

Abstract
Considerable progress has been made recently in defining the interactions of linker histones (H1s) within nucleosomes. Major advancements include atomic-resolution structures of the globular domain of full-length H1s in the context of nucleosomes containing full-length linker DNA. While these studies have led to a detailed understanding of the interactions and dynamics of H1 globular domains in the canonical on-dyad nucleosome binding pocket, more information regarding the intrinsically disordered N-terminal and C-terminal domains is needed. In this review, we highlight studies supporting our current understanding of the structures and interactions of the N-terminal, globular and C-terminal domains of linker histones within the nucleosome.
Keywords: Nucleosome, Linker histone, Chromatosome, H1
Introduction
Nucleosomes not only represent the initial level of packaging of the eukaryotic genome, but also are a critical node of connectivity in condensed chromatin structures. Each nucleosome contains a nucleosome core region, linker DNA, and in metazoans, most are bound by one molecule of linker histone (H1). [1–3]) (see Box 1). H1s stabilize the compaction of nucleosome arrays into higher order chromatin structures and are essential proteins in higher organisms [4]. H1s bind to the DNA surface that crosses the nucleosomal dyad, as well as to the entering/exiting linker DNA to stabilize DNA wrapping, and bring the two linker DNA strands closer together to form a stem-like structure [2,3,5]. The resulting decreased entry-exit angle of nucleosomal linker DNAs might contribute to a unique zigzag folding pattern of nucleosomes within oligonucleosome arrays which further facilitates chromatin compaction and gene regulation [3,6–9]. Although the detailed structure of the protein, including the H1 globular domain, and DNA organization within the nucleosome core has been determined at high resolution [1,10], many questions remain regarding how the disordered NTD and CTD of H1 interacts with the nucleosome and linker DNA.
Definitions of Basic Chromatin Terms.
Nucleosome core: The portion of the nucleosome consisting of the core histone octamer and the ~147 ± 1-2 bp* of DNA in tight association with the core histones.
Nucleosome core particle: Entity produced by micrococcal nuclease (MNase) digestion of native or reconstituted chromatin, containing 147 ± ~10 bp** of DNA and the core histone octamer.
Chromatosome: Entity produced by MNase digestion of native or reconstituted chromatin containing ~ 168 ± ~10** bp DNA (roughly symmetrically distributed to either end of the nucleosome core region), the core histone octamer, and one molecule of full-length linker histone (ref 59). (NB: The term “Chromatosome” should not be used to indicate a full nucleosome bound by a linker histone; continued MNase digestion of chromatosomes results in nucleosome core particles.)
Nucleosome: The complete basic repeating subunit of chromatin, typically contains ~200 ± 40 bp DNA, a nucleosome core, variable lengths of linker DNA and, in many instances, a single molecule of a linker histone. Thus a nucleosome may or may not contain H1.
*As defined by Luger et al., 1997, in the absence of sequence effects, the nucleosome core must contain an odd number of base pairs as the nucleosome dyad passes through a central base pair. The noted variance in total DNA within the core region is due to sequence-dependent variations in DNA twist [Muthurajan, 2003]
**The estimate of variance in DNA fragment sizes is due to the small variance in DNA twist noted above, plus a larger variance due to the sequence-dependent DNA cleavage activity of MNase.
In higher eukaryotes, H1s have a tripartite structure composed of a short (20-35 residue) N-terminal domain (NTD), followed by a conserved ~80 residue trypsin-resistant central globular domain (GD) and by a long intrinsically disordered ~100 residue C-terminal domain (CTD), which accounts for about half of the H1 sequence [2] (Figure 1).
Figure 1. Tripartite structure of metazoan linker histone.
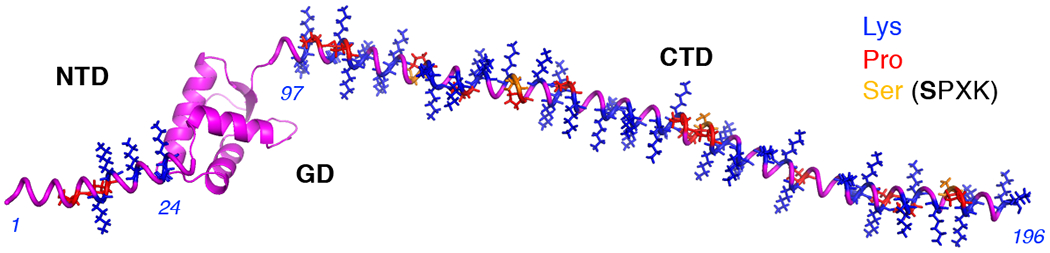
Model of H1 showing the N-terminal domain (NTD), globular domain (GD), and the C-terminal domain (CTD). Model of the GD of built from 5NL0 (Xenopus H1.0b) [3]. The disordered regions are based on the FASTA of H1.0b. Images rendered using PyMol version 1.8 and VMD version 1.9.4. The location of Lysines (Lys), prolines (Pro) and serines within SPXK motifs (Ser) are indicated. Italicized numerals indicate residues delineating domains.
