

Abstract
Programmable site-specific nucleases, such as the clustered regularly interspaced short palindromic repeat (CRISPR)/ CRISPR-associated protein 9 (Cas9) ribonucleoproteins (RNPs), have allowed creation of valuable knockout mutations and targeted gene modifications in Chlamydomonas (Chlamydomonas reinhardtii). However, in walled strains, present methods for editing genes lacking a selectable phenotype involve co-transfection of RNPs and exogenous double-stranded DNA (dsDNA) encoding a selectable marker gene. Repair of the dsDNA breaks induced by the RNPs is usually accompanied by genomic insertion of exogenous dsDNA fragments, hindering the recovery of precise, scarless mutations in target genes of interest. Here, we tested whether co-targeting two genes by electroporation of pairs of CRISPR/Cas9 RNPs and single-stranded oligodeoxynucleotides (ssODNs) would facilitate the recovery of precise edits in a gene of interest (lacking a selectable phenotype) by selection for precise editing of another gene (creating a selectable marker)—in a process completely lacking exogenous dsDNA. We used PPX1 (encoding protoporphyrinogen IX oxidase) as the generated selectable marker, conferring resistance to oxyfluorfen, and identified precise edits in the homolog of bacterial ftsY or the WD and TetratriCopeptide repeats protein 1 genes in ∼1% of the oxyfluorfen resistant colonies. Analysis of the target site sequences in edited mutants suggested that ssODNs were used as templates for DNA synthesis during homology directed repair, a process prone to replicative errors. The Chlamydomonas acetolactate synthase gene could also be efficiently edited to serve as an alternative selectable marker. This transgene-free strategy may allow creation of individual strains containing precise mutations in multiple target genes, to study complex cellular processes, pathways, or structures.
A transgene-free strategy allows precise editing of genes lacking a selectable phenotype by electroporation of CRISPR/Cas9 ribonucleoproteins and single-stranded oligodeoxynucleotide templates.
Introduction
The green alga Chlamydomonas (Chlamydomonas reinhardtii) has gained recognition as a model organism for the study of diverse organelles and physiological processes and has also been used in explorative work related to biofuel, nutraceutical, and pharmaceutical recombinant protein production (Rosales-Mendoza et al., 2012; Jinkerson and Jonikas, 2015; Scranton et al., 2015; Salomé and Merchant, 2019). The ability to genetically manipulate the Chlamydomonas genome allows the investigation of gene function as well as the pursuit of innovative biotechnological applications. A variety of methods have been developed to disrupt nuclear genes in this alga, including chemical-, UV light-, gamma irradiation-, and insertional mutagenesis, the latter involving cell transfection with exogenous DNA that integrates randomly into the genome (Jinkerson and Jonikas, 2015; Picariello et al., 2020). Moreover, a genome-wide mutant library has been recently generated using insertional mutagenesis (Li et al., 2016, 2019). However, all these methods result in random alterations to the genome sequence, and mutations in a desired gene may not be obtained or may not affect gene function.
Targeted strategies for nuclear gene inactivation have also been explored in Chlamydomonas. Post-transcriptional gene silencing by RNA interference, involving transgenic strains expressing artificial microRNAs (Molnar et al., 2009; Zhao et al., 2009) or double-stranded RNAs (Rohr et al., 2004; Kim and Cerutti, 2009), seems to have variable knockdown efficiency, depending on the target gene, and may potentially cause unintended off-target effects. Homologous recombination (HR)-mediated gene disruption has also been attempted, but a major drawback is the low frequency of HR between exogenous DNA and a nuclear gene of interest (Sodeinde and Kindle, 1993; Gumpel et al., 1994; Zorin et al., 2009; Plecenikova et al., 2013; Sizova et al., 2013; Jinkerson and Jonikas, 2015; Jiang et al., 2017). Promisingly, recent studies have successfully used sequence-specific nucleases (SSNs) to achieve targeted gene disruption (Sizova et al., 2013; Jiang et al., 2014; Baek et al., 2016; Shin et al., 2016; Ferenczi et al., 2017; Greiner et al., 2017; Jiang and Weeks, 2017; Shamoto et al., 2018; Guzmán-Zapata et al., 2019; Angstenberger et al., 2020; Cazzaniga et al., 2020; Dhokane et al., 2020; Kang et al., 2020; Kim et al., 2020; Park et al., 2020; Picariello et al., 2020). SSNs cause DNA double-strand breaks (DSBs) at specific sites, which can be repaired by alternative cellular mechanisms resulting in mutations or precise sequence changes. DSB repair by error-prone nonhomologous end-joining (NHEJ), including canonical and alternative pathways, may result in insertions/deletions (i.e. indels) and/or missense/nonsense mutations at the target sites (Rodgers and McVey, 2016; Gallagher and Haber, 2018; Scully et al., 2019; Capdeville et al., 2020; Gallagher et al., 2020). Alternatively, homology directed repair (HDR) in the presence of template donor DNA, which can also occur by several pathways, may result in precise sequence changes (Rodgers and McVey, 2016; Gallagher and Haber, 2018; Paix et al., 2017; Scully et al., 2019; Capdeville et al., 2020; Gallagher et al., 2020).
Most current work on nuclear gene targeting in Chlamydomonas has focused on the RNA-programmable site-specific nucleases, such as the clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated protein 9 (Cas9) system from Streptococcus pyogenes (Makarova et al., 2011; Jinek et al., 2012; Jeon et al., 2017; Swarts and Jinek, 2018), which rely on Watson–Crick base pairing for DNA recognition. Several reports have demonstrated that both CRISPR/Cas9 and CRISPR/Cas12a (formerly Cpf1), belonging to types II-A and V-A of the CRISPR–Cas systems (Makarova et al., 2011; Swarts and Jinek, 2018), are useful for targeted gene disruption in Chlamydomonas (Jiang et al., 2014; Baek et al., 2016; Shin et al., 2016; Ferenczi et al., 2017; Greiner et al., 2017; Jiang and Weeks, 2017; Shamoto et al., 2018; Guzmán-Zapata et al., 2019; Angstenberger et al., 2020; Cazzaniga et al., 2020; Dhokane et al., 2020; Kang et al., 2020; Kim et al., 2020; Park et al., 2020; Picariello et al., 2020). In this organism, the CRISPR/Cas systems have been implemented as a transgenic method, with components expressed either transiently from plasmids (Jiang et al., 2014; Guzmán-Zapata et al., 2019) or constitutively from genome-integrated constructs (Greiner et al., 2017; Jiang and Weeks, 2017; Park et al., 2020), or as a transgene-free approach, by introducing into cells preassembled CRISPR/Cas9 (or CRISPR/Cas12a) ribonucleoproteins (RNPs) by electroporation (Baek et al., 2016; Shin et al., 2016; Ferenczi et al., 2017; Greiner et al., 2017; Shamoto et al., 2018; Angstenberger et al., 2020; Dhokane et al., 2020; Cazzaniga et al., 2020; Kim et al., 2020; Picariello et al., 2020) or by using a cell penetrating peptide (Kang et al., 2020). Published methods using RNA-programmable site-specific nucleases differ in efficiency, applicable genes/strains, and/or experimental details but, in general, targeted disruption of nuclear genes in Chlamydomonas and in the related alga Volvox carteri has become a feasible approach (Baek et al., 2016; Shin et al., 2016; Ferenczi et al., 2017; Greiner et al., 2017; Jiang and Weeks, 2017; Shamoto et al., 2018; Guzmán-Zapata et al., 2019; Ortega-Escalante et al., 2019; Angstenberger et al., 2020; Cazzaniga et al., 2020; Dhokane et al., 2020; Kang et al., 2020; Kim et al., 2020; Park et al., 2020; Picariello et al., 2020).
In contrast, precise gene editing (i.e. precise nucleotide changes in a target genomic sequence) triggered by RNA-programmable SSNs remains fairly inefficient or limited to specific strains/experimental approaches (Ferenczi et al., 2017; Greiner et al., 2017; Jiang and Weeks, 2017). As reported for somatic cells of land plants (Capdeville et al., 2020), a major constraint is that, in Chlamydomonas, DSB repair by error-prone NHEJ pathways appears to be much more efficient than HR in the presence of donor DNA (Sizova et al., 2013; Greiner et al., 2017). As described for some mammalian cell lines (Shy et al., 2016; Mitzelfelt et al., 2017), only a small subset of the population, in asynchronously grown Chlamydomonas, may be capable of HDR, possibly associated with being in a certain phase of the cell cycle (Angstenberger et al., 2020). Additionally, the cell wall appears to pose a substantial barrier for the introduction of macromolecules into Chlamydomonas cells (Jeon et al., 2017; this work). Electroporation of CRISPR/Cas RNPs into cell wall-less mutant strains or after removal of the cell wall by autolysin treatment often resulted in gene editing (disruption) frequencies of a few percentage (relative to the number of electroporated cells; Baek et al., 2016; Ferenczi et al., 2017; Shamoto et al., 2018; Angstenberger et al., 2020; Picariello et al., 2020). However, using similar methods with walled strains resulted in gene editing (disruption) frequencies several orders of magnitude lower (Shin et al., 2016; Greiner et al., 2017; Kang et al., 2020; this work). In the absence of a selectable phenotype for a gene of interest, a common approach has been to electroporate walled strains with CRISPR/Cas RNPs and DNA coding for a selectable marker (e.g. an antibiotic resistance gene), sometimes with the addition of template DNA to elicit HDR. Chlamydomonas colonies surviving on the selective agent, due to nuclear integration and expression of the selectable transgene, were then screened for editing of a desired endogenous gene in labor-intensive efforts (Shin et al., 2016; Greiner et al., 2017; this work). Unfortunately, in many cases, the target gene contained insertions of the exogenous marker DNA or other DNA fragments, preventing the recovery of precisely edited genes for study (Greiner et al., 2017; this work).
To overcome the outlined problems, we explored whether selection for precise editing of an endogenous gene in Chlamydomonas, which would effectively select for cells taking up the editing components and capable of carrying out HDR, may facilitate the recovery of precise, scarless edits in another gene of interest, when both genes were simultaneously targeted by co-electroporation of CRISPR/Cas9 RNPs and template donor DNAs. We used single-stranded oligodeoxynucleotides (ssODNs) as donor DNA because prior work demonstrated their usefulness, based on design simplicity and commercial availability, as templates in the repair of CRISPR/Cas-induced DSBs in Chlamydomonas (Ferenczi et al., 2017; Greiner et al., 2017; Jiang and Weeks, 2017; Sizova et al., 2021) and land plants (Shan et al., 2013; Svitashev et al., 2015; Sauer et al., 2016).
The enzyme protoporphyrinogen oxidase (Protox), encoded by the PPX1 gene in Chlamydomonas, oxidizes protoporphyrinogen IX to protoporphyrin IX in the biosynthetic pathway of heme and chlorophyll (Duke et al., 1991; Randolph-Anderson et al., 1998). Inhibition of this enzyme causes accumulation of protoporphyrinogen IX, which is nonenzymatically oxidized to protoporphyrin IX and eventually leads to membrane peroxidation and cell lethality in the light (Duke et al., 1991; Ha et al., 2004). A single base-pair mutation (G→A, causing a valine-389 to methionine substitution) within the protein coding sequence results in the Protox enzyme being resistant to inhibition by porphyric herbicides such as oxyfluorfen (Randolph-Anderson et al., 1998; Brueggeman et al., 2014). Electroporation of Chlamydomonas cells with a CRISPR/Cas9 (PPX1) RNP and an ssODN donor (designed to introduce the G->A mutation in PPX1 during HDR) led to the isolation of colonies precisely edited in PPX1 by selection on oxyfluorfen containing medium. Interestingly, co-targeting PPX1 (as the selectable marker) and FTSY (encoding a signal recognition particle receptor protein, denoted as FtsY in bacteria, which is required for the integration of light-harvesting complex proteins into thylakoid membranes) or WD and TetratriCopeptide repeats protein 1 (WDTC1; encoding a conserved protein with antiadipogenic functions in several eukaryotes) allowed the recovery of precisely edited mutants in the latter genes. Optimizing this approach of simultaneously editing a selectable marker and any gene of interest may prove to be a viable strategy for introducing precise sequence changes in the genome of Chlamydomonas and, potentially, of other microalgae.
Results
Targeted disruption of the FTSY gene
The FTSY gene encodes a component of the chloroplast signal recognition particle-dependent pathway, which is required for insertion of light-harvesting chlorophyll a/b-binding proteins into the thylakoid membranes (Aldridge et al., 2009; Kirst et al., 2012). In Chlamydomonas, null FTSY mutants showed diminished ability to assemble some proteins of the light harvesting complex II, resulting in lower chlorophyll content than in the wild-type (WT; Kirst et al., 2012; Baek et al., 2016; Kim et al., 2020). Hence, mutant colonies displayed a pale green phenotype. Because of this easily identifiable phenotype, we initially attempted to edit exon 4 of the FTSY gene (Figure 1A), using a CRISPR/Cas9 protocol-dependent on square-wave electroporation as described by Greiner et al. (2017).
Figure 1.
