

Abstract
BACKGROUND
To shed light on the earliest events in oncogenesis, there is growing interest in understanding the mutational landscapes of normal tissues across ages. In the last decade, next-generation sequencing of human tissues has revealed a surprising abundance of cells with what would be considered oncogenic mutations.
AIMS
We performed meta-analysis on previously published sequencing data on normal tissues to categorize mutations based on their presence in cancer and showcase the quantity of cells with cancer-associated mutations in cancer-free individuals.
METHODS AND RESULTS
We analyzed sequencing data from these studies of normal tissues to determine the prevalence of cells with mutations in three different categories across multiple age groups: 1) mutations in genes designated as drivers, 2) mutations that are in the Cancer Gene Census (CGC), and 3) mutations in the CGC that are considered pathogenic. As we age, the percentage of cells in all three levels increase significantly, reaching over 50% of cells having oncogenic mutations for multiple tissues in the older age groups. The clear enrichment for these mutations, particularly at older ages, likely indicates strong selection for the resulting phenotypes. Combined with an estimation of the number of cells in tissues, we calculate that most older, cancer-free individuals possess at least a 100 billion cells that harbor at least one oncogenic mutation, presumably emanating from a fitness advantage conferred by these mutations that promotes clonal expansion.
CONCLUSIONS
These studies of normal tissues have highlighted the specific drivers of clonal expansion and how frequently they appear in us. Their high prevalence throughout cancer-free individuals necessitates reconsideration of the oncogenicity of these mutations, which could shape methods of detection, prevention and treatment of cancer, as well as of the potential impact of these mutations on tissue function and our health.
Keywords: somatic evolution, mutational landscape, cancer evolution, life history theory
Graphical Abstract
Body map representations of each age group showing the fractions of cells with mutations in the CGC that are considered pathogenic for each tissue. The opacity of red color is proportional to the fraction of cells with cancer-associated mutations.
INTRODUCTION
Cancer evolves through the positive selection of somatic mutations in cells that occur over time. These mutations result in fitness advantages that promotes clonal expansion. Importantly, the fitness impact of oncogenic mutations has been shown to be context dependent, with contexts like old age engendering selection for adaptive oncogenic mutations.1,2 Clonal evolution resulting in cancer thus involves sequential selection for adaptive mutations that often involve deactivation of tumor suppressor genes and activation of oncogenes. In cancer samples, these mutations have been well documented in databases such as The Cancer Genome Atlas (TCGA), Integrative Onco Genomics (IntOGen), and the Catalogue of Somatic Mutations in Cancer (COSMIC) in hopes of establishing mutational patterns. However, cancer is the final stage of clonal evolution. Therefore, to understand how somatic evolution leads to oncogenesis and establish methods to better detect and prevent cancers, investigations at earlier stages of the process (i.e. in normal tissue) are merited.
Towards fuller understanding of multi-hit models of carcinogenesis and to serve as a complement to genomic analysis of cancers, sequencing of normal tissues began as early as the 1990s.3,4 Given that multiple mutations in the same cell lead to oncogenesis, there must exist precancerous clones that should be detectable in cancer-free individuals. Additionally, an inability to determine which mutations are responsible for oncogenesis – either due to the prevalence of passenger mutations in tumor samples or an incomplete set of cancer genes to probe – has led to a flurry of investigations of normal tissues to gain insight on clonal expansions that can transform into cancer.4–9 Many of the clones found in normal tissues have genetic alterations that persist in cancers, reinforcing the idea that these clonal expansions can be early steps to oncogenesis. On the other hand, knowing that about 40% of people develop cancers, and such cancers almost always start from a single oncogenically-initiated cell, it is clear that the vast majority of these expansions will not become malignant or threaten the host. Many of these clones may be driven by mutations with low carcinogenic potential or may have limited evolution to a cancer due to various cell intrinsic and extrinsic hurdles.10,11 To our knowledge, these data of oncogenic mutational processes in normal tissues have not been compiled in such a way to provide a comprehensive understanding of the clonal mutational landscape across tissues, including categorization of mutations based on their presence in cancer and their likelihood of disrupting protein function.
When investigating oncogenesis throughout the human body, it is important to consider a life history perspective – how natural selection has shaped our tissues to maximize survival and reproductive success and the limits of these selective pressures at older ages when we are less likely to reproduce.12 In this light, we can better appreciate why some clonal expansions may be more tolerated than others and the age-dependence of this tolerance. Due in large part to the fact that our lifespan has increased significantly in recent centuries, age has become the biggest cancer risk factor and is a crucial component to investigating the transition from clonal evolution to oncogenesis.1,13–15 Attention must also be paid to the fact that different turnover rates and mutation rates exist across epithelial tissues and the hematopoietic system, which when combined with very different microenvironment-driven selective pressures, leads to differing clonal evolution routes to cancer. Therefore, providing insight on the somatic mutational landscape and its potential to progress to cancer throughout the human body across age is critically important.
As we age, random somatic mutations accumulate through DNA replication and the regular assault of cell intrinsic and extrinsic mutagens, leading to a clock-like accumulation of mutations that is less dependent on cell division rates than initially believed16. This age-dependent increase in mutations contributes to the relationship between age and the number of detectable clonal expansions. In addition, selective pressures acting on clones with potential driver mutations change as we age (a gene or mutation is considered a “driver” when demonstrated to be under positive selection during cancer evolution and to contribute to the cancer phenotype). At older ages, these clones can have an increased competitive advantage due to changes in the tissue microenvironment and cell-intrinsic fitness decline.13 Still, we lack a good understanding of what promotes clonal expansions, the impact of these expansions on tissue function and overall health, and the cancer risks that these clonal expansions confer. In fact, there is evidence pointing to the progression of disease absent of environmental pressures.17 With this in mind, determining the prevalence and which clonal expansions and oncogenic mutations are in individuals (independent of the presence of cancer) across ages can significantly add to our understanding of physiological decline and the altered risk of cancer and other illnesses.
With the expanse of available technological capabilities, different methodologies have been used to investigate the mutational patterns of normal tissues throughout the human body. This motivated us to determine the proportions and numbers of cells in the average human body possessing oncogenic mutations in different tissues across the lifespan. Here, we combined information from the array of different methods across different tissues to highlight the prevalence of three different levels of clonal expansion throughout the human body across ages. We estimated the frequency and number of cells with mutations in genes under positive selection (level 1), of cells with mutations in COSMIC’s Cancer Gene Census (CGC) (level 2), and finally of cells with a mutation that is designated as pathogenic in the CGC (level 3), providing common criteria across studies to allow comparisons of mutational landscapes across tissues. We show that by the time a person is middle-aged, many of their tissues are dominated by clones initiated by driver mutations and mutations common in cancer. With this improved understanding of clonal expansions across tissues with age, we are better equipped to address questions critical for understanding cancer evolution and tissue changes with age.
METHODS
Using the Cancer Gene Census
The Cancer Gene Census (CGC) from the Catalogue of Somatic Mutations in Cancer (COSMIC) is a compilation of data for genes implicated in the evolution of cancer.18 To quantify the presence of mutations associated with cancer in tissues from cancer-free individuals, this database was used to identify CGC genes under positive selection (level 1), CGC curated mutations implicated in cancers (level 2), and CGC curated mutations considered to be protein-altering (“pathogenic”; level 3). For a gene to be in the CGC, the gene must be functionally involved in oncogenic transformation and have somatic mutations that affect gene function commonly seen in cancer samples.18 Mutations are included in the CGC if they consistently occur in these cancer genes in studies deemed to be of good quality. It is worth noting that the CGC uses the Functional Analysis through Hidden Markov Model (FATHMM) algorithm to predict whether mutations are pathogenic or not.19 In this sense, pathogenic means that the nucleotide change has a critical effect on protein function, and thus is likely to impact phenotype, which could enable clonal expansion if the mutation confers a selective advantage. Downloads for the CGC genes and their mutations were accessed on 4/25/2020.
For level 1, the genes that are considered to be positively selected were largely determined using dNdScV method20 by the authors of the studies from which data were analyzed. For report on mutations in blood used for analysis,21 the method used for calculating driver mutations predates the dNdScV methodology of determining driver genes. Considering that cases of clonal hematopoiesis are consistently associated with mutations in DNMT3A, TET2, ASXL1, JAK2, TP53, IDH1, and IDH2,22 these were the genes chosen for level 1 in the blood. For level 2, single-nucleotide variants (SNVs) that shared the chromosome, genomic position, and nucleotide change identical to that of a coding mutation in COSMIC’s CGC were added. SNVs in level 2 that were designated pathogenic by COSMIC’s FATHMM prediction constituted level 3. Insertions and deletions (indels) were included in both level 2 and level 3 if they occurred in the exonic coding regions of genes in the CGC. This assumes that all indels in exonic coding regions are damaging to protein function. Despite the fact that indels may be inconsequential, indels cause frameshifts and disordered secondary structures that are readily disruptive to protein function while contributing to genotypic and phenotypic diversity that can drive clonal evolution.6 Therefore, we suspect detectable clones with indel mutations in CGC genes are very likely to have oncogenic potential. To ensure all mutations were in exonic coding regions, we generated a Browser Extensible Data (BED) file (GENCODE V36) using the UCSC Table Browser and filtered all mutations for these regions.
Determining Cell Fractions for Each Level
To calculate the average variant allele frequency (VAF) for an age group in a given level, we divided the summation of the VAFs of mutations in that level by the number of samples sequenced in that age group. To achieve the cell fraction in a given level, we multiplied this value by two unless stated otherwise. This assumes that the mutations are heterozygous and that there is no change in ploidy. Considering the stochasticity of somatic mutations, the likelihood of the same mutation occurring on the second chromosome is low. This is validated by the fact that heterozygosity predominates in the somatic mutations in the studies we analyzed. Additionally, loss of heterozygosity and copy number alterations make up a small fraction of the somatic mutations found throughout the literature. A flow chart of the determination of the cell fractions with the incorporation of the CGC is shown in Figure 1.
Figure 1: Flow chart outlining computational pipeline.
