iPSC-derived forebrain organoid models to investigate mechanisms of FTD neurodegeneration, but several outstanding questions remain. Given the low 4R tau expression in these organoid models, additional effects of the mutant 4R protein may be missed. The generalizability of these findings to other MAPT mutations and to sporadic tauopathies remains to be elucidated. It would be of interest to include microglia in the model, given their contribution to tau pathology, spread, and synaptic degeneration (Vogels et al., 2019). Our data characterizing glutamatergic signaling in tau-V337M organoids require additional validation by comprehensive electrophysiological assessment to confirm our hypothesized mechanism. Future studies are needed to determine the relative contribution of the MAPT mutation-associated changes we identified.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Sally Temple (sallytemple@neuralsci.org).
Materials availability
Human iPSC lines used in this study are available from the Tau Consortium via the Neural Stem Cell Institute (https://www.neuralsci.org/tau).
Data and code availability
The bulk and single cell RNA-sequencing data generated as part of this study are available at the Gene Expression Omnibus (GEO: GSE171345) and the output of the analyses are available upon request. Previously unpublished code for nELAVL binding cargo enrichment and the pseudobulk network analysis can be found in Data S1. All other code used in this study have been previously published, and the pipelines described in the Method Details.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Human iPSC lines were obtained from the Tau Consortium cell line collection (https://www.neuralsci.org/tau) (Karch et al., 2019): GIH6-1-C1Δ1E11 (WT/WT), GIH6-1-C1ΔA02 (V337M/WT), GIH7-C2Δ2B12 (WT/WT), GIH7-C2Δ2F02 (WT/WT), GIH7-C2Δ2A01 (V337M/WT), ND32951A.15Δ1B06 (WT/WT), and ND32951A.15Δ1B09 (V337M/WT), from the NeuraCell core facility (Neural Stem Cell Institute, NY, USA) and from the NINDS Repository ND03231 (WT/WT); Table S1 (all female lines, except ND03231 (male)). The iPSCs were maintained in mTeSR1 medium using a feeder-free culture protocol in six-well plates (Corning) coated with growth factor-reduced Matrigel. iPSCs were cultured at 37°C and 5% CO2 with daily feeding of 2 mL mTeSR per well. To ensure consistency and obtain high quality organoid production, cultures were grown to no higher than 80% confluency. Passaging of iPSC colonies was carried out using Dispase (StemCell Technologies) to the desired split ratio.
METHOD DETAILS
Organoid production
Organoids were generated at the NeuraCell core facility (Neural Stem Cell Institute, NY, USA) as previously described (Yoon et al., 2019) with minor modifications. When iPSC cultures reached ~80% confluency, the medium was aspirated and wells rinsed twice with DMEM/F12.2 mL of Accutase (StemCell Tech.) was added per 6-well and incubated for ~10 minutes at 37°C, 5% CO2 until cells detached from the dish. Using a 1000 μL pipette, gentle trituration was performed to achieve a single cell suspension, which was transferred to a 50 mL conical tube. Cells were washed with DMEM/F12 three times. Following, cells were counted using a hemo-cytometer and resuspended at approximately 3 million cells/mL in spheroid formation medium (SFM) consisting of E8 medium (Life Technologies) plus 10 μM ROCK inhibitor Y-27632 (Tocris). To prepare the AggreWell800 plate (StemCell Tech.), wells were rinsed once with Anti-Adherence Rinsing Solution (StemCell Tech, 07010) and then centrifuged at 2,000 RCFs for 5 minutes in a swinging bucket rotor fitted with a plate holder to remove any air bubbles from the microwells, verified by checking under a microscope. The rinse solution was aspirated and replaced with 2 mL of DMEM/F12, centrifuged again and finally replaced with 0.5 mL of SFM per AggreWell. Next, 1 mL of cell suspension was added per well, with gentle pipetting to evenly distribute cells at ~10,000 cells per microwell. The AggreWell plate was centrifuged at 100 RCF for 3 minutes and then incubated at 37°C and 5% CO2 overnight to generate spheroids. The next day (day 0 of differentiation), spheroids were transferred from the AggreWells into a conical tube containing ~10 mL DMEM/F12 for each AggreWell of spheroids . Spheroids settled to the bottom of the tube, then the supernatant was gently aspirated and replaced with differentiation Medium A, which is comprised of E6 medium supplemented with 2.5 μM dorsomorphin (DM), 10 μM SB431542, and 2.5 μM XAV-939; 1 mL of medium was added for each AggreWell of spheroids. Spheroids were gently mixed and 1 mL of the suspension was added per ultra-low attachment 10 cm plate (Corning, 3262) containing 9 mL of Medium A and incubated at 37°C and 5% CO2 for 48 hours. Plates were fed daily by gently aspirating medium from the plates and replacing with Medium A, achieving ~65% medium exchange from day two until day five. On the sixth day, the medium was changed to neural medium (NM), consisting of Neurobasal-A plus B-27 supplement without vitamin A, GlutaMax and Anti-A and supplemented with 20 ng/mL EGF plus 20 ng/mL FGF2. NM plus EGF/FGF2 (Medium B) was changed daily for 10 days then every other day for 9 days with ~60% media exchanges. On day 25, the medium was replaced with NM supplemented with 20 ng/mL BDNF and 20 ng/mL NT3 (Medium C) with ~65% media feeds every other day. From day 43 onward, the medium was changed every 3 to 4 days using NM without added growth factors with 15-20 mL per dish and ~75% medium changes. Throughout the culture period, organoids that fused together were separated by cutting with a disposable scalpel (McKesson non-safety scalpels, 1626). For experiments in Figures 6A, 7A–7H, 7J, 7K, and S7A–S7F, iPSCs were grown and patterned as described above with minor modifications, culturing in ultra-low attachment 96 well U-bottom plates (S-BIO, MS9096SZ).
Organoids used in Figure 7I on the ND03231 line and one pair in 7E (ND06/09) were generated in the Ichida lab (USC, CA, USA) using the same basic method as described above (Paşca et. al., 2015) with minor modifications. In brief, human iPSCs were grown to ~70% confluence and dissociated to single cells with Accutase (Stem Cell Technologies) and 10,000 cells/well were seeded into 96-well U-bottom low-attachment plates (Corning) in mTeSR1 (Stem Cell Technologies) +10 μM ROCK inhibitor (Y-27632, Tocris) for spheroid formation at 37°C and 5% CO2. Fresh mTeSR1 was replenished after 24 hours without ROCK inhibitor. After spheroid formation, neural induction was achieved through 5 days of daily medium changes using pre-warmed DMEM/F12 (Corning) containing 20% KnockOut Serum (GIBCO), 1 mM non-essential amino acids (GIBCO), 1X Glutamax, and 0.1 mM β-Mercaptoethanol (Sigma), supplemented with 10 μM dorsomorphin (Cayman) and 10 μM SB-431542 (Cayman). Spheroids were then transferred to neural medium (Neurobasal-A (Life Technologies) with B27 supplement without vitamin A (Life Technologies), 2% Pen/Strep (Corning), and 1X GlutaMax (Life Technologies)) with 20 ng/mL FGF2 (Peprotech) and 20 ng/mL EGF (Peprotech), with daily medium changes until day 15, then medium changes every other day until day 25. Spheroids were transferred to 6-well low-attachment plates (Corning) on day 20. To promote differentiation of neural progenitors into neurons, FGF2/EGF were replaced with 20 ng/mL BDNF (R&D) and 20 ng/mL NT3 (Peprotech) from days 25-43. From day 43 onward, medium changes were done every four days using neural medium without added growth factors.
Fixation and frozen sectioning of organoids
Organoids were fixed overnight at 4°C in 4% paraformaldehyde rinsed three times in PBS, allowed to sink in 30% sucrose in PBS. Organoids were then placed in cryomolds (Tissue Tek, 4565) with OCT compound (Tissue Tek, 4583), snap frozen and stored at −80°C. Organoids were cryostat sectioned sequentially at 20 μm thickness using a Leica cryostat (model CM3050S). Sections were placed on microscope glass slides, dried overnight and stored at −20°C for subsequent immunohistochemistry.
Immunohistochemistry of organoid frozen sections
Slides were thawed to room temperature, and lines drawn to partition the edges of sections on the glass slides using a PAPpen (ImmEdge Pen; Vector Labs H4000). The sections were rehydrated, blocked, permeabilized and immunostained with primary antibodies: PAX6 (BioLegend, 1:200), SOX2 (Santa Cruz, 1:100), β-tubulin III (Sigma, 1:1000), FOXG1 (Takara, 1:500), SOX10 (Santa Cruz, 1:100), Ki67 (Thermo Fisher, 1:200), Nestin (Millipore, 1:200), GFAP (Millipore, 1:400), S100beta (DAKO Potts, 1:400), CTIP2 (Abcam, 1:500), SATB2 (Abcam, 1:50), MAP2AB (Abcam, 1:1000-2000), NeuN (Millipore, 1:200), TBR1(Abcam, 1:500), BRN2 (Millipore, 1:500), MEF2C (Novis, 1:200), AT8 (Invitrogen, 1:1000), PHF1 (gift from Peter Davies,1:1000), Total Tau (DAKO Potts, 1:200), DA9 (gift from Peter Davies, 1:1000), ELAVL4 (Santa Cruz, 1:200), TIA1 (Santa Cruz, 1:100), G3BP1 (Protein Tech, 1:200), VGLUT1 (gift from Susan Morton, Tom Jessell, 1:16,000), Calbindin1 (Swant, 1:10,000), Homer1 (SYSY, 1:250) and Synapsin1 (Millipore, 1:100). Primary antibodies were incubated overnight at 4C, washed three times with PBS and then incubated with corresponding Alexa Fluor conjugated secondary antibody (1:333-1000) for 1 hour at room temperature. Sections were coverslipped and imaged using fluorescence and confocal microscopy (Zeiss AXIO Observer.Z1; Zeiss 780).
