

Abstract
Background:
Cancer vaccines targeting nonmutated proteins elicit limited Type I T-cell responses and can generate T-regulatory and Type II T-cells. Class II epitopes that selectively elicit Type I or Type II cytokines can be identified in nonmutated cancer associated proteins. In mice, a T-helper I selective IGFBP-2 N-terminus vaccine generated high levels of IFN-Ɣ secreting T-cells, no regulatory T-cells and significant anti-tumor activity. We conducted a Phase I trial of Th1 selective IGFBP-2 vaccination in patients with advanced ovarian cancer.
Methods:
Twenty-five patients were enrolled. The IGFBP-2 N-terminus plasmid-based vaccine was administered monthly for three months. Toxicity was graded by NCI criteria and antigen specific T-cells measured by IFN-Ɣ/IL-10 ELISPOT. T-cell diversity and phenotype were assessed.
Results:
The vaccine was well tolerated, 99% of adverse events graded 1 or 2, and generated high levels of IGFBP-2 IFN-Ɣ secreting T-cells in 50% of patients. Both Tbet+ CD4 (p=0.04) and CD8 (p=0.007) T-cells were significantly increased in immunized patients. There was no increase in GATA3+ CD4 or CD8, IGFBP-2 IL-10 secreting T-cells or T-regulatory cells. A significant increase in T-cell clonality occurred in immunized patients (p=0.03, pre vs. post vaccine) and studies showed the majority of patients developed epitope spreading within IGFBP-2 and/or to other antigens. Vaccine non-responders were more likely to have pre-existent IGFBP-2 specific immunity and demonstrated defects in CD4 T-cells, upregulation of PD-1 and downregulation of genes associated with T-cell activation, after immunization.
Conclusions:
IGFBP-2 N-terminus Th1 selective vaccination safely induces Type I T-cells without evidence of regulatory responses.
INTRODUCTION
Type I T-cells are needed for tumor eradiation (1). Both Type I cytokine secreting CD4 T-cells as well as cytolytic CD8 T-cells are required for a sustained anti-tumor immune response. Most ovarian cancer patients do not have high levels of Type I T-cells in their tumors. Indeed, tumor infiltrating CD4 and CD8 T-cells are found at much lower levels in ovarian cancer than in other solid tumors (2). There are several mechanisms involved in limiting Type I T-cells in ovarian cancer. The immune microenvironment is dominated by M2 macrophage and T-regulatory cells secreting cytokines, such as IL-10, which prevent the recruitment and proliferation of Type I T-cells (3). Ovarian cancer generally has a low mutational burden and few neoantigens that might stimulate a CD8 T-cell response (4). Finally, most identified tumor antigens in ovarian cancer are nonmutated and associated with the development of Type II T-cells and humoral immune responses (5).
IGFBP-2 is a protein that is overexpressed in most high-grade ovarian cancers and is associated with increased metastases and cell invasion (6). One study of over 400 ovarian cancers demonstrated that IGFBP-2 was overexpressed (2+,3+) in 74% of high-grade tumors, 73% of samples derived from Stage III, and 82% of samples from Stage IV disease (7). IGFBP-2 is also a human tumor antigen as evidenced by significant IGFBP-2 specific antibody levels in the serum of patients with epithelial cancers (8–10). We found, within the native protein sequence of IGFBP-2, class II interacting T-cell epitopes that preferentially elicit either Type I selective (IFN-Ɣ secreting) or Type II selective (IL-10 secreting) antigen specific immune responses in both cancer patients and volunteer donors (11). The Type II selective epitopes have a higher functional avidity for antigen than the Type I epitopes, are more common, and cluster in the C-terminus of the protein (11). In contrast, the Type I selective epitopes cluster in the N-terminus of the protein. We constructed plasmid-based vaccines composed of the N-terminus and the C-terminus domains of IGFBP-2. In mice, the Type I and Type II cytokine selectivity of the constructs was preserved with N-terminus immunized mice generating high levels of IGFBP-2 specific Type I T-cells with little to no Type II cytokine secretion and C-terminus vaccinated mice generating Type II selective IGFBP-2 specific responses. In a tumor challenge, vaccination with the N-terminus significantly inhibited tumor growth, the C-terminus vaccine had no impact on tumor growth, but admixing the N and C terminus vaccines abrogated the anti-tumor effect of the Type I selective N-terminus immunization (11). Vaccines targeting nonmutated proteins have been limited by the generation of vaccine induced T-regulatory cells and Type II T-cells preventing the development of high levels of tumor antigen specific Type I T-cells (12, 13). We hypothesize constructing vaccines directed against nonmutated tumor antigens by including only Type I selective epitopes will allow the generation of high levels of Type I T-cells with no induction of regulatory responses.
We constructed an IGFBP-2 N-terminus plasmid-based vaccine for clinical use and conducted a clinical trial of immunization in patients with advanced stage ovarian cancer. We questioned whether the vaccine was safe, immunogenic, and generated Type I selective immunity without the elaboration of IL-10 secreting T-cells or T-regulatory cells.
MATERIALS AND METHODS
Participants.
Twenty-five patients with advanced stage or recurrent ovarian cancer were enrolled between March 2012 and January 2015 after written informed consent was obtained (NCT01322802). The University of Washington Cancer Consortium granted institutional review board approval. Eligible patients had been treated to complete remission with standard therapies including primary debulking surgery. A CA-125 level within normal limits for the testing laboratory had to have been documented 90 days prior to enrollment. Eligible patients were at least 28 days from cytotoxic or steroid therapy, had no contraindication to receiving sargramostim, and had no history of uncontrolled autoimmunity, diabetes, or significant heart disease.
Study design.
The study was conducted in accordance with the ethical guidelines outlined in the US Common Rule. This single arm phase I study had a primary objective to assess the safety of a Th1 selective polyepitope plasmid-based vaccine, pUMVC3-hIGFBP-2 (1-163), encoding the N-terminus of IGFBP-2 including an evaluation of long-term toxicity. Toxicity was evaluated by Common Terminology Criteria for Adverse Events (AE) v3.0. Long-term toxicity data was collected via yearly review of the medical record. Secondary objectives included (1) the immunogenicity of vaccination, (2) the development of epitope spreading or a broadening of the immune response to other antigens and (3) the level of T-regulatory cells generated during vaccination. Patients received pUMVC3-hIGFBP2 (1-163) (100 mcg) vaccine admixed with rhuGM-CSF (100 mcg) intradermally monthly for three months. A 100 mcg dose was chosen based on a dose-escalation study with a HER2 intracellular domain vaccine encoded in the same plasmid as we used with IGFBP-2 N-terminus (14). Blood was drawn at baseline, prior to each vaccine for toxicity monitoring, and at months four (1 month after vaccination) and nine (6 months after vaccination) from enrollment for toxicity and immunologic evaluation. Targeted enrollment was 22 patients with up to five additional replacement patients to complete the month four toxicity analysis. A sample size of 22 patients would ensure that if no toxicities occur the probability of such an occurrence is at least 90% if the true toxicity rate, i.e. any Grade 3 or 4 toxicity, is 10% or more. For the immunologic response rate, 22 would allow 80% confidence that the estimated immune response rate is within at least 0.14 of the true immunologic response rate. Twenty-three patients completed the vaccine regimen and toxicity evaluation, 21 completed all blood collection time points, and 20 patients completed immunologic analyses (Suppl. Fig. 1).
