

Abstract
Hematopoietic stem cell gene therapy is emerging as a promising therapeutic strategy for many diseases of the blood and immune system. However, several individuals who underwent gene therapy in different trials developed hematological malignancies caused by insertional mutagenesis. Preclinical assessment of vector safety remains challenging because there are few reliable assays to screen for potential insertional mutagenesis effects in vitro. Here we demonstrate that genotoxic vectors induce a unique gene expression signature linked to stemness and oncogenesis in transduced murine hematopoietic stem and progenitor cells. Based on this finding, we developed the surrogate assay for genotoxicity assessment (SAGA). SAGA classifies integrating retroviral vectors using machine learning to detect this gene expression signature during the course of in vitro immortalization. On a set of benchmark vectors with known genotoxic potential, SAGA achieved an accuracy of 90.9%. SAGA is more robust and sensitive and faster than previous assays and reliably predicts a mutagenic risk for vectors that led to leukemic severe adverse events in clinical trials. Our work provides a fast and robust tool for preclinical risk assessment of gene therapy vectors, potentially paving the way for safer gene therapy trials.
Keywords: gene therapy, insertional mutagenesis, genotoxicity, machine learning, gene expression, support vector machine, integrating viral vectors, preclinical risk assessment, safety assay gene therapy, in vitro assay
Graphical abstract
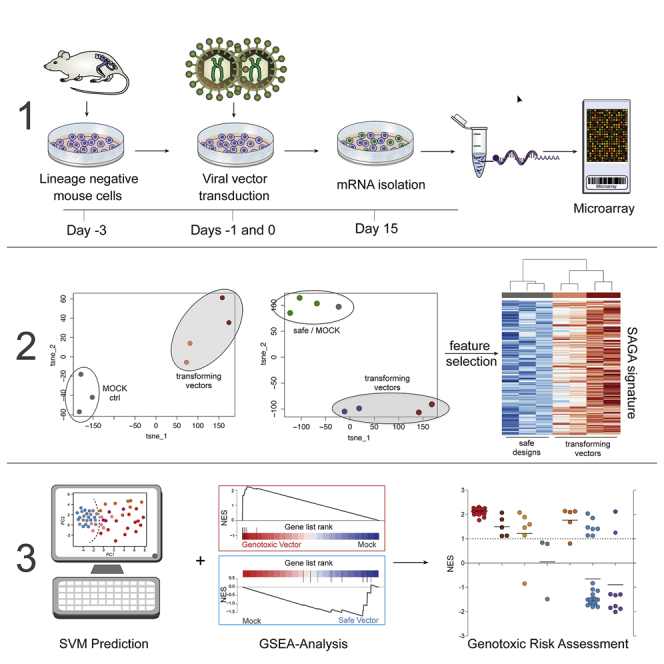
Rothe et al. use machine learning to detect gene expression signatures elicited by genotoxic gene therapy vectors for preclinical risk assessment.
Introduction
Hematopoietic stem cell gene therapy with retroviral vectors has demonstrated effectiveness in clinical trials for treatment of monogenetic diseases.1 However, transplantation of genetically modified hematopoietic stem cells led to myelodysplastic syndromes and leukemias in some gene therapy trials.2, 3, 4 These severe adverse events (SAEs) were caused by integration of the provirus in the vicinity of proto-oncogenes, such as MECOM and LMO2, which were subsequently upregulated by the strong viral promoter and enhancer sequences.5 Research efforts toward safer gene therapy led to removal of the long terminal repeat (LTR) enhancer elements in first-generation vectors. Instead, the field now mostly uses internal promoters in self-inactivating (SIN) retroviral vector designs.6, 7, 8 However, safety tests of integrating retro- and lentiviral vectors remain a bottleneck for transition from basic research to clinical application. Tumor-prone mice can be used to assess the mutagenic potential of integrating vectors, but these models are laborious, require large numbers of animals, and suffer from long readout times.9 Another commonly required safety analysis is the integration site pattern of a vector and a screen for clonal dominance in mouse models. However, judging the results of integration site studies regarding clonal dominance versus normal clonal fluctuation in mouse models can be difficult10 and suffers from poor predictability of the clinical occurrence of SAEs. Hence, efficient and reliable in vitro assays to screen for insertional mutagenesis are instrumental for clinical vector development. We previously developed the in vitro immortalization (IVIM) assay to quantify the risk of vector-induced cellular transformation.11 In this assay, murine hematopoietic progenitor cells are expanded after transduction with retroviral vectors. Following limiting dilution, non-immortalized cells stop proliferating, whereas insertional mutants give rise to clonal outgrowth. The incidence of vector-induced immortalization can be used to quantify and compare the mutagenicity of different vector types. Although the IVIM assay mainly detects mutants with insertions near the Mecom (also known as Evi1) locus, it reliably uncovers the ability of a given vector to activate neighboring proto-oncogenes. Therefore, IVIM results have been accepted by regulatory agencies in Europe, the United States, Canada, and Australia as part of the preclinical safety assessment for gene therapy vectors.12, 13, 14, 15 However, the IVIM assay is limited in terms of sensitivity and suffers from considerable inter-assay variability.
Transformation of healthy hematopoietic progenitors to preleukemic cells and leukemia is linked to specific gene expression programs that cause dysregulation of stemness pathways, growth, and perturbed differentiation.16, 17, 18 By creating a resource of transcriptional responses to vector integration, we show that integrating genotoxic vectors activate a gene expression program linked to transformation, stemness, and cancer de-differentiation. We hypothesized that this transcriptional signature can be exploited to create better predictors of vector-induced genotoxicity. To this end, we develop the surrogate assay for genotoxicity assessment (SAGA) classifier, which uses machine learning to detect dysregulation of this gene expression signature in transduced murine hematopoietic stem and progenitor cells (HSPCs). We compare results from the IVIM assay and SAGA for a variety of integrating benchmark vectors, including the three gammaretroviral vectors that triggered leukemias in clinical trials. The molecular readout of SAGA enhances sensitivity and reproducibility and eliminates the need to rely on the variable replating phenotype of the IVIM assay, reducing assay duration. In addition, we provide the SAGA analysis pipeline as a freely available R package.
Results
Cell culture-based assays for in vitro genotoxicity prediction
For IVIM and SAGA, murine lineage-negative (Lin−) HSPCs are transduced with high multiplicities of infection (MOIs) to reach at least 3 vector copies per cell (Figure 1A). After transduction, bulk cultures are expanded for 15 days in myeloid differentiation-promoting medium. On day 8 after transduction, cells are diluted to increase proliferative selection pressure until day 15. For the IVIM assay, cells are replated at low density and cultured for another 14 days before microscopic and enzymatic detection of growing insertional mutants. The results of the IVIM assay showed that non-transduced mock control cells rarely proliferated under limiting dilution conditions (11 of 124 assays positive), whereas cells transduced with a gammaretroviral vector with strong spleen focus-forming virus (SFFV) promoter/enhancer elements (LTR.RV.SFFV) showed a high incidence of insertional mutants (153 of 204 assays positive). These results are congruent with clinical data because this vector design led to myeloid malignancy in clinical trials for chronic granulomatous disease because of insertional activation of EVI1 and PRDM16. We tested a variety of gammaretroviral, alpharetroviral, and lentiviral vectors with different designs and mutagenic potential in both assays, including several vectors that have been or are currently used in clinical trials (Figures 1B and 1C; Table S1). The IVIM assay identified the potential hazard of two other vectors with clinically demonstrated genotoxicity: LTR.RV.MLV.IL2RG (also known as MFGγC), which caused leukemia in X-SCID studies,2 and LTR.RV.MPSV.WASP (also known as CMMP-WASP), which led to several primary and secondary leukemias in individuals with Wiskott-Aldrich syndrome (WAS).4,19 However, the replating frequencies and, hence, the power of the IVIM assay to uncover the mutagenic potential was substantially lower for vectors that contain weaker LTRs or SIN lentiviral vectors with SFFV as an internal promoter. We summarized the outcome of 502 IVIM assays in a receiver operating characteristic (ROC) curve (Figure 1D) to determine the predictive power of the replating phenotype as a proxy for vector mutagenicity. Overall, the IVIM assay showed a low false negative rate (specificity, 88.1%) and a sensitivity of 68.8%, reaching an overall area under the receiver operating curve (AUROC) of 0.827 (Figure 1D). Next we evaluated the results separately for mock control cells against the strongly transforming vector LTR.RV.SFFV (Figure 1E, red curve; sensitivity, 74.6%; AUROCLTR.RV.SFFV, 0.88) or other vectors with known mutagenicity (Figure 1E, black curve; sensitivity, 54.2%; AUROCother, = 0.74). Hence, the IVIM assay has significant predictive power to detect vector-induced immortalization in mouse hematopoietic cells, but for vectors other than the LTR.RV.SFFV, the IVIM assay suffers from low sensitivity and has to be repeated many times to obtain a reliable prediction of vector safety.
