

Abstract
Rationale:
Vascular smooth muscle cells (SMCs) exhibit remarkable plasticity and can undergo dedifferentiation upon pathological stimuli associated with disease and interventions.
Objective:
Although epigenetic changes are critical in SMC phenotype switching, a fundamental regulator that governs the epigenetic machineries regulating the fate of SMC phenotype has not been elucidated.
Methods and Results:
Using SMCs, mouse models, and human atherosclerosis specimens, we found that focal adhesion kinase (FAK) activation elicits SMC dedifferentiation by stabilizing DNA methyltransferase 3A (DNMT3A). FAK in SMCs is activated in the cytoplasm upon serum stimulation in vitro or vessel injury and active FAK prevents DNMT3A from nuclear FAK-mediated degradation. However, pharmacological or genetic FAK catalytic inhibition forced FAK nuclear localization, which reduced DNMT3A protein via enhanced ubiquitination and proteasomal degradation. Reduced DNMT3A protein led to DNA hypomethylation in contractile gene promoters, which increased SMC contractile protein expression. RNA sequencing identified SMC contractile genes as a foremost upregulated group by FAK inhibition from injured femoral artery samples compared to vehicle group. DNMT3A knockdown in injured arteries reduced DNA methylation and enhanced contractile gene expression supports the notion that nuclear FAK-mediated DNMT3A degradation via E3 ligase TRAF6 drives differentiation of SMCs. Furthermore, we observed that SMCs of human atherosclerotic lesions exhibited decreased nuclear FAK, which was associated with increased DNMT3A levels and decreased contractile gene expression.
Conclusions:
This study reveals that nuclear FAK induced by FAK catalytic inhibition specifically suppresses DNMT3A expression in injured vessels resulting in maintaining SMC differentiation by promoting the contractile gene expression. Thus, FAK inhibitors may provide a new treatment option to block SMC phenotypic switching during vascular remodeling and atherosclerosis.
Keywords: Atherosclerosis, Epigenetics, Restenosis, Smooth Muscle Proliferation and Differentiation, Vascular Biology
Graphical Abstract
INTRODUCTION
Atherosclerosis and coronary artery diseases are the major cause of death and mortality in developed countries. A major player in vascular pathogenesis are vascular smooth muscle cells (SMCs), which have the unique ability to dedifferentiate from mature contractile cells into highly proliferative synthetic cells, and under certain pathological stimuli transdifferentiate into myofibroblasts and macrophage-like cells.1–3 This phenotypic switching is associated with an increase in cell proliferation capacity, wound healing, and reduction in SMC-specific contractile gene expression via transcriptional regulation. Excessive proliferation of SMCs is a clinical problem that contributes to neointimal hyperplasia and atherosclerosis.4–6 Current treatment strategies to restore blood flow through atherosclerotic vessels include angioplasty procedures, stenting, or vein graft surgery.7–9 However, these procedures are associated with vessel injury, resulting in high recurrences of restenosis. Although drug-eluting stents have helped to provide local drug delivery, the risk of late-stent thrombosis and their limited use to larger arteries are still drawbacks. In addition, diabetes-associated atherosclerosis in peripheral arteries or transplant arteriopathies exhibit much higher rates of occlusion following intervention.10, 11 Size and location of the affected artery still remain as major problems facing long-term treatments to prevent restenosis.4, 9, 12, 13 Therefore, there is a need to better understand the mechanism underlying SMC phenotype switching to develop better treatment options for vessel-narrowing diseases.
SMC contractile gene expression is regulated by various SMC-specific transcription factors, such as serum response factor (SRF) and myocardin, which bind CArG boxes (CC(A/T)6GG) in contractile gene promoters. Recent studies have also suggested that DNA methylation regulates SMC phenotype switching and vascular remodeling.14 DNA methylation is predominantly associated with gene silencing,15, 16 is catalyzed by DNA methyltransferases (DNMTs), and typically occurs at the 5 position of cytosine (5-mC) in CG dinucleotides of CpG islands. Many SMC contractile gene promoters are unmethylated in vivo, thus allowing SRF and myocardin to bind CArG boxes.17–19 The process that mediates DNA methylation in the contractile gene has not been fully elucidated in regard to SMC-specific gene transcription.20
There are three DNMTs that can methylate cytosine: DNMT1, DNMT3A and DNMT3B. DNMT1 is a key enzyme in preserving DNA methylation during DNA replication.21 On the other hand, DNMT3A and DNMT3B are de novo DNA methylation enzymes that regulate gene expression. Demethylation of 5-mC to 5-hydroxymethylcytosine (5-hmC) is mediated by Ten-eleven translocation (TET) enzymes. Of the three TET enzymes, TET2 was shown to be highly expressed in SMCs, and increased expression of TET2 promotes SMC differentiation.22 In addition to DNA methylation, histone modifications are connected to DNA methylation state and also play an important role in SMC-specific contractile gene transcription and SMC plasticity.23
Focal adhesion kinase (FAK) is an integrin-associated protein tyrosine kinase that promotes cell migration and proliferation.24 During SMC dedifferentiation, increased secretion of matrix and growth factors alters integrin signaling and leads to aberrant FAK activation promoting FAK-mediated SMC migration and proliferation.25–30 A recent study revealed that FAK is predominantly localized within the nuclei of SMCs in healthy arteries, and that wire injury induces FAK activation and cytoplasmic relocalization.31 Nuclear FAK inhibits SMC proliferation and hyperplasia, in part, through decreasing GATA4 and subsequent inhibition of GATA4-mediated cyclin D1 transcription.31 However, as GATA4 expression did not correlate to changes in contractile gene expression, we investigated a potential parallel mechanism through which nuclear FAK regulates SMC phenotypic switching.
In the present study, we investigated the molecular mechanism of how FAK regulates DNA methylation and SMC contractile gene expression, determining SMC’s fate during SMC remodeling. Using pharmacological and genetic FAK inhibition, and human atherosclerosis samples, we found that nuclear FAK decreases DNMT3A enzyme stability and DNA methylation leading to increased contractile protein expression and maintaining SMCs differentiated.
METHODS
Detailed Materials and Methods and the Major Resources Table can be found in the Online Supplement.
Data Availability.
The sequencing data in this manuscript have been deposited in the Gene Expression Omnibus under accession number GSE183143.
RESULTS
FAK inhibition promote SMC-specific contractile gene expression in vitro.
After a few passages in serum containing medium, aortic SMCs isolated from C57BL6 mice dedifferentiate and take on a synthetic hypertrophic appearance (Figure 1A). Interestingly, we found that pharmacological inhibition of FAK catalytic activity using a FAK inhibitor (FAK-I, VS-4718) reverted SMCs to an elongated spindle-like morphology (Figure 1A). In vivo, vehicle-treated mice showed increased neointima formation following wire injury with SMCs exhibiting a characteristic synthetic phenotype with smaller nuclei and larger cellular areas (Online Figure IA). However, FAK-I-treated mice had reduced neointima formation and SMCs maintained a differentiated phenotype as observed by elongated nuclei and a spindle-like cell morphology (Online Figure IA). While FAK-I-treated mice showed increased lumen area due to decreased neointimal area compared to vehicle-treated injured group, there was no difference in arterial perimeter or medial area between the two groups (Online Figure IA). As FAK-I reduced neointima formation and promoted a contractile morphology in SMCs, we next evaluated if FAK-I altered SMC-specific contractile gene expression. As early as 1 day, FAK-I increased expression of several contractile proteins including MYH11, CALD1, ACTA2, CNN1, and TAGLN (Figure 1B). FAK inhibition was verified by pY397 FAK immunoblotting (Figure 1B and Online Figure IIA). Analysis of contractile gene mRNA levels revealed that FAK-I promoted contractile gene expression through increased transcription (Figure 1C). To evaluate if FAK-I-induced spindle like morphology in SMCs correlated with changes in contractile gene expression, we calculated cell aspect ratio (cell long and short axis ratio)32 using differential interference contrast images and TAGLN fluorescence intensity (Online Figure III). SMCs treated with FAK-I showed a more elongated spindle-like morphology (aspect ratio > 2) compared to vehicle treated (Figure 1D and Online Figure III). Next, we found that the fluorescence intensity of TAGLN was significantly higher in FAK-I-treated spindle-liked SMCs than in cuboidal-like cells (Figure 1E and Online Figure III). We further looked at FAK inhibition effect on SMC phenotype by performing haptotaxis and chemotaxis assays. FAK-I reduced SMC migration in both fibronectin Boyden chamber haptotaxis assays and platelet-derived growth factor (PDGF)-stimulated scratch would assays (Online Figure IV). As we previously demonstrated that FAK-I reduces SMC proliferation through regulation of GATA4 stability,33 we next tested if GATA4 levels regulated contractile gene expression. GATA4 knockdown or overexpression in SMCs had no effect on contractile gene expression (Online Figure V), indicating that FAK-I induces a contractile SMC phenotype in a GATA4-independent manner.