Evaluation of T-cell responses.
All samples for each patient were cryopreserved under GLP conditions, thawed and analyzed simultaneously to ensure comparability (15). IFN-γ and IL-10 ELISPOT assays were performed as previously described (11, 16, 17). Ten mcg/mL of IGFBP-2 class II binding peptides contained in the vaccine were used in a pool: p8-22 (PALPLPPPLLPLLP), p17-31 (LLPLLPLLLLLLGAS), p67-81 (VAAVAGGARMPCAEL), p99-113 (EACGVYTPRCQGLR), p109-123 (GQGLRCYPHPGSELP), p121-135 (ELPLQALVMGEGTCE) (11). Tetanus toxoid (0.5 U/mL) and phytohemaglutinin (2.5 mcg/mL) were positive controls. Each antigen was assessed in four replicates of 2x105 /well. Data are presented as IFN-γ or IL-10 spots per well corrected for background (cSPW). Data are reported as the maximum cSPW determined either at four months or nine months after enrollment. Patients were considered to be a “responder” if the post-vaccination cSPW was greater than two standard deviations (SD) above the pre-vaccination value and a “non-responder” if the post-vaccination cSPW was within two SD above the pre-vaccination value. Two SD is equivalent to a p value of 0.05 in that there is a 95% probability that the values are statistically significant (18). Patients were considered to have pre-existent immunity to IGFBP-2 if, at baseline, the mean antigen-specific SPWs were statistically different from no antigen wells (16).
Intra- and intermolecular epitope spreading were determined via IFN-γ ELISPOT using 10 mcg/mL of IGFBP-2 C-terminal epitopes in a pool: p121-135 (ELPLQALVMGEGTCE), p164-178 (NHVDSTMNMLGGGGS), p190-204 (ELAVFREKVTEQHRQ), p213-227 (LGLEEPKKLRPPPAR), p235-249 (DQVLERISTMRLPDE), p251-265 (GPLEHLYSLHIPNCD), p266-280 (KHGLYNLKQCKMSLN), p291-305 (PNTGKLIQGAPTIRG) and p307-321 (PECHLFYNEQQEARG) or IGF-IR class II binding peptides used in a pool: p1196-1210 (WSFGVVLWEIATLAE), p1242-1256 (FELMRMCWQYNPKMR), p1332-1355 (GVLVLRASFDERQPYAHMNGGRKN) (17).
Flow cytometry.
Cryopreserved PBMC collected at enrollment and four months after enrollment were thawed and washed according our published methods (19), then incubated with 100 mcg of 10% normal mouse serum in PBS at room temperature for 30 minutes to block non-specific binding. After washing, the cells were stained with fluorochrome-conjugated monoclonal antibodies for phenotyping analyses. We performed intercellular staining for detecting Th1/Th2 transcription factors and T-regulatory cells. The cells were surface-stained with CD3 PE-Cy5 and CD4 APC Cy7. After washing, the cells were stained with Tbet PE-Cy7, Gata 3 Alexa Fluor 647, and FOXP3 Alex 488 according to eBioscience FOXP3 staining protocol. The percent of Tbet, Gata3 and FOXP3 positive cells were analyzed among CD3+CD4+ and CD3+CD8+ T cells. Additional markers analyzed included: CD3 FITC, CD8 PE-Cy7, CD69 PE-Cy5, CD279 (PD-1) PE, CD28 PE-CF594, HLA-DR PE-Cy7, CD3 FITC and CD4 APC. All of the antibodies were purchased from Biolegend or BD Biosciences. After washing the cells with PBS/1% FBS, data acquisition was performed on a FACS Canto flow cytometer (BD Biosciences) and was analyzed using the FlowJo software (RRID:SCR_008520).
T-cell receptor sequencing.
T-cell receptor beta chain CDR3 regions were sequenced from PBMC collected at enrollment and four months after enrollment by ImmunoSeq (Adaptive Biotechnologies), with primers annealing to V and J segments, resulting in amplification of rearranged VDJ segments from each cell. Clonality values were obtained through the ImmunoSeq Analyzer software. Clonality was measured as 1−(entropy)/log2 (number of productive unique sequences), with entropy considering the clone frequency. Morisita Overlap, a population overlap metric relating to the dispersion of clones in the samples, was calculated using the ImmunoSeq Analyzer software (Adaptive Technologies).
Analysis of gene expression in CD4+ T-cells.
FACS sorting was performed on PBMC collected at enrollment and four months after enrollment using the Aria Cell Sorter (BD Biosciences) to purify activated CD4 cells (Live CD3+CD4+CD25+CD127hi). Frozen cells were sent to NanoString Technologies for RNA extraction and profiling using the PanCancer IO 360 Panel Gene Expression Panel and analyzed on the nCounter MAX Analysis System (NanoString Technologies). Samples were analyzed using the Advanced Analysis Module of the nSolver™ software (NanoString Technologies). Samples were normalized against positive controls and selected housekeeping genes using the geometric mean. Ideal normalization genes were determined automatically by selecting those that minimize the pairwise variation statistic. Differential expression to identify specific targets was performed, and p-values were adjusted using the Benjamini-Hochberg procedure. Data are presented as differential expression (log2 fold change) of PBMC at enrollment and four months after enrollment.
Statistical analysis.
For ELISPOT and flow cytometry, statistical analysis was performed using GraphPad Prism version 8 (GraphPad Software; RRID:SCR_002798). Data were compared using a paired t test (two-tailed) or the Wilcoxon matched-pairs signed rank test.
RESULTS
Immunization with the IGFBP-2 N-terminus vaccine was associated with minimal toxicity.
Patient characteristics are shown in Table 1. The median age of the patient population was 63 years, the median time from diagnosis was 25 months, and the median time from treatment was 7 months. Fifty six percent of patients had only one prior chemotherapy regimen while 44% had undergone two or more prior regimens prior to enrollment.
