

Abstract
Objective
Heightened inflammation, dysregulated immunity, and thrombotic events are characteristic of hospitalized COVID‐19 patients. Given that platelets are key regulators of thrombosis, inflammation, and immunity they represent prime candidates as mediators of COVID‐19‐associated pathogenesis. The objective of this study was to understand the contribution of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) to the platelet phenotype via phenotypic (activation, aggregation) and transcriptomic characterization.
Approach and Results
In a cohort of 3915 hospitalized COVID‐19 patients, we analyzed blood platelet indices collected at hospital admission. Following adjustment for demographics, clinical risk factors, medication, and biomarkers of inflammation and thrombosis, we find platelet count, size, and immaturity are associated with increased critical illness and all‐cause mortality. Bone marrow, lung tissue, and blood from COVID‐19 patients revealed the presence of SARS‐CoV‐2 virions in megakaryocytes and platelets. Characterization of COVID‐19 platelets found them to be hyperreactive (increased aggregation, and expression of P‐selectin and CD40) and to have a distinct transcriptomic profile characteristic of prothrombotic large and immature platelets. In vitro mechanistic studies highlight that the interaction of SARS‐CoV‐2 with megakaryocytes alters the platelet transcriptome, and its effects are distinct from the coronavirus responsible for the common cold (CoV‐OC43).
Conclusions
Platelet count, size, and maturity associate with increased critical illness and all‐cause mortality among hospitalized COVID‐19 patients. Profiling tissues and blood from COVID‐19 patients revealed that SARS‐CoV‐2 virions enter megakaryocytes and platelets and associate with alterations to the platelet transcriptome and activation profile.
Keywords: COVID‐19, megakaryocytes, platelets, thrombosis
ESSENTIALS
-
•
Thrombosis, inflammation, and dysregulated immunity are characteristic of hospitalized COVID‐19 patients.
-
•
COVID‐19 hospital admission platelet count, size, and maturity associate with critical illness and mortality.
-
•
COVID‐19 platelets are hyperreactive and have a unique transcriptomic profile.
-
•
Anti‐platelet therapy use in hospitalized COVID‐19 patients may reduce adverse clinical outcomes.
Alt-text: Unlabelled Box
1. INTRODUCTION
The severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) is responsible for the coronavirus disease (COVID‐19) global pandemic. COVID‐19 is a complex disease with a broad spectrum of clinical presentations, ranging from asymptomatic to severe respiratory failure with multi‐organ failure and death. Microvascular thrombosis of the pulmonary vasculature and other vascular beds is frequently observed in COVID‐19.1., 2. Patients infected with SARS‐CoV‐2 are at heightened risk for both venous and arterial thrombosis, which further contribute to organ failure and all‐cause mortality.3., 4. Autopsy and clinical data from our group and others highlight the role of megakaryocytes and platelets in the pathogenesis of COVID‐19.1., 5., 6.
Thrombocytopenia is a common laboratory abnormality in critically ill patients7 and is associated with poor clinical outcomes, including death.7., 8. Thrombocytopenia has been reported in patients hospitalized with COVID‐19, and lower platelet counts are associated with worse clinical outcomes.9 A meta‐analysis of 31 studies with 7613 participants noted a lower platelet count in severe COVID‐19 infection and was associated with a 3‐fold increase in the risk of developing severe COVID‐19.10 However, there is uncertainty on the different cut‐offs of platelet count in COVID‐19 and the platelet count trajectory during hospitalization.
Other indices of platelets, including platelet size and platelet maturity, are associated with increased platelet activity and clinical events.11., 12., 13. Larger platelets are more metabolically active and have a greater prothrombotic potential,14 with increased mean platelet volume (MPV) associated with increased morbidity and all‐cause mortality.15 Immature platelet fraction (IPF) reflects the number of circulating young platelets, and increased IPF is associated with morbidity and mortality in multiple disease states.16., 17., 18.
Platelets are shed into circulation by megakaryocytes, and during this process, megakaryocytes distribute their transcriptome into platelets. Once in the circulation, platelets can respond to local and systemic conditions and affect other cell types, including monocytes and endothelial cells.19., 20., 21. Platelet activity is associated with the platelet ribonucleic acid (RNA) expression profiles, and platelet‐viral interactions alter the platelet transcriptome.22 Consistent with increased platelet activity (increased P‐selectin and PAC‐1 (activated GP IIb/IIIa) expression), one study reports that platelets isolated from COVID‐19 patients are hyperreactive and have an altered transcriptomic signature compared to disease‐free controls.6 Several genome‐wide association studies (GWAS) have identified genetic regulators of platelet count and platelet size.23 In addition, recent studies have found that platelet immaturity has a distinct platelet transcriptome.24., 25. Thus, transcriptomic profiling of platelets isolated from patients infected with SARS‐CoV‐2 may provide insight into platelet count, size, and maturity during COVID‐19.
In this study, we analyzed platelet indices from the complete blood count (CBC) data from NYU Langone Health (NYULH), a multihospital health system in New York City, to investigate platelet indices at presentation and trajectory during hospitalization. Following adjustment for demographics, clinical risk factors, medication use, and biomarkers of inflammation and thrombosis, platelet count, size, and immaturity are all associated with increased critical illness and all‐cause mortality. Additionally, we demonstrate SARS‐CoV‐2 present in platelets of hospitalized COVID‐19 patients and provide evidence that COVID‐19 platelets are reprogrammed toward a prothrombotic phenotype. In vitro studies with megakaryocytes highlight that the transcriptomic reprogramming mediated by SARS‐CoV‐2 is distinct from the coronavirus responsible for the common cold (CoV‐OC43). Our findings support investigating the use of anti‐platelet therapy in COVID‐19 patients.
2. MATERIALS AND METHODS
The data that support the findings of this study are available from the corresponding author upon reasonable request.
2.1. Study population
We identified adults age ≥18 years with a positive real‐time polymerase chain reaction (RT‐PCR) COVID‐19 test between March 1, 2020 and June 26, 2020 who were admitted to NYULH. Patients hospitalized with a positive PCR test for COVID‐19 were eligible for this retrospective, observational study if ≥1 CBC was measured during hospital admission (n = 3915). Follow‐up was complete through June 26, 2020. Identifiable patient‐level data from this project are not available to the public. The study was approved by the NYU Grossman School of Medicine Institutional Review Board (IRB) and performed with a waiver of informed consent.
2.2. Data collection
Data were obtained from the electronic health record (Epic Systems), which is an integrated electronic health record (EHR), including all inpatient and outpatient visits in the health system.
At all four NYULH inpatient facilities, routine CBC for individuals with suspected or confirmed diagnoses of COVID‐19 was included in COVID‐19‐‐specific admission order sets in the EHR at the time of hospital admission. At all NYULH sites, CBC assay was measured using SysmexR XN‐Series of automated hematology analyzers (TOA Medical Electronics). The initial platelet indices and all platelet indices measured during hospital admission were recorded for all eligible patients. The measures analyzed were platelet count, mean platelet volume (MPV), and IPF (%).
The platelet count was stratified into low (thrombocytopenia; <149 × 109/L), normal (150–400 × 109/L), and high (>401 × 109/L). We conducted sensitivity analyses using different severities of thrombocytopenia (<49 × 109/L, 50–100 × 109/L, and 101–150 × 109/L). Both MPV and IPF were stratified into tertiles based on values obtained at baseline.
2.3. Study endpoint & variables
Demographic variables included age, sex, race/ethnicity (defined as non‐Hispanic White, non‐Hispanic Black, Hispanic, Asian, multiracial/other, and unknown), smoking status, and body mass index (BMI). Pre‐existing comorbidities included hypertension, hyperlipidemia, coronary artery disease (CAD), heart failure, diabetes, chronic kidney disease, and atrial fibrillation. Prior medication information included statins, beta‐blockers, angiotensin‐converting enzyme inhibitor (ACE‐I) or angiotensin receptor blocker (ARB), and oral anticoagulants. Critical illness was defined as mechanical ventilation or transfer to the intensive care unit.