Immunofluorescence image analysis of frozen sections
Fluorescence image analysis of immunostained frozen sections was performed using a custom macro developed for ImageJ software. Low-power images were used to visualize across organoid sections. Individual thresholds were set to measure CTIP2 and DAPI cell counts within each image. CTIP2/DAPI values were calculated and normalized to the 2-month samples within an individual organoid batch and cell line. Boxplots generated contain counts from at least three organoids per batch and across three different organoid batches. G3BP1 quantification was performed on frozen IHC sections and batch processed for G3BP1 intensity relative to DAPI using a custom imageJ macro. Each organoid measurement was comprised of four high power images. Intensity measurements were normalized within each isogenic line pair to the WT control. Data was collected for 3 organoids per cell line from two individual organoid batches.
Immunohistochemistry of organoid paraffin sections
Organoids were fixed in 10% neutral buffered formalin and embedded in paraffin blocks at the Neuropathology Brain Bank CoRE (NPBB) at the Icahn School of Medicine at Mount Sinai. Sections (4-5 μm) were prepared from organoid blocks, mounted on positively charged slides and baked overnight at 70°C. IHC was performed on a Benchmark XT autostainer platform (Ventana) according to the manufacturer’s protocol with reagents and antibodies acquired from the same lot. Antigen retrieval was done using CC1 (citric acid buffer, Roche Diagnostics, Basel Switzerland) for 1 hour. All primary antibodies were diluted in Antibody dilution buffer (ABD24, Roche Diagnostics). Primary antibodies were incubated for either 16 minutes (GFAP, Ventana, 760-4345, 1:10), 24 minutes (MAP2, Abcam, ab32454, 1:50, LAMP1, Abcam, ab24170, 1:500), 32 minutes (ALDH1L1, Abcam, ab56777, 1:100), 40 minutes (Iba1, Invitrogen, PA5-27436, 1:100; p62, BD Transductions, 610832,1:50) or 44 minutes (NeuN, Abcam, ab104224, 1:100) at 37°C. Primary antibodies were visualized using either the Ultraview or Optiview detection kit (Roche Diagnostics) as an indirect biotin-free system to detect primary antibodies according to manufacturer’s directions. All slides were counterstained with hematoxylin for 8 minutes and coverslipped. Slides were visualized using a Nikon Eclipse brightfield microscope with a Nikon DS-Fi3 camera and NIS-Elements software. For every set of slides stained, a neuropathologically confirmed severe AD case and healthy control case was included as a batch control.
Immunohistochemistry semi-quantitative and digital quantitative morphometric analysis
Pathological changes assessed using semiquantitative analysis were scored by two researchers (JFC and KW) blinded to mutational and control conditions using a semiquantitative scoring system as follows: no staining (0), low staining (1), moderate staining (2) or high staining (3). For unbiased digital quantitative assessment, slides were imaged using an Aperio CS2 (Leica Biosystems, Wetzlar Germany) digital slide scanner at the Department of Pathology at Mount Sinai. Individual organoids were manually segmented into individual annotations on whole slide images (WSI) sections, batch-processed and analyzed in QuPath (version 0.2.3; https://QuPath.github.io/). All analysis was batch-processed using a custom analysis workflow and cell-based analysis was applied to individual organoids using the following methodology. Neuronal density was measured by the number of NeuN+ cells detected using a positive cell detection classifier based on thresholded values for 3,3′-Diaminobenzidine (DAB) intensity and detected object area. Pixel-level analysis for p62 staining was measured by the percentage of positive pixels detected using a positive pixel classifier based on thresholded values for DAB intensity. All quantitative values were normalized to area.
Bulk RNA sequencing
Organoid preparation and RNA extraction
Individual organoids were collected and stored in RNAlater (QIAGEN) at −80°C from each time point across 3 batches of differentiation. RNA was then extracted in a single batch using the KingFisher Flex (ThermoFisher Scientific) automated system and MagMAX mirVana Total RNA isolation kit (ThermoFisher Scientific). The resulting RNA was submitted to GeneWiz for 150bp unstranded paired-end poly-A RNA sequencing on the Illumina platform.
Data alignment and analysis
Paired-end RNA sequencing reads were trimmed for Illumina TruSeq adapters and aligned to the human hg38 genome using the STAR aligner (Dobin et al., 2013) for quantification of gene-level read counts (FastQC v0.11.8 used for quality control of reads, Cutadapt v2.5 for adaptor trimming, STAR v2.5.2b for read alignment). Genes were filtered by expression abundance (counts per million, CPM, > 0.1 in 5 or more samples), and library sizes were estimated by the trimmed mean of M-values normalization (TMM) method (Liao et al., 2014) with the R edgeR package v3.26.8. Differential gene expression between MAPT mutation organoids and isogenic controls was predicted by linear mixed model analysis using the variancePartition package (v1.14.1) in the R programming environment (Hoffman and Roussos, 2021) to account for sample correlations within isogenic lines and differentiation batches. The false discovery rate (FDR) of the differential expression test was estimated using the Benjamini-Hochberg (BH) method (Reiner et al., 2003). Functional Gene Set Enrichment Analysis (GSEA) was performed using the Molecular Signatures Database (MSigDB) Gene Ontology (GO) annotations of canonical pathway, biological process, cellular compartment and molecular function, with 10,000 permutations to establish FDR values (implemented with the R GOtest package v1.0.8; https://rdrr.io/github/mw201608/GOtest/). Additional pathway analyses were carried out using Ingenuity Pathway Analysis (QIAGEN Inc.; https://digitalinsights.qiagen.com/products/ingenuitypathway-analysis). LeafCutter v0.2.7 (Li et al., 2018) was used to identify differentially spliced intron clusters between tau-V337M and tau-V337V organoids at each time point using default settings. Analysis of GO enrichment terms for differentially spliced genes was carried out using g:Profiler (https://biit.cs.ut.ee/gprofiler/gost).
qRT-PCR
RNA was isolated from organoids using the RNeasy kit (QIAGEN) and reverse transcribed using the High Capacity RNA-to-cDNA kit (ThermoFisher Scientific). qRT-PCR for MAPT 4R and 3R isoforms was carried out using SybrGreen mastermix with the following primers: 4R forward 5′-CGGGAAGGTGCAGATAATTAA-3′, 3R forward 5′-AGGCGGGAAGGTGCAAATA-3′, 4R3R reverse 5′-GCCACCTCCTGGTTTATGATG-3′. The 4R:3R ratio was calculated using the ΔΔCt method to normalize 4R expression to 3R expression within the same sample. Statistical significance was determined by the appropriate one-way ANOVA and Tukey post hoc testing.
Cell hashing single-cell RNA-seq
Organoid dissociation and cell hashing
Organoids were incubated in Papain (Worthington) for 90-120 minutes at 37°C with gentle shaking (300 rpm) and regular pipetting with a glass Pasteur pipette to obtain single cell suspensions. Cells were pelleted and resuspended in ice-cold PBS before being passed through a 40 ~m filter and kept on ice. Viability and cell number was determined by Trypan Blue staining and counting using the Countess system (ThermoFisher Scientific). 50-100,000 cells per organoid were submitted to the New York Genome Center (NYGC) for hashtag-oligonucleotide (HTO) antibody binding, library preparation and sequencing as previously described (Stoeckius et al., 2017, 2018).
Single-cell RNA-seq data QC and alignment
Data QC and alignment was carried out at the NYGC. Briefly, analysis up to the counting of UMIs per cell per gene was carried out using Drop-Seq tools (Drop-seq tools v1.0, McCarroll Lab), and reads were aligned to the human hg38 genome using the STAR aligner (Dobin et al., 2013). HTOs were identified and counted using Cite-Seq Count (https://hoohm.github.io/CITE-seq-Count/), and the samples were de-multiplexed using HTODemux (Butler et al., 2018). Following processing at NYGC, the data was further processed using Seurat 3.0 dev (Butler et al., 2018; Stuart et al., 2019) to filter out cells with < 200 detectable genes, < 500 reads and > 20% mitochondrial rate. We retrieved 8000-10,000 high quality cells per lane with a read depth of ~25,000 reads per cell.
Data analysis
Cells were annotated using the SingleR package (Aran et al., 2019), with custom reference libraries constructed from human fetal neocortical single-cell RNA sequencing data (Polioudakis et al., 2019) and bulk adult astrocyte RNA-sequencing data (Zhang et al., 2016). The top 3,000 most variable genes at each time point were used as input for cell type annotation. Data from different sequencing runs, batches and ages were integrated and scaled using SCTransform (Hafemeister and Satija, 2019), while regressing out the following covariates; percentage of mitochondrial genes, the number of cells collected per parent organoid, the number of genes per cell and the number of reads per cell. Data was also corrected for differentiation batch. Using Seurat, principal components analysis (PCA) was carried out using the top 3,000 most variable genes, and data reduction was performed with UMAP (McInnes et al., 2018). For glutamatergic neuron and astrocyte analyses, individual cell types were isolated from each cell line and integrated across each time point using the same covariates as described above. Data from each cell type was then re-scaled and reduced for each isogenic pair. Clusters were identified using the default resolution factor, and mutant-cell enriched clusters were identified as those with a log2FC > 0.5 greater proportion of V337M cells compared to V337V cells. The same parameters were kept for all analyses. Differential gene expression analysis was carried out on raw count data between mutant-enriched clusters and remaining clusters using the MAST model with appropriate covariates. Significantly differentially expressed genes were submitted to pathway analysis in IPA, and pathways shared between all isogenic pairs and altered in the same direction were identified. Cells in V337M and V337V organoids were separately ordered in pseudotime using Monocle3 (Qiu et al., 2017; Trapnell et al., 2014) using the top 3,000 most variable genes. Differential expression between V337M and V337V glutamatergic neurons was carried out using a custom analysis as described previously (Kanton et al., 2019), using a spline function with 6 degrees of freedom. Genes were clustered based on trajectory using the R package pheatmap (Kolde, 2019) using the ward.D2 method, and genes with differential expression trajectories were submitted for GO enrichment using g:Profiler (https://biit.cs.ut.ee/gprofiler/gost).