Figure 1.

IVIM and SAGA assays to detect vector genotoxicity in vitro
(A) Workflow of the in vitro genotoxicity assays. (B) Vector designs used in this study. Indicated are the various promoters and transgenes tested in our study (for details, see Table S1). (C) Replating frequencies (RFs) of different IVIM samples (n = 502) measured in 68 IVIM assays. Each dot represents one individual sample. RFs above Q1 (Q1 = 0.75 quantile of the RF for LTR.RV.SFFV) are counted as positive assays. LOD, limit of detection. Above the graph, the ratios of assays with RFs above and below Q1 are shown. Differences in the incidence of positive and negative assays relative to mock- or LTR.RV.SFFV-transduced cells were analyzed by Fisher’s exact test with Benjamini-Hochberg correction (∗p < 0.05, ∗∗∗p < 0.001; NS, not significant). Bars indicate mean RF. (D) Receiver operating characteristic (ROC) of the IVIM assay for samples (n = 502) with known activity in the IVIM assay. (E) Same as in (D) with separate curves for strongly transforming vectors (LTR.RV.SFFV) and mock controls (red curve) and weakly transforming vectors, safe vectors, and mock controls (black curve) for which the classification based on repeated testing in the IVIM assay was known.
Transforming vectors impose an oncogenic gene expression signature
We hypothesized that gene expression changes induced by transforming vectors might be a more accurate and sensitive predictor of vector-induced genotoxicity than the occurrence of a poorly defined clonal outgrowth after long periods of in vitro culture. Therefore, we analyzed the transcriptome from HSPC bulk cultures on day 15 after transduction. t-distributed stochastic neighbor embedding (t-SNE)20 and heatmaps of single assays (Figures 2A–2D) revealed that transduction with transforming vectors (based on the IVIM assay) imposed a distinct gene expression signature in HSPCs, clearly distinguishing them from the mock samples. In contrast, SIN vectors with weaker internal promoters, such as the EF1α-short (EFS) promoter, clustered with the non-transduced mock controls (Figures 2C and 2D). Importantly, gene expression changes were linked to but independent of the full immortalization phenotype in the IVIM assay. Cultures that were transduced with transforming vectors but did not immortalize in the IVIM assay (indicated as closed circles in Figure 2A) still showed similar gene expression changes compared with LTR.RV.SFFV immortalized samples (Figures 2A and 2B). The transformation-associated gene expression changes were observed across different vector genera as transforming gammaretroviral, lentiviral, and alpharetroviral vectors clustered together (Figures 2C and 2D; Figure S1A). The most consistently dysregulated genes (absolute log2FC > 1.0, Padj. < 10−5; Table S2, tabs 1 and 2) in samples transduced with transforming vectors included stem cell-associated genes (Aldh1a1, Smim5, and Ifitm6), proto-oncogenes (Zbtb16, Sox4, Pdgfrb, and Fgf3), stem cell transcription factors or their target genes (Spns2, Ces2g, and Myct1), and myeloid markers (Mpo and Cebpe). This oncogenic signature was detected as early as day 4 after transduction with transforming vectors (Figures S1B and S1C) and across three gene expression platforms (microarrays, qPCR, and RNA sequencing [RNA-seq]; Figures S1D and S1E). Gene set enrichment analysis (GSEA) showed upregulation of gene sets linked to positive cell cycle regulation, hematopoietic stem cells, erythroid/megakaryocytic differentiation, and Evi1 target genes21 in samples transduced with transforming vectors compared with mock controls and safe vector designs (Figures 2E, 2F, 2H, and 2I; Table S3, tab 2). Interestingly, samples transduced with safe vector designs displayed a similar enrichment of cell cycle gene sets and genes linked to erythroid/megakaryocytic differentiation compared with mock controls (Figure 2G). However, in contrast to transforming vectors, they showed downregulation of stemness and Evi1 target genes and a reduction of myeloid gene sets (Figures 2G and 2J; Table S3, tab 8). We hypothesized that the accelerated cell cycle and upregulation of genes associated with non-myeloid lineages might be a sign that assay progression and myeloid differentiation were generally delayed in transduced samples, independent of the vector type. Therefore, we compared early mock samples from day 8 with mock samples from day 15. Indeed, non-transduced HSPCs from this early time point displayed a similar enrichment of cell cycle, erythroid, and megakaryocytic gene sets but no upregulation of Evi1 target genes (Figure 2K; Figure S1F; Table S3, tab 14), reflecting incomplete myeloid differentiation and faster proliferation at that time. Most importantly, only genotoxic vectors upregulated hematopoietic stem cell transcriptional programs and Evi1 target genes compared with mock and non-transforming vectors. The upregulation of myeloid differentiation genes and stemness programs by transforming vectors (Figures 2E and 2F) underscores that differentiation and transformation are not mutually exclusive, as described for Evi1-driven leukemogenesis.22 By probing more than 8,000 gene sets from the MSigDB collection,22 we sought to obtain a more global view of the biological processes and pathways altered by transforming vectors. We found that transforming vectors triggered an early transcriptional signature that already included several “hallmarks of cancer,”23 including upregulation of gene sets linked to DNA replication, stemness, cancer de-differentiation, and therapeutic resistance and an enrichment of interferon signaling genes (Figure 2L; Table S3, tab 11). These data demonstrate that integrating vectors with the propensity to transform hematopoietic cells induce a unique oncogenic gene expression signature that distinguishes them from non-transforming vectors.
Figure 2.

Transforming vectors impose an oncogenic gene expression signature in murine HSPCs
(A) t-distributed stochastic neighbor embedding (t-SNE) of three mock samples (gray) and 4 samples transduced with LTR.RV.SFFV (red) from one SAGA assay (ID 120411) using all 36,226 annotated probes. (B) Hierarchical clustering of the samples shown in (A) based on the most variable probes (top 1%). (C) t-SNE of a second SAGA assay (ID 150128, 36,226 annotated probes). (D) Hierarchical clustering of the samples shown in (C) based on the most variable probes (top 1%). (E) Gene set enrichment analysis (GSEA) of hematopoiesis-associated gene sets (Table S3, tab 1) of samples transduced with IVIM-transforming vectors versus mock controls. Plotted are normalized enrichment scores (NESs) against the false discovery rate (FDR). The enrichment cutoff (FDR < 0.1) is indicated by the dashed line. (F) GSEA of IVIM-transforming vectors against IVIM-safe vectors. (G) GSEA of samples transduced with IVIM-safe vectors against mock controls. (H–K) GSEA plots for EVI1 target genes20 for the contrasts (H) transforming versus mock, (I) transforming versus safe, (J) safe versus mock, (K) mock day 8 versus mock day 15. (L) Enrichment map of highly upregulated (FDR < 0.005) gene sets from MSigDB in samples transduced with transforming vectors compared with mock control and safe samples.
