

Abstract
The ongoing outbreak of COVID‐19 caused by SARS‐CoV‐2 has resulted in a serious public health threat globally. Nucleocapsid protein is a major structural protein of SARS‐CoV‐2 that plays important roles in the viral RNA packing, replication, assembly, and infection. Here, we report two crystal structures of nucleocapsid protein C‐terminal domain (CTD) at resolutions of 2.0 Å and 3.1 Å, respectively. These two structures, crystallized under different conditions, contain 2 and 12 CTDs in asymmetric unit, respectively. Interestingly, despite different crystal packing, both structures show a similar dimeric form as the smallest unit, consistent with its solution form measured by the size‐exclusion chromatography, suggesting an important role of CTD in the dimerization of nucleocapsid proteins. By analyzing the surface charge distribution, we identified a stretch of positively charged residues between Lys257 and Arg262 that are involved in RNA‐binding. Through screening a single‐domain antibodies (sdAbs) library, we identified four sdAbs targeting different regions of nucleocapsid protein with high affinities that have future potential to be used in viral detection and therapeutic purposes.
Keywords: crystal structure, nanobodies, nucleocapsid protein, SARS‐CoV‐2
Through screening a naive single‐domain antibodies (sdAbs) library, we identified several sdAbs targeting different regions of the nucleocapsid (N) protein of SARS‐CoV‐2. Using X‐ray crystallography and biophysical methods, we revealed the structures of C‐terminal domain (CTD) of N‐protein and characterized its interactions with the identified sdAbs.

Abbreviations
- ASU
asymmetric unit
- CTD
C‐terminal domain
- FLN
full‐length N‐protein
- IDL
intrinsically disordered linker
- IPTG
β‐D‐1‐thiogalactopyranoside
- NC
NTD + CTD
- NLC
NTD + IDL + CTD
- NTD
N‐terminal domain
- RMSD
root mean square deviation
Introduction
COVID‐19, an infectious disease caused by a severe acute respiratory syndrome coronavirus SARS‐CoV‐2, has infected more than 170 million people and caused the death of 3.7 million [1, 2, 3]. Due to the outbreak of COVID‐19, WHO has declared a public health emergency of international concern. Since SARS‐CoV‐2 is newly emerged virus, there is no effective drug specifically targeting this type of virus. There is an urgent need to understand the fundamental biology of SARS‐CoV‐2 and develop efficient detection and effective therapeutic methods accordingly.
As a beta‐coronavirus (βCoV), SARS‐CoV‐2 shares four main structural proteins with other coronaviruses: spike (S), envelope (E), membrane (M), and nucleocapsid (N) proteins [4, 5]. Among them, the N‐protein is abundantly expressed during infection with high immunogenicity [6]. The main role of N‐protein is to associate with the genomic RNA to form a ribonucleoprotein (RNP) complex, also called capsid [7]. It also has role in viral replication, assembly, and infection [8, 9]. In addition, through its double stranded RNA binding activity, the N‐protein also functions as a viral RNA silencing suppressor (VSR) by counteracting host RNA‐mediated antiviral responses [10]. Because of its high abundance, it can also induce strong postinfectious immune responses, which makes it as a good target for diagnostic purpose and for vaccine development [11, 12]. The N‐protein consists of two independently folded domains, the N‐terminal domain (NTD) (residues 44–180), and the CTD (residues 255–362), connected by an intrinsically disordered linker (IDL) (residues 181–254). In addition, two disordered regions are positioned to the sides of NTD and CTD, called N‐arm (residues 1–43) and C‐tail (residues 363–419) [13] (Fig. 1A). It is proposed that the NTD is responsible for RNA binding, while CTD is involved in RNA binding and oligomerization, and the IDL regulates the RNA binding activity of N‐protein by affecting the interaction between the NTD and the CTD. The structures of N‐NTD and N‐CTD from several coronaviruses have been solved [14, 15, 16, 17, 18]. However, because of the high flexibility of the disordered regions and the complicated oligomerization assembly, the structure of the full‐length N‐protein (FLN) remains unknown [19].
Fig. 1.
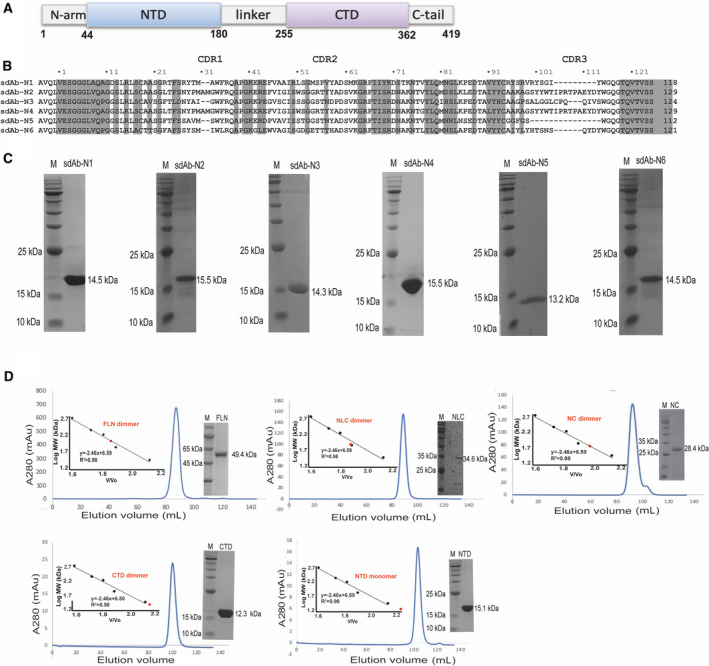
Purification of the full‐length and truncated N‐proteins, and N‐protein targeting sdAbs. (A) A schematic picture of N‐protein domain organization (B) Sequence alignment of the six nonrepetitive sdAbs. The conserved residues are highlighted and the three CDR regions are indicated by the dashed boxes. (C) 15% SDS/PAGE showing protein marker (PM) in the left lanes and purified sdAbs in the right lanes. (D) Elution profile of the full‐length and truncated N‐proteins by SEC using a Superdex 200 16/600 column (GE Healthcare, Marlborough, MA, USA). The inset shows the plotted standard curve for this column and the representative 15% SDS/PAGE showing the purified N‐proteins.
