

Abstract
Protein neddylation is catalyzed by a three-enzyme cascade, namely an E1 NEDD8-activating enzyme (NAE), one of two E2 NEDD8 conjugation enzymes and one of several E3 NEDD8 ligases. The physiological substrates of neddylation are the family members of cullin, the scaffold component of cullin RING ligases (CRLs). Currently, a potent E1 inhibitor, MLN4924, also known as pevonedistat, is in several clinical trials for anti-cancer therapy. Here we report the discovery, through virtual screening and structural modifications, of a small molecule compound HA-1141 that directly binds to NAE in both in vitro and in vivo assays and effectively inhibits neddylation of cullins 1–5. Surprisingly, unlike MLN4924, HA-1141 also triggers non-canonical endoplasmic reticulum (ER) stress and PKR-mediated terminal integrated stress response (ISR) to activate ATF4 at an early stage, and to inhibit protein synthesis and mTORC1 activity at a later stage, eventually leading to autophagy induction. Biologically, HA-1141 suppresses growth and survival of cultured lung cancer cells and tumor growth in in vivo xenograft lung cancer models at a well-tolerated dose. Taken together, our study has identified a small molecule compound with the dual activities of blocking neddylation and triggering ER stress, leading to growth suppression of cancer cells.
Key words: Autophagy, Cullin RING ligase, ER stress, mTORC1, Neddylation, Small molecule inhibitor
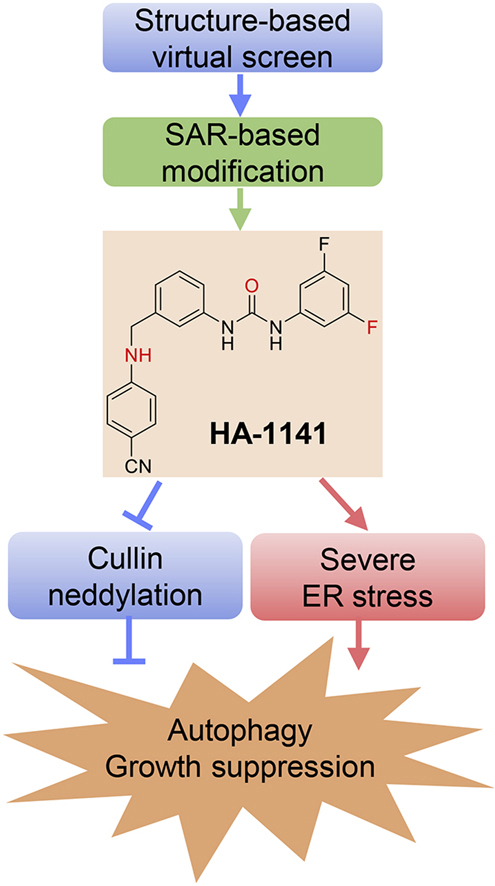
Graphical abstract
We identified HA-1141 through virtual screen and SAR-based modification. HA-1141 has dual activities in blocking cullin neddylation and in triggering severe ER stress to a) induce autophagy and b) suppress growth of cancer cells both in vitro and in vivo.
1. Introduction
Cell survival relies on protein homeostasis, in which proteins must be synthesized in proper amounts, folded with high fidelity, and then degraded in a timely fashion after functional execution. In response to external or internal cues, cells have developed several defensive mechanisms to either restore cellular homeostasis or commit to cell death. For example, the mTOR signal is activated for protein synthesis when growth-arrested cells were supplied with nutrients1; the endoplasmic reticulum (ER) stress response is triggered to reduce the level of defective proteins2; and the ubiquitin‒proteasome system (UPS) and autophagy are activated for timely protein degradation3.
The ER stress response is an evolutionarily conserved process, known as the unfolded protein response (UPR), that is activated in response to the accumulation of misfolded proteins in a cellular effort to maintain or restore ER homeostasis2. The canonical ER stress response is mediated by activation of ER chaperone protein binding-immunoglobulin protein BIP/GRP78 and three traditional UPR sensor proteins, including inositol-requiring enzyme 1α (IRE1α) (PRKR)-like endoplasmic reticulum kinase (PERK), and activating transcription factor 6 (ATF6), leading to sequential activation of the P-eIF2α/ATF4/CHOP axis, followed by integrated stress response (ISR) to recover from stress or to lead to cell death via apoptosis4. In the event of severe ER stress, cytotoxic autophagy is eventually induced for cell death via activating ATF4 activity and blocking the mTORC1 function5.
Autophagy is a highly conserved process, characterized by the lysosomal degradation of intracellular components that are engulfed in double-membrane vesicles known as autophagosomes6. Autophagy is usually considered a cellular survival mechanism by recycling building blocks and energy for cellular renovation and homeostasis6. However, under some stress conditions, such as ER stress, autophagy can cause massive cell death7. In this sense, autophagy-mediated cell death has emerged as an alternative mechanism to kill cancer cells. Several anti-cancer molecules, such as ABTL0812, trigger cancer cell death via an ER-stress-coupled, autophagy-dependent mechanism5.
Protein neddylation is one type of posttranslational modification. It is catalyzed by a three-enzyme cascade, namely a NEDD8-activating enzyme (NAE/E1), a NEDD8-conjugating enzyme (UBE2M or UBE2F/E2), and a NEDD8 ligase (E3), leading to covalent conjugation of a ubiquitin-like molecule NEDD8 to a protein substrate to regulate its function/activity8. The cullin family members, the scaffold components of cullin-RING ubiquitin ligases (CRLs), are the best-characterized physiological substrates of neddylation9. Several other non-cullin proteins, known to play important roles in regulation of mRNA translation, were recently reported as neddylation substrates10,11. Accumulating data from a variety of studies have shown that neddylation modification is over-activated in multiple types of human cancers including lung cancer and is frequently associated with a worse overall patient survival, and neddylation has been validated as an attractive anticancer target12. Indeed, a small molecule neddylation E1 inhibitor, MLN4924, also known as pevonedistat, has been tested in many clinical trials for anticancer therapy (https://clinicaltrials.gov/)13. Apart from MLN4924, most known NAE inhibitors have been discovered by in silico approaches and possess less favorable potency and selectivity profiles, with activity in the micromolar range12,14.
Our laboratory has previously validated that neddylation E2 UBE2F15 and E3 SAG16 are promising anti-lung cancer targets. To identify selective inhibitors of UBE2F, we conducted a computer-aided virtual screening for small molecules that have potential to disrupt the UBA3‒UBE2F binding based upon the co-crystal structure17, followed by structure‒activity-based chemical modifications. Here we report the discovery of such a small molecule compound, designated HA-1141, which has the dual activities of blocking cullin neddylation and triggering ER stress, eventually leading to growth suppression of cancer cells. Mechanistic analysis revealed that in addition to binding and inhibiting neddylation E1 NAE, the compound also triggers severe non-canonical ER stress and ISR with early induction of ATF4 and subsequent inactivation of mTORC1 and inhibition of protein synthesis. This is the first small molecule that has dual activity in neddylation inactivation and ER stress induction with anti-cancer activity.
2. Materials and methods
2.1. Molecular modelling
The crystal structure of human UBE2F in complex with UBA3 (PDB entry: 3FN1) was obtained from RCSB Protein Data Bank and utilized in molecular dynamic (MD) simulations and virtual screening. All MD simulations were carried out in AMBER 1118. The general AMBER force field (gaff) was used for the proteins, and all missing atoms were added using the “tleap” program. Cl− counter ions were added to neutralize the charge in the system. The proteins were placed into a periodic TIP3P water box. The Particle Mesh Ewald (PME) algorithm was used to handle the long-range electrostatics.
The system was first subjected to a three-stage minimization by the “sander” program19, which has been described in our previous work20. The system was then gradually heated up from 0 to 300 K over a period of 50 ps with 2.0 kcal/mol/Å2 restrain. Subsequently, 50 ps MD simulations were carried out at 300 K with the same restrain and 50 ps MD without any restrain. Finally, 16 ns NPT MD simulation with the target temperature of 300 K and the target pressure of 1 atm were carried out. The SHAKE algorithm was used to restrain all bonds involving hydrogen atoms18, and the time step was set to 2.0 fs. Based on the trajectories produced by MD simulation, the contribution of each residue to UBE2F‒UBA3 binding was calculated by MM/GBSA decomposition analysis21, where two hot-spots around residues F56 and V30 were identified as being critical for UBE2F and UBA3 binding and utilized for virtual screening campaigns.
Preparation of the protein structures for virtual screening was performed using the Protein Preparation wizard module in Schrödinger 9.0. All water molecules were removed from the system, and a restrained partial minimization was carried out with the root-mean-square deviation (RMSD) value set to 0.3 Å. The protein grid boxes for docking were generated based on three binding sites (i.e., F56 and V30 pockets, as well as the catalytic active site of UBE2F). The scaling factors for van der Waals radii were set to 1.0 and the maximum partial atomic charge was set to 0.25.
A screening library of about 240,000 compounds was obtained from Specs chemical library and prepared with the LigPrep module. Protonated states were generated at pH = 7.0 ± 2.0. The Epik state penalties were included in docking scores. Default settings were used for the other parameters. The Glide module in Schrödinger was used to perform docking simulations. All the compounds were docked into the binding sites of UBE2F and the binding affinities were scored and ranked by standard precision (SP). The top-ranked 50,000 compounds from each screening were submitted to Glide XP docking and 2000 top-ranked molecules were obtained. ADMET properties of these compounds were subsequently predicted by ACD/ADME package22 to remove those that failed to fulfill the following rules: (1) logP/logD (pH = 7.0) < 5.5; (2) violation of Lipinski's rules of five < 2; (3) violation of Opera's rules of drug-likeness < 3; (4) functional groups without toxic, reactive, or otherwise undesirable moieties defined by the REOS rules. The top 200 molecules were then clustered based on the Tanimoto distance calculated from the FCFP_4 fingerprints using Find Diverse Molecule module in Discovery Studio 2.523. Finally, 30 top-ranked compounds were obtained from each screening.