Table 1.
Patient Characteristics
| Characteristic | Median (range) | No. of Patients | % |
|---|---|---|---|
| Age, years | 63 (29-83) | ||
|
| |||
| Initial disease status | |||
| IIB | 2 | 8% | |
| IIIC | 16 | 64% | |
| IV | 7 | 28% | |
|
| |||
| Histology | |||
| Serous | 22 | 88% | |
| Squamous | 1 | 4% | |
| MMMT/Carcinocarcoma | 1 | 4% | |
| Undifferentiated | 1 | 4% | |
|
| |||
| Time from diagnosis (months) | 25 (9-252) | ||
|
| |||
| Time from treatment (months) | 7 (2-55) | ||
|
| |||
| Prior chemotherapy regimens | |||
| 1 | 14 | 56% | |
| 2 | 8 | 32% | |
| 3 | 2 | 8% | |
| >3 | 1 | 4% | |
Pathologic subtypes were derived from the patient’s original pathology reports in the medical record
Two hundred and three adverse events (AE) were collected during the course of the study. Ninety-nine percent of AE were grades 1 and 2, with one grade 3 transient lymphopenia reported (Table 2 and Suppl. Table 1). The most commonly reported possibly, probably or definitely related AEs were injection site reaction (17%), fatigue (14%) flu like symptoms temporally related to vaccination (7%), and arthralgia and decreased lymphocyte count (6%) (Table 2). Three patients developed asymptomatic transient elevations in autoimmune serologies, all grade 1 or 2, which resolved without intervention during the course of the study.
Table 2.
Adverse Events
| Possibly, Probably, or Definitely Related |
All AEs |
|||
|---|---|---|---|---|
| Most Common | No. | % of Related AEs | No. | % of All AEs |
| Injection site reaction | 24 | 17% | 24 | 12% |
| Fatigue | 19 | 14% | 24 | 12% |
| Flu like symptoms | 10 | 7% | 11 | 5% |
| Lymphocyte count decreased | 9 | 6% | 11 | 5% |
| Arthralgia | 8 | 6% | 8 | 4% |
| Anemia | 7 | 5% | 9 | 4% |
| Hypokalemia | 5 | 4% | 9 | 4% |
| Platelet count decreased | 5 | 4% | 8 | 4% |
| Headache | 5 | 4% | 5 | 4% |
| White blood cell decreased | 5 | 4% | 5 | 3% |
|
| ||||
| AE Gradings | ||||
| 1 | 121 | 86% | 172 | 85% |
| 2 | 19 | 13% | 29 | 14% |
| 3 | 1 | 1% | 1 | 0% |
| 4 | 0 | 0% | 1 | 0% |
| 5 | 0 | 0% | 0 | 0% |
The IGFBP-2 N-terminus vaccine was immunogenic and selectively generated T-helper 1 (Th1) but not T-helper 2 (Th2) immunity.
Nonmutated tumor antigen vaccines have been shown to elicit both Th1 and Th2 responses (20). The IGFBP-2 N-terminus vaccine was specifically designed to be Th1 selective so we questioned to what level Type I (antigen specific IFN-ɣ secretion) and Type II (antigen specific IL-10 secretion) T-cells might be boosted (11). The median antigen-specific IFN-ɣ response before the first vaccine was 168 (range 0-1095) cSPW / 106 PBMC and the median response generated after vaccine was 301 (range 0-1065) cSPW / 106 PBMC (Fig. 1A). Ten (50%) of 20 patients significantly augmented immunity and were considered to be responders. Three (15%) patients did not augment IGFBP-2 Th1 immunity, including the one patient who had greater than 3 previous lines of treatment, and seven patients (35%) who had a decrease in antigen-specific immunity with immunization, all considered non-responders. Of note, the presence of pre-existent IGFBP-2 Th1 immunity was associated with lack of response to vaccination as compared to those patients without pre-existent IGFBP-2 responses (p=0.07). Thirteen (65%) of 20 patients demonstrated a pre-existing IGFBP-2-specific IFN-ɣ response. Only three (23%) of those patents with pre-existing immunity generated significant antigen-specific immune responses after vaccination. The median antigen-specific IL-10 response prior to immunization was 0 (range 0-370) cSPW/106 PBMC and the median response generated after vaccine was 0 (range 0-438) cSPW/106 PBMC. Twenty (100%) of 20 patients did not induce a statistically significant IGFBP-2 IL-10 specific T-cell response (Fig. 1B).
Figure 1. The IGFBP-2 N-terminus vaccine was immunogenic and selectively generated Th1 but not Th2 immunity.
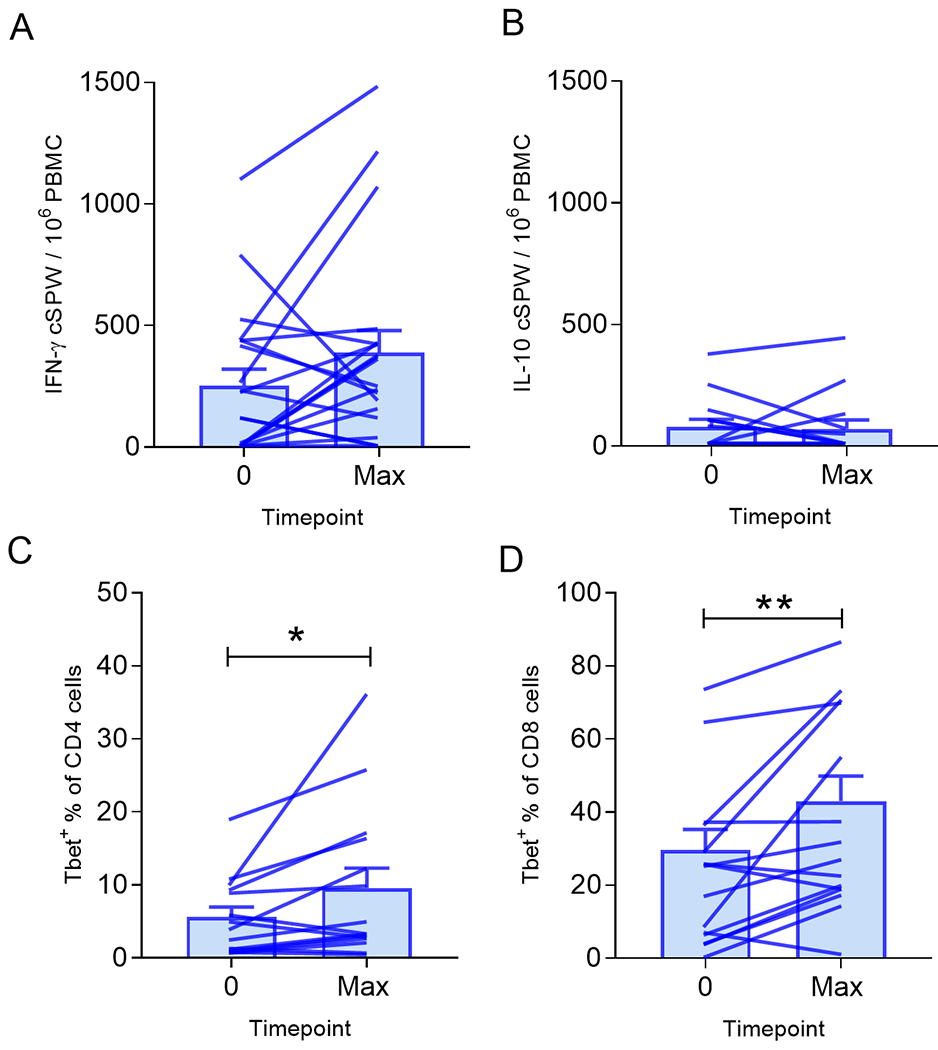
Mean (±SEM) corrected spots per well (cSPW) in 1x106 PBMC for (A) IFN-γ or (B) IL-10. Mean (±SEM) percent (C) Tbet+CD4+ or (D) Tbet+CD8+. Lines connect values from each patient determined pre-treatment (0) and at the maximum response (Max). n=20 patients; *p<0.05, **p<0.01.