Antibodies targeting the key proteins of coronaviruses, such as SARS‐CoV‐1, MERS‐CoV, and SARS‐CoV‐2, have been proven to be useful for diagnosis and treatment purposes [20, 21, 22, 23]. Compared to conventional antibodies, single‐domain antibodies (sdAbs), which were initially discovered from the llama peripheral blood, generally confer increased affinity and specificity for the antigen [24]. Due to the natural loss of light chain, sdAbs contains only a single variable domain (VHH) rather than two variable domains (VH and VL) observed in traditional antibodies, which constitute the antigen binding fragment (FAB) [24]. Interestingly, despite of the smaller size, VHHs cloned and expressed alone have comparable or even higher structural stability and binding activity to antigen compared to FABs [25]. sdAbs also have several additional advantages. For example, sdAbs are less subject to steric hindrance, which may prevent the binding of larger conventional antibodies [26, 27] and are easy to be constructed in the multivalent forms with high thermal stability [28]. So far, a series of neutralizing sdAbs against the RBD domain of SARS‐CoV‐1 and SARS‐CoV‐2 S proteins have been developed for the prevention and therapeutic purposes [29, 30].
Because the N‐protein of SARS‐CoV‐2 is essential for viral RNP formation and genome replication, it has emerged as an important drug target. Blocking its RNA binding or dimerization properties has proven as a good strategy for the development of antiviral drugs [16, 31, 32]. In addition, because of its native high abundance, the N‐protein is also suitable for developing antibodies used for rapid and accurate detection of virus. Here, we reported two crystal structures of SARS‐CoV‐2 N‐CTD at resolutions of 2.0 Å and 3.1 Å, respectively. Our structures reveal the key residues involved in dimer formation and RNA‐binding. In addition, we developed a series of sdAbs targeting the N‐protein of SARS‐CoV‐2 that have the potential to be used for virus detection and therapeutic purposes.
Results
Screening and production of N‐protein targeting sdAbs
We screened a naive llama single‐domain antibody library with a capacity of 109 cfu·µg−1. After three rounds of panning, several N‐protein specific sdAbs were enriched. 96 phage plaques from the library were analyzed by ELISA, and 94 of them showed high absorbance values, proving positive in binding. After sequencing, 59 effective sdAbs sequences were obtained. Based on the diversity of amino acid sequences, 6 nonrepetitive sequences were finally classified (Fig. 1B). Six positive sdAbs were recombinantly expressed in the periplasm of Escherichia coli and purified to homogeneity using affinity and size‐exclusion chromatography (Fig. 1C). The full‐length and four truncated versions of N‐proteins, including NTD + IDL + CTD (NLC), NTD + CTD (NC), NTD, and CTD, were also expressed in E. coli. The FLN was purified by a three‐step purification protocol, including the affinity, ion exchange, and size‐exclusion chromatography (SEC) steps, while the four truncated N‐proteins were purified by a five‐step one, including an additional TEV cleavage and post‐TEV affinity purification steps (Fig. 1D). In order to remove nucleic acids bound to N‐protein, the additional nuclease was added after cell lysis. According to the SEC results, all the constructs containing CTD form dimer in solution. In contrast, NTD by itself forms monomer in solution (Table 1). This supports that CTD functions as a dimerization domain as shown in our crystal structure.
Table 1.
Estimated molecular weights (MWs) and oligomeric forms of N‐protein constructs as determined by SEC using a Superdex 200 16/600 column.
| Construct | Elution volume (ml) | V/V0 | Estimated MW (kDa) | Estimated oligomeric form |
|---|---|---|---|---|
| FLN | 87.42 | 1.86 | 103.4 | 2.1 |
| NLC | 88.58 | 1.89 | 87.2 | 2.5 |
| NC | 92.27 | 1.96 | 61.6 | 2.2 |
| CTD | 100.16 | 2.13 | 26.5 | 2.1 |
| NTD | 103.10 | 2.20 | 19.0 | 1.2 |
sdAbs bind to different regions of N‐protein
We characterized the interactions between sdAbs and N‐protein using isothermal titration calorimetry (ITC). We first tested their binding to the FLN. Four out of six sdAbs showed clear binding. The Kd values of positive sdAb‐N2, sdAb‐N3, sdAb‐N5, and sdAb‐N6 are 1.75 µm, 4.37 µm, 3.97 µm, and 3.53 µm, respectively (Fig. 2). Next, we tested the binding of these four sdAbs with NLC protein. Only sdAb‐N2 and sdAb‐N3 showed positive results with Kd values of 2.24 µm and 1.09 µm, respectively (Fig. 3), indicating that the binding of sdAbs‐N5 and sdAb‐N6 requires the presence of the N‐arm or C‐tail of N‐protein. Subsequently, we tested the binding of sdAb‐N2 with NC, NTD, and CTD separately. sdAb‐N2 illustrated the clear binding with NC and CTD but not with NTD (Fig. 4A–C). The binding affinity between sdAb‐N2 and CTD (Kd = 2.38 µm) is the same to those binding with the FLN (Kd = 1.75 µm), NLC (Kd = 2.24 µm), and NC (Kd = 1.77 µm), suggesting CTD itself forms the major binding site for sdAbs‐N2. We further analyzed the thermodynamics parameters of these molecular bindings. All the interactions of sdAb‐N2 are mainly entropy‐driven and involve endothermic enthalpy (Table 2). The N values for these interactions are closed to 0.5, suggesting a 2 : 1 binding ratio between N‐protein and sdAb‐N2. This is consistent with the ratio of band intensities of N‐protein and sdAb‐N2 shown by SDS/PAGE following the SEC (Fig. 4D). In contrast, sdAb‐N3 does not bind with either NTD or CTD (Fig. 5), suggesting that in this case the linker region has the opposite effect and contributes to the binding with sdAb‐N3. These results suggest that the hydrophobic effect is the most prominent driving force for sdAb‐N2 binding. In contrast, the bindings of the other three sdAbs are mainly enthalpy‐driven with the reduction of entropy (Table 2), indicating more contribution from the specific interactions such as H‐bonds and electrostatic interactions.
Fig. 2.
