

Abstract
Antiviral monoclonal antibody (mAb) discovery enables the development of antibody‐based antiviral therapeutics. Traditional antiviral mAb discovery relies on affinity between antibody and a viral antigen to discover potent neutralizing antibodies, but these approaches are inefficient because many high affinity mAbs have no neutralizing activity. We sought to determine whether screening for anti‐SARS‐CoV‐2 mAbs at reduced pH could provide more efficient neutralizing antibody discovery. We mined the antibody response of a convalescent COVID‐19 patient at both physiological pH (7.4) and reduced pH (4.5), revealing that SARS‐CoV‐2 neutralizing antibodies were preferentially enriched in pH 4.5 yeast display sorts. Structural analysis revealed that a potent new antibody called LP5 targets the SARS‐CoV‐2 N‐terminal domain supersite via a unique binding recognition mode. Our data combine with evidence from prior studies to support antibody screening at pH 4.5 to accelerate antiviral neutralizing antibody discovery.
Keywords: antibody discovery, COVID‐19, SARS‐CoV‐2, yeast display
1. INTRODUCTION
Monoclonal antibodies (mAbs) have been the fastest growing class of therapeutics over the past 30 years, with around 100 therapeutic mAbs approved for clinical use in the United States. 1 mAbs are used for a variety of indications including autoimmune diseases, cancer, genetic deficiencies, and other applications, and in particular antiviral antibodies can be useful for the prevention and treatment of viral infections. Major clinical indications for antiviral mAbs include human immunodeficiency virus (HIV‐1; ibalizumab), respiratory syncytial virus infection (palivizumab) and severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) infection, or its associated coronavirus disease (COVID‐19) (e.g., casirivimab/imdevimab, bamlanivimab); several other anti‐infective antibodies have been used or are currently in clinical trials to prevent or treat numerous other viral infections (e.g., yellow fever virus [YFV], 2 HIV‐1, 3 Ebola virus, 4 and others). While binding to a viral surface antigen is a common feature to all these clinical mAbs, the most promising antiviral antibodies for clinical use show potent viral neutralization, or the ability to prevent viral infection in tissue culture. Increased neutralization potency, often quantified in vitro as a 50% inhibitory concentration (IC50) or an 80% inhibitory concentration (IC80), is often closely correlated with higher clinical efficacy.
mAbs are often discovered from mining the unique B cells of humans or animal models exposed to viral protein antigens, either after infection or immunization. B cells contain both a variable heavy (VH) and variable light (VL) genes, and both must be sequenced together to characterize and discover the antibody as a potential mAb drug. Traditional hybridoma technologies and lower throughput methods like single B‐cell cloning after limiting dilution and/or flow cytometric cell sorting (FACS) have led to numerous successful mAb products over the years 5 , 6 ; these techniques rely on screening for binding antigen‐binding activity of the antibodies encoded by B cells, followed by laborious mAb expression, purification, and neutralization assays with an anticipation that some fraction of the viral antigen‐binding antibodies will neutralize. While binding is required for neutralization, it is often not directly correlated to neutralization potency. 7 Tremendous recent progress has been made in developing viral antigens that better replicate viral structures, thereby increasing the potential to identify neutralizing antibodies, but still only a fraction of the antibodies recovered have strong neutralization activity and associated therapeutic potential.
Antibody discovery technologies have proliferated from the earliest limiting dilution single cell studies using 96‐well plates into faster and more efficient techniques. One approach employs droplet‐based screening platforms and yeast display of the paired VH:VL repertoire from millions of B cells for functional antibody mining. 8 , 9 Other approaches apply microfluidic chambers and microcapillary tubes for direct isolation and screening of antibody secreting cells without B‐cell immortalization and library generation, 10 or droplet assays with antibody‐secreting cells. 11 Increasingly common is the pairing of FACS‐based antigen binding sorts of primary B cells with off‐the‐shelf transcriptome‐based kits for larger scale sequencing of mAbs that bind to synthetic antigens. 12 Still, most extant antibody discovery technologies rely on binding affinity of antibodies to antigen(s) at serological pH conditions (pH 7.4), due to the viability requirements for sorted B cells, followed by a subsequent and laborious screening process of those binding hits in search of neutralization activity. Improved methods to more rapidly select for mAb clones with a higher probability of neutralizing activity will benefit the biochemical engineering community and reduce costs while enhancing the speed of potent antiviral mAb discovery.
In our prior work, we noticed that potent neutralizing antibodies targeting SARS‐CoV‐2 appeared to bind more tightly than non‐neutralizing antibodies at non‐serological pH (4.5–6.5). 13 Performing antibody screening at pH 4.5 may provide stringency to antibody selection from maintained binding outside of the protein‐friendly pH 7.4 of serum. Many viral families, including coronaviruses, can enter cells via endosomal pathways where structural changes to the viral fusion machinery can occur as the endosomal pH gradually reduces from serological pH 7.4 to pH 4.5–5.5 14 , 15 ; therefore, endosomal pH (4.5–6.5) screening could help enrich for antibodies targeting certain conformations of viral fusion proteins. In particular, SARS‐CoV‐2 can enter cells via multiple pathways depending on the availability of proteases on the host cell surface, 16 , 17 and endosomal entry pathways are also used by other flaviviruses including as dengue and yellow fever virus, and by filoviruses like Ebola virus. 18 , 19 , 20 , 21
Based on the above information, we hypothesized that screening mAb libraries at pH 4.5 could preferentially identify the antiviral neutralizing antibodies targeting a potentially immunoevasive conformation of SARS‐CoV‐2 with an all‐down orientation of the receptor binding domain (RBD) 15 in an antibody library. Here, we screened for SARS‐CoV‐2 neutralizing antibodies from a COVID‐19 convalescent individual via yeast display screening at endosomal pH (4.5–5.5), and also at pH 7.4. We isolated and characterized a panel of resulting antibodies, and found the novel and potently neutralizing antibody LP5 targeting the N‐terminal domain (NTD) on the trimer spike glycoprotein. These data, combined with knowledge collected in prior studies, 13 reveal that endosomal pH screening techniques can provide strong selective pressure that helps to enrich screening hits for neutralizing antibodies targeting SARS‐CoV‐2.
2. MATERIALS AND METHODS
2.1. Human sample collection
The peripheral blood mononuclear cell (PBMC) sample used in the study was collected at Columbia University Irving Medical Center in New York, New York. Patient recruitment and sample collection for this study occurred between March 25, 2020 and April 7, 2020. Informed consent was obtained prior to sample collection under IRB #AAAS9517.
2.2. SARS‐CoV‐2 antigen probe generation for yeast display screening and ELISA experiments
The antigen probes used for yeast screening and ELISA experiments were produced as previously described. 22 Briefly, the DNA sequences encoding SARS‐CoV‐2 spike glycoprotein with two stabilizing proline mutations in S2 (S2P‐Trimer) 23 and RBD were cloned in a mammalian expression vector and an AVI tag was placed after the coding sequence for biotinylation. The probes were produced by transient transfection of 293 Freestyle cells as previously described, 24 followed by affinity purification, tag‐cleavage and biotinylation, and finally column purified.