We further analyzed the modulation of Th1 (Tbet) and Th2 (Gata3) cells by evaluating circulating CD4 and CD8 T-cells before and after vaccination. There were significantly increased CD4+Tbet+ cells in the peripheral blood of most patients after vaccination (mean 9.6%, range 0.7-36.4%) as compared to pre-vaccination (mean 5.6%, range 1-19.2%; p=0.04; Fig. 1C). There was no significant difference in CD4+Gata3+ levels in PBMC post-immunization (mean 2.5%, range 0.6-7.3%) as compared to pre-vaccination (mean 2.6%, range 0.4-7.6%; p=0.34). There were significantly increased CD8+Tbet+ cells in the peripheral blood after vaccination as well (mean 43.0%, range 6.3-92.1%) as compared to pre-vaccination (mean 29.6%, range 9-42.6%; p=0.01; Fig. 1D). There was no significant difference in CD8+Gata3+ levels in PBMC post-immunization (mean 1.9%, range 0.3-6.8%) as compared to pre-vaccination (mean 1.7%, range 0.4-6.4%; p=0.45).
The majority of patients developed intra- and/or intermolecular epitope spreading after IGFBP-2 N-terminus vaccination.
An increase in T-cell clonality after vaccination has been associated with improved survival of cancer patients (21). After vaccination, there was a significant increase in T-cell receptor beta (TCRβ) clonality in our population (p=0.03; Fig. 2A). The mean clonality prior to immunization was 0.063 (range, 0.023-0.207) and after vaccination, the mean clonality was 0.079 (range, 0.012-0.234). The development of multiple clonal populations could be an indication of epitope spreading, so we evaluated T-cell responses to the C-terminus of IGFBP-2 as a measure of intramolecular epitope spreading and to IGF-IR as an assessment of intermolecular epitope spreading.
Figure 2. The majority of patients developed intra- and/or intermolecular epitope spreading after IGFBP-2 N-terminus vaccination.
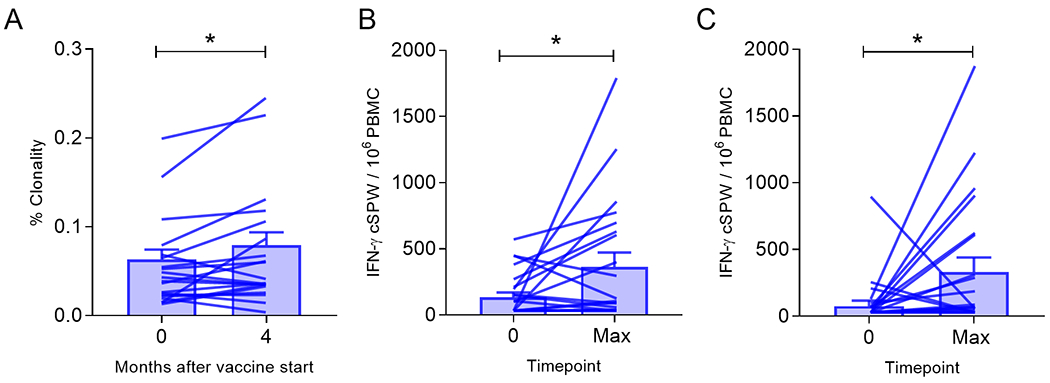
(A) Mean (±SEM) percent T-cell clonality. Mean (±SEM) corrected IFN-γ spots per well (cSPW) in 1x106 PBMC stimulated with (B) the C-terminal epitopes of IGFBP-2 or (C) IGF-IR epitopes. Connected points represent the response determined pre-treatment (0) and at the maximum response (Max) generated for each patient. *p<0.05.
Patients generated a significantly increased IFN-ɣ response to the C-terminal epitopes of IGFBP-2 after N-terminus vaccination (p=0.03; Fig. 2B). The median antigen-specific response before immunization was 71 (range 0-536) cSPW / 106 PBMC and the median response generated after vaccine was 83 (range 0-1747) cSPW / 106 PBMC. Ten (50%) of 20 patients significantly augmented C-terminus Th1 immunity, five of these patients also developed Th1 responses to the N-terminus. Patients also generated a significantly increased IFN-ɣ response to IGF-IR epitopes after vaccination (p=0.02; Fig. 2C). The median IGF-IR-specific response pre-immunization was 2.8 (range 0-869) cSPW / 106 PBMC and the median response generated after vaccine was 52 (range 0-1847) cSPW / 106 PBMC. Fourteen (70%) of 20 patients significantly augmented immunity. Six (30%) of 20 patients developed statistically significant immune responses to both of these antigens, the C-terminus and IGF-IR, after IGFBP-2 N-terminus immunization. Of note, there is only 8% sequence homology between IGFBP-2 (NCBI ascension number: AAA36048.1) and IGF-IR (NCBI ascension number: NP_000866.1).
T-cells from IGFBP-2 N-terminus vaccine non-responders exhibit functional defects.
