

Abstract
Hypertension is a common comorbid condition in patients with diabetes. The pathogenesis of hypertension in diabetes has not been fully clarified. Primary tubular hyperreabsorption may contribute, which may be counteracted by glomerular hyperfiltration in the early diabetic kidney. In this study, we hypothesize that in early diabetes, the macula densa neuronal nitric oxide synthase (NOS1)-derived nitric oxide (NO) production is enhanced, which blunts tubuloglomerular feedback (TGF) response, promotes glomerular hyperfiltration and maintains normal blood pressure; conversely, insufficient NO generation by the macula densa induces hypertension by lowering glomerular filtration rate (GFR) and thus inhibiting natriuresis. To test this hypothesis, we examined the changes of macula densa NOS1 expression and phosphorylation as well as NO production, TGF response, GFR, sodium excretion and blood pressure in a murine model of leptin receptor-deficient (db/db) diabetes with or without macula densa-specific NOS1 deletion. We found that db/db mice presented reduced fractional renal sodium excretion (FENa) and only a small increase in blood pressure, associated with upregulated expression and activity of macula densa NOS1, inhibited TGF response, and glomerular hyperfiltration. Genetic knockout of macula densa NOS1 restored the TGF response and attenuated glomerular hyperfiltration in db/db mice, but also further reduced FENa and substantially increased blood pressure. In conclusion, the present study demonstrates that in the early stage of leptin receptor-deficient diabetes, the upregulation of macula densa NOS1 inhibits TGF and increases GFR, which counteracts renal sodium retention and limits the rise in blood pressure.
Keywords: hypertension, diabetes, renal hemodynamics, tubuloglomerular feedback, macula densa, nitric oxide
INTRODUCTION
Over 34.2 million Americans - 10.5% of the population have diabetes. The rate of hypertension is more than twice in patients with diabetes than those without diabetes.1, 2 Diabetic patients with hypertension exhibit an increased risk of cardiovascular disease and an exacerbation of other diabetic complications.2-4 Although the pathogenesis of hypertension in diabetes has not been fully clarified, one possible reason is the primary proximal tubular hyperreabsorption, which may be offset by glomerular hyperfiltration in the early diabetic kidney.5
The pathophysiology of diabetic glomerular hyperfiltration has been incompletely understood and several mechanisms have been proposed, primarily including vascular and tubular theories. According to the vascular hypothesis, glomerular hyperfiltration results from the imbalance between vasoconstrictive and vasodilatory factors in diabetes.6-8 The tubular thesis describes that the tubular growth as well as the upregulation of sodium-glucose cotransporter 2 (SGLT2) in diabetic kidney enhance proximal tubular reabsorption, which reduces the NaCl delivery to the macula densa and thereby increases glomerular filtration rate (GFR) via attenuation of tubuloglomerular feedback (TGF).5, 9-11 The tubular hypothesis proposes that the increase in GFR in diabetes serves to normalize early distal NaCl delivery and thereby renal NaCl excretion.5 We have recently identified a new mechanism for the hyperglycemia-induced glomerular hyperfiltration: the increase in luminal glucose load at the macula densa during hyperglycemia is sensed through sodium-glucose cotransporter 1 (SGLT1) and upregulates neuronal nitric oxide synthase (NOS1)-derived nitric oxide (NO) generation, which inhibits the TGF responsiveness and thus promotes the elevation of GFR (macula densa SGLT1-NOS1-TGF pathway).12-15 However, the long-term significance of this new mechanism in the control of hemodynamics in diabetes remains to be fully determined.
In the present study, we investigate the significance of macula densa NOS1 in the regulation of TGF, GFR, sodium excretion, and blood pressure in leptin receptor-deficient (db/db) mice, a model of type 2 diabetes. We hypothesize that in early diabetes, the macula densa NOS1-derived NO production is enhanced, which blunts TGF response, promotes glomerular hyperfiltration and maintains normal blood pressure; conversely, insufficient NO generation by the macula densa induces hypertension by lowering GFR and thus inhibiting natriuresis. To test this hypothesis, a set of sophisticated techniques are employed (including microperfusion of juxtaglomerular apparatus (JGAs) in vitro, micropuncture of renal tubules in vivo, clearance kinetics of plasma FITC-sinistrin, and radio-telemetry blood pressure monitoring) to determine macula densa NOS1 expression and phosphorylation as well as NO production, TGF response, GFR, fractional excretion of sodium (FENa) and blood pressure in db/db mice with or without macula densa–selective NOS1 deletion.
METHODS
All data, analytical methods, and study materials16, 17 are available from the corresponding author on reasonable request.
Experimental Animals
All animal experiments were conducted in accordance with the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals with all animal protocols approved by the Institutional Animal Care and Use Committee at the University of South Florida Morsani College of Medicine. As DBA/2J genetic background enhances the diabetic phenotypes including hyperglycemia as well as glomerular hyperfiltration in db/db mice,18, 19 db/db mice on C57BLKS/J genetic background (including 71% C57BL/6J and 25% DBA/2J; #000642, Jackson Laboratories) were used as diabetic models and db/+ mice on C57BLKS/J genetic background (#000642, Jackson Laboratories) were used as non-diabetic controls. For additional studies, MD-NOS1KO (NKCC2cre/NOS1flox/flox) mice, which we generated on C57BL/6J genetic background,20 were first backcrossed to C57BLKS/J mice (#000662, Jackson Laboratories) for 4 generations and then crossed to db/+ mice to generate db/db MD-NOS1KO mice. Littermate db/db NOS1flox/flox mice were used as diabetic controls and littermate db/+ MD-NOS1KO as well as db/+ NOS1flox/flox mice were used as non-diabetic controls. All the mice used in the experiments were males at the age of 8-12 weeks and housed individually at 23°C on a 12:12-h light-dark cycle, with food and water ad libitum (#2918; Envigo, Indianapolis, IN).
Statistical Analysis
Statistical analysis was performed using Prism 8 (GraphPad Software; San Diego, CA). The effects of interest were tested using t-test, two-way analysis of variance (ANOVA), or two-way repeated measures ANOVA followed by Sidak multiple comparisons test when appropriate. Data were presented as a mean ± SD, and a p-value <0.05 was considered statistically significant.
RESULTS
The Development of Glomerular Hyperfiltration in db/db Mice is Associated with Upregulated Macula Densa NOS1 and Inhibited TGF.
Blood glucose concentration was significantly higher in db/db mice (e.g., 473±30 mg/dl at 12 weeks of age) than in db/+ mice (149±20 mg/dl at 12 weeks of age) (Figure 1A). GFR was ~60% higher in db/db mice (e.g., 407±13 μl/min at 12 weeks of age) than in db/+ mice (251±12 μl/min at 12 weeks of age) (Figure 1B).12, 20, 21
Figure 1. Blood glucose, GFR and TGF responsiveness in db/db mice.
