

Abstract
Objective.
Intracortical brain interfaces are an ever evolving technology with growing potential for clinical and research applications. The chronic tissue response to these devices traditionally has been characterized by glial scarring, inflammation, oxidative stress, neuronal loss, and blood-brain barrier disruptions. The full complexity of the tissue response to implanted devices is still under investigation.
Approach.
In this study, we have utilized RNA-sequencing to identify the spatiotemporal gene expression patterns in interfacial (within 100μm) and distal (500μm from implant) brain tissue around implanted silicon microelectrode arrays. Naïve, unimplanted tissue served as a control.
Main Results.
The data revealed significant overall differential expression (DE) in contrasts comparing interfacial tissue vs naïve (157 DE genes), interfacial vs. distal (94 DE genes), and distal vs. naïve tissues (21 DE genes). Our results captured previously characterized mechanisms of the foreign body response, such as astroglial encapsulation, as well as novel mechanisms which have not yet been characterized in the context of indwelling neurotechnologies. In particular, we have observed perturbations in multiple neuron-associated genes which potentially impact the intrinsic function and structure of neurons at the device interface. In addition to neuron-associated genes, the results presented in this study identified significant DE in genes which are associated with oligodendrocyte, microglia, and astrocyte involvement in the chronic tissue response.
Significance.
The results of this study increase the fundamental understanding of the complexity of tissue response in the brain and provide an expanded toolkit for future investigation into the bio-integration of implanted electronics with tissues in the central nervous system.
Keywords: Neural Prostheses, RNA-Sequencing, Tissue Response, Inflammation, Michigan Probe
1. Introduction
Microelectrodes implanted in the nervous system are rapidly evolving technologies with ever-increasing applications in clinical and research settings. By recording from, and/or stimulating neuronal populations, it is possible to interface the nervous system with assistive devices or modulate neuronal activity to treat neurological disease and injury. Re-animation of a patient’s limbs following spinal cord injury, treatment of the medication-resistant motor symptoms of Parkinson’s disease, and interruption of seizure activity in intractable epilepsy are examples of the potential clinical applications of implantable neurotechnologies[1]–[15]. Likewise, the commercial value of neural prosthetics has been highlighted by the recent investment of private companies in “next-generation” electrode designs and the development of novel closed-loop neural interface systems[16], [17].
As advances in neurotechnology continue, the biological response to implanted electrodes in the brain is an on-going challenge to progress in the field[18]–[21]. Vascular disruption and microglial activation are early responses to implantation, where the extension of microglial processes toward the device has been observed within minutes of insertion[22]. An astroglial scar subsequently encapsulates the interface and further separates the electrode from nearby neuronal populations. The glial response is reportedly accompanied by a ~40% loss of neuronal somata within the first 100 microns of the electrode surface in comparison to a stab wound control[23], [24]. These observations, in combination with other early reports[21], [25], [26], provided motivation for the design of next-generation neural interfaces with improved biological integration[18], [27], [28]. However, questions remain regarding the relationship between the biological impacts of electrodes, design, and their long-term performance[18]. Reports of poor signal fidelity and loss of neuronal signals in tissue with no apparent neuronal loss or glial scarring suggest additional complexity in the underlying relationship between the tissue response and device performance[19].
In recent years, new factors have been identified as potential contributors to the biological response to implanted devices. Insertion of electrode arrays damages cellular populations and the extracellular matrix, and disruption of the blood brain barrier (BBB) generates disruptive debris and initiates downstream cytokine signaling cascades[21]. Both in vivo imaging and gene expression studies have confirmed vascular damage and BBB disruption resulting from implanted electrodes, where insertional trauma is evident in the downregulation of genes associated with tight junctions and adherens junctions[29]–[32]. New research also implicates oligodendrocytes and NG2 cells as dynamic players in the response to an indwelling foreign body. Literature has shown that much like other cell types, oligodendrocytes and NG2 cells are affected by BBB disruption, inflammation, and the traumatic injury caused by device insertion[22], [33]. Device insertion causes direct mechanical damage to oligodendroglia and myelin structure as well as secondary damage through inflammatory mechanisms. Increased permeability of the BBB following insertion exposes the cortical environment to inflammatory plasma proteins and debris which can recruit myelin-targeting immune cells which create further damage [33]. Likewise, Bedell et al. have identified numerous differentially expressed (DE) genes at the device interface involved in neuroinflammatory cascades known to contribute to glial scarring and cell death [31], [34]. These recent reports indicate that the biological response to implanted electrode arrays remains incompletely understood, motivating the search for additional biomarkers of device-tissue interaction.
Here, we report the results of sequencing the transcriptome of tissue collected both within 100 microns (“near”, or “interfacial”) and ~500 microns (“far”, or “distal”) from Michigan-style electrode arrays implanted into rat motor cortex. We compared their profiles to the transcriptome of naïve, unimplanted animals. Tissue was collected at time points designed to capture the initial insertion injury (24 hours), early reactivity (1 week), and chronic responses (6 weeks). We detected the expression of >1,000 genes per condition, where >100 were significantly differentially expressed in near-device versus naïve tissue and >90 genes were DE in near-device versus far tissue. Interestingly, >20 genes were DE in tissue 500 μm from the device versus naïve tissue. A description of symbols and reported roles for DE genes are found in Table 1. A selected subset of detected and DE genes identified in this study are discussed which either validate existing understandings of tissue response in the brain or expand upon contemporary reports with additional mechanisms in the context of implanted devices. Complete raw results from the data analysis can be found in supplementary files (1–15). By reporting RNA-sequencing on tissue samples captured at multiple distances and time points, we extend current understanding of the spatiotemporal profile of gene expression surrounding implanted electrode arrays in the brain. The data reinforce observations and hypotheses described in literature while unmasking previously-unreported effects of implanted devices on gene expression.
Table 1.
Genes detected in this study through RNA-sequencing that are associated with tissue response to implanted devices as well as neurodegenerative disease.
| Gene Symbol | Gene name | Role | Reference |
|---|---|---|---|
| Ap2a1 | Adaptor Related Protein Complex 2 Alpha 1 Subunit | Blood brain barrier integrity | [30] |
| Aqp4 | Aquaporin 4 | Water movement, cell adhesion, synaptic plasticity, and cellular migration | [96], [128] |
| Arc | Activity Regulated Cytoskeleton Associated Protein | Synaptic plasticity and dendritic spine maintenance | [69], [70] |
| Aox1 | Aldehyde Oxidase 1 | Oxidative stress | [30] |
| Apoe | Apolipoprotein E | Construction of lipoprotein and lipid transport | [92], [93], [118] |
| Bcl2 | B-cell lymphoma 2 | Apoptosis inhibition. NFκB pathway | [129] |
| Best1 | Bestrophin 1 | GABA / glutamate permissible channel dependent on astrocyte identity. Astrocyte enriched | [95] |
| Bsn | Bassoon Presynaptic Cytomatrix Protein | Presynaptic vesicle release | [60], [62] |
| C1qa | Complement C1q A chain | Innate immune response, promotes phagocytosis and synapse pruning | [107], [114] |
| C1qb | Complement C1q B chain | Innate immune response, promotes phagocytosis and synapse pruning | [107], [114] |
| C1qc | Complement C1q C chain | Innate immune response and promoter of phagocytosis and synapse pruning | [107], [114] |
| C3 | Complement C3 | Innate immune response and promoter of phagocytosis and synapse pruning | [31], [107], [113], [130] |
| Cacna1i | Calcium Voltage-Gated Channel Subunit Alpha1 | Voltage gated calcium activity and plasticity | [78], [79] |
| Cacng3 | Calcium Voltage-Gated Channel Auxiliary Subunit Gamma 3 | Voltage gated calcium activity and plasticity | [131] |
| Camk2a | Calcium/calmodulin-dependent protein kinase II alpha | Calcium signalling intermediate protein. Essential for neuronal function | [74], [132] |
| Ccl3 | C-C Motif Chemokine Ligand 3 | Immune response chemotaxis and regulation of cellular BBB transmigration | [29], [133] |
| Cd68 | CD antigen | Microglial immune response activation molecule | [134], [135] |
| Cdh5 | Cadherin 5 | BBB stability and barrier transmigration | [136] |
| Cldn5 | Claudin 5 | BBB stability and barrier transmigration | [137] |
| Clint1 | Clathrin Interactor 1 | Synaptic vesicle formation and neurotransmitter recycling | [138], [139] |
| Cltb | Clathrin Light Chain B | Synaptic vesicle formation and neurotransmitter recycling | [138], [139] |
| Cnksr2 | Connector enhancer of kinase suppressor of Ras 2 | Synaptic protein assembly | [140] |
| Cnp | 2′,3′-Cyclic Nucleotide 3′ Phosphodiesterase | Oligodendrocyte surface protein | [141] |
| Col4a1 | Collagen alpha-1(IV) | Fibrosis. Glial scar component | [87] |
| Csf1r | Colony stimulating factor 1 receptor | Cytokine response. Macrophage, microglia, and phagocyte differentiation and survival. | [110], [111] |
| Ctsb | Cathepsin B | Cysteine protease. EMC degradation, apoptosis, clathautophagy, and glia induced cell death | [109] |
| Ctsl | Cathepsin L | Cysteine protease. EMC degradation, apoptosis, autophagy, and glia induced cell death | [142] |
| Cx3cr1 | CX3C chemokine receptor 1 | Cytokine signalling | [31], [143] |
| Cxcl1 | C-X-C Motif Chemokine Ligand 1 | Cytokine signalling | [31], [144] |
| Cxcl2 | C-X-C Motif Chemokine Ligand 2 | Cytokine signalling, inflammation, and BBB transmigration | [58] |
| Cyfip2 | Cytoplasmic FMR1 Interacting Protein 2 | Regulations of mRNA translation at the synapse. Synapse maintenance | [68], [145] |
| Dctn1 | Dynactin subunit 1 | Microtubule motor and axonal transport protein | [146], [147] |
| Dock8 | Dedicator of cytokinesis 8 | Microglial immune response activation molecule | [73] |
| Dusp1 | Dual Specificity Phosphatase 1 | Inflammation | [30] |
| Gabbr1 | Gamma-Aminobutyric Acid Type B Receptor Subunit 1 | Inhibits post synaptic potentials | [73] |
| Gabbr2 | Gamma-Aminobutyric Acid Type B Receptor Subunit 2 | Inhibits post synaptic potentials | [33], [148] |
| Galc | Galactosylceramidase | Myelin component. Enriched in oligodendrocytes | [149] |
| Gfap | Glial fibrillary acidic protein | Intermediate filament protein. Enriched in reactive astrocytes | [116], [150] |
| Gpnmb | Transmembrane glycoprotein NMB | Immune response regulation | [116], [150] |
| Gpx1 | Glutathione Peroxidase 1 | Oxidative stress | [30] |
| Gpx4 | Glutathione Peroxidase 4 | Oxidative stress | [30] |
| Hmox1 | Heme Oxygenase 1 | Oxidative stress | [30] |
| IL1a | Interleukin 1 Alpha | Cytokine signalling in immune response | [151] |
| Il1b | Interleukin 1 beta | NF-kB effector in immune response | [151] |
| Il1ra | Interleukin 1 Receptor Antagonist | Il1a and Il1b inhibition | [151] |
| Il6r | Interleukin 6 Receptor | NF-kB effector in immune response | [31], [151] |
| IL1a | Interleukin 1 Alpha | Cytokine signalling. Immune response | [151] |
| Kif5a | Kinesin Family Member 5A | Anterograde transport | [56], [57], [152] |
| Kif5b | Kinesin Family Member 5B | Anterograde transport | [54], [152]–[154] |
| Kif5c | Kinesin Family Member 5C | Anterograde transport | [155], [156] |
| Lcn2 | Lipocalin 2 | Inflammatory response. Secretion via astrocytes promote neuron death | [30], [157] |
| Map2 | Microtubule Associated Protein 2 | Neuronal Cytoskeleton | [67], [158] |
| Map4 | Microtubule Associated Protein 4 | Neuronal Cytoskeleton | [158] |
| Mapt | Microtubule Associated Protein Tau | Neurogenesis microtubule assembly protein; essential for neurodevelopment and recovery | [158], [159] |
| Mbp | Myelin basic protein | Myelin sheath adhesion protein enriched in oligodendrocytes | [120] |
| Mfsd2a | Major Facilitator Superfamily Domain Containing 2a | Causes BBB instability and barrier diffusion for lipids | [160] |
| Mmp2 | Matrix Metallopeptidase 2 | Extracellular matrix lattice protein; may negatively impact myelination | [29], [161] |
| Mmp9 | Matrix Metallopeptidase 9 | Extracellular matrix protein degrader; plays role in neural tissue structuring | [29], [162] |
| Mpv17 | Mitochondrial Inner Membrane Protein | Oxidative stress | [30] |
| Ncan | Neurocan, CSPG3(Chondroitan sulfate proteoglycan 3) | Reactive astrocyte adhesion molecule. Inhibits neurite outgrowth | [163] |
| Ncf1 | Neutrophil Cytosol Factor 1 | Oxidative stress | [30] |
| Nefh | Neurofilament heavy | Neuronal cytoskeleton intermediate filament protein | [50] |
| Nefl | Neurofilament light | Neuronal cytoskeleton intermediate filament protein | [50] |
| Nefm | Neurofilament medium | Neuronal cytoskeleton intermediate filament protein | [50] |
| Nes | Nestin | Neuroepithelial intermediate filament protein. Type IV intermediate filament | [90] |
| Nos1 | Neuronal Nitric-Oxide Synthase 1 | Oxidative stress | [30] |
| Nos2 | Inducible Nitric-Oxide Synthase | Oxidative stress | [30] |
| Nptxr | Neuronal Pentraxin Receptor | Synaptic regulation and plasticity | [164], [165] |
| Nrgn | Neurogranin | Dendritic spine maintenance and plasticity | [76]–[78] |
| Ocln | Occludin | Regulator of BBB stability and barrier diffusion | [29], [166] |
| Olig2 | Oligodendrocyte Transcription Factor 2 | Regulates CNS development via multiple pathways. Oligodendrocyte marker | [33], [167] |
| Plp1 | Proteolipid protein 1 | Myelin sheath adhesion and maintenance | [168] |
| Ptbp1 | Polypyrimidine Tract-Binding Protein 1 | Alternative splicing of genes involved with multiple cellular processes | [97], [99], [100] |
| Ptprz1 | Protein Tyrosine Phosphatase Receptor Type Z1 | PI3K-AKT pathway. Oligodendrocyte differentiation | [117], [118], [169] |
| Prdx1 | Peroxiredoxin 1 | Oxidative stress | [30] |
| Prdx2 | Peroxiredoxin 2 | Oxidative stress | [30] |
| Prdx3 | Peroxiredoxin 3 | Oxidative stress | [30] |
| Rgs5 | Regulator of G-protein signalling 5 | Marker of activated pericytes | [29], [170] |
| Rbfox3 | RNA Binding Fox 3 | Mature Neuron Marker (NeuN). Neuronal differentiation | [171], [172] |
| Rtn1 | Reticulon 1 | Neuron enriched. Cellular trafficking | [126], [127] |
| S100b | S100 calcium binding protein B | Calcium binding protein; Reactive astrocyte marker | [31] |
| Scara3 | Scavenger Receptor Class A Member 3 | Oxidative stress | [30] |
| Shh | Sonic Hedgehog | Astrocyte-endothelium gliovascular subunit maintenance | [30] |
| Snap25 | Synaptosome Associated Protein 25 | Presynaptic terminal regulation | [65] |
| Sod1 | Superoxide Dismutase 1 | Oxidative stress. Superoxide radical degradation | [30] |
| Sod2 | Superoxide Dismutase 2 | Oxidative stress. Superoxide radical degradation | [30] |
| Sod3 | Superoxide Dismutase 3 | Oxidative stress. Superoxide radical degradation | [30] |
| Sox2 | SRY-Box Transcription Factor 2 | Stem cell maintenance and differentiation | [173] |
| Stxbp1 | Syntaxin Binding Protein 1 | Synaptic vesicle regulation | [174], [175] |
| Syn1 | Synapsin 1 | Neurotransmitter release | [59], [176] |
| Tf | Transferrin | Iron transport and sequestration | [118], [119] |
| Tjp1 | Tight Junction Protein 1 | Regulator of BBB barrier transmigration | [29], [166] |
| Tjp2 | Tight Junction Protein 2 | Regulator of BBB barrier transmigration | [29], [136] |
| Tlr2 | Toll-Like Receptor 2 | Inflammation. TLR pathway | [30], [31] |
| Tlr4 | Toll-Like Receptor 4 | Inflammation. TLR pathway | [30], [31] |
| Tmem119 | Transmembrane Protein 119 | Inflammation. Microglial biomarker | [177] |
| Tnfrsf1a | Tumor necrosis factor receptor superfamily member 1A | Inflammation. TNF pathway | [31], [178] |
| Tnfrsf1b | Tumor necrosis factor receptor superfamily member 1B | Inflammation. TNF pathway | [31], [178] |
| Trem2 | Triggering Receptor Expressed on Myeloid Cells 2 | Encourages microglia survival via apoptosis inhibition and proliferation | [115], [179] |
| Tfrc | Transferrin Receptor | Transferrin uptake | [30] |
| Vim | Vimentin | Intermediate filament protein found in mesenchymal cells; plays a role in migration, attachment, and signalling | [85], [90] |
2. Methods
2.1. Surgical Implantation of Silicon Electrodes
Single shank “Michigan”-style probes (16-channel A1x16-3mm, 15μm wide, 703μm2 site size, 100μm site spacing, Neuronexus Technologies) were stereotaxically implanted in the motor cortex (M1) of male Sprague-Dawley rats (aged 12-14 weeks)[35]. Animals were isoflurane-anesthetized and a craniotomy was performed over M1 (+3.0 mm anterior posterior, 2.5 medio lateral from Bregma), dura was resected, and the probe was stereotaxically inserted to a depth of 2mm from the cortical surface[36]. A dental cement headcap was used to secure bilateral implants to two stainless steel bone screws. Bupivacaine and Neosporin were applied topically to the area around the incision to minimize discomfort and infection risk, and meloxicam was administered via injection for post- operative pain management. Devices remained implanted in M1 for the duration of designated time points (1 day, 1 week, and 6 weeks). All surgical procedures described were approved by the Michigan State University Animal Care and Use Committee.