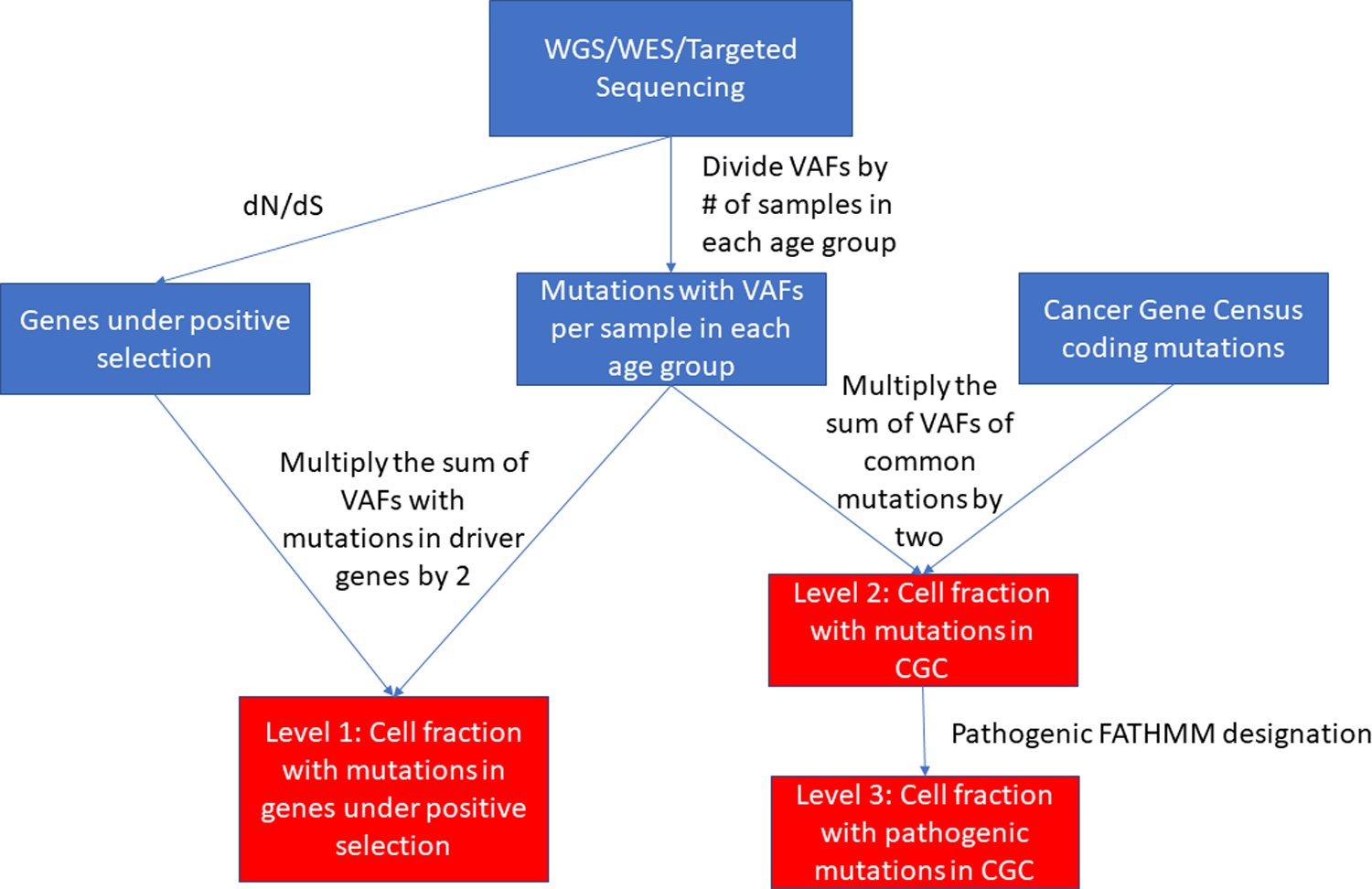
The flow chart shows the calculation of the fractions of cells with mutations in genes under positive selection (level 1), with mutations in the CGC (level 2), and with CGC mutations considered pathogenic (level 3) for each tissue for each age group.
There are some exceptions to the calculation above based on the available data from the literature. In the esophagus, Martincorena et al. showed that TP53 had clones that evolved within larger clones, and NOTCH1 mutations had biallelic inactivation.23 Therefore, the VAFs for the mutations in these genes were not doubled when determining the cell fractions for this tissue. Additionally, consistent with the high VAFs consistently observed in individual colonic crypts and endometrial glands, such units are known to experience clonal dominance by a single stem cell every few years (often by drift, although a driver mutation can bias such dominance).24,25 Therefore, the cell fraction designated to a level in these tissues was calculated by doubling the largest VAF for each crypt or gland if the VAF was below 0.5 and assuming the crypt or gland was clonal for that mutation with a VAF at or above 0.5.
Determining Cell Counts for Each Level
Using our calculated cell fractions and estimations of the total number of epithelial cells in tissues from previous work26,27, we determined the number of cells in each level for a 70 kg human. For the bladder and esophagus, the cell counts of their epithelia were not available; therefore, we made our own estimations of cell counts using an estimated surface area of the epithelial layers divided by the surface area of the cells in those layers.23,28–33 More details of the calculation of cells in the epithelia of the bladder and esophagus are described in the Supporting Information. We were unable to obtain reasonable estimates for the endometrium, and thus this tissue is not included.
Code
The code created for these analyses to determine the fractions of mutations in the different levels (as outlined in Figure 1) and to separate out mutations found in exonic coding regions can be found at https://github.com/edjevans/Normal_Tissue_Analysis.
RESULTS
We analyzed the mutational data from multiple studies that found detectable clones in the bladder34, blood21, colon35, endometrium36, esophagus23, liver37, lung38, neuron39, and skin40. While for some tissues like blood41,42, bladder43, and esophagus44, there were other studies showing similar mutational patterns, we selected studies based on coverage per sample, distributions of samples across ages, and data availability. The fraction of a particular tissue analyzed per sample varied widely across these studies and can be divided into three groups based on how that determines the ability to detect the mutations, as depicted in Figure 2. For blood, a small fraction (on the order of milliliters from the human body that has 5 liters of blood on average) was sampled, representing a tiny fraction of the total hematopoietic system that continuously intermixes throughout the entire organism. Given the low mutation rate of hematopoietic stem and progenitor cells and the relatively high presence of artificial mutations from whole exome sequencing (WES) and whole genome sequencing (WGS) in the blood due to the very small sampling fraction, the detectable mutations were filtered in the original report for previously seen somatic mutations in 160 hematologic cancer genes to provide high confidence in all identified mutations.21 For bladder, esophagus, liver and skin, small microdissections of the epithelia were analyzed, representing nucleated cell numbers from a few hundred in the bladder and liver up to 105 in the esophagus and skin being analyzed per sample. Single cells were analyzed in the lung after expansion in vitro and in neurons. For the colon and endometrium, single crypts and glands (respectively), which are known to represent clonal sweeps by drift processes and originate from a single stem cell24,25, were analyzed. Thus, for these four tissues, mutations analyzed were clonal. For the non-clonal tissues (blood, bladder, esophagus, liver and skin), it is important to keep in mind that the detection of a mutation requires that the cells with the mutation reach some minimal frequency in the analyzed sample (typically, 2–5%), which can occur by either positive selection or drift.
Figure 2.
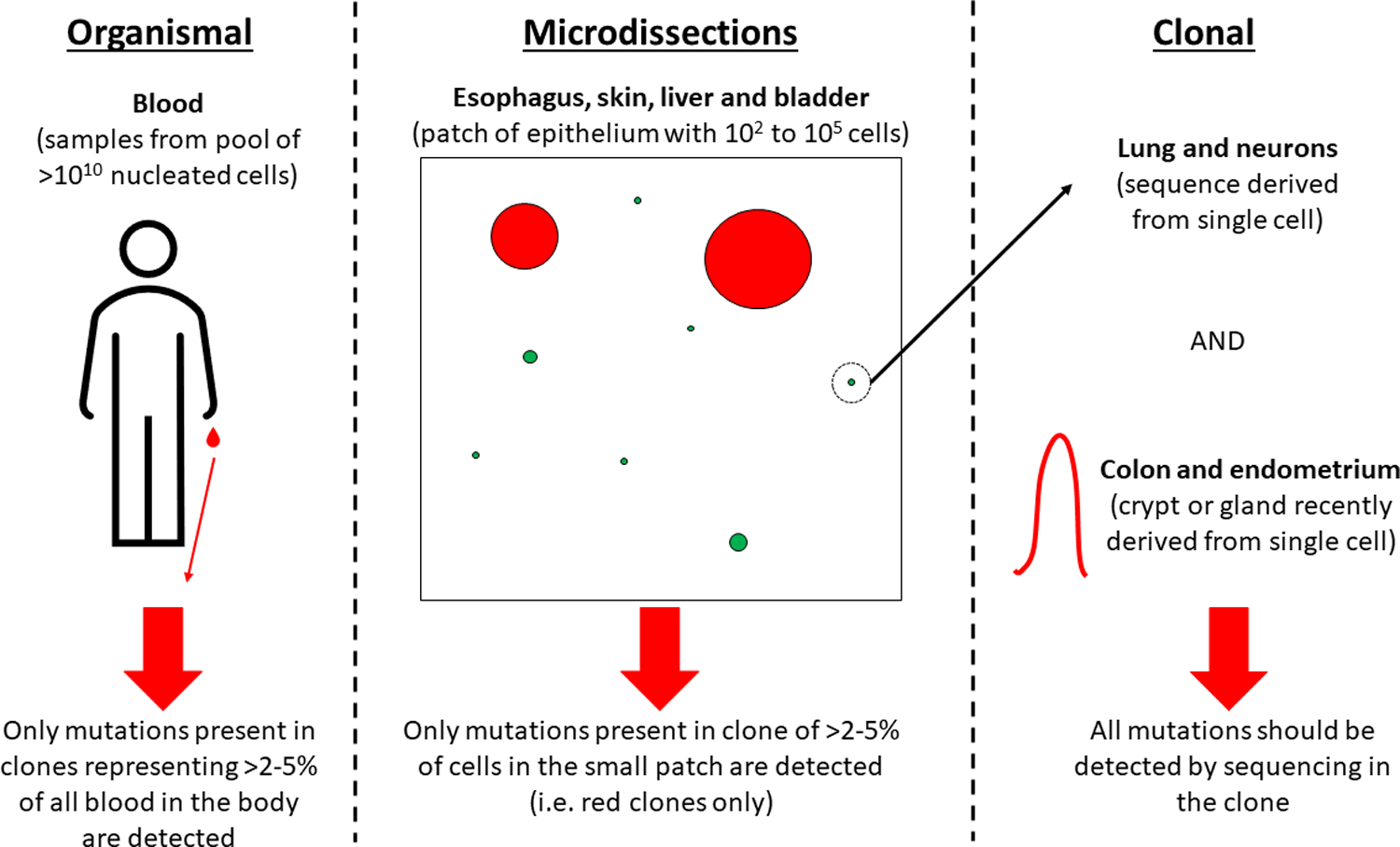
Illustration showing how tissue sampling can affect the ability to detect mutations.