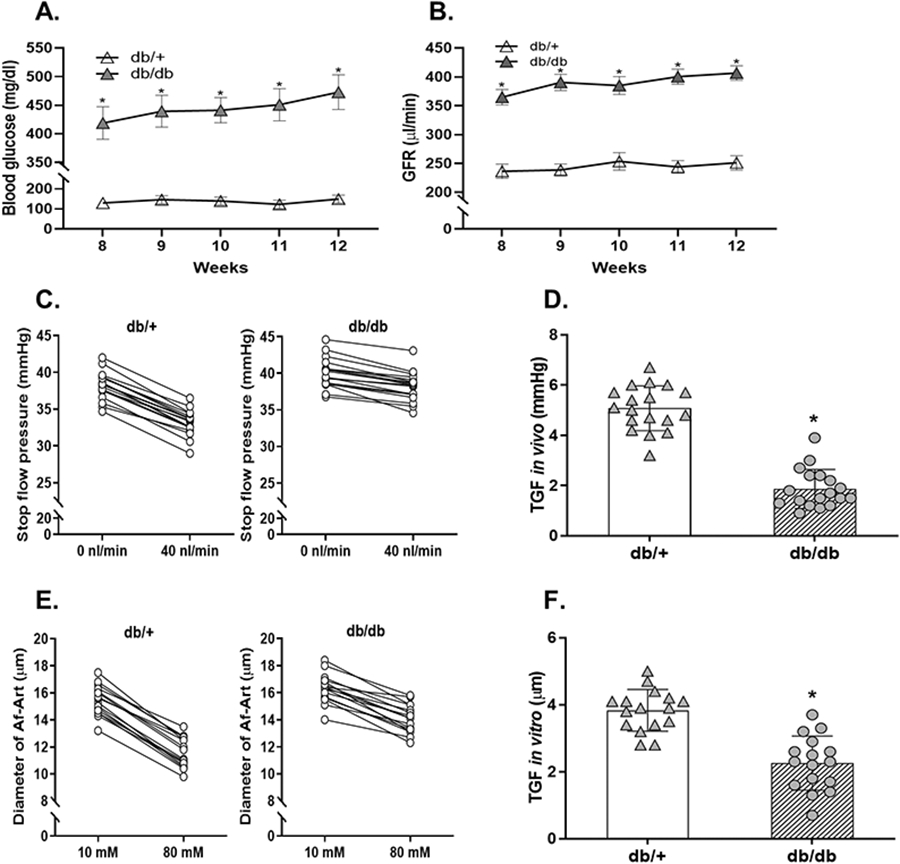
(A) The blood glucose concentration in db/db and db/+ mice. n=12; *P<0.01 vs db/+. (B) The GFR in db/db and db/+ mice. n=10; *P<0.01 vs db/+. (C and D) The TGF response in vivo in db/db and db/+ mice. n=18 tubules/6 mice per group; *P<0.01 vs db/+. (E and F) The TGF response in vitro in Akita and WT mice. n=16 nephrons/group; *P<0.01 vs db/+, *P<0.01 vs db/+. Statistical difference in (A and B) was calculated by two-way repeated measures ANOVA followed by Sidak multiple comparisons test. Statistical difference in (C-F) was calculated by t-test.
To determine the changes in TGF responsiveness during the early stage of diabetes, we measured the TGF response in vivo with micropuncture in db/db and db/+ mice at 12 weeks of age.12, 20-22 In db/+ mice, when tubular perfusion rate of ATF was increased from 0 to 40 nl/min, Psf fell from 38.2±1.8 to 33.1±1.7 mmHg, and thereby the TGF response in vivo, as indicated by the ΔPsf, was 5.1±0.9 mmHg (Figure 1C). In db/db mice, the TGF response in vivo, as indicated by the ΔPsf, was 1.8±0.7 mmHg (Figure 1C), which was ~62% less compared with db/+ mice (Figure 1D).
To eliminate systemic confounding factors such as hormones and sympathetic activity, we also measured the TGF response in vitro in isolated and double perfused JGAs.12, 20-22 In db/+ mice, when NaCl concentration in tubular perfusate was increased from 10 to 80 mM, the diameter of Af-Art decreased from 15.4±1.1 to 11.6±1.0 μm, and thereby the TGF response in vitro, as indicated by the change in Af-Art diameter, was 3.8±0.6 μm (Figure 1E). In db/db mice, the TGF response in vitro, as indicated by the change in Af-Art diameter, was 2.2±0.8 μm (Figure 1E), which was ~40% less compared with db/+ mice (Figure 1F).
To determine the changes in macula densa NO production during the early stage of diabetes, we measured the TGF-induced NO generation in the macula densa of isolated and perfused JGAs with DAF-2 DA in db/db and db/+ mice at 12 weeks of age.12, 20-22 In db/+ mice, when NaCl concentration in tubular perfusate was switched from 10 to 80 mM, macula densa NO production increased by 37.2±8.1% from 109.9±12.2 to 150.4±13.3 units/min. In db/db mice, when NaCl concentration in tubular perfusate was switched from 10 to 80 mM, macula densa NO generation increased by 71.0±15.7% from 113.4±10.4 to 192.8±12.5 units/min, which was significantly greater than in db/+ mice (Figure 2A).
Figure 2. Macula densa NOS1 expression and activity in db/db mice.
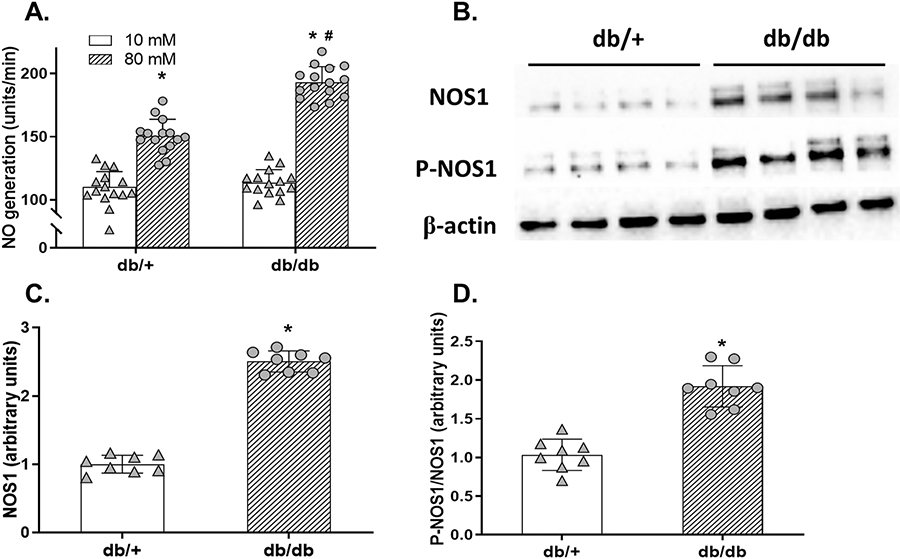
(A) The TGF-induced NO generation at the macula densa in db/db and db/+ mice. n=15; *P<0.01 vs 10 mM, #P<0.01 vs db/+. (B) The immunoblots of renal cortical NOS1, P-NOS1, and loading control of β-actin in db/db and db/+ mice. (C and D) The renal cortical NOS1 and P-NOS1/NOS1 levels in db/db and db/+ mice. n=8; *P<0.01 vs db/+. Statistical difference in (A) was calculated by two-way ANOVA followed by Sidak multiple comparisons test. Statistical difference in (C-D) was calculated by t-test.
