

Abstract
Background
Vascular endothelial cell proliferation, migration, and network formation are key proangiogenic processes involving the prototypic immediate early gene product, Egr‐1 (early growth response‐1). Egr‐1 undergoes phosphorylation at a conserved Ser26 but its function is completely unknown in endothelial cells or any other cell type.
Methods and Results
A CRISPR/Cas9 strategy was used to introduce a homozygous Ser26>Ala mutation into endogenous Egr‐1 in human microvascular endothelial cells. In the course of generating mutant cells, we produced cells with homozygous deletion in Egr ‐1 caused by frameshift and premature termination. We found that Ser26 mutation in Egr‐1, or Egr‐1 deletion, perturbed endothelial cell proliferation in models of cell counting or real‐time growth using the xCELLigence System. We found that Ser26 mutation or Egr‐1 deletion ameliorated endothelial cell migration toward VEGF‐A165 (vascular endothelial growth factor‐A) in a dual‐chamber model. On solubilized basement membrane preparations, Ser26 mutation or Egr‐1 deletion prevented endothelial network (or tubule) formation, an in vitro model of angiogenesis. Flow cytometry further revealed that Ser26 mutation or Egr‐1 deletion elevated early and late apoptosis. Finally, we demonstrated that Ser26 mutation or Egr‐1 deletion increased VE‐cadherin (vascular endothelial cadherin) expression, a regulator of endothelial adhesion and signaling, permeability, and angiogenesis.
Conclusions
These findings not only indicate that Egr‐1 is essential for endothelial cell proliferation, migration, and network formation, but also show that point mutation in Ser26 is sufficient to impair each of these processes and trigger apoptosis as effectively as the absence of Egr‐1. This highlights the importance of Ser26 in Egr‐1 for a range of proangiogenic processes.
Keywords: CRISPR/Cas9, early growth response‐1, endothelial cell, extracellular signal‐regulated kinase‐1
Subject Categories: Basic Science Research, Angiogenesis
Nonstandard Abbreviations and Acronyms
- CK2
casein kinase 2
- EGF
epidermal growth factor
- Egr‐1
early growth response‐1
- ERK1
extracellular signal‐regulated kinase‐1
- FBS
fetal bovine serum
- FGF‐2
fibroblast growth factor‐2
- HMEC‐1
human microvascular endothelial cells
- PDGF
platelet‐derived growth factor
- Ser26
serine residue in early growth response‐1 at position 26
- VE‐cadherin
vascular endothelial cadherin
- VEGF‐A165
vascular endothelial growth factor‐A
Clinical Perspective
What Is New?
We used a CRISPR/Cas9 strategy to introduce a homozygous Ser26>Ala mutation into endogenous Egr‐1 in human vascular endothelial cells, and in the course of this exercise generated cells with homozygous deletion in Egr‐1 caused by frameshift and premature termination.
Ser26 mutation in Egr‐1, or Egr‐1 deletion, perturbed serum‐inducible endothelial cell proliferation, migration toward VEGF‐A165 (vascular endothelial growth factor‐A), network (or tubule) formation on solubilized basement membrane preparations, elevated endothelial early and late apoptosis, and increased VE‐cadherin (vascular endothelial cadherin) expression.
Mutant and deletion cells had similar ability to proliferate, migrate, form networks, undergo apoptosis, and regulate VE‐cadherin.
What Are the Clinical Implications?
Our findings not only indicate that Egr‐1 is essential for endothelial cell proliferation, migration, and network formation, but show that point mutation in Ser26 is sufficient to impair each of these endothelial processes and trigger apoptosis as effectively as the absence of Egr‐1 itself.
This underlines the potential importance of Ser26 in Egr‐1 for a range of proangiogenic processes.
Egr‐1 (early growth response‐1) is the product of an immediate early gene also known as Krox24 and Tis8. It is a transcription factor that belongs to the Cys2His2 class of zinc finger proteins and typically recognizes the DNA binding motif, GCG(G/T)GGGCG. 1 When bound to DNA, Egr‐1 can alter gene expression through mechanisms dependent on coactivators and corepressors. Egr‐1 is typically poorly expressed in growth‐quiescent cells but is rapidly and transiently expressed in response to extracellular agonists including growth factors and hormones, 2 , 3 , 4 hypoxia, 5 cytokines, 2 , 6 , 7 shear stress, 8 and a range of other agonists and stresses. 9 , 10 , 11 , 12 , 13 , 14 , 15 Egr‐1 regulates several hundred genes in vascular endothelial cells, including genes controlling angiogenesis 16 and a range of other biological processes. 17 , 18 , 19 Our recent work in the international FANTOM5 (Functional Annotation of the Mammalian Genome 5) consortium revealed that Egr‐1 is among the most dynamic of all early response genes across a broad and diverse range of human cell types and stimuli. 20
The angiogenic process involves proliferation, migration, and network (also called tubule) formation by vascular endothelial cells and is regulated by factors such as VEGF‐A165 (vascular endothelial growth factor‐A) and FGF‐2 (fibroblast growth factor‐2), 21 which bind and activate receptors on the surface of endothelial cells and initiate and promote the growth and survival of new blood vessels. Previously, we demonstrated that Egr‐1 supports FGF‐2–dependent angiogenesis in a murine model. 2 We also found that VEGF‐A165 stimulates FGF‐2 expression in an Egr‐1–dependent manner. 2 Egr‐1 is controlled by ERK1 (extracellular signal‐regulated kinase‐1), which controls endothelial cell proliferation, migration, and angiogenesis. 22
Although it is well known that Egr‐1 is phosphorylated, 23 we recently defined for the first time a specific amino acid in Egr‐1 that undergoes phosphorylation. In vascular endothelial cells, the principal cells mediating angiogenesis, we discovered that ERK1 can phosphorylate a highly conserved serine residue in Egr‐1 at position 26 (Ser26). 24 However, no study has yet identified a functional role for Ser26 nor any other phosphorylated residue in Egr‐1 in any biological process. Understanding the importance of this residue in Egr‐1 would provide important insights on how Egr‐1–dependent gene‐regulatory networks cross‐communicate and function, and in turn, how this may impact biological processes including angiogenesis.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Cell Culture
Human microvascular endothelial cells (HMEC‐1) originally isolated from foreskin were obtained from the American Type Culture Collection (Rockville, MD) and grown in MCDB131 medium (Invitrogen, Waltham, MA) at pH 7.4 with 10% FBS, hydrocortisone (1 µg/mL), epidermal growth factor (10 ng/mL), L‐glutamine (2 mmol/L), and penicillin/streptomycin (5 U/mL). Cells were routinely passaged with 0.05% trypsin/5 mmol/L EDTA and grown in a humidified atmosphere of 5% CO2 at 37°C.
Generation of CRISPR/Cas9 HMEC‐1 Cell Lines, Wild‐Type, Mutant Ser26, and Deletion
CRISPR/Cas9 cell lines were generated with the approval of the University of New South Wales Gene Technology Research Committee. PX458 (pSpCas[BB]‐2A‐GFP) plasmid was a gift from Feng Zhang (Addgene plasmid no. 48138), and the protocol of CRISPR/Cas9 point mutation was adopted from Yik et al. 25 Five micrograms of PX458 was digested with BbsI (25 U) with New England Biolabs (Ipswich, MA) buffer 2.1 in a total volume of 40 µL, whereby digestion reaction was performed by incubating at 37°C for 1 hour. BbsI was heat inactivated at 65°C for 20 minutes before dephosphorylation by CIP (5 U) (New England Biolabs) at 37°C for 1 hour. Phosphatase was heat inactivated at 65°C for 20 minutes. Linearized and dephosphorylated plasmid was purified using Promega Wizard SV gel and polymerase chain reaction (PCR) Clean‐Up System (Promega, Madison, WI).
Forward and reverse single‐guide RNA oligonucleotides were designed following the design protocol from Ran et al. 26 Briefly, human Egr‐1 sequence (NM 001964.3) was loaded into a CRISPR/Cas9 single‐guide RNA guide design platform (http://crispr.mit.edu), and guides were selected according to the score by reverse likelihood of off‐target binding and if silent protospacer adjacent motif mutation could be obtained. Guide single‐guide RNA oligonucleotides (forward 5′‐CAC CGA GTG AGG AAA GGA TCC GAA C‐3′; reverse 5′‐AAA CGT TCG GAT CCT TTC CTC ACT C‐3′) (Sigma‐Aldrich, St. Louis, MO) were phosphorylated separately with T4 polynucleotide kinase (New England Biolabs) before annealing. Extra nucleotides (in bold) were added to facilitate ligation into the PX458 plasmid. The annealed oligonucleotide was ligated into the PX458/BbsI/CIP‐treated vector plasmid using the Quick T4 DNA ligase (New England Biolabs) and then transformed into XL‐10 Gold ultracompetent cells (Agilent, Santa Clara, CA). Plasmids generated from transformants were screened by Sanger’s capillary sequencing method.