2.4. Statistical analysis
Continuous variables are shown using mean (standard deviation [SD]) and median (interquartile range [IQR]) and compared using the non‐parametric Kruskal‐Wallis test for all non‐normally distributed data. Categorical variables are reported as frequency rates and percentages and compared by Chi‐square tests. The longitudinal trajectory of the platelet indices was created using polynomial regression fitting, and are plotted per day of hospitalization within 28 days of admission. Analyses were performed with and without imputation using the mean of all the labs collected within 28 days of admission. Trajectory plots were stratified by death and critical illness.
Logistic regression models were generated to estimate the odds of the study endpoints, adjusted for demographics, clinical comorbidities, vital signs at presentation, and baseline medications. Covariates in the multivariable models included age; sex; race; BMI; tobacco use; race‐ethnicity; hypertension; hyperlipidemia; chronic kidney disease; prior heart failure; asthma or chronic obstructive pulmonary disease; CAD; cancer; diabetes; temperature; SpO2; and initial laboratory results for lymphocyte count, ferritin and C‐reactive protein (CRP), D‐dimer, creatinine, troponin, and procalcitonin. Statistical analyses were performed using statistical software R (R Foundation for Statistical Computing), with packages ggplot2, and base R. Statistical tests are two‐sided, and P‐values <.05 were considered statistically significant.
2.5. Platelet RNA sequencing
2.5.1. Population
A cohort of eight hospitalized COVID‐19 patients (n = 8) were recruited from NYULH between May 11 and 21, 2020. SARS‐CoV‐2 infection was confirmed by RT‐PCR, in accordance with current standards. All COVID‐19 patients and control donors were recruited under study protocols approved by the NYULH IRB. Each study participant or their legal authorized representative gave written informed consent for study enrollment in accordance with the Declaration of Helsinki. For COVID‐19 patients, enrollment criteria included age greater than 18, hospital admission, positive SARS‐CoV‐2 testing, and informed consent. COVID‐19 patients were monitored until discharge or death.
2.5.2. Platelet isolation
After an initial 2 cc discard, blood was collected into tubes containing 3.2% sodium citrate. Blood was allowed to rest for 10 min prior to isolation of platelet rich plasma by centrifugation at 200 g for 10 min. Platelets were isolated and purified by incubation with microbeads to deplete leukocytes and red blood cells (EasySep™ Human CD45 Depletion Kit II and EasySep™ RBC Depletion Reagent). Isolated platelets lysed in 500 uL of trizol and stored at −80°C. RNA was isolated with Direct‐zol RNA microspin columns (Zymo Research), and quality and quantity were determined with a Bioanalyzer 2100 (Agilent Technologies). Sequencing libraries were barcoded and prepared using the Clontech SMART‐Seq HT with Nxt HT kit (Takara Bio USA), and libraries sequenced single end on a Illumina NovaSeq 6000.
2.5.3. Data processing
Refer to Appendix S1 in supporting information. Data are accessible via GSE176480.
2.5.4. Megakaryocyte and SARS‐CoV‐2 and CoV‐OC43 treatment
Refer to Appendix S1.
2.5.5. Primary megakaryocyte culture and flow cytometry
Purified peripheral blood CD34+ cells were obtained from the New York Blood Center. Cells were cultured in StemSpan SFEM II (Stem Cell) supplemented with stem cell factor (100 ng/ml, R&D Systems) and thrombopoietin (TPO, 50 ng/ml, R&D Systems) for 5 days. Cells were cultured with 50 ng/ml TPO only from days 5–11. On day 11, surface expression of ACE2, CD147, DC‐SIGN, L‐SIGN, CLEC10A, CLEC4G, and ASGR1 was determined by flow cytometry. DC‐SIGN (Bioglend, Cat # 330106), L‐SIGN (eBioscience, Cat # 17–2999–42), CLEC10A (Biolegend, Cat # 354705), and CLEC4G (R&D Systems, Cat # FAB2947A) antibodies were conjugated to APC, CD147 (Biolegend, Cat # 306212) was conjugated to PE and used at a final concentration of (1:100). ACE‐2 (Novus, Cat #NBP2‐80038) and ASGR1 (R&D Systems, Cat # MAB4394) antibodies were incubated with the cells at 1:100, prior to washing and staining with a mouse‐anti‐APC secondary (1:100), prior to analysis. Flow cytometry was performed on a Miltenyi MACSquant 10.
2.5.6. Megakaryocyte and platelet immunohistochemistry, transmission electron microscopy, and in situ hybridization
Refer to Appendix S1.
2.5.7. Platelet aggregation
After an initial 2 cc discard, blood was collected into tubes containing 3.2% sodium citrate. Blood was allowed to rest for 10 min prior to isolation of platelet rich plasma by centrifugation at 200 g for 10 min.
Aggregation was measured using a light transmission aggregometry (Helena AggRAM) in response to ADP (1 µM), epinephrine (0.1 µM), and collagen (0.2 µM) at 37°C under stirred conditions as previously described.26
3. RESULTS
3.1. Admission platelet indices (count, size, and maturity) associate with increased critical illness and all‐cause mortality
Biomarkers of platelet activation associate with critical illness and death in patients with COVID‐19,5 and viruses are known to directly influence platelet count and size.27., 28. To investigate the consequence of SARS‐CoV‐2 infection on platelet count, size, and maturity, and the outcomes of critical illness and death, we analyzed CBCs collected from 3915 patients hospitalized with COVID‐19 within the NYULH system between March 2, 2020 and June 25, 2020. The median age was 65 years (IQR, 53–77), 42.9% were female, and 36.8% were White. A total of 1678 (38.5%) had diabetes, 580 (13.3%) had cancer, and 994 (22.8%) had CAD (Table 1 ). Overall, 1415 (36%) experienced critical illness, and 948 (29%) were discharged to hospice or died at a median of 10 (IQR, 5–22) days after hospital admission.
TABLE 1.
Population characteristics
| Overall population (n = 3915) |
|
|---|---|
| Age, median (IQR) | 64 (52–77) |
| Sex | |
| Female, n (%) | 1544 (42.9) |
| Race | |
| White, n (%) | 1576 (40.2) |
| African American, n (%) | 580 (14.8) |
| Hispanic, n (%) | 837 (21.4) |
| Asian, n (%) | 277 (7.1) |
| Other, n (%) | 456 (11.6) |
| Unknown or declined, n (%) | 189 (4.8) |
| Comorbidities | |
| BMI, median (IQR) | 28.4 (25–33) |
| Asthma or chronic obstructive pulmonary disease, n (%) | 1045 (26.7) |
| CKD, n (%) | 1023 (26.1) |
| Coronary artery disease, n (%) | 968 (24.7) |
| Current smoker, n (%) | 203 (5.2) |
| Diabetes, n (%) | 1635 (41.8) |
| Hyperlipidemia, n (%) | 2164 (55.3) |
| Hypertension, n (%) | 2683 (68.5) |
| Malignancy, n (%) | 536 (13.7) |
| Congestive heart failure, n (%) | 756 (19.3) |
| Vitals | |
| Temperature oC, median (IQR) | 37.1 (36.8–37.6) |
| SpO2%, median (IQR) | 95 (93–97) |
| Labs | |
| C‐reactive protein mg/L, median (IQR) | 109 (51.7–171) |
| D‐dimer ng/ml, median (IQR) | 402 (243–766) |
| Lymphocytes 103/ul, median (IQR) | 0.9 (0.6–1.3) |
| Creatinine mg/dL, median (IQR) | 0.98 (0.80–1.39) |
| Ferritin ng/ml, median (IQR) | 709 (333–1406) |
| Troponin I ng/ml, median (IQR) | 0.03 (0.02–0.1) |
| Procalcitonin ng/ml, median (IQR) | 0.14 (0.06–0.40) |
| Platelet indices | |
| Platelet count 103/ul, median (IQR) | 205 (159–267) |
| Mean platelet volume fL, median (IQR) | 10.5 (9.9–11.2) |
| Immature platelet fraction %, median (IQR) | 3.3 (2.1–5.0) |
Abbreviations: BMI, body mass index; CKD, chronic kidney disease; IQR, interquartile range.
