

Abstract
The coronavirus disease 2019 (COVID‐19) pandemic demonstrates the importance of generating safe and efficacious vaccines that can be rapidly deployed against emerging pathogens. Subunit vaccines are considered among the safest, but proteins used in these typically lack strong immunogenicity, leading to poor immune responses. Here, a biomaterial COVID‐19 vaccine based on a mesoporous silica rods (MSRs) platform is described. MSRs loaded with granulocyte‐macrophage colony‐stimulating factor (GM‐CSF), the toll‐like receptor 4 (TLR‐4) agonist monophosphoryl lipid A (MPLA), and SARS‐CoV‐2 viral protein antigens slowly release their cargo and form subcutaneous scaffolds that locally recruit and activate antigen‐presenting cells (APCs) for the generation of adaptive immunity. MSR‐based vaccines generate robust and durable cellular and humoral responses against SARS‐CoV‐2 antigens, including the poorly immunogenic receptor binding domain (RBD) of the spike (S) protein. Persistent antibodies over the course of 8 months are found in all vaccine configurations tested and robust in vitro viral neutralization is observed both in a prime‐boost and a single‐dose regimen. These vaccines can be fully formulated ahead of time or stored lyophilized and reconstituted with an antigen mixture moments before injection, which can facilitate its rapid deployment against emerging SARS‐CoV‐2 variants or new pathogens. Together, the data show a promising COVID‐19 vaccine candidate and a generally adaptable vaccine platform against infectious pathogens.
Keywords: antibodies, COVID‐19, cytotoxic T‐cells, mesoporous silica rods, monophosphoryl lipid A (MPLA), recombinant proteins, SARS‐CoV‐2, vaccines
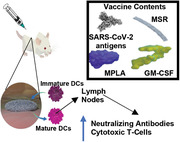
Mesoporous silica rods (MSRs) scaffolds are used as a platform to induce adaptive immunity against SARS‐CoV‐2 antigens. Sustained delivery of GM‐CSF, MPLA, and a variety of SARS‐CoV‐2 subunit immunogens from the MSR vaccine recruite immune cells into the scaffolds and consistently induce antigen‐specific long‐lasting antibodies, cytotoxic T lymphocyte responses (CTL), and viral neutralization in vitro.
1. Introduction
In late December 2019, a cluster of pneumonia cases of unknown etiology was announced in Wuhan, China. In January 2020, the Chinese Center for Disease Control and Prevention (CDC) identified that a novel coronavirus, known as SARS‐CoV‐2, was the causative agent of a new respiratory tract disease,COVID‐19. COVID‐19 rapidly advanced from an epidemic to a pandemic, and as of May 6, 2021, the World Health Organization (WHO) has reported over 154 million confirmed cases, resulting in 3.2 million deaths worldwide.[ 1 ] While refined clinical management and therapies can improve the outcomes for patients with severe disease,[ 2 , 3 ] preventative measures are necessary to halt disease transmission, reduce COVID‐19‐related deaths and hospitalizations, and avoid the development of long‐term sequelae that are being increasingly reported in a fraction of recovered patients.[ 4 ] Safe, efficacious, and rapidly deployable vaccines are imperative to prevent SARS‐CoV‐2 infections and alleviate restrictive public health measures that have been necessary to ward off disease transmission.
Currently approved COVID‐19 vaccines in the United States are based on viral vectors (Johnson & Johnson), which require complex manufacturing processes[ 5 , 6 ] or lipid‐nanoparticle (LNP)‐encapsulated mRNA (Moderna, Pfizer‐BioNtech), which exhibit low stability. Preexisting immunity to viral vectors due to prior infections can limit the efficacy of viral‐based vaccines.[ 7 ] Although this can be mitigated by using highly immunogenic vectors (Ad26 in Johnson and Johnson), animal‐derived vectors (ChAdOx1 in AstraZenaca), or using different vectors for prime and boost (rAd26 and rAd5 in Sputnik V), the antivector immunity generated by these COVID‐19 vaccines will likely limit the use of these same vectors in future vaccine iterations, both to new SARS‐COV‐2 variants and other infectious diseases. By contrast, subunit vaccines based on structural viral proteins can be easily produced at a large scale, and exhibit relatively high stability. Although both viral vector and recombinant proteins require cell culture in bioreactors for at‐scale production, subunit proteins can be achieved in non‐mammalian expression systems and bypass the need for large amounts of materials with human infectious potential. For example, Novavax, the only approved SARS‐CoV‐2 protein vaccine uses insect cells and a baculovirus vector to produce the Spike protein found in their formulation, lowering biosafety risk and requirements.[ 8 ] SARS‐CoV‐2 contains many protein target candidates, including the nucleocapsid (N) protein, the S protein, and the RBD that is required for ACE2 receptor binding and viral entry into host cells. While protein antigens are generally poorly immunogenic, they would be easier to distribute and store than current mRNA vaccines. For example, Novavax has developed a protein antigen‐based coronavirus vaccine that shows effectiveness against the original coronavirus, the B.1.1.7 variant, and the B.1.351 variant to differing levels, but the duration of protection is unclear.[ 9 , 10 ]
Resident dendritic cells (DCs) process the antigenic components of vaccines and present them in the context of major histocompatibility complex (MHC) molecules to prime antigen‐specific T and B cells in lymph nodes.[ 11 ] DCs need to be properly activated in order to achieve optimal antigen presentation. This is often facilitated by adjuvants such as alum, which improve DC activation, although alum skews toward Th2 responses.[ 12 , 13 ] Recently, a mixture of alum and inactivated SARS‐CoV‐2 was tested as a potential vaccine in mice, rats, and macaques. Antibodies against SARS‐CoV‐2 spike proteins, nucleocapsid proteins, RBD, and viral neutralizing antibodies, were detected. However, no significant T cell response was observed.[ 14 ] Past work has demonstrated the importance of co‐delivering protein antigens and adjuvants to DCs for optimal antigen presentation, and the lack of spatiotemporal control in their delivery in conventional approaches may limit success. Vaccine formulations that enable precise control over the delivery of various SARS‐CoV‐2 antigens and adjuvants to DCs will likely potentiate humoral and cellular responses against SARS‐CoV‐2.
Biomaterial carriers of antigens and adjuvants can obviate delivery issues of typical vaccine strategies, amplify immunomodulatory effects, integrate the synergistic effect of different molecules, and importantly, home and manipulate immune cells in vivo.[ 15 , 16 ] We have developed a macroscale materials‐based system based on MSRs that functions as a vaccine by mimicking immune reactions that occur during local infection. Following administration, the MSR forms a defined microenvironment with micro‐sized pores, enabling DC recruitment by exploiting the cells’ migratory response to chemoattractants. The large size and high aspect ratio of MSRs allow the formation of persistent scaffolds with the microporous structure for the homing of a large number of immune cells.[ 17 ] Both antigen and adjuvant can be released to the concentrated DCs. Such vaccines can be easily formulated by simply mixing highly porous MSRs with any bio‐actives of interest, freeze‐drying, and storing them in powder‐form until ready to reconstitute for injection. Our MSR‐based vaccines generate both high antibody titers, and robust CTL responses. A single injection elicits persistent germinal center B‐cell activity (>3 weeks), highly elevated antibody responses (>12 months), superior memory B‐cell generation, and potent CTL responses in the context of cancer immunotherapy and infectious diseases.[ 18 , 19 , 20 ]
In this study, we explored the use of MSRs as a basis for COVID‐19 vaccination by incorporating various protein antigens from the SARS‐CoV‐2 virus (Figure 1 ). MSRs sustainably release GM‐CSF, a potent cytokine that leads to the accumulation of large numbers of APCs at the vaccine site. The FDA‐approved adjuvant, MPLA was simultaneously released to promote maturation of the accumulated APCs, and their persistent trafficking to the draining lymph nodes to present antigens to the B and T cells. We hypothesized this vaccine could induce potent anti‐SARS‐CoV‐2 adaptive responses and we predict its modularity could offer an advantage to respond to new variants.
Figure 1.
