

Abstract
Background
The Epidermal Growth Factor Receptor (EGFR) ligand, Amphiregulin (AREG), is a key proliferative effector of estrogen receptor signaling in breast cancer and also plays a role in other malignancies. AREG is a single-pass transmembrane protein proteolytically processed by TACE/ADAM17 to release the soluble EGFR ligand, leaving a residual transmembrane stalk that is subsequently internalized.
Methods
Using phage display, we identified antibodies that selectively recognize the residual transmembrane stalk of cleaved AREG. Conjugation with fluorescence labels and monomethyl auristatin E (MMAE) was used to study their intracellular trafficking and anti-cancer effects, respectively.
Results
We report the development of an antibody-drug conjugate (ADC), GMF-1A3-MMAE, targeting an AREG neo-epitope revealed following ADAM17-mediated cleavage. The antibody does not interact with uncleaved AREG, providing a novel means of targeting cells with high rates of AREG shedding. Using fluorescent dye conjugation, we demonstrated that the antibody is internalized by cancer cells in a manner dependent on the presence of cell surface cleaved AREG. Antibodies conjugated with MMAE were cytotoxic in vitro and induced rapid regression of established breast tumor xenografts in immunocompromised mice. We further demonstrate that these antibodies recognize the AREG neo-epitope in formalin-fixed, paraffin-embedded tumor tissue, suggesting their utility as a companion diagnostic for patient selection.
Conclusions
This ADC targeting AREG has potential utility in the treatment of breast and other tumors in which proteolytic AREG shedding is a frequent event.
Keywords: ADC, amphiregulin, estrogen receptor, breast cancer, targeted delivery
INTRODUCTION
Breast cancer is the second most frequently diagnosed cancer in women and is classified into clinical subtypes by the expression status of the estrogen and progesterone receptors (ER, PR) and the presence or absence of HER2 amplification. ER+ tumors comprise approximately 70% of all breast tumors (1). Patients with ER+ disease receive treatment with endocrine targeting therapies such as tamoxifen and aromatase inhibitors with significant initial success; however, the emergence of ER independent or endocrine therapy-resistant disease is problematic (2). As such, the development of alternative therapeutics for endocrine-insensitive disease is of critical importance.
Amphiregulin (AREG) is a transmembrane protein which, following TACE/ADAM17-dependent cleavage (3), releases a soluble EGFR ligand domain, which promotes the proliferation of normal and malignant cells. The inhibition of ADAM17 with protease inhibitors prevents AREG shedding and effectively interrupts this EGFR activating signal (4). AREG is transcriptionally regulated by estrogen during normal mammary gland development (5) and in breast cancer (6) and is a required effector for estrogen’s proliferation signal in both settings (6,7). High AREG expression levels are correlated to ER-alpha expression in breast cancer (4,6) and, when overexpressed experimentally, AREG promotes EGF signaling self-sufficiency (8). Although our primary focus has been on the role of AREG in breast cancer, we note that it is expressed in several other cancer types (9) so the potential utility of therapeutic antibody-drug conjugates (ADCs) extends to other malignancies.
ADCs consist of an antibody against a specifically targeted epitope that is used to deliver locally high concentrations of a toxic payload to a cell of interest. The payload can be toxins or chemicals that induce DNA damage, disrupt cytoskeletal integrity or interfere with the process of DNA replication. The value of ADCs is the specific delivery to a tumor of drugs that may be too toxic for patients or have complicating side effects if given at a dose high enough for tumor eradication. At least 34 unique ADCs are in various stages of clinical trials in the USA for treating solid tumors, with only three being Food and Drug Administration approved for clinical use in breast cancer at the time of publication: two targeting HER2 (trastuzumab emtansine, trastuzumab deruxtecan) and sacituzumab govitecan, which targets Trop-2 in metastatic triple-negative breast cancer patients (10). ADC development in breast cancer has mostly been directed toward HER2+ disease, with relatively little attention given to ER+ disease due to a paucity of targets. Thus, the identification of novel epitopes to be targeted in the context of ER+ disease is of value.
Given the strong requirement for AREG expression and proteolytic cleavage in ER+ breast cancer (6), we postulated that the residual cell-surface stalk resulting from AREG cleavage could serve as a neo-epitope for ADC development. Here, we describe the identification of rabbit monoclonal antibodies that selectively recognize cleaved, cell-associated AREG and the evaluation of their utility as ADCs in preclinical breast cancer models.
MATERIALS AND METHODS
Cell lines and culture conditions
Human breast cancer cell line MCF7 was obtained from American Type Culture Collection (Manassas, VA, USA). The cell line was cultured in high-glucose Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (Gibco, Life Technologies, Carlsbad, CA, USA) at 37°C in 5% CO2. The MCF7 fulvestrant-resistant subline (MCF7-F) was kindly provided by Dr Kenneth P. Nephew and cultured as previously described (11). Cell lines were authenticated by STR profiling.