Among 3915 patients with platelet count data at hospital presentation, 2894 (73%) had a normal platelet count (150–400 × 109/L), 795 (20%) had thrombocytopenia (<150 × 109/L), and 226 (7%) had an elevated platelet count (>400 × 109/L). Patients with lower platelet counts were older; more likely to be male; and more likely to have comorbidities, including chronic kidney disease, CAD, and heart failure (Table S1 in supporting information). Critical illness and all‐cause mortality were increased in patients with thrombocytopenia (Figure 1A ). Compared to those with a normal platelet count, patients with thrombocytopenia were more likely to experience critical illness (adjusted odds ratio [adjOR] 1.4, 95% confidence interval [CI] 1.1–1.7, P = .0002) and all‐cause mortality (adjOR 1.6, 95% CI 1.3–1.9, P = .0002, Figure 1A). When thrombocytopenia was further stratified into severe <50 × 109/L, moderate 50–100 × 109/L, and mild 100–150 × 109/L, the association between platelet count and adverse events was most pronounced in subjects with a platelet count <50 × 109/L (Table S2 in supporting information). Interestingly, there was a trend toward less critical illness in those with elevated platelet counts >400 × 109/L, but this did not reach statistical significance compared to a normal platelet count (adjOR 0.75, 95% CI 0.53–1.05, P = .10). Platelet count trajectory by critical illness and all‐cause mortality is presented in Figure 2A and Figure S1 in supporting information. Platelet count increased during the course of hospitalization and peaked at approximately 8 days into hospitalization.
FIGURE 1.

Critical illness and all‐cause mortality stratified by platelet count, mean platelet volume (MPV; fL), and immature platelet fraction (IPF; %). A, Stratification based on low, normal, and elevated platelet count at hospital presentation and adjusted odds ratio for critical illness and all‐cause mortality based on platelet count. B, Stratification based on tertile of MPV at hospital presentation and adjusted odds ratio for critical illness and all‐cause mortality based on MPV. C, Stratification based on tertile of IPF at hospital presentation and adjusted odds ratio for critical illness and all‐cause mortality based on IPF. Covariates in the multivariable models included age, sex, race, body mass index, tobacco use, race‐ethnicity, hypertension, hyperlipidemia, chronic kidney disease, prior heart failure, asthma or chronic obstructive pulmonary disease, coronary artery disease, cancer, diabetes, temperature, SpO2, and initial laboratory results for lymphocyte count, ferritin and C‐reactive protein, D‐dimer, creatinine, troponin, and procalcitonin
FIGURE 2.

Trajectory of platelet indices during 28 days of hospitalization for patients with and without critical illness and all‐cause mortality. A, Platelet count trajectory, (B) mean platelet volume (MPV) trajectory, and (C) immature platelet fraction (IPF) trajectory. Yes = yes critical illness or all‐cause mortality, No = no critical illness or all‐cause mortality
Median MPV was 10.5 fL (IQR, 9.9–11.2). When stratified into tertiles, patients with a higher MPV were older; more frequently African American; and more likely to have CAD, diabetes, and heart failure (Table S3 in supporting information). Critical illness and all‐cause mortality were highest in patients with a higher MPV (Figure 1B). After adjustment for demographics, comorbidities, prior medications, and baseline laboratory values, patients in the highest MPV tertile had higher odds of critical illness (adjOR 1.5, 95% CI 1.3–1.8) and all‐cause mortality (adjOR 1.3, 95% CI 1.1–1.5) compared to lowest MPV tertile (Figure 1B). Because MPV inversely correlates with platelet count, we excluded subjects with a low platelet count in a sensitivity analysis and found that subjects in the highest MPV tertile had higher odds of critical illness and all‐cause mortality (Figure S2A–B in supporting information). The trajectory of MPV during hospitalization stratified by critical illness and all‐cause mortality is presented in Figure 2B. In contrast to patients without an event, MPV increases in patients who were critical ill or died.
Immature platelet fraction (IPF) was collected beginning on April 23, 2020, and was available for 427 patients. Median IPF was 7.4% (IQR, 4.9–10.5). Stratified into tertiles, there were no significant differences between the three tertiles in age or comorbidities (Table S4 in supporting information). Critical illness was more common in patients with the highest IPF tertile; however, mortality was not significantly different between groups (Figure 1C). After adjustment for demographics, comorbidities, prior medications, and baseline laboratory values, patients belonging to the highest tertile of IPF had higher odds of critical illness (adjOR 2.4, 95% CI 1.4–4.2) and a trend toward death (adjOR 2.1, 95% CI 0.9–5.0) versus the lowest tertile (Figure 1C). After excluding individuals with a low platelet count in a sensitivity analysis, subjects in the highest IPF tertile had higher odds of critical illness (Figure S2A–B). The trajectory of IPF during hospitalization stratified by critical illness and all‐cause mortality is presented in Figure 2 and Figure S1.
3.2. Megakaryocytes actively internalize SARS‐CoV‐2
In the setting of viral infections, platelets can internalize viral particles, including influenza, human immunodeficiency virus (HIV), hepatitis C virus, and dengue, resulting in platelet activation.27., 29. To explore the interaction between SARS‐CoV‐2 and platelets, we first assessed whether megakaryocytes, the precursor of platelets, can internalize SARS‐CoV‐2. Autopsy data from COVID‐19 patients revealed megakaryocytes in the bone marrow, with morphology consistent with active platelet production (Figure 3A ). Furthermore, in one autopsy in which the bone marrow was examined within 10 days of COVID‐19 symptom onset, megakaryocytes with SARS‐CoV‐2 engulfed virions were found (Figure 3B). Additionally, by in situ hybridization, we find evidence of replicating SARS‐CoV‐2 within a lung megakaryocyte from a deceased COVID‐19 patient (Figure S3 in supporting information).
FIGURE 3.

Severe acute respiratory syndrome coronavirus 2 virions are present in both megakaryocytes and platelets of COVID‐19 patients. A, H&E staining and CD61 immunohistochemistry of bone marrow of a COVID‐19 patient. B, Transmission electron microscopy of bone marrow of a COVID‐19 patient, identified SARS‐CoV‐2 viruses are circled in red. C, Transmission electron microscopy of platelets from three COVID‐19 patients; dash frame is enlarged area of corresponding image. D, Amplification plots of SARS‐CoV‐2‐specific real‐time quantitative polymerase chain reaction (RT‐qPCR) of Meg01s treated with media control (n = 2) or SARS‐CoV‐2 (n = 3) for 6 h. E, SARS‐CoV‐2‐‐specific RT‐qPCR of Meg01s treated with media control (n = 2) or SARS‐CoV‐2 (n = 3) for 6 h at 37°C. F, Expression of ACE2, CD147, DC‐SIGN, L‐SIGN, CLEC10A, CLEC4G, and ASGR1 mRNA in platelets from COVID‐19 patients and matched controls as determined by RNASeq. G, Surface expression of CD147 as determined by flow cytometry of CD34±derived megakaryocytes. Data are expressed as mean ± standard error of the mean, ****P < .001
Consistent with the observation in megakaryocytes and a previous report in platelets,30 we find viral particles present in approximately 39% of circulating platelets from three COVID‐19–positive patients, indicative that SARS‐CoV‐2 is either transferred from megakaryocytes to platelets or actively engulfed by circulating platelets (Figure 3C, Table S5 in supporting information). The presence of SARS‐CoV‐2 in platelets did not appear to alter their morphology (Table S6 in supporting information). In further support of viral entry, exposure of Meg01 cells with authentic SARS‐CoV‐2 for 6 h results in the detection of SARS‐CoV‐2 RNA (Figure 3D–E). Conversely, Meg01 exposure with a coronavirus responsible for the common cold (CoV‐OC43)31 does not lead to detectable levels of CoV‐OC43 mRNA (data not shown).
To understand the mechanism by which megakaryocytes take up SARS‐CoV‐2 several potential entry receptors of SARS‐CoV‐2 were profiled: ACE2, DC‐SIGN, L‐SIGN, CLEC10A, CLEC4G, CD147 and ASGR1.32 At the level of the platelet transcriptome, only CD147 was found to be expressed (Figure 3F), and consistent with this, only CD147 was found to be expressed on the surface of primary megakaryocytes (Figure 3G, Figure S4 in supporting information).
