

Summary
Stress erythropoiesis and chronic inflammation in subjects with sickle cell disease (SCD) may have an impact on the bone marrow (BM) haematopoietic stem and progenitor cell (HSPC) quality and yield necessary for effective autologous, ex vivo HSPC gene therapy. BM from 19 subjects with SCD and five volunteers without SCD (non-SCD) was collected in different anticoagulants and processed immediately (day 0) or the following day (day 1). Inflammatory, contamination and aggregation markers within the mononuclear layer, and CD34, CD45 and Glycophorin-A (GPA) expression on HSPCs after CD34+ selection were analysed by conventional and imaging flow cytometry. Compared to non-SCD BM, multiple markers of inflammation, contamination (red cells, P < 0·01; platelets, P < 0·01) and aggregates (platelet/granulocytes, P < 0·01; mononuclear/red cells, P < 0·01) were higher in SCD BM. Total CD34+ cell count was lower in SCD BM (P < 0·05), however CD34+ count was higher in SCD BM when collected in acid citrate dextrose-A (ACDA) versus heparin (P < 0 05). Greater than 50% of CD34+ HSPCs from SCD BM are CD34dim due to higher erythroid lineage expression (P < 0·01) as single cell CD34+CD45+GPA+ (P < 0·01) and CD34+CD45‒GPA+ (P < 0·01) HSPCs. SCD BM is characterized by increased inflammation, aggregation and contamination contributing to significant differences in HSPC quality and yield compared to non-SCD BM.
Keywords: bone marrow, gene therapy, haematopoietic stem and progenitor cell, inflammation, sickle cell disease
Sickle cell disease (SCD) encompasses a group of disorders characterized by a single point mutation (c.20A>T) in the β-globin gene (HBB). Whether inherited in a homozygous state or with another HBB mutation, this monogenic change produces abnormal red blood cells (RBCs) with many downstream effects and devastating clinical complications. While hydroxycarbamide (HC) or chronic RBC transfusions (CT) may alleviate severe symptoms of SCD in some patients, allogeneic haematopoietic stem cell transplantation (HSCT) is the only currently available curative option for patients with SCD. Studies report an overall survival of >95% with a myeloablative regimen using human leucocyte antigen (HLA)-matched sibling donors in the paediatric population (Walters et al, 1996) and >90% disease-free survival using a non-myeloablative regimen in adults (Hsieh et al, 2014), however less than 15% of patients with SCD have an appropriately matched HLA donor (Walters et al, 2001). The lack of suitable haematopoietic stem and progenitor cell (HSPC) donors for most patients with SCD, in addition to the morbidity and mortality associated with allogeneic HSCT, makes genetic therapy, modifying a patient’s autologous HSPCs, an attractive long-term solution. In the most advanced gene therapy model currently in clinical trials, autologous HSPCs are transduced with an integrating lentiviral vector (LVV) expressing normal β-globin and are transplanted back into the patient after myeloablative conditioning (NCT02151526, NCT02140554). Bone marrow (BM) is currently the primary source of autologous HSPCs in people with SCD given potentially fatal complications with the use of standard HSPC mobilization using granulocyte colony-stimulating factor (G-CSF; filgrastim) in these patients, therefore the characterization of BM from subjects with SCD is needed (Abboud et al, 1998; Adler et al, 2001; Fitzhugh et al, 2009).
Although clinically heterogenous, SCD is a chronic inflammatory disease characterized by a baseline steady state leucocytosis, thrombocytosis, and elevated C-reactive protein and erythrocyte sedimentation rate (Krishnan et al, 2010; Hatzipantelis et al, 2013; Zhang et al, 2016). Unlike in other haemoglobinopathies such as β-thalassaemia, HSPC quality and yield in SCD may be significantly impacted by this inflammatory environment in addition to the stress erythropoiesis associated with chronic anaemia. Interim results in gene therapy clinical trials in subjects with either β-thalassaemia or SCD utilizing the same lentiviral system show that subjects with SCD have a lower median CD34+ cell collection using BM as the HSPC source, lower median peripheral blood (PB) vector copy number (VCN) [number of LVV copies in mononuclear (MN) cells found in the PB after transplantation], and overall lower median vector-derived haemoglobin (Hb) (normal β-globin expression) (Kanter et al, 2016, 2017; Cavazzana et al, 2017; Kwiatkowski et al, 2017; Thompson et al, 2018). As globin expression occurs downstream in the HSPC lineage, in addition to similar erythropoietic demands as a patient with β-thalassaemia, it could be hypothesized that HSPCs from a patient with SCD at steady state are equivalent and should be equally amenable to genetic modification techniques and long-term survival post transplantation. The difficulty in obtaining high cell doses and a robust engraftment of transduced HSPCs in SCD patients suggests, however, that the unique inflammatory BM environment associated with SCD may have significant impacts on HSPCs (Kanter et al, 2016, 2017; Cavazzana et al, 2017; Kwiatkowski et al, 2017). A patient with SCD is said to be in steady state when there is absence of infection, acute clinical symptoms or overt vaso-occlusive crisis for at least three months (Bookchin & Lew, 1996), yet imaging studies of BM suggests that even during steady state, the BM of patients with SCD appears patchy, probably due to hyperplasia of the haematopoietic marrow in addition to repeated episodes of infarction and fibrosis, and is markedly different from those in matched healthy controls and those with non-sickle haemoglobinopathies (Rao et al, 1989; Mankad et al, 1990; Aguilar et al, 2005). This patchiness worsens during vaso-occlusive crisis and with age, and suggests steady state BM from patients with SCD is uniquely different.
For ex vivo HSPC gene therapy to be successful in patients with SCD, a sufficient quantity and quality of HSPCs need to undergo modification to ensure sustained production of the gene of interest at levels capable of overcoming the pathogenic phenotype. To be a onetime cure, gene modification must occur in the most immature, long term engrafting HSPCs (LT-HSPCs), defined by two unique capabilities, lifelong self-renewal and multilineage differentiation. HSPCs are characterized by their distinct surface immunophenotype (CD34bright CD45RA‒CD90+CD38‒Lin‒ cells), which changes throughout the haematopoietic differentiation process as HSPCs mature to produce all blood cells, first becoming short-term HSPC (CD34dim/CD38+ cells) that maintain full-lineage differentiation potential but have lost the self-renewal capacity of HSPCs (Eaves, 2015). Most CD34+ cells are lineage-restricted progenitors and true HSPCs remain rare, representing less than 0·01% of all cells in the BM and ~1 to 3% of all CD34+ cells (Walasek et al, 2012). Studies have shown that modification of bulk HSPCs with lentiviral transduction can be inefficient because the majority of CD34+ cells are short-term progenitors with a limited post-transplant lifespan (Masiuk et al, 2017; Zonari et al, 2017). Furthermore, most HSPCs are in a quiescent state but require cytokine stimulation in vitro for efficient lentiviral transduction (Case et al, 1999), resulting in the potential loss of in vivo long-term repopulating stem cell function as a result of cell proliferation (Peters et al, 1996; Takatoku et al, 2001). However, in the absence of further perturbation of the HSPC population (for example in the thalassaemia setting) these stem cells are amenable to transduction, engraftment and disease correction (Thompson et al, 2018). In contrast, the inflammatory BM of subjects with SCD may affect the quality of HSPCs from subjects with SCD compared to those from healthy, or non-SCD, BM.
The characterization of BM-derived HSPCs from patients with SCD is required to maximize the likelihood of success of gene therapy protocols for these patients, particularly as promising investigations are occurring regarding HSPC purification and gene transfer efficiency (Masiuk et al, 2017; Zonari et al, 2017), editing strategies (Breda et al, 2016; Dever et al, 2016; DeWitt et al, 2016; Hoban et al, 2016; Ye et al, 2016; Uchida et al, 2017a) and transplant conditioning (Chhabra et al, 2016) that relies on high quality HSPCs for success. We therefore sought to characterize the immunophenotype of CD34+ HSPCs from subjects with SCD (SCD BM) compared to that of healthy volunteers (non-SCD BM) in addition to characterizing the inflammatory microenvironment in the BM within the MN layer. We hypothesized that collection type (El Habbal et al, 1995; Müller-Steinhardt et al, 1998), storage (Müller-Steinhardt et al, 1998; Tong et al, 2016) and delays in processing (Harding et al, 2007) may further impact CD34+ recovery, and was therefore investigated as a strategy to maximize LT-HSPC recovery.
Methods
Study sample
Bone marrow was collected from subjects with SCD (HbSS genotype) or healthy non-SCD volunteers. Subjects with SCD who were being evaluated to undergo HSCT at the National Institutes of Health (NIH) consented to have additional research BM collected at the end of a scheduled BM evaluation under Protocol 03-H-0015. No subject with SCD underwent a separate BM procedure solely for this study. Healthy, African American persons over the age of 18 years who did not have SCD or sickle cell trait and were not on steroids or chronic non-steroidal anti-inflammatories, were consented under Protocol 08-H-0156-T to undergo a screening history and physical with blood work to confirm eligibility prior to undergoing a BM aspirate. All subjects had a full blood count (with reticulocyte count for subjects with SCD) and Hb electrophoresis done prior to BM evaluation. Information regarding HC use and recent transfusion history was collected for subjects with SCD, however it is noted that long-term transfusion history was not always complete in these subjects. Subjects with SCD were not in crisis at the time of BM aspirate.
Sample collection and timing
Bone marrow was collected in two syringes and immediately transferred to either a heparin or acid citrate dextrose-A (ACDA) collection tube, respectively. Approximately 10 ml of BM per anticoagulant was processed immediately (day 0) with duplicate samples stored at 2–8°C and processed the following day (day 1).
MN isolation and CD34+ cell purification
The MN layer was isolated via density gradient centrifugation using Ficoll-Paque Plus (GE Healthcare, Pittsburgh, PA, USA). In the first phase of this study (n = 9), the MN layer was isolated for inflammatory and contamination markers as described below. In the second phase (n = 10), CD34+ cell selection and analyses were done in addition to analysing inflammatory and contamination markers to assess if increased BM inflammation may alter CD34+ cell quality. CD34+ cell selection was performed using a magnetic microbead CD34+ selection kit (Miltenyi Biotec, Inc., Bergisch Gladbach, Germany) according to the company’s protocol. Briefly, the MN layer was incubated with anti-CD34 microbeads at 2–8°C for 30 min, then passed through a positive selection immunomagnetic column three times. Total MN cell count and CD34+ cell count was performed before and after CD34+ selection, respectively. CD34+ purity was assessed by flow cytometry using allophycocyanin (APC)-conjugated anti-human CD34 antibody (clone 581; BD Biosciences, San Jose, CA, USA) on a FACSCanto II (BD Biosciences) (Figure S1).
Cell staining
Cells were incubated for 30 min at 4°C in the dark with fluorescent-labelled antibodies against inflammatory markers or non-MN cells. All antibodies were obtained from Biolegend, San Diego, CA, USA unless otherwise specified. Inflammatory markers investigated the global microenvironment, some with overlap and possible co-expression on a single population, and included phycoerythrin (PE)/Cyanin 7 (PE-Cy7)-conjugated anti-CD11b (clone CBRM1/5), fluorescein isothiocyanate (FITC)-conjugated anti-CD62L (clone DREG-56), APC-conjugated anti-CD62P (clone AK4), PE-conjugated anti-CD35 (clone E11), and APC-Cy7-conjugated anti-CD36 (clone 5–271). Non-MN cells, including red cells, neutrophils and platelets, were identified using FITC-conjugated anti-glycophorin A (GPA) (clone H1264), APC-conjugated anti-CD66b (clone G10F5), and PE-conjugated anti-CD41/61 (clone A2A9/6), respectively. CD45 expression was measured with either Texas red (Tex-Red)-conjugated anti-CD45 (clone HI30; Thermo Fisher, Grand Island, NY, USA) or PE-conjugated anti-CD45 (clone HI30; BD Biosciences), whereas CD34+ cells were identified using APC-conjugated anti-CD34 (clone 581; BD Biosciences). CD34+-selected HSPCs were stained for CD34, CD45, and GPA expression.