2.3. Single B‐cell emulsion‐based capture and paired heavy:light variable antibody genes amplicon generation and yeast antibody library generation
B cells were isolated from cryopreserved PBMCs of Donor NYP01 as previously described. 8 Briefly, PBMC sample was depleted of non‐B cells and enriched for CD27+ antigen‐experienced B cells using microbeads designed for negative selection (EasySep Human B cell enrichment kit w/o CD43 depletion, STEMCELL Technologies) and positive selection (CD27 Human Microbeads, Miltenyi Biotec), respectively. CD27+ antigen‐experienced B cells were stimulated in vitro for 5 days to enhance the transcription of antibody genes. 8 , 25 Single B cells were captured using a flow‐focusing device in single‐cell emulsion droplets consisting of lysis buffer and oligo(dT)‐coated magnetic beads for single‐cell mRNA capture. 8 , 26 Collected beads with single‐cell mRNA were re‐emulsified and an overlap extension reverse transcription polymerase chain reaction (RT‐PCR) was performed to link the heavy and light chains via a linker with NcoI and NheI restriction sites using a Superscript III RT‐PCR kit (Thermo Fisher Scientific) into a ~850 bp amplicon. 9 The resulting cDNA samples were prepared for next‐generation sequencing (NGS) by 2 × 300 bp Illumina MiSeq sequencing to determine the diversity of the sample. In addition to the sequencing of the paired amplicon, sequencing for the separate VH and VL chains were performed to mine NGS data for complete variable region sequences.
Paired VH:VL cDNA libraries were PCR amplified from the paired amplicon using a set of primers 9 to add AscI and NotI restriction sites for cloning into yeast display vectors pCT‐VHVL‐K1 (for heavy chains with kappa light chains) and pCT‐VHVL‐L1 (for heavy chains with lambda light chains) via restriction‐digestion, ligation, and transformation of the ligated product into electrocompetent Escherichia coli. The transformed product was further maxiprepped, digested with NcoI and NheI, ligated with the bidirectional promoter and transformed into electrocompetent E. coli. The resulting transformed libraries were maxiprepped and PCR amplified to generate inserts with homologous regions for transformation along with predigested vectors backbone with AscI and NotI restriction enzymes into electrocompetent AWY101 9 yeast strain using a high efficiency yeast transformation method. 27 Transformed libraries were passaged twice in the synthetic defined media with casamino acids (SDCAA: 20 g/L dextrose, 6.7 g/L yeast nitrogen base, 5 g/L casamino acids, 8.56 g/L NaH2PO4.H2O, and 10.2 g/L Na2HPO4.7H2O) media to ensure single plasmid in each yeast cell, 27 and were subsequently analyzed by FACS for antibody discovery.
2.4. Affinity screening of yeast antibody libraries via FACS
Yeast antibody libraries encoding natively paired VH:VL genes were grown in SDCAA media with 2 g/L dextrose and 20 g/L galactose (SGDCAA) for 36 h to induce Fab expression for surface display. In the first round of screening, we collected 3 × 107 Fab‐expressing yeast cells and washed them twice with ice‐cold staining buffer (phosphate‐buffered saline [PBS] with 0.5% BSA and 2 mM EDTA). Libraries were stained with 20 nM SARS‐CoV‐2 and 100 nM RBD biotinylated antigen probes preconjugated to SA‐PE (Streptavidin, R‐Phycoerythrin Conjugate, Thermo Fisher Scientific) in a 4:1 molar ratio (antigen:fluorophore) using the protocol previously described 28 and a monoclonal anti‐FLAG‐flourescein isothiocyanate (FITC) conjugated antibody was added as a marker for Fab expression (clone M2‐FITC, Sigma‐Aldrich). Samples were stained in dark and at 4°C on a rocking platform for 30 min, after which they were washed thrice with the staining buffer and resuspended in staining buffer for sorting kept in dark on ice until sorting. As sorting was done at three different pH values (4.5, 5.5, and 7.4), we prepared separate staining buffers and sheath buffers for the preparation and sorting of samples at their respective pH.
Sorting was performed on a SONY MA900 cell sorter using associated software for sort data analysis. Sort gates were drawn for sorting as previously described. 9 , 29 Fab expression and antigen binding were detected using the FITC and PE channels, respectively. Sorted yeast cells were grown in SDCAA (pH 4.5) to avoid bacterial contamination and expanded for 24–48 h at 30°C, 225 rpm. In subsequent rounds of sorting and affinity titrations, expanded samples in SDCAA were inoculated at an OD600 0.2 in SGDCAA. In addition to collecting the antigen binding yeast cells, we collected yeast cells expressing Fab on yeast surface (referred as VL+) during the first round of screening by staining presort or input libraries with anti‐FLAG‐FITC conjugated mAb.
2.5. Bioinformatic analysis of sorted yeast library NGS data
Yeast samples after each round of FACS were miniprepped using the protocol described previously. 9 , 30 , 31 Briefly, 4 × 107 sorted yeast cells were expanded in SDCAA, centrifuged, and miniprepped to collect plasmid DNAs. Heavy and light chain antibody genes were amplified from miniprepped plasmid DNA samples using primers targeting the yeast display vector backbone. 9 Further rounds of PCR were used to add unique sample barcodes and Illumina adapters for analysis on a 2 × 300 bp MiSeq platform. Raw Illumina sequencing data in FASTQ format were quality filtered and analyzed as described previously. 9 , 13 , 29 , 32 Clone enrichment across each round of sorting was tracked based on CDR‐H3 amino acid sequences using enrichment ratios (ERs), which were calculated by determining the prevalence of a CDR‐H3 amino acid sequence in each round of sorting divided by its prevalence in the Fab‐expressing, or VL+, reference library. 29 , 32 We used both Round 2 and Round 3 data for selecting antibody clones, as previously reported. 13 , 29 , 32
2.6. Antibody protein expression and purification
The antibody coding heavy and light chain variable region genes were cloned into mammalian expression vectors for expression as IgG1. 9 The heavy and light chain genes were cotransfected into Expi293 mammalian cell line using an ExpiFectamine 293 Transfection Kit (Thermo Fischer Scientific) and cultured for 6 days in 37°C shaker at 125 RPM and 8% CO2. At 6 days post transfection, cultures were centrifuged, supernatant collected, and IgGs purified using protein G resin (GenScript). If necessary, antibody proteins were concentrated using an Amicon Ultra‐4 Centrifugal 30 kDa filtration unit (MilliporeSigma) and stored at 4°C.
2.7. Epitope mapping by ELISA
SARS‐CoV‐2 spike, RBD, and NTD antigens were coated onto ELISA plates at 2 μg/ml in PBS at room temperature (RT) for 2 h. ELISA plates were washed and coated with 100 μl of blocking buffer (5% w/v non‐fat dry milk, 0.1% v/v Tween‐20 in PBS) at RT for 2 h. Purified antibodies were serially diluted using blocking buffer and incubated for 1 h at RT. Plates were washed three times with washing buffer (0.05% v/v Tween‐20 in PBS) and incubated with a 1:2000 dilution of anti‐human kappa light chain detection antibody (A18853, Invitrogen) in blocking buffer at RT for 1 h. Plates were washed with washing buffer and 100 μl Super AquaBlue ELISA substrate (Thermo Fisher Scientific) was added to each well and incubated before reactions were stopped with 1 M oxalic acid. Absorbance was measured at 405 nm using a BioTek Synergy H1 plate reader. All ELISA experiments were performed in triplicate, except ELISAs against RBD were performed in duplicate.