The N-terminal Domain
The NTD exhibits greater sequence divergence both across species and between different H1 subtypes than the GD, but it contains two distinct subdomains, one with an N-terminal region enriched in hydrophobic residues, devoid of basic amino acid residues, and another region closer to the GD that is enriched in basic amino acid residues and contains an ARKS sequence similar to that found in the H3 tail domain [11,12]. Indeed, the NTD is acetylated and phosphorylated like the core histone tails [13–16]. While the NTD is not required for higher-order chromatin structure formation, replacing it with that of another H1 variant, or point mutants within the NTD appears to affect H1-nucleosome binding affinities [17–19].
The amino acid composition of the H1 NTD is characteristic of an intrinsically disordered region (IDR). H1 NTD peptides are unstructured in aqueous solution but acquire α-helical structure in the presence of helix-stabilizing solvents or upon interaction with DNA [20]. Molecular dynamics simulations indicate that NTDs undergo a disorder-to-order transition upon nucleosome binding, with extents of α-helical structure correlating with the nucleosome binding of the H1 [21]. Unfortunately, the dynamic nature of the NTD precludes a detailed picture of H1 NTD secondary structure in H1-nucleosome complexes [3,22,23].
The Globular Domain
The ~80 amino acid residue globular domain is required and sufficient for structure-specific binding of nucleosomes but is deficient in promoting chromatin condensation [4,24,25]. Interestingly, the H1 GD alone protects an additional 20 base pair (bp) linker DNA from micrococcal nuclease digestion beyond the 147 bp core DNA protected by the histone octamer [24]. The GD is more conserved among subtypes than either the N- or C-terminal domains, and unlike these domains, it adopts a stable ‘winged’ helix-turn-helix DNA binding motif [26,27] that interacts with the exposed minor groove at the nucleosome dyad, and with the two linker DNAs [3,22,23].
Indeed, multiple structural studies have shown that a wide variety of H1 subtypes interact with the DNA at the nucleosomal dyad and with both linker DNAs, including H1.5 [5], Xenopus H1.0 and B4 [3,7], GH5 [22], and human H1.0, H1.4 and H1.10 [23,28,29]. In contrast, NMR data suggested that the sole H1 from Drosophila containing four ‘stabilizing’ mutations in the GD and lacking the NTD binds in a position off the nucleosome dyad, bridging the central wrap and one linker DNA [30]. Moreover, a later NMR analysis found that replacing five residues in the free GD of H5 with the corresponding residues in Drosophila H1 shifts the binding mode to off-dyad [31], and an early cryo-EM study also reported a distinct off-dyad binding mode for human H1.4 when in complex with a condensed 12-nucleosome array [32]. However, the 12-nucleosome array was fixed by glutaraldehyde, which appears to affect H1 binding [28], and all high-resolution structures of full-length H1s bound to nucleosomes show GDs located in only the on-dyad position [23,33]. Moreover, a recent MD simulation indicates the GD of Drosophila H1 has a significant enthalpic preference for on-dyad vs off-dyad binding, which may compensate for the greater entropic penalty for constraint of linker DNA predicted to occur upon GD binding at the dyad site [34]. However, the CTD contributes substantially to linker DNA organization, and helps constrain the dynamics of the GD [5,35,36]. Thus, taken together, current data strongly suggest the on-dyad position is preferential in the context of the full-length H1s.
The C-terminal domain (CTD)
H1 CTD is required for high affinity nucleosome binding of H1s both in vitro and in vivo, and is essential for chromatin condensation [17,24,37,38]. FRAP (fluorescence recovery after photobleaching) experiments indicate H1.5 and H1.4 have the highest nucleosome binding affinity, while H1.3 and H1.0 have intermediate affinity, and H1.1 and H1.2 have the lowest affinity [17]. Interestingly, the binding affinities seem correlated with the length of H1 CTD, which decreases in the order of H1.5, H1.4, H1.3, H1.2 and H1.1. However, H1.0 is an exception, as it exhibits intermediate binding affinity, but has the highest positive charge density in the CTD (42 basics residues in 97 amino acids). While the primary sequences of H1 CTDs are not well conserved among H1 isoforms, the amino acid compositions are similar across species and subtypes, with about 40% of CTD residues consisting of basic amino acids, mostly lysine, amounting to a net positive charge of 30 to 50 |e| [39], consistent with the function of shielding the negatively charged DNA in condensed chromatin [40,41]. These positively charged residues are evenly distributed across the CTD with about 70% of the Lys residues in doublets. The CTDs of mammalian H1 isoforms contain one or more copies of the consensus sequence, (S/T)-P-X-(K/R), where the first residue can be phosphorylated by cyclin-dependent kinases (CDKs) (here, X represents any amino acid). H1 CTDs also include a significant fraction of residues represented by alanine and proline, and an almost complete lack of acidic and aromatic residues, characteristic of IDRs [42]. Indeed, scrambling the primary sequence of the H1 CTD did not affect its chromatin condensing function, indicating that the amino acid composition, rather than the specific sequence, is the basis for its chromatin condensing ability [37]. Although the H1 CTD exists as a random coil in aqueous solution, numerous studies indicate that CTD peptides can adopt folded structures in the presence of secondary structure stabilizers or upon interaction with DNA [43,44]. Importantly, the CTD undergoes a significant compaction consistent with a disorder-to-order transition upon binding nucleosomes [45,46]. However, the fully compact state may still retain the dynamic properties of the disordered peptide [23,47]. The intrinsic disorder within the H1 CTD and the entropic cost of ordering this domain upon binding allow fine control over its DNA binding affinity, balance of extensive charge neutralization, and permit rapid and promiscuous binding to its physiological targets.