Flow cytometry and colony-forming unit analysis
Data were analysed by conventional and imaging flow cytometry, the latter confirming post-CD34+ cell selection flow data and demonstrating antibody intensity as a characterization of HSPC heterogeneity and progenitor lineage. Analytic flow cytometry was performed on an LSR II Fortessa instrument (BD Bioscience, San Jose, CA, USA) while imaging flow cytometry was performed on an Amnis Imagestream Mark II (MilliporeSigma, Darmstadt, Germany). The viable MN cell population was gated by forward scatter. A narrow versus wide gate, comparing forward scatter area versus height, established a singlet versus doublet cell population. To further understand if what was considered a doublet on conventional flow cytometry was a true aggregate versus co-expression of lineage markers, imaging flow cytometry elucidated single cell co-expression (singlet) versus aggregation (doublet) between HSPCs (CD34+) with white blood cells (WBCs) (CD45+), RBCs (GPA+) or platelets (CD41+/61+). Finally, a colony-forming unit (CFU) assay was performed on a single SCD BM sample. CD34+ cells selected after magnetic bead separation were labelled with CD34 and GPA and sorted based upon GPA expression. Sorted CD34+GPA‒ and CD34+GPA+ cells were plated in methylcellulose media for colony-forming assays for 14 days after which eythroid CFU (CFU-E)/erythroid burst-forming unit (BFU-E), granulocyte-macrophage CFU (CFU-GM), and mixed lineage CFU (CFU-GEMM) colonies were measured.
Statistical analysis
Analyses compared results for SCD BM versus non-SCD BM on day 0, on day 1, and combined day 0 + day 1 data. Analysis was also done to compare processing conditions for SCD BM only for day of processing (day 0 versus day 1) by combining data (heparin + ACDA) for a given day of processing, and for anticoagulant collection (heparin versus ACDA) by combining data for each day of processing (day 0 + day 1) for a given anticoagulant.
Mean values with standard deviations are reported for haematological data and cell counts. In all remaining studies, the median values with ranges are listed. Pairwise comparison was performed by unpaired t test or ANOVA for multiple group comparison. Two group comparisons by Wilcoxon rank sum test was performed when the assumption of normality was not met. A P-value of <0·05 was considered statistically significant except for analyses with multiple comparisons within a sample where Bonferroni correction was applied and a P-value of <0·01 was considered statistically significant.
Results
Subjects with SCD have lower haemoglobin and higher WBC count at baseline compared to non-SCD subjects, and demonstrate a need for treatment optimization in this SCD cohort
Bone marrow was collected from 24 subjects; 19 subjects with SCD and five healthy volunteers (non-SCD) (Table I). Median age for the entire cohort was 29 years (range 20– 41). The median age of subjects with SCD was 29 (range 20–41) versus 22 in non-SCD subjects (range 21–32) (not significant, ns). Subjects with SCD had a significantly lower mean total haemoglobin (85 ± 12 vs. 137 ± 16 g/l, P < 0·01), higher WBC count (9·1 ± 2·8 vs. 6·1 ± 2·3 ×109/l, P < 0·05) and higher fetal Hb percentage (%HbF) (7·7 ± 5·7 vs. <1·0 ± 2·3%, P < 0·01) compared to non-SCD subjects. Mean percentage of sickle Hb (%HbS) in subjects with SCD was 61% (SD ± 27%), with nine subjects having received an RBC transfusion at some point in the previous 3 months.
Table I.
Patient demographics and mean peripheral blood (PB) haematological parameters in SCD and non-SCD subjects.
| SCD subgroups |
|||||
|---|---|---|---|---|---|
| Non-SCD | SCD (all) | Off hydroxycarbamide | On hydroxycarbamide | Chronic transfusions | |
| Subjects (n) | 5 | 19 | 4 | 12 | 3 |
| Gender (male, %) | 2 (40) | 12 (63) | 4 (100) | 6 (50) | 2 (67) |
| Age in years (median) | 22 | 31 | 32 | 29 | 29 |
| WBC count (×109/l) | 6.1 ± 2.3 | 9.1 ± 2.8* | 10.1 ± 2.6 | 9.2 ± 3.1 | 7.5 ± 1.0 |
| Hb (g/l) | 137 ± 16 | 85 ± 12** | 80 ± 90 | 85 ± 9.0 | 97 ± 21 |
| MCV (fl) | 85 ± 10 | 95 ± 11 | 85 ± 11* | 99 ± 9 | 96 ± 7 |
| Platelet count (×109/l) | 295 ± 54 | 353 ± 105 | 314 ± 24 | 382 ± 107 | 289 ± 148 |
| ARC (×109/l) | N/A | 334 ± 146 | 317 ± 78 | 338 ± 136 | 341 ± 283 |
| ANC (×109/l) | 3.616 ± 1.86 | 5.056 ± 2074 | 5.040 ± 2.592 | 5.18 ± 2.254 | 4.626 ± 0.85 |
| % HbS | N/A | 60.9 ± 27 | 80 ± 18.8 | 63.5 ± 23.2 | 27.0 ± 17.8* |
| % HbA | 96.7 ± 3.7 | 28.7 ± 30.1** | 9.5 ± 16.3 | 22.6 ± 22.8 | 59.5 ± 18.4** |
| % HbF | <10 ± 23 | 7.7 ± 5.7** | 4.6 ± 3.0 | 9.5 ± 6.5 | 5.8 ± 3.5 |
ANC, absolute neutrophil count; ARC, absolute reticulocyte count; BM, bone marrow; CT, chronic transfusions; Hb, haemoglobin; HbA, adult haemoglobin; HbF, fetal haemoglobin; HbS, sickle haemoglobin; MCV, mean corpuscular volume; SCD, sickle cell disease; WBC, white blood cell count.
P < 0·05,
P < 0·01.
Most subjects with SCD were on HC (n = 12, 63%) whereas only three subjects (16%) had recently been transfused over a period of several months and were considered to be on CT (Table I). While there was a relatively higher mean corpuscular volume (MCV) for subjects with SCD on HC compared to those on CT or off HC (P ≤ 0·05), there were no significant differences in mean total WBC, absolute neutrophil count (ANC) or %HbF between groups. As expected, there was a lower mean %HbS for subjects with SCD on CT compared to those on and off HC (P < 0·05), however there were no differences in Hb or absolute reticulocyte count (ARC) for patients on CT. The lack of significant difference in %HbF and ANC in the subjects on HC indicate suboptimal HC dosing or response, whereas the lack of significant difference in total Hb and ARC in subjects on CT indicated that the transfusions had not been successful in suppressing stress erythropoiesis in these patients. Given similarities in many baseline characteristics for subjects treated with of either HC or CT for the subjects with SCD evaluated here, all subsequent data and analyses for subjects with SCD were combined into one group to compare to non-SCD subjects.
SCD BM yields lower total CD34+ cell count compared to non-SCD BM, with CD34+ cell count notably higher in SCD BM when collected in ACDA versus heparin
While total MN cell count from SCD BM (n = 10) and non-SCD BM (n = 5) was similar (51 ± 71 vs. 62 ± 23 ×109/l, ns), total CD34+ cell count from SCD BM was significantly lower (0·21 ± 0·55 vs. 0·45 ± 0·12 ×109/l, P ≤ 0·01) (Fig 1, Table SI). When comparing SCD versus non-SCD BM for each processing condition there were no significant differences in total MN cell count; however, there was a consistently lower total CD34+ cell count in SCD BM regardless of anticoagulant or day of processing, which was significant in ACDA (P ≤ 0·05) and processing day 0 (P ≤ 0·05). Similarly, when comparing processing conditions for SCD BM alone, neither condition (heparin versus ACDA or day 0 versus day 1) showed a difference in total MN cell count; however, processing SCD BM in ACDA yielded a higher CD34+ cell count compared to heparin (0·25 ± 0·25 vs. 0·16 ± 0·71 × 109/l, P < 0·05) (Fig 2, Table SII).
Fig 1.
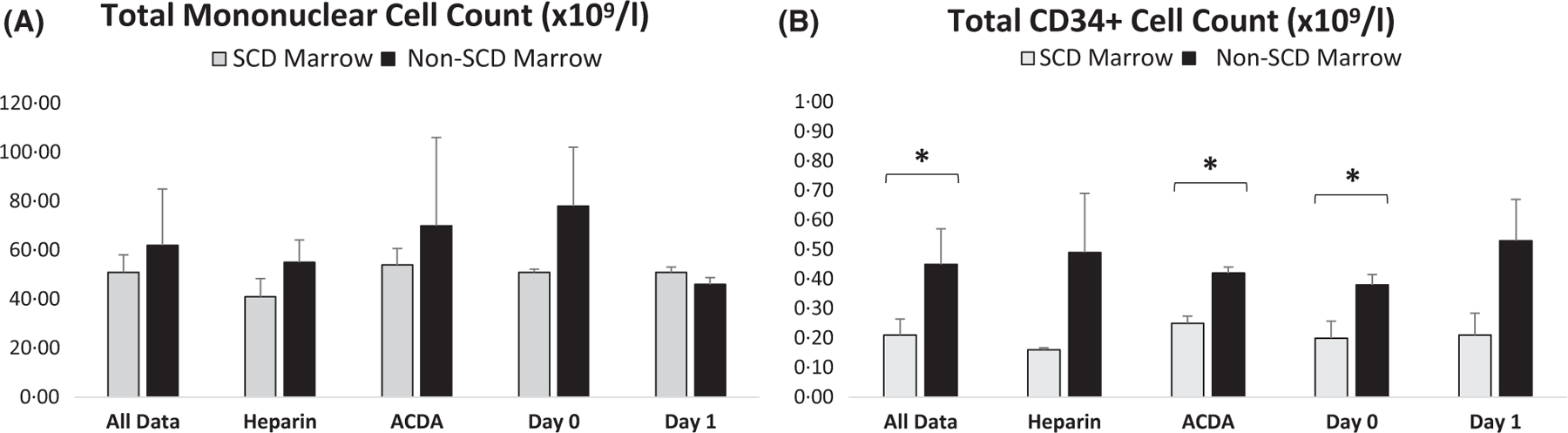
Total mononuclear and CD34+ cell count are lower in SCD BM compared to non-SCD BM. (A) Comparison of SCD BM (n = 10) versus non-SCD BM (n = 5) total mononuclear cell count combing all data, or comparing various methods of processing for all samples including collection in heparin, ACDA, or processing immediately or 1 day after collection. (B) For all data and each processing condition, CD34+ cell count is lower in SCD BM (n = 10) compared to non-SCD marrow (n = 5). *P < 0·05. ACDA, acid citrate dextrose-A; BM, bone marrow; SCD, sickle cell disease.
Fig 2.