2.2. Cell lines and chemicals
Human lung cancer cell lines H358, H1299 and H2170 were obtained from ATCC (American Type of Cell Collection, Manassas, VA, USA). Non-immortalized MEF cells were cultured and maintained by our laboratory. Cells were cultured in RPMI 1640 medium (for H358 and H2170) or DMEM medium (for H1299 and MEF), supplemented with 10% fetal bovine serum (extra 1% non-essential amino acids for MEF) and incubated in a 5% CO2 humidified chamber at 37 °C. MLN4924 (#B1036) and tunicamycin (#B7417) was purchased from ApexBio. Chloroquine (CQ, #C6628), chlorhexidine (CHX, #C7698) and N-acetyl-l-cysteine (NAC, #A7250) were purchased from Sigma. MG132 (#HY-13259) was purchased from MedChem Express.
2.3. Western blotting
Cells were lysed in a RIPA buffer containing both protease inhibitors and phosphatase inhibitors. Lysates were separated in SDS-PAGE gels, transferred to PVDF membranes and probed with the primary antibodies overnight at 4 °C. The membranes were then incubated with suitable HRP-conjugated second antibodies (Jackson). Blots were visualized using standard chemical luminescence methodology. Antibodies used are listed in Supporting Information Table S1.
2.4. Constructs and protein purification
NEDD8 and enzymes for the in-vitro neddylation assay were prepared as described previously24. Briefly, NEDD8 terminating at Gly 76, APPBP1, UBE2M, UBE2F were cloned into a home-made variant of pET-28b vector fused with a His6-SUMO tag at the N-terminus. UBA3 and SAG were cloned into the vector pGEX-6p-1 with GST-fusion expression (GE Healthcare). His-RBX1-CUL1CTD with mutations of L421E, V451E, V452K, and Y455K at CUL1 residues 411–776 to increase solubility were prepared as described previously24. CUL5CTD with the mutations of L407E, L439K, V440K to increase solubility was cloned into the home-made variant of pRSFDuet vector fused with a His6 tag24. The constructs were expressed or co-expressed (GST-UBA3 and His6-SUMO-APPBP1, His6-CUL5CTD and GST-SAG, His6-CUL1CTD and RBX1) in BL21 (DE3) Escherichia coli (TransGen Biotech) and then purified by Ni-NTA agarose beads (QIAGEN). Glutathione Sepharose 4B beads (GE Healthcare) were applied after treatment with Ulp1 to remove His6-SUMO tag of APPBP1 and GST-tag of SAG. Finally, all proteins were purified by gel-filtration chromatography and stored at −80 °C.
2.5. Cellular thermal shift assay (CETSA)
CETSA was performed according to the reported method25. Briefly, cells were treated with DMSO, HA-1141 or MLN4924 for 24 h, harvested and washed with cold PBS for three times, heated at the indicated temperatures for 3 min in a thermal cycler (BIO-RAD, T100TM Thermal Cycler) followed by addition of 30 μL of PBS with proteasome inhibitor and lysed using three cycles of freeze-thawing in liquid nitrogen. The lysates were then centrifuged at 14,000 rpm for 30 min at 4 °C and the protein levels of UBA3, NAE1 and UBE2F in equal amounts of the supernatant were examined by Western blots. Results were representative of three independent experiments.
2.6. The in vitro thermal shift assay (TSA)
Purified UBA3/NAE1 (0.5 μg) was incubated with DMSO or HA-1141 (final concentration of 100 μmol/L) for 10 min at room temperature in 50 μL of buffer containing 25 mmol/L HEPES (pH = 7.4), 1 mmol/L DTT and 150 mmol/L NaCl, then heated at the indicated temperature for 3 min in a BIO-RAD, T100TM Thermal Cycler. The resulting mixtures were centrifuged at 14,000 rpm for 30 min at 4 °C, and the soluble components were examined by Western blotting.
2.7. The in vitro E2-NEDD8 thioester assay
The reaction mixture contains 500 nmol/L NEDD8, 25 nmol/L UBA3/APPBP1, and 200 nmol/L UBE2M or UBE2F in a Tris-HCl (pH = 7.4)-adjusted buffer containing 0.1 mg/mL BSA, 0.5 mmol/L DTT and 5 mmol/L MgCl2. Indicated compounds were incubated in the mixture (final DMSO ≤1%) for 15 min at 4 °C before addition of 20 μmol/L ATP and incubation for 1 min at 37 °C. The 4 × SDS loading buffer (without DTT) was added to quench the reaction. Final samples were subjected to Western blotting analysis.
2.8. The in vivo E2-NEDD8 thioester assay
Cells were treated with HA-1141 at different concentrations, DMSO or MLN4924 as control for 24 h, and lysed in RIPA buffer containing both protease inhibitors and phosphatase inhibitors. To analyze the E2-NEDD8 thioester levels, lysates were fractionated by non-reducing SDS-PAGE and immunoblotted with antibodies against UBE2M or UBE2F.
2.9. The in vitro cullin neddylation assay
The reaction mixture contains 200 nmol/L NEDD8, 25 nmol/L UBA3/APPBP1, 300 nmol/L UBE2M or UBE2F, and 200 nmol/L E3 complex (RBX1‒CUL1CTD or SAG‒CUL5CTD) in a Tris-HCl (pH = 7.4)-adjusted buffer containing 0.1 mg/mL BSA, 0.5 mmol/L DTT and 5 mmol/L MgCl2. Indicated compounds were incubated in the mixture (final DMSO ≤1%) for 10 min at 25 °C before addition of 20 μmol/L ATP and incubation for 30 min at 37 °C. The 4 × SDS loading buffer (without DTT) was added to quench the reaction. The samples were subjected to Western blotting analysis.
2.10. Quantitative RT-PCR
Total RNA was extracted from cells using the Trizol reagent (Invitrogen, CA, USA) and were reverse-transcribed into complementary DNAs (cDNAs) with the PrimeScript™ RT reagent Kit (Perfect Real Time; Takara Biotechnology, Dalian, China). Quantitative RT-PCR analysis was performed according to the manufacturer's instruction for SYBR® Premix Ex Taq™ (Tli RNaseH Plus; Takara Biotechnology). The primer sequences are listed in Supporting Information Table S2.
2.11. Ribosome profiling, RNA isolation, and RT-PCR
Cells were seeded in 150 mm dishes at 1 × 107 cells/dish and cultured overnight. Cells were then treated with DMSO, HA-1141 (20 μmol/L) for 24 h, followed by 100 μg/mL cycloheximide for 30 min prior to harvest. Cells were washed in ice-cold PBS containing 100 μg/mL cycloheximide three times, and then lysed in polysome lysis buffer [20 mmol/L HEPES-KOH (pH = 7.5), 5 mmol/L MgCl2, 150 mmol/L KCl, 1 mmol/L DTT, 0.5% Triton X-100, 0.5% sodium deoxybioate and 100 μg/mL cycloheximide]. Lysates were normalized by RNA content using NanoDrop 2000 (Thermo Scientific) and layered onto 11 mL 10%–50% sucrose density gradients (20 mmol/L HEPES-KOH, 5 mmol/L MgCl2, 150 mmol/L KCl, 1 mmol/L DTT, 100 μg/mL cycloheximide, 50 U/mL RNAse inhibitor). Gradients were generated in an SW-41Ti rotor at 36,000 rpm at 4 °C for 3 h, and then sampled using an Auto Gradient Fractionator (Biocomp GM108-2&PGF152-2, Canada) with constant monitoring of optical density (OD) at 260 nm. A series of fractions of 200 μL was collected throughout, and RNA was extracted using a Trizol reagent (Invitrogen, CA, USA) and precipitated with isopropanol and glycogen (Thermo Fisher Scientific). cDNAs were prepared and quantitative RT-PCR was performed as described above.
2.12. Transfection and siRNAs
Cells were transfected using the Transfection GenMute™ siRNA Transfection Reagent (SignaGen Laboratories, USA) according to the manufacturer's protocol. The siRNA sequences for ATF4 were reported previously5. The primer sequence for the scrambled control and PKR are as follows:
siCon: 5′-UUCUCCGAACGUGUCACGUTT-3′,
siPKR-1: 5′-GCTGAACTTCTTCATGTATGT-3′,
siPKR-2: 5′-GAGGCGAGAAACTAGACAAAG-3′.
2.13. Reactive oxygen species (ROS) detection
Total intracellular ROS was determined with dichlorofluorescin diacetate (DCFH-DA, Beyotime). Briefly, after the compound treatment, cells were washed with PBS and incubated with 2 μmol/L (for H2170) or 0.1 μmol/L (for H358) DCFH-DA at 37 °C for 15 min. Cells were again washed 3 times with PBS and analyzed by flow cytometry (CytoFLEX LX, Beckman Coulter, USA).
2.14. Immunofluorescence staining
After the compound treatment, cells were fixed with cold methanol for 7 min at −20 °C, washed with PBS buffer three times and blocked with PBS buffer containing 2.5% BSA and 0.1% Triton X-100 for 1 h at room temperature. Cells were incubated with primary antibody at 4 °C overnight, washed three times with PBS and then incubated with appropriate secondary antibody conjugated to Alexa 488 (Molecular Probes) for 1 h at room temperature. DNA was stained with DAPI. Slides were examined under a Nikon A1X60 microscope (Nikon A1-Ti, Tokyo, Japan) for punctate vesicle structures of LC3 and images were processed with NIS-elements software (Nikon A1-Ti). The percentage of cells undergoing autophagy was determined. The results were plotted in a bar graph with the mean ± SEM from three independent experiments.
2.15. Transmission electron microscopy (TEM)
Cells were fixed in 2.5% glutaraldehyde in PBS overnight, then post-fixed in 1% OsO4 for 1 h followed by 2% uranyl acetate. After ethanol and acetone dehydration and embedding in polybed 812 resin (Sigma), thin sections (70 nm) were post-stained with 2% uranyl acetate followed by 0.3% lead citrate. The photos of sample sections were taken using a TECNAI 10 transmission electron microscope (FEI Company, Hillsboro, OR, USA) at 120 kV.
2.16. Cell viability and colony-forming assay
Cells were plated in 96-well plates and treated with HA-1141 as indicated. Cell proliferation was determined using the CCK8 according to the manufacturer's protocol. For the colony-forming assay, H1299 cells were seeded into 35 mm dishes (200 cells/dish) in triplicate, treated with HA-1141, or DMSO for 6 days with drug-containing fresh medium replacement once. The colonies were fixed, stained and counted under an inverted microscope (Olympus, Tokyo, Japan). Colonies with 50 cells or more were counted.