2.8. SARS‐CoV‐2 pseudovirus neutralization assays
Recombinant Indiana vesicular stomatitis virus (rVSV) was used for generating SARS‐CoV‐2 pseudovirus as previously described. 33 Human embryonic kidney (HEK) 293 T cells were grown to 80% confluency and transfected with SARS‐CoV‐2 WT, B.1.17, or B.1.351 spike gene using FuGENE 6 (Promega). 34 Cells were cultured overnight at 37°C with 5% CO2. VSV‐G pseudotype ΔG‐luciferase (G* ΔG‐luciferase, Kerafast) was used to infect the cells in Dulbecco's modified Eagle medium (DMEM) at a multiplicity of infection (MOI) of 3 for 1 h. Cells were washed three times with 1× Dulbecco's PBS without calcium and magnesium. DMEM supplemented with 2% fetal bovine serum, 100 IU/ml of penicillin and 100 μg/ml of streptomycin were added to the cells and cultured overnight. The supernatant was collected and clarified by centrifugation at 300 × g for 10 min before storing at −80°C.
SARS‐CoV‐2 pseudovirus neutralization was performed as previously described. 33 , 34 Briefly, neutralization was assessed by incubating rVSV expression SARS‐CoV‐2 pseudovirus with serial dilutions of purified antibody. Neutralization was determined by quantifying the reduction in luciferase gene activity.
2.9. SARS‐CoV‐2 authentic virus neutralization assay
Authentic virus neutralization for the purified antibodies was assessed as previously described. 33 Briefly, purified mAbs were serially diluted and incubated with infectious SARS‐CoV‐2 (BEI Resources) for 1 h at 37°C. The antibody–virus mixture was then transferred onto a monolayer of Vero‐E6 cells and incubated for 72 h. Cytopathic effects caused by virus infection were visually observed and scored in a blind fashion by two independent observers.
2.10. Protein expression and purification for cryo‐electron microscopy (EM)
Recombinant SARS‐CoV‐2 S2P spike was produced as previously described. 23 , 24 Briefly, protein was expressed in HEK 293 Freestyle cells (Invitrogen) in suspension culture using serum‐free media (Invitrogen) by transient transfection using polyethyleneimine (Polysciences). Four days after transfection, the secreted protein was purified by nickel affinity chromatography using Ni‐NTA IMAC Sepharose 6 Fast Flow resin (GE Healthcare) followed by size exclusion chromatography on a Superdex 200 column (GE Healthcare) in 10 mM Tris, 150 mM NaCl, pH 7.4.
mAb LP5 was digested using a previously published protocol 33 to generate the Fab fragment. Briefly, IgG was digested with immobilized papain at 37 °C for 3 h in 50 mM phosphate buffer, 120 mM NaCl, 30 mM cysteine, 1 mM EDTA, pH 7. The resulting Fab was purified from Fc by affinity chromatography using protein A resin. Fab purity was analyzed by SDS‐PAGE and buffer‐exchanged into 10 mM Tris, 150 mM, pH 7.4 for cryo‐EM experiments.
2.11. Structure determination via cryo‐EM
For the SARS‐CoV‐2 complex with LP5 Fab, SARS‐CoV‐2 spike at a final concentration of 1–2 mg/ml was incubated with a ninefold molar excess of LP5 for 60 min on ice, in a buffer containing 10 mM sodium acetate, 150 mM NaCl, 0.005% (w/v) n‐Dodecyl β ‐D‐maltoside, pH 4.5. Cryo‐EM grids were prepared by applying 2.5 μl of sample to a freshly glow‐discharged carbon‐coated copper grid (CF 1.2/1.3300 mesh). The sample was vitrified in liquid ethane using a Vitrobot Mark IV with a wait time of 30 s, a blot time of 3 s, and a blot force of 0.
Data were collected on a Titan Krios electron microscope operating at 300 kV, equipped with a Gatan K3 direct electron detector and energy filter, using the SerialEM software package. 35 A total electron fluence of 41.92 e−/Å 2 was fractionated over 60 frames, with a total exposure time of 3.0 s. A magnification of 81,000× resulted in a pixel size of 1.07 Å, and a defocus range of −0.8 to −2.5 μm was used. All processing was done using cryoSPARC 36 v3.2.0. Raw movies were aligned and dose‐weighted using patch motion correction, and the contrast transfer function (CTF) was estimated using patch CTF estimation. Micrographs were picked using a blob picker, and a particle set was selected using 2D and 3D classification. Selected particle picks were manually curated for a small randomized subset of approximately 300 micrographs and used to train a Topaz neural network. This network was then used to pick particles from the remaining micrographs, which were extracted with a box size of 448 pixels.
The final consensus 3D reconstruction was obtained using homogenous refinement with C3 symmetry. The interface between NTD and LP5 Fab was locally refined by using a mask that included NTD and the variable domains of the Fab. Half maps were provided to Resolve Cryo‐EM tool in Phenix to support manual model building.
SARS CoV‐2 S2P spike density was modeled using PDB entry 6XM5 as initial template. The initial model for LP5 Fab variable region was obtained using the SAbPred server. 37
Automated and manual model building were iteratively performed using real space refinement in Phenix 38 and Coot, 39 respectively. For 2–7, ISOLDE 40 v1.1 was also used to interactively refine the structure. Half maps were provided to Resolve Cryo‐EM tool in Phenix to support manual model building. Geometry validation and structure quality assessment were performed using EMRinger 41 and MolProbity. 42 Map‐fitting cross correlation (Fit‐in‐Map tool) and figures preparation were carried out using PyMOL and UCSF Chimera 43 and Chimera X. 44 A summary of cryo‐EM data collection, reconstruction, and refinement statistics is shown in Table S1.
3. RESULTS
3.1. Natively paired VH:VL library generation from an individual convalescent for COVID‐19
PBMCs were collected from a patient who recovered from COVID‐19 for subsequent antibody screening via yeast Fab display. First, we generated natively paired VH:VL gene amplicons using a single‐cell gene capture and overlap extension RT‐PCR technology 9 , 13 (Figure 1A). Briefly, antigen‐experienced B cells were enriched by CD27+ selection and stimulated in vitro to enhance transcription of antibody genes. Single B cells were captured inside emulsion droplets for mRNA capture, and a subsequent overlap extension RT‐PCR was used to transform the separate VH and VL genes into a single, physically linked VH:VL cDNA amplicon. VH:VL cDNAs were cloned into a yeast display vector to enable functional screening via FACS and NGS. 9 , 13 Prior studies have shown that the majority of anti‐SARS‐CoV‐2 neutralizing antibodies derive from mAbs encoded by the IgG heavy chain isotype, and we therefore focused on the IgG repertoire in this study. 45 , 46 We generated two different libraries for yeast cloning and screening: IgG/VK for antibodies encoding kappa light chains, and IgG/VL for antibodies with lambda light chains.
FIGURE 1.