Figure 1. FAK inhibition alters SMC morphology and promotes SMC specific contractile gene expression.

(A-E) FAK-I (VS-4718, 2.5 μM) was treated in SMCs for 2 days. (A) FAK catalytic inhibition in SMCs changes the cell morphology. Percent of spindle-like cells from total cells (arrows) were numerated from three independent fields (n=3). (B) Shown are representative immunoblots of lysates of SMCs treated with or without FAK-I for active FAK (pY397 FAK), total FAK, MYH11, CALD1, ACTA2, CNN1, TAGLN, and GAPDH as loading control. Relative intensity for blots were indicated relative to GAPDH (n=3). (C) mRNA levels of SMC contractile genes were measured after FAK-I treatment (n=6). (D) Analysis of cell aspect ratio (cell long/short axis ratio) of SMCs after FAK-I treatment (n=40). (E) Fluorescence intensity of TAGLN staining in SMCs with cell aspect ratio below or above 2 (n=46 vehicle and n=44 FAK-I). (F-H) Isolated FAK-/WT (FAK-WT) and FAK-/KD (FAK-KD) SMCs. (F) Percent of spindle-like cells from total cells (arrows) were plotted (n=3). (G) Shown are representative immunoblots of lysates of FAK-WT or FAK-KD SMCs for active FAK (pY397 FAK), total FAK, MYH11, ACTA2, CNN1, TAGLN, and GAPDH as loading control. Relative intensity for blots were indicated relative to GAPDH (n=3). (H) mRNA of SMC contractile genes was measured from FAK-WT or FAK-KD SMCs (n=4). (I and J) Mice were treated with vehicle or FAK-I (VS-4718, 50 mg/kg) twice daily orally following wire injury for 2 weeks. RNA sequencing data from femoral artery RNA samples were analyzed. Volcano plots of transcriptome profiles sham vs. injured (n=3) and injured vs. injured+FAK-I (n=3) were plotted. Blue dots represent genes significantly upregulated and downregulated (adjusted P value<0.05). Red dots: SMC-specific contractile genes. Gray dots: genes with no significant changes. Data are mean±SEM. P values were determined using t-test (A, C, D, F, and H), 2-way ANOVA with Bonferroni multiple comparisons test (E; P values adjusted for 6 comparisons).
To verify if these changes were specific to FAK catalytic activity, we next evaluated changes in SMC morphology and contractile gene expression using FAK kinase-dead (KD) SMCs.31 FAK-KD SMCs demonstrated a more spindle-like morphology compared to FAK wild-type (WT) both in vitro and in injured vessels in vivo (Figure 1F and Online Figure IB). SMC-specific FAK-KD mice showed reduced neointimal hyperplasia and also maintained a differentiated SMC morphology upon vessel injury unlike FAK-WT mice (Online Figure IB). FAK-KD SMCs also exhibited increased expression of contractile genes compared to FAK-WT (Figure 1G and H, Online Figure IIB). To determine if increased contractile gene expression observed in FAK-KD SMCs resulted in increased contractility, we performed a collagen gel contraction assay. FAK-KD SMCs showed a higher level of contraction ability in collagen gel compared to FAK-WT (Online Figure VI), suggesting that FAK activity promotes a dedifferentiated SMC phenotype which can be reversed by inhibition of FAK catalytic activity.
FAK activity regulates SMC contraction-related pathway upon vessel injury.
To further characterize the role of FAK activity in alterations to the transcriptional landscape during SMC phenotypic switching, we performed whole transcriptome RNA-Seq analysis with mouse femoral artery samples following wire injury with or without FAK-I. The differentially expressed genes (DEGs) between experimental groups with Log2 fold change higher than 1 or lower than −1, and adjusted P value (FDR) less than 0.05 were analyzed (Supplemental Table I and II). Overall, there were 3539 DEGs between vehicle-sham and vehicle-injured groups, with 2510 genes upregulated and 1029 genes downregulated (Figure 1I and Supplemental Table I). Comparing vehicle-injured and FAK-I-injured groups, 3546 DEGs were identified with 1357 genes upregulated and 2198 genes down regulated (Figure 1J and Supplemental Table II). KEGG pathway analysis identified upregulation of cytokine-cytokine receptor interaction and cell adhesion molecules in vehicle-injured compared to vehicle-sham mice (Online Figure VIIA). We also found that vascular smooth muscle contraction and focal adhesion pathways were downregulated following injury (Online Figure VIIA). Conversely, FAK-I-treated injured arteries showed upregulation of vascular smooth muscle contraction and ECM-receptor interaction pathways, but downregulation of cytokine-cytokine receptor interaction and chemokine signaling pathway compared to vehicle-injured (Online Figure VIIB). Vehicle-injured mice showed significant downregulation of SMC-specific contractile genes, such as Myh11, Acta2, Cald1, Cnn1, and Tagln, compared to vehicle-sham mice (Figure 1I and Online Figure VIIC). In contrast, FAK-I treated mice showed significant enrichment of SMC-specific contractile genes compared to vehicle-injured mice (Figure 1J and Online Figure VIID). As SMCs have been shown to transdifferentiate and express markers over other cell types, we looked at changes in different lineage markers following wire injury and FAK inhibition. While wire injury increased expression of mesenchymal stem cell (Ly6a/Sca1), macrophage (Lgal3), and chondrocyte (Sox9) lineage markers compared to sham controls, FAK-I treatment had similar levels to sham control (Online Figure VIIE). Wire injury also increased expression of the genes associated with a synthetic phenotype including Mmp2, Mmp14, Mmp13, Spp1, and Col1a1 in vehicle, but not FAK-I treated femoral arteries (Online Figure VIIC and D). Interestingly, we found that the expression level of contractile genes in FAK-I treated arteries was restored to that of sham control (Online Figure VIII). Together, these results establish a role for FAK activity in promoting SMC dedifferentiation into a synthetic phenotype during vascular remodeling.
FAK inhibition reduces DNMT3A expression and causes DNA hypomethylation in SMCs.
During SMC phenotypic switching, several genes including contractile genes are regulated transcriptionally via epigenetic mechanisms.34, 35 One of the best-studied epigenetic models in SMC phenotypic switching is increased DNA methylation, which is linked to promoter silencing of contractile genes.36 Since both genetic and pharmacological FAK inhibition increased SMC contractile genes at a transcriptional level, we next investigated if FAK inhibition altered DNA methylation. Dot blotting of SMC genomic DNA (gDNA) for 5-methylcytosine (5-mC) demonstrated that both pharmacological and genetic FAK inhibition decreased 5-mC levels (Figure 2A), which was associated with activation of gene transcription. Changes in 5-mC levels were further verified using immunofluorescence. While vehicle-treated SMCs showed high levels of 5-mC, FAK-I treated cells showed decreased 5-mC (Figure 2B and Online Figure IX). As FAK inhibition altered global 5-mC levels in SMCs, we tested if FAK-I treatment decreased 5-mC levels within the promoters of SMC contractile genes. We used standard bisulfite sequencing to evaluate changes in methylation status of CpG sites surrounding CArG box regions of Tagln and Myh11 promoters in SMCs (Figure 2C and Online Figure X).36 Analysis of nine bisulfite sequencing clones of Tagln and Myh11 promoters showed that both pharmacological and genetic FAK inhibition significantly decreased the percentage of methylated CpG sites (Figure 2C and Online Figure X). These data suggest that FAK inhibition promotes contractile gene expression through decreased methylation within contractile gene promoters in SMCs.
Figure 2. FAK inhibition in SMCs causes DNA hypomethylation through reduced DNMT3A expression.