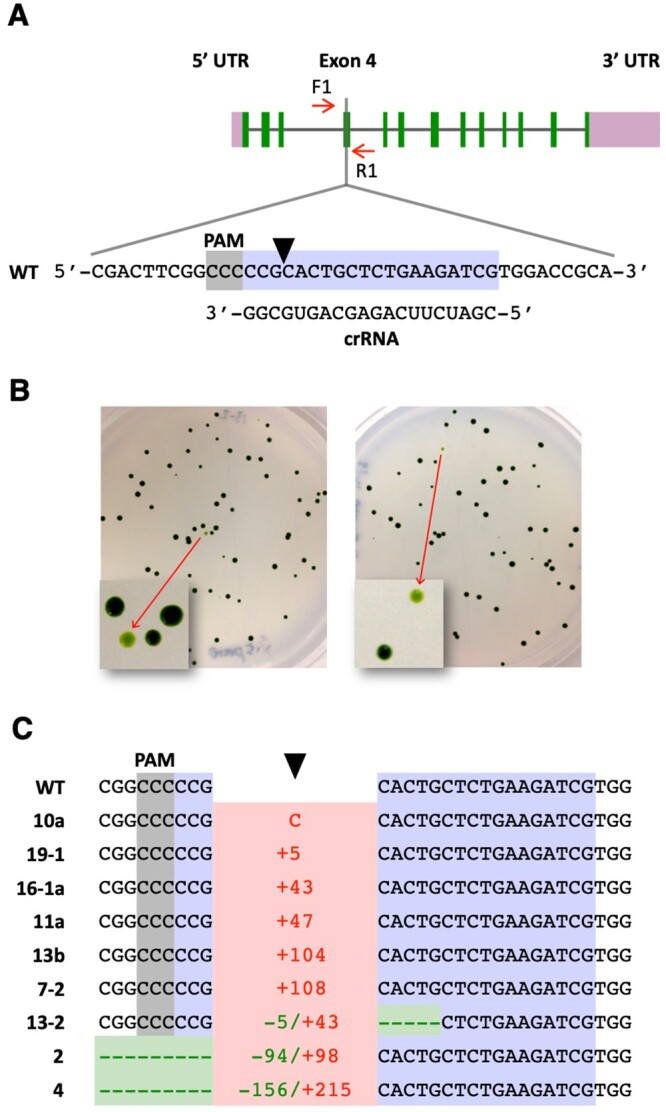
Targeted disruption of the FTSY gene. A, Schematic of the FTSY gene showing the target (highlighted in bluish purple) and reverse complement PAM (highlighted in gray) sequences. A black arrowhead indicates the Cas9 cleavage site. Short red arrows indicate the primers used for PCR analyses. B, Cells co-transfected with the CRISPR/Cas9 (FTSY) RNP and dsDNA encoding the aphVIII transgene were spread on TAP agar plates containing paromomycin. Representative plates show some pale green colonies as a consequence of FTSY gene disruption. C, DNA sequences of pale green colonies, indicating alterations at the FTSY target site relative to the WT. Insertions, indicating type (C, cytosine) or number of base pairs, are depicted in red. Deletions, indicating type or number of base pairs, are depicted in green. Complete sequences are shown in Supplemental Figure S2.
Cells of the walled CC-5415 (g1) strain were electroporated with a CRISPR/Cas9 (FTSY) RNP and a double-stranded DNA (dsDNA) PCR fragment containing a transgene expressing the Streptomyces rimosus aphVIII gene, which confers resistance to the antibiotic paromomycin (Sizova et al., 2001). After electroporation, cells were selected on agar plates containing paromomycin. Antibiotic-resistant colonies displaying a pale green phenotype (Figure 1B) were assumed to be FTSY null mutants for calculation of gene disruption frequencies (Supplemental Table S1). However, we note that the reported values are an approximation to the actual frequency of targeted gene alterations since some CRISPR/Cas9-induced mutations in FTSY may not affect protein function and the corresponding colonies would not be detectable as pale green. Conversely, some pale green colonies may be caused by random insertion of the selectable transgene into genes, other than FTSY, associated with chlorophyll synthesis or photosynthetic complex assembly. Yet, all pale green colonies examined by PCR amplification of the target site (Supplemental Table S1) either showed indels at the Cas9 cleavage site (Figure 1C), consistent with disruption of FTSY gene function, or lacked a detectable PCR product, likely due to large DNA insertions and/or rearrangements at the target site, as previously reported common occurrence associated with CRISPR/Cas9 gene editing in Chlamydomonas (Shin et al., 2016; Greiner et al., 2017; Kang et al., 2020; Picariello et al., 2020).
In agreement with the results of Greiner et al. (2017), heat shocking the cells prior to electroporation increased more than five-fold the number of recovered pale green colonies (Supplemental Table S1, Experiments 2 and 3). To achieve precise FTSY gene editing, in several experiments, we also added to the electroporation mix a ssODN (Supplemental Table S2), overlapping the Cas9 cleavage site and designed to serve as template for HDR. Precise repair by homology directed mechanisms of the DSB caused by CRISPR/Cas9 (FTSY) RNP would introduce stop codons within the FTSY coding sequence, create a new restriction enzyme site (NheI) for genotypic analyses, and destroy the Cas9 protospacer adjacent motif (PAM; Supplemental Figure S1). However, out of 21 pale green colonies examined by PCR amplification of the targeted FTSY exon, 12 lacked a detectable PCR product, and 9 showed indels at the target site (Figure 1C and Supplemental Figure S2). We did not recover any colony precisely edited by HDR in any of these experiments, underlining the difficulty in obtaining precise gene editing in walled Chlamydomonas strains with current methodological approaches. Although, one colony exhibited apparent integration of donor DNA by HR on one side of the DSB whereas the repair was carried out by NHEJ on the other side, resulting in the insertion of a 104-bp DNA fragment (Supplemental Figure S2, colony 13b; see “Discussion”).
We also examined uptake of the CRISPR/Cas9 (FTSY) RNP after electroporation of the walled g1 strain of Chlamydomonas. For these experiments, we assembled the RNP with trans-activating CRISPR RNA (tracrRNA) conjugated to the ATTO 550 fluorophore (Banas et al., 2020). Four hours after electroporation, cells were examined by fluorescence microscopy to visualize uptake and intracellular localization of the labeled RNP. The tracrRNA-ATTO 550 electroporated alone localized predominantly in the nucleus (Supplemental Figure S3A), as previously reported for single-stranded DNA oligonucleotides (Jiang et al., 2017). In contrast, the assembled tracrRNA-ATTO 550-Cas9 RNP was distributed in the cytosol and the nucleus with some preferential perinuclear accumulation (Supplemental Figure S3A). However, examining over a thousand individual cell images revealed that <1% of the cells had taken up enough RNP to be detectable by fluorescence microscopy (Supplemental Figure S3B), suggesting that electroporation of CRISPR/Cas9 RNPs into walled Chlamydomonas strains is quite inefficient.
Precise editing of the PPX1 gene
With the goal of developing a co-editing approach in Chlamydomonas, we next attempted to modify an endogenous gene amenable to being converted (via precise mutagenesis) to a selectable marker gene. As already mentioned, a single point mutation (G→A) in PPX1 exon 10 (Figure 2A), causing a single amino acid substitution (Val389Met) in the Protox enzyme, confers resistance to porphyric herbicides in Chlamydomonas (Randolph-Anderson et al., 1998; Brueggeman et al., 2014). Cells of the g1 strain were electroporated with a CRISPR/Cas9 (PPX1) RNP and an ssODN (Supplemental Table S2), overlapping the Cas9 cleavage site and designed to serve as template for HDR. Precise DSB repair by homology-directed mechanisms would introduce the G→A mutation within the PPX1 coding sequence as well as a nearby, functionally silent, C→T mutation (Figure 2A; Supplemental Figure S4). Electroporated cells were selected on agar plates containing oxyfluorfen. By using the protocol developed by Greiner et al. (2017), we recovered several oxyfluorfen-resistant colonies (Figure 2B). Moreover, the frequency of CRISPR/Cas9 (PPX1) RNP-induced mutants, particularly when including a heat shock treatment (Supplemental Table S3, Experiments 1 and 2), was well above the frequency of spontaneous PPX1 mutation to herbicide resistance, which has been reported to be <1 × 10−8 (Randolph-Anderson et al., 1998). However, the overall number of oxyfluorfen-resistant colonies was quite low.
Figure 2.
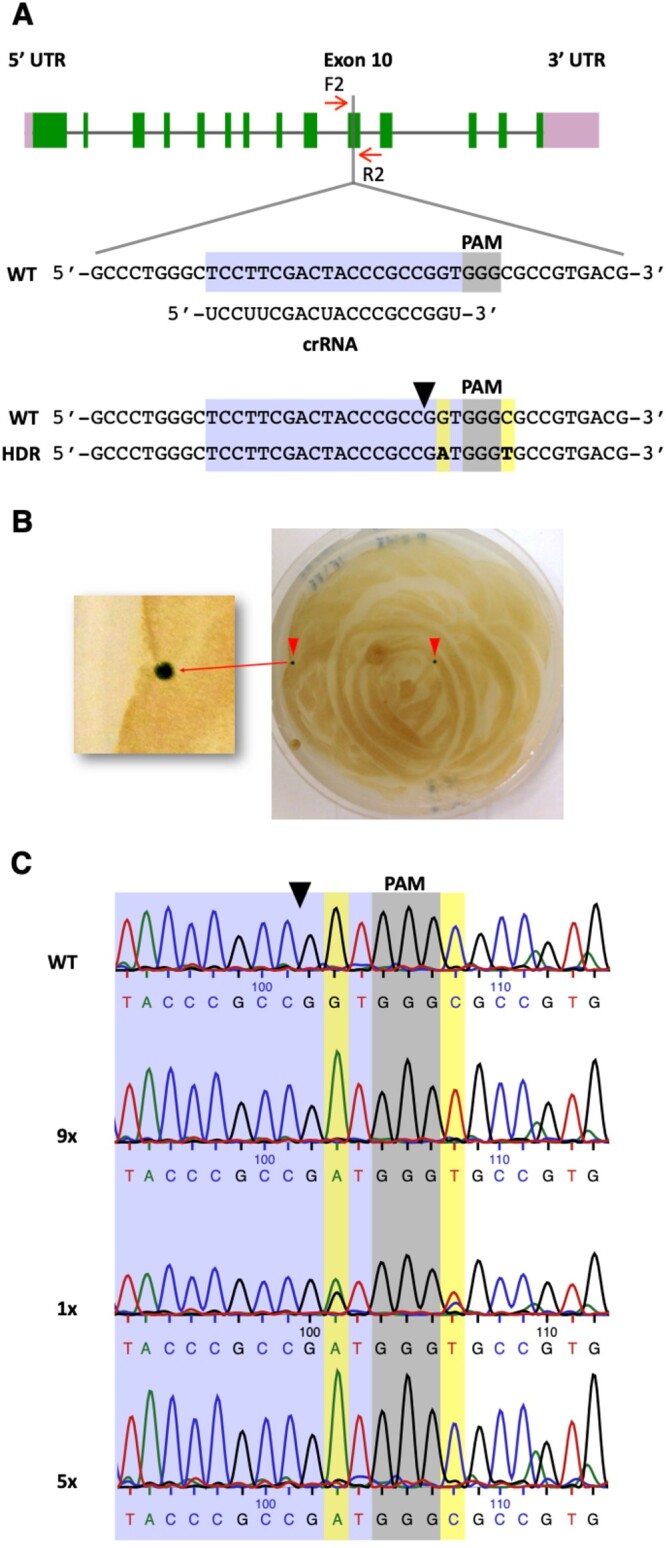
Precise editing of the PPX1 gene. A, Schematic of the PPX1 gene showing the target (highlighted in bluish purple) and PAM (highlighted in gray) sequences. Other symbols are as described under Figure 1A. HDR of the DSB, using as template the transfected PPX1 ssODN, is expected to introduce two base pair changes (highlighted in yellow) into the genome (bottom sequence). B, Cells co-transfected with the CRISPR/Cas9 (PPX1) RNP and ssODN donor DNA were spread on TAP agar plates containing oxyfluorfen. A representative plate shows two green oxyfluorfen resistant colonies as a consequence of PPX1 gene editing. C, Representative sequencing chromatograms of the WT, fully edited (9×, containing both G→A and C→T changes) and, likely, partly edited (5×, containing only the G→A change) colonies. The chromatogram of an apparently mixed colony (1×), displaying a heterozygous (i.e. WT and fully edited) sequence, is also shown. Figures followed by an X indicate the number of colonies of each DNA type examined by sequencing.
To enhance the recovery of PPX1 edited colonies, we introduced several modifications to the Greiner et al. (2017) method, including changes to some electroporation parameters, the amounts of Cas9 nuclease and ssODN donor used in the electroporation mix, as well as the timing of the heat shock treatment, which was performed after (rather than before) cell electroporation. Incubating Chlamydomonas cells at 35°C, during the recovery period after RNP electroporation, has also been recently reported to increase the frequency of edited colonies (Dhokane et al., 2020). All of our modifications have been incorporated into an optimized protocol described under “Materials and Methods”. This modified protocol increased approximately five-fold the number of recovered oxyfluorfen-resistant colonies (Supplemental Table S3, Experiments 3 and 4). Fifteen of these colonies were examined by PCR amplification of the target site and sequencing of the PCR products. Nine colonies displayed the expected (i.e. G→A and C→T) sequence changes, one colony appeared to be a mixture between WT and a properly edited clone, and five colonies showed only the G→A change, which is solely necessary to confer herbicide resistance (Figure 2C). Thus, about two-thirds of the examined colonies were fully consistent with editing by HDR, using as template the electroporated ssODN donor. However, we surmise that the remaining examined colonies were also edited by homology-directed mechanisms, since we have not recovered any spontaneous herbicide-resistant mutant in any of our negative control experiments (Supplemental Table S3). As described in other eukaryotes (Gallagher and Haber, 2018; Paix et al., 2017; Boel et al., 2018; Gallagher et al., 2020), we expected that Cas9-induced DSBs in Chlamydomonas would be repaired by the single-strand template repair (SSTR) mechanism (see “Discussion”). Following this model (Supplemental Figure S4), the PPX1 ssODN would be used as a homologous template for DNA synthesis primed by the 3′ ending strand on the left side of the cleavage site. If a very short stretch of DNA (at most six nucleotides), next to the Cas9 cleavage site, is copied by DNA synthesis, only the G→A change would be introduced into the genome of some cells. Alternatively, a longer stretch of DNA may be copied (including both G→A and C→T modifications) but after annealing of the newly synthesized strand with the complementary WT strand, heteroduplex DNA may be formed and mismatch repair mechanisms may correct the C→T change back to the original sequence (Harmsen et al., 2018; Gallagher et al., 2020). Either of these alternative modes of DNA repair would explain the recovery of colonies showing only the G→A edit.