2.17. Tumor xenograft studies
The liver microsomal metabolic stability test was conducted by Shanghai ChemPartner Co., Ltd. Female BALB/c nude mice (Shanghai Slac laboratory animal Co., Ltd., China) were housed and handled in accordance with the Guide for the Care and Use of Laboratory Animals. Mice (5–6 weeks) were inoculated with 5 × 106 H358 or H2170 cells subcutaneously in both flank, and tumor growth was monitored with caliper measurements. Tumor volumes were calculated as length × width × width × 0.52. When the mean tumor volume reached ~100 mm3, animals were dosed via intraperitoneal (i.p.) injection once a day, 5 days per week for three or 2 weeks with solvent control (20% DMSO + 50% PEG400 + 30% PBS) or HA-1141 (25 mg/kg).
2.18. H&E staining and immunohistochemical staining
Vital organs, including heart, liver, spleen, lung and kidney were collected and analyzed by H&E staining after treatment with HA-1141 or solvent control for 2 weeks.
Tumor tissues obtained from the mice bearing H358 or H2170 xenografts were collected for immunohistochemical analysis. Briefly, antigen retrieval was performed with 1 mmol/L EDTA buffer (pH = 9.0) at 95 °C for 20 min. After quenching endogenous peroxidases with 3% H2O2 and blocking non-specific binding with 1% bovine serum albumin buffer, sections were incubated overnight at 4 °C with antibodies against Ki67 (1:500 dilution), ATF4 (1:100 dilution), CHOP (1:200 dilution), P-S6 (1:50 dilution) or P21 (1:50 dilution). The sections were treated with HRP conjugated secondary antibody for 30 min at room temperature and stained with 0.05% 3,3-diaminobenzidine tetrahydrochloride (DAB). Slides were photographed with a Digital Pathological Slide Scanner (KF-RPO-005-EX, Ningbo Konfoong Bioinformation Tech Co., Ltd., China). The photographs were analyzed with the Image-Pro Plus 6.0 software (Media Cybernetics, Inc., Silver Spring, MD, USA).
2.19. Statistical analysis
The two-tailed Student's t-test was performed. Results are expressed as mean ± SEM from three independent assays. P < 0.05 was considered statistically significant.
3. Results
3.1. Discovery of a small molecule that disrupts the UBE2F‒UBA3 binding
Our recent study showed that UBE2F was overexpressed in lung cancer tissues and was associated with poor patient survival, and UBE2F knockdown suppressed growth and survival of lung cancer cells, indicating UBE2F was an attractive lung cancer target15. In an effort to identify small molecule inhibitors of UBE2F, we employed structure-based virtual screening using the Specs chemical library (http://www.specs.net/) containing a total of 240,000 compounds with the goal of identifying small molecules that have the potential to disrupt UBE2F‒UBA3 binding17. In the first step, molecular dynamics simulation and binding free energy analysis were performed to identify hot-spots on the interaction interface between UBE2F and UBA3 that could be employed as binding sites for virtual screening, and two hot-spots (i.e., regions surrounding residues of F56 and V30) were found to be critical for UBE2F and UBA3 binding (Fig. 1A and B). The virtual screening also included the catalytic active site of UBE2F (C116). Based on the three potential binding pockets, 30 top-ranked compounds were selected from each screening campaign, and a total of 90 compounds were identified and assessed in cell-based Western blotting for the ability to inhibit cullin-5/CUL5 neddylation. Among the identified compounds, the compound Ui5 that binds to the F56 pocket exhibited the highest activity (Supporting Information Figs. S1A and S1B). We tested structural analogues of Ui5 and one of its analogues, IF22, was chosen as the starting structure for further structure–activity relationship (SAR) optimizations (Fig. S1C).
Figure 1.

Structure-based virtual screening and structural modifications of the candidates. (A) Determination of “hot-spot” residues. F56 and V30 pockets (indicated by yellow arrows), two hydrophobic areas of UBE2F protein were determined as ‘hot-spot’ to interact with UBA3 (shown as green) by molecular dynamics simulations and free energy decomposition. (B) Interaction spectrum of UBE2F binding to UBA3. MM/GBSA binding free energy decomposition supported by the mm_pbsa program in Amber 11 was carried out on a per-amino acid basis. Four components (i.e., van der Waals, electrostatic contributions, and polar and non-polar desolvation contributions) were included in the prediction of interactions between UBE2F and UBA3. The GB model (igb = 2) was used to calculate the polar contribution of desolvation. The non-polar contribution of the desolvation free energy was computed by SASA based on the ICOSA technique. (C)‒(F) The optimization to identify compound Ui5-8-11-4-1/HA-1141.
After carefully analyzing the structural features of hit compound (IF22), the molecular scaffold was divided into three parts which included section 1, section 2 and section 3 for optimization. We commenced the first-round structural optimization for section 1 and section 3 (Fig. 1C). At the beginning, section 2 and section 3 of IF22 were unchanged while the 2,4-dichlorophenyl group in section 1 was replaced with 3,5-dimethylphenyl,4-methylphenyl, 2,4-difluorophenyl, 4-fluorophenyl, 4-aminocarbonylphenyl, 4-methoxyphenyl, 4-acetaminophenyl to give compounds Ui5-1 to Ui5-4, Ui5-6, Ui5-15 and Ui5-20. Upon modification of section 3 with various aromatic rings and heterocycles, we obtained thirteen analogues of IF22, including Ui5-7 to Ui5-14, Ui5-16 to Ui5-19, Ui5-5. The biochemical activities of these twenty compounds were evaluated by Western blotting in two lung cancer cell lines H358 and H2170 with inhibition of CUL5 neddylation and accumulation of NOXA, a CRL5 substrate15 as readouts. To our delight, we found that Ui5-5 inhibited neddylation of CUL5 and induced NOXA accumulation in both lung cancer cell lines, and Ui5-8 induced marked NOXA accumulation with little effect on neddylation of CUL5 (Fig. S1D).
We then continued to explore the analogues of Ui5-5 and Ui5-8, respectively (Fig. 1D). By introducing various aromatic rings to replace the 2,4-dichlorophenyl group in section 1 of Ui5-5, the compounds Ui5-5-1 to Ui5-5-14 were obtained. However, none of these Ui5-5 analogues showed inhibitory activity in CUL5 neddylation, nor caused NOXA accumulation (not shown). At the same time, this optimization strategy was also applied for Ui5-8 and the compounds Ui5-8-1 to Ui5-8-10 were obtained. At this stage, the dihydrazide group of Ui5-8 was replaced with urea, benzoylhydrazone, oxadiazole and ketone via a bioisosteric strategy, yielding compounds Ui5-8-11 to Ui5-8-15. The Western blotting showed that Ui5-8-11 was more effective than Ui5-8 in blocking CUL5 neddylation (Fig. S1E).
To further improve the activity, another round of optimization was conducted by varying section 1 of Ui5-8-11 (Fig. 1E). Specifically, 2-chlorophenyl, 4-chlorophenyl, 4-methylphenyl, 2-cyanophenyl, 4-methylaminocarbonylphenyl, 3,4-methylendioxyphenyl, 2-methylpyridinyl, pyrimidinyl, γ-amide, pyrrolidine, piperidine, 4-piperidinyl piperidine, morpholine, 4-acetylpiperazine, 4-methyl piperazine and piperazine were used to replace the 2,4-dichlorophenyl group, and 16 compounds, Ui5-8-11-1 to Ui5-8-11-16 were obtained. The compound Ui5-8-11-4 was found to be the best in inhibition of CUL5 neddylation and accumulation of NOXA (Fig. S1F).
To gain more potency, a fourth round of optimization was carried out. Twelve compounds of Ui5-8-11-4-1 to Ui5-8-11-4-12 were obtained by using rational drug design strategies and the Linpiski's rules (Fig. 1F). Gratifyingly, replacement of the O-atom in Ui5-8-11-4 with an N-atom produced Ui5-8-11-4-1 which was the most potent compound among this series of analogues in the inhibition of neddylation of all six cullins in H358 and H2170 cells (Fig. S1G). All compounds were prepared according to the procedure presented in Supporting Information.
For the remaining study, we focused on compound Ui5-8-11-4-1, designated as HA-1141, to evaluate its biochemical activity, its mechanism of action, and biological function.
3.2. HA-1141 selectively binds to NAE E1 to inhibit neddylation of all cullins, but fails to cause accumulation of CRL substrates
The interactions between UBA3 and HA-1141 were investigated by Glide XP docking based on the crystal structure of 3FN1 (Section 2.1.). As shown in Fig. 2A, the amino acids Gln 442, Thr405, Thr403, Asn 410, Leu 456 and Thr454 in UBA3 were involved in the binding to HA-1141. The interactions of Gln442, Thr405 and Thr403 were found to be most critical for the binding between UBA3 and HA-1141. Two hydrogen bonds were formed with amino acids Thr403 and Thr405, and a halogen bond was also observed between Gln442 and a fluorine atom in HA-1141.
Figure 2.

HA-1141 binds with UBA3, inhibits cullin neddylation but decreases the levels of CRL substrates. (A) Schematic representation of the binding structure of UBA3/HA-1141 complex predicted by Glide XP docking simulation. Important amino acids of UBA3 are colored in yellow, and HA-1141 is shown in cyan. Hydrogen bonds between UBA3 and HA-1141 are highlighted pink, and halogen bonds are colored red. (B–D) Enhanced thermal stability of UBA3/NAE1. (B) Enhanced thermal stability of UBA3/NAE1 proteins in cells. H358 cells were treated with HA-1141 (20 μmol/L), MLN4924 (1 μmol/L) or DMSO for 24 h, followed by heating at the indicated temperatures for 3 min. Protein levels were analyzed by Western blotting analysis. (C) Dose-dependent enhancement of thermal stability of UBA3/NAE1 in cells. H358 were treated with HA-1141 at different concentrations for 24 h and then heated at 57 °C for 3 min. Protein levels of UBA3/NAE1 were analyzed by Western blotting. (D) Enhancement of thermal stability of purified UBA3/NAE1. Purified UBA3/NAE1 proteins (0.5 μg) were incubated with HA-1141 (100 μmol/L) or DMSO for 10 min at room temperature in 50 μL of buffer and then heated at the indicated temperature for 3 min. The mixtures were then centrifuged at 20,000×g for 25 min at 4 °C and the soluble components were examined by Western blotting. Shown is the mean ± SEM (n = 3). ∗∗∗P < 0.001, ∗∗P < 0.01, ∗P < 0.05, NS, no significance vs. control. (E) The in-vitro inhibition of E2-NEDD8 thioester. HA-1141 or MLN4924 (100 μmol/L) were incubated in neddylation reaction buffer (Section 2), followed by Western blotting analysis. (F) Inhibition of neddylation of cullins in vitro. HA-1141 or MLN4924 (100 μmol/L) were incubated in neddylation reaction buffer (Section 2), followed by Western blotting analysis. (G, H) Inhibition of cullin neddylation with depletion of CRL substrates. H358 were treated with HA-1141 or MLN4924 (0.3 μmol/L) for 24 h, or with 20 μmol/L of HA-1141 for the indicated time followed by Western blotting, using the indicated antibodies. N8, NEDD8.