HMEC‐1 were transfected for 24 hours with PX458/human Egr‐1 guide and the donor Egr‐1m26 oligonucleotide (5′‐ATG GCC GCG GCC AAG GCC GAG ATG CAG CTG ATG TCC CCG CTG CAG ATC TCT GAT CCG TTC GGA TCC TTT CCT CAC GCG CCC ACC ATG GAC AAC TAC CCT AAG CTG GAG GAG ATG ATG CTG CTG AGC AAC GGG GCT CCC CAG TTC CTC GGC‐3′) (Sigma‐Aldrich) in the presence of lipofectamine (Invitrogen) and Opti‐MEM medium (Invitrogen). CRISPR/Cas9 control cells were only transfected with pX458/human Egr‐1 plasmid without the donor oligonucleotide. Cells were sorted for GFP using BD FACS Aria II (BD Biosciences San Jose, CA) and green fluorescent protein‐positive single cells were seeded into a 96‐well plate containing HMEC‐1 growth medium (MCDB 131; Invitrogen) supplemented with 10% FBS, 10 ng/mL epidermal growth factor (Sigma‐Aldrich), 1 μg/mL hydrocortisone (Sigma‐Aldrich), 2 mmol/L L‐glutamine (Thermo Fisher Scientific, Waltham, MA), 5 U/mL penicillin–streptomycin (Thermo Fisher Scientific). Cells at almost 50% confluency were treated with Accutase (Thermo Fisher Scientific) to lift the cells and seeded into wells of a 6‐well plate. At 80% confluency, the cells were seeded in triplicate in the 6‐well plate. Cells from the 2 wells were frozen, whereas genomic DNA was extracted from cells in the third well.
Genomic DNA Extraction
Genomic DNA was extracted using the PureLink genomic kit (Invitrogen). Briefly, ≈5×106 CRISPR/Cas9 cell types were resuspended with 200 µL 1×PBS before addition of proteinase K and RNaseA. An equal volume (200 µL) of PureLink genomic lysis/binding buffer was added, mixed well, and incubated for 10 minutes at 55°C. Ethanol was added to the lysate and loaded into the spin columns. The columns were centrifuged at 10 000g for 1 minute at 22°C. The columns were washed as recommended with Wash Buffer 1 then with Wash Buffer 2 before DNA elution. Genomic DNA was eluted using sterile milliQ water and measured using a Nanodrop spectrophotometer. Aliquots of genomic DNA were stored at −20°C until further analysis.
CRISPR/Cas9 Screening
PCR‐based screening of the CRISPR/Cas9/Egr‐1m26 and control was performed using 50 ng of genomic DNA with primers (Egr‐1 fwd139: 5′‐CTG CAC GCT TCT CAG TGT TC‐3′ and Egr‐1 rvr565: 5′‐AAA GAC TCT GCG GTC AGG TG‐3′) (Sigma‐Aldrich). Amplification was done with Platinum SuperFi DNA polymerase (Thermo Fisher Scientific) with initial denaturation of 98°C for 30 seconds. Thirty‐five cycles of 98°C for 10 seconds, 58°C for 10 seconds, and 72°C for 30 seconds were performed before further extension of 72°C for 5 minutes. The PCR reaction mixtures were submitted to the Ramaciotti Centre for Genomics (University of New South Wales) as core prep with EXOSAPIT clean‐up before Sanger sequencing. Alternatively, the amplified 427 bp fragment was purified in 1.5% agarose gel in 1× Tris‐acetate‐EDTA buffer and further isolated using the Promega Wizard SV gel and PCR Clean‐Up System (Promega). The purified PCR fragment was sequenced to confirm mutation of Ser26 to Ala26 (M26), control (wild‐type) and Egr‐1 deletion cell types.
Copy Number Analysis
To test whether human Egr‐1 alleles had been homozygously modified in CRISPR/Cas9 mutant clones, quantitative PCR was done to calculate the copy number of Egr‐1 alleles. Quantitative PCR was performed using 10 ng of genomic DNA with 1× QuantiFast SYBR together with final concentration of 0.8 µmol/L primers in a total volume of 25 µL. Primers used were human Egr‐1CFWD 5‐ACT ACC CTA AGC TGG AGG AGA‐3′, human Egr‐1CREV 5‐GTG TCC GCC TGA GGG TTG A‐3′, human SP1F115 5′‐AAT CGA GGA AGT GGA GGC AA‐3′, and human SP1R302 5‐GGA GTC AAG GTA GCT GCA GA‐3′, with human Sp1 as a reference gene. Amplification was done in LightCycler 480 Instrument II (Roche Diagnostics, Castle Hill, NSW, Australia) with the following conditions: predenaturation at 95°C for 5 minutes, 50 cycles of 95°C for 10 seconds, 58°C for 10 seconds, and 72°C for 30 seconds with a single acquisition. Melt curve settings were 1 cycle of 95°C for 10 seconds, 60°C for 1 minute, and continuous at 97°C. Cycle threshold or crossing point values were calculated automatically, and values from the CRISPR/Cas9 mutants were subtracted from those of the Sp1 reference. The difference was plugged in equation 1/2^(SP1‐EGR1). The obtained value was normalized to the CRISPR/Cas9 wild‐type and multiplied by 2. Values between 1.5 and 2.5 were considered to have retained both alleles of Egr‐1 and therefore to be homozygously modified if the Sanger sequencing trace for the CRISPR/Cas9 mutants showed a single peak at the sites of mutation.
Conventional PCR
Amplification of longer human Egr‐1 fragment was done using 50 ng genomic DNA with AccuPrime Pfx Supermix and final primer concentration of 0.2 µM. Primers used were human Egr‐1DFWD 5′‐CTT CAA CCC TCA GGC GGA CA‐3′ and human Egr‐1EREV 5′‐GCC ACA TGT GAG AGT ACG GT‐3′. Amplifications were performed in a PCR Thermal Cycler (Applied Biosystems, Foster City, CA) with an initial denaturation of 95°C for 5 minutes, 35 cycles of 95°C for 15 seconds, 58°C for 30 seconds, and 68°C for 2.5 minutes. The final PCR reactions were loaded into 0.8% agarose gels with 1× Tris‐borate‐EDTA running buffer. Gels were viewed and photographed using Molecular Imager Gel Doc XR (Bio‐Rad Laboratories, Hercules, CA).
Western Blotting
CRISPR/Cas9 clones/cell types were seeded into 6‐well plates, and at 80% to 90% confluency, cells were incubated in serum‐free medium arrested for 24 hours. The cells were treated with a medium containing 5% FBS for 1 hour, then washed twice with cold 1× PBS before lysing with 1× RIPA buffer. Total protein samples were resolved by electrophoresis with denaturing SDS‐polyacrylamide gels for 1 hour at 100 V. Proteins were transferred to Millipore Immobilon polyvinylidene difluoride membranes (Millipore, Burlington, MA) before incubation with 5% nonfat skim milk. Membranes were incubated with Egr‐1 antibodies (Cell Signaling, Beverly, MA; cat. no. cs4153s) at a dilution of 1:1000. Mouse alpha tubulin antibodies (Sigma‐Aldrich; cat. no. T5168) were used at a dilution of 1:40000 in 5% nonfat skim milk. Detection was achieved with horseradish peroxidase–linked secondary antibodies (1:2000 in 5% skim milk) and chemiluminescence (Perkin‐Elmer, Waltham, MA).
Cell Adherence
Equal numbers of viable CRISPR/Cas9 clones/cell types (2×105 cells/well) in 10% FBS MCDB131 complete medium were seeded into 96‐well plates and incubated for 2 hours. Adherent cells were trypsinized and counted using a Countess II automatic cell counter (Thermo Fisher Scientific).
Endothelial Proliferation by Cell Counting
CRISPR/Cas9 clones/cell types (3×103 viable cells/well) were seeded in 96‐well plates. After 48 hours, cells were serum‐deprived for 24 hours in MCDB131 medium that contained 10 ng/mL EGF (epidermal growth factor) (Sigma‐Aldrich) and 1 µg/mL hydrocortisone (Sigma‐Aldrich), then further incubated in a medium containing 5% FBS, 10 ng/mL EGF (Sigma‐Aldrich), and 1 µg/mL hydrocortisone (Sigma‐Aldrich). Cells were counted, and percent viability (Trypan blue exclusion) 72 hours after serum stimulation was determined using a Countess II automatic cell counter (Thermo Fisher Scientific). Forty‐microliter cell suspension was mixed with an equal volume of 0.4% Trypan blue (Invitrogen; cat. no. T10282), and 10 µL was loaded into Countess cell‐counting chamber slides (Invitrogen; cat. no. C10283).
Cell Proliferation Using the xCELLigence System
The xCELLigence system (Roche Diagnostics) enables continuous, quantitative, and label‐free assessment of cell growth. 27 CRISPR/Cas9 cell types (2×103 viable cells/well) were seeded in 96‐well E‐plates in complete MCDB131 medium containing 10% FBS, EGF (10 ng/mL; Sigma‐Aldrich), and hydrocortisone (1 µg/mL) and inserted into the xCELLigence RTCA station (Roche Diagnostics). Cell growth was automatically monitored every 15 minutes by the system. Cell index, a unitless parameter that reports the impedance of electron flow caused by adherent cells, represents a quantitative measure of cell growth in each well.
