

Abstract
Endothelin-1 (ET-1) is elevated in patients with obesity; however, its contribution to the pathophysiology related to obesity is not fully understood. We hypothesized that high ET-1 levels cause dyslipidemia, inflammation, and insulin resistance within the adipose tissue of obese mice. To test this hypothesis, male C57BL/6J mice were fed either normal diet (NMD) or high-fat diet (HFD) for 8 weeks followed by 2 weeks of treatment with either vehicle, atrasentan (ETA receptor antagonist, 10 mg/kg/day) or bosentan (ETA/ETB receptor antagonist, 100 mg/kg/day). Atrasentan and bosentan lowered circulating non-esterified free fatty acids and triglycerides seen in HFD mice, while atrasentan-treated mice had significantly lower liver triglycerides compared with non-treated HFD mice. ET-1 receptor blockade significantly improved insulin tolerance compared with insulin-resistant HFD mice and lowered expression of genes in epididymal white adipose tissue (eWAT) associated with insulin resistance and inflammation. Flow cytometric analyses of eWAT indicated that HFD mice had significantly higher percentages of both CD4+ and CD8+ T cells compared with NMD mice, which was attenuated by treatment with atrasentan or bosentan. Atrasentan treatment also abolished the decrease in eosinophils seen in HFD mice. Taken together, these data indicate that ETA and ETA/ETB receptor blockade improves peripheral glucose homeostasis, dyslipidemia and liver triglycerides, and also attenuates the pro-inflammatory immune profile in eWAT of mice fed HFD. These data suggest a potential use for ETA and ETA/ETB receptor blockers in the treatment of obesity-associated dyslipidemia and insulin resistance.
Introduction
Obesity is a disease that affects over 40 percent of the population of the United States. It dramatically increases the risk of mortality due to cardiovascular disease [1]. Obesity is associated with alterations in lipid metabolism, as well as the development of insulin resistance and metabolic syndrome. One potential contributor to the pathophysiology of obesity is increased endothelin-1 (ET-1), both circulating and at the tissue level [2]. ET-1 is a vasoactive peptide that is produced mainly by vascular endothelial cells but is also produced by several other cell types including adipocytes and immune cells. Two ET-1 receptor subtypes exist in mammalian species, ETA and ETB. The ET-1 gene and its cognate receptors arose during the development of vertebrates and play a key role in development of the jaw and enteric nervous system in vertebrates. Post-developmentally, ET-1 plays a crucial role in physiology, including regulation of blood pressure and vascular tone. Pathophysiologically, ET-1 promotes inflammation in various diseases including hypertension, kidney disease, and diabetes [3]. Data from our laboratory indicate that patients undergoing bariatric surgery have a 20% reduction in circulating ET-1 six months following vertical T-1 levels observed in patients with obesity. Furthermore, patients with higher circulating ET-1 had higher levels of macrophage chemoattractant protein-1, a marker of inflammation, in visceral adipose [4]. Results from two clinical trials suggest that high levels of ET-1 promote dyslipidemia in patients with diabetic nephropathy [5,6]; however, the mechanisms are not yet understood. Therefore, ET-1 appears to play a major role in pathophysiology of obesity, and ET-1 receptors may be attractive targets to attenuate cardiovascular risk in patients with obesity.
Obesity causes a shift to a pro-inflammatory immune cell profile [2], especially observed in visceral adipose tissue [7–9]. In addition, visceral adipose tissue abundance is highly correlated to cardiovascular disease risk, whereas other depots, such as subcutaneous adipose, have little or no correlation [10]. In white adipose, obesity causes tissue to become hypoxic and immune cell populations become dysregulated, leading to an increase in macrophages, an increase in T lymphocytes and a decrease in eosinophils, among others [11]. This shift in immune cell population promotes dysfunction within the adipose tissue, contributing to pathophysiology related to obesity. This includes insulin resistance at the level of the adipocyte, as well as peripherally, due to alterations in circulating adipokines, including insulin-sensitizing adipokines such as adiponectin and adipsin. Given the importance of immune cells in modulating adipocyte function, more studies are needed to identify methods to target and improve inflammation within the adipose tissue.
A strong relationship between ET-1 and inflammation has been established in several models of disease, including chronic kidney disease and sickle cell disease [12]. ET-1 receptor antagonism has been shown to reduce pro-inflammatory cytokines such as tumor necrosis factor α (TNF-α). Given the relationship among ET-1, obesity and inflammation, we hypothesize that up-regulation of ET-1 within the epididymal white adipose tissue (eWAT) of obese mice promotes an inflammatory immune cell profile which contributes to dyslipidemia and insulin resistance. The goal of the current study is to determine if ET-1 receptor antagonism attenuates dyslipidemia, insulin resistance, and adipose tissue inflammation in the eWAT of mice chronically fed a Western diet.
Materials and methods
Animals
Male C57Bl/6J mice were purchased from Jackson Laboratories at 7 weeks of age and housed at the University of Mississippi Medical Center Animal Facility under controlled light conditions (12-h light/12-h dark). Male mice were used for the present study because in pilot experiments, female mice did not become insulin resistant after 8 weeks of high fat feeding (Supplementary Figure S2), which is consistent with other studies showing that female mice are protected from HFD-induced metabolic syndrome compared with males. Mice were habituated to the animal facility for 1 week upon arrival and fed ad libitum. At 8 weeks of age, mice were randomized and individually housed into four groups; normal diet-fed (NMD; 12.6% kcal fat, 30% kcal carbohydrate, Envigo, TD.05230) (n=7), high-fat diet-fed (HFD; 45% kcal fat, 42% kcal carbohydrate, Envigo TD.88137) (n=7), HFD atrasentan (HFD Atr) (n=5), and HFD bosentan (HFD Bos) (n=6) for 8 weeks. Mice were then treated for 2 weeks with either vehicle (0.1% ethanol), atrasentan (10 mg/kg/day) (PepTech), or bosentan (100 mg/kg/day) (Alomone) administered through the drinking water. Atrasentan is a selective ETA receptor antagonist, and bosentan is a non-selective antagonist of both ETA and ETB receptors. Doses were based on previous publications showing effectiveness [13,14]. Water intake was measured prior to experiments and during administration of treatment. Dosing was adjusted based on weekly water intake and growth. Food and water intake were measured everyday until the end of treatment. Treatment was continued for a third week while insulin and glucose tolerance experiments were performed, with mice continued on their respective diets. Mice were killed after a 6-h fast in clean cages to minimize coprophagia, and tissues were collected at the end of week 11. All assays were conducted on the same mice and on the same day. All protocols were approved by the Institutional Animal Care and Use Committee at the University of Mississippi Medical Center.
Cell culture
3T3-L1 preadipocytes (ATCC) were plated at a density of 40000/cm2 on 24-well plates and differentiation was induced at 2 days post-confluence by culturing cells in differentiation medium (33 μM biotin, 0.1 μM dexamethasone, 1 μM insulin, 200 μM indomethacin, 17 μM pantothenic acid, 10 μg/ml triiodothyronine, 1% FCS in DMEM/F12) for 3 days. Then, cells were cultured with DMEM/F12 for 6 days post-differentiation, after which they were treated in either normoxia (21% O2, 5% CO2) or hypoxia (1% O2, 5% CO2) for 6 h with or without Hif1a inhibitor IDF-11774 (company) at concentrations of 1, 25, and 50 μM. Cells were then lysed in Tri Reagent (Zymo R2050) and RNA was extracted using Direct-Zol RNA Microprep Kit (Zymo R2052).
Body composition analysis
Lean mass, fat mass, and total water composition was measured using Echo MRI (4-in-1 EchoMRI-900™, Echo Medical System, Houston, TX) at weeks 0, 4,8 and 10 while on diets.
Liver triglyceride content
Livers were weighed and then homogenized in 5% NP-40 (Sigma–Aldrich). Hepatic triglyceride content was determined using commercially available colorimetric triglyceride quantification kit (BioVision, K622–100). Absorbance was read at a wavelength of 570 nm (BioTek® Synergy H1) and triglyceride concentration was determined by standard curve and normalized to liver weight.
Insulin and glucose tolerance
Mice were fasted for 6 h prior to intraperitoneal insulin tolerance test (ITT) and oral glucose tolerance test (OGTT). Glucose was measured using a glucometer (Accu-Chek® Guide) via tail vein. For ITT, insulin (0.75 IU/kg of lean mass) was injected into the peritoneal cavity. For OGTT, 2 g/kg of dextrose was administered via oral gavage needle (22 gauge). Glucose was measured at baseline or time 0, 15, 30, 45, 60, and 120 min following insulin injection or glucose bolus. Tests were performed 3 days apart and similar to guidelines set forth by Benede-Ubieto et al. [15] Since only a small drop of blood was taken at each time point, 3 days rather than a week was allowed for recovery. Area under the curve was calculated using GraphPad Prism statistical software. Baseline was set at time = 0 for individual mouse.