Dataset preparation for classifier development
Having shown that genotoxic vectors impose a specific stemness-related gene expression profile, we sought to develop a machine learning algorithm distinguishing transforming vectors from safe designs. We started with the full dataset consisting of 169 SAGA microarrays with 39,428 probes each, resulting in more than 6 million data points. Similar to other cell culture assays analyzed with high-throughput methods,24 we observed systematic differences between assays because of different primary cell material, reagent lots, instruments, time, and personnel (“batch effects”). These batch effects caused clustering of samples by assay and processing date after normalization (Figure 3A). Thus, we first implemented a robust normalization and batch effect correction method to reduce the unwanted variation between individual SAGAs. We found that a combination of quantile normalization25 (Figures S2A and S2B) followed by ComBat26 effectively normalized and removed batch effects, allowing us to analyze all samples within a common gene expression space where transforming and safe/mock groups formed two different but overlapping groups (Figures 3B and 3C). Importantly, both methods are capable of “add-on adjustment” of new test batches, allowing cross-batch predictive modeling by leaving training data and classification rules fixed when new test data are adjusted.27 For the classifier development phase, the jointly normalized and batch-corrected gene expression matrix was reduced to samples with known properties in the IVIM assay (transforming/non-transforming/mock, n = 152; Figure 3D). We split the dataset into 10 different training and test sets, with the training sets comprised of 70% and corresponding test sets comprised of 30% of the samples. Development of models was performed on the training sets using repeated cross-validation to assess model performance during feature selection and hyperparameter tuning (Figure 3D; Figure S3). The test sets were then used to assess the performance of the final model fit and to control for overfitting of the classifier; for instance, because of feature selection bias or hyperparameter tuning.28,29
Figure 3.

Development phase of an SVM classifier to predict genotoxicity
(A–C) Data preprocessing. (A) t-SNE representation of all 169 SAGA assays after quantile normalization using all 39,428 probes. The coloring scheme encodes individual SAGA assays. (B) t-SNE of the 169 SAGA-samples after quantile normalization and ComBat correction using the same color key as in (A). (C) t-SNE plot as in (B) with the samples color coded according to vector properties in the IVIM assay. IVIM positive, transforming vectors; IVIM negative, nontransforming vectors; mock, untransduced controls; unknown, IVIM data inconclusive. (D) Scheme of classifier development during the development phase. The complete raw dataset was quantile normalized and batch corrected. The dataset was split 10 times into training (70% of samples) and test sets (30% of samples). Feature selection by SVM-RFE and SVM-GA was performed by further splitting the training sets using repeated cross-validation and monitoring prediction performance using the hold-out samples. Tuning of the SVM was performed at each step of the feature selection routines using nested cross-validation. An SVM with radial kernel was trained on the training set reduced to the optimal predictors found by SVM-RFE and SVM-GA and used to predict the test set. (E and F) Performance profile of SVM-RFE: accuracy on the hold-out samples plotted against the number of remaining probes during SMV-RFE for a representative training set (split 7). (G) Performance profile of SVM-GA: accuracy on the hold-out samples plotted against generation of the GA for training set 7. (H and I) Estimates of the prediction accuracy for the full models (H), RFE models (I), and GA models (J) using the test set (x axis) or repeated cross-validation (y axis). The horizontal and vertical bars represent the 95% confidence intervals using the test set and resampling approach, respectively.
Genetic algorithm-enhanced feature selection for prediction of genotoxicity
Initial testing of several machine learning approaches revealed that support vector machines (SVMs) offered a good classification performance on our dataset. Because the performance and computational cost of SVMs are negatively influenced by non-informative predictors, we performed feature selection on the training sets to find smaller predictor subsets with higher predictive power. A second aim for feature selection was to reduce the number of predictors as far as possible for potential later transfer of SAGA to other technical platforms that offer higher sample throughput but can interrogate fewer predictors. Starting with all 36,226 annotated probes, an unsupervised filtering step was applied to remove all probes with little or no variation across the training set, leaving a median of 1,195 probes (Figure 3D; Figure S3; Table S4, tab 1). Next we performed recursive feature elimination (SVM-RFE),30 which ranks all predictors according to their individual predictive power and then iteratively removes the least important predictors. The performance profiles across the different predictor subset sizes showed performance maxima between 3 and 45 predictors for the different training sets (median, 23 predictors; Figures 3E and 3F; Table S4, tab 1). On average, SVM-RFE removed more than 98% of the probes from the dataset, which resulted in a slight but significant boost in prediction performance, as measured by cross-validation (median accuracy full models, 89.0%; median accuracy RFE models, 90.8%; median Ppaired, 0.038; Table S4, tab 1). On the separate test sets, the median accuracy for the full models was 90.0%, whereas the SVM-RFE models achieved a median accuracy of 91.1% (Table S4, tabs 1 and 2). Notably, the SVM-RFE models required less than a tenth of the computation times of the full models because of the smaller number of predictors.
SVM-RFE is a greedy algorithm that is effective in eliminating large numbers of less important probes, but it does not perform an exhaustive search to find the best combination of retained predictors. We hypothesized that an optimal combination of probes with high predictive power would allow us to further reduce the number of required predictors while maintaining or even increasing prediction performance. To this end, we next employed a genetic algorithm (GA) to find the best combination of probes retained by SVM-RFE. GAs search for the best solution in a given feature space, guided by evolutionary principles.31 GAs have been shown to efficiently find optimal or near-optimal solutions for complex optimization problems, including feature selection.29 We implemented the GA together with support vector machine-based modeling (SVM-GA) and used cross-validation to asses predictive performance during the feature selection process. For SVM-GA, a population of 40 candidate solutions (individuals) was initially created from random subsets of the most informative probes found by the preceding SVM-RFE step. The predictor subsets with the highest fitness (prediction performance) of each generation had the best chances to survive and produce the next generation of predictor subsets by random crossover and mutation, producing more and more optimized probe combinations over time (Figure 3D; Figure S3). We performed feature selection using SVM-GA for all training/test set splits where SVM-RFE had retained more than 10 predictors. This ensured that the GA could choose from a sufficient number of predictors to create the initial population of predictor subsets. In three of four cases where SVM-RFE alone had arrived at less than 10 predictors, cross-validation accuracy (median accuracy full models, 89.0%; median accuracy RFE models, 90.7%; median Ppaired, 0.024) and test set accuracies (median accuracy full models, 90.0%; median accuracy RFE models, 92.2%; Table S4, tab 1) were already improved, making a second round of feature selection unnecessary. The cross-validation performance over the 40 generations of SVM-GA was aggregated into a performance profile demonstrating the progress of the algorithm and to choose the optimal generation for the entire dataset (Figure 3G). The algorithm further reduced the median number of predictors from 36 to 14 for the six training/test set splits subjected to SVM-GA. Importantly, cross-validation accuracy and test set accuracy showed improved predictive power compared with the full and SVM-RFE models (median cross-validation accuracy SVM-GA, 92.5%; median cross-validation accuracy full model, 89.0%; median test set accuracy SVM-GA, 93.3%; median test set accuracy full model, 90.0%; Figures 3H–3J; Table S4, tabs 1 and 2).
Because the performance of the final SAGA classifier, which was built on all 152 samples available, could only be estimated via cross-validation, we assessed whether our cross-validation strategy was trustworthy or whether it produced overly optimistic results because of feature selection bias or overfitting. Therefore, we plotted the accuracy estimates from cross-validation within the training sets against the test set accuracies (Figures 3H–3J). The performance estimates obtained by cross-validation and the test set accuracies showed good agreement, with the median of both estimates for the different splits (red dots in Figures 3H–3J) being located near the identity line. The wider confidence intervals of the accuracies calculated from single test sets demonstrated the higher uncertainty of performance estimates obtained from a test set of limited size compared with properly implemented resampling.28 Next we defined SAGA as the compound classifier obtained on a training set when SVM-RFE retained less than 10 predictors for this training set and used SVM-RFE followed by SVM-GA otherwise.