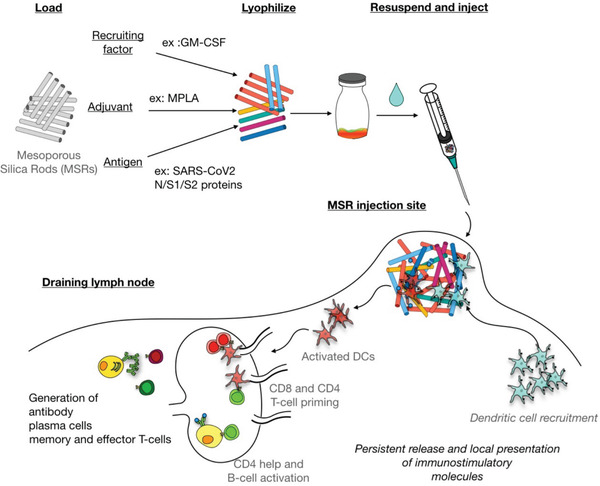
Schematics for the use of MSRs for vaccination. MSRs are loaded with a recruiting factor, adjuvant, and antigen by adsorption, before being lyophilized. The vaccine can be stored in powder form until ready to be administered. After reconstitution in an aqueous buffer, MSR vaccine can be injected subcutaneously and create a scaffold for DC recruitment and homing, allowing their local activation and antigen presentation. Recruited and activated DCs migrate to the draining lymph node where they participate in the generation of an adaptive immune response against the desired antigens by priming cognate T‐cells. B‐cell, activation, in conjunction with DC‐induced CD4+ T cells participate in the generation of antigen‐specific B‐cell response.
2. Results
2.1. Formulation of a COVID‐19 Vaccine Using Mesoporous Silica Rods
Our approach, consisting of MSRs, GM‐CSF, and adjuvants, is illustrated in Figure 1. Both domains of the Spike protein (S1 and S2) as well as the N‐protein were first absorbed into the MSRs. Of note, very low amounts of protein (1 µg each) were utilized, as this could increase the number of vaccine doses that could be rapidly deployed in response to a pandemic. GM‐CSF was incorporated as an APC‐recruiting factor and the FDA‐approved TLR‐4 agonist MPLA was used as an adjuvant (Figure 2a). We previously showed that GM‐CSF and subunit antigens on MSR persist at the scaffold site for days to weeks.[ 17 , 19 ] All biologics in this SARS‐CoV‐2 MSR vaccine demonstrated a slow release upon rehydration following lyophilization and storage. Over the course of a week, a small fraction of GM‐CSF and MPLA (<5% of the total load within 8 days) spontaneously released from the MSR, suggesting a high affinity for the particles. On the other hand, antigens were released at a higher rate with 30–65% of the load releasing 8 days after loading (Figure 2b‐d). Scanning Electron Microscopy (SEM) images confirmed that high aspect ratio MSRs (Figure 2e; Table S1, Supporting Information) loaded with these components (N/S1/S2 MSR vaccine) became heavily infiltrated with immune cells following subcutaneous injection (Figure 2f). Consistent with previous reports,[ 17 , 19 ] the subcutaneous nodules formed by the MSR vaccine grew over time, peaking in size on day 7 before slowly resorbing over the next 3 weeks. MSR scaffolds were undetectable 4–5 weeks after injection (Figure 2g). By contrast, nodules formed by MSR injection alone (no GM‐CSF, antigen, nor MPLA) demonstrated a modest change in size (Figure 2g; Figure S1, Supporting Information), suggesting superior immune cell recruitment in the fully formulated vaccine.
Figure 2.
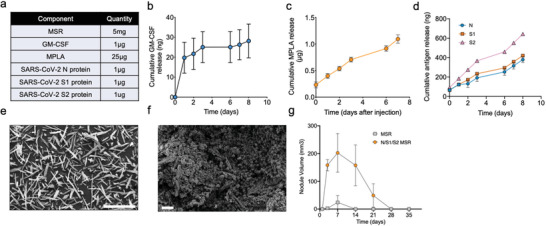
MSR vaccine formulated with SARS‐CoV‐2 antigens attracts immune cells and sustainably releases vaccine adjuvants. a) Composition of SARS‐CoV‐2 MSR vaccine. Cumulative in vitro release of b) GM‐CSF (n = 3), c) MPLA (n = 2), and d) N, S1, and S2 antigens from MSRs (n = 3). Data represent mean ± SD. SEM images of e) unloaded MSRs before injection and f) SARS‐CoV‐2 MSR vaccine explanted 7 days after injection into the skin of BALB/c mice. Scalebar = 200µm and 20µm, respectively. g) Scaffold volume over time (n = 3). Data represent mean and SD.
2.2. MSR Vaccine Generates High Antibody Titers Against SARS‐CoV‐2 Antigens
To test the ability of the MSR vaccine to generate SARS‐CoV‐2 specific antibodies, we immunized BALB/c mice with the full N/S1/S2 MSR or a Sham version containing the MSR, GM‐CSF, and MPLA but no antigen. Mice received a second immunization 4 weeks after the initial vaccination. enzyme‐linked immunoassay (ELISA) serum titer analysis showed a high IgG antibody response to all three incorporated antigens, with no observable rise in antibodies in the serum of Sham‐injected mice (Figure 3a). Peak IgG concentrations were 1.1 ± 0.3 × 106 ng mL−1, 3 ± 2 × 106 ng mL−1, and 1.0 ± 0.5 × 106 ng mL−1 for anti‐N, anti‐S2, and anti‐S1, respectively compared to an average reading of 2.7 ± 0.7 × 102 ng mL−1 in Sham serum. Nine months after the first shot (266 days), IgG responses against N, S1, and S2 proteins had dropped from peak values but remained significantly elevated.
Figure 3.

Two doses of N/S1/S2 MSR vaccine generate persistently high antibody responses to SARS‐CoV‐2 antigens. Six to 8 weeks old BALB/c mice were immunized on day 0 and boosted on day 28 with MSRs loaded with 1 µg GM‐CSF, 25 µg MPLA, 1 µg N protein, 1 µg S1 protein, and 1 µg S2 protein (N/S1/S2 MSR, n = 10) or a Sham vaccine containing GM‐CSF and MPLA only (n = 10). Serum antibody concentration against the full‐length N protein, the S1, S2, and RBD were measured over time by ELISA. Total IgG (n = 10; a), IgG1 (n = 8; b), and IgG2a (n = 8; c) are reported. The dotted line indicates peak concentration. P‐values represent the results of Friedman test with post hoc Wilcoxon for repeated measures comparing all timepoints to day 0 concentration.
Immunization led to a balanced humoral response as evidence by the induction of both IgG1 (Th2‐skewed) and IgG2a (Th1‐skewed) antibodies (Figure 3b,c). Broader isotype analysis using Simoa technology [ 21 ] confirmed the prevalence of IgG1 and IgG2a responses (Figure S2, Supporting Information) as well as high IgG2b responses (Figure S3, Supporting Information). However, little to no IgM and IgA were detected (Figure S3, Supporting Information). Importantly, high antibody titers were induced against the Receptor Binding Domain (RBD) of the S protein (Figure 3c,d). RBD‐specific IgG1 and IgG2a concentrations were already significantly higher than baseline before boosting on day 28, and peak value of 3 ± 2 × 104 ng mL−1 and 10 ± 7 × 103 ng mL−1 were observed 3.6 and 4.6 months after the second injection, respectively. There was a decrease in anti‐RBD IgG1 and IgG2a concentrations 9 months after immunization, but these concentrations were still significantly higher than pre‐vaccination levels.
Overall, these data indicate that a two‐dose regimen of the MSR vaccine induces robust and long‐lasting antibody against SARS‐CoV‐2 antigens.
2.3. MSR Vaccine Generates Functional Adaptive Immunity Against SARS‐CoV‐2 Antigen
We then sought to investigate whether the immune response generated by the MSR vaccine could elicit functional protection upon antigen recognition. Using a pseudovirus neutralization assay,[ 22 , 23 ] we observed that the serum of all MSR immunized mice could effectively decrease in vitro viral infection 1 month after the second dose (Figure 4a). By contrast, the serum of Sham immunized mice had no neutralizing activity. Interestingly, the neutralization titers induced by the MSR vaccine continued to increase up to 4.5 months after the first dose was administered. All vaccinated animals had titers with significant neutralizing activity at the last timepoint tested (169 days after immunization) compared to the baseline and Sham.