To determine the changes in macula densa NOS1 expression and phosphorylation during the early stage of diabetes, we measured the protein abundances of NOS1 and P-NOS1 in the renal cortex, where most of the NOS1 comes from the macula densa,12, 23, 24 in db/db and db/+ mice at 12 weeks of age (Figure 2B). NOS1 levels (Figure 2C) as well as P-NOS1/NOS1 ratios (Figure 2D) were 2.50±0.15-fold and 1.85±0.26-fold higher, respectively, in db/db mice compared with db/+ mice.
These results demonstrate that the development of glomerular hyperfiltration in the early stage of leptin receptor deficiency (db/db)-induced diabetes is associated with upregulation of macula densa NOS1 as well as inhibition of TGF.
The Development of a Diabetic Mouse Model with Deletion of Macula Densa NOS1.
The diabetic mouse model with deletion of macula densa NOS1, db/db MD-NOS1KO, was generated by crossing the db/db mouse line with the MD-NOS1KO mouse line. Blood glucose concentration was not significantly different in db/db MD-NOS1KO mice compared with db/db NOS1flox/flox mice, which indicates that the leptin receptor deficiency (db/db)-induced diabetic condition was not affected by the genetic recombination with the MD-NOS1KO mouse line (Figure 3A). Double-immunofluorescence staining of NOS1 and NKCC2 in the kidney slices of db/db MD-NOS1KO mice confirmed the absence of NOS1 expression in the macula densa,12, 20, 25 which indicates that the deletion of macula densa NOS1 was not affected by the genetic recombination with the db/db mouse line (Figure 3B).
Figure 3. Characterization of db/db MD-NOS1KO mouse model.

(A) The blood glucose concentration in db/+ NOS1flox/flox, db/db NOS1flox/flox, db/+ MD-NOS1KO and db/db MD-NOS1KO mice. n=10; *P<0.01 vs db/+. (B) The double immunofluorescence staining of NOS1 and NKCC2 in the kidney slices of db/db NOS1flox/flox and db/db MD-NOS1KO mice. Red: NOS1; Green: NKCC2; Blue: nucleus; Arrow: macula densa. Statistical difference in (A) was calculated by two-way repeated measures ANOVA followed by Sidak multiple comparisons test.
Selective Deletion of Macula Densa NOS1 Attenuates Diabetes-Induced Inhibition of TGF and Elevation of GFR in db/db mice.
To determine the significance of macula densa NOS1 in the inhibition of TGF during the early stage of diabetes, we measured the TGF response in vivo with micropuncture in diabetic (db/db) and non-diabetic (db/+) MD-NOS1KO as well as NOS1flox/flox mice at 12 weeks of age. In NOS1flox/flox mice, the TGF response in vivo, as indicated by ΔPsf, was ~55% less in the diabetic group (2.0±0.8 mmHg) compared with the non-diabetic group (4.8±0.9 mmHg) (Figures 4A and 4B). In MD-NOS1KO mice, the TGF response in vivo (which was already enhanced) was only ~15% less in the diabetic group (6.6±0.8 mmHg) compared with the non-diabetic group (7.8±1.0 mmHg) (Figures 4A and 4B). The inhibition of TGF in vivo during the early stage of leptin receptor deficiency (db/db)-induced diabetes was significantly less in the MD-NOS1KO mice than the NOS1flox/flox mice (Figure 4B).
Figure 4. Effect of macula densa NOS1 deletion on TGF responsiveness as well as GFR in db/db mice.
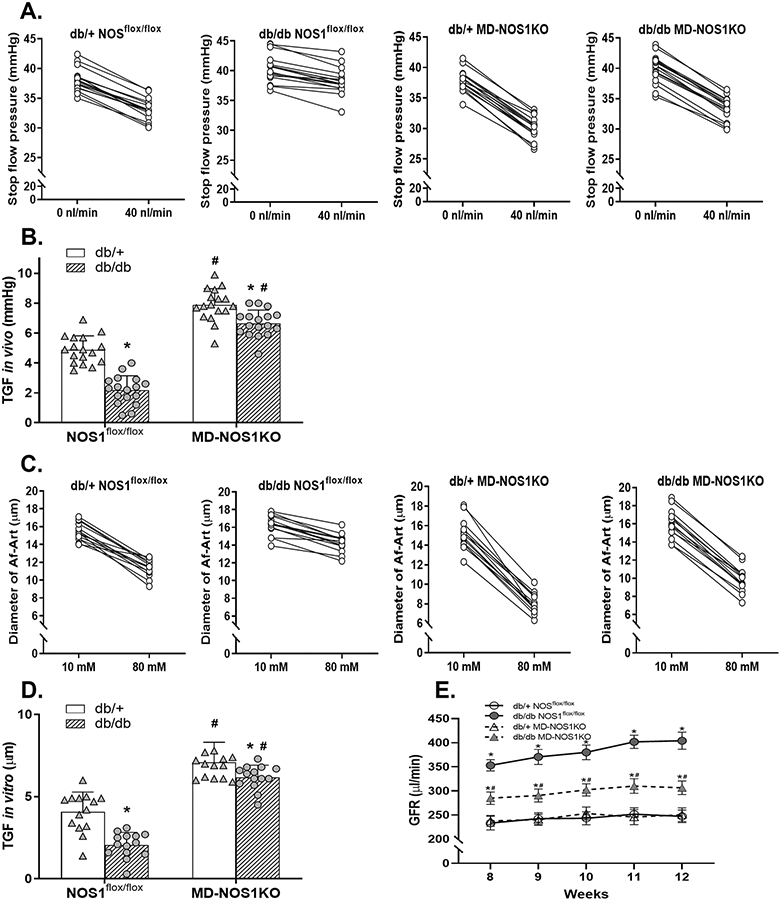
(A and B) The TGF response in vivo in db/+ NOS1flox/flox, db/db NOS1flox/flox, db/+ MD-NOS1KO and db/db MD-NOS1KO mice. n=17 tubules/5 mice per group; *P<0.01 vs db/+; #P<0.01 vs NOS1flox/flox. (C and D) The TGF response in vitro in db/+ NOS1flox/flox, db/db NOS1flox/flox, db/+ MD-NOS1KO and db/db MD-NOS1KO mice. n=14 nephrons/group; *P<0.05 vs db/+; #P<0.01 vs NOS1flox/flox. (E) The GFR in db/+ NOS1flox/flox, db/db NOS1flox/flox, db/+ MD-NOS1KO and db/db MD-NOS1KO mice. n=10; *P<0.01 vs db/+; #P<0.01 vs NOS1flox/flox. Statistical difference in (A and C) was calculated by t-test. Statistical difference in (B and D) was calculated by two-way ANOVA followed by Sidak multiple comparisons test. Statistical difference in (E) was calculated by two-way repeated measures ANOVA followed by Sidak multiple comparisons test.
