

Abstract
BACKGROUND AND AIMS:
Multiple direct-acting antiviral (DAA) regimens are available to treat HCV genotype 1 infection. However, comparative effectiveness from randomized controlled trials of DAA regimens is unavailable.
APPROACH AND RESULTS:
We conducted a pragmatic randomized controlled trial (NCT02786537) to compare the effectiveness of DAAs for HCV genotype 1a or 1b on viral response, safety, tolerability, and medication nonadherence. Adults with compensated liver disease, HCV genotype 1, not pregnant or breastfeeding, and with health insurance likely to cover ledipasvir/sofosbuvir (LDV/SOF) were recruited from 34 US viral hepatitis clinics. Participants were randomized (± ribavirin) to LDV/SOF, elbasvir/grazoprevir (EBR/GZR), and paritaprevir/ritonavir/ombitasvir+dasabuvir (PrOD; treatment arm stopped early). Primary outcomes included sustained viral response at 12 weeks (SVR12), clinician-recorded adverse events, patient-reported symptoms, and medication nonadherence. Between June 2016 and March 2018, 1,609 participants were randomized. Among 1,128 participants who received ≥1 dose of EBR/GZR or LDV/SOF (± ribavirin), SVR12 was 95.2% (95% CI, 92.8%–97.6%) and 97.4% (95% CI, 95.5%–99.2%), respectively, with a difference estimate of 2.2% (−0.5% to 4.7%), falling within the “equivalence” interval (−5% to 5%). While most (56%) participants experienced adverse events, few were serious (4.2%) or severe (1.8%). In the absence of ribavirin, discontinuations due to adverse events were rare. Patient-reported symptoms and medication nonadherence were similar. Study limitations were dropout due to insurance denial and loss to follow-up after treatment, limiting the ability to measure SVR12.
CONCLUSIONS:
This pragmatic trial demonstrated high SVR12 for participants treated with EBR/GZR and LDV/SOF with few adverse effects. Overall, the two regimens were equivalent in effectiveness. The results support current HCV guidelines that do not distinguish between ribavirin-free EBR/GZR and LDV/SOF.
HCV chronically infects more than 2.4 million people in the United States and 70 million people globally, placing these individuals at risk for cirrhosis, liver failure, HCC, and death.(1) Compared to those with ongoing infection, persons who achieve HCV cure have markedly lower risk of liver disease complications, and the incidence rate of HCV-related death in the United States has declined with the uptake of curative HCV treatment.(2) Accordingly, the World Health Organization endorsed the elimination of hepatitis C as a public health threat by 2030, which will require the diagnosis and treatment of most people living with HCV and, if achieved, will reduce HCV-related mortality by 65% over the next 10 years.(3) In this context, multiple direct-acting antiviral (DAA) regimens are available for the treatment of hepatitis C including the most common strain in the United States and world, HCV genotype 1.
Recommended DAA regimens for persons who have not been previously treated combine two drugs that target different steps in the HCV life cycle to create effective antiviral regimens, including HCV nonstructural protein 5A (NS5A) inhibitors (elbasvir [EBR], ledipasvir [LDV], velpatasvir) and NS3 protease inhibitors (grazoprevir [GZR]) or NS5B polymerase inhibitors (sofosbuvir [SOF]).(4) Although recommended as first-line therapy for HCV genotype 1 infection, these combinations may have different safety, tolerability, and efficacy in some patient subpopulations. Despite the widespread use of these and other DAA regimens, evidence of comparative effectiveness from randomized controlled trials is unavailable.
We conducted a comparative effectiveness study, designed as a randomized, pragmatic clinical trial (The PRIORITZE Study, NCT02786537) of the effectiveness of three DAA regimens for treatment of HCV genotype 1a or 1b on (1) sustained viral response at 12 weeks posttreatment (SVR12), (2) clinician-recorded adverse events (AEs), (3) patient-reported symptoms and functional well-being, and (4) patient-reported medication nonadherence.
Methods
STUDY DESIGN
PRIORITIZE was a multicenter, randomized, pragmatic clinical trial comparing three DAA regimens: LDV/SOF (Harvoni; Gilead Sciences, Foster City, CA); EBR/GZR (Zepatier; Merck and Co, Whitehouse Station, NJ); paritaprevir/ritonavir/ombitasvir + dasabuvir (PrOD; Viekira Pak/Viekira XR; AbbVie Pharmaceuticals, Abbott Park, IL) (phase 1 only). Consistent with prescribing information and guidelines, ribavirin (RBV) could be added to any regimen at the discretion of the treating clinician. Treatment assignment was open label, and efficacy outcomes were unblinded.
We planned to randomize 3,750 participants in a 1:1:1 ratio. In January 2017, in anticipation of a US guideline(4) recommendation that PrOD + RBV was to become nonpreferred for genotype 1a infection, randomization to PrOD was discontinued (defining the end of phase 1). In phase 2, enrollees were randomized 1:1 to LDV/SOF or EBR/GZR.
At the start of phase 2, a blinded update of the sample size estimation accounted for the loss of one arm and the low prevalence (~15%) of cirrhosis in phase 1. The revised enrollment target was 1,600 enrolled participants. In both phases, randomization was stratified by cirrhosis status and genotype 1 subtype (a or b). This report is limited to outcomes from LDV/SOF and EBR/GZR in phases 1 and 2. Because the regimen was discontinued in the United States and no longer recommended in other regions, comparisons to PrOD in phase 1 are available in the Supporting Information.
STUDY COORDINATION
The Data Coordinating Center used a Research Data-Capture (REDCap) system to conceal the randomized allocation of treatment assignments, to validate patient eligibility, and to capture and manage the study data. The Clinical Coordinating Center used the operational infrastructure of the HCV-TARGET Research Network(5) for standardized, centralized chart data abstraction and targeted data monitoring. All survey instruments were administered by phone interview or by a secure, web-based link into the REDCap system. Participants received $40 remuneration for completing each survey session.
RECRUITMENT
Adult participants (>18 years) with chronic HCV genotype 1a or 1b infection who presented for initial antiviral treatment were invited to participate if, in their clinician’s opinion, therapy with any of the study regimens was appropriate. The 34 study sites were selected from those participating in the HCV-TARGET network. Site clinicians were generally experienced with the management of patients with chronic HCV infection based on their involvement in HCV-TARGET (Supporting Table S1). Individuals were excluded for inability to provide written informed consent, current or historical evidence of hepatic decompensation (variceal bleeding, HE, or ascites) unless this was prior to successful liver transplant, Child-Turcotte-Pugh stage B or C cirrhosis, pregnancy or breastfeeding, or health insurance that did not permit LDV/SOF which was not provided by the study. Individuals with HIV coinfection, organ transplantation, substance use disorder (past or current), and other medical or psychiatric conditions were eligible.