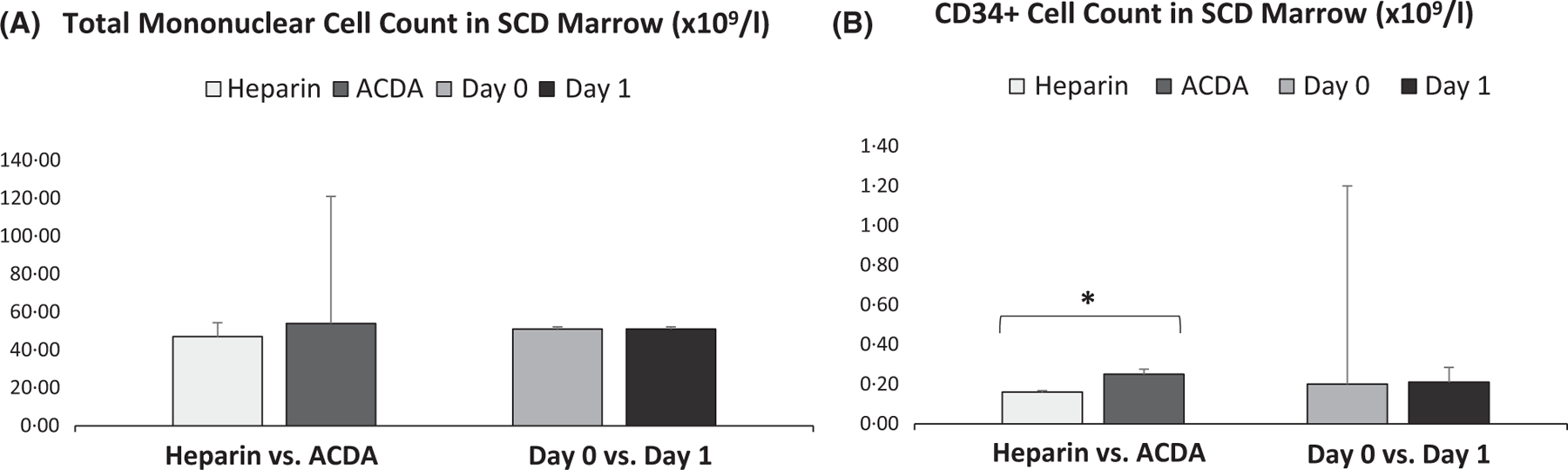
Total mononuclear cell count is similar between anticoagulants or day of processing in SCD BM while CD34+ cell count is higher when SCD BM is collected in ACDA. (A) Comparison of methods of collection or day of processing for SCD BM total mononuclear cell count (n = 10 for each condition). (B) Whereas day of processing did not influence total CD34+ cell count in SCD BM, CD34+ cell count was higher when SCD BM is collected in ACDA versus heparin (n = 10 for each condition). *P < 0·05. ACDA, acid citrate dextrose-A; BM, bone marrow; SCD, sickle cell disease.
CD34+-selected HSPCs from SCD BM demonstrate lower purity compared to those from non-SCD BM
Imaging flow cytometry performed on CD34+-selected HSPCs demonstrated greater than 50% of CD34+ cells from SCD BM were CD34dim [average 56·8% (range 43–69%)], regardless of anticoagulant used (Fig 3). There were significantly fewer CD34bright HSPCs in SCD BM compared to non-SCD BM [average 43·2% (range 33–55%) vs. 95·8% (range 94–98%), P < 0·01], in addition to overall lower CD45 expression [89·9% (range 30·0–98·8%) vs. 97·2% (range 75·6–99·9%), P ≤ 0·05] that remained lower in SCD BM regardless of anticoagulant or day of processing.
Fig 3.
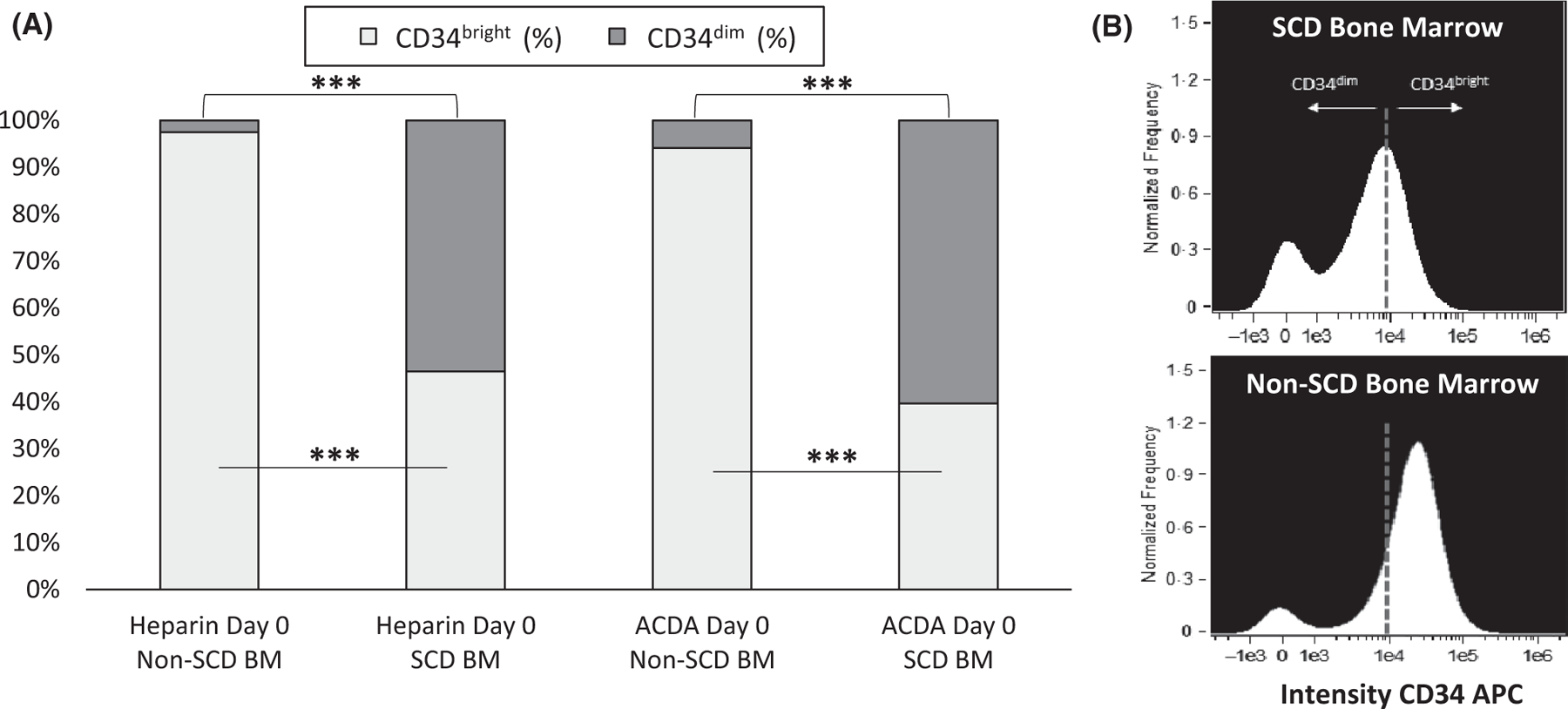
Imagestream data show lower purity in CD34+ cells in subjects with SCD. Image flow cytometry performed on CD34+-selected HSCs collected in two different anticoagulants (heparin and ACDA) and processed immediately after collection demonstrated a significantly lower proportion of CD34bright HSCs in SCD marrow (n = 10) compared to non-SCD marrow (n = 5). (A) Greater than 50% of SCD HSCs are characterized as CD34dim. (B) ImageStream histogram plot displays two populations of CD34+ antibody intensity corresponding to CD34bright and CD34dim populations. ***P < 0·001. ACDA, acid citrate dextrose-A; APC, allophycocyanin; BM, bone marrow; SCD, sickle cell disease.
Early erythroid lineage expression on CD34+ HSPCs from SCD BM contributes to lower purity
CD34+-selected HSPCs from SCD BM showed significantly higher GPA expression than HSPCs from non-SCD BM [32·8% (range 13·7–54·7%) vs. 11·9% (range 1·3–19·1%), P < 0·01) (Fig 4A–F). GPA expression trended higher in HSPCs derived from SCD BM if processing was delayed [30·5% (range 13·7–53·5%) vs. 33·5% (range 13·9–54·7%) on day 1, ns] and was similar between the two different anti-coagulants. Most GPA expression was as co-expression on single CD34+GPA+ cells (singlet), which was higher in SCD BM compared to non-SCD BM regardless of anticoagulant or day of processing (Fig 4C, E). The remaining GPA expression came from GPA+ RBCs attached to CD34+ HSPCs (doublets), which were higher in SCD BM compared to non-SCD BM if processing was delayed by 1 day in both heparin [7·6% [range 2·6–17·1%) vs. 1·3% (range 0·3–5·6%), P ≤ 0·01] and ACDA [7·6 (range 2·8–22·6%) vs. 1·8 (range 1·7–8·0%), P ≤ 0·05] (Fig 4E).
Fig 4.