Intersection of differentially spliced genes with nELAVL-target genes
We obtained a list of genes previously demonstrated to be bound by ELAVL in human brain using RNA immunoprecipitation RNA-seq (RIP-seq) (Scheckel et al., 2016). The nELAVL-RIP-seq genes were intersected with the genes we previously identified to be differentially spliced. Using the ‘hypeR’ package, we then explored the functions of the bound genes by calculating the GO:BP enriched categories followed by semantic analysis with the ‘rrvgo’ package. Code for this analysis can be found in Data S1.
Pseudobulk expression network analysis
To determine differentially expressed genes over time and between treatments, we utilized an algorithm that uses an iterative process to sample and mix the counts from each cell to simulate bulk sequencing and test for differential gene expression (Data S1). Then using the probabilities of each gene being identified as differentially expressed, we used Bayesian inference of a binomial proportion to identify genes likely to be significant. Genes with a Bayes factor greater than 10,000 were selected for inclusion in the network analysis. A network was generated by using STRINGdb to identify interactions between genes and then communities were identified using multi-level modularity optimization algorithm available in the ‘igraph’ package. After discarding communities with less than 20 genes, we performed a GO:BP enrichment analysis of genes in each community followed by a semantic similarity analysis to summarize the results.
Immunoblotting for autophagy-lysosomal markers
Organoids were extracted in lysis buffer (50 mM Tris pH 7.6, 1 mM EDTA, 150 mM NaCl, 1% Triton X-100, phosphatase and protease inhibitors (Protease inhibitor cocktail, Millipore-Sigma, P2714)) and incubated on ice for 5 minutes. Lysates were then centrifuged at 14,000 xg for 10 minutes at 4°C, and the resulting supernatant was saved for analysis. Total protein levels were assayed by bicinchoninic acid assay (BCA) assay (Thermo-Fisher). Standard sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed in 4%–12% Criterion Tris-HCl gels (Bio-Rad) with 10 μg of total protein loaded in each well. Samples were boiled in Laemmli sample buffer prior to electrophoresis. Gels were transferred to PVDF (EMD Millipore,IPVH00010) and the resulting immunoblots were probed with the primary antibodies diluted in PBS-T (1X PBS+0.1% (v/v) Tween-20) with 5% non-fat milk overnight: LAMP1 (Abcam 1:1000), Cathepsin D (gift from Dr. Stuart Kornfeld 1:2000), LC3 I/II (Novus Biologicals, 1:5000), p62 (Abnova 1:2000), and Ubiquitin (Cell Signaling Technology 1:1000) diluted in PBS-T (1X PBS+0.1% (v/v) Tween-20) with 5% BSA. Antibodies were visualized with SuperSignal West Pico Chemiluminescent Substrate (Thermo) or Lumigen ECL Ultra (TMA-6) according to manufacturer’s instructions. Immunoblots were exposed on a Syngene G:Box iChemi XT Gel Documentation System and imaged using GeneSnap software according to manufacturer’s instructions. Band intensity was analyzed using GeneTools, normalized against total protein by Ponceau S staining of each blot, and band intensity was expressed relative to the normalized control within each blot.
Immunoblotting analysis of tau and P-tau
Organoids were removed from the cell culture media, placed in microcentrifuge tubes and rapidly frozen on dry ice. The organoids were lysed using RIPA buffer (50 mM Tris, 150 mM sodium chloride, 0.5% sodium deoxycholate, 2% SDS, 1% nonidet P-40, Boston Bioproducts) containing 1X concentration of HALT protease and phosphatase inhibitors (Thermo Fisher Scientific). Briefly, the RIPA buffer was added to the tubes containing organoids and incubated on ice for 20 min followed by manual disruption and lysis using a wide bore pipette tip. Next, the lysate was sonicated in a water bath at 4°C for 20 min (20 s on, 20 s off) followed by centrifugation at 14,000 ×g at 4°C. The supernatant was collected, and protein concentration was determined by Bicinchoninic Acid (BCA) assay (Thermo Fisher). A total of 5 μg protein was separated by SDS-PAGE (7% Tris-acetate gel) according to standard methods. Proteins were transferred from the gel onto Polyvinylidene fluoride (PVDF) membranes (EMD Millipore) using standard procedures. Membranes were blocked in 5% (w/v) bovine serum albumin (BSA) (Sigma) in Tris-buffered saline with 0.1% Tween-20 (TBST, Boston Bio-Products), incubated overnight with primary antibody at 4°C: Total Tau HT7 (Thermo Fisher, MN1000, 1:1000), Total Tau Tau5 (Thermo Fisher, AHB0042, 1:1000) Phospho-Tau Ser396 (Thermo Fisher, 44752G, 1:1000), 4R Tau RD4 (EMD Millipore, 05804, 1:1000), G3BP1 (BD Biosciences, 611126, 1:2000), GAPDH (Cell Signaling Technology, 2118s, 1:5000). The following day, the blots were incubated with the corresponding horseradish peroxidase (HRP)-linked secondary antibody: goat anti-rabbit HRP linked (Cell Signaling Technology, 7074s, 1:4000) or goat anti-mouse HRP linked (Cell Signaling Technology, 7076s, 1:4000). Blots were developed with SuperSignal Chemiluminescent Substrate (ThermoFisher) according to manufacturer’s instructions, exposed to autoradiographic film (LabScientific by ThermoFisher), and scanned on an Epson Perfection V800 Photo Scanner. Protein bands’ densitometry (pixel mean intensity in arbitrary units, a.u.) was measured with ImageJ and normalized to the respective internal control (GAPDH) band.
Longitudinal tracking of neuron survival in 3D
Cerebral organoids were transduced with lentivirus (with 1:1000 polybrene) encoding Synapsin1::eGFP for 5 days and fixed in Matrigel (Corning) prior to experimental use. Glutamate (L-Glutamic acid monosodium salt hydrate; 5 mM Millipore) was added to the medium on day zero during the survival time course, and replenished after one, two, four, and six days. Continuous z stacks spanning 150 μM from the organoid surface were captured daily using a Zeiss AxioZoom.v16 wide-field upright fluorescent microscope. Post-processing and image alignment were performed using ImageJ software (NIH). Live cell staining was performed by incubation with 1:500 DRAQ5 (Biolegend, 424101) for 15 minutes at room temperature, per manufacturer instructions. Detection of live neurons was assessed by co-localization of SYN::eGFP and DRAQ5.
Antisense Oligonucleotide Treatment
Organoids were treated with 10 μM ASO for 48 hours prior to longitudinal tracking. ASO sequences were custom synthesized (IDT) and the following sequences were used: Negative control- 5′- mG*mC*mG*mA*mC*T*A*T*A*C*G*C*G*C*A*mA*mU*mA*mU*mG–3′. PIKFYVE ASO 5′- mC*mC*mU*mU*mG*G*C*A*G*T*T*T*T*A*C*mA*mC*mA*mG*mG –3′. PIKFYVE expression was determined by qRT-PCR using BioRad iTaq Universal SYBR Green Supermix and the following primers: human PIKFYVE forward: 5′-AGGAGCTGCCTTCTATGCAA-3′, human PIKFYVE reverse: 5′-CCGTGGGCCTCTTTTGTTGA-3′, human 18S forward: 5′-CTCAACACGGGAAACCTCAC-3′, human 18S reverse: 5′-CGCTCCACCAACTAAGAACG-3′. PIKFYVE expression was normalized to 18S using the ddCT method.
Immunofluorescence and quantification of glutamate-treated organoids
Organoids were fixed in 4% paraformaldehyde (EMS) for 1 hour at 4°C, cryopreserved in a 30% sucrose (Sigma) solution overnight and sectioned at 12 μm. Sections were permeabilized in 1X Tris buffered saline (TBS, Sigma) + 0.1% Triton X-100 (Sigma), blocked in 1X TBS + 5% donkey serum (Jackson), and incubated with primary antibodies in blocking solution overnight: SATB2 (Abcam, ab51502; 1:500), NeuN (Abcam, ab177487l 1:200), MAP2 (Aves, MAP 1:2000). Sections were subsequently washed and mounted with Vectashield (Vector Labs). Whole-organoid tile scans were taken using a Zeiss LSM800 confocal microscope at 20X magnification. Optimized automated nuclei counting was performed using ImageJ (NIH).