SMCs were treated with FAK-I (VS-4718, 2.5 mM). (A) Dot blots for 5-mC using genomic DNA (gDNA) from SMCs treated with/without FAK-I for 2 days (top) or FAK-WT and -KD SMCs (bottom). Methylene blue (MB) staining was used to visualize total DNA loading (n=4). (B) SMCs were treated with FAK-I for 24 h. Immunofluorescence staining for 5-mC is shown (n=4). The images on the right show magnifications of the areas framed in the merge. DAPI was used for nuclei staining. Scale bars, 20 μm. (C) CpG methylation of Tagln (top) and Myh11 (bottom) promoters was evaluated by bisulfite sequencing of gDNA isolated from SMCs treated with/without FAK-I for 2 days or FAK-WT and -KD SMCs. Black circles: methylated CpGs. Empty circles: unmethylated CpG (n=9). Shown are representative immunoblots of (D) SMCs with/without FAK-I or (E) FAK-WT and FAK-KD lysates for active FAK (pY397 FAK), total FAK, DNMT3A, DNMT3B, DNMT1, and GAPDH or actin as loading control. Relative intensity for blots were indicated relative to GAPDH or actin (n=3). (F and G) DNMT mRNA levels were measured from SMCs with/without FAK-I treatment for 2 days, or FAK-WT and -KD SMCs using RT-qPCR (n=3). (H) Human brachiocephalic arteries were excised postmortem, and frozen sections were made for immunostaining. Representative immunostainings for DNMT3A, ACTA2, and DAPI are shown (n=5). Merge: Green, red, and DAPI (blue). Uncropped images are shown in Online Figure XA Scale bars: 20 μm. Data are mean±SEM.
As 5-mC is catalyzed by DNA methyltransferases (DNMTs), we tested if FAK inhibition reduced expression of DNMTs. Of three DNMTs known to methylate cytosine at CpG sites, FAK inhibition specifically reduced DNMT3A, but not DNMT1 or DNMT3B expression (Figure 2D and Online Figure IIA). Consistent with pharmacological FAK inhibition, FAK-KD SMCs had decreased DNMT3A expression compared to FAK-WT (Figure 2E and Online Figure IIB). Although FAK-I decreased DNMT3A expression, neither pharmacological nor genetic FAK inhibition altered mRNA expression of Dnmt3a, Dnmt3b, or Dnmt1 (Figure 2F and 2G). Together, these data suggest that FAK inhibition may decrease DNMT3A expression through a post-transcriptional mechanism, resulting in decreased DNA methylation and increased contractile gene expression.
DNMT3A and 5-mC levels are increased in human atherosclerotic lesions.
As silencing of SMC contractile genes has been shown to occur in vascular diseases including atherosclerosis, we next evaluated if there was any correlation between DNMT3A expression and disease state in human specimen. Immunostaining of healthy brachiocephalic arteries (BCAs), renal arteries, and the aortic arch showed low DNMT3A levels (Figure 2H and Online Figure X). However, DNMT3A expression was increased within atherosclerotic lesions of these arteries (Figure 2H and Online Figure XI), suggesting that DNMT3A may play a role in atherosclerosis disease progression. Staining for 5-mC also revealed increased DNA methylation with in human atherosclerotic lesions, and this was associated with decreased expression of ACTA2 (Figure 2H and 3A, Online Figure XI and XII). Interestingly, we observed abundant DNMT3B and DNMT1 in both healthy and diseased BCAs (Online Figure XIII), indicating that DNMT3A may play a critical role in SMCs during atherosclerosis progression. We next analyzed expression of DNMT in different human atherosclerosis cohort studies. Comparison of the gene signature between non-atherosclerotic arterial wall and the atherosclerotic arterial wall from the Stockholm Atherosclerosis Gene Expression study37 revealed no significant difference in DNMT3A and DNMT3B, and increased expression of DNMT1 in atherosclerotic compared to normal vessels (Online Figure XIVA). A similar observation was obtained when we analyzed the data from comparing severe plaque (Stage IV-V) to adjacent areas with early disease with no macroscopic disease (Stage I-II) from the human carotid endarterectomy sample data38(Online Figure XIV). Together, these data suggest DNMT3A protein stability may be increased in atherosclerotic plaques, which increases DNA methylation and silences contractile gene expression.
Figure 3. Nuclear FAK regulates DNMT3A levels and induces SMC contractile gene expression.

(A) Human brachiocephalic arteries were excised postmortem, and frozen sections were made for immunostaining. Representative immunostaining for 5-mC, ACTA2, and DAPI (n=5). Uncropped images are shown in Online Figure XIA. Scale bars: 20 μm. (B and C) SMCs were treated with FAK-I (VS-4718, 2.5 μM). (B) Immunostaining for FAK and DNMT3A or FAK and DNMT3B in SMCs treated with/without FAK-I for 12 h is shown (n=4). Scale bars, 20 μm. (C) FAK −/− SMCs stably expressing either FLAG-FAK-WT or –NLM (nonnuclear localizing mutant) were treated with/without FAK-I for 24 h. Representative immunoblots are shown for pY397 FAK, FAK, DNMT3A, DNMT3B, DNMT1, MYH11, CALD1, ACTA2, CNN1, TAGLN, and GAPDH as loading control. Blots were quantified relative to GAPDH (n=3). (D) Human brachiocephalic arteries were excised postmortem, and frozen sections were made for immunostaining. Representative immunostainings for FAK, pY397 FAK, and DAPI (n=5). White arrows: nuclear FAK. Red arrows: cytoplasmic FAK. Merge: Green, red, and DAPI (blue). Uncropped images are shown in Online Figure XVI Scale bars: 20 μm.
FAK activation and nuclear localization regulates DNMT3A expression.
We have shown that FAK catalytic inhibition triggers FAK nuclear localization and influences the stability of several nuclear factors by direct interaction.31, 39, 40 Indeed, immunostaining demonstrated that in vehicle-treated SMCs, FAK is mainly localized in the cytosol with high levels of DNMT3A in the nucleus (Figure 3B and Online Figure XV). Upon FAK-I treatment, decreased active pY397 FAK was associated with increased FAK nuclear localization and loss of DNMT3A, but not DNMT3B expression (Figure 3B and Online Figure XV and XVI), matching what we saw from immunoblotting (Figure 2D). As DNMT3A is a nuclear protein and FAK inhibition promotes FAK nuclear localization, we next examined whether FAK nuclear localization was required for FAK-I-mediated loss of DNMT3A in SMCs. To address this question, we stably expressed FLAG-tagged FAK-WT (FLAG-FAK-WT) and FAK nonnuclear-localizing mutant (NLM) in FAK−/− SMCs (Figure 3C and Online Figure XVII).31 FAK-I reduced DNMT3A levels in FLAG-FAK-WT SMCs and increased the expression of various SMC contractile proteins (Figure 3C and Online Figure XVII). However, FAK-I failed to reduce DNMT3A levels and increase contractile gene expression in FLAG-FAK-NLM SMCs (Figure 3C and Online Figure XVII), suggesting that FAK nuclear localization to promote SMC differentiation. As we previously reported that vascular injury promotes FAK activation and cytoplasmic localization in mouse femoral arteries,33 we next examined changes in FAK activity and localization in healthy and diseased human specimen. We observed that FAK is mainly in the nucleus and inactive within SMCs of healthy BCAs, renal arteries, and aortic arches (Figure 3D and Online Figure XVIII and XIX). However, FAK showed increased activity and cytoplasmic localization within atherosclerotic lesions (Figure 3D and Online Figure XVIII and XIX). The increased DNMT3A protein expression we observed in atherosclerotic lesions could be due to increased FAK cytoplasmic localization (Figure 2H and Online Figure XI and XIX), thus promoting SMC dedifferentiation during atherosclerosis progression. To further evaluate a connection between FAK and DNMT3A in human SMCs, we treated human coronary artery SMCs (hCASMCs) with FAK-I. Similar to mouse SMCs, FAK-I reduced DNMT3A, but not DNMT1 or DNMT3B, and elevated contractile gene expression in hCASMCs (Figure 3E and Online Figure XX). To get a better insight into potential mechanisms that promote FAK cytoplasmic localization and activation in SMCs during vascular disease, we investigated integrin and extracellular matrix signaling and their effect on FAK. As increased extracellular matrix (ECM) production and degradation plays a key role in SMC proliferation and migration in vascular disease and FAK is closely linked to integrin-ECM signaling, we evaluated the effect different ECM components had on FAK activation. SMCs plated onto plastic, poly-L-lysine, and laminin showed weak FAK activation after 1 h of adhesion (Online Figure XXI). However, FAK showed increased activation in SMCs attached to gelatin, collagen I, and fibronectin (Online Figure XXI). In addition, increased FAK activation was associated with elevated DNMT3A expression (Online Figure XXI). These observations indicate that integrin-ECM signaling associated with vascular disease promotes FAK activation and cytoplasmic retention, leading to increased DNMT3A expression in SMCs.
FAK interaction with DNMT3A and TRAF6 promotes specific DNMT3A turnover via ubiquitination.