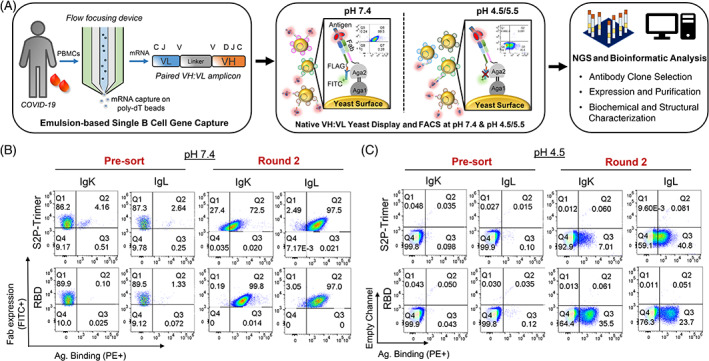
Functional analysis and screening of natively paired variable heavy:variable light (VH:VL) antibody repertoires from a convalescent COVID‐19 patient. (A) Natively paired VH:VL antibody repertoires were generated from a COVID‐19 convalescent donor for functional screening via yeast display. Left: Single B‐cell capture using a flow‐focusing device generating microdroplets consisting of a single B cell, lysis buffer, and oligo‐dT magnetic beads. Single cells are emulsified and mRNA is captured onto magnetic beads used as template for overlap extension RT‐PCR reactions to produce linked antibody VH and VL cDNAs; Center: Natively paired VH:VL cDNA amplicon libraries are cloned into a yeast display vector and expressed as Fabs for flow cytometric cell sorting (FACS) analysis at pH 7.4 and pH 4.5/5.5. Fab expression at pH 7.4 is detected by an anti‐FLAG FITC‐conjugated mAb; antigen binding is detected via PE‐conjugated antigen probes. Fab expression is not measured at pH 4.5/5.5 due to markedly reduced binding affinity between FLAG expression tag. Screening of yeast‐displayed natively paired VH:VL libraries against the SARS‐CoV‐2 spike glycoprotein with two stabilizing proline mutations in S2 domain (S2P‐Trimer) and RBD antigens is shown via FACS at (B) pH 7.4 and (C) pH 4.5. IgK stands for antibodies encoding kappa light chains, and IgL for antibodies with lambda light chains. Substantial enrichment in antigen binding was observed after three rounds of sorting both at pH 7.4 and 4.5 compared to the presort/input libraries. See Figure S1 for sorting and enrichment at pH 5.5, and for an evaluation of expression and binding at pH 7.4 of yeast libraries sorted at pH 4.5/5.5
3.2. In vitro screening for anti‐SARS‐CoV‐2 antibodies via yeast display
We used FACS and NGS 9 to identify the set of anti‐SARS‐CoV‐2 antibodies, mapping antigen recognition at multiple pH values. Linked VH:VL amplicons encoding natively paired heavy and light chain antibody genes were expressed on the yeast surface in a Fab format, with lambda and kappa light constant regions for the IgG/VL and IgG/VK libraries, respectively. Expression was controlled using a galactose‐inducible bidirectional promoter, and yeast cells were incubated with two different fluorescently labelled antigens: a trimeric SARS‐CoV‐2 spike glycoprotein, and RBD. 22 The RBD comprises several potent known vulnerable epitopes on the SARS‐CoV‐2 spike. 33 , 47 , 48 , 49
We performed yeast screening by incubating Fab‐expressing yeast with SARS‐CoV‐2 antigen probes at three different pH values: 4.5, 5.5, and 7.4. The first three rounds of screening enriched libraries for binding against the SARS‐CoV‐2 trimeric glycoprotein (20 nM) and RBD (100 nM) (Figures 1 and S1). Yeast antibody library sorting at pH 4.5/5.5 poses a challenge for the detection of Fab expression on yeast surface, as at pH 4.5 the binding affinity between the FLAG expression tag and its detecting antibody is markedly reduced. 50 To circumvent this issue, we sorted yeast without the use of the FLAG expression tag at pH 4.5/5.5, and then we confirmed that binding yeast were expressing the Fab/FLAG tag by staining with antigen at pH 4.5/5.5, and subsequently washing and changing the pH to 7.4 for labelling of the FLAG tag (Figure S1). After three rounds of FACS and yeast culture in between sorts to enrich for antigen‐binding Fabs in the yeast libraries, we observed that yeast sorted at endosomal pH showed strong expression and binding at pH 7.4 (Figure S1B,C).
3.3. Mining of NGS sequences and selection of mAb clones for characterization
We analyzed the plasmid DNAs encoded by yeast libraries across all sort conditions using 2 × 300 bp Illumina MiSeq NGS analysis to quantitatively track the prevalence of each antibody clone in the libraries across screening rounds. 9 , 13 , 29 , 32 , 50 We followed each clone in the libraries by CDR‐H3 amino acid sequences throughout the screening at all three pH values, calculating an ER for each clone (see Section 2). 9 , 13 , 29 , 50 We used an ER cutoff of 10 to select clones for synthesis and characterization, and we chose five antibody sequences (LP1–LP5) that were enriched across various screening conditions for further analyses (Figure 2; Table 1).
FIGURE 2.
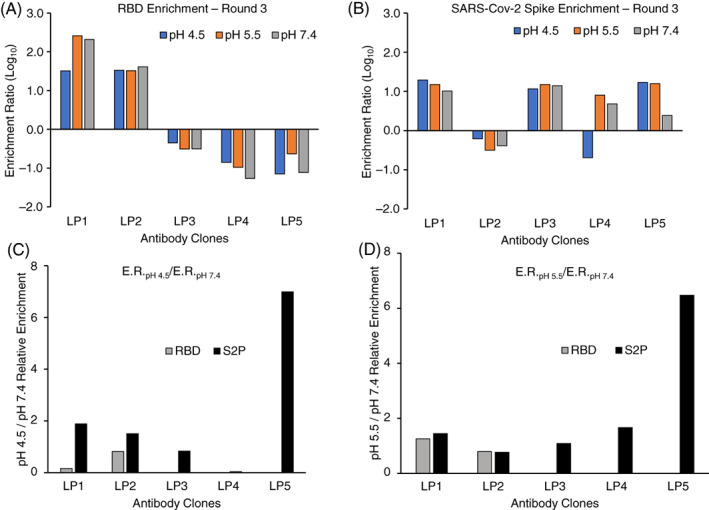
Enrichment ratios (ERs) of selected antibody clones from sorted yeast libraries. ERs were calculated after sorting against (A) receptor binding domain (RBD) and (B) SARS‐CoV‐2 stabilized (S2P) spike trimer after three rounds of sorting, for all three sorted pH values (4.5, 5.5, and 7.4). Ratio of ER values for antibody clones sorted at (C) pH 4.5 and (D) pH 5.5 calculated against pH 7.4. The ratio of ER values against RBD is omitted for LP3, LP4, and LP5 in (C) and (D) because those three mAbs do not target an RBD epitope
TABLE 1.