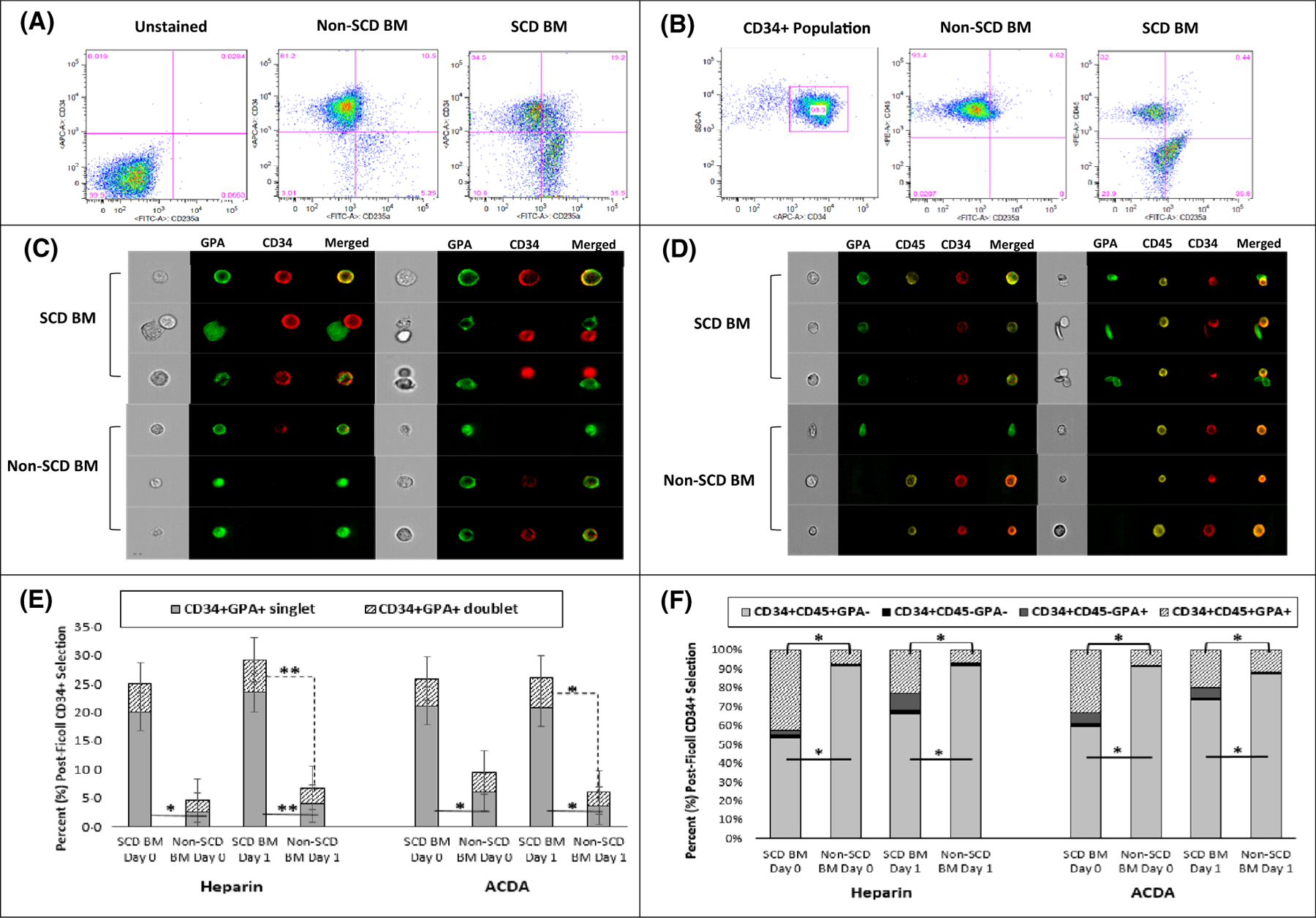
CD34+ selected HSCs from subjects with SCD are characterized by higher erythroid lineage expression. Conventional and image flow cytometry compared BM derived CD34+-selected HSCs stained with CD34, CD45, and GPA from subjects with SCD compared to healthy volunteers (non-SCD). (A) CD34+-selected HSCs in SCD are characterized by higher GPA expression and red cell contamination. (B) After gating on the CD34+ population, there is a larger and distinct CD34+CD45—GPA+ population in SCD BM compared to non-SCD BM. (C) Image and convention flow cytometry (E) demonstrate significantly more CD34+ and GPA+ co-expression on single HSCs (singlet) in SCD (n = 10) compared to non-SCD BM (n = 5), regardless of anticoagulant or day of processing, in addition to increased aggregates (doublets) of CD34+ HSCs aggregated with GPA+ red cells in SCD marrow that worsen with a delay in processing. (D) Image flow cytometry demonstrates a majority of GPA expression in the subject with SCD is from single cell CD34+CD45+GPA+ and CD34+CD45—GPA+ HSCs in addition to aggregates of CD34+ cells with GPA+ red cells, often sickled red cells. A majority of GPA expression in normal marrow is derived from red cell contamination in the post-Ficoll mononuclear layer and rarely as co-expression on a single cell. (F) Regardless of anticoagulant or processing day, there is a significantly lower percentage of CD34+CD45+GPA— HSCs in SCD marrow (n = 10) compared to non-SCD marrow (n = 5) due to increased percentage of SCD CD34+CD45+ and CD34+CD45— HSCs co-expressing early erythroid lineage. CD235a: GPA; *P < 0·05 **P < 0·01. BM, bone marrow; HSCs, haematopoietic stem cells; GPA, glycophorin A; SCD, sickle cell disease.
Imaging flow cytometry confirmed that most of the GPA expression in CD34+-selected HSPCs from subjects with SCD was from single cell CD34+CD45+GPA+ and CD34+CD45–GPA+ cells with occasional aggregates of CD34+ cells with GPA+ RBCs, often sickle RBCs (Fig 4D). Compared to non-SCD BM, SCD BM had significantly fewer CD34+45+GPA– HSPCs [61·5% (range 12·4–85·8%) vs. 89·2% (range 77·1–94·0%), P < 0·01], given higher CD34+CD45‒GPA+ [4·8% (range 0–53·3%) vs. 0·3% (range 0–3·6%), P < 0·01] and CD34+CD45+GPA+ [21·6% (range 13·3–51·0%) vs. 7·8% (range 1·0–16·1%), P < 0·01] cells (Fig 4F). There was a trend toward higher CD34+CD45—GPA+ HSPCs in SCD BM if processing was delayed [3·7% (range 0–5·3%) vs. 7·2% (range 0·2–53·3%) on day 1, ns].
Higher inflammation, contamination, and aggregation within the MN layer in SCD BM may contribute to lower HSPC purity
Compared to non-SCD BM, the MN cell layer in SCD BM contained a significantly higher expression of inflammatory markers, including activated neutrophils [CD11b: 11·3% (range 2·7–32·6%) vs. 3·2% (range 0·2–14·6%), P < 0·01], scavenger monocytes/sickle red cell adhesion receptor [CD36: 25·7% (range 11·4–49·9%) vs. 11·1% (range 2·6–29·3%), P < 0·01], activated complement regulator [CD35: 18·0% (range 1·0–53·5%) vs. 1·1% (range 0·1–26·1%), P < 0·01], and P-selectin [CD62P: 17·5% (range 6·5–39·0%) vs. 7·0% (range 0·8–22·2%), P < 0·01] (Fig 5, Table SIII). The post-Ficoll selected MN layer in SCD BM was contaminated with a significantly higher percentage of platelets [CD41: 14·7% (range 1·5–49·1%) vs. 2·1% (range 0·3–9·1%), P < 0·01], RBCs [GPA: 20·0% (range 5·2–74·3) vs. 7·4% (range 1·1–48·5%), P < 0·01], and aggregates of platelets/granulocytes [CD41+CD66b+: 1·6% (range 0·1–24·5%) vs. 0·4% (range 0–4·0%), P < 0·01] and leucocytes/RBCs (CD45+GPA+: 7·6% (range 1·0–37·4%) vs. 3·9% (range 0–27·0%), P < 0·01). Significant differences in inflammatory and contamination markers between SCD BM and non-SCD BM remained regardless of day of processing as there remained a significantly higher percentage of CD11b, CD36, CD35, P-selectin, CD41, CD41+CD66b+, and CD45+GPA+ expression on day 0 and day 1 in SCD BM (Table SIII).
Fig 5.

Makers of inflammation and contamination are higher in SCD BM compared to non-SCD BM. Aliquots (20 ml) of BM from subjects with SCD (n = 19) and normal volunteers (non-SCD, n = 5) was collected in either heparin or acid citrate dextrose-A and processed immediately (day 0) or stored at 4°C and processed the following day (day 1). The mononuclear layer was isolated via Ficoll density gradient centrifugation, cells were stained with antibodies against inflammatory markers (CD36, CD35, CD11b, CD62L, CD62P) or non-mononuclear cells (GPA, CD66b, CD41/61), and analysed by conventional flow cytometry. Analysis compares SCD marrow to non-SCD marrow on day 0, on day 1, and combined day 0 + day 1 data. A P-value of <0·01 was considered statistically significant. **P < 0·01, ***P < 0·001. BM, bone marrow; GPA, glycophorin A; SCD, sickle cell disease.
Delayed processing contributes to higher inflammation and contamination within the MN layer in SCD BM, with higher RBC contamination noted when BM is collected in heparin compared to ACDA
In SCD BM alone, there were few major differences in expression of inflammatory or contamination markers in SCD BM when comparing anticoagulants (heparin versus ACDA) or day of processing (day 0 versus day 1) (Fig 6A, B). There was a trend toward higher GPA expression in heparin versus ACDA [25·7% (range 5·3–68·3%) vs. 14·8% (range 5·2–74·3%), ns], which worsened with a delay in processing [18·2% (range 5·2–62·0%) vs. 24·4% (range 6·6–74·3%) on day 1, ns], and was noticeable in the post-Ficoll MN layer (Fig 7). In addition to higher GPA expression with delayed processing, there was a trend for higher expression with delayed processing for CD41 [14·7% (range 1·5–49·1%) vs. 15·5% (range 3·1–41·6%) on day 1, ns], CD11b [10·2% (range 2·7–32·6%) vs. 12·2% (range 2·9–29·0%) on day 1, ns], CD36 [21·4% (range 11·4–49·9%) vs. 28·0% (range 12·0–46·2%) on day 1, ns], CD62P [14·6% (range 6·5–39·0%) vs. 19·0% (range 7·6–38·6%) on day 1, ns], and CD45+GPA+ aggregates [7·4% (range 2·9–37·4%) vs. 7·8% (range 1·0–27·4%) on day 1, ns). Comparisons for inflammatory and contamination markers in SCD BM in heparin day 0 versus day 1 and ACDA day 0 versus day 1 are shown in Fig 6C and are listed in Table SIV.
Fig 6.
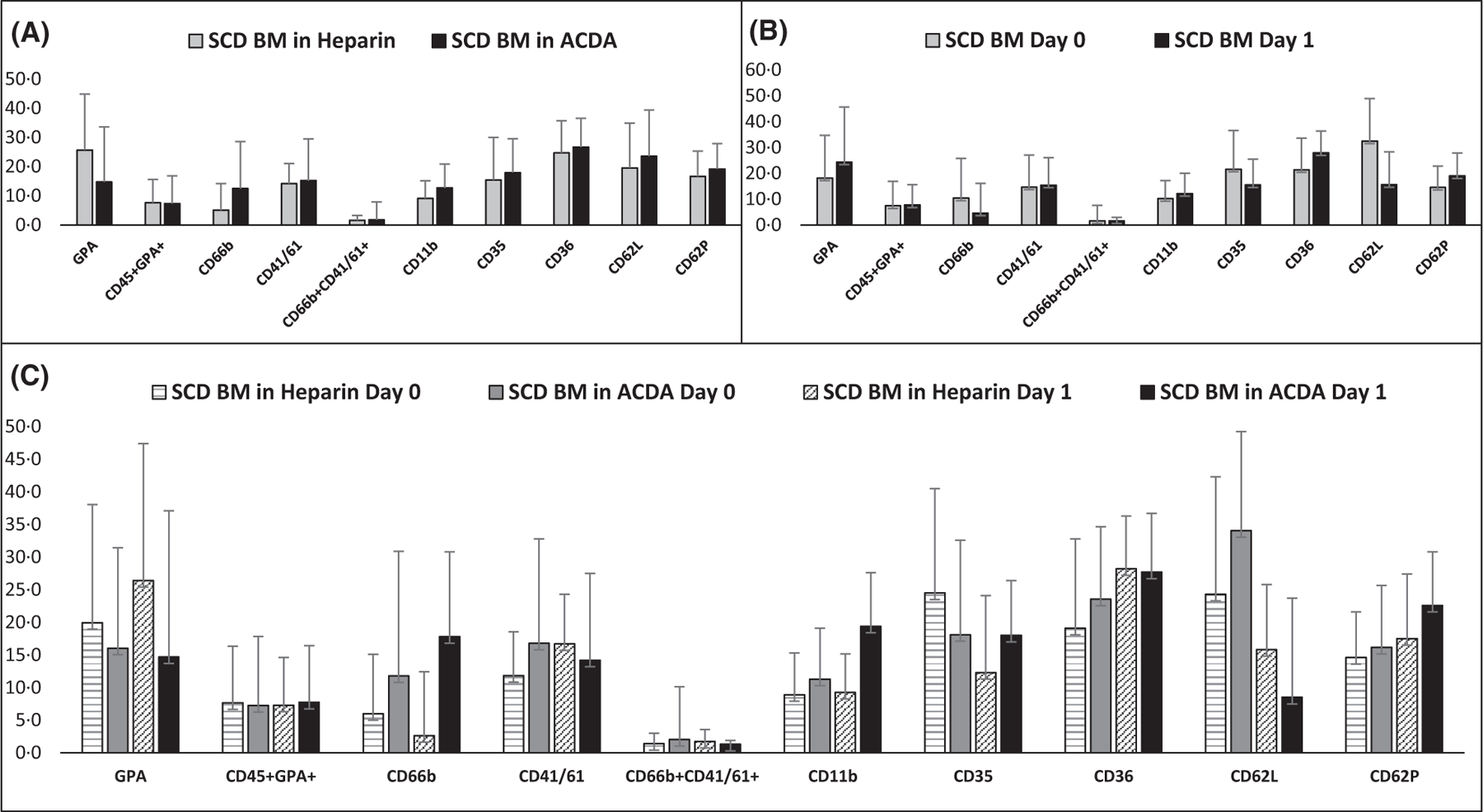
Makers of inflammation and contamination in SCD BM are elevated regardless of anticoagulant or day of processing. (A) Comparison of SCD BM drawn in heparin versus ACDA using combined data from day 0 + day 1 (n = 19 in each condition). (B) Comparison of SCD marrow processed immediately or on day one using combined data from heparin + ACDA (n = 19 in each condition). (C) Flow cytometry results for each anticoagulant and processing day (n = 19 in each condition). A P-value of <0·01 was considered statistically significant. ACDA, acid citrate dextrose-A; BM, bone marrow; SCD, sickle cell disease.