DRUG REGIMENS
EBR/GZR was provided with no prior authorization or cost to participants through a centralized pharmacy; LDV/SOF was provided by prescription and, when applicable, subject to insurance authorization procedures performed by clinical site and the Kroger Specialty Pharmacy. Regardless of the source of medication, participants were treated according to standard practice which, while not protocol-mandated, generally followed US prescribing information and HCV guidelines.(4) All participants were offered free, real-time testing for the presence of NS5A resistance-associated substitutions (RASs) at amino acid positions 28, 30, 31, and 93 (Laboratory Corporation of America, Burlington, NC). LDV/SOF was administered as one tablet (90/400 mg) daily for 8 or 12 weeks at clinician discretion. EBR/GZR was administered as one tablet (50/100 mg) daily for 12 weeks; participants infected with NS5A-resistant genotype 1a received 16 weeks of EBR/GZR plus twice-daily RBV dosed according to body weight.
OUTCOME MEASURES
Effectiveness
The primary outcome was undetectable HCV RNA at 12 weeks after the completion of treatment, a binary indicator of SVR12. Values were missing for participants who did not return for HCV RNA testing and for randomized participants who did not start treatment.
Safety and Tolerability
AEs were defined as any new symptom or event recorded in the medical record regardless of whether it was related to HCV therapy. AEs were further coded according to the Medical Dictionary for Regulatory Activities. Serious AEs were defined as any AE causing death, requiring hospitalization, or meeting criteria for expedited reporting per the US Food and Drug Administration. Nonserious AE severity was coded as mild (requiring no concomitant or only over-the-counter therapies), moderate (requiring prescription therapy or HCV treatment dose adjustment), or severe (requiring HCV therapy discontinuation or blood transfusion). On-treatment AE causal relationships were coded as “related” to HCV therapy based on the contemporaneousness of the event to drug administration unless the event was clearly noted in the submitted medical records as “not related” to the HCV treatment regimen.
Patient-Reported Outcome Survey
We collected patient-reported outcomes (PROs) to evaluate functional well-being and symptoms that have been frequent in phase 3 clinical trials of DAAs, specifically headache, fatigue, and nausea. These were assessed on three occasions: pretreatment, early on-treatment, and late on-treatment. Headache was evaluated by the Headache Impact Test (HIT-6).(6,7) Fatigue was evaluated using the Patient-Reported Outcomes Measurement Information System (PROMIS) Fatigue short form 8a,(8,9) and nausea was assessed with the PROMIS Nausea/Vomiting four-item short form.(10) Raw scores were transformed to standardized PROMIS T scores. Functional well-being was assessed on the same occasions using the HCV-PRO instrument.(11) Based on stakeholder input from the PRIORITZE Patient Engagement Group during the study design phase, we did not ask information about active substance use.
Medication Adherence
We used the Voils Medication Adherence Survey (VMAS),(12,13) which asked the participant three questions about the past 7 days of treatment (early and late on-treatment occasions). Participants responded using a 5-point ordinal scale of missed dosing, from 1 (none of the time) to 5 (all the time). On each occasion participants were coded as being nonadherent if any response was >1.
STATISTICAL ANALYSES
Our analyses and reporting are consistent with guidance from the CONSORT statement,(14) the American Statistical Association,(15–18) and the International Committee of Medical Journal Editors.(19) The main model-based analysis results are presented in this report as point estimates and CI estimates of population parameters. Intent-to-treat estimation was modified (mITT) due to missing values for enrollees who did not start treatment or did not return for SVR12 evaluations. To cope with missing data, we followed the approach of White et al.: (1) attempt to avoid missing data, (2) use nonmissing data to perform a main analysis specified a priori that is valid under plausible assumptions about causes of missing data, (3) use sensitivity analyses to guide trust in the main results by evaluating their robustness/fragility to reasonable perturbations of assumptions and methods used, and (4) account for all randomized enrollees in at least one sensitivity analysis.(20) For the main analyses we anticipated and assumed mechanisms that caused missing data are ignorable. In our sensitivity analyses, baseline characteristics of all randomized enrollees played an important role in our investigation of potential selection biases; e.g., we used inverse-probability weighting, multiple imputation of outcomes, as-treated analyses, and variations on model assumptions. In the main analyses, we modeled the assigned DAA regimen, cirrhosis status, genotype 1 subtype, and specified covariates (sex, age group, treatment-naive status, race) along with terms representing interactions of regimen with cirrhosis, subgenotype, and race.
SVR12 effectiveness
Due to the small number of participants not achieving SVR12, the main logistic regression model for effectiveness represented the probability of achieving SVR12 as a function of just three variables: assigned DAA regimen, cirrhosis status, and subtype. Fitted using Firth’s penalized likelihood method,(21) this model provided point and interval estimates of the regimen-specific proportions (P1, P2) and their difference (P1 – P2) in the target population. We performed a superiority test of the null hypothesis “(P1 – P2) = 0 in the target population” and an equivalence test of the null hypothesis “|P1 – P2| ≥ 5% in the target population.” It is plausible that both null hypotheses are false (i.e., 0% < |P1 – P2| < 5%). For hypothesis generation regarding heterogeneity of treatment effects (HTE), (1) covariates were added to the model one at a time to avoid overfitting bias, (2) an overfitted logistic model with least absolute shrinkage and selection operator penalized likelihood estimation was explored, and (3) unadjusted SVR12 frequencies for subgroups were examined.
Safety and Tolerability
Clinically recorded AEs, laboratory abnormalities, and reasons for discontinuation were tabulated for each regimen “as-treated.”
PROs
To characterize and compare the regimens, we used a linear mixed-effects model to estimate means and mean changes from baseline to on-treatment for each of four PRO measures: HIT-6 score, PROMIS fatigue T score, PROMIS nausea T score, and HCV-PRO. In these constrained longitudinal data analysis models,(22) the baseline score was treated as one of the longitudinal outcomes, and mean response was represented as a function of the as-randomized regimen and the a priori covariates and interactions described. Therefore, an on-treatment mean increase from baseline would be evidence of participants becoming more symptomatic during treatment. The models were also used to explore subgroup differences and HTE. Sensitivity analyses included analysis of residuals and comparison to analyses that used inverse-probability-of-missing weighting or did not assume treatment assignment had no effect on scores at baseline.
Medication Nonadherence
We used a generalized logistic regression model for repeated binary measures, which represented the probability of patient-reported nonadherence as a function of regimen, cirrhosis status, genotype, and VMAS survey occasion (early, late) during treatment. As-treated and as-randomized (mITT) treatment effects were estimated separately. Point and interval estimates of population proportions (P1, P2) and difference (P1 – P2) were obtained, and a superiority test was conducted.
Nonadherence and SVR12
For hypothesis generation about potential association between the rare cases of non-SVR12 and the rare cases of medication nonadherence, we examined frequencies and obtained point and interval estimates of the Pearson correlation coefficient.
INFORMED CONSENT
This study complied with the US Department of Health and Human Services Federal Policy for the Protection of Human Research Subjects. It relied on the National Institutes of Health interpretation of the Common Rule. The study observed site institutional review board and federal requirements for protection of human subjects and their health information. All participants provided written informed consent before screening for enrollment.
