

SUMMARY
NLRP6 is important in host defense by inducing functional outcomes including inflammasome activation and interferon production. Here we show that NLRP6 undergoes liquid-liquid phase separation (LLPS) upon interaction with dsRNA in vitro and in cells, and that an intrinsically disordered poly-lysine sequence (K350–354) of NLRP6 is important for multivalent interactions, phase separation and inflammasome activation. Nlrp6-deficient or Nlrp6K350−354A mutant mice show reduced inflammasome activation upon mouse hepatitis virus or rotavirus infection, and in steady state stimulated by intestinal microbiota, implicating NLRP6 LLPS in anti-microbial immunity. Recruitment of ASC via helical assembly solidifies NLRP6 condensates, and ASC further recruits and activates caspase-1. Lipoteichoic acid, a known NLRP6 ligand, also promotes NLRP6 LLPS, and DHX15, a helicase in NLRP6-induced interferon signaling, co-forms condensates with NLRP6 and dsRNA. Thus, LLPS of NLRP6 is a common response to ligand stimulation, which serves to direct NLRP6 to distinct functional outcomes depending on the cellular context.
Keywords: NLRP6, RNA, Liquid-Liquid Phase Separation, LLPS, Inflammasome
Graphical Abstract
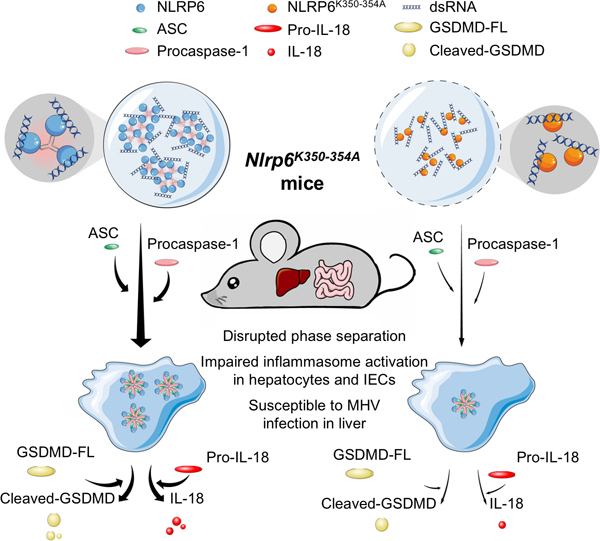
In brief:
dsRNA induces phase separation of NLRP6 for inflammasome activation
INTRODUCTION
The innate immune system coordinates a wide array of germline-encoded pattern recognition receptors (PRRs) to sense pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) (Broz and Dixit, 2016; Evavold and Kagan, 2019). Canonical inflammasomes are cytosolic multi-protein complexes involved in innate immunity. They assemble mainly through nucleotide-binding domain (NBD) and leucine-rich repeat containing (NLR) proteins, such as NLRP1, NLRP3, NAIP, and NLRP6. Upon activation, these sensor proteins recruit pro-caspase-1 directly or indirectly through the adaptor ASC or NLRC4, which leads to the dimerization and activation of caspase-1. The subsequent caspase-mediated processing of pro-interleukin (IL)-1β, pro-IL-18, and the pore-forming protein gasdermin D (GSDMD) results in the maturation and release of these cytokines, as well as pyroptotic cell death (Lieberman et al., 2019; Xia et al., 2020).
NLRP6 plays important roles in host defense by undertaking a plethora of seemingly unrelated inflammasome-dependent and -independent functions. In intestinal epithelial cells, NLRP6 has been reported to sense microbiota-associated metabolites to form an ASC-dependent inflammasome for downstream IL-18 release and secretion of anti-microbial peptides (Levy et al., 2015). Meanwhile, NLRP6 has been shown to engage viral RNA via the RNA helicase DHX15 to restrict infection by enteric viruses through the interferon pathway (Wang et al., 2015). In sentinel goblet cells, NLRP6 has been shown to regulate mucus secretion in an inflammasome- and autophagy-dependent manner to prevent invasion by enteric bacteria (Birchenough et al., 2016; Wlodarska et al., 2014), although this conclusion has been challenged (Volk et al., 2019). In myeloid cells, NLRP6 suppresses the inflammatory response by negatively regulating nuclear factor-kappa B (NF-κB) signaling (Anand et al., 2012). Genetic analyses have identified NLRP6 to be an inflammasome sensor for lipoteichoic acid (LTA), a cell-wall component of Gram-positive bacteria (Hara et al., 2018). Biochemical studies have shown that Escherichia coli-expressed NLRP6 interacts with lipopolysaccharide (LPS), an outer-membrane component of Gram-negative bacteria (Leng et al., 2020). However, whether any of these potential ligands engage NLRP6 directly is not clear.
Several inflammasomes including the NLRP6 inflammasome form specks in cells upon activation (Broz and Dixit, 2016; Ghimire et al., 2018; Hara et al., 2018; Magupalli et al., 2020). These specks are dependent on the pyrin domain (PYD) and the caspase recruitment domain (CARD) of the ASC adaptor, which oligomerize using helical symmetry (Li et al., 2018; Lu et al., 2014) to form ordered assemblies to recruit and activate caspase-1 (Lu et al., 2016). An established paradigm is that in order to promote adaptor oligomerization, an NLR sensor forms a disk-like assembly (Hu et al., 2015; Sharif et al., 2019; Tenthorey et al., 2017; Yang et al., 2018; Zhang et al., 2015) to bring their PYDs or CARDs into proximity for nucleating the PYD or CARD filaments.
Here, we provide biochemical and cellular evidence that double-stranded (ds) RNA, which is produced by many viruses, can interact directly with NLRP6 to form a dynamic, liquid-like condensed phase (also known as liquid–liquid phase separation, or LLPS), rather than an ordered assembly. Physiologically, mutation of a positively charged region of NLRP6 important for LLPS compromised dsRNA-induced formation of NLRP6 puncta, GSDMD cleavage, and cell death, and impaired anti-microbial defense in mice. In an overarching paradigm, we revealed that LTA also induced NLRP6 LLPS, and that DHX15 co-localized with NLRP6 in dsRNA-induced liquid condensate. These findings suggest that LLPS of NLRP6 may endow the versatility of NLRP6 to respond to multiple ligands and/or to diversify the downstream pathway and shed lights on the sensory and activation mode of PRRs in inflammasomes as well as other immune signaling pathways.
RESULTS
NLRP6 Interacts with dsRNA Directly
Sequence analyses revealed high isoelectric points of 8.4 and 8.8 for human and mouse NLRP6, respectively, suggesting that NLRP6 is conserved as a positively charged protein. To identify direct ligands of NLRP6, we first tested the ability of insect cell-expressed, maltose-binding protein (MBP)-tagged NLRP6 to bind nucleic acids. MBP-NLRP6 formed complexes with the long-dsRNA analog high molecular weight (HMW) polyinosinic-polycytidylic acid (poly I:C), as well as with a short (20-bp) dsRNA in gel shift assays (Figure 1A). MBP-removed, untagged NLRP6 exhibited monomeric and filamentous forms (Shen et al., 2019) (Figure S1A), both of which interacted with HMW poly I:C and 20-bp dsRNA, which suggested that neither the tag nor the oligomeric form altered the interaction radically (Figure S1B, C).
Figure 1. NLRP6 Interacts with dsRNA in Vitro.
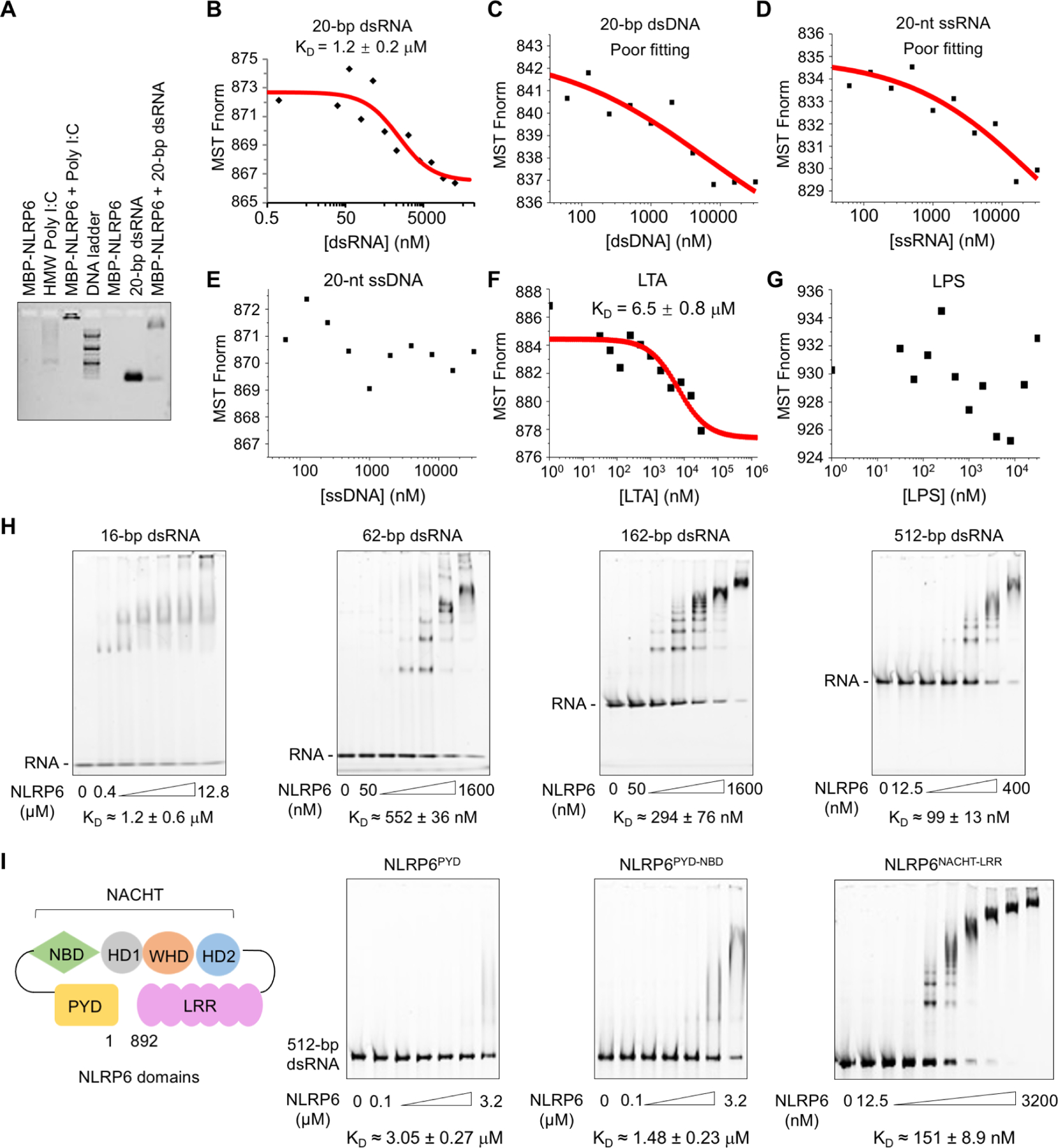
(A) Agarose gel-shift assay of HMW poly I:C and 20-bp dsRNA with MBP-tagged NLRP6. HMW poly I:C alone ran as a smear.
(B–E) Measurements of dissociation constants (KD) of NLRP6 with different types of nucleic acids by microscale thermophoresis (MST). Each KD was derived from the binding response as a function of the GFP-NLRP6 concentration. Errors in KD represent fitting errors.
(F, G) KD measurements of NLRP6 with different ligands (LTA, LPS) by MST.
(H, I) Electrophoretic mobility shift assays (EMSAs) of MBP-NLRP6 with dsRNA of different lengths at 2.5 ng/μL (H) and of different MBP-NLRP6 truncations with 512-bp dsRNA at 2.5 ng/μL (I). The MBP-NLRP6 concentrations were in two-fold dilutions and the first and last concentrations are shown. KD was derived from the free dsRNA Intensity as a function of the MBP-NLRP6 concentration. Errors in KD represent fitting errors.
See also Figure S1
We further measured the binding dissociation constants (KD) of monomeric, green fluorescent protein (GFP)-tagged NLRP6 (GFP-NLRP6) with 20-bp dsRNA and dsDNA, and 20-nt ssRNA and ssDNA using microscale thermophoresis (MST). NLRP6 bound 20-bp dsRNA preferentially with a KD of ~1.2 μM, but did not interact (or interacted weakly) with other forms of nucleic acids (Figure 1B–E, S1D). The robust interaction of NLRP6 with dsRNA was confirmed by quantifying the intensity of 20-bp dsRNA with increasing concentrations of NLRP6 in an electrophoretic mobility shift assay (EMSA), which revealed the somewhat higher affinity of monomeric NLRP6 for dsRNA than filamentous NLRP6 (Figure S1E, F). We also used MST to measure the binding of GFP-NLRP6 to other previously implicated NLRP6 ligands or modulators: LTA from Gram-positive bacteria (Hara et al., 2018), LPS from Gram-negative bacteria (Leng et al., 2020) and microbiota-associated metabolites taurine and spermine (Levy et al., 2015). Only LTA exhibited relatively robust direct binding with NLRP6 (KD = 6.5 μM) (Figure 1F, G, S1G, H).
We wished to ascertain whether dsRNA recognition by NLRP6 is dependent on length. We undertook EMSA and found that longer dsRNA exhibited increased binding affinity relative to shorter dsRNA, with a KD of ~99 nM for 512-bp dsRNA (Figure 1H). NLRP6 has an N-terminal PYD, a central NBD-containing NACHT domain and a C-terminal LRR domain (Figure 1I). EMSA between 512-bp dsRNA and NLRP6 recombinant proteins containing various domains indicated that the NACHT-LRR region dominates the interaction of NLRP6 with dsRNA, whereas the PYD appeared not to play an important role (Figure 1I). Among different NLR proteins, only NLRP6, but not NLRC4∆CARD or NLRP3∆PYD, interacted with 20-bp dsRNA and poly I:C (Figure S1I). Thus, NLRP6 alone displayed strong and selective binding to dsRNA, with further increased affinity for longer dsRNA, and NLRP6 may act as a specific PRR for various dsRNA species from pathogens.
dsRNA Induces LLPS of NLRP6 in vitro and in Cells
We noticed that NLRP6 solutions became turbid upon mixing with various RNA species, and wondered if phase separation had occurred. To determine this, we expressed and purified MBP-tagged NLRP6-mCherry, mixed it with fluorescein amidite (FAM)-labeled 60-bp dsRNA, and added tobacco etch virus protease (TEV) to remove MBP. After 45 min, we observed droplet formation by fluorescence microscopy between NLRP6-mCherry and dsRNA, with the two species showing almost complete overlap (Figure 2A). Similarly, recombinant MBP-NLRP6 without the mCherry tag also induced droplet formation with FAM-labeled 60-bp dsRNA in a time-dependent manner with or without TEV for removing the MBP tag (Figure S2A).
Figure 2. dsRNA-Induced LLPS of NLRP6 in Vitro and in Cells.
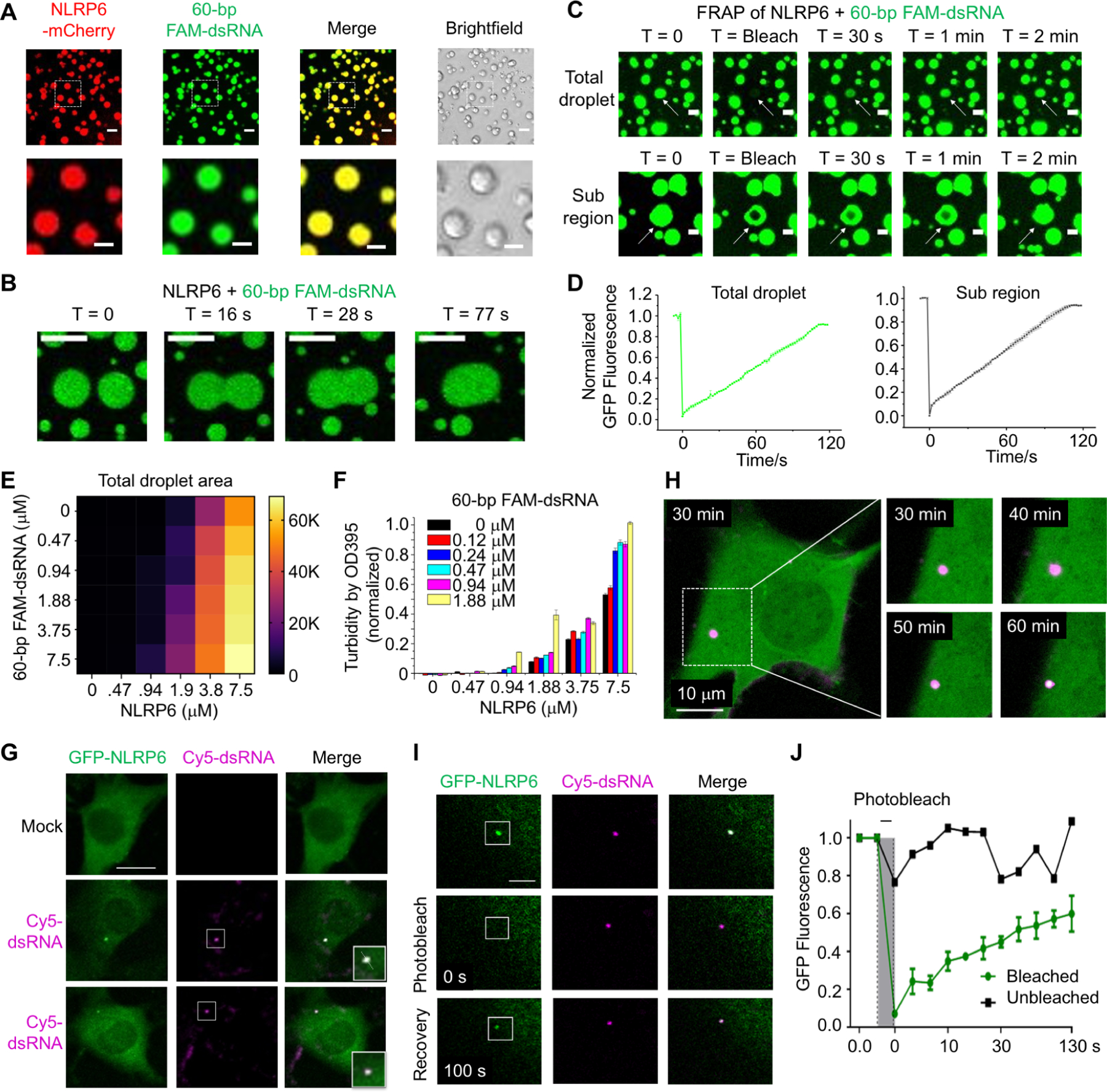
(A) Images of phase-separated droplets from purified NLRP6-mCherry (10 μM) and 60-bp FAM-labeled dsRNA (10 μM) (upper panel, scale bar: 5 μm) and their zoomed subregions (lower panel, scale bar: 3 μm). Incubation time: 45 min.