RIP for MAPT transcript with anti-ELAVL4
RIP was performed using the Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Sigma Aldrich, 17-700) according to manufacturers’ instructions. 30 organoids at 1.5 months obtained from Neuracell core facility were pooled and used as starting material for each RIP. Briefly, protein A/G beads were blocked with 2% BSA (ThermoFisher, AM2616) at room temperature for 1 hour before overnight incubation with 10 μg mouse monoclonal anti-ELAVL4 primary antibody (Santa Cruz, sc-28299) or a negative control mouse IgG antibody (MAGNA-RIP kit, Sigma Aldrich). 10% of the starting material (organoid lysate) was used as input control not subjected to RIP. RNA was purified using TRIzol (ThermoFisher) and RNA Clean and Concentrator (Zymo, R1014) kits, with DNase treatment. cDNA synthesis was performed with the Superscript VILO kit (ThermoFisher, 11754050). PCR and qRT-PCR were performed using GoTaq Mastermix (Promega, M7123) and POWER SYBR Green Master Mix (ThermoFisher, 4367659) respectively. PCR products were run on a 2% agarose gel. The following primers were used:
MAPT 3′UTR forward: 5′-ccttgatgtcttgagagcgc-3′, reverse: 5′-cctaaagtcccaggtctgca-3′
MAPT exon 13 forward: 5′-AGAGTCCAGTCGAAGATTGGGTC-3′, reverse: 5′- GGGTTTCAATCTTTTTATTTCCTCC-3′
MAPT exon 11-12 forward: 5′-tagcaacgtccagtccaagt-3′, reverse: 5′- tgttgcctaatgagccacac-3′
Calm3 forward: 5′-cgcctcttccatccatgtct-3′, reverse: 5′- catccagtcaacgccacaat-3′
Electron microscopy
Organoids were fixed overnight in 2% glutaraldehyde and buffered with 0.1 M sodium cacodylate (pH 6.9). After 3 × 10 min buffer washes with sodium cacodylate, organoids were post-stained with 2% osmium tetroxide followed by 2 × 10 min washes with ddH20 and en bloc staining with saturated uranyl acetate for 1 hr in the dark. Organoids were subsequently washed 2 × 10 min in ddH20 and dehydrated in an increasing ethanol series. Finally, organoids were infiltrated and embedded in Epon 812 resin and polymerized at 70°C for 48 hr. One-micron toluidine-stained sections were prepared for light microscopic orientation and 80 nm ultrathin sections stained with lanthanum and lead citrate were examined with a Hitachi 7650 transmission electron microscope at 80kV.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantifiable phenotypes by immunohistochemistry were compared between mutants and controls at 2, 4 and 6 months using a Mann-Whitney U test (Figure 2C; NeuN, 4 months: nmutant = 24, nisogenic = 25, Mann-Whitney U = 175, p = 0.0119, 6 months: nmutant = 26, nisogenic = 34, Mann-Whitney U = 274, p = 0.0117; Figure S2B, MAP2, 6 months, nmutant = 29, nisogenic = 33, Mann-Whitney U = 303.5, p = 0.0060). All statistical analysis and data visualization were performed using GraphPad Prism v8.4.3 (La Jolla, CA). Western blot analysis of autophagy and lysosomal pathway markers (Figures 2G–2I) was performed for three technical replicates and 3 biological replicates, and statistical differences were determined using an unpaired Student’s t test, *p < 0.05, ***p < 0.001 (non-significant for p > 0.05); Statistical analysis and data visualization were performed using GraphPad Prism v9.0.0 (La Jolla, CA). Statistical analysis for tau protein levels and phosphorylation (Figures 2J–2L) were performed using two-tailed unpaired Student’s t test, *p < 0.05, **p < 0.01, ***p < 0.001, with n = 9 per group from three independent experiments; Statistical significance was determined at p < 0.05; All tests were performed using GraphPad Prism v8 (La Jolla, CA). Statistical analysis of CTIP2-positive neurons via immunofluorescence was performed using one-way ANOVA with Tukey test post hoc, *p < 0.05, ****p < 0.0001; Neurons were analyzed across 3 unique organoid batches, and number of organoids analyzed per sample are reported in Figure 3G (n = number of organoids/biological replicates). Quantification of G3BP1 by IHC image analysis (Figure 6D) of mutant organoids was normalized to the respective isogenic control and statistical analysis was performed across all pairs with a two-tailed unpaired Student’s t test. Protein levels of G3BP1 (Figure 6F) in V337M organoids relative to the isogenic controls were determined for n = 6 per group from two independent experiments and statistical significance was calculated by unpaired Student’s t test **p ≤ 0.01 (GraphPad Prism v8, La Jolla, CA). Statistical analysis of neuronal survival in mutant and corrected isogenic control organoids using Log-rank (Mantel-Cox) tests is as follows for Figures 7B: nisogenic = 80, nmutant = 80, Chi-sq = 3.723, p = 0.0537; Figure 7D: nisogenic = 100, nmutant = 100, Chi-sq = 28.95, p < 0.0001; Figure S7D: nisogenic+glutamate = 120, nisogenic+3i = 120, Chi-sq = 7.869, p = 0.005; Figure 7G: nmutant+glutamate = 120, nmutant+3i = 120, Chi-sq = 27.88, p < 0.0001, nmutant+apilimod = 120, Chi-sq = 18.09, p < 0.0001; Figure 7J: nmutant+nc = 120, nmutant+ASO = 120, Chi-sq = 23.23, p < 0.0001; Figure 7K: nisogenic+nc = 120, nisogenic+ASO = 120, Chi-sq = 0.3814, p = 0.5369. Survival % at day 7 across independent experiments in Figure 7E was performed using two-tailed unpaired Student’s t test: nV337V = 4, nV337M = 4, p = 0.0436). Analysis of relative mRNA expression through qRT-PCR in organoids transduced with ASO in Figure 7I (nnc = 6, nASO = 6, p = 0.0095) were performed using two-tailed unpaired Student’s t test; Significance was defined at p < 0.05; Statistical analysis and data visualization was performed using GraphPad Prism v9.0.0 (La Jolla, CA).
The analysis pipelines for bulk and single-cell RNA sequencing data are described in the Method details. 241 individual organoids were collected and submitted for bulk RNA-sequencing across all cell lines, three time points and three differentiation batches. Read quality was assessed using MultiQC (https://multiqc.info/). Two organoids were excluded from further analysis due to inconsistent positioning away from the remaining samples by principal components analysis, resulting in a final dataset consisting of 239 organoids (V337M n = 99, V337V n = 140). All presented differential expression, GSEA, pathway enrichment and splicing analyses p values were adjusted for multiple test correction. For single cell RNA-sequencing, 348 individual organoids were processed (V337M n = 151, V337V n = 197), of which 339 passed quality control, determined by samples containing detectable cells with clear HTO identities. Additional filtering is described in Method details. The resulting dataset for analysis included 379,064 cells (V337M n = 161,706, V337V n = 217,358). All reported p values were adjusted for multiple test correction; Significance was defined as adjusted p < 0.05.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| ALDH1L1 (mouse monoclonal IgG3) | Abcam | ab56777; RRID:AB_940204 |
| AT8 (mouse monoclonal IgG1) | Invitrogen | MN1020: RRID:AB_223647 |
| β-tubulin III (mouse monoclonal IgG2b) | Sigma | T-8660; RRID:AB_477590 |
| BRN2 (mouse monoclonal IgG1) | Millipore | MAB-D51; RRID:AB_11204531 |
| Calbindin 1(rabbit) | Swant | cb-38a |
| Cathepsin D (rabbit polyclonal) | Gift from Dr. Stuart Kornfeld lab | RRID:AB_2314096 |
| CTIP2 (rat monoclonal IgG2a) | Abcam | ab18465; RRID:AB_2064130 |
| ELAVL4 (HuD-E1) (mouse IgG2a) | SantaCruz | sc-28299; RRID:AB_627765 |
| FOXG1 (rabbit polyclonal) | Takara | M227; RRID:ZFIN_ZDB-ALT-980203-55 |
| G3BP (mouse monoclonal IgG1) | BD Biosciences | 611126; RRID:AB_398437 |
| G3BP1 (rabbit polyclonal) | Protein Tech | 13057-2-AP; RRID:AB_2232034 |
| GAPDH (rabbit monoclonal) | Cell Signaling Technology | 2118S; RRID:AB_561053 |
| GFAP (mouse monoclonal IgG1) | Millipore | MAB3402; RRID:AB_94844 |
| GFAP, clone EP672Y (rabbit) | Ventana | 760-4345 |
| Homer1 (mouse monoclonal IgG1) | SYSY | 160011; RRID:AB_2795016 |
| Iba1 (rabbit polyclonal) | Invitrogen | PA5-27436; RRID:AB_2544912 |
| Ki67 (rat monoclonal IgG2a) | Thermo Fisher | 14-5698-82; RRID:AB_10854564 |
| LAMP1 (rabbit polyclonal) | Abcam | 24170; RRID:IMSR_CMMR:PST24170 |
| LC3I/II (rabbit polyclonal) | Novus Biologicals | NB100-2331; RRID:IMSR_CMMR:PST24170 |
| MAP2 (chicken) | Aves labs | MAP; RRID:AB_2313549 |
| MAP2 (rabbit polyclonal) | Abcam | ab32454; RRID:AB_776174 |
| MAP2ab (chicken polyclonal) | Abcam | ab5392; RRID:AB_2138153 |
| MEF2C (rabbit polyclonal) | Novus | NBP2-17260 |
| Nestin (mouse monoclonal IgG1) | Millipore | MAB5326; RRID:AB_11211837 |