Precise co-editing of the PPX1 and FTSY genes
If in asynchronously grown Chlamydomonas only a small subpopulation of cells is receptive to DSB repair by homology-directed mechanisms, selection for an HDR-based editing event (such as PPX1 editing) may facilitate the recovery of simultaneous HDR-mediated edits at other loci. To test this hypothesis, we attempted co-editing of PPX1 and FTSY by co-electroporation of the respective CRISPR/Cas9 RNPs and template ssODN donors. Electroporated cells were selected on agar plates containing oxyfluorfen. Out of three independent experiments, we isolated nine (4.0%) pale green colonies, with alterations in the FTSY gene, among 223 oxyfluorfen-resistant colonies (Supplemental Table S4). However, only three (1.3%) of these colonies displayed changes in the FTSY sequence (Figure 3A) consistent with precise, scarless editing by HDR. In these cases, a new NheI restriction enzyme site, as shown for a subset of these colonies (Figure 3B), and the designed stop codons within the FTSY coding sequence were incorporated at the intended site (Figure 3C, colonies 10-1b, 8-1, and 6-1). The other six colonies showed indels at or near the expected Cas9 cleavage site (Figure 3C; Supplemental Figure S5), although FTSY in one of these colonies did appear to be repaired by homology-directed mechanisms but including an unexpected single base-pair deletion (Figure 3C, colony 3-2), possibly resulting from replicative errors (see “Discussion”).
Figure 3.
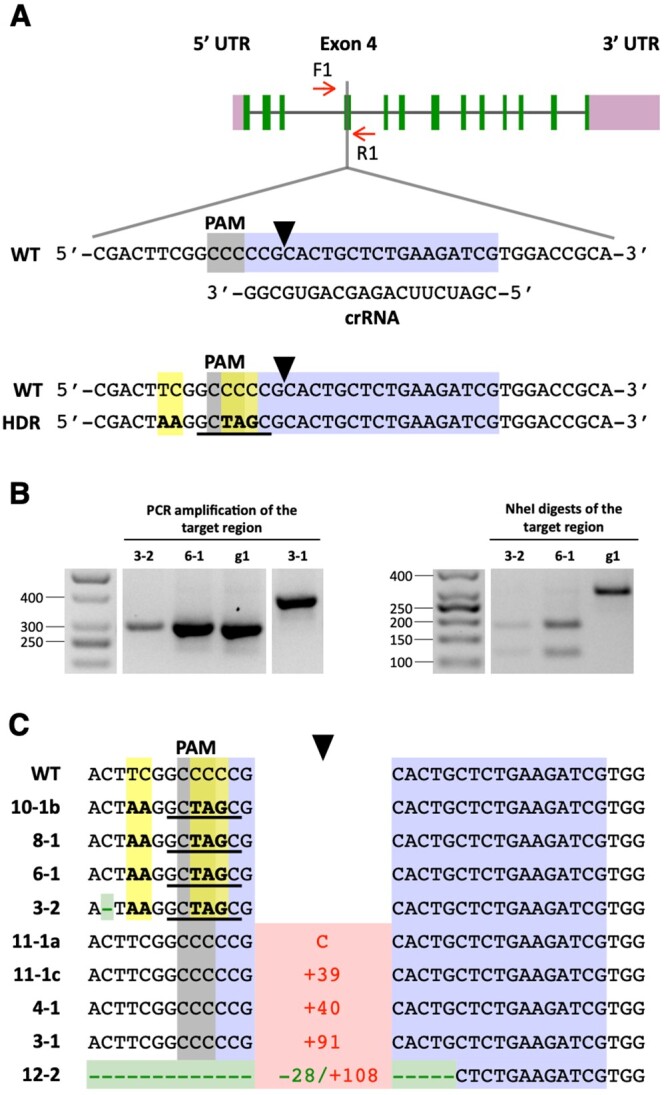
Co-editing of the FTSY gene in colonies selected for precise PPX1 editing. A, Schematic of the FTSY target region in exon 4. Color schemes and symbols are as described under Figure 1A. HDR of the DSB, using as template the transfected FTSY ssODN, is expected to introduce five base pair changes (highlighted in yellow) into the genome (bottom sequence). These sequence changes insert two stop codons (i.e. TAA and TAG) in the coding frame, destroy the PAM site, and create a new NheI restriction enzyme site (underlined in black). B, The FTSY target region of selected oxyfluorfen-resistant, pale green colonies was amplified by PCR (with primers F1 and R1) and the PCR products digested with NheI. The panels show representative reverse images of agarose resolved PCR products stained with ethidium bromide. The sizes of molecular weight markers are indicated in base pairs. g1, WT strain. C, DNA sequences of oxyfluorfen-resistant, pale green colonies, indicating alterations at the FTSY target site relative to the WT. Insertions, indicating type (C, cytosine) or number of base pairs, are depicted in red. Deletions, indicating type or number of base pairs, are depicted in green. Base substitutions are highlighted in yellow. Complete sequences for colonies exhibiting FTSY indels are shown in Supplemental Figure S5.
As discussed above, in our initial attempts at precise FTSY editing, by supplying a ssODN as template for HDR together with dsDNA encoding a selectable marker, out of 21 examined pale green colonies, none showed precise HDR. In contrast, when selecting first for PPX1 edited colonies on medium containing oxyfluorfen, out of nine examined pale green colonies, three showed precise FTSY HDR and an additional one showed FTSY sequence changes consistent with repair by homology-directed mechanisms, although including an unintended single base-pair deletion. Thus, in comparison to current gene editing methodology for walled Chlamydomonas strains, our observations suggest that a CRISPR/Cas9 RNP co-targeting strategy does facilitate the isolation of colonies precisely edited in a gene of interest lacking a selectable phenotype.
Precise co-editing of the PPX1 and WDTC1 genes
WDTC1 is a conserved eukaryotic polypeptide, containing WD40 and tetratricopeptide repeat domains. In flies and mammals, loss of function of WDTC1 results in an increase in adipocytes, fat accumulation, and obesity (Häder et al., 2003; Groh et al., 2016). The Arabidopsis thaliana homolog, Altered Seed Germination 2 (ASG2), also appears to have antiadipogenic functions, since asg2 knockout mutants produce seeds that have greater weight and increased fatty acid content than the WT (Ducos et al., 2017). The Chlamydomonas WDTC1 gene has not been characterized in any detail and the phenotype(s), if any, of a null mutant is unknown. Therefore, it provided a good system for testing the practicality of our co-targeting approach, because edited colonies in the gene of interest needed identification by bulk PCR analyses.
Cells of the g1 strain were co-electroporated with CRISPR/Cas9 RNPs and template ssODN donors targeting PPX1 and WDTC1. Electroporated cells were then selected on agar plates containing oxyfluorfen. Precise repair of the DSB caused by CRISPR/Cas9 (WDTC1) RNP, using a ssODN (Supplemental Table S2) as a homologous template, would eliminate the WDTC1 start codon (precluding synthesis of a full-length protein) and create a new BssHII restriction enzyme site for genotypic analyses (Figure 4A; Supplemental Figure S6A). Out of two independent experiments, we isolated nine (2.3%) colonies with alterations in the WDTC1 gene, among 395 oxyfluorfen-resistant colonies examined by PCR (Supplemental Table S5). However, only three (0.8%) of these colonies displayed changes in the WDTC1 sequence (Figure 4A) consistent with precise, scarless editing by HDR. In these cases, the new BssHII restriction enzyme site was incorporated into the genome, as shown for a subset of these colonies (Figure 4B), and the WDTC1 sequence around the start codon (i.e. AATG) was replaced with the intended sequence (i.e. GCGC; Figure 4C, colonies 3-15b, 1A-37, and 3B-48). In one colony, all changes expected for HDR did occur but we also observed unintended substitution of two base pairs (Figure 4C, colony 2B-19), possibly the result of errors during DNA synthesis (see “Discussion”). The remaining five colonies showed insertions at the expected Cas9 cleavage site (Figure 4C; Supplemental Figure S7).
Figure 4.
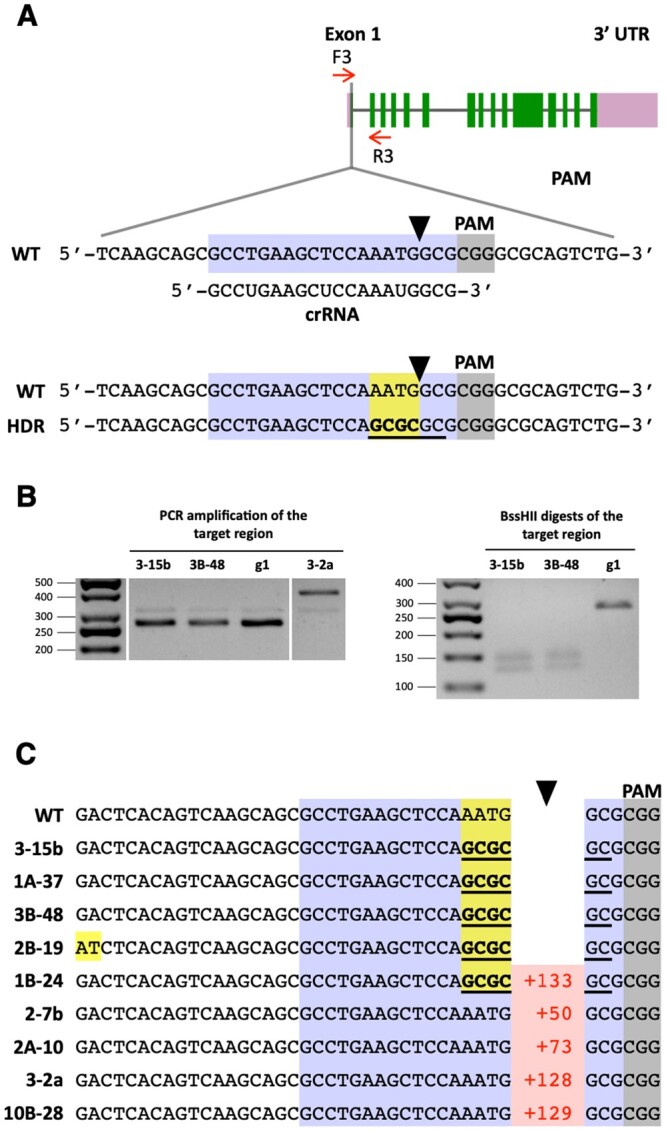
Co-editing of the WDTC1 gene in colonies selected for precise PPX1 editing. A, Schematic of the WDTC1 gene showing the target (highlighted in bluish purple) and PAM (highlighted in gray) sequences. Other symbols are as described under Figure 1A. HDR of the DSB, using as template the transfected WDTC1 ssODN, is expected to introduce four base-pair changes (highlighted in yellow) into the genome (bottom sequence). These sequence changes destroy the WDTC1 start codon and create a new BssHII restriction enzyme site (underlined in black). B, The WDTC1 target region of selected oxyfluorfen-resistant colonies was amplified by PCR (with primers F3 and R3) and the PCR products digested with BssHII. The panels show representative reverse images of agarose resolved PCR products stained with ethidium bromide. The sizes of molecular weight markers are indicated in base pairs. g1, WT strain. C, DNA sequences of oxyfluorfen resistant colonies showing alterations at the WDTC1 target site relative to the WT. Insertions, indicating number of base pairs, are depicted in red. Base substitutions are highlighted in yellow. Complete sequences for colonies exhibiting insertions at the WDTC1 gene are shown in Supplemental Figure S7.
For comparison purposes, we also attempted precise WDTC1 editing by electroporating g1 cells with CRISPR/Cas9 (WDTC1) RNP, a dsDNA PCR fragment containing the aphVIII transgene and the WDTC1 ssODN. Cells were selected on paromomycin containing medium. Out of 96 paromomycin resistant colonies examined by PCR, 2 (2.1%) showed alterations in the WDTC1 gene. However, both colonies had insertions of the aphVIII transgene at the Cas9 cleavage site, consistent with DSB repair by NHEJ mechanisms (Supplemental Figure S8, colonies 20 and 63). Although, in one case, SSTR appears to have started correctly, using the WDTC1 ssODN as a homologous repair template (see “Discussion”), but the event was resolved by NHEJ on the left side of the cleavage site (Supplemental Figure S8, colony 63). Thus, as for the FTSY gene, we did not recover any colony showing precise editing of WDTC1 by using established gene editing methodology for walled Chlamydomonas strains.