When considering the level of immunity generated in responders, the fold-change in IGFBP-2 specific IFN-ɣ secreting T-cells was striking, mean, 178 (range, 33-425) as compared to non-responders, mean, −0.01 (range, 0.05-1.3; p<0.0001; Fig. 3A). We questioned whether we could determine any characteristics in the T-cells between populations which would predict a lack of response to the immunizing antigen after vaccination. We evaluated the Morisita index to assess TCRβ sequences pre- and post-vaccine to determine similarities in the clonal repertoire between the populations (Fig. 3B). There was a significant decrease in the index in responders (mean, 0.83, range 0.654-0.969) as compared to the non-responders (mean, 0.926; range, 0.866-0.992; p=0.04; Fig. 3B) indicating responders employed different TCRβs than non-responders.
Figure 3. T-cells from IGFBP-2 N-terminus vaccine non-responders exhibit functional defects.
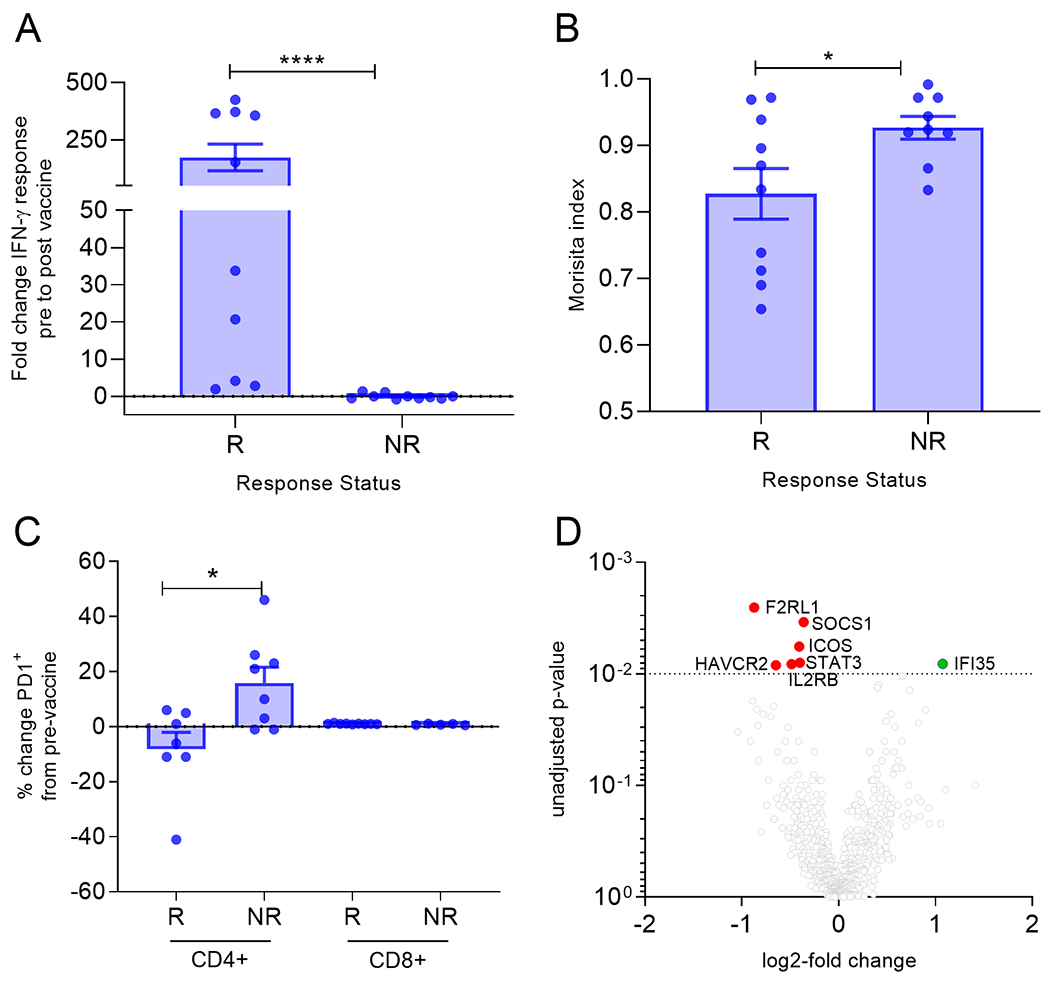
(A) Mean (±SEM) fold change in IFN-γ pre-vaccine to max response for responders (R) and non-responders (NR). (B) Mean (±SEM) Morisita index for the TCRβ in patients with the indicated response status. (C) Mean (±SEM) percent change of PD1+ T-cells cells from pre-vaccine in patients with the indicated response status. (D) Volcano plot of log2 fold change of the genes that were differentially expressed in non-responders. Horizontal dotted line: p=0.05. *p<0.05, ****p<0.0001.
Evaluating different T-cell subsets, we did not observe a significant difference in levels of FOXP3+ T-regulatory cells, or activated CD28+ or CD69+ T-cell subsets in the PBMC before as compared to after vaccination in responders or non-responders (all p>0.05). IGFBP-2 N-terminus responders, however, had significantly fewer PD1+CD4+ cells (mean decrease 8%, range; decrease 41%-increase 1%) post-vaccination as compared to those who had no or a decreased response to IGFBP-2 (non-responder; mean; increase 20%; range, 1% decrease to 46% increase; p=0.02; Fig. 3C). There was no difference in PD1+CD8+ levels after vaccination in either group (Fig. 3C).
PBMC were sorted for the CD4+CD127+CD25− subset, which included IL-2-producing naive and central memory T-helper cells, and a panel of genes associated with T-cell function and immunity was analyzed. Differential expression analyses from pre- and post-vaccination gene signatures from non-responders revealed a decrease in expression of genes associated with T-cell activation (F2RL1, SOCS1, STAT3, ICOS, IL2RB) and an increase in the gene IFI35, which is a negative regulator of interferon (Fig. 3D). There was no difference in expression of these same genes in responding patients pre- and post-vaccination.
IGFBP-2 N-terminus vaccination did not induce long-term toxicity.
There is a concern that immunity to nonmutated tumor antigens or the use of DNA-based vaccines may be associated with longer term toxicities including autoimmunity (22). The median follow-up for the study population was 43 months (range 12-84). Three adverse events were reported in long term follow-up. One patient developed an elevated TSH (5.52) which resolved. A second patient developed breast cancer and a third patient experienced a lichen planus flare. No other toxicities were reported. An additional concern was immunization with the entire N-terminus domain of an oncogene. For this reason, we evaluated disease outcome. The median progression free survival was 12 months with 8 of 23 patients (34%) showing no progression at the time of this report (Fig. 4A). The median overall survival was 48 months (Fig. 4B). Twelve of 23 (52%) patients are still alive a median of 52 months after entry to the study (range, 49-84 months).