MTT Assay
The MTT assay provides a measure cellular metabolic activity and a surrogate measure of proliferation. CRISPR/Cas9 clones/cell types (2×103 viable cells/well) were seeded in 96‐well plates and grown for 48 hours. Cells were incubated in serum‐free medium for 24 hours before serum stimulation (5% FBS) for 48 hours. Ten microliters of MTT Labelling Reagent (Sigma‐Aldrich) were added to each well (containing 100 µL of medium), and the plates were incubated for 4 hours at 37°C before addition of 100 µL of the solubilization solution. Overnight incubation was performed to ensure complete solubilization. Spectrophotometric absorbance of the samples was measured at 550 nm, with 690 nm reference wavelength.
Endothelial Dual‐Chamber Migration
CRISPR/Cas9 cell types (4×104 viable cells/well) in MCDB131 containing 10% FBS with complete growth supplements were seeded into the upper chamber of 24‐well plates fitted with Millicell cell culture inserts (Millipore; cat. no. PI8P01250,). After 24 hours, the medium was changed to 0.1% FBS serum, and the cells were left undisturbed for 24 hours. VEGF‐A165 (25 ng/mL) (Sigma‐Aldrich; cat. no. V7259) in 0.1% FBS was added to the lower chamber. After 24 hours, medium from the upper chamber was removed, and nonmigrated cells and excess liquid were removed with a cotton swab. The cells were fixed in 70% ethanol for 10 minutes, and membranes were dried for 10 to 15 minutes. Filters were placed on slides and mounting medium (Fluoroshield with DAPI; Sigma; cat. no. 6057) was added. Specimens were visualized with an EVOS FL microscope. Images were taken at 10× magnification.
Endothelial Network Formation
CRISPR/Cas9 cell types (4×104 viable cells/well) in MCDB131 containing 1% FBS and 50 ng/mL FGF‐2 were added to 96‐well plates coated overnight at 4°C, with 100 µL of growth factor–reduced reconstituted basement membrane matrix (Matrigel; Corning, Corning, NY; cat. no. 354230). Tubular network formation was observed after 6 hours and photographed under 40× using an Olympus (Tokyo, Japan) CKX41 microscope. The number of tubules formed was counted using ImageJ (National Institutes of Health, Bethesda, MD).
Apoptosis/Necrosis Assay by Flow Cytometry
CRISPR/Cas9 cell types (3×105 viable cells/well) were seeded into 6‐well plates in MCDB131 containing 10% FBS. After 72 hours, the culture medium was removed, and the cells were washed with PBS. Accutase (STEMCELL Technologies, Cambridge, MA; cat. no. 07920) was used to detach the cells. The cells were washed and centrifuged at 300g for 5 minutes and resuspended at 1×106 cells/mL in 500 µL of 1× binding buffer (annexin V‐fluorescein isothiocyanate (FITC) Apoptosis Staining/Detection Kit; Abcam, Cambridge, UK; cat. no. ab14085). The cells were transferred to 12×75‐mm tubes and annexin V‐FITC/propidium iodide was added and incubated for 5 minutes at 22°C, protected from light. Stained cell suspensions were analyzed by flow cytometry using a BD LSRFortessa X20 (BD Biosciences).
Vascular Endothelial Cadherin Expression
CRISPR/Cas9 cell types were grown in 25‐cm2 flasks in MCDB131 containing 10% FBS for 24 hours. The cells were washed in PBS, and Accutase (STEMCELL Technologies; cat. no. 07920) was used to detach the cells. The cells were washed and spun at 300g for 5 minutes, then resuspended (0.5×106 cells in 100 µL)in 4% formaldehyde for 15 minutes at 22°C, and permeabilized in cold 90% methanol for 10 minutes on ice. After washing in PBS, the cells were incubated with 100 µL diluted VE‐cadherin (vascular endothelial cadherin) antibody (Cell Signaling Technology, Danvers, MA; cat. no. 2500) for 1 hour at 22°C. After washing again, the cells were incubated in 100 µL of diluted fluorochrome‐conjugated secondary antibody (goat anti‐rabbit immunoglobulin G, Alexa Fluor 488 [Thermo Fisher Scientific; cat. no. A‐11008]) for 30 minutes at 22°C. Stained cell populations were identified by gating preparations with or without antibody and analyzed by flow cytometry using a BD LSRFortessa X20 (BD Biosciences).
Statistical Analysis
Statistical analysis was performed as stated in the figure legends using GraphPad (San Diego, CA) Prism software, and significance was indicated by asterisk(s). Normality was assessed using Kolmogorov‐Smirnov and Shapiro‐Wilk test. If distribution was not normal, a Mann‐Whitney or Kruskal‐Wallis test was performed as appropriate. Normally distributed data were analyzed by Student t test or 1‐way ANOVA, as appropriate. Plotted data are expressed as mean±SEM. An “n” indicates biological triplicates rather than technical triplicates. Differences were considered significant when P≤0.05. Where indicated, *P≤0.05, **P<0.01, ***P<0.001, and ****P<0.0001 are provided.
Results
Generation and Validation of CRISPR/Cas9 HMEC‐1 Clones
With the aim of elucidating the functional importance of Ser26 in human endothelial cells, a CRISPR/Cas9 strategy was used to introduce into HMEC‐1, a point mutation in the codon encoding Ser26, changing it to Ala (Figure 1A). Screening by sequencing genomic DNA identified 3 separate clones each for wild‐type and mutant (Ser26>Ala). We also identified 3 clones in which a premature stop codon effectively caused deletion in Egr‐1. Forward and reverse sequencing of Egr‐1 gave well‐defined sequences for Egr‐1 in wild‐type (Figure S1A through S1C), mutant (Figure S2A through S2C), and deletion (Figure S3A through S3C). Quantitative PCR–based copy number analysis confirmed that the Ser26>Ala mutation was homozygous in mutant cells (Figure S4) and produced amplicons of correct size, suggesting no downstream insertion and/or deletion (Figure S5). Translation of nucleotide sequences and alignment with the human Egr‐1 protein sequence (P18146) (Figure 1B) using MultAlin (http://multalin.toulouse.inra.fr/multalin/) revealed 100% homology for wild‐type and only a Ser26>Ala change in mutant cells. The 4‐nucleotide deletion in the 3 deletion clones resulted in a frame shift causing premature termination (Figure S3).
Figure 1. Generation and validation of CRISPR/Cas9 clones.
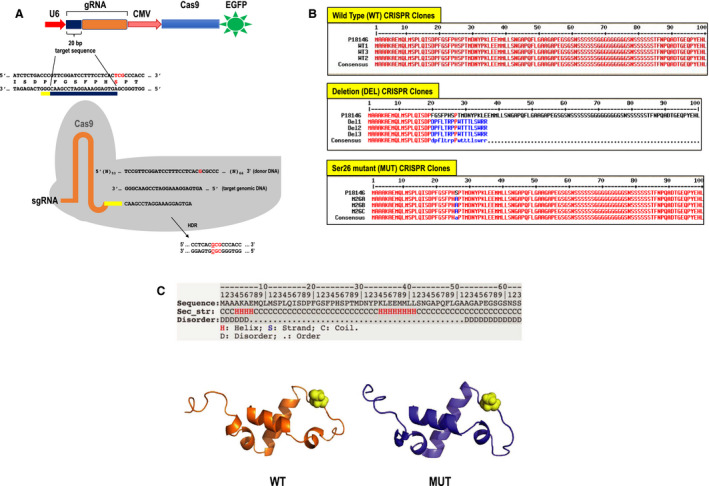
A, CRISPR/Cas9 strategy to introduce Egr‐1 mutation. Using CRISPR/Cas9 single‐guide RNA (sgRNA) guide design platform (http://crispr.mit.edu), a guide was selected that gave a high score and where protospacer adjacent motif (PAM) silent mutation was possible. A target on the reverse strand (indicated by the thick black line) was chosen and annealed oligonucleotides cloned into the PX458 (digested with BbsI). The generated PX‐458‐Egr‐1 was transfected into human microvascular endothelial cells together with the donor nucleotide to introduce a silent PAM mutation and point mutation of serine residue in Egr‐1 at position 26 (Ser26)>Ala. This CRISPR/Cas9 system generated indels by nonhomologous end joining and the desired mutation via homology‐directed repair (HDR). Transformants were sorted by fluorescence‐activated cell sorting, and single cells with green fluorescent protein were grown and later screened. The target sequence (blue), not including the PAM site (yellow), was inserted into the guide RNA (gRNA). sgRNA contains the custom‐designed sequence fused to the scaffold RNA sequence. Sequences of the target genomic DNA and donor oligonucleotide were shown. B, Alignment of amino acid sequences of different CRISPR/Cas9 clones with that of human Egr‐1 (P18146). Multiple sequence alignment with hierarchical clustering was done using MultAlin software (http://multalin.toulouse.inra.fr/multalin/). C, In silico structure prediction by trRosetta of the amino‐terminal region of Egr‐1. The amino‐terminus of the protein starts at the helix. The first 63 amino acid residues of Egr‐1 were entered into trRosetta. Ser26 (yellow) is predicted to lie in a loop region between 2 helices. Wild‐type (WT) (with Ser26) and mutant (MUT) (Ala26) Egr‐1 sequence were run through the trRosetta program, and images were generated (using Pymol software). The WT model (shown in orange) was aligned structurally with the MUT model (blue). Residue 26 is highlighted in yellow. CVM indicates cytomegalovirus; Cas9, caspase 9; DEL, deletion; EGFP, enhanced green fluorescent protein; Egr‐1, early growth response‐1; and U6, the U6 promoter.