For these nine tissues, we were able to find papers that measured normal tissue samples across ages and that enabled us to make estimations on mutations in genes under positive selection (level 1), mutations that were in the CGC (level 2), and those that are in the CGC and also pathogenic (level 3). From these data, we determined the fraction of cells in each level, as shown in Figure 3, with each age group indicated by a different color. Asterisks reflect a cell fraction of 0 with a mutation in any level, even though a sufficient number of samples were analyzed for that age group. In most of the studies, the determination of genes with driver mutations (under positive selection) have been calculated in the original reports. Most of these reports did not specifically query for CGC and CGC/pathogenic variants, and our goal was to use these common criteria across all studies in order to better compare mutational landscapes across tissues. It is worth noting that some studies such as for the colon accounted for specific driver mutations, while we quantified all mutations within a driver gene, which is the cause for discrepancies in the percentages of mutations under positive selection between our analysis and the original reports.
Figure 3. Prevalence of cancer-associated mutations in tissues across age groups.
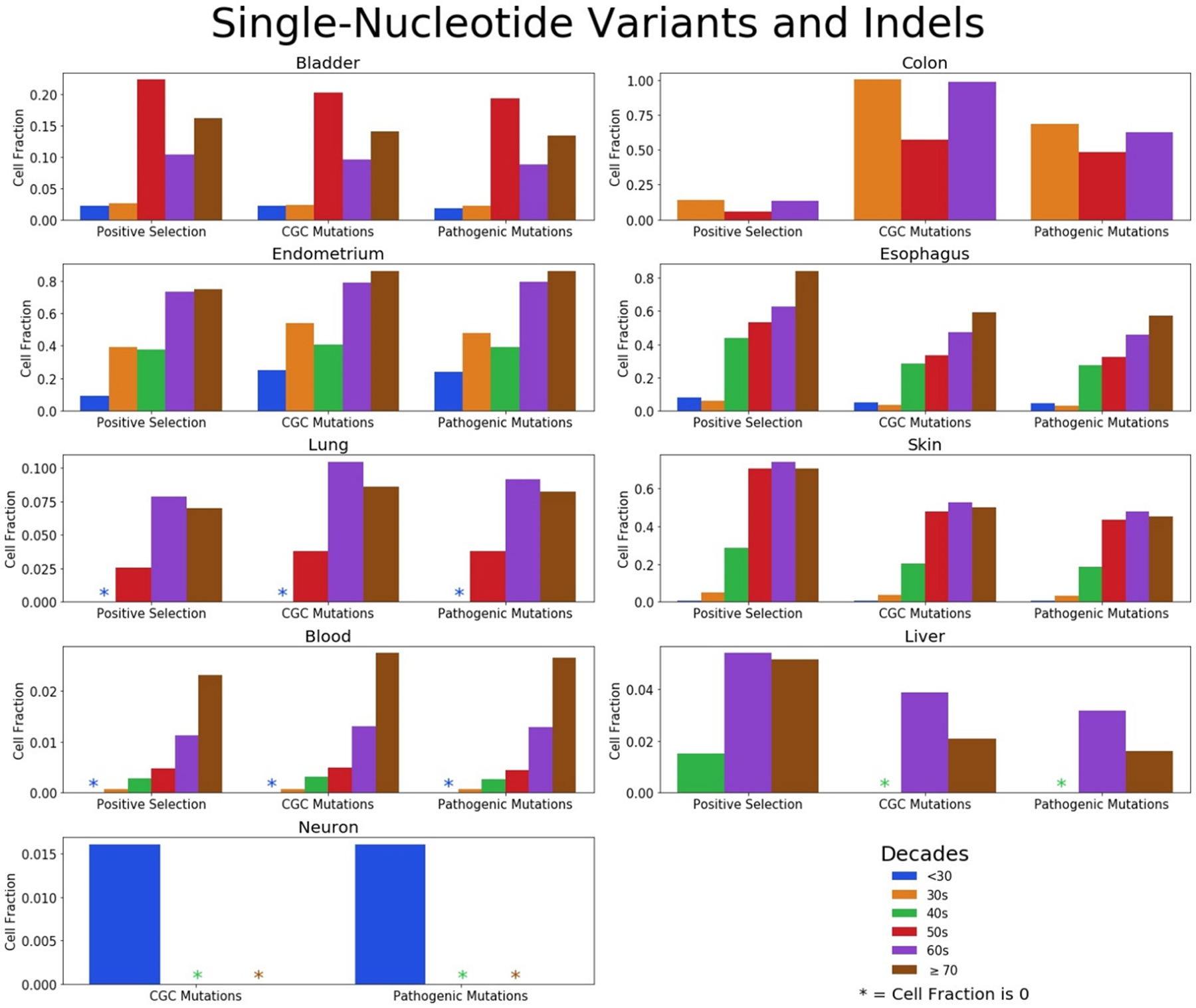
Fractions of cells with mutations (SNV and indels) in genes under positive selection (level 1), mutations cataloged in the CGC (level 2), and CGC mutations that are considered pathogenic (level 3) divided into age groups for the bladder, colon, endometrium, esophagus, lung, skin, blood, liver, and neurons. Note differences in the scale of the y-axes for the different tissues.
There are several trends that can be gleaned from these analyses. We observed that certain tissues have much larger fractions of cells bearing putative oncogenic mutations than others. For the colon, endometrium, esophagus, and skin, at least half of the cells have pathogenic mutations in the oldest cohorts. The high turnover rate of these tissues may be a contributing factor. It is important to reinforce that epithelial cells in an endometrial gland or colonic crypt are relatively clonal, as described above. As such, it may be “easier” for a driver mutation to dominate the underlying small stem cell pools for each gland or crypt, relative to the case for other tissues, perhaps accounting for the large fraction of these endometrial and colonic clones bearing oncogenic mutations. In contrast, lung epithelial cells and neurons exhibit a much lower frequency of oncogenic variants, even though these studies also involved sequencing of individual clones, although through isolation and analyses of individual cells. This indicates that the presence of a high fraction of oncogenic mutations in the colon and endometrium is not simply the consequence of the analysis of clonal populations. Of note, turnover rates for neurons and the lung are much lower than for blood, skin, colon and endometrium.45
On the contrary, the blood and liver have notably low fractions with cataloged mutations. A sample of blood has cells that originate from many cell lineages and is a minuscule volume relative to the total volume throughout the human body, as discussed earlier and shown in Figure 2. Therefore, to be able to detect a mutation in a blood sample, strong selection is required to overcome the relatively small sample size and the dilution from other cell types; the variant clone needs to dominate the entire hematopoietic system, not just in one small area of epithelium like the tissues microdissected for sequencing. In addition, the variant for a blood clone would need to originate in a very early progenitor like a hematopoietic stem cell in order to contribute to all lineages and to make a detectable contribution to overall hematopoiesis. For the microdissected tissues, the clone only needs to dominate within a population of 102 to 105 cells, depending on the tissue fraction analyzed. Such a variant could occur within a more committed progenitor and need not have the capacity to dominate the entire organ. As for the liver, only a very small fraction of cells with cataloged driver or cancer-associated mutations were evident, whether in regeneration nodules, those diseased with cirrhosis, or disease-free liver.37,46 More work is needed to determine why this is.
We also observed that as humans get older, the percentage of mutated cells increase across all three levels. Although mutation accumulation has largely been associated with cancer and tissue decline, the idea that mutations accumulate as we get older has been proposed as early as the 1950s,47 which we now know contribute to numerous clones in the normal human body9. However, there are some younger age groups for some tissues with notably higher cell fractions, particularly the 50s in the bladder and the 30s in the colon. All individuals who provided bladder samples in their 50s were heavy smokers, had higher alcohol consumption, or both, whereas other age groups had individuals who abstained from consistent use.34 Although there were not increases in smoking mutational signatures associated with smoking seen in this work, this does not preclude smoking’s effect on the tissue microenvironment that may enable clonal expansions. In the colon, there are millions of crypts in each individual, enabling the opportunity to accumulate a large amount of data per person. However, this can limit biological replicates and hinder robustness of the data if crypts are not procured from multiple individuals per age group. Notably, the crypts that make up the data in the 50s age group come from targeted sequencing of one individual with multiple samples, which limits conclusions that can be drawn across age groups for this tissue. Finally, the endometrial cell fractions from those in their 30s is inexplicably higher than those in their 40s. In this case, the disruption of the trend cannot be simply explained by the number of samples analyzed or procuring samples from few individuals.
In the analyses of neurons, the authors used whole-genome sequencing (WGS) on single cells to investigate neurodegeneration (the focus was not on oncogenesis).39 It is worth noting that a very low number of neurons are considered for each age group (<10) such that the numbers did not suffice to determine genes with mutations under positive selection. Of the over 7000 SNVs in the coding regions of genes, we show that only one was in the CGC and that this mutation is considered pathogenic. There were no indels found in the neurons. This could be due to low limits of detection and small sample sizes because indels have been identified in neurons with extremely sensitive techniques in another study.16
We made separate cell fraction plots for SNVs and indels, as shown in Figures 4 and 5, respectively, to highlight SNV and indel contributions to clonal expansions in normal tissues. Considering that the numbers of SNVs dwarf those of the indels in the mutation data, the trends of the SNV cell fraction largely mimic that of the total mutation cell fractions, especially in level 1 where the summation is based on occurrence in certain genes. Although SNVs are more common in most tissues, indels are more likely than SNVs to alter protein function. Indels in cells of the liver and bladder contribute to the clonal expansions at a fairly high rate compared to other tissues. We also noticed that the cell fractions of those in their 60s appear to be higher than that of those in their 70s in some tissues, which is largely due to the diminished quantity of indels from those in their 70s. This is pronounced in the endometrium where the total and SNV cell fractions for the 60s is lower than the 70s, but the opposite is true for the indels in this tissue. Still, this could be due to the small numbers of indels observed in the 60s and 70s age groups (37 and 15, respectively) across few individuals for this tissue. An additional caveat is that different individuals are being studied in the different age groups, with their many differences in lifestyles, genetics, and other factors.