2.2. Tissue Extraction and Slide Preparation
At the terminal time point, animals were deeply anesthetized with sodium pentobarbital, perfused with 4% paraformaldehyde transcardially, and the brains were extracted. Following graded sucrose protection (5-20%) and cryo-embedding, the brains were sliced via cryostat (Leica) as 20μm thick sections and mounted on Superfrost™ Plus slides (Fisher Scientific). Six tissue sections (n = 6) were collected for analysis at each timepoint (24 hours, n = 3 rats; 1 week, n = 5 rats; 6 weeks, n = 4 rats) in addition to six samples collected from naïve, unimplanted rats. Depth of collection spanned the implant shank (~600-1700 μm from cortical surface). The nature of the tissue collection along the implantation depth did not allow for analysis of the full volume of tissue or tissue proximity to different electrode materials (e.g. recording sites versus bulk material).
2.3. Laser Capture Microscopy (LCM) for Tissue Collection
Tissue near the implant injury, or ‘interfacial’ (within 100μm) was extracted using laser capture microscopy (LCM) (Zeiss Palm MicroBeam IV). Distal tissue of an approximately equivalent total surface area was extracted from ~500μm away from the implant site to assess distance-dependent effects. These samples were collected and pooled from four smaller sections obtained at locations equidistant from the implant site. Using similar collection methods, control tissue from naïve brains was used to compare implanted tissue to unimplanted tissue. Settings were optimized by using excess tissue to calibrate laser strength and focus, allowing for efficient collection of tissue while avoiding any apparent heat damage to either the slide or the tissue. This process was repeated for each laser capture session.
2.4. RNA Extraction and Sequencing
RNA was extracted from LCM-collected tissue using a specialized RNAstorm extraction kit (Cell Data Sciences). cDNA library preparation and RNA sequencing was carried out by the University of Michigan Advanced Genomics Core. cDNA libraries were prepped using a Takara SMART-stranded kit and subsequently subjected to 150 paired-end cycles on the NovaSeq-6000 platform (Illumina). Sequencing adapters were trimmed using Cutadapt (v2.3). FastQC[37] (v0.11.8) was used to ensure the quality of data. Reads are mapped to the reference genome Rattus_norvegicus.Rnor_6.0.9, using STAR[38] (v2.6.1b) and assigned count estimates to genes with RSEM[39] (v1.3.1). Alignment options follow ENCODE standards for RNA-seq[40]. FastQC is used in an additional post-alignment step to ensure that only high-quality data gets used for expression quantitation and differential expression.
2.5. Differential Expression of RNA
Data was pre-filtered to remove genes with 0 counts in all samples. Differential gene expression analysis is performed using DESeq2[41], using a negative binomial generalized linear model (thresholds: linear fold change >1.5 or <−1.5, Benjamini-Hochberg FDR (Padj) <0.05). Functional analysis, including candidate pathways activated or inhibited in comparison(s) and GO-term enrichments[42], are performed using iPathway Guide (Advaita)[43], [44]. While the nature of our tissue preparation and retrieval is prone to degradation, duplication, and low yield, these conditions were consistent across samples and were not expected to influence any sample cohort preferentially. Likewise, a review of data via principal component analysis did not reveal outliers associated with specific section depths. As such, genes identified as DE are expected to represent effects related to the presence of the device.
3. Results & Discussion
3.1. Differential Gene Expression in Interfacial, Distal, and Naïve Tissues
In comparison to traditional immunohistochemistry or analysis of gene expression through quantitative polymerase chain reaction (qPCR), RNA-sequencing simultaneously assesses thousands of genes while obviating the need to pre-select a limited number of biomarkers of interest [45], [46]. Traditionally, device-tissue interaction has been assessed through quantitative immunohistochemistry or qPCR, while a more recent approach by Bedell et. al. profiled a broader set of genes associated specifically with neuroinflammatory cascades[31]. To the best of our knowledge, our data is the first to report sequencing analysis of the whole transcriptome of tissue collected surrounding implanted Michigan-style electrode arrays in rat motor cortex. The data revealed differential gene expression as a function of time and distance from implanted devices (figures (1) and (2)). Overall, 157 genes were detected as significantly DE in interface versus naïve, 94 genes were detected as significantly DE in near versus far, and 21 genes were detected as significantly DE in far versus naïve (figure 1). The majority of DE genes were upregulated in near versus naïve and near versus far tissue, while a shift toward downregulation was observed in far versus naïve tissue (figure 1). We observed the highest number of DE genes at the interface relative to naïve tissue following implantation (157 DE genes at 24 hours) and fewer DE genes over time post-implantation (62 DE genes at 1-week, 26 DE genes at 6-weeks), likely reflecting a pronounced impact of insertional trauma. Contrasts in distal versus naïve tissue followed an opposite time course, with 1 DE gene at 24-hours, 5 DE genes at 1-week, and 5 DE genes 6-weeks post implantation. The identification of DE genes in distal tissue collected 500 microns from the device versus naïve control tissue suggests that implanted electrode arrays affect tissue beyond the proximal device interface.
Figure 1: RNA-sequencing of cortical tissue reveals spatiotemporal gene expression at the device interface.
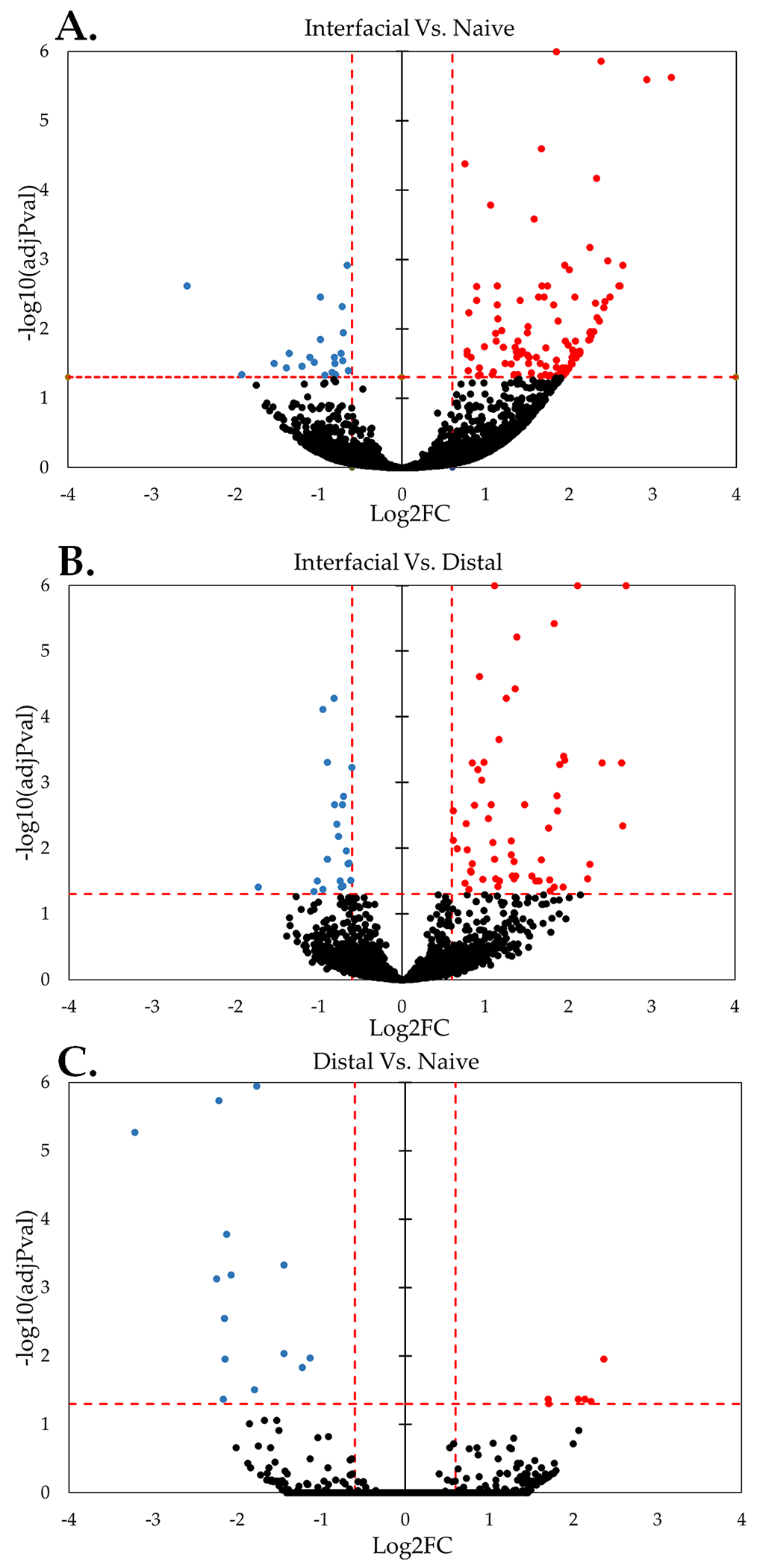
Volcano plots illustrate overall DE of genes at near-device relative to naïve tissue ((A) 157 DE genes), near relative to far tissue ((B) 94 DE genes), and far relative to naïve tissue ((C) 21 DE genes). “Overall” expression represents the group comparisons of samples pooled across time points. Significance was thresholded at Log2FC ≥ 0.6 and P ≤ 0.05 (dashed red lines). (Red: Upregulated DE, Blue: Downregulated DE, Black: Detected not DE)
Figure 2: Transcriptomic analysis of interfacial and distal tissue at the device interface.
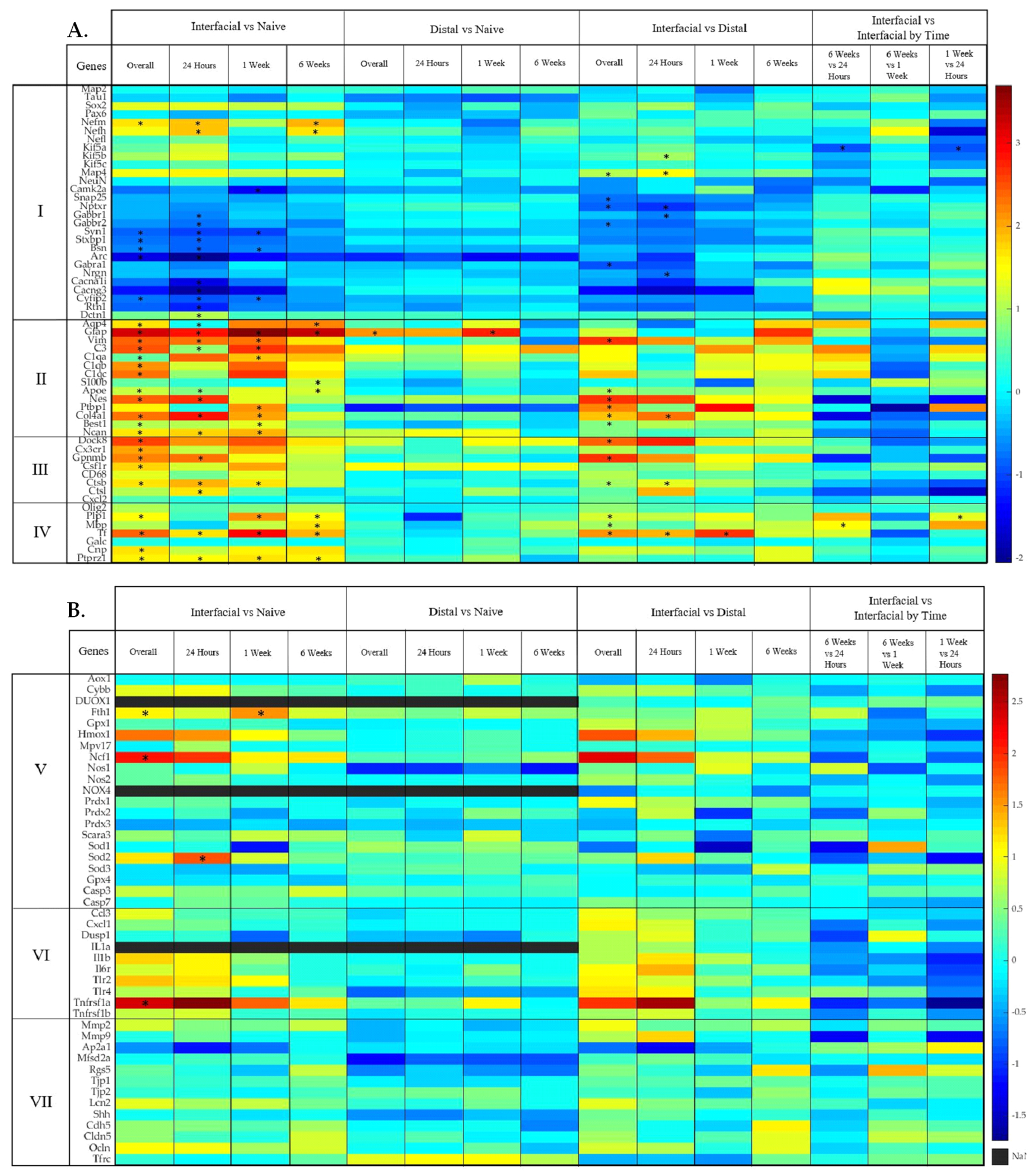
(A) Representative heatmap of differential gene expression for each contrast for previously characterized cell types and their known roles in tissue response to implanted devices. (I) neurons, (II) astrocytes, (III) microglia, and (IV) oligodendrocytes. (B) Representative heatmap showing differential gene expression of each contrast in our analysis for (V) oxidative stress, (VI) inflammation, and (VII) blood-brain barrier. Color bar indicates Log2FC. “NaN” indicates non-detection. Significance was thresholded at Log2FC ≥ 0.6 and P ≤ 0.05. Asterisks* denote statistically significant differentially expressed genes.
As described in following sections, our results validate foundational and contemporary literature while also providing new observations of patterns of spatiotemporal gene expression surrounding devices. The DE genes discussed in this study have been grouped into known associations of cellular expression and interactions. We observed DE of glial and neuronal genes that have not been characterized in the context of implanted electrode arrays. While the majority of these genes reinforce mechanisms of neuronal loss, synaptic pruning, and reactive gliosis, our data also revealed a minority of genes which are associated with protective and regenerative effects, suggesting novel therapeutic targets.