In Silico Modeling of the Ser26>Ala Mutation
Ser26 resides near the amino terminus of Egr‐1. In silico modeling of this region of the protein using trRosetta 28 predicts the N‐terminus of Egr‐1 and comprises a small globular domain of few short regions of helix. Ser26 resides in a solvent‐exposed position in a loop linking 2 helical regions together (Figure 1C). Beyond this region, from approximately residue 60, Egr‐1 is predicted to contain long disordered random coils with stretches of glycines and serines that suggest the amino‐terminal region (containing Ser26) is isolated by a flexible tether from later regions such as the zinc fingers. Because a Ser26>Ala mutation is a mild mutation, in a loop, and solvent exposed, it is not expected the mutation perturbs the wild‐type protein structure in this region (Figure 1C).
Effects of Ser26 Mutation and Egr‐1 Deletion on Serum‐Inducible Egr‐1 Expression
Egr‐1 is a serum‐inducible immediate early gene product. 12 , 29 Western blotting showed that Egr‐1 (≈75 kDa) is expressed in growth‐quiescent wild‐type and mutant cells exposed to medium containing 5% FBS for 1 hour (Figure 2). The induction of Egr‐1 was more intense in wild‐type cells than mutant cells. In contrast, Egr‐1 was not detected in deletion cells (Figure 2).
Figure 2. Induction by serum of Egr‐1 in CRISPR/Cas9 clones.
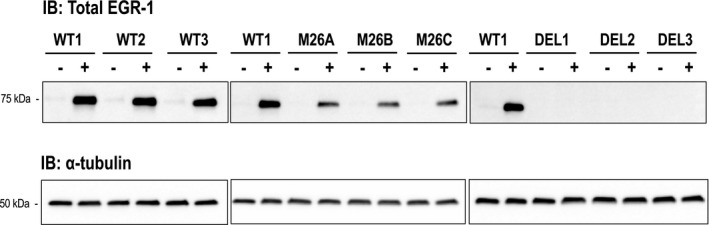
Growth‐quiescent wild‐type (WT) (WT1, WT2, WT3), mutant (M) (M26A, M26B, M26C), and deletion (DEL) (DEL1, DEL2, DEL3) cells were treated with (+) or without (−) 5% FBS for 1 hour. Upper panels show total Egr‐1 protein and lower panels correspond to α‐tubulin. Approximate molecular weight markers are shown. Immunoblots are representative of 2 biologically independent experiments. Egr‐1 indicates early growth response‐1; and IB, immunoblotting.
Ser26 Mutation and Egr‐1 Deletion Perturbs Endothelial Cell Proliferation
Cell proliferation assays were performed by stimulating growth with serum for 72 hours (Figure 3A). There was an 11‐fold increase in the number of wild‐type cells after this time. In contrast, proliferation of mutant cells or deletion cells increased by 2.6‐fold, indicating that mutation of Ser26 or the absence of Egr‐1 perturbed endothelial cell counts (Figure 3A). There was no significant difference in proliferation rate between mutant and deletion cells (Figure 3A). There was no difference in cell viability by Trypan blue exclusion after 72 hours (Figure 3B). To exclude the possibility that cell counts were merely because of impaired attachment to the well surface among the cell types (although equal numbers of cells were seeded per well), we quantified the attached cells 2 hours after seeding. Figure 3C (left) demonstrates no difference in the extent of cell attachment among cell types, whereas Figure 3C (right) shows no difference in cell viability (by dye exclusion) after this time.
Figure 3. Effect of Ser26 mutation and Egr‐1 deletion on endothelial cell total cell counts.
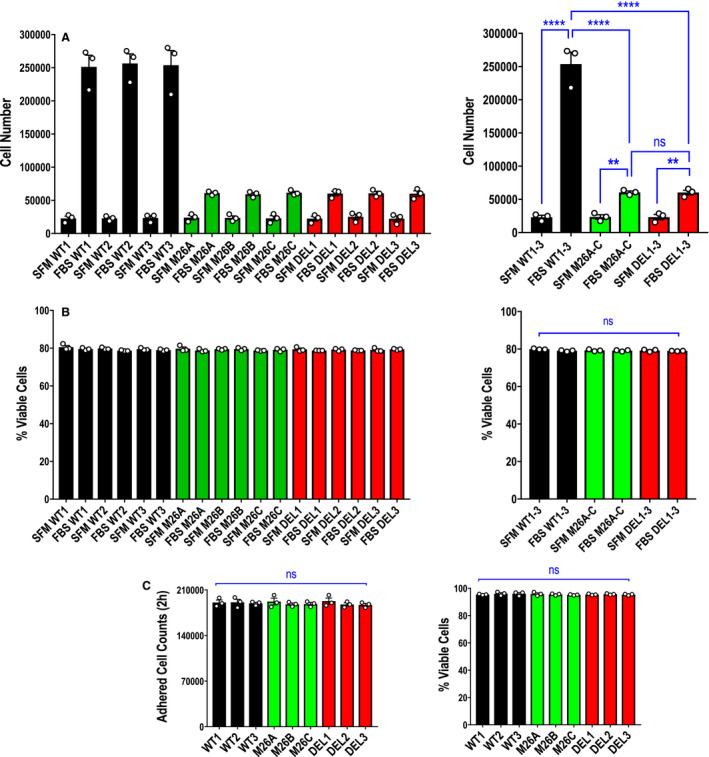
A, Proliferation assays were performed with CRISPR/Cas9 clones WT1, WT2, WT3, M26A, M26B, M26C, DEL1, DEL2, and DEL3 using the Countess II automated cell counter. Data represent the means of the means of 3 biologically independent experiments±SEM. Left, means of individual experiments. Right, combined means. Significance was assessed by 1‐way ANOVA. **P<0.01; ****P<0.0001. B, Viability of WT1, M26A, and DEL1 cells (from A) was determined after 72 hours when cell counts were quantified using the Countess II automatic cell counter with Trypan blue exclusion. Data represent the means of the means of 3 biologically independent experiments±SEM. Left, means of individual experiments. Right, combined means. Significance was assessed by Kruskal‐Wallis test. C, Assessment of adherent cell counts (left) and viability (Trypan blue exclusion; right) of 3 clones per cell type 2 hours after 2×105 cells were seeded per well using the Countess II automatic cell counter. Data represent the means of the means of 3 biologically independent experiments±SEM. Significance was assessed by Kruskal‐Wallis test or 1‐way ANOVA. SFM denotes serum free medium; FBS denotes medium containing fetal bovine serum. DEL indicates deletion; Egr‐1, early growth response‐1; M, mutant; ns, not significant; and WT, wild‐type.
We performed further experiments using the xCELLigence system, in which wild‐type, mutant, and deletion cells were seeded into E‐plates, and growth was followed over time. There was no statistical difference between mutant and deletion growth, and both cell types grew slower than wild‐type cells (Figure 4A). Similar results were obtained using the MTT assay, which provides a colorimetric assessment of metabolic activity (Figure 4B).
Figure 4. Effect of Ser26 mutation and Egr‐1 deletion on real‐time endothelial cell growth and metabolic activity.
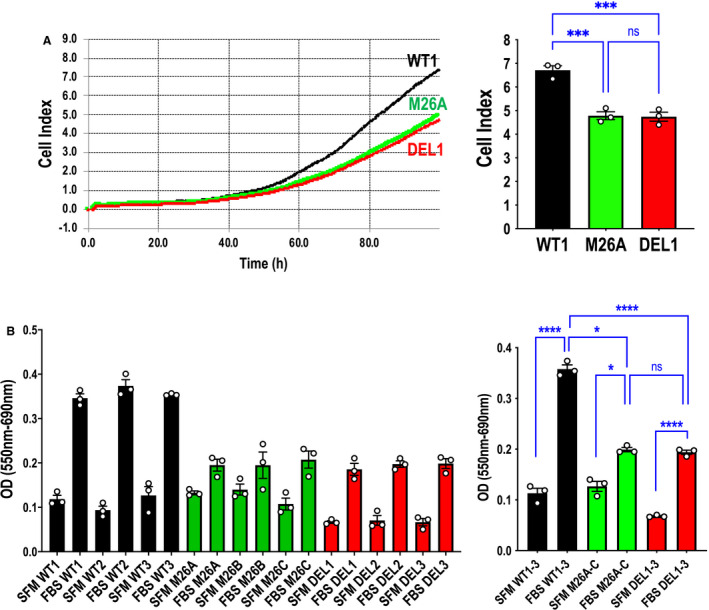
A, Cell growth assays were performed using the xCELLigence system and WT1, M26A, and DEL1 cells. Data represent the means of the means of 3 biologically independent experiments±SEM. Left, representative xCELLigence real‐time profile. Right, combined means 96 hours after seeding. Significance was assessed by 1‐way ANOVA. ***P<0.001. B, MTT assays were performed with CRISPR/Cas9 clones WT1, WT2, WT3, M26A, M26B, M26C, DEL1, DEL2, and DEL3. Data represent the means of the means of 3 biologically independent experiments±SEM. Left, means of individual experiments. Right, combined means. Significance was assessed by Mann‐Whitney or t test. *P<0.05. ****P<0.0001. SFM denotes serum free medium; FBS denotes medium containing fetal bovine serum. DEL indicates deletion; Egr‐1, early growth response‐1; M, mutant; ns, not significant; WT, wild‐type; and OD, optical density.