List of selected antibody clones via yeast display functional screening of natively paired variable heavy:variable light (VH:VL) antibody repertoire of a COVID‐19 convalescent individual
| mAb | CDRH3 | CDRL3 | Heavy chain V‐gene | Light chain V‐gene |
|---|---|---|---|---|
| LP1 | CARLQQANYFDYW | CQQYNSYAWTF | VH5‐51 | VK1‐5 |
| LP2 | CARGGLVPDSSGAFDIW | CCSYAGSSTFVVF | VH1‐46 | VL2‐23 |
| LP3 | CARDVSITIFGVEDNWFDPW | CNSRDSSGNHRVF | VH4‐59 | VL3‐19 |
| LP4 | CATSGPLGLERHNWLDPW | CSSYVGSNLVIF | VH1‐24 | VL2‐8 |
| LP5 | CARADYGDFFFDYW | CQSADSSATYWVF | VH3‐33 | VL2‐35 |
We synthesized the VH and VL regions of LP1‐5 mAbs and cloned them into human IgG1 expression plasmids for transient transfection in Expi293F human cells and purification as full‐length IgG for functional evaluation. ELISA studies demonstrated that all five mAbs bound to SARS‐CoV‐2 spike glycoprotein, with two mAbs (LP1 and LP2) targeting the RBD antigen. The remaining three mAbs (LP3‐5) targeted the NTD of SARS‐CoV‐2 (Figure 3). Because LP2 and LP3 bind spike antigens in ELISA assays but do not neutralize pseudoviruses, it is likely that LP2 and LP3 target non‐neutralizing epitopes on the spike protein.
FIGURE 3.
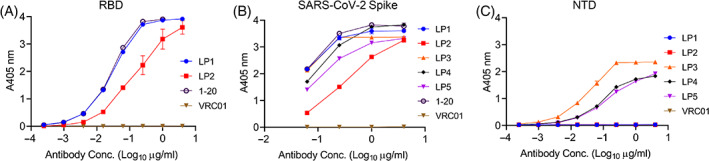
Binding profiles of selected purified mAbs via ELISA. Binding against (A) receptor binding domain (RBD), (B) SARS‐CoV‐2 stabilized (S2P) spike trimer and (C) NTD of spike trimer, all at pH 7.4. Triplicates of binding were done for spike trimer and NTD, and duplicates were performed for RBD. Data are shown as mean ± SEM
3.4. Potently neutralizing SARS‐CoV‐2 function was associated with preferential enrichment at pH 4.5
We assessed the neutralization potency of each mAb against a SARS‐CoV‐2 pseudovirus based on VSV expressing the SARS‐CoV‐2 spike surface glycoprotein. 33 We first assayed the neutralization activity of the five mAbs against an early SARS‐CoV‐2 spike gene variant (Wuhan‐Hu‐1, China, or WT). Out of the five clones tested, LP5 showed the strongest neutralization activity with an IC50 <0.005 μg/ml (Figure 4A); LP1 also showed some neutralization activity (Figure 4A). Based on pseudovirus neutralization assay performance, we selected mAb LP5 for further neutralization studies.
FIGURE 4.
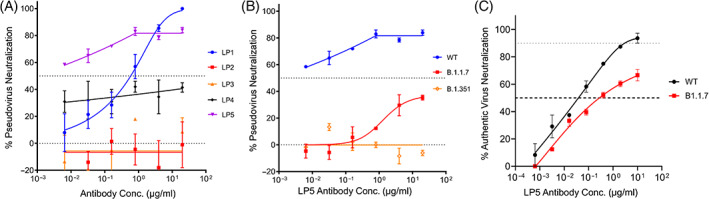
Neutralization profiles of five selected mAbs. (A) Pseudovirus neutralization against the SARS‐CoV‐2 WT strain. (B) Pseudovirus neutralization compared for mAb LP5 against the WT strain, the UK spike variant (B.1.1.7), and the SA spike variant (B.1.351). (C) Authentic virus neutralization against the WT strain and the UK spike variant (B.1.1.7)
We found that, out of the five antibody clones isolated from libraries here, LP5 showed the highest ratio of enrichment against the SARS‐CoV‐2 spike protein in our reduced pH library sorts (pH 4.5/5.5) compared to the library sorts at pH 7.4 (Figure 2C,D). mAb LP5 was also found to be the most potent antibody in neutralization assays (Figure 4). Our prior studies also revealed a similar association with binding to the spike at pH 4.5 with potent neutralizing activity against the conserved ACE2 binding site on the RBD domain. 13 Taken together, these data suggested that reduced pH sorts against the SARS‐CoV‐2 spike antigen may help to provide efficient selection of potent neutralizing antibodies.
To determine if LP5 would recognize recently emerged SARS‐CoV‐2 variants of concern (VOCs), 51 we next assessed potential cross‐neutralization activity of LP5 against the United Kingdom (UK) spike variant (also known as 20I/501Y.V1, VOC 202012/01, or B.1.1.7) and the South African (SA) spike variant (20H/501Y.V2 or B.1.351) using the VSV‐pseudovirus assay. We observed that the LP5 showed only 30% neutralization against the UK variant and did not neutralize the SA variant (Figure 4B). We also evaluated the neutralization activity of LP5 against SARS‐CoV‐2 WT and UK variant authentic viruses. LP5 neutralized an authentic SARS‐CoV‐2 WT virus with an IC50 0.038 μg/ml and showed over 60% neutralization of the UK variant, with an IC50 of 2.00 μg/ml against the UK strain (Figure 4C). Based on its neutralization performance features and strong recognition at endosomal pH, we selected LP5 for additional characterization and structural determination.
3.5. LP5 Cryo‐EM structure demonstrates a novel SARS‐CoV‐2 recognition mechanism for anti‐NTD antibodies
LP5 is encoded by the heavy and light chain V‐genes VH3‐33 and VL2‐35, respectively (Table 1). Several groups have reported other potent anti‐NTD neutralizing antibodies from COVID‐19 convalescent individuals that are also encoded by the VH3‐33 heavy chain gene, 52 , 53 , 54 suggesting a possible convergent antibody recognition mechanism for related anti‐NTD neutralizing antibodies. We assessed the antibody:antigen bound state for mAb LP5 with the SARS‐CoV‐2 spike glycoprotein via cryo‐EM. We collected single‐particle data using Titan Krios microscope, yielding a cryo‐EM reconstruction to an overall resolution of 4.46 Å (Figures 5A and S2). A single conformational state with three Fabs per trimer, with three RBD “down” conformation, was identified.
FIGURE 5.