(B) Time-lapse micrographs of merging droplets formed by MBP-NLRP6 (15 μM) and 60-bp FAM-labeled dsRNA (15 μM). Data are representative of 10 merging droplets. Scale bar: 2 μm.
(C) FRAP of FAM-dsRNA in a total NLRP6-dsRNA droplet (top) and a region (bottom). MBP-NLRP6 and dsRNA are both at 15 μM. Data are representative of three independent experiments. White arrows indicate the target droplets for FRAP. Scale bar: 1 μm.
(D) Quantitative FRAP curves generated form (C). Error bars were from three independent experiments.
(E) Phase diagram of MBP-NLRP6 with 60-bp FAM-dsRNA. Total area (μm2) occupied by droplets in each image (128,010 μm2) was quantified into a 6 × 6 matrix. Brightfield images related to Fig. S3C were used for quantification.
(F) Phase diagram of MBP-NLRP6 with 60-bp FAM-dsRNA from the OD395 turbidity assay.
(G) Representative live-cell images of NLRP6-dsRNA specks after transfection of 45-bp Cy5-dsRNA into MEFs stably expressing GFP-NLRP6. Zoomed images indicate NLRP6-RNA puncta (boxes). Scale bar: 5 μm. These images are representative of ≥ 10 cells.
(H) Time-lapse micrographs of NLRP6 (green) and dsRNA (purple) speck formation, showing merging of NLRP6 into the dsRNA droplet evidenced by the whiter color as a function of time after Cy5-dsRNA transfection due to more green color in the purple speck. Scale bar:10 μm.
(I) Images of an NLRP6-dsRNA speck before and after photobleaching (white box: bleach site), representative of ≥ 3 cells. Scale bar: 5 μm.
(J) Quantification of FRAP data in (I), shown as the mean ± SD of n = 3 NLRP6-dsRNA puncta. The bleaching event is marked at t = 0 s.
The droplets of NLRP6 and dsRNA were highly dynamic because we could capture fusion between droplets in < 80 s (Figure 2B). We undertook fluorescence recovery after photobleaching (FRAP) on a full droplet and on a region of a single droplet. Both droplets showed full recovery within 120 s after photobleaching, which indicated fast exchange of materials with the environment and confirmation of the liquid state (Figure 2C, D). Consistent with the EMSA experiments, PYD-truncated NLRP6 (NLRP6∆PYD) formed droplets with dsRNA, whereas PYD-truncated NLRP3 (NLRP3∆PYD) did not (Figure S2B). We also used the phase-separation assay to map the different domains of NLRP6: the NACHT domain was an important region for droplet formation with dsRNA (Figure S2C). However, the truncated NLRP6 protein containing the NACHT domain formed irregularly shaped condensates that were most likely solid (Figure S2C, D), and negative staining electron microscopy (EM) of the PYD-NACHT construct showed filamentous assemblies (Figure S2E), suggesting that the multivalency provided by full-length NLRP6 is required for LLPS formation by NLRP6.
We compared the property of the NLRP6–dsRNA LLPS under different conditions. Titrations of the protein and RNA concentrations indicated the different critical points for formation of the condensed protein–RNA phase in vitro, and the protein concentration was a more important factor in triggering phase separation (Figure S3A, B). The interdependence of these factors was confirmed by the measured phase diagram on NLRP6 and dsRNA concentrations (Figure 2E, S3C). Importantly, while NLRP6 formed droplets on its own at a high protein concentration, it required dsRNA to form droplets when its concentration was low (Figure S3D). We next used a turbidity assay that measures OD at 395 nm and confirmed the dependence of LLPS on both NLRP6 and dsRNA concentrations (Figure 2F). The NLRP6–dsRNA droplets also favored lower salt and higher temperature (up to 37 °C) (Figure S3E, F), and the crowding reagent polyethylene glycol 3350 (PEG3350) enhanced droplet formation by decreasing the required NLRP6 and RNA concentrations (Figure S3G).
We investigated dsRNA-induced LLPS of NLRP6 in cells. We first co-transfected NLRP6-mCherry and fluorescein isothiocyanate (FITC)-HMW poly I:C in HBL-1 cells, which led to large co-localized specks, whereas transfection of NLRP6-mCherry alone did not lead to speck formation and transfected FITC-HMW poly I:C alone formed much smaller speckles (Figure S3H). We then generated a mouse embryonic fibroblast (MEF) cell line that stably expressed GFP-tagged mouse NLRP6 (GFP-mNLRP6). Upon transfection of 45-bp Cyanine 5 (Cy5)-labeled dsRNA, we observed punctum formation of NLRP6 (green) that colocalized with the dsRNA (magenta) with 1 to 3 puncta per cell (Figure 2G, S3I). When tracked by live-cell imaging, NLRP6 and dsRNA foci appeared less than 1 h after dsRNA transfection, with dsRNA foci first and NLRP6 merging into the foci as a function of time (Figure 2H, S3J, Movie S1A–B). MEFs stably expressing GFP also formed dsRNA-alone puncta upon dsRNA transfection, likely due to the transfection condition as NLRP6 was required for dsRNA punctum formation in vivo (see below). We undertook FRAP on GFP-mNLRP6 foci in MEF cells (Figure 2I, Movie S2), and NLRP6 droplets showed robust recovery in ~130 s (Figure 2J), which supported the dynamic, liquid-like nature of these foci.
Identification of a Poly-Lys Region in NLRP6 that is Critical for Multivalent Interaction and Phase Separation
We showed previously that upon removal of the MBP tag, recombinant MBP-NLRP6 formed long helical filaments with PYD in the center and disordered NACHT-LRR domains at the periphery (Shen et al., 2019). Therefore, we investigated if NLRP6 contains intrinsically disordered regions (IDRs) that may contribute to the multivalency required to drive phase separation (Boeynaems et al., 2018; Du and Chen, 2018; Wu and Fuxreiter, 2016). IDR prediction on mouse and human NLRP6 with the Predictor of Natural Disordered Regions (PONDR) program revealed six regions with a high tendency for disorder (Figure 3A and S4A). This phenomenon was in contrast to the low PONDR scores on human NLRC4 and NLRP3 (Figure S4B, C).
Figure 3. IDRs and Polybasic Regions of NLRP6 are Required for Phase Separation and Inflammasome Activation.
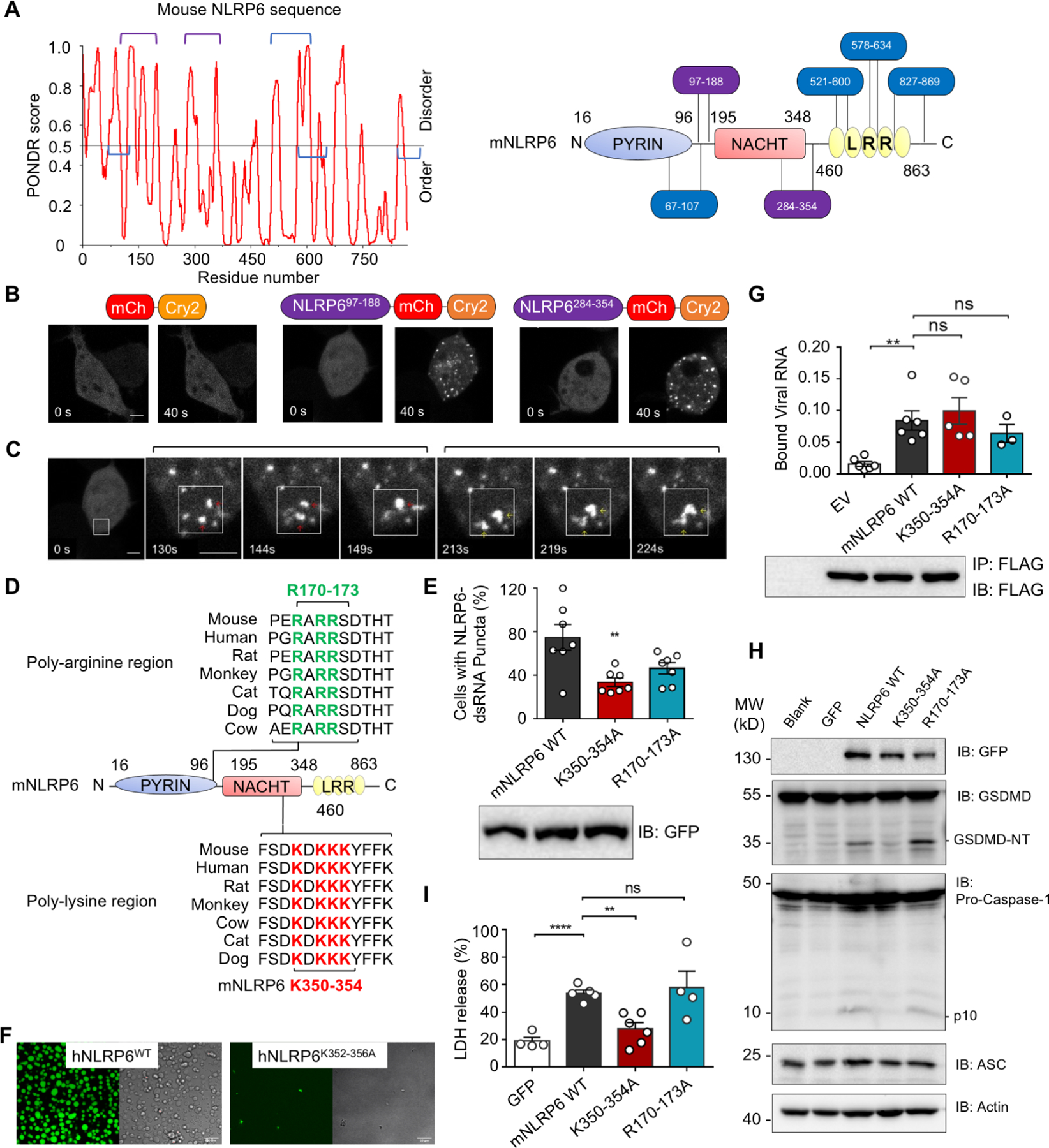
(A) Graph plotting intrinsic disorder (by PONDR VSL2) in mNLRP6 (left) and the locations of the IDR segments (right).
(B) Time-lapse images of 293T cells expressing NLRP6-optoIDR constructs containing indicated mNLRP6 IDRs (residues 97–188 or 284–354) linked to mCherry (red) and Cry2 (orange). mCherry-Cry2 fusion alone was used as a control. Cells were subjected to laser excitation every 2 s for the indicated time. Scale bar: 5 μm.
(C) Time-lapse images (0 s – 224 s) of an 293T cell expressing NLRP6-optoIDR (residues 97–188) subjected to laser excitation every 2 s for the times indicated. Two sequential droplet fusion events within the region highlighted by the white box are shown. Scale bar: 5μm.
(D) Conservation of polybasic sequences in NLRP6 IDRs across homologs, aligned using Mega X.
(E) Percentage of cells with NLRP6-dsRNA foci upon co-transfection of dsRNA and NLRP6 WT or mutants, representative of ≥ 3 independent experiments.
(F) In vitro phase-separation assay of WT MBP-hNLRP6 and MBP-hNLRP6 K352–356A (15 μM) with 60-bp FAM-dsRNA (15 μM). Scale bars: 10 μm.
(G) qPCR of viral RNA bound by FLAG-tagged mouse NLRP6 and its mutants expressed in EMCV-infected HEK293T cells.
(H) Immunoblots of GSDMD, caspase-1, ASC and actin in iBMDMs transduced with lentiviruses expressing NLRP6 or its mutants.
(I) Pyroptotic cell death indicated by release of lactate dehydrogenase (LDH) in culture supernatants 24 h post infection by WT or mutant NLRP6-containing lentiviruses.
See also Figure S4
To ascertain whether the predicted IDRs could form LLPS in cells, we used an optogenetic platform (optoDroplets) involving designed optoIDR constructs in which the predicted IDRs from mNLRP6 were fused to mCherry, and the photolyase homology region of Cry2 (a light-sensitive protein that self-associates upon blue light exposure to facilitate LLPS) (Sabari et al., 2018; Shin et al., 2017). Two out of the six predicted mNLRP6 IDRs, IDR (97–188) and IDR (284–354), promoted speck formation in cells upon overexpression and light stimulation (Figure 3B and S4D, Movie S3A–B). These specks exhibited liquid-like properties shown by droplet fusion (Figure 3C, Movie S3A). When the corresponding human NLRP6 IDR1 (86–190) and IDR2 (286–356) were expressed as an MBP fusion, purified, labeled with TAMRA dye, and treated with TEV to remove the MBP tag, they all formed droplets in vitro when the crowding agent PEG3350 (5%) was added to mimic the cellular environment (Figure S4E). Taken together, these results suggested that the predicted IDRs in human and mouse NLRP6 could undergo LLPS, and may assist dsRNA-induced LLPS of full-length NLRP6.
We selectively chose poly-basic regions (poly-lysine or poly-arginine) within these IDRs (Figure 3D), constructed mutations to alanine on evolutionarily conserved lysine or arginine residues (170–173 and 350–354 of mNLRP6), and generated MEF cell lines stably expressing these mutants. Upon dsRNA transfection, K350–354A mutation significantly weakened the dsRNA-induced phase separation of NLRP6 (Figure 3E and S4F). The corresponding K352–356A mutation on hNLRP6 abolished dsRNA-induced formation of NLRP6 droplets in vitro (Figure 3F). However, the K350–354A mutant did not impair the binding of NLRP6 to viral RNA (Figure 3G), but reduced overexpression-induced LLPS of IDR (284–354) (Figure S4G, H, Movie S4), which was probably due to its influence on multivalent NLRP6–NLRP6 interactions shown by immunoprecipitation (Figure S4I). Of note, the K350–354 region is within the NACHT domain of NLRP6 (Figure 1I), which in other NLRs such as NLRC4 is involved in self oligomerization (Diebolder et al., 2015; Hu et al., 2015; Tenthorey et al., 2017; Yang et al., 2018; Zhang et al., 2015).
Lentiviral transduction of wild-type (WT) Nlrp6 into mouse immortalized bone marrow-derived macrophages (iBMDMs) caused activation of the NLRP6 inflammasome as shown by caspase-1 activation and GSDMD cleavage (Figure 3H). When Nlrp6 mutant genes were transduced into iBMDMs, the K350–354A mutation, but not the R170–173A mutation, impaired GSDMD and caspase-1 cleavage (Figure 3H). These results were cross validated by the lactate dehydrogenase (LDH) release assay that measures pyroptotic cell death (Figure 3I). Thus, NLRP6 multivalent interactions are important regulators for RNA-induced activation of the NLRP6 inflammasome and the K350–354A mutation abolished inflammasome activation by its deficient ability to phase separate.