Ser26 Mutation and Egr‐1 Deletion Ameliorates Endothelial Cell Migration
Knockdown studies have demonstrated a key role for Egr‐1 in cell migration. 30 , 31 To determine the importance of Ser26 in endothelial migration, we used a dual‐chamber model in which cells traveled from the upper chamber toward VEGF‐A165 as a chemoattractant in the lower chamber. There was a 1.9‐fold increase in the number of wild‐type cells migrating toward VEGF‐A165. In contrast, there was a minor increase in migration (1.2‐fold) by mutant or deletion cells. There was no difference in migration between mutant and deletion cells (Figure 5A and 5B).
Figure 5. Effect of Ser26 mutation and Egr‐1 deletion on endothelial migration.
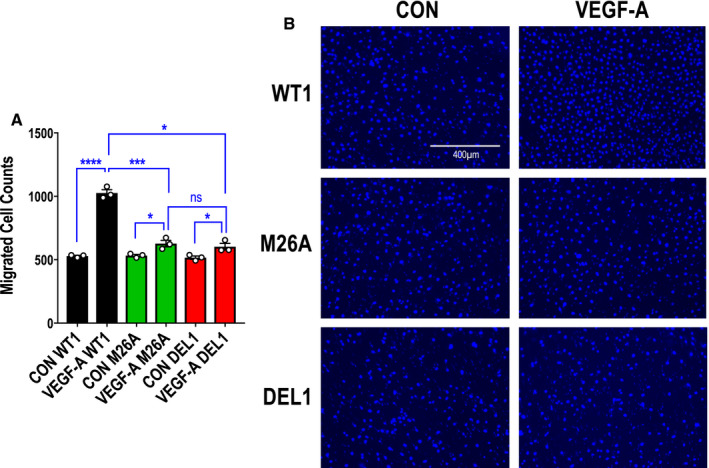
A, Migration assays were performed with CRISPR/Cas9 clones WT1, M26A, and DEL1 using a dual‐chamber model containing VEGF‐A165 (vascular endothelial growth factor‐A) (25 ng/mL) in the lower chamber. Data represent the means of the means of 3 biologically independent experiments±SEM. Significance was assessed by Mann‐Whitney or t test. *P≤0.05; ***P<0.001; ****P<0.0001. B, Representative DAPI‐stained nuclei in each condition. The cells were photographed under 10× magnification. CON indicates control cells (in 0.1% FBS) in the model without VEGF‐A165; DEL, deletion; Egr‐1, early growth response‐1; M, mutant; ns, not significant; and WT, wild‐type.
Ser26 Mutation and Egr‐1 Deletion Prevents Endothelial Network Formation
Network (or tubule) formation on solubilized basement membrane preparations is a widely used in vitro model of angiogenesis. 32 Six hours after seeding each cell type, wild‐type cells formed spontaneous networks on Matrigel (with FGF‐2). However, mutant and deletion cells formed networks 56% and 83% less efficiently than wild‐type cells, respectively. Again, there was no difference in network formation between mutant and deletion cells (Figure 6A and 6B).
Figure 6. Effect of Ser26 mutation and Egr‐1 deletion on endothelial network formation on Matrigel.
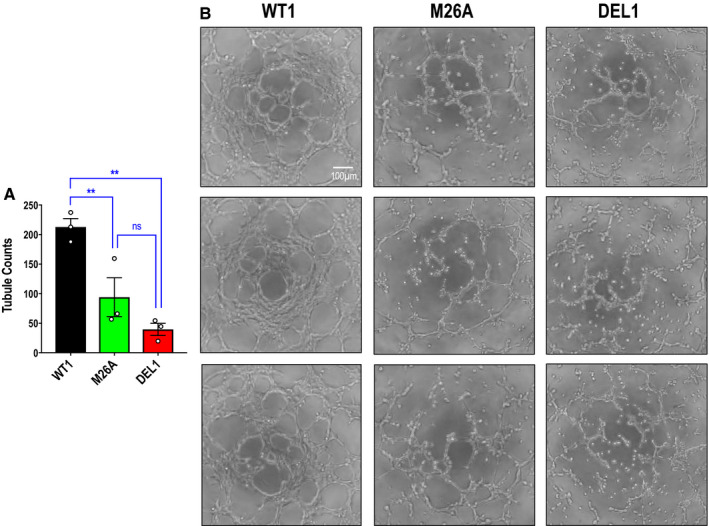
A, Network formation assays were performed with CRISPR/Cas9 clones WT1, M26A, and DEL1 in MCDB131 containing 1% FBS and 50 ng/mL FGF‐2 (fibroblast growth factor‐2) on a bed of Matrigel. Data represent the means of the means of 3 biologically independent experiments±SEM. Significance was assessed by 1‐way ANOVA. **P<0.01. B, Representative network formation. The cells were photographed under 40× magnification. Three separate fields are shown for each cell type. DEL indicates deletion; Egr‐1; early growth response‐1; M, mutant; ns, not significant; and WT, wild‐type.
Ser26 Mutation and Egr‐1 Deletion Elevates Endothelial Early and Late Apoptosis
We performed flow cytometry to determine levels of apoptosis among cell types. We were surprised to detect dramatic differences in early (annexin V‐FITC+/PI‐) and late (annexin V‐FITC+/PI+) apoptosis when comparing wild‐type cells with mutant or deletion cells at 72 hours (Figure 7A and 7B). This contrasts with Trypan blue exclusion studies showing no difference among cell types (Figure 3B).
Figure 7. Effect of Ser26 mutation and Egr‐1 deletion on endothelial early and late apoptosis.
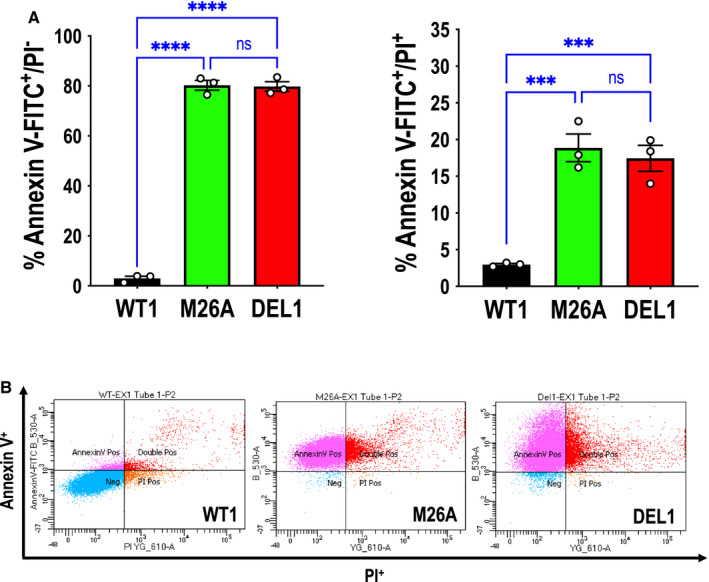
A, Annexin V‐FITC+/PI‐ (left) or annexin V‐FITC+/PI+ (right) staining comparing CRISPR/Cas9 clones WT1, M26A, and DEL1 in MCDB131 containing 10% FBS. Data represent the means of the means of 3 biologically independent experiments±SEM. Significance was assessed by 1‐way ANOVA. ***P<0.001; ****P<0.0001. B, Representative annexin V‐FITC+/PI‐ and annexin V‐FITC+/PI+ staining with WT1, M26A, and DEL1 cells by flow cytometry. Annexin V‐FITC+/PI‐ and annexin V‐FITC+/PI+ reflect early and late apoptosis, respectively. DEL indicates deletion; Egr‐1, early growth response‐1; M, mutant; ns, not significant; and WT, wild‐type.
Ser26 Mutation and Egr‐1 Deletion Elevates VE‐Cadherin Expression
VE‐cadherin is a well‐established regulator of endothelial adhesion and signaling, permeability, and angiogenesis. 33 Because VE‐cadherin is negatively regulated by Egr‐1, 16 we hypothesized that levels of this master regulator of endothelial function would increase in HMEC‐1 deficient in Egr‐1. Flow cytometry revealed that VE‐cadherin expression is weakly expressed in wild‐type–1 cells, consistent with prior studies using HMEC‐1, 34 , 35 but considerably higher in deletion‐1 and mutant‐1 cells (Figure 8), indicating an inverse proportional relationship between Egr‐1 (and Ser26 in Egr‐1) and VE‐cadherin expression.
Figure 8. Effect of Ser26 mutation and Egr‐1 deletion on VE‐cadherin (vascular endothelial cadherin) expression.