To further validate the molecular docking findings, we carried out a cellular thermal shift assay (CETSA)25 to assess direct binding to UBA3 in human lung cancer cells. As shown in Fig. 2B and Supporting Information Fig. S2A, cellular UBA3 protein was largely degraded at 54 °C in H358 or H2170 cells treated with solvent control DMSO. The thermal stability of UBA3 protein was clearly enhanced by HA-1141 at 57 and 60 °C, while it had no such effect on UBE2F. In this assay we used the well-characterized NAE inhibitor MLN4924 as a control, and found that HA-1141 stabilized UBA3 more efficiently at higher temperature than MLN4924, suggesting that HA-1141 bound directly to UBA3, which differs from MLN4924, known to form a NAE-catalyzed adduct with NEDD8 or a NEDD8-AMP mimetic in-situ to indirectly stabilize UBA326. Since UBA3 and NAE1 formed a heterodimer in cells, the thermal stability of NAE1 was also enhanced by HA-1141. In addition, HA-1141 stabilized UBA3 and NAE1 proteins in a dose-dependent manner in both H358 (Fig. 2C) and H2170 cells (Fig. S2B). Finally, in an in-vitro thermal stability assay, HA-1141 stabilized purified UBA3 and NAE1 protein at 60, 63 or 66 °C (Fig. 2D).
Next, we explored whether HA-1141 could inhibit neddylation as a new NAE inhibitor. In an in-vitro neddylation assay, using MLN4924 as a positive control, HA-1141 blocked formation of thioester of NEDD8-UBE2M and NEDD8-UBE2F in a dose-dependent manner (Fig. 2E). This inhibitory effect was confirmed in cell-based assay (Fig. S2C), which clearly indicated that HA-1141 targeted NAE E1 rather than UBE2F E2.
Previous studies have demonstrated that UBE2F knockdown blocked only CUL5 neddylation, while NAE inhibitors blocked neddylation of CUL1‒515,27. HA-1141 inhibited neddylation of both CUL1 and CUL5 in dose-dependent manner in an in vitro cullin neddylation assay (Fig. 2F). Moreover, HA-1141 inhibited neddylation of all cullins in H358 (Fig. 2G and H) and in H2170 (Figs. S2D and S2E) in a dose- and time-dependent manner, with NAE inhibitor MLN4924 included as a positive control. In both cell lines, HA-1141 effectively inhibits neddylation of CUL1, CUL2, CUL3, CUL4a, CUL4b and CUL5 at the concentration of 10 μmol/L, the same dose that enhanced thermal stability of UBA3/NAE1 (Fig. 2C, Fig. S2B). It is worth noting that HA-1141 appears to have better activity in a cell-based assay than in a test-tube-based biochemical assay. However, there are many differences in the way that these two types of assays were conducted, including the compound incubation time (24 h vs. 10–30 min) and the amounts of enzymes used to directly interact with the compound. Consistent with this caution, a previous study also reported a similar discrepancy between cell-based and test-tube based assays when a natural product-like inhibitor of NAE was tested28.
Surprisingly, however, while MLN4924 caused accumulation of typical substrates of CRLs, including CUL1 substrates ATF4, DEPTOR and P21, CUL2 substrate HIF1α, CUL3 substrate NRF2, CUL4 substrate CDT1 and CUL5 substrate NOXA, HA-1141 caused a reduction of most substrates tested (Fig. 2G, Fig. S2D). The time-course study showed that HA-1141 indeed stabilized almost all substrates at early time points, but reduced their levels at later time points (at 16 and 24 h) in H358 cells (Fig. 2H) and in H2170 (Fig. S2E).
3.3. HA-1141 reduces the level of CRL substrates by inhibiting translation
To investigate the underlying mechanism behind this unexpected finding, we first measured the mRNA levels of these substrates after compound treatment. Some of transcripts (e.g., P21, NOXA) were strikingly increased, whereas the others (e.g., CDT1) were remarkably decreased, and still the others (DEPTOR, NRF2) showed little change (Fig. 3A, Supporting Information Fig. S3A). We next explored whether HA-1141 caused substrate reduction via ubiquitin-proteasome or lysosome degradation systems and found that treatment with neither proteasome inhibitor MG132, lysosome inhibitor CQ, nor their combination could rescue substrates reduction by HA-1141 (Fig. 3B, Fig. S3B). Thus, reduction of substrate proteins by HA-1141 was not due to reduced transcription (except CDT1), nor enhanced degradation.
Figure 3.

HA-1141 inhibits mRNA translation. (A) HA-1141 does not decrease mRNA levels of most CRL substrates. H358 cells were treated with HA-1141 (20 μmol/L) or MLN4924 (0.3 μmol/L) for 24 h followed by qRT-PCR analysis (n = 3). Shown are the mean ± SEM. ∗∗P < 0.01, ∗P < 0.05, NS, no significance vs. DMSO control. (B) CQ and MG132 cannot rescue protein decreases caused by HA-1141. H358 cells were treated with HA-1141 (20 μmol/L) or DMSO for 24 h and CQ (50 μmol/L) or MG132 (10 μmol/L) was added to the medium 5 h before harvesting, followed by Western blotting analysis. (C)‒(H) HA-1141 inhibits general mRNA translation. (C) A representative profile of mRNA translation. H358 cells were treated with DMSO or HA-1141 (20 μmol/L) for 24 h, followed by ribosome profiling via a sucrose gradient. (D)‒(H) The RT-PCR results with the indicated mRNAs (n = 3). Shown are the mean ± SEM. ∗∗P < 0.01, ∗P < 0.05, NS, no significance vs. DMSO control.
We then focused our attention on mRNA translation using the ribosome profiling assay to directly compare the distribution of cellular RNA species bound with mono-ribosomes (not translated) or polyribosomes (actively translated) after compound treatment. H358 cells were treated with HA-1141, along with DMSO control for 24 h, and cell lysates were prepared and layered onto a sucrose density gradient for ribosome profiling. Compared with DMSO, treatment with HA-1141 led to a sharp increase in 80S ribosomes and significant reduction of polysomes (Fig. 3C). To further confirm whether impaired mRNA translation would lead to reduced translation of a few mRNAs of interest, we carried out quantitative RT-PCR from the samples of ribosome fractions (fractions 2–5 vs. 1), and found that translation of mRNAs encoding P21, CDT1 or NOXA was inhibited, although no statistical significance was found for P21 due to high variation (Fig. 3D–F). On the other hand, the translation of ATF4, a substrate of CRL1 and an active response protein of ER stress and ISR, and translation of CHOP, a protein downstream of ATF4 and a direct effector of ISR was not significantly altered (Fig. 3G‒H).
3.4. HA-1141 induces non-canonical ER stress and ISR
We next examined ATF4, a known substrate of CRL1, and also a known response protein to ER stress. Unlike MLN4924, which caused ATF4 accumulation at the later time points (16–24 h), HA-1141 caused its rapid accumulation at 2 h, which lasted up to 12 h (Fig. 2H, Fig. S2E), suggesting early accumulation of ATF4 was not the consequence of CRL1 inactivation.
Given that HA-1141 inhibits protein synthesis by blocking mRNA translation and ER stress could also induce translation arrest via activation of PEAK kinase to trigger eIF2α phosphorylation, a core biomarker of ISR4, we then focused our study on the possible induction of ER stress by HA-1141. We used tunicamycin as a positive control for ER stress induction, and MLN4924 as a positive control of neddylation/CRL inactivation. While MLN4924 had no effect, HA-1141, like tunicamycin, caused both dose- and time-dependent induction of mRNAs encoding ATF4, CHOP and BIP, three proteins known to be induced upon ER stress in H358 lung cancer cells (Fig. 4A‒F), and to a lesser extent in H2170 cells (Supporting Information Fig. S4A‒S4F). Thus, HA-1141 had unexpected activity in addition to inactivating NAE.
Figure 4.

HA-1141 triggers non-canonical ER stress, ISR and ROS production. (A–F) HA-1141 triggers ER stress in a dose- and time-dependent manner. H358 cells were exposed to different concentration of HA-1141, tunicamycin (12 μmol/L) or MLN4924 (0.3 μmol/L) for 24 h or treated with HA-1141 (20 μmol/L) for the indicated periods of time. The relative mRNA level was analyzed by RT-PCR analysis (n = 3). Shown are the mean ± SEM. ∗∗∗P < 0.001, ∗∗P < 0.01, ∗P < 0.05, NS, no significance vs. control. Tun, tunicamycin, MLN, MLN4924. (G, H) HA-1141 induces non-canonical ER stress and ISR. H358 cells were treated with HA-1141 or tunicamycin at the indicated concentrations for 24 h, or with HA-1141 (20 μmol/L) or tunicamycin (3 μmol/L) for the indicated periods of time, followed by Western blotting analysis. (I) PKR knockdown rescues P-eIF2α induced by HA-1141. H358 cells were transfected with the indicated siRNAs for 72 h and then treated with DMSO or HA-1141 (20 μmol/L) for 24 h, followed by Western blotting analysis. (J) HA-1141 induces ROS production. H358 cells were treated with HA-1141 for 0.5 h and then labeled with DCFH-DA (0.1 μmol/L) for 15 min at 37 °C, followed by FACS analysis (n = 3). Shown are the mean ± SEM. ∗P < 0.05, ∗∗P < 0.01 vs. control. (K) NAC rescues upregulation of ATF4 by HA-1141. H358 cells were pre-treated with NAC (10 mmol/L) for 2 h and then co-incubated with HA-1141 (20 μmol/L) or DMSO for 12 h, followed by Western blotting with the indicated antibodies. N8, NEDD8.