Restriction of a Coronavirus Infection in the Liver by Phase Separation of NLRP6 and Inflammasome Activation
To facilitate determination of expression of endogenous NLRP6, we generated a Flag-Nlrp6 knock-in mouse line using the method described previously (Wang et al., 2015). We found that endogenous Flag-NLRP6 showed high expression in intestinal epithelial cells (IECs) and hepatocytes (Figure 4A). Meanwhile, by reverse transcription-quantitative real-time polymerase chain reaction (RT-qPCR), Nlrp9b showed exclusive expression in IECs, Nlrp3 exhibited the highest expression in Kupffer cells, and Nlrp6 was detected in both IECs and hepatocytes (Figure 4B and S5A), suggesting that the NLRP6 inflammasome may have a unique role in hepatocytes.
Figure 4. Physiological Role of NLRP6 Phase Separation in Anti-RNA Virus Defense in Hepatocytes.
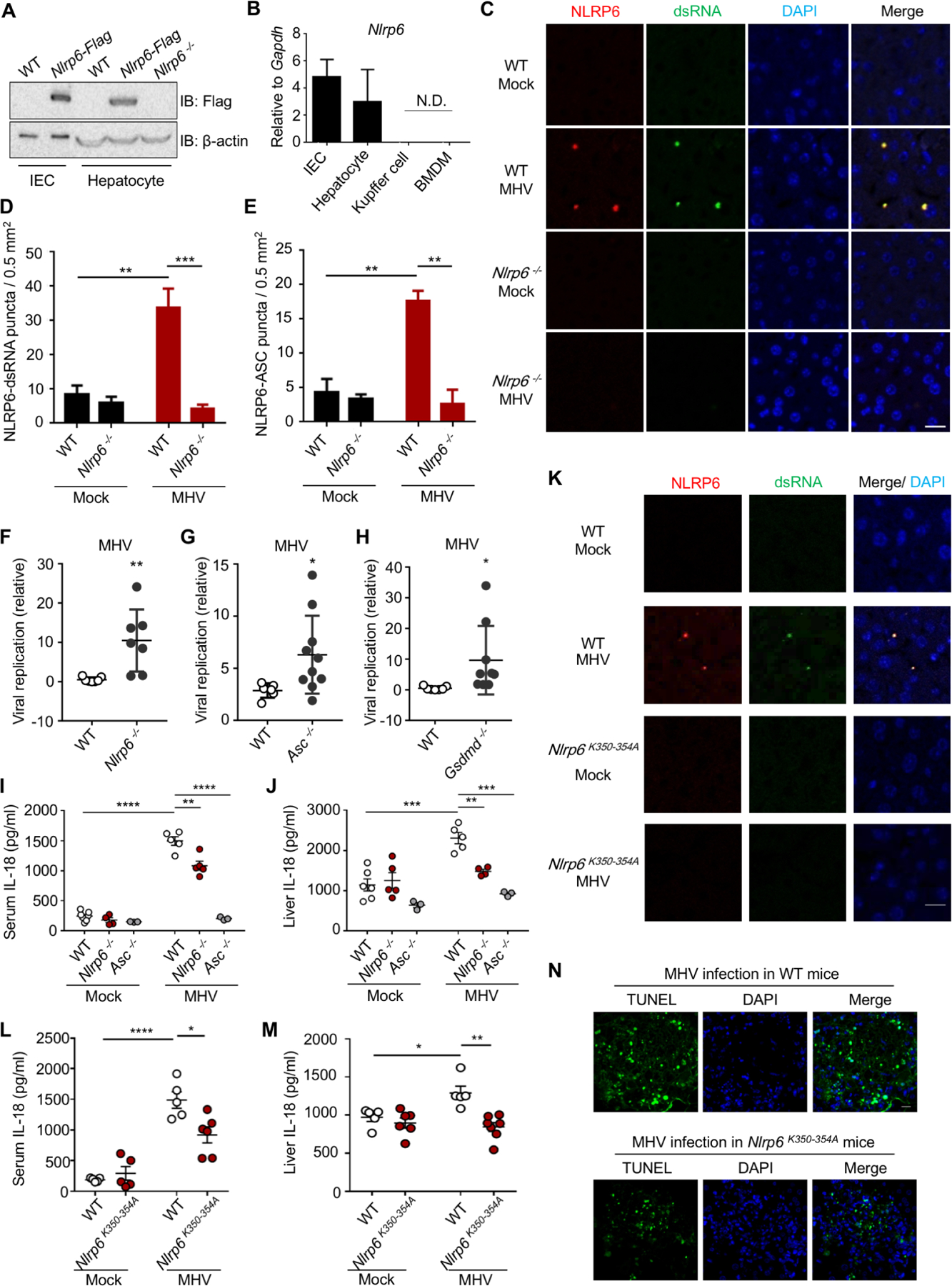
WT, Nlrp6−/− or Nlrp6K350−354A knock-in mice were infected with MHV (i.p.) and analyzed as indicated (C–N).
(A) Immunoblot of NLRP6 in primary hepatocytes or intestinal epithelial cells (IECs) from Nlrp6-Flag knock-in mice.
(B) qPCR assays for Nlrp6 mRNA levels in different cells. N.D.: not detectable.
(C) Fluorescence immunohistochemistry staining of dsRNA (J2) and NLRP6 in liver sections from uninfected or MHV-infected WT and Nlrp6−/− mice. Nuclei were stained with DAPI.
(D, E) Quantification of NLRP6-dsRNA (D) and NLRP6-ASC (E) puncta in liver sections. Data are mean ± SEM from 6 randomly sampled regions.
(F-H) Viral replication in the liver 3 days after MHV infection in Nlrp6−/− (F), Asc−/− (G) and Gsdmd−/− mice (H) in comparison to WT mice.
(I, J) Serum (I) and liver-explant supernatant (J) IL-18 levels determined by ELISA from WT, Nlrp6−/− and Asc−/− mice 3 days post-infection.
(K) Fluorescence immunohistochemistry staining of dsRNA (J2) and NLRP6 in liver sections from uninfected or MHV-infected WT and Nlrp6K350−354A mice. Nuclei were stained with DAPI.
(L, M) Serum (L) and liver-explant supernatant (M) IL-18 levels in WT and Nlrp6K350−354A mice 3 days post-infection.
(N) TUNEL staining of MHV-infected WT and Nlrp6K350−354A mice 3 days after infection. Scale bar: 20 μm.
To test the role of NLRP6 in the liver, we used the MHV strain A59, which is a positive-sense ssRNA virus that belongs to the coronavirus family. Upon MHV infection, we observed inducible endogenous NLRP6–dsRNA puncta and endogenous NLRP6–ASC puncta in MHV-infected mouse liver sections in comparison to the non-infection control and the NLRP6−/− mouse control (Figure 4C–E, S5B). Unlike the situations in vitro (Figure S3J), these dsRNA-containing puncta were dependent on NLRP6 (Figure 4C–D). We then used RT-qPCR to measure viral replication in mouse livers three days after MHV infection. We found a significantly enhanced viral replication in Nlrp6−/−, Asc−/− or Gsdmd−/− mice in comparison with that in WT mice (Figure 4F–H), which suggested an important role of the NLRP6 inflammasome in the clearance of MHV in hepatocytes. Furthermore, the concentrations of IL-18 in the sera and supernatants of liver explants of Nlrp6−/− mice infected with MHV were substantially lower than littermate WT control mice, whereas the Nlrp6 deficiency did not affect basal IL-18 levels (Figure 4I, J). By contrast, Asc−/− mice exhibited reduced IL-18 levels with and without MHV infection (Figure 4I, J). MHV also induced more pyroptosis shown by GSDMD cleavage in liver homogenates (Figure S5C), and more cell death shown by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining in liver sections in WT mice than in Nlrp6−/− mice (Figure S5D). Previous studies proposed that MHV can activate the NLRP3 inflammasome in macrophages, likely by ROS generation upon infection (Guo et al., 2015; Zheng et al., 2020), which provided complementary NLR activation in a different cell type.
To validate the physiological role of LLPS in vivo, we constructed knock-in mice bearing the K350–354A mutation in Nlrp6 to disrupt the region critical for multivalent interaction and phase separation. The NLRP6 protein levels were not affected by the K350–354A mutation in liver and small intestine (Figure S5E, F). When challenged by MHV infection, mice carrying the K350–354A mutation in Nlrp6 did not show puncta in liver sections, like those of NLRP6−/− mice or uninfected control mice (Figure 4K, S5G), and exhibited reduced IL-18 secretion in the sera and in the supernatants of liver explants (Figure 4L, M), reduced GSDMD cleavage in liver homogenates (Figure S5H), and reduced TUNEL staining in liver explants (Figure 4N, S5I), in comparison with WT littermate control mice. These results suggested that NLRP6 and its ability to form dsRNA-dependent LLPS are important in inflammasome activation and anti-MHV immune response in the liver.
The RNA helicase DHX15 was shown to associate with NLRP6 to induce interferon and interferon stimulated genes in IECs (Wang et al., 2015). To verify the role of Dhx15, we infected mice that had liver-specific deletion of Dhx15 with MHV, and found identical viral replication in the liver and equivalent GSDMD cleavage or IL-18 levels in the serum and the liver as the WT mice (Figure S6A–D), suggesting that MHV activates the NLRP6 inflammasome independent of DHX15. We further found decreased mRNA levels of Ifnb, Ifit2 and Isg15 in Nlrp6K350−354A mice upon MHV infection relative to WT mice (Figure S6E). These results suggest that the impaired phase separation in Nlrp6K350−354A mice affected both inflammasome activation and the induction of interferons and interferon-stimulated genes.
Involvement of NLRP6 Phase Separation and Inflammasome Activation in the Intestine
Within the small intestine, we found that Nlrp6 had high expression in the jejunum (proximal small intestine) whereas Nlrp9b showed high expression in the ileum (distal small intestine) (Figure 5A, B). WT mice, but not Nlrp6−/− mice, showed robust caspase-1 and GSDMD processing in duodenum/jejunum explants at a steady state, likely due to stimulation by the intestinal microbiome (Figure 5C, D). The concentrations of IL-18 in the supernatants of duodenum/jejunum explants of Nlrp6−/− mice were lower than those in WT littermate control mice, and this difference was more substantial for the duodenum explants than for the jejunum explants (Figure 5E). For the LLPS-defective Nlrp6K350−354A knock-in mice, we consistently detected less GSDMD cleavage in their duodenum/jejunum explants (Figure 5F), and lower IL-18 concentration in the culture supernatants of the explants, in comparison with the WT mice (Figure 5G). These data suggest the importance of NLRP6-mediated phase separation and inflammasome activation in maintaining intestinal homeostasis in mice.
Figure 5. The Physiological Role of NLRP6 Phase Separation in IECs in Vivo.
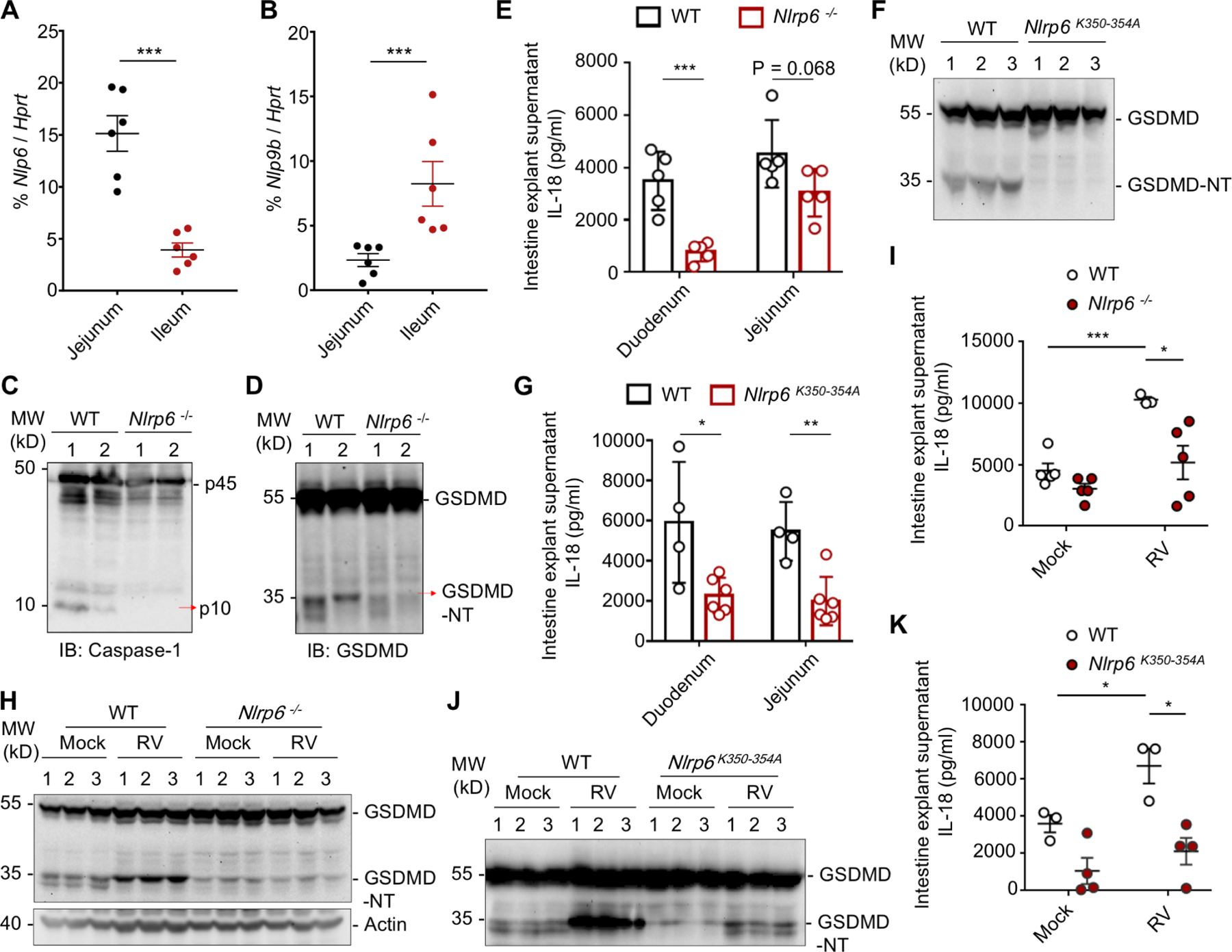
(A, B) qPCR assays for Nlrp6 (A) and Nlrp9b (B) mRNA levels in different locations of the intestine relative to the Hprt control. Mean ± SEM for 6 mice is displayed for each group.
(C, D) Immunoblotting of IEC lysates of multiple WT and Nlrp6−/− mice for caspase-1 (C) and GSDMD (D).
(E) IL-18 levels of intestine explant supernatant of WT and Nlrp6−/− mice determined by ELISA. Mean ± SEM for 5 mice is displayed for each group.
(F) Immunoblotting of IEC lysates of 3 WT and K350–354A mutant mice each for GSDMD processing.
(G) IL-18 levels of intestine explant supernatant of WT and K350–354A mutant mice determined by ELISA. Mean ± SEM for 4 WT and 6 mutant mice is displayed for each group.
(H) Immunoblot of 3 small intestine homogenates each from mock or RV-infected WT and Nlrp6−/− mice 2 days post RV infection to detect cleaved GSDMD.
(I) Intestine-explant supernatant IL-18 levels in WT and Nlrp6−/− mice 2 days post-infection.
(J) Immunoblot of 3 small intestine homogenates each from mock or RV-infected WT and K350–354A mutant mice 2 days post RV infection to detect cleaved GSDMD.
(K) Intestine-explant supernatant IL-18 levels in WT and K350–354A mutant mice 2 days post-infection.
To test the role of LLPS in enterovirus infection, we infected Nlrp6−/− and Nlrp6K350−354A mice with the rotavirus (RV) EC strain, which is a dsRNA virus that belongs to the reoviridae family (Shi et al., 2019). Two days after infection, we detected less GSDMD cleavage in jejunum explants (Figure 5H) and less IL-18 in the supernatants of jejunum explants of Nlrp6−/− mice (Figure 5I), in comparison with WT mice. We also consistently observed lower GSDMD cleavage and IL-18 concentration in jejunum explants of Nlrp6K350−354A mice compared with littermate WT mice upon RV challenge (Figure 5J, K). Considering the high expression of NLRP9 (Figure 5B) and the role of the NLRP9 inflammasome in anti-RV immune response in the ileum (Zhu et al., 2017), we propose that NLRP6 and its phase separation might cooperate with NLRP9 to defend against RV in a regional specific manner.
ASC Solidifies RNA-induced NLRP6 Phase Separation to Promote Inflammasome Activation
NLRP6 can recruit the downstream adaptor protein ASC to form a speck that acts as a supramolecular organization center (SMOC) (Kagan et al., 2014). To reconstitute formation of the NLRP6 inflammasome in vitro, we prepared recombinant ASC and caspase-1 for co-incubation with NLRP6 and dsRNA. Carboxytetramethylrhodamine (TAMRA)-labeled ASCPYD (ASCPYD-TAMRA) colocalized with NLRP6 and 60-bp FAM-dsRNA droplets (Figure 6A, B). Despite the round morphology, these ASC-containing droplets were not dynamic and the fluorescence of FAM-dsRNA or ASCPYD-TAMRA did not recover after photobleaching (Figure 6C), unlike the dsRNA−NLRP6 droplets (Figure 2D). As controls, dsRNA did not form LLPS with ASCPYD (Figure S7A) and NLRP6∆PYD−dsRNA droplets did not recruit ASCPYD (Figure 6D). The W53E mutant of NLRP6, which is defective in formation of PYD filaments (Shen et al., 2019), also formed droplets with dsRNA (Figure 6E), validating that PYD was not required for LLPS. However, the W53E mutant was defective in formation of NLRP6 full-length filaments (Figure 6F), indicating that PYD–PYD interactions are required for NLRP6 to form filaments, but are not required for NLRP6 to form LLPS. For reconstitution of the full inflammasome, TAMRA-labeled full-length ASC and mTurquoise2-labeled full-length caspase-1 (C285A catalytic mutant) co-localized with NLRP6−dsRNA droplets (Figure 6G). These results implied that these liquid droplets could serve as a SMOC for recruiting downstream molecules in inflammasome signaling.