ROLE OF THE FUNDING SOURCES
The study was funded by the Patient-Centered Outcomes Research Institute. Merck Sharp & Dohme Corp. provided EBR/GZR and funds for HCV NS5A RAS testing. Kroger Specialty Pharmacy provided centralized pharmacy services. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Results
Between June 2016 and March 2018, 1,609 participants were enrolled at 34 US centers. Of these randomized participants, 1,128 received at least one dose of EBR/GZR (n = 700) or LDV/SOF (n = 428) (Fig. 1) and were followed longitudinally. Failure to initiate therapy was more frequent among participants randomized to LDV/SOF (41%, 298 of 726 participants) than EBR/GZR (4%, 29 of 729 participants). Of the 726 enrollees randomized to LDV/SOF, 150 (21%) experienced insurance denial of treatment, and another 168 (23%) did not start for diverse reasons, including the burden of the prior authorization process; 408 were treated with LDV/SOF, and 20 (with insurance denials) were treated with EBR/GZR. Insurance denials of LDV/SOF were primarily by state Medicaid programs (75% of the 150 denials).(23) The 1,128 treated participants were more commonly men (60%) than women (40%), the mean age was 55 years, approximately 42% were Black, 17% had cirrhosis, and 3% had HIV coinfection. NS5A RASs at key amino acid positions (28, 30, 31, or 93) were detected in 9.9% and 12.6% of participants receiving EBR/GZR and LDV/SOF, respectively. RBV was coadministered more frequently with EBR/GZR (56 participants, 8%) compared to LDV/SOF (15 participants, 3.5%) (Table 1). Of the 1,128 participants who began treatment, 91% completed therapy and 85% returned for SVR12 evaluation with similar proportions for each DAA regimen. The proportion of participants lost to follow-up was ~16% for both regimens (LDV/SOF, n = 69, 16.1%; EBR/GZR, n = 114, 16.3%). Discontinuation due to AEs (n = 22) or lack of efficacy (n = 1) was rare.
FIG. 1.
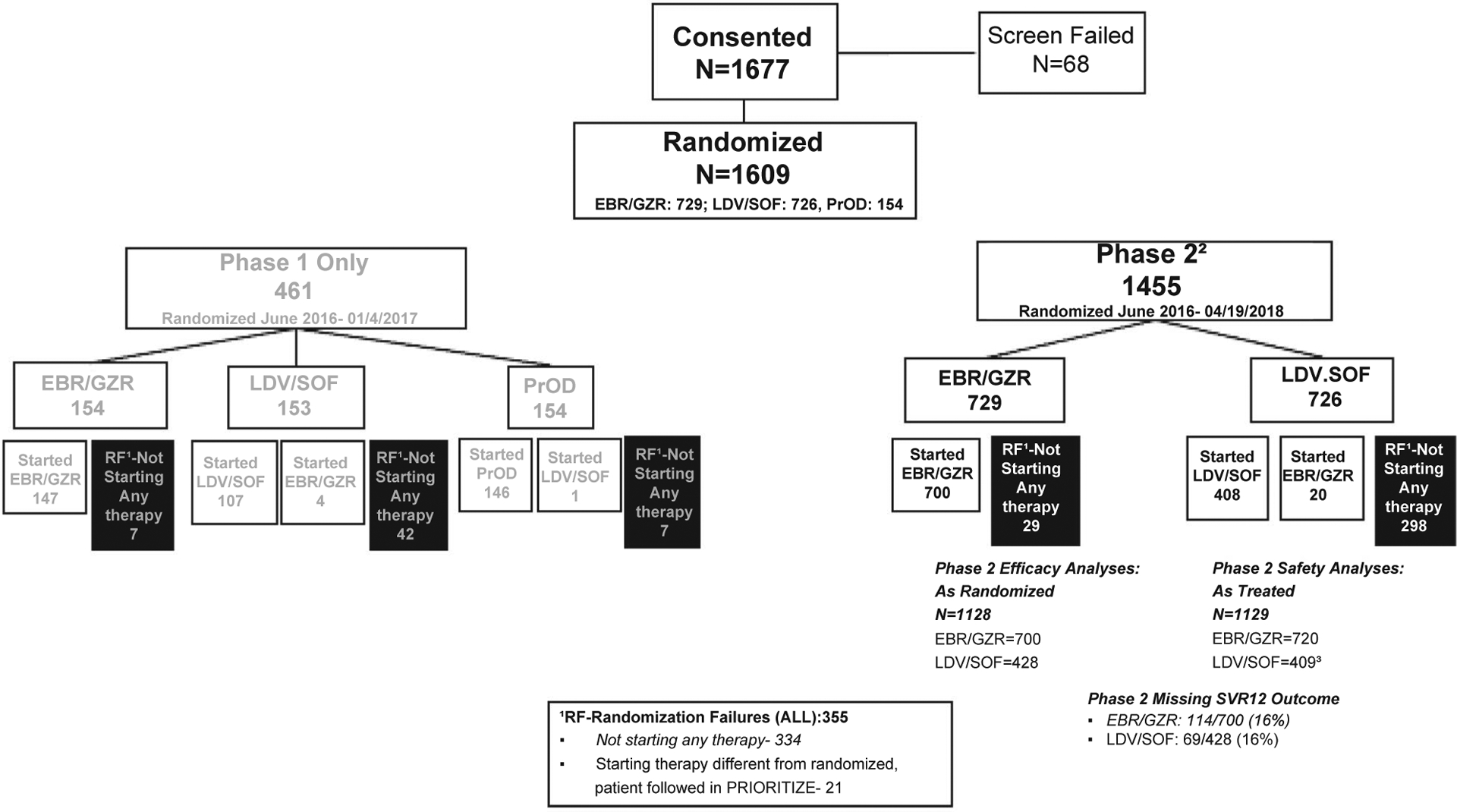
Consort diagram–PRIORITIZE study flowchart. 1Randomization Failures. 2Includes ALL EBR.GZR & LDV/SOF Patients from Phase 1. 3Includes 1 PrOD patient treated with LDV/SOF. Abbreviations: EBR/GZR, elbasvir/grazoprevir; LDV/SOF, ledipasvir/sofosbuvir; PrOD, paritaprevir/ritonavir/ombitasvir+dasabuvir.
TABLE 1.