We then made a head-to-head comparison between HA-1141 and tunicamycin on triggering phosphorylation of IRE1α, PERK, and eIF2α, three classic biomarkers for ER stress. Indeed, HA-1141 caused a dose-dependent induction of P-IRE1α, occurring as early as 30 min post-treatment, but to a greater extent than tunicamycin. Unexpectedly and opposite to tunicamycin, which caused P-PERK induction, HA-1141 caused a dose-dependent reduction of P-PERK at 8 h post treatment. Furthermore, HA-1141 caused a dose-dependent induction of P-eIF2α, which occurred at much later stage (12- and 24-h post treatment) to a much greater extent than with tunicamycin (Fig. 4G, H, Fig. S4G and S4H). All these data strongly suggest that HA-1141 triggers ER stress via a non-canonical pathway.
We next determined the levels of other downstream proteins responsive to canonical ER stress. While both compounds caused rapid induction of both ATF4 and CHOP in a sequential order, HA-1141 caused a remarkable reduction of both ATF4 and CHOP at a late stage of treatment (particularly 24 h), when the level of P-eIF2α was extremely high (Fig. 4G, H, Fig. S4G and S4H). Another striking difference between the two compounds was that unlike tunicamycin, which caused a remarkable increase of ER chaperone protein BIP starting at 4 h and lasting to 24 h of treatment, HA-1141 had no effect on BIP protein levels (Fig. 4G, H, Fig. S4G and S4H), although it did increase BIP mRNA (Fig. 4C, Fig. S4C). These data again support the suggestion that ER stress induced by HA-1141 was via a non-canonical pathway.
It is known that severe ER stress, such as that induced by prolonged HA-1141 treatment (for 24 h), could induce terminal ISR29, which shuts off general mRNA translation4. Four protein kinases, PERK, PKR (double-stranded RNA-dependent protein kinase), GCN2 (eIF-2-alpha kinase GCN2) and HRI (heme-regulated inhibitor) are known to phosphorylate eIF2α to mediate ISR, thus affecting protein synthesis4. We have shown that while tunicamycin triggered PERK activation, HA-1141 caused a dose-dependent inactivation of PERK, particularly after a 24 h treatment (Fig. 4G, H, Figs. S4G and S4H). Interestingly, while tunicamycin had no effect on the total or phosphorylated levels of the other three kinases, GCN2, PKR, and HRI, HA-1141 treatment caused both dose- and time-dependent activation of PKR, starting as early as 30–60 min, without affecting GCN2 nor HRI (Fig. 4G, H, Figs. S4G and S4H). Importantly, in our assay system, PKR appeared to be responsible, at least in part, for eIF2α phosphorylation, since PKR knockdown partially blocked eIF2α phosphorylation induced by HA-1141 (Fig. 4I, Fig. S4I), suggesting that the PKR-eIF2α axis modulated ISR, eventually leading to the inhibition of protein synthesis.
Finally, we investigated a possible underlying mechanism by which HA-1141 caused early upregulation of ATF4 and PKR phosphorylation. A few studies have reported that during ER stress the unfolded protein response promotes the production of ROS in the endoplasmic reticulum to trigger oxidative stress30, and that ATF4 can be induced and stabilized during oxidative stress31,32. We found that HA-1141 indeed caused a massive and dose-dependent increase in the ROS levels within 30 min of exposure (Fig. 4J, Fig. S4J). ROS appeared to be responsible for the increase of ATF4, but not P-PKR, since ROS scavenger NAC only partially abrogated ATF4 induction by HA-1141 (Fig. 4K, Fig. S4K). Taken together, our study demonstrates that HA-1141 is a non-canonical inducer of ER stress and ISR, which activates P-eIF2α at a late stage to block mRNA translation, leading to reduced protein synthesis.
3.5. HA-1141 inactivates mTORC1 to induce autophagy in a manner dependent on ATF4
It has been well-established that protein synthesis is activated by the mTOR signaling pathway1 and oxidative stress is an important factor that links bidirectional crosstalk between ER stress and the mTOR pathway33. We investigated the effect of HA-1141 on mTOR activity. While the compound had a minor inhibitory effect on mTOR auto-phosphorylation on S2448, it caused remarkable inactivation of mTORC1, as evidenced by elimination of phosphorylation of 4E-BP1, S6K1 and S6 in a dose-dependent manner in both lung cancer cell lines (Fig. 5A, Fig. S5A). The time-course study showed that HA-1141 caused early stage (2–4/8 h) activation, but late stage (12/24 h) inactivation of mTORC1, and late stage (8/12 h) activation of mTORC2 (as reflected by increased P-AKT; Fig. 5B, Supporting Information Fig. S5B). Interestingly, tunicamycin also caused inactivation of mTORC1 (but not mTORC2), particularly at the later time points (8–24 h) with a high compound dose (12 μmol/L; Fig. 5A and B, Supporting Information Figs. S5A and S5B).
Figure 5.

HA-1141 inactivates mTORC1 to induce autophagy in an ATF4-dependent manner. (A, B) HA-1141 inactivates mTORC1. H358 cells were treated with HA-1141 or tunicamycin at the indicated concentrations for 24 h, or with HA-1141 (20 μmol/L) or tunicamycin (3 μmol/L) for the indicated periods of time, followed by Western blotting analysis. (C) Autophagy measured by appearance of punctate vesicle structure. H358 were treated with HA-1141 at the indicated concentration for 24 h before photography under a fluorescent microscope (left panels). Cells with punctate vesicle structures of LC3 were counted in five independent areas and data are plotted in a bar graph (right panel). Shown are the mean ± SEM (n = 3), size bar, 10 μm ∗∗∗P < 0.001 vs. control. (D) Detection of autophagosomes by TEM: H358 cells were treated with HA-1141 (20 μmol/L) for 24 h, along with a DMSO vehicle control, followed by TEM analysis. Autophagosomes are indicated by arrows. Direct magnification: × 30,000, size bar, 1 μm. (E) Time-dependent LC3-Ⅱ conversion: H358 cells were treated with HA-1141 (20 μmol/L) or tunicamycin (3 μmol/L) for the indicated time followed by Western blotting analysis. (F, G) ATF4 knockdown decreases punctate vesicle structure by HA-1141. H358 cells were transfected with the indicated siRNAs for 60 h and then treated with DMSO or HA-1141 (20 μmol/L) for 24 h before photography (F). Cells with punctate vesicle structures of LC3 were counted in five independent areas and the data are plotted in a bar graph (G). Shown are the mean ± SEM (n = 3), size bar, 3 μm ∗∗∗P < 0.001 (H) ATF4 knockdown partially rescues autophagy. H358 cells were transfected with the indicated siRNAs for 60 h and then treated with DMSO or HA-1141 (20 μmol/L) for 8 h, followed by Western blotting analysis.
Since the mTOR pathway is a well-known negative regulator of autophagy, mTORC1 inactivation would then be expected to induce autophagy34. Indeed, HA-1141 treatment significantly induced autophagy in a dose-dependent manner, as evidenced by an increasing number of autophagic punctate vesicle structures (Fig. 5C, Fig. S5C). More autophagosomes were detected by electron microscopy in both lung cancer cell lines upon HA-1141 exposure (Fig. 5D, Fig. S5D). At the biochemical level, HA-1141 caused a time-dependent conversion of LC3-I to LC3-II (Fig. 5E, Fig. S5E), a widely used biomarker of autophagy35. As a classic ER stress inducer, tunicamycin also induced obvious autophagy in our assay setting, as evidenced by a time-dependent conversion of LC3-I to LC3-II (Fig. 5E, Fig. S5E). Mechanistically, ATF4 appeared to mediate, at least in part, HA-1141 induced autophagy, since ATF4 knockdown significantly reduced the number of autophagosomes (Fig. 5F and G) and reduced the levels of both LC-I and LC3-II (Fig. 5H).
Given the similar effects on ER stress induction and mTORC1 inactivation of HA-1141 and tunicamycin, we next determined whether these activities were associated with neddylation inactivation. While HA-1141 reduced conjugation of NEDD8 to various cellular proteins in a dose- and time-dependent manner, tunicamycin, used at the highest concentration, had no effect (Fig. 6A and B), indicating that these two activities were independent of neddylation inactivation.
Figure 6.

HA-1141 inhibits neddylation and induces ISR independently. (A, B) HA-1141 inhibits neddylation dose- and time-dependently. Cells were treated with HA-1141, tunicamycin (12 μmol/L) or MLN4924 (0.3 μmol/L) for 24 h, or with HA-1141 (20 μmol/L) for the indicated periods of time. Neddylation was measured using antibody against N8. (C) HA-1141, but not MLN4924, induces ER stress/ISR. Cells were treated with HA-1141 (20 μmol/L) or MLN4924 (0.3 μmol/L) for the indicated periods of time, followed by Western blotting analysis. (D) HA-1141 induces ISR in MEF cells. Non-immortalized MEF cells were treated with HA-1141 (20 μmol/L) or tunicamycin (3 μmol/L) for indicated h, followed by Western blotting analysis. (E) NAC rescues upregulation of ATF4 by HA-1141. Non-immortalized MEF cells were pretreated with NAC (10 mmol/L) for 2 h and then co-incubated with HA-1141 (20 μmol/L) for 24 h, followed by Western blotting analysis. SE, short exposure. LE, long exposure. N8, NEDD8.
One previous study claimed that MLN4924 activated ER stress-associated apoptosis involving CHOP induction in human chondrosarcoma36. However, CHOP is downstream of ATF4, a known substrate of CRL1, which accumulates upon MLN4924 treatment37. In this study, we have shown that in lung cancer cells, MLN4924 had no effect on induction of mRNA of ATF4, CHOP and BIP, three well-characterized biomarkers of ER stress (Fig. 4A‒C, Fig. S4A‒S4C)38. To further evaluate whether MLN4924 induces ER stress, we directly compared the effect of MLN4924 and HA-1141 on several ER stress-responsive proteins. As shown, while MLN4924 induced only ATF4 and CHOP at a late stage (16/24 h) via inactivation of CRL1, as expected, HA-1141 induced ATF4 and CHOP at an early-stage (2 h, Fig. 6C). Furthermore, HA-1141 induced remarkable phosphorylation of eIF2α, the core marker of ISR, at late-stage, while MLN4924 had no such effect (Fig. 6C). Thus, MLN4924 is not a potent inducer of ER stress/ISR. Taken together, HA-1141 has dual activities: 1) as an E1 inhibitor to inactivate neddylation and CRLs, and 2) as a non-canonic inducer of ER stress/ISR to inactivate mTORC1 and protein synthesis, leading to autophagy induction.