Antibodies
Rabbit monoclonal IgG antibodies specific to cleaved AREG were identified in contracted screening (Oak Biosciences Inc., Sunnyvale, CA, USA) using a recombinant Rabbit naïve phage display library (scFv format, VK-linker-VH) screening platform after four rounds of panning and ELISA validation. For panning, phage were selected against a peptide modeling the ADAM17-cleaved cell-associated epitope of AREG (12), represented by the N-terminal region of THSMIDSSLSKI, and counter-selected against a peptide spanning this region (SMKTHSMIDSSLSKIAC), representing uncleaved AREG. Three candidate clones—GMF-1A3, GMF-3A3 and GMF-3E4—were reformatted as IgG and selectivity for cleaved AREG was confirmed by ELISA. GMF-1A3, referred to hereafter as simply “1A3”, was the antibody chosen for further characterization in this study.
Immunohistochemistry
A breast disease spectrum tissue micro-array was purchased from US Biomax Inc. (Derwood, MD, USA). The slides were deparaffinized in xylene and rehydrated by serial incubations in graded ethanol and then in water in a Histo-Tek® SL Slide Stainer (Sakura Finetek USA, Inc., Torrance, CA, USA). Antigen retrieval was performed in a steamer by boiling slides in a container of citrate buffer (pH 6.0) for 20 min, which was then removed for 15 min of cooling on the benchtop. Slides were washed in 1× Wash buffer (Dako Agilent, Santa Clara, CA, USA) and endogenous peroxidase was quenched by incubating with Dako Dual Endogenous Enzyme Block (S2003) for 10 min. Slides were washed in 1× Wash buffer, blocked (5% rabbit/10% goat serum in PBS), and immunostained with goat anti-AREG antibody (15 μg/mL; AF262, R&D Systems) or rabbit anti-cleaved AREG 1A3 antibody (10 μg/mL) overnight at 4°C. Slides were washed four times in 1× Wash buffer, followed by the incubation for 45 min at room temperature in 1:100 dilution of rabbit anti-goat immunoglobulins/HRP (Dako, P0160 or ready-to-use goat anti-rabbit HRP labeled polymer (Dako, K4003). The slides were washed twice in 1× Wash buffer and the color was developed with 3,3-diaminobenzidine tetrahydrochloride substrate chromogen system (K3468, DAKO, Santa Clara, CA, USA). Sections were washed with water and counterstained with hematoxylin, rinsed with water, dehydrated by serial ethanol washes to 100%, cleared and mounted in Permount (Thermo Fisher Scientific). The staining intensity was assessed semi-quantitatively using a four-point scale (negligible = 0, low = 1, medium = 2, high = 3) by two investigators working independently on blinded samples. Discordant scores were resolved by joint review.
Internalization and intracellular localization analysis of anti-cleaved-AREG 1A3 antibody
In order to study internalization, 1A3 was conjugated with pHrodo iFL Red dye using pHrodo iFL Red Microscale Protein Labeling Kit (P36014, Thermo Fisher Scientific). MCF7 cells were seeded in 12 mm Nunc Glass Bottom Dish, at a density of 1 × 105 cells/dish. After allowing the cells to attach overnight, the cells were incubated for 60 min with 1 mg/mL Hoechst dye for nuclei staining and LysoTracker® Green DND-26 (Thermo Fisher Scientific) for lysosomal labeling. The cells were then incubated with 1A3-pHrodo (2 μg/mL) for 20 min at 37°C. After washing the wells with PBS, the cells were supplemented with 1× live-cell imaging solution (Molecular probes, Thermo Fisher Scientific). Fluorescence images were acquired at 30 min intervals for 2.5 h with the Olympus 1X71 inverted fluorescence microscope (Olympus, Shinjuku, Tokyo, Japan). TAPI-2 (20 μM)—pre-treatment was performed overnight, with DMSO serving as a control.
Preparation and characterization of ADCs
Monomethyl auristatin E (MMAE) conjugated 1A3 antibody (1A3-VC-PAB-MMAE) was prepared using a kit (CM11409) from CellMosaic Inc. (Woburn, MA, USA) following the manufacturer’s instructions. Maleimide-activated valine-citrulline p-aminobenzylcarbamate (VC-PAB) MMAE was coupled directly to the antibody after reduction through alkylation (13). The specificity of 1A3 and 1A3-VC-PAB-MMAE to peptides representing human cleaved-AREG was confirmed by immunoblotting. Briefly, transferrin (TRF) conjugated peptides representing cleaved and uncleaved AREG (200 ng) were electrophoresed on SDS-PAGE gels, transferred to PVDF membrane and probed with 1 μg/mL of 1A3 and 1A3-VC-PAB-MMAE. Horseradish peroxidase-conjugated goat anti-rabbit IgG was used. Antibody binding was detected with enhanced chemiluminescence (ECL; Amersham, Arlington Heights, IL, USA). Furthermore, the characterization and purity of 1A3-VC-PAB-MMAE were analyzed using hydrophobic interaction chromatography (HIC) and size-exclusion chromatography (Cell Mosaic).