Flow cytometric analyses of adipose tissue
Given the high correlation between visceral adipose tissue abundance, inflammation and cardiovascular disease risk in obesity [10], immune cell populations in eWAT were assessed. eWAT is the most abundant visceral adipose tissue depot in mice. eWAT was homogenized in GentleMACS Octo Dissociator (Miltenyi Biotec). Briefly, ∼1g of adipose tissue was minced into ∼5 mm pieces and placed in a GentleMACS C (Miltenyi Biotec) tube with 10 ml RPMI medium containing 200 U/ml DNase and 10 mg/ml collagenase IV. The sample was homogenized using the manufacturer’s program designed for adipose tissue. The homogenate was then filtered through a 70-μm filter into a 50-ml tube and allowed to settle for 10 min to allow the adipocytes to separate from the stromal vascular fraction (SVF). The lower SVF was removed and transferred to a 15-ml tube and centrifuged for 10 min at 400×g. The resulting cell pellet was resuspended in 3 ml of 1× PharmLyse (BD Biosciences) and incubated for 5 min at room temperature to lyse erythrocytes. Ten milliliters of 1× PBS, 2% FCS was added to the cells to wash followed by centrifugation at 350×g for5 min. Cells were then used for flow cytometry.
Briefly, cells were washed and resuspended in 1× PBS, 2% FCS, and 0.9% sodium azide at a concentration of 1 × 107 cells/ml. A total of 1 × 106 cells (100 μl) were aliquoted into a flow cytometry tube and incubated with 0.25 μg of anti-mouse CD32/CD16 (FcR block, BD Biosciences) for 5 min on ice. Cells were then stained with either isotype control antibodies or antibodies shown in Table 1 for 30 min on ice protected from light. All antibodies were diluted 1:200 in 1× PBS, 2% FCS, and 0.09% sodium azide. Samples were analyzed on an LSR II flow cytometer (BD Biosciences), and a total of 20000 CD45+ cells were acquired for each sample. Data were analyzed using FCS Express 7 Software (De Novo Software).
Table 1.
Monoclonal antibodies used for flow cytometry
| Antibody | Supplier | Clone |
|---|---|---|
| CD32 | BD Biosciences | 2.4G2 |
| CD45 | BD Biosciences | 30-F11 |
| CD3 | BD Biosciences | 145–2C11 |
| CD4 | BD Biosciences | GK1.5 |
| CD8a | BD Biosciences | 53–6.7 |
| CD45R | BD Biosciences | RA3–682 |
| NK1.1 | BD Biosciences | PK136 |
| CD11b | BD Biosciences | M1/70 |
| F4/80 | BioLegend | BM8 |
| Siglec-F | BD Biosciences | E50–2440 |
Digital droplet PCR gene expression
Mouse tissue was collected, snap-frozen in liquid nitrogen, and stored at −80° C. Tissue was homogenized in TRI reagent® (Zymo, R2050–1-200) in 2 ml lysing matrix D tubes (MP, 6913500). Total RNA was isolated from eWAT using Direct-zol™ RNA MiniPrep Kit (Zymo, R2052). One microgram of RNA was converted into cDNA with iScript Reverse Transcription kit (Bio-Rad Laboratories, Hercules, CA). Next, gene expression was carried out by droplet PCR. The PCR was set up by manufacturer’s recommendations using ddPCR probes Supermix (no dUTP) and 1 μl of TaqMan primer/probes (Applied Biosystems) and cDNA from 50 ng or RNA. To determine the effect of ET-1 receptor blockade on expression of several adipokines and PPARG (a major regulator of adiponectin expression), which are all altered in obesity, PCR was carried out using the following respective primer/probe sequences: Adiponectin (AdipoQ; Mm00456425_m1), Cfd/Adipsin (Mm01143935_g1), Leptin (Mm00434759_m1), Resistin (Mm00445641_m1), and Pparg (Mm00440940_m1). To characterize ET-1 and ET-1 receptor in adipose tissue of obese mice, the following primer/probes were used for PCR: Edn1 (Mm00438656_m1), EdnrA (Mm01243722_m1), EdnrB (Mm00432989_m1). In order to assess if adipose tissue is hypoxic in the current model of diet-induced obesity, Hif1α (Mm00468869 m1) was measured. To determine the effect of ET-1 receptor blockade on cytokine expression, the following primers were used: Il12b (Mm01208835 m1), Il-6 (Mm00446190_m1), Tnf (Mm00443258_m1), Il-10 (Mm01288386_m1), Csf1 (Mm00432686_m1) from 50 ng of total RNA. The reaction mix was separated into nanodroplets using the automated droplet generator (Bio-Rad). PCR was carried out for 40 cycles per manufacturer’s instructions. Droplets were counted using the QX200 Droplet Reader and data were analyzed and copy count calculated using QuantaSoft software.
Biochemical analysis
Blood was collected under anesthesia via cardiac puncture using a 22g × 1 needle, placed in 1.5-ml tubes coated with EDTA (0.5 M), and immediately placed on ice. Blood was spun at 400×g for 15 min at 4° C to separate plasma. Blood chemistry (ALT, CHOL, HDL, LDL, NEFA, TRIG) was analyzed via Vet Axcel® chemistry analyzer (Alfa Wasserman). Plasma insulin and adiponectin concentrations were measured by mouse enzyme-linked immunoassay (ELISA) (Crystal Chem, 90080, 80569) according to the manufacturer’s protocols.
Randomization and statistics
Although it is not possible to blind researchers from diet of mice, samples were blinded for all assays. All data are expressed as mean ± SEM. Data were tested for statistical significance by one-way ANOVA for one variable datasets or two-way repeated measure ANOVA (ITT and OGTT). Tukey’s post-hoc test used to compare groups. P<0.05 was considered statistically significant. All graphs and statistical analyses were performed using GraphPad Prism.
Results
ET-1 receptor blockade does not significantly affect body weight or fat mass of HFD-fed mice
C57BL/6J mice were fed NMD or HFD for 10 weeks. As expected, male mice fed NMD had significantly lower body weight, fat mass, and lean mass compared with HFD-fed mice. Female mice on HFD gained less body weight and fat mass over the course of the experiment compared with male mice (Supplementary Figure S2A,B). Further, there were no significant differences in fasting blood glucose, glucose tolerance, or insulin tolerance (Supplementary Figure S2D–F) between NMD- and HFD-fed females; therefore, we proceeded with only male mice. There were no differences in body weight, lean mass, or fat mass between HFD, HFD+Atr, and HFD+Bos mice throughout the experimental protocol (Figure 1A–C). In addition, there were no significant differences in total body water during treatment between any group (Figure 1D). Finally, there were no detectable differences in caloric or water intake among all four treatment groups throughout the duration of the treatment, indicating that the differences in any endpoints were independent of caloric intake and hydration status (Figure 1E,F).
Figure 1. ET-1 receptor blockade does not significantly affect body weight or fat mass of HFD-fed mice.
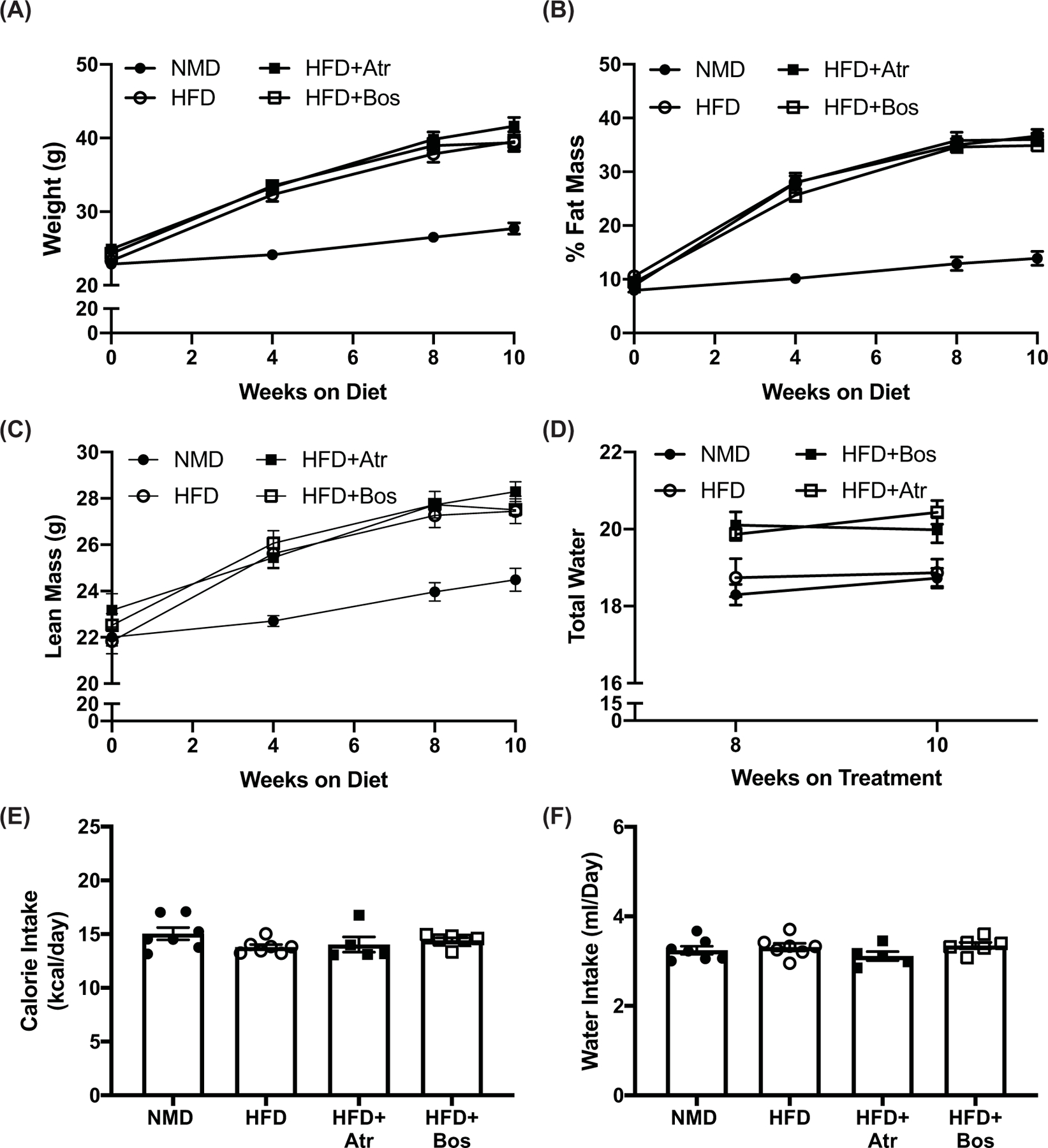
Weight (A), percent fat mass (B), and lean mass (C) of NMD (n=7), HFD (n=7), HFD+Atr (n=5), and HFD+Bos (n=6) mice fed NMD or HFD, measured on weeks 0, 4, 8, and 10. Total water was measured through weeks 8 and 10 (D). Food (kcal/day) (E) and water intake (ml/day) (F) was measured daily. (A–D) were analyzed by two-way ANOVA, and panels (E,F) were analyzed by one-way ANOVA with post-hoc Tukey’s test between individual groups. Data are expressed as mean ± SEM.