The H1 CTD may serve as a nexus for signaling in chromatin. The CTD inhibits epigenetic posttranslational modification of the H3 tail domain in chromatin, and modulates its interactions with linker DNA [48]. In addition, H1 CTD structure is linked to the compaction state of the chromatin fiber at the earliest levels of chromatin folding, as extended ‘beads on a string’ structures transition to contacting zig-zag arrays [49]. H1 CTD structure is dependent upon linker trajectory, suggesting a mechanism by which chromatin structure and CTD structure may be coupled [49]. Of note, H1 CTD structure is distinct in oligo-nucleosomes compared to mononucleosomes and can be altered by binding of constituents to the linker DNA. [49]. Importantly, several chromatin modifications associated with transcriptional permissibility or other functional chromatin states appear to impinge on the H1 CTD. HMGNs, architectural transcription factors that bind to the nucleosome core, are enriched in active regions of chromatin, however the mechanism by which they promote H1 exchange and transcription remain unclear [50]. Interestingly, HMGNs bind to H1-bound nucleosomes, abrogate H1-dependent stabilization of compact nucleosomal arrays, and alter H1 CTD conformation in mononucleosomes [51]. In addition, transcription-associated acetylation in the H3 tail domain alters H1 CTD conformation in manner that appears independent from changes in linker DNA conformation [52]. Finally, cell-cycle dependent phosphorylation of H1 CTD directly alters the extent of H1 CTD condensation [53,54]. Evidence suggests that the effect of phosphorylation may be through structural changes rather than changes in charge [53,54]. This notion is supported by the fact that the CTD is most heavily phosphorylated in mitosis where the chromatin is in its most compact state.
H1-nucleosome structures
Obtaining high-resolution structures of H1-bound nucleosomes by cryo-EM has been challenging due to highly dynamic binding of linker histones to nucleosomes and the flexibility of the linker DNA arms [3,55]. Of note, binding of a single-chain antibody fragment to the core histone surfaces of the nucleosome was found to stabilize dissociation and increase resolution in cryo-EM [23,56], but may alter the dynamic of histones and DNA, and cause allosteric changes in structure [57,58]. Nevertheless, several high-resolution (3-5 Å) H1-nucleosome structures containing full-length linker histones and nucleosomes with full-length linker DNA have been elucidated by both X-ray crystallography and cryo-EM [3,7,23] (Table 1), including a recent crystal structure of an H1-containing 355 bp dinucleosome which showed one copy of H1 bound in the on-dyad position and a second H1 positioned in between the two nucleosomes [29]. In addition, informative structures containing partial H1 molecules, and/or partial segments of linker DNA, have also appeared [3,22]. (Of note, no structures of a true “chromatosome” [59] have yet appeared (See Table 1)). All high-resolution structures with full-length proteins show very similar on-dyad binding for the GD, and asymmetric interactions with the two linker DNAs (Figure 2) [3,22,23]. The GD makes stronger interactions with the linker DNA that faces its third helix (α3), thereby coining the term “α3-linker”. This helix represents a critical DNA-binding interface in other DNA-interacting winged-helix proteins as well [60]. The second linker, the “L1-linker”, faces the first loop region, L1, which separates helices α1 and α2. This loop contains a critical Arginine, R42 in Xenopus H1.0, that is well-conserved across H1 subtypes [3]. The dyad interface is broader and more complex as spanned by the charged and polar residues of the β2-L3-β3 segment as well as contributions from some of the N-terminal end residues of helix α2. Residues of the dyad interface are generally well-conserved, albeit with exceptions. For example, N110 of human H1.10, a position generally occupied by a serine in most isoforms, permits a firm dyad-binding as seen in cryo-EM and verified by molecular dynamics [23].
Table 1.