Figure 4.

N/S1/S2 MSR vaccine induces adaptive immunity with anti‐SARS‐CoV‐2 activity. Six to 8 weeks old BALB/c mice were immunized with MSRs loaded with 1 µg GM‐CSF, 25 µg MPLA, 1 µg N protein, 1 µg S1 protein, and 1 µg S2 protein (N/S1/S2 MSR vaccine, n = 10) or a Sham vaccine containing GM‐CSF and MPLA only (n = 10). All animals received a second injection on day 28 and serum was used to determine neutralization titers against a SARS‐CoV‐2 pseudovirus load over 180 days (a). The solid line represents mean, P‐values represent the results of Friedman test with post hoc Wilcoxon for repeated measures comparing all timepoints to day 0 values. Splenocytes of animals having received a single injection on day 0 were restimulated with peptides spanning the S‐protein on day 28 and CTLs were analyzed for IFN‐γ production by Flow Cytometry. Kruskal–Wallis test with post hoc Dunn test. **P < 0.01.
As antigen‐specific cytotoxic CD8+ T‐cells can help rapidly clear host cells that have become virally infected,[ 24 , 25 ] we tested whether splenic T‐cells from immunized mice could become activated upon antigen re‐encounter. After a single injection of N/S1/S2 MSR, the proportion of IFN‐γ secreting CD8+ T‐cells in response to S‐antigen was significantly increased in the vaccine groups compared to naïve animals (Figure 4b). Together, these data suggest that the MSR vaccine can generate an adaptive immune response with both prophylactic and therapeutic potential.
MSR‐based vaccines can be fully formulated and stored in a powder form or manufactured as a lyophilized “Shell” solely loaded with the recruiting factor and adjuvant. In that case, the antigen could be simply added at the rehydration step, right before immunization, which could allow stockpiling of vaccine and rapid adaptation to new antigen targets (Figure S4, Supporting Information). When the MSR vaccine was formulated in such “add‐in‐time” manner with N/S1/S2, a prime‐boost vaccine‐induced similarly persistent antibodies with high neutralization activity, demonstrating the feasibility of this approach (Figure 5 ).
Figure 5.
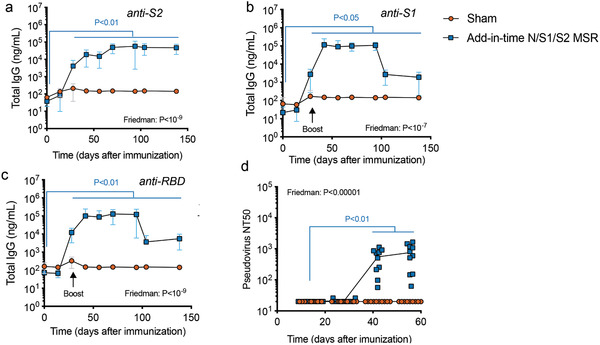
Add‐in‐time MSR vaccine induces persistent humoral response with neutralization potential. Six to 8 weeks old BALB/c mice were immunized on day 0 and boosted on day 28 with MSRs loaded with 1 µg GM‐CSF, 25 µg MPLA with 1 µg N protein, 1 µg S1 protein, and 1 µg S2 protein added minute injections (N/S1/S2 MSR vaccine, n = 10) or a Sham vaccine containing GM‐CSF and MPLA only (n = 10). Serum IgG concentration against the S2 domain (a), S1 domain (b), and RBD (c) were measured over time by ELISA. P‐values represent the results of Friedman test with post hoc Wilcoxon for repeated measures comparing all timepoints to day 0 concentration. d) Neutralization titer – line represents data mean (n = 10). P‐values represent the results of Friedman test with post hoc Wilcoxon for repeated measures comparing all timepoints to day 14 titer.
2.4. Single‐Dose of MSR Vaccine Against Various S Protein Domains Induces Neutralizing Activity
A SARS‐CoV‐2 vaccine that induces neutralizing antibodies with a single‐dose would help ensure patient compliance and quickly deploy vaccines to a greater number of people in emergency situations. Therefore, we tested if the MSR vaccine could elicit persistent antibodies in a single dose context. The RBD of the S1 domain is the functional binder of the ACE2 receptor on human cells. RBD contains many CD4 Th epitopes and could focus the antibody response to a neutralizing region of the protein, making it an attractive immunogen.[ 26 , 27 ] However, some reports have shown it to be less immunogenic than the full S protein.[ 28 , 29 , 30 ] Therefore, we explored the ability of the MSR vaccine incorporating RBD alone or in combination with domains of the S protein to provide RBD immunogenicity. We also tested whether a fivefold higher antigen dose would improve antibody titers (Figure 6a).
Figure 6.
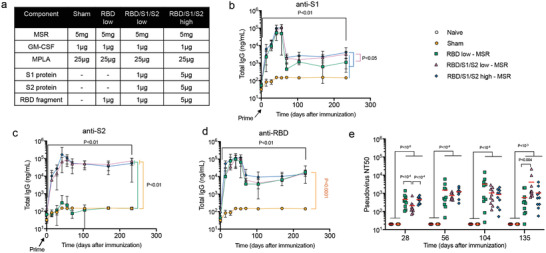
Single‐shot MSR vaccine formulations induce anti‐SARS‐CoV‐2 activity. Six to 8 weeks old BALB/c mice were single immunized with vaccine formulations shown in (a) on day 0 (n = 10). Total S1‐ (b), S2‐ (c), and RBD‐specific (d) serum IgG was measured using ELISA. P‐values (black) represent the results of Wilcoxon signed‐rank test between day 0 and day 233 for each group (in all 4 groups, P < 0.01). In (b–d), all groups were compared against each other at the final timepoint using Kruskal–Wallis test with P < 0.05 for (b), P < 0.01 for (c), and P < 0.0001 for (d), the P values of the post hoc analysis of the comparisons at day 233 are indicated in orange. The neutralization potential of immunized sera was assessed against SARS‐CoV‐2 pseudovirus. e) Kruskal–Wallis test P‐values were P < 1 × 10−7, 10−4, 10−4, and 10−5 for days 28, 56, 104, and 135, respectively. Post hoc Conover test P‐values for the comparison of each group with each other are depicted in the plot.
All of the MSR vaccine formulations induced antibody generation against incorporated antigens up to 8 months after a single dose (Figure 6b‐d). As expected, incorporation of RBD alone did not elicit any detectable IgG against the S2 domain (Figure 6c). However, all formulations showed comparable levels of anti‐RBD antibodies, regardless of dose or antigen type. By contrast, anti‐S1 IgG concentrations were higher in the 5 µg antigen dose and in S1‐containing formulations compared to RBD‐alone, presumably due to a decreased diversity of S1‐specific epitopes in the RBD vaccine. Peak concentrations were observed between 1 to 2 months after vaccination before stabilizing about an order of magnitude lower.
Functionally, all of these MSR formulations induced significant neutralization serum in all animals as early as 1 month post‐vaccination, and the neutralization activity increased over the following 2 months. Neutralization Titers were still high over 4 months after immunization with an average NT50 over 5 × 102 for all groups (limit of detection is 20; Figure S5, Supporting Information). Together, these findings demonstrate that MSR vaccines directed at fragments of the S‐protein can induce persistent humoral response with protective potential following a vaccine single‐dose.