ET-1 receptor blockade improves dyslipidemia in HFD-fed mice
Fasting plasma NEFA and triglyceride concentrations were increased in HFD-fed mice compared with NMD-fed mice (P=0.001 and P<0.0001, respectively). The increase in triglycerides and NEFA was attenuated in HFD-fed mice treated with atrasentan (P=0.005 and P=0.0003, respectively) or bosentan (P=0.06 and P=0.001, respectively; Figure 2A,B). Similar to the circulating lipid profile, hepatic triglyceride content was significantly increased in HFD-fed mice compared with NMD (P<0.001). Atrasentan-treated mice had lower hepatic triglyceride content compared with HFD-fed mice treated with vehicle (P=0.009, Figure 2C), while bosentan treatment had no significance difference in hepatic triglycerides (P=0.09). Total cholesterol was increased in HFD-fed mice compared with NMD-fed mice (P<0.0001), with no significant effect of treatment with either atrasentan or bosentan (Figure 2D). There were no differences in HDL levels between NMD-fed mice and HFD-fed groups. There was, however, a significant increase in HDL in HFD+Bos (P=0.03) treated mice compared with HFD vehicle, but no statistical difference between HFD and HFD+Atr (P=0.08; Figure 2E). LDL levels were higher all in all HFD-fed mice groups compared with NMD-fed mice (Figure 2F).
Figure 2. ET-1 receptor blockade improves dyslipidemia in HFD-fed mice.

Plasma analysis of Cholesterol (A), Triglycerides (B), HDL (C), LDL (D), and NEFA (E) in NMD (n=7), HFD (n=7), HFD+Atr (n=5), and HFD+Bos (n=6) mice after 10 weeks on diets and 2 weeks of treatment with atrasentan or bosentan. Liver triglycerides were measured among all groups and normalized to liver weight (F). Data were analyzed by one-way ANOVA with Tukey’s post-hoc test for individual groups. (E,F) were analyzed by two-way ANOVA. Data are expressed as mean ± SEM. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
ET-1 receptor blockade reduces blood glucose and improves insulin tolerance in HFD-fed mice
We next determined whether ET-1 receptor blockade improved glucose and insulin tolerance in HFD-fed mice. After a 6-h fast, HFD-fed mice exhibited significant hyperglycemia compared with NMD-fed mice (P<0.0001), an effect that was attenuated in mice treated with atrasentan or bosentan (P=0.07 and P=0.02, respectively; Figure 3A). In addition, fasting plasma insulin concentration was increased in HFD fed mice compared with NMD (P=0.004), and there was no significant effect of treatment with atrasentan or bosentan (P=0.33 and P=0.81, respectively; Figure 3B). As expected, HFD-fed mice had significantly impaired glucose tolerance compared with NMD-fed mice evidenced by a significant increase in AUC (P=0.004; Figure 3C,D). Treatment with atrasentan improved glucose tolerance with a significant reduction in AUC (P=0.04; Figure 3C,D). Similarly, insulin tolerance was significantly impaired in HFD-fed mice compared with NMD (P=0.001; Figure 3E,F). Treatment with atrasentan or bosentan improved insulin tolerance compared to HFD mice indicated by a significant increase in AUC (P<0.0001 and P=0.01, respectively; Figure 3E,F).
Figure 3. ET-1 receptor blockade reduces blood glucose and improves insulin tolerance in HFD-fed mice.
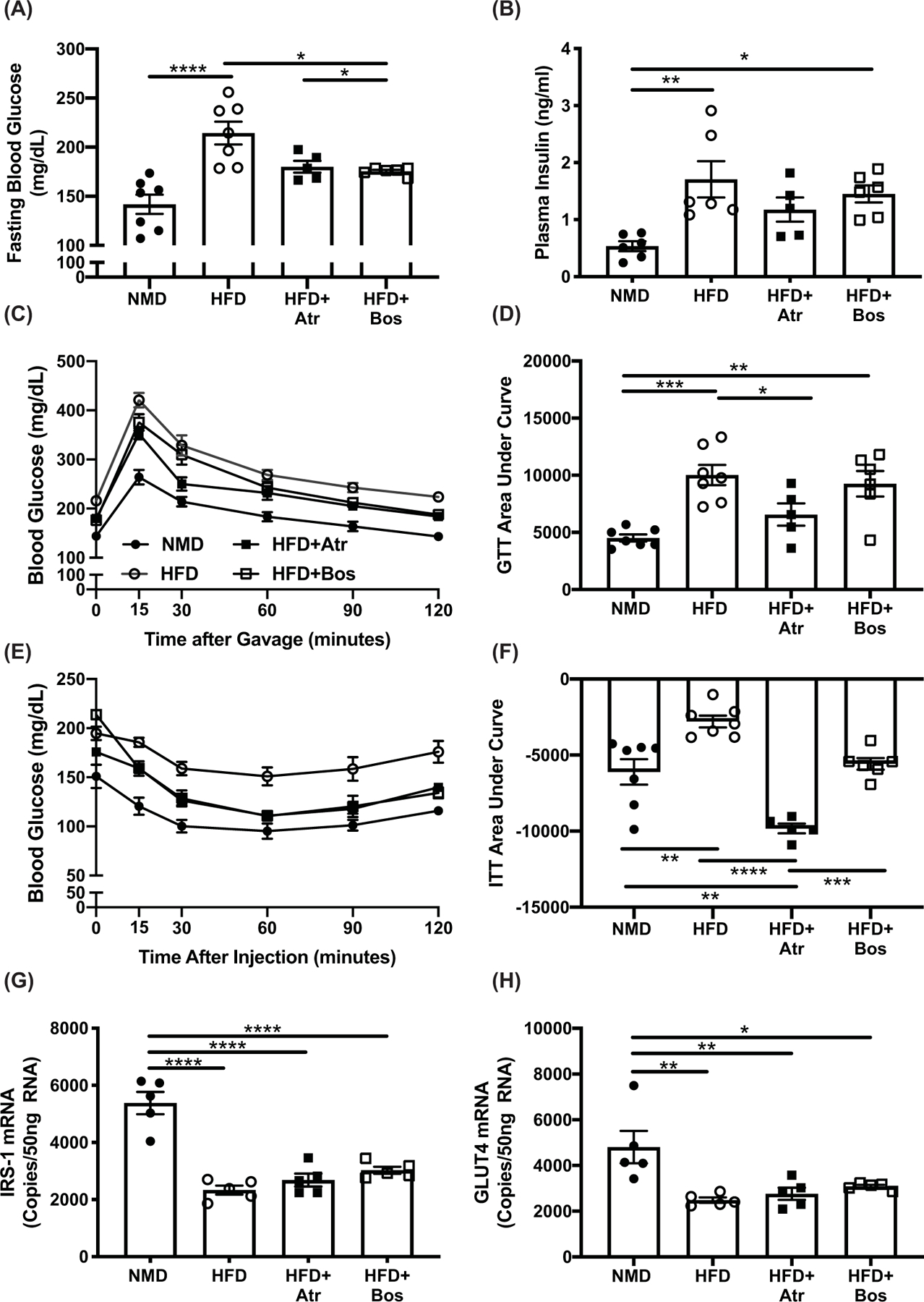
In vivo glucose homeostasis was determined by OGTT (A) and IPITT (C) for of NMD (n=7), HFD (n=7), HFD+Atr (n=5), and HFD+Bos (n=6) mice after 10 weeks on diet and 2 weeks of treatment with atrasentan or bosentan. Area under the curve for plasma insulin (B) and GTT (D). Fasting blood glucose was measured after a 6-h fast (E). Plasma insulin was measured via ELISA (n=5 per group). Data in (A,B,D,F–H) were analyzed by one-way ANOVA with Tukey’s post-hoc test for individual groups. Statistical analysis for panels (A,B,D,F–H) were done by one-way ANOVA with post-hoc Tukey’s test between individual groups, and panels (C,E) were analyzed by two-way ANOVA with repeated measures. Data are expressed as mean ± SEM. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
ET-1 is up-regulated in adipose tissue via Hif1α
ET-1 expression has been shown to increase in hypoxic environments, which occurs in the adipose tissue of patients with obesity and rodent models of obesity. In the current study, Hif1α mRNA, a marker of hypoxia [16], was significantly increased in eWAT of HFD-fed mice compared with NMD-fed mice (P<0.0001). Interestingly, the increase in Hif1α mRNA was attenuated by 49% in mice treated with atrasentan (P=0.002; Figure 4A). In addition, ET-1 mRNA in eWAT was increased from 711 to 1086 copy counts/50 ng RNA in response to diet-induced obesity (P=0.04; Figure 4B). Atrasentan exacerbated the increase in ET-1 expression, although there was no detectable difference between vehicle and bosentan-treated HFD-fed mice (P=0.95; Figure 4B). Surprisingly, protein content had a negative correlation with mRNA expression, in that HFD-fed mice had significantly lower eWAT ET-1 protein content compared with NMD mice, and atrasentan treatment reduced ET-1 content even more. This is likely due to increased ET-1 binding to ETB receptors. ETA receptor expression was significantly reduced in HFD-fed and atrasentan-treated mice compared with NMD mice, while bosentan-treated mice had no detectable difference in ETA mRNA expression compared with NMD mice (Figure 4D). There were no significant differences in ETB mRNA expression among all groups (Figure 4E), although eWAT had higher gene expression levels of ETB mRNA compared with ETA mRNA in all groups.