Figure 4: Prevalence of cancer-associated single-nucleotide variants in tissues across age groups.
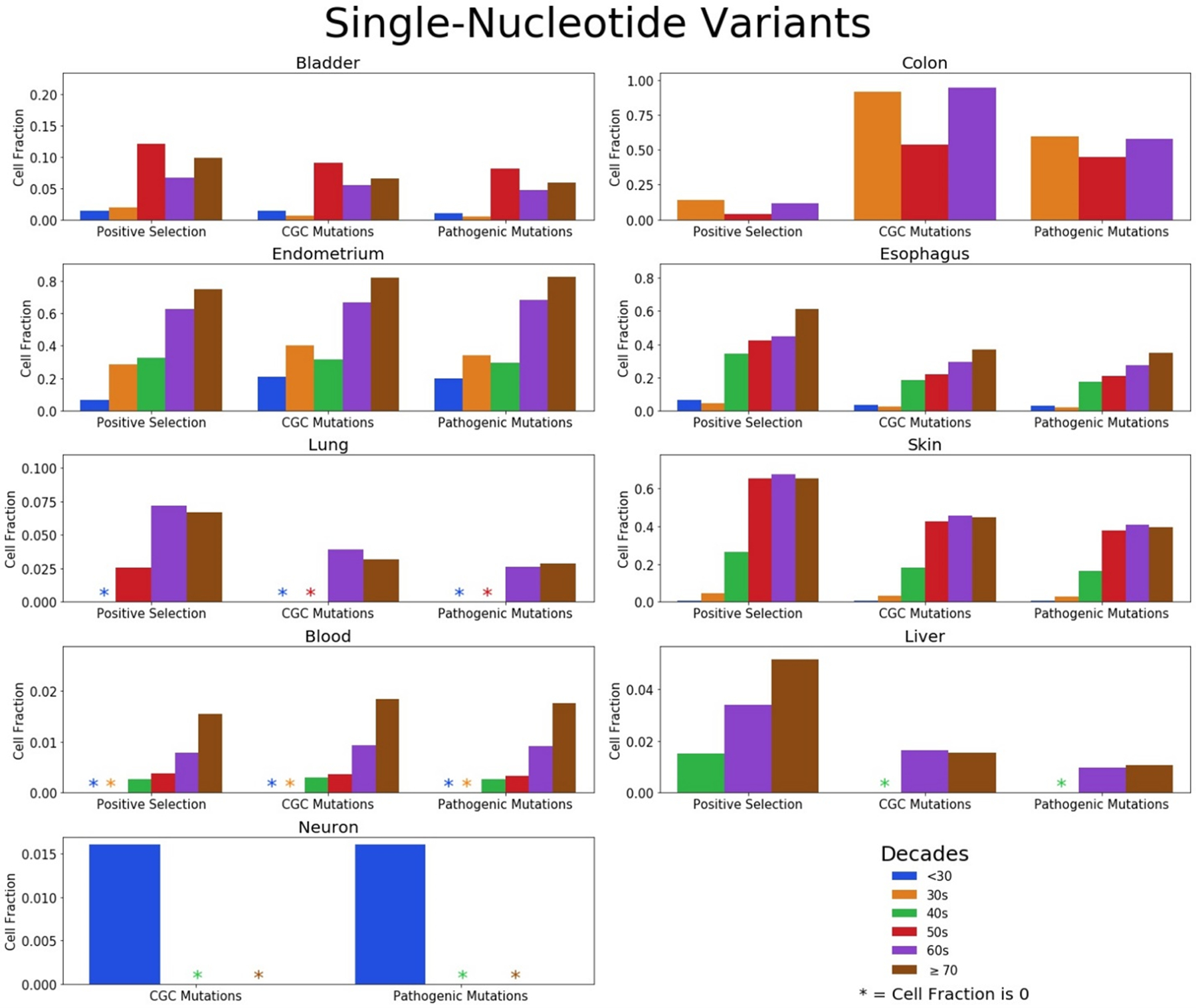
Fractions of cells with SNVs in genes under positive selection (level 1), mutations in the CGC (level 2), and CGC mutations considered pathogenic (level 3) divided in age groups for the bladder, colon, endometrium, esophagus, lung, skin, blood, liver, and neurons. Note differences in the scale of the y-axes for the different tissues.
Figure 5: Prevalence of cancer-associated insertions and deletions in tissues across age groups.
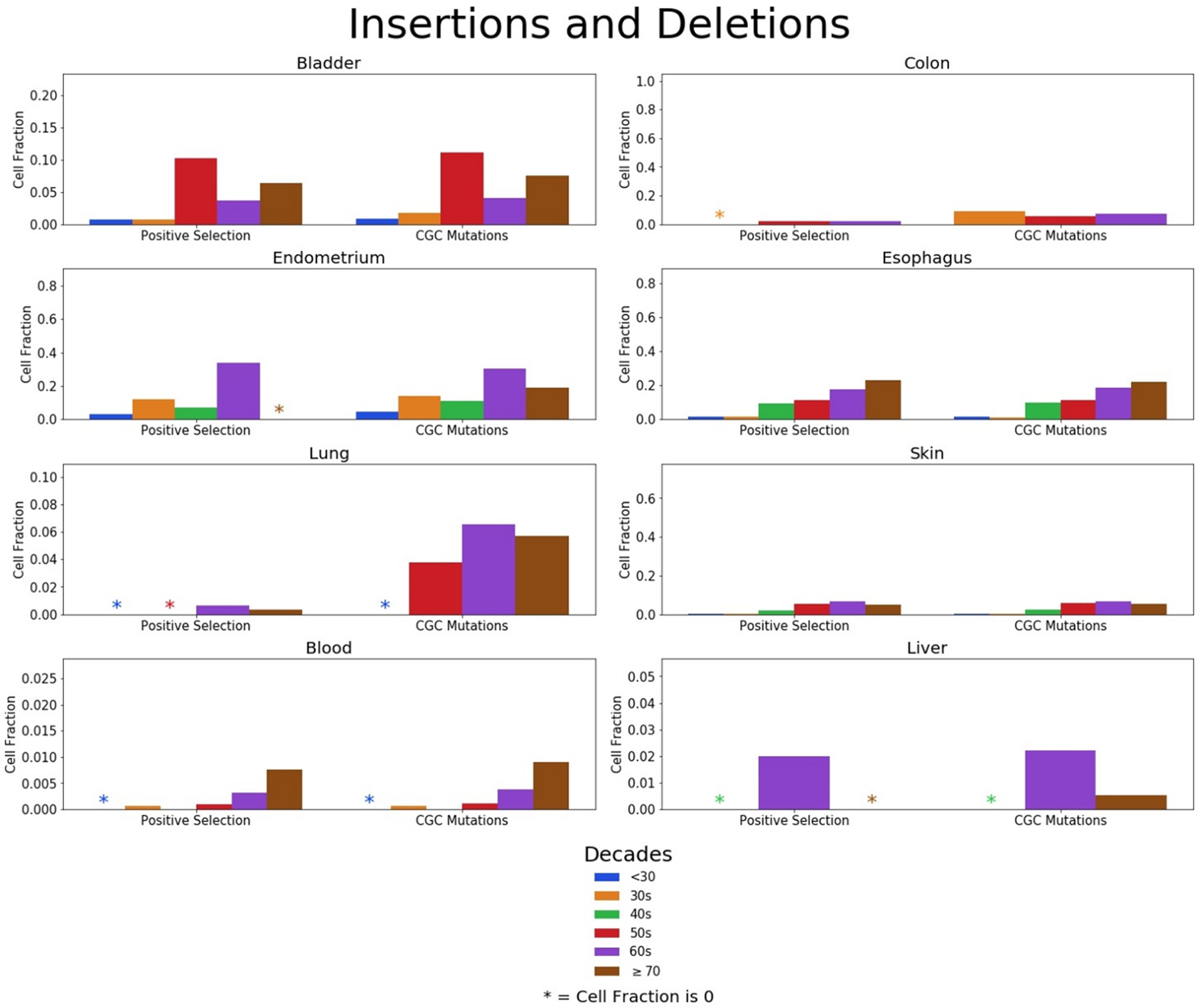
Fractions of cells with indels in genes under positive selection (level 1) and indels in CGC genes (level 2 and 3) divided in age groups for the bladder, colon, endometrium, esophagus, lung, skin, blood, and liver. Note differences in the scale of the y-axes for the different tissues.
From the cell fractions of level 3 of Figure 3, we created body maps for each age group, representing the prevalence of pathogenic mutations through the entire body across tissues (Figure 6). We readily observe the increase in mutational proportions (indicated by the intensity of red) as we progress from the youngest to the oldest age groups throughout all tissues. Note that the red seen in the <30 age group for the neuron is from the lone pathogenic mutation observed in the analysis. This figure also highlights the fact that there are data missing from several age groups; there is not a single age group where all of the tissues analyzed are represented. It is worth noting that this is merely a depiction tissues analyzed, not true representations of how they exist in the human body, most notably for the neuron and endometrium. For example, neurons exist throughout the human body, not just the brain. Additionally, the depiction of the endometrium as the uterus with fallopian tubes is to distinguish the endometrium from the many tissues in this area and not to indicate that the cell fractions presented in this study extend throughout this part of reproductive system.
Figure 6: Body map representations of each age group showing the fractions of cells with mutations in the CGC that are considered pathogenic for each tissue.