Figure 4. Progression free and overall survival after immunization.
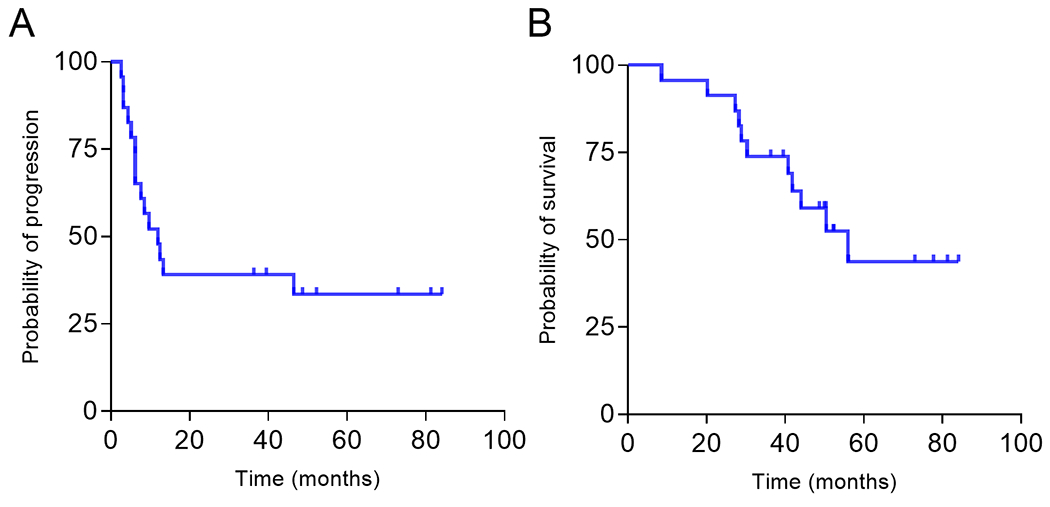
Kaplan-Meier curve for (A) progression free survival and (B) overall survival.
DISCUSSION
Vaccines targeting CD4 T-cells can be designed to be universal to most HLA-DR types by identifying T-cell epitopes that bind promiscuously across multiple HLA-DR alleles (23). We use a multi-algorithm approach for the identification of pan-HLA-DR binding epitopes based on a scoring system which prioritizes high binding affinity across the 15 most common MHC class II alleles (10). We further introduced functional screening to identify epitopes which selectively induce Type I cytokine secretion in response to an antigen such as IGFBP-2 (24). IGFBP-2 is upregulated in many common solid tumors and promotes oncogenic pathways such as epithelial to mesenchymal transformation, cell migration, and metastasis (25). Vaccination with IGFBP-2 epitopes designed to elicit antigen specific Th1 cells inhibits tumor growth in mouse models via the generation of both tumor specific CD4+ and CD8+ T-cells (10). Data presented here demonstrates Th1 selective immunity is safely generated against IGFBP-2, that epitope spreading is induced in a majority of patients and is the predominant type of immune response elicited, and that pre-existent immunity and specific T-cell functional defects may be associated with a lack of T-cell response to the immunizing antigen.
Immunization with the IGFBP-2 N-terminus vaccine elicited Th1 selective immunity with no evidence of the generation of T-regulatory cells, which have been reported to be stimulated after vaccination with nonmutated tumor antigens. Vaccine induced T-regulatory cells will suppress immune responses (26). We have shown that within the natural sequence of nonmutated tumor antigens are CD4 T-cell epitopes that will elicit Type I or Type II antigen specific cytokine secretion (11). Inclusion of the Type II epitopes in a vaccine significantly reduces vaccine efficacy. The IGFBP-2 N-terminus vaccine was designed to be enriched for Th1 selective epitopes with Type II cytokine inducing epitopes edited from the vaccine construct (11). As a result, the median level of immunity achieved after vaccination was 1:3,300 PBMC being an IFN-Ɣ secreting IGFBP-2 specific T-cell. These levels are consistent with those achieved with mutated antigen vaccines. In a study of neoantigen vaccines deployed for the treatment of melanoma, immunization with numerous mutated antigenic peptides could produce T-cell levels as high as 1:2,000 PBMC responding to a neoantigen after immunization (27). Despite high levels of unopposed Type I T-cells generated with IGFBP-2 N-terminus vaccination, there was no evidence of autoimmunity either in short-or long-term follow-up. Abnormal overexpression of a nonmutated protein in oncogenesis enhances the immunogenicity of that protein (28). Subdominant epitopes, not present when the protein is expressed at basal levels, become unmasked with overexpression and are present only on the tumor (29). It may be for this reason, vaccines targeting overexpressed nonmutated antigens have been associated with minimal toxicity (30).
The majority of vaccinated patients demonstrated evidence of epitope spreading; a broadening of the immune response to new epitopes within an antigen or to other antigens not included in the vaccine. Even patients who did not show statistically significant immune response to the immunizing antigen, IGFBP-2, could develop epitope spreading. Tbet expressing CD8 T-cells were also elevated after Th1 selective immunization, suggesting proliferation of activated non-exhausted cytolytic T-cells (31). Epitope spreading reflects enhanced cross-priming due to activation of antigen presenting cells by tumor trafficking type I T-cells (32). Type I cytokines released by the T-cells increase the efficiency and efficacy of antigen processing by innate immune cells. Epitope spreading occurring in most patients suggests that at least some vaccine induced Th1 cells could home to the tumor in the majority of vaccinated patients despite the inability to measure significant differences in the levels of IGFBP-2 Th1 at the prescribed time-points. The development of epitope spreading is associated with improved clinical outcomes after various forms of cancer immunotherapy. One report of the use of a dendritic cell-based vaccine targeting the MART-1 antigen suggested that favorable clinical outcomes after vaccination were not related to the levels of immunity achieved with the immunizing antigen, but rather to the development of epitope spreading to other melanoma associated antigens (33). A study of adoptive transfer of EBV specific T-cells for the treatment of lymphoma demonstrated that evidence of epitope spreading to non-viral tumor associated antigens was only seen in those patients achieving a clinical response (34). A limitation of our study is that the small number of patients preclude our ability to significantly associate the development of epitope spreading with disease outcome. The assessment of epitope spreading, however, could be an important biomarker of response and will be evaluated in Phase II trials of the vaccine.