Baseline characteristics of treated participants as randomized to EBR/GZR and LDV/SOF
| EBR/GZR | LDV/SOF | |
|---|---|---|
| Participants who started treatment, n | 700 | 428 |
| Age, years mean (range) | 53.4 (18.0–83.0) | 56.4 (21.0–82.0) |
| Sex, n (%) | ||
| Female | 293 (41.9) | 163 (38.1) |
| Male | 407 (58.1) | 265 (61.9) |
| Race, n (%) | ||
| White | 349 (49.9) | 225 (52.6) |
| Black | 295 (42.1) | 182 (42.5) |
| Other | 56 (8.0) | 21 (4.9) |
| HCV genotype 1 subtype, n (%) | ||
| 1a | 540 (77.1) | 317 (74.1) |
| 1b | 160 (22.9) | 111 (25.9) |
| Cirrhosis, n (%) | ||
| Yes | 112 (16.0) | 80 (18.7) |
| No | 588 (84.0) | 348 (81.3) |
| NS5A RAS, n (%) | ||
| RAS at any 28/30/31/93 | 69 (9.9) | 54 (12.6) |
| RAS at 28 only | 28 (4.0) | 14 (3.3) |
| RAS at 30 only | 4 (0.6) | 2 (0.5) |
| RAS at 31 only | 8 (1.1) | 14 (3.3) |
| RAS at 93 only | 21 (3.0) | 14 (3.3) |
| RBV administration, n (%) | ||
| Yes | 56 (8.0) | 15 (3.5) |
| No | 644 (92.0) | 413 (96.5) |
| HIV coinfection, n (%) | ||
| Yes | 22 (3.1) | 13 (3.0) |
| HCC history, n (%) | ||
| Yes | 4 (0.6) | 2 (0.5) |
| Type of health insurance, n (%) | ||
| Medicaid | 331 (47.3) | 113 (26.4) |
| Medicare | 98 (14.0) | 90 (21.0) |
| Commercial | 231 (33.0) | 186 (43.5) |
| Other | 40 (5.7) | 39 (9.1) |
| Platelets (1,000/mL), mean (range) | 222 (39.0–645.0) | 216 (67.0–343.0) |
| Duration of therapy, n (%) | ||
| 8 weeks (42–69 days) | 28 (4) | 110 (26) |
| 12 weeks (70–97 days) | 562 (80) | 275 (64) |
| 16 weeks (98–125 days) | 78 (11) | 9 (2.1) |
| Other Durations | 32 (4.5) | 34 (7.9) |
| Alcohol/tobacco use, n (%) | ||
| Current | 245 (35.0)/348 (49.7) | 159 (37.1)/168 (39.3) |
| Former | 166 (23.7)/193 (27.6) | 99 (23.1)/150 (35.0) |
| Never | 265 (37.9)/134 (19.1) | 157 (36.7)/94 (22.0) |
| Unknown | 24 (3.4)/26 (3.6) | 13 (3.0)/16 (3.7) |
Of the 428 randomized to LDV/SOF, 20 experienced insurance denial and were then treated with EBR/GZR instead.
SVR12 EFFECTIVENESS
Primary Results
In the mITT analysis, the adjusted proportions (adjusted for cirrhosis and genotype) who achieved SVR12 for EBR/GZR and LDV/SOF were 95.2% (92.8%–97.6%) and 97.4% (95.5%–99.2%), respectively. The difference of 2.2% (−0.5% to 4.7%) was within the prespecified equivalence range (±5%). The superiority test was inconclusive (P = 0.0930). Among the 945 participants with known SVR12 outcomes, only 40 (4.2%) did not achieve SVR12 including 5.1% (30 of 586 participants; nonresponse, 11; relapse, 18; breakthrough, 1) and 2.7% (10 of 359 participants; nonresponse, 10; relapse, 7) of those randomized to EBR/GZR and LDV/SOF, respectively (Supporting Table S2).
Exploratory Results
Exploratory analyses of HTE based on unadjusted SVR12 proportions (Table 2) suggested the following. (1) SVR12 proportions were similar for Black and non-Black participants treated with EBR/GVR and LDV/SOF. (2) Participants with NS5A RASs treated with either DAA regimen may have lower SVR12 proportions compared to those without NS5A RASs; the difference was 9.3% (95% CI, −0.3% to 18.2%) for LDV/SOF and 8.8% (95% CI, −0.3% to 7.9%) for EBR/GZR. The SVR12 rates for patients with specific NS5A RASs at positions 28, 30, 31, and 91 are presented by DAA regimen in Supporting Table S3a (patients with HCV genotype 1a infection) and Table S3b (patients with HCV genotype 1b infection). (3) Among women, the SVR12 proportions between DAA regimens differed by 5.1% (95% CI, 2.4%–7.7%) in favor of LDV/SOF. (4) Among participants with genotype 1a infection, the SVR12 proportions between DAA regimens differed by 3.5% (95% CI, 0.1%–6.4%) favoring LDV/SOF. (5) The difference in SVR between regimens among participants without cirrhosis was 3.4% (95% CI, 0.5%–6.1%) favoring LDV/SOF, whereas among those with cirrhosis the difference was −2.8% (95% CI, −11.5% to 4.0%) favoring EBR/GZR 3.
TABLE 2.
Exploration of subgroup differences based on unadjusted SVR12 frequencies
| Subpopulation | Counts | EBR/GZR* | Counts | LDV/SOF* | EBR/GZR vs. LDV/SOF |
|---|---|---|---|---|---|
| Percentage (CI) | Percentage (CI) | Difference (CI) | |||
| Overall | 556/586 | 94.9 (92.8–96.5)† | 349/359 | 97.2 (94.9–98.7) | −2.3 (−4.8 to 0.4) |
| With RBV | 40/46 | 87.0 (73.7–95.1) | 14/15 | 93.3 (68.1–99.8) | −6.4 (−20.1 to 17.8) |
| Without RBV | 516/540 | 95.6 (93.5–97.1) | 335/344 | 97.4 (95.1–98.8) | −1.8 (−4.2 to 0.9) |
| Black | 253/266 | 95.1 (91.8–97.4) | 153/159 | 96.2 (92.0–98.6) | −1.1 (−5.0 to 3.6) |
| Non-Black | 303/320 | 94.7 (91.6–96.9) | 196/200 | 98.0 (95.0–99.5) | −3.3 (−6.6 to 0.3) |
| Prior treatment | 56/62 | 90.3 (80.1–96.4) | 44/45 | 97.8 (88.2–99.9) | −7.5 (−17.5 to 3.2) |
| No prior treatment | 500/524 | 95.4 (93.3–97.0) | 305/314 | 97.1 (94.6–98.7) | −1.7 (−4.2 to 1.2) |
| Male | 312/329 | 94.8 (91.9–96.7) | 212/222 | 95.5 (91.9–97.5) | −0.7 (−4.3 to 3.4) |
| Female | 244/257 | 94.9 (91.5–97.0) | 137/137 | 100 (97.3–100) | −5.1 (−7.7 to −2.4) |
| Genotype 1a | 419/445 | 94.2 (91.6–96.0) | 254/260 | 97.7 (95.1–98.9) | −3.5 (−6.4 to −0.1) |
| Genotype 1b | 137/141 | 97.2 (92.9–98.9) | 95/99 | 96.0 (90.1–98.4) | 1.2 (−3.7 to 7.3) |
| Cirrhosis | 92/95 | 96.8 (91.0–99.3) | 63/67 | 94.0 (85.4–98.3) | 2.8 (−4.0 to 11.5) |
| No cirrhosis | 464/491 | 94.5 (92.1–96.3) | 286/292 | 97.9 (95.6–99.2) | −3.4 (−6.1 to −0.5) |
| NS5a RAS | 47/54 | 87.0 (75.1–94.6) | 42/47 | 89.4 (76.9–96.5) | −2.3 (−15.3 to 11.3) |
| No NS5a RAS | 485/506 | 95.8 (93.7–97.4) | 286/290 | 98.6 (96.5–99.6) | −2.8 (−5.0 to −0.2) |
As assigned by randomization.
95% CIs were computed using the Wilson score method.