3.3. Platelets from COVID‐19 patients are hyperreactive and have a unique transcriptome
Because platelets are affected by viral entry,33 we recruited a cohort of eight hospitalized COVID‐19 patients and ten age, sex, and race/ethnicity controls with similar risk factor profiles (demographics in Table S7 in supporting information). While platelet count was similar between the groups, MPV was increased in subjects with COVID‐19 (Figure 4A–B ). Platelet aggregation in response to submaximal platelet agonists (ADP, 1 uM; epinephrine, 0.1 uM; and collagen, 0.2 uM) revealed that platelets from COVID‐19 patients were hyperreactive compared to controls (Figure 4C–D). However, in our cohort, we found a degree of heterogeneity associated with platelet count and activity within the COVID‐19 population.
FIGURE 4.

Platelets from COVID‐19 patients are hyperreactive. A, Platelet count, (B) mean platelet volume (MPV) in a cohort of eight COVID‐19 patients and nine controls. C, Representative light transmission aggregometry (LTA) plots following platelet exposure to ADP (1 uM), epinephrine (0.1 uM), collagen (0.2 uM), and (D) quantification of platelet aggregation in controls and COVID‐19 patients. Data are expressed as mean ± standard error of the mean, n = 6‐9/grp, * P < .01, **P < .005
To investigate the potential mechanism(s) as to how SARS‐CoV‐2 may alter the platelet phenotype, we performed RNA sequencing of RNA isolated from highly purified platelets26 from eight COVID‐19 patients and ten age, sex, and race/ethnicity controls without COVID‐19. Principal component analysis demonstrates a clear separation in the platelet transcriptomic profile of platelets isolated from subjects with COVID‐19 versus controls (Figure 5A ). We analyzed the platelet transcriptomic data using DESeq234 and found 2049 discriminating transcripts (Padj <.05; 1078 upregulated [log2FC >1] and 971 downregulated [log2FC < −1]) between the COVID‐19 patients and controls (Figure 5B–C). Congruent with a previous report,6 we find a significant overlap between platelet transcripts differentially regulated in our COVID‐19 New York cohort compared to a COVID‐19 patient population in Utah (R = 0.86, P = 2.2 × 10−16, Figure S5A in supporting information). Of note, in our cohort, the COVID‐19 platelet transcriptome was not found to be very different between those who survived (n = 3) versus those who died (n = 5; 35 DE transcripts adjusted P‐value <.05 |log2fc| >1). Additionally, alterations to the platelet transcriptome are largely independent of aspirin use (Figure S5B). To investigate the direct viral effect on the platelet phenotype, megakaryocytes were exposed to SARS‐CoV‐2 and CoV‐OC43. Incubation of Meg01 cells with SARS‐CoV‐2 resulted in a significant increase in the proinflammatory transcripts previously identified in our study and the Utah cohort, LGALS3BP and S100A9, relative to vehicle‐treated cells (Figure 5D, red bars). Conversely, exposure to CoV‐OC43 did not result in changes to LGALS3BP and S100A9 mRNA transcription (Figure 5D, blue bars), indicating that direct SARS‐CoV‐2‐megakaryocyte interactions can induce our observed COVID‐19 platelet signature transcriptome. Notably, CD147 antibody‐mediated blocking significantly reduced the expression of SARS‐CoV‐2, S100A9, and S100A8 on megakaryocytes following incubation with SARS‐CoV‐2 (Figure S6A–C in supporting information). These data indicate that megakaryocytes and platelets actively take up SARS‐CoV‐2 virions, likely via an ACE‐2‐independent mechanism.
FIGURE 5.

Platelets from COVID‐19 patients have a unique transcriptome. A) Principal component analysis from top varied genes, (B) heat map of significantly differentially expressed platelet transcripts (adjusted P‐value <.05, |log2fc| >1) from COVID‐19 patients and control donors. Red indicates increased relative expression, and blue indicates decreased relative expression, and (C) volcano plot (red dots: adjusted P‐value <.05, dark red: log2fc >1; blue dots: adjusted P‐value <.05, dark blue < −1) (D) LGALS3BP, and S100A9 mRNA expression in Meg‐01 cells treated with either media control, SARS‐CoV‐2, or CoV‐OC43 for 24 h. SARS‐CoV‐2 incubations performed at 37°C, CoV‐OC43 incubations performed at 33°C. E, Gene set enrichment analysis of Hallmark pathways, displayed pathways represent those with an adjusted P < .05. F, Weighted correlation network analysis (WGCNA), boxplots of eigengene values representing relative coexpression of gene sets on a per sample basis. G, Heatmap of correlation metrics for eigengene values versus percentage aggregation to ADP (1 uM), epinephrine (0.1 uM), and collagen (0.2 uM) on a per patient basis. Data are expressed as mean ± standard error of the mean, *P < .05, **P < .01
Gene set enrichment analysis (GSEA) of Hallmark pathways found that platelets from COVID‐19 patients are enriched in metabolic pathways, including oxidative phosphorylation, fatty acid metabolism, and glycolysis, in addition to enrichment of the apical junction pathway (Figure 5E). Additionally, enrichment in the platelet degranulation pathway was found by gene ontology analysis (Figure S7 in supporting information). Using weighted correlation network analysis (WGCNA),35 eigengene values were derived to represent the relative coexpression of gene sets on a per‐sample basis. Comparing the eigengene values of these genesets between platelets from COVID‐19 patients and controls, the platelet metabolic pathways and the apical junction pathway were enriched in COVID‐19 platelets, and tumor necrosis factor alpha (TNFα) and KRAS signaling significantly decreased (Figure 5F). To understand the relationship between the transcriptional changes and the increased platelet activity seen in patients with COVID‐19, we correlated each subject’s eigengene score with their platelet aggregation to ADP, epinephrine, and collagen. The resulting correlations demonstrate that differential expression of these specific metabolic pathways is directly associated with an increased platelet response to stimulus (Figure 5G).
3.4. The COVID‐19 platelet transcriptome is characterized by genes linked to platelet count, size, and immaturity
Because platelet count, size, and maturity were associated with adverse outcomes; we used our transcriptomic data to shed light on platelet indices in COVID‐19. To identify potential mediators of platelet count and platelet size mediated by SARS‐CoV‐2, we compared our RNA‐seq results to previously published GWAS focused on platelet count and activity.23 Of the 44 genes previously identified to regulate platelet count, 16 were differentially expressed (padj <.1) in platelets isolated from COVID‐19 patients relative to controls (Figure 6A ). Glucokinase regulator (GCKR) was enriched in platelets of COVID‐19 patients, and flap structure‐specific endonuclease 1 (FEN1) was suppressed relative to platelets from control subjects. Among the 30 genes previously identified to regulate MPV, 15 were differentially expressed (padj <.1) in COVID‐19 versus controls (Figure 6B). Coagulation factor II thrombin receptor (F2R), phosphatidylinositol‐4,5‐bisphosphate 3‐kinase catalytic subunit gamma (PIK3CG), and ring finger protein 145 (RNF145) were the top differentially expressed transcripts in platelets from COVID‐19 patients that have previously been identified to regulate platelet size. Within the COVID‐19 cohort, five patients subsequently died. We found genes differentially regulated in COVID‐19 versus controls that regulate platelet count and size to be the same comparing COVID‐19 patients who later died to controls (Figure 6A–B). Treatment of Meg‐01 cells with SARS‐CoV‐2 leads to a significant increase in the expression of GCKR, BAZ2A, and ITPK1 (Figure 6C, red bars), indicating that direct viral‐‐megakaryocyte interactions may influence the expression of transcripts linked to platelet count and size. Conversely, no such change is observed following the treatment of Meg‐01s with an alternative coronavirus, CoV‐OC43. Altogether, these data suggest that SARS‐CoV‐2 modulates platelet count and size by modulating megakaryocytes and platelet gene expression.
FIGURE 6.