Phenotypic characterization of WDTC1 edited mutants
To gain insight on WDTC1 function in Chlamydomonas, we examined growth and the accumulation of nonpolar lipids in a subset of edited mutants. We chose to analyze two precise HDR edited mutants (i.e. 3-15b and 3B-48) and three insertional mutants (i.e. 3-2a, 20, and 63). Semi-quantitative reverse transcriptase (RT)-PCR analyses of exon–exon junctions close to the beginning (i.e. exon2/exon3) or to the end (i.e. exon 13/exon14) of the WDTC1 coding sequence (Figure 5A) revealed that the HDR edited mutants had similar or slightly higher WDTC1 transcript abundance than the WT, when cells were grown under either mixotrophic or phototrophic conditions (Figure 5, B and C). In contrast, the insertional mutants displayed reduced WDTC1 transcript abundance, albeit to different and somewhat variable degrees, relative to the WT (Figure 5, B and C).
Figure 5.
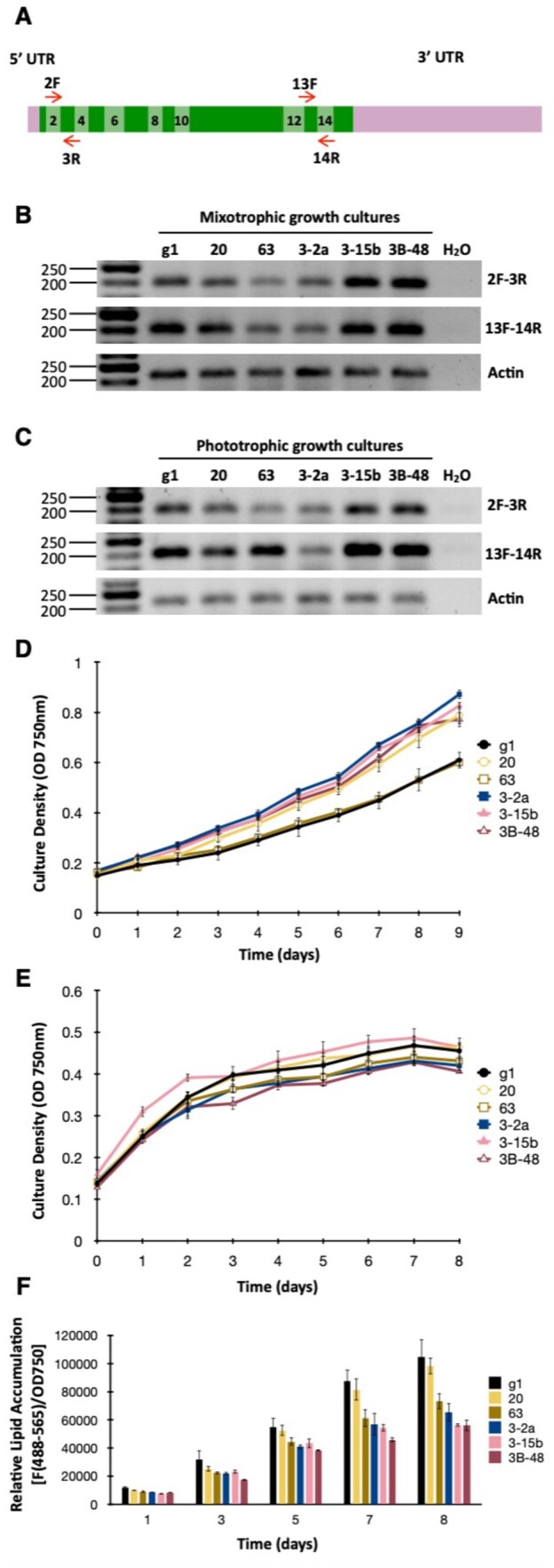
Phenotypic characterization of WDTC1 mutants generated by CRISPR/Cas9 editing. A, Schematic of the WDTC1 transcript. Exons are depicted as boxes in two alternating shades of green and even exons are numbered. Short red arrows indicate the primers used for semi-quantitative RT-PCR analyses. B, WDTC1 transcript abundance examined by semiquantitative RT-PCR in cells grown under mixotrophic conditions. The panels show representative reverse images of agarose resolved RT-PCR products stained with ethidium bromide. Amplification of the Actin transcript was used for normalization purposes. The sizes of molecular weight markers are indicated in base pairs. g1, WT strain. C, WDTC1 transcript abundance examined by semiquantitative RT-PCR in cells grown under phototrophic conditions. D, Growth of the indicated strains under phototrophic conditions in nutrient replete minimal medium. Values shown are the average of three biological replicates ± sd. E, Growth of the indicated strains under mixotrophic conditions in nitrogen deprived medium (TAP-N). Values shown are the average of three biological replicates ± sd. F, Nonpolar lipid accumulation in the indicated strains cultured under mixotrophic conditions in TAP medium lacking nitrogen. Nonpolar lipid content was estimated by staining with the lipophilic fluorophore Nile Red and measuring fluorescence (excitation at 488 nm; emission at 565 nm) in a multiwell plate reader. Nile Red fluorescence was normalized to cell density (determined as absorbance at 750 nm) and expressed in arbitrary units. Values shown are the average of three biological replicates ± sd.
Interestingly, under phototrophic conditions, four of the edited mutants appeared to grow at a somewhat faster rate than the WT (Figure 5D), whereas the growth of insertional mutant 63 was nearly identical to that of the WT. Since WDTC1 homologs have been implicated in antiadipogenic functions in several eukaryotes (Häder et al., 2003; Groh et al., 2016; Ducos et al., 2017), we also examined the performance of the Chlamydomonas mutants with respect to triacylglycerol biosynthesis. This alga has been shown to accumulate significant amounts of triacylglycerol when subject to nitrogen deprivation (Siaut et al., 2011; Msanne et al., 2012; Kim et al., 2018). Thus, we first analyzed the growth/survival of the strains in the absence of nitrogen (in medium supplemented with acetate as a carbon source). Under these conditions, none of the edited mutants differed substantially in growth from the WT (Figure 5E). To evaluate neutral lipid accumulation, Chlamydomonas cells were examined by fluorometry after staining with the nonpolar lipid fluorophore Nile Red (Msanne et al., 2012). As expected, the WT strain showed substantial accumulation of nonpolar lipids, over time, under nitrogen starvation in the presence of acetate (Figure 5F). However, four of the Chlamydomonas WDTC1 edited mutants showed reduced nonpolar lipid accumulation relative to the WT (Figure 5F); only insertional mutant 20 behaved similarly to the WT strain.
Given that an antiadipogenic role of WDTC1 was not supported by our experiments, defining the function(s) of WDTC1 in Chlamydomonas will require further work. However, in the context of CRISPR/Cas9-generated mutants, we observed that the phenotypes of the two HDR edited mutants (i.e. 3-15b and 3B-48) and of the insertional mutant isolated during co-editing with PPX1 (i.e. 3-2a) were consistently similar in all the analyses that we performed. In contrast, the aphVIII insertional mutants (i.e. 20 and 63) showed divergent behavior under certain conditions. For instance, in comparison to the other edited mutants, mutant 63 differed in growth under phototrophic conditions whereas mutant 20 differed in nonpolar lipid accumulation under nitrogen deprivation. Since the WDTC1 start codon or the proper translation frame were disrupted in all five examined mutants, these idiosyncratic phenotypic variations cannot be explained based exclusively on loss-of-function of the WDTC1 gene. In Southern blot analyses of mutants 20 and 63, using the aphVIII coding sequence as a probe, we only detected single genomic insertions of the selectable transgene (Supplemental Figure S9); and the fragment sizes were consistent with those predicted by transgene integration at the WDTC1 Cas9 cleavage site (Supplemental Figure S8). However, genomic insertion of a very short segment of the aphVIII coding sequence or of a fragment from the regulatory sequences controlling transgene expression would not be detected by this approach. While the actual reason(s) for the described phenotypic discrepancies remains elusive, our observations emphasize the need to carefully examine CRISPR/Cas9-generated insertional mutants since it is conceivable that integration of short dsDNA fragments at off-target sites in the Chlamydomonas genome may cause unintended phenotypic alterations.
Precise editing of the ALS1 gene, an alternative selectable marker
For studies of groups of genes involved in specific enzymatic pathways or cellular functions, the ability to sequentially produce in the same strain (i.e. in the same genetic background) precise mutations in multiple genes of interest can be a powerful tool. However, with the strategy, we put forward in the present communication, each additional round of mutagenesis with a CRISPR/Cas9 RNP and an ssODN donor targeting a specific gene would require the simultaneous targeting of a different gene that is capable of being converted into a selectable marker. We have already shown that the acetolactate synthase (ALS1) gene is an attractive target for CRISPR/Cas9 RNP-directed mutagenesis since a single amino acid change from lysine 257 to threonine (caused by a single base-pair substitution, changing codon AAG to ACG) results in strong resistance to the herbicide sulfometuron methyl (SMM; Kovar et al., 2002; Jiang and Weeks, 2017). To test the efficiency of ALS1 as a selectable marker gene as well as an alternative transfection protocol relying on exponential-wave electroporation, cells of the walled CC-124 strain were electroporated with a CRISPR/Cas9 (ALS1) RNP and a ssODN (Supplemental Table S2), overlapping the Cas9 cleavage site and designed to serve as template for HDR. Precise DSB repair by homology-directed mechanisms would introduce the A→C mutation within the ALS1 coding sequence as well as a nearby, functionally silent, C→T mutation that creates a new EcoRV restriction enzyme site for genotypic analyses (Figure 6A; Supplemental Figure S10). Electroporated cells were selected on agar plates containing SMM.
Figure 6.
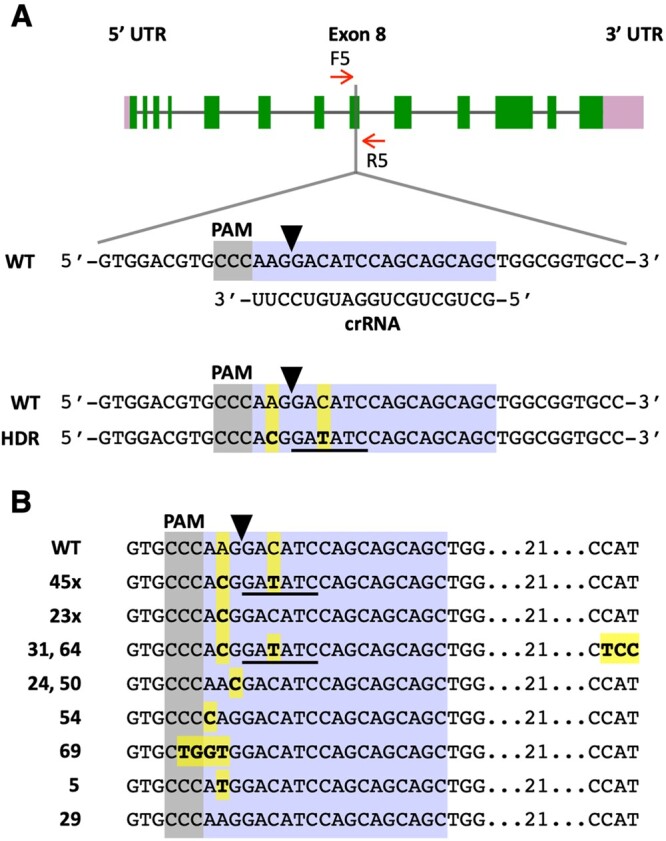
Precise editing of the ALS1 gene. A, Schematic of the ALS1 gene showing the target (highlighted in bluish purple) and reverse complement PAM (highlighted in gray) sequences. A black arrowhead indicates the Cas9 cleavage site. HDR of the DSB, using as template the transfected ALS1 ssODN, is expected to introduce two base pair changes (highlighted in yellow) into the genome (bottom sequence). These sequence changes cause an amino acid substitution (K257T) in the encoded ALS enzyme and create a new EcoRV restriction enzyme site (underlined in black). B, DNA sequences of SMM resistant colonies showing alterations at the ALS1 target site relative to the WT. Base substitutions are highlighted in yellow. Figures followed by an X (i.e. 45× and 23×) indicate the number of colonies of each DNA type analyzed by sequencing (complete sequences for each colony are shown in Supplemental Figure S11). Other numbers indicate the colony names.
Out of 483 SMM resistant colonies obtained in the treatment with CRISPR/Cas9 (ALS1) RNP and ssODN donor, 76 were examined by PCR amplification of the target site and sequencing of the PCR products (Supplemental Table S6). Forty-seven (61.8%) colonies displayed the expected (i.e. A→C and C→T) sequence changes, although in two cases (i.e. colonies 31 and 64) with additional sequence alterations (Figure 6B; Supplemental Figure S11A; Supplemental Table S6). Twenty-three (30.2%) colonies only showed the A→C change, which is necessary to confer herbicide resistance (Figure 6, B; Supplemental Figure S11, B; Supplemental Table S6). These results are similar to those obtained in experiments with the PPX1 gene (see above) and suggest that ∼92% of the SMM-resistant colonies are consistent with editing by HDR (Supplemental Table S6), using as homologous template the electroporated ssODN donor. Five additional colonies showed mutations causing substitution of lysine 257 for alternative amino acids (other than threonine; Figure 6B, colonies 24, 50, 54, 69, and 5; Supplemental Table S6), which apparently also confer resistance to SMM. In two colonies, a single nucleotide change occurred at the Cas9 cleavage site (Figure 6B, colonies 24 and 50; Supplemental Table S6), possibly as a result of errors during NHEJ. The other three colonies displayed single or a few nucleotide substitutions close to the cleavage site (Figure 6B, colonies 54, 69, and 5; Supplemental Table S6) but the mechanism(s) responsible for these changes is not clear. One colony out of the 76 examined had a WT target sequence (Figure 6B, colony 29), suggesting a potential spontaneous mutation conferring SMM resistance elsewhere in the ALS1 gene.