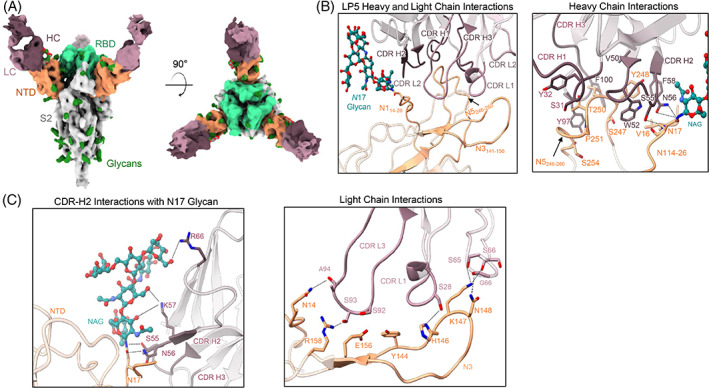
Structural analysis of the N‐terminal domain (NTD)‐directed neutralizing antibody LP5. (A) Cryo‐electron microscopy (cryo‐EM) reconstructions for SARS‐CoV‐2 spike complexes with LP5. The NTD is shown in orange, receptor binding domain (RBD) in green, glycans in dark green, with the LP5 antibody heavy chains in magenta and light chains in gray. (B) Expanded view of LP5 interactions with NTD showing the overall interface (left), and recognition by the LP5 heavy chain (right), (C) CDR‐H2 recognition of the SARS‐CoV‐2 N17 glycan (left), and recognition of NTD by the LP5 light chain (right). NTD regions N1 (14–26), N3 (residues 141–156) and N5 (residues 246–260) are shown. All hydrogen bonds (distance ~3.2 Å) are represented as dashed lines
The structure of LP5 with the SARS‐CoV‐2 spike revealed that LP5 binds within the NTD antigenic supersite, targeting a similar region as some other previously reported neutralizing antibodies 52 , 54 (Figure 5A). The CDR‐H2 makes the dominant contribution to LP5 recognition by interacting with the NTD loop region N1 (14–26), while the CDR‐L3 interacts with the NTD loops N3 (141–156) and N5 (246–260) (Figure 5B; left and right panels). The SARS‐CoV‐2 N17 glycan stacks against the LP5 CDR‐H2, forming hydrogen bonds and hydrophobic interactions between LP5 and the NTD (Figure 5C, left panel); the CDR‐H2 of LP5 forms an extensive hydrogen bond network with the SARS‐CoV‐2 N17 glycan on NTD. Ser55HC and Asn56HC also interact with both the backbone and side chain of Asn17NTD. The light chain also contributed to NTD recognition through the CDR‐L1 and ‐L3, predominantly via hydrogen bonds involving the hydroxyl groups of Ser28LC in the CDR‐L1 and Ser92LC in the CDR‐L3, as well as the framework residues Ser64LC and Ser65LC (Figure 5C, right panel), while Lys57HC interacts with the first two N‐acetylglucosamine units of the glycan. The framework residue Arg66HC also stabilizes the hydroxyl group of a branched mannose residue via a mechanism that has not been reported previously (Figures 5B and S1F). These data highlighted the unique interactions that enabled potent viral recognition of antibody LP5.
4. DISCUSSION
Antibody discovery technologies have seen tremendous growth in recent years. However, the identification of neutralizing antibodies has remained a time‐consuming, inefficient, and slow step in antiviral mAb discovery. Here, we applied endosomal pH (4.5) screening via yeast surface display to identify a novel and potent SARS‐CoV‐2 neutralizing antibody LP5 that binds to the supersite region on the NTD of the trimeric spike glycoprotein. This yeast display screening method described here enhances the speed of neutralizing antibody discovery by highlighting a subset of antibodies as candidate neutralizers using endosomal pH antigen screening. Based on the current data and other recent reports, 13 endosomal pH conditions have been shown to preferentially select for the most potent SARS‐CoV‐2 neutralizing antibodies in mAb panels and libraries. Antigen recognition at endosomal pH has been supported both by the current study (targeting the RBD and the spike protein), and also in a prior study characterizing antibodies against the SARS‐CoV‐2 RBD. 13 These data suggest that endosomal pH screening could be a broadly useful tool to help preferentially identify potent antiviral antibodies and enhance screening efficiency while reducing the costs of antibody discovery campaigns. We do not anticipate that every antibody that binds to viral antigens at endosomal pH will potently neutralize the virus; however, our data collected to date have revealed that most non‐neutralizing antibodies do not bind to viral spike antigens to the same extent as the most potent neutralizing antibodies can recognize spike antigens at endosomal pH. 13 Screening for potent neutralizing antibodies at low pH will be a potential growth area for antibody discovery, and future studies will build on this initial report.
Based on the date and geographic location of sample collection in this study (New York, USA, March/April 2020), the COVID‐19 convalescent donor was most likely infected with an early, founder‐like virus strain. 33 Similar to data observed in other studies, 34 LP5 shows cross‐reactive neutralization against the UK variant of concern (B.1.1.7), albeit with reduced potency. LP5 also shows some hallmarks of genetic and functional convergence with other reported anti‐NTD recognition antibodies. LP5 is encoded by the VH3‐33 heavy chain V‐gene, which has also been observed in other potent anti‐NTD supersite targeting mAb structures. 52 , 53 , 54 LP5, however, shows a slightly different mode of epitope recognition compared to its class members, as the unique glycan interactions from Lys57HC and Arg66HC have not been reported in other anti‐NTD reported antibody structures to date.
In this study, we tested the capacity of endosomal pH screening to identify SARS‐CoV‐2 neutralizing antibodies, but we anticipate that endosomal pH screening can be useful to discover neutralizing antibodies against other viruses like YFV, dengue, rabies virus, and Ebola virus, which all leverage endosomal entry pathways to infect cells. 19 , 20 , 21 , 55 , 56 , 57 , 58 Flamand et al. reported that maintaining the rabies virion at low pH converted the spike to a more neutralization sensitive form. 59 , 60 Screening at endosomal pH could help simulate the environment within endosomes that is conducive to viral fusion, including replicating potential structural changes that viral fusion proteins undergo after endocytosis when the intraendosomal pH is reduced. We note that yeast display is particularly suited for screening at pH 4.5–5.5, as yeast grows very well in the endosomal pH range. Phage display screening at pH 4.5–5.5 should also work effectively. Mammalian cell screening platforms are likely not compatible with endosomal pH screening due to the need to maintain high cell viability for most mammalian antibody screening platforms.
LP5 showed potent neutralization against the SARS‐CoV‐2 founder virus strain, but it had diminished neutralization activity towards the UK variant (B.1.1.7) and was unable to neutralize the SA variant strain (B.1.351). The reduction in neutralization against the UK variant could be due to the deletion ΔY144 in the NTD supersite region, which is proximal to the LP5 binding epitope. 34 , 61 The lack of neutralization activity against the SA variant may have been caused by the SA variant's genetic deletion (Δ242–Δ244) and/or R246I substitution in the supersite region of the NTD. These changes can result in complete loss of NTD recognition in anti‐NTD mAbs, leading to the resistance towards neutralization observed widely for the SA variant. 34 , 62 We expect that follow‐up screening studies with the SA variant, potentially at endosomal pH, can help to identify additional potent antibodies targeting this variant of concern.
We anticipate that endosomal pH screening as described here can help accelerate the identification of potent neutralizing antibodies against SARS‐CoV‐2 and other viral disease threats. While our current display platform has a limited ability to incorporate surface expression tags at pH 4.5, we will address this issue in future studies by the use of covalent tag‐based detection of antibody gene expression that is not affected by endosomal pH. 50 We look forward to continued implementation of this simple and effective approach to rapidly downselect antibody libraries for potent antiviral neutralization activity and to enhance the efficiency of antibody discovery against emergent and established viral disease threats.
Supporting information
Appendix S1: Supporting Information
ACKNOWLEDGMENTS
We thank Jennifer Hackett for help with sequencing and Matias Gutiérrez González for help with bioinformatic analysis. This work was supported by the University of Kansas Departments of Chemical Engineering and Pharmaceutical Chemistry, the Intramural Research Program of the Vaccine Research Center, National Institute of Allergy and Infectious Diseases, the Jack Ma Foundation, COVID‐19 Fast Grants, the American Lung Association, and by NIH grants DP5OD023118, P20GM103418, R01AI141452, R21AI143407, and R21AI144408.