To eliminate systemic confounding factors, we also measured the TGF response in vitro in isolated and double perfused JGAs. In NOS1flox/flox mice, the TGF response in vitro, as indicated by the change of Af-Art diameter, was ~50% less in the diabetic group (2.0±0.7 μm) compared with the non-diabetic group (4.1±1.2 μm) (Figures 4C and 4D). In MD-NOS1KO mice, the TGF response in vitro (which was already enhanced) was only ~ 12% less in the diabetic group (6.2±0.7 μm) compared with the non-diabetic group (7.1±1.2 μm) (Figures 4C and 4D). The inhibition of TGF in vitro during the early stage of leptin receptor deficiency (db/db)-induced diabetes was significantly less in the MD-NOS1KO mice than the NOS1flox/flox mice (Figure 4D).
To determine the significance of macula densa NOS1 in the development of glomerular hyperfiltration during the early stage of diabetes, we measured GFR by assessing the clearance kinetics of plasma FITC-sinistrin in db/db and db/+ MD-NOS1KO as well as NOS1flox/flox mice once a week from 8 to 12 weeks of age. In NOS1flox/flox mice, GFR was ~64% higher in the diabetic group (e.g., 404±18 μl/min at 12 weeks of age) than in the non-diabetic group (247±13 μl/min at 12 weeks of age). In MD-NOS1KO mice, GFR was only ~22% higher in the diabetic group (e.g. 306±14 μl/min at 12 weeks of age) than in the non-diabetic group (251±14 μl/min at 12 weeks of age). The increase of GFR during the early stage of leptin receptor deficiency (db/db)-induced diabetes was significantly less in the MD-NOS1KO mice than the NOS1flox/flox mice (Figure 4E).
These results demonstrate that selective deletion of NOS1 from the macula densa attenuates the inhibition of TGF as well as the development of glomerular hyperfiltration in the early stage of leptin receptor deficiency (db/db)-induced diabetes.
Deletion of Macula Densa NOS1 Impairs Natriuresis and Increases Blood Pressure in db/db Mice.
To determine the significance of macula densa NOS1 in the regulation of sodium excretion during the early stage of diabetes, we measured FENa by testing the concentrations of sodium and creatinine in blood and urine in diabetic (db/db) and non-diabetic (db/+) MD-NOS1KO as well as NOS1flox/flox mice at 12 weeks of age (Table 1).15, 26, 27 In NOS1flox/flox mice, FENa was 20.6±9.2% less in the diabetic group (0.058±0.007%) compared with the non-diabetic group (0.074±0.018%). In MD-NOS1KO mice, FENa was 45.8±10.6% less in the diabetic group (0.036±0.005%) compared with the non-diabetic group (0.069±0.017%). Moreover, there were no significant differences in FENa between the non-diabetic NOS1flox/flox and MD-NOS1KO mice, while it was 36.3±8.7% less in the diabetic MD-NOS1KO mice compared with the diabetic NOS1flox/flox mice (Figure 5A).
Table 1.
Fractional excretion of Na+ (FENa) in diabetic (db/db) and non-diabetic (db/+) MD-NOS1KO as well as NOS1flox/flox mice.
| Groups | UNa (mmol/l) |
PCr (mg/dl) | PNa (mmol/l) |
UCr (mg/dl) | FENa (%) |
|---|---|---|---|---|---|
| db/+ NOS1flox/flox | 125.5±12.0 | 0.040±0.002 | 145.2±1.6 | 47.891±6.848 | 0.074±0.018 |
| db/+ MD-NOS1KO | 122.1±20.0 | 0.043±0.003 | 144.4±1.6 | 52.696±6.129 | 0.069±0.017 |
| db/db NOS1flox/flox | 30.5±9.1* | 0.020±0.003* | 145.0±2.0 | 7.366±1.848* | 0.058±0.007* |
| db/db MD-NOS1KO | 16.3±9.3*# | 0.025±0.002*# | 143.9±1.1 | 7.981±4.909* | 0.036±0.005*# |
UNa, urinary Na+ concentration; PCr, plasma creatinine concentration; PNa, plasma Na+ concentration; UCr, urinary creatinine concentration.
P<0.05 vs db/+
P<0.01 vs NOS1flox/flox.
Figure 5. Effect of macula densa NOS1 deletion on natriuresis as well as blood pressure in db/db mice.
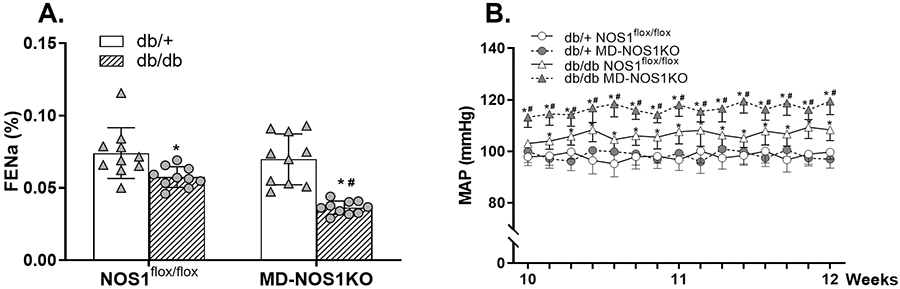
(A) The FENa in db/+ NOS1flox/flox, db/db NOS1flox/flox, db/+ MD-NOS1KO and db/db MD-NOS1KO mice. n=10; *P<0.05 vs db/+; #P<0.01 vs NOS1flox/flox. (B) The MAP in db/+ NOS1flox/flox, db/db NOS1flox/flox, db/+ MD-NOS1KO and db/db MD-NOS1KO mice. *P<0.05 vs db/+; #P<0.05 vs NOS1flox/flox. Statistical difference in (A) was calculated by two-way ANOVA followed by Sidak multiple comparisons test. Statistical difference in (B) was calculated by two-way repeated measures ANOVA followed by Sidak multiple comparisons test.
To determine the significance of macula densa NOS1 in the control of blood pressure during the early stage of diabetes, we continuously measured MAP with radio telemetry system in diabetic (db/db) and non-diabetic (db/+) MD-NOS1KO as well as NOS1flox/flox mice from 10 to 12 weeks of age.28 In NOS1flox/flox mice, MAP was 108.3±4.2 mmHg in the diabetic group, which was 8.7±2.4% higher than in the non-diabetic group (99.7±2.8 mmHg). In MD-NOS1KO mice, MAP was 119.4±5.2 mmHg in the diabetic group, which was 23.1±3.5% higher than in the non-diabetic group (97.0±3.5 mmHg). Moreover, there were no significant differences in MAP between the non-diabetic NOS1flox/flox and MD-NOS1KO mice, while it was 10.2±3.0% higher in the diabetic MD-NOS1KO mice compared with the diabetic NOS1flox/flox mice (Figure 5B).