Binding affinity of 1A3 and 1A3-VC-PAB-MMAE by ELISA
Maxisorp flat bottom 96-well plates (Thermo Fisher Scientific) were coated with 5 μg/mL cleaved-AREG peptide at 4°C overnight. After washing thrice with PBST (PBS with 0.05% Tween20) and blocking for 2 h at RT with 5% non-fat dry milk in PBST, various concentrations of 1A3 and 1A3-VC-PAB-MMAE (0.001, 0.01, 0.1, 1 μg/mL) were added to the wells and incubated for 2 h at RT. The wells were washed four times with PBST and were incubated at RT with Goat anti-rabbit IgG (H + L) Cross-adsorbed -HRP conjugate secondary Ab (ThermoFisher G-21234) for 1 h. One-Step™ Turbo TMB-ELISA Substrate Solution (Thermo Fisher Scientific) was added to the wells for color development, incubated for 15 min and the reaction was stopped by adding 2 M H2SO4. The absorbance was measured at 450 nm using VersaMax Microplate Reader (Molecular Devices LLC, San Jose, California,USA).
In vitro antitumor studies
MTT assay: The cytotoxic effect of 1A3 and 1A3-VC-PAB-MMAE antibodies on MCF7 and MCF7-F breast cancer cells were determined using an MTT (3-(4,5-Dimethylthiazol2-yl)-2,5-Diphenyltetrazolium Bromide) assay. The cells were seeded at a density of 5000 cells/well in 96-well plates. Approximately 24 h after cell seeding, the cells were treated with varying doses (1.56, 3.125, 6.25, 12.5, 25, 50, 100 and 200 nM) of MMAE, 1A3 and 1A3-VC-PAB-MMAE. Untreated cells were used as control. After incubating for 72 h, the medium was aspirated and 50 μL of serum-free media and 50 μL of MTT solution (5 mg/mL solution in PBS) was added into each well; the cells were then incubated for 3 h at 37°C. The formazan crystals formed were dissolved with 4 mM HCl, 0.1% NP40 in isopropanol. The absorbance was measured at 590 nm using VersaMax Microplate Reader (Molecular Devices LLC, San Jose, California, USA). The assays were performed in triplicate.
Clonogenic assay
MCF7 breast cancer cells were plated in 48-well plates, allowed to attach overnight and treated with 12.5, 25 or 50 nM of 1A3-VC-PAB-MMAE for 72 h. A vehicle treated well served as control. After removing the ADC-containing medium, cells were washed using PBS, trypsinized and plated at a density of 1000 cells/well in 12-well plates. The cells were cultured for 14 days, with fresh medium addition every 3 days. The colonies were stained with 0.5% crystal violet solution. The number of colonies per well was counted.
In vivo antitumor activity studies
The animal experiments were approved by the Institutional Animal Care and Use Committee of University of Wisconsin La Crosse (La Crosse, Wisconsin, USA). Four-week-old female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice (NSG; The Jackson Laboratory) were acclimated for 1 week before the experiments. For MCF7 breast cancer xenograft model, the mice were implanted subcutaneously with 0.72-mg 17β-estradiol 90-day release tablets (Innovative Research of America) 3 days prior to cell injection. The endocrine-resistant MCF7-F xenograft model did not require 17β-estradiol pellets. Firefly luciferase expressing MCF7 and MCF7-F cells (2 × 106 cells in a 1:1 mixture of serum-free DMEM and Matrigel) were injected orthotopically into the fourth inguinal mammary fat pad of each mouse. Once the tumors were well established, each cell line cohort was divided into three groups (n = 5). Mean tumor volume at the start of treatment was 111 mm3 for the MCF7 cohorts and 119 mm3 for the MCF7-F cohorts. The mice were treated by intraperitoneal injection every 4 days for a total of six doses with unconjugated or MMAE conjugated anti-cleaved-AREG 1A3 antibody (5 mg/kg), or vehicle control (PBS). Tumor volumes were calculated from caliper data by using the formula: Volume (mm3) = (Length x Width2)/2. Mice were imaged weekly with the in vivo FX PRO imaging system (Carestream Molecular Imaging, Woodbridge, CA, USA) after injecting luciferin (150 mg/kg body weight). Tumor volume and body weight were monitored once aweek.
Statistical analysis
Statistical analyses were performed using Graph Pad Prism 7 (San Diego, CA, USA) software. Comparisons between groups were made using unpaired t-test (two groups) or one-way/two-way analysis of variance (three or more groups) with Bonferroni (for intergroup comparisons) or Dunnett’s (when comparing relative to a single control) multiple comparison tests. Spearman’s rank order coefficient correlation was used to analyze full length vs cleaved AREG IHC results. P values < 0.05 were considered as significant and are indicated by asterisks in figures (****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05).
RESULTS
Candidate antibody identification
The EGFR-activating signaling domain of AREG is inducibly released by ADAM17-mediated cleavage and the residual transmembrane stalk is internalized by endocytosis (Fig. 1A). We previously reported the N-terminal sequence of this residual transmembrane AREG fragment (12). Reasoning that antibodies selectively recognizing this sequence in its cleaved but not uncleaved conformation might provide useful therapeutic reagents, we used phage display to isolate rabbit VK-linker-VH scFVs with these binding characteristics (Fig. 1B). After IgG reformatting of the most favorable hits, we obtained three monoclonal rabbit IgG antibodies that selectively recognized a peptide representing the neo-epitope of the cell membrane-bound ADAM17-cleavage product of human AREG (Fig. 1C). We pursued further study with 1A3, the candidate antibody which exhibited the highest binding ratio of cleaved AREG to intact AREG as determined by ELISA.
Figure 1.