Platelet transcriptomics and comparative analyses with established gene signatures. A, ‐log10(adj pvalue) for genes associated with platelet count with red indicating genes positively differentially expressed between COVID‐19 and control, and blue indicating genes negatively differentially expressed between COVID‐19 and control. Heatmap above shows raw scaled gene counts across all samples and genes. B, ‐log10(adj pvalue) for genes associated with mean platelet volume with red indicating genes positively differentially expressed between COVID‐19 patients and control, and blue indicating genes negatively differentially expressed between COVID‐19 patients and control. Heatmap above shows raw scaled gene counts across all samples and genes. C, GCKR, BAZ2A, and ITPK1 mRNA expression in Meg‐01 cells treated with either media control, SARS‐CoV‐2, or CoV‐OC43 for 24 h. SARS‐CoV‐2 incubations performed at 37°C, CoV‐OC43 incubations performed at 33°C. D–E, Scatterplots of fold change differences comparing reticulated platelets (RP) versus mature platelets (MP), and COVID‐19 patients versus control. Size and color of dots indicative of the respective P‐value of the fold change; all genes shown have adjusted P‐value <.05 for both RP versus MP comparison and COVID‐19 patients versus controls. Squares indicate number of genes in each quadrant. Correlation of fold changes shown on plot
Immature platelet fraction is an excellent proxy for reticulated (younger) platelets. Immature platelets are larger, contain a higher amount of RNA, and can produce various proteins typical for platelet activation (e.g., GPIIb/IIIa, P‐selectin).36 Reticulated or newly formed platelets have a distinct prothrombotic transcriptomic profile.25 To understand if platelets isolated from patients with COVID‐19 had a transcriptomic profile characteristic of immature prothrombotic platelets, we compared our RNAseq results to a publically available dataset comparing immature (reticulated) versus mature platelets.25 We found a significant overlap between transcripts differentially expressed between reticulated versus mature platelets and platelets from COVID‐19 versus controls (Figure 6D, R = 0.38, P = 3.7 × 10−10, Table S8 in supporting information). Overall, 211/256 transcripts were differentially expressed in the same direction, of which 167 were increased and 44 decreased. Not surprisingly, we find a stronger association between the transcriptome of COVID‐19 patients who died versus controls and between reticulated versus mature platelets (Figure 6E, R = 0.44, P = 1.9 × 10−12, Table S9 in supporting information), suggesting that COVID‐19 patients who died have a more prothrombotic platelet profile.
4. DISCUSSION
Platelets are increasingly recognized as key mediators of inflammation and immune dysfunction.21., 37. Patients with COVID‐19 are at heightened risk of critical illness and all‐cause mortality secondary to a heightened inflammatory state and altered immune phenotype.38., 39., 40. Thus, it is not surprising that platelets contribute to the pathophysiology of COVID‐19 and play a significant role in the development of COVID‐19‐‐associated complications. We report that SARS‐CoV‐2 infects bone marrow megakaryocytes and show for the first time that virions are present in circulating platelets. Additionally, we provide evidence that SARS‐CoV‐2 induces transcriptomic changes to megakaryocytes, which are unique relative to CoV‐OC43, a coronavirus responsible for the common cold.31 In a large cohort of hospitalized COVID‐19 patients, we find that markers of platelet activity (platelet size and platelet maturity) are significantly associated with both critical illness and all‐cause mortality, even after adjusting for demographics, comorbidities, medication, and clinical laboratory values, including biomarkers of inflammation and thrombosis (e.g., D‐dimer, CRP).
Consistent with previous data, we found increased platelet activity in patients infected with SARS‐CoV‐2.6., 41., 42., 43. Exploration of the platelet transcriptome revealed a significant difference between platelets isolated from COVID‐19 patients relative to controls, characterized by enrichment in metabolic pathways, including oxidative phosphorylation and glycolysis and an immature platelet phenotype (Figure 7 ). Differentially expressed platelet pathways identified by GSEA between patients with versus without COVID‐19 correlate with platelet aggregation suggesting these pathways identify an activated platelet phenotype. These data indicate key differences in the platelet activity profiles of those with and without COVID‐19, despite a degree of activity heterogeneity within the COVID‐19 population, and suggest that activation mechanisms may contribute to adverse events. While our current study cannot distinguish between the direct effects of virus‐‐megakaryocyte/platelet interactions versus systemic inflammation in COVID‐19, herein, we present data highlighting that the direct infection of megakaryocytes with SARS‐CoV‐2 uniquely alters the transcriptome providing evidence that SARS‐CoV‐2 is a contributor to the altered platelet phenotype observed in the setting of COVID‐19. Results from our study warrant further investigation into the mechanistic and pathogenic role of platelets in COVID‐19 and highlight the potential beneficial role of anti‐platelet therapy in hospitalized COVID‐19 patients.
FIGURE 7.
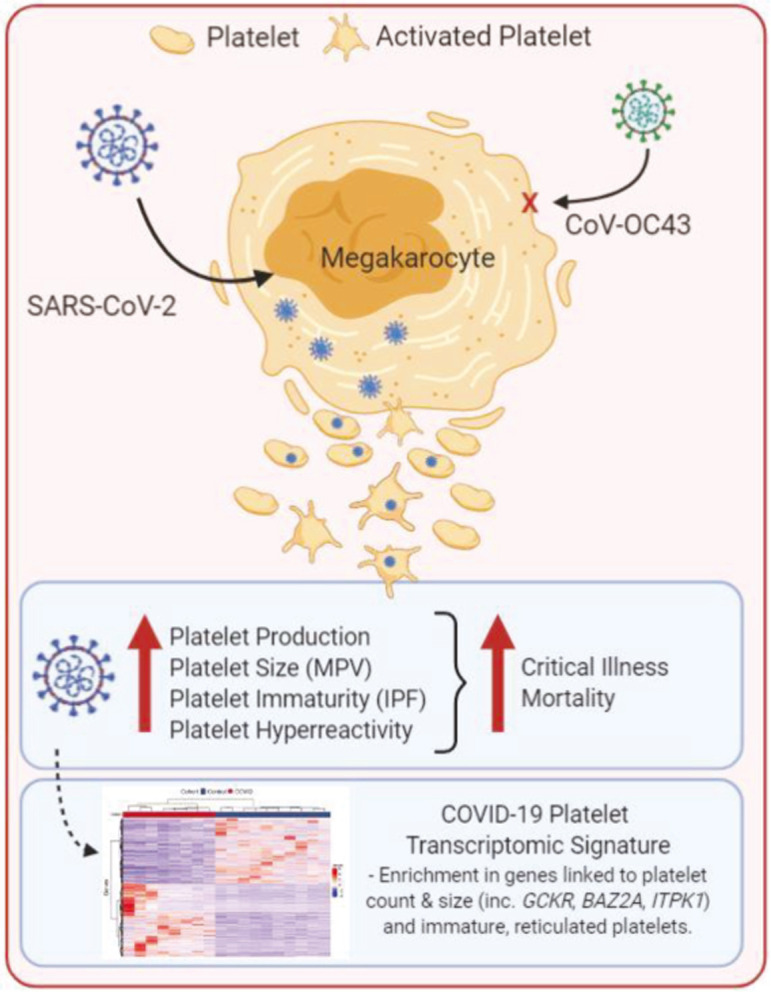
Summary of platelet activation by SARS‐CoV‐2
Autopsy data demonstrate extensive micro‐ and macro‐platelet thrombi in multiple tissues.1., 2. We previously reported that both arterial and venous thrombosis was significantly associated with all‐cause mortality even after adjustment for demographics and clinical risk factors.4 While endothelial cells,44 monocyte/macrophages,45 neutrophils,46 and T cells47 are hypothesized to play a role in COVID‐19, the role of platelets is largely understudied. In addition to their primary role in thrombosis, platelets mediate key aspects of inflammatory and immune processes,21., 37. including effects on endothelial cell activation,19., 48. macrophage polarization,21 and neutrophil extracellular trap formation.