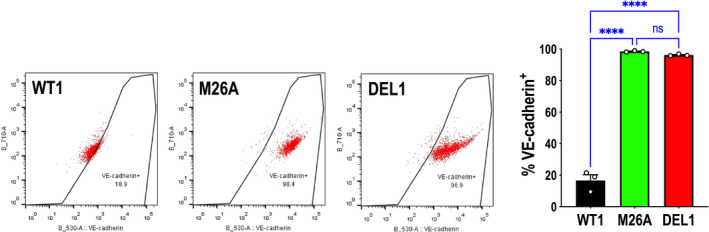
VE‐cadherin staining comparing CRISPR/Cas9 clones WT1, M26A, and DEL1 in MCDB131 containing 10% FBS. Data represent the means of the means of 3 biologically independent experiments±SEM. Significance was assessed by 1‐way ANOVA. ****P<0.0001. The figure shows representative VE‐cadherin+ staining with WT1, M26A, and DEL1 cells by flow cytometry. DEL indicates deletion; M, mutant; ns, not significant; and WT, wild‐type.
Discussion
Egr‐1 regulates the expression of hundreds of genes in vascular endothelial cells 16 and is pivotal in the control of a diverse range of biological processes in this and other cell types. 17 For example, our own work has shown that Egr‐1 is activated by vascular injury, 15 fluid shear stress, 8 phorbol esters, 36 and growth factors such as FGF‐2, 24 and controls wound repair in vitro and injured arteries in rats 13 , 37 and pigs. 12 Egr‐1 also controls tumor angiogenesis in mice and corneal neovascularization in rats. 2 Egr‐1 physically interacts with CREB1 (cAMP responsive element binding protein 1) and controls proangiogenic growth factors such as VEGF‐A165, PGF (placental growth factor), HB‐EGF (heparin‐binding EGF‐like growth factor) and PDGF (platelet‐derived growth factor), 38 sustains high levels of phosphatase of activated cells 1, which mediates reactive oxygen species–dependent T‐cell function, 39 positively regulates the superoxide dismutase 2 promoter, 40 recruits the DNA demethylase TET1 to remove methylation marks and activate downstream genes, 41 mediates estrous cycle–dependent chromatin and transcriptional change, 42 interacts with the receptor tyrosine kinase colony stimulating factor‐1 receptor, which controls monocyte/macrophage generation, 43 and regulates stem/progenitor cell survival. 44 This recent work reiterates that Egr‐1 serves a critical role in a wide range of biological processes in multiple cell types. 17 , 19 However, our understanding of how cellular processes are influenced by Egr‐1 phosphorylation, a critical epigenetic switch that can dramatically influence the activity of a protein, 45 is unclear. We recently identified a highly conserved serine residue in Egr‐1 undergoing phosphorylation in human endothelial cells. 24 This article investigates the functional importance of Ser26 in Egr‐1.
Here we used the CRISPR/Cas9 system to create a germline deletion in Egr‐1 in HMEC‐1 to show that this transcription factor is not only critical for endothelial cell proliferation, migration, and network formation, but that a single‐point mutation in Ser26 is sufficient to impair each of these cellular processes as effectively as the complete absence of Egr‐1. HMEC‐1 are transformed human endothelial lines, with characteristics akin to primary endothelial cells. 46 There was no difference in the ability of mutant and deletion cells to proliferate (whether basal growth or after serum induction), migrate in response to VEGF‐A165, or form networks on a solubilized basement membrane, because both of these cell types have reduced ability compared with wild‐type cells. These are important findings because although on the one hand they confirm the reliance of endothelial cell proliferation, migration, and network formation on Egr‐1, our data also indicate the reliance upon Ser26 in Egr‐1 for each of these processes. This follows our demonstration that Egr‐1 is inducibly expressed by serum in wild‐type and mutant cells but not in deletion cells exposed to serum. Reduced inducible Egr‐1 expression in mutant cells in response to FBS (Figure 2) may be caused by Egr‐1 autoregulation with mutant Egr‐1 noting that the Egr‐1 promoter contains nucleotide recognition elements for Egr‐1. 6 .
Wild‐type, mutant, and deletion cells were used in experimental models involving multiple concentrations of FBS, some involving prior serum deprivation, to investigate effects on inducible Egr‐1 expression or cell phenotype. Mutant and deletion cells showed similar effects (relative to wild‐type) despite our use of serum prestarvation then 5% FBS (Countess, Figure 3A and 3B; MTT, Figure 4B), continuous 10% FBS without prestarvation (Countess, Figure 3C; xCELLigence, Figure 4A; apoptosis, Figure 7), or reduced FBS/agonist exposure (migration toward VEGF‐A165, Figure 5; network formation with FGF‐2, Figure 6). This shows that the functional importance of Ser26 in Egr‐1 is not confined to a particular experimental condition, and that the integrity of this highly conserved 24 residue in Egr‐1 is as crucial as the presence of Egr‐1 itself.
Although no difference in cell viability was detected among cell types using Trypan blue exclusion, flow cytometric analysis revealed that both early and late apoptosis increased when Ser26 was mutated or Egr‐1 was deleted. The Trypan blue exclusion method is commonly used to evaluate cytotoxicity in experimental investigations where dead or dying cells absorb dye into the cytoplasm because of loss of membrane integrity, with live cells remaining unstained, but this is less precise than flow cytometry. 47 Recent studies have found that Trypan blue can alter the morphology of dead cells, which causes dead cells to disappear and leads to overestimation of viability, a phenomenon not observed with propidium iodide staining. 48 We recently showed that suppression of Egr‐1 expression can lead to apoptosis and reduced mitochondrial membrane potential. 9 Our findings suggest that Egr‐1, and in particular Ser26 in Egr‐1, has protective effects preventing apoptosis, consistent with proproliferative, angiogenic, and reparative roles we previously described. 2 , 14 , 30 , 49
Limitations in this study include our use of an immortalized endothelial cell line, making uncertain as to whether identical mechanisms are operational in primary endothelial cells. HMEC‐1 have nonetheless been used by many groups and retain many phenotypic, morphologic, and functional properties of normal human microvascular endothelial cells. 50 , 51 A further limitation may be the apparent absence of a naturally occurring polymorphism or mutation in Egr‐1 at Ser26 and its link with disease, vascular or otherwise. Conversely, the lack of a polymorphism or mutation at Ser26 and correlation with disease could indicate how critically important this highly conserved residue 24 is to the structure and function of Egr‐1.
Wild‐type, mutant, and deletion cells provide a new living resource for future research to gain further insights on how Egr‐1 phosphorylation is controlled, how it can influence interactions with binding partners, and how it regulates other genes. For example, Egr‐1 binds RelA, CBP, p300, or NAB1/2. 17 It also interacts and competes with Sp1 in the promoter regions of PDGF‐A, PDGF‐B, transforming growth factor‐β1, urokinase‐type plasminogen activator and tissue factor, 15 and other promoters as observed by Huang et al, 52 Thottassery et al, 53 Nebbaki et al, 54 Snyder et al, 55 and Su et al. 56 Egr‐1 phosphorylation by CK2 (casein kinase 2) can reduce DNA‐binding affinity and transcriptional activity 57 and results in less avid binding to Sp1 increasing colony‐stimulating factor expression. 58 Our findings show that Ser26 in Egr‐1, like Egr‐1 itself, negatively controls VE‐cadherin expression in line with prior work showing that Egr‐1 suppresses VE‐cadherin in endothelial 16 and other cell types. 59 Wild‐type, mutant, and deletion cells may therefore help better understand how Egr‐1 phosphorylation links into gene‐regulatory networks and alters the cell phenotype.
Sources of Funding
This work was supported by grants from the National Health and Medical Research Council of Australia (Program, Fellowship) and National Heart Foundation of Australia (Vanguard) (Prof Khachigian).
Disclosures
None.
Supporting information
Figures S1–S5
Acknowledgments
The authors are grateful to A/Prof K. Quinlan and Dr G. Martyn (School of Biotechnology and Biomolecular Sciences, University of New South Wales Science) for advice on the generation of CRISPR cell lines. The authors acknowledge Dr K. Michie from the Structural Biology Facility (University of New South Wales Mark Wainwright Analytical Centre (which is funded in part by the Australian Research Council Linkage Infrastructure, Equipment and Facilities Grant: ARC LIEF 190100165) for assistance with the structural analysis of the Egr‐1, Dr E. Johansson Beves and C. Brownlee (Flow Cytometry Facility, University of New South Wales Mark Wainwright Analytical Centre) for assistance with fluorescence‐activated cell sorting analysis, and Dr J. Chan (Ramaciotti Centre for Genomics, University of New South Wales) for sequencing analysis. The authors also thank Prof I. Dawes for critical review of the article. Author contributions: Prof Khachigian conceptualized this project, interpreted data, directed experiments, and wrote the article. Drs Santiago and Li planned and performed experiments, analyzed and interpreted data, and wrote the article.
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.020521
For Sources of Funding and Disclosures, see page 13.