SAFETY AND TOLERABILITY
The percentage of participants treated with EBR/GZR or LDV/SOF with any AE was 56% (628 of 1,129 participants). Among those patients experiencing any AE, most had AEs coded as related to HCV treatment (601 of 628 patients with AEs). Participants taking DAAs plus RBV reported more AEs than those not taking DAAs alone. The most common AEs recorded in the medical record were fatigue (19%), headache (16%), and nausea (9.1%). Anemia (27%), dizziness (10%), and insomnia (10%) were observed almost exclusively in participants prescribed RBV. Severe AEs were observed in 1.8% (n = 23) of 1,129 participants. Serious AEs occurred in 4.2% (42 participants with 53 events), of which two were considered treatment-related, chest pain and flare of HBV infection.
Liver-related AEs were rare, occurring in 7 patients (0.6%) with eight events (0.4%) among 1,129 participants. Hepatic decompensation was not observed in the 102 participants with cirrhosis who received the HCV protease inhibitor GZR (part of the EBR/GZR regimen); however, one patient without cirrhosis treated with EBR/GZR had HBV reactivation with evidence of trace ascites. Early discontinuation of treatment due to an AE occurred in 22 participants, and the incidence was similar in participants treated with LDV/SOF (7 of 409, 1.7%) and EBR/GZR (15/720, 2.1%). Six participants treated with EBR/GZR or LDV/SOV died during the study, with five deaths during treatment; none were treatment-related (Table 3).
TABLE 3.
All AEs With Prevalence Exceeding 10% By Treatment Regimen*
| EBR/GZR | LDV/SOF | Overall | |||||||
|---|---|---|---|---|---|---|---|---|---|
| RBV | No RBV | All | RBV | No RBV | All | RBV | No RBV | All | |
| (56) | (664) | (720)† | (15) | (394) | (409)‡ | (71) | (1,058) | (1,129) | |
| No. patients—any AE | 46 (82%) | 361 (54%) | 407 (57%) | 8 (53%) | 213 (54%) | 221 (54%) | 54 (76%) | 574 (54%) | 628 (56%) |
| Fatigue | 18 (32%) | 105 (16%) | 123 (17%) | 5 (33%) | 82 (21%) | 87 (21%) | 23 (32%) | 187 (18%) | 210 (19%) |
| Headache | 12 (21%) | 94 (14%) | 106 (15%) | 1 (6.7%) | 68 (17%) | 69 (17%) | 13 (18%) | 162 (15%) | 175 (16%) |
| Nausea | 12 (21%) | 54 (8.1%) | 66 (9.2%) | 3 (20%) | 34 (8.6%) | 37 (9.0%) | 15 (21%) | 88 (8.3%) | 103 (9.1%) |
| Insomnia | 6 (11%) | 26 (3.9%) | 32 (4.4%) | 1 (6.7%) | 10 (2.5%) | 11 (2.7%) | 7 (9.9%) | 36 (3.4%) | 43 (3.8%) |
| Dizziness | 7 (13%) | 15 (2.3%) | 22 (3.1%) | 0 (0%) | 11 (2.8%) | 11 (2.7%) | 7 (9.9%) | 26 (2.5%) | 33 (2.9%) |
| Dyspnea | 8 (14%) | 9 (1.4%) | 17 (2.4%) | 0 (0%) | 11 (2.8%) | 11 (2.7%) | 8 (11%) | 20 (1.9%) | 28 (2.5%) |
| Anemia | 17 (30%) | 2 (0.3%) | 19 (2.6%) | 2 (13%) | 0 (0%) | 2 (0.5%) | 19 (27%) | 2 (0.2%) | 21 (1.9%) |
| Diarrhea | 3 (5.4%) | 45 (6.8%) | 48 (6.7%) | 2 (13%) | 19 (4.8%) | 21 (5.1%) | 5 (7.0%) | 64 (6.0%) | 69 (6.1%) |
Treatment-emergent AEs from treatment start to ≤31 days post–end of treatment.
As-treated population.
Includes 20 patients randomized to LDV/SOF and treated with EBR/GZR.
Includes 1 patient randomized to PrOD and treated with LDV/SOF.
PROs
In terms of mean changes in PROs from baseline, symptoms did not worsen during treatment with either regimen, and the mean scores for measures of symptoms (headache, nausea, and fatigue) and overall functioning and well-being improved during treatment relative to baseline (Fig. 2). For functional well-being, the difference favored LDV/SOF: −4.3 (−8.4 to −0.3), P = 0.0354.
FIG. 2.
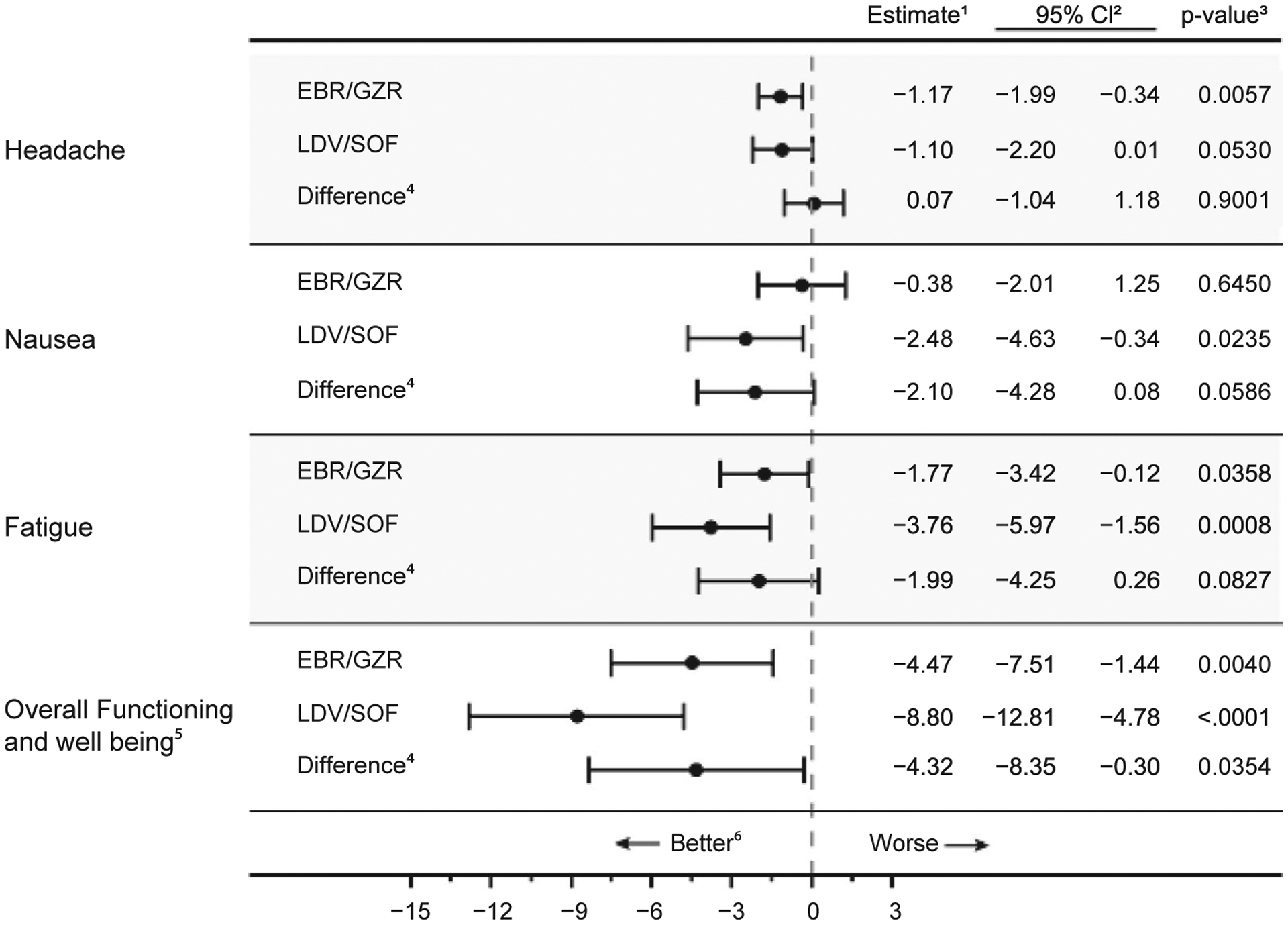
Mean change in PRO scores from baseline to on-treatment. ¹The estimates of mean change and difference were obtained from a constrained longitudinal linear mixed-effects model that treated the baseline score as one of the outcomes. The model expressed mean score as a function of DAA regimen, cirrhosis status, HCV genotype, sex, age, race, and previous treatment status. 295% confidence interval estimate. ³p-value for a test of the null hypothesis “the parameter is zero in the target population”. 4Difference of the mean change for LDV/SOF minus the mean change for EBR/GZR. 5The scale for function and well-being is reversed (= 100-HCV-PRO) for directional consistency with symptom scores. 6The scare for “Headache” is the HIT-6 score. The scare for “Nausea” is the PROMIS® Nausea Short Form T-score. The scale for “Fatigue” is the PROMIS® Fatigue Short Form T-score. Negative values for mean change represent improvement, while negative values for “Difference” indicate the LDV/SOF performed better than EBR/GZR.
MEDICATION NONADHERENCE