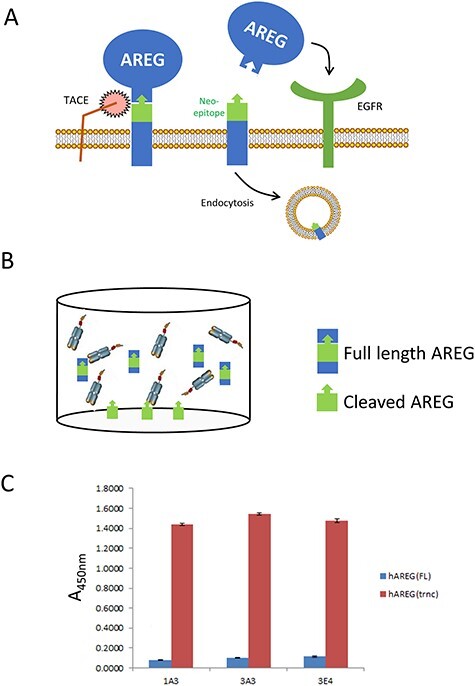
Isolation of antibodies that selectively recognize a peptide representing cleaved AREG. A. Schematic representation of the AREG cleavage and EGFR activation process, one consequence of which is the internalization of the small transmembrane fragment that remains cell-associated after TACE/ADAM17-mediated cleavage. B. Schematic representation of phage panning strategy using a peptide representing the cleaved AREG neo-epitope adhered to the well and a peptide representing the non-cleaved AREG region in the liquid phase. C. ELISA analysis of the three lead candidate rabbit IgG molecules demonstrating their relative affinities for peptides representing the full length and cleaved/truncatedAREG.
Internalization of 1A3 antibody is dependent on AREG cleavage
Having demonstrated that the 1A3 antibody had the requisite selectivity profile against synthetic peptides (Fig. 1C), we then evaluated whether it could recognize endogenous cleaved AREG in its cellular context and, further, if it could be used to internalize a cargo. Conjugation with pHrodo, a dye that fluoresces in acidic environments, was used to evaluate both cell binding and internalization. We treated MCF7 cells with Hoechst and Lysotracker to visualize the nuclei and lysosomes, respectively, and then exposed them to 1A3-pHrodo. Red fluorescence, indicating the presence of 1A3 pHrodo in acidic endosomes, was evident after 30 min of 1A3-pHrodo treatment and increased in intensity over time (Fig. 2A). The colocalization of pHrodo and Lysotracker signals also increased throughout the time course, demonstrating that 1A3 was trafficked to the lysosome after internalization, a subcellular destination necessary for linker cleavage and release of the active drug (14).
Figure 2.
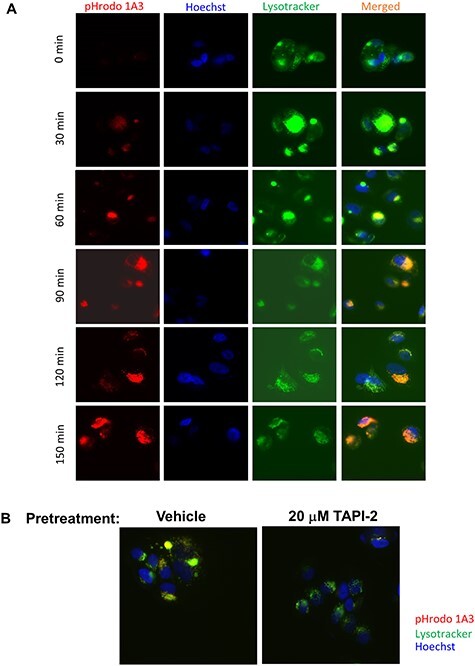
1A3 recognizes the cleaved neo-epitope in its cellular context and can be used to internalize a fluorescent cargo. A. MCF-7 cells labeled with Hoechst 33342 (blue) and Lysotracker (green) were treated with 1A3-pHrodo (red) and monitored with live-cell imaging (40×). Note the appearance of yellow in the merged image indicating trafficking of the antibody to lysosomes. B. MCF-7 cells were pretreated with TAPI-2 overnight to prevent AREG cleavage, labeled with Hoechst and Lysotracker, and imaged as in A (40×). Merged images are shown. Comparative absence of red and yellow signal in the TAPI-2 treated cells shows failure to internalize 1A3-pHrodo.
To determine whether ADAM17-mediated proteolytic generation of this neo-epitope was required for 1A3 internalization, we pretreated cells with the ADAM17 inhibitor, TAPI-2. This prevented the development of fluorescence after cells were incubated with 1A3-pHrodo treatment (Fig. 2B), from which we conclude that internalization of 1A3-pHrodo requires the generation of the neo-epitope by proteolytic cleavage.