High-resolution structures of nucleosomes bound by H1 or partial H1 molecules.
| Protein | Organism | Protein | Method | Reference | GD location |
|---|---|---|---|---|---|
| Full-length nucleosome with full-length H1 | |||||
| H1.0 | Xenopus | Full length | cryoEM | Bednar et al., 2017 | On-dyad |
| H1.0 | Xenopus | Full length | crystal structure | Bednar et al., 2017 | On-dyad |
| H1.0 | Human | Full length | cryoEM | Zhou et al., 2021 | On-dyad |
| H1.4 | Human | Full length | cryoEM | Zhou et al., 2021 | On-dyad |
| H1.10 | Human | Full length | cryoEM | Zhou et al., 2021 | On-dyad |
| GH1.10-ncH1.4 | Human | Full length | cryoEM | Zhou et al., 2021 | On-dyad |
| Partial H1/partial DNA | |||||
| H1.5del50 | Human | 50 residue CTD- deletion |
cryoEM | Bednar et al., 2016 | On-dyad |
| GH5 | Chicken | GD of H5 | crystal structure | Zhou, 2015 | On-dyad |
Figure 2. H1 globular domain located in the dyad position within a 197 bp nucleosome bound by full-length H1.0b.
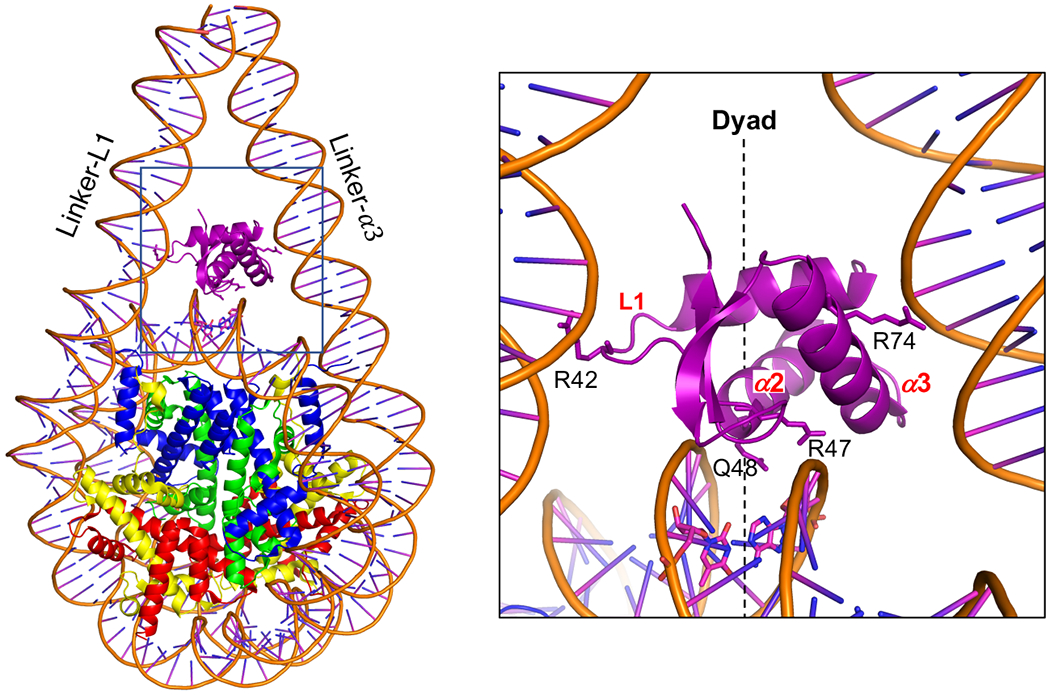
(5NLO) [3]. Left: Nucleosome, rotated ~40° about the vertical (dyad) axis from normal for clarity. Note that the NTD and CTD were not included in the model due to disorder. Right, blowup of up the area in the box (right) showing details of the globular domain orientation and interactions with both linker DNAs. Images prepared as in Figure 1, see text for details.
While interactions of the NTD and CTD are, as yet, not well-defined due to their highly dynamic nature (see above), hints of CTD location are gleaned from a comparison of cryo-EM structures of full-length H1 and a CTD deletion, where the H1 CTD appears to be associated with the L1 linker DNA [3]. In the absence of H1, the DNA linkers are highly flexible likely due to “breathing” of the histone-DNA interactions near NCP exit (Bednar et al, 2017). Binding of linker histones and localization of the CTD stabilizes the most convergent linker DNA conformation and draws them closer together to form a stem-like structure [3,23,35]. Hydroxyl radical footprinting suggests that the CTD aligns along one side of the linker DNA arms in the center of the DNA stem structure [5,33,35]. Indeed, the C-terminus of H1 was found to be responsible for decreasing nucleosome linker flexibility and the formation of the stem structure of the linkers [Meyer, 2011; Bednar, 2017). In the stem structure, the angles between the dyad and the linkers showed a very narrow distribution.
Outlook
Of immediate interest is fixing the location(s) and interactions of the H1 NTD and CTD. Questions include which basic residues (all?) interact with DNA, where the primary interactions occur and to what extent these domains adopt secondary and tertiary structures and equilibrate with intrinsically disordered states. Other questions include whether linker histones on adjacent nucleosomes orient at all with respect to each other, and to what extent the H1 CTDs on adjacent nucleosomes interact or cooperate in the formation of condensed chromatin. What are the structural differences between CTDs of H1 isotypes? Moreover, how exactly do chromatin modifications including H3 tail acetylation, CTD phosphorylation, and HMGN binding modulate CTD structure? Finally, given the influence of chromatin modifiers and posttranslational modifications on H1 structure, it will be important to understand how CTD structure and linker DNA trajectory are coupled and to what extent this coupling influences chromatin folding.