3.2. Differential Expression of Neuron-Associated Genes
Foundational literature has described a “kill-zone” at the device interface where neuronal density declines over time, as evidenced by a loss of neuronal cell bodies and neurofilaments in interfacial tissue. Our data did not reveal a statistically significant reduction in the neuronal nuclear marker NeuN (Rbfox3) near the device, but we did observe decreases in the expression of several genes associated with neuronal structure and synaptic function in excitatory pyramidal neurons (e.g., CaMKIIa) (figures (3) and (4)), which may reflect a simple loss of neurons from the local population. An alternative explanation is that altered gene expression occurs within individual neurons, potentially as a result of structural or functional remodeling in the neuronal network. Our recent observations have revealed significant loss of dendritic arbors and spine density locally to implanted electrodes [47], supporting an at-least partial role for plasticity to contribute to the observed gene expression results. The data also showed increased expression of neuronal cytoskeleton-associated genes (figure 3), which is not explainable by broad-based neuronal loss. Potential reasons for the apparent decoupling of synapse and cytoskeleton-associated genes include: (1) a separation of damage-associated effects on local neurons and dendritic arbors versus long-range connections from axons of passage, and/or (2) cycles of persistent repair and damage within individual neurons at the interface, potentially related to pulsatile micromotion of brain tissue relative to the device. Review of the data set revealed novel observations of neuronal genes associated with cytoskeletal remodeling, intracellular signaling, synaptic structure and intrinsic excitability surrounding implanted electrodes, revealing new mechanisms and potential targets to improve integration.
Figure 3: Differential expression of genes associated with the neuronal synaptic architecture at the device interface relative to naïve tissue.
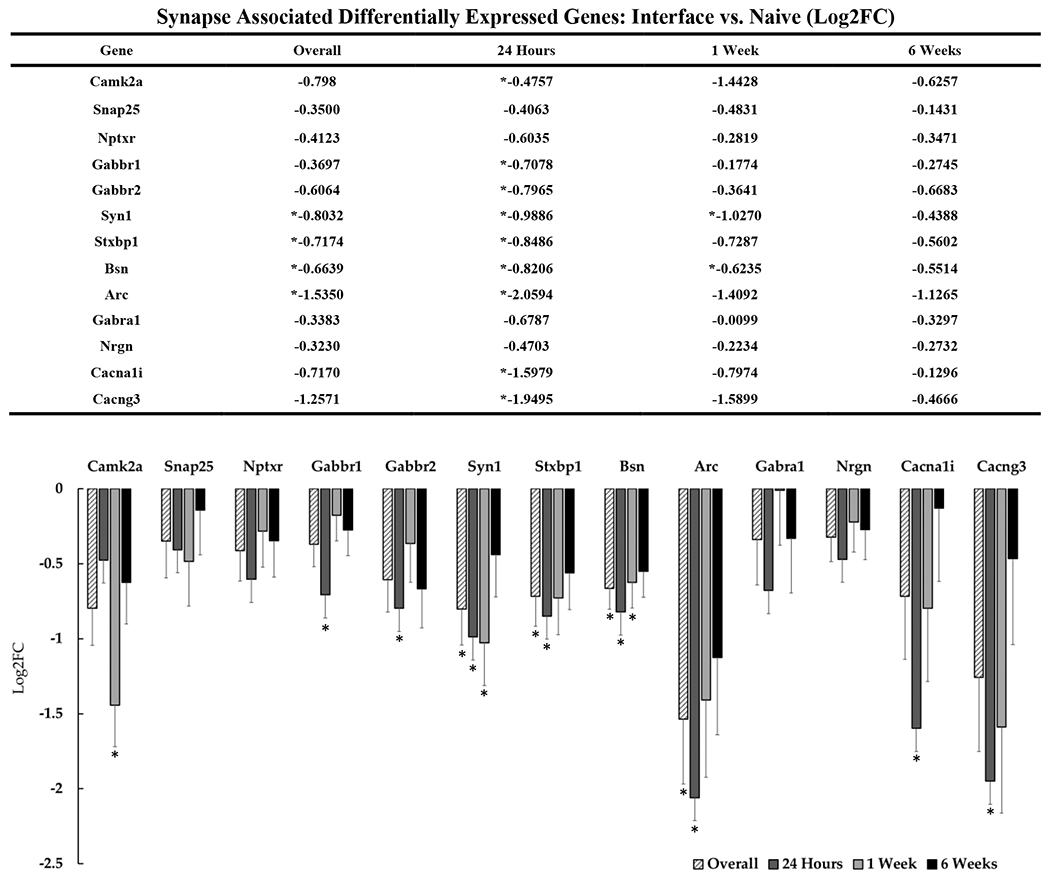
The table and representative graphs that illustrate the downregulation of synaptic associated genes at 24 hours, 1-week and 6-weeks post-implantation. “Overall” expression represents the group comparisons of samples pooled across time points. Significance was thresholded at Log2FC ≥ 0.6 and P ≤ 0.05. Asterisks (*) denote statistically significant differentially expressed genes.
Figure 4: Differential expression of genes associated with the cytoskeletal architecture of neurons at the device interface relative to naïve tissue.
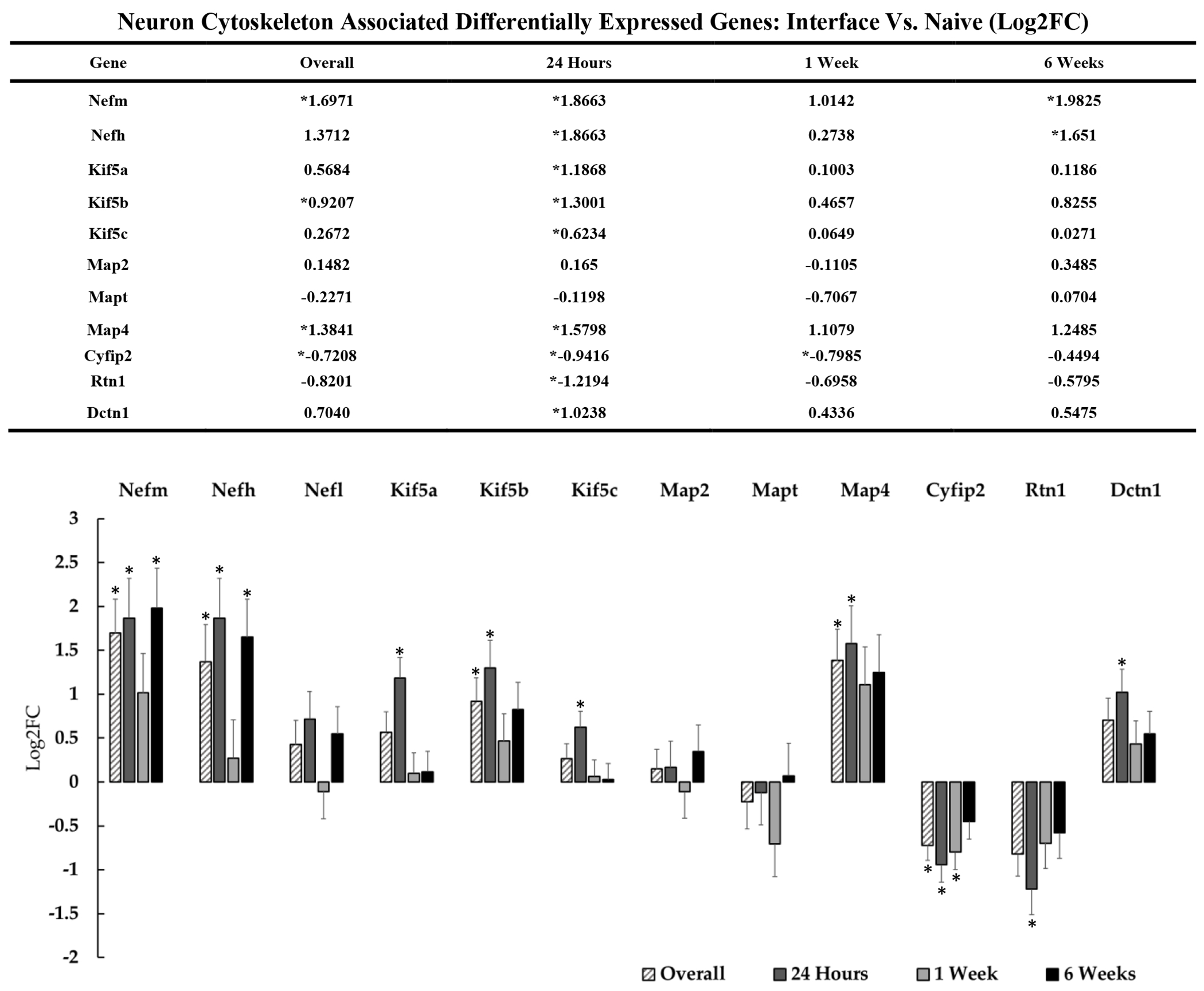
The table and representative graphs above show fluctuations in neuronal genes associated with cytoskeleton and motor proteins at 24 hours, 1-week and 6-weeks post-implantation. “Overall” expression represents the group comparisons of samples pooled across time points. Significance was thresholded at Log2FC ≥ 0.6 and P ≤ 0.05. Asterisks* denote significantly differentially expressed genes.
3.2.1. Neuronal Structure: Cytoskeletal Genes
Previous descriptions of the tissue response to indwelling electrodes have been characterized by a loss of neurofilament protein at the device interface[24]. We did not observe significant reductions in expression in any of the isoforms of neurofilament protein, but rather an apparent upregulation of Nefh, Nefm, and Map4 throughout the duration of device implantation out to 6-week timepoint (figure 4). This observation has been corroborated by recent histological studies where neurofilament protein was found to be elevated above control tissue over time [48], [49]. Accumulation of neurofilament at sites of injury is known to be associated with neuronal pathology as well as the dysfunction of axonal transport mechanisms[50]. In accordance with altered axonal transport, we detected DE of kinesins at the device interface. The kinesin superfamily and dynein transport proteins play an essential role in axo-dendritic transport of synaptic vesicles, cytoskeletal proteins, and mitochondria[51]. These motor proteins have also been shown to play a role in the transport of post-synaptic density (PSD) proteins such as Snap-25, Syntaxin-1, and Bsn, which were also DE at the device interface[52]. Upregulation of Kif5a, Kif5b, and Kif5c at the device interface relative to naïve tissue was significant at 24-hours post-implantation. Kif5a and Kif5c are neuron-specific kinesins. Kif5b is expressed ubiquitously in many cell types and is known to play a role in ion channel and mitochondrial transport in neurons, which can be disrupted in states of injury[53], [54]. Upregulation of neuronal kinesins is associated with changes in mitochondrial trafficking during injury, but it is unclear if this response is adaptive and neuroprotective or a driver of neurodegeneration [55]. Dctn1, a microtubule motor component of the dynein complex, was also upregulated coincidentally with the observed kinesins at the 24-hour timepoint. It is currently unknown whether upregulation of axonal transport proteins is adaptive for neuronal survival or results in axonopathy[56], but dysfunction of Dctn1, kinesins, and related proteins are known to be highly associated with neurodegenerative disease [56], [57].
3.2.2. Neuronal Function: Synapse-Associated Genes
In addition to neuronal cytoskeletal perturbations, we observed significant downregulation of several synapse-associated genes in interfacial tissue, particularly during the first week post-implantation (including CaMKIIa, Syn1, Stxbp1, Bsn, Arc, Gabbr1/2, Cacna1i and Cacng3) (figure 3)[58]. Several of these genes are associated with regulating vesicular release. For example, synapsins are known to play key functions in synaptic formation and plasticity through their role in chaperoning synaptic vesicles during cytoskeletal transport[59]. Syn1, which is downregulated in our analysis at 24 hours and 1 week post-implantation, has been reported to play roles in neurite outgrowth, synapse formation, and synapse maturation[59]. Bsn is a protein component of the presynaptic skeleton that is well-known for its role in vesicle loading at synaptic ribbons in the auditory system [60], and it was also downregulated during the first week post-implantation in our data. Bsn has been reported to be expressed in the cortex, although its function in that location has yet to be fully characterized[61]. Bsn has been reported to play a role in inflammatory pathologies such as multiple sclerosis, where it has been reported to contribute to neurodegeneration via upregulation and somatic Bsn accumulation[62]. The observed downregulation of Bsn and other genes associated with synaptic release is potentially another indicator of neuronal loss, or perhaps an indirect adaptive mechanism to preserve neuronal health.
We also observed acute and overall downregulation of Stxbp1 at the interface relative to both naïve and distal tissue. Stxbp1 binds synaptic vesicle at the pre-synapse and is a protein that has been reported to be essential for the exocytosis of neurotransmitter release[63], [64]. Studies where Stxbp1 is dysfunctional has been shown to eliminate neurotransmitter release in affected neurons[63]. Likewise, while not statistically significant, we observed consistent downregulation of Snap-25 at the device interface relative to distal tissue. Snap-25 is known to interact with Stxbp1 in their roles for docking pre-synaptic vesicles, regulation of Ca2+ channels, and in some cases, post-synaptic spine development and neuronal survival[65], [66]. Taken together, the decreased expression of these genes indicates a decline in synaptic transmission surrounding the device, likely due to neuronal loss and/or loss of local dendrites and spines[47] on residual neurons, both of which have been observed at the device interface[67].
Genes associated with dendritic spine formation, function, and maintenance also were significantly DE at the device interface. We found that Cyfip2 is downregulated overall, at 24-hours, and 1-week post implantation at the interface relative to naïve tissue. Cyfip2 is enriched in neurons and has been reported to play roles in mRNA translation at the synapse as well as the structural maintenance of the pre-synapse, and the maturity of dendritic spines[68]. Reduced Cyfip2 has been implicated in the progression of Alzheimer’s disease but has yet to be investigated in the context of implanted electrodes[68]. Arc is a highly regulated neuronal specific protein and its mRNA levels are directly controlled by neuronal activity, specifically via N-Methyl-D-aspartic acid receptors receptors[69], [70]. The Arc gene is widely expressed in the brain and has been directly implicated in its role in synaptic plasticity at the post-synapse by modulating the formation of dendritic spines and the recruitment and maintenance of AMPAr[69]. Arc is best characterized as a player in behavior and learning, but has also been identified in M1 following motor learning tasks[71]. Arc is also suspected to bind dynamin in its role as an intermediate-early gene which we also found to be downregulated at the interface[69]. We have observed downregulation of Arc expression overall and at 24 hours post-implantation. Loss of Arc at the post-synapse in the event of injury has been shown to exacerbate neuronal injury and even lead to neuronal death through endoplasmic reticulum stress and necroptosis[72]. Because loss of Arc has been implicated in the decline of neuronal health, this gene may find use as a novel biomarker for evaluating device-tissue integration.
Many of the DE synapse-associated genes identified in this study are known to be driven by calcium-based mechanisms. Gabbr1 is the primary component of the metabotropic G-protein coupled receptor for GABAB1. Gabbr2 (gpr51) combines with GABA-B1 as a heterodimer to form functional GABA-B receptors and inhibits high voltage activated Ca2+ channels as a driver of inhibitory post-synaptic potentials [73]. We have observed downregulation of both Gabbr1 and Gabbr2 24-hours post-implantation. If these downregulations are not solely a product of neuronal loss at the interface, downregulation of Gabbr1 and Gabbr2 could potentially be indicators of neuronal excitotoxicity and increased calcium influx at early stages in the tissue response. We observed later downregulation of the calcium/calmodulin-dependent protein kinase CaMKIIa which was significant one week following insertion. CaMKIIa is a gene that has been found to be necessary for neuronal function and long-term potentiation through its interaction with post-synaptic proteins in response to calcium influx[74], [75]. Nrgn has been reported to bind calmodulin (CaM) at the post-synapse and facilitate the generation of active CamKII required for long-term potentiation (LTP)[76]. We observed overall downregulation in Nrgn in interfacial vs. distal contrasts with pronounced changes at 24-hours post-implantation. Ngrn knockout studies have shown a marked decline in intracellular Ca2+ and increased incidence of long-term depression (LTD) of neuronal synapses[76]–[78]. Nrgn is a neuronal protein that is highly expressed in cortex, specifically in the post-synapse in dendritic spines[77], [78]. Cacna1i and Cacng3 are both neuronal low voltage-activated calcium channel components which are downregulated at the device interface 24 hours post-implantation. Cacna1i encodes the pore forming subunit of the CaV 3.3 ion channel in subsets of neurons such as GABAergic neurons in the thalamic reticular nucleus (TRN). In TRN neurons, the Cav3.3 ion channel is activated by transient membrane hyperpolarization as a mediator of rebound burst firing in oscillatory neuronal activity [79]. Cacng3 codes for a calcium channel γ3 auxiliary subunit that is also known as a transmembrane AMPA regulatory protein (TARP)[80], [81]. Both Cacna1i and Cacng3 have been reported to play roles in neuronal plasticity and in the development of epilepsy[79], [80].