Figure 6. ASC Solidifies NLRP6-dsRNA Phase Separation.
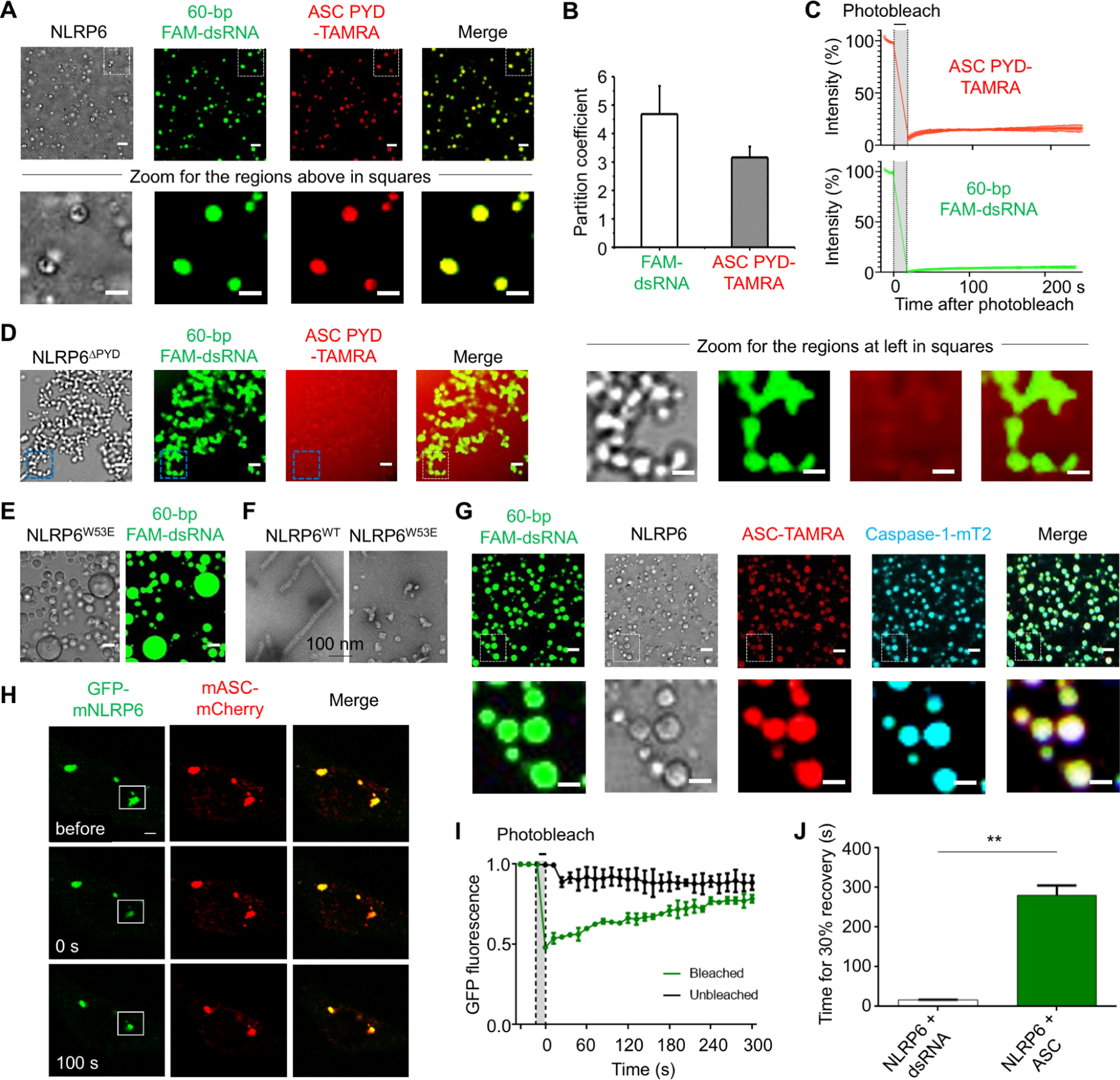
MBP-NLRP6, 60-bp FAM-dsRNA, MBP-ASC PYD-TAMRA and caspase-1-mTurquoise2 (mT2) were at 15 μM, 15 μM, 7.5 μM and 7.5 μM, respectively, and TEV was added at time 0 to cleave the MBP tags. Incubation time and temperature were 45 min and 37 °C, respectively. Unless otherwise noted, scale bars are 5 μm except for zoomed images which have scale bars of 3 μm.
(A) Colocalization of ASC PYD in NLRP6-dsRNA droplets (upper panel) and the zoomed images (lower panel).
(B) Partition coefficients calculated from (A). Error bars indicate three independent experiments.
(C) Quantified FRAP experiments of the droplets formed by FAM-dsRNA, NLRP6 and ASC PYD-TAMRA, showing lack of recovery.
(D) Lack of recruitment of ASC PYD into NLRP6∆PYD-dsRNA droplets (left panel) and the zoomed images (right panel).
(E) LLPS formed by dsRNA and the PYD filament-defective MBP-NLRP6 W53E mutant.
(F) Negative-stain electron microscopy of MBP-removed NLRP6 and NLRP6 W53E proteins. Scale bar: 100 nm.
(G) Colocalization of ASC and caspase-1 in NLRP6-dsRNA droplets (upper panel) and the zoomed images (lower panel).
(H) Representative images of an NLRP6-ASC punctum before and after photobleaching in MEFs, among ≥ 10 cells. The white box indicates the bleaching site.
(I) A plot of quantified FRAP data for NLRP6-ASC puncta as a function of time. Bleaching event occurred at t = 0 s. Shown are the mean ± SD for n = 3 NLRP6-ASC puncta.
(J) Time taken to reach recovery of 30% fluorescence intensity for data in (H) for comparison with NLRP6-dsRNA puncta. Shown are the mean ± SD for n = 3 NLRP6-ASC puncta and NLRP6-dsRNA puncta.
See also Figure S7
To verify the role of ASC in the phase-separation process in cells, we constructed a stable cell line co-expressing GFP-mNLRP6 and mCherry-tagged mouse ASC (mASC-mCherry) and examined the intracellular localization of NLRP6 and ASC. Exogenously co-expressed NLRP6 and ASC formed specks even without dsRNA; upon photobleaching, the GFP fluorescence recovered slowly (Figure 6H, I, Movie S5A–B). In comparison with NLRP6-dsRNA specks (Figure 2J), ASC overexpression significantly prolonged the recovery process after photobleaching (by ~20-fold) (Figure 6J). Together with the FRAP data on the in vitro assembled NLRP6 inflammasome condensate (Figure 6C), this observation is consistent with NLRP6−ASC puncta possessing ordered filaments to solidify the NLRP6 inflammasome (Li et al., 2018; Lu et al., 2016; Lu et al., 2014; Shen et al., 2019), and suggests that the initial NLRP6−dsRNA clusters at a liquid-like state may serve as “microreactors” to recruit downstream signaling molecules.
Formation of LLPS as an Integrating Signal for Multiple Biological Functions
It is intriguing that NLRP6 is implicated in multiple systems of host defense. We wondered whether its ability to form LLPS may contribute to its functional versatility. We first checked bacterial LTA (Hara et al., 2018), the only other ligand for which we could detect robust binding in vitro (Figure 1F). Mixing NLRP6 with an increasing molar ratio of LTA induced droplet formation, similar to that observed in NLRP6–dsRNA LLPS (Figure 7A, B). Determination of the phase diagram revealed the dependence of LLPS on both LTA and NLRP6 concentrations (Figure 7B, S7B, C). This conclusion was confirmed by the OD 395 nm turbidity assay (Figure 7C). By contrast, an anion polymer, poly(sodium 4-styrenesulfonate), did not show concentration dependent enhancement of NLRP6 LLPS (Figure S7D). We then observed that LTA-TAMRA induced NLRP6 LLPS after transfection of LTA to MEFs stably expressing GFP-mNLRP6 (Figure 7D). When tracked by live-cell imaging, LTA and dsRNA foci appeared less than 1 h after LTA transfection, with LTA foci first and NLRP6 merging into the foci as a function of time (Figure 7E, F, Movie S6A–B).
Figure 7. NLRP6-dsRNA LLPS Integrates Different Signaling Pathways.
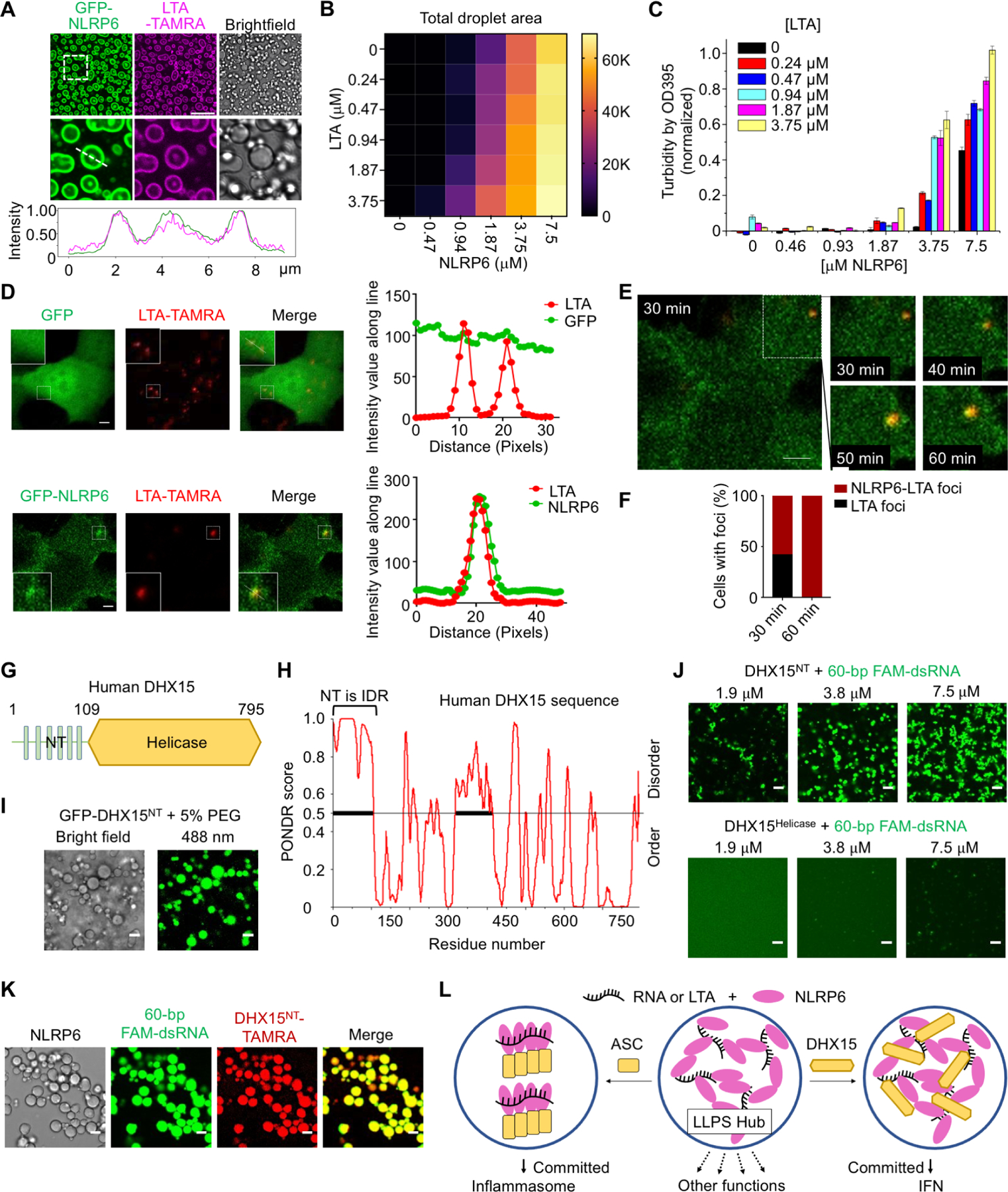
(A) Images of phase-separated droplets from purified GFP-NLRP6 (15 μM) and TAMRA-labeled LTA (3.75 μM) (upper panel), the zoomed images (middle panel) and intensity traces along the line in the middle panel (lower panel). Scale bar: 20 μm.
(B) Phase diagram of MBP-NLRP6 with LTA-TAMRA. Total area (μm2) occupied by droplets in each image (128,010 μm2) was quantified into a 6×6 matrix. Brightfield images related to Fig. S7B were used for the quantification.
(C) Phase diagram of MBP-NLRP6 with LTA-TAMRA from the OD 395 turbidity assay.
(D) Representative live cell images of NLRP6-LTA speck formation after transfection of LTA-TAMRA into MEFs stably expressing GFP or GFP-NLRP6, among ≥ 10 cells. Zoomed images indicate NLRP6-RNA puncta (boxes). Scale bar: 5μm. Right panel: Plot of the colocalization of GFP with LTA or GFP-NLRP6 with LTA.
(E) A time-lapse micrograph of NLRP6 (green) and LTA-TAMRA (red) speck formation at 30 min after LTA-TAMRA transfection (Left panel) and the zoomed images of the speck at 30–60 min after transfection (right panel). Scale bar: 5 μm.
(F) Quantification of NLRP6-LTA foci or LTA foci at time point 30 min and 60 min.
(G) Domain architecture of human DHX15.
(H) Graph plotting intrinsic disorder (by PONDR VSL2) in human DHX15.
(I) LLPS by GFP-DHX15-NT (residues 1–109; 30 μM) with the crowding reagent 5% PEG3350. Scale bar: 5 μm.
(J) LLPS of DHX15-NT, but not DHX15-Helicase (residues 107–795), when mixed with 60-bp FAM-dsRNA at the indicated concentrations (1:1 molar ratio). Scale bar: 5 μm.
(K) Colocalization of DHX15-NT in NLRP6-dsRNA droplets. TAMRA-labeled MBP-DHX15-NT was at 7.5 μM and TEV was added at time 0 to cleave MBP tags. Scale bar: 5 μm.
(L) A brief model to hypothesize how NLRP6 LLPS may serve as a hub to integrate and regulate multiple signaling pathways.
See also Figure S7
We then looked at DHX15 (Wang et al., 2015), which has an IDR at the N-terminal region (NT) as assessed by the PONDR program (Figure 7G, H). The DHX15 NT, but not the helicase domain, formed droplets in the presence of the crowding agent PEG3350 (Figure 7I) or in the presence of dsRNA (Figure 7J). Furthermore, DHX15 and NLRP6 co-localized in the same dsRNA-induced droplets (Figure 7K). These data suggested an NLRP6 signaling paradigm in which multiple stimuli can induce NLRP6-containing condensates to allow sensing of diverse upstream signals, and multiple signaling proteins may co-assemble with NLRP6 condensates to diverge downstream signals. Mechanisms that regulate the differential signal transduction in cells remain to be elucidated.
DISCUSSION
Phase separation has revolutionized cell biology in recent years (Alberti et al., 2019; Babu, 2016; Boeynaems et al., 2018; Lafontaine et al., 2020; Lyon et al., 2020; Tauber et al., 2020). However, despite the vast number of examples of “structured” supramolecular assemblies in immune signaling (Shi et al., 2020), only a few examples of phase separation and its role in the immune system have been shown. These examples include the validated role of clustering of signaling molecules by phase separation in signal transduction of T-cell receptors (Su et al., 2016), and the dsDNA-induced LLPS of cyclic GMP-AMP synthase (cGAS) for enhancing production of cyclic guanosine adenosine monophosphate (cGAMP), and subsequent activation of stimulator of interferon genes (STING) and type-I interferon (Du and Chen, 2018). Here, we extended understanding of the role of LLPS in the immune system by discovering three important aspects of NLRP6 function centered on its ability to form LLPS.