Figure 4. ET-1 is up-regulated in adipose tissue via Hif1α.
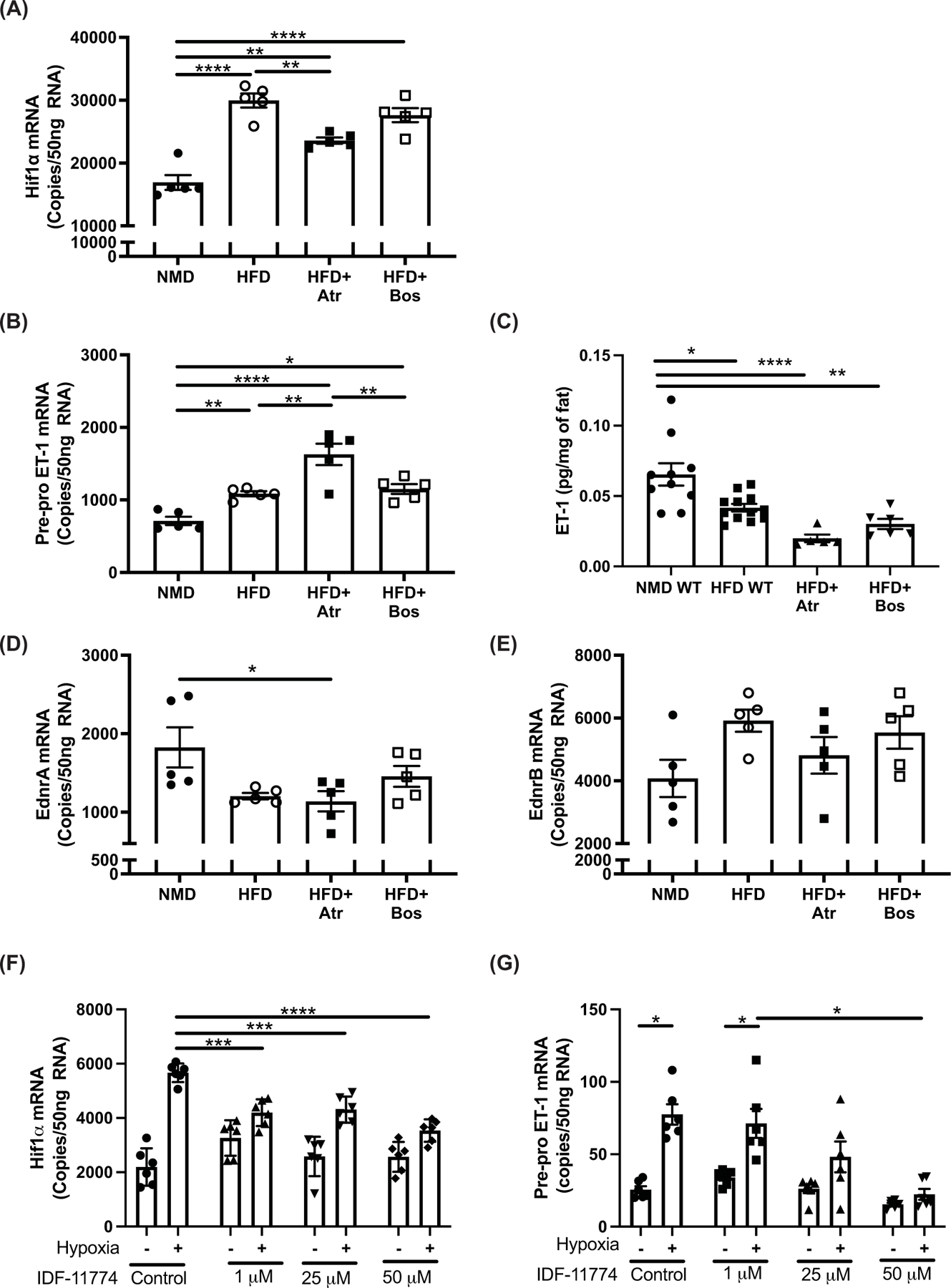
(A) Hif1α mRNA, (B) Pre-pro ET-1 mRNA, (C) ET-1 protein, (D) EdnrA mRNA, and (E) EdnrB mRNA from eWAT of mice after 10 weeks on diet and 2 weeks of treatment with atrasentan or bosentan were determined via ddPCR (n=5 per group). (F) Hif1α and (G) Pre-pro ET-1 gene expression from 3T3-L1 adipocytes exposed to 6 h of normoxia or hypoxia in the presence of Hif1α inhibitor IDF-117744 (1, 25, 50 μM) (n=6). Data from (A–E) were analyzed by one-way ANOVA with Tukey’s post-hoc test for individual groups. (F,G) were analyzed by two-way ANOVA. Data are expressed as mean ± SEM. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
To determine if ET-1 is elevated in response to hypoxia in adipocytes, 3T3-L1 fibroblast cells were differentiated into adipocytes and exposed to hypoxia. Hypoxia induced a three-fold increase in Hif1α mRNA (Figure 4F) and this was associated with four-fold increase in ET-1 mRNA. The increase in ET-1 was attenuated in a dose-dependent manner when cells were pretreated with the Hif1α inhibitor IDF-11774 (Figure 4G), suggesting that Hif1α drives the increase in ET-1 expression in the adipose of obese mice.
ET-1 receptor blockade improves hypoadiponectinemia in HFD-fed mice
Circulating plasma adiponectin was significantly decreased in HFD compared with NMD mice (P<0.0001), whereas treatment with atrasentan or bosentan attenuated the decrease in plasma adiponectin (P=0.02 for both treatments; Figure 5A). Gene expression analysis of eWAT indicates a decrease in Pparg, a transcription factor that promotes adiponectin production, and AdipoQ mRNA. Further, there was a reduction in Adipsin/Cfd and Retn, adipokines that are associated with improved insulin sensitivity and/or release, and an increase in Lep expression in HFD mice compared with NMD mice. Even though there was an increase in circulating adiponectin in atrasentan or bosentan-treated mice compared with HFD mice, there was no significant differences in eWAT gene expression of any adipokines (Figure 5B–F).
Figure 5. ET-1 receptor blockade improves hypoadiponectinemia in mice fed a HFD.