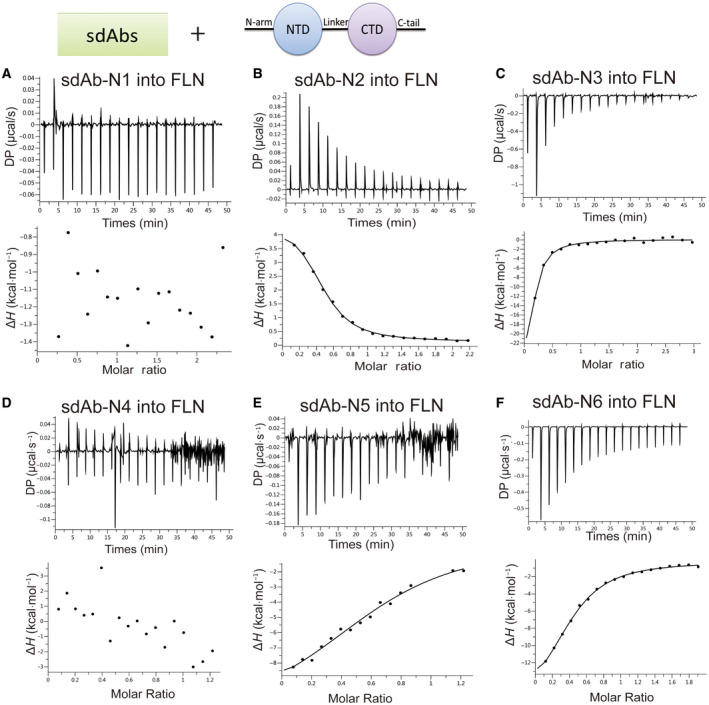
sdAbs bind to the FLN. ITC binding isotherms show the interactions between six sdAbs (A–F) titrated into FLN.
Fig. 3.
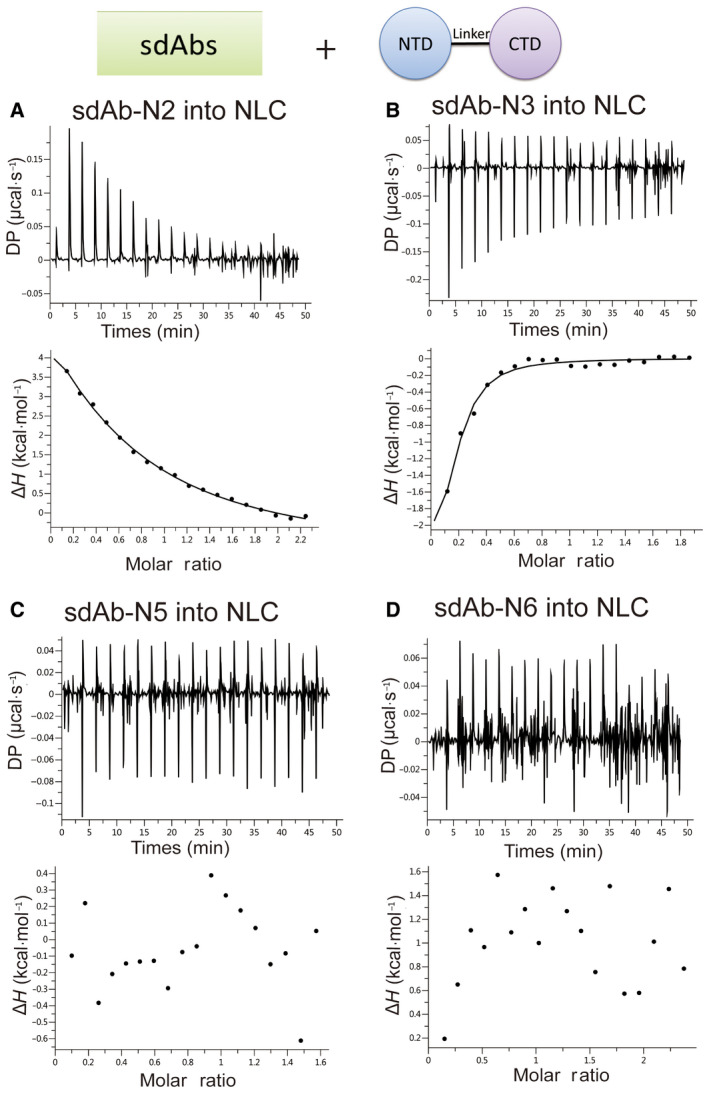
sdAbs bind to the NLC. ITC binding isotherms show the interactions between four sdAbs (A–D) titrated into NLC.
Fig. 4.
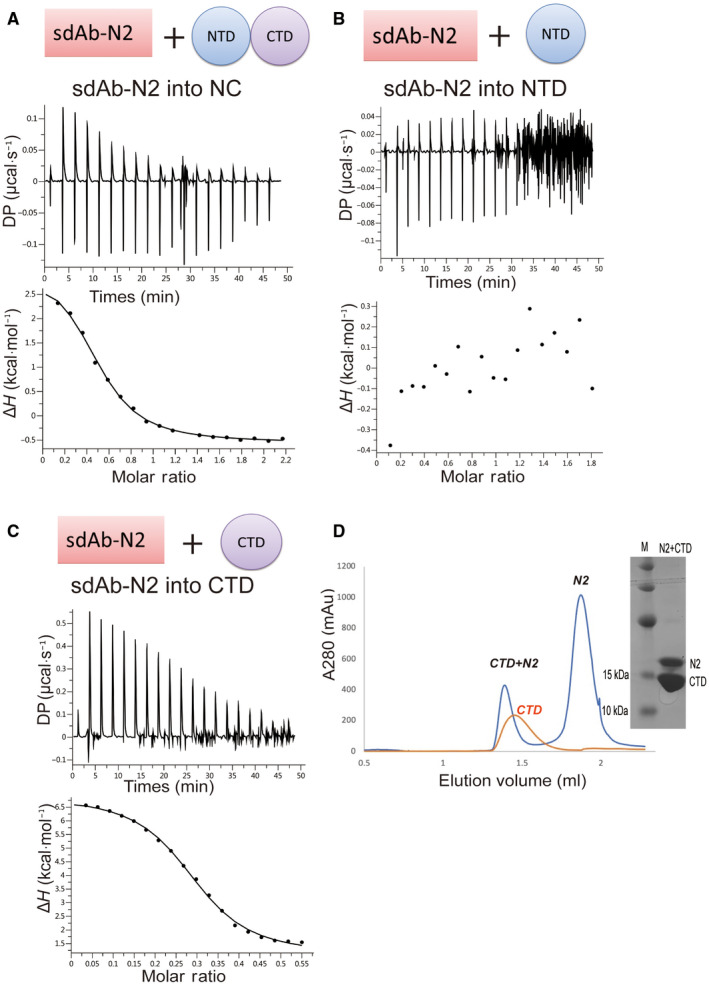
sdAb‐N2 binds to the truncated N‐proteins. ITC binding isotherms show the interactions between sdAb‐N2 titrated into NTD+CTD (A) or NTD (B) or CTD (C). (D) Elution profile of the CTD+N2 complex and CTD alone by SEC using a Superdex 75 3.2/300 column and the representative 15% SDS/PAGE.