3. Discussion
Here, we demonstrate the use of a modular biomaterial platform based on MSR as a potential COVID‐19 vaccine. One of the potential benefits of this technology is its plug‐and‐play versatility that allows one to quickly incorporate various antigens and readily scale manufacturing. These capabilities could allow for the rapid development of new vaccines targeting SARS‐CoV‐2 variants as they emerge. Synthetic amorphous silica is known to have good biocompatibility, supporting its development as a versatile platform for clinical applications.[ 31 , 32 ] We have previously reported high cell viability inside and in the vicinity of MSR scaffolds in vivo and no systemic cytotoxicity was found by histological analysis of major organs.[ 19–17 ] Swelling and mild redness in some animals are the sole adverse effects we could observe and mice did not show overt signs of pain or discomfort, supporting the overall biocompatibility of the use of MSRs for vaccination. Further, GM‐CSF and MPLA used in the MSR vaccine have a track record of safe use in humans local release of these agents from a biomaterial vaccine allows small quantities to be used (e.g., 1 µg of GM‐CSF per vaccine), minimizing concerns regarding systemic side‐effects or toxicity.[ 33 , 34 , 35 ] As with all recombinant protein therapies, there is a concern for anti‐GM‐CSF autoantibodies development, which could cause toxicity or decrease the efficacy of the technology for multiple uses. However, GM‐CSF has been used in numerous clinical trials including cancer vaccine applications with a favorable safety profile.[ 33 ] Moreover, while anti‐GM‐CSF have been detected in the context of melanoma, they have not been associated with a decrease in vaccine efficacy.[ 36 ] Previous work using a different biomaterial system delivering GM‐CSF and tumor antigens showed that GM‐CSF was required to maximize vaccine efficacy against acute myeloid leukemia (AML). It would be important to confirm the role of GM‐CSF in the generation of anti‐SARS‐CoV‐2 by MSR vaccines in future experiments.[ 37 ]
We show that the MSR vaccines consistently induced high, persistent (8+ months) humoral responses against all protein antigens tested, included the N protein as well as the S1, S2, and RBD of the S‐ protein. Antibody affinity maturation depends on GC formation and prolonged antigen presentation.[ 38 , 39 ] A recent study showed that modification of HIV immunogens with phosphoserines improved antigen loading onto Alum adjuvant and prolonged their bioavailability, augmenting both GC activity and the magnitude of the humoral response generated. Similarly, MSR vaccines enable the slow release of both SARS‐CoV‐2 antigens and the MPLA adjuvant. Together with our previous finding that MSR can prolong GC reactions,[ 19 ] this likely contributed to the high titer observed here. Vaccines generated both Th2‐skewed (IgG1) and Th1‐skewed (IgG2a, IgG2b) antibody responses. Moreover, we could detect CTL responses, which rely on the generation of Th1 responses. The finding of a balanced Th1/Th2 response is encouraging, as preponderant Th2 responses are associated with a risk of vaccine‐enhanced respiratory disease (VAERD) in respiratory syncytial virus (RSV) and measles morbillivirus (MeV).[ 40 , 41 ] Moreover, disease‐enhancement associated with an Alum‐based (Th2‐skewing) SARS‐CoV‐2 vaccine has been reported in preclinical models.[ 42 ] Further characterization of the Th response (CD4 T‐cell compartment) will be required to evaluate the balance of Th1/Th2 induced by the MSR vaccine. Isotype switching, Th1‐skewing, and efficient CTL generation are all dependent on TLR stimulation on B‐cells and APCs.[ 43 , 44 ] The CTL response described in this vaccine, while in range with other reports, can likely be enhanced by using other adjuvants than MPLA.[ 45 ] We have previously shown that a combination of the toll‐like receptor 9 (TLR‐9) ligand CpG‐ODN and polyethyleneimine (PEI) on MSRs could boost CTL responses in the context of peptide cancer neoantigens.[ 18 ] Due to its modular nature, the MSR vaccine can be easily adapted to incorporate other TLR‐agonists, and future investigation could focus on screening adjuvants to reduce SARS‐CoV‐2‐specific Th2 and increase Th1 and CTL responses.
All vaccines tested induced high neutralizing antibodies against a SARS‐CoV‐2 pseudovirus 1–2 months after the first shot. Interestingly, neutralization titers raised faster in the RBD‐containing vaccines compared to N/S1/S2 MSR formulations. This could be due to: 1. decreased competition of B‐cell clones specific for SARS‐CoV‐2 antigens with no or low neutralization potential—that is, N‐specific or S2‐specific clones or: 2. improved clonal‐selection towards neutralizing RBD‐specific antibodies. MSR vaccines formulated with only 1µg of each antigen produced high neutralization titers, and a fivefold higher dose did not further improve. By comparison, other preclinical studies using RBD as the immunogen have reportedly used 5–50 µg.[ 29 , 30 , 46 ] Interestingly, 1 µg RBD in the MSR vaccine generated neutralizing titers after a single dose whereas 25 µg RBD formulated with the potent clinically approved adjuvant MF59 did not induce any neutralization unless a booster shot was administered.[ 29 ] Our finding that RBD and Spike‐based vaccines could induce similar levels of anti‐RBD and neutralization antibodies confirms that, when adequately presented, RBD is a potent immunogen that can elicit comparable functional antibodies to full‐length proteins.
The need for only low amounts of antigen, and a single‐dose immunization regimen are highly desirable to enable the timely production and deployment of vaccines in an emergency context. Further, a vaccine that can be shipped and stored without the need for cold‐chain maintenance could reach remote or low‐income areas more easily by eliminating the need for refrigeration equipment during transportation as well as at the vaccine administration site. Compared to mRNA vaccines and other vaccine technologies based on viral vectors, the MSR vaccine is fully manufactured at room temperature and its final lyophilized form retains bioactivity upon rehydration and injection. Mesoporous silica has been widely used to load and stabilize therapeutic proteins in powder form and protect them from thermal degradation[ 47 , 48 ] and it was recently demonstrated that desilicated tuberculosis and tetanus protein antigens could resist high‐temperature denaturation.[ 49 , 50 ] However, further testing is warranted to test the ability of MSR vaccines to induce high levels of neutralizing antibodies following long‐term storage at room temperature.
MSR vaccines have significant potential to induce long‐term protection against COVID‐19, as neutralizing titers correlate with protection against SARS‐CoV‐2 infection in preclinical hamster and primate models.[ 51 , 52 ] SARS‐CoV‐2 variants have already emerged that can evade neutralization by convalescent serum and significantly decrease the viral neutralization capacity of vaccinated individuals.[ 53 , 54 , 55 ] Thus, it becomes increasingly probable that COVID‐19 vaccines formulated with new antigenic variants will need to be deployed in the future. Here, we showed that pre‐made Shell vaccines can be pre‐manufactured with GM‐CSF and MPLA and reconstituted “in‐time” with soluble antigens. This could allow stockpiling of the MSR vaccine and its adaptation to new immunogens as they become available.
4. Conclusion
We have developed a preclinical COVID‐19 vaccine based on MSR. MSR vaccines could slowly release immunostimulatory factors GM‐CSF, MPLA, and protein antigens, and elicited antigen‐specific cytotoxic T‐cells and consistently high SARS‐CoV‐2 neutralizing titers. Potent neutralization was observed using both S‐protein larger domains S1 and S2 and the less immunogenic Receptor Binding Domain as the antigen. Both a prime‐boost and single‐dose regimen induced persistent antibody responses (8+ months) and neutralization. These data present MSR as a promising vaccine platform and warrants its further testing in animal models of viral challenge.
5. Experimental Section
Vaccine Components
To generate MSR (≈46 µm × 4.5 µm), the surfactant, P123 (4 g; Mn ≈5800, Sigma–Aldrich, USA), was dissolved in 1.6 m hydrochloric acid (150 g; HCl), then stirred with tetraethyl orthosilicate (8.6 g; 98%, Sigma–Aldrich, USA) at 40 °C for 20 h, followed by aging at 100 °C for 24 h. The surfactant was then extracted by refluxing the particles for 24 h in 1% HCl in 70% ethanol. The resulting MSR particles were filtered, washed with 70% ethanol, and dried. MSR morphology was measured and determined using optical microscopy and scanning electron microscopy. Pore volume, pore size, and surface area were analyzed by N2 adsorption/desorption isotherms. GM‐CSF was purchased from Peprotech, USA (315‐03). MPLA derived from Salmonella minnesota R595 (vac‐mpla) was purchased from Invivogen. SARS‐CoV‐2 antigens included spike protein subunits, S1 (40591‐V08H), S2 (40590‐V08B), and N‐protein (40588‐V08B) purchased from Sino Biological, USA, and the receptor‐binding domain (RBD) protein (NR‐52306) was purchased from BEI resources, USA.