| NeuN (mouse monoclonal IgG1) | Millipore | MAB377; RRID:AB_2298767 |
| NeuN (mouse monoclonal IgG2b) | Abcam | 104224; RRID:Addgene_104224 |
| NeuN (rabbit monoclonal) | Abcam | Ab177487; RRID:AB_2532109 |
| p62 (mouse monoclonal) | Abnova | H00008878-M01; RRID:AB_437085 |
| p62 Ick ligand (mouse monoclonal) | BD Transductions | 610832; RRID:AB_398151 |
| PAX6 (rabbit polyclonal) | BioLegend | 901301; RRID:AB_2565003 |
| PHF-1 (mouse IgG1) | Gift from Dr. Peter Davies | N/A |
| Phospho-Tau Ser396 (rabbit polyclonal) | Thermo Fisher | 44752G; RRID:AB_2533745 |
| S100beta (rabbit polyclonal) | DAKO Potts | Z0311; RRID:AB_10013383 |
| SATB2 (mouse monoclonal IgG1) | Abcam | ab51502; RRID:AB_882455 |
| SOX10 (A-2) (mouse monoclonal IgG1) | Santa Cruz | sc-365692; RRID:AB_10844002 |
| SOX2 (E4) (mouse monoclonal IgG1) | Santa Cruz | sc-365823; RRID:AB_10842165 |
| Synapsin1 (rabbit polyclonal) | Millipore | AB1543; RRID:AB_2200400 |
| TAU (4 Repeat Isoform RD4) (mouse monoclonal IgG) | EMD Millipore | 05-804; RRID:AB_310014 |
| TBR1 (rabbit polyclonal) | Abcam | ab31940; RRID:AB_2200219 |
| TIA1 (mouse monoclonal IgG1) | SantaCruz | sc-166247; RRID:AB_2201545 |
| Total TAU (rabbit polyclonal) | DAKO Potts | A-0024; RRID:AB_10013724 |
| Total TAU DA9 (mouse IgG1) | Gift from Peter Davies | N/A |
| Total TAU, HT7 (mouse monoclonal IgG1) | Thermo Fisher | MN1000; RRID:AB_2314654 |
| Total TAU, TAU5 (mouse monoclonal IgG1) | Thermo Fisher | AHB0042; RRID:AB_1502093 |
| Ubiquitin (rabbit polyclonal) | Cell Signaling Technology | 3933S; RRID:AB_2180538 |
| VGLUT1 (guinea pig) | Gift from Susan Morton, Tom Jessell | N/A |
| Goat anti-chicken IgG-Alexa 647 | Invitrogen | A-21449; RRID:AB_2535866 |
| Goat anti-guinea pig-Alexa 647 | Jackson | 106-605-003; RRID:AB_2337446 |
| Goat anti-guinea pig-Cy3 | Jackson | 106-165-003; RRID:AB_2337423 |
| Goat anti-mouse HRP linked | Cell Signaling Technology | 7076s; RRID:AB_330924 |
| Goat anti-mouse IgG1-Alexa 488 | Invitrogen | A-21121; RRID:AB_2535764 |
| Goat anti-mouse IgG1-Alexa 546 | Invitrogen | A-21123; RRID:AB_141592 |
| Goat anti-mouse IgG1-Alexa 647 | Invitrogen | A-21240; RRID:AB_141658 |
| Goat anti-mouse IgG2a-Alexa 488 | Invitrogen | A-21131; RRID:AB_141618 |
| Goat anti-mouse IgG2b-Alexa 546 | Invitrogen | A21143; RRID:AB_1500891 |
| Goat anti-rabbit HRP linked | Cell Signaling Technology | 7074s; RRID:AB_2099233 |
| Goat anti-rabbit-Alexa 488 | Jackson | 111-545-144; RRID:AB_2338052 |
| Goat anti-rabbit-Alexa 647 | Jackson | 111-605-144; RRID:AB_2338078 |
| Goat anti-rabbit-Cy3 | Jackson | 111-165-045; RRID:AB_2338003 |
| OptiView DAB IHC Detection Kit | Roche | 760-700; RRID:AB_2833075 |
| ultraView Universal DAB Detection Kit | Roche | 760-500; RRID:AB_2753116 |
| Chemicals, peptides, and recombinant proteins | ||
| DMEM/F12 | Corning | 10-090-CV |
| Essential 6 medium (E6) | Life Technologies | A1516401 |
| Essential 8 medium (E8) | Life Technologies | A1517001 |
| mTeSR1 medium | StemCell Technologies | 85850 |
| Neurobasal-A ([−] L-glutamine) | Life Technologies | 10888-022 |
| Accutase | StemCell Technologies | 7922 |
| Antibiotic-Antimycotic (Anti-A) 100X | Life Technologies | 15240-062 |
| B-27 supplement without vitamin A | Life Technologies | 12587010 |
| Dispase | StemCell Technologies | 07923 |
| GlutaMAX Supplement | Life Technologies | 3505-061 |
| KnockOut Serum Replacement | Thermo | 10828028 |
| Matrigel | Corning | 356231 |
| Anti-Adherence Rinsing Solution | StemCell Technologies | 07010 |
| MEM Non-essential amino acids 100X | Thermo | 11140050 |
| Penicillin-streptomycin 50X | Corning | 30-001-CI |
| (+)-MK-801 (hydrogen maleate) | Cayman | 10009019 |
| Apilimod | AChemBlock | O33822 |
| CNQX | Cayman | 14618 |
| Dorsomorphin | Tocris | 3093 |
| Dorsomorphin | Cayman | 11967 |
| L-Glutamic acid monosodium salt hydrate | Millipore | G1626 |
| Nimodipine | Cayman | 14573 |
| SB-431542 | Tocris | 1614 |
| SB-431542 | Cayman | 13031 |
| XAV-939 | Tocris | 3748 |
| Y-27632 | Tocris | 1254 |
| β-mercaptoethanol | Sigma | M6250 |
| human BDNF | Peprotech | 450-02 |
| human BDNF | R&D | 248-BDB |
| human EGF | Peprotech | AF-100-15 |
| human NT3 | Peprotech | 450-03 |
| rhFGF2 | R & D systems | 233-FB |
| rhFGF2 | Peprotech | 100-18B |
| Protease Inhibitor cocktail | Sigma | P2714 |
| Deposited data | ||
| Bulk RNA-seq data from 2-, 4- and 6-month organoids | GEO: GSE171345 | |
| Single-cell RNA-seq data from 2-, 4- and 6-month organoids | GEO: GSE171345 | |
| Experimental models: Cell lines | ||
| ND32951A.15Δ1B06 (V337V) | Tau Consortium | ND-B06 |
| ND32951A.15Δ2B09 (V337M) | Tau Consortium | ND-B09 |
| GIH-6-C1-ΔA02 (V337M) | Tau Consortium | GIH6-A02 |
| GIH-6-C1Δ1E11 (V337V) | Tau Consortium | GIH6-E11 |
| GIH-7-C2-Δ2A01 (V337M) | Tau Consortium | GIH7-A01 |
| GIH-7-C2-Δ2B12 (V337V) | Tau Consortium | GIH7-B12 |
| GIH-7-C2-Δ2F02 (V337V) | Tau Consortium | GIH7-F02 |
| ND03231 (Population Control) | NINDS Biorepository | 3231 |
| Software and algorithms | ||
| QuPath | Bankhead et al. (2017) | https://qupath.github.io/ |
| Seurat 3.0 (dev) | Stuart et al. (2019); Butler et al. (2018) | https://satijalab.org/seurat/ |
| Monocle3 | Trapnell et al. (2014); Qiu et al. (2017) | https://cole-trapnell-lab.github.io/monocle3/ |
| SingleR | Aran et al. (2019) | https://bioconductor.org/packages/devel/bioc/vignettes/SingleR/inst/doc/SingleR.html |
| Script for differential gene expression over pseudotime trajectories | Kanton et al. (2019) | N/A |
| ImageJ | https://imagej.nih.gov/ij/index.html | |
| Other | ||
| GW17-18 cortical scRNA-seq data | Polioudakis et al. (2019) | http://solo.bmap.ucla.edu/shiny/webapp/ |
| Adult astrocyte bulk RNaseq data | Zhang et al. (2016) | https://www.brainrnaseq.org/ |
| ELAVL RIP-seq data | Scheckel et al. (2016) | N/A |
Highlights.
Tau and P-tau accumulation and autophagy disruption in tau-V337M organoids
Accelerated synaptic maturation and loss of glutamatergic cortical-layer neurons
Altered ELAVL4 expression, dysregulated splicing, accelerated synaptic maturation
Rescue of susceptibility to glutamatergic toxicity by PIKFYVE inhibitor apilimod
ACKNOWLEDGMENTS
We are grateful to the Tau Consortium and Rainwater Charitable Foundation for supporting this project, CurePSP for protocol development support, and the New York Genome Center for scRNA-seq expertise. We thank the research subjects and their families for their generous participation, including in ARTFL (U54NS092089) and LEFFTDS (U01AG045390), and support staff at participating sites. We thank Shawn Sutton, Brian Unruh, Isabel Tian, Khadijah Onanuga, and Nicholas St. John at the NeuraCell core facility (https://www.neuracell.org/) for organoid production; the ISMMS Neuropathology Brain Bank & Research Core for technical assistance and Aaron Bell for sample preparation; and the Hope Center for Neurological Disorders and the Departments of Neurology and Psychiatry at Washington University School of Medicine for equipment access. Elements of Figures 1A and 2B and the graphical abstract were created with BioRender. This work was supported by the Tau Consortium; the Rainwater Charitable Foundation (to C.M.K., J.F.C., S.J.H., J.K.I., A.M.G., and S.T.); NIH AG046374 (to C.M.K.); CurePSP (to K.R.B.); the MGH Research Scholars Program (to S.J.H.); the Association for Frontotemporal Degeneration (AFTD; to M.C.S. and K.R.B.); the BrightFocus Foundation (to K.R.B.); the Farrell Family Alzheimer’s Disease Research Fund; NIH NS110890 (to C.M.K.); NIH R01AG054008 and R01NS095252 (to J.F.C.); NIH/NINDS R35 NS097277 (to S.T.); NIH/NIA R01 AG056293 (to S.T. and C.M.K.); NIH 2R01NS097850; Department of Defense W81XWH-21-1-0168, W81XWH-20-1-0424, and W81XWH-21-1-0131;the Harrington Discovery Institute; CIRM, the ALS Association; the John Douglas French Alzheimer’s Foundation; ADDF, the New York Stem Cell Foundation (to J.K.I.); an Amgen postdoctoral fellowship (to J.D.L.); and NIH NINDS/NIA F31NS117075 (to J.E.B.). Neuracell received support from the Empire State Stem Cell Fund (NYSTEM) through New York State Department of Health contract C029158. Opinions expressed here are solely those of the authors and do not necessarily reflect those of the Empire State Stem Cell Board, the NewYork State Department of Health, or the State of New York.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2021.07.003.