Finally, we extended our observation made in lung cancer cells to non-immortalized mouse embryonic fibroblasts (MEFs) with particular focus on the P-eIF2α/ATF4/CHOP axis. Both HA-1141 and tunicamycin induced transient phosphorylation of eIF2α between 0.5 and 1 h, followed by sequential induction of ATF4 and CHOP, consistent with a previous report of tunicamycin induction of ER stress/ISR in MEF cells39. However, in the case of PKR, HA-1141 caused PKR activation at later time points (8–24 h), whereas tunicamycin caused its inactivation, starting at 2 h of treatment (Fig. 6D). Thus, the two compounds differ in modulation of ER/ISR stresses in MEFs. More importantly, we found that HA-1141 induction of ATF4 also can be blocked by the ROS scavenger NAC in MEFs (Fig. 6E), suggesting a general phenomenon of ROS activation of ATF4. It is worth noting that HA-1141 also inactivated neddylation of CUL1 and CUL5 in MEFs, which cannot be blocked by NAC, indicating a ROS-independent effect.
3.6. HA-1141 inhibits growth of cancer cells both in vitro and in vivo
We next determined the anticancer activity of HA-1141. Using a cell proliferation assay, we found that the IC50 values of the compound were within 2–6 μmol/L in various lung cancer cell lines when cells were seeded in 96-well plates at low density after 72 h of treatment (Fig. 7A). The IC50 values increased to ~20 μmol/L after 24 h of treatment when cells were seeded at a much higher density (Supporting Information Fig. S6A). Importantly, growth suppression by HA-1141 can be partially rescued by NAC, a typical ROS scavenger (Fig. S6B), suggesting that HA-1141-induced growth suppression of cancer cells was mediated at least in part through ROS induction. A clonogenic survival assay also showed a dose-dependent inhibition of colony formation in H1299 lung cancer cells (Fig. 7B). Note that both H358 and H2170 cells failed to form colonies under monolayer culture even in the absence of compounds.
Figure 7.

HA-1141 inhibits tumor growth both in vitro and in vivo. (A) IC50 determination in multiple lung cancer cell lines. H358, H1299 or H2170 cells were seeded in triplicate in 96-well plates and treated with HA-1141 at the indicated concentrations for 72 h, followed by CCK8 proliferation analysis. Shown is the mean ± SEM from three independent experiments. (B) Colony-forming assay. H1299 cells were seeded 200 per dish in 35 mm dishes in triplicate and treated with HA-1141 or DMSO for 6 days. The representative dishes are shown (left), and the data are plotted in a bar graph (right). Shown are the mean ± SEM (n = 3), ∗∗P < 0.01, ∗P < 0.05 vs. control. (C, D) Tumor volumes and weights. H358 or H2170 cells were injected 5 × 106 per flank in both flanks of nude mice. Solvent control (n = 5 for H358, n = 6 for H2170) or 25 mg/kg of HA-1141 (n = 6) was injected i.p. when tumor volume reached ~100 mm3 once a day, 5 days per week for three or 2 weeks. Tumor volumes were measured every other day. Tumor weights were measured on the last day. ∗∗∗P < 0.001, ∗∗P < 0.01. (E) IHC staining. Three tumor tissues derived from H358 xenograft mice for each group were collected for immunohistochemical staining. Five random areas of each tumor were photographed and then quantified. Shown is the mean ± SEM (n = 3). ∗∗P < 0.01, ∗P < 0.05, NS, no significance, size bar: 100 μm.
Finally, before the in vivo anti-tumor assay, a liver microsomal metabolic stability test was conducted for HA-1141, which showed a t1/2 of 49 min (Supporting Information Fig. S7A). Two xenograft models were established in nude mice using H358 or H2170 cells, respectively, to determine anti-tumor activity of HA-1141. Briefly, HA-1141 was administered at a dose of 25 mg/kg by i.p. injection once a day for 3 weeks (2 weeks for H2170 xenografts to reduce mouse suffering), starting when tumor size reached ~100 mm3. The tumor volume was examined every other day and a growth curve plotted. The results showed that HA-1141 significantly inhibited in vivo tumor growth in both models (Fig. 7C), leading to remarkable reduction of tumor size and weight (Fig. 7D) without visible toxicity, as measured by body weight (Fig. S7B) and H&E staining of several organs, including heart, liver, spleen, lung and kidney (Fig. S7C). As shown by IHC staining, HA-1141 significantly reduced the number of Ki67+ cells in H358 xenograft mice (Fig. 7E), but to a lesser extent in H2170 xenograft mice (Fig. S7D). In both xenograft models, HA-1141, in general, increased the levels of ATF4 and CHOP, but decreased the levels of P-S6 without much effect on P21, although some of these changes did not reach statistically significant levels due to large individual variation of tumor samples (Fig. 7E, Fig. S7D). Nevertheless, these results strongly suggest that HA-1141 induced ER stress and inhibited mTORC1 activity in vivo as well. Finally, HA-1141 appeared to induce autophagy in vivo as well, as tumors from H2170 xenograft mice with compound treatment, as compared to vehicle control, showed higher levels of LC3Ⅱ, accompanied by increased levels of ATF4 and decreased levels of P-S6 (Fig. S7E). Thus, HA-1141 has anti-tumor activity, as demonstrated in both in vitro cell culture and in vivo xenograft models.
4. Discussion
The discovery of MLN4924, the first potent NAE/E1 inhibitor, opened a new era in targeting the neddylation pathway for cancer therapy27. Impressive anti-cancer activity from numerous pre-clinical studies in a variety of human cancers has advanced MLN4924 into a wealth of clinical trials, used as a single agent or in combination with conventional anticancer drugs13. However, given that MLN4924-mediated inactivation of neddylation E1 blocks all neddylation modifications, MLN4924 suffers relatively high cytotoxicity40. Furthermore, several studies have shown that MLN4924 also has some unexpected “off-target” effects, including activation of the EGFR signal pathway, promoting glycolysis-energy metabolism, and altering inflammation and immune responses41. It is, therefore, rather urgent to discover neddylation pathway inhibitors downstream of E1 for better selectivity with anticipated lower cytotoxicity.
In mammalian cells, two family members of E2 neddylation conjugating enzyme UBE2M and UBE2F were identified. UBE2M couples with RBX1 E3 to promote neddylation of CUL1‒4, whereas UBE2F “teams” up with SAG/RBX2 E3 to neddylate CUL5 only17. Our recent studies have validated UBE2F as an attractive anti-lung cancer target15 and this study was undertaken to discover small molecule inhibitors of UBE2F. With structure-based virtual screening and subsequent SAR optimization, we identified HA-1141 as an E1 inhibitor that selectively bound to UBA3 E1, but not UBE2F E2. The following lines of evidence support this conclusion: 1) A docking study supported HA-1141‒UBA3 binding, and CETSA and TSA assays showed that HA-1141 stabilizes UBA3; 2) both in-vitro and in-vivo neddylation assays showed that HA-1141 abrogated thioester formation of both UBE2M and UBE2F and inhibited neddylation of CUL1 and CUL5; 3) Western blotting showed that HA-1141 inhibits neddylation of all six cullins in a dose- and time-dependent manner. Thus, HA-1141 is a neddylation E1 inhibitor, but with much lower potency than MLN4924.
Unlike MLN4924, which caused substantial and prolonged accumulation of CRL substrates, HA-1141 caused substrate accumulation at an early stage, but substrate depletion at a late stage of treatment. We pursued this unexpected finding and found that HA-1141 did not hinder mRNA transcription nor enhance protein degradation; rather, it inhibited general mRNA translation to reduce global protein synthesis by triggering unmitigated non-canonical ER stress and PKR-mediated terminal ISR.
The ER is a central organelle where proteins undergo chaperone-assisted folding to acquire their appropriate conformation, which is a highly error-prone process2. ER stress is provoked when misfolded or unfolded proteins accumulate in the ER lumen. Three ER transmembrane proteins operate as sensors of ER stress: PERK, IRE1α, and ATF6, which are sequestered by chaperone BIP/GRP78 under proteostasis conditions2. During canonical ER stress, BIP is occupied by increased misfolded or unfolded proteins and dissociates from the sensors, which triggers PERK activation via auto-phosphorylation to phosphorylate eukaryotic translation initiation factor 2 subunit-α (eIF2α), leading to general attenuation of protein synthesis but enhanced translation of specific mRNAs, a process known as ISR2,4. The best-characterized protein downstream of ISR is ATF4, a key transcription factor that activates the expression of genes involved in redox homeostasis, amino acid metabolism, protein synthesis, apoptosis and autophagy2,4,39,42.
The following lines of evidence support our conclusion that HA-1141 induced unmitigated non-canonical ER stress: 1) IRE1α phosphorylation, a well characterized upstream response protein of ER stress, was triggered immediately after HA-1141 treatment; 2) HA-1141 induced dose- and time-dependent upregulation of the mRNA levels of ATF4, CHOP and BIP, three classical ER stress markers; 3) contrary to canonical ER stress, PERK phosphorylation was reduced rather than increased, and ATF4 induction occurred much earlier than eIF2α phosphorylation; 4) though BIP mRNA was elevated, BIP protein did not accumulate to improve folding capacity, indicating unmitigated ER stress.
Canonically, persistent or severe ER stress would eventually trigger ISR by the PERK/eIF2α/ATF4/CHOP axis, in which ATF4 transactivates pro-apoptotic genes, like CHOP, NOXA and many other genes to cope with the stress or lead to cell death4,39,42. In contrast to tunicamycin, which triggers canonical ER stress with PERK activation, followed by a moderate increase of P-eIF2α, HA-1141 inactivated PERK, but triggered an enormous increase of P-eIF2α, leading to translation arrest.