Assessing the predictive performance of SAGA
In the classifier development phase, the test samples were selected by random sampling from a jointly preprocessed gene expression matrix to obtain test sets of sufficient size and with the same class distributions as the training set. However, this approach did not fully reflect the later test scenario, where a new test set is to be predicted by SAGA using a preprocessed and fixed training set and classifier. To realistically assess the predictive performance of SAGA with unseen data in the absence of external validation data, we employed a jack-knife, leave-one-batch-out approach. The SAGA dataset was comprised of 19 individual SAGAs (batches). For each iteration, one complete batch was set aside as an independent test set (Figure 4A). The remaining 18 batches were used as the training set to which the preprocessing and feature selection pipeline developed above was applied (Figure 4A; Figure S3). On median, 10 optimal predictors were derived from the training sets (Table S5, tab 1), and an SVM was trained on the training set reduced to the optimal predictors (Figure 4B). Next, the batch that served as the independent test set was add-on normalized and add-on batch corrected. This adjusted the test set to the training set without altering the latter (Figure 4C), ruling out data leakage from the test samples into the training set or the classifier.32,33 Finally, the add-on adjusted test set was reduced to the optimal predictors, and the class labels were predicted. We repeated this procedure for all 19 batches and aggregated the prediction results over the 19 iterations (Figures 4D–4I; Figure S4; Table S5, tabs 1 and 2). Compared with the IVIM assay, SAGA outperformed the IVIM assay in terms of AUROC (AUROCSAGA, 0.940; AUROCIVIM, 0.827; PDELONG < 10−4; Figure 4D), overall accuracy (accuracySAGA, 90.9%; accuracyIVIM, 76.9%; Figure 4D), and the area under the precision recall curve (AUPRCSAGA, 0.944; AUPRCIVIM, 0.89; Figure 4G). Specifically, SAGA had a markedly higher sensitivity (87.7%) compared with the IVIM assay (68.8%). Most importantly, SAGA detected the genotoxicity of strongly transforming vectors (accuracySAGA, 97.1%; accuracyIVIM, 80.8%; Figures 4E and 4H) and vectors with weaker transforming potential with higher predictive power than the IVIM assay (accuracySAGA, 88.9%; accuracyIVIM, 78.5%; Figures 4F and 4I). The negative predictive value of SAGA was much higher than that of the IVIM assay (negative predictive value [NPV]SAGA, 0.91; NPVIVIM, 0.67; Table S5, tab 2); therefore, there is much higher confidence to classify a vector as “safe” for clinical use when using SAGA than IVIM. The number of predictors at each step and the classification performance metrics over the 19 iterations of the leave-one-batch-out approach are summarized in Figures S4A–S4D. To assess the stability of the feature selection process, we quantified how often individual predictors were included in the set of optimal predictors in each of the 19 iterations (Table 1; Table S5, tab 3). We found a high degree of overlap between the sets of optimal predictors found during the different iterations with a core set of highly potent predictors, such as Naip1 and Itih5, which were included in most of the sets (Table 1). These predictors also showed a high degree of overlap with the features found for the random test sets during the development phase (Table 1; Table S4, tab 3), as well as with the final list of 11 optimal features (Table 1; Table S4, tab 5) obtained on the complete SAGA dataset (described below).
Figure 4.

Estimation of model performance via the leave-one-batch-out approach
(A) Scheme of the leave-one-batch-out approach used to estimate SAGA performance. Details are given in the main text. (B) PCA representation of training set 01 reduced to the 8 optimal predictors derived from the training set and used to train the SVM. (C) Projection of add-on adjusted test set 01 samples into the PCA plot spanned by training set 01. (D–I) Aggregated prediction results over 19 iterations for the leave-one-batch-out approach versus a conventional IVIM assay. (D) AUC-ROC for all vector genera. (E) AUC-ROC for strongly transforming LTR.RV.SFFV vectors. (F) AUC-ROC for non-LTR.RV.SFFV vectors. (G) AUC-PRC for all vector genera. (H) AUC-PRC for strongly transforming LTR.RV.SFFV vectors. (I) AUC-PRC for non-LTR.RV.SFFV vectors.
Table 1.
Top 20 predictors most often selected in the leave-one-batch-out approach and random test set approach used during the development phase
| Leave-one-batch-out approach |
Random test set approach |
|||||
|---|---|---|---|---|---|---|
| Probe ID | Gene symbol | Times selected (of 19) | Variable importance (AUC) | Probe ID | Gene symbol | Times selected (of 10) |
| A_51_P289392 | Naip1∗ | 16 | 94.49 | A_55_P2077048 | Itih5∗ | 10 |
| A_55_P2077048 | Itih5∗ | 15 | 98.06 | A_55_P2024155 | Zbtb16∗ | 8 |
| A_51_P106059 | Traf4∗ | 14 | 94.65 | A_51_P289392 | Naip1∗ | 7 |
| A_66_P135106 | Slco3a1∗ | 12 | 94.18 | A_66_P135106 | Slco3a1∗ | 7 |
| A_55_P1987984 | Zfpm1 | 12 | 93.64 | A_55_P2018929 | Spns2∗ | 7 |
| A_55_P2018929 | Spns2∗ | 11 | 96.98 | A_66_P122559 | Myct1 | 5 |
| A_55_P2024155 | Zbtb16∗ | 9 | 100.00 | A_51_P334942 | Aldh1a1 | 4 |
| A_51_P486121 | Aff3∗ | 8 | 96.59 | A_55_P2108248 | Art4∗ | 4 |
| A_55_P2057587 | Arx | 8 | 95.15 | A_55_P1987984 | Zfpm1 | 4 |
| A_55_P1976882 | 4930519L02Rik | 7 | 95.00 | A_55_P2136426 | Prss57 | 3 |
| A_55_P2108248 | Art4∗ | 7 | 95.62 | A_52_P6828 | Xk | 3 |
| A_52_P56682 | Sla2∗ | 6 | 95.77 | A_55_P1976882 | 4930519L02Rik | 2 |
| A_51_P177171 | Tie1∗ | 6 | 93.49 | A_55_P2472735 | A530032D15Rik | 2 |
| A_51_P334942 | Aldh1a1 | 5 | 90.31 | A_51_P486121 | Aff3∗ | 2 |
| A_52_P73475 | Fam78a | 5 | 93.76 | A_55_P2057587 | Arx | 2 |
| A_52_P162957 | Frat2∗ | 5 | 93.52 | A_52_P73475 | Fam78a | 2 |
| A_52_P68221 | Gria3 | 5 | 92.75 | A_52_P162957 | Frat2∗ | 2 |
| A_51_P115626 | Shank3 | 5 | 94.49 | A_52_P68221 | Gria3 | 2 |
| A_55_P2146034 | Abca4 | 4 | 91.82 | A_52_P663904 | Lhfpl1 | 2 |
Tabulated are the number of times a predictor was included in the list of optimal predictors for a given training set and the global variable importance (AUC-ROC) of each individual predictor computed on the complete dataset of 152 SAGA samples. Indicated with asterisk (∗) are 11 optimal predictors of the final SAGA classifier. Complete lists can be found in Table S4, tab 3; Table S5, tab 3; and Table S6, tab 4.
Construction of the final SAGA model
Next, using the pipeline developed above, we built the final SAGA classifier on the entire set of available samples (n = 152) for use as the training set in the SAGA R package. After variance-based filtering, 1,243 features (Table S6, tabs 1 and 2) were supplied to SVM-RFE (Figure 5A), which retained 20 predictors (Table S6, tab 3). The following SVM-GA step found an optimal combination of 11 predictors after 14 iterations for the complete dataset (Figure 5B; Table S6, tab 4). Principal-component analysis of the SAGA dataset reduced to these 11 probes showed a clear separation of IVIM-transforming vectors against mock controls and IVIM-neutral vectors (Figure 5C), whereas a separation of classes was not discernible when the dataset was reduced to 11 randomly selected probes (Figure 5D). Finally, we queried transcriptome data from the Immunological Genome Consortium34 to determine which cell types of the hematopoietic system expressed the 20 most important predictors found by SVM-RFE. A majority of these predictors were highly expressed in the most immature hematopoietic stem cells (Figure 5E), whereas the predictors that were retained after unsupervised filtering and used as input into the feature selection process showed no such association with HSCs (Figure S5). The remaining genes, such as Frat2 and Traf4, were mainly associated with the lymphoid lineage, whereas none of these genes was expressed in mature granulocytes, a route of differentiation normally supported by the cytokine conditions used for the IVIM assay and SAGA. The most important predictors to detect genotoxic vectors were linked to stemness, differentiation arrest, and non-myeloid cell fates reflecting early steps of leukemogenesis that precede full cellular transformation and leukemia.35
Figure 5.