References
- 1. Crosby SD, Puetz JJ, Simburger KS, Fahrner TJ, Milbrandt J. The early response gene NGFI‐C encodes a zinc finger transcriptional activator and is a member of the GCGGGGGCG (GSG) element‐binding protein family. Mol Cell Biol. 1991;11:3835–3841. DOI: 10.1128/MCB.11.8.3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fahmy RG, Dass CR, Sun LQ, Chesterman CN, Khachigian LM. Transcription factor EGR‐1 supports FGF‐dependent angiogenesis during neovascularization and tumor growth. Nature Med. 2003;9:1026–1032. DOI: 10.1038/nm905. [DOI] [PubMed] [Google Scholar]
- 3. Day FL, Rafty LA, Chesterman CN, Khachigian LM. Angiotensin II (ATII)‐inducible platelet‐derived growth factor A‐chain gene expression is p42/44 extracellular signal‐regulated kinase‐1/2 and Egr‐1‐dependent and mediated via the ATII type 1 but not type 2 receptor. induction by ATII antagonized by nitric oxide. J Biol Chem. 1999;274:23726–23733. DOI: 10.1074/jbc.274.34.23726. [DOI] [PubMed] [Google Scholar]
- 4. Delbridge GJ, Khachigian LM. Fgf‐1‐induced PDGF a‐chain gene expression in vascular endothelial cells involves transcriptional activation by Egr‐1. Circ Res. 1997;81:282–288. [DOI] [PubMed] [Google Scholar]
- 5. Yan SF, Mackman N, Kisiel W, Stern DM, Pinsky DJ. Hypoxia/hypoxemia‐induced activation of the procoagulant pathways and the pathogenesis of ischemia‐associated thrombosis. Arterioscler Thromb Vasc Biol. 1999;19:2029–2035. DOI: 10.1161/01.ATV.19.9.2029. [DOI] [PubMed] [Google Scholar]
- 6. Wang B, Chen J, Santiago FS, Janes M, Kavurma MM, Chong BH, Pimanda JE, Khachigian LM. Phosphorylation and acetylation of histone H3 and autoregulation by early growth response 1 mediate interleukin 1beta induction of early growth response 1 transcription. Arterioscler Thromb Vasc Biol. 2010;30:536–545. [DOI] [PubMed] [Google Scholar]
- 7. Grimbacher B, Aicher WK, Peter HH, Eibel H. TNF‐alpha induces the transcription factor Egr‐1, pro‐inflammatory cytokines and cell proliferation in human skin fibroblasts and synovial lining cells. Rheumatol Int. 1998;17:185–192. [DOI] [PubMed] [Google Scholar]
- 8. Khachigian LM, Anderson KA, Halnon NJ, Resnick N, Gimbrone MA Jr, Collins T. Egr‐1 is activated in endothelial cells exposed to fluid shear stress and interacts with a novel shear‐stress response element in the PDGF a‐chain promoter. Arterioscl Thromb Vasc Biol. 1997;17:2280–2286. [DOI] [PubMed] [Google Scholar]
- 9. Billah M, Ridiandries A, Rayner BS, Allahwala UK, Dona A, Khachigian LM, Bhindi R. Egr‐1 functions as a master switch regulator of remote ischemic preconditioning‐induced cardioprotection. Basic Res Cardiol. 2019;115:3. DOI: 10.1007/s00395-019-0763-9. [DOI] [PubMed] [Google Scholar]
- 10. Bhindi R, Fahmy RG, McMahon AC, Khachigian LM, Lowe HC. Intracoronary delivery of dnazymes targeting human Egr‐1 reduces infarct size following myocardial ischaemia reperfusion. J Pathol. 2012;227:157–164. DOI: 10.1002/path.2991. [DOI] [PubMed] [Google Scholar]
- 11. Bhindi R, Khachigian LM, Lowe HC. Dnazymes targeting the transcription factor Egr‐1 reduce myocardial infarct size following ischemia‐reperfusion in rats. J Thromb Haemost. 2006;4:1479–1483. DOI: 10.1111/j.1538-7836.2006.02022.x. [DOI] [PubMed] [Google Scholar]
- 12. Lowe HC, Fahmy RG, Kavurma MM, Baker A, Chesterman CN, Khachigian LM. Catalytic oligodeoxynucleotides define a key regulatory role for early growth response factor‐1 in the porcine model of coronary in‐stent restenosis. Circ Res. 2001;89:670–677. [DOI] [PubMed] [Google Scholar]
- 13. Santiago FS, Lowe HC, Kavurma MM, Chesterman CN, Baker A, Atkins DG, Khachigian LM. New DNA enzyme targeting Egr‐1 mRNA inhibits vascular smooth muscle proliferation and regrowth factor injury. Nature Med. 1999;5:1264–1269. [DOI] [PubMed] [Google Scholar]
- 14. Santiago FS, Lowe HC, Day FL, Chesterman CN, Khachigian LM. Early growth response factor‐1 induction by injury is triggered by release and paracrine activation by fibroblast growth factor‐2. Am J Pathol. 1999;154:937–944. DOI: 10.1016/S0002-9440(10)65341-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Khachigian LM, Lindner V, Williams AJ, Collins T. Egr‐1‐induced endothelial gene expression: a common theme in vascular injury. Science. 1996;271:1427–1431. DOI: 10.1126/science.271.5254.1427. [DOI] [PubMed] [Google Scholar]
- 16. Fu M, Zhu X, Zhang J, Liang J, Lin Y, Zhao L, Ehrengruber MU, Chen YE. Egr‐1 target genes in human endothelial cells identified by microarray analysis. Gene. 2003;315:33–41. DOI: 10.1016/S0378-1119(03)00730-3. [DOI] [PubMed] [Google Scholar]
- 17. Khachigian LM. Egr‐1 in the pathogenesis of cardiovascular disease. J Mol Med. 2016;94:747–753. DOI: 10.1007/s00109-016-1428-x. [DOI] [PubMed] [Google Scholar]
- 18. Ramadas N, Rajaraman B, Kuppuswamy AA, Vedantham S. Early growth response‐1 (EGR‐1)–a key player in myocardial cell injury. Cardiovasc Hematol Agents Med Chem. 2014;12:66–71. DOI: 10.2174/1871525713666150123152131. [DOI] [PubMed] [Google Scholar]
- 19. Khachigian LM. Early growth response‐1 in cardiovascular pathobiology. Circ Res. 2006;98:186–191. DOI: 10.1161/01.RES.0000200177.53882.c3. [DOI] [PubMed] [Google Scholar]
- 20. Arner E, Daub CO, Vitting‐Seerup K, Andersson R, Lilje B, Drabløs F, Lennartsson A, Rönnerblad M, Hrydziuszko O, Vitezic M, et al. Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science. 2015;347:1010–1014. DOI: 10.1126/science.1259418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Munoz‐Chapuli R. Evolution of angiogenesis. Int J Dev Biol. 2011;55:345–351. DOI: 10.1387/ijdb.103212rm. [DOI] [PubMed] [Google Scholar]
- 22. Srinivasan R, Zabuawala T, Huang H, Zhang J, Gulati P, Fernandez S, Karlo JC, Landreth GE, Leone G, Ostrowski MC. Erk1 and Erk2 regulate endothelial cell proliferation and migration during mouse embryonic angiogenesis. PLoS One. 2009;4:e8283. DOI: 10.1371/journal.pone.0008283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cao XM, Koski RA, Gashler A, McKiernan M, Morris CF, Gaffney R, Hay RV, Sukhatme VP. Identification and characterization of the egr‐1 gene product, a DNA‐binding zinc finger protein induced by differentiation and growth signals. Mol Cell Biol. 1990;10:1931–1939. DOI: 10.1128/MCB.10.5.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Santiago FS, Sanchez‐Guerrero E, Zhang G, Zhong L, Raftery M, Khachigian LM. Extracellular signal‐regulated kinase‐1 phosphorylates early growth response‐1 at serine 26. Biochem Biophys Res Commun. 2019;510:345–351. DOI: 10.1016/j.bbrc.2019.01.019. [DOI] [PubMed] [Google Scholar]
- 25. Yik JJ, Crossley M, Quinlan KGR. Chapter 15: genome editing of erythroid cell culture model systems. In: Lloyd JA, ed. Erythropoeisis: Methods and Protocols, Methods and Molecular Biology. Springer; 2018:245–257. [DOI] [PubMed] [Google Scholar]
- 26. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR‐Cas9 system. Nat Protoc. 2013;8:2281–2308. DOI: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li Y, Alhendi MN, Yeh MC, Elahy M, Santiago FS, Deshpande N, Wu B, Chan E, Inam S, Prado‐Lourenco L, et al. Thermostable small molecule inhibitor of angiogenesis and vascular permeability that suppresses a pERK‐FosB/∆FosB‐VCAM‐1 axis. Sci Adv. 2020;6:eaaz7815. DOI: 10.1126/sciadv.aaz7815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang J, Anishchenko I, Park H, Peng Z, Ovchinnikov S, Baker D. Improved protein structure prediction using predicted interresidue orientations. Proc Natl Acad Sci USA. 2020;117:1496–1503. DOI: 10.1073/pnas.1914677117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fahmy RG, Khachigian LM. Antisense Egr‐1 RNA driven by the CMV promoter is an inhibitor of vascular smooth muscle cell proliferation and regrowth after injury. J Cell Biochem. 2002;84:575–582. DOI: 10.1002/jcb.10057. [DOI] [PubMed] [Google Scholar]
- 30. Abdel‐Malak NA, Mofarrahi M, Mayaki D, Khachigian LM, Hussain SN. Early growth response‐1 regulates angiopoietin‐1‐induced endothelial cell proliferation, migration, and differentiation. Arterioscler Thromb Vasc Biol. 2009;29:209–216. DOI: 10.1161/ATVBAHA.108.181073. [DOI] [PubMed] [Google Scholar]