Previous work by Eles et al. reports increased calcium-based activity as a direct result of device implantation-based trauma, which appeared to normalize by 1-month post-insertion. Insertion-driven Ca2+ influx can activate cellular mechanisms that contribute to axonal blebbing, axon transport disruption, neurite degeneration, synaptic degradation, and neuron death[82]. Early downregulation of Gabbr1 and Gabbr2 may facilitate early calcium influx and promote excitotoxicity. Decreased expression of a cluster of calcium-related genes at the one-week time point potentially could be an adaptive response following electrode insertion-driven Ca2+ influx to reduce Ca2+ driven activity. Future work will need to explore these mechanisms.
It is possible that monitoring synaptic-associated genes could serve as useful indicators of neuronal health and function in surviving populations. Downregulation of Syn1 may point to potential synaptic dysfunction and axonal disruptions in local neurons. For example, the significant downregulation of Bsn at 24 hours and 1 week post implantation could indicate a decline of neuronal populations or possibly indicate early synaptic dysfunction or neuronal loss at the device interface [61]. The observed downregulation of Stxbp1 overall and at 24 hours post-implantation may reflect early neuronal damage and loss of neuronal processes. Further investigation is required to determine whether these genes are related to adaptive mechanisms in individual neurons and/or neuronal loss, and assess their suitability as novel biomarkers for neuronal responses to implanted electrodes.
3.3. Differential Expression of Astrocyte Related Genes
3.3.1. Astroglial Scar-Associated Genes
Astrogliosis is considered to be a significant component of the fibrotic glial “scar” that forms over time around indwelling devices. This scar is believed to impede signal acquisition, segregate neuronal populations from insertion insult, and interfere with the exchange of ions and soluble factors[83]. We detected multiple DE genes associated with astrocytic activity around implanted devices (figure 5). Activated astrocytes at the device interface are commonly characterized through the progressive increase of GFAP and vimentin[21], [23], [24], [84], [85]. Our analysis confirmed a significant upregulation of these genes near the device interface and, in the case of Gfap, radiating out to tissue ~500 microns distal to the device (far versus naïve, log2FC = 2.219, padj= 0.042). Complementary to these previously-reported effects, we detected DE of additional genes potentially associated with glial scar formation. Col4a1, which astrocytes are known to secrete at sites of injury and inflammation, was significantly upregulated at the device interface overall and specifically at 24-hours and 1-week post-implantation[86], [87]. Ncan (chondroitin sulfate proteoglycan 3, CSPG3) was also upregulated through 1-week post-implantation at the device interface and is reportedly expressed by activated astrocytes in the fibrotic scar following traumatic brain injury[88]. Similarly to other reported CSPGs, Ncan is implicated in the failure of neural regeneration in the central nervous system (CNS) via interference of neuronal adhesion molecules and cadherins. At the device interface, the intermediate filament nestin (Nes) is upregulated overall and at 24-hours. Nestin is commonly associated with multiple cell types such as neural progenitors[89], but because Nes is strongly upregulated in proliferating reactive astrocytes[90], at the device interface this may indicate the transition of local astrocytes to reactive states.
Figure 5: Differential expression of genes associated with astrocyte activity at the device interface relative to naïve tissue.
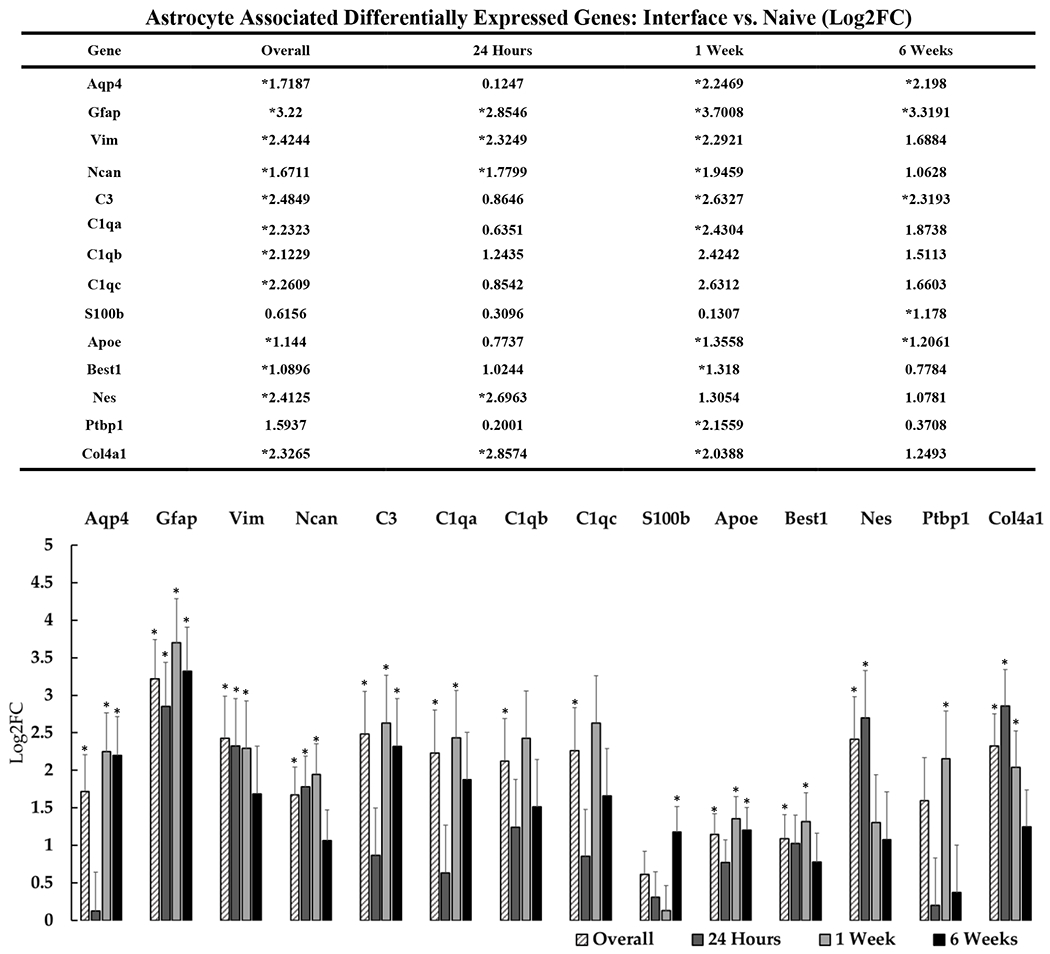
The table and representative graphs that outline the general upregulation of astrocyte associated genes at 24 hours, 1-week and 6-weeks post-implantation. “Overall” expression represents the group comparisons of samples pooled across time points. Significance was thresholded at Log2FC ≥ 0.6 and P ≤ 0.05. Asterisks (*) denote statistically significant differentially expressed genes.
3.3.2. Homeostatic Support and Repair
Astrocytes are also well known for their ability to communicate with neurons in the cortex and provide homeostatic support[83]. As such, open questions remain regarding the beneficial versus detrimental impacts of reactive astrocytes surrounding devices[91]. Our data unmasked modulation of genes potentially associated with a neuroprotective or reparative role. For example, we observed significant upregulation of Apolipoprotein E (Apoe), which has been associated with reactive astrocytes as well as neurons in inflammatory states[92]–[94]. Apoe plays a key role in positive cellular processes, but increased presence of Apoe is most commonly reported as a constituent of inflammatory tissue response which is common during neurodegeneration[92]–[94]. We also observed upregulation of Bestrophin-1 (Best1), which is an ion channel that is highly expressed in astrocytes in the brain and is permeable to both glutamate and GABA[95]. Under normal conditions, Best1 is localized to astrocytic processes where it favors glutamate release to maintain neuronal synapses. Under pathological conditions, Best1 is redistributed to the astrocytic soma and takes on the role of GABAergic release, which is known to suppress synaptic transmission and neuronal excitability[95]. At the device interface, this mechanism could potentially work to counteract neuronal excitotoxicity during the initial inflammatory phase of the tissue response created by BBB breach, microglial activation, and insertion-driven calcium influx. Modifying the excitatory/inhibitory tone of surrounding brain tissue has been previously proposed as a candidate protective mechanism to preserve neuronal tissue surrounding devices, albeit at the likely expense of signal generation[35], [49].
We observed significant upregulation of Aqp4 overall and at 1- and 6-weeks post-implantation. Aqp4 is essential for cellular water homeostasis in the brain and is abundantly expressed in astrocytic end-feed; its upregulation in astrocytes has been proposed to be involved in cell swelling during injury and ischemia. Aqp4 can also influence astrocyte-neuron communication as an adhesion molecule that is involved during cellular migration, neuromodulation, and neuronal plasticity. The complete extent to which Aqp4 is involved in the tissue response to brain injury is still unclear, but increased expression is strongly correlated with glial scar formation and inflammation[96]. Ptbp1 was strongly upregulated at the device interface relative to distal tissue and at 1-week post-implantation relative to naïve tissue. Ptbp1 is an RNA-binding protein which has been implicated in alternative splicing and the regulation of numerous cellular processes in the brain[97]–[99]. Recently, Ptbp1 has been shown to suppress pro-neural genes, and shRNA knockdown of Ptbp1 in midbrain converted astrocytes into functional dopaminergic neurons within the nigrostriatal region of the mouse brain[100]. It is still unclear if Ptbp1 upregulation at the interface correlates with increased astrocyte density, but due to the recently demonstrated potential for repair, this gene is a promising target for future investigation.
3.4. Differential Expression of Genes Associated with Microglia and Inflammation
Microglia have long been implicated in the tissue response to implanted electrode arrays as well as in neurodegenerative disease[101], [102]. In healthy cortical tissue, microglia play a supportive role in a variety of cellular processes such as synapse formation and maintenance, disposal of cellular debris, pruning of nonfunctional synapses, and promotion of oligodendrocyte precursor cell (OPC) survival and differentiation. Following insult to cortical tissue, microglia become activated, causing them to proliferate, migrate to sites of injury, produce inflammatory cytokines, upregulate lytic enzymes and assume a pathological phenotype[91], [103]–[108]. Activated microglia are documented to lose the ability to support healthy processes such as maintaining functioning synapses. Cytokines secreted by activated microglia can drive neurons into a state of excitotoxicity and neurodegeneration, potentially exacerbating an environment that is already unfriendly for neurons at the interface. In our data, we observed expected upregulation of genes typically associated with microglial reactivity (figure 6), particularly at early time points (e.g., Cx3cr1, Csf1r).
Figure 6: Differential expression of genes associated with inflammation and microglial activity at the device interface relative to naïve tissue.
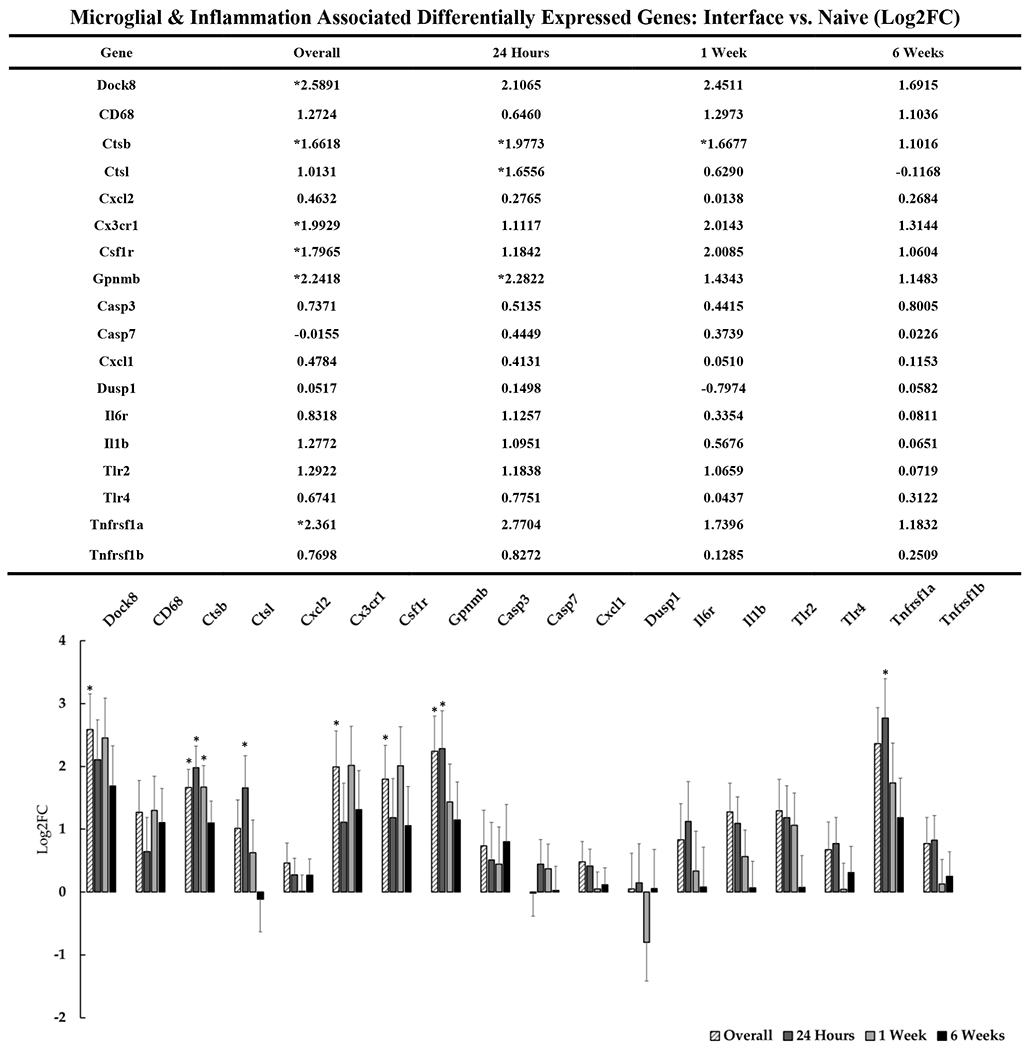
The table and representative graphs that show the generalized upregulation of microglial and inflammation associated genes at 24 hours, 1-week and 6-weeks post-implantation. “Overall” expression represents the group comparisons of samples pooled across time points. Significance was thresholded at Log2FC ≥ 0.6 and P ≤ 0.05. Asterisks (*) denote statistically significant differentially expressed genes.
The upregulation of lysosomal Ctsl also appears to validate microglial-driven inflammation at the interface. Knockdown studies have provided evidence that Cstl is associated with phagocytotic microglia and contributes to neuronal cell death[109]. We observed acute upregulation of Ctsl at 24-hours post-implantation. In the context of indwelling devices, upregulation of Ctsl could act as a marker for microglial activation and may contribute to inflammatory neuronal damage. Similarly to Ctsl, Ctsb is highly expressed in pro-inflammatory microglia and plays a role in degradation of extracellular matrix proteins and can contribute to neuronal damage[110]–[112]. Tnfrsf1a, as a known activator of inflammatory microglial pathways NF–κB and MAPK, is also modestly upregulated at the device interface. Ptprc (CD45) is associated with infiltrating leukocytes and is known to be expressed in microglia as well, so it is possible that upregulation of this gene suggests the presence of general macrophage-like activity. However, it is likely that Ptprc expression is being driven by the local microglial population. In general, our data confirms the expected presence of activated microglia at the device interface, while identifying the perturbation of previously unreported genes related to these cells.
We also observed a cluster of gene expression associated with the complement cascade relevant to microglial function, which is well documented in pathological states where cellular debris and apoptotic cell bodies are present[101], [107], [113], [114]. C3 and C1q bind the membrane of apoptotic cell bodies and synapses as a marker for pruning by local microglia[101], [105]. The upregulation of C1q and C3 have been reported to destabilize functional synapses[107]. Additionally, the secretion of C1q from microglia is associated with the induction neurotoxic ‘A1’ reactive astrocytes, which in turn stimulates C3 expression as a key biomarker of A1-astrocytes[91]. High complement levels during pathological states can lead to ‘over-pruning’ of synapses and myelin which can, in turn, lead to excessive loss of neuronal connectivity [108], [115]. Thus, genes associated with microglial-mediated inflammation and the complement cascade are candidate targets for restoration of lost neuronal network connectivity surrounding devices.