Acknowledgements
This work was supported by National Institutes of Health Grants R01GM052426 (to JJH) and was benefiting from the 2232 International Fellowship for Outstanding Researchers Program of TÜBİTAK [Project No: 118C354] (the financial support received from TÜBİTAK does not mean that the content of the publication is approved in a scientific sense by TÜBİTAK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ: Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389:251–260. [DOI] [PubMed] [Google Scholar]
- 2.Cutter AR, Hayes JJ: A brief review of nucleosome structure. FEBS Lett 2015, 589:2914–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.••.Bednar J, Garcia-Saez I, Boopathi R, Cutter AR, Papai G, Reymer A, Syed SH, Lone IN, Tonchev O, Crucifix C, et al. : Structure and Dynamics of a 197 bp Nucleosome in Complex with Linker Histone H1. Mol Cell 2017, 66:384–397 e388. [DOI] [PMC free article] [PubMed] [Google Scholar]; First high-resolution structural analysis of a full-length H1 bound to a nucleosome. Cryo-EM and X-ray crystallography reveal the structure of Xenopus H1.0 bound to a 197 bp 601 nucleosome as well as human H1.5ΔC50 and hydroxyl radical footprints of several other H1s. The results show on-dyad binding and interactions with both DNA linker segments, with stronger interactions between the α3 helix and α3-linker. However, density ascribed to the CTD was found primarily associated with the L1 linker DNA.
- 4.Cutter AR, Hayes JJ: Linker histones: novel insights into structure-specific recognition of the nucleosome. Biochem Cell Biol 2017, 95:171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Syed SH, Goutte-Gattat D, Becker N, Meyer S, Shukla MS, Hayes JJ, Everaers R, Angelov D, Bednar J, Dimitrov S: Single-base resolution mapping of H1-nucleosome interactions and 3D organization of the nucleosome. Proc Natl Acad Sci U S A 2010, 107:9620–9625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bednar J, Horowitz RA, Grigoryev SA, Carruthers LM, Hansen JC, Koster AJ, Woodcock CL: Nucleosomes, linker DNA, and linker histone form a unique structural motif that directs the higher-order folding and compaction of chromatin. Proc. Natl. Acad. Sci. USA 1998, 95:14173–14178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.••.Garcia-Saez I, Menoni H, Boopathi R, Shukla MS, Soueidan L, Noirclerc-Savoye M, Le Roy A, Skoufias DA, Bednar J, Hamiche A, et al. : Structure of an H1-Bound 6-Nucleosome Array Reveals an Untwisted Two-Start Chromatin Fiber Conformation. Mol Cell 2018, 72:902–915 e907. [DOI] [PubMed] [Google Scholar]; A 9.7 Å crystal structure, cryo-EM data, and biochemical analyses of a 6-nucleosome array with H1 revealing a two-start array at an intermediate level of folding, with stacked nucleosomes and the H1 globular domain bound at the dyad position.
- 8.Healton SE, Pinto HD, Mishra LN, Hamilton GA, Wheat JC, Swist-Rosowska K, Shukeir N, Dou Y, Steidl U, Jenuwein T, et al. : H1 linker histones silence repetitive elements by promoting both histone H3K9 methylation and chromatin compaction. Proc Natl Acad Sci U S A 2020, 117:14251–14258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.•.Willcockson MA, Healton SE, Weiss CN, Bartholdy BA, Botbol Y, Mishra LN, Sidhwani DS, Wilson TJ, Pinto HB, Maron MI, et al. : H1 histones control the epigenetic landscape by local chromatin compaction. Nature 2021, 589:293–298. [DOI] [PMC free article] [PubMed] [Google Scholar]; Healton et al. and Willcockson et al., employ regulated H1 knockout cells, whole-genome analyses, and in-vitro biochemical experiments to uncover the central role H1 plays in remodeling chromatin by determining epigenetic patterning, influencing chromatin condensation, and regulating gene expression.