- 31. Alhendi AMN, Patrikakis M, Daub CO, Kawaji H, Itoh M, de Hoon M , Carninci P, Hayashizaki Y, Arner E, Khachigian LM. Promoter usage and dynamics in vascular smooth muscle cells exposed to fibroblast growth factor‐2 or interleukin‐1beta. Sci Rep. 2018;8:13164. DOI: 10.1038/s41598-018-30702-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Montesano R, Orci L, Vassalli P. In vitro rapid organization of endothelial cells into capillary‐like networks is promoted by collagen matrices. J Cell Biol. 1983;97:1648–1652. DOI: 10.1083/jcb.97.5.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Harris ES, Nelson WJ. Ve‐cadherin: at the front, center, and sides of endothelial cell organization and function. Curr Opin Cell Biol. 2010;22:651–658. DOI: 10.1016/j.ceb.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Evangelista K, Franco R, Schwab A, Coburn J. Leptospira interrogans binds to cadherins. PLoS Negl Trop Dis. 2014;8:e2672. DOI: 10.1371/journal.pntd.0002672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Oostingh GJ, Schlickum S, Friedl P, Schon MP. Impaired induction of adhesion molecule expression in immortalized endothelial cells leads to functional defects in dynamic interactions with lymphocytes. J Invest Dermatol. 2007;127:2253–2258. DOI: 10.1038/sj.jid.5700828. [DOI] [PubMed] [Google Scholar]
- 36. Khachigian LM, Williams AJ, Collins T. Interplay of Sp1 and Egr‐1 in the proximal PDGF‐a promoter in cultured vascular endothelial cells. J Biol Chem. 1995;270:27679–27686. [DOI] [PubMed] [Google Scholar]
- 37. Lowe HC, Chesterman CN, Khachigian LM. Catalytic antisense DNA molecules targeting Egr‐1 inhibit neointima formation following permanent ligation of rat common carotid arteries. Thromb Haemost. 2002;87:134–140. DOI: 10.1055/s-0037-1612956. [DOI] [PubMed] [Google Scholar]
- 38. Kant S, Craige SM, Chen K, Reif MM, Learnard H, Kelly M, Caliz AD, Tran KV, Ramo K, Peters OM, et al. Neural JNK3 regulates blood flow recovery after hindlimb ischemia in mice via an Egr1/Creb1 axis. Nat Commun. 2019;10:4223. DOI: 10.1038/s41467-019-11982-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dan L, Liu L, Sun Y, Song J, Yin Q, Zhang G, Qi F, Hu Z, Yang Z, Zhou Z, et al. The phosphatase PAC1 acts as a T cell suppressor and attenuates host antitumor immunity. Nat Immunol. 2020;21:287–297. DOI: 10.1038/s41590-019-0577-9. [DOI] [PubMed] [Google Scholar]
- 40. Wang X, Lu J, Xie W, Lu X, Liang Y, Li M, Wang Z, Huang X, Tang M, Pfaff DW, et al. Maternal diabetes induces autism‐like behavior by hyperglycemia‐mediated persistent oxidative stress and suppression of superoxide dismutase 2. Proc Natl Acad Sci USA. 2019;116:23743–23752 DOI: 10.1073/pnas.1912625116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sun Z, Xu X, He J, Murray A, Sun MA, Wei X, Wang X, McCoig E, Xie E, Jiang XI, et al. Egr1 recruits TET1 to shape the brain methylome during development and upon neuronal activity. Nat Commun. 2019;10:3892. DOI: 10.1038/s41467-019-11905-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jaric I, Rocks D, Greally JM, Suzuki M, Kundakovic M. Chromatin organization in the female mouse brain fluctuates across the oestrous cycle. Nat Commun. 2019;10:2851. DOI: 10.1038/s41467-019-10704-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bencheikh L, Diop MK, Rivière J, Imanci A, Pierron G, Souquere S, Naimo A, Morabito M, Dussiot M, De Leeuw F, et al. Dynamic gene regulation by nuclear colony‐stimulating factor 1 receptor in human monocytes and macrophages. Nat Commun. 2019;10:1935. DOI: 10.1038/s41467-019-09970-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pellicano F, Park L, Hopcroft LEM, Shah MM, Jackson L, Scott MT, Clarke CJ, Sinclair A, Abraham SA, Hair A, et al. hsa‐mir183/EGR1‐mediated regulation of E2F1 is required for CML stem/progenitor cell survival. Blood. 2018;131:1532–1544. DOI: 10.1182/blood-2017-05-783845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schmidt C, Beilsten‐Edmands V, Robinson CV. Insights into eukaryotic translation initiation from mass spectrometry of macromolecular protein assemblies. J Mol Biol. 2016;428:344–356. DOI: 10.1016/j.jmb.2015.10.011. [DOI] [PubMed] [Google Scholar]
- 46. Majewska A, Wilkus K, Brodaczewska K, Kieda C. Endothelial cells as tools to model tissue microenvironment in hypoxia‐dependent pathologies. Int J Mol Sci. 2021;22:520. DOI: 10.3390/ijms22020520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tennant JR. Evaluation of the trypan blue technique for determination of cell viability. Transplantation. 1964;2:685–694. DOI: 10.1097/00007890-196411000-00001. [DOI] [PubMed] [Google Scholar]
- 48. Chan LL, Rice WL, Qiu J. Observation and quantification of the morphological effect of trypan blue rupturing dead or dying cells. PLoS One. 2020;15:e0227950. DOI: 10.1371/journal.pone.0227950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gousseva N, Kugathasan K, Chesterman CN, Khachigian LM. Early growth response factor‐1 mediates insulin‐inducible vascular endothelial cell proliferation and regrowth after injury. J Cell Biochem. 2001;81:523–534. DOI: . [DOI] [PubMed] [Google Scholar]
- 50. Ades EW, Candal FJ, Swerlick RA, George VG, Summers S, Bosse DC, Lawley TJ. HMEC‐1: establishment of an immortalized human microvascular endothelial cell line. J Invest Dermatol. 1992;99:683–690. DOI: 10.1111/1523-1747.ep12613748. [DOI] [PubMed] [Google Scholar]
- 51. Muñoz‐Vega M, Massó F, Páez A, Carreón‐Torres E, Cabrera‐Fuentes HA, Fragoso JM, Pérez‐Hernández N, Martinez LO, Najib S, Vargas‐Alarcón G, et al. Characterization of immortalized human dermal microvascular endothelial cells (HMEC‐1) for the study of HDL functionality. Lipids Health Dis. 2018;17:44. DOI: 10.1186/s12944-018-0695-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Huang RP, Fan Y, Ni Z, Mercola D, Adamson ED. Reciprocal modulation between Sp1 and Egr‐1. J Cell Biochem. 1997;66:489–499. DOI: . [DOI] [PubMed] [Google Scholar]
- 53. Thottassery JV, Sun D, Zambetti GP, Troutman A, Sukhatme VP, Schuetz EG, Schuetz JD. Sp1 and Egr‐1 have opposing effects on the regulation of the RatPgp2/mdr1b gene. J Biol Chem. 1999;274:3199–3206. DOI: 10.1074/jbc.274.5.3199. [DOI] [PubMed] [Google Scholar]
- 54. Nebbaki SS, El Mansouri FE, Afif H, Kapoor M, Benderdour M, Duval N, Pelletier JP, Martel‐Pelletier J, Fahmi H. Egr‐1 contributes to il‐1‐mediated down‐regulation of peroxisome proliferator‐activated receptor gamma expression in human osteoarthritic chondrocytes. Arthritis Res Ther. 2012;14:R69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Snyder R, Thekkumkara T. Interplay between EGR1 and SP1 is critical for 13‐cis retinoic acid‐mediated transcriptional repression of angiotensin type 1A receptor. J Mol Endocrinol. 2013;50:361–374. DOI: 10.1530/JME-12-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Su T, Liu P, Ti X, Wu S, Xue X, Wang Z, Dioum E, Zhang Q. ΗΙF1alpha, egr1 and sp1 co‐regulate the erythropoietin receptor expression under hypoxia: an essential role in the growth of non‐small cell lung cancer cells. Cell Commun Signal. 2019;17:152. DOI: 10.1186/s12964-019-0458-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jain N, Mahendran R, Philp R, Guy GR, Tan YH, Cao X. Casein kinase II associates with Egr‐1 and acts as a negative modulator of its DNA binding and transcription activities in NIH 3T3 cells. J Biol Chem. 1996;271:13530–13536. DOI: 10.1074/jbc.271.23.13530. [DOI] [PubMed] [Google Scholar]
- 58. Srivastava S, Weitzmann MN, Kimble RB, Rizzo M, Zahner M, Milbrandt J, Ross FP, Pacifici R. Estrogen blocks M‐CSF gene expression and osteoclast formation by regulating phosphorylation of Egr‐1 and its interaction with Sp‐1. J Clin Invest. 1998;102:1850–1859. DOI: 10.1172/JCI4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mourad‐Zeidan AA, Melnikova VO, Wang H, Raz A, Bar‐Eli M. Expression profiling of Galectin‐3‐depleted melanoma cells reveals its major role in melanoma cell plasticity and vasculogenic mimicry. Am J Pathol. 2008;173:1839–1852. DOI: 10.2353/ajpath.2008.080380. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S5