While the majority of our observations of microglial-associated genes suggest mechanisms associated with synaptic pruning, neurotoxicity and inflammation, upregulation of Gpnmb overall and at the 24-hour timepoint may suggest a more complex interplay of protective and detrimental effects. Gpnmb, which has been discussed in the context of Alzheimer’s disease, may suggest that there are microglia- mediated mechanisms which work to attenuate the inflammatory response of reactive astrocytes through CD44 receptor action[116]. Gpnmb is a transmembrane glycoprotein that has been reported to be expressed in microglia and macrophages in the brain and are reported to play roles in neurodegenerative states. Gpnmb has been shown to bind astrocytic CD44 to attenuate astrocyte driven inflammation and provide neuroprotection in neurodegenerative disease. As such, it has been suggested as a potential therapeutic target against neuroinflammation[116].
3.5. Differential Expression of Oligodendrocytes Associated Genes
Oligodendrocytes are well-known for their role in myelination of axonal fibers in the brain, but they also provide metabolic and trophic support directly to neurons. While previous studies have often focused on microglia and astrocytes as the primary glial players in the tissue response to implanted electrode arrays, more recent studies have explored the role of oligodendrocytes and their progenitors (OPCs) in device-tissue interaction. Our data identified several DE genes associated with oligodendrocytes and OPCs (figure 7). Interestingly, Ptprz1 was found to be upregulated overall and at every timepoint out to 6-weeks post-implantation. Ptprz1 is enriched in OPCs and is believed to play a role in the maintenance OPCs in an undifferentiated state[117]. Upregulation of Ptprz1 by itself doesn’t necessary imply that OPCs are being locked into an undifferentiated state, but it would allow for more binding sites for associated substrate molecules which have been shown to directly inhibit OPC differentiation into mature oligodendrocytes.
Figure 7: Differential expression of genes associated with oligodendrocytes at the device interface relative to naïve tissue.
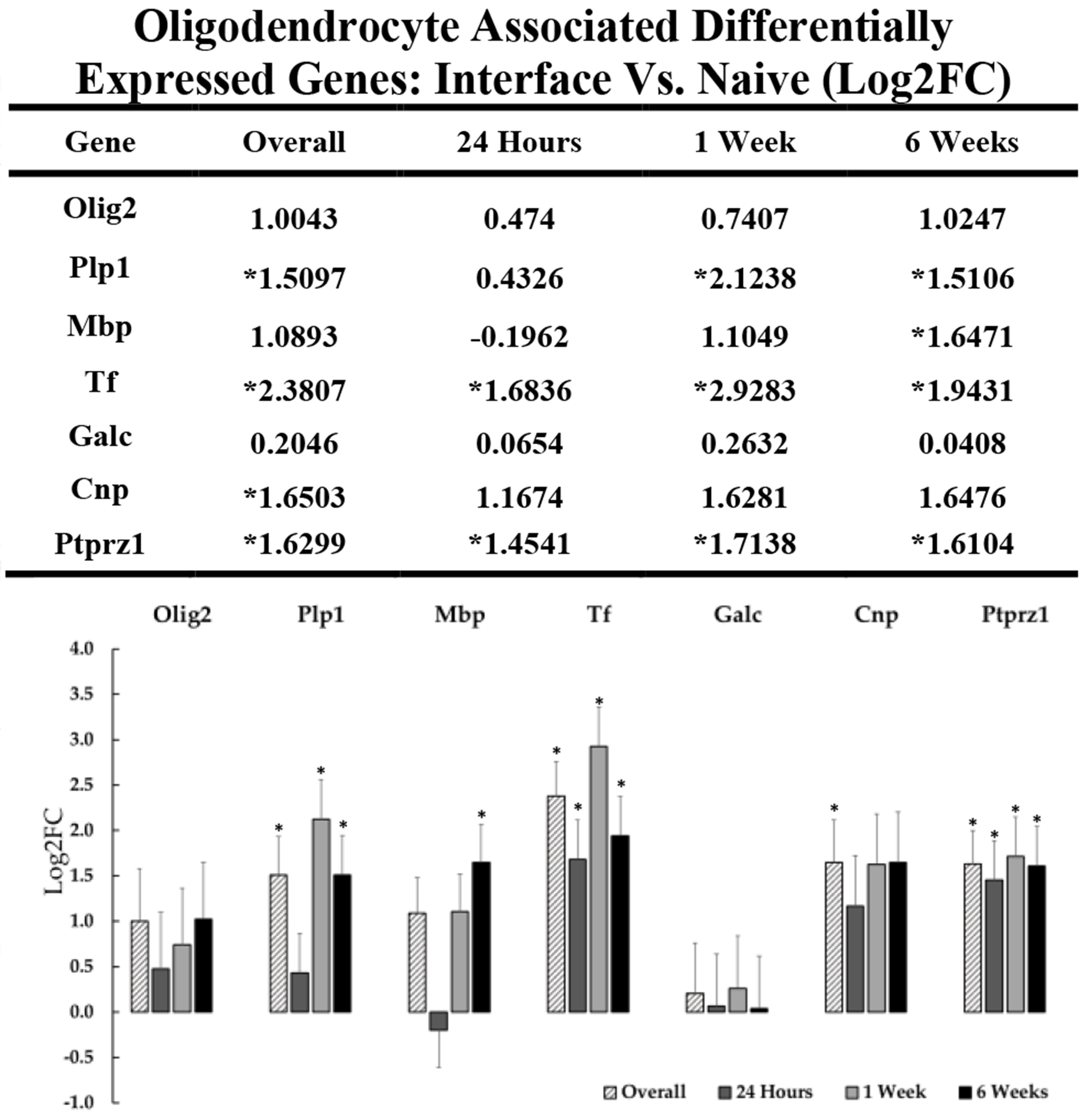
The table and representative graphs illustrate the upregulation of key genes associated with oligodendrocytes at 24 hours, 1-week and 6-weeks post-implantation. “Overall” expression represents the group comparisons of samples pooled across time points. Significance was thresholded at Log2FC ≥ 0.6 and P ≤ 0.05. Asterisks (*) denote statistically significant differentially expressed genes.
Oligodendrocytes are one of the few cell types in the brain to express transferrin (Tf) post developmentally[118], [119], and it is notable that Tf is upregulated at all timepoints throughout the six week implantation period in comparison to naïve tissue. It is possible that the chronic upregulation of iron sequestering proteins such as Tf and possibly Fth1 reflect the increased metabolic demands of oligodendrocytes, which may result from chronic cycles of damage and repair presented by a fixed, indwelling microelectrode array. Oligodendrocytes are known to be susceptible to oxidative damage due to their relatively high basal metabolic requirements to produce and maintain myelination while providing trophic support to nearby cellular populations. These demands may be further exacerbated in the injury zone of the device interface. As with other reactive glia, there may be a combination of reparative and degenerative effects of these cells at the interface.
Myelin is largely comprised of structural proteins Plp1 and Mbp, and the expression of these genes is directly linked to axonal myelin construction[120], [121]. It is possible that upregulation of these genes reflects a need for myelin regeneration and repair, or alternatively, the formation of damage-associated “myelinosomes.” Myelinosomes have been recently reported to be frequently targeted by microglia and invasive macrophages for phagocytosis[122], likely via the complement system. It has been reported that high prevalence and upregulation of Plp1 is directly linked to microglial activation and inflammation, and myelinosomes may contribute to persistent microglial inflammation at the interface[106]. The need for remyelination after myelinosome pruning may be one explanation for the upregulation of Plp1 and Mbp at the device interface over the duration of implantation. Plp1 overexpression also has been reported to directly influence activation of inflammatory microglia, so there is some uncertainty as to whether the upregulation of Plp1 at the device interface is regenerative or inflammatory[104], [106]. In the context of Alzheimer’s disease, states of chronic inflammation can drive OPCs into a proinflammatory state over long periods of time (out to 18 months)[123], [124], but we have not seen evidence in this 6-week dataset of that particular phenotype of oligodendrocyte. The chronic upregulation of oligodendrocyte and myelin specific genes such as Plp1 and Mbp at the device interface in our data supports the need to further understand the role of oligodendrocytes in device-tissue integration, which is an emerging line of inquiry recently pursued by Kozai and colleagues[22], [122], [125].
3.5. Differential Expression of Genes Associated with BBB and Oxidative Stress
Recent literature has begun to explore the role of BBB integrity as a component of the tissue response to indwelling electrodes. Insertion of devices in most cases causes ischemic insult through direct contact with vasculature. Transient rupture of the BBB causes an influx of circulatory cell types, plasma proteins, and extracellular iron, thus exacerbating the existing immune response[29]. Increased permeability of the BBB disrupts cortical homeostasis and is known to result in the upregulation of matrix metalloproteases, antioxidant activity, and genes that control regeneration of the neurovascular unit[29], [30]. BBB disruption and the associated oxidative stress that follows has been typically observed at 48 and 72 hours post implantation, with one report suggesting no significant upregulation of these genes within 24 hours of device insertion[30]. We detected numerous genes associated with blood brain barrier (figure 8) and oxidative stress (figure 9), but few of them were flagged as statistically significant DE. It is possible that we did not observe significant DE in genes associated with vascular trauma and associated pathways because the 24-hour time point was not a sufficient duration to reveal effects. While we did detect many genes associated with oxidative stress, neurovascular unit and inflammation, most of these effects were not statistically significant. However, we detected significant upregulation of the antioxidant Sod2 at the 24-hour timepoint, which may be related to acute oxidative stress following device insertion. Ncf1 was also found to be generally upregulated overall at the device interface. Ncf1 is enriched in phagocytic cells such as microglia and is upregulated as a part of the innate immune response. Upregulation of Ncf1 may also be a signifier of infiltrating neutrophils following BBB breach caused by device insertion. Ncf1 upregulation is known to directly increase the levels of reactive oxygen species (ROS) in the extracellular environment and may contribute to cellular damage at the device interface[30]. In addition to generators of oxidative stress, we observed upregulation of protective mechanisms which are responsive to oxidative stress in the brain. Fth1 has recently been characterized as a protectant against oxidative stress following device insertion[30]. We observed upregulation of Fth1 overall and also at 1-week post-implantation. Fth1 upregulation could be a sign of increased extracellular heme degradation due to increased BBB permeability related to device insertion and micromotion. Likewise, Rtn1 downregulation is notable since the reticulon protein family has been reported to play involvement in neuronal apoptotic pathways in injury and disease[126], [127]. Rtn1 upregulation following injury has been implicated in activation of apoptosis in neurons as a result of endoplasmic reticulum stress through the Bcl2 pathway[126]. Neuronal oxidative stress and cell death has been suggested at the device interface, but we have observed a marked downregulation of Rtn1 at the device interface, which could imply potentially compensatory activation of neuroprotective mechanisms in surviving neurons.
Figure 8: Differential expression of genes associated with blood brain barrier integrity at the device interface relative to naïve tissue.
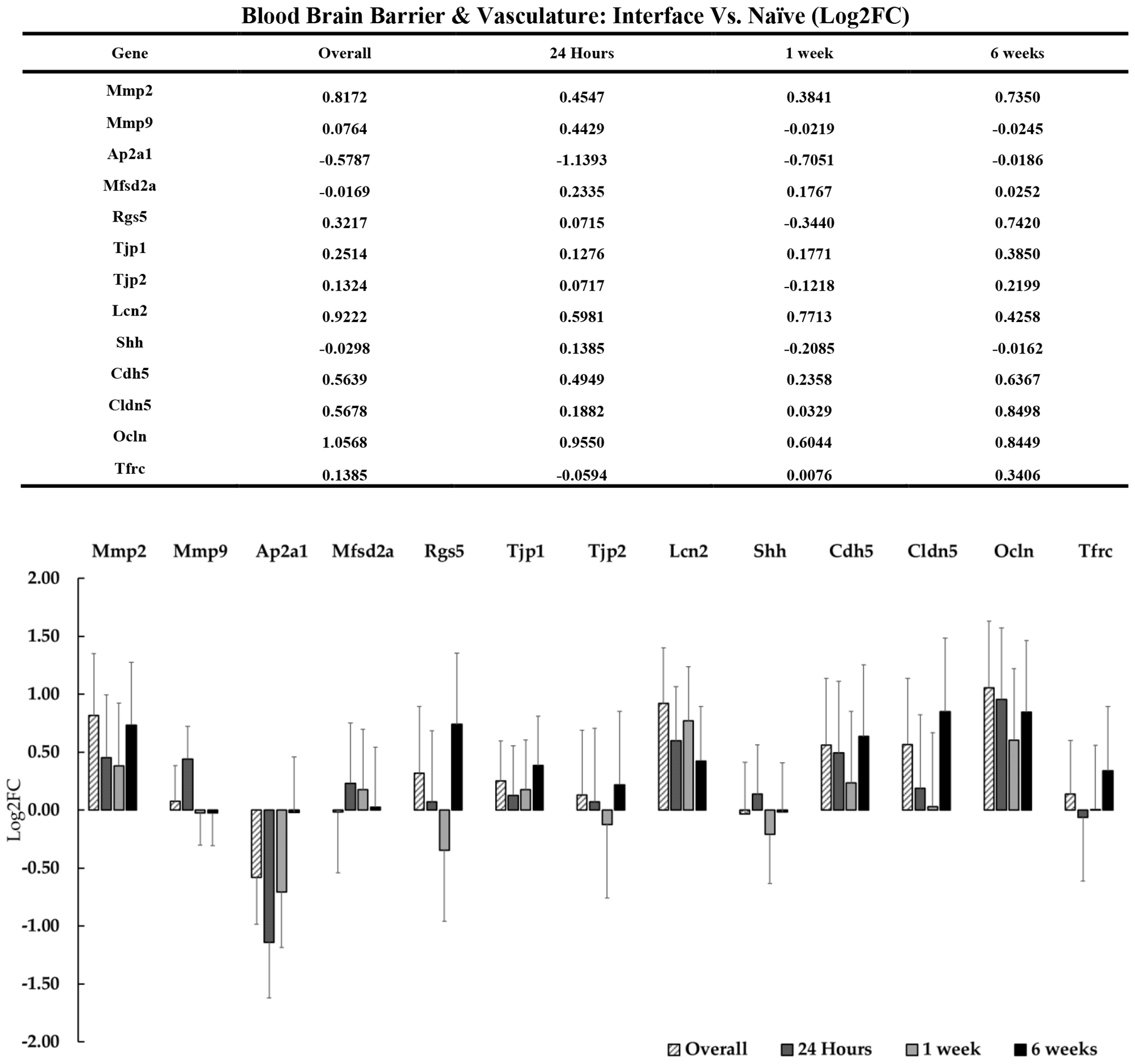
The table and representative graphs illustrate fluctuations of blood brain barrier associated genes at 24 hours, 1-week and 6-weeks post-implantation. “Overall” expression represents the group comparisons of samples pooled across time points. Significance was thresholded at Log2FC ≥ 0.6 and P ≤ 0.05. No genes were identified as significantly DE in this group.
Figure 9: Differential expression of genes associated with oxidative stress at the device interface relative to naïve tissue.