Fig 7.
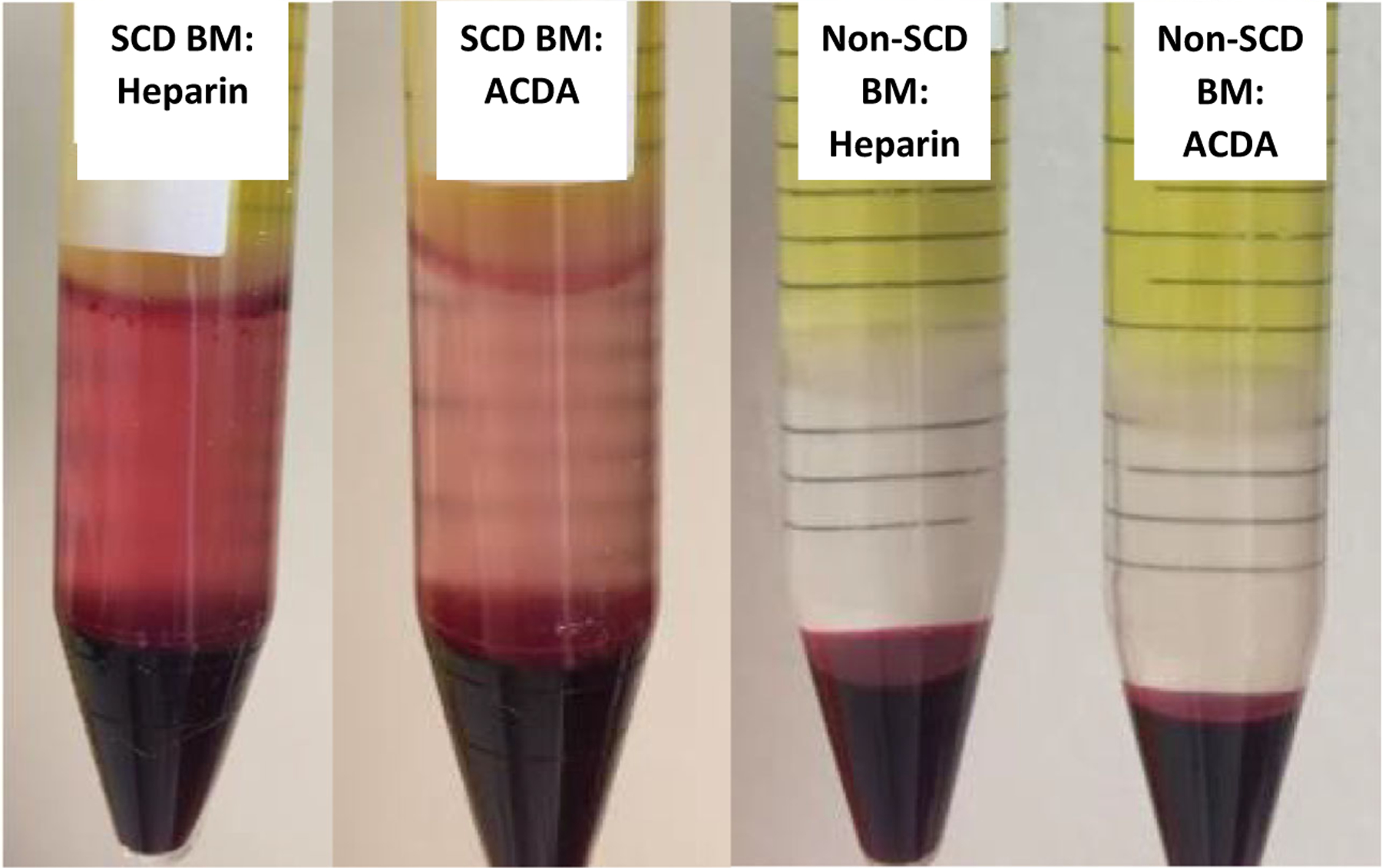
Red cell contamination in the mononuclear layer of SCD BM. Glycophorin A expression was higher in SCD BM (n = 19) compared to non-SCD BM (n = 5), with red cell contamination noticeably higher in the post-Ficoll mononuclear layer in SCD BM, and in heparin > ACDA in SCD BM. ACDA, acid citrate dextrose-A; BM, bone marrow; SCD, sickle cell disease.
Discussion
Compared to non-SCD BM, SCD BM is characterized by (i) a lower total CD34+ cell count, (ii) CD34 signal intensity that is a majority CD34dim, (iii) fewer CD34+45+GPA‒ HSPCs given higher erythroid (GPA) marking as evidenced by co-expression on CD34+CD45+GPA+ and CD34+CD45‒GPA+ cells, (iv) increased aggregates of CD34+CD45+ HSPCs attached to erythrocytes, (v) significantly higher inflammatory cells within the MN layer and (vi) greater contamination with platelets, RBCs, aggregated platelets/granulocytes and leucocytes/RBCs within the MN layer that worsens with a delay in processing. In SCD BM, ACDA is a superior anticoagulant for BM collection, given that collection in heparin had a lower total CD34+ cell count and higher GPA expression that worsened with delays in processing.
The MN layer from SCD BM contained higher expression of activated neutrophils (CD11b), monocyte scavenger/sickle RBC adhesion receptor (CD36), activated complement regulator (CD35), and P-selectin (CD62P) compared to non-SCD BM. RBC sickling (Hebbel, 1991), vascular endothelial cell activation with inflammatory cytokine secretion (Hebbel, 1997, 2000; Sultana et al, 1998; Wun et al, 2002; Kaul et al, 2009; Zhang et al, 2016) and continual oxidative stress (Chirico & Pialoux, 2012) contribute to SCD pathogenesis and an abnormal baseline steady state. Even at steady or non-crisis state, subjects with SCD have significantly elevated plasma levels of various endothelial factors, adhesion molecules and pro-inflammatory cytokines compared to healthy controls, including elevated endothelin 1, soluble vascular cell adhesion molecule 1, soluble P-selectin, tumour necrosis factor-a, interleukin (IL) 6, IL1β, granulocyte-macrophage colony-stimulating factor, IL3 and prostaglandin E2 (Francis & Haywood, 1992; Croizat, 1994; Graido-Gonzalez et al, 1998; Lanaro et al, 2009; Hatzipantelis et al, 2013). Despite data supporting that steady state BM is a useful HSPC source for SCD gene therapy, with similar transduction efficiency to mobilized PB CD34+ cells in the Rhesus competitive repopulation model (Uchida et al, 2017b), steady state BM in subjects with SCD is characterized in this study by increased inflammation, activation and adhesion within the MN layer regardless of anticoagulant or day of processing compared to non-SCD subjects, and probably contributes to sub-optimal HSPC quality and yield observed in this study.
The MN layer from SCD BM was more likely to contain trapped RBCs, platelets, granulocyte/platelet and leucocyte/RBC aggregates and activated granulocytes, suggesting increased inflammation, activation and adhesion contribute to poor MN purity in SCD BM. Many studies characterize neutrophils from subjects with SCD as exhibiting an activated phenotype characterized by lower expression of L-selectin and higher CD11b membrane expression (Lard et al, 1999; Lum et al, 2004), consistent with elevated CD11b expression that worsened with time in this study. Lower L-selectin expression for subjects with SCD on day 1 compared to day 0 suggests continued neutrophil activation ex vivo that worsens with delays in processing. Compared to the MN layer from fresh blood, increased granulocyte contamination of the MN layer has been reported within 6–8 h after venepuncture and room temperature storage (2-fold increase), increased 11-fold by 24–26 h, and worsened 84-fold if stored in refrigerated conditions for 22–26 h (McKenna et al, 2009). Although HC has been found to inhibit neutrophil recruitment and suppress neutrophil activation (Saleh et al, 1999; Benkerrou et al, 2002), subjects in this study appeared not to be well optimized on HC, preventing the study of this effect on neutrophils in the MN layer or the ability to dampen the effects seen with delayed processing time. Furthermore, increased platelet activation is well known in subjects with SCD under steady state (Wun et al, 1997; Inwald et al, 2000; Villagra et al, 2007) leading to significantly more aggregates of platelets bound to erythrocytes, monocytes and neutrophils compared to controls (Wun et al, 1997; Zarbock et al, 2007; Frelinger et al, 2014). These platelet/monocyte aggregates activate monocytes, are significantly higher in subjects with SCD compared to healthy controls (Polanowska-Grabowska et al, 2010) and occur in a P-selectin dependent manner (Belcher et al, 2000). In this study, there was higher monocyte activation (CD11b, CD36), platelet/neutrophil aggregates and higher P-selectin levels that worsened with time in subjects with SCD compared to non-SCD subjects. Similarly, aggregates of monocytes with RBCs were demonstrated to be higher in the MN layer of BM from subjects with SCD, consistent with data suggesting that aggregates of monocytes with mature erythrocytes and reticulocytes are 10-fold higher in subjects with SCD compared to control subjects (Chaar et al, 2010).
Studies investigating the effects of anticoagulant, cell separation, storage and transportation have shown better preservation of cell viability and/or function in blood samples that are manipulated less (i.e. processed immediately on site), collected in specific anticoagulants, or stored in specific conditions when processing is delayed (El Habbal et al, 1995; Müller-Steinhardt et al, 1998; Harding et al, 2007; McKenna et al, 2009; Safaya et al, 2012; Kim et al, 2013; Tong et al, 2016). In this study, BM from subjects with SCD demonstrated a trend toward higher GPA expression within the MN layer when BM was collected in heparin versus ACDA, which worsened when processing was delayed. Further, CD34+-selected HSPCs had significantly higher adherent RBCs (doublets) compared to non-SCD BM, which worsened more significantly in heparin than ACDA by day 1. Consistent with our results, other studies have demonstrated increased RBC contamination within the PB MN layer in heparin compared to ACDA (Tong et al, 2016), and that storage of whole blood beyond 18 h increases erythrocyte and granulocyte contamination (Kim et al, 2013), specifically if stored at 4°C, by decreasing the granulocyte density (McKenna et al, 2009; Safaya et al, 2012). Platelet/granulocyte aggregation trended higher on day 1 in both SCD and non-SCD BM, with elevated CD11b in SCD BM probably contributing to higher aggregates overall in this study. Upregulation of the neutrophil adhesion molecule CD11b leads to increased platelet-monocyte aggregation in samples collected in heparin (El Habbal et al, 1995) and, for every 10 min of delay prior to processing, platelet-monocyte aggregates increase significantly (Harding et al, 2007). Although heparin is commonly used to prevent clotting, enhance Ficoll MN separation, and has potent anti-inflammatory activity (Downing et al, 1998; Mousavi et al, 2015), ACDA is possibly a preferred anticoagulant because dextrose increases cell survival and maintains cell functionality, giving superior preservative qualities compared to EDTA or heparin (Kim et al, 2013). This suggestion of superior cell survival in ACDA is consistent with an overall higher CD34+ cell count in this study when SCD BM was collected in ACDA versus heparin. Overall, delays in processing affect PB MC yield, with storage at 4°C giving a 35–40% decrease in yield by 24 h and a 60–66% decrease by 48 h (Tong et al, 2016), further suggesting earlier cell processing is important.