Vaccine Manufacture
Prime vaccines were made by adding MSRs (5 mg) to a sterile scintillation vial followed by GM‐CSF (1 µg), MPLA (25 µg), and SARS antigens (1 µg of S1, S2, and N; 1 µg of S1, S2, and RBD; 5 µg of S1, S2, and RBD; 1 µg of RBD). Following, water (0.5 mL) for injection (WFI, HyClone) was added to the vial, and the resulting slurry was vortexed and mixed overnight (≈15 h) using a HulaMixer (Thermo Fisher Scientific) set at 45 rpm. Afterward, the mixture was frozen at ‐80 °C then lyophilized for a minimum of 48 h. Boost vaccines (1 µg of S1, S2, and N) were made the same way as described above. Sham vaccines were made as described above, except no antigen was added. For the shell vaccine, MPS (5 mg) was added to a vial followed by GM‐CSF (1 µg) and MPLA (25 µg). Then WFI (0.5 mL) was added to the vial, and the resulting slurry was vortexed and mixed overnight (≈15 h) using a HulaMixer set at 45 rpm. Next, the mixture was frozen at ‐80 °C and lyophilized for a minimum of 48 h. Antigens (1 µg of S1, S2, and N) were added to shell vaccines and allowed to mix 30 min before immunization. Naïve mice were unvaccinated.
Vaccine Administration and Serum Collection
Eight‐week‐old BALB/c female mice (n = 10 per group) were used for the duration of the study (over 1 year). All the animals were acclimated for 72 h according to the Harvard University Animal Care and Use Committee (IACUC) guidance. Vaccines were administered by subcutaneous injection (18G needle) in the flank in a final volume of 250 µL in WFI. Animals received a second immunization (boost vaccine) on day 28. PBS (1×) was used as the unvaccinated control (naïve). All the mice were bled from the submandibular vein into serum collection tubes. The whole blood was allowed to clot at room temperature for 30 min and to remove the clot, then was centrifuged at 15 000 rpm for 15 min. The resulting serum was aliquoted and stored frozen (‐80 °C). One group of mice received the boost vaccine 4 weeks after the primary immunization, with the same dose as the initial vaccine (1 µg of S1, S2, and N). Mice were maintained under specific pathogen‐free conditions at Harvard University, and all experiments were conducted in accordance with animal use guidelines and protocols approved by IACUC.
MSR Nodule Size
Mice were injected subcutaneously with SARS‐CoV‐2 MSR vaccine (1 µg of S1, S2, and N). Nodule size was quantified over time by measuring the nodule length and width using a caliper. Volume was calculated as (L × W × W/2).
ELISA for SARS‐CoV‐2 Antibody Response
To evaluate the antibody response against SARS‐CoV‐2 proteins, polystyrene 96‐well high‐bind plates (Santa Cruz Biotechnology, USA) were coated with 1 µg mL–1 of S1, S2, N, and RBD proteins in phospate buffered saline (PBS), and standard curves were applied to each plate. Standard Curves were created by 100 µL of stock with a concentration of 50 ng mL–1 mouse IgG standard, and serially diluted twofold. Completed plates were incubated overnight at 4 °C. The following day, the plates were washed and blocked with blocking buffer (1% BSA in PBS‐Tween (PBST)) overnight at 4 °C. The following day, plates were washed again in 1× PBST and sera previously diluted in blocking buffer was added at 1:100, 1:1000, and 1:10 000 (100 µL each) to the plates and incubated at for 1.5 h at 450 rpm, 22 °C. After sera incubation, plates were washed and secondary antibodies peroxidase‐AffiniPure F(ab’)2 Fragment Goat anti‐mouse IgG (111‐035‐006, Jackson ImmunoResearch, USA), Peroxidase‐AffiniPure F(ab’)2 fragment goat anti‐mouse IgG1 and peroxidase‐AffiniPure F(ab’)2 fragment goat anti‐mouse IgG2a secondary antibodies were diluted at 1:20 000 in blocking buffer and added to each well for 1 h at 450 rpm and 22 °C. Plates were washed, and TMB Ultra ELISA solution (100 µL; 34 028, Thermo Fisher Scientific) was added to each well and statically incubated under aluminum foil for 15 min. The reaction was quenched using 1 m sulfuric acid (50 µL) and measured at OD 450 nm using a plate reader (BioTek Synergy HTX). Sera samples were run in duplicate. All plate washes were done in triplicate with 1× PBST (200 µL) using a BioTek microplate washer. Standard curve OD readouts on each plate were used to create a 4PL‐sigmoidal curve to interpolate sample data, which was analyzed, processed, and presented using GraphPad Prism 8 (version 8.1.2).
Quantitation of SARS‐CoV‐2 Proteins, GM‐CSF, and MPLA Release
To quantify the release of bioactives from MSR vaccines, individual components were loaded onto MSRs, lyophilized, then reconstituted in 1 mL PBS. The supernatant was periodically collected to determine the amount released over time using a standard of known concentrations for each component. MPLA concentration was measured by High Perfomance Liquid Chromatography‐Size Exclusion Column (HPLC–SEC) using an Xterra C18 column in TEAA/Acetonitrile buffers on an Agilent 1260 II. GM‐CSF samples were run using R&D Duoset Mouse GM‐CSF kit (DY415) according to the manufacturer's instructions. SARS‐CoV‐2 protein samples (S1, S2, N, and RBD) were measured by ELISA using an anti‐his tag HRP‐conjugated antibody.
SARS‐CoV‐2 Plaque reduction Neutralization Assay
NR‐596 Vero E6 cells were plated in six‐well plates (Corning R CellBIND) overnight to achieve confluency. Sera from vaccinated animals were serially diluted in Dulbecco's PBS (Gibco) using twofold dilutions in a 96‐well polypropylene deep well plate (Thermo Scientific). SARS‐CoV‐2 (isolate USA‐WA1/2020) was diluted in DMEM (Gibco), supplemented with 2% FBS (Gibco) and antibiotic‐antimycotic (Gibco), to a concentration of 1000 plaque‐forming units per mL (pfu mL–1), sufficient for a number of samples, and mixed 1:1 with the diluted samples in the deep well plate. Wells containing only DMEM with 2% FBS and antibiotic‐antimycotic were included as a negative control, and positive controls of 1000 pfu mL–1 SARS‐CoV‐2 incubated with Dulbecco's PBS alone were included. Deep well plates were then incubated at 37 °C for 1 h. Following this incubation, media was removed from the six‐well plates and each dilution (200 µL) was added to the appropriate wells in triplicate. Plates were incubated at 37 °C, 5% CO2 for 1 h, with gentle rocking approximately every 10–15 min. After 1 h, the cell monolayers were overlaid with a 1:1 mixture of 2.5% Avicel RC‐591 microcrystalline cellulose and carboxymethylcellulose sodium (2 mL) (DuPont Nutrition and Biosciences) and 2× MEM (Temin's modification, Gibco), supplemented with 2× antibiotic‐antimycotic, 2× GlutaMAX (Gibco), and 10% FBS, and plates were incubated for 2 days at 37 °C and 5% CO2. Following 2 days incubation, plates were fixed with 10% neutral buffered formalin, removed from biocontainment, stained with 0.2% aqueous gentian violet (RICCA Chemical), and plaques were counted. To determine the IC50 values, GraphPad Prism v.8.4.3 software was used and the nonlinear regression analysis, “log(inhibitor) versus response,” was performed.