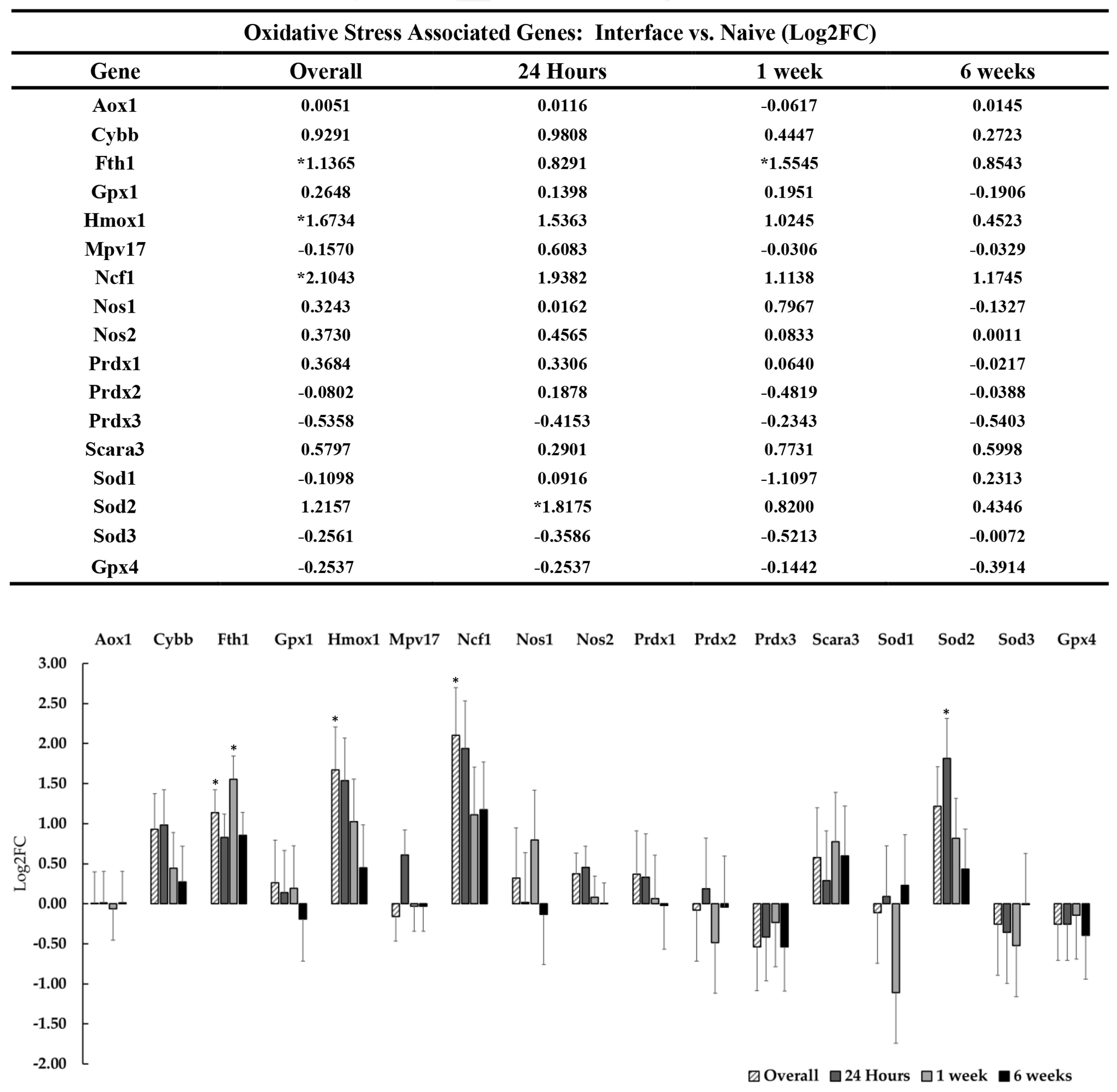
The table and representative graphs illustrate fluctuations of oxidative stress associated genes at 24 hours, 1-week and 6-weeks post-implantation. “Overall” expression represents the group comparisons of samples pooled across time points. Significance was thresholded at Log2FC ≥ 0.6 and P ≤ 0.05. Asterisks (*) denote significant differentially expressed genes.
4. Conclusion and Perspectives
This study has expanded current understanding of the complexity of the biological impacts of electrodes implanted in the brain. The data validate previous observations while identifying novel genes associated with the tissue response to implanted cortical devices. While we present and discuss selected genes in this initial report, we have provided the comprehensive raw data set, which includes many additional statistically significant DE genes, as supplementary files (1–15).
In addition to expected DE of genes associated with astrocytic fibrosis, inflammation, and glial activation, our transcriptional analysis has highlighted new DE genes at the device interface which may be contributing to performance outcomes. The observed upregulation of neuronal cytoskeletal genes in parallel with downregulation of synapse associated genes leads to new questions regarding the neuronal response at the device interface (i.e., plasticity versus loss). Changes in neuronal kinesins, pre- and post-synaptic proteins, and myelin structural proteins are all implicated in injury and neurogenerative disease, and the impact that these effects have in surviving neuronal populations at the device interface is the subject of future work. The coincident and persistent upregulation of Tf, Plp1, and Mbp support evidence that oligodendrocytes play a role in the tissue response. It is possible that neuronal injury and inflammation leads to increased generation of myelin associated proteins and a subsequent increase in oligodendrocyte metabolism required to maintain myelinated axons at the electrode interface. We also observed the expression of multiple genes which may contribute to a positive, adaptive function. For instance, potential astrocyte-driven neuronal hypo-excitability via Best1 may provide neuroprotective benefits immediately post-implantation, but prolonged neuronal inhibition may contribute to signal loss or instability over time. It is likely that the DE of genes at the device interface represent a spectrum of tissue response effects, both protective as well as detrimental to local interfacial tissue. The fibrotic scar that forms around the device is essential to re-establish the BBB and cortical homeostasis, but prolonged presence of an Ncan rich glial scar may prove to be detrimental for long-term device integration. Many of the DE genes are expressed in multiple cell types and may play multi-functional roles in the tissue response, which warrants additional investigations to determine cell-type specificity and downstream outcomes of gene expression effects.
We have also identified a small number (21) of DE genes in distal tissue to implanted devices relative to naïve tissue. We observed downregulation of the cholesterol synthesis intermediate lanosterol synthase (Lss) and 7SK RNA. Additionally, we observed upregulation of Gfap, Tensin3, collagen type IV, neural precursor cell expressed, neural precursor expressed developmentally down-regulated 9 (Nedd9), and the Hsp70 co-chaperone Hsp40 in distal tissue. Upregulation of Gfap is expected, but the DE of Lss and Nedd9 lead to questions regarding a novel role of these genes in the context of implanted electrode arrays and brain injury. The presence of DE genes in distal tissue suggests that future work should explore distal gene expression, as it raises new questions about an influence of the tissue response on the broader network generating the local field potential.
Many of the DE neuronal genes discussed in this study have been previously implicated in neurodegenerative diseases such as multiple sclerosis, Alzheimer’s disease, and Parkinson’s disease. The possibility that mechanisms are conserved between device-based tissue response and neurodegenerative disease may allow for insights to be shared between these fields of research. Future work will explore gene ontology and pathway analysis to contextualize newly identified DE genes surrounding devices.
Significant questions for further investigation remain, such as: (1) how do the changes in gene expression influence the interplay between affected cell types at the device interface and their contribution to the overall tissue response, (2) to what extent is this observed DE being driven by fluctuations in individual cells versus changes in cell populations at the device interface, (3) do the observed fluctuations in gene expression drive significant alterations in protein expression, and (4) which, if any, of these genes are useful biomarkers of signal quality? Finally, the observation that genes are DE in tissue 500 microns away from the device relative to unimplanted tissue indicates that the tissue response to the implanted electrode array may extend further than previously thought.
Future work may extend on the current observations by assessing chronic time-points beyond 6-weeks and performing focused analysis of gene expression localized to electrode sites. Likewise, assessing the relationship between recording quality and gene expression remains an important area of future work. Nonetheless, identification of genes associated with multiple cell types and processes at the device interface provides an expanded toolkit for evaluation of the tissue-device interface. This study has opened new avenues to investigate how the DE genes identified contribute to tissue response, creating opportunities for intervention and improved chronic performance.
Supplementary Material
Acknowledgements
The authors thank Rebecca Tagett and the Bioinformatics Core of the University of Michigan Medical School’s Biomedical Research Core Facilities for analysis support. Steven Suhr of BiomiLab, LLC provided helpful feedback and discussions. Kaleb Howard assisted with initial study planning.
This research is supported by the National Institutes of Health (R01-NS107451S1) and a National Science Foundation CAREER award.
References
- [1].Laxton AW et al. , “A phase I trial of deep brain stimulation of memory circuits in Alzheimer’s disease,” Ann. Neurol, vol. 68, no. 4, pp. 521–534, October. 2010. [DOI] [PubMed] [Google Scholar]
- [2].Kuhn J et al. , “Deep brain stimulation of the nucleus basalis of Meynert in Alzheimer’s dementia,” Mol. Psychiatry, vol. 20, no. 3, pp. 353–360, March. 2015. [DOI] [PubMed] [Google Scholar]
- [3].Whiting DM et al. , “Lateral hypothalamic area deep brain stimulation for refractory obesity: a pilot study with preliminary data on safety, body weight, and energy metabolism,” J. Neurosurg, vol. 119, no. 1, pp. 56–63, July. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ho AL, Sussman ES, Pendharkar AV, Azagury DE, Bohon C, and Halpern CH, “Deep brain stimulation for obesity: rationale and approach to trial design,” Neurosurg. Focus, vol. 38, no. 6, p. E8, June. 2015. [DOI] [PubMed] [Google Scholar]
- [5].Moore DR and Shannon RV, “Beyond cochlear implants: awakening the deafened brain,” Nat. Neurosci, vol. 12, no. 6, pp. 686–691, June. 2009. [DOI] [PubMed] [Google Scholar]
- [6].Lim HH and Lenarz T, “Auditory midbrain implant: Research and development towards a second clinical trial,” Hear. Res, vol. 322, no. 1, pp. 212–223, April. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lewis PM, Ackland HM, Lowery AJ, and Rosenfeld JV, “Restoration of vision in blind individuals using bionic devices: A review with a focus on cortical visual prostheses,” Brain Res, vol. 1595, pp. 51–73, January. 2015. [DOI] [PubMed] [Google Scholar]
- [8].Smit JV et al. , “Deep brain stimulation in tinnitus: Current and future perspectives,” Brain Res, vol. 1608, pp. 51–65, 2015. [DOI] [PubMed] [Google Scholar]
- [9].Collinger JL et al. , “High-performance neuroprosthetic control by an individual with tetraplegia,” Lancet, vol. 381, no. 9866, pp. 557–564, February. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Aflalo T et al. , “Decoding motor imagery from the posterior parietal cortex of a tetraplegic human,” Science (80-. )., vol. 348, no. 6237, pp. 906–910, May 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bouton CE et al. , “Restoring cortical control of functional movement in a human with quadriplegia,” Nature, vol. 533, no. 7602, pp. 247–250, May 2016. [DOI] [PubMed] [Google Scholar]
- [12].Sun FT, Morrell MJ, and Wharen RE, “Responsive cortical stimulation for the treatment of epilepsy,” Neurotherapeutics, vol. 5, no. 1, pp. 68–74, January. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Quinn EJ et al. , “Beta oscillations in freely moving Parkinson’s subjects are attenuated during deep brain stimulation,” Mov. Disord, vol. 30, no. 13, pp. 1750–1758, November. 2015. [DOI] [PubMed] [Google Scholar]
- [14].Grahn PJ et al. , “A neurochemical closed-loop controller for deep brain stimulation: toward individualized smart neuromodulation therapies,” Front. Neurosci, vol. 8, no. 8 JUN, p. 169, June. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Crowell AL, Riva-Posse P, Garlow SJ, and Mayberg HS, “Toward an Understanding of the Neural Circuitry of Major Depressive Disorder Through the Clinical Response to Deep Brain Stimulation of Different Anatomical Targets,” Curr. Behav. Neurosci. Reports, vol. 1, no. 2, pp. 55–63, June. 2014. [Google Scholar]
- [16].Hanson TL, Diaz-Botia CA, Kharazia V, Maharbiz MM, and Sabes PN, “The ‘sewing machine’ for minimally invasive neural recording,” bioRxiv, no. 1, p. 578542, 2019. [Google Scholar]
- [17].Musk E, “An integrated brain-machine interface platform with thousands of channels,” J. Med. Internet Res, vol. 21, no. 10, p. e16194, October. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Thompson CH, Riggins TE, Patel PR, Chestek CA, Li W, and Purcell E, “Toward guiding principles for the design of biologically-integrated electrodes for the central nervous system,” J. Neural Eng, vol. 17, no. 2, p. 021001, March. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Michelson NJ et al. , “Multi-scale, multi-modal analysis uncovers complex relationship at the brain tissue-implant neural interface: New emphasis on the biological interface,” J. Neural Eng, vol. 15, no. 3, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wellman SM et al. , “A Materials Roadmap to Functional Neural Interface Design,” Adv. Funct. Mater, vol. 28, no. 12, pp. 1–38, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kozai TDY, Jaquins-Gerstl AS, Vazquez AL, Michael AC, and Cui XT, “Brain tissue responses to neural implants impact signal sensitivity and intervention strategies,” ACS Chem. Neurosci, vol. 6, no. 1, pp. 48–67, January. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wellman SM and Kozai TDY, “In vivo spatiotemporal dynamics of NG2 glia activity caused by neural electrode implantation,” Biomaterials, vol. 164, pp. 121–133, May 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Biran R, Martin DC, and Tresco PA, “The brain tissue response to implanted silicon microelectrode arrays is increased when the device is tethered to the skull,” J. Biomed. Mater. Res. Part A, vol. 82A, no. 1, pp. 169–178, July. 2007. [DOI] [PubMed] [Google Scholar]
- [24].Biran R, Martin DC, and Tresco PA, “Neuronal cell loss accompanies the brain tissue response to chronically implanted silicon microelectrode arrays,” Exp. Neurol, vol. 195, no. 1, pp. 115–126, September. 2005. [DOI] [PubMed] [Google Scholar]
- [25].Ghane-Motlagh B and Sawan M, “Design and Implementation Challenges of Microelectrode Arrays: A Review,” Mater. Sci. Appl, vol. 04, no. 08, pp. 483–495, 2013. [Google Scholar]
- [26].Seymour JP and Kipke DR, “Neural probe design for reduced tissue encapsulation in CNS,” Biomaterials, vol. 28, no. 25, pp. 3594–3607, September. 2007. [DOI] [PubMed] [Google Scholar]
- [27].Rivnay J, Wang H, Fenno L, Deisseroth K, and Malliaras GG, “Next-generation probes, particles, and proteins for neural interfacing,” Sci. Adv, vol. 3, no. 6, p. e1601649, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Thompson CH, Zoratti MJ, Langhals NB, and Purcell EK, “Regenerative Electrode Interfaces for Neural Prostheses,” Tissue Eng. Part B Rev, vol. 22, no. 2, pp. 125–135, April. 2016. [DOI] [PubMed] [Google Scholar]
- [29].Bennett C, Samikkannu M, Mohammed F, Dietrich WD, Rajguru SM, and Prasad A, “Blood brain barrier (BBB)-disruption in intracortical silicon microelectrode implants,” Biomaterials, vol. 164, no. 3, pp. 1–10, May 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bennett C et al. , “Neuroinflammation, oxidative stress, and blood-brain barrier (BBB) disruption in acute Utah electrode array implants and the effect of deferoxamine as an iron chelator on acute foreign body response,” Biomaterials, vol. 188, pp. 144–159, January. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bedell HW, Schaub NJ, Capadona JR, and Ereifej ES, “Differential expression of genes involved in the acute innate immune response to intracortical microelectrodes,” Acta Biomater, vol. 102, no. xxxx, pp. 205–219, January. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kozai TDY et al. , “Reduction of neurovascular damage resulting from microelectrode insertion into the cerebral cortex using in vivo two-photon mapping,” J. Neural Eng, vol. 7, no. 4, p. 046011, August. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wellman SM, Cambi F, and Kozai TD, “The role of oligodendrocytes and their progenitors on neural interface technology: A novel perspective on tissue regeneration and repair,” Biomaterials, vol. 183, no. August, pp. 200–217, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ereifej ES et al. , “Implantation of Neural Probes in the Brain Elicits Oxidative Stress,” Front. Bioeng. Biotechnol, vol. 6, no. FEB, February. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Salatino JW, Kale AP, and Purcell EK, “Alterations in Ion Channel Expression Surrounding Implanted Microelectrode Arrays in the Brain,” bioRxiv, p. 518811, 2019. [Google Scholar]
- [36].Purcell EK, Thompson DE, Ludwig KA, and Kipke DR, “Flavopiridol reduces the impedance of neural prostheses in vivo without affecting recording quality,” J. Neurosci. Methods, vol. 183, no. 2, pp. 149–157, October. 2009. [DOI] [PubMed] [Google Scholar]
- [37].Andrews S, “No Title,” FastQC: a quality control tool for high throughput sequence data. [Online]. Available: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- [38].Dobin A et al. , “STAR: ultrafast universal RNA-seq aligner,” Bioinformatics, vol. 29, no. 1, pp. 15–21, January. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Li B and Dewey CN, “RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome,” Bioinforma. Impact Accurate Quantif. Proteomic Genet. Anal. Res, February. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Dobin A, “No Title.” [Online]. Available: https://github.com/alexdobin/STAR/blob/master/doc/STARmanual.pdf.
- [41].Love MI, Huber W, and Anders S, “Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2,” Genome Biol, vol. 15, no. 12, p. 550, December. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].“No Title.” [Online]. Available: http://www.geneontology.org/.