It's well established that four kinases, PERK, GCN2, PKR, and HRI converge on phosphorylation of eIF2α to activate ISR4. While PERK is predominantly activated during ER stress, GCN2 mainly responds to amino acid deprivation and HRI to heme deficiency. Many other stresses can activate these kinases, including ultraviolet light, viral infection and oxidative stress for GCN2, and oxidative and mitochondrial stress, heat shock, and cytosolic protein aggregation for HRI4. PKR is an antiviral kinase, being activated potently by long stretches of perfect dsRNA as well as RNAs having structural defects such as bulges and internal loops43. Among these four kinases, we found by a rescue experiment that PKR was responsible for HA-1141 induction of eIF2α phosphorylation. The exact mechanism by which HA-1141 causes early activation of PKR is unknown at the present time but certainly deserves a future investigation, given the fact that PKR is mainly activated by viral dsRNA and RNA that mimics dsRNA, as well as by degradation of circular RNAs44, and that DNA-demethylating agents could induce viral mimicry by endogenous transcripts to yield anticancer activity45.
Although both HA-1141 and tunicamycin induced ISR with their own distinct features, ATF4 induction was not the consequence of ISR considering much earlier induction of ATF4 than P-eIF2α. A number of studies have shown that ATF4 and its down-stream CHOP were the main executors responsible for ISR-related cell death5,39. How ISR switches between pro-survival and pro-death in response to ER stress has been a long-standing enigma. Our study clearly demonstrates that HA-1141 triggered substantial induction of ATF/CHOP at an early stage and induction of P-eIF2α at a late stage, likely contributing to reduced protein synthesis and ISR-mediated cell death.
How is ATF4 induced at such an early stage upon HA-1141 treatment? It is unlikely due to inactivation of CRL1, since ATF4 accumulation is a late event, as demonstrated by MLN4924 treatment. We found that it is likely due to ROS at least in part, based upon the observations that 1) ROS was induced by HA-1141 within 30 min of treatment prior to ATF4 induction and 2) ROS scavenger NAC blocked ATF4 induction by HA-1141. Indeed, several studies have reported that during ER stress the unfolded protein response promotes the production of ROS in the endoplasmic reticulum to trigger oxidative stress30, and that ATF4 can be induced and stabilized during oxidative stress31,32. Oxidative stress has also been shown to be an important factor that links ER stress and the mTOR pathway33. ATF4 has been shown to either enhance mTOR activity by increasing availability of amino acids via autophagy and to inhibit mTOR activity by inducing the expression of the mTOR repressors SESN2, DDIT4, and REDD142. In our study, we did observe an early-stage activation and late-stage inactivation of mTORC1 activity by HA-1141.
ATF4 is reported to induce autophagy in response to ER stress5,46, whereas mTOR is a well-established autophagy negative regulator34. We observed massive autophagy after HA-1141 treatment in lung cancer cells. Many protein post-translational modifications, including phosphorylation, ubiquitination, and acetylation are reported to be crucial to the regulation of autophagy47. Neddylation controls activities of cullin-RING ligases to participate in the process of autophagy as well48. It's well accepted that autophagy plays a double-edged role in tumorigenesis, in which it suppresses tumor growth at the early stage but promotes tumor survival at the late stage6. Specially, during conventional cancer therapy including chemotherapy and radiation, induction of autophagy appears to be a side-effect that serves as a mechanism for drug resistance6. We have recently shown that the neddylation inhibitor MLN4924 induced protective autophagy by modulating the HIF1-REDD1-TSC1-mTORC1-DEPTOR axis, and blockage of autophagy by genetic or pharmacological approaches enhanced MLN4924-induced cell killing via apoptosis49. However, there is extensive evidence in a variety of experimental tumor models that autophagy can also have a cytotoxic function50. Specifically, several recent studies showed that a number of unconventional agents induced cytotoxic autophagy by triggering ER stress5,51,52. Our study fits the following working model: HA-1141, on one hand, binds to and inactivates NAE leading to inactivation of CRLs and transient accumulation of CRL substrates. One the other hand, HA-1141 also triggers unmitigated non-canonical ER stress and severe ISR in a manner dependent of the PKR-ATF4 axis to inhibit protein translation as well as to inactivate the mTORC1 pathway, leading to massive autophagy. It appears that here ATF4 serves as linker to connect two events: blocking neddylation and triggering ER stress, since ATF4 is stabilized by inactivation of CRL1 and induced by ER stress (Fig. 8).
Figure 8.
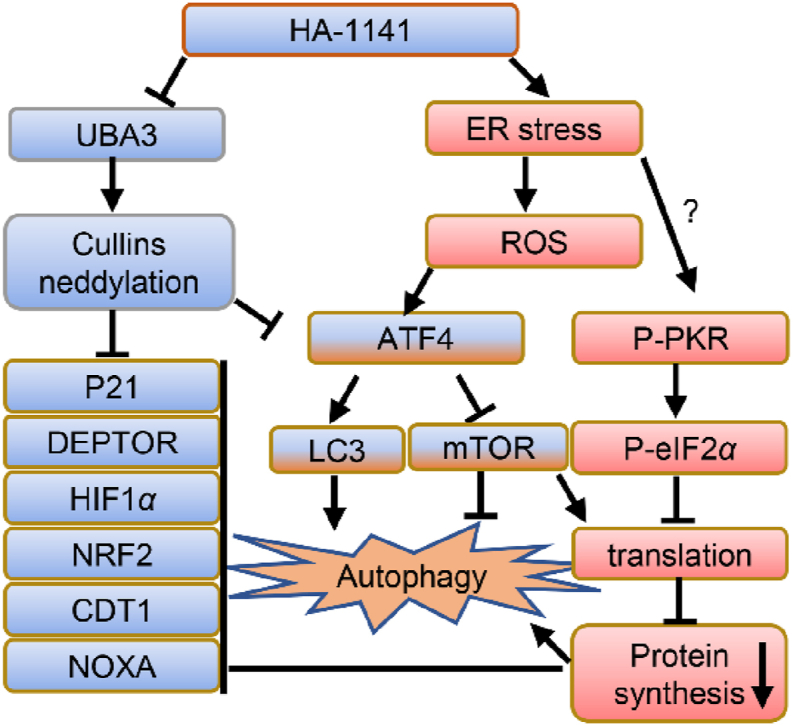
Working model. HA-1141, on one hand, binds to UBA3 to inactivate NEDD8 E1, leading to neddylation inhibition of all cullins to cause accumulation of CRL substrates at early time points. On the other hand, HA-1141 triggers non-canonical ER stress, ISR and produces ROS to inactivate mTORC1 and inhibit protein synthesis, leading to reduction of CRL substrates and induction of autophagy at a later stage. See text for details.
5. Conclusions
We have identified HA-1141, a small molecule with a unique and distinct chemical structure, to have dual activities in blocking cullin neddylation and in triggering non-canonical ER stress and severe ISR. HA-1141 showed impressive anti-cancer activity in both in vitro cell culture and in vivo xenograft lung cancer models. To the best of our knowledge, this is the first-in-class small molecule with such dual activities. Further optimization of HA-1141 to make it more soluble and potent in its dual activities would certainly provide a sound opportunity for future development of a novel class of anti-cancer drugs with a unique mechanism of action.
Acknowledgments
We gratefully acknowledge the National Key R&D Program of China (2016YFA0501800 to Yi Sun) for financial support. We thank the staff members at the Center of Cryo Electron Microscopy at the Zhejiang University School of Medicine for their assistance in transmission electron microscopy and the staff members from the core facilities and the Morphological Platform at Zhejiang University School of Medicine for their assistance in data collection and technical support.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting information to this article can be found online at https://doi.org/10.1016/j.apsb.2021.07.012.
Contributor Information
Sunliang Cui, Email: slcui@zju.edu.cn.
Yi Sun, Email: yisun@zju.edu.cn.
Author contributions
Yi Sun: conceptualization, methodology, supervision, writing-reviewing and editing. Sunliang Cui: methodology, supervision, writing-reviewing and editing. Tingjun Hou: software, supervision, writing-reviewing and editing. Xiufang Xiong: methodology, supervision. Peichen Pan: software, writing-original draft preparation. Lei Xu: software. Chaorong Wang: investigation, writing-original draft preparation. Qing Yu: methodology. Tiantian Xu: investigation. Yanan Li: investigation, writing-original draft preparation.