Retention of epitope selectivity following MMAE conjugation
Using MMAE, we generated an ADC from 1A3 in order to assess the cytotoxicity in vitro and in vivo. To evaluate whether 1A3-MMAE retained the ability to selectively recognize the AREG neo-epitope after conjugation, immunoblots with 1A3 or 1A3-MMAE were performed with AREG peptides representing either an intact ADAM17-cleavage sequence (FL AREG) or the fragment that remains cell-bound after cleavage by ADAM17 (Trunc AREG). Both 1A3 and 1A3-MMAE specifically recognized the truncated AREG and also, as expected, did not react with the peptide representing uncleaved AREG (Fig. 3A). A capture ELISA using the cleaved AREG peptide also exhibited no substantial difference in binding between the unconjugated 1A3 and 1A3-MMAE (Fig. 3B). HIC-HPLC (Fig. 3C) of 1A3-MMAE (red) exhibited increased retention time relative to the unconjugated 1A3 (blue). Based on the ultraviolet light absorbance ratio (A248/A280) of the 1A3-MMAE antibody (0.715) and the equation derived by Hamblett et al. (15), the estimated drug antibody ratio (DAR) was 3.98. This placed our ADC within the range of several previously investigated ADCs with DARS between 2 and 6 that was strikes a balance between ADC solubility with an effective degree of payload delivery (15–18).
Figure 3.
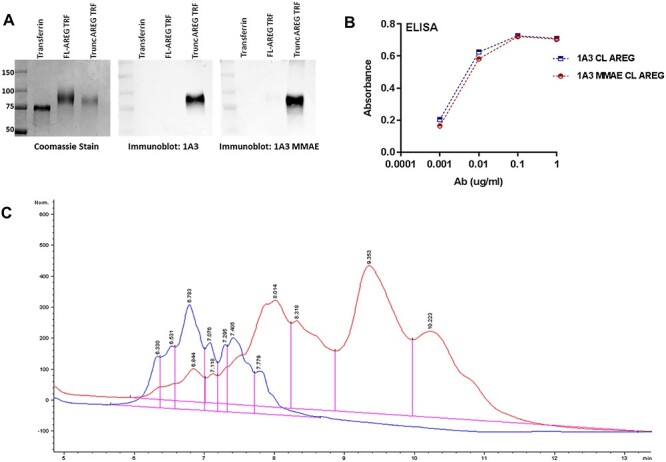
Conjugation of 1A3 with MMAE does not disrupt target recognition. A. SDS-PAGE of TRF, and TRF conjugated AREG peptides including the cleavage site (FL-AREG) or a mimic of cleaved AREG (Trunc AREG). Samples were blotted with 1A3 or 1A3-MMAE antibody. B. ELISA comparing binding of 1A3 and 1A3-MMAE to a cleaved AREG peptide C. HIC-HPLC profile of unconjugated 1A3 antibody (blue) and the 1A3-MMAE conjugate (red).
1A3-MMAE is cytotoxic in breast cancer cell lines
Treating MCF7 and Fulvestrant-resistant MCF7 (MCF7-F) with 1A3-MMAE resulted in considerable cytotoxicity, as measured by MTT assay (Fig. 4A and B). Importantly, unconjugated 1A3 did not induce cell death, whereas free MMAE potently killed the cells. Clonogenic assays with 25 or 50 nM 1A3-MMAE also showed a dose-dependent effect on survival in both MCF7 and MCF7-F cell lines visually (Fig. 4C) and when quantified as number of colonies relative to the control (Fig. 4D). As MMAE acts to prevent tubulin polymerization, we evaluated microtubule integrity in cells exposed to 1A3-MMAE. Cells that received free MMAE or 1A3-MMAE exhibited tubulin disruption, whereas control treatments with PBS, unconjugated 1A3 or IgG-MMAE did not (Fig. 4E).
Figure 4.
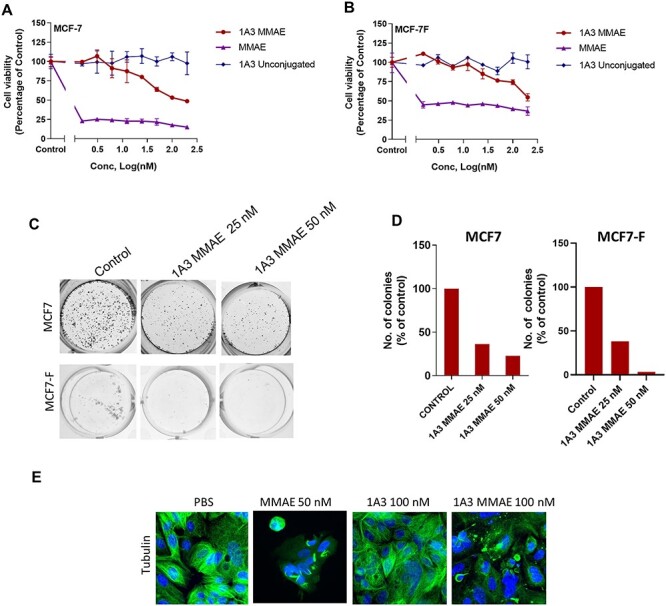
In vitro 1A3-MMAE cytotoxicity in MCF-7 (A) and MCF-7F (B) cells measured by MTT assay after treatment with 1A3-MMAE, 1A3 or MMAE. C. Clonogenic growth of MCF-7 or MCF-7F cells after exposure to 1A3-MMAE and replating. D. The quantification of clonogenic growth assays in C. E. Tubulin staining of MCF-7 cells after 48 h treatment with MMAE, 1A3, 1A3-MMAE (40× magnification).