Activated monocytes, leucocytes, platelets, plasma proteins and endothelium are contributors to end organ damage in SCD (Wun et al, 1997; Saleh et al, 1999; Villagra et al, 2007; Polanowska-Grabowska et al, 2010; Mousavi et al, 2015; Sankaran & Weiss, 2015; Zhang et al, 2016), and create a microenvironment in major organ systems, such as the BM, that alters homeostatic balance. Whereas the total number of MN cells collected from the BM of subjects with and without SCD were comparable, the total number of CD34+ cells after selection was lower in subjects with SCD compared to non-SCD subjects. Of those CD34+ cells, more than 50% from SCD BM were CD34dim, suggesting multilineage differentiation into lineage-restricted progenitors that have lost their long-term repopulating capacity. This was confirmed by increased GPA expression on CD34+-selected HSPCs and in the CFU assay (Figure S2), consistent with previous results demonstrating a unique population of GPA+CD34+ cells in SCD that represent an accelerated erythroid differentiation pathway that has not down-regulated CD34 antigen expression, predominantly generates F cells and has lower clonogenicity and erythroid expansion potential (Luck et al, 2004). Multiple cytokines, including stem cell factor (SCF), thrombopoietin, Flt-3 ligand, IL11, IL3, IL6 and GM-CSF, or combinations of these, have been studied in in vitro HSPC expansion protocols of mouse and human cells to support the HSPC survival, proliferation and maintenance during culture (Bhatia et al, 1997; Miller & Eaves, 1997; Audet et al, 2002). Of these, SCF, GM-CSF, IL3 and IL6 are known to be elevated in steady state in selected subjects with SCD (Croizat & Nagel, 1999; Hebbel, 2000; Pathare et al, 2004) and probably drive HSPC differentiation toward the erythroid lineage in the setting of chronic haemolytic anaemia and erythroid demand. As has been described, stress erythropoiesis in SCD accelerates erythropoiesis and thus CD34+/CD38— HSPCs expressing GPA may represent the “stress progenitor” population (Luck et al, 2004), further supporting an altered HSPC population in subjects with SCD, as seen in this study as increased lineage-restricted (GPA+) progenitors with a majority CD34dim population.
In contrast to the successes observed in clinical studies of LentiGlobin LVV-mediated gene therapy in transfusion-dependent β-thalassaemia (HGB-204, NCT01745120; HGB-205, NCT02151526) (Cavazzana et al, 2017; Kwiatkowski et al, 2017; Thompson et al, 2018), initial high-level results for the multi-centre study of LentiGlobin trial in severe SCD (HGB-206 Group A, NCT02140554) showed suboptimal production of vector-derived haemoglobin (HbAT87Q) (Kanter et al, 2016). In these subjects, BM was used as a source of HSPCs while mobilized HSPCs were used in thalassaemia patients. Despite nearly identical transplantation strategies utilizing autologous HSPCs transduced with the LentiGlobin BB305 vector, lower median cell dose [HGB-204: 8·1 × 106 CD34+ per kg; HGB-205 (Thal): 10·5 × 106 CD34+ per kg; HGB-206: 2·1 × 106 CD34+ per kg], decreased median drug product (DP) VCN (HGB-204: 0·7; HGB-205: 1·3; HGB-206: 0·6), decreased PB VCN at follow-up range 15–42 months [HGB-204: 0·1–1·0; HGB-205 (Thal): 0·3–3·5] and 3·7–12·7 months (HGB-206: 0·05–0·13), and lower HbAT87Q levels [HGB-204 (non-β0/β0): 11–96 g/l; HGB-205 (Thal non-β0/β0): 66–100 g/l; HGB-206: 4·0–24 g/l] were reported for patients with SCD, suggesting that the unique inflammatory BM environment and stress erythropoiesis associated with SCD may have significant impacts on HSPC quality, yield and modification efficacy. Here we demonstrate a lower CD34+ cell count, with >50% of SCD HSPCs characterized as CD34dim. CD34dim cells have less colony-forming potential than CD34bright HSPCs (Rundberg Nilsson et al, 2016), and in vivo transplant studies in NOD/SCID/IL2Rγnull (NSG) mice demonstrate that sorted CD34dim cells do not engraft into NSG mice (Wang et al, 2003). Preliminary data in xenograft models suggests at least 15–20% of all engrafted HSPCs must express the therapeutic globin gene to overcome the pathological phenotype, though higher requirements may be necessary (Persons et al, 2001). The HGB-206 study of LentiGlobin in SCD is now evaluating the hypothesis that a period of regular RBC transfusions prior to cell collection, combined with refinements to the gene therapy manufacturing process, may increase transduction of HSPCs and improve engraftment of transduced HSPCs. The study protocol has also been modified to incorporate a new, plerixafor-only mobilization process that has recently shown acceptable safety in patients with SCD (Cavazzana et al, 2017; Tisdale et al, 2017; Boulad et al, 2018; Lagresle-Peyrou et al, 2018), which will allow evaluation of mobilization and apheresis compared to direct BM harvest as a source of HSPCs for SCD gene therapy. Very early, high-level results appear promising (Kanter et al, 2017).
One of the major limitations of this study is our inability to compare HSPCs from subjects with SCD off HC versus on HC versus on CT. Despite a rise in MCV, the equivalent WBC and ANC of subjects on HC in this study suggest these subjects are not optimally dosed, not compliant or not responsive to therapy. Although not optimized, most subjects in this study were on HC, which may have contributed to the overall lower CD34+ cell counts, given previous reports of lower CD34+ cell counts in both the PB and BM in subjects with SCD treated with HC (Uchida et al, 2017b). As for subjects on CT, equivalent ARC for subjects who came to the NIH with a recent transfusion history suggested a history of only short-term transfusion(s) and therefore ineffective suppression of stress erythropoiesis normally associated with CT. Given the ability to suppress ineffective erythropoiesis with CT and data suggesting efficient plerixafor mobilization of HSPC in subjects with SCD after exchange transfusion (Lagresle-Peyrou et al, 2018), characterizing HSPCs from subjects maintained on CT is critical. Comparison of HSPCs from subjects with SCD on and off CT, and defining the time period a subject must be on CT to effectively improve HSPC quality and yield, should be investigated given the implications as a potential requirement prior to autologous gene therapy treatment. Further limitations include a lack of comprehensive characterization of CD34+ HSPCs, including CD38, CD45RA or CD90 expression as further markers of primitive HSPCs, a comparison with plerixafor-mobilized HSPCs from subjects with SCD, given preliminary data suggesting superior cell yield and a lower CD34dim population, and a lack of direct evidence that the CD34dim population from SCD subjects will not engraft long-term.
Conclusions
Challenges in autologous ex vivo HSPC gene therapy for SCD include overcoming a hypoxic and inflamed BM, stress erythropoiesis and a low percentage of LT-HSPCs. Options that may maximize success for subjects with SCD include collection in ACDA with early cell processing to maximize HSPC recovery, targeting a highly purified selection of HSPCs, such as CD34bright HSPCs to maximize transduction and engraftment strategies, and probably includes a period of CT prior to HSPC collection to reduce haematopoietic demand and, potentially, HSPC quality. Even with these interventions, however, BM as a source of HSPCs from subjects with SCD may ultimately prove to be sub-optimal compared to plerixafor-mobilized HSPCs, given early published results. If the early promise of plerixafor-mobilized product is maintained, there will no longer be a need or justification to use HSPCs collected from BM from subjects with SCD, given the suboptimal quality and yield, in addition to an overall increased risk to patients of anaesthesia and pain associated with BM harvests compared to the ease of peripheral mobilization.
For BM or plerixafor-mobilized HSPCs to be an adequate, efficient, and sustainable source of HSPCs for autologous gene therapy in SCD, there must be safe and efficient gene transfer into a sufficient number of LT-HSPCs capable of lifelong engraftment, with sustained production of the gene of interest at levels capable of overcoming the pathogenic HbSS phenotype. Here we demonstrate a significant difference in steady state BM for subjects with SCD compared to non-SCD subjects, with increased inflammation, aggregation and contamination contributing to significant differences in HSPC quality and yield, as evidenced by a majority CD34dim population and lineage-restricted progenitors that are may be incapable of long-term engraftment that worsen with time. As advances are being made to improve gene addition efficiency, editing strategies and transplant conditioning, suppression of inflammation and stress erythropoiesis, combined with optimized cell collection procedures and early cell processing may be critical for maximal HSPC recovery necessary for successful gene therapy in SCD.
Supplementary Material
Fig S1. Sorting efficiency CD34 positive versus negative fraction.
Fig S2. Colony forming assay comparing CD34+GPA+ versus CD34+GPA‒ fractions.
Table SI. Total MN and CD34+ cell count are lower in SCD bone marrow compared to non-SCD bone marrow
Table SII. Total MN cell count is similar between anticoagulants or day of processing in SCD bone marrow while CD34+ cell count is higher when SCD bone marrow is collected in ACDA.
Table SIII. Makers of inflammation and contamination are higher in SCD bone marrow compared to non-SCD bone marrow.
Table SIV. Makers of inflammation and contamination in SCD bone marrow are elevated regardless of anticoagulant or day of processing.
Acknowledgements
The authors acknowledge Anna Conrey, NP, Wynona Coles, Mary Link, RN, and Matthew Hsieh, MD for their help recruiting subjects and obtaining samples used in this study, and Neal Jeffries, PhD for statistical guidance.
Footnotes
Disclosure
ML is a former and FJP is a current employee and shareholder of Bluebird Bio, Inc.
Supporting Information
Additional supporting information may be found online in the Supporting Information section at the end of the article.