- 10.Davey CA, Sargent DF, Luger K, Maeder AW, Richmond TJ: Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 a resolution. J. Mol. Biol 2002, 319:1097–1113. [DOI] [PubMed] [Google Scholar]
- 11.Bohm L, Crane-Robinson C: Proteases as structural probes for chromatin: the domain structure of histones. Biosci. Rep 1984, 4:365–386. [DOI] [PubMed] [Google Scholar]
- 12.Pan C, Fan Y: Role of H1 linker histones in mammalian development and stem cell differentiation. Biochim Biophys Acta 2016, 1859:496–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kamieniarz K, Izzo A, Dundr M, Tropberger P, Ozretic L, Kirfel J, Scheer E, Tropel P, Wisniewski JR, Tora L, et al. : A dual role of linker histone H1.4 Lys 34 acetylation in transcriptional activation. Genes Dev 2012, 26:797–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daujat S, Zeissler U, Waldmann T, Happel N, Schneider R: HP1 binds specifically to Lys26-methylated histone H1.4, whereas simultaneous Ser27 phosphorylation blocks HP1 binding. J Biol Chem 2005, 280:38090–38095. [DOI] [PubMed] [Google Scholar]
- 15.Fischle W, Franz H, Jacobs SA, Allis CD, Khorasanizadeh S: Specificity of the chromodomain Y chromosome family of chromodomains for lysine-methylated ARK(S/T) motifs. J Biol Chem 2008, 283:19626–19635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hergeth SP, Dundr M, Tropberger P, Zee BM, Garcia BA, Daujat S, Schneider R: Isoform-specific phosphorylation of human linker histone H1.4 in mitosis by the kinase Aurora B. J Cell Sci 2011, 124:1623–1628. [DOI] [PubMed] [Google Scholar]
- 17.Hendzel MJ, Lever MA, Crawford E, Th’ng JP: The C-terminal domain is the primary determinant of histone H1 binding to chromatin in vivo. J Biol Chem 2004, 279:20028–20034. [DOI] [PubMed] [Google Scholar]
- 18.Vyas P, Brown DT: N- and C-terminal domains determine differential nucleosomal binding geometry and affinity of linker histone isotypes H1(0) and H1c. J Biol Chem 2012, 287:11778–11787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oberg C, Belikov S: The N-terminal domain determines the affinity and specificity of H1 binding to chromatin. Biochem Biophys Res Commun 2012, 420:321–324. [DOI] [PubMed] [Google Scholar]
- 20.Vila R, Ponte I, Collado M, Arrondo JL, Jimenez MA, Rico M, Suau P: DNA-induced alpha-helical structure in the NH2-terminal domain of histone H1. J Biol Chem 2001, 276:46429–46435. [DOI] [PubMed] [Google Scholar]
- 21.•.Sridhar A, Orozco M, Collepardo-Guevara R: Protein disorder-to-order transition enhances the nucleosome-binding affinity of H1. Nucleic Acids Res 2020, 48:5318–5331. [DOI] [PMC free article] [PubMed] [Google Scholar]; Molecular dynamics simulations of H1.0, H1.1 and H1.2 show transition from a completely unstructured state to adopting varying degrees of helical structure in a nucleosome environment that correlates with the experimentally determined binding affinities for each H1.
- 22.Zhou BR, Jiang J, Feng H, Ghirlando R, Xiao TS, Bai Y: Structural Mechanisms of Nucleosome Recognition by Linker Histones. Mol Cell 2015, 59:628–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.••.Zhou BR, Feng H, Kale S, Fox T, Khant H, de Val N, Ghirlando R, Panchenko AR, Bai Y: Distinct Structures and Dynamics of Chromatosomes with Different Human Linker Histone Isoforms. Mol Cell 2021, 81:166–182 e166. [DOI] [PMC free article] [PubMed] [Google Scholar]; Comprehensive analysis of H1.0, H1.4 and H1.10 (H1.x) bound to 197 bp 601 nucleosomes stabilized by the binding of a single-chain antibody fragment (scFv) showing all globular domains bound to the dyad position. NMR analyses showed generally disordered CTD, with select residues exhibiting some degree of reduced mobility and partial ordering of the H2A and H3 tail domains.
- 24.Allan J, Mitchell T, Harborne N, Bohm L, Crane-Robinson C: Roles of H1 domains in determining higher order chromatin structure and H1 location. J. Mol. Biol. 1986, 187:591–601. [DOI] [PubMed] [Google Scholar]
- 25.Lu X, Hansen JC: Identification of specific functional subdomains within the linker histone H10 C-terminal domain. J Biol Chem 2004, 279:8701–8707. [DOI] [PubMed] [Google Scholar]
- 26.Ramakrishnan V, Finch JT, Graziano V, Lee PL, Sweet RM: Crystal structure of globular domain of histone H5 and its implications for nucleosome binding. Nature 1993, 362:219–224. [DOI] [PubMed] [Google Scholar]
- 27.Cerf C, Lippens G, Ramakrishnan V, Muyldermans S, Segers A, Wyns L, Wodak SJ, Hallenga K: Homo- and heteronuclear two-dimensional NMR studies of the globular domain of histone H1: full assignment, tertiary structure, and comparison with the globular domain of histone H5. Biochemistry 1994, 33:11079–11086. [DOI] [PubMed] [Google Scholar]
- 28.Zhou BR, Jiang J, Ghirlando R, Norouzi D, Sathish Yadav KN, Feng H, Wang R, Zhang P, Zhurkin V, Bai Y: Revisit of Reconstituted 30-nm Nucleosome Arrays Reveals an Ensemble of Dynamic Structures. J Mol Biol 2018, 430:3093–3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.•.Adhireksan Z, Sharma D, Lee PL, Davey CA: Near-atomic resolution structures of interdigitated nucleosome fibres. Nat Commun 2020, 11:4747. [DOI] [PMC free article] [PubMed] [Google Scholar]; Near atomic-resolution crystal structures of continuous oligo-nucleosomes assembled from dinucleosomes with cohesive ends; inclusion of human H1.0 shows on-dyad binding of the globular domain, while a second H1 is perched between nucleosomes in a novel bridging fashion in the crystals.