To decipher how FAK regulates DNMT3A, but not DNMT3B or DNMT1, we performed endogenous FAK-DNMT interaction studies in SMCs. While FAK immunoprecipitation (IP) upon FAK-I treatment increased FAK-DNMT3A association (Figure 4A and Online Figure XXIIA), and this association was further increased when SMCs were treated with the proteasomal inhibitor MG132 in addition to FAK-I (Figure 4A). However, DNMT3B and DNMT1 failed to associate with FAK under these conditions (Figure 4A), suggesting that FAK specially associates with DNMT3A. Immunoblotting of DNMT3A IP in SMCs showed that FAK-I increased ubiquitination of DNMT3A, which was further enhanced by MG132 co-treatment (Figure 4B and Online Figure XXIIA). To confirm the observation that FAK inhibition decreased DNMT3A protein stability, SMCs were treated with a protein synthesis inhibitor cycloheximide (CHX). While CHX slowly reduced protein expression of DNMT3A, co-treatment with FAK-I accelerated turnover of DNMT3A, but not DNMT3B or DNMT1 (Figure 4C and Online Figure XXIIB). We further verified FAK-I-mediated post-translational regulation of DNMT3A protein stability using actinomycin D (ActD), a transcriptional inhibitor. While ActD treatment alone failed to reduce DNMT3A levels, FAK-I still decreased DNMT3A protein expression in the presence of ActD (Online Figure XXIIC). DNMT3B or DNMT1 levels remained unchanged under cotreatment of ActD and FAK-I (Online Figure XXIIC), suggesting that FAK specifically regulates DNMT3A. As nuclear FAK has been shown to act as a scaffold that promotes protein degradation by recruiting a target protein and its E3 ligases (e.g., p53 and Mdm-2, GATA4 and CHIP),39, 40 we screened for a potential E3 ligase for FAK-mediated DNMT3A ubiquitination and degradation. Based on known E3 ligases for DNMT3A, we focused on the E3 ligases TRAF6, UHRF1, and UHRF2.41, 42 We found that overexpression of Myc-TRAF6, but not HA-UHRF1 and HA-UHRF2, significantly reduced DNMT3A expression in 293T cells (Online Figure XXIIIA). Thus, we further tested whether TRAF6 might be the E3 ligase that mediates DNMT3A ubiquitination via nuclear FAK in SMCs. Immunoblotting of endogenous FAK IP revealed that TRAF6, DNMT3A, and FAK form a ternary complex only upon FAK inhibition, and this association was further increased by MG132 (Figure 4D and Online Figure XXIIA). Although both DNMT3A and DNMT3B contain highly conserved amino acid sequences in their PWWP domains, ADD (cysteine-rich) domains, and carboxyl-terminal catalytic domains (Online Figure XXIIIB), the homology between the two proteins at the N-terminal variable region is only 28%.43, 44 As DNMT3A contains a unique 70 amino acid N-terminal domain not present in DNMT3B, we predicted that FAK associates with this unique N-terminal region (Online Figure XXIIIB). To determine whether the DNMT3A N-terminal domain associates with FAK, we overexpressed Myc-tagged DNMT3A full-length (WT), N-term (1–70), or N-term deletion constructs (ΔN-term, 71–912) (Figure 4E and Online Figure XXIIIB). As expected, the 70 amino acid N-term of DNMT3A was critical for FAK-DNMT3A association (Figure 4E and Online Figure XXIIIC).
Figure 4. FAK interacts with DNMT3A and promotes DNMT3A turnover via ubiquitination and proteasomal degradation.

(A-D) SMCs were treated with FAK-I (VS-4718, 2.5 μM). (A, B, and D) SMCs were treated with FAK-I only or together with MG132 (20 μM) for 6 h. Lysates were immunoprecipitated with control IgG, anti-FAK (A), anti-DNMT3A (B) or anti-TRAF6 (D), and subjected to immunoblotting with indicated antibodies (n=3). (C) SMCs were treated with cycloheximide (CHX, 50 μg/ml) with/without FAK-I for indicated times. DNMT3A blots were measured and plotted (n=3), also see Online Figure XXB. (E) 293T cells were transfected with Myc-tagged DNMT3A mutants and subjected to immunoprecipitation with anti-Myc antibody and subjected to immunoblotting with indicated antibodies (n=3). (F) Mice were treated with MG132 (0.3 mg/kg) by intraperitoneal injection at 24, 6, and 1 h before euthanasia. Isolated aorta lysates were immunoprecipitated with anti-FAK antibody and subjected to immunoblotting with indicated antibodies (n=6). Uncropped images are shown in Online Figure XXIC. (G) Representative immunoblots of shDNMT3A and shDNMT3B RNA expressing SMCs for DNMT3A, DNMT3B, DNMT1, pY397 FAK, FAK, CALD1, ACTA2, CNN1, and GAPDH as loading control. pY397 FAK blots were quantified relative to total FAK and others to GAPDH (n=3). Data are mean±SEM. P values were determined using 2-way ANOVA with Bonferroni multiple comparisons test (C; P values adjusted for 5 comparisons).
As nuclear FAK appears to regulate DNMT3A protein stability, we treated mice with MG132 to investigate FAK-DNMT3A association in vivo. Mice were treated with MG132 (0.3 mg/kg) at 24, 6, and 1 h prior to euthanasia. Whole aortas were then lysed subjected to FAK IP. Immunoblotting of aorta lysates revealed that MG132 treatment increased DNMT3A levels but had no effect on DNMT1 and DNMT3B (Figure 4F and Online Figure XXIC). Further, DNMT3A was only detected in FAK IP from mice treated with MG132 (Figure 4F and Online Figure XXIIIC). However, we failed to detect DNMT1 and DNMT3B association with FAK under any condition, further supporting that nuclear FAK specifically regulates DNMT3A.
Both DNMT3A and DNMT3B can mediate de novo DNA methylation. However, we observed nuclear FAK affected only DNMT3A expression. To determine the importance of either DNMT3A or DNMT3B expression in SMC contractile protein expression, we evaluated the effect of DNMT3A and DNMT3B knockdown by lentiviral shRNA in SMCs. While loss of DNMT3A was associated with increased expression of SMC contractile genes in cultured SMCs, DNMT3B did not affect contractile gene expression (Figure 4G and Online Figure XXIV). There were no significant differences in pY397 FAK levels in both DNMT3A and 3B shRNA expressing cells (Figure 4G and Online Figure XXIV), confirming that only DNMT3A expression is responsible for contractile gene expression. Interestingly, we observed that knockdown of DNMT3A by lentiviral shRNA reduced SMCs proliferation (Online Figure XXV). These data suggest that nuclear FAK promotes DNA hypomethylation by specifically reducing DNMT3A stability via ubiquitination and proteasomal degradation, thus increasing SMC contractile gene expression and SMC differentiation.
Vascular injury upregulates DNMT3A expression leading to phenotypic switching in vivo.
To understand the effect of FAK activity on DNMT3A expression in SMC phenotype switching in vivo, we performed femoral arterial wire injury in mice. Vessel injury caused neointimal hyperplasia in control mice, but not in FAK-I treated mice (Online Figure IA). We found that DNMT3A expression was not observed in uninjured arteries but was increased in injured arteries, particularly within the neointima area (Figure 5 and Online Figure XXVI and XXVII). Elevated DNMT3A levels were associated with increased cytoplasmic active pY397 FAK (Figure 5 and Online Figure XXVI and XXVII). However, in FAK-I treated mice, FAK remained inactive and in the nucleus, with a significant decrease in DNMT3A expression compared to vehicle-treated mice (Figure 5 and Online Figure XXVI and XXVII). Upregulation of DNMT3A expression following wire injury was associated with increased 5-mC staining compared to sham controls (Figure 5 and Online Figure XXVI and XXVII). However, FAK-I treated mice showed very little 5-mC staining compared to vehicle (Figure 5 and Online Figure XXVI and XXVII), consistent with our in vitro data (Figure 2). Although DNMT3A protein was increased in control after vascular injury and reduced upon FAK inhibition, there was no significant difference in Dnmt3a, Dnmt3b, and Dnmt1 mRNA expression in artery samples following wire injury in all conditions (Online Figure XXVIIIA and B). From analysis of hit counts from RNA-seq data, we noted that DNMT3A levels appear to be much higher than DNMT3B in mouse arteries (Online Figure XXVIIIC), suggesting that DNMT3A may be a major de novo DNA methylation enzyme in SMCs during vascular remodeling.
Figure 5. Pharmacological FAK inhibition reduced injury-induced upregulation of DNMT3A and 5-mC preventing neointima formation in mice.

Mice were treated with vehicle or FAK-I (VS-4718, 50 mg/kg) twice daily following wire injury for 2 weeks. It is noted that FAK localization or activity exhibited an inverse relation with DNMT3A expression. Shown are representative immunofluorescence stainings of frozen section from vehicle or FAK-I-treated femoral arteries for FAK, pY397 FAK, DNMT3A, 5-mC, and ACTA2. Red, green, and blue (DAPI) were merged (n=4). Dashed line in image marks the elastic lamina. White line indicates endothelial layer. L, lumen. Scale bars: x20, 100 μm; or x60, 20 μm.