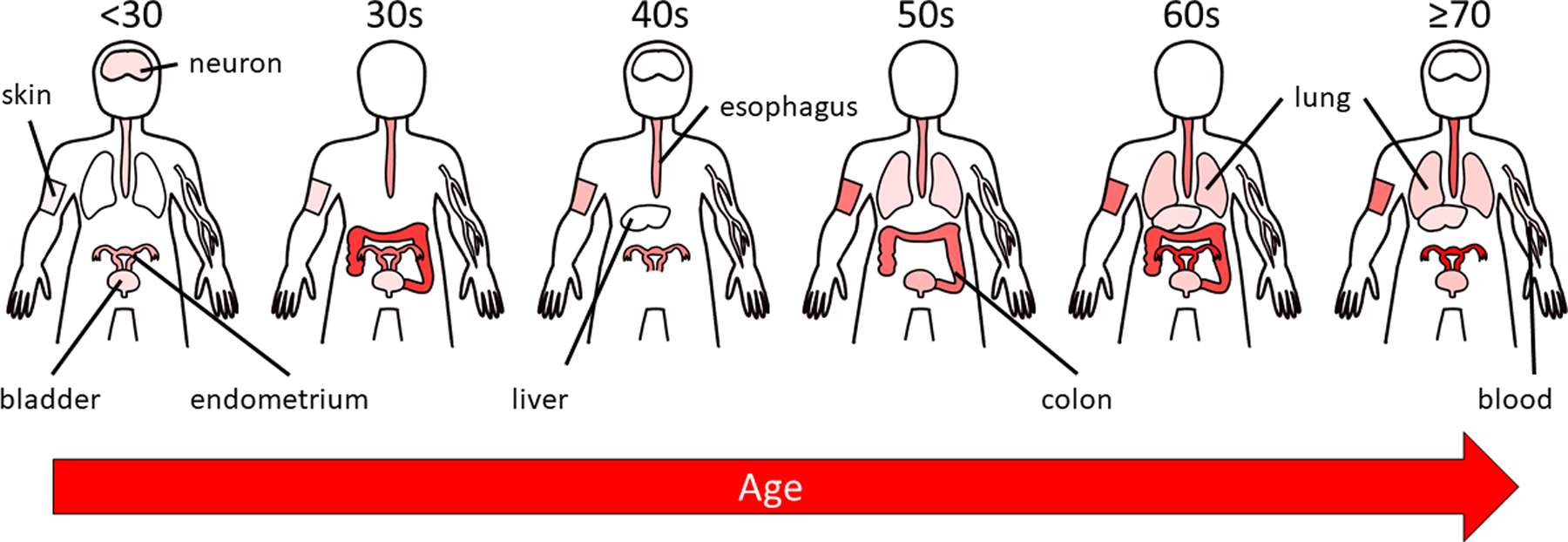
The opacity of red color is proportional to the fraction of cells with cancer-associated mutations. For epithelial tissues, this only tracks the epithelial components of the tissue. Note that the weak signal evident in the brain (“neuron”) of the 30s figure is based on a single variant, and thus should not be overinterpreted. If an organ or tissue is not depicted for a particular age group, then that age group was not represented for that tissue.
Additionally, we wanted to compare the occurrence of protein-altering SNVs and indels across ages in normal tissues to chance expectations, which entails using mutations solely in coding regions of the exome. For tissues where the cell populations analyzed were recently derived from a single stem cell (endometrial glands and colonic crypts) or were analyzed using single-cell techniques (lung and neuron), the vast majority of mutations observed (typically thousands per cell) will be passenger mutations, present due to their random occurrence and not due to selection, even when a particular driver mutation or two is present in the sampled clone. Therefore, a ratio of the total number of pathogenic CGC SNVs in the exome from these tissues by the total number of SNVs in the exome observed can be used to calculate the threshold (0.008) for which the prevalence exceeds expectation by chance alone for the SNVs in the other tissues where selection would be likely necessary for clonal expansion and thus detection. Also, we calculated a ratio of the number of pathogenic mutations in the CGC by the total number of mutations possible in the exome (exome size X 3), which can also be used as an alternative threshold (0.003) for overrepresentation of pathogenic mutations. For the SNVs in these latter tissues for each age group, we calculated a ratio of the number of SNVs found in the tissues analyzed with WGS or whole-exome sequencing that is considered pathogenic in the CGC by the total number of SNVs in that age group, as shown in Table 1. For all the age groups across all tissues except the liver, the values exceeded the threshold by at least an order of magnitude where SNVs were observed. For blood, the original WES data was filtered for specific variants in 160 genes in myeloid malignancies.21 Considering the low mutation rate of blood cells and the presence of artifacts when conducting WGS, this was done to eliminate false positive and germline mutations. This comes at the expense of observing potential passenger mutations. Due to this prefiltration for leukemia-associated variants, we could not use these data for an assessment of enrichment for either SNVs or indels. Finally, we note that the WGS data of the skin was not readily accessible for accurate calculations.
Table 1:
Ratios of pathogenic CGC SNVs to total SNVs for each tissue with non-clonal samples separated by age groups.
| Bladder | Esophagus | Liver | ||
|---|---|---|---|---|
| <30 | 0.158 | 0.189 | No Data | |
| 30s | 0.058 | 0.186 | No Data | |
| 40s | No Data | 0.337 | No Variants | |
| 50s | 0.253 | 0.289 | No Data | |
| 60s | 0.263 | 0.360 | 0.005 | |
| ≥70 | 0.189 | 0.399 | 0.003 | |
| Colon | Endometrium | Lung | Neuron | |
| 0.020 | 0.032 | 0.001 | 0.006 | |
| Four clonal tissues | ||||
| 0.008 | ||||
Additionally, we wanted to know if the pathogenic CGC SNVs were occurring in tissues by chance alone. Only tissues that were analyzed with WGS can be effectively used for this analysis. Therefore, the blood (which filtered for variants in 160 genes commonly mutated in hematologic cancers), skin, and esophagus are excluded from the analysis. Considering that there are approximately 260k pathogenic mutations and there are 9.3×109 possibilities (the length of the genome times 3 potential nucleotide changes) for SNVs, we determine that approximately 1 in every 36k mutations is pathogenic by chance alone. Using the total number of SNVs found in the original studies, we calculated the number of pathogenic CGC mutations by chance and used the chi-squared test to compare the number of pathogenic CGC SNVs observed to the expected number of pathogenic CGC SNVs, as shown in Table 2. With an alpha value of 0.02 (98% probability of accurately rejecting the null hypothesis of the values being similar), all of the tissues except the neurons have a statistically different amount of observed mutations as opposed to those by chance. In the neurons, the single pathogenic CGC SNV observed aligns with what is to be expected, but it is difficult to determine the biological or statistical significance of this given the small number of pathogenic CGC mutations. However, there were significantly more pathogenic CGC SNVs observed in the bladder, colon and endometrium than expected by chance; Interestingly, the opposite is true for the lung and liver. This may be related to divergent turnover rates and tolerance of damaging mutations across tissues. Based on the low (<1) number of stem cell divisions per cell per year,17 the lung and the liver stem cells largely remain in the epithelia for long periods of time without renewal and, consequently, may be less tolerant of mutations that affect protein function. On the other hand, the epithelia of the colon and endometrium renew multiple times each year, which may enable greater tolerance. The skin and esophagus also renew frequently. Coupling this with the constant exposure of mutation-causing ultraviolet radiation to the skin, we suspect that the skin and esophagus would have an overrepresentation of pathogenic CGC SNVs as well. Further analysis on these tissues could reinforce a correlation between stem cell turnover and the presence of pathogenic mutations.
Table 2. The number of pathogenic mutations observed in WGS, pathogenic mutations expected by chance alone, and total number of SNVs observed in the original study.
Chi-square values above 5.412 indicate at least a 98% probability of accurately rejecting the null hypothesis of the observed and expected number of pathogenic CGC mutations being similar.
| Observed | By Chance | Total WGS SNVs | Chi-square | |
|---|---|---|---|---|
| Bladder | 32 | 4 | 152457 | 179.72 |
| Colon | 135 | 1 | 21074 | 30561.06 |
| Endometrium | 139 | 9 | 307945 | 1967.38 |
| Liver | 5 | 23 | 830147 | 14.36 |
| Lung | 13 | 25 | 904842 | 6.04 |
| Neurons | 1 | 1 | 22706 | 0.21 |
For the indels, we made an analogous calculation as done with the SNVs in Table 1, obtaining a ratio of the number of indels in coding regions of CGC genes by the total number of indels observed for each age group across tissues (Table 3). We calculated the threshold by obtaining the ratio of genes in the CGC by the total number of protein-coding genes (0.035). This assumes that the coding regions of genes in the CGC do not have a statistically different number of base pairs from the rest of the protein-coding genes. Additionally, we calculated the ratio of the number of indels in the coding region of CGC genes by the total number of indels detected for the clonal tissues (0.079) as an additional means of comparison. Like the SNVs, indels are observed more frequently than chance expectations across of the bladder and esophagus but not the liver. Overall, the values are remarkably higher than the analogous calculations for SNVs (and both thresholds). Thus, the detection of an indel in the coding region of a CGC gene appears to reflect strong positive selection for these events. Considering the inherent damage to a protein that indels generate, especially through frameshifts, it is not surprising to see higher ratios of pathogenic mutations versus that seen for SNVs in the same tissues.
Table 3:
Ratios of indels in CGC genes to total number of indels for each tissue with non-clonally analyzed tissues separated by age groups.
| Bladder | Esophagus | Liver | ||
|---|---|---|---|---|
| <30 | 0.650 | 0.767 | No Data | |
| 30s | 0.913 | 0.773 | No Data | |
| 40s | No Data | 0.891 | No Variants | |
| 50s | 0.880 | 0.821 | No Data | |
| 60s | 0.875 | 0.908 | 0.156 | |
| ≥70 | 0.839 | 0.934 | 0.077 | |
| Colon | Endometrium | Lung | Neuron | |
| 0.085 | 0.185 | 0.042 | No Variants | |
| Four clonal tissues | ||||
| 0.079 | ||||
Although there is not a direct correlation between positive selection and the number of mutations observed in the CGC, their relative abundances are informative. When driver mutations and genes dominate a tissue, that is reflected by a high cell fraction for level 1 mutations. However, if they have few pathogenic mutations, this may be indicative of a tissue landscape that is protected from cancerous transformation. In the skin and esophagus, NOTCH1 mutations predominate, leading to high cell fractions for genes under positive selection.23,40,44 However, many of the individual NOTCH1 mutations seen in the normal skin and esophagus are not considered pathogenic in the CGC database, despite the clear signature of strong positive selection. Thus, these mutations enable clonal expansion, apparently without being harmful on the organismal level, and may in fact be protective from more deleterious (malignant) mutations4,48.