Plasma adiponectin was analyzed via ELISA after 10 weeks on diet and 2 weeks of treatment with atrasentan or bosentan (A). Gene expression of AdipoQ, Pparγ, Adipsin/Cfd, Retn, and Leptin from eWAT of mice after 10 weeks on diet and 2 weeks of treatment with atrasentan or bosentan were determined via ddPCR (B–F) (n=5 per group). Data were analyzed by one-way ANOVA with Tukey’s post-hoc test for individual groups. Data are expressed as mean ± SEM. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
ET-1 receptor blockade attenuates increase in pro-inflammatory cells in eWAT in HFD-fed mice
To determine the effect of ET-1 receptor blockade on adipose tissue inflammation, flow cytometry was performed on the stromal fraction following eWAT dissociation. Representative gating strategies are available in Supplementary Figure S1. HFD-fed mice had a significant increase in both CD4+ T cells (P<0.0001) and CD8+ T cells (P=0.001) in eWAT compared with NMD-fed mice. Bosentan treatment significantly attenuated the increase in the CD4+ T cell population percentage (P=0.002; Figure 6A), while atrasentan and bosentan treatment attenuated the increase in the CD8+ T cell population percentage (P=0.01 and P=0.02, respectively; Figure 6B). In addition, the percentage of eosinophils was significantly reduced in obese mice (P=0.003). The reduction in eosinophils was attenuated in mice administered atrasentan and bosentan (P=0.004 and P=0.14; Figure 6C). There were no differences in the percentages of NK1.1+ NK cells between NMD and HFD mice, but both atrasentan- and bosentan-treated mice had significantly lower levels of NK cells (P=0.006 and P=0.008, respectively vs. HFD; Figure 6D). There were no differences in the percentages of CD45R+ B cells or CD11b+F4/80+ macrophages among the groups (Figure 6E,F). HFD mice had increased gene expression levels of Tnfα (P=0.0006), IL-12b (P=0.001), Il-10 (P=0.001), Il-6 (P=0.04), and Il-1r (P=0.003) in eWAT compared with NMD mice. Treatment with atrasentan or bosentan attenuated the increase in expression of Tnfα (P=0.05 and P=0.0001, respectively) and IL-12b (P=0.04 for both treatments; Figure 6G,H), while the increased expression for Il-10 was only significantly attenuated by atrasentan treatment (Figure 6I). There were no significant differences in gene expression for Il-6 and Il-1r between treated and HFD-fed mice (Supplementary Figure S2).
Figure 6. ET-1 receptor blockade attenuates increase in pro-inflammatory cells in eWAT in HFD-fed mice.
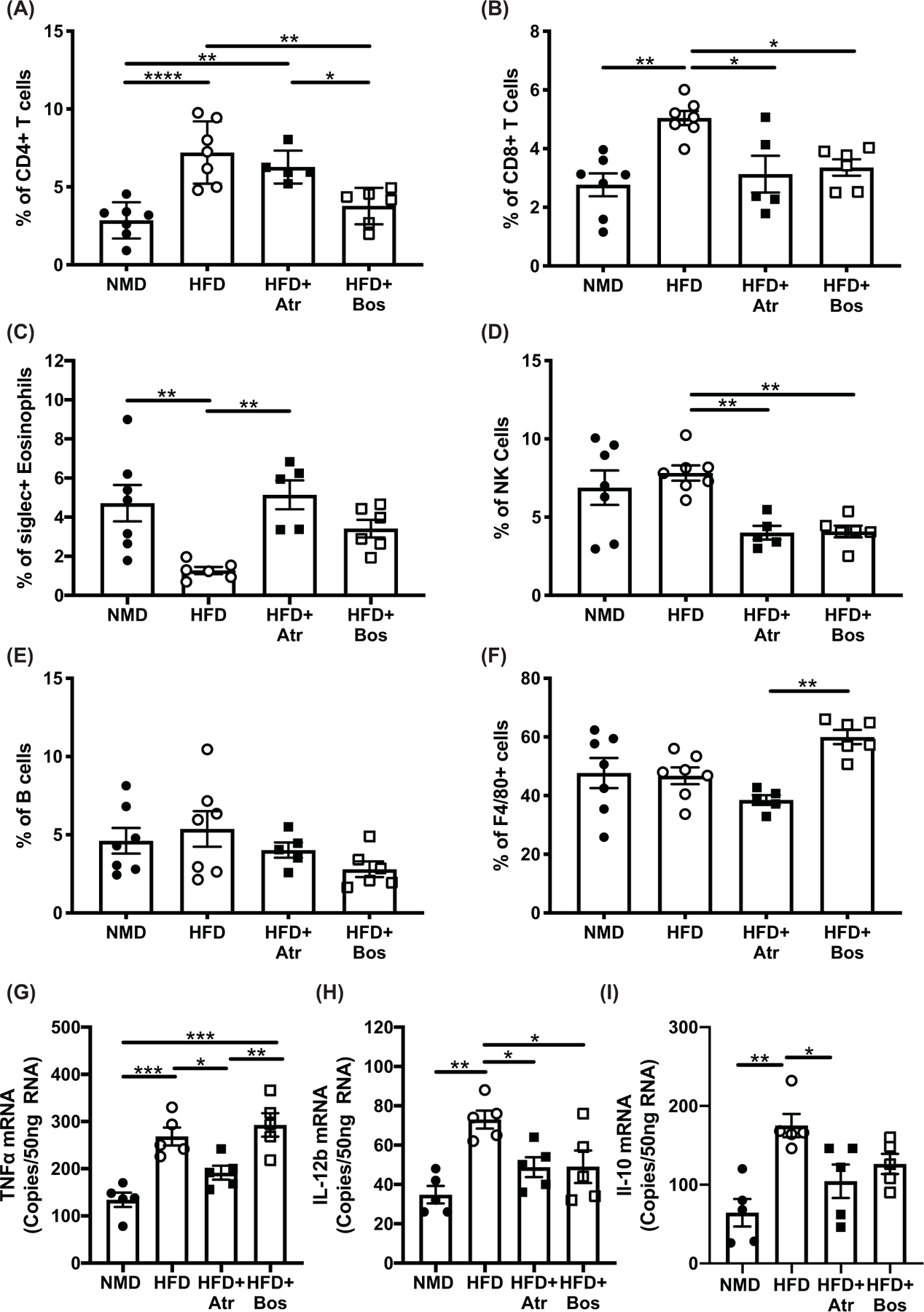
Percent of total stromal fraction cells of CD4+, CD8+, eosinophils, NK, B, and F480+ (A–F) (n=5 per group) obtained by flow cytometry from eWAT of mice after 10 weeks on diet and 2 weeks of treatment with atrasentan or bosentan. Gene expression of Tnfα (G), Il-12b (H), and Il-10 (I) from eWAT of mice after 10 weeks on diet and 2 weeks of treatment with atrasentan or bosentan were determined via ddPCR (n=5 per group). Data were analyzed by one-way ANOVA with Tukey’s post-hoc test for individual groups. Data are expressed as mean ± SEM. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Discussion
Data from animal experiments and clinical trials suggest that ET-1 promotes several pathophysiological conditions related to obesity including dyslipidemia, insulin resistance, and inflammation [17,18]. In this study, male mice fed a HFD were treated with the ETA receptor blocker atrasentan or the non-selective ETA/ETB blocker bosentan for 2 weeks. The major findings of the present study are that pharmacological blockade of either ETA or both the ETA and ETB receptor improved glucose handling, insulin sensitivity, dyslipidemia, adipokine levels and eWAT inflammation in HFD-fed mice, independent of changes in body weight or body composition. Overall, these results support the hypothesis that high ET-1, whether circulating or within the adipose tissue, promotes and exacerbates insulin resistance, dyslipidemia, and eWAT inflammation in obesity.
Overweight and obese states are associated with higher circulating levels of ET-1 and increased ET-1 activity via ETA receptor activation as evidenced by impairments in flow-mediated dilation in a cohort of mixed male and female subjects [19,20]. Sex differences in ET-1 production in obesity has not been extensively studied, although 17β-estradiol administration reduces circulating ET-1 in post-menopausal women, suggestive of possible sex differences in obesity associated increases in ET-1 [21]. One potential source for elevated ET-1 in obesity is the adipose tissue which becomes hypoxic as adipose tissue expands, which could result in the up-regulation of ET-1 [22,23]. Our group recently reported that circulating ET-1 concentration is reduced following drastic weight loss in patients who underwent vertical sleeve gastrectomy, suggesting the adipose tissue as the source of elevated ET-1 in obesity [24]. In support of this concept, HFD-fed mice in the present study had a significant increase in Hif1-α mRNA in eWAT as compared with NMD-fed mice, and this was associated with a significant increase in ET-1 production. Further, hypoxia induced a four-fold increase in ET-1 mRNA of 3T3-L1 adipocytes, and this was attenuated with pretreatment of a Hif1-α inhibitor. These data suggest that the most likely source of the increase in ET-1 in patients with obesity is the adipose tissue due to the relatively hypoxic environment. Although the adipocyte is likely partly responsible, identifying the relative contribution of other cell types, such as endothelial cells, will require experiments using tissue-specific knockout animals.
A major finding of the current study is that ETA or dual ETA/ETB receptor blockade significantly attenuated the increase in triglycerides and free fatty acids in plasma of HFD-fed mice. Two recent clinical trials aimed at determining if treatment with an ETA receptor antagonist improves or delays the onset of diabetic nephropathy also showed efficacy in reducing circulating lipids [17], although this was not specifically done in subjects with obesity. Results from the Reducing Residual Albuminuria in Subjects with Diabetes and Nephropathy (RADAR) trial indicate that atrasentan significantly reduces circulating triglycerides and LDL cholesterol [25]. This was followed up with similar results in a study published by Farrah et al. showing sitaxentan, an ETA receptor antagonist, significantly lowered circulating triglycerides and LDL cholesterol [26]. Taken together, the data suggest an important role for ET-1 in promoting dyslipidemia in obesity and suggest that treatment with an ETA receptor antagonist may be beneficial to reduce cardiovascular risk in patients with obesity.