Table 2.
Thermodynamic parameters of binding between sdAbs and N‐proteins.
| sdAb | Construct of N‐protein | No. of sites (N) | Kd (µm) | ΔH (kcal·mol−1) | TΔS (kcal·mol−1) | ΔG (kcal·mol−1) |
|---|---|---|---|---|---|---|
| sdAb‐N1 | FLN | ‐ | No binding | ‐ | ‐ | ‐ |
| sdAb‐N2 | FLN | 0.457 | 1.75 ± 0.179 | 4.44 ± 0.146 | −12.3 | −7.86 |
| NLC | 0.530 | 2.24 ± 0.307 | 4.40 ± 0.206 | −12.1 | −7.71 | |
| NC | 0.495 | 1.77 ± 0.217 | 3.66 ± 0.147 | −11.5 | −7.85 | |
| CTD | 0.292 | 2.38 ± 0.243 | 5.85 ± 0.141 | −13.5 | −7.67 | |
| NTD | ‐ | No binding | ‐ | ‐ | ‐ | |
| sdAb‐N3 | FLN | 0.066 | 4.37 ± 1.68 | −80 ± 99.8 | 72.7 | −7.31 |
| NLC | 0.157 | 1.09 ± 0.485 | −2.90 ± 0.558 | −5.18 | −8.14 | |
| CTD | ‐ | No binding | ‐ | ‐ | ‐ | |
| NTD | ‐ | No binding | ‐ | ‐ | ‐ | |
| sdAb‐N4 | FLN | ‐ | No binding | ‐ | ‐ | ‐ |
| sdAb‐N5 | FLN | 0.697 | 3.97 ± 2.42 | −11.7 ± 3.22 | 4.3 | −7.37 |
| NLC | ‐ | No binding | ‐ | ‐ | ‐ | |
| sdAb‐N6 | FLN | 0.416 | 3.53 ± 0.50 | −17.8 ± 1.26 | 10.4 | −7.44 |
| NLC | ‐ | No binding | ‐ | ‐ | ‐ |
Fig. 5.
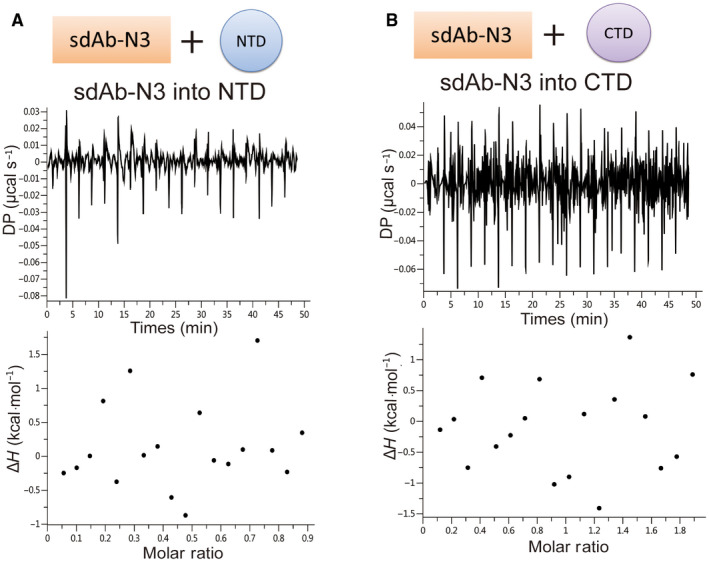
sdAb‐N3 binds with neither NTD nor CTD. ITC binding isotherms show the interactions between sdAb‐N3 titrated into NTD (A) or CTD (B).
The crystal structures of SARS‐CoV‐2 N‐protein CTD
We screened the crystals of SARS‐CoV‐2 N‐protein CTD in the absence and presence of a CTD‐targeting sdAb (sdAb‐N2) and solved their crystal structures individually (Table 3). Regardless of the presence of sdAb‐N2, both structures only contain CTD, however, their crystal packings and space groups are very different. In one structure determined at 2.0 Å, there are two CTDs in the asymmetric unit (ASU), while in the other structure determined at 3.1 Å, twelve CTDs are found in a single ASU (Fig. 6A). In order to examine the quaternary structure in solution, purified CTDs were subjected to size‐exclusion chromatography. It elutes as expected for a dimer with or without sdAb‐N2 (Fig. 6B). Both structures show an interface with extensive interactions between two CTD monomers, implicating a stable native dimeric structure (Fig. 6C). These interactions are mainly contributed by the residues from two β‐strands, including Ile320, Met322 from β1 and Thr329, Trp330, Tyr333, Ile337, Lys338 from β2, which buries a total surface area of ~ 2500 Å2. These residues are all conserved between the N‐proteins of SARS‐CoV‐1 and SARS‐CoV‐2, but partially different in MERS (Fig. 6D). Strands β1 and β2 form a β‐hairpin motif, which is swapped between two monomers to form extensive intersubunit interactions. In contrast, the other inter‐CTD interfaces observed in dodecameric structure are much smaller, burying only ~ 100–400 Å2, suggesting that they are probably only present iN‐protein crystals. Therefore, we focus our analysis on the dimeric units from both structures. Two dimeric structures are very similar to each other. The root mean square deviation (RMSD) between the CTD (dimer) and CTD (dodecamer) chain AB is 0.4 Å for 212 Cα atoms. The RMSD values between different dimeric units of dodecameric CTD are in the same ball park. Each monomer contains five ɑ‐helices, two β‐strands, two 310‐helices, and several connecting loops (Fig. 6A). The analysis of the surface charge distribution of the dimeric CTDs reveals a positively charged pocket, constituted by a stretch of positively charged residues between Lys257 and Arg262 conserved among SARS‐CoV‐1, SARS‐CoV‐2, and MERS (Fig. 6E). It has been shown that the mutations of these conserved positive residues can weaken the binding of RNA in SARS‐CoV‐1, SARS‐CoV‐2, and MERS [33, 34, 35, 36], suggesting their common important role in RNA‐binding.