COVID‐19 Mouse Serological Single Molecule Array (Simoa)
COVID‐19 mouse serological Simoa assays for IgG, IgG1, IgG2a, IgG2b, IgM, and IgA against four viral antigen proteins S1, S2, N, and RBD were developed and validated. The Simoa assays were performed on the HD‐X Analyzer (Quanterix) in an automated three‐step assay format according to the manufacturer's instructions and as previously reported.[ 21 ] Mouse serum samples were diluted 4000‐fold (or 500‐fold for IgM and IgA assays) in Homebrew Detector/Sample Diluent (Quanterix). The four prepared SARS‐CoV‐2 antigen‐conjugated capture beads were mixed and diluted in Bead Diluent, with a total of 500 000 beads per reaction (125 000 each of Nucleocapsid, S1, S2, and RBD beads). Biotinylated anti‐mouse immunoglobulin antibodies were diluted in Homebrew Detector/Sample Diluent to the final concentrations of IgG (Abcam ab97033): 1 ng mL–1, IgG1 (Abcam ab99604): 1 ng mL–1, IgG2a (Bethyl Laboratories A90‐107B): 400 ng mL–1, IgG2b (BioLegend 406 704): 200 ng mL–1, IgM (BioLegend 406 504), and IgA (Bethyl Laboratories A90‐103B). Streptavidin‐b‐galactosidase (SβG) concentrate (Quanterix) was diluted to 30 × 10–12 m in SβG Diluent (Quanterix). In the first step of each assay, the capture beads (25 µL) were incubated with diluted mouse sera (100 µL) for 15 min. After the incubation, six wash steps were performed with System Wash Buffer 1 (Quanterix). In the second step, the beads were resuspended in the respective biotinylated anti‐mouse immunoglobulin antibody (100 µL) and incubated for 5.25 min, and then washed six times with System Wash Buffer 1. In the third step, the beads were resuspended in SβG (100 µL), incubated for 5.25 min, and washed six times with System Wash Buffer 1. The beads were then resuspended in RGP (25 µL) and loaded into the microwell array. Following the bead loading, the microwell array was sealed with oil and imaged in five optical channels. Average Enzyme per Bead (AEB) values were calculated by the HD‐X Analyzer software. All samples were measured in duplicates.
T Cell Stimulation and Intracellular Cytokine Staining
Spleens from vaccinated mice were collected 4 weeks post‐prime. Mononuclear single‐cell suspensions from whole mouse spleens were generated. Splenocytes from each mouse were seeded at 106 per well in a 96‐well plate in R10 media (RPMI‐1640 supplemented with Pen‐Strep antibiotic, 10% HI‐FBS, Glutamax) under three conditions: no peptide stimulation, and stimulation with two spike peptide pools (JPT product PM‐WCPV‐S‐1) at a final concentration of 2 µg mL–1 each peptide for 24 h at 37 °C. Following stimulation, protein transport inhibitor cocktail was added, and cells were incubated for additional 4 h at 37 °C. Following centrifugation, cells were washed with 1× PBS prior to staining with LIVE/DEAD Fixable Blue Dead Cell Stain (Invitrogen) and mouse Fc Block for 15 min at room temperature. Cells were then washed with staining buffer (100 µL; PBS supplemented with 2% HI‐FBS) and resuspended with a surface stain cocktail containing the following antibodies purchased from Biolegend: CD4 FITC (GK1.5), CD8 Amcyan (53‐6.7), CD62L PE/Cy7(MEL‐14), CD44 PE (IM7), and CD3 APC/Cy7. After 30 min, cells were washed with staining buffer and fixed and permeabilized using the Biolegend Cytofix/Cytoperm fixation/permeabilization solution kit according to manufacturer instructions. Cells were washed in perm/wash solution and stained by intracellular staining (20 min at 4 °C) using a cocktail of the following antibodies purchased from Biolegend: IFN‐γ APC and TNF‐α Pac Blue in 1× perm/wash buffer. Finally, cells were washed in perm/wash solution and resuspended in staining buffer. All the samples were analyzed using a BD LSR FORTESSA flow cytometer instrument. An acquisition gate was established based on forward scatter and side scatter parameters that included only lymphocyte populations and excluded dead cells and debris. Analysis was performed using FlowJo software, version 10.6.2 according to the gating strategy outlined in Figure S6, Supporting Information.
Pseudo Virus Neutralization Assay
The SARS‐CoV‐2 pseudoviruses expressing a luciferase reporter gene were generated in an approach similar to described previously.[ 51 ] Briefly, the packaging construct psPAX2 (AIDS Resource and Reagent Program), luciferase reporter plasmid pLenti‐CMV Puro‐Luc (Addgene), and Spike protein‐expressing pcDNA3.1‐ SARS CoV‐2 SΔCT were co‐transfected into HEK293T cells with calcium phosphate. The supernatants containing the pseudo‐type viruses were collected 48 h post‐transfection; pseudo‐type viruses were purified by filtration with a 0.45 µm filter. To determine the neutralization activity of the antisera from vaccinated animals, HEK293T‐hACE2 cells were seeded in 96‐well tissue culture plates at a density of 1.75 × 104 cells/well overnight. Twofold serial dilutions of heat‐inactivated serum samples were prepared and mixed with pseudo virus (50 µL). The mixture was incubated at 37 °C for 1 h before adding to HEK293T‐hACE2 cells. Forty‐eight hours after infection, cells were lysed in Steady‐Glo Luciferase Assay (Promega) according to the manufacturer's instructions. SARS‐CoV‐2 neutralization titers were defined as the sample dilution at which a 50% reduction in relative light units was observed relative to the average of the virus control wells.
Scanning Electron Microscopy
Samples were fixed in 10% formalin for 24 h and washed extensively in 1× PBS. Samples were dehydrated by soaking them in various solutions, going sequentially from 50% ethanol: water to 70%, 80%, 90% ×2, and finally 100% ×2 ethanol. Each soak lasted for 15–20 min. After the final wash, samples were soaked for 15mn with a solution of hexamethyldisilazane:ethanol (HMDS:EtOH), going sequentially from 1:2 to 2:1 molar ratios and then to 100% HMDS. Finally, samples were transferred to a fresh 100% HMDS solution and let dry overnight. Dehydrated samples were mounted and sputter‐coated with platinum/palladium (Pt/Pd: 80/20) before imaging with an Ultra55 Field Emission Scanning Electron Microscope (FESEM).
Statistical Analysis
All data processing, data management, and statistical analysis for all plots and tables were performed using R (3.6.0). Graphs were plotted with GraphPad Prism (8.3.0). All variables are continuous. Unless otherwise noted, data represent mean ± SD. Sample sizes are noted in figure legends. Normality of all the dependent variables was assessed using graphical methods and the Shapiro–Wilk test for normality, considering 5% as the level of significance, all groups are random samples of the population. Consequently, comparisons between day 0 and every other time point in Figures 3a–c, and 4a and between day 14 and every other time point in Figure 5 were identified using the non‐parametric Friedman test[ 56 ] followed by post hoc analysis using paired Wilcoxon signed‐rank test with false discovery rate adjustment to control family‐wise type I error. Statistically significant differences between groups 4 weeks post‐immunization (day 28) in Figure 4b, at the final time point (day 233) in Figure 6b–d and at different time points (Figure 6e) were identified by implementing non‐parametric Kruskal–Wallis test [ 57 ] followed by post hoc analysis using Conover test with false discovery rate adjustment to control family‐wise type I error.[ 50 , 51 ] Whereas comparisons between day 0 and only day 233 in Figure 6b–d were performed using the Wilcoxon signed‐rank test. Results were deemed as statistically significant when the null hypothesis could be rejected with >95% confidence (P < 0.05). Bars represent the mean and standard deviation in all figures.
Conflict of Interest
Several authors are inventors on patent applications related to this technology (E.D, D.J.M); Novartis, sponsored research (D.J.M); Immulus, equity (D.J.M.), Attivare Therapeutics, equity (E.D., D.J.M., F.L., B.S.). David Walt has a financial interest in Quanterix Corporation, a company that develops an ultra‐sensitive digital immunoassay platform. He is an inventor of the Simoa technology, a founder of the company and also serves on its Board of Directors. Dr. Walt's interests were reviewed and are managed by BWH and Partners Healthcare in accordance with their conflict‐of‐interest policies.