Hepatic steatosis and liver dysfunction are commonly associated with obesity [27,28]. ETA receptor blockade significantly reduced HFD-induced hepatic triglyceride levels, whereas ETA/ETB receptor blockade modestly reduced hepatic triglycerides by an average of 33%. It has been previously shown that ETA receptor blockade with ambrisentan has antifibrotic effects in the liver [29], and ETA/ETB receptor blockade with bosentan (50 mg/kg) has hepatoprotective effects in diabetic rats [30]; however, to our knowledge, we are the first to show that endothelin receptor blockade is protective against hepatic triglyceride accumulation in a model of diet-induced obesity. Though not fully understood, the beneficial effects of ET receptor blockade in HFD-induced dyslipidemia may be due to increased hepatic blood flow, which would allow more efficient transport of insulin and insulin-sensitizing adipokines, such as adiponectin/adipsin, to the liver [31–34]. This would in turn act to increase fatty acid oxidation and utilization of lipids, thereby decreasing hepatic lipid accumulation.
Numerous studies suggest a direct involvement of ET-1 signaling in the pathogenesis of hyperglycemia and insulin resistance [18,35,36]; however, the contribution of ET-1 in promoting insulin resistance in obesity has yet to be tested. Our data indicate that pharmacological blockade with either an ETA specific or dual ETA/ETB receptor antagonist improves insulin-mediated glucose uptake and fasting blood glucose in rodents. Several potential receptor- and tissue-dependent mechanisms exist by which ET-1 may promote insulin resistance. One of the most likely mechanisms is through actions at the level of the adipose tissue. First, ET-1 mediates vascular function by acting as a potent vasoconstrictor via ETA receptor activation [3]. This contributes to hypoxia observed in the adipose tissue, because treatment with either atrasentan or bosentan partially attenuated the increase in Hif1-α expression in eWAT of HFD-fed mice. A second potential mechanism by which ET-1 may promote insulin resistance is through activation of the ETB receptor. van Harmelen et al. demonstrated that chronic activation of the ETB receptor decreases insulin’s ability to inhibit lipolysis in human primary adipocytes, in addition to demonstrating a significantly higher ETB to ETA receptor ratio in human primary adipocytes [37]. Gene expression data from this study also support a higher ETB to ETA receptor ratio in the eWAT of mice, which suggests that ET-1 signaling in eWAT is primarily through the ETB receptor, although protein expression analysis would be needed to confirm. Further, our lab recently showed that rats lacking functional ETB receptors have reduced fasting blood glucose and improved glucose and insulin tolerance [18]. Taken together, the current data are inconclusive on whether there is an improvement in insulin sensitivity at the level of the adipocyte following ET-1 receptor blockade, and more studies are warranted to definitively answer this question. A third potential mechanism is through a reduction in circulating FFAs which are thought to cause insulin resistance in all insulin-sensitive tissues, although there is still debate about whether levels seen in obesity cause insulin resistance [38]. Blockade of the ETA receptor decreased circulating FFAs, possibly through a decrease in basal adipocyte lipolysis [39,40]. In summary, activation of ET-1 receptors most likely causes insulin resistance through multiple mechanisms at several different insulin-sensitive sites.
Individuals with metabolically abnormal obesity exhibit deleterious changes in circulating adipokines including increased circulating leptin and reduced adiponectin and adipsin [41], all of which are emulated in rodent models of diet-induced obesity, including the current study. Adiponectin is an insulin-sensitizing adipokine whose circulating levels inversely correlate with obesity and insulin resistance [42–44]. In fact, hypoadiponectinemia is thought to be related to increased ET-1 production, although this has yet to be formally demonstrated [45]. Here we showed that treatment with both atrasentan and bosentan attenuated the hypoadiponectinemia induced by high-fat feeding and improved adiponectin gene expression in eWAT, with ETA/ETB receptor blockade showing the greatest improvements in gene expression (21% increase vs HFD). Studies in humans and rodents show that increasing either circulating or adipocyte specific levels of adiponectin are sufficient to ameliorate obesity-induced insulin resistance [46,47], suggesting another contributing factor by which ET-1 receptor blockade improves in circulating lipids and insulin sensitivity in the current model of diet-induced obesity. In addition, Adipsin gene expression in eWAT was increased with both ETA and ETA/ETB receptor blockade. Adipsin is an adipokine, also known as compliment factor D, that is highly expressed in white adipose tissue. Circulating adipsin levels in humans correlate negatively with insulin resistance, and in mice, adipsin improves β-cell function in HFD-induced insulin resistance [48,49]. These data suggest that high circulating or adipose tissue levels of ET-1 during obesity could impair adiponectin and adipsin production by adipocytes thereby exacerbating peripheral insulin resistance in obesity.
Elevated visceral adipose in humans is a major contributor to cardiovascular risk in patients with obesity [10]. As adipose expands, the total number of immune cells increases with the tissue, and the cells adopt a more pro-inflammatory phenotype. The immune cell profile within adipose tissue plays an integral role in regulating lipid metabolism and storage at the level of the adipocyte [50]. Briefly, lean adipose primarily is composed of M2-like anti-inflammatory macrophages, eosinophils, regulatory T cells, and iNKT cells, among others. In an obese state, however, the cells are markedly different, and are primarily M1-like macrophages, CD4+ Th1 and Th17 cells, and CD8+cytotoxic T lymphocytes (CTLs) [51,52]. In the present study, we measured relative percentages of immune cell populations in the eWAT, an abundant visceral adipose depot in mice. We observed increase percentages of both CD4+ and CD8+ T cells in the eWAT in mice administered HFD, which has been previously shown by other laboratories [45]. CD4+ and CD8+ T cell percentages were significantly lower in mice that received bosentan, while atrasentan caused a significant decrease in CD8+ T cell percentages. CD4+Th1 cells secrete IFN-γ, which can disrupt insulin signaling in the adipose and could lead to or exacerbate insulin resistance systematically [53]. TNF-α, which has been widely studied in obesity [54,55], is also secreted by Th1 cells, among other cell types. Expression of TNF-α was elevated in eWAT of mice fed HFD, but was decreased in mice that received atrasentan. Congruous with our results, previous studies have shown that blockade of the ETA receptor lowered TNF-α concentrations in humans and rats [56,57], although the exact mechanism is yet to be determined. Further studies are needed to characterize the Th subset dynamics in adipose tissue in response to endothelin antagonism. CD8+ CTLs infiltrate the adipose in obesity and accumulate, along with macrophages, in crown-like structures around dying adipocytes. These cells are important in the recruitment of macrophages, and their depletion decreases pro-inflammatory cytokine production and improves insulin sensitivity [58]. We also detected increased interleukin (IL) 12 (IL-12) and IL-10 mRNA expression in eWAT of HFD-fed mice. IL-12 is a pro-inflammatory cytokine, which is important for the polarization of CTL to an activated Tc1 phenotype [59]. IL-10 is principally known as an anti-inflammatory cytokine that decreases the activity and/or expression of various pro-inflammatory cytokines and chemokines [60]. IL-10 is up-regulated in white adipose tissue during obesity, where it may havea deleterious role since knockdown of IL-10 in mouse adipose tissue protects from obesity and promotes thermogenesis [61].
A seeming contradiction observed in the current study is the apparent negative correlation between ET-1 mRNA and ET-1 peptide content in eWAT. Obesity increased ET-1 mRNA in eWAT, and ETA blockade increased ET-1 mRNA further, while protein content was proportionally lower. One potential reason is the increase in ET-1 receptor expression, especially that of the ETB receptor. ETB receptor content in the lungs is high compared with most other tissues and is responsible for clearing ET-1 from the circulation by internalizing bound ET-1. This inverse relationship between ET-1 mRNA and protein levels has been observed previously in obese mice. In fact, obese mice fed a HFD had significantly lower peptide levels in the lung, even in mice that overexpress ET-1 in vascular endothelial cells [62]. This is thought to be from up-regulation of ETB receptors in the lungs leading to increased clearance. We speculate the same phenomenon in the current study.
Eosinophils are necessary for proper adipocyte maturation and contribute to insulin sensitivity and glucose tolerance through the release of anti-inflammatory cytokines, such as IL-4, that promote macrophage polarization toward the M2 phenotype [63,64]. It has been previously shown that eosinophil cell numbers significantly decrease in adipose tissue in response to HFD feeding [65]. Endothelin antagonism reversed the decrease in eosinophils in HFD-fed mice. Endothelin receptors are expressed on immune cells [66], including T cells and polymorphonuclear cells such as eosinophils [67], so it is unknown whether the alterations in immune cells in the adipose are an indirect result of changes in adipokine expression and glucose handling or due to direct effects of the antagonist on immune cells.