DISCUSSION
The present study demonstrated the significance of the macula densa NOS1 in the regulation of TGF, GFR, sodium excretion, and blood pressure during the early stage of leptin receptor-deficient diabetes. We found (1) that FENa was lesser in db/db versus db/+ mice along with only a small increase in blood pressure, consistent with a proposed theory of primary tubular hyperreabsorption,5 and this was associated with an upregulated expression and activity of macula densa NOS1, an inhibited TGF response, and glomerular hyperfiltration; and (2) that deletion of macula densa NOS1 not only limited the inhibition of TGF response and attenuated glomerular hyperfiltration in db/db mice, but also further reduced FENa and substantially increased blood pressure. These data support the concept that in the early stage of diabetes, macula densa NOS1-derived NO blunts TGF and increases GFR, which facilitates renal NaCl excretion in the face of primary tubular hyperreabsorption and thereby limits the need for pressure natriuresis.
We have recently identified a new mechanism, the macula densa SGLT1-NOS1-TGF pathway, for the acute hyperglycemia–induced glomerular hyperfiltration.12 The macula densa is a group of specialized epithelial cells located at the distal end of the TAL, serving as a sensor of the tubular fluid.29, 30 The TGF describes a fundamental and intrinsic mechanism in the control of renal hemodynamics wherein an increase of NaCl delivery to the macula densa promotes the release and formation of ATP and/or adenosine, which constricts the Af-Art and induces an inhibition of single-nephron GFR.31-34 NOS1 is the predominant NOS isoform expressed in the macula densa23, 24 and the NO derived from macula densa NOS1 buffers or attenuates the TGF responsiveness via a cGMP-dependent signaling pathway.20, 24, 35, 36 Furthermore, several previous studies from our group have also demonstrated that the macula densa NOS1-mediated modulation of TGF is an important mechanism in the long-term control of GFR, salt-water balance and blood pressure.20, 22, 37 In particular, the mice with selective deletion of NOS1 from the macula densa exhibit augmented TGF responses, blunted increases in GFR, urine flow, and sodium excretion in response to acute volume expansion, and salt-sensitive hypertension.20 However, the significance of the macula densa NOS1-mediated TGF mechanism in the pathogenesis of glomerular hyperfiltration as well as hypertension in diabetes had not been determined, which was examined in the present study using a murine model of leptin receptor deficiency (db/db)-induced type 2 diabetes.
In the present study, we found that both the TGF response in vitro measured by microperfusion and TGF response in vivo measured by micropuncture were inhibited in db/db mice compared with db/+ mice, which is consistent with the results in previous studies that the TGF response, as indicated by ΔPsf, proximal-distal differences of single nephron GFR, or homeostatic efficiency of TGF, was attenuated or reset in streptozotocin-induced diabetic rats,9-11, 38, 39 Akita mice40 as well as db/db mice.41, 42 Additionally, we found that NOS1 expression as well as phosphorylation at Ser1417 (which activates NOS1 via cAMP-dependent protein kinase)43-45 in the renal cortex, where most of the NOS1 comes from the macula densa,12, 23, 24 were significantly upregulated in db/db mice compared with db/+ mice. In accordance with the changes in expression and phosphorylation, the activity of NOS1 in the macula densa, as indicated by the TGF-induced NO production, was also found to be markedly enhanced in db/db mice compared with db/+ mice, which is consistent with the previous findings that NO production, as indicated by plasma concentration and urinary excretion of nitrite/nitrate as well as cGMP, was increased in diabetic animals.46-48 These results demonstrate that the development of glomerular hyperfiltration in the early stage of diabetes is associated with upregulation of macula densa NOS1 as well as inhibition of TGF responsiveness.
Next, we examined whether the upregulated macula densa NOS1 and the inhibited TGF are causal factors or consequences of diabetic glomerular hyperfiltration. Recently, our group generated the MD-NOS1KO mouse model (NKCC2cre/NOS1flox/flox) by crossing a NKCC2cre line with a NOS1flox/flox line.20 As the NOS1flox/flox mouse line49 (kindly provided by Dr. Paul Huang) targets the exon 6, a common exon for all the splice variants of NOS1, the excision by NKCC2 promoter-driven Cre recombinase results in complete ablation of all the splice variants of NOS1 in the macula densa. Thus, in the present study, this transgenic mouse model was utilized to examine the significance of the macula densa NOS1-mediated TGF mechanism in the development of glomerular hyperfiltration as well as the pathogenesis of hypertension in diabetes. Diabetic MD-NOS1KO mice were generated by crossing MD-NOS1KO mouse line with db/db mouse line, and further characterization showed that neither the blood glucose nor the NOS1 expression in the macula densa was affected by this genetic recombination. We found that the MD-NOS1KO enhanced the TGF response to a similar extent in db/+ and db/db mice; nevertheless, MD-NOS1KO only affected GFR and MAP in db/db mice but not in db/+ mice. We propose that this is in part due to the enhanced macula densa NOS1 as well as tubular sodium retention (reduced FENa) in the diabetic kidney, which makes GFR and MAP more sensitive to the functional role and influence of macula densa NOS1. These findings are consistent with the observations in previous studies with pharmacological inhibition of NOS1 or NOS. For example, selective NOS1 inhibition induced a greater reduction in single-nephron GFR or whole-body GFR in diabetic animals than non-diabetics,41, 48, 50, 51 and GFR was not further decreased by non-selective NOS inhibitors in the presence of a NOS1 inhibitor.50 Similarly, chronic administration of L-NAME resulted in a greater increase in blood pressure in diabetic versus non-diabetic rats.52-54
In addition to the macula densa NOS1-mediated TGF mechanism, the role of SGLT1 in the control of GFR and blood pressure in the early diabetic kidney has also been recently examined by our group in two murine models of type 1 diabetes, namely streptozotocin-treated and insulin deficient (Akita) mice with genetic KO of SGLT1.15 These studies demonstrate that the absence of SGLT1 mitigates the upregulation of macula densa NOS1, attenuates glomerular hyperfiltration, and increases systolic blood pressure in diabetes.15 All the above evidences indicate that the macula densa SGLT1-NOS1-TGF pathway determines GFR, natriuresis and blood pressure in early diabetes.
Hypertension is a common comorbid condition in diabetes, affecting approximately 30% of patients with type 1 diabetes and 50% to 80% of patients with type 2 diabetes.12, 55-57 Moreover, hypertension substantially raises the risk of diabetic complications, including cardiovascular disease, neuropathy, retinopathy, and nephropathy. The reasons for the high prevalence of hypertension in diabetes have been extensively investigated but not fully elucidated yet. Based on the findings in the present study, we speculate that people who upregulate macula densa NOS1 in response to diabetes manifest glomerular hyperfiltration accompanied with normal blood pressure or only little blood pressure increase. Other individuals, potentially in African American58, 59 or elderly people,60-62 however, may have lower macula densa NOS1 expression or blunted NOS1 activity. These individuals thus might not be able to generate sufficient NO in response to diabetes, which may limit glomerular hyperfiltration and promote the development of hypertension.
Besides its effect on the TGF response, the macula densa NOS1-derived NO also affects renin release in granular cells, a rate-limiting step of renin-angiotensin-aldosterone system (RAAS). Various in vitro studies in the isolated and perfused JGAs have demonstrated that macula densa NOS1 promotes the synthesis and secretion of renin in granular cells.63-66 In addition, KO of SGLT1 in Akita mice is associated with reduced renal renin mRNA expression.15 Moreover, it is generally found that the plasma renin activity is normal67, 68 or inhibited69-71 in early diabetes. Thus, changes in RAAS are unlikely to contribute to the development of hypertension in the diabetic mice with macula densa NOS1 or SGLT1 deletion. On the contrary, a suppression of RAAS likely occurs to compensate for the sodium and fluid retention in the diabetic MD-NOS1KO and SGLT1 KO mice.