SMC-specific FAK-KD prevent vessel injury-induced DNMT3A upregulation.
As FAK-I can affect FAK activity in multiple cell types in vivo, we assessed if SMC-specific FAK activity is responsible for DNMT3A expression in SMCs. Similar to FAK-I treated mice, wire injury using SMC-specific FAK-WT and FAK-KD mice31 significantly prevented neointima formation in SMC-specific FAK-KD mice (Online Figure IB). In sham controls, we observed low levels of DNMT3A in both FAK-WT and -KD mice with abundant nuclear FAK (Figure 6 and Online Figure XXIX and XXV). Wire injury increased DNMT3A expression in FAK-WT mice, where it was correlated with high levels of active pY397 FAK in the cytoplasm. However, wire injury did not increase DNMT3A expression in FAK-KD mice, where FAK was inactive and retained in the nucleus (Figure 6 and Online Figure XXIX and XXV). We did not observe any differences in DNMT3B levels between FAK-WT and FAK-KD mice in both sham control and injured groups (Figure 6 and Online Figure XXIX and XXV). Additionally, FAK-KD mice failed to show increased 5-mC staining following wire injury (Figure 6 and Online Figure XXIX and XXV). These results support the notion that SMC phenotypic switching during vascular injury requires FAK activation and cytoplasmic localization to promote DNMT3A expression.
Figure 6. SMC-specific genetic FAK-KD mice did not upregulate injury-induced DNMT3A and 5-mC expression protecting from neointima formation.

Representative immunofluorescence staining of FAK-WT and FAK-KD femoral arteries 2 weeks postinjury for FAK, pY397 FAK, DNMT3A, DNMT3B, 5-mC and ACTA2 (n=4). FAK-KD in SMCs from the injured group exhibited a low DNMT3A and 5-mC as observed in sham. Merge 1, green and red; Merge 2, green, red, and blue (DAPI) were merged. Dashed line in image marks the elastic lamina. White line indicates endothelial layer. L, lumen. Scale bars: 20 μm.
Upregulated DNMT3A upon wire injury reduced contractile gene expression via increased 5-mC levels and decreased active histone mark.
To gain more comprehensive insights into DNMT3A expression in SMC contractile gene expression during vascular remodeling, we performed immunoblots with artery lysates from 2 weeks postinjury. Wire injury increased both active pY397 FAK and DNMT3A expression, but there were no significant differences in DNMT1 and DNMT3B levels (Figure 7A and Online Figure XXXIA). Mice treated with FAK-I did not increase DNMT3A following wire injury, and this was associated with maintaining normal levels of SMC contractile proteins compared to sham controls (Figure 7A and Online Figure XXXIA). To verify that these changes were specific to FAK activity in SMCs, we next examined changes in DNMT3A and SMC contractile genes in FAK-WT and FAK-KD mice. Similar to FAK-I experiments, FAK-KD mice showed no significant difference in DNMT3A, pY397 FAK, SMC contractile genes following wire injury (Figure 7B and Online Figure XXXIB). Meanwhile, FAK-WT mice showed elevated DNMT3A and pY397 FAK, which was accompanied by decreased SMC contractile gene expression (Figure 7B and Online Figure XXXIB). As FAK inhibition following wire injury maintained the low DNMT3A expression observed in normal artery, we further investigated if these results were consistent with changes in DNA methylation using genomic DNA isolated from femoral arteries of 2 weeks postinjury. Wire injury significantly increased the levels of 5-mC when compared to sham controls (Figure 7C). However, FAK-I treatment reduced wire injury-induced 5-mC levels compared to vehicle-treated mice (Figure 7C).
Figure 7. FAK inhibition blocks wire injury-induced upregulation of DNMT3A and 5-mC levels and maintains expression of SMC-specific contractile genes.

(A) Mice were treated with vehicle or FAK-I (VS-4718, 50 mg/kg) twice daily for 2 weeks following wire injury. Representative immunoblots of femoral artery lysates from (A) FAK-I treated or (B) FAK-WT and -KD mice 2 weeks following injury for pY397 FAK, FAK, DNMT3A, DNMT3B, DNMT1, MYH11, CALD1, ACTA2, CNN1, TAGLN and GAPDH as loading control (n=4). pY397 FAK blots were quantified relative to total FAK, and other blots to GAPDH. (C) Representative dot blots for 5-mC using gDNA isolated from femoral arteries with or without FAK-I treatment 2 weeks postinjury (n=3). The methylene blue (MB) staining of 150 ng total genomic DNA was used as loading control. (D) SMCs were treated with either vehicle or FAK-I for 24 h. Chromatin immunoprecipitation (ChIP) on Myh11, Tagln, and Acta2 promoters was performed with the indicated histone and SRF antibodies (n=3). Data are mean±SEM. P values were determined using t-test (D).
In addition to DNA methylation, histone modifications have been reported to regulate SMC cell phenotype and contractile gene expression. There is now emerging evidence that DNA modification eventually affects the acetylation/methylation states on accompanying histones in chromatin by recruiting histone modifying machineries.45, 46 To evaluate whether FAK inhibition alters histone modifications in cultured SMCs, we performed chromatin immunoprecipitation (ChIP) assays with several antibodies specific to histone modifications. FAK inhibition increased active histone marks including H4 acetylation (H4ac) and H3K27ac surrounding the CArG-boxes of Myh11, Tagln, and Acta2 promoters of SMCs (Figure 7D and Online Figure XXXIIA), which correlated with increased contractile protein expression (Figure 1). Additionally, FAK-I reduced inhibitory H3K9 trimethylation (H3K9me3) within these promoters (Figure 7D). Total H3 and H4 content were revealed no significant difference by FAK-I within SMC CArG boxes (Online Figure XXXIIB). Importantly, FAK inhibition increased binding of SRF, a key transcription factor for contractile gene expression, to the CArG boxes of SMC contractile genes (Figure 7D). Taken together, our data demonstrate that nuclear FAK induced by FAK inhibition promotes SMC differentiation by reducing DNA methylation and increasing active histone modifications, resulting in increased SMC contractile gene expression through enhanced SRF binding.
DNMT3A knockdown blocks wire injury-induced neointimal hyperplasia and maintains SMC contractile gene expression.
To explore the role of DNMT3A expression in neointima formation and contractile gene expression in vivo, we knocked down DNMT3A using shRNA (shDNMT3A) in femoral artery. Lentivirus encoding mCherry and DNMT3A or scramble control (shScr) were mixed with Pluronic F-127 and pasted to the outside of femoral arteries immediately following surgery. Arteries were collected 2 weeks post injury and analyzed. Neointimal hyperplasia was significantly decreased in shDNMT3A mice compared to shScr (Online Figure XXXIIIA). Lentiviral delivery was verified through mCherry expression within femoral arteries (Figure 8) and DNMT3A mRNA expression (Online Figure XXXIIIB). DNMT3A knockdown blocked wire injury-induced upregulation of DNMT3A, but there was no significant difference DNMT3B in all conditions (Figure 8 and Online Figure XXXIV and XXXV). Additionally, DNMT3A knockdown blocked wire injury-induced increases in 5-mC levels (Online Figure XXXIIIC and XXXVI). Even though shDNMT3A reduced neointimal hyperplasia, wire injury still increased FAK cytoplasmic localization and activation (Online Figure XXXVI), further supporting the notion that increased DNMT3A stability following wire injury is critical for neointimal hyperplasia formation.
Figure 8. In vivo knock-down of DNMT3A in femoral artery reduces wire injury-induced neointima formation.

Femoral arteries were coated with either scramble shRNA (shScr) or DNMT3A shRNA (shDNMT3A) lentivirus coexpressing mCherry immediately following wire injury. Representative immunostainings of 2-week post-injury samples for ACTA2, DNMT3A and DNMT3B are shown (n=4). Green, mCherry (red) and DAPI (blue) were merged. mCherry was used to verify lentiviral infection. Dotted line in image marks the elastic lamina. White line indicates endothelial layer. L, lumen. Scale bars: x20, 100 μm; or x60, 20 μm.