Conclusions
The current findings suggest an important role for ET-1 in promoting cardiometabolic risk in patients suffering from obesity. Currently, three ET-1 receptor antagonists have been approved for patients with pulmonary hypertension; however, there have been no clinical trials to determine their efficacy in reducing cardiovascular risk in obesity. It should be noted that these studies were carried out on male mice because of their earlier susceptibility to HFD-induced IR and dyslipidemia compared with females, and that further studies on the effectiveness of ET-1 receptor antagonists in ameliorating IR and dyslipidemia in obese female mice should be evaluated. Given their efficacy in reducing blood pressure, improving dyslipidemia and insulin sensitivity, and inflammation, ET-1 antagonists could prove beneficial in reducing obesity-related cardiovascular disease.
Supplementary Material
Clinical perspectives.
ET-1 is up-regulated in patients with and animal models of obesity; however, its contribution to the pathophysiology related to obesity is unknown.
Blockade of ET-1 receptors improved insulin resistance, dyslipidemia, and adipose tissue inflammation in a mouse model of diet-induced obesity.
ET-1 antagonists, which are currently FDA approved for the use in patients with pulmonary hypertension, could prove beneficial in reducing obesity-related cardiovascular disease.
Acknowledgments
Funding
This work was supported by the National Institutes of Health [grant numbers R01 DK124327, R00 HL127178 (to Joshua S. Speed), K99HL146888 (to Erin B. Taylor), F31DK125035 (to Osvaldo Rivera-Gonzalez)]; and the UMMC Department of Physiology and Biophysics [grant number P20 GM104357].
Abbreviations
- CTL
cytotoxic T lymphocyte
- ET-1
endothelin-1
- eWAT
epididymal white adipose tissue
- HFD
high-fat diet
- IL
interleukin
- ITT
insulin tolerance test
- NMD
normal diet
- OGTT
oral glucose tolerance test
- SVF
stromal vascular fraction
- TNF-α
tumor necrosis factor α
Footnotes
Data Availability
All data will be made available upon request.
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
CRediT Author Contribution
Osvaldo Rivera-Gonzalez: Conceptualization, Data curation, Formal analysis, Funding acquisition, Validation, Investigation, Visualization, Methodology, Writing—original draft, Project administration. Natalie A. Wilson: Data curation, Formal analysis, Investigation, Writing—review and editing. Laura E. Coats: Data curation, Investigation, Writing—review and editing. Erin B. Taylor: Conceptualization, Resources, Data curation, Software, Formal analysis, Supervision, Funding acquisition, Validation, Investigation, Visualization, Methodology, Writing—review and editing. Joshua S. Speed: Conceptualization, Resources, Data curation, Software, Formal analysis, Supervision, Funding acquisition, Validation, Investigation, Visualization, Methodology, Writing—original draft, Project administration, Writing—review and editing.
References
- 1.Reaven GM (2008) Insulin resistance: the link between obesity and cardiovascular disease. Endocrinol. Metab. Clin. N. Am 37, 581–601, vii-viii [DOI] [PubMed] [Google Scholar]
- 2.Quante M, Dietrich A, ElKhal A and Tullius SG (2015) Obesity-related immune responses and their impact on surgical outcomes. Int. J. Obes. (Lond.) 39, 877–883 [DOI] [PubMed] [Google Scholar]
- 3.Davenport AP, Hyndman KA, Dhaun N, Southan C, Kohan DE et al. (2016) Endothelin. Pharmacol. Rev 68, 357–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jenkins HN, Williams LJ, Dungey A, Vick KD, Grayson BE et al. (2019) Elevated plasma endothelin-1 is associated with reduced weight loss post vertical sleeve gastrectomy. Surg. Obes. Relat. Dis 15, 1044–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lam H-C, Chu C-H, Wei M-C, Keng H-M, Lu C-C et al. (2006) The effects of different doses of atorvastatin on plasma endothelin-1 levels in type 2 diabetic patients with dyslipidemia. Exp. Biol. Med 231, 1010–1015 [PubMed] [Google Scholar]
- 6.Seligman B, Biolo A, Polanczyk CA, Gross JL and Clausell N (2000) Increased plasma levels of endothelin 1 and von willebrand factor in patients with type 2 diabetes and dyslipidemia. Diabetes Care 23, 1395–1400 [DOI] [PubMed] [Google Scholar]
- 7.Lolmède K, Duffaut C, Zakaroff-Girard A and Bouloumié A (2011) Immune cells in adipose tissue: Key players in metabolic disorders. Diabetes Metab 37, 283–290 [DOI] [PubMed] [Google Scholar]
- 8.Duffaut C, Zakaroff-Girard A, Bourlier V, Decaunes P, Maumus M et al. (2009) Interplay between human adipocytes and t lymphocytes in obesity: Ccl20 as an adipochemokine and t lymphocytes as lipogenic modulators. Arterioscler. Thromb. Vasc. Biol 29, 1608–1614 [DOI] [PubMed] [Google Scholar]
- 9.Lee B-C, Kim M-S, Pae M, Yamamoto Y, Eberle D et al. (2016) Adipose natural killer cells regulate adipose tissue macrophages to promote insulin resistance in obesity. Cell Metab 23, 685–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berg AH and Scherer PE (2005) Adipose tissue, inflammation, and cardiovascular disease. Circ. Res 96, 939–949 [DOI] [PubMed] [Google Scholar]
- 11.Wu H and Ballantyne CM (2020) Metabolic inflammation and insulin resistance in obesity. Circ. Res 126, 1549–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kohan DE and Barton M (2014) Endothelin and endothelin antagonists in chronic kidney disease. Kidney Int 86, 896–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vercauteren M, Trensz F, Pasquali A, Cattaneo C, Strasser DS et al. (2017) Endothelin eta receptor blockade, by activating etb receptors, increases vascular permeability and induces exaggerated fluid retention. J. Pharmacol. Exp. Ther 361, 322–333 [DOI] [PubMed] [Google Scholar]
- 14.Wessale JL, Adler AL, Novosad EI, Calzadilla SV, Dayton BD et al. (2002) Pharmacology of endothelin receptor antagonists abt-627, abt-546, a-182086 and a-192621: ex vivo and in vivo studies. Clin. Sci. (Lond.) 103, 112S–117S [DOI] [PubMed] [Google Scholar]
- 15.Benede-Ubieto R, Estevez-Vazquez O, Ramadori P, Cubero FJ and Nevzorova YA (2020) Guidelines and considerations for metabolic tolerance tests in mice. Diabetes Metab. Syndr. Obes 13, 439–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seo JB, Riopel M, Cabrales P, Huh JY, Bandyopadhyay GK et al. (2019) Knockdown of ant2 reduces adipocyte hypoxia and improves insulin resistance in obesity. Nat. Metab 1, 86–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Zeeuw D, Coll B, Andress D, Brennan JJ, Tang H et al. (2014) The endothelin antagonist atrasentan lowers residual albuminuria in patients with type 2 diabetic nephropathy. J. Am. Soc. Nephrol 25, 1083–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rivera-Gonzalez OJ, Kasztan M, Johnston JG, Hyndman KA and Speed JS (2020) Loss of endothelin type b receptor function improves insulin sensitivity in rats. Can. J. Physiol. Pharmacol 98, 604–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferri C, Bellini C, Desideri G, Di Francesco L, Baldoncini R et al. (1995) Plasma endothelin-1 levels in obese hypertensive and normotensive men. Diabetes 44, 431–436 [DOI] [PubMed] [Google Scholar]
- 20.Weil BR, Westby CM, Van Guilder GP, Greiner JJ, Stauffer BL et al. (2011) Enhanced endothelin-1 system activity with overweight and obesity. Am. J. Physiol. Heart Circ. Physiol 301, H689–H695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Webb CM, Ghatei MA, McNeill JG and Collins P (2000) 17beta-estradiol decreases endothelin-1 levels in the coronary circulation of postmenopausal women with coronary artery disease. Circulation 102, 1617–1622 [DOI] [PubMed] [Google Scholar]
- 22.Ye J (2009) Emerging role of adipose tissue hypoxia in obesity and insulin resistance. Int. J. Obes. (Lond.) 33, 54–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu J, Discher DJ, Bishopric NH and Webster KA (1998) Hypoxia regulates expression of the endothelin-1 gene through a proximal hypoxia-inducible factor-1 binding site on the antisense strand. Biochem. Biophys. Res. Commun 245, 894–899 [DOI] [PubMed] [Google Scholar]
- 24.Jenkins HN, Williams LJ, Dungey A, Vick KD, Grayson BE et al. (2019) Elevated plasma endothelin-1 is associated with reduced weight loss post vertical sleeve gastrectomy. Surg. Obes. Relat. Dis 15, 1044–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kohan DE, Heerspink HJL, Coll B, Andress D, Brennan JJ et al. (2015) Predictors of atrasentan-associated fluid retention and change in albuminuria in patients with diabetic nephropathy. Clin. J. Am. Soc. Nephrol 10, 1568–1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Farrah TE, Anand A, Gallacher PJ, Kimmitt R, Carter E et al. (2019) Endothelin receptor antagonism improves lipid profiles and lowers pcsk9 (proprotein convertase subtilisin/kexin type 9) in patients with chronic kidney disease. Hypertension 74, 323–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.James O and Day C (1999) Non-alcoholic steatohepatitis: another disease of affluence. Lancet North Am. Ed 353, 1634–1636 [DOI] [PubMed] [Google Scholar]