First, we found that dsRNA can promote NLRP6 to form LLPS (Figure 7L), which is distinct from the classical paradigm for activation of an NLR inflammasome involving structured supramolecular assemblies. As first demonstrated for the NAIP–NLRC4 inflammasome, disk-like oligomerization by the NACHT and LRR domains of an NLR promotes the helical oligomerization of PYD or CARD of the NLR for recruitment of downstream molecules (Diebolder et al., 2015; Hu et al., 2015; Tenthorey et al., 2017; Yang et al., 2018; Zhang et al., 2015). The apparent intrinsic structural disorder of NLRP6 may be the reason for its adoption of LLPS rather than an ordered assembly. However, despite not being an ordered supramolecular assembly, the liquid condensate increases the local concentration of the PYD of NLRP6 to allow it to form a platform to recruit ASC through filamentous PYD–PYD interactions, and ASC in turn recruits caspase-1 through filamentous CARD–CARD interactions. Perhaps because of the formation of ordered PYD–PYD interactions, ASC recruitment reduced the dynamic property of the dsRNA–NLRP6 condensate, likely leading to a commitment to assembly and activation of the inflammasome. We demonstrated the role of phase separation not only in cells, but also physiologically in mice upon viral infections or in homeostatic activation of the NLRP6 inflammasome in the intestine. Of note, NLRP6 expression is increased in the lungs of pneumonia patients (Ghimire et al., 2018), which may help to explain more severe COVID-19 in patients with underlying lung disease.
Intriguingly, the mutual dependence of dsRNA and NLRP6 in the phase separation has not been fully consistent. In vitro, dsRNA did not form LLPS without NLRP6 at all concentrations tested, while NLRP6 could form LLPS at high, but not at low concentrations without dsRNA. However in cells, RNA puncta can occur upon RNA transfection in the absence of NLRP6 with unclear reasons, and NLRP6 often appeared to merge into these RNA puncta. We speculate that this is due to the fusion of RNA with the liposome used for RNA transfection. Nonetheless, in the physiological setting of MHV-infection of the liver, speck formation only occurred when both virus (RNA) and WT NLRP6 were present, and Nlrp6−/− or Nlrp6K350−354A mutant mice showed reduced RNA or ASC specks. Thus, both the biochemical data and the in vivo data support our conclusion that NLRP6 and dsRNA undergo LLPS upon interaction.
Second, we revealed that NLRP6 might be a “pattern” binder, rather than a specific binder, in its recognition of ligands, which extended understanding of NLRP6 as an innate immune receptor. Using quantitative in vitro binding assays, we showed that dsRNA is a direct and strong ligand for NLRP6, and that among the NLRP6 ligands reported previously, only LTA from Gram-positive bacteria showed robust affinity to NLRP6. It is intriguing what molecular signature NLRP6 recognizes in dsRNA and LTA. Conversely, metabolites from commensal microbiota showed very weak binding to NLRP6. This finding is consistent with the observation that only millimolar concentrations of such metabolites can stimulate the NLRP6 inflammasome (Levy et al., 2015). One way to rationalize the versatility of NLRP6 is to use an analogy with the NLRP3 inflammasome, which is activated by diverse stimuli that induce cytosolic K+ efflux (Munoz-Planillo et al., 2013). For NLRP6, any stimuli that promote NLRP6-dependent LLPS may “set the stage” for NLRP6-mediated downstream effects.
Third, we propose that LLPS may help to recruit other molecules to diversify NLRP6 signaling (Figure 7L). In this context, we showed that DHX15 (a helicase in an enteric virus-induced and NLRP6-dependent interferon pathway) (Wang et al., 2015) co-formed LLPS with dsRNA and NLRP6. We propose that the droplets formed by NLRP6, ligands, and other signaling molecules might be an intermediate state in the signal transduction pathway, which serves as a checkpoint to direct to different outcomes (be it inflammasome activation, interferon response and/or NF-κB inhibition) and may depend on the spatial and temporal distribution of downstream molecules. In support of this hypothesis, we observed that RNA-induced NLRP6 LLPS preceded PYD-induced inflammasome assembly, and that once NLRP6 engaged ASC, the droplets underwent “maturation” and solidification, perhaps to commit to downstream activation of the inflammasome (Figure 7L). For inflammasome-mediated caspase-1 activation, the “solid” filamentous interactions between the caspase recruitment domains (CARDs) of ASC and caspase-1, which also help to solidify the phase separation, need to happen to bring the caspase domains into proximity for their dimerization and activation. Thus, while the identities of other signaling molecules that can be recruited to NLRP6 LLPS require further investigation, our studies provide a framework for understanding how NLRP6 signal transduction may be integrated, diversified and regulated by the LLPS mechanism.
Limitations of the Study
The current study provides important insights on the role of LLPS as a central hub that regulates NLRP6-directed host defense pathways. However, it is still unclear if dsRNA or other ligands induce or promote NLRP6 LLPS, and if NLRP6 is also able to form an ordered oligomers like NLRC4 from the liquid droplets. How does NLRP6 interact with its ligands? What is the molecular architecture of NLRP6-containing condensates? Do they change with the recruitment of different signaling components and what are the functional consequences? And how is RNA-NLRP6 LLPS terminated? We expect that examination of these and other inquiries will not only provide more details and depth to understanding NLRP6 signaling, but also open new therapeutic opportunities in inflammasome regulation in general.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
All information and requests for further resources and reagents should be directed to and will be fulfilled by the Lead Contact, Hao Wu, wu@crystal.harvard.edu
Materials Availability
All requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact. All reagents will be made available on request after completion of a Materials Transfer Agreement.
Data and code availability
This study did not generate/analyze unique datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
The HBL1 cell line was a kind gift from Professor Ari M. Melnick (Cornell University, New York, NY, USA). Cells were cultured in RPMI 1640 medium (61870127; Thermo Fisher Scientific) with 10% fetal bovine serum (FBS; TMS-013-B; Millipore), 1 mmol/L sodium pyruvate (11360070; Thermo Fisher Scientific), and 1% penicillin/streptomycin (15140163; Thermo Fisher Scientific). For live-cell confocal microscopy, cells were grown in microwell dishes (catalog number, P35G-1.5–14-C; MatTek). Immortalized BMDM, MEF, L2 and HEK293T cells and mouse primary hepatocytes were grown in DMEM (HyClone) supplemented with 10% FBS (Clark), penicillin (100 U/mL; Gibco) and streptomycin (100 mg/mL; Gibco). All cells were maintained at 37 °C in an atmosphere of 5% CO2.
Mice
Wild-type, Nlrp6−/−, Asc−/−, Gsdmd−/−, Dhx15f/f alb-cre, Dhx15f/f, Nlrp63xflag and Nlrp6K350−354A male mice (aged 6–8 weeks) were housed in a state-of-the-art animal facility at the University of Science and Technology of China. The mice were maintained in a strict 12 hour light cycle under SPF condition.
METHOD DETAILS
Cloning, Expression and Purification of Proteins
The human full-length NLRP6 (1–892) and NLRP6∆PYD (111–892) were cloned into the pFastBac-HTB vector to generate constructs with a TEV protease cleavable His6-MBP tag at the N-terminus. The NLRP6-mCherry construct was generated through Gibson assembly into the same pFastBac-HTB-His6-MBP vector with a linker of “GSGSGSGS” between NLRP6 and the C-terminal mCherry. The GFP-NLRP6 construct was generated through Gibson assembly with the “GSGSGSGS” linker between the N-terminal GFP and NLRP6. The human full-length caspase-1C285A (1–404)-mTurquoise2 construct was cloned into pFastBac-HTB with the “GSGSGSGS” linker between caspase-1 and mTurquoise2. Human NLRP6 W53E and K352–356A (352KDKKK356 to 352ADAAA356) mutants were created using the QuikChange™ Site-Directed Mutagenesis Kit (Stratagene). All the insect-cell expression constructs were transformed into E. coli DH10Bac cells for the production of recombinant bacmids. Upon transfection to obtain recombinant baculoviruses, proteins were expressed in Sf9 cells. After 48 hours of infection at 27 °C, cells were harvested and lysed by sonication. For NLRP6-related constructs, cells were lysed in Tris-HCl (20 mM) at pH 7.4, NaCl (500 mM) and imidazole (20 mM). For the caspase-1-mTurquoise2 construct, cells were lysed in Tris-HCl (20 mM) at pH 7.4, NaCl (150 mM) and imidazole (20 mM). The supernatants of the lysates were incubated with Ni-NTA resin and washed with lysis buffer containing imidazole (20 mM). Target proteins were eluted with lysis buffer containing imidazole (300 mM). Ni-NTA eluates were injected into a Superdex 200 column to collect the purified fractions. The MBP-tagged NLRP3∆PYD (134–1034) and the Sumo-tagged NLRC4∆CARD (90–1024) constructs were expressed similarly as described previously (Sharif et al., 2019; Zhang et al., 2015).
For gel-shift assays, NLRP6PYD (1–106), NLRP6PYD+NBD (1–348) and NLRP6PYD+NACHT (1–564) were cloned into the pDB-His-MBP vector and expressed in E. coli at 18 °C with IPTG (Isopropyl β-D-1-thiogalactopyranoside, 0.1 mM) induction overnight. The Ni-NTA purification and gel-filtration buffers were the same as those used for NLRP6 full-length constructs.
For the phase-separation assay, MBP-tagged ASCPYD (1–91) and MBP-tagged full-length ASC (1–197) constructs with an engineered C-terminal “LPETGG” sequence for sortase labeling were expressed in E. coli BL21(DE3) and purified by Ni-affinity chromatography and gel-filtration chromatography. The labeling reaction contained calcium-independent sortase (30 μM), MBP-ASCPYD (50 µM) and fluorophore (GGG-TAMRA; 500 μM) (Guimaraes et al., 2013). The mixture was incubated on ice overnight and passed through a Superdex 200 column in Tris (20 mM) at pH 7.4, NaCl (150 mM) and TCEP (tris(2-carboxyethyl)phosphine, 1 mM). hNLRP6 IDR (86–190) and hNLRP6 IDR (286–356) were cloned into the pDB-His-MBP vector and expressed in E. coli at 18 °C with induction using IPTG (0.1 mM) overnight. The MBP tag was cleaved by TEV at time 0 of phase-separation assays. The GFP-Dhx15NT (1–109) construct was cloned through Gibson assembly with the “GSGSGSGS” linker between the N-terminal GFP and Dhx15NT into the pFastBac-HTB vector. MBP-tagged Dhx15NT (1–109) was cloned into the pFastBac-HTB-His6-MBP vector. Dhx15Helicase (107–795) was cloned into the pFastBac-HTB vector with an N-terminal His6 tag.
Spinning-Disk Confocal Microscopy
HBL-1 cells were transfected at 60% confluency with 5 μg of pCDNA3.1-human NLRP6 for 24 hours followed by transfection with 10 μg of fluorescein-labeled poly I:C (tlrl-picf; Invivogen) for an additional 6 hours using FuGENE 6 (E2691; Promega) according to manufacturer guidelines. Transfected HBL-1 cells were placed in a 35-mm petri dish (10-mm microwell, number 1.5 cover glass; catalog number P35G-1.5–14-C; MatTek) and mounted in a 20/20 microscope stage chamber (Bionomic Technologies) warmed to 37 °C. Dulbecco’s modified Eagle’s medium (DMEM) without phenol red (21063–045; Thermo Fisher Scientific) was used during image acquisition, with a layer of mineral oil on top of the medium in order to prevent evaporation. All images were collected with a spinning disk confocal (Yokogawa) on a Ti inverted microscope (Nikon) equipped with Plan Apo phase-3 oil-immersion 40× (1.3 numerical aperture) lenses. The Perfect Focus System (Nikon) was in place for the continuous maintenance of focus. Transfected HBL-1 cells were labeled with poly I:C fluorescein and excited with the 491 nm line (selected with a 488/10 filter, HQ480/40x; Chroma) from a 100 mW cobalt diode laser and collected with a quadruple bandpass dichroic mirror (Di01-T405/488/568/647; Semrock) and a 525/50 emission filter (ET525/50m; Chroma). Transfected HBL-1 cells with human NLRP6-mCherry were excited with a 561 nm line (selected with a 500/20 filter, ET500/20x; Chroma) from 200 mW cobalt diode laser and collected with a quadruple bandpass dichroic mirror (Di01T405/488/568/647; Semrock) and a 620/60 emission filter (ET620/60m; Chroma).
For imaging phase-separated droplets, 96 glass-bottom well plates (glass diameter, 5 mm; number 1.5 Coverslip, uncoated; P96G-1.5–5-F; MatTek) were coated with 5% bovine serum albumin (BSA) in 1× DPBS (Dulbecco’s phosphate-buffered saline; 14190250; Thermo Fisher Scientific) overnight. The following day, we discarded the incubated BSA solution, added the protein and RNA components, and incubated for 45 minutes at 37 °C before imaging. Phase-separated droplets were imaged using the same confocal microscope and settings mentioned above, with 60-bp FAM-labeled dsRNA excited with the 491 nm line, and TAMRA-labeled full-length ASC excited with the 561 nm line (emission filter, ET605/70m; Chroma). Full-length caspase-1-mTurquoise2 droplets were excited with the 442 nm line (selected with 436/20x; Chroma) with a triple-band pass dichroic mirror (Di01-T442/514/647; Semrock) and a 480/40 emission filter (ET480/40m; Chroma). Images were acquired with an ORCA ER-cooled charge-coupled device camera (Hamamatsu) controlled with MetaMorph 7 (Molecular Devices). Images were collected using an exposure time of 700 ms and 2×2 binning, with illumination light shuttered between acquisitions. At each time point, z-series optical sections were collected with a step size of 0.5 μm for the indicated time intervals, using a ProScan™ focus motor (Prior). Gamma, brightness, and contrast were adjusted on displayed images (identically for comparative image sets) using MetaMorph 7. Imaging power was at 15% and images were analyzed using ImageJ (US National Institutes of Health). Time-lapse images of phase separation were captured using the Time-Lapse Acquisition tool of the Ti inverted confocal microscope (Nikon) every 20 seconds over 1 hour.
EMSA
For agarose gel shifts, MBP-NLRP6 (5 μM) was mixed with 20-bp dsRNA at a 1:1 molar ratio, and with HMW poly I:C at a 200:1 molar ratio. The mixtures were incubated on ice for 1 hour. Then, MBP-NLRP6 alone, 20-bp dsRNA and HMW poly I:C alone, and protein−RNA mixtures were loaded onto 0.8% agarose gel (homemade, Tris-acetate-EDTA as the gel buffer) and ran for 45 minutes on ice. NLRP6 filament form/monomer form gel shift was done under similar conditions. The NLRP6, NLRP3, or NLRC4 gel-shift assay was performed similarly to the one performed for the MBP-NLRP6 except for the final protein concentration of 3 μM. For titrating the binding affinity of 20-bp dsRNA with NLRP6-monomer (no tag) and NLRP6 filaments, the concentration of 20-bp dsRNA was 5 ng/μL. The concentrations of NLRP6-monomer (no tag) were 0, 3 nM, 6 nM, 11 nM, 23 nM, 46 nM, 92 nM, 183 nM, 366 nM, 732 nM, 1465 nM and 2930 nM. The concentrations of the NLRP6-filaments were 0, 73 nM, 146 nM, 291 nM, 582 nM, 1165 nM, 2330 nM and 4660 nM. The band intensity was measured through ImageJ. The curve and fitting were prepared in Origin 8.0. For other EMSAs involving RNA length-dependent binding and NLRP6 construct-dependent binding, dsRNAs (2.5 ng/µL) were incubated with MBP-NLRP6 proteins for 30 minutes at room temperature. The MBP-NLRP6 concentrations were in two-fold dilutions. The experiments were performed on NativePAGE™ 3–12% Bis-Tris Protein Gel (Invitrogen). Gels were stained with SybrGold™ stain (Life Technologies). The band intensity was measured through Gel-Doc™ (Bio-Rad Laboratories). The intensity data was subsequently used to plot the binding curve and calculate the KD values employing Origin 8.0 (OriginLab).
| Nucleic Acids in the current study | Sequences (5’−3’) |
|---|---|
| 20bp dsRNA | GACCUUCUGAUCAGAAGGUC (palindromic sequence, self-annealed in Hao Wu Lab) |
| 20bp dsDNA | GACCTTCTGATCAGAAGGTC (palindromic sequence, self-annealed in Hao Wu Lab) |
| 20nt ssRNA | AACUCAUCUCGACCUUCUGA |
| 20nt ssDNA | AACTCATCTCGACCTTCTGA |
| 16bp dsRNA | AAAUAAGCGCUUAUUU (palindromic sequence, self-annealed in Hao Wu Lab) |
| Cy5–45bp-dsRNA(+) | Cy5-AAACAAAAACAAAACAAACAACACAACAAACAAAACAAAAACAAA |
| Cy5–45bp-dsRNA(−) | UUUGUUUUUGUUUUGUUUGUUGUGUUGUUUGUUUUGUUUUUG UUU |
| 5’ 6-FAM™-dsRNA-60bp (+) | 5’ 6-FAM™-AUCACACACCCAGCGCUGGACGGCCACCCAC AACCUCCCAAGGAACUCAUCUCGACCUUC |
| 5’ 6-FAM™-dsRNA-60bp (−) | GAAGGUCGAGAUGAGUUCCUUGGGAGGUUGUGGGUGGCCGUC CAGCGCUGGGUGUGUGAU |
Electron Microscopy
NLRP6WT (Filament sample, 0.2 mg/mL), NLRP6W53E (0.04 mg/mL) and NLRP6K350−354A (0.04 mg/mL) were placed onto a glow-discharged copper grid (Electron Microscopy Sciences) coated with a layer of thin carbon for 1 minute, stained with 2% uranyl formate for 40 seconds, and air-dried. The grids were imaged on a G2 Spirit BioTWIN electron microscope (Tecnai) and recorded with an AMT 2k charge-coupled device camera (Harvard Medical School Electron Microscopy Facility).