- [43].Draghici S et al. , “A systems biology approach for pathway level analysis,” Genome Res, vol. 17, no. 10, pp. 1537–1545, September. 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zhang ZH et al. , “A Comparative Study of Techniques for Differential Expression Analysis on RNA-Seq Data,” PLoS One, vol. 9, no. 8, p. e103207, August. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Zhang Y et al. , “An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex,” J. Neurosci, vol. 34, no. 36, pp. 11929–11947, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Byron SA, Van Keuren-Jensen KR, Engelthaler DM, Carpten JD, and Craig DW, “Translating RNA sequencing into clinical diagnostics: opportunities and challenges,” Nat. Rev. Genet, vol. 17, no. 5, pp. 257–271, May 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gregory B et al. , “Structural and Functional Changes of Pyramidal Neurons in Primary Motor Cortex at the Site of an Implanted Microelectrode Array,” Soc. Neurosci. Present, 2021. [Google Scholar]
- [48].Kozai TDY et al. , “Chronic tissue response to carboxymethyl cellulose based dissolvable insertion needle for ultra-small neural probes,” Biomaterials, vol. 35, no. 34, pp. 9255–9268, 2014. [DOI] [PubMed] [Google Scholar]
- [49].Salatino JW, Winter BM, Drazin MH, and Purcell EK, “Functional remodeling of subtype-specific markers surrounding implanted neuroprostheses,” J. Neurophysiol, vol. 118, no. 1, pp. 194–202, July. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Perrot R, Berges R, Bocquet A, and Eyer J, “Review of the multiple aspects of neurofilament functions, and their possible contribution to neurodegeneration,” Mol. Neurobiol, vol. 38, no. 1, pp. 27–65, 2008. [DOI] [PubMed] [Google Scholar]
- [51].Millecamps S and Julien JP, “Axonal transport deficits and neurodegenerative diseases,” Nat. Rev. Neurosci, vol. 14, no. 3, pp. 161–176, 2013. [DOI] [PubMed] [Google Scholar]
- [52].Szodorai A et al. , “APP anterograde transport requires Rab3A GTPase activity for assembly of the transport vesicle,” J. Neurosci, vol. 29, no. 46, pp. 14534–14544, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lin MY and Sheng ZH, “Regulation of mitochondrial transport in neurons,” Exp. Cell Res, vol. 334, no. 1, pp. 35–44, May 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Su YY et al. , “KIF5B promotes the forward transport and axonal function of the voltage-gated sodium channel Nav1.8,” J. Neurosci, vol. 33, no. 45, pp. 17884–17896, November. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Chang DTW and Reynolds IJ, “Mitochondrial trafficking and morphology in healthy and injured neurons,” Prog. Neurobiol, vol. 80, no. 5, pp. 241–268, December. 2006. [DOI] [PubMed] [Google Scholar]
- [56].Hares K et al. , “Overexpression of Kinesin Superfamily Motor Proteins in Alzheimer’s Disease,” J. Alzheimer’s Dis, vol. 60, no. 4, pp. 1511–1524, 2017. [DOI] [PubMed] [Google Scholar]
- [57].Xia C-H et al. , “Abnormal neurofilament transport caused by targeted disruption of neuronal kinesin heavy chain KIF5A,” J. Cell Biol, vol. 161, no. 1, pp. 55–66, April. 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Dutta R and Sarkar SR, “Role of Dynein and Dynactin (DCTN-1) in Neurodegenerative Diseases,” Neurophysiol. Rehabil, vol. 2, no. 1, pp. 53–58, December. 2019. [Google Scholar]
- [59].Cesca F, Baldelli P, Valtorta F, and Benfenati F, “The synapsins: Key actors of synapse function and plasticity,” Prog. Neurobiol, vol. 91, no. 4, pp. 313–348, 2010. [DOI] [PubMed] [Google Scholar]
- [60].Frank T et al. , “Bassoon and the synaptic ribbon organize Ca2+ channels and vesicles to add release sites and promote refilling,” Neuron, vol. 68, no. 4, pp. 724–738, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Richter K et al. , “Presynaptic cytomatrix protein bassoon is localized at both excitatory and inhibitory synapses of rat brain,” J. Comp. Neurol, vol. 408, no. 3, pp. 437–448, April. 1999. [DOI] [PubMed] [Google Scholar]
- [62].Schattling B et al. , “Bassoon proteinopathy drives neurodegeneration in multiple sclerosis,” Nat. Neurosci, vol. 22, no. 6, pp. 887–896, 2019. [DOI] [PubMed] [Google Scholar]
- [63].Verhage M et al. , “Synaptic assembly of the brain in the absence of neurotransmitter secretion,” Science (80-. )., vol. 287, no. 5454, pp. 864–869, 2000. [DOI] [PubMed] [Google Scholar]
- [64].Toonen RFG et al. , “Munc18–1 expression level control synapse recovery by regulating readily releasable pool size,” Proc. Natl. Acad. Sci. U. S. A, vol. 103, no. 48, pp. 18332–18337, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Kádková A, Radecke J, and Sørensen JB, “The SNAP-25 Protein Family,” Neuroscience, vol. 420, pp. 50–71, 2019. [DOI] [PubMed] [Google Scholar]
- [66].Antonucci F, Corradini I, Fossati G, Tomasoni R, Menna E, and Matteoli M, “SNAP-25, a Known presynaptic protein with emerging postsynaptic functions,” Front. Synaptic Neurosci, vol. 8, no. MAR, pp. 1–9, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Hayashi N, Oohira A, and Miyata S, “Synaptic localization of receptor-type protein tyrosine phosphatase ζ/β in the cerebral and hippocampal neurons of adult rats,” Brain Res, vol. 1050, no. 1–2, pp. 163–169, 2005. [DOI] [PubMed] [Google Scholar]
- [68].Tiwari SS et al. , “Alzheimer-related decrease in CYFIP2 links amyloid production to tau hyperphosphorylation and memory loss,” Brain, vol. 139, no. 10, pp. 2751–2765, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Korb E and Finkbeiner S, “Arc in synaptic plasticity: From gene to behavior,” Trends Neurosci, vol. 34, no. 11, pp. 591–598, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Li L, Carter J, Gao X, Whitehead J, and Tourtellotte WG, “The Neuroplasticity-Associated Arc Gene Is a Direct Transcriptional Target of Early Growth Response (Egr) Transcription Factors,” Mol. Cell. Biol, vol. 25, no. 23, pp. 10286–10300, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Hosp JA, Mann S, Wegenast-Braun BM, Calhoun ME, and Luft AR, “Region and task-specific activation of Arc in primary motor cortex of rats following motor skill learning,” Neuroscience, vol. 250, pp. 557–564, 2013. [DOI] [PubMed] [Google Scholar]
- [72].Chen T, Zhu J, Wang Y-H, and Hang C-H, “Arc silence aggravates traumatic neuronal injury via mGluR1-mediated ER stress and necroptosis,” Cell Death Dis, vol. 11, no. 1, p. 4, January. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Zai G, King N, Wong GWH, Barr CL, and Kennedy JL, “Possible association between the gamma-aminobutyric acid type B receptor 1 (GABBR1) gene and schizophrenia,” Eur. Neuropsychopharmacol, vol. 15, no. 3, pp. 347–352, 2005. [DOI] [PubMed] [Google Scholar]
- [74].Kool MJ et al. , “CAMK2-dependent signaling in neurons is essential for survival,” J. Neurosci, vol. 39, no. 28, pp. 5424–5439, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Takemoto-Kimura S et al. , “Calmodulin kinases: essential regulators in health and disease,” J. Neurochem, vol. 141, no. 6, pp. 808–818, 2017. [DOI] [PubMed] [Google Scholar]
- [76].Huang KP, Huang FL, Jäger T, Li J, Reymann KG, and Balschun D, “Neurogranin/RC3 enhances long-term potentiation and learning by promoting calcium-mediated signaling,” J. Neurosci, vol. 24, no. 47, pp. 10660–10669, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Díez-Guerra FJ, “Neurogranin, a link between calcium/calmodulin and protein kinase C signaling in synaptic plasticity,” IUBMB Life, vol. 62, no. 8, pp. 597–606, 2010. [DOI] [PubMed] [Google Scholar]
- [78].Zhabotinsky AM, Camp RN, Epstein IR, and Lisman JE, “Role of the neurogranin concentrated in spines in the induction of long-term potentiation,” J. Neurosci, vol. 26, no. 28, pp. 7337–7347, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Ghoshal A et al. , “Effects of a patient-derived de novo coding alteration of CACNA1I in mice connect a schizophrenia risk gene with sleep spindle deficits,” Transl. Psychiatry, vol. 10, no. 1, pp. 1–12, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Kato AS, Gill MB, Yu H, Nisenbaum ES, and Bredt DS, “TARPs differentially decorate AMPA receptors to specify neuropharmacology,” Trends Neurosci, vol. 33, no. 5, pp. 241–248, May 2010. [DOI] [PubMed] [Google Scholar]
- [81].Templin JS, Bang SJ, Soiza-Reilly M, Berde CB, and Commons KG, “Patterned expression of ion channel genes in mouse dorsal raphe nucleus determined with the Allen Mouse Brain Atlas,” Brain Res, vol. 1457, no. 1, pp. 1–12, May 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Eles JR, Vazquez AL, Kozai TDY, and Cui XT, “In vivo imaging of neuronal calcium during electrode implantation: Spatial and temporal mapping of damage and recovery,” Biomaterials, vol. 174, pp. 79–94, August. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Salatino JW, Ludwig KA, Kozai TDY, and Purcell EK, “Glial responses to implanted electrodes in the brain,” Nat. Biomed. Eng, vol. 1, no. 11, pp. 862–877, November. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Szarowski DHH et al. , “Brain responses to micro-machined silicon devices.,” Brain Res, vol. 983, no. 1–2, pp. 23–35, September. 2003. [DOI] [PubMed] [Google Scholar]
- [85].Adolf A et al. , “Release of astroglial vimentin by extracellular vesicles: Modulation of binding and internalization of C3 transferase in astrocytes and neurons,” Glia, vol. 67, no. 4, pp. 703–717, 2019. [DOI] [PubMed] [Google Scholar]
- [86].Skorupa A et al. , “Angiogenin induces modifications in the astrocyte secretome: Relevance to amyotrophic lateral sclerosis,” J. Proteomics, vol. 91, pp. 274–285, 2013. [DOI] [PubMed] [Google Scholar]
- [87].Liesi P and Kauppila T, “Induction of type IV collagen and other basement-membrane-associated proteins after spinal cord injury of the adult rat may participate in formation of the glial scar,” Exp. Neurol, vol. 173, no. 1, pp. 31–45, 2002. [DOI] [PubMed] [Google Scholar]
- [88].Heindryckx F and Li JP, “Role of proteoglycans in neuro-inflammation and central nervous system fibrosis,” Matrix Biol, vol. 68–69, pp. 589–601, August. 2018. [DOI] [PubMed] [Google Scholar]
- [89].Bernal A and Arranz L, “Nestin-expressing progenitor cells: function, identity and therapeutic implications,” Cell. Mol. Life Sci, vol. 75, no. 12, pp. 2177–2195, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Tamagno I and Schiffer D, “Nestin expression in reactive astrocytes of human pathology,” J. Neurooncol, vol. 80, no. 3, pp. 227–233, 2006. [DOI] [PubMed] [Google Scholar]
- [91].Liddelow SA et al. , “Neurotoxic reactive astrocytes are induced by activated microglia,” Nature, vol. 541, no. 7638, pp. 481–487, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Tzioras M, Davies C, Newman A, Jackson R, and Spires-Jones T, “Invited Review: APOE at the interface of inflammation, neurodegeneration and pathological protein spread in Alzheimer’s disease,” Neuropathol. Appl. Neurobiol, vol. 45, no. 4, pp. 327–346, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Rebeck GW, “The role of APOE on lipid homeostasis and inflammation in normal brains,” J. Lipid Res, vol. 58, no. 8, pp. 1493–1499, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Iwata A, Browne KD, Chen XH, Yuguchi T, and Smith DH, “Traumatic brain injury induces biphasic upregulation of ApoE and ApoJ protein in rats,” J. Neurosci. Res, vol. 82, no. 1, pp. 103–114, 2005. [DOI] [PubMed] [Google Scholar]
- [95].Oh SJ and Lee CJ, “Distribution and function of the Bestrophin-1 (Best1) channel in the brain,” Exp. Neurobiol, vol. 26, no. 3, pp. 113–121, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Ikeshima-Kataoka H, “Neuroimmunological implications of AQP4 in astrocytes,” Int. J. Mol. Sci, vol. 17, no. 8, pp. 1–16, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Vuong JK, Lin CH, Zhang M, Chen L, Black DL, and Zheng S, “PTBP1 and PTBP2 Serve Both Specific and Redundant Functions in Neuronal Pre-mRNA Splicing,” Cell Rep, vol. 17, no. 10, pp. 2766–2775, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Pan J, Ma N, Yu B, Zhang W, and Wan J, “Transcriptomic profiling of microglia and astrocytes throughout aging,” J. Neuroinflammation, vol. 17, no. 1, pp. 1–19, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Georgilis A et al. , “PTBP1-Mediated Alternative Splicing Regulates the Inflammatory Secretome and the Pro-tumorigenic Effects of Senescent Cells,” Cancer Cell, vol. 34, no. 1, pp. 85–102.e9, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Qian H et al. , “Reversing a model of Parkinson’s disease with in situ converted nigral neurons,” Nature, vol. 582, no. 7813, pp. 550–556, June. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Presumey J, Bialas AR, and Carroll MC, “Complement System in Neural Synapse Elimination in Development and Disease,” in Advances in Immunology, 1st ed., vol. 135, Elsevier Inc., 2017, pp. 53–79. [DOI] [PubMed] [Google Scholar]
- [102].Li Q and Barres BA, “Microglia and macrophages in brain homeostasis and disease,” Nat. Rev. Immunol, vol. 18, no. 4, pp. 225–242, 2018. [DOI] [PubMed] [Google Scholar]
- [103].Hong S, Dissing-Olesen L, and Stevens B, “New insights on the role of microglia in synaptic pruning in health and disease,” Curr. Opin. Neurobiol, vol. 36, pp. 128–134, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Shimizu T et al. , “The balance between cathepsin C and cystatin F controls remyelination in the brain of Plp1-overexpressing mouse, a chronic demyelinating disease model,” Glia, vol. 65, no. 6, pp. 917–930, 2017. [DOI] [PubMed] [Google Scholar]
- [105].Kettenmann H, Kirchhoff F, and Verkhratsky A, “Microglia: New Roles for the Synaptic Stripper,” Neuron, vol. 77, no. 1, pp. 10–18, 2013. [DOI] [PubMed] [Google Scholar]
- [106].Tatar CL, Appikatla S, Bessert DA, Paintlia AS, Singh I, and Skoff RP, “Increased Plp1 gene expression leads to massive microglial cell activation and inflammation throughout the brain,” ASN Neuro, vol. 2, no. 4, pp. 219–231, August. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Zabel MK and Kirsch WM, “From development to dysfunction: Microglia and the complement cascade in CNS homeostasis,” Ageing Res. Rev, vol. 12, no. 3, pp. 749–756, June. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Keren-Shaul H et al. , “A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease,” Cell, vol. 169, no. 7, pp. 1276–1290.e17, 2017. [DOI] [PubMed] [Google Scholar]
- [109].Xu S, Zhang H, Yang X, Qian Y, and Xiao Q, “Inhibition of cathepsin L alleviates the microglia-mediated neuroinflammatory responses through caspase-8 and NF-κB pathways,” Neurobiol. Aging, vol. 62, pp. 159–167, 2018. [DOI] [PubMed] [Google Scholar]
- [110].Luo CL et al. , “Cathepsin B contributes to traumatic brain injury-induced cell death through a mitochondria-mediated apoptotic pathway,” J. Neurosci. Res, vol. 88, no. 13, pp. 2847–2858, 2010. [DOI] [PubMed] [Google Scholar]
- [111].Hook G, Jacobsen JS, Grabstein K, Kindy M, and Hook V, “Cathepsin B is a new drug target for traumatic brain injury therapeutics: Evidence for E64d as a promising lead drug candidate,” Front. Neurol, vol. 6, no. SEP, pp. 1–27, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Kingham PJ and Pocock JM, “Microglial secreted cathepsin B induces neuronal apoptosis,” J. Neurochem, vol. 76, no. 5, pp. 1475–1484, 2001. [DOI] [PubMed] [Google Scholar]