While the macula densa NOS1 is the focus of the present study, we are aware that the ablation of macula densa NOS1 does not completely block the inhibition of TGF or the rise of GFR in diabetic mice, and other mechanisms, in particular, the SGLT2-NaCl pathway, also contribute. We propose that the macula densa SGLT1-NOS1 and proximal tubule SGLT2-NaCl pathways are separate mechanisms operating in parallel in the diabetic kidney that additively promote glomerular hyperfiltration and maintain normal blood pressure, consistent with the findings in our recent studies.15 In addition, the diabetes-associated proximal tubular hyperreabsorption increases GFR in part through the physiology of the TGF mechanism. This helps to restore NaCl and fluid delivery to the distal nephron and urine, and thereby helps to maintain volume status. Preventing hyperfiltration without affecting the primary tubular hyperreabsorption may thus cause NaCl and fluid retention and increase blood pressure. In this regard, because SGLT2 is responsible for the majority of the primary hyperreabsorption in the diabetic kidney, SGLT2 inhibition lowers hyperfiltration secondarily to lowering tubular hyperreabsorption, and thus NaCl and fluid are not retained.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What is new?
Using a variety of sophisticated techniques and a novel macula densa–specific neuronal nitric oxide synthase (NOS1) knockout model, we identified a new mechanism of blood pressure regulation in diabetes, wherein the upregulation of macula densa NOS1-derived nitric oxide (NO) generation in the early diabetic kidney inhibits tubuloglomerular feedback (TGF) and increases glomerular filtration rate (GFR), which counteracts renal sodium retention and thereby limits the rise in blood pressure.
What is relevant?
Hypertension is a common comorbid condition in patients with diabetes. However, the pathogenesis of hypertension in diabetes has not been fully clarified.
Summary
The macula densa NOS1 through its effect on TGF and GFR plays a critical role in the control of natriuresis and blood pressure in a murine model of leptin receptor-deficient diabetes.
PERSPECTIVES.
The present study demonstrates that the macula densa NOS1 through its effect on TGF and GFR plays a critical role in the control of sodium excretion and blood pressure in a murine model of leptin receptor-deficient diabetes. These findings not only establish a novel pathophysiological mechanism in the early diabetic kidney but may also have implications for the development of hypertension in diabetic patients and its therapy.
SOURCES OF FUNDING
This work was supported by ASN Post-doctoral Fellowship and ASN Transition to Independence Award (to JW), ASN Post-doctoral Fellowship and ASN Transition to Independence Award (to JZ), National Institute of Health Grants DK112042 and AG061296 (to VV), and National Institute of Health Grants DK099276, DK098582 and HL137987 (to RL).
Footnotes
DISCLOSURES
None.
REFERENCE
- 1.Gillespie CD, Hurvitz KA, Centers for Disease C, Prevention. Prevalence of hypertension and controlled hypertension - united states, 2007-2010. MMWR Suppl. 2013;62:144–148 [PubMed] [Google Scholar]
- 2.Colosia AD, Palencia R, Khan S. Prevalence of hypertension and obesity in patients with type 2 diabetes mellitus in observational studies: A systematic literature review. Diabetes Metab Syndr Obes. 2013;6:327–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hypertension in diabetes study (hds): I. Prevalence of hypertension in newly presenting type 2 diabetic patients and the association with risk factors for cardiovascular and diabetic complications. J Hypertens. 1993;11:309–317 [DOI] [PubMed] [Google Scholar]
- 4.Sowers JR, Epstein M, Frohlich ED. Diabetes, hypertension, and cardiovascular disease: An update. Hypertension. 2001;37:1053–1059 [DOI] [PubMed] [Google Scholar]
- 5.Vallon V, Thomson SC. The tubular hypothesis of nephron filtration and diabetic kidney disease. Nat Rev Nephrol. 2020;16:317–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bank N Mechanisms of diabetic hyperfiltration. Kidney Int. 1991;40:792–807 [DOI] [PubMed] [Google Scholar]
- 7.Levine DZ. Can rodent models of diabetic kidney disease clarify the significance of early hyperfiltration? Recognizing clinical and experimental uncertainties. Clin Sci. 2008;114:109–118 [DOI] [PubMed] [Google Scholar]
- 8.Anderson S, Vora JP. Current concepts of renal hemodynamics in diabetes. J Diabetes Complicat. 1995;9:304–307 [DOI] [PubMed] [Google Scholar]
- 9.Thomson SC, Deng A, Bao D, Satriano J, Blantz RC, Vallon V. Ornithine decarboxylase, kidney size, and the tubular hypothesis of glomerular hyperfiltration in experimental diabetes. J Clin Invest. 2001;107:217–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vallon V, Richter K, Blantz RC, Thomson S, Osswald H. Glomerular hyperfiltration in experimental diabetes mellitus: Potential role of tubular reabsorption. J Am Soc Nephrol. 1999;10:2569–2576 [DOI] [PubMed] [Google Scholar]
- 11.Thomson SC, Rieg T, Miracle C, Mansoury H, Whaley J, Vallon V, Singh P. Acute and chronic effects of sglt2 blockade on glomerular and tubular function in the early diabetic rat. Am J Physiol Regul Integr Comp Physiol. 2012;302:R75–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang J, Wei J, Jiang S, Xu L, Wang L, Cheng F, Buggs J, Koepsell H, Vallon V, Liu R. Macula densa sglt1-nos1-tubuloglomerular feedback pathway, a new mechanism for glomerular hyperfiltration during hyperglycemia. J Am Soc Nephrol. 2019;30:578–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carney EF. A novel sglt1-mediated mechanism of hyperglycaemia-induced hyperfiltration. Nat Rev Nephrol. 2019;15:388. [DOI] [PubMed] [Google Scholar]