Table 3.
Data collection and refinement statistics for the SARS‐CoV‐2 N‐CTD.
| Crystal | CTD (dimer) | CTD (dodecamer) |
|---|---|---|
| PDB ID | 7F2B | 7F2E |
| λ for data collection (Å) | 1.540562 | 0.9795 |
| Data collection | ||
| Space group | P1 | R3 |
| Cell dimension (Å) | ||
| a, b, c (Å) | 36.89, 37.21, 42.84 | 102.54, 102.54, 389.68 |
| α, β, γ, (°) | 78.68, 74.65, 65.46 | 90.00, 90.00, 120.00 |
| Total number of reflections observed | 69791 (7142) | 134323 (11777) |
| Number of unique reflections observed | 13449 (1358) | 27734 (2756) |
| Resolution limits | 28.70‐2.00 (2.07‐2.00) | 24.84‐3.10 (3.21‐3.10) |
| Rmerge | 0.062 (0.226) | 0.147 (0.589) |
| CC1/2 | 0.989 (0.475) | 0.989(0.837) |
| Average I/σ(I) | 25.26 (10.00) | 11.23 (2.46) |
| Completeness of data (%) | 99.94 (99.93) | 99.57 (99.31) |
| Data redundancy | 2.6 (2.7) | 4.8 (4.2) |
| Copies in the ASU | 2 | 12 |
| Refinement | ||
| Resolution limits | 28.70‐2.00 Å | 24.58‐3.10 Å |
| Number of reflections used | 13447 (1358) | 27675 (2754) |
|
Rfactor/Rfree (10% data) 7F2B Rfactor/Rfree (5% data) 7F2E |
0.157/0.214 | 0.268/0.297 |
| RMSD in bond‐lengths (Å) | 0.006 | 0.002 |
| RMSD in bond angles (°) | 0.80 | 0.43 |
| Number of atoms in the refined structure | ||
| Protein | 1648 | 8730 |
| Ligands | 32 | 20 |
| Solvent | 189 | 2 |
| Ramachandran plot (%) | ||
| Most favored | 98.08 | 93.72 |
| Additionally allowed | 1.92 | 5.44 |
| Average B‐factor (Å2) | 17.52 | 51.16 |
Values in parentheses refer to the highest resolution shell.
Fig. 6.
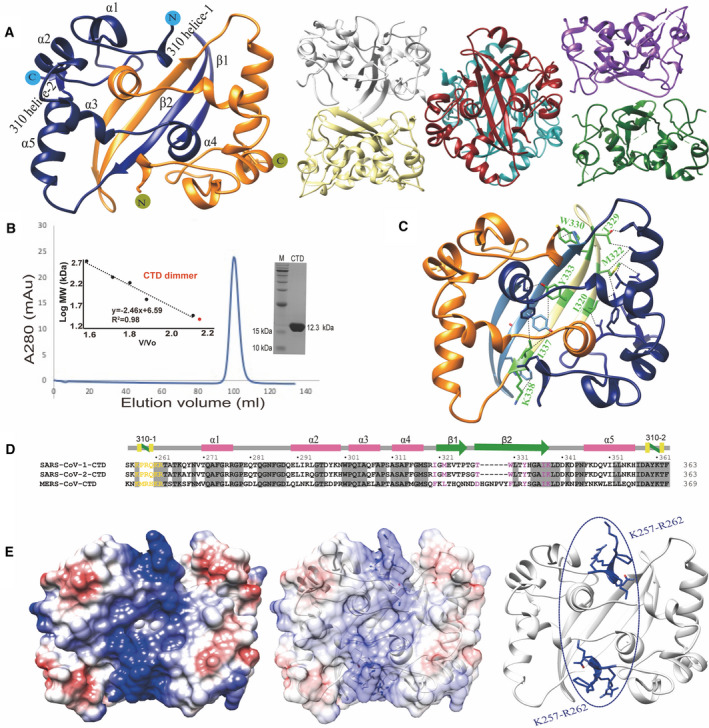
Crystal structures of N‐protein CTD. (A) The crystal structures of N‐protein CTD with 2 monomers in ASU (left) and 12 monomers in ASU (right). (B) Elution profile of N‐protein CTD by SEC using a Superdex 200 16/600 column (GE Healthcare). The inset shows the plotted standard curve for this column and the representative 15% SDS/PAGE showing the purified CTD protein. (C) The crystal structure of dimeric CTD shows the residues involved in dimer formation. (D) Sequence alignment of CTDs among SARS‐CoV‐1, SARS‐CoV‐2, and MERS. The residues involved in RNA‐binding are colored in yellow and involved in dimerization are colored in pink. (E) Surface view of dimeric CTD shows a positive binding pocket consisted of a stretch of positively charged residues between Lys257 and Arg262.
Discussion
The COVID‐19 pandemic has caused a historic impact on global health and the economy of society. Antibodies targeting the key structural proteins of SARS‐CoV‐2 have been proven to be effective in detecting and combating the virus. However, the precise selection of proper epitope is crucial for the development of antibody. Antibodies targeting structurally adjacent areas could have opposite effects depending on the conformational changes they induce. Thus, the understanding of the fundamental biology and underlying working mechanism of the key proteins of SARS‐CoV‐2 is crucial for the design of antibodies with high potency and specificity. In this study, we solved the crystal structures of SARS‐CoV‐2 N‐protein CTD and proved that it forms a dimer in solution form under physiological condition as determined by SEC. In contrast, CTD was crystallized in two different oligomeric forms, dimer, and dodecamer, probably due to the difference in crystallization condition. Dimeric CTD was crystallized under a condition at pH 6.2, while the dodecameric form was crystallized under pH 4.5. The condition of dimeric CTD is relatively closer to the physiological one, which agrees with our SEC result obtained at pH 7.4, suggesting dimer is more likely the physiological form of CTD in solution. On the other hand, the other inter‐CTD interfaces observed in dodecameric structure are much smaller compared to the dimer interface, also supporting the dimeric form is the minimal stable form of CTD. The key residues involved in dimer formation were identified and found conserved among different coronaviruses. Based on the surface charge analysis, we propose that a positively charged surface area is important for RNA‐binding. Recently the crystal structures of SARS‐CoV‐2 N‐protein CTD have also been reported by some other groups, which show a similar structure [18, 36].