1A3-MMAE treatment causes regression of breast cancer xenografts
To test in vivo efficacy of 1A3-MMAE, human breast cancer cell lines expressing luciferase were injected orthotopically into NSG mice (one tumor in each inguinal mammary gland). Once the average tumor size reached approximately 100–125 mm3, mice were randomized to one of three treatment groups: PBS, 1A3 (5 mg/kg) or 1A3-MMAE (5 mg/kg). Mice received intraperitoneal injections every 4 days for six total injections and were monitored for tumor progression or regression. Similar experiments were performed with MCF7 (Fig. 5, left panels) and MCF7-F (Fig. 5, right panels) xenografts. For both cell lines, caliper measurements demonstrated strong tumor shrinkage in response to 5 mg/kg 1A3-MMAE (Fig. 5A). Neither the PBS nor unconjugated 1A3 treated groups showed a disruption in tumor growth with continued, sometimes rapid increases in tumor volume relative to the 1A3-MMAE group.
Figure 5.
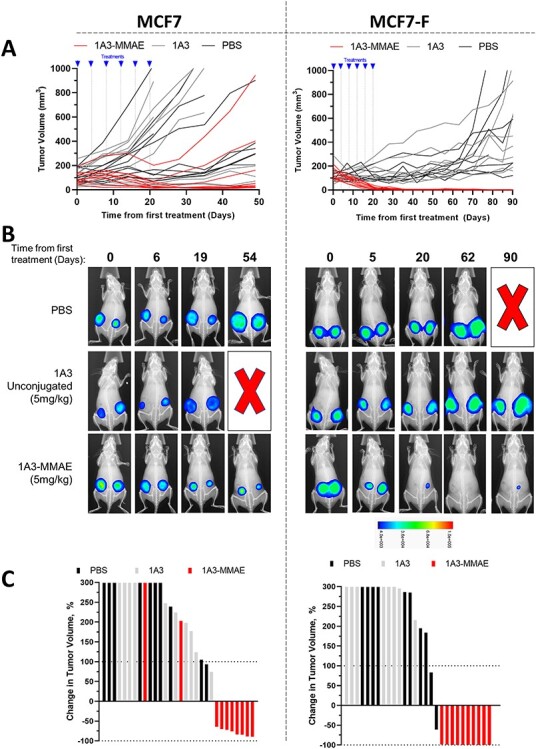
In vivo anti-tumor efficacy in MCF7 and MCF7-F cell lines. A. Caliper measurements of MCF-7 (left) and MCF7-F (right) orthotopic xenografts after treatment initiation. Each curve represents an individual tumor. Treatment times for the six treatments are indicated. B. Representative in vivo luminescence of MCF7 (left panels) and MCF7-F (right panels) xenograft tumors from A during the course of treatment. A red X indicates an animal euthanized due to tumor burden. The heatmap indicates the intensity range of the luminescence signal. C. Waterfall plot showing maximum response of the MCF7 (left) and MCF7-F (right) xenograft tumors to the treatment course. The Y-axis is truncated at 300%.
Longitudinal luciferase imaging of established xenografts and their response to treatments (Fig. 5B) showed changes in tumor size that matched volume trends seen with caliper measurements. One representative animal is shown from each cell line and treatment group. The PBS and unconjugated 1A3 groups exhibited tumor growth evident by increased lesion diameter and increasing signal intensity (color change). In contrast, tumors in the 1A3-MMAE treated groups both decreased in both volume and signal. Notably, in the MCF7-F/1A3-MMAE image, one can see the tumor signal decreasing below the limit of detection for imaging, and with continued follow-up, 70 days after the end of dosing, the re-appearance of a lesion.
To evaluate overall efficacy, we determined the maximum response as defined by the maximum change in tumor volume relative to pre-treatment measurements, depicted as waterfall plots for MCF7 and MCF7-F (Fig. 5C). Of the MCF7 xenografts, tumor volume increased in 2/10 and decreased in 8/10 dosed with 5 mg/kg 1A3-MMAE. As the 5 mg/kg dose of 1A3-MMAE successfully cleared the tumors to sub-detectable levels in all 10 of the MCF7-F xenografts, we continued follow-up in this cohort to determine whether this was curative or if recurrence was possible. After a 204 day follow-up period, 10/10 tumors had recurred in mice treated with 5 mg/kg 1A3-MMAE; however, only one tumor treated with 1A3-MMAE returned to a size seen prior to treatment, with an average time to recurrence of 73 days after initial tumor clearance determined by luciferase imaging.
Evaluation of 1A3 utility as a companion diagnostic
We performed immunohistochemistry on a breast tumor tissue micro-array to obtain an estimate of the prevalence of the target in breast cancer and to determine whether 1A3 staining had potential for use as a treatment selection biomarker. In general, samples with high AREG expression usually exhibited high-cleaved AREG intensity (Fig. 6A and B). This indicates that (1) AREG-high tumors generally actively cleave AREG, (2) the recycling kinetics in tumors are not so rapid as to preclude detection of the epitope by these antibodies and (3) these antibodies may have additional utility as companion diagnostics. Among ER-positive (n = 88) and ER-negative (n = 50) tumors evaluated, the proportions of tumors exhibiting medium/high intensity staining with 1A3 were essentially equivalent (69.3% v 70%, respectively). Although we have previously reported that AREG tends to be more highly expressed in ER-positive breast tumors (6), there is clearly a substantial fraction of ER-negative tumors which express and cleave AREG in sufficient quantities to be considered therapeutic candidates.