In Vitro Phase Separation Assay
One-hundred microliters of a BSA solution (50 mg/mL) were added to a glass-bottom 96-well plate (MatTek) and left overnight. This incubation solution was discarded before starting the assay. For the phase separation of different constructs of NLRP6 (MBP-NLRP6, MBP-NLRP6-mCherry, MBP-NLRP6 mutants or domain and IDR truncations) and RNA, MBP-NLRP6 (15 μM) was mixed with 60-bp dsRNA (15 μM) either with or without TEV (0.5 mg/mL) added at time 0 to reach a final buffer concentration of Tris-HCl (20 mM), pH 7.4, NaCl (110 mM), TCEP (0.5 mM), and BSA (1 mg/mL). For the phase-separation assay of the in vitro-reconstituted inflammasome, MBP-NLRP6 (15 μM) was mixed with 60-bp dsRNA (15 μM), MBP-ASC-TAMRA (7.5 μM) and caspase-1-mTurquoise2 (7.5 μM), with TEV (0.5 mg/mL) added at time point 0. The final reaction buffer was Tris-HCl (20 mM) at pH 7.4, NaCl (115 mM), BSA (0.32 mg/mL), and TCEP (0.5 mM). The phase-separation assays for the dependency of salt, temperature, and protein/RNA concentrations were undertaken under similar conditions.
Phase Diagram of NLRP6 with RNA or LTA
Before the phase diagram experiments, we added 50 mg/ml of BSA solution into the glass-bottom 96-well plate (MatTek) for overnight pre-incubation and discarded the BSA before adding the sample. For the phase diagram of MBP-NLRP6 with 60-bp FAM-dsRNA, we created a 6×6 reaction matrix with 6 concentration points of MBP-NLRP6 in X axis (0, 0.47 μM, 0.94 μM, 1.88 μM, 3.75 μM, 7.5 μM) and 6 concentration points of 60-bp FAM-dsRNA in Y axis (0, 0.47 μM, 0.94 μM, 1.88 μM, 3.75 μM, 7.5 μM). The final buffer components were Tris-HCl (20 mM) at pH 7.4, NaCl (156 mM), TCEP (0.6 mM), and BSA (1 mg/mL). The conditions for the phase diagram of MBP-NLRP6 and LTA-TAMRA were similar except that LTA-TAMRA had 6 concentration points of 0, 0.24 μM, 0.47 μM, 0.94 μM, 1.88 μM, 3.75 μM. After adding all the components into the wells, the plate was incubated for 45 minutes at 37 °C. Images were obtained using an ImageXpress Micro Confocal (Molecular Devices) using a 40X objective (S Plan Fluor, NA=0.6)
Quantification of NLRP6−RNA and NLRP6−LTA Phase Diagrams
We used the parameter “area occupied by droplets” to quantify the tendency of phase separation. Brightfield images were processed using MetaXpress software. Briefly, a local minimum intensity filter was used to get rid of bright edges around particles followed by inverting the image and segmentation of particles based on intensity and size. This was used to calculate the total area occupied by particles per field and averaged over 9 fields per treatment.
Turbidity Assay
Turbidity assay used the same plate and the same pre-treatment as for the in vitro phase separation assay. We also prepared the 6×6 reaction matrix with 6 concentration points of MBP-NLRP6 (0, 0.47μM, 0.94 μM, 1.88 μM, 3.75 μM, 7.5 μM). Orthogonally, we prepared 6 concentration points of 60-bp FAM-dsRNA (0, 0.12μM, 0.24 μM, 0.47μM, 0.94 μM, 1.88 μM), 6 concentration points of LTA-TAMRA (0, 0.24 μM, 0.47 μM, 0.94 μM, 1.88 μM, 3.75 μM), 6 concentration data points of Poly(sodium 4-styrenesulfonate) (0, 0.47μM, 0.94 μM, 1.88 μM, 3.75 μM, 7.5 μM). The final buffer components in each well included Tris-HCl (20 mM) at pH 7.4, NaCl (156 mM), TCEP (0.6 mM), and BSA (1 mg/mL). The reaction system was incubated at 37 °C for 30 minutes, and absorbance at 395 nm (OD395) was monitored at 37 °C with a SpectraMax M5e (Molecular Devices) plate reader.
Labeling and Phase Separation Assay of Different IDRs
His6-MBP-tagged hNLRP6 IDR1 (86–190) and hNLRP6 IDR2 (286–356) were purified with Ni-NTA following the manufacturer’s instructions, and exchanged to a buffer containing 20 mM HEPES at pH 7.4, 50 mM NaCl, 100 mM Sodium Bicarbonate, and 1 mM TCEP using gel filtration. For labeling, we prepared a 40 mM stock of BODIPY™ TMR-X NHS Ester (Succinimidyl Ester) in DMSO. Then we gradually added the dye stock into prepared hNLRP6 IDRs solution and mixed them thoroughly to reach 1:4 molar ratio of the IDR proteins and the dye. After a reaction of 1.5 hours under room temperature and in the dark, the sample was spun at 15,000 rpm for 10 minutes to remove potential precipitants and collect the supernatant. The supernatant was quenched with 2.5 mM Glycine (Neutral pH) for 30 minutes. The quenched solution was injected to a Superdex 200 column for the second round gel filtration (buffer: 20 mM HEPES at pH 7.4, 150 mM NaCl, 1 mM TCEP). The monomeric peak of MBP-IDRs-TAMRA were collected for in vitro phase separation assay. The reaction system of phase separation assay consists of 15 μM MBP-IDR1 or IDR2, 5% PEG3350, 20 mM HEPES at pH 7.4, 105 mM NaCl, 0.6 mM TCEP and 1 mg/mL BSA. Images were acquired after 30 minutes of incubation under 37 °C.
Labeling of LTA
Commercial purified LTA Staphylococcus aureus from Invivogen (Cata. Code: tlrl-pslta) was prepared as 5 mg/mL stock into toxin-free water following the manufacturer’s instruction, and labeled following a procedure described previously (Levels et al., 2003). Briefly, we transferred 500 µL 5 mg/mL LTA into a 15 mL Falcon tube, diluted to 1.25 mL, and sonicated it with 70% power for 10 minutes (Branson sonicator) on ice. Then we generated the labeling reaction system by mixing 1.25 mL 200 mM Sodium Bicarbonate with 1.25 mL sonicated LTA solution. The LTA concentration was 1 mg/mL (or 125 µM). After preparing a 40 mM stock of BODIPY™ TMR-X NHS Ester (Succinimidyl Ester) in DMSO (recommended by manufacturer), we added the dye into the prepared 1 mg/mL LTA solution gradually to reach the molar ratio of LTA and dye of 1:4. After a reaction of 1.5 hours under room temperature and in the dark, the sample was centrifuged at 15,000 rpm for 10 minutes to remove potential precipitants and collect the supernatant. The experiments were quenched with 2.5 mM Glycine (neutral pH) for 30 minutes. At the final step, we performed gel filtration of the quenched solution over a Superdex 75 column and collected the void fractions. The concentration of LTA-TAMRA at the peak fraction was approximately 592 ng/μL.
In Vitro FRAP
FRAP experiments were undertaken using an A1R+ confocal microscope (Nikon) at 37 °C. For FRAP of NLRP6−dsRNA droplets, spots of ~2 μm diameter in ~10 μm droplets were photobleached with 80% laser power for 1 second using a 491 nm laser. Time-lapse images were acquired over 10 minutes after bleaching with 10 s intervals.
Measurement of Binding Affinity by MST
The binding affinity between GFP-NLRP6 and different ligands was measured at 25 °C in a binding buffer containing Tris-HCl (20 mM) at pH 7.5, NaCl (150 mM), and TCEP (1 mM) by MST using a MONOLITH NT.115 system (NanoTemper Technologies). Briefly, 10 μL of GFP-NLRP6 (1 μM) was mixed with 10 μL of twofold serial-diluted ligands to reach a final protein concentration of 500 nM. Data were plotted by the MO.Affinity Analysis (NanoTemper Technologies).
Calculation of Partition Coefficient
The partition coefficient was determined by calculating the mean fluorescence intensity for pixels spanning particles divided by the mean fluorescence intensity for background pixels.
Virus Expansion
Mouse hepatitis virus A59 (MHV-A59) was expanded in murine L2 cells to 3×106 of 50% tissue culture infective dose (TCID50)/mL. Rotavirus EC stock was prepared by infecting 5-day-old B6 mice, and harvesting crude centrifugation-clarified intestinal homogenate as previously described (Zhu et al., 2017).
Construction of Expression Plasmids and Transfection
Mouse Nlrp6 (National Center for Biotechnology Information accession number, NM_133946.2, NP_598707) and mouse Asc (National Center for Biotechnology Information accession number, NM_023258.4, NP_075747.3) were cloned into the pLVX vector. Cloned pLVX plasmids were co-transfected with Δ8.9 and vsvg viral packaging plasmids using PEI transfection reagent (23966–1; Polysciences). The virus was produced in HEK293T cells. For transductions, MEFs were plated 1 day before transduction, and seeded at 2×105/mL (suspension) target cell in a 12-well plate. Viral media were added to cells for 24 hours, at which point cells were expanded in normal media for imaging or propagation.
Viral infection
Mice (6–8 weeks) were infected (i.p.) with 104 or 106 50% TCID50 of MHV-A59. Mice were sacrificed by CO2 anoxia 3 days post-infection. For all rotavirus infection, 6-week-old mice were orally inoculated by gavage with 1 50% diarrhea dose (DD50) of EC virus in 100 μl PBS. Virus infection in Nlrp6−/− mice were conducted using co-housed littermate control mice.
Reagents and Antibodies
Primary antibodies were mouse anti-capase-1 (AG-20B-0042; Adipogen), mouse anti-FLAG (F1804; Sigma–Aldrich), mouse anti-actin (66009–1-Ig; ProteinTech), anti-ASC (67824; Cell Signaling Technology), anti-GFP (BM3883; Boster Biotechnology), and anti-GSDMD (209845; Abcam). Mouse IL-18 enzyme-linked immunosorbent assay (ELISA) kit was purchased from Invivogen (BMS618). An LDH Cytotoxicity kit was obtained from ThermoFisher (C20300).
Isolation of IECs and Primary Mouse Hepatocytes
Small intestines were thrice excised and flushed thoroughly with PBS. They were turned inside out and cut into ~1 cm sections and then transferred into RPMI with 2 mM EDTA, and shaken for 20 minutes at 37 °C. Supernatants were collected through a 100 mm cell strainer in order to obtain single-cell suspensions. Cells were collected as the IEC fraction which contains epithelial cells (~90%). Single-cell suspension was used for further analysis.
For isolation of primary mouse hepatocytes, the liver was perfused sequentially with two solutions. Solution A was the original solution (NaCl (136 mM), KCl (5.3 mM), KH2PO4 (0.4 mM), Na2HPO4 (0.3 mM), tricine (25 mM), EGTA (0.5 mM)). Solution B was DMEM (Hyclone) supplemented with 0.075% collagenase I (Biosharp). After perfusion, the liver was excised and shaken in DMEM with 0.075% type-I collagenase for 5 minutes. Perfused liver tissue was dispersed gently in DMEM containing 2% FBS. Then, the suspension was centrifuged at 50 × g for 3 minutes at room temperature. The collected pellet was separated by a 50% Percoll solution with centrifugation at 400 × g for 10 minutes at room temperature.
Immunoprecipitation and RNA-binding Assays
FLAG-immunoprecipitation was done according to manufacturer’s instructions. Briefly, HEK293T cells were transfected with expression plasmids using PEI. At 24 hours post-transfection, HEK293T cells were lysed in lysis buffer (50 mM Tris-HCl, pH 7.4, 0.5% NP-40, 2 mM EDTA and 150 mM NaCl, with complete protease inhibitors). Lysates were cleared by centrifugation and then incubated for 2 hours with M2 agarose beads (Sigma–Aldrich). After four washes with lysis buffer and two washes with Tris-buffered saline, proteins bound to M2 beads were eluted using 3× FLAG peptides (Sigma–Aldrich). In cases of protein–RNA binding, 24 hours after transfection of plasmid DNA, HEK293T cells were infected with Encephalomyocarditis virus (EMCV) for 20 hours. Then, the cell lysates were subjected to FLAG-IP and the final 3× FLAG eluates were used for RNA purification using the RNeasy Mini Kit (Qiagen). The purified RNA was reverse-transcribed and EMCV D3 genes were quantified by SYBRGreen™ PCR (Vazyme).
TUNEL Staining
Tissues were fixed in 4% paraformaldehyde and embedded in paraffin. TUNEL+ cells were detected with an in situ cell-death detection kit (AP 11684809910; Roche Life Science) according to manufacturer protocols. Fluorescence changes in stained sections were examined under a light microscope (TissueFAXS PLUS; TissueGnostics).
In Vivo Speck Assay by Fluorescent Immunohistochemistry
Fluorescent immunohistochemistry staining was performed on 4 μm-thick formalin-fixed, paraffin-embedded (FFPE) liver tissue sections. FFPE slides were deparaffinized by immersion in 100% xylene twice for 5 minutes and then rehydrated twice in fresh 100% ethanol, 95% ethanol, 70% ethanol, and 50% ethanol for 3 minutes each. Sections were washed twice in double distilled H2O and then permeabilized with 0.1% Triton X-100. Heat-induced antigen retrieval (HIER) was performed by heating sections in 1 mM EDTA at 95 °C in a pressure cooker for 20 minutes and was cooled for 20 minutes at room temperature. Sections were incubated for 12 hours at 4 °C in blocking buffer (5% goat serum + 0.3% Triton X-100 + 3% BSA diluted in 1× PBS) and subsequently incubated with primary antibody diluted 1:200 in blocking buffer for 2 hours at room temperature. Sections were washed in 0.1% Triton X-100 in 1× PBS three times for 10 minutes, incubated with secondary antibody at 1:200 for 1 hour at room temperature and washed again with 1× PBS–Tween. All processed slides were mounted in antifade mounting medium with DAPI (Beyotime cat # P0131).
Acquisition and Analyses of Images
Cells were grown on glass plates (Nest) and placed on heated (37 °C) microscope stage supplemented with warmed (37 °C), humidified air. They were imaged using the Airyscan™ detector on an LSM880 confocal microscope (Zeiss) enclosed in a 37 °C incubation chamber. ZEN (black edition) v2.3 (Zeiss) was used for acquisition. Images were acquired with a 63× oil objective.
Cellular FRAP Assays
FRAP was done on the LSM880 Airyscan™ microscope with a 488 nm laser. Bleaching was undertaken over a ~1 µm radius using 100% laser power, and images were collected every 2 s. Fluorescence intensity was measured using ZEN (black edition) v2.3 (Zeiss).
OptoIDR Assays
HEK293T cells were transfected with optoIDR plasmids (IDR-mCherry-Cry2) (Shin et al., 2017). At 24 hours post-transfection, cells were plated at 400,000 cells per 35 mm glass-bottom dish. After another 24 hours, imaging was done on an LSM 880 confocal microscope (Zeiss). Unless indicated otherwise, droplet formation was induced with 488 nm light pulses every 2 s for the duration of the imaging as indicated, and images were taken every 2 s. Fluorescence from mCherry was excited with 561 nm light.
Live-Cell Imaging
Cells were grown on chambered cover glass to an appropriate density. Cells were transfected with Cy5-labeled RNA using Lipofectamine 2000 (Thermo Fisher). Live-cell images were captured after 30 min using an LSM880 confocal microscope with a 63× oil objective. Images were analyzed by ZEN (black edition) v2.3 (Zeiss).