Construction of the final SAGA classifier
(A) Performance profile of the SVM-RFE procedure for the complete set of 152 samples. The filled circle represents the predictor subset with the highest performance comprised of the 20 most important predictors. (B) Performance profile of the SVM-GA procedure for the complete set of 152 samples over 40 generations of the GA. (C) Principal-component analysis (PCA) of 152 samples with known IVIM activity on the 11 optimal probes found by SVM-GA. (D) PCA of 152 samples with known IVIM activity on 11 randomly selected probes of 36,226 annotated probes. (E) Heatmap representing expression of the 20 genes with the highest predictive power from SVM-RFE across murine hematopoiesis.34 The boxplot below the heatmap represents the expression of genes in each column relative to the expression of all genes. LT-HSC, long-term HSC; ST-HSC, short-term HSC; MPP, multipotent progenitor; Mac/MF, macrophage; Mo, monocyte; Gran/GN, granulocyte.
SAGA-GSEA
A critical step in the SAGA-SVM procedure is correct estimation and correction of batch effects to project the new samples into a common gene expression space together with the training samples. This can be error prone for assays with few samples, a profoundly skewed class distribution, or particularly severe batch effects. For these cases, we sought to implement a more robust classifier that can be used within each individual SAGA independently (Figure 6A). We first examined whether the predictors found by our feature selection approach could be used in GSEA to discriminate genotoxic from safe vector designs. Indeed, we observed strong enrichment of the 11 optimal predictors from the final SAGA classifier in transforming vectors compared with mock controls (Figure 6B), whereas this signature was coordinately downregulated in safe vector designs compared with mock samples (Figure 6C). To estimate the predictive performance of this approach, we performed SAGA-GSEA within each of the leave-one-batch-out iterations by using the optimal predictors found for each of the training sets by our feature selection routine as a gene set for GSEA. We then examined the enrichment of the optimal predictor gene sets by performing GSEA for each sample against the mock controls in the left-out batches, yielding an AUC-ROC of 0.91 over all iterations (Figure 6D). To determine the optimal normalized enrichment score (NES) cutoff, we performed a ROC analysis, yielding an NES of greater than 1.7 as the ideal cutoff point for the complete dataset (Figure 6D). However, we found that this was confounded by inclusion of many samples of the strongly genotoxic LTR.RV.SFFV vector, which served as positive control and displayed very strong enrichment of the predictor gene sets. Therefore, we determined the cutoff again on the dataset without LTR.RV.SFFV samples, for which a NES of greater than 1.3 was the optimal threshold (Figure 6D). Using this NES cutoff, SAGA-GSEA outperformed the IVIM assay, albeit with a lower specificity than with the SVM-based SAGA classifier (AUROCGSEA, 0.91; AUROCIVIM, 0.827; PDELONG, 0.005; accuracyGSEA, 84.8%; accuracyIVIM, 76.9%; AUPRCGSEA, 0.91; AUPRCIVIM, 0.89; Table S7, tab 1; Figures 6E and 6F). Similar to SAGA-SVM, we used the 11 final predictors (Table S6, tab 4) that were derived from the complete dataset as a GSEA gene set for the final SAGA-GSEA classifier. The optimal NES cutoff for this 11-predictor gene set was determined by ROC analysis on the complete dataset after exclusion of the LTR.RV.SFFV samples (NES > 1.0; Figure 6G). LTR-driven gammaretroviral, SIN lentiviral, or alpharetroviral vectors with strong promoters and transforming properties in the IVIM assay showed a mean NES between 1.22 and 2.15 (Figure 6H), whereas potentially safer vector architectures with weaker internal promoters did not or only rarely showed this enrichment (Figure 6H; Table S7, tab 2). SAGA-GSEA presents an alternative classifier that circumvents the caveats of cross-batch prediction when correct add-on adjustment is difficult to achieve but critically depends on the integrity of the mock samples.
Figure 6.

SAGA-GSEA
(A) t-SNE representation of gene expression data from three independent SAGAs without batch correction. (B) GSEA plot for the 11 optimal predictors from the final classifier for LTR.SFFV.EGFP (sample X4991) versus mock from IVIM 3 (shown in A). (C) GSEA plot for the 11 optimal predictors for SIN.LV.EFS (sample X4997) versus mock from IVIM 3. (D) AUC-ROC aggregated from the leave-one-batch-out approach for all vector genera (red) and without strongly transforming LTR.RV.SFFV vectors (gray). The points on the curve indicate the best NES cutoff. (E) AUC-ROC for all vector genera (same curve as in D) versus AUC-ROC of the IVIM assay. (F) AUC-PRC aggregated from the leave-one-batch-out approach for all vector genera versus IVIM. (G) AUC-ROC using the 11 optimal predictors from the final classifier on all IVIM batches for all vector genera (red) and without strongly transforming LTR.RV.SFFV vectors (gray). The point on the curve indicates the best NES cutoff. (H) SAGA-GSEA results for all tested vectors. Plotted are the NESs of the 11-probe gene set from the final classifier over the different vector genera. The dashed line denotes NES ≥ 1.0, indicating evidence of genotoxicity as determined from the ROC analysis (Figure 6G) for genotoxic vectors when the strongly transforming LTR.SFFV samples were disregarded. Above the graph, mean NES values are shown for each vector type. The level of evidence whether the NES is significantly different from the positive control is indicated (ns = not significant, ∗p < 0.05, ∗∗∗p < 0.001; p values were calculated using a Kruskal-Wallis test with Dunn’s post hoc test).
Discussion
One important bottleneck for gene therapy is the necessity to assess potential safety risks. Since its inception,11, the IVIM assay has become the de facto gold standard in vitro assay for risk assessment of gene therapy vectors, and multiple groups have used the IVIM assay to test their vector constructs.12, 13, 14, 15 Here we show that the IVIM assay uncovers the genotoxic potential of vectors that caused SAEs in clinical trials for CGD3 (LTR.RV.SFFV), X-SCID2 (LTR.RV.MLV), and WASP4 (LTR.RV.MPSV). However, transformation potential is affected by vector design, integration sites, vector copy number, the transgene itself, and the disease background. In some contexts, such as ADA-SCID, even LTR-driven vector designs seemed to have an acceptable safety profile,36 although one individual treated with Strimvelis developed T cell leukemia linked to an insertional event.37 Consequently, most gene therapy trials now use SIN vectors. IVIM assays for the mutagenic vector design SIN.LV.SFFV revealed the genotoxic risk in only 40% of assays. Hence, even though the IVIM has an excellent specificity because of its low sensitivity, it has to be repeated multiple times to produce an informative and reliable result. We developed SAGA as a robust, standardized pipeline that efficiently identifies genotoxic vectors with higher accuracy by coupling a shortened IVIM assay with a molecular readout. By performing gene expression profiling on murine hematopoietic progenitors transduced with vectors with known IVIM properties, we show that only genotoxic vectors upregulate a specific gene expression signature that is reminiscent of immature HSC transcriptional programs, myeloid differentiation, and early transformation. One challenge was to identify a set of optimal predictors from the highly dimensional predictor space that allow precise classification to keep computational costs low and make the model more interpretable and as a potential starting point for possible later transfer of SAGA to simpler technical platforms. However, even with efficient feature reduction using RFE, the predictor space to be explored to find the best combination of features retained after SVM-RFE is vast. For instance, finding the best combination of 10–15 of 30 retained predictors would require building and testing over 500 million models. Instead of a complete search of the predictor space, we show here that Darwinian natural selection embedded in a GA can be used efficiently for a guided search of the predictor space. Thus, harnessing principles of population biology for complex optimization tasks is a powerful approach, as shown before for optimization of a gene expression-based classifier using particle swarm optimization.38 However, because of the nature of the GA, the solution represents a local optimum for each training set, and it cannot be excluded that better solutions may exist. Initializing the GA with different random seeds yielded slightly different lists of optimal predictors. However, the solutions mostly differed by only one or two predictors, indicating that the feature selection procedure was stable and the GA found a near-optimal solution. Importantly, as more samples are added to the dataset, improved solutions will be found, and SAGA will continue to evolve.