Conflicts of interest
The authors declare no competing interests.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Saxton R.A., Sabatini D.M. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168:960–976. doi: 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hetz C., Zhang K., Kaufman R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol. 2020;21:421–438. doi: 10.1038/s41580-020-0250-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pohl C., Dikic I. Cellular quality control by the ubiquitin−proteasome system and autophagy. Science. 2019;366:818–822. doi: 10.1126/science.aax3769. [DOI] [PubMed] [Google Scholar]
- 4.Costa-Mattioli M., Walter P. The integrated stress response: from mechanism to disease. Science. 2020;368:eaat5314. doi: 10.1126/science.aat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Munoz-Guardiola P., Casas J., Megias-Roda E., Sole S., Perez-Montoyo H., Yeste-Velasco M., et al. The anti-cancer drug ABTL0812 induces ER stress-mediated cytotoxic autophagy by increasing dihydroceramide levels in cancer cells. Autophagy. 2021;17:1349–1366. doi: 10.1080/15548627.2020.1761651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dikic I., Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19:349–364. doi: 10.1038/s41580-018-0003-4. [DOI] [PubMed] [Google Scholar]
- 7.Deegan S., Saveljeva S., Gorman A.M., Samali A. Stress-induced self-cannibalism: on the regulation of autophagy by endoplasmic reticulum stress. Cell Mol Life Sci. 2013;70:2425–2441. doi: 10.1007/s00018-012-1173-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watson I.R., Irwin M.S., Ohh M. NEDD8 pathways in cancer, sine quibus non. Cancer cell. 2011;19:168–176. doi: 10.1016/j.ccr.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 9.Deshaies R.J. SCF and Cullin/Ring H2-based ubiquitin ligases. Annu Rev Cell Dev Biol. 1999;15:435–467. doi: 10.1146/annurev.cellbio.15.1.435. [DOI] [PubMed] [Google Scholar]
- 10.Jayabalan A.K., Sanchez A., Park R.Y., Yoon S.P., Kang G.Y., Baek J.H., et al. Neddylation promotes stress granule assembly. Nat Commun. 2016;7:12125. doi: 10.1038/ncomms12125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiong X., Cui D., Bi Y., Sun Y., Zhao Y. Neddylation modification of ribosomal protein RPS27L or RPS27 by MDM2 or NEDP1 regulates cancer cell survival. Faseb J. 2020;34:13419–13429. doi: 10.1096/fj.202000530RRR. [DOI] [PubMed] [Google Scholar]
- 12.Yu Q., Jiang Y., Sun Y. Anticancer drug discovery by targeting cullin neddylation. Acta Pharm Sin B. 2020;10:746–765. doi: 10.1016/j.apsb.2019.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou L., Zhang W., Sun Y., Jia L. Protein neddylation and its alterations in human cancers for targeted therapy. Cell Signal. 2018;44:92–102. doi: 10.1016/j.cellsig.2018.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barghout S.H., Schimmer A.D. E1 enzymes as therapeutic targets in cancer. Pharmacol Rev. 2021;73:1–58. doi: 10.1124/pharmrev.120.000053. [DOI] [PubMed] [Google Scholar]
- 15.Zhou W., Xu J., Li H., Xu M., Chen Z.J., Wei W., et al. Neddylation E2 UBE2F promotes the survival of lung cancer cells by activating CRL5 to degrade NOXA via the K11 linkage. Clin Cancer Res. 2017;23:1104–1116. doi: 10.1158/1078-0432.CCR-16-1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H., Tan M., Jia L., Wei D., Zhao Y., Chen G., et al. Inactivation of SAG/RBX2 E3 ubiquitin ligase suppresses KrasG12D-driven lung tumorigenesis. J Clin Invest. 2014;124:835–846. doi: 10.1172/JCI70297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang D.T., Ayrault O., Hunt H.W., Taherbhoy A.M., Duda D.M., Scott D.C., et al. E2-RING expansion of the NEDD8 cascade confers specificity to cullin modification. Mol Cell. 2009;33:483–495. doi: 10.1016/j.molcel.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hornak V., Abel R., Okur A., Strockbine B., Roitberg A., Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Case D.A., Cheatham T.E., 3rd, Darden T., Gohlke H., Luo R., Merz K.M., Jr., et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pan P., Yu H., Liu Q., Kong X., Chen H., Chen J., et al. Combating drug-resistant mutants of anaplastic lymphoma kinase with potent and selective Type-I1/2 inhibitors by stabilizing unique DFG-Shifted loop conformation. ACS Cent Sci. 2017;3:1208–1220. doi: 10.1021/acscentsci.7b00419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hou T., Li N., Li Y., Wang W. Characterization of domain-peptide interaction interface: prediction of SH3 domain-mediated protein−protein interaction network in yeast by generic structure-based models. J Proteome Res. 2012;11:2982–2995. doi: 10.1021/pr3000688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.ACD/ADME suite 5.0. Toronto, Canada: Advanced Chemistry Development Inc.; 2011.
- 23.Discovery studio 2.5 guide. San Diego: Accelrys Inc.; 2009. Available from: http://www.accelrys.com
- 24.Wu K., Chong R.A., Yu Q., Bai J., Spratt D.E., Ching K., et al. Suramin inhibits cullin-RING E3 ubiquitin ligases. Proc Natl Acad Sci U S A. 2016;113:E2011–E2018. doi: 10.1073/pnas.1601089113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martinez Molina D., Jafari R., Ignatushchenko M., Seki T., Larsson E.A., Dan C., et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 2013;341:84–87. doi: 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- 26.Brownell J.E., Sintchak M.D., Gavin J.M., Liao H., Bruzzese F.J., Bump N.J., et al. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol Cell. 2010;37:102–111. doi: 10.1016/j.molcel.2009.12.024. [DOI] [PubMed] [Google Scholar]
- 27.Soucy T.A., Smith P.G., Milhollen M.A., Berger A.J., Gavin J.M., Adhikari S., et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 28.Leung C.H., Chan D.S., Yang H., Abagyan R., Lee S.M., Zhu G.Y., et al. A natural product-like inhibitor of NEDD8-activating enzyme. Chem Commun (camb) 2011;47:2511–2513. doi: 10.1039/c0cc04927a. [DOI] [PubMed] [Google Scholar]
- 29.Rzymski T., Harris A.L. The unfolded protein response and integrated stress response to anoxia. Clin Cancer Res. 2007;13:2537–2540. doi: 10.1158/1078-0432.CCR-06-2126. [DOI] [PubMed] [Google Scholar]
- 30.Malhotra J.D., Miao H., Zhang K., Wolfson A., Pennathur S., Pipe S.W., et al. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc Natl Acad Sci U S A. 2008;105:18525–18530. doi: 10.1073/pnas.0809677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lange P.S., Chavez J.C., Pinto J.T., Coppola G., Sun C.W., Townes T.M., et al. ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo. J Exp Med. 2008;205:1227–1242. doi: 10.1084/jem.20071460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koditz J., Nesper J., Wottawa M., Stiehl D.P., Camenisch G., Franke C., et al. Oxygen-dependent ATF-4 stability is mediated by the PHD3 oxygen sensor. Blood. 2007;110:3610–3617. doi: 10.1182/blood-2007-06-094441. [DOI] [PubMed] [Google Scholar]
- 33.Appenzeller-Herzog C., Hall M.N. Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol. 2012;22:274–282. doi: 10.1016/j.tcb.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 34.Kim Y.C., Guan K.L. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 2015;125:25–32. doi: 10.1172/JCI73939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mizushima N., Yoshimori T., Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu M.H., Lee C.Y., Huang T.J., Huang K.Y., Tang C.H., Liu S.H., et al. MLN4924, a protein neddylation inhibitor, suppresses the growth of human chondrosarcoma through inhibiting cell proliferation and inducing endoplasmic reticulum stress-related apoptosis. Int J Mol Sci. 2018;20:72. doi: 10.3390/ijms20010072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen P., Hu T., Liang Y., Li P., Chen X., Zhang J., et al. Neddylation inhibition activates the extrinsic apoptosis pathway through ATF4-CHOP-DR5 axis in human esophageal cancer cells. Clin Cancer Res. 2016;22:4145–4157. doi: 10.1158/1078-0432.CCR-15-2254. [DOI] [PubMed] [Google Scholar]
- 38.Abdullahi A., Stanojcic M., Parousis A., Patsouris D., Jeschke M.G. Modeling acute ER stress in vivo and in vitro. Shock. 2017;47:506–513. doi: 10.1097/SHK.0000000000000759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Han J., Back S.H., Hur J., Lin Y.H., Gildersleeve R., Shan J., et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 2013;15:481–490. doi: 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shah J.J., Jakubowiak A.J., O'Connor O.A., Orlowski R.Z., Harvey R.D., Smith M.R., et al. Phase i study of the novel investigational NEDD8-activating enzyme inhibitor pevonedistat (MLN4924) in patients with relapsed/refractory multiple myeloma or lymphoma. Clin Cancer Res. 2016;22:34–43. doi: 10.1158/1078-0432.CCR-15-1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mao H., Sun Y. Neddylation-independent activities of MLN4924. Adv Exp Med Biol. 2020;1217:363–372. doi: 10.1007/978-981-15-1025-0_21. [DOI] [PubMed] [Google Scholar]
- 42.Wortel I.M.N., van der Meer L.T., Kilberg M.S., van Leeuwen F.N. Surviving stress: modulation of ATF4-mediated stress responses in normal and malignant cells. Trends Endocrinol Metab. 2017;28:794–806. doi: 10.1016/j.tem.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hull C.M., Bevilacqua P.C. Discriminating self and non-self by RNA: roles for RNA structure, misfolding, and modification in regulating the innate immune sensor PKR. Acc Chem Res. 2016;49:1242–1249. doi: 10.1021/acs.accounts.6b00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu C.X., Li X., Nan F., Jiang S., Gao X., Guo S.K., et al. Structure and degradation of circular RNAs regulate PKR activation in innate immunity. Cell. 2019;177:865–880. doi: 10.1016/j.cell.2019.03.046. e21. [DOI] [PubMed] [Google Scholar]
- 45.Roulois D., Loo Yau H., Singhania R., Wang Y., Danesh A., Shen S.Y., et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell. 2015;162:961–973. doi: 10.1016/j.cell.2015.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luhr M., Torgersen M.L., Szalai P., Hashim A., Brech A., Staerk J., et al. The kinase PERK and the transcription factor ATF4 play distinct and essential roles in autophagy resulting from tunicamycin-induced ER stress. J Biol Chem. 2019;294:8197–8217. doi: 10.1074/jbc.RA118.002829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Antonioli M., Di Rienzo M., Piacentini M., Fimia G.M. Emerging mechanisms in initiating and terminating autophagy. Trends Biochem Sci. 2017;42:28–41. doi: 10.1016/j.tibs.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 48.Lu G., Wang L., Zhou J., Liu W., Shen H.M. A destiny for degradation: interplay between Cullin-RING E3 ligases and autophagy. Trends Cell Biol. 2021;31:432–444. doi: 10.1016/j.tcb.2021.01.005. [DOI] [PubMed] [Google Scholar]
- 49.Zhao Y., Xiong X., Jia L., Sun Y. Targeting Cullin-RING ligases by MLN4924 induces autophagy via modulating the HIF1-REDD1-TSC1-mTORC1-DEPTOR axis. Cell Death Dis. 2012;3 doi: 10.1038/cddis.2012.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sharma K., Le N., Alotaibi M., Gewirtz D.A. Cytotoxic autophagy in cancer therapy. Int J Mol Sci. 2014;15:10034–10051. doi: 10.3390/ijms150610034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tang B., Li Q., Zhao X.H., Wang H.G., Li N., Fang Y., et al. Shiga toxins induce autophagic cell death in intestinal epithelial cells via the endoplasmic reticulum stress pathway. Autophagy. 2015;11:344–354. doi: 10.1080/15548627.2015.1023682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Salazar M., Carracedo A., Salanueva I.J., Hernandez-Tiedra S., Lorente M., Egia A., et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J Clin Invest. 2009;119:1359–1372. doi: 10.1172/JCI37948. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.