From the fractions of cells calculated for level 3, we also estimated the number of cells with pathogenic CGC mutations in a human body, as shown in Figure 7. Considering that all tissues are not represented across the age groups, we condensed the data into three age groups for this analysis, grouping the age groups of <30 and 30s, 40s and 50s, and 60s and ≥70. We took the average cell counts if the data was available for both original age groups and used data from one age group if the other was not available. There are approximately 3 trillion nucleated cells in the average human body27, but we only accounted for approximately half of the total cells in the construction of Figure 7 due to missing tissues (i.e. various non-hematopoietic and non-epithelial cell types) and an inability to get accurate cell counts for the endometrial epithelium. Even still, we can see that cell counts reach over 130 billion cells for the oldest age groups. In fact, the youngest age group of cancer-free individuals still has more than 36 billion cells with pathogenic CGC mutations. These results highlight the surprising abundance of cells with cancer-associated mutations in our tissues and raises numerous questions concerning the impact of these mutations on cancer risk, tissue aging, and immune surveillance, as well as the evolved strategies to either limit or tolerate such mutations.
Figure 7. Counts of cells with mutations in the CGC and considered pathogenic for each tissue across the six age groups.
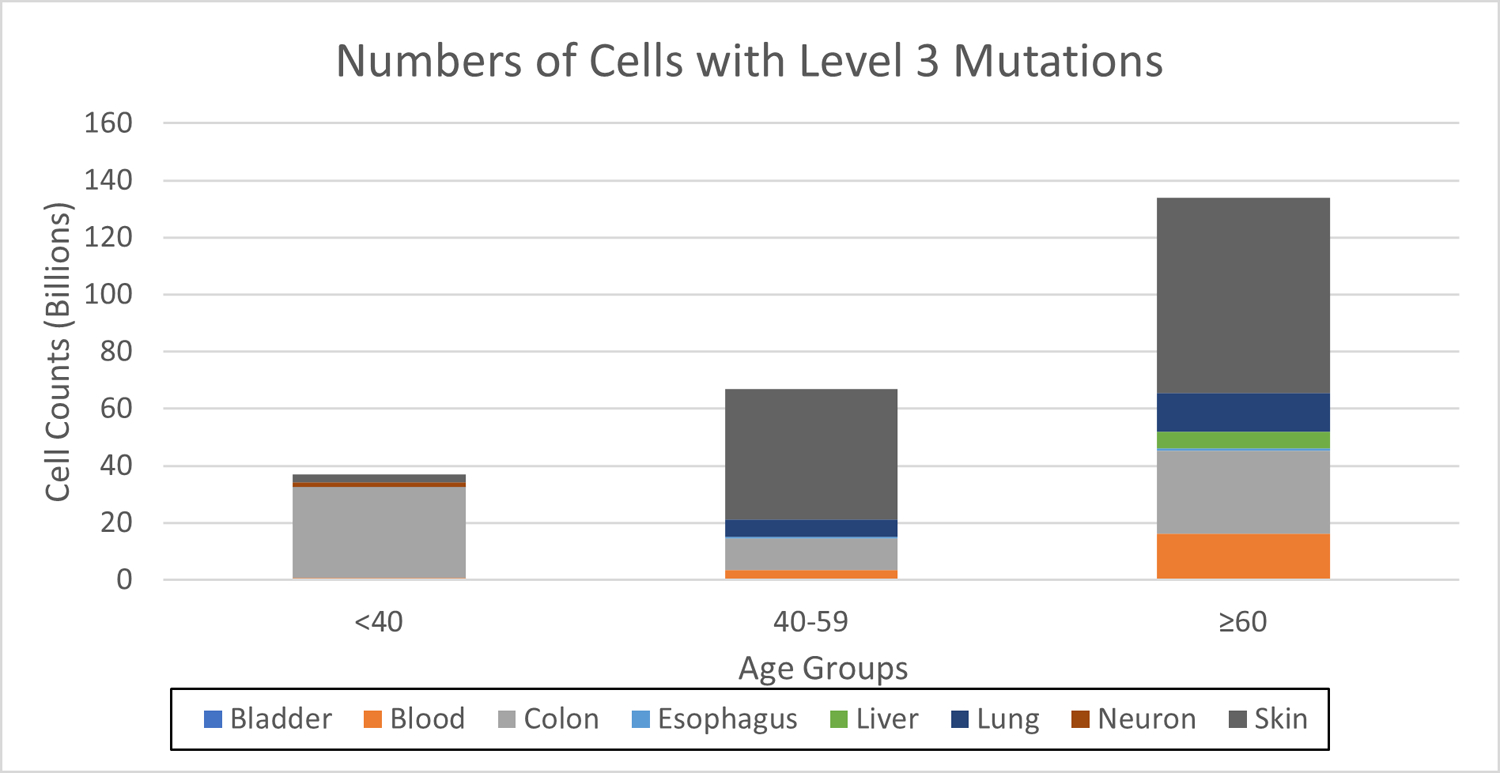
The endometrium is not included.
DISCUSSION
In this paper, we have cataloged and analyzed the striking prevalence of mutations known to be associated with cancers throughout many of our tissues and how this prevalence changes with age. These results have important implications for understanding evolved mechanisms of tissue maintenance and tumor suppression, immune tolerance (including for malignant growths), aging and tissue decline, and cancer risk and the influences of aging and exposures.
The abundance of potentially oncogenic mutations in histologically normal tissue leads to more questions than answers. Mutations in genes under positive selection (Level 1) highlight the presence of clones that can lead to various disruptions in tissue function and diseases, including cancer. For the original studies except for the blood, genes under positive selection were determined using a dN/dS algorithm. Therefore, clonal expansions of the mutations in level 1 are largely not from neutralizing cell competition or a decline in diversity but due to increased fitness. However, some studies determined whether specific mutations were under positive selection, whereas we incorporate all mutations from entire genes that either had positively-selected mutations or that were considered under positive selection, leading to higher estimations for Level 1 mutations for the colon. We estimate that about 10% of crypts in the colon have positively selected mutations for older age groups. However, in the work conducted by Lee-six et al. they derived an estimate of 1% by limiting their analysis to specific mutations associated with colorectal cancers.49 Given the fact that clones detected in the blood are likely to be selected for, we suspect that the percentage of cells with mutations under positive selection would align with the number of individuals considered to have clonal hematopoiesis. According to the study by Jaiswal et al.21, approximately 10% of individuals 70 and older have clonal hematopoiesis. Accounting for the fact that we have diploid cells and that the median VAF for these individuals was approximately 10%, we estimate that approximately 2% of cells in the blood have detectable variants for this age group. This is consistent with our fraction of cells with mutations under positive selection for those aged 70 and older. Still, we recognize that this number would be much higher for blood if more sensitive detection methods were used.
COSMIC is an extensive database of information cataloging somatic mutations in human cancers. Particularly, mutations in its Cancer Gene Census (Level 2) have been observed in human cancer samples. Observing these cancer-associated mutations in cancer-free tissues may indicate that initiating clones remain dormant in our tissues for years before additional events (e.g. additional mutations or decline in the tissue microenvironment) leads to transformation. As could be the case with level 1 mutations, these mutation-driven clonal expansions may also contribute to altered tissue function. Considering that some mutations found in cancer can be neutral passenger mutations, even if in known driver genes, we must be cognizant of the mutations that can actually alter protein function (level 3). This can be used to distinguish between mutations that persist through a fitness increase as opposed to through neutral cell competition. Still, there is not a significant decrease in cell fractions between level 2 and level 3 across the tissues, except for the colon, indicating that most of the mutations in normal tissues that are also found in COSMIC are likely to disrupt protein function. As for the colon, its crypts are more subjected to neutral drift due to the small number of stem cells maintaining each crypt,50 which can account for non-pathogenic CGC mutations being more prevalent in this tissue. On the other hand, this decrease in cell fraction from level 2 to level 3 is not seen so dramatically in the endometrium, which is made up glands with similar clonal dynamics to that of the colonic crypts. This divergence may be attributable to differences between these tissues that are currently not appreciated. All in all, the presence of so many pathogenic CGC mutations without a cancer diagnosis across ages (reaching well over 100 billion cells with pathogenic CGC mutations in the oldest age group) encourages us to investigate alternative effects of these mutations.
Although many of the pathogenic CGC mutations described in this study do clearly contribute to cancer evolution, we have to recognize that natural selection has acted to limit the damaging impacts of mutations. Life history theory attempts to explain the various strategies that species evolve to maximize their reproductive success under the influences of their environment with its extrinsic hazards and resource limitations.12 Factors such as number of offspring, size, parental investment, lifespan and senescence all play a role in reproductive success. For most of human history, our survival has been limited due to predation, starvation or illness, reducing the chances of contributing to subsequent generations with each passing year of adulthood. While humans still have reproductive value even beyond the age of final reproduction, given the importance of child-rearing even as grandparents51,52, the odds of making it to older ages were relatively low. Therefore, from an evolutionary perspective, investments in tissue maintenance wane at older ages (beginning at least by 40 years) as the odds of contributing to future generations declined. Technology has evolved much faster than the human body, leading to extended lifespans well into the 70s and lifestyles to which we are not yet adapted.