We further evaluated the effect of DNMT3A knockdown in vivo on SMC contractile gene expression. By performing whole transcriptome RNA-Seq analysis of shDNMT3A expressing femoral artery samples upon injury, we found that a total of 4323 DEGs were identified in shDNMT3A samples, with 1661 being upregulated and 2662 being downregulated compared to injured controls (Supplemental Table III). KEGG pathway analysis showed that vascular smooth muscle contraction was one of the top upregulated pathways by shDNMT3A (Online Figure XXXIIID). Conversely, cytokine and chemokine signaling, and cell adhesion molecule pathways were among the top downregulated pathways in shDNMT3A arteries (Online Figure XXXIIID). Several SMC-specific contractile genes including Myh11, Acta2, Cald1, Cnn1, and Tagln were upregulated in shDNMT3A arteries compared to injured controls (Online Figure XXXIIIE). These findings support the notion that increased DNMT3A expression following vascular injury is required for increased DNA methylation and suppression of SMC-specific contractile genes.
DISCUSSION
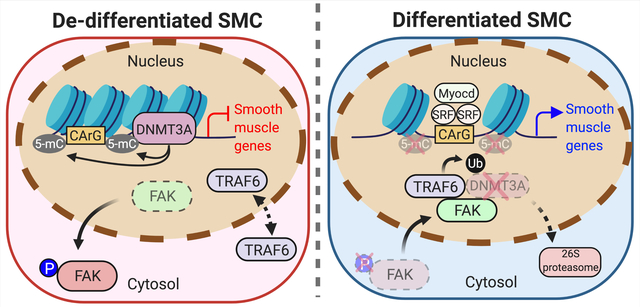
In this study, we demonstrated that the nuclear FAK-DNMT3A axis regulates SMC phenotypic switching and determines SMC’s fate to differentiate or dedifferentiate. It appears that in healthy mouse arteries, FAK naturally resides within the nucleus of SMCs to maintain a differentiated state by destabilizing DNMT3A protein and keeping DNA methylation low. In injured arteries, increased active cytoplasmic FAK and DNA methylation, results in SMC dedifferentiation. Of clinical importance, forced nuclear localization of FAK induced by pharmacological FAK catalytic inhibition or genetic FAK kinase-dead mutant during vessel injury promoted SMC differentiation by reducing DNMT3A expression and DNA methylation, which increased SMC contractile gene expression. Our RNA-Seq data further demonstrated that genes primarily associated with SMC contraction were downregulated and inflammation-associated genes were upregulated following wire injury but were blocked by FAK-I treatment. Importantly, we observed similar patterns in DNMT3A expression and FAK activation and localization within SMCs of both healthy and atherosclerotic human arteries. These data show that the paradigm of FAK activation and localization and DNMT3A expression may be evolutionarily conserved between mice and humans, and that forcing FAK back to the nucleus could prove beneficial in treating various vascular remodeling-associated disease in humans.
The study also established the role of FAK activation in the cytoplasm in promoting SMC dedifferentiation and synthetic phenotypes during vascular remodeling via modulating DNA methylation through DNMT3A. Interestingly, vascular injury upregulated only DNMT3A protein, but not DNMT3B or DNMT1. Although DNMTs are structurally similar, endogenous IP demonstrated that nuclear FAK interacted only with DNMT3A, but not DNMT3B or DNTM1, to promote its ubiquitination and proteasomal degradation via E3 ligase TRAF6. Additionally, using FAK nonnuclear localizing mutant (FAK-NLM), we demonstrated that FAK nuclear localization is required to promote SMC differentiation following FAK-I treatment. This revealed that FAK inhibition promotes SMC differentiation through regulation of DNMT3A stability by increased FAK nuclear localization, but not through inhibition of FAK cytoplasmic signaling.
Further, knockdown of the de novo DNA methyltransferases DNMT3A and DNMT3B revealed that DNMT3A, but not DNMT3B, is responsible for silencing SMC contractile genes. This could either be simply because DNMT3B expression levels are very low compared to that of DNMT3A in SMCs, or DNMT3A and DNMT3B may have differential regulation for various gene promoters (e.g., contractile gene promoters) which is currently not known. The study found the new mechanism that lowering DNMT3A specifically by nuclear FAK decrease DNA methylation to enhance gene expression related to SMC differentiation.
FAK inhibition not only reduced 5-mC levels, but also promoted an active chromatin environment as observed by elevated active histone marks (H4ac, H3K27ac) within contractile gene promoters to drive SMC differentiation. Active SMC contractile genes were further verified by enhanced binding of SRF, a transcription factor critical for contractile gene expression. Further characterization of alterations on the histone landscape by FAK inhibition still needs to be done and may identify a new molecular mechanism regulating SMC phenotype switching by FAK.
Finding a new therapy that halts SMC dedifferentiation and forces a contractile phenotype could prove promising in the treatment of occlusive vascular diseases. Several cardiovascular diseases associated with atherosclerosis, such as myocardial infarction and stroke, result from rupture of unstable plaques. It is thought that stabilization of plaques through thickening of the fibrous cap with contractile SMCs could prove beneficial in reducing myocardial infarctions and strokes. Our study has demonstrated that FAK activity is elevated in human atherosclerosis specimen and that FAK-I could force SMC differentiation in vitro and in mice. Although blocking SMC dedifferentiation in atherosclerosis would be beneficial to reduce intimal thickening, FAK inhibitors also affect SMC proliferation and migration, which may reduce SMC investment into the cap region and could adversely affect the formation of a proper fibrous cap. Further studies evaluating the effect of FAK on SMC function in atherosclerosis will be needed. However, our study does demonstrate that FAK inhibitors could prove to be a potential therapeutic option to block restenosis following percutaneous transluminal angioplasty procedures. While advances in drug coated balloons and stents have increased patency following intervention, they still have their limitations.47–49 A lot of these limitations have been observed when treating peripheral artery disease. Due to the mechanical nature of the leg, stents can become cracked, promote artery kinking, and result in pseudoaneurysms.50–52 As such, a systemically deliverable drug that can block SMC restenosis following percutaneous transluminal angioplasty could prove significantly beneficial to patients.
In conclusion, nuclear FAK appears to be a fundamental regulator of SMC differentiation by regulating master enzymes in DNA methylation and increased cytoplasmic localization of FAK allows SMCs to undergo phenotypic switching. SMC phenotypic switching is a key phenomenon underlying several vessel narrowing diseases such as atherosclerosis. While interventional therapies can reopen vessels following occlusion during atherosclerosis, they often result in restenosis. The changes in FAK subcellular localization in healthy and diseased arteries from both mice and human further support the notion that forced FAK nuclear localization through pharmacological FAK inhibitors could prove beneficial in blocking vascular remodeling.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What is Known?
Smooth muscle cell (SMC) dedifferentiation promotes SMC proliferation and reduces contractile gene expression.
Focal adhesion kinase (FAK) catalytic inhibition reduces SMC proliferation in part via the GATA4-cyclin D axis.
While FAK is inactive in the nucleus of SMCs in healthy vessels, FAK is active in the cytoplasm in injured vessels.
What New Information Does This Article Contribute?
FAK catalytic inhibition reduces DNA methylation of contractile gene promoters in SMCs and maintains differentiated status of SMCs.
Nuclear FAK specifically suppresses DNA methyltransferase 3A (DNMT3A) protein stability by promoting DNMT3A ubiquitination.
Of clinical importance, the role of FAK/DNMT3A regulation of SMC phenotypic switching is correlated with human atherosclerosis specimens.
Although epigenetic changes are critical in SMC phenotypic switching, the fundamental epigenetic machinery regulating the fate of SMC phenotype has not been elucidated. We report the molecular mechanism of how FAK regulates DNA methylation and SMC contractile gene expression, determining SMC’s fate during SMC remodeling. We found that DNMT3A, but not DNMT3B, is responsible for silencing SMC contractile genes during vascular remodeling. FAK inhibition forced FAK nuclear localization which mediated DNMT3A ubiquitination and proteasomal degradation via E3 ligase TRAF6. RNA sequencing identified SMC contractile genes as one of most significantly upregulated groups by FAK inhibition from injured artery samples compared to vehicle group. Importantly, we observed similar patterns in DNMT3A expression and FAK activation and localization within SMCs of both healthy and atherosclerotic human arteries. These data suggest that FAK activation and localization and DNMT3A expression are evolutionarily conserved between mice and humans. Overall, this study has significant translational value for the use FAK inhibitors of to block SMC phenotypic switching during vascular remodeling and atherosclerosis.
Acknowledgments
SOURCES OF FUNDING
This work was supported by American Heart Association 16GRNT30960007 (to S Lim), National Institutes of Health R01CA190688 (to E Ahn), R01HL136432 (to S Lim), R01HL158875 (to S Lim), and 2019 College of Medicine Intramural Grant (to S Lim).