RT-qPCR
Total RNA was extracted and purified from IECs, hepatocytes or FACS sorted cells using trizol reagent (TaKaRa) according to the manufacturer’s instructions. Reverse transcription (RT) procedure was performed according to the instruction of TaKaRa Advantage RT-for-PCR kit. After RT, qPCR (SYBR premix EX Taq, TaKaRa) was performed using the CFX384 Real-Time System (Bio-Rad). RT-qPCR primers are listed below. Data were analyzed using the Sequence Detection Software according to the ΔCt method. All results were normalized to Hprt or Gapdh quantified in parallel amplification reactions.
| RT-qPCR primers | Sequence | IDENTIFIER |
|---|---|---|
| Gapdh-Forward | TGAGGCCGGTGCTGAGTATGTCG | N/A |
| Gapdh-Reverse | CCACAGTCTTCTGGGTGGCAGTG | N/A |
| Nlrp3-Forward | ATTACCCGCCCGAGAAAGG | N/A |
| Nlrp3-Reverse | TCGCAGCAAAGATCCACACAG | N/A |
| Nlrp6-Forward | CTCGCTTGCTAGTGACTACAC | N/A |
| Nlrp6-Reverse | AGTGCAAACAGCGTCTCGTT | N/A |
| Nlrp9b-Forward | GCGGGCTCATCTGGCTATG | N/A |
| Nlrp9b-Reverse | TCGATTTTCGTCCAGGATATTGG | N/A |
| MHV-Forward | TATAAGAGTGATTGGCGTCC | N/A |
| MHV-Reverse | GAGTAATGGGGAACCACACT | N/A |
| Ifnb-Forward | TCCTGCTGTGCTTCTCCACCACA | N/A |
| Ifnb-Reverse | AAGTCCGCCCTGTAGGTGAGGTT | N/A |
| Ifit2-Forward | AGTACAACGAGTAAGGAGTCACT | N/A |
| Ifit2-Reverse | AGGCCAGTATGTTGCACATGG | N/A |
| Isg15-Forward | GGTGTCCGTGACTAACTCCAT | N/A |
| Isg15-Reverse | TGGAAAGGGTAAGACCGTCCT | N/A |
QUANTIFICATION AND STATISTICAL ANALYSIS
The sample size chosen for our animal experiments in this study was estimated based on our prior experience of performing similar sets of experiments. All animal results were included and no method of randomization was applied. For all the bar graphs, data were expressed as mean ± SEM, and FRAP data were shown as mean ± SD. Statistical analyses were performed with a standard two-tailed unpaired Student’s t-test or paired Student’s t-test using GraphPad Prism 5. To compare two non-parametric data sets, a Mann–Whitney U-test was used. P values < 0.05 were considered significant. *, ** , *** and **** represent p < 0.05, 0.01, 0.001 and 0.0001, respectively. The sample sizes (biological replicates), specific statistical tests used, and the main effects of our statistical analyses for each experiment were detailed in each figure legend. In-vitro FRAP curve, phase diagram plot, turbidity assay, partition coefficient plot, and EMSA plot were shown as mean ± SD.
ADDITIONAL RESOURCES
This study did not include pre-registered samples.
Supplementary Material
(A) Gel-filtration profile of human NLRP6-FL upon removal of the MBP tag (left) and the corresponding SDS-PAGE of the fractions (upper right). The filament and monomeric NLRP6 peaks are labeled. The third peak with * is the MBP peak.
(B) Agarose gel-shift assay of the monomeric form of NLRP6 with poly I:C and 20-bp dsRNA.
(C) Agarose gel-shift assay of the filament form of NLRP6 with poly I:C and 20-bp dsRNA.
(D) Two additional independent MST measurements of GFP-NLRP6 binding to 20-bp dsRNA to complement that in Figure 1B. Fitting errors in KD are shown.
(E) Binding curve generated from the EMSA of 20-bp dsRNA with NLRP6 monomer. Error bars indicate three independent experiments. Fitting error in KD is shown.
(F) Binding curve generated from the EMSA of 20-bp dsRNA with NLRP6 filaments. Error bars indicate three independent experiments. Fitting error in KD is shown.
(G, H) KD measurement of NLRP6 with different ligands (taurine, spermine) by MST. Error in KD represents fitting error.
(I) Recombinant MBP-NLRP6, MBP-NLRP3∆PYD, Sumo-NLRC4∆CARD proteins (left) and their agarose gel-shift assays with 20-bp dsRNA (middle) and HMW poly I:C (right).
Unless noted otherwise, MBP-NLRP6 proteins and 60-bp FAM-dsRNA were at 15 μM, incubation time and temperature were 45 min and 37 °C, respectively. Scale bar: 5 μm.
(A) Time-dependent formation of NLRP6-dsRNA LLPS without (top) and with TEV cleavage (bottom) to remove the MBP tag.
(B) Phase-separation assay of MBP-NLRP6 FL, MBP-NLRP6∆PYD and MBP-NLRP3∆PYD (protein concentrations: 15 μM) with 60-bp FAM-labeled dsRNA (15 μM). Incubation time: 45 min. Scale bar: 5 μm.
(C) dsRNA-dependent LLPS of different NLRP6 truncation constructs. Top: without dsRNA. Bottom: with dsRNA. TEV was added at time 0 to cleave the MBP tag. Zoomed images showed irregular shaped condensates. Scale bar for zoomed images: 3 μm
(D) Quantified FRAP experiment of the condensates formed by FAM-dsRNA and NLRP6PYD+NACHT, showing lack of recovery.
(E) Negative stain EM from the in vitro phase separation sample of 60-bp FAM-dsRNA with NLRP6PYD+NACHT.
Unless noted otherwise, MBP-NLRP6 proteins and FAM-60-bp dsRNA were at 15 μM, incubation time and temperature were 45 min and 37 °C, respectively, and TEV was not added to remove the MBP tag. Unless otherwise noted, scale bars are 5 μm.
(A) RNA concentration-dependence of MBP-NLRP6−dsRNA LLPS.
(B) Protein concentration-dependence of MBP-NLRP6−dsRNA LLPS.
(C) Phase diagram of MBP-NLRP6 and 60-bp FAM-dsRNA. The left panel shows the brightfield image. The concentrations are from 2-fold dilution series from 7.5 µM. The right panel is the heat map generated from the FAM channel. Color bar: FAM fluorescence intensity units.
(D) Effects of 60-bp dsRNA on NLRP6 LLPS at low (1.875 µM, top panel) and high (7.5 µM, lower panel) NLRP6 concentrations, extracted from the phase diagram in (C) and replotted. At a low NLRP6 concentration, RNA greatly promoted NLRP6 LLPS.
(E) Temperature-dependence of MBP-NLRP6−dsRNA LLPS.
(F) Salt-concentration dependence of MBP-NLRP6−dsRNA LLPS.
(G) Effect of the crowding reagent 5% PEG3350 on lowering the concentrations required for NLRP6−dsRNA LLPS. TEV was added at time 0 to cleave the MBP tag.
(H) Co-transfection of NLRP6-mcherry and FITC-HMW poly I:C into HBL-1 cells, showing speck formation.
(I) Colocalization of GFP-NLRP6 and Cy5-dsRNA in a speck in Figure 2G.
(J) Quantification of Figure 2H for the fraction of cells with dsRNA foci and dsRNA-NLRP6 foci at time points 30 min and 60 min. n=3, error bar not shown in this quantification mode.
(A–C) Graphs plotting intrinsic disorder (by PONDR VSL2) for human NLRP6 (A), human NLRC4 (B) and human NLRP3 (C). PONDR score (y-axis) and residue position (x-axis) are shown.
(D) Time-lapse images of 293T cells expressing NLRP6-optoIDR constructs containing indicated NLRP6 IDRs linked to mCherry (red) and Cry2 (orange). Cells were subjected to laser excitation every 2 s for the indicated time. Scale bar: 5 μm.
(E) Formation of droplets in vitro by purified human NLRP6 IDR fragments labeled with TAMRA. Scale bar: 5 μm
(F) Representative images of MEFs expressing GFP-mNLRP6 or its poly-lysine region mutant K350–354A after transfection of 45-bp Cy5-dsRNA. Individual cells and NLRP6–dsRNA puncta (Boxes) are enlarged. Scale bar: 10 μm
(G) Images of 293T cells expressing NLRP6 IDRs with the indicated poly-lysine or poly-arginine mutations linked to mCherry-Cry2. Cells were subjected to laser excitation every 2 s for the indicated time. Scale bar: 5 μm.
(H) Quantification for the number of puncta in (G). Data are representative of ≥ 3 independent experiments.
(I) Immunoblots showing disruption of NLRP6 self-association by the K350–354A mutation.
WT and NLRP6 mutant mice were infected with MHV and analyzed in B-I.
(A) qPCR assays for Nlrp9b and Nlrp3 mRNA levels in IECs, hepatocytes, Kupffer cells and BMDM. N.D. denotes not detectable.
(B) Fluorescence immunohistochemistry staining of ASC and NLRP6 in liver sections from uninfected or MHV-infected WT and Nlrp6−/− mice. Nuclei were stained with DAPI.
(C) Immunoblot of 3 liver homogenates each from mock or MHV-infected WT and Nlrp6−/− mice 3 days post MHV infection to detect cleaved GSDMD. Numbers (1–3) each indicate a different mouse.
(D) TUNEL staining of MHV-infected WT and Nlrp6−/− mice 3 days after infection. Scale bar: 20 μm. Right panel is the quantification of TUNEL-positive cells. Numbers represent mean ± SEM for 3 mice/group.
(E, F) Immunoblots of NLRP6 and actin in liver (E) and small intestine (F) lysates of WT and K350–354A mutant mice.
(G) Quantification of NLRP6−dsRNA puncta. Data are mean ± SEM from 6 randomly sampled regions in liver sections.
(H) Immunoblot of multiple liver homogenates from mock or MHV-infected WT and Nlrp6 K350–354A mice 3 days post MHV infection to detect cleaved GSDMD. Numbers (1–3) indicate different mice.
(I) Quantification of TUNEL-positive cells in Figure 4N. Numbers represent mean ± SEM for 3 mice/group.
(A) Viral replication in liver of Dhx15f/f alb-cre and WT(Dhx15f/f) mice on day 3 after MHV infection.
(B) Immunoblot of multiple liver homogenates from mock or MHV-infected Dhx15f/f and Dhx15f/f alb-cre mice 3 days post high-dose MHV infection. Numbers indicate different mice. SE: short exposure. LE: long exposure.
(C, D) Serum (C) and liver explant supernatant (D) IL-18 levels in Dhx15f/f and Dhx15f/f alb-cre mice at day 3 post high-dose injection. IL-18 levels were determined by ELISA.
(E) qPCR assays for Ifnb, Ifit2 and Isg15 mRNA levels in WT and Nlrp6K350−354A mice 3 days post MHV infection.
(A) Lack of LLPS by ASC PYD and dsRNA. TAMRA-labeled MBP-ASC-PYD was at 7.5 μM and TEV was added at time 0 to cleave MBP tags.
(B) Phase diagram of MBP-NLRP6 and LTA-TAMRA. The left panel shows the brightfield image. The right panel is the heat map generated from the TAMRA channel. Scale bar: 5 μm Color bar: TAMRA fluorescence intensity units.
(C) Effects of LTA on NLRP6 LLPS at low (1.875 µM. top panel) and high (7.5 µM, lower panel) NLRP6 concentrations, extracted from the phase diagram in (C) and replotted. At a low NLRP6 concentration, LTA greatly promoted NLRP6 LLPS.
(D) Phase diagram of MBP-NLRP6 with poly(sodium 4-styrenesulfonate) using OD 395 turbidity assay, showing no promotion of NLRP6 LLPS.
Time-lapse imaging of MEFs stably expressing GFP-NLRP6 (green) upon transfection of 45-bp Cy5-dsRNA (purple) showing speck formation and fusion of NLRP6 into dsRNA puncta. The movie starts at 30 min post dsRNA transfection and lasts 19.35 min (26 frames) with 46.5 s/frame.
(A) GFP-NLRP6 channel alone (Left).
(B) GFP-NLRP6 channel overlaid with Cy5-dsRNA channel (Right).
Time-lapse imaging of an NLRP6-dsRNA speck before and after photobleaching. Bleaching of NLRP6 starts at 6 s and the movie lasts 2.55 min (123 frames) with1.24 s/frame.
Time-lapse imaging of 293T cells expressing NLRP6-optoIDR constructs containing NLRP6 IDRs from amino acids 97–188 (A) or 284–354 (B) fused to mCherry (red) and Cry2 (orange). After 24 h post-transfection, cells were subjected to laser excitation every 2 s for the duration of imaging and the movie starts at 40 s.
(A) The movie lasts 4.90 min (2.85 s/frame) (Left)
(B) The movie lasts 4.50 min (2.60 s/frame) (Right)
Time-lapse imaging of 293T cells expressing NLRP6-optoIDR constructs containing the NLRP6 IDR (284–354) mutant K350–354A fused to mCherry (red) and Cry2 (orange). At 24 h post-transfection, cells were subjected to laser excitation every 2 s for the duration of imaging and the movie starts at 40 s. The movie lasts 4.68 min (3.38 s/frame)
Time-lapse imaging of MEFs stably expressing GFP-NLRP6 (green) and mCherry-ASC (red) showing an NLRP6-ASC speck before and after photobleaching. Bleaching of NLRP6 starts at 26.9 s and the movie lasts 4.87 min (40 frames) with 7.3 s/frame.
(A) GFP-NLRP6 channel alone (Left).
(B) GFP-NLRP6 channel overlaid with mCherry-ASC channel (Right).
Time-lapse imaging of MEFs stably expressing GFP-NLRP6 (green) upon transfection of LTA-TAMRA (red) showing speck formation and fusion of NLRP6 into LTA puncta. The movie starts at 30 min post LTA transfection and lasts 33.01 min (172 frames) with 11.5 s/frame.
(A) GFP-NLRP6 channel alone (Left).
(B) GFP-NLRP6 channel overlaid with LTA-TAMRA channel (Right).
Highlights.