DECLARATION OF INTERESTS
J.D.L. employee, Amgen. A.M.G.: Scientific Advisory Board (SAB), Denali Therapeutics (2015-2018); Genetic SAB, Pfizer (2019); SAB, Genentech; consultant, GSK, AbbVie, Biogen, and Eisai. S.J.H.: SAB, Rodin Therapeutics, Frequency Therapeutics, Psy Therapeutics, Vesigen Therapeutics, and Souvien Therapeutics; inventor, patent 6,475,723. S.T.: president, StemCultures; cofounder, LUXA Biotech; SAB, Sana Biotechnology, Blue Rock Therapeutics, and Vita Therapeutics; inventor, patent 16/331,063. J.K.I.: cofounder, AcuraStem and Modulo Bio; SAB, Spinogenix. Named companies were not involved in this project.
REFERENCES
- Akamatsu W, Fujihara H, Mitsuhashi T, Yano M, Shibata S, Hayakawa Y, Okano HJ, Sakakibara S, Takano H,Takano T, et al. (2005). The RNA-binding protein HuD regulates neuronal cell identity and maturation. Proc. Natl. Acad. Sci. USA 102, 4625–4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P, and Kedersha N (2008). Stress granules: the Tao of RNA triage. Trends Biochem. Sci 33, 141–150. [DOI] [PubMed] [Google Scholar]
- Apicco DJ, Zhang C, Maziuk B, Jiang L, Ballance HI, Boudeau S, Ung C, Li H, and Wolozin B (2019). Dysregulation of RNA Splicing in Tauopathies. Cell Rep. 29, 4377–4388.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, et al. (2019). Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol 20, 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt T (2002). Dysregulation of neuronal differentiation and cell cycle control in Alzheimer’s disease. J. Neural Transm Suppl., 77–85. [DOI] [PubMed] [Google Scholar]
- Ash PEA, Lei S, Shattuck J, Boudeau S, Carlomagno Y, Medalla M, Mashimo BL, Socorro G, Al-Mohanna LFA, Jiang L, et al. (2021). TIA1 potentiates tau phase separation and promotes generation of toxic oligomeric tau. Proc. Natl. Acad. Sci. USA 118, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atlas R, Behar L, Sapoznik S, and Ginzburg I (2007). Dynamic association with polysomes during P19 neuronal differentiation and an untranslated-region-dependent translation regulation of the tau mRNA by the tau mRNA-associated proteins IMP1, HuD, and G3BP1. J. Neurosci. Res 85, 173–183. [DOI] [PubMed] [Google Scholar]
- Bain HDC, Davidson YS, Robinson AC, Ryan S, Rollinson S, Richardson A, Jones M, Snowden JS, Pickering-Brown S, and Mann DMA (2019). The role of lysosomes and autophagosomes in frontotemporal lobar degeneration. Neuropathol. Appl. Neurobiol 45, 244–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG, et al. (2017). QuPath: Open source software for digital pathology image analysis. Sci. Rep 7, 16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beagle AJ, Darwish SM, Ranasinghe KG, La AL, Karageorgiou E, and Vossel KA (2017). Relative Incidence of Seizures and Myoclonus in Alzheimer’s Disease, Dementia with Lewy Bodies, and Frontotemporal Dementia. J. Alzheimers Dis 60, 211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar L, Marx R, Sadot E, Barg J, and Ginzburg I (1995). cis-acting signals and trans-acting proteins are involved in tau mRNA targeting into neurites of differentiating neuronal cells. Int. J. Dev. Neurosci 13, 113–127. [DOI] [PubMed] [Google Scholar]
- Bendiske J, and Bahr BA (2003). Lysosomal activation is a compensatory response against protein accumulation and associated synaptopathogenesis–an approach for slowing Alzheimer disease? J. Neuropathol. Exp. Neurol 62, 451–463. [DOI] [PubMed] [Google Scholar]
- Benussi A, Padovani A, and Borroni B (2015). Phenotypic heterogeneity of monogenic frontotemporal dementia. Front. Aging Neurosci 7, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benussi A, Alberici A, Buratti E, Ghidoni R, Gardoni F, Di Luca M, Padovani A, and Borroni B (2019). Toward a glutamate hypothesis of frontotemporal dementia. Front. Neurosci 13, 304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaduri A, Andrews MG, Mancia Leon W, Jung D, Shin D, Allen D, Jung D, Schmunk G, Haeussler M, Salma J, et al. (2020). Cell stress in cortical organoids impairs molecular subtype specification. Nature 578, 142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird T, Knopman D, VanSwieten J, Rosso S, Feldman H, Tanabe H, Graff-Raford N, Geschwind D, Verpillat P, and Hutton M (2003). Epidemiology and genetics of frontotemporal dementia/Pick’s disease. Ann. Neurol 54 (Suppl 5), S29–S31. [DOI] [PubMed] [Google Scholar]
- Bodea LG, Eckert A, Ittner LM, Piguet O, and Götz J (2016). Tau physiology and pathomechanisms in frontotemporal lobar degeneration. J. Neurochem 138 (Suppl 1), 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolognani F, Contente-Cuomo T, and Perrone-Bizzozero NI (2010). Novel recognition motifs and biological functions of the RNA-binding protein HuD revealed by genome-wide identification of its targets. Nucleic Acids Res. 38, 117–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borroni B, Stanic J, Verpelli C, Mellone M, Bonomi E, Alberici A, Bernasconi P, Culotta L, Zianni E, Archetti S, et al. (2017). Anti-AMPA GluA3 antibodies in Frontotemporal dementia: a new molecular target. Sci. Rep 7, 6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borroni B, Benussi A, Premi E, Alberici A, Marcello E, Gardoni F, Di Luca M, and Padovani A (2018). Biological, Neuroimaging, and Neurophysiological Markers in Frontotemporal Dementia: Three Faces of the Same Coin. J. Alzheimers Dis 62,1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie D (2008). Ionotropic glutamate receptors & CNS disorders. CNS Neurol. Disord. Drug Targets 7, 129–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright F, Werry EL, Dobson-Stone C, Piguet O, Ittner LM, Halliday GM, Hodges JR, Kiernan MC, Loy CT, Kassiou M, and Kril JJ (2019). Neuroinflammation in frontotemporal dementia. Nat. Rev. Neurol 15, 540–555. [DOI] [PubMed] [Google Scholar]
- Bronicki LM, and Jasmin BJ (2013). Emerging complexity of the HuD/ELAVl4 gene; implications for neuronal development, function, and dysfunction. RNA 19, 1019–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Xu Y, Cheung AK, Tomlinson RC, Alcázar-Román A, Murphy L, Billich A, Zhang B, Feng Y, Klumpp M, et al. (2016). PIKfyve, a class III PI-kinase, is the target of the small molecular IL12/23 inhibitor apilimod and a new player in toll-like receptor signaling. Chem. Biol 20, 912–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camp JG, Badsha F, Florio M, Kanton S, Gerber T, Wilsch-BrÄuninger M, Lewitus E, Sykes A, Hevers W, Lancaster M, et al. (2015). Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc. Natl. Acad. Sci. USA 112, 15672–15677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clare R, King VG, Wirenfeldt M, and Vinters HV (2010). Synapse loss in dementias. J. Neurosci. Res 88, 2083–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santis R, Alfano V, de Turris V, Colantoni A, Santini L, Garone MG, Antonacci G, Peruzzi G, Sudria-Lopez E, Wyler E, et al. (2019). Mutant FUS and ELAVL4 (HuD) Aberrant Crosstalk in Amyotrophic Lateral Sclerosis. Cell Rep. 27, 3818–3831.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBoer EM, Azevedo R, Vega TA, Brodkin J, Akamatsu W, Okano H, Wagner GC, and Rasin MR (2014). Prenatal deletion of the RNA-binding protein HuD disrupts postnatal cortical circuit maturation and behavior. J. Neurosci 34, 3674–3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich M, Hallmann AL, Reinhardt P,Araúzo-Bravo MJ, Korr S, Röpke A, Psathaki OE, Ehling P, Meuth SG, Oblak AL, et al. (2015). Distinct Neurodegenerative Changes in an Induced Pluripotent Stem Cell Model of Frontotemporal Dementia Linked to Mutant TAU Protein. Stem Cell Reports 5, 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallini C, Bassell GJ, and Rossoll W (2012). The ALS disease protein TDP-43 is actively transported in motor neuron axons and regulates axon outgrowth. Hum. Mol. Genet 21, 3703–3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faravelli I, Costamagna G, Tamanini S, and Corti S (2020). Back to the origins: Human brain organoids to investigate neurodegeneration. Brain Res. 1727, 146561. [DOI] [PubMed] [Google Scholar]