Author Contributions
F.L., M.O.D., and C.Y. contributed equally to this work. F.L. and M.J.C. designed the experiments that were directed by E.J.D., D.H.B., A.G., D. R.W., and D.J.M. Vaccine manufacture and in vitro experiments were performed and analyzed by F.L., B.T.S., J.Y., M.J.C., C.Y., M.O.D., H.I., C.C., T.G., A.H., R.J, and C.M.T. In vivo work was performed by F.L. and C.Y. Statistical analysis was performed by N.D. The manuscript was written by F.L, M.O.D, and C.Y. All authors critically reviewed.
Supporting information
Supporting Information
Acknowledgements
The authors want to thank Dr. Mohan Karkada, Dr. Hua Wang, David Zhang and Miguel Sobral for helpful discussions and valuable input. This work was supported by the Wyss Institute for Biologically Inspired Engineering.
Langellotto F., Dellacherie M. O., Yeager C., Ijaz H., Yu J., Cheng C.‐A., Dimitrakakis N., Seiler B. T., Gebre M. S., Gilboa T., Johnson R., Storm N., Bardales S., Graveline A., White D., Tringides C. M., Cartwright M. J., Doherty E. J., Honko A., Griffiths A., Barouch D. H., Walt D. R., Mooney D. J., A Modular Biomaterial Scaffold‐Based Vaccine Elicits Durable Adaptive Immunity to Subunit SARS‐CoV‐2 Antigens. Adv. Healthcare Mater. 2021, 10, 2101370. 10.1002/adhm.202101370
Data Availability Statement
Research data are not shared.
References
- 1.World Health Organization. WHO Coronavirus (COVID‐19) Dashboard. Available at: https://covid19.who.int/
- 2. Gupta R. K., Harrison E. M., Ho A., Docherty A. B., Knight S. R., Van Smeden M., Abubakar I., Lipman M., Quartagno M., Pius R., Buchan I., Carson G., Drake T. M., Dunning J., Fairfield C. J., Gamble C., Green C. A., Halpin S., Hardwick H. E., Holden K. A., Horby P. W., Jackson C., Mclean K. A., Merson L., Nguyen‐Van‐Tam J. S., Norman L., Olliaro P. L., Pritchard M. G., Russell C. D., Scott‐Brown J., et al. Lancet Respir. Med. 2021, 9, 349.33444539 [Google Scholar]
- 3. Wynants L., Sotgiu G., Lancet Respir. Med. 2021, 9, 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Logue J. K., Franko N. M., Mcculloch D. J., Mcdonald D., Magedson A., Wolf C. R., Chu H. Y., JAMA Netw. Open 2021, 4, e210830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ura T., Okuda K., Shimada M., Vaccines 2014, 2, 624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Matthews J. T., Egg‐Based Production of Influenza Vaccine: 30 Years of Commercial Experience, in The Bridge, The National Academies Press, Washington, DC: 2006. [Google Scholar]
- 7. Fausther‐Bovendo H., Kobinger G. P., Hum. Vaccin. Immunother. 2014, 10, 2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bangaru S., Ozorowski G., Turner H. L., Antanasijevic A., Huang D., Wang X., Torres J. L., Diedrich J. K., Tian J. ‐. H., Portnoff A. D., Patel N., Massare M. J., Yates J. R., Nemazee D., Paulson J. C., Glenn G., Smith G., Ward A. B., Science (80‐.). 2020, 370, 1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.NovaVax. Press Release: Novavax COVID‐19 Vaccine Demonstrates 89.3% Efficacy in UK Phase 3 Trial. 2021.
- 10. Keech C., Albert G., Cho I., Robertson A., Reed P., Neal S., Plested J. S., Zhu M., Cloney‐Clark S., Zhou H., Smith G., Patel N., Frieman M. B., Haupt R. E., Logue J., Mcgrath M., Weston S., Piedra P. A., Desai C., Callahan K., Lewis M., Price‐Abbott P., Formica N., Shinde V., Fries L., Lickliter J. D., Griffin P., Wilkinson B., Glenn G. M., N. Engl. J. Med. 2020, 383, 2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Randolph G. J., Angeli V., Swartz M. A., Nat. Rev. Immunol. 2005, 5, 617. [DOI] [PubMed] [Google Scholar]
- 12. Li H., Willingham S. B., Ting J. P.‐Y., Re F., J. Immunol. 2008, 181, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Flach T. L., Ng G., Hari A., Desrosiers M. D., Zhang P., Ward S. M., Seamone M. E., Vilaysane A., Mucsi A. D., Fong Y., Prenner E., Ling C. C., Tschopp J., Muruve D. A., Amrein M. W., Shi Y., Nat. Med. 2011, 17, 479. [DOI] [PubMed] [Google Scholar]
- 14. Gao Q., Bao L., Mao H., Wang L., Xu K., Yang M., Li Y., Zhu L., Wang N., Lv Z., Gao H., Ge X., Kan B., Hu Y., Liu J., Cai F., Jiang D., Yin Y., Qin C., Li J., Gong X., Lou X., Shi W., Wu D., Zhang H., Zhu L., Deng W., Li Y., Lu J., Li C., Wang X., Yin W., Zhang Y., Qin C., Science (80‐.). 2020, 369, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moon J. J., Huang B., Irvine D. J., Adv. Mater. 2012, 24, 3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Irvine D. J., Swartz M. A., Szeto G. L., Nat. Mater. 2013, 12, 978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim J., Li W. A., Choi Y., Lewin S. A., Verbeke C. S., Dranoff G., Mooney D. J., Nat. Biotechnol. 2015, 64. 10.1038/nbt.3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li A. W., Sobral M. C., Badrinath S., Choi Y., Graveline A., Stafford A. G., Weaver J. C., Dellacherie M. O., Shih T.‐Y.u, Ali O. A., Kim J., Wucherpfennig K. W., Mooney D. J., Nat. Mater. 2018, 528. 10.1038/s41563-018-0028-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dellacherie M. O., Li A., Lu B. Y., Verbeke C. S., Gu L., Stafford A. G., Doherty E. J., Mooney D. J., Adv. Funct. Mater. 2020, 2002448. 10.1002/adfm.202002448. [DOI] [Google Scholar]
- 20. Super M., et al. Modular biomaterials vaccine technology protects against multiple pathogens and septic shock. bioRxiv 2020.02.25.964601, 2020. 10.1101/2020.02.25.964601 bioRxiv. [DOI]
- 21. Norman M., Gilboa T., Ogata A. F., Maley A. M., Cohen L., Busch E. L., Lazarovits R., Mao C. ‐. P., Cai Y., Zhang J., Feldman J. E., Hauser B. M., Caradonna T. M., Chen B., Schmidt A. G., Alter G., Charles R. C., Ryan E. T., Walt D. R., Nat. Biomed. Eng. 2020, 4, 1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yu J., Tostanoski L. H., Peter L., Mercado N. B., Mcmahan K., Mahrokhian S. H., Nkolola J. P., Liu J., Li Z., Chandrashekar A., Martinez D. R., Loos C., Atyeo C., Fischinger S., Burke J. S., Slein M. D., Chen Y., Zuiani A., Lelis F. J. N., Travers M., Habibi S., Pessaint L., Van Ry A., Blade K., Brown R., Cook A., Finneyfrock B., Dodson A., Teow E., Velasco J., et al. Science (80‐.). 2020, 369, 806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang Z. ‐. Y., Kong W. ‐. P., Huang Y., Roberts A., Murphy B. R., Subbarao K., Nabel G. J., Nature 2004, 428, 561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen Z., John Wherry E., Nat. Rev. Immunol. 2020, 20, 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schmidt M. E., Varga S. M., Front. Immunol. 2018, 9, 678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Low J. S., Vaqueirinho D., Mele F., Foglierini M., Jerak J., Perotti M., Jarrossay D., Jovic S., Perez L., Cacciatore R., Terrot T., Pellanda A. F., Biggiogero M., Garzoni C., Ferrari P., Ceschi A., Lanzavecchia A., Sallusto F., Cassotta A., Science (80‐.) 