N‐protein is relatively conserved among different coronaviruses. The sequence identity of N‐proteins among SARS‐CoV‐1, SARS‐CoV‐2, and MERS is more than 50%. Thus, the antibodies developed to target SARS‐CoV‐2 N‐protein could also bind N‐proteins of other coronaviruses with similar high affinities. This has an advantage in therapeutics because these antibodies could be used to treat multiple diseases, but meanwhile it also has a disadvantage in disease diagnosis due to the lack of specificity. In this work, we developed a series of sdAbs targeting different regions of SARS‐CoV‐2 N‐protein, which would add multiple new tools with distinct functions to the existing toolbox. The thermodynamics parameters obtained from the ITC experiments can lead to the optimization of the positive sdAbs in future.
The structure‐based design is a useful strategy to improve the binding properties of antibodies. To obtain the complex structure between N‐protein and sdAbs, we co‐purified CTD with sdAb‐N2, which presented as a complex on SEC with a 2:1 binding ratio. However, eventually the co‐crystals produced using the complex protein contained only CTD protein. We re‐loaded the complex protein on SEC after storage, which showed two peaks at the elution volumes for the dissociated individual components, reflecting the relatively low stability of the complex. When the affinity is not high enough, there would be a mixture of CTD dimer and CTD dimer+N2 during crystallization. If CTD dimer is easier to crystallize compared to the complex, it might shift the equilibrium, further dissociate the complex, and prevent the complex from crystallizing. It is also possible that the binding of N2 induces a conformational change of CTD dimer, making the structure of CTD dimer in complex less favorable for crystal packing. The directed evolution in combination with the structure‐based design would further improve the affinity and specificity of our sdAbs that make them practical candidates for the antiviral therapy and diagnosis purpose.
Materials and methods
sdAbs naive library screening
A phage display sdAbs naive library with a capacity of 109 cfu·µg−1 was used to screen the sdAbs. After three rounds of biopanning, sdAbs targeting to SARS‐CoV‐2 N‐protein were enriched. For each round of biopanning, the antigen was coated on the immune tubes with the coating of 5% nonfat milk as control, after which phage library was added. The phages were incubated with N‐protein for 1 h and washed with PBST (0.05% Tween 20+ PBS) buffer. The bound phages were eluted by digesting with 1 mL 0.25 mg·mL−1 trypsin and amplified in E. coli SS320 cells cultured in 2xYT media. 96 individual clones from the third round of panning were picked for ELISA verification. The positive clones were sequenced.
The cloning, expression, and purification of sdAbs
sdAbs were cloned into the pET22b vector, which contains a C‐terminal His‐tag and a N‐terminal pelB signal peptide. E. coli BL21 (DE3) cells (NEB) were used to express protein. The cells were grown at 37 °C in 2xYT media supplemented with 100 μg·mL−1 ampicillin and induced with 0.2 mm β‐D‐1‐thiogalactopyranoside (IPTG) when OD600 reached 0.6–0.8. The cells were grown at 25 °C for another 16 h and harvested by centrifugation (8000 g for 10 min at 4 °C). The cells were resuspended with a hypertonic solution (30 mm Tris, 20% w/v sucrose, 1 mm EDTA, pH 8.8) and incubated for 20 min at 4 °C. After centrifugation at 12 000 g at 4 °C for 30 min, the pellets were resuspended with a hypotonic solution (5 mm MgSO4), put on ice for 20 min, and then centrifuged at 12 000 g at 4 °C for 20 min. The supernatant was loaded onto a 10 mL HisTrap HP column (GE Healthcare) pre‐equilibrated with buffer A (10 mm HEPES, pH 7.4, 250 mm KCl). The protein was eluted using buffer A supplemented with 150 mm imidazole and concentrated using Amicon concentrators (3K MWCO from Millipore, Darmstadt, Germany). The concentrated protein was injected on a Superdex 200 16/600 gel filtration column (GE Healthcare) and eluted with buffer A [37].
The cloning, expression, and purification of the full‐length and truncated N‐proteins
The FLN was cloned into pET28a vector, which contains a N‐terminal His‐tag, a T7 tag and a thrombin cleavage site. Four truncated N‐protein versions were cloned into pET28HMT vector containing a N‐terminal His‐tag, an MBP‐tag, and a TEV cleavage site. Plasmids were transformed into E. coli BL21 (DE3) cells (NEB) for expression. The growth condition is same as the one for sdAbs except that the used induction temperature was 30 °C. The cells were lysed by sonication in a lysis buffer (10 mm HEPES, pH 7.4, 250 mm KCl, 0.03 mg·mL−1 DNase I, 0.3 mg·mL−1 lysozyme, 1 mm phenylmethanesulfonyl fluoride). The soluble fraction was collected after centrifugation at 12 000 g for 20 min at 4 °C. To remove the nucleotide, 0.03 mg·mL−1 DNase and RNase were added into the soluble fraction and incubated for 3 h at 20 °C. The soluble fraction was filtered through a 0.22‐μm filter. For the FLN, it was first purified by HisTrap HP column (GE Healthcare) using the same protocol as the one for sdAbs, and further purified by an SP Sepharose high efficiency column (GE Healthcare) using a linear gradient of 20–500 mm KCl in elution buffer (20 mm Tris, pH 6.8). Finally, the protein was injected on a Superdex 200 16/600 gel filtration column (GE Healthcare) and eluted with buffer A. For the truncated versions, the eluted protein from HisTrap HP column was first digested with TEV protease overnight and was further purified by an amylose resin column (New England Biolabs, Ipswich, MA, USA) and a TALON column (GE Healthcare) to remove the fusion tags. The protein from the flow‐through of TALON was collected and purified by a SP Sepharose high efficiency column (GE Healthcare) using the same protocol for the FLN. Finally, the protein was purified by a Superdex 200 16/600 gel filtration column (GE Healthcare). The protein samples were concentrated to 10 mg·mL−1 before stored at −80 °C. Analytical gel filtration column, Superdex 75 3.2/300, was used to determine the solution form of CTD‐N2 complex.
Isothermal titration calorimetry
The purified sdAbs and N‐proteins were dialyzed in a buffer containing 10 mm HEPES, pH 7.4, and 150 mm KCl, at 4 °C overnight. Titrations consisted of 20 injections of 2 μL of sdAbs into the cell solution containing N‐proteins at a 10‐fold lower concentration. Typical concentrations for the titrant were between 100 and 400 μm depending on the affinity. The reference cell was filled with water. Experiments were performed at 25 °C and a stirring speed of 750 rpm on a PEAQ‐ITC instrument (Malvern, Worcestershire, UK).