From the perspective of life history theory, we can rationalize that the particularly large increases in cells with cancer-associated mutations at latter ages occur when contributions to future generations were particularly unlikely.53 Such a pattern is most clearly evident for the blood system and lung. Thus, the potential negative impact of these expansions, whether to tissue health or to cancer risk, is delayed till ages where we were either less likely to still be alive or to contribute to future generations. Still, it is also clear that clonal expansions likely driven by cancer-associated mutations are evident in some of our tissues (e.g. endometrium and esophagus) during earlier periods such as our 30s and 40s when humans clearly had (and have) high reproductive value. Notably, the risk of death from cancers or other causes is quite low in these periods, indicating that these expansions are less damaging than we might otherwise have thought. In all, we can speculate that natural selection has favored mechanisms, from effective DNA repair to tissue maintenance strategies that limit mutation-driven clonal expansions, which are “good enough” to support an effective strategy for reproductive success among humans. Given tradeoffs, such as resource investments that can either be allocated to reproductive success in youth versus tissue maintenance as we age, such strategies are not perfect and do not last forever. Moreover, we can also appreciate that while natural selection has disfavored somatic evolution that reduces our fitness during periods of a lifespan where reproductive success was likely, this is not to say that natural selection has disfavored all somatic evolution (even beyond the somatic evolution that generates our adaptive immune system). Some somatic evolution, even during youth, may not be disfavored if it either does not reduce tissue health (i.e. it is essentially neutral in this regard) and may even contribute to increased tissue function as we age or in response to damage (as discussed below). Some clonal expansions, even those driven by mutations associated with cancers, may actually reduce cancer risk in some tissues,4,48 as proposed for Notch1 mutations in the esophagus as we age or NFKBIZ mutations in the colon of individuals with inflammatory bowel disease54–56. Similarly, selection for mutations in cirrhotic liver may contribute to improved liver function and regeneration.37,46
So then, why do we experience these cancer-associated mutation driven clonal expansions in our tissues? First, the mutations have to occur. We know that mutations occur throughout our life, given the huge number of cell divisions required to generate and maintain a human body populated by trillions of cells.57,58 In addition, the clock-like accumulation of mutations has been shown to be to a large extent independent of cell cycling, occurring at similar rates in postmitotic tissues.16 Thus, while DNA repair mechanisms are quite effective, given the cell intrinsic and extrinsic insults to our cellular genomes, mutations accumulate with each passing year (roughly 20 mutations per cell per year). While there are numerous evolved mechanisms to eliminate cells with mutations that could contribute to malignancies, from apoptosis to senescence to immune elimination,59 these mechanisms are clearly imperfect.
We should also consider the implications of the frequent occurrence of clones driven by mutations, many of which would be expected to generate new immune epitopes. We are unaware of any study that has systematically analyzed variants expanded in normal tissues to ask whether there is observable underrepresentation of those that are predicted to generate new epitopes for presentation by major histocompatibility complex proteins. Still, such an analysis for somatic mutations in the bladder did not observe evidence for immune editing.34 While underrepresentation of such immunogenic epitopes might indicate immune-mediated elimination of cells bearing these mutations, it is perhaps more likely that the frequent presence of oncogenic mutations in our normal tissues necessitates immune tolerance to new epitopes.60 Otherwise our tissues would likely be under immune attack, given the ubiquitous presence of these mutations. We can further speculate that this tolerance, while necessary to avoid autoimmune attack, may be a cost that is manifested in greater tolerance to malignancies. Thus, the immune tolerant state of cancers could at least in part result from the tolerance to cancer-associated mutations that necessarily resulted from somatic mosaicism throughout our tissues, further bolstered by the evolution of additional suppressive mechanisms within a particular tumor or cancer.
The accumulation of mutations that results in clonal mosaicism throughout tissues is a major hallmark of aging.61 While unexamined thus far (as the focus has been on oncogenesis), these genetic alterations in important genes could potentially result in changes in tissue homeostasis and a decline in tissue function. As described in the original reports, mutations in genes that are important for tissue maintenance are commonly observed. A notable example is high cell fractions of NOTCH1 mutations in the esophagus23,44 and skin40,62. NOTCH1 is integral in cell-fate determination, ranging in involvement from cell proliferation to cell lineage commitment to apoptosis.50,63 Despite the high mutation burden in this important gene, the tissues were determined to be histologically and morphologically normal. However, this does not mean that tissue function is not altered by the presence of these clones, even if they do not directly contribute to oncogenesis. This is seen in the blood of healthy individuals where mutations in epigenetic regulators increase the risk of cardiovascular illness and immune dysfunction.41 This begs the question as to how, or if, tissue function is affected by the presence of these clones and the roles these clones play in aging and the onset of disease. Considering that many of these genes are essential to proper tissue function and mutations are commonly observed in cancer samples, it is logical to suspect that these clones are both harmful and potential precursors to cancer. On the other hand, some of these mutations could be protective against more deleterious mutations that could readily disrupt tissue function and evolve into cancer48, which suggests that expansions of these clones may even be beneficial later in life. In fact, NOTCH1 mutations are more prevalent in normal esophagus than they are in esophageal cancer,23,44 indicating that cancers selectively initiate from cells without these Notch1 mutations.
In addition to the mutations that occur endogenously through aging, mutations also accumulate exogenously through exposures to mutagens. With the advancement of next-generation sequencing, mutational signatures have been established that enable us to surmise the origin of the mutation whether it is from aging, smoking, or ultraviolet radiation.64,65 Therefore, cancer risk can be associated with the nature and frequency of certain mutations in normal tissues. For example, the prevalent occurrence of G to T transitions in lung cancers of smokers has enabled the identification of specific carcinogens responsible for heightened cancer risk.66 For the lung, the authors observed these specific mutational signatures in some subjects who were smokers, despite the fact that signatures associated with age were even more prevalent.38 However, a fraction of lung epithelial cells analyzed in smokers and even more commonly in former smokers exhibited mutational landscapes similar to never-smokers, highlighting the plethora of known and unknown factors that lead to the complexity of understanding mutational and clonal dynamics. Even so, this information could facilitate efforts to determine who is more susceptible to cancer, so preventative measures can be applied prior to oncogenesis or the decline in tissue function. In particular, the presence of a fraction of lung cells with “normal” mutational patterns even in smokers, and the numerical rebound of these cells in former smokers, may lead to efforts to specifically favor these cells particularly post-smoking cessation.
All in all, we have leveraged numerous studies to analyze clonal mutational landscapes in normal tissues across ages and observed that many of the mutations responsible for these clones occur in cancer contexts. This creates more questions than answers as some tissues are dominated by mutations that are associated with cancers. With some cancer-free individuals harboring over 100 billion cells with oncogenic mutations, the mutational patterns that distinguish benign or even protective states from damaging and/or malignant states, and the relevant factors that determine the fortunately rare transitions from an oncogenically-initiated clone to one that forms an aggressive cancer, remain elusive.
LIMITATIONS
For the analyses described above, it is important to note the limitations of our methods and the data, and alternative interpretations for the results. As mentioned previously, all tissues throughout the human body across all age groups are not represented in our analyses. There are numerous other studies that focused on somatic mutations in normal skin67,68, blood42 (reviewed in reference41), esophagus44, and a variety of tissues4,5,7,8,69 that we did not include in our analysis. These reports provided additional insight on mutation patterns and in many cases corroborated our calculations, but we acknowledge that different techniques and limits of detection can lead to modest variations in outputs. Even for the tissues used for our analysis, all age groups are not covered. Although excellent studies have been conducted on tissues we did not cover such as the prostate70, skeletal muscle71, and brain72, more samples from cancer-free individuals across age groups will be needed for the type of analysis done in this study. Furthermore, the sensitivity for some methods do not enable the detection of clones with VAFs much lower than 1–5% (depending on the study). Therefore, the many clones with VAFs below the level of detection are not accounted for, leading to an underestimation of applicable cell fractions. For these reasons, we did not attempt to compare VAFs for the mutations in the different categories (e.g. CGC level 2 and level 3), given that detection of the variants were only possible for non-clonal tissues if they exceeded some threshold.
It is also worth noting the limited range of demographics covered. When calculating the number of cells with oncogenic mutations, estimations in given tissues were based on a 70 kg male, which may deviate significantly from many people, most notably females. Furthermore, subjects in some of these studies are dominated by those of European ancestry. Considering that research is now showing that ancestry can play a critical role in susceptibility to certain illnesses,73–75 it is not farfetched to believe clonal expansions in normal tissues could differ depending on genetic ancestry. These limitations highlight the fact that there is much to investigate concerning the understanding of the mutational landscape in cancer-free cells and how they alter the susceptibility to cancers and other diseases.
Beyond the availability of data, there are limitations in our analysis. When looking across normal tissues, we cannot control for lifestyle choices made by the individuals who donated samples. A notable example of this is seen in the bladder for those in their 50s where all were smokers and drinkers, which may have led to higher cell fractions with cancer-associated mutations. Not only do we have limited knowledge of the lifestyle of the subjects, but there is minimal information about how choices like diet and exercise affect the clonal landscape in normal tissues. Additionally, the CGC uses FATHMM as a variant effect predictor (VEP), but there are many other options that may represent the quantity of pathogenic mutations differently. A compiler that incorporates many algorithms for protein-altering mutations could lead to more robust determinations about the effects of variants.
Supplementary Material
ACKNOWLEDGEMENTS
These studies were supported by grants from the American Association for Cancer Research/Johnson&Johnson (18–90-52-DEGR), the Veteran’s Administration (1 I01 BX004495), and the National Institute of Aging (R01AG066544 and R01AG067584) to J.D. and support from the National Cancer Institute Ruth L. Kirschstein National Research Service Award T32-CA190216 to E.J.E. The contents of this study are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health. We thank the multiple researchers whose mutational data were analyzed here for providing additional data to us in formats that facilitated our analyses – Drs. Iñigo Martincorena, Luiza Moore, Henry Lee-Six, Peter Campbell, Phillip Jones, Siddhartha Jaiswal, and Benjamin Ebert. We are particularly grateful to Dr. Phillip Jones for his guidance with the calculations of cell numbers for the esophagus. We also thank Michael DeGregori for artwork in Figure 6.
Footnotes
CODE AVAILABILITY
The code created for these analyses can be found at https://github.com/edjevans/Normal_Tissue_Analysis.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to disclose.
DATA AVAILABILITY
To conduct this analysis, we obtained the subject data that had the ages and number of samples for each individual and the mutation data for each tissue through the supplemental information of the original work or associated GitHub accounts. We procured any missing information directly from the original authors.