Nonstandard Abbreviations and Acronyms:
- 5-mC
5-methylcytosine
- ACTA2
α-smooth muscle actin
- CNN1
calponin 1
- CALD1
Caldesmon
- DNMT
DNA methyltransferase
- FAK
focal adhesion kinase
- FAK-I
FAK inhibitor
- gDNA
genomic DNA
- hCASMC
human coronary artery smooth muscle cell
- KD
kinase-dead
- MYH11
myosin heavy chain 11
- NLM
nonnuclear localizing mutant
- pY397
autophosphorylation at tyrosine 397
- RT-qPCR
real-time quantitative polymerized chain reaction
- shRNA
short hairpin RNA
- SMC
smooth muscle cell
- TAGLN
transgelin, smooth muscle protein 22 a
- TRAF6
TNF receptor associated factor 6
- WT
wild-type
Footnotes
DISCLOSURES
None, the authors have no conflicts to disclose.
SUPPLEMENTAL MATERIALS
Expanded Materials and Methods
Data Supplement Figures I–XXXVI
Data Supplement Tables I–III
Full Unedited Blots
Publisher's Disclaimer: This article is published in its accepted form. It has not been copyedited and has not appeared in an issue of the journal. Preparation for inclusion in an issue of Circulation Research involves copyediting, typesetting, proofreading, and author review, which may lead to differences between this accepted version of the manuscript and the final, published version.
REFERENCES
- 1.Newman AAC, Serbulea V, Baylis RA, Shankman LS, Bradley X, Alencar GF, Owsiany K, Deaton RA, Karnewar S, Shamsuzzaman S, Salamon A, Reddy MS, Guo L, Finn A, Virmani R, Cherepanova OA and Owens GK. Multiple cell types contribute to the atherosclerotic lesion fibrous cap by PDGFRbeta and bioenergetic mechanisms. Nat Metab. 2021;3:166–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AA, Greene ES, Straub AC, Isakson B, Randolph GJ and Owens GK. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21:628–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pan H, Xue C, Auerbach BJ, Fan J, Bashore AC, Cui J, Yang DY, Trignano SB, Liu W, Shi J, Ihuegbu CO, Bush EC, Worley J, Vlahos L, Laise P, Solomon RA, Connolly ES, Califano A, Sims PA, Zhang H, Li M and Reilly MP. Single-Cell Genomics Reveals a Novel Cell State During Smooth Muscle Cell Phenotypic Switching and Potential Therapeutic Targets for Atherosclerosis in Mouse and Human. Circulation. 2020;142:2060–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gomez D and Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovascular research. 2012;95:156–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nguyen AT, Gomez D, Bell RD, Campbell JH, Clowes AW, Gabbiani G, Giachelli CM, Parmacek MS, Raines EW, Rusch NJ, Speer MY, Sturek M, Thyberg J, Towler DA, Weiser-Evans MC, Yan C, Miano JM and Owens GK. Smooth muscle cell plasticity: fact or fiction? Circulation research. 2013;112:17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AA, Greene ES, Straub AC, Isakson B, Randolph GJ and Owens GK. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nature medicine. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clowes AW, Reidy MA and Clowes MM. Mechanisms of stenosis after arterial injury. Laboratory investigation; a journal of technical methods and pathology. 1983;49:208–15. [PubMed] [Google Scholar]
- 8.Newby AC. An overview of the vascular response to injury: a tribute to the late Russell Ross. Toxicology letters. 2000;112–113:519–29. [DOI] [PubMed] [Google Scholar]
- 9.Chaabane C, Otsuka F, Virmani R and Bochaton-Piallat ML. Biological responses in stented arteries. Cardiovascular research. 2013;99:353–63. [DOI] [PubMed] [Google Scholar]
- 10.Kornowski R, Mintz GS, Kent KM, Pichard AD, Satler LF, Bucher TA, Hong MK, Popma JJ and Leon MB. Increased restenosis in diabetes mellitus after coronary interventions is due to exaggerated intimal hyperplasia. A serial intravascular ultrasound study. Circulation. 1997;95:1366–9. [DOI] [PubMed] [Google Scholar]
- 11.Mitchell RN and Libby P. Vascular remodeling in transplant vasculopathy. Circulation research. 2007;100:967–78. [DOI] [PubMed] [Google Scholar]
- 12.Li JJ, Nie SP, Zhang CY, Gao Z, Zheng X and Guo YL. Is inflammation a contributor for coronary stent restenosis? Medical hypotheses. 2007;68:945–51. [DOI] [PubMed] [Google Scholar]
- 13.Drachman DE and Simon DI. Restenosis: Intracoronary Brachytherapy. Current treatment options in cardiovascular medicine. 2002;4:109–118. [DOI] [PubMed] [Google Scholar]
- 14.Findeisen HM, Gizard F, Zhao Y, Qing H, Heywood EB, Jones KL, Cohn D and Bruemmer D. Epigenetic regulation of vascular smooth muscle cell proliferation and neointima formation by histone deacetylase inhibition. Arterioscler Thromb Vasc Biol. 2011;31:851–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Newell-Price J, Clark AJ and King P. DNA methylation and silencing of gene expression. Trends Endocrinol Metab. 2000;11:142–8. [DOI] [PubMed] [Google Scholar]
- 16.Schubeler D Function and information content of DNA methylation. Nature. 2015;517:321–6. [DOI] [PubMed] [Google Scholar]
- 17.Madsen CS, Regan CP and Owens GK. Interaction of CArG elements and a GC-rich repressor element in transcriptional regulation of the smooth muscle myosin heavy chain gene in vascular smooth muscle cells. J Biol Chem. 1997;272:29842–51. [DOI] [PubMed] [Google Scholar]
- 18.McDonald OG, Wamhoff BR, Hoofnagle MH and Owens GK. Control of SRF binding to CArG box chromatin regulates smooth muscle gene expression in vivo. J Clin Invest. 2006;116:36–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McDonald OG and Owens GK. Programming smooth muscle plasticity with chromatin dynamics. Circ Res. 2007;100:1428–41. [DOI] [PubMed] [Google Scholar]
- 20.Rozenberg JM, Tesfu DB, Musunuri S, Taylor JM and Mack CP. DNA methylation of a GC repressor element in the smooth muscle myosin heavy chain promoter facilitates binding of the Notch-associated transcription factor, RBPJ/CSL1. Arterioscler Thromb Vasc Biol. 2014;34:2624–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Denis H, Ndlovu MN and Fuks F. Regulation of mammalian DNA methyltransferases: a route to new mechanisms. EMBO Rep. 2011;12:647–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu R, Jin Y, Tang WH, Qin L, Zhang X, Tellides G, Hwa J, Yu J and Martin KA. Ten-eleven translocation-2 (TET2) is a master regulator of smooth muscle cell plasticity. Circulation. 2013;128:2047–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salmon M, Gomez D, Greene E, Shankman L and Owens GK. Cooperative binding of KLF4, pELK-1, and HDAC2 to a G/C repressor element in the SM22alpha promoter mediates transcriptional silencing during SMC phenotypic switching in vivo. Circ Res. 2012;111:685–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Calalb MB, Polte TR and Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol. 1995;15:954–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown XQ, Bartolak-Suki E, Williams C, Walker ML, Weaver VM and Wong JY. Effect of substrate stiffness and PDGF on the behavior of vascular smooth muscle cells: implications for atherosclerosis. J Cell Physiol. 2010;225:115–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taylor JM, Mack CP, Nolan K, Regan CP, Owens GK and Parsons JT. Selective expression of an endogenous inhibitor of FAK regulates proliferation and migration of vascular smooth muscle cells. Mol Cell Biol. 2001;21:1565–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sayers RL, Sundberg-Smith LJ, Rojas M, Hayasaka H, Parsons JT, Mack CP and Taylor JM. FRNK expression promotes smooth muscle cell maturation during vascular development and after vascular injury. Arterioscler Thromb Vasc Biol. 2008;28:2115–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bae YH, Mui KL, Hsu BY, Liu SL, Cretu A, Razinia Z, Xu T, Pure E and Assoian RK. A FAK-Cas-Rac-lamellipodin signaling module transduces extracellular matrix stiffness into mechanosensitive cell cycling. Sci Signal. 2014;7:ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mui KL, Bae YH, Gao L, Liu SL, Xu T, Radice GL, Chen CS and Assoian RK. N-Cadherin Induction by ECM Stiffness and FAK Overrides the Spreading Requirement for Proliferation of Vascular Smooth Muscle Cells. Cell Rep. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klein EA, Yin L, Kothapalli D, Castagnino P, Byfield FJ, Xu T, Levental I, Hawthorne E, Janmey PA and Assoian RK. Cell-cycle control by physiological matrix elasticity and in vivo tissue stiffening. Current biology : CB. 2009;19:1511–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jeong K, Kim JH, Murphy JM, Park H, Kim SJ, Rodriguez Y, Kong H, Choi C, Guan JL, Taylor JM, Lincoln TM, Gerthoffer WT, Kim JS, Ahn EE, Schlaepfer DD and Lim SS. Nuclear Focal Adhesion Kinase Controls Vascular Smooth Muscle Cell Proliferation and Neointimal Hyperplasia Through GATA4-mediated Cyclin D1 Transcription. Circ Res. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao Y, Zang G, Yin T, Ma X, Zhou L, Wu L, Daniel R, Wang Y, Qiu J and Wang G. A novel mechanism of inhibiting in-stent restenosis with arsenic trioxide drug-eluting stent: Enhancing contractile phenotype of vascular smooth muscle cells via YAP pathway. Bioact Mater. 2021;6:375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jeong K, Kim JH, Murphy JM, Park H, Kim SJ, Rodriguez YAR, Kong H, Choi C, Guan JL, Taylor JM, Lincoln TM, Gerthoffer WT, Kim JS, Ahn EE, Schlaepfer DD and Lim SS. Nuclear Focal Adhesion Kinase Controls Vascular Smooth Muscle Cell Proliferation and Neointimal Hyperplasia Through GATA4-Mediated Cyclin D1 Transcription. Circ Res. 2019;125:152–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu R, Leslie KL and Martin KA. Epigenetic regulation of smooth muscle cell plasticity. Biochim Biophys Acta. 2015;1849:448–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Owens GK, Kumar MS and Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. [DOI] [PubMed] [Google Scholar]
- 36.Rozenberg JM, Tesfu DB, Musunuri S, Taylor JM and Mack CP. DNA methylation of a GC repressor element in the smooth muscle myosin heavy chain promoter facilitates binding of the Notch-associated transcription factor, RBPJ/CSL1. Arterioscler Thromb Vasc Biol. 2014;34:2624–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hagg S, Skogsberg J, Lundstrom J, Noori P, Nilsson R, Zhong H, Maleki S, Shang MM, Brinne B, Bradshaw M, Bajic VB, Samnegard A, Silveira A, Kaplan LM, Gigante B, Leander K, de Faire U, Rosfors S, Lockowandt U, Liska J, Konrad P, Takolander R, Franco-Cereceda A, Schadt EE, Ivert T, Hamsten A, Tegner J and Bjorkegren J. Multi-organ expression profiling uncovers a gene module in coronary artery disease involving transendothelial migration of leukocytes and LIM domain binding 2: the Stockholm Atherosclerosis Gene Expression (STAGE) study. PLoS Genet. 2009;5:e1000754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ayari H and Bricca G. Microarray analysis reveals overexpression of IBSP in human carotid plaques. Adv Med Sci. 2012;57:334–40. [DOI] [PubMed] [Google Scholar]
- 39.Lim ST, Miller NL, Chen XL, Tancioni I, Walsh CT, Lawson C, Uryu S, Weis SM, Cheresh DA and Schlaepfer DD. Nuclear-localized focal adhesion kinase regulates inflammatory VCAM-1 expression. J Cell Biol. 2012;197:907–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lim ST, Chen XL, Lim Y, Hanson DA, Vo TT, Howerton K, Larocque N, Fisher SJ, Schlaepfer DD and Ilic D. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol Cell. 2008;29:9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu J, Qin B, Moyer AM, Nowsheen S, Liu T, Qin S, Zhuang Y, Liu D, Lu SW, Kalari KR, Visscher DW, Copland JA, McLaughlin SA, Moreno-Aspitia A, Northfelt DW, Gray RJ, Lou Z, Suman VJ, Weinshilboum R, Boughey JC, Goetz MP and Wang L. DNA methyltransferase expression in triple-negative breast cancer predicts sensitivity to decitabine. J Clin Invest. 2018;128:2376–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jia Y, Li P, Fang L, Zhu H, Xu L, Cheng H, Zhang J, Li F, Feng Y, Li Y, Li J, Wang R, Du JX, Li J, Chen T, Ji H, Han J, Yu W, Wu Q and Wong J. Negative regulation of DNMT3A de novo DNA methylation by frequently overexpressed UHRF family proteins as a mechanism for widespread DNA hypomethylation in cancer. Cell Discov. 2016;2:16007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okano M, Xie S and Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet. 1998;19:219–20. [DOI] [PubMed] [Google Scholar]
- 44.Xie S, Wang Z, Okano M, Nogami M, Li Y, He WW, Okumura K and Li E. Cloning, expression and chromosome locations of the human DNMT3 gene family. Gene. 1999;236:87–95. [DOI] [PubMed] [Google Scholar]
- 45.Cedar H and Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009;10:295–304. [DOI] [PubMed] [Google Scholar]
- 46.Lachner M and Jenuwein T. The many faces of histone lysine methylation. Curr Opin Cell Biol. 2002;14:286–98. [DOI] [PubMed] [Google Scholar]
- 47.Diamantopoulos A and Katsanos K. Treating femoropopliteal disease: established and emerging technologies. Semin Intervent Radiol. 2014;31:345–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ho KJ and Owens CD. Diagnosis, classification, and treatment of femoropopliteal artery in-stent restenosis. J Vasc Surg. 2017;65:545–557. [DOI] [PubMed] [Google Scholar]
- 49.Somitsu Y, Ikari Y, Ui K, Nakamura M, Hara K, Saeki F, Degawa T, Tamura T, Yabuki S and Yamaguchi T. Elastic recoil following percutaneous transluminal coronary angioplasty and Palmaz-Schatz stent implantation. J Invasive Cardiol. 1995;7:165–72. [PubMed] [Google Scholar]
- 50.Dos Reis JMC, Kudo FA and do Carmo Bastos M. Fracture of a popliteal nitinol stent and pseudoaneurysm: a case report and review of the literature. J Surg Case Rep. 2019;2019:rjz312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsuji Y, Kitano I, Iida O, Kajita S, Sawada K and Nanto S. Popliteal pseudoaneurysm caused by stent fracture. Ann Vasc Surg. 2011;25:840 e5–8. [DOI] [PubMed] [Google Scholar]
- 52.Adlakha S, Sheikh M, Wu J, Burket MW, Pandya U, Colyer W, Eltahawy E and Cooper CJ. Stent fracture in the coronary and peripheral arteries. J Interv Cardiol. 2010;23:411–9. [DOI] [PubMed] [Google Scholar]
- 53.Kobayashi M, Inoue K, Warabi E, Minami T and Kodama T. A simple method of isolating mouse aortic endothelial cells. J Atheroscler Thromb. 2005;12:138–42. [DOI] [PubMed] [Google Scholar]
- 54.Ray JL, Leach R, Herbert JM and Benson M. Isolation of vascular smooth muscle cells from a single murine aorta. Methods Cell Sci. 2001;23:185–8. [DOI] [PubMed] [Google Scholar]
- 55.Slack-Davis JK, Martin KH, Tilghman RW, Iwanicki M, Ung EJ, Autry C, Luzzio MJ, Cooper B, Kath JC, Roberts WG and Parsons JT. Cellular characterization of a novel focal adhesion kinase inhibitor. J Biol Chem. 2007;282:14845–52. [DOI] [PubMed] [Google Scholar]
- 56.Ngo P, Ramalingam P, Phillips JA and Furuta GT. Collagen gel contraction assay. Methods Mol Biol. 2006;341:103–9. [DOI] [PubMed] [Google Scholar]
- 57.Nam D, Ni CW, Rezvan A, Suo J, Budzyn K, Llanos A, Harrison D, Giddens D and Jo H. Partial carotid ligation is a model of acutely induced disturbed flow, leading to rapid endothelial dysfunction and atherosclerosis. Am J Physiol Heart Circ Physiol. 2009;297:H1535–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gaspar-Maia A, Alajem A, Polesso F, Sridharan R, Mason MJ, Heidersbach A, Ramalho-Santos J, McManus MT, Plath K, Meshorer E and Ramalho-Santos M. Chd1 regulates open chromatin and pluripotency of embryonic stem cells. Nature. 2009;460:863–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim JH, Baddoo MC, Park EY, Stone JK, Park H, Butler TW, Huang G, Yan X, Pauli-Behn F, Myers RM, Tan M, Flemington EK, Lim ST and Ahn EY. SON and Its Alternatively Spliced Isoforms Control MLL Complex-Mediated H3K4me3 and Transcription of Leukemia-Associated Genes. Mol Cell. 2016;61:859–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data in this manuscript have been deposited in the Gene Expression Omnibus under accession number GSE183143.