Crystallization, data collection, and structure determination
Protein crystals were grown at 18 °C using the hanging‐drop method. The crystals of CTD dodecamer (35 mg·mL−1) were grown in 0.1 M phosphate citrate, pH 4.5, and 40% PEG300. Diffraction data were collected on BL18U1 at Shanghai Synchrotron Radiation Facility (SSRF) [38] to a resolution of 3.1 Å. The crystals of CTD dimer (15 mg·mL−1) were set up in the presence of sdAb‐N2 and grown in 0.1 M potassium phosphate, pH 6.2, 0.2 M sodium chloride, and 52% PEG 200. Diffraction data from a single crystal was collected by in‐house X‐ray diffraction machine (Rigaku MicroMax‐007 HF) to a resolution of 2.0 Å. The datasets were indexed, integrated, and scaled using HKL [39]. Molecular replacements were performed using PHENIX [40, 41]. The model was built in COOT and refined by PHENIX [40]. UCSF Chimera was used to conduct all the structural analysis and generate structural figures [42].
Conflict of interest
The authors declare no conflict of interest.
Author contributions
ZH. J designed methodology. CL, YW. C, HJ done the experiments. ZY supervised the project and wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We thank J. Xu from the Instrument Analytical Center of the School of Pharmaceutical Science and Technology at Tianjin University for assisting in using the in‐house X‐ray diffraction machine, and the staff at the beamline BL18U1 at Shanghai Synchrotron Radiation Facility. Funding for this research was provided by the National Natural Science Foundation of China (no. 32022073 and 31972287 to Z.Y.), and the Natural Science Foundation of Tianjin (no. 19JCYBJC24500 to Z.Y.).
Zhenghu Jia, Chen Liu, Yuewen Chen and Heng Jiang contribute equally to this article
Contributor Information
Boqing Zhang, Email: jacky.zhang@beronigroup.com.
Zhiguang Yuchi, Email: yuchi@tju.edu.cn.
Data accessibility
The atomic coordinates and structure factors for CTD dimer (PDB ID 7F2B) and DBM CTD dodecamer (PDB ID 7F2E) have been deposited in the RCSB Protein Data Bank.
References
- 1. Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, Wang W, Song H, Huang B, Zhu N et al. (2020) Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395, 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL et al. (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R et al. (2020) A Novel Coronavirus from Patients with Pneumonia in China, 2019. N Engl J Med 382, 727–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brian DA & Baric RS (2005) Coronavirus genome structure and replication. Curr Top Microbiol Immunol 287, 1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fehr AR & Perlman S (2015) Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol 1282, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burbelo PD, Riedo FX, Morishima C, Rawlings S, Smith D, Das S, Strich JR, Chertow DS, Davey RT & Cohen JI (2020) Sensitivity in detection of antibodies to nucleocapsid and spike proteins of severe acute respiratory syndrome coronavirus 2 in patients with coronavirus disease 2019. J Infect Dis 222, 206–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McBride R, van Zyl M & Fielding BC (2014) The coronavirus nucleocapsid is a multifunctional protein. Viruses 6, 2991–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zuniga S, Cruz JL, Sola I, Mateos‐Gomez PA, Palacio L & Enjuanes L (2010) Coronavirus nucleocapsid protein facilitates template switching and is required for efficient transcription. J Virol 84, 2169–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cong Y, Ulasli M, Schepers H, Mauthe M, V'Kovski P, Kriegenburg F, Thiel V, de Haan CAM & Reggiori F (2020) Nucleocapsid protein recruitment to replication‐transcription complexes plays a crucial role in coronaviral life cycle. J Virol 94, e01925‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mu J, Xu J, Zhang L, Shu T, Wu D, Huang M, Ren Y, Li X, Geng Q, Xu Y et al. (2020) SARS‐CoV‐2‐encoded nucleocapsid protein acts as a viral suppressor of RNA interference in cells. Sci China Life Sci 63, 1413–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xiang F, Wang X, He X, Peng Z, Yang B, Zhang J, Zhou Q, Ye H, Ma Y, Li H et al. (2020) Antibody detection and dynamic characteristics in patients with coronavirus disease 2019. Clin Infect Dis 71, 1930–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ni L, Ye F, Cheng ML, Feng Y, Deng YQ, Zhao H, Wei P, Ge J, Gou M, Li X et al. (2020) Detection of SARS‐CoV‐2‐specific humoral and cellular immunity in COVID‐19 convalescent individuals. Immunity 52, 971–977.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chang CK, Hou MH, Chang CF, Hsiao CD & Huang TH (2014) The SARS coronavirus nucleocapsid protein–forms and functions. Antiviral Res 103, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yu IM, Oldham ML, Zhang J & Chen J (2006) Crystal structure of the severe acute respiratory syndrome (SARS) coronavirus nucleocapsid protein dimerization domain reveals evolutionary linkage between corona‐ and arteriviridae. J Biol Chem 281, 17134–17139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Saikatendu KS, Joseph JS, Subramanian V, Neuman BW, Buchmeier MJ, Stevens RC & Kuhn P (2007) Ribonucleocapsid formation of severe acute respiratory syndrome coronavirus through molecular action of the N‐terminal domain of N protein. J Virol 81, 3913–3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lin SY, Liu CL, Chang YM, Zhao J, Perlman S & Hou MH (2014) Structural basis for the identification of the N‐terminal domain of coronavirus nucleocapsid protein as an antiviral target. J Med Chem 57, 2247–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Papageorgiou N, Lichiere J, Baklouti A, Ferron F, Sevajol M, Canard B & Coutard B (2016) Structural characterization of the N‐terminal part of the MERS‐CoV nucleocapsid by X‐ray diffraction and small‐angle X‐ray scattering. Acta Crystallogr D 72, 192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Peng Y, Du N, Lei Y, Dorje S, Qi J, Luo T, Gao GF & Song H (2020) Structures of the SARS‐CoV‐2 nucleocapsid and their perspectives for drug design. EMBO J 39, e105938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cong