Precise co-editing of the PPX1 and FTSY genes in cells treated with autolysin to remove their cell walls
The cell wall appears to pose a significant barrier for the introduction of macromolecules into Chlamydomonas (Jeon et al., 2017), and this may be the reason for the poor uptake of CRISPR/Cas9 RNPs by electroporated walled cells (Supplemental Figure S3). Indeed, the efficiency of targeted insertional mutagenesis induced by electroporation of CRISPR/Cas9 RNPs was improved substantially after removal of the cell wall by autolysin treatment (Picariello et al., 2020). Thus, we also attempted co-editing of PPX1 and FTSY in the g1 strain treated with autolysin prior to delivery of the respective CRISPR/Cas9 RNPs and template ssODN donors by co-electroporation. As before, electroporated cells were selected on agar plates containing oxyfluorfen (Figure 7A). In one experiment, the number of oxyfluorfen-resistant colonies (indicative of PPX1 precise editing) obtained with autolysin-treated cells increased by approximately five-fold relative to those obtained with walled cells (Supplemental Table S7, Experiment 1). However, in a replicate experiment with a less effective autolysin preparation, treated cells only displayed ∼20% increase in the number of recovered oxyfluorfen-resistant colonies (Supplemental Table S7, Experiment 2). In both experiments, the proportion of pale green colonies (with a presumably edited FTSY gene) relative to the number of oxyfluorfen resistant colonies remained between ∼4% and 6%, independently of whether the RNPs were introduced into walled or autolysin treated cells (Supplemental Table S7 and Figure 7A). In the first experiment, we also verified FTSY editing in most pale green colonies by PCR amplification of the target region followed by assessment of PCR product size and its cleavage by the NheI endonuclease (indicative of precise editing that creates a new restriction enzyme site). Many of the tested pale green colonies (∼3% of the oxyfluorfen-resistant colonies) showed a PCR product of the expected size digestable by NheI (Figure 7B; Supplemental Table S7). The rest of the examined pale green colonies showed PCR products of abnormal size, mainly larger than expected (Figure 7C), suggesting repair by NHEJ with insertion of additional sequences (Supplemental Table S7). Thus, our limited set of experiments suggest that cell wall removal does enhance the overall efficiency of CRISPR/Cas9-mediated editing in WT Chlamydomonas, as previously reported (Picariello et al., 2020), presumably by improving the delivery of electroporated RNPs. The co-editing efficiency of an unselectable gene of interest (i.e. FTSY edited colonies as percentage of PPX1 edited colonies) remained at a similar rate to that observed in walled cells. However, the effectiveness of autolysin preparations appears to be quite variable and, as suggested by Picariello et al. (2020), it may be necessary to test for successful cell wall removal by treatment with detergent before using cells for CRISPR/Cas9 mediated editing.
Figure 7.

Co-editing of the FTSY and PPX1 genes in g1 cells treated with autolysin prior to RNPs electroporation. A, Cells co-transfected with the CRISPR/Cas9 (FTSY) RNP, CRISPR/Cas9 (PPX1) RNP and the corresponding ssODN donor DNAs were spread on TAP agar plates containing oxyfluorfen. Representative plates (7A and 8B) are shown after 15 d of incubation under continuous light. To facilitate the identification of pale green colonies (expected to be FTSY edited), images were manipulated to reduce shadows and increase brightness (as shown on the right). Colonies identified as pale green are indicated by black arrowheads. B, The FTSY target region of selected pale green colonies was amplified by PCR (with primers F1 and R1, Figure 3A) and the PCR products digested with NheI. The panel shows a representative reverse image of agarose resolved DNA fragments stained with ethidium bromide. g1, WT strain. C, For a subset of pale green colonies, amplification of the FTSY target region revealed PCR products of larger size, suggestive of insertions at the Cas9 cleavage site, relative to the WT sequence. The panel shows a representative reverse image of agarose resolved DNA fragments stained with ethidium bromide.
Discussion
As mentioned earlier, targeted disruption of nuclear genes, mediated by RNA-programmable SSNs, has become a practical method in Chlamydomonas (Baek et al., 2016; Shin et al., 2016; Ferenczi et al., 2017; Greiner et al., 2017; Jiang and Weeks, 2017; Shamoto et al., 2018; Guzmán-Zapata et al., 2019; Angstenberger et al., 2020; Cazzaniga et al., 2020; Dhokane et al., 2020; Kang et al., 2020; Kim et al., 2020; Park et al., 2020; Picariello et al., 2020). In contrast, precise gene editing still occurs at relatively low frequencies or it is limited to specific (cell wall-less) strains or experimental approaches (Ferenczi et al., 2017; Greiner et al., 2017; Jiang and Weeks, 2017). In Chlamydomonas, as observed in land plants and other eukaryotes (Gallagher and Haber, 2018; Kan et al., 2017; Paix et al., 2017; Boel et al., 2018; Richardson et al., 2018; Sansbury et al., 2019; Capdeville et al., 2020; Gallagher et al., 2020), the repair of DSBs induced by CRISPR/Cas9 RNPs likely occurs by several distinct pathways, partly determined by the repair machinery expressed in each cell, the complexity of the DSB, and the nature of the DNA molecules involved in the repair. As described for some mammalian cell lines (Shy et al., 2016; Mitzelfelt et al., 2017), only a small subset of the population, in asynchronously grown Chlamydomonas, may be capable of HDR, possibly associated with being in a certain phase of the cell cycle (Angstenberger et al., 2020). In addition, the cell wall appears to pose a substantial barrier for the introduction of macromolecules into Chlamydomonas cells (Jeon et al., 2017; this work). Indeed, when precise editing of walled strains is attempted by co-electroporation of a CRISPR/Cas9 RNP, a PCR fragment or a plasmid encoding a transgene expressing an antibiotic resistance gene (for selection of the small fraction of cells taking up the editing components) and a donor DNA (to be used as homologous template for HDR), most Chlamydomonas cells appear to repair the induced DSBs by NHEJ pathways, based on the sequence analyses of recovered colonies (Greiner et al., 2017; Sizova et al., 2021; this work). Such repair often involves the undesirable incorporation of intact or frequently jumbled antibiotic resistance gene sequences at the site of Cas9 DNA cleavage (Greiner et al., 2017; Sizova et al., 2021; this work).
Given these constraints, we tested whether co-targeting two genes by electroporation of CRISPR/Cas9 RNPs and template ssODN donors would facilitate the recovery of precise edits in a gene of interest after selecting for precise editing of the other gene (corresponding to a selectable marker). Conceptually, this strategy is expected to select for cells capable of taking up the editing components and expressing the machinery required for HDR after transfection. We first chose PPX1, encoding Protox, as a selectable marker since a single base-pair change (G→A) within the protein coding sequence confers resistance to porphyric herbicides such as oxyfluorfen (Randolph-Anderson et al., 1998; Brueggeman et al., 2014). Selecting electroporated cells on oxyfluorfen containing medium allowed the recovery of colonies edited in two co-targeted genes, namely FTSY and WDTC1. On average, between 2% and 5% of oxyfluorfen-resistant colonies showed sequence alterations in the co-targeted genes, and at least one third of these colonies showed precise, scarless editing mediated by HDR. These observations suggest that our co-editing approach does facilitate the isolation of colonies precisely edited in genes of interest in walled Chlamydomonas. Moreover, the co-editing approach avoids the incorporation of exogenous selectable marker gene fragments at CRISPR/Cas9 cleavage sites, which makes it difficult to obtain scarless gene edits, as observed in previous gene editing protocols (Shin et al., 2016; Greiner et al., 2017; Kang et al., 2020; Picariello et al., 2020) and once more documented in this study (Figure 1 and Supplemental Figure S2).
The heart of the scheme presented here is to expedite the recovery of a specific mutation in any Chlamydomonas gene, in the absence of a selectable phenotype for that mutation. Selection of the desired mutant is based on the ability to simultaneously deliver into cells two pairs of components: a CRISPR/Cas9 RNP and ssODN donor designed to modify the target gene of interest along with a second pair consisting of CRISPR/Cas9 (PPX1) RNP and a ssODN donor designed to convert the PPX1 gene into a selectable marker gene. Our isolation of oxyfluorfen-resistant colonies containing precise, scarless mutations in the FTSY and WDTC1 genes is proof of concept for this approach. Since each additional mutation that an investigator wishes to incorporate into a strain requires another gene that can be converted to a selectable marker, we also demonstrated that the ALS gene could serve as an additional selectable marker. Preliminary experiments indicate that the argininosuccinate lyase (ARG7) gene, of the arginine auxotroph arg7-8, could also be used as a selectable marker since conversion to the WT sequence can create easily selected autotrophic cells (Jiang and Weeks, 2017; Jiang et al., 2017). Other endogenous genes could potentially be edited by single base-pair substitutions to become selectable markers, such as those encoding phytoene desaturase, conferring resistance to the bleaching herbicide norflurazon (Brueggeman et al., 2014; Suarez et al., 2014), and cytosolic ribosomal protein S14 (CRY1), conferring resistance to the antibiotics cryptopleurine and emetine (Nelson et al., 1994; Neupert et al., 2009). These examples of endogenous genes with demonstrated or potential utility as selectable markers suggest it may soon be possible to attempt the creation of Chlamydomonas cell lines containing precise mutations in multiple target genes, directed at aiding the study of complex cellular functions or metabolic pathways of academic or biotechnological interest. We also note that our strategy is “transgene-free” since, in the instances of precise gene editing, exogenous DNA is not expected to be incorporated into the genome, although potential off-target effects mediated by CRISPR/Cas9 RNPs remain to be examined.
If the presence of transgenes is not a concern, the Hegemann laboratory (Sizova et al. 2013; Greiner et al., 2017. Sizova et al., 2021) demonstrated that an antibiotic resistance gene (i.e. an aphVIII transgene), which had been deliberately inactivated by a simple sequence modification, can be introduced by transformation into the Chlamydomonas nuclear genome. Using this transgenic strain, they subsequently employed CRISPR/Cas9-mediated editing to modify the mutant aphVIII gene and restore antibiotic resistance. A similar approach, using inactivated versions of the many antibiotic resistance genes effective in Chlamydomonas, has the potential to greatly broaden the spectrum of editable genes available for selection purposes in co-targeting experiments.
Sequence analyses of the recovered Chlamydomonas colonies provided some insights regarding the mechanism of DSB repair. We note that beyond the alterations reported in close proximity to the Cas9 cleavage site, we did not observe any other changes in ∼125–150 bp of flanking DNA sequences on either side of the expected cleavage site. In mammalian cells, DNA repair of various lesions with donor ssODNs can occur through two main pathways: synthesis-dependent strand annealing or single-strand DNA incorporation, the latter involving the physical integration of the ODNs into the genome (Kan et al., 2017). However, in both animals and fungi, the repair of Cas9-induced DSBs with homologous ssODNs appears to occur predominantly by the synthesis-dependent pathway, often referred to as SSTR (Radecke et al., 2006; Gallagher and Haber, 2018; Kan et al., 2017; Paix et al., 2017; Boel et al., 2018; Harmsen et al., 2018; Richardson et al., 2018; Sansbury et al., 2019; Gallagher et al., 2020). In SSTR, ssODNs are used for the synthesis of complementary DNA, rather than being integrated into the genome, and the process is polarity sensitive and dependent, unlike other homologous repair mechanisms, on the Fanconi anemia pathway (Gallagher and Haber, 2018; Kan et al., 2017; Paix et al., 2017; Boel et al., 2018; Harmsen et al., 2018; Richardson et al., 2018). As shown diagrammatically in Supplemental Figure S1, for the proposed repair of FTSY, a DSB generated by Cas9 is expected to be resected to yield 3′-overhangs on both sides of the DSB. However, only one of the 3′-overhangs can pair with the homologous ssODN donor, which confers polarity to the repair, and primes the synthesis of a complementary DNA strand. Bridging of the DSB is eventually accomplished when the newly synthesized strand is displaced from the ssODN donor and anneals with the complementary strand at the locus. Finally, DNA polymerases and ligases complete the DSB repair. As proposed in mammalian cells (Kan et al., 2017; Harmsen et al., 2018), the likely erosion of the ends of 3′-overhangs would allow the introduction of edits very close to the DSB by de novo synthesis of both DNA strands (i.e. by gap filling; Supplemental Figure S1), avoiding the formation of heteroduplex DNA and interference from DNA mismatch repair mechanisms.
In land plants, ssODNs have been used for precise gene editing, as templates for the repair of DSBs generated by CRISPR/Cas9, but very few edited plants/calli have been analyzed by sequencing precluding any inference on the repair mechanism (Shan et al., 2013; Svitashev et al., 2015; Sauer et al., 2016). In our current work with Chlamydomonas, several lines of evidence, taken together, support the interpretation that SSTR is responsible for the observed HDR. First, synthesis of a DNA strand complementary to a ssODN donor begins at the DSB and, since repair DNA polymerases are not very processive (Rodgers and McVey, 2016), nucleotide changes closer to the DSB are more likely to be incorporated into the genome (Paquet et al., 2016; Kan et al., 2017; Paix et al., 2017; Boel et al., 2018). Additionally, since the locus strand complementary to the newly synthesized strand may often be somewhat eroded at its 3′-end (Dorsett et al., 2014; Harmsen et al., 2018), the generation of heteroduplex DNA (which could be corrected back to the WT sequence by DNA mismatch repair mechanisms) may be avoided closer to the DSB. These predictions are consistent with the greater incorporation efficiency of G→A (closer to the DSB) versus C→T (more distant from the DSB) in edited PPX1 (Figure 2C). Second, SSTR is unidirectional since only one of the 3′-overhangs at the DSB can pair with the ssODN donor to synthesize a complementary strand (Gallagher and Haber, 2018; Paix et al., 2017; Boel et al., 2018). In Chlamydomonas repair polarity is best supported by the analysis of colonies where one side of the DSB appears to show integration of donor DNA by HR (i.e. a crossover) and the other side by NHEJ. Close inspection of the sequences at the target site in these colonies (for both FTSY and WDTC1) indicates strict polarity (Supplemental Figure S2, colony 13b; Supplemental Figure S5, colony 4-1; Supplemental Figure S7, colony 2A-10; Supplemental Figure S8, colony 63). The DSB side with apparent integration by a crossover event is always the side that would prime DNA synthesis using the ssODN donor as template. A more plausible explanation is that SSTR started correctly, with the synthesis of a DNA strand complementary to the ssODN donor, but the repair was eventually resolved by NHEJ resulting in templated insertions.