Figure 6.
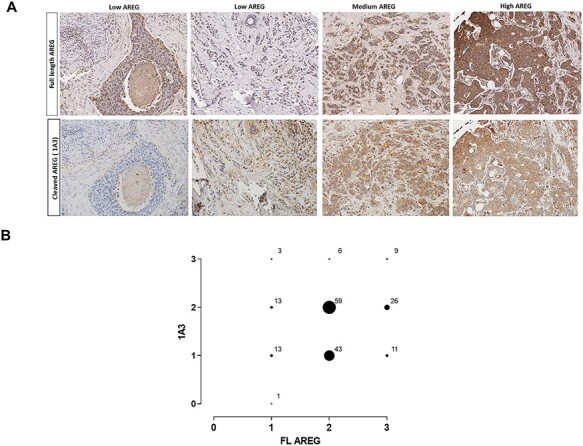
Evaluation of 1A3 antibody utility as a companion diagnostic. A. Representative examples of immunohistochemical staining intensities observed (×20 magnification) in a human breast tissue array using an antibody to FL AREG (top row), or 1A3 targeting cleaved AREG (bottom row). B. Cross-comparison of intensity scores for FL and cleaved AREG (1A3) from the breast tissue array.
DISCUSSION
In this study, we establish that the cell-associated transmembrane stalk of AREG that is generated by ADAM17-mediated cleavage is a viable therapeutic target in breast cancer. We identified three rabbit monoclonal antibodies that very selectively recognized this neo-epitope after cleavage, and which did not interact with full-length (FL) AREG. In vitro and in vivo experiments demonstrated anti-tumor efficacy of MMAE-ADCs. In all experiments, the naked antibody was ineffective, demonstrating that it is the target-mediated MMAE internalization that underlines the therapeutic effect. This work establishes in principle that antibodies with this target recognition profile may have anti-cancer utility, but further engineering to incorporate human sequences would be required to recruit the full range of effector functions in humans and prevent likely anti-rabbit immune responses against the ADC itself (19).
Identifying an appropriate target antigen is critical for efficacy and tolerability of ADC dosing in patients; ideally one would choose a tumor-specific antigen rather than a tumor-associated antigen to decrease the likelihood of on-target, off-tumor effects. Here, by demonstrating the feasibility of designing an ADC against a transient protein cleavage product, our study suggests that developing ADCs against targets that display different epitopes in some manner dependent on their state of activation/processing may generally provide even more selective ways to target relevant cell populations in vivo. Human and mouse AREG have some amino acid sequence differences in the region of the neo-epitope making it unlikely that our antibody recognizes endogenous mouse AREG. Accordingly, although we did not observe weight loss, lethargy, or breathing difficulty not attributed to tumor burden in any treated animals, we cannot draw any inference from these data about the potential for toxicity in humans.
Our two series of in vivo experiments differed in one important respect. MCF7 growth required supplementation with slow-release estrogen pellets, whereas the endocrine-resistant MCF7-F cell line grows independently of estrogen and these animals were not supplemented. Although both groups of tumors responded well to 1A3-MMAE, the MCF7-F tumors experienced more pronounced responses. Whether this can be explained by the difference in hormonal milieu, a greater dependence on AREG in this endocrine-resistant cell line or some other reason is unclear. Nevertheless, the strong response observed in the endocrine-resistant line suggests that these ADCs may warrant evaluation in this patient population for whom there are fewer effective treatments available.
Another anti-AREG therapeutic antibody strategy was recently reported (20) in which antibodies recognizing the shed AREG signaling domain can be used to stoichiometrically bind and sequester AREG released by ovarian cancer cells, thereby blocking autocrine signaling. The approach outlined in our study has the potential advantage of directly killing the AREG releasing cells; however, given that off-target toxicities with an ADC (if they occur) are likely to be more severe, this study offers a useful alternative strategy for targeting AREG, as well as establishing ovarian cancer as another malignancy in which this EGFR ligand may play an importantrole.
An appropriate companion diagnostic is critical to the successful development of a targeted therapeutic. We have shown that the 1A3 antibody efficiently detects cleaved AREG in formalin-fixed, paraffin-embedded breast cancer specimens (Fig. 6). Importantly, this series demonstrated that while cleaved AREG is quite commonly expressed in ER+ tumors, it is also found in ER- tumors. Accordingly, although our experimental focus has been on ER+/AREG+ breast tumors, given broader expression of AREG in breast tumors (Fig. 6) and in both HER2-amplified (21) and non-amplified (4) ER-negative breast cancer cell lines, it is reasonable to speculate that the potential utility may extend to ER−/AREG+ tumors or, indeed, to AREG-high tumors in other tissue types (9).
In conclusion, this study demonstrates the feasibility of generating selective ADCs against transient neo-epitopes generated by post-translational processes like proteolytic cleavage. We present a novel anti-AREG ADC with potential utility in breast cancer treatment.