Some patients were not effectively immunized against IGFBP-2 N-terminus. Non-responders were more likely to have a pre-existent immune response to IGFBP-2 at the start of the trial. Ovarian cancer patients have been shown to have pre-existent immune responses directed against many antigens expressed in the tumor (5). Presumably these T-cells were antigen educated in the tumor microenvironment and exposed to chronic antigen stimulation. Although the presumption is that pre-existent immune responses would be significantly boosted by immunization, there is some evidence in animal models that an existing response is negatively modulated by vaccination. One study vaccinated mice with established melanoma tumors with an antigen specific vaccine (35). Pre-existing intratumoral T-cells could initially proliferate in response to vaccination, but over time became less functional with subsequent immunizations. An initial vaccine boost could explain why we observed a higher rate of epitope spreading than IGFBP-2 specific Th1 immunity. The time course of our immune monitoring could have missed an initial vaccine induced immune response that was diminished by continuing immunization in some patients. Vaccination may have driven the existing antigen specific T-cells to an exhausted phenotype as evidenced by upregulation of PD-1. We have shown that upregulation of PD-1 on peripheral blood CD4 T-cells after immune modulation of chest wall tumors with the topical toll-like receptor-7 agonist imiquimod is associated with lack of clinical response to the immune therapy (19). The T-cells derived from non-responders showed evidence of other functional defects. The CD4+ T-cells had downregulated genes associated with T-cell activation as compared to vaccine responders. Upregulation of SOCS1 is associated with a high magnitude Type I T-cells response which was lacking in these patients (36). Expression of ICOS and F2RL1 (PAR-2) stimulates T-cell activation and enhanced effector function (37, 38). Signaling through IL-2 receptor beta increases proliferation of CD8 effector T-cells (39) and STAT 3 is important for inflammatory T-cell differentiation (40). One upregulated differentially expressed gene between vaccine responders and non-responders was IFI35 and encodes interferon inducible protein 35 which functions to attenuate the interferon response (41). This constellation of differentially expressed genes highlights T-cells that are not capable of activation and tissue destruction.
Data shown in this report demonstrate that vaccines targeting nonmutated tumor antigens, such as IGFBP-2, can be constructed to generate high level Type I immune responses without significant evidence of self-regulation. Studies described here also highlight potential biomarkers that may be useful for correlation to clinical outcome, such as epitope spreading. We have also identified biomarkers which may predict patients that will respond to the immunizing antigen, such as evidence of significant pre-existent immunity. Phase II studies will address the therapeutic efficacy of Th1 selective IGFBP-2 vaccination in advanced ovarian cancer.
Supplementary Material
Translational Relevance.
Th1 selective cancer vaccines can be developed to target overexpressed nonmutated tumor associated antigens. A Th1 selective vaccine directed against IGFBP-2 generated high levels of IGFBP-2 specific Th1 in patients with advanced ovarian cancer as well as intra- and intermolecular epitope spreading.
ACKNOWLEDGEMENTS
The work was supported by NIH-P50 CA083636 (funded the conduct of the study; collection, management, analysis, and interpretation of the data), NIH-UL1 TR002319 (supported the facility where the patients were treated during the conduct of the study), the Helen B. Slonaker Endowed Professor for Cancer Research for M. Disis (supported data analysis and manuscript preparation) and The Ovarian Cancer Research Program, Early Investigator Award, OC130304 and Department of Defense, W81XWH-14-1-0161 for J. Liao (supported the clinical conduct of the study, data analysis, and manuscript preparation).
Conflict of Interest:
MLD is a stockholder in EpiThany and receives grant support from Celgene, EMD Serono, Pfizer, Janssen, and Precigen. JBL received research funding through his institution from Merck, Forty-Seven, Sanofi, Harpoon Therapeutics, Laekna Therapeutics, Sumitomo Dainippon Pharma Oncology, and Precigen.
REFERENCES
- 1.Disis ML. Immune regulation of cancer. J Clin Oncol. 2010;28:4531–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gokuldass A, Draghi A, Papp K, Borch TH, Nielsen M, Westergaard MCW, et al. Qualitative analysis of tumor-infiltrating lymphocytes across human tumor types reveals a higher proportion of bystander CD8(+) T cells in non-melanoma cancers compared to melanoma. Cancers (Basel). 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Batchu RB, Gruzdyn OV, Kolli BK, Dachepalli R, Umar PS, Rai SK, et al. IL-10 Signaling in the tumor microenvironment of ovarian cancer. Adv Exp Med Biol. 2021;1290:51–65. [DOI] [PubMed] [Google Scholar]
- 4.Martin SD, Brown SD, Wick DA, Nielsen JS, Kroeger DR, Twumasi-Boateng K, et al. Low mutation burden in ovarian cancer may limit the utility of neoantigen-targeted vaccines. PLoS One. 2016;11:e0155189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goodell V, Salazar LG, Urban N, Drescher CW, Gray H, Swensen RE, et al. Antibody immunity to the p53 oncogenic protein is a prognostic indicator in ovarian cancer. J Clin Oncol. 2006;24:762–8. [DOI] [PubMed] [Google Scholar]
- 6.Lee EJ, Mircean C, Shmulevich I, Wang H, Liu J, Niemisto A, et al. Insulin-like growth factor binding protein 2 promotes ovarian cancer cell invasion. Mol Cancer. 2005;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang H, Rosen DG, Fuller GN, Zhang W, Liu J. Insulin-like growth factor-binding protein 2 and 5 are differentially regulated in ovarian cancer of different histologic types. Mod Pathol. 2006;19:1149–56. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Y, Ying X, Han S, Wang J, Zhou X, Bai E, et al. Autoantibodies against insulin-like growth factorbinding protein-2 as a serological biomarker in the diagnosis of lung cancer. Int J Oncol. 2013;42:93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Y, Jiang T, Zhang J, Zhang B, Yang W, You G, et al. Elevated serum antibodies against insulin-like growth factor-binding protein-2 allow detecting early-stage cancers: evidences from glioma and colorectal carcinoma studies. Ann Oncol. 2012;23:2415–22. [DOI] [PubMed] [Google Scholar]