- 14.Carlstrom M The other glucose transporter, sglt1-also a potential trouble maker in diabetes? Journal of the American Society of Nephrology. 2019;30:519–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song PN, Huang W, Onishi A, Patel R, Kim YC, van Ginkel C, Fu YL, Freeman B, Koepsell H, Thomson S, Liu RS, Vallon V. Knockout of na+-glucose cotransporter sglt1 mitigates diabetes-induced upregulation of nitric oxide synthase nos1 in the macula densa and glomerular hyperfiltration. Am J Physiol-Renal. 2019;317:F207–F217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang S, Wang X, Boone J, Wie J, Yip KP, Zhang J, Wang L, Liu R. Application of hanging drop technique for kidney tissue culture. Kidney Blood Press Res. 2017;42:220–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Watanabe H, Belyea BC, Paxton RL, Li M, Dzamba BJ, DeSimone DW, Gomez RA, Sequeira-Lopez MLS. Renin cell baroreceptor, a nuclear mechanotransducer central for homeostasis. Circ Res. 2021;129:262–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gartner K Glomerular hyperfiltration during the onset of diabetes mellitus in two strains of diabetic mice (c57bl/6j db/db and c57bl/ksj db/db). Diabetologia. 1978;15:59–63 [DOI] [PubMed] [Google Scholar]
- 19.Sharma K, McCue P, Dunn SR. Diabetic kidney disease in the db/db mouse. Am J Physiol Renal Physiol. 2003;284:F1138–1144 [DOI] [PubMed] [Google Scholar]
- 20.Lu Y, Wei J, Stec DE, Roman RJ, Ge Y, Cheng L, Liu EY, Zhang J, Hansen PB, Fan F, et al. Macula densa nitric oxide synthase 1beta protects against salt-sensitive hypertension. J Am Soc Nephrol. 2016;27:2346–2356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wei J, Zhang J, Jiang S, Wang L, Persson AEG, Liu R. High-protein diet-induced glomerular hyperfiltration is dependent on neuronal nitric oxide synthase beta in the macula densa via tubuloglomerular feedback response. Hypertension. 2019;74:864–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang J, Zhu J, Wei J, Jiang S, Xu L, Qu L, Yang K, Wang L, Buggs J, Cheng F, Tan X, Liu R. New mechanism for the sex differences in salt-sensitive hypertension: The role of macula densa nos1beta-mediated tubuloglomerular feedback. Hypertension. 2020;75:449–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mundel P, Bachmann S, Bader M, Fischer A, Kummer W, Mayer B, Kriz W. Expression of nitric oxide synthase in kidney macula densa cells. Kidney Int. 1992;42:1017–1019 [DOI] [PubMed] [Google Scholar]
- 24.Wilcox CS, Welch WJ, Murad F, Gross SS, Taylor G, Levi R, Schmidt HH. Nitric oxide synthase in macula densa regulates glomerular capillary pressure. Proc Natl Acad Sci U S A. 1992;89:11993–11997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang J, Qu L, Wei J, Jiang S, Xu L, Wang L, Cheng F, Jiang K, Buggs J, Liu R. A new mechanism for the sex differences in angiotensin ii-induced hypertension: The role of macula densa nos1beta-mediated tubuloglomerular feedback. Am J Physiol Renal Physiol. 2020;319:F908–F919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wei J, Zhang J, Wang L, Jiang S, Fu L, Buggs J, Liu R. New mouse model of chronic kidney disease transitioned from ischemic acute kidney injury. Am J Physiol Renal Physiol. 2019;317:F286–F295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang J, Wang X, Wei J, Wang L, Jiang S, Xu L, Qu L, Yang K, Fu L, Buggs J, Cheng F, Liu R. A two-stage bilateral ischemia-reperfusion injury-induced aki to ckd transition model in mice. Am J Physiol Renal Physiol. 2020;319:F304–F311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang J, Qu HY, Song J, Wei J, Jiang S, Wang L, Wang L, Buggs J, Liu R. Enhanced hemodynamic responses to angiotensin ii in diabetes are associated with increased expression and activity of at1 receptors in the afferent arteriole. Physiol Genomics. 2017;49:531–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bell PD, Lapointe JY, Peti-Peterdi J. Macula densa cell signaling. Annu Rev Physiol. 2003;65:481–500 [DOI] [PubMed] [Google Scholar]
- 30.Barajas L Anatomy of the juxtaglomerular apparatus. Am J Physiol. 1979;237:F333–343 [DOI] [PubMed] [Google Scholar]
- 31.Schnermann J, Persson AEG, Agerup B. Tubuloglomerular feedback - nonlinear relation between glomerular hydrostatic pressure and loop of henle perfusion rate. Journal of Clinical Investigation. 1973;52:862–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun D, Samuelson LC, Yang T, Huang Y, Paliege A, Saunders T, Briggs J, Schnermann J. Mediation of tubuloglomerular feedback by adenosine: Evidence from mice lacking adenosine 1 receptors. Proc Natl Acad Sci U S A. 2001;98:9983–9988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Castrop H Mediators of tubuloglomerular feedback regulation of glomerular filtration: Atp and adenosine. Acta Physiol (Oxf). 2007;189:3–14 [DOI] [PubMed] [Google Scholar]
- 34.Thomson S, Bao D, Deng A, Vallon V. Adenosine formed by 5'-nucleotidase mediates tubuloglomerular feedback. J Clin Invest. 2000;106:289–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kovacs G, Komlosi P, Fuson A, Peti-Peterdi J, Rosivall L, Bell PD. Neuronal nitric oxide synthase: Its role and regulation in macula densa cells. J Am Soc Nephrol. 2003;14:2475–2483 [DOI] [PubMed] [Google Scholar]
- 36.Ren YL, Garvin JL, Carretero OA. Role of macula densa nitric oxide and cgmp in the regulation of tubuloglomerular feedback. Kidney Int. 2000;58:2053–2060 [DOI] [PubMed] [Google Scholar]
- 37.Wang X, Chandrashekar K, Wang L, Lai EY, Wei J, Zhang G, Wang S, Zhang J, Juncos LA, Liu R. Inhibition of nitric oxide synthase 1 induces salt-sensitive hypertension in nitric oxide synthase 1alpha knockout and wild-type mice. Hypertension. 2016;67:792–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vallon V, Osswald H. Dipyridamole prevents diabetes-induced alterations of kidney function in rats. Naunyn Schmiedebergs Arch Pharmacol. 1994;349:217–222 [DOI] [PubMed] [Google Scholar]
- 39.Vallon V, Blantz RC, Thomson S. Homeostatic efficiency of tubuloglomerular feedback is reduced in established diabetes mellitus in rats. Am J Physiol. 1995;269:F876–883 [DOI] [PubMed] [Google Scholar]
- 40.Faulhaber-Walter R, Chen L, Oppermann M, Kim SM, Huang Y, Hiramatsu N, Mizel D, Kajiyama H, Zerfas P, Briggs JP, Kopp JB, Schnermann J. Lack of a1 adenosine receptors augments diabetic hyperfiltration and glomerular injury. J Am Soc Nephrol. 2008;19:722–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Levine DZ, Iacovitti M, Robertson SJ, Mokhtar GA. Modulation of single-nephron gfr in the db/db mouse model of type 2 diabetes mellitus. Am J Physiol Regul Integr Comp Physiol. 2006;290:R975–981 [DOI] [PubMed] [Google Scholar]
- 42.Levine DZ, Iacovitti M, Robertson SJ. Modulation of single-nephron gfr in the db/db mouse model of type 2 diabetes mellitus. Ii. Effects of renal mass reduction. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1840–1846 [DOI] [PubMed] [Google Scholar]