dsRNA induces liquid–liquid phase separation (LLPS) of NLRP6 in vitro and in cells
ASC solidifies LLPS of NLRP6 and RNA to promote inflammasome activation
Poly-Lysine region in NLRP6 is critical for multivalent interaction and LLPS
Phase separation of NLRP6 promotes inflammasome activation in intestine and liver
ACKNOWLEDGEMENTS
We thank Xin Mu and Sun Hur for providing dsRNA samples for the EMSA experiments, Harry Leung, Jennifer Waters and Talley Lambert for help with confocal microscopy, Maria Ericsson for help with transmission electron microscopy, Deying Guo and Liu Cao for providing MHV-A59 and L2 cells, Yuwei Wu for helping with hepatocyte isolation, Qi Ha for help with animal experiments, Xiong Ji for providing the optoIDR system, Tengchuan Jin for help purifying LTA, and Jiazhi Hu for helpful discussion. This work was supported by grants from the National Key R&D Program of China (2018YFA0508000 to S.Z.), Strategic Priority Research Program of the Chinese Academy of Sciences (XDB29030101 to S.Z.), National Natural Science Foundation of China (81822021, 91842105, 31770990, 82061148013, 81821001 to S.Z.), US National Institutes of Health (HD087988 and Al124491 to H.W.), and Cancer Research Institute (Irvington Postdoctoral Fellowship to C.S.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alberti S, Gladfelter A, and Mittag T (2019). Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell 176, 419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand PK, Malireddi RK, Lukens JR, Vogel P, Bertin J, Lamkanfi M, and Kanneganti TD (2012). NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature 488, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu MM (2016). The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochem Soc Trans 44, 1185–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchenough GM, Nystrom EE, Johansson ME, and Hansson GC (2016). A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science 352, 1535–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeynaems S, Alberti S, Fawzi NL, Mittag T, Polymenidou M, Rousseau F, Schymkowitz J, Shorter J, Wolozin B, Van Den Bosch L, et al. (2018). Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol 28, 420–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz P, and Dixit VM (2016). Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 16, 407–420. [DOI] [PubMed] [Google Scholar]
- Diebolder CA, Halff EF, Koster AJ, Huizinga EG, and Koning RI (2015). Cryoelectron Tomography of the NAIP5/NLRC4 Inflammasome: Implications for NLR Activation. Structure 23, 2349–2357. [DOI] [PubMed] [Google Scholar]
- Du M, and Chen ZJ (2018). DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 361, 704–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evavold CL, and Kagan JC (2019). Inflammasomes: Threat-Assessment Organelles of the Innate Immune System. Immunity 51, 609–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghimire L, Paudel S, Jin L, Baral P, Cai S, and Jeyaseelan S (2018). NLRP6 negatively regulates pulmonary host defense in Gram-positive bacterial infection through modulating neutrophil recruitment and function. PLoS Pathog 14, e1007308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimaraes CP, Witte MD, Theile CS, Bozkurt G, Kundrat L, Blom AE, and Ploegh HL (2013). Site-specific C-terminal and internal loop labeling of proteins using sortase-mediated reactions. Nat Protoc 8, 1787–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Yang C, Diao B, Huang X, Jin M, Chen L, Yan W, Ning Q, Zheng L, Wu Y, et al. (2015). The NLRP3 Inflammasome and IL-1beta Accelerate Immunologically Mediated Pathology in Experimental Viral Fulminant Hepatitis. PLoS Pathog 11, e1005155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara H, Seregin SS, Yang D, Fukase K, Chamaillard M, Alnemri ES, Inohara N, Chen GY, and Nunez G (2018). The NLRP6 Inflammasome Recognizes Lipoteichoic Acid and Regulates Gram-Positive Pathogen Infection. Cell 175, 1651–1664 e1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Zhou Q, Zhang C, Fan S, Cheng W, Zhao Y, Shao F, Wang HW, Sui SF, and Chai J (2015). Structural and biochemical basis for induced self-propagation of NLRC4. Science 350, 399–404. [DOI] [PubMed] [Google Scholar]
- Kagan JC, Magupalli VG, and Wu H (2014). Supramolecular organizing centres: location-specific higher-order signalling complexes that control innate immunity. Nat Rev Immunol 14, 821–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafontaine DLJ, Riback JA, Bascetin R, and Brangwynne CP (2020). The nucleolus as a multiphase liquid condensate. Nat Rev Mol Cell Biol. [DOI] [PubMed]
- Leng F, Yin H, Qin S, Zhang K, Guan Y, Fang R, Wang H, Li G, Jiang Z, Sun F, et al. (2020). NLRP6 self-assembles into a linear molecular platform following LPS binding and ATP stimulation. Sci Rep 10, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levels JH, Abraham PR, van Barreveld EP, Meijers JC, and van Deventer SJ (2003). Distribution and kinetics of lipoprotein-bound lipoteichoic acid. Infect Immun 71, 3280–3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy M, Thaiss CA, Zeevi D, Dohnalova L, Zilberman-Schapira G, Mahdi JA, David E, Savidor A, Korem T, Herzig Y, et al. (2015). Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 163, 1428–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Fu TM, Lu A, Witt K, Ruan J, Shen C, and Wu H (2018). Cryo-EM structures of ASC and NLRC4 CARD filaments reveal a unified mechanism of nucleation and activation of caspase-1. Proc Natl Acad Sci U S A 115, 10845–10852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman J, Wu H, and Kagan JC (2019). Gasdermin D activity in inflammation and host defense. Sci Immunol 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu A, Li Y, Schmidt FI, Yin Q, Chen S, Fu TM, Tong AB, Ploegh HL, Mao Y, and Wu H (2016). Molecular basis of caspase-1 polymerization and its inhibition by a new capping mechanism. Nature structural & molecular biology 23, 416–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, Schroder GF, Fitzgerald KA, Wu H, and Egelman EH (2014). Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 156, 1193–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon AS, Peeples WB, and Rosen MK (2020). A framework for understanding the functions of biomolecular condensates across scales. Nat Rev Mol Cell Biol. [DOI] [PMC free article] [PubMed]
- Magupalli VG, Negro R, Tian Y, Hauenstein AV, Di Caprio G, Skillern W, Deng Q, Orning P, Alam HB, Maliga Z, et al. (2020). HDAC6 mediates an aggresome-like mechanism for NLRP3 and pyrin inflammasome activation. Science 369, eaas8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, and Nunez G (2013). K(+) Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity 38, 1142–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC, et al. (2018). Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif H, Wang L, Wang WL, Magupalli VG, Andreeva L, Qiao Q, Hauenstein AV, Wu Z, Nunez G, Mao Y, et al. (2019). Structural mechanism for NEK7-licensed NLRP3 inflammasome activation. Nature 570, 338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen C, Lu A, Xie WJ, Ruan J, Negro R, Egelman EH, Fu TM, and Wu H (2019). Molecular mechanism for NLRP6 inflammasome assembly and activation. Proc Natl Acad Sci U S A 116, 2052–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M, Zhang P, Vora SM, and Wu H (2020). Higher-order assemblies in innate immune and inflammatory signaling: A general principle in cell biology. Curr Opin Cell Biol 63, 194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z, Zou J, Zhang Z, Zhao X, Noriega J, Zhang B, Zhao C, Ingle H, Bittinger K, Mattei LM, et al. (2019). Segmented Filamentous Bacteria Prevent and Cure Rotavirus Infection. Cell 179, 644–658 e613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin Y, Berry J, Pannucci N, Haataja MP, Toettcher JE, and Brangwynne CP (2017). Spatiotemporal Control of Intracellular Phase Transitions Using Light-Activated optoDroplets. Cell 168, 159–171 e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X, Ditlev JA, Hui E, Xing W, Banjade S, Okrut J, King DS, Taunton J, Rosen MK, and Vale RD (2016). Phase separation of signaling molecules promotes T cell receptor signal transduction. Science 352, 595–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauber D, Tauber G, and Parker R (2020). Mechanisms and Regulation of RNA Condensation in RNP Granule Formation. Trends Biochem Sci 45, 764–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenthorey JL, Haloupek N, Lopez-Blanco JR, Grob P, Adamson E, Hartenian E, Lind NA, Bourgeois NM, Chacon P, Nogales E, et al. (2017). The structural basis of flagellin detection by NAIP5: A strategy to limit pathogen immune evasion. Science 358, 888–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk JK, Nystrom EEL, van der Post S, Abad BM, Schroeder BO, Johansson A, Svensson F, Javerfelt S, Johansson MEV, Hansson GC, et al. (2019). The Nlrp6 inflammasome is not required for baseline colonic inner mucus layer formation or function. J Exp Med 216, 2602–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Zhu S, Yang L, Cui S, Pan W, Jackson R, Zheng Y, Rongvaux A, Sun Q, Yang G, et al. (2015). Nlrp6 regulates intestinal antiviral innate immunity. Science 350, 826–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodarska M, Thaiss CA, Nowarski R, Henao-Mejia J, Zhang JP, Brown EM, Frankel G, Levy M, Katz MN, Philbrick WM, et al. (2014). NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 156, 1045–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, and Fuxreiter M (2016). The Structure and Dynamics of Higher-Order Assemblies: Amyloids, Signalosomes, and Granules. Cell 165, 1055–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S, Hollingsworth L.R.t., and Wu H (2020). Mechanism and Regulation of Gasdermin-Mediated Cell Death. Cold Spring Harb Perspect Biol 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Yang F, Wang W, Lin G, Hu Z, Han Z, Qi Y, Zhang L, Wang J, Sui SF, et al. (2018). Structural basis for specific flagellin recognition by the NLR protein NAIP5. Cell Res 28, 35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Chen S, Ruan J, Wu J, Tong AB, Yin Q, Li Y, David L, Lu A, Wang WL, et al. (2015). Cryo-EM structure of the activated NAIP2-NLRC4 inflammasome reveals nucleated polymerization. Science 350, 404–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M, Williams EP, Malireddi RKS, Karki R, Banoth B, Burton A, Webby R, Channappanavar R, Jonsson CB, and Kanneganti TD (2020). Impaired NLRP3 inflammasome activation/pyroptosis leads to robust inflammatory cell death via caspase-8/RIPK3 during coronavirus infection. J Biol Chem 295, 14040–14052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Ding S, Wang P, Wei Z, Pan W, Palm NW, Yang Y, Yu H, Li H-B, Wang G, et al. (2017). Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature 546, 667–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Gel-filtration profile of human NLRP6-FL upon removal of the MBP tag (left) and the corresponding SDS-PAGE of the fractions (upper right). The filament and monomeric NLRP6 peaks are labeled. The third peak with * is the MBP peak.
(B) Agarose gel-shift assay of the monomeric form of NLRP6 with poly I:C and 20-bp dsRNA.
(C) Agarose gel-shift assay of the filament form of NLRP6 with poly I:C and 20-bp dsRNA.
(D) Two additional independent MST measurements of GFP-NLRP6 binding to 20-bp dsRNA to complement that in Figure 1B. Fitting errors in KD are shown.
(E) Binding curve generated from the EMSA of 20-bp dsRNA with NLRP6 monomer. Error bars indicate three independent experiments. Fitting error in KD is shown.
(F) Binding curve generated from the EMSA of 20-bp dsRNA with NLRP6 filaments. Error bars indicate three independent experiments. Fitting error in KD is shown.
(G, H) KD measurement of NLRP6 with different ligands (taurine, spermine) by MST. Error in KD represents fitting error.
(I) Recombinant MBP-NLRP6, MBP-NLRP3∆PYD, Sumo-NLRC4∆CARD proteins (left) and their agarose gel-shift assays with 20-bp dsRNA (middle) and HMW poly I:C (right).
Unless noted otherwise, MBP-NLRP6 proteins and 60-bp FAM-dsRNA were at 15 μM, incubation time and temperature were 45 min and 37 °C, respectively. Scale bar: 5 μm.
(A) Time-dependent formation of NLRP6-dsRNA LLPS without (top) and with TEV cleavage (bottom) to remove the MBP tag.
(B) Phase-separation assay of MBP-NLRP6 FL, MBP-NLRP6∆PYD and MBP-NLRP3∆PYD (protein concentrations: 15 μM) with 60-bp FAM-labeled dsRNA (15 μM). Incubation time: 45 min. Scale bar: 5 μm.
(C) dsRNA-dependent LLPS of different NLRP6 truncation constructs. Top: without dsRNA. Bottom: with dsRNA. TEV was added at time 0 to cleave the MBP tag. Zoomed images showed irregular shaped condensates. Scale bar for zoomed images: 3 μm
(D) Quantified FRAP experiment of the condensates formed by FAM-dsRNA and NLRP6PYD+NACHT, showing lack of recovery.
(E) Negative stain EM from the in vitro phase separation sample of 60-bp FAM-dsRNA with NLRP6PYD+NACHT.
Unless noted otherwise, MBP-NLRP6 proteins and FAM-60-bp dsRNA were at 15 μM, incubation time and temperature were 45 min and 37 °C, respectively, and TEV was not added to remove the MBP tag. Unless otherwise noted, scale bars are 5 μm.
(A) RNA concentration-dependence of MBP-NLRP6−dsRNA LLPS.
(B) Protein concentration-dependence of MBP-NLRP6−dsRNA LLPS.
(C) Phase diagram of MBP-NLRP6 and 60-bp FAM-dsRNA. The left panel shows the brightfield image. The concentrations are from 2-fold dilution series from 7.5 µM. The right panel is the heat map generated from the FAM channel. Color bar: FAM fluorescence intensity units.
(D) Effects of 60-bp dsRNA on NLRP6 LLPS at low (1.875 µM, top panel) and high (7.5 µM, lower panel) NLRP6 concentrations, extracted from the phase diagram in (C) and replotted. At a low NLRP6 concentration, RNA greatly promoted NLRP6 LLPS.
(E) Temperature-dependence of MBP-NLRP6−dsRNA LLPS.
(F) Salt-concentration dependence of MBP-NLRP6−dsRNA LLPS.
(G) Effect of the crowding reagent 5% PEG3350 on lowering the concentrations required for NLRP6−dsRNA LLPS. TEV was added at time 0 to cleave the MBP tag.
(H) Co-transfection of NLRP6-mcherry and FITC-HMW poly I:C into HBL-1 cells, showing speck formation.
(I) Colocalization of GFP-NLRP6 and Cy5-dsRNA in a speck in Figure 2G.
(J) Quantification of Figure 2H for the fraction of cells with dsRNA foci and dsRNA-NLRP6 foci at time points 30 min and 60 min. n=3, error bar not shown in this quantification mode.
(A–C) Graphs plotting intrinsic disorder (by PONDR VSL2) for human NLRP6 (A), human NLRC4 (B) and human NLRP3 (C). PONDR score (y-axis) and residue position (x-axis) are shown.
(D) Time-lapse images of 293T cells expressing NLRP6-optoIDR constructs containing indicated NLRP6 IDRs linked to mCherry (red) and Cry2 (orange). Cells were subjected to laser excitation every 2 s for the indicated time. Scale bar: 5 μm.
(E) Formation of droplets in vitro by purified human NLRP6 IDR fragments labeled with TAMRA. Scale bar: 5 μm
(F) Representative images of MEFs expressing GFP-mNLRP6 or its poly-lysine region mutant K350–354A after transfection of 45-bp Cy5-dsRNA. Individual cells and NLRP6–dsRNA puncta (Boxes) are enlarged. Scale bar: 10 μm
(G) Images of 293T cells expressing NLRP6 IDRs with the indicated poly-lysine or poly-arginine mutations linked to mCherry-Cry2. Cells were subjected to laser excitation every 2 s for the indicated time. Scale bar: 5 μm.
(H) Quantification for the number of puncta in (G). Data are representative of ≥ 3 independent experiments.
(I) Immunoblots showing disruption of NLRP6 self-association by the K350–354A mutation.
WT and NLRP6 mutant mice were infected with MHV and analyzed in B-I.
(A) qPCR assays for Nlrp9b and Nlrp3 mRNA levels in IECs, hepatocytes, Kupffer cells and BMDM. N.D. denotes not detectable.
(B) Fluorescence immunohistochemistry staining of ASC and NLRP6 in liver sections from uninfected or MHV-infected WT and Nlrp6−/− mice. Nuclei were stained with DAPI.
(C) Immunoblot of 3 liver homogenates each from mock or MHV-infected WT and Nlrp6−/− mice 3 days post MHV infection to detect cleaved GSDMD. Numbers (1–3) each indicate a different mouse.
(D) TUNEL staining of MHV-infected WT and Nlrp6−/− mice 3 days after infection. Scale bar: 20 μm. Right panel is the quantification of TUNEL-positive cells. Numbers represent mean ± SEM for 3 mice/group.
(E, F) Immunoblots of NLRP6 and actin in liver (E) and small intestine (F) lysates of WT and K350–354A mutant mice.
(G) Quantification of NLRP6−dsRNA puncta. Data are mean ± SEM from 6 randomly sampled regions in liver sections.
(H) Immunoblot of multiple liver homogenates from mock or MHV-infected WT and Nlrp6 K350–354A mice 3 days post MHV infection to detect cleaved GSDMD. Numbers (1–3) indicate different mice.
(I) Quantification of TUNEL-positive cells in Figure 4N. Numbers represent mean ± SEM for 3 mice/group.
(A) Viral replication in liver of Dhx15f/f alb-cre and WT(Dhx15f/f) mice on day 3 after MHV infection.
(B) Immunoblot of multiple liver homogenates from mock or MHV-infected Dhx15f/f and Dhx15f/f alb-cre mice 3 days post high-dose MHV infection. Numbers indicate different mice. SE: short exposure. LE: long exposure.
(C, D) Serum (C) and liver explant supernatant (D) IL-18 levels in Dhx15f/f and Dhx15f/f alb-cre mice at day 3 post high-dose injection. IL-18 levels were determined by ELISA.
(E) qPCR assays for Ifnb, Ifit2 and Isg15 mRNA levels in WT and Nlrp6K350−354A mice 3 days post MHV infection.
(A) Lack of LLPS by ASC PYD and dsRNA. TAMRA-labeled MBP-ASC-PYD was at 7.5 μM and TEV was added at time 0 to cleave MBP tags.
(B) Phase diagram of MBP-NLRP6 and LTA-TAMRA. The left panel shows the brightfield image. The right panel is the heat map generated from the TAMRA channel. Scale bar: 5 μm Color bar: TAMRA fluorescence intensity units.
(C) Effects of LTA on NLRP6 LLPS at low (1.875 µM. top panel) and high (7.5 µM, lower panel) NLRP6 concentrations, extracted from the phase diagram in (C) and replotted. At a low NLRP6 concentration, LTA greatly promoted NLRP6 LLPS.
(D) Phase diagram of MBP-NLRP6 with poly(sodium 4-styrenesulfonate) using OD 395 turbidity assay, showing no promotion of NLRP6 LLPS.
Time-lapse imaging of MEFs stably expressing GFP-NLRP6 (green) upon transfection of 45-bp Cy5-dsRNA (purple) showing speck formation and fusion of NLRP6 into dsRNA puncta. The movie starts at 30 min post dsRNA transfection and lasts 19.35 min (26 frames) with 46.5 s/frame.
(A) GFP-NLRP6 channel alone (Left).
(B) GFP-NLRP6 channel overlaid with Cy5-dsRNA channel (Right).
Time-lapse imaging of an NLRP6-dsRNA speck before and after photobleaching. Bleaching of NLRP6 starts at 6 s and the movie lasts 2.55 min (123 frames) with1.24 s/frame.
Time-lapse imaging of 293T cells expressing NLRP6-optoIDR constructs containing NLRP6 IDRs from amino acids 97–188 (A) or 284–354 (B) fused to mCherry (red) and Cry2 (orange). After 24 h post-transfection, cells were subjected to laser excitation every 2 s for the duration of imaging and the movie starts at 40 s.
(A) The movie lasts 4.90 min (2.85 s/frame) (Left)
(B) The movie lasts 4.50 min (2.60 s/frame) (Right)
Time-lapse imaging of 293T cells expressing NLRP6-optoIDR constructs containing the NLRP6 IDR (284–354) mutant K350–354A fused to mCherry (red) and Cry2 (orange). At 24 h post-transfection, cells were subjected to laser excitation every 2 s for the duration of imaging and the movie starts at 40 s. The movie lasts 4.68 min (3.38 s/frame)
Time-lapse imaging of MEFs stably expressing GFP-NLRP6 (green) and mCherry-ASC (red) showing an NLRP6-ASC speck before and after photobleaching. Bleaching of NLRP6 starts at 26.9 s and the movie lasts 4.87 min (40 frames) with 7.3 s/frame.
(A) GFP-NLRP6 channel alone (Left).
(B) GFP-NLRP6 channel overlaid with mCherry-ASC channel (Right).
Time-lapse imaging of MEFs stably expressing GFP-NLRP6 (green) upon transfection of LTA-TAMRA (red) showing speck formation and fusion of NLRP6 into LTA puncta. The movie starts at 30 min post LTA transfection and lasts 33.01 min (172 frames) with 11.5 s/frame.
(A) GFP-NLRP6 channel alone (Left).
(B) GFP-NLRP6 channel overlaid with LTA-TAMRA channel (Right).
Data Availability Statement
This study did not generate/analyze unique datasets or code.