Third, repair DNA polymerases, such as those involved in the synthesis of a strand complementary to the ssODN donor, are error prone and cause base pair substitutions and frameshift mutations (Rodgers and McVey, 2016; Gallagher and Haber, 2018; Richardson et al., 2018; Gallagher et al., 2020). This is consistent with the recovery of several Chlamydomonas colonies where the intended nucleotide modifications in the FTSY, WDTC1, or ALS1 genes did occur, but additional sequence changes typical of replicative errors (such as single base-pair deletions or substitutions) were also observed (Figure 3C, colony 3-2; Figure 4C, colony 2B-19; Figure 6B, colonies 31 and 64). Similar findings have already been reported for CRISPR/Cas12a editing with ssODN templates in cell wall-less Chlamydomonas strains (Ferenczi et al., 2017). Fourth, ssODN-directed DNA synthesis is prone to template switching between donor DNA molecules, an event apparently dependent on regions of microhomology (Rodgers and McVey, 2016; Paix et al., 2017; Boel et al., 2018). This appears to have happened at least in one case in Chlamydomonas, partly copying two molecules of WDTC1 ssODN donor DNA (Supplemental Figure S6B and Supplemental Figure S7, colony 1B-24). In addition, DNA polymerase theta (POLQ) has recently been demonstrated to be required for the repair of CRISPR/Cas-induced DSBs using ssODNs as donor DNA in Chlamydomonas (Sizova et al., 2021). This polymerase participates in a microhomology-mediated DSB repair pathway, referred to as theta mediated end joining and characterized by several features consistent with our observations. The repair is error-prone, it often involves “templated insertions,” which result from DNA synthesis initiated from the free 3′-end of a resected DSB and template switching is fairly common (Schimmel et al., 2019; Brambati et al., 2020; Sizova et al., 2021; Zahn et al., 2021).
Thus, our current strategy involving ssODNs as donor DNA will largely be useful for the introduction of relatively short edits, for instance to investigate the function of specific amino acid residues in proteins expressed in their endogenous context. In mammalian cells, it has been proposed that Cas9-induced DSB repair using ssODN donors mimics some substrate of the Fanconi anemia pathway, such as a stalled replication fork (Richardson et al., 2018). This diverts DSB repair through SSTR, which results in short conversion tracts prone to replicative errors, as also appears to be the case in Chlamydomonas.
In summary, our co-editing approach allows the recovery of precise sequence edits of genes of interest at a practical frequency, often requiring the PCR analysis of little more than 100 herbicide-resistant colonies (at least for the genes currently tested). The methods described are likely applicable to most WT, autolysin-treated WT, or wall-less strains of Chlamydomonas and, potentially, other microalgal species. The co-targeting strategy employed in sequential rounds of gene editing also offers the possibility to create one cell line with precise gene modifications in multiple genes that affect particular metabolic pathways or cellular mechanisms.
Materials and methods
Strains, culture conditions and lipid accumulation
Chlamydomonas (C. reinhardtii) strains CC-620 (nit1, nit2, mt+), CC-621 (nit1, nit2, mt-), g1 (CC-5415; nit1, agg1, mt+), CC-124 (nit1, nit2, agg1, mt-; Chlamydomonas Resource Center, https://www.chlamycollection.org), or derived edited mutants were used in all reported experiments. Unless noted otherwise, cultures were incubated under continuous illumination (150 µmol m−2 s−1 photosynthetic photon flux density) on an orbital shaker (190 rpm) at 25°C and ambient CO2 levels. For transfection experiments, cells were precultured, after inoculation from one-week-old plates, to an optical density of ∼0.4 at 750 nm in Tris–Acetate–Phosphate (TAP) medium (Harris, 1989) supplemented with 1 μg mL−1 cyanocobalamin (vitamin B12), to enhance Chlamydomonas thermal tolerance (Xie et al., 2013). A one-tenth aliquot was then transferred into fresh medium and grown to middle logarithmic phase (∼1 to 2 × 106 cells mL−1). Prior to electroporation, cells were collected by centrifugation and resuspended in TAP medium containing 40 mM sucrose and 1 μg mL−1 cyanocobalamin to a final density of ∼2.2 × 108 cells mL−1. For growth experiments, cells were pre-cultured as described before and then inoculated into minimal (Sueoka, 1960) medium (phototrophic conditions) or TAP medium (mixotrophic conditions). Culture growth was examined by measuring daily absorbance at 750 nm. For lipid accumulation analyses, cells were inoculated into TAP medium lacking nitrogen and 200 μL-aliquots were tranferred daily to a multi-well plate and mixed with Nile Red (Sigma, 72,485) to a final concentration of 1 μg mL−1 (Msanne et al., 2012). Nile Red fluorescence (excitation at 488 nm; emission at 565 nm) was measured in a multi-well plate reader (Synergy H1, Biotek), normalized to cell density (determined as absorbance at 750 nm) and expressed in arbitrary units (Kim et al., 2018). Autolysin preparation by mating of CC-620 and CC-621 and cell treatment of the g1 strain prior to electroporation were carried out as previously described (Picariello et al., 2020).
crRNA, tracrRNA, ssODN donors, and aphVIII transgenic DNA
The 19- or 20-nt guide sequences, corresponding to the target-specific protospacer regions (Supplemental Table S8, were designed using the Cas-Designer (www.rgenome.net/cas-designer) or Chopchop (https://chopchop.cbu.uib.no) websites. Each CRISPR RNA (crRNA) was synthesized as a custom, chemically modified 35- or 36-nt oligo (containing 16 additional, common nucleotides for annealing to the tracrRNA) by Integrated DNA Technologies (IDT). We used a commercially available trans-acting CRISPR RNA (tracrRNA; IDT, 1072534 Alt-R® CRISPR-Cas9 tracrRNA). ssODN donors (Supplemental Table S2) were designed overlapping the CRISPR/Cas9 cleavage site, but with different lengths of homology arms (from 30 to 70 nt), and synthesized as Ultramer DNA Oligos (IDT). The paromomycin resistance cassette (aphVIII transgene) was amplified by PCR from the pSI103 plasmid (Sizova et al., 2001).
Preparation of CRISPR/Cas9 RNPs
CRISPR/Cas9 crRNA and tracrRNA (each at ∼53 μM final concentration) in 1.2× NEB 3.1 buffer (New England Biolabs, B7203S) were annealed by placing a microcentrifuge tube in a beaker with water heated to 96°C and then allowed to cool slowly to room temperature. To assemble the RNP complex, one volume of Alt-R® S.p.Cas9 Nuclease V3 (IDT, 1081059) was mixed with 3.5 volumes of annealed crRNA/tracrRNA and incubated at 37°C for 20 min. In this mixture, the guide RNA is present at approximately three-fold molar excess relative to the Cas9 protein and, if saturation binding is achieved, the RNP final concentration would be ∼13.6 μM.
Chlamydomonas transfection
The initial experiments (Supplemental Tables S1 and S3) were carried out following the method of Greiner et al. (2017). In order to improve the editing frequency, we subsequently introduced several modifications that resulted in an optimized protocol. For single gene editing experiments, 2.3 μL of the CRISPR/Cas9 (FTSY) RNP [or the CRISPR/Cas9 (WDTC1) RNP] were mixed with 1.5 μL of the PCR product (∼1.0 μg of dsDNA in TE buffer) encoding the aphVIII transgene (±225 pmol of the corresponding ssODN donor). For co-editing experiments, the CRISPR/Cas9 (PPX1) RNP and the CRISPR/Cas9 (FTSY) RNP or the CRISPR/Cas9 (WDTC1) RNP were first mixed in a 1–3 ratio (i.e. a three-fold molar excess of the RNP targeting the unselected gene of interest). Three microliters of the combined RNPs were then added to 1.5 μL of pre-mixed (in TE buffer) PPX1 (at 50 μM) and FTSY (or WDTC1; at 150 μM) ssODN donors. For each electroporation, an aliquot of 36 μL of cells (∼7.9 × 106 cells mL−1), resupended in TAP medium containing 40 mM sucrose and 1 μg mL−1 cyanocobalamin, was mixed with 3.8 μL of the RNP/aphVIII dsDNA/ssODN or 4.5 μL of the combined RNPs/ssODNs and placed in an electroporation cuvette with a 2 mm gap. Transfection was performed with a NEPA21 electroporator (Nepa Gene Co.), with adjusted parameters relative to a published protocol (Yamano et al., 2013). Prior to elecroporation the impedance was adjusted to 0.25–0.28 kΩ by adding more resuspended cells or withdrawing from the mixture in the cuvette (stepwise, 3 μL at a time). Electroporation was carried out by using two 6-ms/250-V poring pulses at 50-ms intervals and a decay rate of 40%, followed by five 50-ms/20-V polarity-exchanged transfer pulses at 50-ms intervals and decay rate of 40%. Electroporation of autolysin treated g1 cells was performed with two 4-ms/200-V poring pulses at 50-ms intervals and a decay rate of 40%, followed by five polarity-exchanged transfer pulses as before. Immediately after electroporation, the cells were diluted in 500 μL of TAP medium containing 40 mM sucrose and 1 μg mL−1 cyanocobalamin and transferred to a 1.5-mL microcentrifuge tube.
Post-electroporation heat shock, cell recovery and plating
Electroporated cells were placed on an orbital shaker (50 rpm) under dim lights for 3 h at room temperature. Cells were then heat shocked at 39°C for 30 min, with gentle agitation, in a water bath. Subsequently, cells were incubated again on the orbital shaker for ∼40 h. After completion of this recovery period, cells from each electroporation were spread on two TAP agar plates containing the appropriate selective agent (12 μg mL−1 paromomycin or 0.18 μM oxyfluorfen). The plates were incubated at room temperature under continuous light (∼100 µmol m−2 s−1 photosynthetically active radiation) until visible colonies appeared (usually about 2 weeks).
Testing the ALS1 gene as a selectable marker
Cells of the WT strain CC-124 were grown in TAP medium, in 5% (v/v) CO2 at 25°C with shaking and continuous light, to mid-log phase and then concentrated to ∼5 × 107 cells mL−1 in TAP medium containing 60 mM sucrose. A mixture consisting of ∼100 pmol Cas9 (New England Biolabs, M0386M) and ∼850 pmol of ALS1 crRNA (Supplemental Table S8 preannealed with an equimolar amount of tracrRNA, in a total volume of 30 µL in NEB Cas9 buffer, was allowed to incubate at 37°C for 30 min. Then, 1 µL (∼800 pmol) of the ALS1 ssODN donor (Supplemental Table S2) was added, just before cell transfection. This mixture was added to 0.5 mL of concentrated cells (∼2.5 × 107 cells) and placed in an electroporation cuvette with a 4-mm gap for 5 min at 16°C. Electroporation was carried out with a Gene Pulser Xcell system (Bio-Rad) set at 750 V, 25 µF, and ∞ resistance (to produce a time constant of 5–6 ms). After electroporation, cells were allowed to rest for 10 min and then diluted into 50 mL of TAP containing 60-mM sucrose and incubated for 24 h in 5% CO2 at 25°C with shaking and light. No post-electroporation heat shock treatment was applied. Cells were eventually concentrated by centrifugation, resuspended in ∼0.2 mL TAP, split into two aliquots and spread on TAP agar plates containing SMM at 5 μM. After incubation for ∼14 d in 5% CO2 at 25°C in continuous light, colonies were picked and restreaked on TAP + SMM plates to isolate single colonies. DNA was extracted from each colony and used for PCR amplification and DNA sequencing of the ALS1 target site.
Genotyping of potentially edited mutants
When targeting the PPX1 gene, a subset of colonies surviving on oxyfluorfen containing medium was examined by colony PCR amplification (Cao et al., 2009) of the target site followed by sequencing of the PCR products. When targeting the FTSY gene, only pale green colonies surviving on paromomycin containing medium or oxyfluorfen containing medium (in case of co-editing with the PPX1 gene) were examined by colony PCR amplification of the target site. All detectable PCR products, those of expected size and those differing in size from the WT amplicon (due to possible insertions or deletions), were examined by sequencing. PCR products of the expected size were also characterized by digestion with the NheI enzyme (Sambrook and Russell, 2001). When targeting the WDTC1 gene, colonies surviving on paromomycin containing medium or oxyfluorfen containing medium (in case of co-editing with the PPX1 gene) were examined by colony PCR amplification of the target site followed by digestion of the PCR product with the BssHII enzyme (Sambrook and Russell, 2001). Only PCR products successfully digested with BssHII or potentially having indels were characterized by sequencing. In all cases, amplification of the Actin gene was used as a positive control. To ensure amplification of the correct DNA fragments from the target genes, in most cases nested polymerase chain reactions were carried out with the primers listed in Supplemental Table S9. The PCR conditions for general amplification were 35 cycles at 94°C for 30 s, at 50°C for 30 s, and at 71°C for 90 s. Aliquots (8 µL) of each PCR were resolved on 1.5% (w/v) agarose gels and visualized by ethidium bromide staining (Sambrook and Russell, 2001).