- 30.Zhou BR, Feng H, Kato H, Dai L, Yang Y, Zhou Y, Bai Y: Structural insights into the histone H1-nucleosome complex. Proc Natl Acad Sci U S A 2013, 110:19390–19395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou BR, Feng H, Ghirlando R, Li S, Schwieters CD, Bai Y: A Small Number of Residues Can Determine if Linker Histones Are Bound On or Off Dyad in the Chromatosome. J Mol Biol 2016, 428:3948–3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song F, Chen P, Sun D, Wang M, Dong L, Liang D, Xu RM, Zhu P, Li G: Cryo-EM study of the chromatin fiber reveals a double helix twisted by tetranucleosomal units. Science 2014, 344:376–380. [DOI] [PubMed] [Google Scholar]
- 33.Roulland Y, Ouararhni K, Naidenov M, Ramos L, Shuaib M, Syed SH, Lone IN, Boopathi R, Fontaine E, Papai G, et al. : The Flexible Ends of CENP-A Nucleosome Are Required for Mitotic Fidelity. Mol Cell 2016, 63:674–685. [DOI] [PubMed] [Google Scholar]
- 34.•.Woods DC, Wereszczynski J: Elucidating the influence of linker histone variants on chromatosome dynamics and energetics. Nucleic Acids Res 2020, 48:3591–3604. [DOI] [PMC free article] [PubMed] [Google Scholar]; Computational estimations of the enthalpic and entropic components of GD binding to on- and off-dyad sites for GH1.0 and a modified version of the Drosophila GD (dGH1). Results indicate strong enthalpic preference for GH1.0 binding to the on-dyad location but less so for dGH1, suggesting that the greater estimated entropic penalty for GD constraint of linker DNA at the dyad site may explain on- and off-dyad binding preferences for GH1.0 and dGH1, respectively.
- 35.Meyer S, Becker N, Syed SH, Goutte-Gattat D, Shukla MS, Hayes J, Angelov D, Bednar J, Dimitrov S, Everaers R: From crystal and NMR structures, footprints and cryo- electron-micrographs to large and soft structures: nanoscale modeling of the nucleosomal stem. Nucleic Acids Res 2011, 39:9139–9154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.•.Wu H, Dalal Y, Papoian GA: Binding Dynamics of Disordered Linker Histone H1 with a Nucleosomal Particle. J Mol Biol 2021, 433:166881. [DOI] [PMC free article] [PubMed] [Google Scholar]; Coarse-grained computational analysis of GH1 and full-length H1 bound to a 193 bp nucleosome indicates that the NTD and CTD greatly restrict the dynamics of the globular domain to dyad binding sites and constrain linker DNA excursions to generate a more compact structure than in the absence of H1.
- 37.Lu X, Hamkalo B, Parseghian MH, Hansen JC: Chromatin condensing functions of the linker histone C-terminal domain are mediated by specific amino acid composition and intrinsic protein disorder. Biochemistry 2009, 48:164–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caterino TL, Hayes JJ: Structure of the H1 C-terminal domain and function in chromatin condensation. Biochem Cell Biol 2011, 89:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Happel N, Doenecke D: Histone H1 and its isoforms: contribution to chromatin structure and function. Gene 2009, 431:1–12. [DOI] [PubMed] [Google Scholar]
- 40.Subirana JA: Analysis of the charge distribution in the C-terminal region of histone H1 as related to its interaction with DNA. Biopolymers 1990, 29:1351–1357. [DOI] [PubMed] [Google Scholar]
- 41.Clark DJ, Kimura T: Electrostatic mechanism of chromatin folding. J. Mol. Biol. 1990, 211:883–896. [DOI] [PubMed] [Google Scholar]
- 42.Hansen JC, Lu X, Ross ED, Woody RW: Intrinsic protein disorder, amino acid composition, and histone terminal domains. J Biol Chem 2006, 281:1853–1856. [DOI] [PubMed] [Google Scholar]
- 43.Clark DJ, Hill CS, Martin SR, Thomas JO: Alpha-helix in the carboxy-terminal domains of histones H1 and H5. EMBO J. 1988, 7:69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roque A, Iloro I, Ponte I, Arrondo JL, Suau P: DNA-induced secondary structure of the carboxyl-terminal domain of histone H1. J Biol Chem 2005, 280:32141–32147. [DOI] [PubMed] [Google Scholar]