- 28.Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L et al. (2016) Global epidemiology of nonalcoholic fatty liver disease—meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84 [DOI] [PubMed] [Google Scholar]
- 29.Okamoto T, Koda M, Miyoshi K, Onoyama T, Kishina M et al. (2016) Antifibrotic effects of ambrisentan, an endothelin-a receptor antagonist, in a non-alcoholic steatohepatitis mouse model. World J. Hepatol 8, 933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Demirci E, Ferah I, Gundogdu C, Ozkanlar S, Baygutalp N et al. (2015) Endothelin receptor inhibition with bosentan delays onset of liver injury in streptozotocin-induced diabetic condition. Drug Res 65, 272–280 [DOI] [PubMed] [Google Scholar]
- 31.Gamberi T, Magherini F, Modesti A and Fiaschi T (2018) Adiponectin signaling pathways in liver diseases. Biomedicines 6, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moore K (2004) Endothelin and vascular function in liver disease. Gut 53, 159–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rockey DC and Chung JJ (1996) Endothelin antagonism in experimental hepatic fibrosis. Implications for endothelin in the pathogenesis of wound healing. J. Clin. Invest 98, 1381–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weil BR, Westby CM, Van Guilder GP, Greiner JJ, Stauffer BL et al. (2011) Enhanced endothelin-1 system activity with overweight and obesity. Am. J. Physiol. Heart Circ. Physiol 301, H689–H695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Polak J, Punjabi NM and Shimoda LA (2018) Blockade of endothelin-1 receptor type b ameliorates glucose intolerance and insulin resistance in a mouse model of obstructive sleep apnea. Front. Endocrinol. (Lausanne) 9, 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu A, Tam B, Yau W, Chan K, Yu S et al. (2015) Association of endothelin-1 and matrix metallopeptidase-9 with metabolic syndrome in middle-aged and older adults. Diabetol. Metab. Syndr 7, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Harmelen V, Eriksson A, Astrom G, Wahlen K, Naslund E et al. (2008) Vascular peptide endothelin-1 links fat accumulation with alterations of visceral adipocyte lipolysis. Diabetes 57, 378–386 [DOI] [PubMed] [Google Scholar]
- 38.Karpe F, Dickmann JR and Frayn KN (2011) Fatty acids, obesity, and insulin resistance: Time for a reevaluation. Diabetes 60, 2441–2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eriksson A, Van Harmelen V, Stenson B, Åström G, Wåhlén K et al. (2009) Endothelin-1 stimulates human adipocyte lipolysis through the et a receptor. Int. J. Obes 33, 67–74 [DOI] [PubMed] [Google Scholar]
- 40.Morigny P, Houssier M, Mouisel E and Langin D (2016) Adipocyte lipolysis and insulin resistance. Biochimie 125, 259–266 [DOI] [PubMed] [Google Scholar]
- 41.Taylor EB (2021) The complex role of adipokines in obesity, inflammation, and autoimmunity. Clin. Sci. (Lond.) 135, 731–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cnop M, Havel PJ, Utzschneider K, Carr D, Sinha M et al. (2003) Relationship of adiponectin to body fat distribution, insulin sensitivity and plasma lipoproteins: evidence for independent roles of age and sex. Diabetologia 46, 459–469 [DOI] [PubMed] [Google Scholar]
- 43.Hu E, Liang P and Spiegelman BM (1996) Adipoq is a novel adipose-specific gene dysregulated in obesity. J. Biol. Chem 271, 10697–10703 [DOI] [PubMed] [Google Scholar]
- 44.Mantzoros CS, Li T, Manson JE, Meigs JB and Hu FB (2005) Circulating adiponectin levels are associated with better glycemic control, more favorable lipid profile, and reduced inflammation in women with type 2 diabetes. J. Clin. Endocrinol. Metab 90, 4542–4548 [DOI] [PubMed] [Google Scholar]
- 45.Nacci C, Leo V, De Benedictis L, Carratu MR, Bartolomeo N et al. (2013) Elevated endothelin-1 (et-1) levels may contribute to hypoadiponectinemia in childhood obesity. J. Clin. Endocrinol. Metab 98, E683–E693 [DOI] [PubMed] [Google Scholar]
- 46.Sugii S, Olson P, Sears DD, Saberi M, Atkins AR et al. (2009) Pparγ activation in adipocytes is sufficient for systemic insulin sensitization. Proc. Natl. Acad. Sci. U.S.A 106, 22504–22509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N et al. (2001) The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat. Med 7, 941–946 [DOI] [PubMed] [Google Scholar]
- 48.Lo JC, Ljubicic S, Leibiger B, Kern M, Leibiger IB et al. (2014) Adipsin is an adipokine that improves β cell function in diabetes. Cell 158, 41–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang J-S, Lee W-J, Lee I-T, Lin S-Y, Lee W-L et al. (2019) Association between serum adipsin levels and insulin resistance in subjects with various degrees of glucose intolerance. J. Endocr. Soc 3, 403–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klöting N and Blüher M (2014) Adipocyte dysfunction, inflammation and metabolic syndrome. Rev. Endocr. Metab. Disord 15, 277–287 [DOI] [PubMed] [Google Scholar]
- 51.Mraz M and Haluzik M (2014) The role of adipose tissue immune cells in obesity and low-grade inflammation. J. Endocrinol 222, R113–R127 [DOI] [PubMed] [Google Scholar]
- 52.Schipper HS, Prakken B, Kalkhoven E and Boes M (2012) Adipose tissue-resident immune cells: Key players in immunometabolism. Trends Endocrinol. Metab 23, 407–415 [DOI] [PubMed] [Google Scholar]
- 53.McGillicuddy FC, Chiquoine EH, Hinkle CC, Kim RJ, Shah R et al. (2009) Interferon γ attenuates insulin signaling, lipid storage, and differentiation in human adipocytes via activation of the jak/stat pathway. J. Biol. Chem 284, 31936–31944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hotamisligil GS, Arner P, Caro JF, Atkinson RL and Spiegelman BM (1995) Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J. Clin. Invest 95, 2409–2415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nieto-Vazquez I, Fernández-Veledo S, Krämer DK, Vila-Bedmar R, Garcia-Guerra L et al. (2008) Insulin resistance associated to obesity: the link tnf-alpha. Arch. Physiol. Biochem 114, 183–194 [DOI] [PubMed] [Google Scholar]
- 56.Chen Y, Hanaoka M, Droma Y, Chen P, Voelkel NF et al. (2010) Endothelin-1 receptor antagonists prevent the development of pulmonary emphysema in rats. Eur. Respir. J 35, 904–912 [DOI] [PubMed] [Google Scholar]
- 57.Ford RL, Mains IM, Hilton EJ, Reeves ST, Stroud RE et al. (2008) Endothelin-a receptor inhibition after cardiopulmonary bypass: cytokines and receptor activation. Ann. Thorac. Surg 86, 1576–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H et al. (2009) Cd8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med 15, 914–920 [DOI] [PubMed] [Google Scholar]
- 59.Yang Q, Li G, Zhu Y, Liu L, Chen E et al. (2011) Il-33 synergizes with tcr and il-12 signaling to promote the effector function of cd8+ t cells. Eur. J. Immunol 41, 3351–3360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Saraiva M and O’garra A (2010) The regulation of il-10 production by immune cells. Nat. Rev. Immunol 10, 170–181 [DOI] [PubMed] [Google Scholar]
- 61.Rajbhandari P, Thomas BJ, Feng A-C, Hong C, Wang J et al. (2018) Il-10 signaling remodels adipose chromatin architecture to limit thermogenesis and energy expenditure. Cell 172, 218.e217–233.e217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baretella O, Chung SK, Xu A and Vanhoutte PM (2017) Paradoxical lack of increase in endothelin-1 levels in obese mice - possible role of endothelin-b receptors. Acta Pharmacol. Sin 38, 1699–1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee EH, Itan M, Jang J, Gu HJ, Rozenberg P et al. (2018) Eosinophils support adipocyte maturation and promote glucose tolerance in obesity. Sci. Rep 8, 9894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang Y, Yang P, Cui R, Zhang M, Li H et al. (2015) Eosinophils reduce chronic inflammation in adipose tissue by secreting th2 cytokines and promoting m2 macrophages polarization. Int. J. Endocrinol 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bolus WR, Kennedy AJ and Hasty AH (2018) Obesity-induced reduction of adipose eosinophils is reversed with low-calorie dietary intervention. Physiol. Rep 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Elisa T, Antonio P, Giuseppe P, Alessandro B, Giuseppe A et al. (2015) Endothelin receptors expressed by immune cells are involved in modulation of inflammation and in fibrosis: relevance to the pathogenesis of systemic sclerosis. J. Immunol. Res 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cui P, Sharmin S, Okumura Y, Yamada H, Yano M et al. (2004) Endothelin-1 peptides and il-5 synergistically increase the expression of il-13 in eosinophils. Biochem. Biophys. Res. Commun 315, 782–787 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.