Madan B, Reddem ER, Wang P, et al. Antibody screening at reduced pH enables preferential selection of potently neutralizing antibodies targeting SARS‐CoV‐2. AIChE J. 2021;67(12):e17440. doi: 10.1002/aic.17440
Bharat Madan and Eswar R. Reddem contributed equally to this study.
Funding information Intramural Research Program of the Vaccine Research Center; National Institute of Allergy and Infectious Diseases, National Institutes of Health, Grant/Award Numbers: R01AI141452, R21AI143407, R21AI144408; National Institute of General Medical Sciences, Grant/Award Number: P20GM103418; NIH Office of the Director, Grant/Award Number: DP5OD023118; American Lung Association; COVID‐19 Fast Grants; Jack Ma Foundation; University of Kansas Departments of Chemical Engineering and Pharmaceutical Chemistry
DATA AVAILABILITY STATEMENT
The Cryo‐EM structure of LP5 in complex with SARS‐CoV‐2 Spike glycoprotein is deposited in the Research Collaboratory for Structural Bioinformatics Protein Data Bank under Protein Data Bank (PDB) ID 7MXP, and in the Electron Microscopy Data Bank (EMDB) at PDBe under ID EMD‐24075.
REFERENCES
- 1. Mullard A. FDA approves 100th monoclonal antibody product. Nat Rev Drug Discov. 2021;20:491‐495. [DOI] [PubMed] [Google Scholar]
- 2. Low JG, Ng JHJ, Ong EZ, et al. Phase 1 trial of a therapeutic anti–yellow fever virus human antibody. N Engl J Med. 2020;383(5):452‐459. [DOI] [PubMed] [Google Scholar]
- 3. Corey L, Gilbert PB, Juraska M, et al. Two randomized trials of neutralizing antibodies to prevent HIV‐1 acquisition. N Engl J Med. 2021;384(11):1003‐1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Levine MM. Monoclonal antibody therapy for Ebola virus disease. N Engl J Med. 2019;381(24):2365‐2366. [DOI] [PubMed] [Google Scholar]
- 5. Burton DR, Hangartner L. Broadly neutralizing antibodies to HIV and their role in vaccine design. Annu Rev Immunol. 2016;34(1):635‐659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Corti D, Lanzavecchia A. Broadly neutralizing antiviral antibodies. Annu Rev Immunol. 2013;31(1):705‐742. [DOI] [PubMed] [Google Scholar]
- 7. Rogers TF, Zhao F, Huang D, et al. Isolation of potent SARS‐CoV‐2 neutralizing antibodies and protection from disease in a small animal model. Science. 2020;369(6506):956‐963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. DeKosky BJ, Kojima T, Rodin A, et al. In‐depth determination and analysis of the human paired heavy‐ and light‐chain antibody repertoire. Nat Med. 2015;21(1):86‐91. [DOI] [PubMed] [Google Scholar]
- 9. Wang B, DeKosky BJ, Timm MR, et al. Functional interrogation and mining of natively paired human VH:VL antibody repertoires. Nat Biotechnol. 2018;36(2):152‐155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Winters A, McFadden K, Bergen J, et al. Rapid single B cell antibody discovery using nanopens and structured light. mAbs. 2019;11(6):1025‐1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gérard A, Woolfe A, Mottet G, et al. High‐throughput single‐cell activity‐based screening and sequencing of antibodies using droplet microfluidics. Nat Biotechnol. 2020;38(6):715‐721. [DOI] [PubMed] [Google Scholar]
- 12. Goldstein LD, Chen Y‐JJ, Wu J, et al. Massively parallel single‐cell B‐cell receptor sequencing enables rapid discovery of diverse antigen‐reactive antibodies. Commun Biol. 2019;2(1):1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Banach BB, Cerutti G, Fahad AS, et al. Paired heavy and light chain signatures contribute to potent SARS‐CoV‐2 neutralization in public antibody responses. bioRxiv. 2021. 10.1101/2020.12.31.424987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang Z‐Y, Huang Y, Ganesh L, et al. pH‐dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through DC‐SIGN. J Virol. 2004;78(11):5642‐5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhou T, Tsybovsky Y, Gorman J, et al. Cryo‐EM structures of SARS‐CoV‐2 spike without and with ACE2 reveal a pH‐dependent switch to mediate endosomal positioning of receptor‐binding domains. Cell Host Microbe. 2020;28(6):867‐879.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hoffmann M, Kleine‐Weber H, Schroeder S, et al. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271‐280.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shang J, Wan Y, Luo C, et al. Cell entry mechanisms of SARS‐CoV‐2. Proc Natl Acad Sci USA. 2020;117(21):11727‐11734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Perera‐Lecoin M, Meertens L, Carnec X, Amara A. Flavivirus entry receptors: an update. Viruses. 2013;6(1):69‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Smit JM, Moesker B, Rodenhuis‐Zybert I, Wilschut J. Flavivirus cell entry and membrane fusion. Viruses. 2011;3(2):160‐171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aleksandrowicz P, Marzi A, Biedenkopf N, et al. Ebola virus enters host cells by macropinocytosis and clathrin‐mediated endocytosis. J Infect Dis. 2011;204(suppl_3):S957‐S967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cruz‐Oliveira C, Freire JM, Conceição TM, Higa LM, Castanho MARB, Da Poian AT. Receptors and routes of dengue virus entry into the host cells. FEMS Microbiol Rev. 2015;39(2):155‐170. [DOI] [PubMed] [Google Scholar]
- 22. Zhou T, Teng I‐T, Olia AS, et al. Structure‐based design with tag‐based purification and in‐process biotinylation enable streamlined development of SARS‐CoV‐2 spike molecular probes. Cell Rep. 2020;33(4):108322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hsieh C‐L, Goldsmith JA, Schaub JM, et al. Structure‐based design of prefusion‐stabilized SARS‐CoV‐2 spikes. Science. 2020;369(6510):1501‐1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wrapp D, De Vlieger D, Corbett KS, et al. Structural basis for potent neutralization of betacoronaviruses by single‐domain camelid antibodies. Cell. 2020;181(5):1004‐1015.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lagerman CE, López Acevedo SN, Fahad AS, Hailemariam AT, Madan B, DeKosky BJ. Ultrasonically‐guided flow focusing generates precise emulsion droplets for high‐throughput single cell analyses. J Biosci Bioeng. 2019;128(2):226‐233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McDaniel JR, DeKosky BJ, Tanno H, Ellington AD, Georgiou G. Ultra‐high‐throughput sequencing of the immune receptor repertoire from millions of lymphocytes. Nat Protoc. 2016;11(3):429‐442. [DOI] [PubMed] [Google Scholar]
- 27. Benatuil L, Perez JM, Belk J, Hsieh C‐M. An improved yeast transformation method for the generation of very large human antibody libraries. Protein Eng Des Sel. 2010;23(4):155‐159. [DOI] [PubMed] [Google Scholar]