Treatment persistence was high for both regimens; 90% of EBR/GZR-treated participants and 92% of LDV/SOF-treated participants completed the planned duration of therapy. The difference of 2% (−1.5% to 5.4%) did not favor either regimen. The regimens were similar in the as-treated estimates of nonadherence: 16% (1%–21%) for LDV/SOF, 20% (12%–23%) for EBR/GZR (P = 0.20). In models controlling for treatment duration and other factors, there was no evidence of an effect of treatment duration on the probability of adherence. These comparisons indicate little or no difference in adherence or persistence for patients prescribed LDV/SOF or EBR/GZR. The proportion of participants who achieved SVR12 was independent of adherence (Supporting Information).
SENSITIVITY ANALYSES
We found a negligible impact of patient dropout (failure to start treatment or failure to return after treatment) based on estimates obtained using inverse probability of missing weighting and estimates using multiple imputation of SVR12 values. The results of other sensitivity analyses of PROs and nonadherence closely approximated the main results.
Discussion
This pragmatic trial is a comparative effectiveness study for oral DAAs for the treatment of chronic HCV infection. We treated 1,128 participants with genotype 1 infection at 34 clinical sites in the United States and randomly assigned them to one of two recommended antiviral regimens. Consistent with observations from efficacy trials and clinical cohorts, SVR12 proportions were high for both DAA regimens (≥95%), and HCV virologic failure was uncommon (<1%). In the absence of ribavirin, the DAA regimens had similar side-effect profiles based on the medical records, and treated participants reported improved symptoms and functional well-being during and after therapy. These findings have important implications for the treatment of HCV infection, globally and by nonspecialists, because the safety and tolerability of RBV-free regimens allow for minimal monitoring during therapy.
The randomized design of our study allowed for direct comparison of the effectiveness and side-effect profile of these two recommended DAA regimens, EBR/GZR and LDV/SOF. The overall SVR12 proportions were similar, with strong evidence that the two regimens are equivalent in the target population. Compared to the registration trial enrollment for these regimens (EBR/GZR, 18%; LDV/SOF, 12%–15%), we enrolled a significantly larger proportion of Black Americans. Approximately 42% of the PRIORITIZE study populations was Black, allowing for greater precision around the estimated SVR12 rate, which was similar for both DAA regimens and the SVR observed in non-Black participants. The high participation rate in the study by Black Americans, a group underrepresented in HCV trials, also provides a roadmap for continued engagement to reduce HCV-related disparities and health inequities, essential for the elimination of hepatitis C as a public health threat.(24,25)
We also observed small differences in the unadjusted SVR12 proportions in important patient subgroups in exploratory analyses conducted for hypothesis generation. For both EBR/GZR and LDV/SOF, the SVR12 rate was lower in participants with NS5A RASs compared to those with wild-type HCV. Because only 1 in 10 patients had evidence of baseline NS5A resistance and the impact of SVR was similar for both DAA regimens, we do not believe that our data support routine RAS testing prior to therapy. We also observed small differences in the unadjusted SVR12 proportions favoring LDV/SOF in women, in those with genotype 1a infection, and in those without cirrhosis. Conversely, these small differences in the unadjusted SVR12 proportions favored EBR/GZR in participants with cirrhosis. These exploratory observations are consistent with reports from uncontrolled trials of each regimen and may serve to guide the selection of HCV treatment regimen by patients and clinicians.(26–29)
Overall, 56% of participants experienced some AEs during treatment; but few were severe (1.8%) or serious (3.7%), and few participants (<2%) discontinued RBV-free treatment due to an AE. As expected, participants who received RBV experienced more AEs unique to RBV use. Clinical and laboratory abnormalities were uncommon with RBV-free DAAs with few treatment-related serious AEs including liver-related events in persons taking HCV protease inhibitors, which has been associated with liver dysfunction and death in participants with moderate to severe liver impairment.(30) In our study population, which did not include persons with decompensated cirrhosis, liver-related AEs were rare. We also measured PROs with surveys to evaluate changes in headache, nausea, and fatigue from baseline, demonstrating that, on average, these symptoms did not worsen during either treatment regimen, consistent with previous studies of PROs.(31) Similarly, functional well-being improved on average during treatment with both regimens and especially in participants receiving LDV/SOF.