References
- Abboud M, Laver J & Blau CA (1998) Granulocytosis causing sickle-cell crisis. Lancet, 351, 959. [DOI] [PubMed] [Google Scholar]
- Adler BK, Salzman DE, Carabasi MH, Vaughan WP, Reddy VV & Prchal JT (2001) Fatal sickle cell crisis after granulocyte colony-stimulating factor administration. Blood, 97, 3313–3314. [DOI] [PubMed] [Google Scholar]
- Aguilar C, Vichinsky E & Neumayr L (2005) Bone and joint disease in sickle cell disease. Hematology/oncology Clinics of North America, 19, 929–941. [DOI] [PubMed] [Google Scholar]
- Audet J, Miller CL, Eaves CJ & Piret JM (2002) Common and distinct features of cytokine effects on hematopoietic stem and progenitor cells revealed by dose-response surface analysis. Biotechnology and Bioengineering, 80, 393–404. [DOI] [PubMed] [Google Scholar]
- Belcher JD, Marker PH, Weber JP, Hebbel RP & Vercellotti GM (2000) Activated monocytes in sickle cell disease: potential role in the activation of vascular endothelium and vaso-occlusion. Blood, 96, 2451–2459. [PubMed] [Google Scholar]
- Benkerrou M, Delarche C, Brahimi L, Fay M, Vilmer E, Elion J, Gougerot-Pocidalo MA & Elbim C (2002) Hydroxyurea corrects the dys-regulated L-selectin expression and increased H (2)O(2) production of polymorphonuclear neutrophils from patients with sickle cell anemia. Blood, 99, 2297–2303. [DOI] [PubMed] [Google Scholar]
- Bhatia M, Bonnet D, Kapp U, Wang JC, Murdoch B & Dick JE (1997) Quantitative analysis reveals expansion of human hematopoietic repopulating cells after short-term ex vivo culture. Journal of Experimental Medicine, 186, 619–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bookchin RM & Lew VL (1996) Pathophysiology of sickle cell anaemia. Hematology/oncology Clinics of North America, 10, 1241–1253. [DOI] [PubMed] [Google Scholar]
- Boulad F, Shore T, van Besien K, Minniti C, Barbu-Stevanovic M, Fedus SW, Perna F, Greenberg J, Guarneri D, Nandi V, Mauguen A, Yazdanbakhsh K, Sadelain M & Shi PA (2018) Safety and efficacy of plerixafor dose escalation for the mobilization of CD34+ hematopoietic progenitor cells in patients with sickle cell disease: interim results. Haematologica, 103, 770–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breda L, Motta I, Lourenco S, Gemmo C, Deng W, Rupon JW, Abdulmalik OY, Manwani D, Blobel GA & Rivella S (2016) Forced chromatin looping raises fetal hemoglobin in adult sickle cells to higher levels than pharmacologic inducers. Blood, 128, 1139–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case SS, Price MA, Jordan CT, Yu XJ, Wang L, Bauer G, Haas DL, Xu D, Stripecke R, Naldini L, Kohn DB & Crooks GM (1999) Stable transduction of quiescent CD34+CD38‒ human hematopoietic cells by HIV-1-based lentiviral vectors. Proceedings of the National Academy of Sciences of the United States of America, 96, 2988–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavazzana M, Hacein-Bey-Abina S, Payen E, Magrin E, Magnani A, Semeraro M, Caccavelli L, Touzot F, Lefrere F, Suarez F, Hermine O, Brousse V, Poirot C, Moshous D, Bourget P, El Nemer W, Bartolucci P, Weber L, Puy H, Meritet JF, Grévent D, Beuzard Y, Chretien S, Lefebvre T, Asmal M, Miller AL, De Montalembert M, Blanche S, Leboulch P & Ribeil JA (2017) Longer term follow-up on the first patients with severe hemoglobinopathies treated with lentiglobin gene therapy. Blood, 130, 4609. [Google Scholar]
- Chaar V, Picot J, Renaud O, Bartolucci P, Nzouakou R, Bachir D, Galactéros F, Colin Y, Le Van Kim C & El Nemer W (2010) Aggregation of mononuclear and red blood cells through an {alpha}4{beta}1-Lu/basal cell adhesion molecule interaction in sickle cell disease. Haematologica, 95, 1841–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhabra A, Ring AM, Weiskopf K, Schnorr PJ, Gordon S, Le AC, Kwon HS, Ring NG, Volkmer J, Ho PY, Tseng S, Weissman IL & Shizuru JA (2016) Hematopoietic stem cell transplantation in immunocompetent hosts without radiation or chemotherapy. Science Translational Medicine, 8, 351ra105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirico EN & Pialoux V (2012) Role of oxidative stress in the pathogenesis of sickle cell disease. IUBMB Life, 64, 72–80. [DOI] [PubMed] [Google Scholar]
- Croizat H (1994) Circulating cytokines in sickle cell patients during steady state. British Journal of Haematology, 87, 592–597. [DOI] [PubMed] [Google Scholar]
- Croizat H & Nagel RL (1999) Circulating cytokines response and the level of erythropoiesis in sickle cell anemia. American Journal of Hematology, 60, 105–115. [DOI] [PubMed] [Google Scholar]
- Dever DP, Bak RO, Reinisch A, Camarena J, Washington G, Nicolas CE, Pavel-Dinu M, Saxena N, Wilkens AB, Mantri S, Uchida N, Hendel A, Narla A, Majeti R, Weinberg KI & Porteus MH (2016) CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature, 539, 384–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWitt MA, Magis W, Bray NL, Wang T, Berman JR, Urbinati F, Heo SJ, Mitros T, Muñoz DP, Boffelli D, Kohn DB, Walters MC, Carroll D, Martin DI & Corn JE (2016) Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Science Translational Medicine, 8, 360ra134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downing LJ, Strieter RM, Kadell AM, Wilke CA, Greenfield LJ & Wakefield TW (1998) Low-dose low-molecular-weight heparin is anti-inflammatory during venous thrombosis. Journal of Vascular Surgery, 28, 848–854. [DOI] [PubMed] [Google Scholar]
- Eaves CJ (2015) Hematopoietic stem cells: concepts, definitions, and the new reality. Blood, 125, 2605–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Habbal MH, Smith L, Elliott MJ & Strobel S (1995) Effect of heparin anticoagulation on neutrophil adhesion molecules and release of IL8: C3 is not essential. Cardiovascular Research, 30, 676–681. [DOI] [PubMed] [Google Scholar]
- Fitzhugh CD, Hsieh MM, Bolan CD, Saenz C & Tisdale JF (2009) Granulocyte colony-stimulating factor (G-CSF) administration in individuals with sickle cell disease: time for a moratorium? Cytotherapy, 11, 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis RB Jr & Haywood LJ (1992) Elevated immunoreactive tumor necrosis factor and interleukin-1 in sickle cell disease. Journal of the National Medical Association, 84, 611–615. [PMC free article] [PubMed] [Google Scholar]
- Frelinger AL III, Jakubowski JA, Brooks JK, Carmichael SL, Berny-Lang MA, Barnard MR, Heeney MM & Michelson AD (2014) Platelet activation and inhibition in sickle cell disease (pains) study. Platelets, 25, 27–35. [DOI] [PubMed] [Google Scholar]
- Graido-Gonzalez E, Doherty JC, Bergreen EW, Organ G, Telfer M & McMillen MA (1998) Plasma endothelin-1, cytokine, and prostaglandin E2 levels in sickle cell disease and acute vaso-occlusive sickle crisis. Blood, 92, 2551–2555. [PubMed] [Google Scholar]
- Harding SA, Din JN, Sarma J, Jessop A, Weatherall M, Fox KA & Newby DE (2007) Flow cytometric analysis of circulating platelet-monocyte aggregates in whole blood: methodological considerations. Thrombosis and Haemostasis, 98, 451–456. [PubMed] [Google Scholar]
- Hatzipantelis ES, Pana ZD, Gombakis N, Taparkou A, Tzimouli V, Kleta D, Zafeiriou DJ, Garipidou V, Kanakoudi F & Athanassiou M (2013) Endothelial activation and inflammation biomarkers in children and adolescents with sickle cell disease. International Journal of Hematology, 98, 158–163. [DOI] [PubMed] [Google Scholar]
- Hebbel RP (1991) Beyond hemoglobin polymerization: the red blood cell membrane and sickle disease pathophysiology. Blood, 77, 214–237. [PubMed] [Google Scholar]
- Hebbel RP (1997) Adhesive interactions of sickle erythrocytes with endothelium. Journal of Clinical Investigation, 100, S83–S86. [PubMed] [Google Scholar]
- Hebbel RP (2000) Blockade of adhesion of sickle cells to endothelium by monoclonal antibodies. New England Journal of Medicine, 342, 1910–1912. [DOI] [PubMed] [Google Scholar]
- Hoban MD, Lumaquin D, Kuo CY, Romero Z, Long J, Ho M, Young CS, Mojadidi M, Fitz-Gibbon S, Cooper AR, Lill GR, Urbinati F, Campo-Fernandez B, Bjurstrom CF, Pellegrini M, Hollis RP & Kohn DB (2016) CRISPR/Cas9-mediated correction of the sickle mutation in human CD34+ cells. Molecular Therapy, 24, 1561–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh MM, Fitzhugh CD, Weitzel R, Link ME, Coles WA, Zhao X, Rodgers GP, Powell JD & Tisdale JF (2014) Nonmyeloablative HLA-matched sibling allogeneic hematopoietic stem cell transplantation for severe sickle cell phenotype. JAMA, 312, 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inwald DP, Kirkham FJ, Peters MJ, Lane R, Wade A, Evans JP & Klein NJ (2000) Platelet and leucocyte activation in childhood sickle cell disease: association with nocturnal hypoxaemia. British Journal of Haematology, 111, 474–481. [DOI] [PubMed] [Google Scholar]
- Kanter J, Walters M, Hsieh MM, Lakshmanan K, Kwiatkowski J, Rammurti TK, von Kalle C, Kuypers FA, Cavazzana M, Leboulch P, Joseney-Antoine M, Asmal M, Thompson AA & Tisdale JF (2016) Initial results from study Hgb-206: interim results from a phase ½ clinical study of lentiglobin gene therapy for severe sickle cell disease. Blood, 128, 1176. [Google Scholar]
- Kanter J, Walters MC, Hsieh M, Krishnamurti L, Kwiatkowski JL, Kamble R, von Kalle C, Joseney-Antoine M, Pierciey FJ Jr, Shi W, Asmal M, Thompson AA & Tisdale JF (2017) Interim results from a phase 1/2 clinical study of lentiglobin gene therapy for severe sickle cell disease. Blood, 130, 527.28611024 [Google Scholar]
- Kaul DK, Finnegan E & Barabino GA (2009) Sickle red cell–endothelium interactions. Microcirculation, 16, 97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DW, Jang YY, Shin MG, Shin JH, Suh SP, Ryang DW, Yoon M, Lee JJ, Kim SK & Cho D (2013) Overnight storage of blood in ACD tubes at 4{degrees}C increases NK cell fraction in peripheral blood mononuclear cells. Annals of Clinical and Laboratory Science, 43, 267–273. [PubMed] [Google Scholar]
- Krishnan S, Setty Y, Betal SG, Vijender V, Rao K, Dampier C & Stuart M (2010) Increased levels of the inflammatory biomarker C-reactive protein at baseline are associated with childhood sickle cell vasocclusive crises. British Journal of Haematology, 148, 797–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski JL, Thompson AA, Rasko J, Hongeng S, Schiller GJ, Anurathapan U, Cavazzana M, Ho PJ, von Kalle C, Kletzel M, Leboulch P, Vichinsky E, Deary B, Asmal M & Walters MC (2017) Clinical outcomes up to 3 years following lentiglobin gene therapy for transfusion-dependent β-thalassemia in the Northstar Hgb-204 study. Blood, 130, 360.28495794 [Google Scholar]
- Lagresle-Peyrou C, Lefrère F, Magrin E, Ribeil JA, Romano O, Weber L, Magnani A, Sadek H, Plantier C, Gabrion A, Ternaux B, elix T, Couzin C, Stanislas A, Tréluyer JM, Lamhaut L, Joseph L, Delville M, Miccio A, André-Schmutz I & Cavazzana M (2018) Plerixafor enables safe, rapid, efficient mobilization of hematopoietic stem cells in sickle cell disease patients after exchange transfusion. Haematologica, 103, 778–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanaro C, Franco-Penteado CF, Albuqueque DM, Saad ST, Conran N & Costa FF (2009) Altered levels of cytokines and inflammatory mediators in plasma and leukocytes of sickle cell anemia patients and effects of hydroxyurea therapy. Journal of Leukocyte Biology, 85, 235–242. [DOI] [PubMed] [Google Scholar]