Semi-quantitative RT-PCR analyses of WDTC1 transcript abundance
Total cell RNA, from cells harvested in the middle of the logarithmic phase, was purified with TRI Reagent (Molecular Research Center), following the manufacturer’s instructions. RT reactions were performed as previously described (Carninci et al., 1998) using Superscript III (Invitrogen, 18980051). The synthesized cDNA was then used as a template in standard PCRs (Sambrook and Russell, 2001) using a number of cycles showing a linear relationship between input RNA and the final product, as determined in preliminary experiments. The PCR conditions for amplification of the Actin control were 25 cycles at 94°C for 30 s, at 58°C for 30 s, and at 71°C for 30 s. The PCR conditions for amplification of the WDTC1 transcript were 30 cycles at 94°C for 30 s, at 60°C for 30 s, and at 71°C for 30 s. Aliquots (8 µL) of each RT-PCR were resolved on 1.5% agarose gels and visualized by ethidium bromide staining (Sambrook and Russell, 2001). All primers used for RT-PCR are listed in Supplemental Table S9.
Examination of CRISPR/Cas9 (FTSY) RNP cellular uptake by fluorescence microscopy
Chlamydomonas cells were electroporated with a commercially available trans-acting CRISPR RNA conjugated to the ATTO 550 fluorophore (IDT, 1075928 Alt-R CRISPR-Cas9 tracrRNA, ATTO 550), either alone or assembled into a CRISPR/Cas9 (FTSY) RNP (using ∼two-fold molar excess of the Cas9 protein relative to the tracrRNA-ATTO 550). Cell were examined for uptake of the transfected macromolecules at 1, 4, and 24 h after electroporation. However, the reported data corresponds to 4 h after electroporation, when the strongest signal was detected. For microscopy analyses, cells from a single electroporation were pelleted by centrifugation, washed twice and eventually resuspended in TAP medium containing 40 mM sucrose, to which a 1/100 volume of iodine (1% in ethanol) was added. Cells were visualized with an EVOS FL Auto Cell Imaging System (ThermoFisher Scientific), using the RFP light cube (Excitation 542/20, Emission 593/40) for ATTO 550 fluorescence and the Cy5 light cube (Excitation 635/18, Emission 692/40) for chlorophyll fluorescence. Pseudo-colors were used to represent signals from the different channels.
Accession numbers
The accession numbers of all genes used in this paper are taken from Phytozome v12.1 (https://phytozome.jgi.doe.gov/pz/portal.html): ALS1, Cre09.g386758; FTSY, Cre05.g241450; PPX1, Cre09.g396300; and WDTC1, Cre10.g425050.
Supplemental data
The following materials are available in the online version of this article.
Supplemental Figure S1. SSTR model for homology directed repair of the FTSY gene, using as complementary template the transfected ssODN.
Supplemental Figure S2. DNA sequences of FTSY disrupted mutants obtained by co-transfection of CRISPR/Cas9 (FTSY) RNP, a dsDNA PCR product encoding the aphVIII transgene and the FTSY ssODN donor.
Supplemental Figure S3. Fluorescence microscopy analysis of the cellular uptake of the CRISPR/Cas9 (FTSY) RNP after electroporation.
Supplemental Figure S4. SSTR model for homology directed repair of the PPX1 gene, using as complementary template the transfected ssODN.
Supplemental Figure S5. DNA sequences of FTSY insertional mutants obtained by co-targeting the PPX1 and FTSY genes for CRISPR/Cas9 editing.
Supplemental Figure S6. SSTR model for homology directed repair of the WDTC1 gene, using as complementary template the transfected ssODN.
Supplemental Figure S7. DNA sequences of WDTC1 insertional mutants obtained by co-targeting the PPX1 and WDTC1 genes for CRISPR/Cas9 editing.
Supplemental Figure S8. DNA sequences of WDTC1 insertional mutants obtained by co-transfection of CRISPR/Cas9 (WDTC1) RNP, a dsDNA PCR product encoding the aphVIII transgene and the WDTC1 ssODN donor.
Supplemental Figure S9. Southern blot analysis of CRISPR/Cas9-induced insertional mutants of the WDTC1 gene.
Supplemental Figure S10. SSTR model for homology-directed repair of the ALS1 gene, using as complementary template the transfected ssODN.
Supplemental Figure S11. DNA sequence of the ALS1 target site in sulfometuron methyl-resistant colonies.
Supplemental Table S1. Efficiency of FTSY targeted gene disruption by electroporation with CRISPR/Cas9 (FTSY) RNP and a PCR product encoding the aphVIII transgene.
Supplemental Table S2. ssODNs electroporated into cells as templates for HDR.
Supplemental Table S3. Efficiency of PPX1 gene editing by electroporation with CRISPR/Cas9 (PPX1) RNP and ssODN donor DNA.
Supplemental Table S4. Efficiency of FTSY gene editing by co-electroporation with CRISPR/Cas9 (FTSY) RNP, CRISPR/Cas9 (PPX1) RNP and the corresponding ssODN donor DNAs.
Supplemental Table S5. Efficiency of WDTC1 gene editing by co-electroporation with CRISPR/Cas9 (WDTC1) RNP, CRISPR/Cas9 (PPX1) RNP, and the corresponding ssODN donor DNAs.
Supplemental Table S6. Mutagenesis of the Chlamydomonas ALS1 gene using CRISPR/Cas9 (ALS1) RNP and ALS1 ssODN donor DNA.
Supplemental Table S7. Efficiency of FTSY gene editing, in cells pre-treated with autolysin, by co-electroporation with CRISPR/Cas9 (FTSY) RNP, CRISPR/Cas9 (PPX1) RNP and the corresponding ssODN donor DNAs.
Supplemental Table S8. CRISPR RNAs (crRNAs) used in the study.
Supplemental Table S9. PCR primers used in the study.
Supplementary Material
Acknowledgments
We thank Samantha Rau, Rakim Ali, Yingshan Li, and Dr Ananya Mukherjee for technical assistance with colony PCR and Southern blotting analyses.
Funding
This work was supported in part by grants from the National Science Foundation (MCB 1616863) and the Gordon and Betty Moore Foundation (Award No. 4968.01) to H.C.
Conflict of interest statement. The authors have no conflict of interest.
D.A.W., M.H.S., D.P.W., and H.C. conceived and designed the research. S.A., X.M., R.B., Z.P.H., M.K., X.W., and D.A.W. performed the experiments. S.A., X.M., D.A.W., M.H.S., D.P.W., and H.C. analyzed the data. S.A., D.P.W., and H.C. wrote the manuscript. All the authors read and approved the article.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://academic.oup.com/plphys/pages/general-instructions) is Heriberto Cerutti (hcerutti1@unl.edu).
References
- Aldridge C, Cain P, Robinson C (2009) Protein transport in organelles: Protein transport into and across the thylakoid membrane. FEBS J 276: 1177–1186 [DOI] [PubMed] [Google Scholar]
- Angstenberger M, de Signori F, Vecchi V, Dall'Osto L, Bassi R (2020) Cell synchronization enhances nuclear transformation and genome editing via Cas9 enabling homologous recombination in Chlamydomonas reinhardtii. ACS Synth Biol 9: 2840–2850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek K, Kim DH, Jeong J, Sim SJ, Melis A, Kim J-S, Jin E, Bae S (2016) DNA-free two-gene knockout in Chlamydomonas reinhardtii via CRISPR-Cas9 ribonucleoproteins. Sci Rep 6: 30620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banas K, Rivera-Torres N, Bialk P, Yoo BC, Kmiec EB (2020) Kinetics of nuclear uptake and site-specific DNA cleavage during CRISPR-directed gene editing in solid tumor cells. Mol Cancer Res 18: 891–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boel A, De Saffel H, Steyaert W, Callewaert B, De Paepe A, Coucke PJ, Willaert A (2018) CRISPR/Cas9-mediated homology-directed repair by ssODNs in zebrafish induces complex mutational patterns resulting from genomic integration of repair-template fragments. Dis Model Mech 11: dmm035352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambati A, Barry RM, Sfeir A (2020) DNA polymerase theta (Polθ) - an error-prone polymerase necessary for genome stability. Curr Opin Genet Dev 60: 119–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brueggeman AJ, Kuehler D, Weeks DP (2014) Evaluation of three herbicide resistance genes for use in genetic transformations and for potential crop protection in algae production. Plant Biotechnol J 12: 894–902 [DOI] [PubMed] [Google Scholar]
- Capdeville N, Merker L, Schindele P, Puchta H (2020) Sophisticated CRISPR/Cas tools for fine-tuning plant performance. J Plant Physiol 257: 153332. [DOI] [PubMed] [Google Scholar]
- Cao M, Fu Y, Guo Y, Pan J (2009) Chlamydomonas (Chlorophyceae) colony PCR. Protoplasma 235: 107–110 [DOI] [PubMed] [Google Scholar]
- Carninci P, Nishiyama Y, Westover A, Itoh M, Nagaoka S, Sasaki N, Okazaki Y, Muramatsu M, Hayashizaki Y (1998) Thermostabilization and thermoactivation of thermolabile enzymes by trehalose and its application for the synthesis of full length cDNA. Proc Natl Acad Sci U S A 95: 520–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazzaniga S, Kim M, Bellamoli F, Jeong J, Lee S, Perozeni F, Pompa A, Jin E, Ballottari M (2020) Photosystem II antenna complexes CP26 and CP29 are essential for nonphotochemical quenching in Chlamydomonas reinhardtii. Plant Cell Environ 43: 496–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhokane D, Bhadra B, Dasgupta S (2020) CRISPR based targeted genome editing of Chlamydomonas reinhardtii using programmed Cas9-gRNA ribonucleoprotein. Mol Biol Rep 47: 8747–8755 [DOI] [PubMed] [Google Scholar]
- Dorsett Y, Zhou Y, Tubbs AT, Chen B-R, Purman C, Lee B-S, George R, Bredemeyer A, Zhao J-Y, Sodergen E, et al. (2014) HCoDES reveals chromosomal DNA end structures with single-nucleotide resolution. Mol Cell 56: 808–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducos E, Vergès V, Dugé de Bernonville T, Blanc N, Giglioli-Guivarc'h N, Dutilleul C (2017) Remarkable evolutionary conservation of antiobesity ADIPOSE/WDTC1 homologs in animals and plants. Genetics 207: 153–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duke SO, Lydon J, Becerril JM, Sherman TD, Lehnen LP, Matsumoto H (1991) Protoporphyrinogen oxidase-inhibiting herbicides. Weed Sci 39: 465–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferenczi A, Pyott DE, Xipnitou A, Molnar A (2017) Efficient targeted DNA editing and replacement in Chlamydomonas reinhardtii using Cpf1 ribonucleoproteins and single-stranded DNA. Proc Natl Acad Sci U S A 114: 13567–13572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher DN, Haber JE (2018) Repair of a site-specific DNA cleavage: old-school lessons for Cas9-mediated gene editing. ACS Chem Biol 13: 397–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher DN, Pham N, Tsai AM, Janto AN, Choi J, Ira G, Haber JE (2020) A Rad51-independent pathway promotes single-strand template repair in gene editing. PLoS Genet 16: e1008689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greiner A, Kelterborn S, Evers H, Kreimer G, Sizova I, Hegemann P (2017) Targeting of photoreceptor genes in Chlamydomonas reinhardtii via zinc-finger nucleases and CRISPR/Cas9. Plant Cell 29: 2498–2518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh BS, Yan F, Smith MD, Yu Y, Chen X, Xiong Y (2016) The antiobesity factor WDTC1 suppresses adipogenesis via the CRL4WDTC1 E3 ligase. EMBO Rep 17: 638–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumpel NJ, Rochaix JD, Purton S (1994) Studies on homologous recombination in the green alga Chlamydomonas reinhardtii. Curr Genet 26: 438–442 [DOI] [PubMed] [Google Scholar]
- Guzmán-Zapata D, Sandoval-Vargas JM, Macedo-Osorio KS, Salgado-Manjarrez E, Castrejón-Flores JL, Oliver-Salvador MDC, Durán-Figueroa NV, Nogué F, Badillo-Corona JA (2019) Efficient editing of the nuclear APT reporter gene in Chlamydomonas reinhardtii via expression of a CRISPR-Cas9 module. Int J Mol Sci 20: 1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha SB, Lee SB, Chung JS, Han SU, Han O, Guh JO, Jeon JS, An G, Back K (2004) The plastidic Arabidopsis protoporphyrinogen IX oxidase gene, with or without the transit sequence, confers resistance to the diphenyl ether herbicide in rice. Plant Cell Environ 27: 79–88 [Google Scholar]
- Häder T, Müller S, Aguilera M, Eulenberg KG, Steuernagel A, Ciossek T, Kühnlein RP, Lemaire L, Fritsch R, Dohrmann C, et al. (2003) Control of triglyceride storage by a WD40/TPR-domain protein. EMBO Rep 4: 511–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmsen T, Klaasen S, van de Vrugt H, Te Riele H (2018) DNA mismatch repair and oligonucleotide end-protection promote base-pair substitution distal from a CRISPR/Cas9-induced DNA break. Nucleic Acids Res 46: 2945–2955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris EH (1989) The Chlamydomonas Sourcebook: A Comprehensive Guide to Biology and Laboratory Use, Ed 1. Academic Press, San Diego: [DOI] [PubMed] [Google Scholar]