Our study findings should be interpreted in the context of several limitations. (1) Due to the economic barriers in accessing HCV treatment in the United States at the time of this study, we provided participants with EBR/GZR sourced directly from a commercial manufacturer, whereas LDV/SOF was sourced externally through commercial or other health insurance. As a result of health insurance denial and other barriers to treatment access (e.g., prior authorization), failure to start treatment was more common in participants randomized to LDV/SOF, resulting in some imbalances in patient characteristics between the treatment arms. Despite this, our sensitivity analyses indicated that these occurrences of pretreatment dropout (as well as posttreatment dropouts) did not induce selection biases. Further, the observation that curative treatment was denied to 41% of patients using health insurance underscores the impact of systemic barriers to HCV elimination. (2) The HCV treatment landscape changed rapidly during our study, leading to discontinuation of one arm (PrOD) and modification of our research plans. (3) Several factors (e.g., declining prevalence of cirrhosis and loss to follow-up during and after treatment) limited the number of non-SVR12 cases to a level (n = 40) that did not support our intended investigation of the HTE across subgroups. To avoid overfitting bias, the logistic model for SVR12 analysis was limited to accounting for only three factors—regimen, cirrhosis, and genotype 1 subtype— and inclusion of subgroup-by-treatment interaction terms was not feasible. (4) Based on feedback from patient stakeholders during the study design phase, we did not collect data related to active substance use, which precludes analysis. (5) We were not able to evaluate the two newer, pangenotypic regimens, glecaprevir/pibrentasvir and SOF/velpatasvir, as they were not available at the time of the study.
The PRIORITIZE study is a large randomized controlled trial to compare oral DAA regimens for the treatment of chronic HCV infection on effectiveness, safety, side effects, and medication adherence in a usual clinical-care setting. Our findings are consistent with observations from controlled phase 3 efficacy trials and uncontrolled cohort studies, demonstrating high SVR12 proportions with few virologic failures and, in the absence of RBV, minimal adverse effects. Our data support HCV guidelines that do not distinguish between RBV-free EBR/GZR or LDV/SOF for the treatment of persons with compensated liver disease due to HCV genotype 1 infection. While this trial had a large, representative population, there was a low proportion of patients with cirrhosis, and there were no patients with decompensated cirrhosis. The findings may not generalize to the larger HCV population including decompensated cirrhosis, younger people injecting drugs, veterans, and those incarcerated.
Future randomized controlled trials should focus on the comparative effectiveness of the recommended pangenotypic DAA regimens SOF/velpatasvir and glecaprevir/pibrentasvir.
Supplementary Material
Acknowledgment:
We extend our sincere gratitude and appreciation to a large community of stakeholders and friends who brought a vision of a pragmatic, comparative effectiveness trial to reality. First and foremost, we are indebted to the HCV Patient Engagement Group members Finton Brown, Lourdes Chaney, Larry Houston, and Kim Thomas. Similarly, we acknowledge the HCV community of nonprofit patient organizations, the National AIDS Treatment Advocacy Project, the National Viral Hepatitis Roundtable, the Hepatitis Education Project, Project Inform, the Hepatitis C Association, the Treatment Action Group, HCV Advocate, and the Caring Ambassadors Program, Inc., who added critical perspective on the HCV treatment landscape and areas of impact. We also specifically thank the two pharmaceutical companies (AbbVie and Merck) that provided millions of dollars in free drug for the study, as well as an open honesty to have their products evaluated in a transparent, comparative process. We also appreciate the partnership of Kroger Specialty Pharmacy which supported the pragmatic trial design by providing real-world pharmacy services from which free drug could be dispensed to study patients. This study could not have been possible without the hundreds of staff and investigators from the University of Florida, the University of North Carolina, and the member site institutions of the HCV-TARGET Network (see Supporting Table S1 for a listing of participating sites and principal investigators). In particular, we acknowledge Lauren Morelli, Dona-Marie Mintz, Damaris Andino, Briana Foerman, Troy Chasteen, Lasheaka McClellan, Ken Bergquist, and Ashley Magee. Lastly and most importantly, we thank the thousands of patients who participated in this trial and provided critical knowledge for the next generation of patients with HCV.
Potential conflict of interest:
Dr. Di Bisceglie consults for Gilead, AbbVie, and UpToDate. Dr. Muir received grants from AbbVie, Gilead, and Merck. Dr. Lok advises and received grants from Gilead and TARGET. She received grants from Bristol-Myers Squibb. Dr. Pearlman consults, is on the speakers’ bureau for, and received grants from Gilead, AbbVie, and Merck. Dr. Nelson received grants from Gilead, AbbVie, and Merck. He owns stock in Target. Dr. Fishbein advises and received grants from Gilead. She owns stock in AbbVie and Merck. Dr. Evon received grants from Gilead and Merck. Dr. Hinestrosa advises and is on the speakers’ bureau for Viiv and Gilead. He is on the speakers’ bureau for and received grants from Merck. He is on the speakers’ bureau for AbbVie and Theratechnologies. Dr. Segal consults for Gilead, McKesson, and Provention Bio. She received grants from Bristol-Myers Squibb. Dr. Sherman advises and received grants from Gilead. He received grants from AbbVie and Merck. Dr. Khalili consults and received grants from Gilead. She received grants from Intercept. Dr. Sulkowski consults for, advises, and received grants from Gilead and AbbVie. Dr. Fried is employed by and owns stock in Target. Dr. Shiffman consults for, advises, is on the speakers’ bureau for, and received grants from Gilead. He consults for, advises, and is on the speakers’ bureau for AbbVie. Dr. Horne advises AbbVie, Gilead, and Merck. Dr. Reddy consults for and received grants from AbbVie, Gilead, Merck, Bristol-Myers Squibb, Grifols, and Mallinckrodt. He consults for Spark, Shionogi, and Dova. He received grants from Conatus, Exact Sciences, and Target.
Supported by a Patient-Centered Outcomes Research Institute (PCORI) award (HPC-1503-27891) and by the National Institutes of Health (K24AA022523, to M.K.; K24DA034621, to M.S.S.). Free drug was provided by AbbVie Pharmaceuticals (Abbott Park, IL) and by Merck and Co (Whitehouse Station, NJ). Kroger Specialty Pharmacy provided in-kind pharmacy services for Merck and AbbVie product.
Abbreviations:
- AE
adverse event
- DAA
direct-acting antiviral
- EBR
elbasvir
- GZR
grazoprevir
- HIT
Headache Impact Test
- HTE
heterogeneity of treatment effects
- LDV
ledipasvir
- mITT
modified intention to treat
- NS5A
nonstructural protein 5A
- PRO
patient-reported outcome
- PrOD
paritaprevir/ritonavir/ombitasvir+dasabuvir
- PROMIS
Patient-Reported Outcomes Measurement Information System
- RAS
resistance-associated substitution
- RBV
ribavirin
- SOF
sofosbuvir
- SVR12
sustained viral response at 12 weeks
Footnotes
Supporting Information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.32053/suppinfo.