- Lard LR, Mul FP, de Haas M, Roos D & Duits AJ (1999) Neutrophil activation in sickle cell disease. Journal of Leukocyte Biology, 66, 411–415. [DOI] [PubMed] [Google Scholar]
- Luck L, Zeng L, Hiti AL, Weinberg KI & Malik P (2004) Human CD34(+) and CD34(+) CD38(–) hematopoietic progenitors in sickle cell disease differ phenotypically and functionally from normal and suggest distinct subpopulations that generate F cells. Experimental Hematology, 32, 483–493. [DOI] [PubMed] [Google Scholar]
- Lum AF, Wun T, Staunton D & Simon SI (2004) Inflammatory potential of neutrophils detected in sickle cell disease. American Journal of Hematology, 76, 126–133. [DOI] [PubMed] [Google Scholar]
- Mankad VN, Williams JP, Harpen MD, Manci E, Longenecker G, Moore RB, Shah A, Yang YM & Brogdon BG (1990) Magnetic resonance imaging of BM in sickle cell disease: clinical, hematologic, and pathologic correlations. Blood, 75, 274–283. [PubMed] [Google Scholar]
- Masiuk KE, Brown D, Laborada J, Hollis RP, Urbinati F & Kohn DB (2017) Improving gene therapy efficiency through the enrichment of human hematopoietic stem cells. Molecular Therapy, 25, 2163–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna KC, Beatty KM, Vicetti Miguel R & Bilonick RA (2009) Delayed processing of blood increases the frequency of activated CD11b+ CD15+ granulocytes which inhibit T cell function. Journal of Immunological Methods, 341, 68–75. [DOI] [PubMed] [Google Scholar]
- Miller CL & Eaves CJ (1997) Expansion in vitro of adult murine hematopoietic stem cells with transplantable lympho-myeloid reconstituting-ability. Proceedings of the National Academy of Sciences of the United States of America, 94, 13648–13653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousavi S, Moradi M, Khorshidahmad T & Motamedi M (2015) Anti-inflammatory effects of heparin and its derivatives: a systematic review. Advances in Pharmacological Sciences, 2015, 507151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller-Steinhardt M, Kirchner H & Klüter H (1998) Impact of storage at 22 degrees C and citrate anticoagulation on the cytokine secretion of mononuclear leukocytes. Vox Sanguinis, 75, 12–17. [PubMed] [Google Scholar]
- Pathare A, Al Kindi S, Alnaqdy AA, Daar S, Knox-Macaulay H & Dennison D (2004) Cytokine profile of sickle cell disease in Oman. American Journal of Hematology, 77, 323–328. [DOI] [PubMed] [Google Scholar]
- Persons DA, Allay ER, Sabatino DE, Kelly P, Bodine DM & Nienhuis AW (2001) Functional requirements for phenotypic correction of murine beta-thalassemia: implications for human gene therapy. Blood, 97, 3275–3282. [DOI] [PubMed] [Google Scholar]
- Peters SO, Kittler EL, Ramshaw HS & Quesenberry PJ (1996) Ex vivo expansion of murine BM cells with interleukin-3 (IL-3), IL-6, IL-11, and stem cell factor leads to impaired engraftment in irradiated hosts. Blood, 87, 30–37. [PubMed] [Google Scholar]
- Polanowska-Grabowska R, Wallace K, Field JJ, Chen L, Marshall MA, Figler R, Gear AR & Linden J (2010) P-selectin-mediated platelet-neutrophil aggregate formation activates neutrophils in mouse and human sickle cell disease. Arteriosclerosis, Thrombosis, and Vascular Biology, 30, 2392–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao VM, Mitchell DG, Rifkin MD, Steiner RM, Burk DL Jr, Levy D & Ballas SK (1989) Marrow infarction in sickle cell anemia: correlation with marrow type and distribution by MRI. Magnetic Resonance Imaging, 7, 39–44. [DOI] [PubMed] [Google Scholar]
- Rundberg Nilsson A, Soneji S, Adolfsson S, Bryder D & Pronk CJ (2016) Human and murine hematopoietic stem cell aging is associated with functional impairments and intrinsic megakaryocytic/erythroid bias. PLoS ONE, 11, e0158369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safaya S, Steinberg MH & Klings ES (2012) Monocytes from sickle cell disease patients induce differential pulmonary endothelial gene expression via activation of NF-kB signaling pathway. Molecular Immunology, 50, 117–123. [DOI] [PubMed] [Google Scholar]
- Saleh AW, Hillen HF & Duits AJ (1999) Levels of endothelial, neutrophil and platelet-specific factors in sickle cell anemia patients during hydroxyurea therapy. Acta Haematologica, 102, 31–37. [DOI] [PubMed] [Google Scholar]
- Sankaran VG & Weiss MJ (2015) Anemia: progress in molecular mechanisms and therapies. Nature Medicine, 21, 221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana C, Shen Y, Rattan V, Johnson C & Kalra VK (1998) Interaction of sickle erythrocytes with endothelial cells in the presence of endothelial cell conditioned medium induces oxidant stress leading to transendothelial migration of monocytes. Blood, 92, 3924–3935. [PubMed] [Google Scholar]
- Takatoku M, Sellers S, Agricola BA, Metzger ME, Kato I, Donahue RE & Dunbar CE (2001) Avoidance of stimulation improves engraftment of cultured and retrovirally transduced hematopoietic cells in primates. Journal of Clinical Investigation, 108, 447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AA, Walters MC, Kwiatkowski J, Rasko JEJ, Ribeil JA, Hongeng S, Magrin E, Schiller GJ, Payen E, Semeraro M, Moshous D, Lefrere F, Puy H, Bourget P, Magnani A, Caccavelli L, Diana JS, Suarez F, Monpoux F, Brousse V, Poirot C, Brouzes C, Meritet JF, Pondarré C, Beuzard Y, Chrétien S, Lefebvre T, Teachey DT, Anurathapan U, Ho PJ, von Kalle C, Kletzel M, Vichinsky E, Soni S, Veres G, Negre O, Ross RW, Davidson D, Petrusich A, Sandler L, Asmal M, Hermine O, De Montalembert M, Hacein-Bey-Abina S, Blanche S, Leboulch P & Cavazzana M (2018) Therapy, gene in patients with transfusion-dependent β-thalassemia. New England Journal of Medicine, 378, 1479–1493. [DOI] [PubMed] [Google Scholar]
- Tisdale JF, Pierciey FJ, Kamble R, Kanter J, Krishnamurti L, Kwiatkowski JL, Thompson AA, Shestopalov I, Bonner M, Joseney-Antoine M, Asmal M & Walters MC (2017) Successful plerixafor-mediated mobilization, apheresis, and lentiviral vector transduction of hematopoietic stem cells in patients with severe sickle cell disease. Blood, 130, 990. [Google Scholar]
- Tong TN, Burke-Murphy E, Sakac D, Pendergrast J, Cserti-Gazdewich C, Laroche V & Branch DR (2016) Optimal conditions for the performance of a monocyte monolayer assay. Transfusion, 56, 2680–2690. [DOI] [PubMed] [Google Scholar]
- Uchida N, Washington KN, Mozer B, Platner C, Ballantine J, Skala LP, Raines L, Shvygin A, Hsieh MM, Mitchell LG & Tisdale JF (2017a) RNA trans-splicing targeting endogenous β-globin pre-messenger RNA in human erythroid cells. Human Gene Therapy Methods, 28, 91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida N, Fujita A, Hsieh MM, Bonifacino AC, Krouse AE, Metzger ME, Donahue RE & Tisdale JF (2017b) BM as a hematopoietic stem cell source for gene therapy in sickle cell disease: evidence from Rhesus and SCD patients. Human Gene Therapy Clinical Development, 28, 136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villagra J, Shiva S, Hunter LA, Machado RF, Gladwin MT & Kato GJ (2007) Platelet activation in patients with sickle disease, hemolysis associated pulmonary hypertension, and nitric oxide scavenging by cell-free hemoglobin. Blood, 110, 2166–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walasek MA, van Os R & de Haan G (2012) Hematopoietic stem cell expansion: challenges and opportunities. Annals of the New York Academy of Sciences, 1266, 138–150. [DOI] [PubMed] [Google Scholar]
- Walters MC, Patience M, Leisenring W, Eckman JR, Scott JP, Mentzer WC, Davies SC, Ohene-Frempong K, Bernaudin F, Matthews DC, Storb R & Sullivan KM (1996) Bone marrow transplantation for sickle cell disease. New England Journal of Medicine, 335, 369–376. [DOI] [PubMed] [Google Scholar]
- Walters MC, Patience M, Leisenring W, Rogers ZR, Aquino VM, Buchanan GR, Roberts IA, Yeager AM, Hsu L, Adamkiewicz T, Kurtzberg J, Vichinsky E, Storer B, Storb R & Sullivan KM (2001) Stable mixed hematopoietic chimerism after bone marrow transplantation for sickle cell anemia. Biology of Blood and Marrow Transplantation, 7, 665–673. [DOI] [PubMed] [Google Scholar]
- Wang J, Kimura T, Asada R, Harada S, Yokota S, Kawamoto Y, Fujimura Y, Tsuji T, Ikehara S & Sonoda Y (2003) SCID-repopulating cell activity of human cord blood-derived CD34— cells assured by intra-bone marrow injection. Blood, 101, 2924–2931. [DOI] [PubMed] [Google Scholar]
- Wun T, Paglieroni T, Tablin F, Welborn J, Nelson K & Cheung A (1997) Platelet activation and platelet erythrocyte aggregates in patients with sickle cell anemia. Journal of Laboratory and Clinical Medicine, 129, 507–516. [DOI] [PubMed] [Google Scholar]
- Wun T, Cordoba M, Rangaswami A, Cheung AW & Paglieroni T (2002) Activated monocytes and platelet-monocyte aggregates in patients with sickle cell disease. Clinical and Laboratory Haematology, 24, 81–88. [DOI] [PubMed] [Google Scholar]
- Ye L, Wang J, Tan Y, Beyer AI, Xie F, Muench MO & Kan YW (2016) Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: an approach for treating sickle cell disease and β-thalassemia. Proceedings of the National Academy of Sciences of the United States of America, 113, 10661–10665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarbock A, Polanowska-Grabowska RK & Ley K (2007) Platelet-neutrophil-interactions: linking hemostasis and inflammation. Blood Reviews, 21, 99–111. [DOI] [PubMed] [Google Scholar]
- Zhang D, Xu C, Manwani D & Frenette PS (2016) Neutrophils, platelets, and inflammatory pathways at the nexus of sickle cell disease pathophysiology. Blood, 127, 801–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zonari E, Desantis G, Petrillo C, Boccalatte FE, Lidonnici MR, Kajaste-Rudnitski A, Aiuti A, Ferrari G, Naldini L & Gentner B (2017) Efficient ex vivo engineering and expansion of highly purified human hematopoietic stem and progenitor cell populations for gene therapy. Stem Cell Reports, 8, 977–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1. Sorting efficiency CD34 positive versus negative fraction.
Fig S2. Colony forming assay comparing CD34+GPA+ versus CD34+GPA‒ fractions.
Table SI. Total MN and CD34+ cell count are lower in SCD bone marrow compared to non-SCD bone marrow
Table SII. Total MN cell count is similar between anticoagulants or day of processing in SCD bone marrow while CD34+ cell count is higher when SCD bone marrow is collected in ACDA.
Table SIII. Makers of inflammation and contamination are higher in SCD bone marrow compared to non-SCD bone marrow.
Table SIV. Makers of inflammation and contamination in SCD bone marrow are elevated regardless of anticoagulant or day of processing.