- Jeon S, Lim JM, Lee HG, Shin SE, Kang NK, Park YI, Oh HM, Jeong WJ, Jeong BR, Chang YK (2017) Current status and perspectives of genome editing technology for microalgae. Biotechnol Biofuels 10: 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Brueggeman AJ, Horken KM, Plucinak TM, Weeks DP (2014) Successful transient expression of Cas9 and single guide RNA genes in Chlamydomonas reinhardtii. Eukaryot Cell 13: 1465–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang WZ, Dumm S, Knuth ME, Sanders SL, Weeks DP (2017) Precise oligonucleotide-directed mutagenesis of the Chlamydomonas reinhardtii genome. Plant Cell Rep 36: 1001–1004 [DOI] [PubMed] [Google Scholar]
- Jiang WZ, Weeks DP (2017) A gene-within-a-gene Cas9/sgRNA hybrid construct enables gene editing and gene replacement strategies in Chlamydomonas reinhardtii. Algal Research 26: 474–480 [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinkerson RE, Jonikas MC (2015) Molecular techniques to interrogate and edit the Chlamydomonas nuclear genome. Plant J 82: 393–412 [DOI] [PubMed] [Google Scholar]
- Kan Y, Ruis B, Takasugi T, Hendrickson EA (2017) Mechanisms of precise genome editing using oligonucleotide donors. Genome Res 27: 1099–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S, Jeon S, Kim S, Chang YK, Kim YC (2020) Development of a pVEC peptide-based ribonucleoprotein (RNP) delivery system for genome editing using CRISPR/Cas9 in Chlamydomonas reinhardtii. Sci Rep 10: 22158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EJ, Cerutti H (2009) Targeted gene silencing by RNA interference in Chlamydomonas. Methods Cell Biol 93: 99–110 [DOI] [PubMed] [Google Scholar]
- Kim J, Lee S, Baek K, Jin E (2020) Site-specific gene knock-out and on-site heterologous gene overexpression in Chlamydomonas reinhardtii via a CRISPR-Cas9-mediated knock-in method. Front Plant Sci 11: 306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Terng EL, Riekhof WR, Cahoon EB, Cerutti H (2018) Endoplasmic reticulum acyltransferase with prokaryotic substrate preference contributes to triacylglycerol assembly in Chlamydomonas. Proc Natl Acad Sci U S A 115: 1652–1657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirst H, García-Cerdán JG, Zurbriggen A, Melis A (2012) Assembly of the light-harvesting chlorophyll antenna in the green alga Chlamydomonas reinhardtii requires expression of the TLA2-CpFTSY gene. Plant Physiol 158: 930–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovar JL, Zhang J, Funke RP, Weeks DP (2002) Molecular analysis of the acetolactate synthase gene of Chlamydomonas reinhardtii and development of a genetically engineered gene as a dominant selectable marker for genetic transformation. Plant J 29: 109–117 [DOI] [PubMed] [Google Scholar]
- Li X, Patena W, Fauser F, Jinkerson RE, Saroussi S, Meyer MT, Ivanova N, Robertson JM, Yue R, Zhang R, et al. (2019) A genome-wide algal mutant library and functional screen identifies genes required for eukaryotic photosynthesis. Nat Genet 51: 627–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhang R, Patena W, Gang SS, Blum SR, Ivanova N, Yue R, Robertson JM, Lefebvre PA, Fitz-Gibbon ST, et al. (2016) An indexed, mapped mutant library enables reverse genetics studies of biological processes in Chlamydomonas reinhardtii. Plant Cell 28: 367–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Aravind L, Wolf YI, Koonin EV (2011) Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR-Cas systems. Biol Direct 6: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitzelfelt KA, McDermott-Roe C, Grzybowski MN, Marquez M, Kuo C-T, Riedel M, Lai S, Choi MJ, Kolander KD, Helbling D, et al. (2017) Efficient precision genome editing in iPSCs via genetic co-targeting with selection. Stem Cell Reports 8: 491–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnar A, Bassett A, Thuenemann E, Schwach F, Karkare S, Ossowski S, Weigel D, Baulcombe D (2009) Highly specific gene silencing by artificial microRNAs in the unicellular alga Chlamydomonas reinhardtii. Plant J 58: 165–174 [DOI] [PubMed] [Google Scholar]
- Msanne J, Xu D, Konda AR, Casas-Mollano JA, Awada T, Cahoon EB, Cerutti H (2012) Metabolic and gene expression changes triggered by nitrogen deprivation in the photoautotrophically grown microalgae Chlamydomonas reinhardtii and Coccomyxa sp. C-169. Phytochemistry 75: 50–59 [DOI] [PubMed] [Google Scholar]
- Nelson JA, Savereide PB, Lefebvre PA (1994) The CRY1 gene in Chlamydomonas reinhardtii: structure and use as a dominant selectable marker for nuclear transformation. Mol Cell Biol 14: 4011–4019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neupert J, Karcher D, Bock R (2009) Generation of Chlamydomonas strains that efficiently express nuclear transgenes. Plant J 57: 1140–1150 [DOI] [PubMed] [Google Scholar]
- Ortega-Escalante JA, Jasper R, Miller SM (2019) CRISPR/Cas9 mutagenesis in Volvox carteri. Plant J 97: 661–672 [DOI] [PubMed] [Google Scholar]
- Paix A, Folkmann A, Goldman DH, Kulaga H, Grzelak MJ, Rasoloson D, Paidemarry S, Green R, Reed RR (2017) Precision genome editing using synthesis-dependent repair of Cas9-induced DNA breaks. Proc Natl Acad Sci U S A 114: E10745–E10754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquet D, Kwart D, Chen A, Sproul A, Jacob S, Teo S, Olsen KM, Gregg A, Noggle S, Tessier-Lavigne M (2016) Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 533: 125–129 [DOI] [PubMed] [Google Scholar]
- Park RV, Asbury H, Miller SM (2020) Modification of a Chlamydomonas reinhardtii CRISPR/Cas9 transformation protocol for use with widely available electroporation equipment. MethodsX 7: 100855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picariello T, Hou Y, Kubo T, McNeill NA, Yanagisawa H-A, Oda T, Witman GB (2020) TIM, a targeted insertional mutagenesis method utilizing CRISPR/Cas9 in Chlamydomonas reinhardtii. PLoS One 15: e0232594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plecenikova A, Mages W, Andrésson ÓS, Hrossova D, Valuchova S, Vlcek D, Slaninova M (2013) Studies on recombination processes in two Chlamydomonas reinhardtii endogenous genes, NIT1 and ARG7. Protist 164: 570–582 [DOI] [PubMed] [Google Scholar]
- Radecke F, Peter I, Radecke S, Gellhaus K, Schwarz K, Cathomen T (2006) Targeted chromosomal gene modification in human cells by single stranded oligodeoxynucleotides in the presence of a DNA double-strand break. Mol Ther 14: 798–808 [DOI] [PubMed] [Google Scholar]
- Randolph-Anderson BL, Sato R, Johnson AM, Harris EH, Hauser CR, Oeda K, Ishige F, Nishio S, Gillham NW, Boynton JE (1998) Isolation and characterization of a mutant protoporphyrinogen oxidase gene from Chlamydomonas reinhardtii conferring resistance to porphyric herbicides. Plant Mol Biol 38: 839–859 [DOI] [PubMed] [Google Scholar]
- Richardson CD, Kazane KR, Feng SJ, Zelin E, Bray NL, Schäfer AJ, Floor SN, Corn JE (2018) CRISPR-Cas9 genome editing in human cells occurs via the Fanconi anemia pathway. Nat Genet 50: 1132–1139 [DOI] [PubMed] [Google Scholar]
- Rodgers K, McVey M (2016) Error-prone repair of DNA double-strand breaks. J Cell Physiol 231: 15–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohr J, Sarkar N, Balenger S, Jeong BR, Cerutti H (2004) Tandem inverted repeat system for selection of effective transgenic RNAi strains in Chlamydomonas. Plant J 40: 611–621 [DOI] [PubMed] [Google Scholar]
- Rosales-Mendoza S, Paz-Maldonado LM, Soria-Guerra RE (2012) Chlamydomonas reinhardtii as a viable platform for the production of recombinant proteins: current status and perspectives. Plant Cell Rep 31: 479–494 [DOI] [PubMed] [Google Scholar]
- Salomé PA, Merchant SS (2019) A series of fortunate events: introducing Chlamydomonas as a reference organism. Plant Cell 31: 1682–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Russell DW (2001) Molecular cloning – A laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor [Google Scholar]
- Sansbury BM, Hewes AM, Kmiec EB (2019) Understanding the diversity of genetic outcomes from CRISPR-Cas generated homology-directed repair. Commun Biol 2: 458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer NJ, Narváez-Vásquez J, Mozoruk J, Miller RB, Warburg ZJ, Woodward MJ, Mihiret YA, Lincoln TA, Segami RE, Sanders SL, et al. (2016) Oligonucleotide-mediated genome editing provides precision and function to engineered nucleases and antibiotics in plants. Plant Physiol 170: 1917–1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimmel J, van Schendel R, den Dunnen JT, Tijsterman M (2019) Templated insertions: a smoking gun for polymerase theta-mediated end joining. Trends Genet 35: 632–644 [DOI] [PubMed] [Google Scholar]
- Scranton MA, Ostrand JT, Fields FJ, Mayfield SP (2015) Chlamydomonas as a model for biofuels and bio-products production. Plant J 82: 523–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R, Panday A, Elango R, Willis NA (2019) DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol 20: 698–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamoto N, Narita K, Kubo T, Oda T, Takeda S (2018) CFAP70 is a novel axoneme-binding protein that localizes at the base of the outer dynein arm and regulates ciliary motility. Cells 7: 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Q, Wang Y, Li J, Zhang Y, Chen K, Liang Z, Zhang K, Liu J, Xi JJ, Qiu JL, Gao C (2013) Targeted genome modification of crop plants using a CRISPR-Cas system. Nat Biotechnol 31: 686–688 [DOI] [PubMed] [Google Scholar]
- Shin SE, Lim JM, Koh HG, Kim EK, Kang NK, Jeon S, Kwon S, Shin WS, Lee B, Hwangbo K, et al. (2016) CRISPR/Cas9-induced knockout and knock-in mutations in Chlamydomonas reinhardtii. Sci Rep 6: 27810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shy BR, MacDougall MS, Clarke R, Merrill BJ (2016) Co-incident insertion enables high efficiency genome engineering in mouse embryonic stem cells. Nucleic Acids Res 44: 7997–8010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siaut M, Cuiné S, Cagnon C, Fessler B, Nguyen M, Carrier P, Beyly A, Beisson F, Triantaphylidès C, Li-Beisson Y, et al. (2011) Oil accumulation in the model green alga Chlamydomonas reinhardtii: characterization, variability between common laboratory strains and relationship with starch reserves. BMC Biotechnol 11: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sizova I, Fuhrmann M, Hegemann P (2001) A Streptomyces rimosus aphVIII gene coding for a new type phosphotransferase provides stable antibiotic resistance to Chlamydomonas reinhardtii. Gene 277: 221–229 [DOI] [PubMed] [Google Scholar]
- Sizova I, Greiner A, Awasthi M, Kateriya S, Hegemann P (2013) Nuclear gene targeting in Chlamydomonas using engineered zinc-finger nucleases. Plant J 73: 873–882 [DOI] [PubMed] [Google Scholar]
- Sizova I, Kelterborn S, Verbenko V, Kateriya S, Hegemann P (2021) Chlamydomonas POLQ is necessary for CRISPR/Cas9-mediated gene targeting. G3 (Bethesda) 11: jkab114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodeinde OA, Kindle KL (1993) Homologous recombination in the nuclear genome of Chlamydomonas reinhardtii. Proc Natl Acad Sci U S A 90: 9199–9203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez JV, Banks S, Thomas PG, Day A (2014) A new F131V mutation in Chlamydomonas phytoene desaturase locates a cluster of norflurazon resistance mutations near the FAD-binding site in 3D protein models. PLoS One 9: e99894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sueoka N (1960) Mitotic replication of deoxyribonucleic acid in Chlamydomonas reinhardtii .Proc Natl Acad Sci U S A 46: 83–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitashev S, Young JK, Schwartz C, Gao H, Falco SC, Cigan AM (2015) Targeted mutagenesis, precise gene editing, and site-specific gene insertion in maize using Cas9 and guide RNA. Plant Physiol 169: 931–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarts DC, Jinek M (2018) Cas9 versus Cas12a/Cpf1: Structure-function comparisons and implications for genome editing. Wiley Interdiscip Rev RNA 9: e1481. [DOI] [PubMed] [Google Scholar]
- Xie B, Bishop S, Stessman D, Wright D, Spalding MH, Halverson LJ (2013) Chlamydomonas reinhardtii thermal tolerance enhancement mediated by a mutualistic interaction with vitamin B12-producing bacteria. ISME J 7: 1544–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano T, Iguchi H, Fukuzawa H (2013) Rapid transformation of Chlamydomonas reinhardtii without cell-wall removal. J Biosci Bioeng 115: 691–694 [DOI] [PubMed] [Google Scholar]
- Zahn KE, Jensen RB, Wood RD, Doublié S (2021) Human DNA polymerase θ harbors DNA end-trimming activity critical for DNA repair. Mol Cell 81: 1534–1547 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhao T, Wang W, Bai X, Qi Y (2009) Gene silencing by artificial microRNAs in Chlamydomonas. Plant J 58: 157–164 [DOI] [PubMed] [Google Scholar]
- Zorin B, Lu Y, Sizova I, Hegemann P (2009) Nuclear gene targeting in Chlamydomonas as exemplified by disruption of the PHOT gene. Gene 432: 91–96 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.