We tested a SIN lentiviral vector for RAG1 and RAG2 deficiency, in which transgene expression is controlled by a strong MND or a weaker PGK promoter.39 The PGK promoter did not display genotoxic potential in IVIM or SAGA. Conversely, the MND promoter constructs differed in their risk profile; vector SIN.LV.MND.RAG1 was determined to be safe by IVIM (0 of 9 positive assays) and SAGA (2 of 9 samples with an NES ≥ 1.0). In contrast, SIN.LV.MND.RAG2 showed a replating phenotype in 2 of 9 IVIM assays and a core set enrichment in 7 of 9 SAGA tests. This underscores the higher sensitivity and predictive potential of SAGA compared with IVIM. Based on these data, the preferred vector for potential treatment of RAG2-SCID is PGK-RAG2 rather than MND-RAG2. Similarly, when we tested an integrase mutant (LTR.RV.SFFV.W390A) of the strongly transforming LTR.RV.SFFV vector with an altered and potentially safer integration profile,40,41 SAGA detected a decrease in the mean NES to 1.35 (compared with 2.15 of the wild-type integrase vector). Thus, by providing a continuous score rather than a digital outcome, SAGA provides a higher resolution of genotoxic risk.
Recently, Zhou et al.42 observed that murine thymocytes transduced with mutagenic vectors show developmental arrest during T-lymphocyte development. The arrested progenitors overexpressed Lmo2, Mef2c, and Prdm16. The transcription factor LMO2 was the most clinically relevant dysregulated proto-oncogene in vector-associated transformation in clinical trials for X-SCID and WASP. Importantly, Lmo2 and Mef2c upregulation was detected in SAGA samples transduced with mutagenic vectors (log2FC Lmo2, 0.67; p = 5 × 10−24; log2FC Mef2c, 0.79; p = 9 × 10−15; moderated t test with BH adjustment; Table S2, tab 1). Hence, SAGA detects perturbation of proto-oncogenes of the lymphoid lineage, something that is beyond the capacity of the conventional IVIM assay. In addition, more work is needed to determine whether SAGA can detect genotoxic potential caused by transformation mechanisms other than cis activation of proto-oncogenes, such as aberrant splicing,43,44 and whether the SAGA principle can be transferred to non-hematopoietic target tissues. In the future, single-cell RNA-seq experiments might help to further fine-tune the SAGA signature. Our work provides insights into the early molecular events of genotoxicity following transduction of hematopoietic cells with integrating vectors and presents a powerful machine-learning approach to prospectively estimate the mutagenic potential of integrating vector systems for gene therapy.
Materials and methods
Study design
The study aimed to develop a gene expression-based diagnostic classifier to distinguish potentially genotoxic gene therapy vectors from safe vector designs. For SAGA, murine Lin− HSPCs were transduced with vectors of interest (Table S1) at high MOIs (target, >3 vector copies per cell) and expanded in myeloid growth-promoting medium (Figure 1A). On day 15, RNA was extracted for microarray analysis on Agilent Whole Mouse Genome 4x44K v.2 microarrays. The data were analyzed using R 3.5.1 and Bioconductor 3.7. All available microarrays (n = 169) were read in, quantile normalized, and batch corrected. 152 SAGA samples with known behavior in the IVIM assay (65 transforming, 55 safe, and 32 mock samples; Supplemental materials and methods; Table S8, tabs 1 and 2) were analyzed for differential expression (Table S2), GSEA (Table S3), and development of the SAGA classifier (Table S4). During classifier development, the jointly preprocessed gene expression matrix of all 152 SAGA samples was split into 10 different training (70% of samples) and test sets (30%) using stratified resampling to ensure comparable class distributions in test and training sets (Figure S3). Development of models was performed on the training sets only using cross-validation to assess model performance during feature selection and nested cross-validation for hyperparameter tuning. The test sets were not used at any point for feature selection or model tuning. Three different feature selection routines were applied to the training data to reduce the number of predictors as far as possible. First, an unsupervised filter was applied to exclude probes showing little variation in the dataset, followed by RFE, which iteratively removes the least important predictors before applying a GA to find a near-optimal combination of predictors retained by the preceding steps. After feature selection, an SVM was trained on the training data reduced to the optimal predictors and used to predict the test sets. For estimation of classifier performance, a jack-knife, leave-one-batch-out procedure was employed by leaving one batch of SAGA assays completely out of the model building process. The complete feature selection and model training pipeline was applied to the remaining batches. After the optimal predictors had been derived from the training set and the classifier had been trained and fixed, the batch that served as the independent test set was add-on normalized, add-on batch corrected, and predicted. The procedure was repeated for each of the 19 experimental SAGA batches, and the predictions results on the left-out batches were aggregated to determine performance. The final SAGA classifier was built on the entire set of available samples (n = 152) for use as the training set in the SAGA R package. For SAGA-GSEA, we used the optimal predictors found by the feature selection routines on each training set for each iteration as a gene set for GSEA. We then examined the enrichment of these optimal predictor gene sets by performing GSEA for each test set sample against the mock controls within the left-out test sets.
Cell culture for IVIM and SAGA
Lin− cells were isolated from tibiae, femora, and iliac crests of 8- to 12-week-old female C57BL/6J animals (Janvier) using the mouse lineage cell depletion kit (Miltenyi Biotec). Cells were frozen in aliquots of 5 × 105 cells in 90% fetal bovine serum (FBS) (PAA Laboratories) and 10% DMSO (Merck). After thawing, one aliquot per assay was cultured for 48 h in StemSpan (STEMCELL Technologies) supplemented with 50 ng/mL rm-SCF, 100 ng/mL rh-Flt-3L, 100 ng/mL rh-interleukin-11 (IL-11), and 20 ng/mL rm-IL-3 (PeproTech). For transduction, 250 μL of viral supernatant (or medium for the mock controls) was preloaded on 24-well suspension plates coated with RetroNectin (TaKaRa) to reach a defined MOI. Following the preloading, 1 × 105 cells were added to the wells in a total volume of 250 μL and incubated overnight. The preloading procedure was repeated for the second round of transduction. For this, suspension cells from the first transduction round were harvested, and cells still bound to RetroNectin were incubated with cell dissociation buffer (Gibco), pelleted, and resuspended in 250 μL of fresh culture medium before being added to the suspension harvest. Subsequently, 750 μL of the cell suspension was added to the wells preloaded for the second transduction. Cells were incubated for 24 h; harvested as described; mixed with 1.6 mL IMDM (Biochrom) containing 10% FBS, 1% penicillin/streptomycin (PAN Biotech), 2 mM glutamine (Biochrom). and cytokines as described above; and seeded onto 12-well suspension plates. On day 4 after (the second round of) transduction, we isolated DNA and/or RNA from 10% and used 2.5% of the cell material for flow cytometry analysis of transgene expression. After feeding the cells with 1.9 mL IMDM containing supplements (IMDM+), cells were incubated for 48 h on 6-well suspension plates before adding another 2.2 mL of medium. On day 8 post transduction (p.t.), samples were diluted (∼1:10) by seeding 1 × 106 cells in 4 mL of IMDM+ in 6-well suspension plates. On days 11 and 13 p.t., cells were given 1.2 mL of IMDM+. For the IVIM replating step, cells were re-seeded on day 15 p.t. at 100 cells/well in 96-well flat-bottom suspension plates. Following 14 days of incubation, plates were screened microscopically for growth of insertional mutants. Afterward, 20 μL of 0.25% thiazolyl blue tetrazolium bromide (Sigma) in DPBS (Pan Biotech) was added to the wells and incubated for 2–3 h at 37°C. Cells were lysed by addition of 100 μL of 20% SDS (Sigma). Plates were set on a shaker overnight at room temperature before absorption was measured at 540 nm with a SpectraMax 340PC (Molecular Devices). After background subtraction, the highest absorption value from the mock plate was used as a threshold to determine positive wells unless the value was higher than the mean absorption value of immortalized wells from a meta-analysis of 22 assays (5.61 times the expression value of a microscopically negative well). In this case, the second-highest mock value was used as a cutoff. Differences in the incidence of positive and negative assays relative to mock or LTR.RV.SFFV.EGFP-transduced cells were analyzed by Fisher’s exact test with the Benjamini-Hochberg multiple testing correction procedure.