Within a large cohort of patients admitted to a large health system in New York City, we find MPV and IPF are significantly associated with adverse outcomes in hospitalized patients with COVID‐19. Importantly, even following the exclusion of patients with thrombocytopenia, a potential driver of platelet size and maturity, this association remains robust. Reticulated (younger/immature) platelets and larger platelets are more metabolically active and have increased thrombotic potential.14 Both MPV and IPF have been found to be associated with increased morbidity and all‐cause mortality11., 12., 13., 15., 16., 17., 18., 49. and can be used as markers of platelet activation and increased risk of thrombosis.50., 51. In addition to potentially providing clinicians with a biomarker for risk stratification of COVID‐19 patients, the association between platelet indices and adverse outcomes provides a rationale for the use of platelet‐directed therapies in COVID‐19.52., 53., 54., 55. Currently, clinical trials are under way to investigate the clinical effect of aspirin (NCT04381936, NCT02735707) and P2Y12 inhibitors (NCT04505774) in hospitalized COVID‐19 patients.
Numerous platelet activation pathways initiate and sustain thrombosis formation.56., 57. Previously we have reported that circulating biomarkers of platelet activity are associated with thrombosis and mortality in patients with COVID‐19.5 Consistent with an activated platelet phenotype, we now report that platelets isolated from COVID‐19 subjects aggregate more readily to various agonists, including ADP, epinephrine, and collagen. These findings complement a previous study that also reports increased platelet aggregation in response to agonists and increased adhesion and spreading of platelets on fibrinogen and collagen in COVID‐19 patients.6., 42. Our RNA‐seq data also provide evidence for metabolically more active platelets in COVID‐19 patients, as noted by enrichment in pathway oxidative phosphorylation and glycolysis, metabolic processes previously linked to a hyperactive platelet phenotype and thrombosis.58 This is further confirmed by the enrichment of genes related to platelet degranulation in platelets from COVID‐19 patients. Consistent with a previous report, we find a high degree of overlap between differentially expressed transcripts in our COVID‐19 platelet transcriptome compared to that found by the Utah group (R = 0.86, P < 2.2 × 10−16).6
In the setting of viral infections, platelets can internalize viral particles, including influenza, HIV, hepatitis C virus, and dengue, resulting in platelet activation.27 Our study is the first to demonstrate SARS‐CoV‐2 in a human megakaryoblastic cell line after incubation in vitro, and by transmission electron microscopy that SARS‐CoV‐2 viral particles are found in circulating platelets of COVID‐19 positive patients. Given recent studies confirming that the lung is a critical site of thrombopoiesis59 and that lung megakaryocytes are key immune modulatory cells,60 it is likely that megakaryocytes and platelets present in the lung at the time of infection are immediate SARS‐CoV‐2 targets. Given that the viral load of SARS‐CoV‐2 peaks around symptom onset or a few days thereafter, we hypothesize megakaryocytes and circulating platelets are early SARS‐CoV‐2 targets;61., 62. however, this remains to be rigorously investigated. Our studies investigating SARS‐CoV‐2, and a coronavirus responsible for the common cold CoV‐OC43, indicate that megakaryocytes actively take up SARS‐CoV‐2, and this interaction induces changes to the megakaryocyte transcriptome, a phenomenon that does not occur with CoV‐OC43; thus, while our study cannot delineate between the impact of viral‐megakaryocyte interactions versus systemic inflammation, these results indicate that SARS‐CoV‐2–megakaryocyte interactions play a role in altering the transcriptome, and subsequently platelet phenotype and function. The origin of the transcriptomic differences mediated by SARS‐CoV‐2 to megakaryocytes and platelets relative to CoV‐OC43 requires further investigation.
We find SARS‐CoV‐2 viral particles in platelets, despite finding no mRNA expression of the documented SARS‐CoV‐2 receptor ACE2 and a serine protease for Spike protein priming, TMPRSS2, consistent with previous platelet sequencing studies.6., 20., 63. However, despite no evidence of mRNA expression, a recent study found that human platelets have ACE2 and TMPRSS2 protein and can internalize SARS‐CoV‐2 in vitro.43 Our profiling of platelets and megakaryocytes for a potential entry receptor identifies CD147 as a likely candidate. We find it highly expressed at the mRNA level and on the surface of megakaryocytes; furthermore, it has been proposed to be a novel entry receptor for SARS‐CoV‐2.64 Consistent with a SARS‐CoV‐2‐‐mediated activated platelet phenotype, ex vivo SARS‐CoV‐2 enhances platelet activation as documented by increased PAC‐1 binding, CD62P expression, alpha granule secretion, and dense granule release. Mice transfused with hACE2 transgenic platelets have enhanced thrombosis formation, further suggesting the prothrombotic potential of platelets infected with SARS‐CoV‐2.43
Further evidence for a hyperreactive platelet phenotype is SARS‐CoV‐2 is our analysis of the COVID‐19 platelet transcriptome relative to the transcriptome analysis of reticulated platelets.25 Comparative transcriptome analysis of young reticulated platelets (RPs) relative to mature platelets found significant enrichment of prothrombotic genes in RPs, providing a biological rationale for the hyperreactive phenotype of RPs. The high degree of association between transcripts differentially regulated in COVID‐19 versus controls, compared to RPs versus mature platelets (R = 0.38, P = 3.7 × 10−10) provides evidence for a younger, prothrombotic platelet phenotype in those with COVID‐19. Further interrogation of the RNA‐seq data and validation of target genes or pathways may lead to the identification of mechanisms that contribute to SARS‐CoV‐2‐‐mediated platelet size, maturity, hyperreactivity, and the increased thrombotic risk in this population. Furthermore, the association between platelet reactivity and additional emerging risk factors that promote the development of adverse events and death, including sex, race, BMI, and how these may amplify platelet hyperreactivity to exacerbate the incidence of thrombotic events remains to be established.
Collectively, our findings highlight the importance of platelet indices at initial hospital presentation to aid as a surrogate biomarker for subsequent critical illness and death. Assessment of platelet size and maturity may aid in the risk stratification of hospitalized COVID‐19 patients and guide therapeutic options and clinical management. Furthermore, our results suggest multiple platelet activation mechanisms likely contribute to adverse events and warrant further investigation into the mechanistic role of platelets in COVID‐19 pathogenesis and highlight the potential role of anti‐platelet therapy. However, whether a more aggressive antithrombotic strategy with platelet‐directed therapies in these patients would alter the critical illness course is unknown and under investigation.
CONFLICTS OF INTEREST
All authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
JB had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Concept and design: JB, TB. Acquisition, analysis, or interpretation of data: JB, TB, SB, MC, YH, FL, JA, SP, AR, KR, KS, HB, IM, VWV, KD. Drafting of the manuscript: JB, TB, SB, HI. Critical revision of the manuscript for important intellectual content: JB, TB, SB, MC, HI, EY, ML, JH, YH, FL, JA, SP, AR, KR, CIR, JW. Statistical analysis: SB, JB, TB, MC. Obtained funding: JB.
ACKNOWLEDGMENTS
We thank NYU Langone Health DART Microscopy Lab, Chris Petzold, and Kristen Dancel‐Manning for their assistance with TEM work, and the Genomic Technology Core for RNA sequencing.
National Institutes of Health1OT2HL156812‐01R35HL144993R21‐AI163924
American Heart Association18CDA34110203AHA
American Society of Hematology18‐A0‐00‐1001884
NYU CTSAUL1TR001445
NYU Cancer Center SupportP30CA016087
NYU Grossman School of Medicine Department of Pathology & Office of Science & Research
Footnotes
Manuscript handled by: Matthew T. Rondina
Final decision: Matthew T. Rondina, 16 September 2021
Funding informationSupport for this study was provided by the National Institutes of Health (1OT2HL156812‐01, R35HL144993 to JB), the American Heart Association (18CDA34110203AHA to TB), the American Society of Hematology (18‐A0‐00‐1001884 to TB). Supported in part by the NYU CTSA grant UL1TR001445, from the National Center for Advancing Translational Sciences (NCATS). NYU Langone Health DART Microscopy Lab and Genomic Technology Core are partially funded by NYU Cancer Center Support Grant NIH/NCI P30CA016087. The views and conclusions contained in this document are those of the authors and should not be interpreted as representing the official policies, either expressed or implied, of the NIH.