- 10.Park KH, Gad E, Goodell V, Dang Y, Wild T, Higgins D, et al. Insulin-like growth factor-binding protein-2 is a target for the immunomodulation of breast cancer. Cancer Res. 2008;68:8400–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cecil DL, Holt GE, Park KH, Gad E, Rastetter L, Childs J, et al. Elimination of IL-10-inducing T-helper epitopes from an IGFBP-2 vaccine ensures potent antitumor activity. Cancer Res. 2014;74:2710–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ebert LM, MacRaild SE, Zanker D, Davis ID, Cebon J, Chen W. A cancer vaccine induces expansion of NY-ESO-1-specific regulatory T cells in patients with advanced melanoma. PLoS One. 2012;7:e48424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nistor GI, Dillman RO. Cytokine network analysis of immune responses before and after autologous dendritic cell and tumor cell vaccine immunotherapies in a randomized trial. J Transl Med. 2020;18:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Disis MLCA, Higgins D, Fintak P, Waisman JR, Reichow J, Slota M, et al. A phase I trial of the safety and immunogenicity of a DNA-based vaccine encoding the HER2/neu (HER2) intracellular domain in subjects with HER2+ breast cancer. J Clin Oncol. 2014;32. [Google Scholar]
- 15.Disis ML, dela Rosa C, Goodell V, Kuan LY, Chang JC, Kuus-Reichel K, et al. Maximizing the retention of antigen specific lymphocyte function after cryopreservation. J Immunol Methods. 2006;308:13–8. [DOI] [PubMed] [Google Scholar]
- 16.Disis ML, Wallace DR, Gooley TA, Dang Y, Slota M, Lu H, et al. Concurrent trastuzumab and HER2/neu-specific vaccination in patients with metastatic breast cancer. J Clin Oncol. 2009;27:4685–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cecil DL, Park KH, Gad E, Childs JS, Higgins DM, Plymate SR, et al. T-helper I immunity, specific for the breast cancer antigen insulin-like growth factor-I receptor (IGF-IR), is associated with increased adiposity. Breast Cancer Res Treat. 2013;139:657–65. [DOI] [PubMed] [Google Scholar]
- 18.Moodie Z, Price L, Gouttefangeas C, Mander A, Janetzki S, Lower M, et al. Response definition criteria for ELISPOT assays revisited. Cancer Immunol Immunother. 2010;59:1489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salazar LG, Lu H, Reichow JL, Childs JS, Coveler AL, Higgins DM, et al. Topical Imiquimod Plus Nab-paclitaxel for Breast Cancer Cutaneous Metastases: A Phase 2 Clinical Trial. JAMA Oncol. 2017;3:969–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–22. [DOI] [PubMed] [Google Scholar]
- 21.Hopkins AC, Yarchoan M, Durham JN, Yusko EC, Rytlewski JA, Robins HS, et al. T cell receptor repertoire features associated with survival in immunotherapy-treated pancreatic ductal adenocarcinoma. JCI Insight. 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rahma OE, Gammoh E, Simon R, Khleif SN. Is the “3+3” dose escalation phase 1 clinical trial design suitable for therapeutic cancer vaccine development? A recommendation for alternative design. Clin Cancer Res. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salazar LG, Fikes J, Southwood S, Ishioka G, Knutson KL, Gooley TA, et al. Immunization of cancer patients with HER-2/neu-derived peptides demonstrating high-affinity binding to multiple class II alleles. Clin Cancer Res. 2003;9:5559–65. [PubMed] [Google Scholar]
- 24.Watt WC, Cecil DL, Disis ML. Selection of epitopes from self-antigens for eliciting Th2 or Th1 activity in the treatment of autoimmune disease or cancer. Semin Immunopathol. 2017;39:245–53. [DOI] [PubMed] [Google Scholar]
- 25.Li T, Forbes ME, Fuller GN, Li J, Yang X, Zhang W. IGFBP2: integrative hub of developmental and oncogenic signaling network. Oncogene. 2020;39:2243–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brezar V, Godot V, Cheng L, Su L, Levy Y, Seddiki N. T-regulatory cells and vaccination “Pay attention and do not neglect them”: Lessons from HIV and cancer vaccine trials. Vaccines (Basel). 2016;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547:217–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goodell V, Waisman J, Salazar LG, de la Rosa C, Link J, Coveler AL, et al. Level of HER-2/neu protein expression in breast cancer may affect the development of endogenous HER-2/neu-specific immunity. Mol Cancer Ther. 2008;7:449–54. [DOI] [PubMed] [Google Scholar]
- 29.Schoenberger SP, Sercarz EE. Harnessing self-reactivity in cancer immunotherapy. Semin Immunol. 1996;8:303–9. [DOI] [PubMed] [Google Scholar]
- 30.Dafni U, Martin-Lluesma S, Balint K, Tsourti Z, Vervita K, Chenal J, et al. Efficacy of cancer vaccines in selected gynaecological breast and ovarian cancers: A 20-year systematic review and meta-analysis. Eur J Cancer. 2021;142:63–82. [DOI] [PubMed] [Google Scholar]
- 31.Granier C, De Guillebon E, Blanc C, Roussel H, Badoual C, Colin E, et al. Mechanisms of action and rationale for the use of checkpoint inhibitors in cancer. ESMO Open. 2017;2:e000213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brossart P The role of antigen spreading in the efficacy of immunotherapies. Clin Cancer Res. 2020;26:4442–47. [DOI] [PubMed] [Google Scholar]
- 33.Butterfield LH, Ribas A, Dissette VB, Amarnani SN, Vu HT, Oseguera D, et al. Determinant spreading associated with clinical response in dendritic cell-based immunotherapy for malignant melanoma. Clin Cancer Res. 2003;9:998–1008. [PubMed] [Google Scholar]
- 34.Bollard CM, Gottschalk S, Torrano V, Diouf O, Ku S, Hazrat Y, et al. Sustained complete responses in patients with lymphoma receiving autologous cytotoxic T lymphocytes targeting Epstein-Barr virus latent membrane proteins. J Clin Oncol. 2014;32:798–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stevens AD, Bullock TNJ. Therapeutic vaccination targeting CD40 and TLR3 controls melanoma growth through existing intratumoral CD8 T cells without new T cell infiltration. Cancer Immunol Immunother. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cecil D, Park KH, Curtis B, Corulli L, Disis MN. Type I T cells sensitize treatment refractory tumors to chemotherapy through inhibition of oncogenic signaling pathways. J Immunother Cancer. 2021;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Solinas C, Gu-Trantien C, Willard-Gallo K. The rationale behind targeting the ICOS-ICOS ligand costimulatory pathway in cancer immunotherapy. ESMO Open. 2020;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Du L, Long Y, Kim JJ, Chen B, Zhu Y, Dai N. Protease activated receptor-2 induces immune activation and visceral hypersensitivity in post-infectious irritable bowel syndrome mice. Dig Dis Sci. 2019;64:729–39. [DOI] [PubMed] [Google Scholar]
- 39.Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol. 2012;12:180–90. [DOI] [PubMed] [Google Scholar]
- 40.Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282:9358–63. [DOI] [PubMed] [Google Scholar]
- 41.Das A, Dinh PX, Panda D, Pattnaik AK. Interferon-inducible protein IFI35 negatively regulates RIG-I antiviral signaling and supports vesicular stomatitis virus replication. J Virol. 2014;88:3103–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.