- Fong H, Wang C, Knoferle J, Walker D, Balestra ME, Tong LM, Leung L, Ring KL, Seeley WW, Karydas A, et al. (2013). Genetic correction of tauopathy phenotypes in neurons derived from human induced pluripotent stem cells. Stem Cell Reports 1, 226–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghetti B, Oblak AL, Boeve BF, Johnson KA, Dickerson BC, and Goedert M (2015). Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol. Appl. Neurobiol 41, 24–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez C, Armijo E, Bravo-Alegria J, Becerra-Calixto A, Mays CE, and Soto C (2018). Modeling amyloid beta and tau pathology in human cerebral organoids. Mol. Psychiatry 23, 2363–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafemeister C, and Satija R (2019). Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman GE, and Roussos P (2021). Dream: powerful differential expression analysis for repeated measures designs. Bioinformatics 37, 192–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomov OC, Sbrissa D, Delvecchio K, Xie Y, Jin JP, Rappolee D, and Shisheva A (2011). The phosphoinositide kinase PIKfyve is vital in early embryonic development: preimplantation lethality of PIKfyve−/− embryos but normality of PIKfyve+/− mice. J. Biol. Chem 286, 13404–13413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ince-Dunn G, Okano HJ, Jensen KB, Park WY, Zhong R, Ule J, Mele A, Fak JJ, Yang C, Zhang C, et al. (2012). Neuronal Elav-like (Hu) proteins regulate RNA splicing and abundance to control glutamate levels and neuronal excitability. Neuron 75, 1067–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iovino M, Agathou S, González-Rueda A, Del Castillo Velasco-Herrera M, Borroni B, Alberici A, Lynch T, O’Dowd S, Geti I, Gaffney D, et al. (2015). Early maturation and distinct tau pathology in induced pluripotent stem cell-derived neurons from patients with MAPT mutations. Brain 138, 3345–3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S, Wen N, Li Z, Dube U, Del Aguila J, Budde J, Martinez R, Hsu S, Fernandez MV, Cairns NJ, et al. ; Dominantly Inherited Alzheimer Network (DIAN); International FTD-Genomics Consortium (IFGC) (2018). Integrative system biology analyses of CRISPR-edited iPSC-derived neurons and human brains reveal deficiencies of presynaptic signaling in FTLD and PSP. Transl. Psychiatry 8, 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung M, and Lee EK (2021). RNA-Binding Protein HuD as a Versatile Factor in Neuronal and Non-Neuronal Systems. Biology (Basel) 10, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanton S, Boyle MJ, He Z, Santel M, Weigert A, Sanchís-calleja F, Guijarro P, Sidow L, Fleck JS, Han D, et al. (2019). Organoid Single-Cell Genomic Atlas Uncovers Human-Specific Features of Brain Development. Nature 574, 418–422. [DOI] [PubMed] [Google Scholar]
- Karch CM, Kao AW, Karydas A, Onanuga K, Martinez R,Argouarch A, Wang C, Huang C, Sohn PD, Bowles KR, et al. ; Tau Consortium Stem Cell Group (2019). A Comprehensive Resource for Induced Pluripotent Stem Cells from Patients with Primary Tauopathies. Stem Cell Reports 13,939–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike M, Tsukada S, Tsuzuki K, Kijima H, and Ozawa S (2000). Regulation of kinetic properties of GluR2 AMPA receptor channels by alternative splicing. J. Neurosci 20, 2166–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolde R (2019). pheatmap: Pretty Heatmaps. R package version 1.0.12. https://rdrr.io/cran/pheatmap/. [Google Scholar]
- Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, and Knoblich JA (2013). Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster MA, Corsini NS, Wolfinger S, Gustafson EH, Phillips AW, Burkard TR, Otani T, Livesey FJ, and Knoblich JA (2017). Guided self-organization and cortical plate formation in human brain organoids. Nat. Biotechnol 35, 659–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester E, Ooi FK, Bakkar N, Ayers J, Woerman AL, Wheeler J, Bowser R, Carlson GA, Prusiner SB, and Parker R (2021). Tau aggregates are RNA-protein assemblies that mislocalize multiple nuclear speckle components. Neuron 109, 1675–1691.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuzy A, Zimmer ER, Dubois J, Pruessner J, Cooperman C, Soucy JP, Kostikov A, Schirmaccher E, Désautels R, Gauthier S, and Rosa-Neto P (2016). In vivo characterization of metabotropic glutamate receptor type 5 abnormalities in behavioral variant FTD. Brain Struct. Funct 221, 1387–1402. [DOI] [PubMed] [Google Scholar]
- Li YI, Knowles DA, Humphrey J, Barbeira AN, Dickinson SP, Im HK, and Pritchard JK (2018). Annotation-free quantification of RNA splicing using LeafCutter. Nat. Genet 50, 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lin W-L, Lewis J, Yen S-H, Hutton M, and Dickson DW (2003). Ultra-structural neuronal pathology in transgenic mice expressing mutant (P301L) human tau. J. Neurocytol 32, 1091–1105. [DOI] [PubMed] [Google Scholar]
- Lin LC, Nana AL, Hepker M, Hwang JL, Gaus SE, Spina S, Cosme CG, Gan L, Grinberg LT, Geschwind DH, et al. (2019). Preferential tau aggregation in von Economo neurons and fork cells in frontotemporal lobar degeneration with specific MAPT variants. Acta Neuropathol. Commun 7, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Wang H, Peterson M, Zhang W, Hou G, and Zhang ZW (2019). N-terminal alternative splicing of GluN1 regulates the maturation of excitatory synapses and seizure susceptibility. Proc. Natl. Acad. Sci. USA 116, 21207–21212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marat AL, and Haucke V (2016). Phosphatidylinositol 3-phosphates-at the interface between cell signalling and membrane traffic. EMBO J. 35, 561–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mclnnes L, Healy J, and Melville J (2018). UMAP: Uniform manifold approximation and projection for dimension reduction. arXiv, ar-Xiv:1802.03426. https://arxiv.org/abs/1802.03426. [Google Scholar]
- Nakamura M, Shiozawa S, Tsuboi D, Amano M, Watanabe H, Maeda S, Kimura T, Yoshimatsu S, Kisa F, Karch CM, et al. (2019). Pathological Progression Induced by the Frontotemporal Dementia-Associated R406W Tau Mutation in Patient-Derived iPSCs. Stem Cell Reports 13, 684–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, and Cuervo AM (2005). Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J. Neuropathol. Exp. Neurol 64, 113–122. [DOI] [PubMed] [Google Scholar]
- Olszewska DA, Lonergan R, Fallon EM, and Lynch T (2016). Genetics of Frontotemporal Dementia. Curr. Neurol. Neurosci. Rep 16, 107. [DOI] [PubMed] [Google Scholar]
- Paşca AM, Sloan SA, Clarke LE, Tian Y, Makinson CD, Huber N, Kim CH, Park J-Y, O’Rourke NA, Nguyen KD, et al. (2015). Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 12, 671–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei W, Huang Z, Wang C, Han Y, Park JS, and Niu L (2009). Flip and flop: a molecular determinant for AMPA receptor channel opening. Biochemistry 48, 3767–3777. [DOI] [PubMed] [Google Scholar]
- Piras A, Collin L, Grüninger F, Graff C, and Rönnbäck A (2016). Autophagic and lysosomal defects in human tauopathies: analysis of post-mortem brain from patients with familial Alzheimer disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol. Commun 4, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polioudakis D, de la Torre-Ubieta L, Langerman J, Elkins AG, Shi X, Stein JL, Vuong CK, Nichterwitz S, Gevorgian M, Opland CK, et al. (2019). A Single-Cell Transcriptomic Atlas of Human Neocortical Development during Mid-gestation. Neuron 103, 785–801.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovitchenko T, Park Y, Page NF, Luo X, Krsnik Z, Liu Y, Salamon I, Stephenson JD, Kraushar ML, Volk NL, et al. (2020). Translational derepression of Elavl4 isoforms at their alternative 5′ UTRs determines neuronal development. Nat. Commun 11, 1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Nguyen HN, Song MM, Hadiono C, Ogden SC, Hammack C, Yao B, Hamersky GR, Jacob F, Zhong C, et al. (2016). Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 165, 1238–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, and Trapnell C (2017). Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravanidis S, Kattan FG, and Doxakis E (2018). Unraveling the pathways to neuronal homeostasis and disease: Mechanistic insights into the role of RNA-binding proteins and associated factors. Int. J. Mol. Sci 19, 1–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A, Yekutieli D, and Benjamini Y (2003). Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics 19, 368–375. [DOI] [PubMed] [Google Scholar]
- Rohrer JD, Guerreiro R, Vandrovcova J, Uphill J, Reiman D, Beck J, Isaacs AM, Authier A, Ferrari R, Fox NC, et al. (2009). The heritability and genetics of frontotemporal lobar degeneration. Neurology 73,1451–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sands BE, Jacobson EW, Sylwestrowicz T, Younes Z, Dryden G, Fedorak R, and Greenbloom S (2010). Randomized, double-blind, placebo-controlled trial of the oral interleukin-12/23 inhibitor apilimod mesylate for treatment of active Crohn’s disease. Inflamm. Bowel Dis 16, 1209–1218. [DOI] [PubMed] [Google Scholar]