- 43.Bredt DS, Ferris CD, Snyder SH. Nitric oxide synthase regulatory sites. Phosphorylation by cyclic amp-dependent protein kinase, protein kinase c, and calcium/calmodulin protein kinase; identification of flavin and calmodulin binding sites. J Biol Chem. 1992;267:10976–10981 [PubMed] [Google Scholar]
- 44.Hurt KJ, Sezen SF, Lagoda GF, Musicki B, Rameau GA, Snyder SH, Burnett AL. Cyclic amp-dependent phosphorylation of neuronal nitric oxide synthase mediates penile erection. Proc Natl Acad Sci U S A. 2012;109:16624–16629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adak S, Santolini J, Tikunova S, Wang Q, Johnson JD, Stuehr DJ. Neuronal nitric-oxide synthase mutant (ser-1412 -> asp) demonstrates surprising connections between heme reduction, no complex formation, and catalysis. Journal of Biological Chemistry. 2001;276:1244–1252 [DOI] [PubMed] [Google Scholar]
- 46.Bank N, Aynedjian HS. Role of edrf (nitric-oxide) in diabetic renal hyperfiltration. Kidney International. 1993;43:1306–1312 [DOI] [PubMed] [Google Scholar]
- 47.Komers R, Allen TJ, Cooper ME. Role of endothelium-derived nitric-oxide in the pathogenesis of the renal hemodynamic-changes of experimental diabetes. Diabetes. 1994;43:1190–1197 [DOI] [PubMed] [Google Scholar]
- 48.Komers R, Lindsley JN, Oyama TT, Allison KM, Anderson S. Role of neuronal nitric oxide synthase (nos1) in the pathogenesis of renal hemodynamic changes in diabetes. Am J Physiol Renal Physiol. 2000;279:F573–583 [DOI] [PubMed] [Google Scholar]
- 49.Gyurko R, Leupen S, Huang PL. Deletion of exon 6 of the neuronal nitric oxide synthase gene in mice results in hypogonadism and infertility. Endocrinology. 2002;143:2767–2774 [DOI] [PubMed] [Google Scholar]
- 50.Thomson SC, Deng AH, Komine N, Hammes JS, Blantz RC, Gabbai FB. Early diabetes as a model for testing the regulation of juxtaglomerular nosi. Am J Physiol-Renal. 2004;287:F732–F738 [DOI] [PubMed] [Google Scholar]
- 51.Komers R, Oyama TT, Chapman JG, Allison KM, Anderson S. Effects of systemic inhibition of neuronal nitric oxide synthase in diabetic rats. Hypertension. 2000;35:655–661 [DOI] [PubMed] [Google Scholar]
- 52.Brands MW, Bell TD, Gibson B. Nitric oxide may prevent hypertension early in diabetes by counteracting renal actions of superoxide. Hypertension. 2004;43:57–63 [DOI] [PubMed] [Google Scholar]
- 53.Majithiya JB, Parmar AN, Trivedi CJ, Balaraman R. Effect of pioglitazone on l-name induced hypertension in diabetic rats. Vasc Pharmacol. 2005;43:260–266 [DOI] [PubMed] [Google Scholar]
- 54.Fitzgerald SM, Brands MW. Hypertension in l-name-treated diabetic rats depends on an intact sympathetic nervous system. Am J Physiol-Reg I. 2002;282:R1070–R1076 [DOI] [PubMed] [Google Scholar]
- 55.de Boer IH, Bangalore S, Benetos A, Davis AM, Michos ED, Muntner P, Rossing P, Zoungas S, Bakris G. Diabetes and hypertension: A position statement by the american diabetes association. Diabetes Care. 2017;40:1273–1284 [DOI] [PubMed] [Google Scholar]
- 56.Landsberg L, Molitch M. Diabetes and hypertension: Pathogenesis, prevention and treatment. Clin Exp Hypertens. 2004;26:621–628 [DOI] [PubMed] [Google Scholar]
- 57.Passarella P, Kiseleva TA, Valeeva FV, Gosmanov AR. Hypertension management in diabetes: 2018 update. Diabetes Spectr. 2018;31:218–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Melikian N, Wheatcroft SB, Ogah OS, Murphy C, Chowienczyk PJ, Wierzbicki AS, Sanders TA, Jiang B, Duncan ER, Shah AM, Kearney MT. Asymmetric dimethylarginine and reduced nitric oxide bioavailability in young black african men. Hypertension. 2007;49:873–877 [DOI] [PubMed] [Google Scholar]
- 59.Leineweber K, Moosmang S, Paulson D. Genetics of no deficiency. Am J Cardiol. 2017;120:S80–S88 [DOI] [PubMed] [Google Scholar]
- 60.Erdely A, Greenfeld Z, Wagner L, Baylis C. Sexual dimorphism in the aging kidney: Effects on injury and nitric oxide system. Kidney Int. 2003;63:1021–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Baylis C Sexual dimorphism of the aging kidney: Role of nitric oxide deficiency. Physiology (Bethesda). 2008;23:142–150 [DOI] [PubMed] [Google Scholar]
- 62.Baylis C Changes in renal hemodynamics and structure in the aging kidney; sexual dimorphism and the nitric oxide system. Exp Gerontol. 2005;40:271–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Greenberg SG, He XR, Schnermann JB, Briggs JP. Effect of nitric oxide on renin secretion. I. Studies in isolated juxtaglomerular granular cells. Am J Physiol. 1995;268:F948–952 [DOI] [PubMed] [Google Scholar]
- 64.He XR, Greenberg SG, Briggs JP, Schnermann JB. Effect of nitric oxide on renin secretion. Ii. Studies in the perfused juxtaglomerular apparatus. Am J Physiol. 1995;268:F953–959 [DOI] [PubMed] [Google Scholar]
- 65.Vargas SL, Toma I, Kang JJ, Meer EJ, Peti-Peterdi J. Activation of the succinate receptor gpr91 in macula densa cells causes renin release. J Am Soc Nephrol. 2009;20:1002–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Peti-Peterdi J, Harris RC. Macula densa sensing and signaling mechanisms of renin release. J Am Soc Nephrol. 2010;21:1093–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Luik PT, Kerstens MN, Hoogenberg K, Navis GJ, Dullaart RP. Low plasma aldosterone despite normal plasma renin activity in uncomplicated type 1 diabetes mellitus: Effects of raas stimulation. Eur J Clin Invest. 2003;33:787–793 [DOI] [PubMed] [Google Scholar]
- 68.Beretta-Piccoli C, Weidmann P, Keusch G. Responsiveness of plasma renin and aldosterone in diabetes mellitus. Kidney Int. 1981;20:259–266 [DOI] [PubMed] [Google Scholar]
- 69.Christlieb AR, Kaldany A, D'Elia JA. Plasma renin activity and hypertension in diabetes mellitus. Diabetes. 1976;25:969–974 [DOI] [PubMed] [Google Scholar]
- 70.Fernandez-Cruz A Jr., Noth RH, Lassman MN, Hollis JB, Mulrow PJ. Low plasma renin activity in normotensive patients with diabetes mellitus: Relationship to neuropathy. Hypertension. 1981;3:87–92 [DOI] [PubMed] [Google Scholar]
- 71.Price DA, Porter LE, Gordon M, Fisher ND, De'Oliveira JM, Laffel LM, Passan DR, Williams GH, Hollenberg NK. The paradox of the low-renin state in diabetic nephropathy. J Am Soc Nephrol. 1999;10:2382–2391 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.