REFERENCES
- 1).Polaris Observatory HCV Collaborators. Global prevalence and genotype distribution of hepatitis C virus infection in 2015: a modelling study. Lancet Gastroenterol Hepatol 2015;2017:161–176. [DOI] [PubMed] [Google Scholar]
- 2).Dang H, Yeo YH, Yasuda S, Huang CF, Iio E, Landis C, et al. Cure with interferon free DAA is associated with increased survival in patients with HCV related HCC from both east and west. Hepatology 2020;71:1910–1922. [DOI] [PubMed] [Google Scholar]
- 3).Hofmeister MG, Rosenthal EM, Barker LK, Rosenberg ES, Barranco MA, Hall EW, et al. Estimating prevalence of hepatitis C virus infection in the United States, 2013–2016. Hepatology 2019;69:1020–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4).AASLD/IDSA HCV Guidance Panel Recommendations for testing, managing, and treating hepatitis C website. Published 2017. Accessed June 13, 2017. http://hcvguidelines.org
- 5).Mishra P, Florian J, Peter J, Vainorius M, Fried MW, Nelson DR, et al. Public–private partnership: targeting real-world data for hepatitis C direct-acting antivirals. Gastroenterology 2017;153:626–631. [DOI] [PubMed] [Google Scholar]
- 6).Kosinski M, Bayliss MS, Bjorner JB, Ware JE Jr, Garber WH, Batenhorst A, et al. A six-item short-form survey for measuring headache impact: the HIT-6. Qual Life Res 2003;12:963–974. [DOI] [PubMed] [Google Scholar]
- 7).Bjorner JB, Kosinski M, Ware JE Jr. Using item response theory to calibrate the Headache Impact Test (HIT™) to the metric of traditional headache scales. Qual Life Res 2003;12:981–100. [DOI] [PubMed] [Google Scholar]
- 8).Cella D, Riley W, Stone A, Rothrock N, Reeve B, Yount S, et al. The Patient-Reported Outcomes Measurement Information System (PROMIS) developed and tested its first wave of adult self-reported health outcome item banks: 2005–2008. J Clin Epidemiol 2010;63:1179–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9).Reeve BB, Hays RD, Bjorner JB, Cook KF, Crane PK, Teresi JA, et al. Psychometric evaluation and calibration of health-related quality of life item banks: plans for the Patient-Reported Outcomes Measurement Information System (PROMIS). Med Care 2007;45(5 Suppl. 1):S22–S31. [DOI] [PubMed] [Google Scholar]
- 10).Spiegel BM, Hays RD, Bolus R, Melmed GY, Chang L, Whitman C, et al. Development of the NIH Patient-Reported Outcomes Measurement Information System (PROMIS) gastrointestinal symptom scales. Am J Gastroenterol 2014;109:1804–1814. Erratum in: Am J Gastroenterol 2015;110:608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11).Anderson RT, Baran RW, Dietz B, Kallwitz E, Erickson P, Revicki DA. Development and initial psychometric evaluation of the hepatitis C virus-patient-reported outcomes (HCV-PRO) instrument. Qual Life Res 2014;23:561–570. [DOI] [PubMed] [Google Scholar]
- 12).Voils CI, King HA, Thorpe CT, Blalock DV, Kronish IM, Reeve BB, et al. Content validity and reliability of a self-report measure of medication nonadherence in hepatitis C treatment. Dig Dis Sci 2019;64:2784–2797. [DOI] [PubMed] [Google Scholar]
- 13).Voils CI, Maciejewski ML, Hoyle RH, Reeve BB, Gallagher P, Bryson CL, et al. Initial validation of a self-report measure of the extent of and reasons for medication nonadherence. Med Care 2012;50:1013–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14).Schulz KF, Altman DG, Moher D; CONSORT Group. CONSORT 2010 statement: updated guidelines for reporting parallel group randomized trials. BMJ 2010;340:c332. [DOI] [PubMed] [Google Scholar]
- 15).Amrhein V, Greenland S, McShane B. Scientists rise up against statistical significance. Nature 2019;567:305–307. [DOI] [PubMed] [Google Scholar]
- 16).It’s time to talk about ditching statistical significance. Nature 2019;567:283. [DOI] [PubMed] [Google Scholar]
- 17).Wasserstein RL, Lazar NA. The ASA statement on p-values: context, process, and purpose. Am Stat 2016;70:129–133. [Google Scholar]
- 18).Wasserstein RL, Schirm AL, Lazar NA. Moving to a world beyond “p < 0.05.” Am Stat 2019;73(Suppl. 1):1–19. [Google Scholar]
- 19).International Committee of Medical Journal Editors. Recommendations for the conduct, reporting, editing and publication of scholarly work in medical journals. http://www.icmje.org/icmje-recommendations.pdf. Published December 2019. Accessed July 9, 2020. [PubMed]
- 20).White IR, Horton NJ, Carpenter J, Pocock SJ. Strategy for intention to treat analysis in randomized trials with missing outcome data. BMJ 2011;342:d40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21).Firth D Bias reduction of maximum likelihood estimates. Biometrika 1993;80:27–38. [Google Scholar]
- 22).Lu K On efficiency of constrained longitudinal data analysis versus longitudinal analysis of covariance. Biometrics 2010;66:891–896. [DOI] [PubMed] [Google Scholar]
- 23).Segal JB, PRIORITIZE HCV Study Group. Formulary restrictions may impact enrollment in pragmatic trials and limit generalizability of findings to vulnerable populations. Clin Trials 2020;17:729–731. [DOI] [PubMed] [Google Scholar]
- 24).Wilder J, Saraswathula A, Hasselblad V, Muir A. A systematic review of race and ethnicity in hepatitis C clinical trial enrollment. J Natl Med Assoc 2016;108:24–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25).US Department of Health and Human Services. Viral hepatitis national strategic plan for the United States: A roadmap to elimination (2021–2025). Washington, DC; 2020. [Google Scholar]
- 26).Zeuzem S, Mizokami M, Pianko S, Mangia A, Han KH, Martin R, et al. NS5A resistance-associated substitutions in patients with genotype 1 hepatitis C virus: prevalence and effect on treatment outcome. J Hepatol 2017;66:910–918. [DOI] [PubMed] [Google Scholar]
- 27).Fox DS, McGinnis JJ, Tonnu-Mihara IQ, McCombs JS. Comparative treatment effectiveness of direct acting antiviral regimens for hepatitis C: data from the Veterans Administration. J Gastroenterol Hepatol 2017;32:1136–1142. [DOI] [PubMed] [Google Scholar]
- 28).O’Brien TR, Lang Kuhs KA, Pfeiffer RM. Subgroup differences in response to 8 weeks of ledipasvir/sofosbuvir for chronic hepatitis C. Open Forum Infect Dis 2014;1:ofu110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29).Zeuzem S, Ghalib R, Reddy KR, Pockros PJ, Ben Ari Z, Zhao Y, et al. Grazoprevir-elbasvir combination therapy for treatment-naive cirrhotic and noncirrhotic patients with chronic hepatitis C virus genotype 1, 4, or 6 infection: a randomized trial. Ann Intern Med 2015;163:1–13. [DOI] [PubMed] [Google Scholar]
- 30).US Food and Drug Administration. FDA warns about rare occurrence of serious liver injury with use of hepatitis C medicines Mavyret, Zepatier, and Vosevi in some patients with advanced liver disease. FDA Drug Safety Communication. https://www.fda.gov/drugs/drug-safety-and-availability/fda-warns-about-rare-occurrence-serious-liver-injury-use-hepatitis-c-medicines-mavyret-zepatier-and. Published August 28, 2019. Accessed August 2, 2020.
- 31).Evon DM, Sarkar S, Amador J, Lok AS, Sterling RK, Stewart PW, et al. Patient-reported symptoms during and after direct-acting antiviral therapies for chronic hepatitis C: the PROP UP study. J Hepatol 2019;71:486–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.