- Sato C, Barthélemy NR, Mawuenyega KG, Patterson BW, Gordon BA, Jockel-Balsarotti J, Sullivan M, Crisp MJ, Kasten T, Kirmess KM, et al. (2018). Tau Kinetics in Neurons and the Human Central Nervous System. Neuron 97, 1284–1298.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheckel C, Drapeau E, Frias MA, Park CY, Fak J, Zucker-Scharff I, Kou Y, Haroutunian V, Ma’ayan A, Buxbaum JD, and Darnell RB (2016). Regulatory consequences of neuronal ELAV-like protein binding to coding and non-coding RNAs in human brain. eLife 5, 1–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seebohm G, Neumann S, Theiss C, Novkovic T, Hill EV, Tavaré JM, Lang F, Hollmann M, Manahan-Vaughan D, and Strutz-Seebohm N (2012). Identification of a novel signaling pathway and its relevance for GluA1 recycling. PLoS ONE 7, e33889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lin S, Staats KA, Li Y, Chang W-H, Hung S-T, Hendricks E, Linares GR, Wang Y, Son EY, et al. (2018). Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med 24, 313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Hung ST, Rocha G, Lin S, Linares GR, Staats KA, Seah C, Wang Y, Chickering M, Lai J, et al. (2019). Identification and therapeutic rescue of autophagosome and glutamate receptor defects in C9ORF72 and sporadic ALS neurons. JCI Insight 5, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiarli AM, Jennings R, Shi J, Bailey K, Davidson Y, Tian J, Bigio EH, Ghetti B, Murrell JR, Delisle MB, et al. (2006). Comparison of extent of tau pathology in patients with frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17), frontotemporal lobar degeneration with Pick bodies and early onset Alzheimer’s disease. Neuropathol. Appl. Neurobiol 32, 374–387. [DOI] [PubMed] [Google Scholar]
- Silva MC, Cheng C, Mair W, Almeida S, Fong H, Biswas MHU, Zhang Z, Huang Y, Temple S, Coppola G, et al. (2016). Human iPSC-Derived Neuronal Model of Tau-A152T Frontotemporal Dementia Reveals Tau-Mediated Mechanisms of Neuronal Vulnerability. Stem Cell Reports 7, 325–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva MC, Nandi GA, Tentarelli S, Gurrell IK, Jamier T, Lucente D, Dickerson BC, Brown DG, Brandon NJ, and Haggarty SJ (2020). Prolonged tau clearance and stress vulnerability rescue by pharmacological activation of autophagy in tauopathy neurons. Nat. Commun 11, 3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn PD, Huang CTL, Yan R, Fan L, Tracy TE, Camargo CM, Montgomery KM, Arhar T, Mok SA, Freilich R, et al. (2019). Pathogenic Tau Impairs Axon Initial Segment Plasticity and Excitability Homeostasis. Neuron 104, 458–470.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, and Goedert M (1996). Comparison of the neurofibrillary pathology in Alzheimer’s disease and familial presenile dementia with tangles. Acta Neuropathol. 92, 42–48. [DOI] [PubMed] [Google Scholar]
- Spina S, Schonhaut DR, Boeve BF, Seeley WW, Ossenkoppele R, O’Neil JP, Lazaris A, Rosen HJ, Boxer AL, Perry DC, et al. (2017). Frontotemporal dementia with the V337M MAPT mutation: Tau-PET and pathology correlations. Neurology 88, 758–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sposito T, Preza E, Mahoney CJ, Setó-Salvia N, Ryan NS, Morris HR, Arber C, Devine MJ, Houlden H, Warner TT, et al. (2015). Developmental regulation of tau splicing is disrupted in stem cell-derived neurons from frontotemporal dementia patients with the 10 + 16 splice-site mutation in MAPT. Hum. Mol. Genet 24, 5260–5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staats KA, Seah C, Sahimi A, Wang Y, Koutsodendris N, Lin S, Kim D, Chang WH, Gray KA, Shi Y, et al. (2019). Small molecule inhibition of PIKFYVE kinase rescues gain- and loss-of-function C9ORF72 ALS/FTD disease processes in vivo. BioRxiv. [Google Scholar]
- Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R, and Smibert P (2017). Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckius M, Zheng S, Houck-Loomis B, Hao S, Yeung BZ, Mauck WM 3rd, Smibert P, and Satija R (2018). Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 19, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumi SM, Bird TD, Nochlin D, and Raskind MA (1992). Familial presenile dementia with psychosis associated with cortical neurofibrillary tangles and degeneration of the amygdala. Neurology 42, 120–127. [DOI] [PubMed] [Google Scholar]
- Tebaldi T, Zuccotti P, Peroni D, Köhn M, Gasperini L, Potrich V, Bonazza V, Dudnakova T, Rossi A, Sanguinetti G, et al. (2018). HuD Is a Neural Translation Enhancer Acting on mTORC1-Responsive Genes and Counteracted by the Y3 Small Non-coding RNA. Mol. Cell 71, 256–270.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiruchinapalli DM, Caron MG, and Keene JD (2008). Activity-dependent expression of ELAV/Hu RBPs and neuronal mRNAs in seizure and cocaine brain. J. Neurochem 107, 1529–1543. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn L (2014). Pseudo-temporal ordering of individual cells reveals dynamics and regulators of cell fate decisions. Nat. Biotechnol 32, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevino AE, Sinnott-Armstrong N, Andersen J, Yoon SJ, Huber N, Pritchard JK, Chang HY, Greenleaf WJ, and Paşca SP (2020). Chromatin accessibility dynamics in a model of human forebrain development. Science 367, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderweyde T, Apicco DJ, Youmans-Kidder K, Ash PEA, Cook C, Lummertz da Rocha E, Jansen-West K, Frame AA, Citro A, Leszyk JD, et al. (2016). Interaction of tau with the RNA-Binding Protein TIA1 Regulates tau Pathophysiology and Toxicity. Cell Rep. 15, 1455–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco S, Kedaigle AJ, Simmons SK, Nash A, Rocha M, Quadrato G, Paulsen B, Nguyen L, Adiconis X, Regev A, et al. (2019). Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature 570, 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheyen A, Diels A, Reumers J,Van Hoorde K, Van den Wyngaert I, van Outryve d’Ydewalle C, De Bondt A, Kuijlaars J, De Muynck L, De Hoogt R, et al. (2018). Genetically Engineered iPSC-Derived FTDP-17 MAPT Neurons Display Mutation-Specific Neurodegenerative and Neurodevelopmental Phenotypes. Stem Cell Reports 11, 363–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogels T, Murgoci AN, and Hromádka T (2019). Intersection of pathological tau and microglia at the synapse. Acta Neuropathol. Commun 7, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Martinez-Vicente M, Krüger U, Kaushik S, Wong E, Mandelkow EM, Cuervo AM, and Mandelkow E (2009). Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum. Mol. Genet 18, 4153–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warmus BA, Sekar DR, McCutchen E, Schellenberg GD, Roberts RC, McMahon LL, and Roberson ED (2014). Tau-mediated NMDA receptor impairment underlies dysfunction of a selectively vulnerable network in a mouse model of frontotemporal dementia. J. Neurosci 34, 16482–16495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Buth JE, Vishlaghi N, Taxidis J, Khakh B, Coppola G, Pearson CA, Gong D, Dai X, Damoiseaux R, et al. (2017). Self-organized cerebral organoids with human specific features predict effective drugs to combat Zika virus infection. Cell Rep. 21, 517–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyn-Vanhentenryck SM, Feng H, Ustianenko D, Duffié R, Yan Q, Jacko M, Martinez JC, Goodwin M, Zhang X, Hengst U, et al. (2018). Precise temporal regulation of alternative splicing during neural development. Nat. Commun 9, 2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray S (2017). Modeling tau pathology in human stem cell derived neurons. Brain Pathol. 27, 525–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wren MC, Zhao J, Liu CC, Murray ME, Atagi Y, Davis MD, Fu Y, Okano HJ, Ogaki K, Strongosky AJ, et al. (2015). Frontotemporal dementia-associated N279K tau mutant disrupts subcellular vesicle trafficking and induces cellular stress in iPSC-derived neural stem cells. Mol. Neurodegener 10, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S-J, Elahi LS, Paşca AM, Marton RM, Gordon A, Revah O, Miura Y, Walczak EM, Holdgate GM, Fan HC, et al. (2019). Reliability of human cortical organoid generation. Nat. Methods 16, 75–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu WH, Kumar A, Peterhoff C, Shapiro Kulnane L, Uchiyama Y, Lamb BT, Cuervo AM, and Nixon RA (2004). Autophagic vacuoles are enriched in amyloid precursor protein-secretase activities: implications for β-amyloid peptide over-production and localization in Alzheimer’s disease. Int. J. Biochem. Cell Biol 36, 2531–2540. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MSB, Li G, et al. (2016). Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89, 37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HL, Hinman MN, Barron VA, Geng C, Zhou G, Luo G, Siegel RE, and Lou H (2011). Hu proteins regulate alternative splicing by inducing localized histone hyperacetylation in an RNA-dependent manner. Proc. Natl. Acad. Sci. USA 108, E627–E635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Hinman MN, Hasman RA, Mehta P, and Lou H (2008). Regulation of neuron-specific alternative splicing of neurofibromatosis type 1 pre-mRNA. Mol. Cell. Biol 28, 1240–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The bulk and single cell RNA-sequencing data generated as part of this study are available at the Gene Expression Omnibus (GEO: GSE171345) and the output of the analyses are available upon request. Previously unpublished code for nELAVL binding cargo enrichment and the pseudobulk network analysis can be found in Data S1. All other code used in this study have been previously published, and the pipelines described in the Method Details.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.