Supporting Information
Fig S1‐S7
Tab S1‐S9
App S1
REFERENCES
- 1.Rapkiewicz A.V., Mai X., Carsons S.E., et al. Megakaryocytes and platelet‐fibrin thrombi characterize multi‐organ thrombosis at autopsy in COVID‐19: a case series. EClinical Medicine. 2020;24:100434. doi: 10.1016/j.eclinm.2020.100434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ackermann M., Verleden S.E., Kuehnel M., et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid‐19. N Engl J Med. 2020;383(2):120–128. doi: 10.1056/NEJMoa2015432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klok F.A., Kruip M.J.H.A., van der Meer N.J.M., et al. Incidence of thrombotic complications in critically ill ICU patients with COVID‐19. Thromb Res. 2020;191:145–147. doi: 10.1016/j.thromres.2020.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bilaloglu S., Aphinyanaphongs Y., Jones S., et al. Thrombosis in hospitalized patients with COVID‐19 in a New York City health system. JAMA. 2020;324(8):799. doi: 10.1001/jama.2020.13372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barrett T.J., Lee A., Xia Y., et al. Biomarkers of platelet activity and vascular health associate with thrombosis and mortality in patients with COVID‐19. Circ Res. 2020;127:945–947. doi: 10.1161/CIRCRESAHA.120.317803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manne B.K., Denorme F., Middleton E.A., et al. Platelet gene expression and function in COVID‐19 patients. Blood. 2020;136(11):1317–1329. doi: 10.1182/blood.2020007214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vanderschueren S., De Weerdt A., Malbrain M., et al. Thrombocytopenia and prognosis in intensive care. Crit Care Med. 2000;28(6):1871–1876. doi: 10.1097/00003246-200006000-00031. [DOI] [PubMed] [Google Scholar]
- 8.Stephan F., Montblanc J.D., Cheffi A., Bonnet F. Thrombocytopenia in critically ill surgical patients: a case‐control study evaluating attributable mortality and transfusion requirements. Crit Care. 1999;3(6):151–158. doi: 10.1186/cc369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang X., Yang Q., Wang Y., et al. Thrombocytopenia and its association with mortality in patients with COVID‐19. J Thromb Haemost. 2020;18(6):1469–1472. doi: 10.1111/jth.14848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang S.Q., Huang Q.‐.F., Xie W.‐.M., et al. The association between severe COVID‐19 and low platelet count: evidence from 31 observational studies involving 7613 participants. Br J Haematol. 2020;190(1):e29–e33. doi: 10.1111/bjh.16817. [DOI] [PubMed] [Google Scholar]
- 11.Briggs C., Kunka S., Hart D., Oguni S., Machin S.J. Assessment of an immature platelet fraction (IPF) in peripheral thrombocytopenia. Br J Haematol. 2004;126(1):93–99. doi: 10.1111/j.1365-2141.2004.04987.x. [DOI] [PubMed] [Google Scholar]
- 12.Kickler T.S., Oguni S., Borowitz M.J. A clinical evaluation of high fluorescent platelet fraction percentage in thrombocytopenia. Am J Clin Pathol. 2006;125(2):282–287. doi: 10.1309/50H8-JYHN-9JWC-KAM7. [DOI] [PubMed] [Google Scholar]
- 13.Jung H., Jeon H.‐.K., Kim H.‐.J., Kim S.‐.H. Immature platelet fraction: establishment of a reference interval and diagnostic measure for thrombocytopenia. Korean J Lab Med. 2010;30(5):451–459. doi: 10.3343/kjlm.2010.30.5.451. [DOI] [PubMed] [Google Scholar]
- 14.Shah B., Valdes V., Nardi M.A., et al. Mean platelet volume reproducibility and association with platelet activity and anti‐platelet therapy. Platelets. 2014;25(3):188–192. doi: 10.3109/09537104.2013.793794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chu S.G., Becker R.C., Berger P.B., et al. Mean platelet volume as a predictor of cardiovascular risk: a systematic review and meta‐analysis. J Thromb Haemost. 2010;8(1):148–156. doi: 10.1111/j.1538-7836.2009.03584.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ibrahim H., Schutt R.C., Hannawi B., et al. Association of immature platelets with adverse cardiovascular outcomes. J Am Coll Cardiol. 2014;64(20):2122–2129. doi: 10.1016/j.jacc.2014.06.1210. [DOI] [PubMed] [Google Scholar]
- 17.Muronoi T., Koyama K., Nunomiya S., et al. Immature platelet fraction predicts coagulopathy‐related platelet consumption and mortality in patients with sepsis. Thromb Res. 2016;144:169–175. doi: 10.1016/j.thromres.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 18.Wu Q., Ren J., Hu D., et al. An elevated percentage of reticulated platelet is associated with increased mortality in septic shock patients. Medicine (Baltimore) 2015;94(19):e814. doi: 10.1097/MD.0000000000000814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nhek S., Clancy R., Lee K.A., et al. Activated platelets induce endothelial cell activation via an interleukin‐1beta pathway in systemic lupus erythematosus. Arterioscler Thromb Vasc Biol. 2017;37(4):707–716. doi: 10.1161/ATVBAHA.116.308126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Campbell R.A., Schwertz H., Hottz E.D., et al. Human megakaryocytes possess intrinsic antiviral immunity through regulated induction of IFITM3. Blood. 2019;133(19):2013–2026. doi: 10.1182/blood-2018-09-873984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barrett T.J., Schlegel M., Zhou F., et al. Platelet regulation of myeloid suppressor of cytokine signaling 3 accelerates atherosclerosis. Sci Transl Med. 2019;11(517):eaax0481. doi: 10.1126/scitranslmed.aax0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Assinger A. Platelets and infection ‐ an emerging role of platelets in viral infection. Front Immunol. 2014;5:649. doi: 10.3389/fimmu.2014.00649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gieger C., Radhakrishnan A., Cvejic A., et al. New gene functions in megakaryopoiesis and platelet formation. Nature. 2011;480(7376):201–208. doi: 10.1038/nature10659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davizon‐Castillo P., Rowley J.W., Rondina M.T. Megakaryocyte and platelet transcriptomics for discoveries in human health and disease. Arterioscler Thromb Vasc Biol. 2020;40(6):1432–1440. doi: 10.1161/ATVBAHA.119.313280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bongiovanni D., Santamaria G., Klug M., et al. Transcriptome analysis of reticulated platelets reveals a prothrombotic profile. Thromb Haemost. 2019;119(11):1795–1806. doi: 10.1055/s-0039-1695009. [DOI] [PubMed] [Google Scholar]
- 26.Dann R., Hadi T., Montenont E., et al. Platelet‐Derived MRP‐14 induces monocyte activation in patients with symptomatic peripheral artery disease. J Am Coll Cardiol. 2018;71(1):53–65. doi: 10.1016/j.jacc.2017.10.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koupenova M., Clancy L., Corkrey H.A., Freedman J.E. Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ Res. 2018;122(2):337–351. doi: 10.1161/CIRCRESAHA.117.310795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koupenova M., Freedman J.E. Platelets: the unsung hero of the immune response. J Thromb Haemost. 2015;13(2):268–270. doi: 10.1111/jth.12806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Banerjee M., Huang Y., Joshi S., et al. Platelets endocytose viral particles and are activated via TLR (Toll‐Like Receptor) signaling. Arterioscler Thromb Vasc Biol. 2020;40(7):1635–1650. doi: 10.1161/ATVBAHA.120.314180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koupenova M., Corkrey H.A., Vitseva O., et al. SARS‐CoV‐2 initiates programmed cell death in platelets. Circ Res. 2021;129(6):631–646. doi: 10.1161/CIRCRESAHA.121.319117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gaunt E.R., Hardie A., Claas E.C.J., et al. Epidemiology and clinical presentations of the four human coronaviruses 229E, HKU1, NL63, and OC43 detected over 3 years using a novel multiplex real‐time PCR method. J Clin Microbiol. 2010;48(8):2940–2947. doi: 10.1128/JCM.00636-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu Q., Liu J., Zhao S., et al. SARS‐CoV‐2 exacerbates proinflammatory responses in myeloid cells through C‐type lectin receptors and Tweety family member 2. Immunity. 2021;54(6):1304–1319 e9. doi: 10.1016/j.immuni.2021.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hottz E.D., Bozza F.A., Bozza P.T. Platelets in immune response to virus and immunopathology of viral infections. Front Med (Lausanne) 2018;5:121. doi: 10.3389/fmed.2018.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guthikonda S., Alviar C.L., Vaduganathan M., et al. Role of reticulated platelets and platelet size heterogeneity on platelet activity after dual antiplatelet therapy with aspirin and clopidogrel in patients with stable coronary artery disease. J Am Coll Cardiol. 2008;52(9):743–749. doi: 10.1016/j.jacc.2008.05.031. [DOI] [PubMed] [Google Scholar]