- 28. Zhou T, Georgiev I, Wu X, et al. Structural basis for broad and potent neutralization of HIV‐1 by antibody VRC01. Science. 2010;329(5993):811‐817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Madan B, Zhang B, Xu K, et al. Mutational fitness landscapes reveal genetic and structural improvement pathways for a vaccine‐elicited HIV‐1 broadly neutralizing antibody. Proc Natl Acad Sci USA. 2021;118(10). https://www.pnas.org/content/118/10/e2011653118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. DeKosky BJ, Lungu OI, Park D, et al. Large‐scale sequence and structural comparisons of human naive and antigen‐experienced antibody repertoires. Proc Natl Acad Sci USA. 2016;113(19):E2636‐E2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Medina‐Cucurella AV, Whitehead TA. Characterizing protein‐protein interactions using deep sequencing coupled to yeast surface display. Methods Mol Biol. 1764;2018:101‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fahad AS, Timm MR, Madan B, et al. Functional profiling of antibody immune repertoires in convalescent Zika virus disease patients. Front Immunol. 2021;12. 10.3389/fimmu.2021.615102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu L, Wang P, Nair MS, et al. Potent neutralizing antibodies against multiple epitopes on SARS‐CoV‐2 spike. Nature. 2020;584(7821):450‐456. [DOI] [PubMed] [Google Scholar]
- 34. Wang P, Nair MS, Liu L, et al. Antibody resistance of SARS‐CoV‐2 variants B.1.351 and B.1.1.7. Nature. 2021;593:130‐135. [DOI] [PubMed] [Google Scholar]
- 35. Mastronarde DN. Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol. 2005;152(1):36‐51. [DOI] [PubMed] [Google Scholar]
- 36. Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA. cryoSPARC: algorithms for rapid unsupervised cryo‐EM structure determination. Nat Methods. 2017;14(3):290‐296. [DOI] [PubMed] [Google Scholar]
- 37. Dunbar J, Krawczyk K, Leem J, et al. SAbPred: a structure‐based antibody prediction server. Nucleic Acids Res. 2016;44(W1):W474‐W478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Adams PD, Gopal K, Grosse‐Kunstleve RW, et al. Recent developments in the PHENIX software for automated crystallographic structure determination. J Synchrotron Radiat. 2004;11(Pt 1):53‐55. [DOI] [PubMed] [Google Scholar]
- 39. Emsley P, Cowtan K. Coot: model‐building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126‐2132. [DOI] [PubMed] [Google Scholar]
- 40. Croll TI. ISOLDE: a physically realistic environment for model building into low‐resolution electron‐density maps. Acta Cryst D. 2018;74(6):519‐530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Barad BA, Echols N, Wang RY‐R, et al. EMRinger: side chain–directed model and map validation for 3D cryo‐electron microscopy. Nat Methods. 2015;12(10):943‐946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Davis IW, Murray LW, Richardson JS, Richardson DC. MOLPROBITY: structure validation and all‐atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004;32(Web Server issue):W615‐W619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605‐1612. [DOI] [PubMed] [Google Scholar]
- 44. Pettersen EF, Goddard TD, Huang CC, et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 2021;30(1):70‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Garcia‐Beltran WF, Lam EC, Astudillo MG, et al. COVID‐19‐neutralizing antibodies predict disease severity and survival. Cell. 2021;184(2):476‐488.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sasisekharan V, Pentakota N, Jayaraman A, Tharakaraman K, Wogan GN, Narayanasami U. Orthogonal immunoassays for IgG antibodies to SARS‐CoV‐2 antigens reveal that immune response lasts beyond 4 mo post illness onset. Proc Natl Acad Sci USA. 2021;118(5). 10.1073/pnas.2021615118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Brouwer PJM, Caniels TG, van der Straten K, et al. Potent neutralizing antibodies from COVID‐19 patients define multiple targets of vulnerability. Science. 2020;369(6504):643‐650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Piccoli L, Park Y‐J, Tortorici MA, et al. Mapping neutralizing and immunodominant sites on the SARS‐CoV‐2 spike receptor‐binding domain by structure‐guided high‐resolution serology. Cell. 2020;183(4):1024‐1042.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pinto D, Park Y‐J, Beltramello M, et al. Cross‐neutralization of SARS‐CoV‐2 by a human monoclonal SARS‐CoV antibody. Nature. 2020;583(7815):290‐295. [DOI] [PubMed] [Google Scholar]
- 50. Jin W, Madan B, Mussman BK, et al. The covalent SNAP tag for protein display quantification and low‐pH protein engineering. J Biotechnol. 2020;320:50‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Abdool Karim SS, de Oliveira T. New SARS‐CoV‐2 variants—clinical, public health, and vaccine implications. N Engl J Med. 2021;384(19):1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McCallum M, Marco AD, Lempp FA, et al. N‐terminal domain antigenic mapping reveals a site of vulnerability for SARS‐CoV‐2. Cell. 2021;184(9):2332‐2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Suryadevara N, Shrihari S, Gilchuk P, et al. Neutralizing and protective human monoclonal antibodies recognizing the N‐terminal domain of the SARS‐CoV‐2 spike protein. Cell. 2021;184(9):2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cerutti G, Guo Y, Zhou T, et al. Potent SARS‐CoV‐2 neutralizing antibodies directed against spike N‐terminal domain target a single supersite. Cell Host Microbe. 2021;29:819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lu X, Xiao H, Li S, et al. Double lock of a human neutralizing and protective monoclonal antibody targeting the yellow fever virus envelope. Cell Rep. 2019;26(2):438‐446.e5. [DOI] [PubMed] [Google Scholar]
- 56. Laureti M, Narayanan D, Rodriguez‐Andres J, Fazakerley JK, Kedzierski L. Flavivirus receptors: diversity, identity, and cell entry. Front Immunol. 2018;9. 10.3389/fimmu.2018.02180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gaudin Y, Ruigrok RW, Knossow M, Flamand A. Low‐pH conformational changes of rabies virus glycoprotein and their role in membrane fusion. J Virol. 1993;67(3):1365‐1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gaudin Y, Tuffereau C, Durrer P, Flamand A, Ruigrok RW. Biological function of the low‐pH, fusion‐inactive conformation of rabies virus glycoprotein (G): G is transported in a fusion‐inactive state‐like conformation. J Virol. 1995;69(9):5528‐5534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Flamand A, Raux H, Gaudin Y, Ruigrok RWH. Mechanisms of rabies virus neutralization. Virology. 1993;194(1):302‐313. [DOI] [PubMed] [Google Scholar]
- 60. Burton DR, Williamson RA, Parren PW. Antibody and virus: binding and neutralization. Virology. 2000;270(1):1‐3. [DOI] [PubMed] [Google Scholar]
- 61. McCarthy KR, Rennick LJ, Nambulli S, et al. Recurrent deletions in the SARS‐CoV‐2 spike glycoprotein drive antibody escape. Science. 2021;371(6534):1139‐1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wibmer CK, Ayres F, Hermanus T, et al. SARS‐CoV‐2 501Y.V2 escapes neutralization by South African COVID‐19 donor plasma. Nat Med. 2021;27(4):622‐625. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information
Data Availability Statement
The Cryo‐EM structure of LP5 in complex with SARS‐CoV‐2 Spike glycoprotein is deposited in the Research Collaboratory for Structural Bioinformatics Protein Data Bank under Protein Data Bank (PDB) ID 7MXP, and in the Electron Microscopy Data Bank (EMDB) at PDBe under ID EMD‐24075.