- 45.Caterino TL, Fang H, Hayes JJ: Nucleosome linker DNA contacts and induces specific folding of the intrinsically disordered h1 carboxyl-terminal domain. Mol Cell Biol 2011, 31:2341–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fang H, Clark DJ, Hayes JJ: DNA and nucleosomes direct distinct folding of a linker histone H1 C-terminal domain. Nucleic Acids Res. 2012, 40:1475–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Borgia A, Borgia MB, Bugge K, Kissling VM, Heidarsson PO, Fernandes CB, Sottini A, Soranno A, Buholzer KJ, Nettels D, et al. : Extreme disorder in an ultrahigh-affinity protein complex. Nature 2018, 555:61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stutzer A, Liokatis S, Kiesel A, Schwarzer D, Sprangers R, Soding J, Selenko P, Fischle W: Modulations of DNA Contacts by Linker Histones and Post-translational Modifications Determine the Mobility and Modifiability of Nucleosomal H3 Tails. Mol Cell 2016, 61:247–259. [DOI] [PubMed] [Google Scholar]
- 49.Fang H, Wei S, Lee TH, Hayes JJ: Chromatin structure-dependent conformations of the H1 CTD. Nucleic Acids Res 2016, 44:9131–9141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang S, Zhu I, Deng T, Furusawa T, Rochman M, Vacchio MS, Bosselut R, Yamane A, Casellas R, Landsman D, et al. : HMGN proteins modulate chromatin regulatory sites and gene expression during activation of naive B cells. Nucleic Acids Res 2016, 44:7144–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murphy KJ, Cutter AR, Fang H, Postnikov Y, Bustin M, Hayes JJ: HMGN1 and 2 Remodel Core And Linker Histone Tail Domains Within Chromatin. Nucleic Acids Res 2017, 45:9917–9930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.•.Hao F, Murphy KJ, Kujirai T, Kamo N, Kato J, Koyama M, Okamato A, Hayashi G, Kurumizaka H, Hayes JJ: Acetylation-modulated communication between the H3 N-terminal tail domain and the intrinsically disordered H1 C-terminal domain. Nucleic Acids Res 2020, 48:11510–11520. [DOI] [PMC free article] [PubMed] [Google Scholar]; Förster resonance energy transfer (FRET) and biochemical experiments show that specific epigenetic modifications within the H3 tail domain directly influence H1 CTD structure within the nucleosome in a way that does not appear to involve repositioning of linker DNA.
- 53.Raghuram N, Strickfaden H, McDonald D, Williams K, Fang H, Mizzen C, Hayes JJ, Th’ng J, Hendzel MJ: Pin1 promotes histone H1 dephosphorylation and stabilizes its binding to chromatin. J Cell Biol 2013, 203:57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lopez R, Sarg B, Lindner H, Bartolome S, Ponte I, Suau P, Roque A: Linker histone partial phosphorylation: effects on secondary structure and chromatin condensation. Nucleic Acids Res 2015, 43:4463–4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Misteli T, Gunjan A, Hock R, Bustin M, Brown DT: Dynamic binding of histone H1 to chromatin in living cells. Nature 2000, 408:877–881. [DOI] [PubMed] [Google Scholar]
- 56.Zhou BR, Yadav KNS, Borgnia M, Hong J, Cao B, Olins AL, Olins DE, Bai Y, Zhang P: Atomic resolution cryo-EM structure of a native-like CENP-A nucleosome aided by an antibody fragment. Nat Commun 2019, 10:2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.•.Boopathi R, Danev R, Khoshouei M, Kale S, Nahata S, Ramos L, Angelov D, Dimitrov S, Hamiche A, Petosa C, et al. : Phase-plate cryo-EM structure of the Widom 601 CENP-A nucleosome core particle reveals differential flexibility of the DNA ends. Nucleic Acids Res 2020, 48:5735–5748. [DOI] [PMC free article] [PubMed] [Google Scholar]; A high -resolution cryo-EM structure of a CENP-A nucleosome core determined by Volta phase-plate imaging. The authors find that one 601 DNA end is well-ordered and bound to the nucleosome surface, while the other is flexible, likely due to asymmetric DNA sequence-dependent effects.
- 58.••.Dogan D, Arslan M, Ulucay T, Kalyoncu S, Dimitrov S, Kale S: CENP-A Nucleosome is a Sensitive Allosteric Scaffold for DNA and Chromatin Factors. J Mol Biol 2021, 433:166789. [DOI] [PubMed] [Google Scholar]; Describes molecular dynamics simulations of the H3- or the CENP-A containing Widom 601 nucleosomes either isolated or bound to the scFv antibody fragments derived from autoimmune mice. Results show that the structure and the dynamics of the CENP-A nucleosomes are altered when bound to the scFv fragments while canonical H3 nucleosomes remain structurally more robust. Core octamers of both nucleosomes undergo asymmetric internal rearrangements that alter antibody affinity due to DNA sequence.
- 59.Simpson RT: Structure of the chromatosome, a chromatin particle containing 160 base pairs of DNA and all the histones. Biochemistry 1978, 17:5524–5531. [DOI] [PubMed] [Google Scholar]
- 60.Gajiwala KS, Burley SK: Winged helix proteins. Curr Opin Struct Biol 2000, 10:110–116. [DOI] [PubMed] [Google Scholar]