- 37.Rolfes V., Ribeiro L.S., Hawwari I., et al. Platelets fuel the inflammasome activation of innate immune cells. Cell Rep. 2020;31(6):107615. doi: 10.1016/j.celrep.2020.107615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou T., Su T.T., Mudianto T., Wang J. Immune asynchrony in COVID‐19 pathogenesis and potential immunotherapies. J Exp Med. 2020;217(10) doi: 10.1084/jem.20200674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mathew D., Giles J.R., Baxter A.E., et al. Deep immune profiling of COVID‐19 patients reveals distinct immunotypes with therapeutic implications. Science. 2020;369(6508):eabc8511. doi: 10.1126/science.abc8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smilowitz N.R., Kunichoff D., Garshick M., et al. C‐reactive protein and clinical outcomes in patients with COVID‐19. Eur Heart J. 2021;42(23):2270–2279. doi: 10.1093/eurheartj/ehaa1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Denorme F., Manne B.K., Portier I., et al. COVID‐19 patients exhibit reduced procoagulant platelet responses. J Thromb Haemost. 2020;18(11):3067–3073. doi: 10.1111/jth.15107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zaid Y., Puhm F., Allaeys I., et al. Platelets can associate with SARS‐Cov‐2 RNA and are hyperactivated in COVID‐19. Circ Res. 2020;127(11):1404–1418. doi: 10.1161/CIRCRESAHA.120.317703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang S., Liu Y., Wang X., et al. SARS‐CoV‐2 binds platelet ACE2 to enhance thrombosis in COVID‐19. J Hematol Oncol. 2020;13(1):120. doi: 10.1186/s13045-020-00954-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Teuwen L.A., Geldhof V., Pasut A., Carmeliet P. COVID‐19: the vasculature unleashed. Nat Rev Immunol. 2020;20(7):389–391. doi: 10.1038/s41577-020-0343-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Merad M., Martin J.C. Pathological inflammation in patients with COVID‐19: a key role for monocytes and macrophages. Nat Rev Immunol. 2020;20(6):355–362. doi: 10.1038/s41577-020-0331-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barnes B.J., Adrover J.M., Baxter‐Stoltzfus A., et al. Targeting potential drivers of COVID‐19: neutrophil extracellular traps. J Exp Med. 2020;217(6) doi: 10.1084/jem.20200652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Le Bert N., Tan A.T., Kunasegaran K., et al. SARS‐CoV‐2‐specific T cell immunity in cases of COVID‐19 and SARS, and uninfected controls. Nature. 2020;584(7821):457–462. doi: 10.1038/s41586-020-2550-z. [DOI] [PubMed] [Google Scholar]
- 48.Marcantoni E., Allen N., Cambria M.R., et al. Platelet transcriptome profiling in HIV and ATP‐binding cassette subfamily C member 4 (ABCC4) as a mediator of platelet activity. JACC Basic Transl Sci. 2017;3(1):9–22. doi: 10.1016/j.jacbts.2017.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anetsberger A., Blobner M., Haller B., et al. Immature platelets as a novel biomarker for adverse cardiovascular events in patients after non‐cardiac surgery. Thromb Haemost. 2017;117(10):1887–1895. doi: 10.1160/TH16-10-0804. [DOI] [PubMed] [Google Scholar]
- 50.Kissova J., Bulikova A., Ovesna P., Bourkova L., Penka M. Increased mean platelet volume and immature platelet fraction as potential predictors of thrombotic complications in BCR/ABL‐negative myeloproliferative neoplasms. Int J Hematol. 2014;100(5):429–436. doi: 10.1007/s12185-014-1673-0. [DOI] [PubMed] [Google Scholar]
- 51.Michiels J.J., Berneman Z., Bockstaele D., et al. Clinical and laboratory features, pathobiology of platelet‐mediated thrombosis and bleeding complications, and the molecular etiology of essential thrombocythemia and polycythemia vera: therapeutic implications. Semin Thromb Hemost. 2006;32(3):174–207. doi: 10.1055/s-2006-939431. [DOI] [PubMed] [Google Scholar]
- 52.Bernlochner I., Goedel A., Plischke C., et al. Impact of immature platelets on platelet response to ticagrelor and prasugrel in patients with acute coronary syndrome. Eur Heart J. 2015;36(45):3202–3210. doi: 10.1093/eurheartj/ehv326. [DOI] [PubMed] [Google Scholar]
- 53.Ibrahim H., Nadipalli S., DeLao T., Guthikonda S., Kleiman N.S. Immature platelet fraction (IPF) determined with an automated method predicts clopidogrel hyporesponsiveness. J Thromb Thrombolysis. 2012;33(2):137–142. doi: 10.1007/s11239-011-0665-7. [DOI] [PubMed] [Google Scholar]
- 54.Stratz C., Bömicke T., Younas I., et al. Comparison of immature platelet count to established predictors of platelet reactivity during thienopyridine therapy. J Am Coll Cardiol. 2016;68(3):286–293. doi: 10.1016/j.jacc.2016.04.056. [DOI] [PubMed] [Google Scholar]
- 55.Guthikonda S., Lev E.I., Patel R., et al. Reticulated platelets and uninhibited COX‐1 and COX‐2 decrease the antiplatelet effects of aspirin. J Thromb Haemost. 2007;5(3):490–496. doi: 10.1111/j.1538-7836.2007.02387.x. [DOI] [PubMed] [Google Scholar]
- 56.Packham M.A. Role of platelets in thrombosis and hemostasis. Can J Physiol Pharmacol. 1994;72(3):278–284. doi: 10.1139/y94-043. [DOI] [PubMed] [Google Scholar]
- 57.Hou Y., Carrim N., Wang Y., Gallant R.C., Marshall A., Ni H. Platelets in hemostasis and thrombosis: novel mechanisms of fibrinogen‐independent platelet aggregation and fibronectin‐mediated protein wave of hemostasis. J Biomed Res. 2015;29:437–444. doi: 10.7555/JBR.29.20150121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kulkarni P.P., Tiwari A., Singh N., et al. Aerobic glycolysis fuels platelet activation: small‐molecule modulators of platelet metabolism as anti‐thrombotic agents. Haematologica. 2019;104(4):806–818. doi: 10.3324/haematol.2018.205724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lefrançais E., Ortiz‐Muñoz G., Caudrillier A., et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature. 2017;544(7648):105–109. doi: 10.1038/nature21706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pariser D.N., Hilt Z.T., Ture S.K., et al. Lung megakaryocytes are immune modulatory cells. J Clin Invest. 2021;131(1) doi: 10.1172/JCI137377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Walsh K.A., Jordan K., Clyne B., et al. SARS‐CoV‐2 detection, viral load and infectivity over the course of an infection. J Infect. 2020;81(3):357–371. doi: 10.1016/j.jinf.2020.06.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.He X., Lau E.H.Y., Wu P., et al. Temporal dynamics in viral shedding and transmissibility of COVID‐19. Nat Med. 2020;26(5):672–675. doi: 10.1038/s41591-020-0869-5. [DOI] [PubMed] [Google Scholar]
- 63.Heffron S.P., Marier C., Parikh M., Fisher E.A., Berger J.S. Severe obesity and bariatric surgery alter the platelet mRNA profile. Platelets. 2019;30(8):967–974. doi: 10.1080/09537104.2018.1536261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Campbell R.A., Boilard E., Rondina M.T. Is there a role for the ACE2 receptor in SARS‐CoV‐2 interactions with platelets? J Thromb Haemost. 2021;19(1):46–50. doi: 10.1111/jth.15156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S7
Tab S1‐S9
App S1