Y, Kriegenburg F, de Haan CAM & Reggiori F (2017) Coronavirus nucleocapsid proteins assemble constitutively in high molecular oligomers. Sci Rep 7, 5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hwang WC, Lin Y, Santelli E, Sui J, Jaroszewski L, Stec B, Farzan M, Marasco WA & Liddington RC (2006) Structural basis of neutralization by a human anti‐severe acute respiratory syndrome spike protein antibody, 80R. J Biol Chem 281, 34610–34616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Walls AC, Xiong X, Park YJ, Tortorici MA, Snijder J, Quispe J, Cameroni E, Gopal R, Dai M, Lanzavecchia A et al. (2019) Unexpected Receptor Functional Mimicry Elucidates Activation of Coronavirus Fusion. Cell 176, 1026–1039 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang L, Shi W, Joyce MG, Modjarrad K, Zhang Y, Leung K, Lees CR, Zhou T, Yassine HM, Kanekiyo M et al. (2015) Evaluation of candidate vaccine approaches for MERS‐CoV. Nat Commun 6, 7712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang L, Shi W, Chappell JD, Joyce MG, Zhang Y, Kanekiyo M, Becker MM, van Doremalen N, Fischer R, Wang N et al. (2018) Importance of neutralizing monoclonal antibodies targeting multiple antigenic sites on the middle east respiratory syndrome coronavirus spike glycoprotein to avoid neutralization escape. J Virol 92, e02002‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hamers‐Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hamers C, Songa EB, Bendahman N & Hamers R (1993) Naturally occurring antibodies devoid of light chains. Nature 363, 446–448. [DOI] [PubMed] [Google Scholar]
- 25. De Vlieger D, Ballegeer M, Rossey I, Schepens B & Saelens X (2019) Single‐domain antibodies and their formatting to combat viral infections. Antibodies 8 (1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Govaert J, Pellis M, Deschacht N, Vincke C, Conrath K, Muyldermans S & Saerens D (2012) Dual beneficial effect of interloop disulfide bond for single domain antibody fragments. J Biol Chem 287, 1970–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rotman M, Welling MM, van den Boogaard ML, Moursel LG, van der Graaf LM, van Buchem MA, van der Maarel SM & van der Weerd L (2015) Fusion of hIgG1‐Fc to 111In‐anti‐amyloid single domain antibody fragment VHH‐pa2H prolongs blood residential time in APP/PS1 mice but does not increase brain uptake. Nucl Med Biol 42, 695–702. [DOI] [PubMed] [Google Scholar]
- 28. Forsman A, Beirnaert E, Aasa‐Chapman MM, Hoorelbeke B, Hijazi K, Koh W, Tack V, Szynol A, Kelly C, McKnight A et al. (2008) Llama antibody fragments with cross‐subtype human immunodeficiency virus type 1 (HIV‐1)‐neutralizing properties and high affinity for HIV‐1 gp120. J Virol 82, 12069–12081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wrapp D, De Vlieger D, Corbett KS, Torres GM, Wang N, Van Breedam W, Roose K, van Schie L, Team VCCR, Hoffmann M et al. (2020) Structural basis for potent neutralization of betacoronaviruses by single‐domain camelid antibodies. Cell 181, 1004–1015 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huo J, Le Bas A, Ruza RR, Duyvesteyn HME, Mikolajek H, Malinauskas T, Tan TK, Rijal P, Dumoux M, Ward PN et al. (2020) Neutralizing nanobodies bind SARS‐CoV‐2 spike RBD and block interaction with ACE2. Nat Struct Mol Biol 27, 846–854. [DOI] [PubMed] [Google Scholar]
- 31. Chang CK, Jeyachandran S, Hu NJ, Liu CL, Lin SY, Wang YS, Chang YM & Hou MH (2016) Structure‐based virtual screening and experimental validation of the discovery of inhibitors targeted towards the human coronavirus nucleocapsid protein. Mol Biosyst 12, 59–66. [DOI] [PubMed] [Google Scholar]
- 32. Zhang X, Tan Y, Ling Y, Lu G, Liu F, Yi Z, Jia X, Wu M, Shi B, Xu S et al. (2020) Viral and host factors related to the clinical outcome of COVID‐19. Nature 583, 437–440. [DOI] [PubMed] [Google Scholar]
- 33. Chen CY, Chang CK, Chang YW, Sue SC, Bai HI, Riang L, Hsiao CD & Huang TH (2007) Structure of the SARS coronavirus nucleocapsid protein RNA‐binding dimerization domain suggests a mechanism for helical packaging of viral RNA. J Mol Biol 368, 1075–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hsin WC, Chang CH, Chang CY, Peng WH, Chien CL, Chang MF & Chang SC (2018) Nucleocapsid protein‐dependent assembly of the RNA packaging signal of Middle East respiratory syndrome coronavirus. J Biomed Sci 25, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Takeda M, Chang CK, Ikeya T, Guntert P, Chang YH, Hsu YL, Huang TH & Kainosho M (2008) Solution structure of the c‐terminal dimerization domain of SARS coronavirus nucleocapsid protein solved by the SAIL‐NMR method. J Mol Biol 380, 608–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang M, He S, Chen X, Huang Z, Zhou Z, Zhou Z, Chen Q, Chen S & Kang S (2020) Structural insight into the SARS‐CoV‐2 nucleocapsid protein C‐terminal domain reveals a novel recognition mechanism for viral transcriptional regulatory sequences. Front Chem 8, 624765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ingham VA, Anthousi A, Douris V, Harding NJ, Lycett G, Morris M, Vontas J & Ranson H (2020) A sensory appendage protein protects malaria vectors from pyrethroids. Nature 577, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang Q‐S, Yu F, Huang S, Sun B, Zhang K‐H, Liu K, Wang Z‐J, Xu C‐Y, Wang S‐S, Yang L‐F et al. (2015) The macromolecular crystallography beamline of SSRF. Nucl Sci Tech 26, 12–17. [Google Scholar]
- 39. Minor W, Cymborowski M, Otwinowski Z & Chruszcz M (2006) HKL‐3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr D 62, 859–866. [DOI] [PubMed] [Google Scholar]
- 40. Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse‐Kunstleve RW et al. (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Crystallogr D 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Emsley P & Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Crystallogr D 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- 42. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC & Ferrin TE (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The atomic coordinates and structure factors for CTD dimer (PDB ID 7F2B) and DBM CTD dodecamer (PDB ID 7F2E) have been deposited in the RCSB Protein Data Bank.