References
- 1.Laconi E, Marongiu F & DeGregori J Cancer as a disease of old age: changing mutational and microenvironmental landscapes. Br. J. Cancer 1–10 (2020). [DOI] [PMC free article] [PubMed]
- 2.Busque L et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat. Genet 44, 1179–1181 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jonason AS et al. Frequent clones of p53-mutated keratinocytes in normal human skin. Proc. Natl. Acad. Sci. U. S. A 93, 14025–14029 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kakiuchi N & Ogawa S Clonal expansion in non-cancer tissues. Nat. Rev. Cancer 1–18 (2021). [DOI] [PubMed]
- 5.García-Nieto PE, Morrison AJ & Fraser HB The somatic mutation landscape of the human body. Genome Biol 20, 298 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoang ML et al. Genome-wide quantification of rare somatic mutations in normal human tissues using massively parallel sequencing. Proc. Natl. Acad. Sci. U. S. A 113, 9846–9851 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yizhak K et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science (80-. ) 364, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martincorena I Somatic mutation and clonal expansions in human tissues. Genome Med 11, 35 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vijg J Somatic mutations, genome mosaicism, cancer and aging. Current Opinion in Genetics and Development vol. 26 141–149 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gatenby RA & Gillies RJ A microenvironmental model of carcinogenesis. Nature Reviews Cancer vol. 8 56–61 (2008). [DOI] [PubMed] [Google Scholar]
- 11.DeGregori J Evolved tumor suppression: Why are we so good at not getting cancer? Cancer Research vol. 71 3739–3744 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hill K & Kaplan H Life history traits in humans: Theory and Empirical Studies. Annu. Rev. Anthropol 28, 397–430 (1999). [DOI] [PubMed] [Google Scholar]
- 13.Rozhok A & DeGregori J A generalized theory of age-dependent carcinogenesis. Elife 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fane M & Weeraratna AT How the ageing microenvironment influences tumour progression. Nat. Rev. Cancer 20, 89–106 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.White MC et al. Age and cancer risk: A potentially modifiable relationship. Am. J. Prev. Med 46, S7–S15 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abascal F et al. Somatic mutation landscapes at single-molecule resolution. Nature 1–6 (2021). [DOI] [PubMed]
- 17.Tomasetti C & Vogelstein B Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science (80-. ) 347, 78–81 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sondka Z et al. The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nature Reviews Cancer vol. 18 696–705 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shihab HA, Gough J, Cooper DN, Day INM & Gaunt TR Predicting the functional consequences of cancer-associated amino acid substitutions. Bioinformatics 29, 1504–1510 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martincorena I et al. Universal Patterns of Selection in Cancer and Somatic Tissues. Cell 171, 1029–1041.e21 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaiswal S et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med 371, 2488–2498 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steensma DP & Ebert BL Clonal hematopoiesis as a model for premalignant changes during aging. Exp. Hematol 83, 48–56 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martincorena I et al. Somatic mutant clones colonize the human esophagus with age. Science 362, 911–917 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clevers H XThe intestinal crypt, a prototype stem cell compartment. Cell vol. 154 274 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Tanaka M et al. Evidence of the monoclonal composition of human endometrial epithelial glands and mosaic pattern of clonal distribution in luminal epithelium. Am. J. Pathol 163, 295–301 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bianconi E et al. An estimation of the number of cells in the human body. Ann. Hum. Biol 40, 463–471 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Sender R, Fuchs S & Milo R Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLOS Biol 14, e1002533 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chalana V, Dudycha S, Yuk J-T & McMorrow G Automatic Measurement of Ultrasound-Estimated Bladder Weight (UEBW) from Three-Dimensional Ultrasound. Rev. Urol 7 Suppl 6, S22–8 (2005). [PMC free article] [PubMed] [Google Scholar]
- 29.Lewis SA Everything you wanted to know about the bladder epithelium but were afraid to ask. Am. J. Physiol. Physiol 278, F867–F874 (2000). [DOI] [PubMed] [Google Scholar]
- 30.Bolla SR & Jetti R Histology, Bladder. StatPearls (StatPearls Publishing, 2019). [PubMed] [Google Scholar]
- 31.Lee J et al. Esophageal Diameter Is Decreased in Some Patients With Eosinophilic Esophagitis and Might Increase With Topical Corticosteroid Therapy. Clin. Gastroenterol. Hepatol 10, 481–486 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Awad ZT et al. Correlations between Esophageal Diseases and Manometric Length: A Study of 617 Patients. J. Gastrointest. Surg 3, 483–488 (1999). [DOI] [PubMed] [Google Scholar]
- 33.Zhang X et al. The microscopic anatomy of the esophagus including the individual layers, specialized tissues, and unique components and their responses to injury. Ann. N. Y. Acad. Sci 1434, 304–318 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Lawson ARJ et al. Extensive heterogeneity in somatic mutation and selection in the human bladder. Science (80-. ) 370, 75–82 (2020). [DOI] [PubMed] [Google Scholar]
- 35.Lee-Six H et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature 574, 532–537 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Moore L et al. The mutational landscape of normal human endometrial epithelium. Nature 580, 640–646 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Brunner SF et al. Somatic mutations and clonal dynamics in healthy and cirrhotic human liver. Nature 574, 538–542 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoshida K et al. Tobacco smoking and somatic mutations in human bronchial epithelium. Nature 578, 266–272 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lodato MA et al. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science (80-. ) 359, 555–559 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fowler JC et al. Selection of Oncogenic Mutant Clones in Normal Human Skin Varies with Body Site. Cancer Discov 11, 340–61 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jaiswal S & Ebert BL Clonal hematopoiesis in human aging and disease. Science vol. 366 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Genovese G et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med 371, 2477–2487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li R et al. Macroscopic somatic clonal expansion in morphologically normal human urothelium. Science (80-. ) 370, 82–89 (2020). [DOI] [PubMed] [Google Scholar]
- 44.Yokoyama A et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature 565, 312–317 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Hogan BLM et al. Repair and regeneration of the respiratory system: Complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell vol. 15 123–138 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu M et al. Somatic Mutations Increase Hepatic Clonal Fitness and Regeneration in Chronic Liver Disease. Cell 177, 608–621.e12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Promislow DEL & Tatar M Mutation and senescence: where genetics and demography meet. Genetica 102/103, 299–314 (1998). [PubMed] [Google Scholar]
- 48.Higa KC & DeGregori J Decoy fitness peaks, tumor suppression, and aging. Aging Cell vol. 18 e12938 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee-Six H et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature 574, 532–537 (2019). [DOI] [PubMed] [Google Scholar]
- 50.Nakamura T, Tsuchiya K & Watanabe M Crosstalk between Wnt and Notch signaling in intestinal epithelial cell fate decision. Journal of Gastroenterology vol. 42 705–710 (2007). [DOI] [PubMed] [Google Scholar]
- 51.Hamilton WD The genetical evolution of social behaviour. I. J. Theor. Biol 7, 1–16 (1964). [DOI] [PubMed] [Google Scholar]
- 52.Brown JS & Athena Aktipis C Inclusive fitness effects can select for cancer suppression into old age. Philos. Trans. R. Soc. B Biol. Sci 370, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rozhok AI & DeGregori J The Evolution of Lifespan and Age-Dependent Cancer Risk. Trends in Cancer 2, 552–560 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nanki K et al. Somatic inflammatory gene mutations in human ulcerative colitis epithelium. Nature 577, 254–259 (2020). [DOI] [PubMed] [Google Scholar]
- 55.Kakiuchi N et al. Frequent mutations that converge on the NFKBIZ pathway in ulcerative colitis. Nature 577, 260–265 (2020). [DOI] [PubMed] [Google Scholar]
- 56.Olafsson S et al. Somatic Evolution in Non-neoplastic IBD-Affected Colon. Cell 182, 672–684.e11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mustjoki S & Young NS Somatic Mutations in “Benign” Disease. N. Engl. J. Med 384, 2039–2052 (2021). [DOI] [PubMed] [Google Scholar]
- 58.Vijg J From DNA damage to mutations: All roads lead to aging. Ageing Research Reviews vol. 68 101316 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lowe SW, Cepero E & Evan G Intrinsic tumour suppression. Nature vol. 432 307–315 (2004). [DOI] [PubMed] [Google Scholar]
- 60.Brodnicki TC Somatic Mutation and Autoimmunity. Cell vol. 131 1220–1221 (2007). [DOI] [PubMed] [Google Scholar]
- 61.López-Otín C, Blasco MA, Partridge L, Serrano M & Kroemer G The hallmarks of aging. Cell vol. 153 1194 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Martincorena I et al. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348, 880–6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Radtke F, Wilson A, Mancini SJC & MacDonald HR Notch regulation of lymphocyte development and function. Nature Immunology vol. 5 247–253 (2004). [DOI] [PubMed] [Google Scholar]
- 64.Van Hoeck A, Tjoonk NH, Van Boxtel R & Cuppen E Portrait of a cancer: Mutational signature analyses for cancer diagnostics. BMC Cancer vol. 19 1–14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Helleday T, Eshtad S & Nik-Zainal S Mechanisms underlying mutational signatures in human cancers. Nature Reviews Genetics vol. 15 585–598 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pfeifer GP et al. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene 21–48, 7435–7451 (2002). [DOI] [PubMed] [Google Scholar]
- 67.Hernando B et al. The effect of age on the acquisition and selection of cancer driver mutations in sun-exposed normal skin. Ann. Oncol 32, 412–421 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tang J et al. The genomic landscapes of individual melanocytes from human skin. Nature 586, 600–605 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li R et al. A body map of somatic mutagenesis in morphologically normal human tissues. bioRxiv 2020.11.30.403436 (2020). [DOI] [PubMed]
- 70.Cooper CS et al. Analysis of the genetic phylogeny of multifocal prostate cancer identifies multiple independent clonal expansions in neoplastic and morphologically normal prostate tissue. Nat. Genet 47, 367–372 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Franco I et al. Somatic mutagenesis in satellite cells associates with human skeletal muscle aging. Nat. Commun 9, 800 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nishioka M, Bundo M, Iwamoto K & Kato T Somatic mutations in the human brain: implications for psychiatric research. Mol. Psychiatry 24, 839–856 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Newman LA et al. Hereditary Susceptibility for Triple Negative Breast Cancer Associated With Western Sub-Saharan African Ancestry. Ann. Surg 270, 484–492 (2019). [DOI] [PubMed] [Google Scholar]
- 74.Carrot-Zhang J et al. Genetic Ancestry Contributes to Somatic Mutations in Lung Cancers from Admixed Latin American Populations. Cancer Discov 11, 591–599 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Terao C et al. Chromosomal alterations among age-related haematopoietic clones in Japan. Nature 1–6 (2020). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
To conduct this analysis, we obtained the subject data that had the ages and number of samples for each individual and the mutation data for each tissue through the supplemental information of the original work or associated GitHub accounts. We procured any missing information directly from the original authors.