ACKNOWLEDGMENTS
This study was supported by the Department of Defense Breast Cancer Research Program (W81XWH-14-1-0294 to P.K.), the American Cancer Society (123001-RSG-12-267-01-TBE to P.K.) and the Gundersen Medical Foundation. K.L. was supported by the Norman L. Gillette, Jr. Breast Cancer Research Fellowship. P.K. holds the Dr Jon and Betty Kabara Endowed Chair of Precision Oncology.
CONFLICT OF INTEREST STATEMENT
K.L., S.S. and PK are inventors on a patent application (US 63/211,356) describing the antibodies used in this manuscript.
DATA AVAILABILITY
The data underlying this article will be shared upon reasonable request to the corresponding author.
References
- 1. Waks, AG, Winer, EP. Breast cancer treatment: a review. JAMA 2019; 321: 288–300. [DOI] [PubMed] [Google Scholar]
- 2. Clarke, R., Tyson, J.J. and Dixon, J.M. (2015) Endocrine resistance in breast cancer--an overview and update. Mol Cell Endocrinol, 418Pt 3, 220–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gschwind, A, Hart, S, Fischer, OMet al. TACE cleavage of proamphiregulin regulates GPCR-induced proliferation and motility of cancer cells. Embo J 2003; 22: 2411–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kenny, PA, Bissell, MJ. Targeting TACE-dependent EGFR ligand shedding in breast cancer. J Clin Invest 2007; 117: 337–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McBryan, J, Howlin, J, Kenny, PAet al. ERalpha-CITED1 co-regulated genes expressed during pubertal mammary gland development: implications for breast cancer prognosis. Oncogene 2007; 26: 6406–19. [DOI] [PubMed] [Google Scholar]
- 6. Peterson, EA, Jenkins, EC, Lofgren, KAet al. Amphiregulin Is a critical downstream effector of estrogen signaling in ERalpha-positive breast cancer. Cancer Res 2015; 75: 4830–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ciarloni, L, Mallepell, S, Brisken, C. Amphiregulin is an essential mediator of estrogen receptor alpha function in mammary gland development. Proc Natl Acad Sci U S A 2007; 104: 5455–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Willmarth, NE, Ethier, SP. Autocrine and juxtacrine effects of amphiregulin on the proliferative, invasive, and migratory properties of normal and neoplastic human mammary epithelial cells. J Biol Chem 2006; 281: 37728–37. [DOI] [PubMed] [Google Scholar]
- 9. Busser, B, Sancey, L, Brambilla, Eet al. The multiple roles of amphiregulin in human cancer. Biochimica et Biophysica Acta 2011; 1816: 119–31. [DOI] [PubMed] [Google Scholar]
- 10. Barroso-Sousa, R, Tolaney, SM. Clinical development of new antibody-drug conjugates in breast cancer: to infinity and beyond. BioDrugs 2021; 35: 159–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fan, M, Yan, PS, Hartman-Frey, Cet al. Diverse gene expression and DNA methylation profiles correlate with differential adaptation of breast cancer cells to the antiestrogens tamoxifen and fulvestrant. Cancer Res 2006; 66: 11954–66. [DOI] [PubMed] [Google Scholar]
- 12. Levano, KS, Kenny, PA. Clarification of the C-terminal proteolytic processing site of human Amphiregulin. FEBS Letters 2012; 586: 3500–2. [DOI] [PubMed] [Google Scholar]
- 13. Sun, MM, Beam, KS, Cerveny, CGet al. Reduction-alkylation strategies for the modification of specific monoclonal antibody disulfides. Bioconjug Chem 2005; 16: 1282–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schrama, D, Reisfeld, RA, Becker, JC. Antibody targeted drugs as cancer therapeutics. Nat Rev Drug Discov 2006; 5: 147–59. [DOI] [PubMed] [Google Scholar]
- 15. Hamblett, KJ, Senter, PD, Chace, DFet al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res 2004; 10: 7063–70. [DOI] [PubMed] [Google Scholar]
- 16. Akaiwa, M, Dugal-Tessier, J, Mendelsohn, BA. Antibody-drug conjugate payloads; study of Auristatin derivatives. Chem Pharm Bull (Tokyo) 2020; 68: 201–11. [DOI] [PubMed] [Google Scholar]
- 17. Lyon, RP, Bovee, TD, Doronina, SOet al. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat Biotechnol 2015; 33: 733–5. [DOI] [PubMed] [Google Scholar]
- 18. Sun, X, Ponte, JF, Yoder, NCet al. Effects of drug-antibody ratio on pharmacokinetics, biodistribution, efficacy, and tolerability of antibody-maytansinoid conjugates. Bioconjug Chem 2017; 28: 1371–81. [DOI] [PubMed] [Google Scholar]
- 19. Peters, C, Brown, S. Antibody-drug conjugates as novel anti-cancer chemotherapeutics. Biosci Rep 2015; 35: e00225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lindzen, M, Ghosh, S, Noronha, Aet al. Targeting autocrine amphiregulin robustly and reproducibly inhibits ovarian cancer in a syngeneic model: roles for wildtype p53. Oncogene 2021; 40: 3665–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schmucker, H, Blanding, WM, Mook, JMet al. Amphiregulin regulates proliferation and migration of HER2-positive breast cancer cells. Cell Oncol (Dordr) 2018; 41: 159–68. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data underlying this article will be shared upon reasonable request to the corresponding author.