SAGA sample processing and bioinformatics
A complete list of SAGA samples with a detailed description of RNA isolation, microarray acquisition, and all bioinformatic procedures, including de novo annotation of microarrays, raw data preprocessing, t-SNE, and principal-component analysis (PCA) visualizations, differential expression analysis, GSEA, description of the classifier development and performance measurements, the SAGA R package, qRT-PCR, and RNA-seq are outlined in the Supplemental materials and methods.
Statistical analysis
The incidence of positive and negative IVIM assays in Figure 1C was analyzed by a Fisher’s exact test with Benjamini-Hochberg correction. The SAGA-GSEA results in Figure 6H were compared using a Kruskal-Wallis test with Dunn’s post hoc test. For a detailed description of the different R package versions and statistical tests used in each bioinformatic step of SAGA, refer to the respective Supplemental material and methods section.
Data and materials availability
All data associated with this paper can be found in the main text or the Supplemental materials. Raw and processed expression data from all experiments have been deposited in the Gene Expression Omnibus under GEO: GSE109391. The R code for the SAGA genotoxicity prediction package and all other computations is available as source code and compiled R package via https://github.com/rothemi/SAGA. For convenience, an Amazon Machine Image running R3.6 and SAGA is available, including test samples from two different SAGA assays.
Acknowledgments
We are grateful to Prof. Christopher Baum (Charité, Berlin) for scientific support and valuable discussions. We thank Anne Galy and Fulvio Mavilio (Genethon, France) for providing vector LV.WASP.WASP. We are grateful to Prof. Christoph Klein (LMU, Munich) for providing the CMMP.WASP plasmid. We thank David A. Williams and Christian Brendel (Boston Children’s Hospital) for vector supernatant SIN.LV.LCR.D12G5. We thank Chris Mason (AVROBIO) for providing the lentiviral supernatants LV2AGA, LV2GBA and pCCL.CTNS. This work was supported by the German Research Foundation (RO 5102/1-1, cluster of excellence REBIRTH [Exc 62/2], and SFB738), the Federal State of Lower Saxony (research project R2N), and the CRACK-IT initiative from the National Centre for the Replacement, Refinement and Reduction of Animals in Research (NC3Rs; NC/C015102/1). Results incorporated in this study received funding from the European Union Horizon 2020 Research and Innovation Program (755170) and the FP7 Project CELL-PID (261387). A. Schwarzer is a fellow of the Clinician-Scientist Program “Young Academy” of Hannover Medical School and received funding from the Else Kröner-Fresenius-Stiftung (2017_A74) and Joachim Herz Stiftung. A.J.T. received funding from The Wellcome Trust (090233/Z/09/Z).
Author contributions
M.R., V.D., B.W., A.L.B., and O.D.-B. performed the experiments. A. Schwarzer, S.R.T., and M.R. designed and analyzed experiments, performed bioinformatic analyses, and wrote the manuscript. S.R.T. programmed the SAGA R package. A.J.T., R.G., and H.B.G. provided materials and revised the manuscript. A. Selich, J.W.S., F.J.T.S., U.M., M.M., and T.C.H. discussed data, helped to adjust experimental designs, and revised the manuscript. A. Schwarzer supervised the study and revised the manuscript.
Declaration of interests
A patent application has been filed under registration number EP3394286A1 (Analytical process for genotoxicity assessment).
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.06.017.
Supplemental information
References
- 1.Morgan R.A., Gray D., Lomova A., Kohn D.B. Hematopoietic Stem Cell Gene Therapy: Progress and Lessons Learned. Cell Stem Cell. 2017;21:574–590. doi: 10.1016/j.stem.2017.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hacein-Bey-Abina S., von Kalle C., Schmidt M., Le Deist F., Wulffraat N., McIntyre E., Radford I., Villeval J.-L., Fraser C.C., Cavazzana-Calvo M., Fischer A. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2003;348:255–256. doi: 10.1056/NEJM200301163480314. [DOI] [PubMed] [Google Scholar]
- 3.Stein S., Ott M.G., Schultze-Strasser S., Jauch A., Burwinkel B., Kinner A., Schmidt M., Krämer A., Schwäble J., Glimm H., et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat. Med. 2010;16:198–204. doi: 10.1038/nm.2088. [DOI] [PubMed] [Google Scholar]
- 4.Braun C.J., Boztug K., Paruzynski A., Witzel M., Schwarzer A., Rothe M., Modlich U., Beier R., Gohring G., Steinemann D., et al. Gene Therapy for Wiskott-Aldrich Syndrome–Long-Term Efficacy and Genotoxicity. Sci. Transl. Med. 2014;6:227ra33. doi: 10.1126/scitranslmed.3007280. [DOI] [PubMed] [Google Scholar]
- 5.Howe S.J., Mansour M.R., Schwarzwaelder K., Bartholomae C., Hubank M., Kempski H., Brugman M.H., Pike-Overzet K., Chatters S.J., de Ridder D., et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Invest. 2008;118:3143–3150. doi: 10.1172/JCI35798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schambach A., Zychlinski D., Ehrnstroem B., Baum C. Biosafety features of lentiviral vectors. Hum. Gene Ther. 2013;24:132–142. doi: 10.1089/hum.2012.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baum C., Schambach A., Bohne J., Galla M. Retrovirus vectors: toward the plentivirus? Mol. Ther. 2006;13:1050–1063. doi: 10.1016/j.ymthe.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 8.Rothe M., Schambach A., Biasco L. Safety of gene therapy: new insights to a puzzling case. Curr. Gene Ther. 2014;14:429–436. doi: 10.2174/1566523214666140918110905. [DOI] [PubMed] [Google Scholar]
- 9.Cesana D., Ranzani M., Volpin M., Bartholomae C., Duros C., Artus A., Merella S., Benedicenti F., Sergi Sergi L., Sanvito F., et al. Uncovering and dissecting the genotoxicity of self-inactivating lentiviral vectors in vivo. Mol. Ther. 2014;22:774–785. doi: 10.1038/mt.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gonin P., Buchholz C.J., Pallardy M., Mezzina M. Gene therapy bio-safety: scientific and regulatory issues. Gene Ther. 2005;12(Suppl 1):S146–S152. doi: 10.1038/sj.gt.3302629. [DOI] [PubMed] [Google Scholar]
- 11.Modlich U., Bohne J., Schmidt M., von Kalle C., Knöss S., Schambach A., Baum C. Cell-culture assays reveal the importance of retroviral vector design for insertional genotoxicity. Blood. 2006;108:2545–2553. doi: 10.1182/blood-2005-08-024976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Punwani D., Kawahara M., Yu J., Sanford U., Roy S., Patel K., Carbonaro D.A., Karlen A.D., Khan S., Cornetta K., et al. Lentivirus Mediated Correction of Artemis-Deficient Severe Combined Immunodeficiency. Hum. Gene Ther. 2017;28:112–124. doi: 10.1089/hum.2016.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang J., Khan A., Au B.C., Barber D.L., López-Vásquez L., Prokopishyn N.L., Boutin M., Rothe M., Rip J.W., Abaoui M., et al. Lentivector Iterations and Pre-Clinical Scale-Up/Toxicity Testing: Targeting Mobilized CD34+ Cells for Correction of Fabry Disease. Mol. Ther. Methods Clin. Dev. 2017;5:241–258. doi: 10.1016/j.omtm.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wolstein O., Boyd M., Millington M., Impey H., Boyer J., Howe A., Delebecque F., Cornetta K., Rothe M., Baum C., et al. Preclinical safety and efficacy of an anti-HIV-1 lentiviral vector containing a short hairpin RNA to CCR5 and the C46 fusion inhibitor. Mol. Ther. Methods Clin. Dev. 2014;1:11–14. doi: 10.1038/mtm.2013.