- [113].Stevens B et al. , “The Classical Complement Cascade Mediates CNS Synapse Elimination,” Cell, vol. 131, no. 6, pp. 1164–1178, 2007. [DOI] [PubMed] [Google Scholar]
- [114].Hammad A, Westacott L, and Zaben M, “The role of the complement system in traumatic brain injury: A review,” J. Neuroinflammation, vol. 15, no. 1, pp. 1–15, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Hickman S, Izzy S, Sen P, Morsett L, and El Khoury J, “Microglia in neurodegeneration,” Nat. Neurosci, vol. 21, no. 10, pp. 1359–1369, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Neal ML, Boyle AM, Budge KM, Safadi FF, and Richardson JR, “The glycoprotein GPNMB attenuates astrocyte inflammatory responses through the CD44 receptor,” J. Neuroinflammation, vol. 15, no. 1, pp. 1–14, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Tanga N et al. , “The PTN-PTPRZ signal activates the AFAP1L2-dependent PI3K-AKT pathway for oligodendrocyte differentiation: Targeted inactivation of PTPRZ activity in mice,” Glia, vol. 67, no. 5, pp. 967–984, 2019. [DOI] [PubMed] [Google Scholar]
- [118].Cahoy JD et al. , “A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function,” J. Neurosci, vol. 28, no. 1, pp. 264–278, January. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Todorich B, Pasquini JM, Garcia CI, Paez PM, and Connor JR, “Oligodendrocytes and myelination: The role of iron,” Glia, vol. 57, no. 5, pp. 467–478, April. 2009. [DOI] [PubMed] [Google Scholar]
- [120].Boggs JM, “Myelin basic protein: A multifunctional protein,” Cell. Mol. Life Sci, vol. 63, no. 17, pp. 1945–1961, September. 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Nave KA, “Myelination and support of axonal integrity by glia,” Nature, vol. 468, no. 7321, pp. 244–252, 2010. [DOI] [PubMed] [Google Scholar]
- [122].Chen K, Wellman SM, Yaxiaer Y, Eles JR, and Kozai TD, “In vivo spatiotemporal patterns of oligodendrocyte and myelin damage at the neural electrode interface,” Biomaterials, vol. 268, no. Iii, p. 120526, November. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Zhou Y et al. , “Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease,” Nat. Med, vol. 26, no. 1, pp. 131–142, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Zhang P et al. , “Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model,” Nat. Neurosci, vol. 22, no. 5, pp. 719–728, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Wellman SM, Li L, Yaxiaer Y, McNamara I, and Kozai TDY, “Revealing Spatial and Temporal Patterns of Cell Death, Glial Proliferation, and Blood-Brain Barrier Dysfunction Around Implanted Intracortical Neural Interfaces,” Front. Neurosci, vol. 13, no. MAY, pp. 1–17, May 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Gong L, Tang Y, An R, Lin M, Chen L, and Du J, “RTN1-C mediates cerebral ischemia/reperfusion injury via ER stress and mitochondria-associated apoptosis pathways,” Cell Death Dis, vol. 8, no. 10, p. e3080, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Shi Q, Ge Y, He W, Hu X, and Yan R, “RTN1 and RTN3 protein are differentially associated with senile plaques in Alzheimer’s brains,” Sci. Rep, vol. 7, no. 1, pp. 1–9, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Yong YX, Li YM, Lian J, Luo CM, Zhong DX, and Han K, “Inhibitory role of lentivirus-mediated aquaporin-4 gene silencing in the formation of glial scar in a rat model of traumatic brain injury,” J. Cell. Biochem, vol. 120, no. 1, pp. 368–379, 2019. [DOI] [PubMed] [Google Scholar]
- [129].Lu G, Kwong WH, Li Q, Wang X, Feng Z, and Yew DT, “Bcl2, Bax, and Nestin in the Brains of Patients With Neurodegeneration and Those of Normal Aging,” J. Mol. Neurosci, vol. 27, no. 2, pp. 167–174, 2005. [DOI] [PubMed] [Google Scholar]
- [130].Lian H, Litvinchuk A, Chiang ACA, Aithmitti N, Jankowsky JL, and Zheng H, “Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of alzheimer’s disease,” J. Neurosci, vol. 36, no. 2, pp. 577–589, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Xia J et al. , “Identification and characterization of human neuronal voltage-gated calcium channel gamma 3 subunit gene,” Chinese Sci. Bull, vol. 45, no. 23, pp. 2172–2176, December. 2000. [Google Scholar]
- [132].Chia PH et al. , “A homozygous loss-of-function camk2a mutation causes growth delay, frequent seizures and severe intellectual disability,” Elife, vol. 7, pp. 1–19, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Chui R and Dorovini-Zis K, “Regulation of CCL2 and CCL3 expression in human brain endothelial cells by cytokines and lipopolysaccharide,” J. Neuroinflammation, vol. 7, pp. 1–12, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Lopez ME, “Combined in situ Hybridization/Immunohistochemistry (ISH/IH) on Free-floating Vibratome Tissue Sections.,” Bio-protocol, vol. 4, no. 18, pp. 1–10, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Hendrickx DAE, van Eden CG, Schuurman KG, Hamann J, and Huitinga I, “Staining of HLA-DR, Iba1 and CD68 in human microglia reveals partially overlapping expression depending on cellular morphology and pathology,” J. Neuroimmunol, vol. 309, no. November 2016, pp. 12–22, 2017. [DOI] [PubMed] [Google Scholar]
- [136].Gaston JD, Bischel LL, Fitzgerald LA, Cusick KD, Ringeisen BR, and Pirlo RK, “Gene Expression Changes in Long-Term in Vitro Human Blood-Brain Barrier Models and Their Dependence on a Transwell Scaffold Materia,” J. Healthc. Eng, vol. 2017, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Jiao H, Wang Z, Liu Y, Wang P, and Xue Y, “Specific role of tight junction proteins claudin-5, occludin, and ZO-1 of the blood-brain barrier in a focal cerebral ischemic insult,” J. Mol. Neurosci, vol. 44, no. 2, pp. 130–139, 2011. [DOI] [PubMed] [Google Scholar]
- [138].Stamm S, Casper D, Dinsmore J, Kaufmann CA, Brosius J, and Helfman DM, “Clathrin light chain B: gene structure and neuron-specific splicing.,” Nucleic Acids Res, vol. 20, no. 19, pp. 5097–103, October. 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Royle SJ and Lagnado L, “Clathrin-Mediated Endocytosis at the Synaptic Terminal: Bridging the Gap Between Physiology and Molecules,” Traffic, vol. 11, no. 12, pp. 1489–1497, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Yang L, Kan EM, Lu J, Wu C, and Ling EA, “Expression of 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNPase) and its roles in activated microglia in vivo and in vitro,” J. Neuroinflammation, vol. 11, no. 1, pp. 1–17, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, and Rüttinger D, “Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy,” J. Immunother. Cancer, vol. 5, no. 1, pp. 1–13, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Hering H and Sheng M, “Activity-Dependent Redistribution and Essential Role of Cortactin in Dendritic Spine Morphogenesis,” J. Neurosci, vol. 23, no. 37, pp. 11759–11769, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Tamura Y et al. , “Association study of the chemokine, CXC motif, ligand 1 (CXCL1) gene with sporadic Alzheimer’s disease in a Japanese population,” Neurosci. Lett, vol. 379, no. 3, pp. 149–151, 2005. [DOI] [PubMed] [Google Scholar]
- [144].Babcock AA, Kuziel WA, Rivest S, and Owens T, “Chemokine expression by glial cells directs leukocytes to sites of axonal injury in the CNS,” J. Neurosci, vol. 23, no. 21, pp. 7922–7930, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Özer I, “Analyzing the Role of CyFIP2 in the Mouse Brain,” p. 160, 2019. [Google Scholar]
- [146].Namekata K, Guo X, Kimura A, Arai N, Harada C, and Harada T, “DOCK8 is expressed in microglia, and it regulates microglial activity during neurodegeneration in murine disease models,” J. Biol. Chem, vol. 294, no. 36, pp. 13421–13433, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Kearney CJ, Randall KL, and Oliaro J, “DOCK8 regulates signal transduction events to control immunity,” Cell. Mol. Immunol, vol. 14, no. 5, pp. 406–411, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Weinstock NI et al. , “Brainstem development requires galactosylceramidase and is critical for pathogenesis in a model of Krabbe disease,” Nat. Commun, vol. 11, no. 1, pp. 1–16, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Burda JE, Bernstein AM, and Sofroniew MV, “Astrocyte roles in traumatic brain injury,” Exp. Neurol, vol. 275, pp. 305–315, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Tanaka H et al. , “The potential of GPNMB as novel neuroprotective factor in amyotrophic lateral sclerosis,” Sci. Rep, vol. 2, pp. 1–11, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Helmy A, De Simoni MG, Guilfoyle MR, Carpenter KLH, and Hutchinson PJ, “Cytokines and innate inflammation in the pathogenesis of human traumatic brain injury,” Prog. Neurobiol, vol. 95, no. 3, pp. 352–372, 2011. [DOI] [PubMed] [Google Scholar]
- [152].Nakajima K, Yin X, Takei Y, Seog DH, Homma N, and Hirokawa N, “Molecular Motor KIF5A Is Essential for GABAA Receptor Transport, and KIF5A Deletion Causes Epilepsy,” Neuron, vol. 76, no. 5, pp. 945–961, December. 2012. [DOI] [PubMed] [Google Scholar]
- [153].Rivera J, Chu PJ, Lewis TL, and Arnold DB, “The role of Kif5B in axonal localization of Kv1 K+ channels,” Eur. J. Neurosci, vol. 25, no. 1, pp. 136–146, January. 2007. [DOI] [PubMed] [Google Scholar]
- [154].Ma H, Cai Q, Lu W, Sheng ZH, and Mochida S, “KIF5B motor adaptor syntabulin maintains synaptic transmission in sympathetic neurons,” J. Neurosci, vol. 29, no. 41, pp. 13019–13029, October. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Iworima DG, Pasqualotto BA, and Rintoul GL, “Kif5 regulates mitochondrial movement, morphology, function and neuronal survival.,” Mol. Cell. Neurosci, vol. 72, pp. 22–33, April. 2016. [DOI] [PubMed] [Google Scholar]
- [156].Willemsen MH et al. , “Involvement of the kinesin family members KIF4A and KIF5C in intellectual disability and synaptic function,” J. Med. Genet, vol. 51, no. 7, pp. 487–494, 2014. [DOI] [PubMed] [Google Scholar]
- [157].Bi F et al. , “Reactive astrocytes secrete lcn2 to promote neuron death,” Proc. Natl. Acad. Sci. U. S. A, vol. 110, no. 10, pp. 4069–4074, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Tokuraku K, Okuyama S, Matsushima K, Ikezu T, and Kotani S, “Distinct neuronal localization of microtubule-associated protein 4 in the mammalian brain,” Neurosci. Lett, vol. 484, no. 2, pp. 143–147, 2010. [DOI] [PubMed] [Google Scholar]
- [159].Dehmelt L and Halpain S, “Protein family review The MAP2 / Tau family of microtubule-associated proteins,” Genome Biol, vol. 6, no. 1, pp. 1–10, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Yang YR et al. , “Mfsd2a (major facilitator superfamily domain containing 2a) attenuates intracerebral hemorrhage-induced blood-brain barrier disruption by inhibiting vesicular transcytosis,” J. Am. Heart Assoc, vol. 6, no. 7, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Walker EJ and Rosenberg GA, “Divergent role for MMP-2 in myelin breakdown and oligodendrocyte death following transient global ischemia,” J. Neurosci. Res, vol. 88, no. 4, pp. 764–773, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Leppert D, Leib SL, Grygar C, Miller KM, Schaad UB, and Holländer GA, “Matrix metalloproteinase (MMP)-8 and MMP-9 in cerebrospinal fluid during bacterial meningitis: Association with blood-brain barrier damage and neurological sequelae,” Clin. Infect. Dis, vol. 31, no. 1, pp. 80–84, 2000. [DOI] [PubMed] [Google Scholar]
- [163].Sajad M, Zargan J, Chawla R, Umar S, and Khan HA, “Upregulation of CSPG3 accompanies neuronal progenitor proliferation and migration in EAE,” J. Mol. Neurosci, vol. 43, no. 3, pp. 531–540, 2011. [DOI] [PubMed] [Google Scholar]
- [164].Lee SJ et al. , “Presynaptic neuronal pentraxin receptor organizes excitatory and inhibitory synapses,” J. Neurosci, vol. 37, no. 5, pp. 1062–1080, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Figueiro-Silva J et al. , “Neuronal pentraxin 1 negatively regulates excitatory synapse density and synaptic plasticity,” J. Neurosci, vol. 35, no. 14, pp. 5504–5521, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Hawkins BT, Lundeen TF, Norwood KM, Brooks HL, and Egleton RD, “Increased blood-brain barrier permeability and altered tight junctions in experimental diabetes in the rat: Contribution of hyperglycaemia and matrix metalloproteinases,” Diabetologia, vol. 50, no. 1, pp. 202–211, 2007. [DOI] [PubMed] [Google Scholar]
- [167].Sauvageot CM and Stiles CD, “Molecular mechanisms controlling cortical gliogenesis,” Curr. Opin. Neurobiol, vol. 12, no. 3, pp. 244–249, 2002. [DOI] [PubMed] [Google Scholar]
- [168].Griffiths I et al. , “Axonal swellings and degeneration in mice lacking the major proteolipid of myelin,” Science (80-. )., vol. 280, no. 5369, pp. 1610–1613, 1998. [DOI] [PubMed] [Google Scholar]
- [169].Kuboyama K, Fujikawa A, Masumura M, Suzuki R, Matsumoto M, and Noda M, “Protein Tyrosine Phosphatase Receptor Type Z Negatively Regulates Oligodendrocyte Differentiation and Myelination,” PLoS One, vol. 7, no. 11, pp. 1–11, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Sladojevic N, Yu B, and Liao JK, “Regulator of G-Protein Signaling 5 Maintains Brain Endothelial Cell Function in Focal Cerebral Ischemia,” J. Am. Heart Assoc, vol. 9, no. 18, p. e017533, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Watts ME, Wu C, and Rubin LL, “Suppression of MAP4K4 Signaling Ameliorates Motor Neuron Degeneration in Amyotrophic Lateral Sclerosis-Molecular Studies Toward New Therapeutics,” J. Exp. Neurosci, vol. 13, pp. 10–13, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Kim KK, Kim YC, Adelstein RS, and Kawamoto S, “Fox-3 and PSF interact to activate neural cell-specific alternative splicing,” Nucleic Acids Res, vol. 39, no. 8, pp. 3064–3078, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Jaffe RB, “Induction of pluripotent stem cells from adult human fibroblasts by defined factors: Commentary,” Obstet. Gynecol. Surv, vol. 63, no. 3, p. 153, 2008. [Google Scholar]
- [174].Irfan M, Gopaul KR, Miry O, Hökfelt T, Stanton PK, and Bark C, “SNAP-25 isoforms differentially regulate synaptic transmission and long-term synaptic plasticity at central synapses,” Sci. Rep, vol. 9, no. 1, pp. 1–14, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Milh M et al. , “Epileptic and nonepileptic features in patients with early onset epileptic encephalopathy and STXBP1 mutations,” Epilepsia, vol. 52, no. 10, pp. 1828–1834, 2011. [DOI] [PubMed] [Google Scholar]
- [176].Gitler D et al. , “Different presynaptic roles of synapsins at excitatory and inhibitory synapses,” J. Neurosci, vol. 24, no. 50, pp. 11368–11380, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Van Wageningen TA, Vlaar E, Kooij G, Jongenelen CAM, Geurts JJG, and Van Dam AM, “Regulation of microglial TMEM119 and P2RY12 immunoreactivity in multiple sclerosis white and grey matter lesions is dependent on their inflammatory environment,” Acta Neuropathol. Commun, vol. 7, no. 1, pp. 1–16, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Paban V et al. , “Molecular gene expression following blunt and rotational models of traumatic brain injury parallel injuries associated with stroke and depression,” J. Transl. Sci, vol. 2, no. 6, 2016. [Google Scholar]
- [179].Carmona S, Zahs K, Wu E, Dakin K, Bras J, and Guerreiro R, “The role of TREM2 in Alzheimer’s disease and other neurodegenerative disorders,” Lancet Neurol, vol. 17, no. 8, pp. 721–730, 2018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.