2021, 372, 1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tai W., He L., Zhang X., Pu J., Voronin D., Jiang S., Zhou Y., Du L., Cell. Mol. Immunol. 2020, 613. 10.1038/s41423-020-0400-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang Y., Wang L., Cao H., Liu C., J. Med. Virol. 2021, 93, 892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mandolesi M., Sheward D. J., Hanke L., Ma J., Pushparaj P., Perez Vidakovics L., Kim C., Àdori M., Lenart K., Loré K., Castro Dopico X., Coquet J. M., Mcinerney G. M., Karlsson Hedestam G. B., Murrell B., Cell Rep. Med. 2021, 2, 100252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tan H. ‐. X., Juno J. A., Lee W. S., Barber‐Axthelm I., Kelly H. G., Wragg K. M., Esterbauer R., Amarasena T., Mordant F. L., Subbarao K., Kent S. J., Wheatley A. K., Nat. Commun. 2021, 12, 1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Croissant J. G., Fatieiev Y., Khashab N. M., Adv. Mater. 2017, 1604634. 10.1002/adma.201604634. [DOI] [PubMed] [Google Scholar]
- 32. Hudson S. P., Padera R. F., Langer R., Kohane D. S., Biomaterials 2008, 29, 4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Arellano M., Lonial S., Biologics 2008, 2, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. N. Engl. J. Med. 2011, 365, 1863.22007715 [Google Scholar]
- 35. Lal H., Cunningham A. L., Godeaux O., Chlibek R., Diez‐Domingo J., Hwang S. ‐. J., Levin M. J., Mcelhaney J. E., Poder A., Puig‐Barberà J., Vesikari T., Watanabe D., Weckx L., Zahaf T., Heineman T. C., N. Engl. J. Med. 2015, 372, 2087.25916341 [Google Scholar]
- 36. Butterfield L. H., Zhao F., Lee S., Tarhini A. A., Margolin K. A., White R. L., Atkins M. B., Cohen G. I., Whiteside T. L., Kirkwood J. M., Lawson D. H., Clin. Cancer Res. 2017, 23, 5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shah N. J., Najibi A. J., Shih T.‐Y.u, Mao A. S., Sharda A., Scadden D. T., Mooney D. J., Nat. Biomed. Eng. 2020, 40. 10.1038/s41551-019-0503-3. [DOI] [PubMed] [Google Scholar]
- 38. Heesters B. A., Van Der Poel C. E., Das A., Carroll M. C., Trends Immunol. 2016, 37, 844. [DOI] [PubMed] [Google Scholar]
- 39. Kurosaki T., Kometani K., Ise W., Nat. Rev. Immunol. 2015, 149. 10.1038/nri3802. [DOI] [PubMed] [Google Scholar]
- 40. Fulginiti V. A., JAMA 1967, 202, 1075. [DOI] [PubMed] [Google Scholar]
- 41. Kim H. W., Canchola J. G., Brandt C. D., Pyles G., Chanock R. M., Jensen K., Parrott R. H., Am. J. Epidemiol. 1969, 89, 422. [DOI] [PubMed] [Google Scholar]
- 42. Bolles M., Deming D., Long K., Agnihothram S., Whitmore A., Ferris M., Funkhouser W., Gralinski L., Totura A., Heise M., Baric R. S., J. Virol. 2011, 85, 12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pulendran B., Arunachalam P. S., O'hagan D. T., Nat. Rev. Drug Discovery 2021, 454. 10.1038/s41573-021-00163-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pone E., J., Front. Biosci. 2011, 2594. 10.2741/4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Corbett K. S., Edwards D. K., Leist S. R., Abiona O. M., Boyoglu‐Barnum S., Gillespie R. A., Himansu S., Schäfer A., Ziwawo C. T., Dipiazza A. T., Dinnon K. H., Elbashir S. M., Shaw C. A., Woods A., Fritch E. J., Martinez D. R., Bock K. W., Minai M., Nagata B. M., Hutchinson G. B., Wu K., Henry C., Bahl K., Garcia‐Dominguez D., Ma L., Renzi I., Kong W. ‐. P., Schmidt S. D., Wang L., Zhang Y.i, et al. Nature 2020, 586, 567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Volpatti L. R., Wallace R. P., Cao S., Raczy M. M., Wang R., Gray L. T., Alpar A. T., Briquez P. S., Mitrousis N., Marchell T. M., Sasso M. S., Nguyen M., Mansurov A., Budina E., Solanki A., Watkins E. A., Schnorenberg M. R., Tremain A. C., Reda J. W., Nicolaescu V., Furlong K., Dvorkin S., Yu S. S., Manicassamy B., LaBelle J. L., Tirrell M. V., Randall G., Kwissa M., Swartz M. A., Hubbell J. A., bioRxiv 2021, 2021.04.08.438884, 10.1101/2021.04.08.438884 bioRxiv. [DOI] [Google Scholar]
- 47. Xu C., Lei C., Yu C., Front. Chem. 2019, 7, 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen Y.‐C., Smith T., Hicks R. H., Doekhie A., Koumanov F., Wells S. A., Edler K. J., Van Den Elsen J., Holman G. D., Marchbank K. J., Sartbaeva A., Sci. Rep. 2017, 7, 46568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Doekhie A., Dattani R., Chen Y.‐C., Yang Y., Smith A., Silve A. P., Koumanov F., Wells S. A., Edler K. J., Marchbank K. J., Elsen J. M. H. V. D., Sartbaeva A., Sci. Rep. 2020. 10.1038/s41598-020-65876-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wahid A. A., Doekhie A., Sartbaeva A., Van Den Elsen J. M. H., Sci. Rep. 2019, 9, 11409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mercado N. B., Zahn R., Wegmann F., Loos C., Chandrashekar A., Yu J., Liu J., Peter L., Mcmahan K., Tostanoski L. H., He X., Martinez D. R., Rutten L., Bos R., Van Manen D., Vellinga J., Custers J., Langedijk J. P., Kwaks T., Bakkers M. J. G., Zuijdgeest D., Rosendahl Huber S. K., Atyeo C., Fischinger S., Burke J. S., Feldman J., Hauser B. M., Caradonna T. M., Bondzie E. A., Dagotto G., et al. Nature 2020, 586, 583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Van Der Lubbe J. E. M., Rosendahl Huber S. K., Vijayan A., Dekking L., Van Huizen E., Vreugdenhil J., Choi Y., Baert M. R. M., Feddes‐De Boer K., Izquierdo Gil A., Van Heerden M., Dalebout T. J., Myeni S. K., Kikkert M., Snijder E. J., De Waal L., Stittelaar K. J., Tolboom J. T. B. M., Serroyen J., Muchene L., Van Der Fits L., Rutten L., Langedijk J. P. M., Barouch D. H., Schuitemaker H., Zahn R. C., Wegmann F., npj Vaccines 2021, 6, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Reynolds C. J., Pade C., Gibbons J. M., Butler D. K., Otter A. D., Menacho K., Fontana M., Smit A., Sackville‐West J. E., Cutino‐Moguel T., Maini M. K., Chain B., Noursadeghi M., Brooks T., Semper A., Manisty C., Treibel T. A., Moon J. C., Valdes A. M., Mcknight Á., Altmann D. M., Boyton R., Science (80‐.). 2021, 1418. 10.1126/science.abh1282. [DOI] [Google Scholar]
- 54. Shen X., Tang H., Pajon R., Smith G., Glenn G. M., Shi W., Korber B., Montefiori D. C., N. Engl. J. Med. 2021, 2352. 10.1056/NEJMc2103740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wu K., Werner A. P., Koch M., Choi A., Narayanan E., Stewart‐Jones G. B. E., Colpitts T., Bennett H., Boyoglu‐Barnum S., Shi W., Moliva J. I., Sullivan N. J., Graham B. S., Carfi A., Corbett K. S., Seder R. A., Edwards D. K., N. Engl. J. Med. 2021, 384, 1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Riffenburgh R. H., Statistics in Medicine (Second Edition), Academic Press, Cambridge, MA: 2006, 10.1016/B978-0-12-088770-5.50072-1. [DOI] [Google Scholar]
- 57. Lantz B., Br. J. Math. Stat. Psychol. 2013, 66, 224. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
Research data are not shared.