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Negre O., Bartholomae C., Beuzard Y., Cavazzana M., Christiansen L., Courne C., Deichmann A., Denaro M., de Dreuzy E., Finer M., et al. Preclinical evaluation of efficacy and safety of an improved lentiviral vector for the treatment of β-thalassemia and sickle cell disease. Curr. Gene Ther. 2015;15:64–81. doi: 10.2174/1566523214666141127095336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krivtsov A.V., Twomey D., Feng Z., Stubbs M.C., Wang Y., Faber J., Levine J.E., Wang J., Hahn W.C., Gilliland D.G., et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–822. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- 17.Eppert K., Takenaka K., Lechman E.R., Waldron L., Nilsson B., van Galen P., Metzeler K.H., Poeppl A., Ling V., Beyene J., et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat. Med. 2011;17:1086–1093. doi: 10.1038/nm.2415. [DOI] [PubMed] [Google Scholar]
- 18.Ng S.W.K., Mitchell A., Kennedy J.A., Chen W.C., McLeod J., Ibrahimova N., Arruda A., Popescu A., Gupta V., Schimmer A.D., et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature. 2016;540:433–437. doi: 10.1038/nature20598. [DOI] [PubMed] [Google Scholar]
- 19.Boztug K., Schmidt M., Schwarzer A., Banerjee P.P., Díez I.A., Dewey R.A., Böhm M., Nowrouzi A., Ball C.R., Glimm H., et al. Stem-cell gene therapy for the Wiskott-Aldrich syndrome. N. Engl. J. Med. 2010;363:1918–1927. doi: 10.1056/NEJMoa1003548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van der Maaten L., Hinton G. Visualizing Data using t-SNE. J. Mach. Learn. Res. 2008;9:2579–2605. [Google Scholar]
- 21.Kustikova O.S., Schwarzer A., Stahlhut M., Brugman M.H., Neumann T., Yang M., Li Z., Schambach A., Heinz N., Gerdes S., et al. Activation of Evi1 inhibits cell cycle progression and differentiation of hematopoietic progenitor cells. Leukemia. 2013;27:1127–1138. doi: 10.1038/leu.2012.355. [DOI] [PubMed] [Google Scholar]
- 22.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 24.Goh W.W.B., Wang W., Wong L. Why Batch Effects Matter in Omics Data, and How to Avoid Them. Trends Biotechnol. 2017;35:498–507. doi: 10.1016/j.tibtech.2017.02.012. [DOI] [PubMed] [Google Scholar]
- 25.Gautier L., Cope L., Bolstad B.M., Irizarry R.A. affy--analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- 26.Johnson W.E., Li C., Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 27.Hornung R., Causeur D., Bernau C., Boulesteix A.-L. Improving cross-study prediction through addon batch effect adjustment or addon normalization. Bioinformatics. 2017;33:397–404. doi: 10.1093/bioinformatics/btw650. [DOI] [PubMed] [Google Scholar]
- 28.Kuhn M., Johnson K. Springer; 2013. Applied Predictive Modeling; p. 67. [Google Scholar]
- 29.Kuhn M., Johnson K. First Edition. Chapman and Hall/CRC; 2019. Feature Engineering and Selection: A Practical Approach for Predictive Models. [Google Scholar]
- 30.Guyon I., Weston J., Barnhill S. Gene Selection for Cancer Classification using Support Vector Machines. Mach. Learn. 2002;46:389–422. [Google Scholar]
- 31.Chatterjee S., Laudato M., Lynch L.A. Genetic algorithms and their statistical applications: an introduction. Comput. Stat. Data Anal. 1996;22:633–651. [Google Scholar]
- 32.Castaldi P.J., Dahabreh I.J., Ioannidis J.P.A. An empirical assessment of validation practices for molecular classifiers. Brief. Bioinform. 2011;12:189–202. doi: 10.1093/bib/bbq073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hornung R., Boulesteix A.L., Causeur D. Combining location-and-scale batch effect adjustment with data cleaning by latent factor adjustment. BMC Bioinformatics. 2016;17:27. doi: 10.1186/s12859-015-0870-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoshida H., Lareau C.A., Ramirez R.N., Rose S.A., Maier B., Wroblewska A., Desland F., Chudnovskiy A., Mortha A., Dominguez C., et al. Immunological Genome Project The cis-Regulatory Atlas of the Mouse Immune System. Cell. 2019;176:897–912.e20. doi: 10.1016/j.cell.2018.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stavropoulou V., Kaspar S., Brault L., Sanders M.A., Juge S., Morettini S., Tzankov A., Iacovino M., Lau I.-J., Milne T.A., et al. MLL-AF9 Expression in Hematopoietic Stem Cells Drives a Highly Invasive AML Expressing EMT-Related Genes Linked to Poor Outcome. Cancer Cell. 2016;30:43–58. doi: 10.1016/j.ccell.2016.05.011. [DOI] [PubMed] [Google Scholar]
- 36.Cicalese M.P., Ferrua F., Castagnaro L., Pajno R., Barzaghi F., Giannelli S., Dionisio F., Brigida I., Bonopane M., Casiraghi M., et al. Update on the safety and efficacy of retroviral gene therapy for immunodeficiency due to adenosine deaminase deficiency. Blood. 2016;128:45–54. doi: 10.1182/blood-2016-01-688226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Strimvelis: risk of lymphoid T-cell leukaemia? React. Wkly. 2020;1830:6. [Google Scholar]
- 38.Best M.G., Sol N., In ’t Veld S.G.J.G., Vancura A., Muller M., Niemeijer A.N., Fejes A.V., Tjon Kon Fat L.A., Huis In ’t Veld A.E., Leurs C., et al. Swarm Intelligence-Enhanced Detection of Non-Small-Cell Lung Cancer Using Tumor-Educated Platelets. Cancer Cell. 2017;32:238–252.e9. doi: 10.1016/j.ccell.2017.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garcia-Perez L., van Eggermond M., van Roon L., Vloemans S.A., Cordes M., Schambach A., Rothe M., Berghuis D., Lagresle-Peyrou C., Cavazzana M., et al. Successful Preclinical Development of Gene Therapy for Recombinase-Activating Gene-1-Deficient SCID. Mol. Ther. Methods Clin. Dev. 2020;17:666–682. doi: 10.1016/j.omtm.2020.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.El Ashkar S., De Rijck J., Demeulemeester J., Vets S., Madlala P., Cermakova K., Debyser Z., Gijsbers R. BET-independent MLV-based Vectors Target Away From Promoters and Regulatory Elements. Mol. Ther. Nucleic Acids. 2014;3:e179. doi: 10.1038/mtna.2014.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.El Ashkar S., Van Looveren D., Schenk F., Vranckx L.S., Demeulemeester J., De Rijck J., Debyser Z., Modlich U., Gijsbers R. Engineering Next-Generation BET-Independent MLV Vectors for Safer Gene Therapy. Mol. Ther. Nucleic Acids. 2017;7:231–245. doi: 10.1016/j.omtn.2017.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou S., Fatima S., Ma Z., Wang Y.-D., Lu T., Janke L.J., Du Y., Sorrentino B.P. Evaluating the Safety of Retroviral Vectors Based on Insertional Oncogene Activation and Blocked Differentiation in Cultured Thymocytes. Mol. Ther. 2016;24:1090–1099. doi: 10.1038/mt.2016.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scholz S., Fronza R., Bartholomae C., Cesana D., Montini E., Kalle C.V., Gil-Farina I., Schmidt M. Lentiviral Vector Promoter is Decisive for Aberrant Transcript Formation. Hum. Gene Ther. 2017;28:875–885. doi: 10.1089/hum.2017.162. [DOI] [PubMed] [Google Scholar]
- 44.Knight S., Bokhoven M., Collins M., Takeuchi Y. Effect of the internal promoter on insertional gene activation by lentiviral vectors with an intact HIV long terminal repeat. J. Virol. 2010;84:4856–4859. doi: 10.1128/JVI.02476-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.